IKTZRNATIONAL IOURNAL OF QUAh'77JM CHEMISTRY. VOL. XXX, 495-508 (1986) On the Red Shift of OH Stretching Region Vibrations in Ice and Water EUGENE S. KRYACHKO Institute for Theoretical Physics, Kiev-130, USSR 252130 Abstracts The nature of the red shift of frequencies of the fundamental modes, Y, and v3, in the OH stretching region of the vibrational spectrum of ice (and, possibly, in water) under H-bonding formation is explained in the framework of the continuum approach for the one-dimensional infinite chain of water molecules rep- resented as dipolar and polarizable OH oscillators vibrating in the definite force field. The explicit expres- sions in the form of the generalized cnoidal nonlinear waves describing these fundamental modes and obtained as the solutions of the nonlinear Klein-Gordon equation of motion, and their red shifts that are consistent with experimental observations, are presented. I. Introduction As is well known [ 11, the diverse spectroscopic methods constitute a powerful tool for studies of the nature of hydrogen bonding. Moreover, the definitions of the hydrogen (or, for short, H-) bonding are mainly of spectroscopic character [ 1,2]. The interpretation of spectra of hydrogen-bonded complexes is nevertheless a highly com- plicated problem that has not yet been resolved completely [3-141. In particular, the problem of theoretical interpretation of changes, which occur in an equilibrium geometry as well as in a vibrational spectrum owing to a transition from isolated monomers (in a gas phase) and dimers to associated liquids and crystals with the cooperative hydrogen bonding, still remain open [12]. For instance, it is common knowledge that a change of nature of stretching vibrations of a group, forming H- bonding, under its formation appears to be the striking peculiarity of a spectroscopic manifestation of H-bonding [ 1-14]. For example, one can observe experimentally the so-called red shifts of frequencies of the symmetric, vl, and the antisymmetnc, v3, fundamental stretching modes under transition from a gas phase to liquid or amor- phous water and ice. These shifts take the following approximate values: Au, and Av3 = -300 cm-' [3,4]. Therefore, the magnitude of the red shift accounts for approximately 8-10% of the corresponding frequencies of monomer (see Table I). As mentioned above, the nature of the red shift has not been elucidated com- pletely. Many authors assumed that its nature could be explained mainly by the formation of water dimers (H20)*. .Namely, a drastic decrease in the stretching vibra- tional frequencies was explained ordinarily by the interaction of a proton H with an oxygen atom O2 in a hydrogen bridge 0, -H * * . 02. This interaction results in a stretching of 0,-H bond and, therefore, in a lowering of the corresponding fre- 0 1986 John Wiley & Sons, Inc. CCC 0020-7608/86/040495- 14$04.00

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

IKTZRNATIONAL IOURNAL OF QUAh'77JM CHEMISTRY. VOL. XXX, 495-508 (1986)
On the Red Shift of OH Stretching Region Vibrations in Ice and Water
EUGENE S. KRYACHKO Institute for Theoretical Physics, Kiev-130, USSR 252130
Abstracts
The nature of the red shift of frequencies of the fundamental modes, Y, and v3, in the OH stretching region of the vibrational spectrum of ice (and, possibly, in water) under H-bonding formation is explained in the framework of the continuum approach for the one-dimensional infinite chain of water molecules rep- resented as dipolar and polarizable OH oscillators vibrating in the definite force field. The explicit expres- sions in the form of the generalized cnoidal nonlinear waves describing these fundamental modes and obtained as the solutions of the nonlinear Klein-Gordon equation of motion, and their red shifts that are consistent with experimental observations, are presented.
I. Introduction
As is well known [ 11, the diverse spectroscopic methods constitute a powerful tool for studies of the nature of hydrogen bonding. Moreover, the definitions of the hydrogen (or, for short, H-) bonding are mainly of spectroscopic character [ 1,2]. The interpretation of spectra of hydrogen-bonded complexes is nevertheless a highly com- plicated problem that has not yet been resolved completely [3-141. In particular, the problem of theoretical interpretation of changes, which occur in an equilibrium geometry as well as in a vibrational spectrum owing to a transition from isolated monomers (in a gas phase) and dimers to associated liquids and crystals with the cooperative hydrogen bonding, still remain open [12]. For instance, it is common knowledge that a change of nature of stretching vibrations of a group, forming H- bonding, under its formation appears to be the striking peculiarity of a spectroscopic manifestation of H-bonding [ 1-14]. For example, one can observe experimentally the so-called red shifts of frequencies of the symmetric, v l , and the antisymmetnc, v3 , fundamental stretching modes under transition from a gas phase to liquid or amor- phous water and ice. These shifts take the following approximate values: A u , and Av3 = -300 cm-' [3,4]. Therefore, the magnitude of the red shift accounts for approximately 8-10% of the corresponding frequencies of monomer (see Table I).
