Organic & Biomolecular Chemistry PAPER Cite this: Org. Biomol. Chem., 2013, 11, 1407 Received 14th August 2012, Accepted 26th December 2012 DOI: 10.1039/c2ob26602d www.rsc.org/obc Comprehensive studies on the tautomerization of glycine: a theoretical study Chang Kon Kim, Byung-Ho Park, Hai Whang Lee and Chan Kyung Kim* The tautomerization process of glycine between the neutral (NE) and zwitterionic (ZW) forms in aqueous solution was explored theoretically using the conductor-like polarizable continuum model (CPCM) by adopting the PAULING cavity model at the B3LYP, MP2 and CCSD levels with the 6-311+G(d,p) basis set. The tautomerization of glycine is unable to be predicted satisfactorily within the equilibrated framework of the CPCM method. Instead, in this study, three plausible non-equilibrated solvation situations were assumed: (S-1) one water molecule attached to the transferring proton in the ZW moves together with the transferring proton; (S-2) one water molecule attached to the transferring proton in the ZWremains motionless at a fixed position near the NH 2 fragment at the TS structure; and (S-3) proton transfer occurs without changing the position of the surrounding water molecules from their initial state, the ZW form, in the eight water clusters. Although the calculation of (S-3) failed, the Gibbs free energies of activation for tautomerization from the ZW to NE, ΔG ≠ (ZW → NE), was well consistent with the experimental findings in the hypothetical non-equilibrated solvation states of (S-1) and (S-2). This suggests that non- equilibrium solvation is essential to explain the observed experimental data. Introduction In metabolism, amino acids play important roles as the build- ing blocks of proteins, and many experimental 1–4 and theoreti- cal studies 5–19 have been reported. Among the amino acids, glycine has attracted considerable attention in theoretical studies 5–18 owing to its small size and the availability of experi- mental data. 1–4 Moreover, it has been reported that the neutral form (NE) of glycine exists in the gas phase but the zwitterionic form (ZW) is unstable in vacuo. 5–18 On the other hand, the ZW of glycine predominates in crystalline or in aqueous solutions. 2,3 Therefore, solvent (or environmental) effects should be included in theoretical studies 20–25 to examine glycine chemistry. 5–18 In previous work, 14 the confor- mations of glycine and inter-conversion processes between them were examined theoretically in the gas phase and in aqueous solution, and it was shown that the conductor-like polarizable continuum model (CPCM) 26,27 adopting the cavity model with explicit hydrogen(s), such as the BONDI 28 or PAULING 29 cavity model, could be applied to aqueous solu- tion. On the other hand, the Gibbs free energies of activation for tautomerization going from the ZW to NE forms, ΔG ≠ (ZW → NE), were underestimated compared to the experimental value, 1,2 even though the ΔG ≠ (ZW → NE) value was fairly consistent with the experimental one relative to any other theoretical studies reported previously, 8,9,12,16 i.e. the experi- mental ΔG ≠ (ZW → NE) value was reported to be 14.6 kcal mol −1 , 1,2 but the ΔG ≠ (ZW → NE) value obtained at the CPCM-CCSD/6-311+G(d,p) level was 9.2 kcal mol −1 . 14 Tortonda et al. reported that the activation energy barrier for tautomeri- zation was only 3.5 kcal mol −1 at the MP2(Full)/6-31+G(d,p) level in aqueous solution using the continuum model. 6,18 On the other hand, this theoretical barrier was too low to compare with the experimental value. 1,2 The activation barrier for the transformation from the NE to ZW for the proton transfer with a single water molecule was reported to be 15.56 kcal mol −1 at the MP2(Full)/6-31+G(d,p) level in the gas phase, which is much higher than the experimentally determined value, 7.3 kcal mol −1 . 6 This experimental value was obtained by con- sidering the ΔG ≠ (ZW → NE) and ΔG o (ZW → NE) values of 14.6 and 7.3 kcal mol −1 , respectively. Similarly, the ΔG ≠ (ZW → NE) value reported by Tuñón et al. was too low (only 5.42 kcal mol −1 ) at the PCM-B3LYP/6-31+G(d,p) level. 17 Therefore, they suggested that the experimental barrier of 7.3 kcal mol −1 corre- sponds to the reorientation of the H-atom in the –COOH group of the neutral conformers. This suggestion appears question- able because the activation barriers for the reorientation of the –COOH group between the NE conformers could be reversely too high compared to the experimental finding of 7.3 kcal mol −1 . In previous work, the highest activation barrier among the inter-conversions between the NE conformers was 13.8 kcal mol −1 . 14 On the other hand, the activation free Department of Chemistry, Inha University, Incheon 402-751, Korea. E-mail: [email protected]; Fax: +82-32-867-5604; Tel: +82-32-860-7684 This journal is © The Royal Society of Chemistry 2013 Org. Biomol. Chem., 2013, 11, 1407–1413 | 1407 Downloaded by University of Oxford on 06 February 2013 Published on 03 January 2013 on http://pubs.rsc.org | doi:10.1039/C2OB26602D View Article Online View Journal | View Issue

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Organic &Biomolecular Chemistry
PAPER
Cite this: Org. Biomol. Chem., 2013, 11,1407
Received 14th August 2012,Accepted 26th December 2012
DOI: 10.1039/c2ob26602d
www.rsc.org/obc
Comprehensive studies on the tautomerization ofglycine: a theoretical study
Chang Kon Kim, Byung-Ho Park, Hai Whang Lee and Chan Kyung Kim*
The tautomerization process of glycine between the neutral (NE) and zwitterionic (ZW) forms in aqueous
solution was explored theoretically using the conductor-like polarizable continuum model (CPCM) by
adopting the PAULING cavity model at the B3LYP, MP2 and CCSD levels with the 6-311+G(d,p) basis set.
