Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von Polyvinylaminen und Hybridmaterialien von der Fakultät für Naturwissenschaften der Technischen Universität Chemnitz genehmigte Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) vorgelegt von: Dipl.-Chem. Isabelle Roth geboren am: 06.04.1976 in: Plauen i. V. eingereicht am: 19.12.2006 Gutachter: Prof. Dr. Stefan Spange Prof. Dr. Klaus Banert Prof. Dr. André Laschewsky Tag der Verteidigung: 05.07.2007 http://archiv.tu-chemnitz.de/pub/2007/0164

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Nucleophile aromatische Substitution
als Werkzeug zur Funktionalisierung von
Polyvinylaminen und Hybridmaterialien
von der Fakultät für Naturwissenschaften der Technischen Universität Chemnitz
genehmigte Dissertation zur Erlangung des akademischen Grades
doctor rerum naturalium
(Dr. rer. nat.)
vorgelegt von: Dipl.-Chem. Isabelle Roth
geboren am: 06.04.1976 in: Plauen i. V.
eingereicht am: 19.12.2006
Gutachter: Prof. Dr. Stefan Spange
Prof. Dr. Klaus Banert
Prof. Dr. André Laschewsky
Tag der Verteidigung: 05.07.2007
http://archiv.tu-chemnitz.de/pub/2007/0164

y≤Ü Zu|wÉ uÇw Åx|Çx YtÅ|Ä|x

Bibliografische Beschreibung, Referat, Schlagwörter 3
Bibliografische Beschreibung
Roth, Isabelle
„Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylaminen und Hybridmaterialien“
206 Seiten, 84 Abbildungen, 37 Schemen, 27 Tabellen, 152 Literaturquellen
Technische Universität Chemnitz, Fakultät für Naturwissenschaften, Institut für
Chemie, Professur Polymerchemie (Prof. S. Spange), Dissertation.
Referat
Die nucleophile aromatische Substitution von ausgewählten aktivierten Fluor-
aromaten mit Polyvinylaminen in Wasser wurde untersucht, um neue chromophore
wasserlösliche Polyelektrolyte zu synthetisieren. Die Synthese solcher π-Elektronen-
push-pull-Systeme (Nitroanilinderivate) in einem Schritt ist eine elegante Möglichkeit
zur Einführung von chromophoren Gruppen direkt am Polymerrückgrat. Die
Löslichkeit der –NO2-gruppentragenden Fluorverbindungen in Wasser wurde durch
Komplexierung mittels 2,6-O-Dimethyl-β-Cyclodextrin vermittelt. Die chromophoren
Einheiten von funktionalisierten Polyvinylaminen weisen interessante optische
Eigenschaften sowohl in Abhängigkeit vom pH-Wert als auch von der Natur des
Lösungsmittels (Solvatochromie) auf. Der quantitative Anteil der chromophoren
Gruppen im Polyvinylamin wurde UV/vis-spektroskopisch und mit Hilfe der NMR-
Spektroskopie ermittelt. Durch konstante Reaktionsbedingungen bei der Synthese
der funktionalisierten Polyvinylamine konnten die Substitutionsgrade untereinander
verglichen und Hinweise zur Reaktivität der jeweiligen Fluoraromaten gegenüber
Polyvinylamin erhalten werden. Ein direkter Zusammenhang zwischen der Struktur
und der Reaktivität des jeweils eingesetzten Fluoraromaten bei der Substitutions-
reaktion mit Polyvinylamin wurde durch kinetische Untersuchungen mit Hilfe der
UV/vis-Spektroskopie nachweisbar. Weiterhin wurde die nucleophile aromatische
Substitution von aktivierten Fluoraromaten mit Polyvinylaminen an der Grenzfläche
Kieselgel/Wasser untersucht, um neue chromophore Hybridmaterialien zu erhalten.
Die Charakterisierung der neuen Materialien erfolgte mit Hilfe der Elementaranalyse,
der UV/vis-Spektroskopie, der Festkörper-NMR-Spektroskopie, der XPS, anhand von
BET-Untersuchungen und elektrokinetischer Messungen.
Schlagwörter
nucleophile aromatische Substitution, Polyvinylamin, Fluoraromat, Cyclodextrin,
π-Elektronen-push-pull-System, Nitroaniline, Kieselgel, Hybridmaterialien