As mentioned above, the nature of the red shift has not been elucidated com- pletely. Many authors assumed that its nature could be explained mainly by the formation of water dimers (H20)*. .Namely, a drastic decrease in the stretching vibra- tional frequencies was explained ordinarily by the interaction of a proton H with an oxygen atom O2 in a hydrogen bridge 0, -H * * . 02. This interaction results in a stretching of 0 , -H bond and, therefore, in a lowering of the corresponding fre-
0 1986 John Wiley & Sons, Inc. CCC 0020-7608/86/040495- 14$04.00
496 KRYACHKO
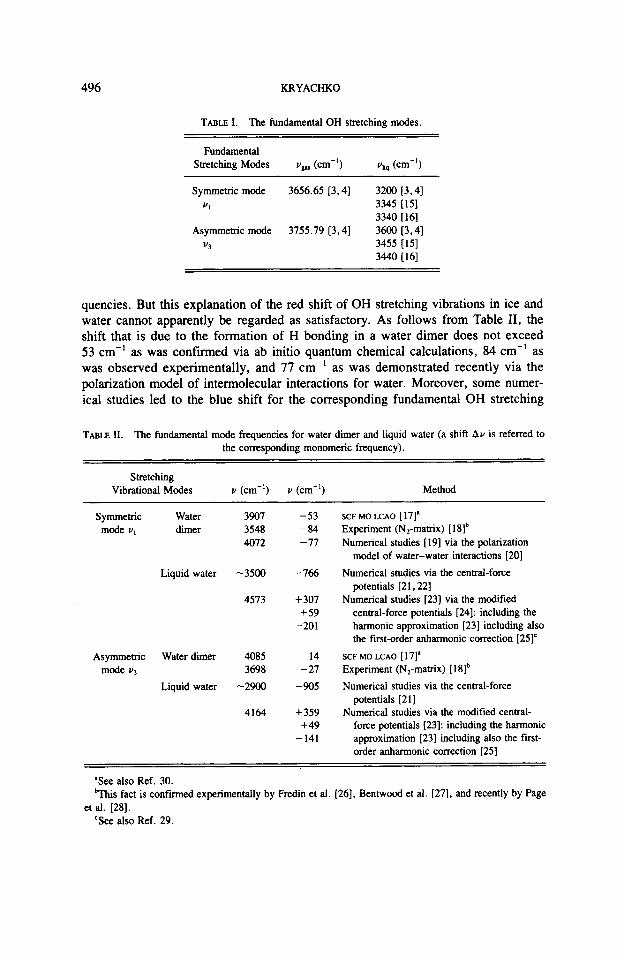
TABLE I . The fundamental OH stretching modes.
Fundamental Stretching Modes vW (cm-I) vliq (cm-')
Symmetric mode 3656.65 13.41 3200 [3,41 VI 3345 [IS]
3340 1161 Asymmetric mode 3755.79 [3,4] 3600 [3,4]
v3 3455 [15] 3440 [I61
quencies. But this explanation of the red shift of OH stretching vibrations in ice and water cannot apparently be regarded as satisfactory. As follows from Table 11, the shift that is due to the formation of H bonding in a water dimer does not exceed 53 cm-' as was confmed via ab initio quantum chemical calculations, 84 cm-' as was observed experimentally, and 77 cm-' as was demonstrated recently via the polarization model of intermolecular interactions for water. Moreover, some numer- ical studies led to the blue shift for the corresponding fundamental OH stretching
TABLE 11. The fundamental mode frequencies for water dimer and liquid water (a shift A v is referred to the corresponding monomeric frequency).
Stretching Vibrational Modes v (cm-I) v (cm-I) Method
Symmetric Water mode vI dimer
Liquid water
Asymmetric Water d m e r
Liquid water mode v,
3907 -53 3548 - 84 4072 - 77
-3500 -766
4573 +307 + 59
-201
4085 - I4 3698 - 27
-2900 -905
4164 +359 + 49 - 141
SCF MO LCAO [ 171' Experiment (N,-matrix) [l8lb Numerical studies 1191 via the polarization
model of water-water interactions [20] Numerical studies via the central-force
potentials [21,22] Numerical studies 123) via the modified
central-force potentials [24]: including the harmonic approximation [23] including also the fmt-order anharmonic correction 1251'
SCF MO LCAO [ 171' Experiment (N,-matrix) [l8Ib Numerical studies via the central-force
Numerical studies via the modified central- potentials 121 J
force potentials [23]: including the harmonic approximation [23] including also the fmt- order anharmonic correction [25]
'See also Ref. 30. bThis fact is confmed experimentally by Fredin et al. [26], Bentwood et al. [27], and recently by Page
'See also Ref. 29. et al. [28].

RED SHIIT OF OH STRETCHING 497
modes (Table 11). Hence, a study of the nature of the red shift of stretching region of the vibrational spectrum of ice and water on a level of the general theory of inter- molecular interactions [31-331 becomes of great importance, at least for creating the more adequate and realistic empirical water-water intermolecular potentials. In this connection the paper by Reimers and Watts [34], where the authors obtained the shift A U ~ , ~ = -300 cm-’ by means of computer simulations of liquid water, is of great interest. At the same time the intermolecular nature of the red shift cannot be eluci- dated via computer simulations. It is worth considering in detail the heuristic approach for interpreting the vibrational spectrum of ice and water proposed and developed with great success by Rice and his collaborators [35-391 and based on the lattice dynamics simulations. This approach by Rice is evidently of the interpretative nature; nevertheless, it is more methodological for elucidating the nature of the vibrational spectrum in ice and water in a comparison with the approach by Reimers and Watts [34]. In conclusion it should be noted that Whalley and his collaborators [40] as- sumed that the intermolecular nature of the OH stretching vibrations in ice and water can be explained mainly on the basis of a strong intermolecular coupling of vibrations of isolated OH oscillators.