The tautomerization of glycine is unable to be predicted satisfactorily within the equilibrated framework
of the CPCM method. Instead, in this study, three plausible non-equilibrated solvation situations were
assumed: (S-1) one water molecule attached to the transferring proton in the ZW moves together with
the transferring proton; (S-2) one water molecule attached to the transferring proton in the ZW remains
motionless at a fixed position near the NH2 fragment at the TS structure; and (S-3) proton transfer occurs
without changing the position of the surrounding water molecules from their initial state, the ZW form,
in the eight water clusters. Although the calculation of (S-3) failed, the Gibbs free energies of activation
for tautomerization from the ZW to NE, ΔG≠(ZW → NE), was well consistent with the experimental
findings in the hypothetical non-equilibrated solvation states of (S-1) and (S-2). This suggests that non-
equilibrium solvation is essential to explain the observed experimental data.
Introduction
In metabolism, amino acids play important roles as the build-ing blocks of proteins, and many experimental1–4 and theoreti-cal studies5–19 have been reported. Among the amino acids,glycine has attracted considerable attention in theoreticalstudies5–18 owing to its small size and the availability of experi-mental data.1–4 Moreover, it has been reported that theneutral form (NE) of glycine exists in the gas phase but thezwitterionic form (ZW) is unstable in vacuo.5–18 On the otherhand, the ZW of glycine predominates in crystalline or inaqueous solutions.2,3 Therefore, solvent (or environmental)effects should be included in theoretical studies20–25 toexamine glycine chemistry.5–18 In previous work,14 the confor-mations of glycine and inter-conversion processes betweenthem were examined theoretically in the gas phase and inaqueous solution, and it was shown that the conductor-likepolarizable continuum model (CPCM)26,27 adopting the cavitymodel with explicit hydrogen(s), such as the BONDI28 orPAULING29 cavity model, could be applied to aqueous solu-tion. On the other hand, the Gibbs free energies of activationfor tautomerization going from the ZW to NE forms, ΔG≠(ZW→ NE), were underestimated compared to the experimentalvalue,1,2 even though the ΔG≠(ZW → NE) value was fairly
consistent with the experimental one relative to any othertheoretical studies reported previously,8,9,12,16 i.e. the experi-mental ΔG≠(ZW → NE) value was reported to be 14.6 kcalmol−1,1,2 but the ΔG≠(ZW → NE) value obtained at theCPCM-CCSD/6-311+G(d,p) level was 9.2 kcal mol−1.14 Tortondaet al. reported that the activation energy barrier for tautomeri-zation was only 3.5 kcal mol−1 at the MP2(Full)/6-31+G(d,p)level in aqueous solution using the continuum model.6,18 Onthe other hand, this theoretical barrier was too low to comparewith the experimental value.1,2 The activation barrier for thetransformation from the NE to ZW for the proton transfer witha single water molecule was reported to be 15.56 kcal mol−1 atthe MP2(Full)/6-31+G(d,p) level in the gas phase, which ismuch higher than the experimentally determined value,7.3 kcal mol−1.6 This experimental value was obtained by con-sidering the ΔG≠(ZW → NE) and ΔGo(ZW → NE) values of 14.6and 7.3 kcal mol−1, respectively. Similarly, the ΔG≠(ZW → NE)value reported by Tuñón et al. was too low (only 5.42 kcalmol−1) at the PCM-B3LYP/6-31+G(d,p) level.17 Therefore, theysuggested that the experimental barrier of 7.3 kcal mol−1 corre-sponds to the reorientation of the H-atom in the –COOH groupof the neutral conformers. This suggestion appears question-able because the activation barriers for the reorientation of the–COOH group between the NE conformers could be reverselytoo high compared to the experimental finding of 7.3 kcalmol−1. In previous work, the highest activation barrier amongthe inter-conversions between the NE conformers was13.8 kcal mol−1.14 On the other hand, the activation free
Department of Chemistry, Inha University, Incheon 402-751, Korea.
E-mail: [email protected]; Fax: +82-32-867-5604; Tel: +82-32-860-7684
This journal is © The Royal Society of Chemistry 2013 Org. Biomol. Chem., 2013, 11, 1407–1413 | 1407
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2D
View Article OnlineView Journal | View Issue

energies for the ZW → NE and NE → ZW conversions werereported to be 16.9 and 8.5 kcal mol−1, respectively, usingempirical valence bond (EVB) molecular dynamics simu-lations, which is in good agreement with the experimentaldata.16 This appears to be fortuitous because the parametersof the EVB method were obtained from the activation barriersthat were overestimated considerably at the Hartree–Fock levelof theory. Therefore, comparisons of the activation barriersbetween the experimental and theoretical studies are unclear.
In this study, the tautomerization of glycine was examinedin more detail to understand the precise mechanism becauseKim et al. were only concerned with a direct intra-molecularproton transfer between ZW and NE.14 On the other hand, thetautomerization process could proceed through many otherpossible processes, i.e. a proton exchange process between thesolvent (water) molecule and the solute (glycine) producing ananionic intermediate (AN) of glycine and hydronium ions, andinter-molecular proton transfer by a participating water mole-cule, as shown in Scheme 1.