Bearbeitungszeitraum und Danksagung 4
Die vorliegende Arbeit wurde von September 2001 bis Juli 2006 an der Professur
Polymerchemie (Institut für Chemie, Fakultät für Naturwissenschaften, Technische
Universität Chemnitz) angefertigt und im Rahmen des Forschungsprojektes
„Nucleophile aromatische Substitution mit Polyaminen in Wasser“ von der DFG
gefördert.
Mein ausdrücklicher Dank gilt Herrn Prof. S. Spange für seine stete Hilfs- und
Diskussionsbereitschaft bei der Bearbeitung dieses sehr interessanten, farbenfrohen
und inhaltlich abwechslungsreichen Themas.
Ich danke der DFG für die finanzielle Unterstützung bei der Anfertigung dieser Arbeit.
Bei der BASF AG, Ludwigshafen, möchte ich mich für die kostenneutral zur
Verfügung gestellten Polyvinylaminlösungen und bei der Wacker Chemie,
Burghausen, für die kostengünstig bereitgestellte Menge an Cyclodextrinen
bedanken.
Insbesondere danke ich Herrn Prof. K. Banert für das mir entgegengebrachte
Vertrauen und die damit verbundene Möglichkeit der selbständigen Messung von
Flüssig-NMR-Spektren an der Professur Organische Chemie (TU Chemnitz, Fakultät
für Naturwissenschaften, Institut für Chemie).
Herrn Prof. R. Holze (TU Chemnitz, Institut für Chemie, Professur Physikalische
Chemie, Arbeitsgruppe Elektrochemie) und seinem wissenschaftlichen Mitarbeiter
Herrn Dr. A. A. Jbarah sowie Herrn Dr. M. Friedrich (Friedrich-Schiller Universität
Jena, Chemisch-Geowissenschaftliche Fakultät, Institut für Anorganische und
Analytische Chemie) danke ich für die Aufnahme von ESR-Spektren.
Mein besonderer Dank gilt Frau Dr. C. Bellmann und Herrn Dr. F. Simon vom
Leibniz-Institut für Polymerforschung Dresden e. V. für die elektrokinetischen
Messungen und die XPS-Untersuchungen und der damit verbundenen steten
Hilfsbereitschaft bei der Interpretation erhaltener Messergebnisse.

Bearbeitungszeitraum und Danksagung 5
Für die elementaranalytischen Untersuchungen gilt mein Dank:
Frau J. Buschmann (TU Chemnitz, Fakultät für Naturwissenschaften, Institut für
Chemie, Professur Organische Chemie unter der Leitung von Prof. K. Banert),
Frau K. Muchina (Friedrich-Schiller Universität Jena, Chemisch-Geowissen-
schaftliche Fakultät, Institut für Organische und Makromolekulare Chemie,
Kompetenzzentrum Polysaccharidforschung unter der Leitung von Prof. T. Heinze)
und Frau R. Ziehn (Martin-Luther-Universität Halle-Wittenberg, Naturwissen-
schaftliche Fakultät II, Fachbereich Chemie, Institut für Organische Chemie,
AG Organische und bioorganische Chemie unter der Leitung von Prof. R. Csuk).
Für die BET-Untersuchungen bedanke ich mich bei Frau Dipl.-Ing. M. Berger
(TU Chemnitz, Institut für Chemie, Professur Technische Chemie unter der Leitung
von Prof. E. Klemm).
Für die elektronenmikroskopischen Aufnahmen danke ich Herrn Dr. S. Schulze und
seiner Mitarbeiterin Frau G. Baumann (TU Chemnitz, Fakultät für Naturwissen-
schaften, Institut für Physik, Professur Analytik an Festkörperoberflächen unter der
Leitung von Prof. M. Hietschold).
Meinen Kolleginnen Frau Dipl.-Chem. S. Anders und Frau Dipl.-Chem. K. Hofmann
möchte ich für die Unterstützung bei der Aufnahme von Flüssig-NMR-Spektren
danken.
Meinen Kollegen Herrn Dr. A. Seifert, Herrn Dipl.-Chem. A. Mehner und Herrn
Dipl.-Chem. R. Lungwitz danke ich für die Aufnahme von Festkörper-NMR-Spektren.
Ich möchte allen an dieser Arbeit direkt und indirekt beteiligten Mitarbeiterinnen und
Mitarbeitern der Professur Polymerchemie für die gute Zusammenarbeit und die
Unterstützung, die mir zuteil wurde, herzlich danken.