This present paper is devoted to-a possible explanation of the red shift occumng in the OH stretching region of the vibrational spectrum in ice and water in the frame- work of the continuum approach for a one-dimensional chain of water molecules (the so-called Bernal-Fowler chain) or, in a stronger sense, of dipolar polarizable OH oscillators. In the context of the present paper, the one-dimensional chain of OH oscillators appears to be the adequate model that is justified and confirmed by the lat- tice dynamics simulations carried out by Rice et al. [35-391, who demonstrated that the lowest fundamental modes of the OH stretching region in ice represent them- selves by the concerted and cooperative ( u I is in-phase and u3 is out-of-phase) motions of the whole system of OH oscillators and can be reproduced, in fact, via computer simulation with the one-dimensional model where the force constants are slightly renormalized [36]. In the case of water, according to Frank and Wen [41], one can assume the existence of “icebergs” in water where fairly lengthy quasi-one- dimensional chains of molecules can be realized. Undoubtedly, the model proposed reflects the cooperative, or collective, nature of hydrogen bonding in ice and water. This idea is originally due to Frank and Wen [41] and has been confirmed strikingly in different aspects [3,30,42-461. It should be noted finally that a coherence in the model presented is defined in a sense by Frohlich [47].
The structure of the present paper is as follows. Section I1 outlines the one-dimen- sional model of water molecules and serves as a justification of this model in view of the general theory of hydrogen bonding. The discussion of the dipole moments and of the potential presented there is classic. In this Section the “continuum approach” non- perturbative treatment for the model proposed that is exploited leads to the general formulation of the Euler-Lagrange equation of motion for the classic field of protonic displacements. This general formulation for the equation of motion is realized numeri- cally for the in-phase coherent motion (ul mode) in Section 111 and for the out-of- phase motion (u3 mode) in Section IV. Section V is devoted to the discussion of results obtained.

498 KRYACHKO
11. One-Dimensional Chain of OH Oscillators: Continuum Approach
Let us consider first a system of two dipoles arranged “head to tail.” Its interaction energy is defined by the following expression:
where pI and p2 are the magnitudes of these dipoles, and R I 2 is the distance between their centers of mass. The dipoles are assumed to be point ones, i.e., the length of each dipole is considerably less than R 1 2 . In particular, one can suggest that this assumption is fulfilled for a system of two OH oscillators arranged as in an ice or water network. Moreover, one can suppose also that each dipole vibrates near its equilibrium positions and presents itself as a polarizable dipole. So, one can represent pl and p2 as follows:
where pIo’ is the equilibrium magnitude of an ith dipole; pll) and pi2’ are its first and second derivatives with respect to its displacement r ( r = 0 corresponds to the equi- librium position), respectively; and Q is its polarizability coefficient: Q = 1.444 A3.
In order to be correct, one can discuss in detail all the approximations accepted in Eq. (2). Firstly, because of the work by Sandorfy and coworkers [48,8], the third- and higher-order perturbation correction terms in the powers of ri are neglected in Eq. (2). Secondly, together with p(I) the electrical anharmonicity, i.e., p”’, is taken into account in Eq. (2) owing to its strong effect on the stretching vibrations of OH oscillators [48,49]. Thirdly, we take into account in Eq. (2) an effect of the induced dipole moment on the H-bonding formation, and, therefore, on its spectral peculiari- ties, as was emphasized by Zundel [6 ] . But naturally, within the framework of the model proposed, it is unable to take into account completely the polarization effect on each OH oscillator under the formation of H-bonding between them. It refers, in particular, to the so-called charge shift (a lone pair of electrons-proton) with the H- bonding to be formed that produces a significant effect on the properties of hydrogen- bonded complexes [50] . Nevertheless, one can take this factor into account by renormalizing in a definite way the dipole moment of each OH oscillator and its derivatives (see Sections I11 and IV).
Let us consider now a one-dimensional chain of the dipolar and polarizable OH oscillators. Each oscillator vibrates in the definite potential field and interacts with the other ones by the dipole-dipole interaction law, Eq. (1). We invoke the following simplifying assumption that the positions of the oxygen atoms are static, or “frozen,” and chosen in such a way that reflects a real network of ice. Taking into account Eq. ( 2 ) , one can represent the total potential energy of such a system in the following form within the framework of the second-order perturbation treatment:

RED SHIFT OF OH STRETCHING 499
where k 2 , k 3 , and k4 are the harmonic and first two anharmonic force constants, respectively, which determine the potential field in which each OH oscillator vi- brates. Replacing an infinite set of discrete variables { r l } by the continuous function r = r ( x , t ) , where the xth axis is directed along the chain under study, and taking into account that
(4a)
(4b)
rlrI+l = I[rf + rf+l - (r l - r1+l)’I
r1r,+, = (1b-f + d + n ) - [ (r l - r l+ i ) + * * * + (ri+n-l - ri+n)12}
ar r,+I(t) = T(X + R e , , ) = r ( x , t ) + R e - ax
and
(4c)
which means the continuum approach* is to be accepted, one can rewrite the expres- sion for the total potential energy in the following way:
( 5 )
where the higher-order terms in the powers of r and r, = &/ax are neglected, and the following notations are used:
V(r , rr) = Vlr + f12r2 + 13r3 + 14r4 + fP(rJ2 + v,,
*The continuum approach for the similar class of nonlinear problems is discussed in detail by Davydov [51] and means a replacement of the discrete infinite set of points by a single function r (x ) , where x is a continuously varied variable. The validity of the continuum approach for the present model is proved in Section 111.