Computational methods
All the structures studied in aqueous solution were fully opti-mized without geometrical constraints using the CPCMmethod26,27 adopting the PAULING cavity model29 at theB3LYP,30,31 MP232 and CCSD33,34 levels with the 6-311+G(d,p)basis set. The optimized structures were then characterized byfrequency calculations at the B3LYP and MP2 levels. The ener-getics were then refined at the CCSD(T)/6-311+G(d,p) levelusing the geometries optimized at the CCSD/6-311+G(d,p)
level. In the CPCM method, non-electrostatic terms are impor-tant because the computed energies depend on the cavity size,which is one of the major components of the non-electrostaticterms.26,27 Therefore, in this study, the calculated Gibbs freeenergies in aqueous solution were obtained using eqn (1),where Gs is the Gibbs free energy of solvation including thenon-electrostatic terms, such as cavitation and dispersion. Ineqn (1), Eel is the electronic energy in the gas phase on the geo-metry optimized at the CPCM calculation, and EZPVE, ETh andS are the zero-point vibration energy, thermal energy andentropy terms, respectively, which were obtained from CPCMcalculations. The CCSD(T) calculations in aqueous solutionwere performed at the gas-phase CCSD(T) level on the geo-metries at the CPCM-CCSD/6-311+G(d,p) or CPCM-B3LYP/6-311+G(d,p) levels, and the Gs values at the CPCM-CCSD levelwere used because the CCSD(T) calculations were not appli-cable to the CPCM method. All the calculations were per-formed with the Int(grid = ultrafine) option using theGaussian 03 program.35
Gðat 298 KÞ ¼ Eel þ EZPVE þ ETh þ PV � TSþ Gs
¼ Eel þ Gcorr þ Gs ð1Þ
Results and discussion
As reported previously,14 the ΔG≠(ZW → NE) values deter-mined using the direct proton transfer process, (A) inScheme 1, were 9.2 and 8.8 kcal mol−1 at the CCSD and CCSD(T) levels with the 6-311+G(d,p) basis set, respectively, inaqueous solution. On the other hand, the calculated valuesappeared to be somewhat lower than the experimental value of14.6 kcal mol−1. Therefore, the possibility that tautomerizationproceeds via two different paths, (B) and (C) in Scheme 1,cannot be excluded. Especially, the path (B) could be a plaus-ible process, because the autoionization processes ofammonia21 and hydrogen halides22 in water are similar topath (B) and these processes were studied extensively. Table 1summarizes the ΔG≠(ZW → NE) values for path (B) calculatedat various levels. Tautomerization via a proton exchangeprocess with AN and hydronium ion as intermediates, path(B), could be excluded because the Gibbs free energy changesfrom the ZW + H2O cluster to AN + H3O
+, ΔGAN–ZW, were muchmore unfavorable than the experimental ΔG≠(ZW → NE)values. For example, the ΔGAN–ZW values were 35.5 and35.7 kcal mol−1 at the CCSD and CCSD(T) levels, respectively.Moreover, the Gibbs free energies of the intermediates, ANand hydronium ion, were also 26.3 and 26.9 kcal mol−1 higher,respectively, than that for the direct intra-molecular protontransfer, path (A), at the CCSD and CCSD(T) levels. Thissuggests that the tautomerization of ZW ⇄ NE in glycinechemistry cannot occur through acid–base equilibrium.
On the other hand, Table 2 shows that the ΔG≠(ZW → NE)values for the inter-molecular proton transfer with a participat-ing water molecule, path (C), 14.8 and 14.0 kcal mol−1 at the
Scheme 1
Paper Organic & Biomolecular Chemistry
1408 | Org. Biomol. Chem., 2013, 11, 1407–1413 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online
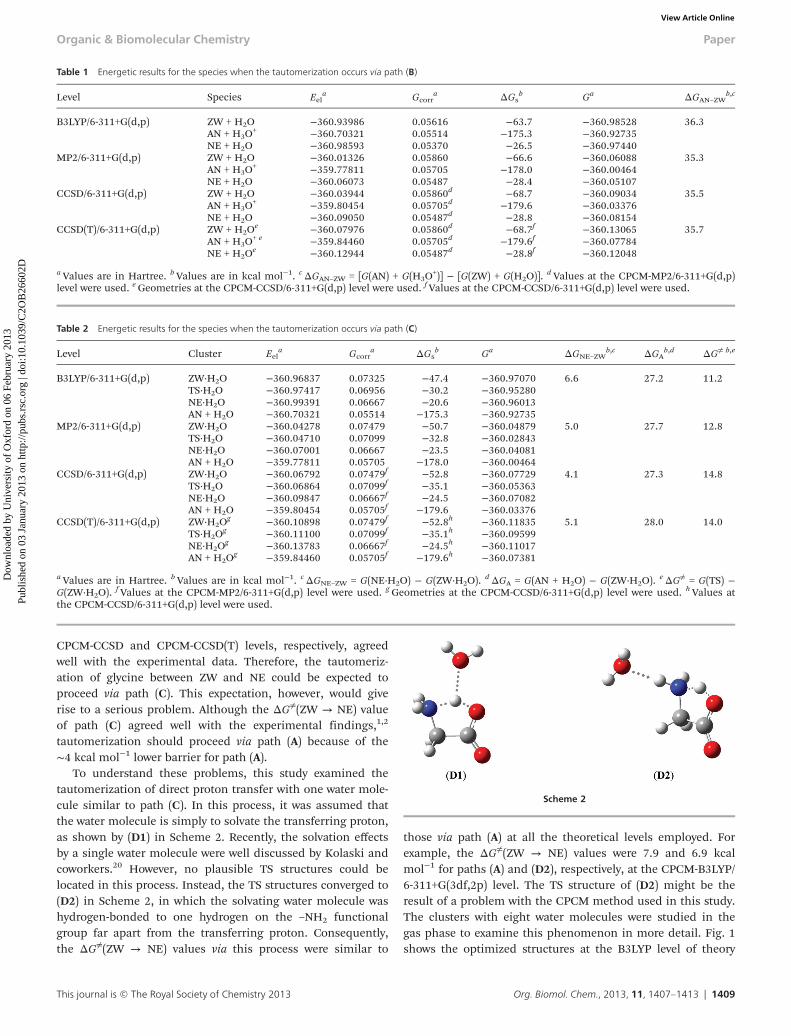
CPCM-CCSD and CPCM-CCSD(T) levels, respectively, agreedwell with the experimental data. Therefore, the tautomeriz-ation of glycine between ZW and NE could be expected toproceed via path (C). This expectation, however, would giverise to a serious problem. Although the ΔG≠(ZW → NE) valueof path (C) agreed well with the experimental findings,1,2
tautomerization should proceed via path (A) because of the∼4 kcal mol−1 lower barrier for path (A).