Inhaltsverzeichnis
6
Inhaltsverzeichnis
Abkürzungen und Symbole ............................ ..........................................................8
1 Einleitung und Zielsetzung...................... ...........................................................11
2 Theoretische Grundlagen......................... ..........................................................17
2.1 Polyelektrolyte ................................................................................................17
2.2 Polyvinylamin (PVAm) ....................................................................................20
2.3 Nucleophile aromatische Substitution von Arylhalogeniden ...........................24
2.4 Cyclodextrine..................................................................................................26
2.5 Solvatochrome Eigenschaften von Nitroanilinen ............................................32
3 Ergebnisse und Diskussion ....................... ........................................................37
3.1 Funktionalisierung von Polyvinylaminen in Wasser ........................................37
3.1.1 Syntheseplanung und Charakterisierungsmöglichkeiten ......................37
3.1.2 Nitrophenyl-funktionalisierte Polyvinylamine mittels..............................40
4-Fluornitrobenzen (4-FNB) ..................................................................40
2-Fluornitrobenzen (2-FNB) ..................................................................42
3.1.3 2-Nitro-4-aminobenzen-funktionalisierte Polyvinylamine mittels ...........44
4-Fluor-3-nitroanilin (4-FNA) .................................................................44
N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA).......................................48
3.1.4 Dinitrophenyl-funktionalisierte Polyvinylamine mittels...........................59
1-Fluor-2,4-dinitrobenzen (Sangers Reagenz)......................................59
1,5-Difluor-2,4-dinitrobenzen (DFDNB) .................................................61
3.1.5 2-Nitro-4-azobenzen-funktionalisiertes Polyvinylamin mittels ...............63
4-Fluor-3-nitroazobenzen (FNAzoB) .....................................................63
3.1.6 2,4´-Dinitrostilben-funktionalisiertes Polyvinylamin mittels....................65
4-Fluor-3,4´-dinitrostilben (FDNstilben) .................................................65
3.1.7 Abhängigkeit der UV/vis-Absorptionseigenschaften vom pH-Wert .......68
3.1.8 Substitutionsgrad funktionalisierter Polyvinylamine ..............................76
4-nitrophenyl-funktionalisiertes PVAm ..................................................77
2-nitrophenyl-funktionalisiertes PVAm ..................................................80
2-nitro-4-aminobenzen-funktionalisiertes PVAm ...................................84
2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PVAm ............87
2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PNMVAm.......89
2,4´-dinitrostilben-funktionalisiertes PVAm............................................91

Inhaltsverzeichnis
7
3.1.9 Kinetische Untersuchungen mittels UV/vis-Spektroskopie....................94
3.1.9.1 Meßprinzip und Auswertung.....................................................94
3.1.9.2 Funktionalisierung von Polyvinylamin (PVAm) mittels..............96
4-Fluornitrobenzen (4-FNB) .....................................................96
2-Fluornitrobenzen (2-FNB) .....................................................98
1-Fluor-2,4-dinitrofluorbenzen (Sangers Reagenz) ................100
1,5-Difluor-2,4-dinitrobenzen (DFDNB)..................................101
4-Fluor-3-nitroanilin (4-FNA) ..................................................103
N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA) .......................104
3.1.9.3 Einfluss des pH-Wertes..........................................................109
3.1.9.4 Einfluss des Molekulargewichtes von PVAm..........................114
3.1.9.5 Einfluss der Ionenstärke (Fremdsalzzugabe) .........................117
3.2 Polymermodifizierung von Kieselgel H (KGH) ..............................................120
3.2.1 Syntheseplanung und Charakterisierungsmöglichkeiten ....................120
3.2.2 UV/vis-spektroskopische Untersuchungen .........................................123
3.2.3 13C-CP-MAS-Festkörper-NMR-spektroskopische Untersuchungen....134
3.2.4 Röntgen-Photoelektronenspektroskopische Untersuchungen (XPS)..137
3.2.5 Elementaranalytische Untersuchungen ..............................................142
3.2.6 BET-(Brunauer-Emmett-Teller)-Untersuchungen................................145
3.2.7 Elektrokinetische Untersuchungen (Zetapotential)..............................149
4 Zusammenfassung und Ausblick .................... ................................................155
5 Experimenteller Teil ............................ ..............................................................162
5.1 Analysenmethoden und verwendete Geräte.................................................162
5.2 Eingesetzte Chemikalien ..............................................................................164
5.2.1 Polyvinylamine....................................................................................164
5.2.2 2,6-O-Dimethyl-β-Cyclodextrin (Cavasol® W7 M) ...............................168
5.2.3 Fluoraromaten.....................................................................................169
5.2.4 Kieselgel H (KGH)...............................................................................170
5.3 Funktionalisierung von Polyvinylaminen mittels Fluoraromaten ...................171
5.4 Kinetische Untersuchungen der Funktionalisierung von PVAm....................172
5.5 Polymermodifizierung von Kieselgel H (KGH) ..............................................175
5.6 Synthesevorschriften und Charakterisierungen ............................................176
6 Literaturverzeichnis ............................ ..............................................................195
7 Anhang.......................................... .....................................................................201