500 KRYACHKO
li = ki + V i , i = 2 , 3 , 4 ; and
It is clear that the approach presented generalizes the reduced vibrational model developed by Rice and coworkers [35 ,36 ] . Given in the framework of the continuum approach, the reduced vibrational model leads to the following nonvanishing terms:
In view of the approach proposed by Sandorfy and his collaborators [ 4 8 , 8 ] , and based on the second-order perturbation treatment, one can naturally assume that the present model seems to be more adequate in comparison with the reduced vibrational model (in the one-dimensional picture).
Therefore, within the framework of the continuum approach, the Hamiltonian of the one-dimensional chain of OH oscillators takes the following form:
where i. = &/at, V(r , rx) = U(r) + @ / 2 ) (rx)2, /3 = mc;, and m is the reduced mass of a single OH dipole. In particular, U(r) itself represents the total potential energy of a given chain of coupled OH oscillators given in the continuum approach. One can demonstrate fairly easy that the Euler-Lagrange equation of motion for the classic field r = dx, t ) of protonic displacements with the Hamiltonian H given by Eq. (12) has the form of the generalized nonlinear Klein-Gordon equation [51]:
2 dU dr m(rrr - corn) + - = 0.
Owing to the Lorentz invariance of Eq. (13), one can introduce the standard vari- able [51]
5 = x - xo - Vr(V > 0) ( 14) and rewrite this equation in the following manner:
dU dr
m(V2 - c:)rc6 + - = o (15)
Multiplying it by rc, taking the expression for U(r) into account, and integrating the equation obtained in the limits from zero to r , one can obtain finally

RED SHIFT OF OH STRETCHING 501
where E is the integration constant with the dimension of energy and s = V/co. Therefore, one obtains the following relation:
Hence, based on the given expression for U(r) with the reasonable magnitudes of its coefficients, one can derive, in principle, the explicit form for the classic field r = r(8) of displacements of protons. Such a program outlined will be realized for the system of OH oscillators under study in the following Sections.
111. Collective Fundamental Mode vI
In order to determine the explicit form for the function r = r(x, f ) , which describes the collective and coherent in-phase stretching vibration of all OH oscillators, one can choose initially the reasonable and correct magnitudes of the force constants, k 2 , k3 , and k4, and the electrical dipole constants, p'", p"), and F ' ~ ) . We based the force constants on the data given by Rice et al. [35-371:
k2 = 7.01 mdyn/A
k3 = -9.555 mdyn/A2
k4 = 15.066 mdyn/A3.
Choosing reasonable magnitudes for the equilibrium dipole moment of an OH oscil- lator and its fmt- and second-order electrical anharmonicities, one can deal with both the computational and experimental data on these quantities for a water dimer, liquid water, and ice [38,52,53,3,4], taking into account the correlations observed between the electrical dipole properties of H 2 0 and HF monomers and the dimers (H20h and HF-H20 [53,54]. In addition, one should take into account the charge shift (a lone pair-proton) occumng under H-bonding formation. Bearing in mind all these argu- ments, we choose therefore the following values:
p(O) = 2.643 D (19)
p") = (4.0 - 0.211) D/A = 3.789 D/A
p(2) = 8.5 D/A2. (21)
(20)
It should be emphasized that we take into account in Eqs. (20) and (21) the contribu- tions provided by the corresponding terms in Eq. (2).
Substituting the values given in Eqs. (18)-(21) into the expressions (6)-( 1 I), one obtains
V1 = -0.252 mdyn
V2 = -0.905 mdyn/A
V3 = -0.248 mdyn/A2
502 KRYACHKO
V4 = -0.342 mdyn/A3
/3 = 1.890 mdyn * 8,
Vo = -0.087 mdyn * A I , = 6.106 mdyn/A
i3 = -9.803 mdyn/A2
L4 = 14.725 mdyn/A3
where Re = 2.76 A, and the contributions provided by the nearest four neighboring OH oscillators, i.e., R - 9-11 A, are taken into account only because of the asymp- totic divergence of the multipole series expansion for the interaction energy in the inverse powers of R [33].