To understand these problems, this study examined thetautomerization of direct proton transfer with one water mole-cule similar to path (C). In this process, it was assumed thatthe water molecule is simply to solvate the transferring proton,as shown by (D1) in Scheme 2. Recently, the solvation effectsby a single water molecule were well discussed by Kolaski andcoworkers.20 However, no plausible TS structures could belocated in this process. Instead, the TS structures converged to(D2) in Scheme 2, in which the solvating water molecule washydrogen-bonded to one hydrogen on the –NH2 functionalgroup far apart from the transferring proton. Consequently,the ΔG≠(ZW → NE) values via this process were similar to
those via path (A) at all the theoretical levels employed. Forexample, the ΔG≠(ZW → NE) values were 7.9 and 6.9 kcalmol−1 for paths (A) and (D2), respectively, at the CPCM-B3LYP/6-311+G(3df,2p) level. The TS structure of (D2) might be theresult of a problem with the CPCM method used in this study.The clusters with eight water molecules were studied in thegas phase to examine this phenomenon in more detail. Fig. 1shows the optimized structures at the B3LYP level of theory
Table 1 Energetic results for the species when the tautomerization occurs via path (B)
Level Species Eela Gcorr
a ΔGsb Ga ΔGAN–ZW
b,c
B3LYP/6-311+G(d,p) ZW + H2O −360.93986 0.05616 −63.7 −360.98528 36.3AN + H3O
+ −360.70321 0.05514 −175.3 −360.92735NE + H2O −360.98593 0.05370 −26.5 −360.97440
MP2/6-311+G(d,p) ZW + H2O −360.01326 0.05860 −66.6 −360.06088 35.3AN + H3O
+ −359.77811 0.05705 −178.0 −360.00464NE + H2O −360.06073 0.05487 −28.4 −360.05107
CCSD/6-311+G(d,p) ZW + H2O −360.03944 0.05860d −68.7 −360.09034 35.5AN + H3O
+ −359.80454 0.05705d −179.6 −360.03376NE + H2O −360.09050 0.05487d −28.8 −360.08154
CCSD(T)/6-311+G(d,p) ZW + H2Oe −360.07976 0.05860d −68.7f −360.13065 35.7
AN + H3O+ e −359.84460 0.05705d −179.6f −360.07784
NE + H2Oe −360.12944 0.05487d −28.8f −360.12048
a Values are in Hartree. b Values are in kcal mol−1. cΔGAN–ZW = [G(AN) + G(H3O+)] − [G(ZW) + G(H2O)].
d Values at the CPCM-MP2/6-311+G(d,p)level were used. eGeometries at the CPCM-CCSD/6-311+G(d,p) level were used. f Values at the CPCM-CCSD/6-311+G(d,p) level were used.
Table 2 Energetic results for the species when the tautomerization occurs via path (C)
Level Cluster Eela Gcorr
a ΔGsb Ga ΔGNE–ZW
b,c ΔGAb,d ΔG≠ b,e
B3LYP/6-311+G(d,p) ZW·H2O −360.96837 0.07325 −47.4 −360.97070 6.6 27.2 11.2TS·H2O −360.97417 0.06956 −30.2 −360.95280NE·H2O −360.99391 0.06667 −20.6 −360.96013AN + H2O −360.70321 0.05514 −175.3 −360.92735
MP2/6-311+G(d,p) ZW·H2O −360.04278 0.07479 −50.7 −360.04879 5.0 27.7 12.8TS·H2O −360.04710 0.07099 −32.8 −360.02843NE·H2O −360.07001 0.06667 −23.5 −360.04081AN + H2O −359.77811 0.05705 −178.0 −360.00464
CCSD/6-311+G(d,p) ZW·H2O −360.06792 0.07479f −52.8 −360.07729 4.1 27.3 14.8TS·H2O −360.06864 0.07099f −35.1 −360.05363NE·H2O −360.09847 0.06667f −24.5 −360.07082AN + H2O −359.80454 0.05705f −179.6 −360.03376
CCSD(T)/6-311+G(d,p) ZW·H2Og −360.10898 0.07479f −52.8h −360.11835 5.1 28.0 14.0
TS·H2Og −360.11100 0.07099f −35.1h −360.09599
NE·H2Og −360.13783 0.06667f −24.5h −360.11017
AN + H2Og −359.84460 0.05705f −179.6h −360.07381
a Values are in Hartree. b Values are in kcal mol−1. cΔGNE–ZW = G(NE·H2O) − G(ZW·H2O).dΔGA = G(AN + H2O) − G(ZW·H2O).
eΔG≠ = G(TS) −G(ZW·H2O).
f Values at the CPCM-MP2/6-311+G(d,p) level were used. gGeometries at the CPCM-CCSD/6-311+G(d,p) level were used. h Values atthe CPCM-CCSD/6-311+G(d,p) level were used.