Abkürzungen und Symbole
8
Abkürzungen und Symbole
Abkürzung Bedeutung
Abb. Abbildung
a. u. arbitrary unit, willkürliche Einheit
BET Brunauer-Emmett-Teller
β-DMCD 2,6-O-Dimethyl-β-cyclodextrin
bzw. beziehungsweise
CDCl3 deuteriertes Chloroform
CD2Cl2 deuteriertes Methylenchlorid, Dichlormethan
CH2Cl2 Methylenchlorid, Dichlormethan
CP Cross Polarization, Kreuzpolarisation
DCE 1,2-Dichlorethan
DFDNB 1,5-Difluor-2,4-dinitrobenzen
DMF N,N-Dimethylformamid
4-DMFNA N,N-Dimethyl-4-fluor-3-nitroanilin
DMSO Dimethylsulfoxid
D2O Deuteriumoxid
EA Elementaranalyse
ESR Elektronen-Spin-Resonanz
EtOH Ethanol
FDNB 1-Fluor-2,4-dinitrobenzen, Sangers Reagenz
FDNStilben 4-Fluor-3,4´-dinitrostilben
4-FNA 4-Fluor-3-nitroanilin
FNAzoB 4-Fluor-3-nitroazobenzen
FNAzoB-2 N-(2´-Nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-4-fluor-3-
nitroanilin
2-FNB 2-Fluornitrobenzen
4-FNB 4-Fluornitrobenzen
Gl. Gleichung
HBA hydrogen bond acceptor
HBD hydrogen bond donor
HCl Salzsäure

Abkürzungen und Symbole
9
Abkürzung Bedeutung
HFiPrOH 1,1,1,3,3,3-Hexafluor-2-propanol
H2O destilliertes Wasser
IEP isoelektrischer Punkt
KCl Kaliumchlorid
KG Kieselgel
KGH Kieselgel H
LM Lösungsmittel
MAS Magic Angle Spinning
MeOH Methanol
NaOEt Natriumethanolat
NaOH Natriumhydroxid
NMR nuclear magnetic resonance, Kernresonanz
N-DNP-FNA N-(2´,4´-Dinitrophenyl)-4-fluor-3-nitroanilin
PNMVAm Poly-N-Methylvinylamin
ppm parts per million
PVAm Polyvinylamin
PVFA Polyvinylformamid
RSB Rotationsseitenbande
s. siehe
sog. so genannt
Tab. Tabelle
TCE 1,1,2,2-Tetrachlorethan
THF Tetrahydrofuran
TFE 1,1,2,2-Tetrafluorethanol
u. a. unter anderem
UV/vis Ultraviolett/visuell
VFA N-Vinylformamid
vgl. vergleiche
XPS Röntgen-Photoelektronen-Spektroskopie
z. B. zum Beispiel

Abkürzungen und Symbole
10
Symbol Bedeutung
A0 erreichte UV/vis-Endabsorption bei t = ∞
A1 Gesamtabsorptionsänderung zwischen t = 0 und t = ∞
α Kamlet-Taft-Lösungsmittelparameter für die Wasserstoffbrücken-
Donor (HBD) Fähigkeit des Lösungsmittels
β Kamlet-Taft-Lösungsmittelparameter für die Wasserstoffbrücken-
Akzeptor (HBA) Fähigkeit des Lösungsmittels
c Konzentration [mol·l-1]
d Schichtdicke der Küvette [cm]
EA Aktivierungsenergie [kJ·mol-1]
ε´ spezifischer Absorptionskoeffizient der chromophoren Einheit
des Polymeren [l·g-1·cm-1]
εModell molarer Absorptionskoeffizient der Modellverbindung
∆G‡ Freie Aktivierungsenthalpie [kJ·mol-1]
∆H‡ Aktivierungsenthalpie [kJ·mol-1]
h Plancksches Wirkungsquantum [6.63 · 10-34 J·s]
[η] Grenzviskositätszahl, Staudinger Index
k Geschwindigkeitskonstante pseudo-erster Ordnung [s-1]
kB Boltzmann-Konstante [1.38 · 10-23 J·K-1]
λ Wellenlänge [nm]
M Molekulargewicht [g·mol-1]
ṽ Wellenzahl [cm-1]
π* empirischer Lösungsmittelparameter nach Kamlet und Taft
für die Dipolarität/Polarisierbarkeit des Lösungsmittels
R Gaskonstante [8.3145 J·K-1·mol-1]
s Substitutionsgrad [mol%]
∆S‡ Aktivierungsentropie [J·K-1·mol-1]
t Reaktionszeit [s]
σ Hammett-Substituenten-Konstante
ζ Zetapotential [mV]