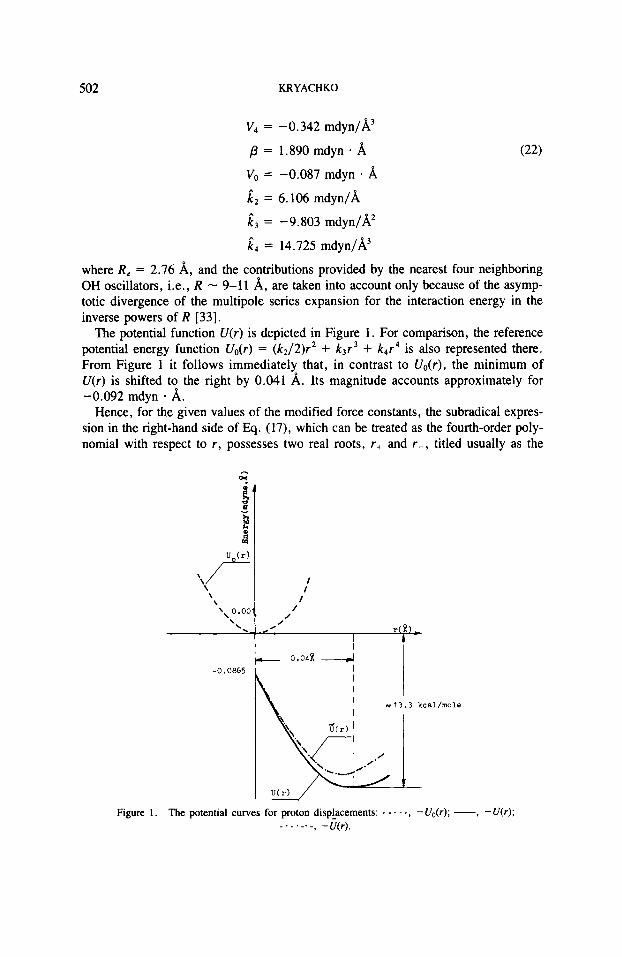
The potential function V(r ) is depicted in Figure 1. For comparison, the reference potential energy function Vo(r) = (k2/2)r2 + k3r3 + k4r4 is also represented there. From Figure 1 it follows immediately that, in contrast to Uo(r) , the minimum of V(r) is shifted to the right by 0.041 A. Its magnitude accounts approximately for -0.092 mdyn A.
Hence, for the given values of the modified force constants, the subradical expres- sion in the right-hand side of Eq. (17), which can be treated as the fourth-order poly- nomial with respect to r , possesses two real roots, r+ and r - , titled usually as the
\ \ '\C.CO
'., \
-C .C865
\ I I I I ~ 1 3 . 3 kcalhole
I I ~ 1 3 . 3 kcalhole
I
Figure 1 . The potential curves for proton displacements: - - - - -, -U,,(r); -, -U(r); , -UW. - . - . - . -

RED SHIFT OF OH STRETCHING 503
turning points with r- < r + , and two complex roots, b t ic, for the integration con- stant E > -0.092 mdyn . A. Therefore, the explicit form for the classic field r(x, t ) becomes as follows
(23) (r+ cos o1 + r- cos e,) + (T+ cos el - r- cos e,) cn@&
r = r(5) = (COS O1 + cos 0,) + (COS - cos 0,) cn(PJ)
where
tg0,,, = (rt - b ) / c . 1 2L4 cos el - cos e, . rn(s' - 1) Po = &- "
The classic field r ( x , t ) = r(5) of protonic coherent in-phase displacements is therefore expressed in terms of an elliptic cosine, i.e., represents itself the general- ized cnoidal wave. *
Assuming the harmonic approach, i.e., f i , = f i , = 0, to be valid,** one can obtain the more simple expression for r(x, t):
(x - xo - V t ) . m(VZ - ci) 1
It is fairly easy to demonstrate (see, for instance, Refs. 55 and 56) that the frequency of vibrations of the classic field rh(x, t ) is given by the following expression:
I
Substituting rn = 0.941 rnp, where rnp is the proton mass, V = 10' cm * s - I [571, and co = 3.401 X lo6 cm * s-I into Eq. (25), one obtains finally the fundamental frequency of the coherent and collective in-phase stretching vibrations of the classic protonic field:
u;"" = 3308.770 cm-I. (26)
Therefore, taking the value for ufas given in Table I into account, one can evaluate the frequency shift,
AuI = ucol1 I - uI = -347.880 cm-I, (27)
i.e., the so called red shift results. In concluding this Section we will easily prove the validity of the continuum approach accepted in the present treatment. As follows from Eq. (24), the width of the excitation region, Ax, takes the following value,
V AX y..ll= 16.045 A ,
1
*The term "cnoidal wave" is introduced for waves whose behavior is described by an elliptic cosine (cn) (see note 12 on p. 305 of Ref. 51).
**The anharmonic effects can be neglected because the harmonic force constant k, changes drastically (by 12.9%), owing to electrical nature effects, as compared with the anharmonic force constants, k 3 and k,, whose changes account about for 2.6% and 2.396, respectively.

504 KRYACHKO
i.e., the inequality A x l R , = 5.8 > 1 is satisfied that confirms the validity of the continuum approach in accordance with the arguments given by Davydov (see pp. 62 and 224 of Ref. 51).
