Orphanet Nomenclature & Orphanet Nomenclature & Knowledge management Ana Rath, Marc Hanauer , Annie Olry , Montserrat Alfaro, Martin Ana Rath, Marc Hanauer , Annie Olry , Montserrat Alfaro, Martin Arles, Charlotte Gueydan, Sylvie Maiella, Charlotte Rodwell [email protected] 5th International Summer School on Rare Disease and Orphan Drug Registries, September 18-22, 2017 Roma

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Orphanet Nomenclature & Orphanet Nomenclature &
Knowledge management
Ana Rath, Marc Hanauer, Annie Olry, Montserrat Alfaro, Martin Ana Rath, Marc Hanauer, Annie Olry, Montserrat Alfaro, Martin
Arles, Charlotte Gueydan, Sylvie Maiella, Charlotte Rodwell
5th International Summer School on Rare Disease and Orphan Drug Registries, September 18-22, 2017
Roma

Orphanet, since 1997
• Mission:
– collect
– integrate
– produce
Added-value data and information on RD
- manually curated,
- expert-reviewed, – produce
– disseminate
• Reference nomenclature, classification and ontology of RD
- expert-reviewed,
- re-usable
www.orpha.net
Literature survey
PubMed queries
…
AnnotationIndexationWriting
ExpertsPatients groupsProfessionals
Completeness
Coherence
Sources survey

Catalog of RD-related services
www.orpha.net

Providing information
Providing data
International, multilingual websiteIntended to a multi-stakeholders audience
2016
www.orpha.netMassive agregated data in a computer-friendly format

Orphanet website users around the world VS Orphadata users
Orphanet
Orphadata
www.orpha.net

Orphanet in the RD landscape
ClinVarMedGen
www.orpha.net

Orphanet International database for RD
www.orpha.net

Interoperability
Research
Registries/Cohorts
Care
Health Information
System (EHRs)
Orphanet central
nomenclature
HGNC
OMIM
ICD10/11
GenesGenes
Terminologies
www.orpha.net
HGNC
OMIM
UniProt
Reactome
Ensembl
Genatlas
IUPHAR
ICD10/11
UMLS
MedDRA
DisabilitiesDisabilitiesPhenotypesPhenotypes
SNOMED

Orphanet RD nomenclature
ORPHA number Preferred label Synonyms
ORPHA:93545 Renal or urinary tract malformation CAKUT
Congenital anomalies of kidney and urinary tract
• The only clinical terminology specific for rare diseases
• Unique, stable ORPHA number
Congenital anomalies of kidney and urinary tract
ORPHA:216 Neuronal ceroid lipofuscinosis NCL
ORPHA:586 Cystic fibrosis CF
Mucoviscidosis
ORPHA:355 Gaucher disease Acid beta-glucosidase deficiency
Glucocerebrosidase deficiency
ORPHA:77259 Gaucher disease type 1 Non-cerebral juvenile Gaucher disease
www.orpha.net
• Unique, stable ORPHA number
• Definitions
• 8 languages (En, Fr, Es, It, Nl, De, Pt, Pl)
• Peer-reviewed publications only (2 cases<RD<1/2000)

RD classification
Organized by medical specialties• How?
Improve information
Epidemiology and statistics studies• Why?
www.orpha.net
Majority of systemic disorders• Particularity

Multi-dimensional
Multi- classification
Multi-hierarchicalMulti-dimensional Multi-hierarchical
www.orpha.net

Logical structure
Group
Category: clinically heterogeneous
Clinical group: clinically homogeneousClinical group: clinically homogeneous
Disorder:
• Disease, clinical syndrome, malformation
syndrome, morphological anomaly, biological
anomaly, particular clinical situation
Subtype:
• Clinical, etiological, histopathological
www.orpha.net
• Every entity is meaningful
• Entities are disjointed
• Parts are added to form the wholes
• Transitivity applies at every level

Rare bone disease Rare genetic disease
Primary bone dysplasia
Rare genetic bone disease
Spondylodysplastic dysplasia
Achondrogenesis
Achondrogenesis type IA
Sulfation-related bone disease
Rare bone disease related to a commongene or pathway defect
Achondrogenesis type IB
Achondrogenesis type II
Hypochondrogenesis