1 Einleitung und Zielsetzung
11
1 Einleitung und Zielsetzung
Polyvinylamin (PVAm) ist ein kommerziell erhältlicher, wasserlöslicher, schwacher
kationischer Polyelektrolyt. Die pro-ionischen Aminogruppen am Polymerrückgrat
können in Abhängigkeit vom pH-Wert als freie Basen oder protoniert vorliegen, so
dass die Ladungsdichte durch Änderung des pH-Wertes gezielt eingestellt werden
kann.1-7 Eine Nachfunktionalisierung von Polyvinylamin (PVAm) über
polymeranaloge Reaktion an der primären Aminogruppe einer PVAm-Einheit eröffnet
vielfältigste Derivatisierungsmöglichkeiten.8-10 So ist die nucleophile aromatische
Substitution von Fluoraromaten, die zusätzlich Elektronenakzeptor-Substituenten
tragen, durch Bildung von π-Elektronen-push-pull-Systemen eine elegante
Möglichkeit zur Einführung von chromophoren Gruppen direkt am Polymerrückgrat
von Polyvinylaminen.9,10
Donor-Akzeptor-substituierte π-Systeme in Form von chromophoren Einheiten von
Polyvinylaminen könnten interessante optische, elektronische und optoelektronische
Eigenschaften für materialwissenschaftliche Anwendungen aufweisen. Im
Zusammenhang mit den Phänomenen der nichtlinearen Optik (NLO) und der
Elektrolumineszenz haben π-Elektronen-push-pull-Systeme in den letzten Jahren
mehr und mehr an Aufmerksamkeit gewonnen. Der push-pull-Effekt hängt neben der
Stärke des Donors und des Akzeptors auch vom konjugierten π-System ab, an dem
die zwitterionische Resonanzstruktur mit para-chinoidem Charakter beteiligt ist.11
Polyvinylamine mit höheren Molekulargewichten sind nur in Wasser gut löslich, was
eine polymeranaloge Umsetzung mit Fluoraromaten schwierig gestaltet, da diese
sehr schlecht oder unlöslich in Wasser sind. Suhr hat 1964 durch kinetische
Untersuchungen der nucleophilen aromatischen Substitution von 4-Fluornitrobenzen
(4-FNB) mit Piperidin gezeigt, dass die Reaktionsgeschwindigkeit mit zunehmender
Polarität des verwendeten Lösungsmittels steigt.12 Die Geschwindigkeitskonstanten
bei der Umsetzung von 4-FNB mit Piperidin sind in Dimethylsulfoxid (DMSO) um
mehr als vier Zehnerpotenzen größer als in Ethanol.12 Polyvinylamine lösen sich
zwar teilweise auch in heißem DMSO und in Alkoholen, jedoch ist DMSO aufgrund
seiner Oxidationseigenschaften nicht geeignet.9,13