IV. Collective Fundamental Mode v3 Let us now consider, within the framework of the given one-dimensional infinite
chain of water molecules under the assumption that the continuum approach is valid, the collective out-of-phase stretching vibration fundamental mode v3. One can sug- gest that this mode corresponds to the collective vibration of all OH oscillators of the infinite one-dimensional chain when two adjacent OH oscillators vibrate in opposite directions coherently [36]. This nature of the collective vibration leads one to take into account the following relations, assuming the continuum approach to be valid:
ri+l(t) = r ( x , t ) - Rerx
rtri+i = i [ ( ~ + ri+i)2 - (r? + ri:t)I (28a)
(28b) etc.
Similarly to Eq. (3, one can obtain
V(r, rJ = Vlr + iL2r2 + L3r3 + L4r4 + i&rx)2 + v,, (29)
where - vi = vj, i = 1,3,4; (30)
Inserting the magnitudes for the force and electrical dipole constants given by Eqs. (18)-(21) into Eqs. (30)-(32), one has
GI = -0.252 mdyn
V2 = -0.335 mdyn/A
V3 = -0.248 mdyn/A2
f4 = -0.342 mdyn/A3
= 1.116 mdyn A L 2 = 6.675 mdyn/A
i3 = -9.803 mdyn/A2
L4 = 14.725 mdyn/A3.
(33)

RED SHIFT OF OH STRETCHING 505
The potential energy function C(r) = VO + (1/2)i2r2 + i 3 r 3 + k4r4 + f I r of the classic field r = r ( x , r ) = r ( t ) , where $ = x - xo - f r , of the collective out-of- phase protonic displacements, is also depicted in Figure 1. Similarly to V(r) , the minimum of &r) is also displaced with respect to that for Vo(r) by 0.038 A, and its magnitude becomes equal approximately to -0.092 mdyn * A.
One obtains that the classic field of the collective out-of-phase protonic displace- ments takes the following explicit form:
- - - - - - (34) (F+ cos aI + F- cos a2) + (F+ cos 8, - F- cos 8,) cn@& r = r(%) =
(COS el + cos e,) + (COS el - cos 0,) cn(P&)
Po - = 0 ” cos iI cos i2 * m(i2 - 1)
where
1 2i4
and F, and 6 t iS are the real and complex roots of the allebraic equation C(r) = i, with the integration constant E satisfying the inequality E > -0.092 mdyn - A; and fi = m& This classic field itself represents the generalized cnoidal wave character- ized by a weak anharmonicity. Assuming the harmonic approach to be valid, one can rewrite Eq. (34) as follows:
Its fundamental frequency us”” is I
Substituting f = 10’ cm * s-l and to = 2.663 X lo6 cm * s-I into Eq. (36), one obtains the magnitude of the fundamental frequency of the collective out-of-phase stretching vibrations of the infinite one-dimensional chain of OH oscillators,
(37) vcoll = 3458.767 cm-I.
Taking into account the value of the monomer frequency vgas given in Table I, one can evaluate the shift Av3.
(38) i.e., the “red” shift for the fundamental mode v3 owing to H-bonding formation is obtained.
hv3 = us”” - v3 = -297.023 cm-‘,
V. Discussion
In the framework of the continuum approach for the classic field of protonic dis- placements, it is shown that in the case G f the collective and coherent (concerted) in- phase stretching vibrations of OH oscillators, the resulting length of the OH bond, rOH, is increased approximately by 0.04 A. This result lies in a good agreement with

506 K R Y A C H K 0
a similar lengthening for rHF obtained by Karpfen and Schuster [12] in the case of H-bonding formation in (HF),, and is in agreement with a similar value for roH in liquid water and ice that is well established [58] and discussed recently by Newton [59] and Watts [60]. Moreover, we also demonstrate that the resulting potential energy field U(r) is varied drastically compared with the reference potential energy field Uo(r) for a single proton displacement. In particular, the minimum of U(r) is lowered, compared with that of Uo(r), by about 13.3 kcal/mol, which is just approxi- mately the energy of H bonding of ice and water [3]. Summarizing all the results obtained, one can conclude that the red shift of v l under the H-bonding formation is the most striking and stringent peculiarity and manifestation of H-bonding formation in ice and water, and probably in other H-bonded complexes, that lies in a complete accordance with the similar statement by Pimentel and McClellan [l].
The “continuum approach,” exploited in the present paper attempts to provide explicit expressions for the classic fields, describing the collective and coherent in- phase and out-of-phase stretching vibrations of the infinite one-dimensional chain of water molecules, and the quite reasonable values for red shifts of the fundamental modes, v 1 and v3, and, moreover, justifies, at least within the framework of the zeroth-order perturbation treatment with respect to ~ 1 - ~ 3 coupling, the study of prop- erties of in-phase and out-of-phase stretching vibrations in a separate Fanner, i.e., as occurring in the uncoupled proton potential energy fields, U(r) and U(r). In conclu- sion, to be correct, the following question arises: What magnitude of the shift Av, is provided by the H-bonding formation in a system of two OH oscillators? The answer is obtained fairly easily: Avl accounts for approximately -64 cm-’, which is consis- tent with the data presented in Table I1 for a water dimer.