Update process
DBs(OMIM,…)
Literaturesurvey
Usersissues
Decision
Disease committee
Verification of prevalence, database and classification
consistency
Orphanet DB verification, literature search, expert advice Qualify the demand
Creation
New entry + infoReject
Modification
Nomenclature Obsolescence Deprecation
Expert advice
Disease committee
Impact on the inventory New Orpha numbers, Status modification
Impact on the classificationNew hierarchy including new orpha numbers
consitency inter- and intra-classification

Interoperability
Research
Registries/Cohorts
Care
Health Information
System (EHRs)
Orphanet central
nomenclature
HGNC
OMIM
ICD10/11
GenesGenes
Terminologies
www.orpha.net
HGNC
OMIM
UniProt
Reactome
Ensembl
Genatlas
IUPHAR
ICD10/11
UMLS
MedDRA
DisabilitiesDisabilitiesPhenotypesPhenotypes
SNOMED

Mappings with other terminologies
Terminology mapped RD
OMIM Manually 4,390
ICD-10 Manually All
Snomed-CT Manually 3,800
GARD Semi-automatically 2,998
Qualifier
E exact mapping (the terms and the concepts are equivalent)
NTBT narrower term maps to a broader term
BTNT broader term maps to a narrower term
W incorrect mapping (two different concepts)
ND not yet decided/unable to decide
GARD Semi-automatically 2,998
UMLS Semi-automatically 2,885
MeSH Semi-automatically 1,763
MedDRA Semi-automatically 1,224
www.orpha.net
ICD10 codes only :
Specific code The term has its own code in the ICD10
Inclusion term The term is included under a ICD10 category and has not its own code
Index term The term is oncluded in ICD10 index and refers to one more general code
Attributed
codeThe term does not exist in ICD10 and a code was attributed by Orphanet

Orpha number Preferred label Synonyms Typology Status ICD-10 Definition/relationship
ORPHA:93545 Renal or urinary tract
malformation
CAKUT Category _ _ _
Congenital anomalies of kidney and
urinary tract
ORPHA:216 Neuronal ceroid lipofuscinosis NCL Clinical group _ E75.4 Yes
Coding perspective
ORPHA:216 Neuronal ceroid lipofuscinosis NCL Clinical group _ E75.4 Yes
ORPHA:586 Cystic fibrosis CF Disease _ E84.0 E84.1
E84.8 E84.9
Yes
Mucoviscidosis
ORPHA:355 Gaucher disease Acid beta-glucosidase deficiency Disease _ E75.2 Yes
Glucocerebrosidase deficiency
ORPHA:1245 BIDS syndrome Amish brittle hair syndrome Disease Deprecated _ moved to Trichothiodystrophy
Trichothiodystrophy type D
ORPHA:77259 Gaucher disease type 1 Non-cerebral juvenile Gaucher disease Subtype _ E75.2 yes
ORPHA:101042 Taussig-Bing syndrome Subtype Obsolete _ Refered to Double outlet right
ventricle with subpulmonary
ventricular septal defect
www.orpha.net
Orpha numbers used to be assigned to a patient within an information system
=
Orpha Code