1 Einleitung und Zielsetzung
12
Bei der nucleophilen aromatischen Substitution von Fluoraromaten mit aliphatischen
Aminen in Alkoholen kann es zum Austausch des Fluorsubstituenten durch
Alkoxidgruppen vom Lösungsmittel kommen. Das zeigte u. a. Suhr 1966 am Beispiel
der Umsetzung von 4-Fluornitrobenzen mit aliphatischen primären Aminen in
Methanol und Bellamy et al. 2004 am Beispiel der Umsetzung von Pikrylchlorid mit
PVAm in einer Glykol/Methanol-Mischung.14,15 Wird auf hochreaktive Fluoraromaten
wie z. B. Fluoranil verzichtet, ist Wasser als polares Reaktionsmedium
unproblematisch, da es aufgrund seiner geringen Nucleophilie nicht mit dem
Fluoraromat reagiert.9,14 Diese Erkenntnis wird u. a. genutzt, um nucleophile
aromatische Substitutionsreaktionen in inversen Micellen zu studieren.16-18 Wasser
als Lösungsmittel bietet sich auch wegen der vergleichsweise niedrigen Kosten und
insbesondere wegen seiner physiologischen Unbedenklichkeit an.
In jüngster Zeit hat vor allem Ritter et al. gezeigt, dass freie radikalische
Polymerisationen auch in Wasser, über Cyclodextrine als Löslichkeitsvermittler
hydrophober Monomere, vonstatten gehen.19 Dieses Konzept der Einkapselung
hydrophober Gastmoleküle in Cyclodextrine, um diese Wirt-Gast-Komplexe in eine
wasserlösliche Form zu überführen, wurde in dieser Arbeit zur Komplexbildung von
ausgewählten Fluoraromaten übernommen. Durch die Bildung wasserlöslicher Wirt-
Gast-Komplexe kann auf organische Lösungsmittel verzichtet und die nucleophile
aromatische Substitution mit Polyvinylaminen kann in Wasser durchgeführt werden.
Eigene, orientierende Untersuchungen zur Funktionalisierung von PVAm mit Hilfe
cyclodextrinkomplexierter Fluoraromaten haben gezeigt, dass 4-Fluornitrobenzen,
2,4-Dinitrofluorbenzen und 1,5-Difluor-2,4-dinitrobenzen gut zur Funktionalisierung
von PVAm in Wasser geeignet sind. Weniger aktivierte Fluoraromaten wie
4-Fluorbenzophenon oder 4,4´-Difluorbenzophenon konnten in Wasser nicht mit
PVAm zur Reaktion gebracht werden.9,10 Um Fluoraromaten zur Funktionalisierung
von Polyvinylaminen in Wasser einsetzen zu können, müssen diese einerseits durch
Substituenten mit –M-Effekt und/oder –I-Effekt für die nucleophile aromatische
Substitution ausreichend aktiviert sein, da die Reaktionstemperatur beim Arbeiten
ohne Überdruck (z. B. Autoklav) vom Siedepunkt des Reaktionsmediums limitiert
wird. Andererseits sollte der Fluoraromat auch nicht zu reaktiv sein, um
Nebenreaktionen mit dem Lösungsmittel Wasser zu vermeiden.

1 Einleitung und Zielsetzung
13
Mit Zunahme der Anzahl elektronenziehender Gruppen wie z. B. –NO2-Gruppen wird
der Fluorsubstituent zur besseren Abgangsgruppe bei der nucleophilen aromatischen
Substitution mit aliphatischen Aminen im Vergleich zu Fluorbenzen.20-22 Daher sollten
sich nitrogruppensubstituierte Fluoraromaten für die polymeranaloge Reaktion mit
Polyvinylaminen gut eignen. Die so entstehenden nitrophenyl-funktionalisierten Poly-
vinylamine mit π-Elektronen-push-pull-Systemen als chromophore Polyvinylamin-
Einheiten lassen interessante optische Eigenschaften erhoffen.
So besitzen z. B. niedermolekulare ortho- und para-Nitroanilinderivate solvatochrome
Eigenschaften. In Abhängigkeit vom Lösungsmittel ändert sich die Lage des UV/vis-
Absorptionsmaximums. Daher haben Kamlet und Taft in den 1970´er Jahren u. a.
Nitroanilinderivate als Lösungsmittelindikatoren herangezogen. Sie unterscheiden bei
den Eigenschaften eines Lösungsmittels zwischen deren Dipolarität/Polarisierbarkeit,
der Fähigkeit des Lösungsmittels als Wasserstoffbrücken-Donor (HBD, hydrogen
bond donor) bzw. als Wasserstoffbrücken-Akzeptor (HBA, hydrogen bond acceptor)
mit dem Farbstoff in Wechselwirkung treten zu können. So konnten mit Hilfe von
Nitroanilinderivaten und anderer solvatochromer Farbstoffe Lösungsmittelskalen für
die sog. Kamlet-Taft-Parameter α (HBD Fähigkeit), β (HBA Fähigkeit) und π*
(Dipolarität/Polarisierbarkeit) von einer Vielzahl an Lösungsmitteln aufgestellt
werden.23-26 Da die solvatochromen Eigenschaften niedermolekularer Nitroanilin-
derivate vor allem von der Dipolarität/Polarisierbarkeit π* der verwendeten Lösungs-
mittel abhängen, werden sie oft auch als solvatochrome π*-Indikatoren bezeichnet
und sind nach wie vor Gegenstand aktueller Forschung.27-30
Auch könnten 2-nitro-1,4-diaminobenzen-funktionalisierte Polyvinylamine
interessante optische Eigenschaften der chromophoren Einheit (Phenylendiamin-
Derivat) in verschiedenen Lösungsmitteln zeigen. Corbett et al. und Griffiths et al.
untersuchten Nitrophenylendiamine und entsprechende N-Alkyl-Derivate und
zeigten, dass die Lage der UV/vis-Absorptionsmaxima von Nitrophenylendiamin-
Derivaten in der Reihenfolge 4-Nitro-1,3-phenylendiamin-Derivate (orange), 4-Nitro-
1,2-phenylendiamin-Derivate (rot) und 2-Nitro-1,4-phenylendiamin-Derivate (violett)
bathochrom verschoben ist.31-35 Die Einführung einer –NO2-Gruppe in 2-Position
setzt dabei die Sensibilität von 1,4-Phenylendiamin-Derivaten gegenüber
Luftsauerstoff herab und in Abhängigkeit des verwendeten Lösungsmittels zeigten
sich solvatochrome Eigenschaften.31,34,35