Acknowledgments
The author is very indebted to Professor G. V. Yukhnevich, Drs. P. I. Holod, and Yu. I. Naberukhin for their help and valuable comments, and Professors A. S. Davy- dov, A.M. Kuznetsov, V. P. Smilga, N. D. Sokolov, J. Ulstrup, and R. 0. Watts, Drs. V. Ya. Antonchenko, L. I. Trakhtenberg, and V. A. Shirokov for their support and fruitful and useful discussions, and to the referees for their valuable comments.
Bibliography
[l] G . C. Pimentel and A. L. MacClellan, The Hydrogen Bond (Freeman, San Francisco, 1960). [2] P. Schuster, in Perspectives in Quantum Chemistry and Biochemistry, B. Pullman, Ed. (Wiley, New
[3] D. Eisenberg and W. Kauzmann, The Structure and Properties of Water (Oxford University Press,
[4] G . V. Yukhnevich, Infrared Spectroscopy of Water (Nauka, Moscow, 1973). [5] N. G . Bakhshiev, Spectroscopy of Intermolecular Interactions (Nauka, Moscow, 1972). [6] G . Zundel, Hydration and Intermolecular Interactions (Academic, New York, 1969). [7] D. HadZi and S . Bratos, in The Hydrogen Bond. Recent Developments in Theory and Experiments,
P. Schuster, G . Zundel, and C. Sandorfy, Eds. (North-Holland, Amsterdam, 1976), Vol. 11, p. 565. [8] C. Sandorfy, in Ref. 7, Vol. II, p. 613. [9] S. Bratos, Croat. Chem. Acta 15, 15 (1982).
York, 1977), Vol. 11, p. 2.
New York, 1969).

RED SHIFT OF OH STRETCHING 507
[ lo] N.D. Sokolov, Croat. Chem. Acta 15, 223 (1982); in Hydrogen Bonding. N. D. Sokolov, Ed.
[ I l l Y. Marechal, Chem. Phys. 79, 69 (1983). [12] A. Karpfen and P. Schuster, Chem. Phys. Lett. 44, 454 (1976). 113) Yu.Ya. Efimov and Yu. 1. Naberukhin, Mol. Phys. 30, 1621 (1975). (141 G. N. Robertson, in Vibrationul Spectroscopy of Molecular Liquids and Solids, S. Bratos and R. M.
[15] W. F. Murphy and H. J. Bernstein, J. Phys. Chem. 76, 1147 (1972). [I61 J. R. Schemer, M. K. Go, and S. Kint, J. Phys. Chem. 78, 1304 (1974). C. I. Ratcliffe and D. E.
[I71 L. A. Curtiss and J. A. Pople, J. Mol. Spectros. 55, 1521 (1970). (181 A. 1. Tursi and E. R. Nixon, J. Chem. Phys. 52, 1521 (1970). [ 191 V. Ya. Antonchenko, E. S. Kryachko, and S. A. Polesja, Preprint ITP-83-96R, Kiev, ITP, 1983. [20] F. H. Stillinger and C. W. David, J. Chem. Phys. 69, 1473 (1978); ibid. 73, 3384 (1980). [21] P. Bopp, W. Dietz, and K. Heinzinger, Z. Naturforsch. Ma, 1424 (1979). [22] A. Rahman, F. H. Stillinger, and F. Lemberg, J. Chem. Phys. 63, 5223 (1975). [23] G. Jancs6 and P. Bopp, 2. Naturforsch. %a, 206 (1983). [24] F. H. Stillinger and A. Rahman, J. Chem. Phys. 68, 666 (1978). [25] P. Bopp, G. Jancs6, and K. Heinzinger, Chem. Phys. Lett. 98, 129 (1983). [26] L. Fredin, B. Nelander, and G. Ribbergard, J. Chem. Phys. 66, 4065 (1977). [27] R. M. Bentwood, A. J. Barnes, and W. J. Orville-Thomas, J. Mol. Spectrosc. 84. 391 (1980). [28] R. H. Page, J. G. Frey, Y.-R. Shen, and Y. T. Lee, Chem. Phys. Lett. 106, 373 (1984). [29] G. Jancs6, P. Bopp, and K. Heinzinger, Chem. Phys. 85, 377 (1984). [30] B. A. Zilles and W. B. Person, J. Chem. Phys. 79, 66 (1983). S. Chin, T. A. Ford, and W. B. Person,
J. Mol. Struct. 1l3, 341 (1984). [31] A. D. Buckingharn, in Ref. 14, p. 1. [32] P. Schuster, in Intermolecular Interactions: From Diatomics to Biopolymers, B. Pullman, Ed.
[33] I. G. Kaplan, Theory of Molecular Interactions (Elsevier, Amsterdam, 1986). [34] J. R. Reimers and R. 0. Watts, Chem. Phys. Lett. 94, 222 (1983); Chem. Phys. 85, 83 (1984). [35] T. C. Sivakumar, D. Schuh, M.G. Sceats, and S. A. Rice, Chem. Phys. Lett. 48, 212 (1977). R.
McGraw, W. G. Madden, S. A. Rice, and M. G. Sceats, Chem. Phys. Lett. 48, 219 (1977). T. C. Sivakumar, S. A. Rice, and M. G. Sceats, J. Chem. Phys. 69, 3468 (1978).