Scheme 2
Organic & Biomolecular Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Org. Biomol. Chem., 2013, 11, 1407–1413 | 1409
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online

with 6-311+G(d,p) and 6-311+G(3df,2p) basis sets. Again, thefully optimized TS structures were similar to (D2), i.e. the sol-vating water molecules were far apart from the transferringproton. Therefore, the TS structure similar to (D2) was theintrinsic nature of the tautomerization of glycine and not aproblem with the CPCM method.
The structures optimized using the CPCM method in thetraditional condition might, in principle, correspond to anequilibrium or at least quasi-equilibrium state. In general, theproton transfer process normally takes place on a very shorttime scale17 but the orientational relaxation time of thesolvent molecules might take place approximately 1000 timeslater than the time for proton transfer. This suggests that thetautomerization TS between the ZW and NE of glycine mightbe in the non-equilibrium state of solvent structures becausetautomerization typically corresponds to proton transfer. Ifthis is true, the strategy considered above could not bedescribed adequately in terms of the equilibrium hypothesis.Kurz and Kurz36 proposed an interesting situation for protontransfer in solution, i.e. the fast protonic motions could beresisted by a force that would not be present if solvent relax-ation was fast. According to their work, if the activatedcomplex has a non-equilibrated environment, the deviation ofa solvent configuration from its equilibrium state could beexpected to be toward a configuration that is appropriate foran internal structure, where the proton is half-transferred.Moreover, they showed that their model is related both to theearlier qualitative suggestions by Schowen et al.,37,38 Ritchie39
and the Marcus theory of proton transfer reactions.40,41 On theother hand, some theoretical studies also used continuummodels for the non-equilibrium solvation effects on protontransfer.42–44 In particular, Tortonda et al.19 showed that theactivation barrier for the NE → ZW conversion of serinechanges substantially in the range of 1.5–5.5 kcal mol−1 whenthe non-equilibrated solvent effects are considered. On theother hand, they concluded that proton transfer in the tauto-merization of serine is extremely favorable with a negligibleactivation barrier, which is similar to that of glycine shown bythe quantum mechanical–molecular mechanics (QM–MM)simulation.45 Instead, to account for the differences in acti-vation barriers between the experimental and theoreticalresults for the tautomerization of amino acids, they suggestedthat the essential activation barrier for producing ZW wouldnot have originated from proton transfer itself but by a confor-mational change between the NE conformers. Although it ispossible that the essential activation barrier for producing ZWoriginated from a conformational change between the NE con-formers, a previous study showed that the activation barriersfor inter-conversions between the NE conformers are muchhigher than the experimental ΔG≠(ZW → NE) values at thesophisticated level of theory, such as CCSD and CCSD(T)levels. Therefore, the suggestion of Tuñón et al. appearsquestionable.
In previous work,14 theoretical results from the CPCMmethod using the PAULING cavity model29 agreed well withthe experimental findings, such as the differences in Gibbsfree energy between ZW and NE of glycine, ΔGZW–NE inaqueous solution.29 The ΔGZW–NE values were defined as theGibbs free energy differences between the ZW and the moststable NE in aqueous solution. Therefore, it is expected thatthe kinetic quantities, such as the ΔG≠(ZW → NE) obtainedusing the same method, are also consistent with the experi-mental findings. On the other hand, as noted above, theΔG≠(ZW → NE) values were underestimated considerably (bymore than 4 kcal mol−1) even at the CPCM-CCSD and CCSD(T)levels using the 6-311+G(d,p) basis set. Nevertheless, thesevalues agreed reasonably well with the experimental data com-pared to those reported previously.8,9,12,16 To understand thereasons for the underestimation, the non-equilibrium sol-vation effects at the TS were examined by assuming three situ-ations: (S-1), one water molecule attached to the transferringproton in the ZW moves together with the transferring proton;(S-2), one water molecule attached to the transferring protonin the ZW remains motionlessly at a fixed position near theNH2 fragment at the TS structure; and (S-3), proton transferoccurred without changing the position of the surroundingwater molecules from its initial state, the ZW form, in theeight water clusters.
(S-1) was assumed to be a non-equilibrated solvent con-figuration because the water molecule attached to the protonmoves while the proton is transferring. To observe thismotion, the bond distance, dO–H, between the transferringproton and oxygen atom on the moving water molecule wasfixed arbitrarily. These structures do not correspond to the
Fig. 1 The optimized geometries of the clusters for (a) the zwitterionic (ZW),(b) transition state (TS), and (c) neutral (NE) forms of the eight attached watermolecules at the B3LYP/6-311+G(d,p) level. The values are in Å, and the valuesin parentheses are at the B3LYP/6-311+G(3df,2p) level.