1 Einleitung und Zielsetzung
14
Die bereits durch Luftsauerstoff eintretende Oxidation von 1,4-Phenylendiamin und
N,N,N,N-Tetramethyl-1,4-phenylendiamin wurde bereits 1879 von Wurster entdeckt,
weshalb diese rot bzw. blau gefärbten Oxidationsprodukte seinen Namen tragen.36-38
Erst Michaelis konnte sicher sagen, dass die Farbigkeit von Wurster´s-Rot und
Wurster´s-Blau auf stabile Radikal-Kationen zurückzuführen ist.39,40 Er fand weiterhin,
dass bei 1,4-Phenylendiaminen eine Methylgruppe in ortho-Stellung zu einer
monomethylierten Aminogruppe zur Destabilisierung des Radikal-Kationen-
Charakters führt.40 So könnte die Funktionalisierung von Polyvinylaminen mit
4-Fluoranilinen, die in ortho-Stellung zum Fluor eine Nitrogruppe tragen, zu
chromophoren Einheiten am PVAm-Rückgrat führen, die wie ihre niedermolekularen
Analoga solvatochrome Eigenschaften besitzen könnten.35
In jüngster Zeit hat sich das Interesse an photoschaltbaren Verbindungen neben der
Entwicklung molekularer Schaltsysteme für die Datenspeicherung auch auf die
supramolekulare Chemie, Nanotechnologie und Optobioelektronik erweitert, um die
Kontrolle über Strukturbildungen und bestimmter Funktionen zu erhalten.41,42 Daher
könnten funktionalisierte Polyvinylamine mit Donor-Akzeptor-substituierten Stilben-
PVAm-Einheiten und Azobenzen-PVAm-Einheiten interessante Eigenschaften
aufweisen, die durch Photoisomerisierung (lichtinduzierte cis/trans-Isomerisierung)
als optische, reversible Schalter und für NLO-Anwendungen Verwendung finden
könnten.43-47
Ziel dieser Arbeit war die weitere Erschließung der nucleophilen aromatischen
Substitution in Wasser als Funktionalisierungsreaktion für wasserlösliche
Polyvinylamine und die Ermittlung grundlegender Struktur-Reaktivitäts-Beziehungen.
Dazu waren neben der Auswahl geeigneter Fluoraromaten als Funktionalisierungs-
reagenzien die Syntheseoptimierung, kinetische Untersuchungen und Struktur-
charakterisierungen bei der Entstehung neuer, chromophorer Polyvinylamine
vorgesehen. Es galt zu untersuchen, wie sich der Einbau der polaren aromatischen
Komponente auf die optischen Eigenschaften und auf das Löslichkeitsverhalten der
Polymere in Wasser und in anderen Lösungsmitteln auswirkt. Zur Bestimmung des
Anteils substituierter chromophorer Polyvinylamin-Einheiten (Substitutionsgrad)
wurde neben 1H-NMR-spektroskopischer Untersuchungen vor allem die UV/vis-
Spektroskopie durch Vergleich mit niedermolekularen Modellverbindungen
herangezogen. Diese, der chromophoren Polyvinylamin-Einheit in etwa strukturell
entsprechenden Modellverbindungen sollten synthetisiert und charakterisiert werden.