HPO annotations of Orphanet Rare Disorders
Around 6,000 rare disorders HPO phenotypes
2787 RD 5008 HPO
57,635 57,635
annotations
Frequencies Diagnostic criteria
Obligate (100%) Pathognomonic sign
Very frequent (99-80%) Diagnostic criterion
Frequent (79-30%)
Occasional (29–5%)
Very rare (1-4%)
Absent 0%
2787 RD 5008 HPOannotationsannotations
www.orpha.net
Absent 0%
Annotated by
Orphanet
Disseminated
by Orphanet
and HPO
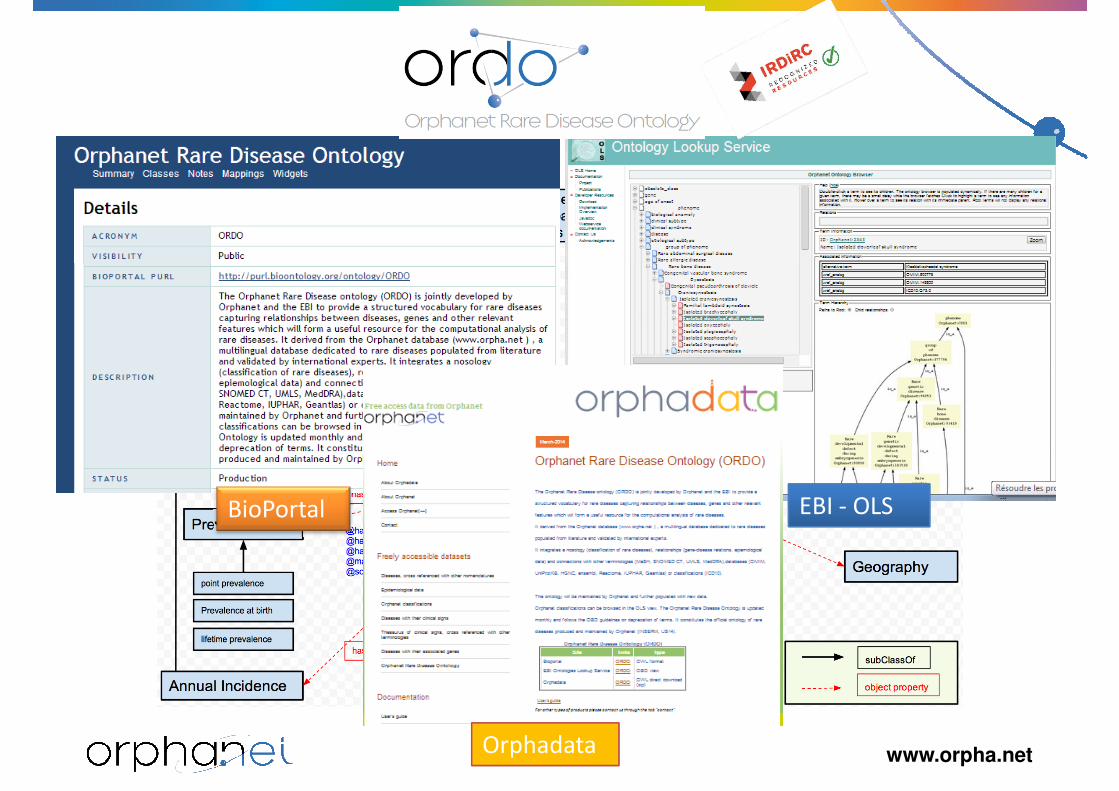
BioPortal EBI - OLS
www.orpha.netOrphadata

To know more about…
Orphanet process
www.orpha.net

• Integrating pieces of a puzzling knowledge
Knowledge
– Around the Orphanet nomenclature and classification
• Providing integrated, re-usable data
– Orphanet Rare Disease Ontology (ORDO)
• Promoting interoperability
disease gene
phenotypedisability
www.orpha.net
– Bridging health and research
• Networking and partnering

Orphanet Curation Platformhttp://curation.orphanet.org
www.orpha.net

What is the Orphanet Curation Platform?
• It is a community-driven curation platform for rare disorders based on
Orphanet’s scientific content.
• It is a pilot project being co-developped by Orphanet and the Garvan
Institute (Australia), with the support of the Orphanet Australia’s country
coordinator.
• Goals:
– Allow users to visually explore the Orphanet scientific data
www.orpha.net
– Enable contributing experts and the Orphanet team to connect in a more dynamic and
efficient manner
– Facilitate communication between the contributing experts and the Orphanet team in
order to maintain the Orphanet database with the most up-to-date information

Orphanet Curation Platform « Key Players »
www.orpha.net

Create an Account
www.orpha.net

Browsing the Classification
www.orpha.net

www.orpha.net

www.orpha.net

Discussing a Suggestion
www.orpha.net
1
2

Life Cycle of a Suggestion
Suggestion posted by a
contributor Suggestion is incorporated
into Orphanet database
Suggestion appears on
Orphanet website
openBeing reviewed by the Orphanet team
Not yet accessedby Orphanet team
Curated by the Orphanet team
Being reassessed by
new
All persons involved in
closed
into Orphanet database Orphanet website
www.orpha.net
openReopen
the Orphanet teamBeing reassessed by the Orphanet team
Discussion of suggestion
between Orphanet team
& expert
All persons involved in
discussion agree on content of
suggestion

Harmonising phenomics information for a better interoperability in the rare diseases field
www.orpha.net

THANK YOU FOR YOUR ATTENTION
Orphanet Nomenclature & Knowledge management.
www.orpha.net
THANK YOU FOR YOUR ATTENTION
Related Documents