Paper Organic & Biomolecular Chemistry
1410 | Org. Biomol. Chem., 2013, 11, 1407–1413 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online
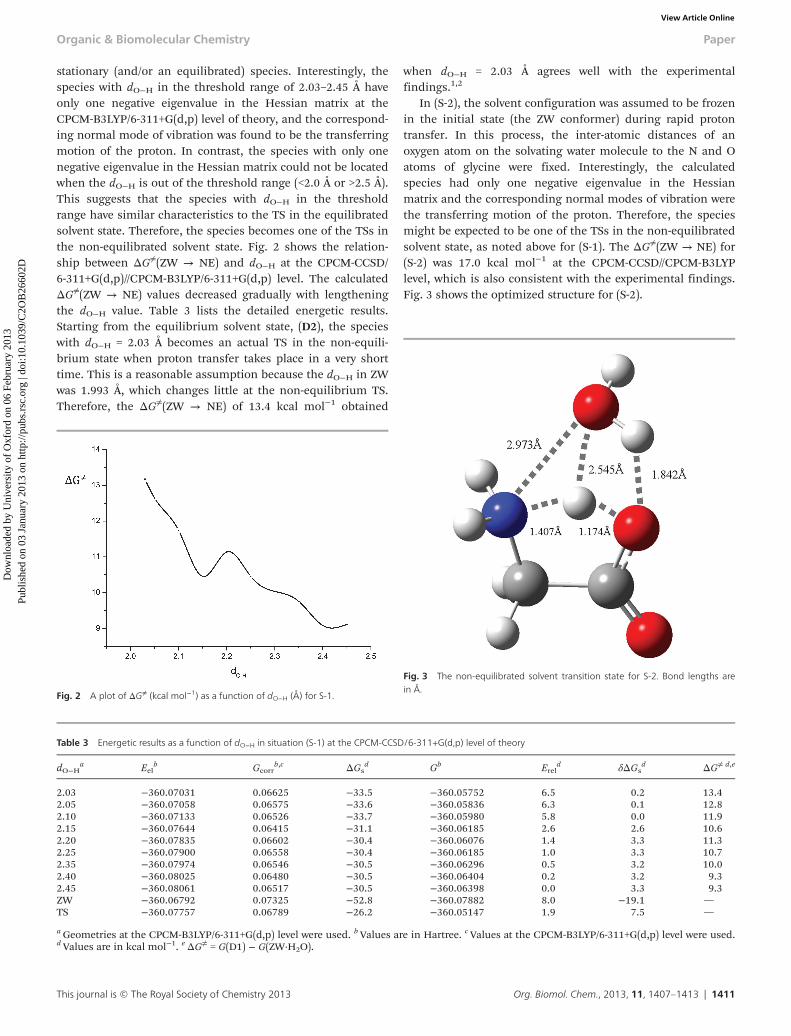
stationary (and/or an equilibrated) species. Interestingly, thespecies with dO−H in the threshold range of 2.03–2.45 Å haveonly one negative eigenvalue in the Hessian matrix at theCPCM-B3LYP/6-311+G(d,p) level of theory, and the correspond-ing normal mode of vibration was found to be the transferringmotion of the proton. In contrast, the species with only onenegative eigenvalue in the Hessian matrix could not be locatedwhen the dO−H is out of the threshold range (<2.0 Å or >2.5 Å).This suggests that the species with dO−H in the thresholdrange have similar characteristics to the TS in the equilibratedsolvent state. Therefore, the species becomes one of the TSs inthe non-equilibrated solvent state. Fig. 2 shows the relation-ship between ΔG≠(ZW → NE) and dO−H at the CPCM-CCSD/6-311+G(d,p)//CPCM-B3LYP/6-311+G(d,p) level. The calculatedΔG≠(ZW → NE) values decreased gradually with lengtheningthe dO−H value. Table 3 lists the detailed energetic results.Starting from the equilibrium solvent state, (D2), the specieswith dO−H = 2.03 Å becomes an actual TS in the non-equili-brium state when proton transfer takes place in a very shorttime. This is a reasonable assumption because the dO−H in ZWwas 1.993 Å, which changes little at the non-equilibrium TS.Therefore, the ΔG≠(ZW → NE) of 13.4 kcal mol−1 obtained
when dO−H = 2.03 Å agrees well with the experimentalfindings.1,2
In (S-2), the solvent configuration was assumed to be frozenin the initial state (the ZW conformer) during rapid protontransfer. In this process, the inter-atomic distances of anoxygen atom on the solvating water molecule to the N and Oatoms of glycine were fixed. Interestingly, the calculatedspecies had only one negative eigenvalue in the Hessianmatrix and the corresponding normal modes of vibration werethe transferring motion of the proton. Therefore, the speciesmight be expected to be one of the TSs in the non-equilibratedsolvent state, as noted above for (S-1). The ΔG≠(ZW → NE) for(S-2) was 17.0 kcal mol−1 at the CPCM-CCSD//CPCM-B3LYPlevel, which is also consistent with the experimental findings.Fig. 3 shows the optimized structure for (S-2).
Fig. 2 A plot of ΔG≠ (kcal mol−1) as a function of dO−H (Å) for S-1.