[36] R. McGraw, W. G. Madden, M. S. Bergren, S. A. Rice, and M. G. Sceats, J. Chem. Phys. 69, 3468, 3483 (1978).
(371 W. G. Madden, M. S. Bergren, R. McGraw, S. A. Rice, and M. G. Sceats, J. Chem. Phys. 69, 3497 (1978). M.G. Sceats, M. Stavola, and S. A. Rice, J. Chem. Phys., 70, 3927 (1979); ibid. 71, 983 (1979); S. A. Rice, and M. G. Sceats, J. Phys. Chem. 85, 1108 (198 1).
[38] M. S. Bergren and S. A. Rice, J. Chem. Phys. 77, 583 (1982). A. C. Belch and S. A. Rice, J . Chem. Phys. 78, 4817 (1983). G. Nielson and S. A. Rice, J. Chem. Phys. 78, 4824 (1983); S. A. Rice, M. S. Bergren, A. C. Belch, and G. Nielson, J. Phys. Chem. 87, 4295 (1983).
(Nauka, Moscow, 1981).
Pick, Eds. (Plenum, New York, 1979). p. 101.
Irish, J. Phys. Chem. 86, 4897 (1982).
(Wiley, Chichester, 1978). p. 363.
[39] S. A. Rice, Top. Current Chem. 60, 109 (1975). [40] J .E. Bertie and E. Whalley, J. Chem. Phys. 40, 1637, 1646 (1964). M. J. Taylor and E. Whalley,
[41] H. S. Frank and W.-Y. Wen, Disc. Faraday SOC. 24, 133 (1957). [42] P. Schuster, A. Karpfen, and A. Beyer, in Molecular Interactions, W. J . Orville-Thomas and H.
[43] F. H. Stillinger, Science 209, 451 (1980). [44] P. Barnes, J. L. Finney, J. D. Nichols, and J. E. Quinn, Nature 282, 459 (1979). J. L. Finney, in Bio-
physics of Water, F. Franks, Ed. (Wiley, Chichester. 1982), p. 73. J. L. Finney and J. M. Goodflow, in Srructure and Dynamics: Nucleic Acids and Proteins. E. Clementi and R. H. Sharma, Eds. (Adenine, New York, 1983). p. 81.
J. Chem. Phys. 40, 1660 (1964).
Ratajczak, Eds. (Wiley, London, 1979), p. 1.
[45) J. E. Del Bene, J. Chem. Phys. 72, 3423 (1980).

508 KRYACHKO
[46] E. S . Campbell and M. Mezei, Mol. Phys. 41, 883 (1980). E. S. Campbell and D. Belford, Theor.
(471 H. Frijhlich, Int. J. Quantum Chem. 23, 1583 (1983). [48] T. Di Paolo, C. Bourderon, and C. Sandorfy, Can. J. Chem. 50, 3161, (1972). C. Sandorfy, Top.
[49] V. M. Chulanovski and S . Ya. Khaikin; Opt. Spectrosc. USSR 23, 709 (1976). S. Ya. Khaikin and
[50] L. C. Allen, J. Amer. Chem. SOC. 97, 6921 (1975). P. A. Kollman, Acc. Chem. Res. 10, 365 (1977). [51] A. S . Davydov, Solitons in Molecular Sysrem (D. Reidel, Amsterdam, 1985). [52] Z. Kecki, J. Sadlej, and A. J. Sadlej, J. Mol. Struct. 88, 71 (1982). J. A. Odutola and T. R. Dyke,
J. Chem. Phys. 72, 5052 (1980). P. W. Fowler, G. Riley, and W. T. Raynes, Mol. Phys. 42, 1463 (1981). P. W. Fowler and W. T. Raynes, Mol. Phys. 43, 65 (1981).
Chim. Acta 61, 295 (1982).
Current Chem. l20, 41 (1984).
V. M. Chulanovski, Opt. Spectrosc. USSR 20, 234 (1966).
(531 B. P. Sheljukhov and G. V. Yukhnevich, Opt. Spectrosc. USSR 41, 404 (1976). [54] Y. Bouteiller, M. Allavena, and J. M. Leclerq, J. Chem. Phys. 73, 2851 (1980). H.-J. Werner and
P. Rosmus, J. Chem. Phys. 73, 2319 (1980). D. Maillard and B. Silvi, Mol. Phys. 40, 933 (1980). J. M. Leclerq, M. Allavena, and Y. Bouteiller, J. Chem. Phys. 78, 4606 (1983).
[55] G. B. Whitham, Linear and Nonlinear Waves (Wiley, New York, 1974). [56] P. L. Bhatnagar, Nonlinear Waves in One-Dimensional Dispersive Systems (Clarendon, Oxford,
[57] P. A. Giguere, J. Chem. Educ. 56, 571 (1979). [58] E. Whalley, Mol. Phys. 28, 1105 (1974). [59] M. D. Newton, Acta Cryst. B39, 104 (1983). [60] R. 0. Watts (private communication). M. F. Mills, J. R. Reimers and R. 0. Watts (unpublished).
1979).
Received December 21, 1984 Accepted for publication February 24, 1986
Related Documents