Table 3 Energetic results as a function of dO−H in situation (S-1) at the CPCM-CCSD/6-311+G(d,p) level of theory
dO−Ha Eel
b Gcorrb,c ΔGs
d Gb Ereld δΔGs
d ΔG≠ d,e
2.03 −360.07031 0.06625 −33.5 −360.05752 6.5 0.2 13.42.05 −360.07058 0.06575 −33.6 −360.05836 6.3 0.1 12.82.10 −360.07133 0.06526 −33.7 −360.05980 5.8 0.0 11.92.15 −360.07644 0.06415 −31.1 −360.06185 2.6 2.6 10.62.20 −360.07835 0.06602 −30.4 −360.06076 1.4 3.3 11.32.25 −360.07900 0.06558 −30.4 −360.06185 1.0 3.3 10.72.35 −360.07974 0.06546 −30.5 −360.06296 0.5 3.2 10.02.40 −360.08025 0.06480 −30.5 −360.06404 0.2 3.2 9.32.45 −360.08061 0.06517 −30.5 −360.06398 0.0 3.3 9.3ZW −360.06792 0.07325 −52.8 −360.07882 8.0 −19.1 —TS −360.07757 0.06789 −26.2 −360.05147 1.9 7.5 —
aGeometries at the CPCM-B3LYP/6-311+G(d,p) level were used. b Values are in Hartree. c Values at the CPCM-B3LYP/6-311+G(d,p) level were used.d Values are in kcal mol−1. eΔG≠ = G(D1) − G(ZW·H2O).
Fig. 3 The non-equilibrated solvent transition state for S-2. Bond lengths arein Å.
Organic & Biomolecular Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Org. Biomol. Chem., 2013, 11, 1407–1413 | 1411
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online

Nonetheless, the non-equilibrated states using one solvent(water) molecule with the arbitrarily fixed distances could notbe the real stationary species on the potential energy surface(PES). Therefore it might be insufficient to explain any non-equilibrated solvation states clearly. To overcome this problem,the clusters with eight water molecules as shown in Fig. 1 wereexamined in aqueous solution. Fig. 1 shows, as is generallyexpected, that the eight solvent water molecules in the ZWform were bound more tightly owing to its zwitterionic charac-ter than those in the NE form. These water molecules shouldreorganize gradually during the tautomerization process ongoing from the ZW form to the NE form (or vice versa), if thetautomerization occurs under an equilibrated solvation state.Unfortunately, the fully optimized TS structure similar to (b) inFig. 1 could not be located in aqueous solution despite numer-ous attempts to find it. The difficulty has arisen due to asudden structural change of solvated water molecules in thevicinity of a plausible TS structure. However, we managed tolocate a second-order saddle point, (D3), with two negativeeigenvalues in the Hessian matrix at the CPCM-B3LYP/6-311+G(d,p) level46 as shown in Fig. 4. Normal mode analysesshow that one negative frequency (−756 cm−1) corresponds to
the proton transfer, but the second one (−78 cm−1) correspondsto the fluctuation of the eight solvent water molecules (seeFig. 4b for normal modes of vibrations). Note that the differ-ence in Gibbs free energy between the NE form and (D3) wasonly 1.9 kcal mol−1. This value was nearly the same as thoseobtained from the CPCM calculations14 and much lower thanthe experimental value of 7.3 kcal mol−1.6 Again, this couldstrongly imply that the tautomerization of glycine proceeds viathe non-equilibrated solvation states. Nevertheless, the clusterswith eight water molecules might be still arbitrary, and thus amore detailed study is in progress in our laboratory.
Finally, in (S-3), structures corresponding to the TS and NEwere inserted into the cavity formed by the eight water mole-cules by removing the ZW in the eight water clusters of theZW form. To perform this operation, the root-mean squaredeviation (RMSD) of the four heavy atoms (N and O atomsdirectly involved in the proton transfer and two carbon back-bone atoms) was minimized and the RMSD-minimized struc-tures were placed inside the cavity formed by the ZW. Asexpected from Fig. 1, the ZW formed the tightest hydrogen-bonded network owing to its zwitterionic character among thethree structures, and inserting the TS structure allowedbumping between the moving proton and the hydrogen atomin the solvent shell (bond distance = 0.937 Å), which makesthis plausible structure highly unstable (ΔG≠(ZW → NE) =102.49 kcal mol−1). This shows that the procedure using eightwater clusters failed to provide a reasonable non-equilibriumdescription.
Conclusions
From the theoretical studies on the tautomerization of glycine,non-equilibrium solvation is essential to explain the observedexperimental data. In the CPCM methodology, the user cannotcontrol the solvent configuration of the continuum model. Theonly way to accomplish this is to introduce ancillary water mole-cule(s) to consider the non-equilibrated solvent states. Thefollowing conclusions were drawn from this study:
(i) The tautomerization of glycine can proceed via inter-mole-cular proton transfer with a participating water molecule, (C) inScheme 1, if the process occurs in an equilibrated solvent state.
(ii) The tautomerization process can occur via direct intra-molecular proton transfer, (S-1) or (S-2) without active catalyticwater molecule(s), if the process occurs in a non-equilibratedsolvent state.
(iii) The proton exchange process, such as (B) in Scheme 1,could not occur because the sum of the energies of the AN +hydronium ion is higher than that of the ZW + H2O.
Acknowledgements
This study was supported by INHA University. One of theauthors (C. K. Kim) thanks Dr. Noham Weinberg for hishelpful discussions.
Fig. 4 The optimized geometry of the clusters for (a) the second-order tran-sition state, (D3), at the B3LYP/6-311+G(d,p) level. Bond lengths are in Å. (b)The displacement vectors corresponding to two imaginary frequencies, ν1and ν2.
Paper Organic & Biomolecular Chemistry
1412 | Org. Biomol. Chem., 2013, 11, 1407–1413 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online

References
1 J. S. Gaffney, R. C. Pierce and L. Friedman, J. Am. Chem.Soc., 1977, 99, 4293.
2 R. Wolfenden, L. Andersson, P. M. Cullis andC. C. B. Southgate, Biochemistry, 1981, 20, 849.
3 G. Wada, E. Tamura, M. Okina and M. Nakamura, Bull.Chem. Soc. Jpn., 1982, 55, 3064.
4 K. A. Sharp, A. Nicholls, R. Friedman and B. Honig, Bio-chemistry, 1991, 30, 9686.
5 R. Ramaekers, J. Pajak, B. Lambie and G. Maes, J. Chem.Phys., 2004, 120, 4182.
6 F. R. Tortonda, J. L. Pascual-Ahuir, E. Silla and I. Tuñón,Chem. Phys. Lett., 1996, 260, 21.
7 A. Fernández-Ramos, Z. Smedarchina, W. Siebrand andM. Z. Zgierski, J. Chem. Phys., 2000, 113, 9714.
8 B. Balta and V. Aviyente, J. Comput. Chem., 2003, 24, 1789.9 B. Balta and V. Aviyente, J. Comput. Chem., 2004, 25, 690.10 C. M. Aikens and M. S. Gordon, J. Am. Chem. Soc., 2006,
128, 12835.11 S. M. Bachrach, J. Phys. Chem. A, 2008, 112, 3722.12 S. Tiwari, P. C. Mishra and S. Suhai, Int. J. Quantum Chem.,
2008, 108, 1004.13 R. M. Balabin, J. Phys. Chem. B, 2010, 114, 15075.14 C. K. Kim, B.-H. Park, H. W. Lee and C. K. Kim, Bull.
Korean Chem. Soc., 2011, 32, 1985.15 D. Yu, D. A. Armstrong and A. Rauk, Can. J. Chem., 1992,
70, 1762.16 N. Okuyama-Yoshida, K. Kataoka, M. Nagaoka and
T. Yamabe, J. Phys. Chem. A, 1998, 102, 285.17 I. Tuñón, E. Silla and M. F. Ruiz-López, Chem. Phys. Lett.,
2000, 321, 433.18 F. R. Tortonda, J.-L. Pascual-Ahuir, E. Silla and I. Tuñón,
J. Chem. Phys., 1998, 109, 592.19 F. R. Tortonda, E. Silla, I. Tuñón, D. Rinaldi and M. F. Ruiz-
López, Theor. Chem. Acc., 2000, 104, 89.20 M. Kołaski, A. A. Zakharenko, S. Karthikeyan and
K. S. Kim, J. Chem. Theory Comput., 2011, 7, 3447.21 S. Karthikeyan, N. Jiten Singh and K. S. Kim, J. Phys. Chem.
A, 2008, 112, 6527.22 S. Odde, B. J. Mhin, S. Lee, H. M. Lee and K. S. Kim,
J. Chem. Phys., 2004, 120, 9524.23 A. Kumar, M. Park, J. Y. Huh, H. M. Lee and K. S. Kim,
J. Phys. Chem. A, 2006, 110, 12484.24 A. C. Olleta and H. M. Lee, J. Chem. Phys., 2006, 124,
024321.25 A. C. Olleta, H. M. Lee and K. S. Kim, J. Chem. Phys., 2007,
126, 144311.26 M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput.
Chem., 2003, 24, 669.27 V. Barone and M. Cossi, J. Comput. Chem., 1998, 19, 404.28 A. Bondi, J. Phys. Chem., 1964, 68, 441.
29 Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005,1, 70.
30 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.31 C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens.
Matter, 1988, 37, 785.32 M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys.
Lett., 1988, 153, 503.33 G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76,
1910.34 E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem.
Phys., 1988, 89, 7382.35 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,
M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr.,T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam,S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi,G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji,M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai,M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross,V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts,R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi,C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma,G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski,S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas,D. K. Malick, A. D. Rabuck, K. Raghavachari,J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford,J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko,P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith,M. A. Al-Laham, C. Y. Peng, A. Nanayakkara,M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen,M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03(Revision D.02), Gaussian, Inc., Wallingford, CT, 2004.
36 J. L. Kurz and L. C. Kurz, J. Am. Chem. Soc., 1972, 94, 4451.37 C. G. Swain, D. A. Kuhn and R. L. Schowen, J. Am. Chem.
Soc., 1965, 87, 1553.38 L. D. Kershner and R. L. Schowen, J. Am. Chem. Soc., 1971,
93, 2014.39 C. D. Richie, J. Am. Chem. Soc., 1969, 91, 6749.40 R. A. Marcus, J. Phys. Chem., 1968, 72, 891.41 A. O. Cohen and R. A. Marcus, J. Phys. Chem., 1968, 72,
4249.42 M. F. Ruiz-López, A. Oliva, I. Tuñón and J. Bertrán, J. Phys.
Chem. A, 1998, 102, 10728.43 M. A. Aguilar and A. Hidalgo, J. Phys. Chem., 1995, 99,
4293.44 J. J. Timoneda and J. T. Hynes, J. Phys. Chem., 1991, 95,
10431.45 I. Tuñón, E. Silla, C. Millot, M. T. C. Martin-Costa and
M. F. Ruiz-López, J. Phys. Chem. A, 1998, 102, 8673.46 The CPCM options were changed to RMIN = 0.5 and OFAC
= 0.8 due to convergence problems in the clustercalculations.
Organic & Biomolecular Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Org. Biomol. Chem., 2013, 11, 1407–1413 | 1413
Dow
nloa
ded
by U
nive
rsity
of
Oxf
ord
on 0
6 Fe
brua
ry 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2OB
2660
2DView Article Online
Related Documents