1 Einleitung und Zielsetzung
15
Die erhaltenen Substitutionsgrade an chromophoren Polyvinylamin-Einheiten und
kinetische Untersuchungen können dabei Aussagen zur Reaktivität von
Polyvinylaminen gegenüber aktivierten Fluoraromaten liefern.
Weiterhin sollte durch kinetische Messungen mit Hilfe der UV/vis-Spektroskopie
untersucht werden, in wie weit das Molekulargewicht von Polyvinylamin, der pH-Wert
und die Ionenstärke der wässrigen Reaktionslösung einen Einfluss bei der
nucleophilen aromatischen Substitution ausgewählter Fluoraromaten hat.
Nach der Auswahl geeigneter Fluoraromaten für die nucleophile aromatische
Substitution mit Polyvinylaminen sollte untersucht werden, ob es möglich ist, die
nucleophile aromatische Substitution auch an der Phasengrenze Feststoff/Wasser
durchzuführen, um eine Oberflächenmodifizierung von anorganischen oxidischen
Trägern zu erreichen. Die Synthese von Hybridmaterialien, die aus einem
anorganischen Kern und einer organischen Polymerhülle bestehen, eröffnen viele
Möglichkeiten in der Materialforschung und sind neben dem akademischen auch von
technischem Interesse.
Eine elegante Möglichkeit Polyelektrolyte auf feste Oberflächen zu fixieren, ist die
Adsorption des Polyelektrolyten aus der Lösung. Dabei spielen vor allem
elektrostatische Wechselwirkungen zwischen Substrat und dem Polymeren eine
wichtige Rolle. Hat die Oberfläche des Substrates eine dem Polyelektrolyten
entgegengesetzte Ladung, so wird die Adsorption des Polyelektrolyten begünstigt
sein. So kann die Adsorption von Polyelektrolyten z. B. zur Flockung kleinster
Partikel aus einer Lösung führen, was in der Papierherstellung, Abwasser- und
Trinkwasseraufbereitung ausgenutzt wird. Auch kann die Agglomeration durch
Polymermodifizierung von Partikeln verhindert werden, wenn sich die Partikel
aufgrund ihrer gleichsinnig geladenen Oberfläche untereinander abstoßen, was zur
Stabilisierung von Dispersionen ausgenutzt wird.1,48
Polyvinylamine eignen sich u. a. zum Aufbringen von Adsorptionsschichten auf
Kieselgelen, auf Metallen, auf Gläsern, auf Cellulose und auf ungeladene
Substrate.9,49-56 Der Vorteil des Einsatzes von Kieselgelen als Träger besteht neben
der thermischen und mechanischen Stabilität auch in der hohen Reinheit, den
einheitlichen Partikelgrößen, den engen Porengrößenverteilungen und den großen
spezifischen Oberflächen mit denen sie kommerziell erhältlich sind.48,57 Auch ist von
Vorteil, dass Kieselgele gegenüber den meisten aggressiven Medien beständig
sind.48

1 Einleitung und Zielsetzung
16
An der Oberfläche von Kieselgel-Partikeln befinden sich Siloxangruppen (Si-O-Si)
und Silanolgruppen (Si-OH).48 Silanolgruppen sind schwache Säuren, wodurch acide
Oberflächenzentren resultieren, die mit einem kationischen Polyelektrolyten wie z. B.
Polyvinylamin in Wechselwirkung treten können. Sind die Wechselwirkungen
zwischen Partikeloberfläche und Polymerem stark genug, können Polyelektrolyte
irreversibel an einer Kieselgeloberfläche fixiert werden. Trägt das Polymerrückgrat
von PVAm zusätzlich funktionelle Gruppen, so können bestimmte Eigenschaften des
Polymeren mit denen von Kieselgel im Hybridmaterial miteinander kombiniert
werden. Das könnte ein relativ einfacher Weg zu interessanten funktionellen
Hybridmaterialien auf PVAm-Basis sein.
Eigene Voruntersuchungen haben gezeigt, dass die nucleophile aromatische
Substitution von Fluoraromaten mit PVAm auch an der Phasengrenze
Feststoff/Wasser zur Oberflächenfunktionalisierung genutzt werden kann. Durch
Umsetzung von 4-Fluornitrobenzen, 2,4-Dinitrofluorbenzen und 1,5-Difluor-2,4-
dinitrobenzen – jeweils adsorbiert an Kieselgel 60 – mit wässriger PVAm-lösung
wurden farbige Kieselgele erhalten, wobei die chromophoren Polymere irreversibel
fixiert blieben.9
Ziel dieser Arbeit war die Herstellung PVAm-basierender, chromophorer
Hybridmaterialien durch Einsatz geeigneter Fluoraromaten für die nucleophile
aromatische Substitution mit PVAm an der Phasengrenze Kieselgel/Wasser. Die
Auswahl geeigneter Fluoraromaten sollte dabei nach Erkenntnissen der
Funktionalisierung von PVAm über die Löslichkeitsvermittlung mittels Cyclodextrin
erfolgen. Die strukturelle Charakterisierung der chromophoren PVAm-Einheiten des
Polymeren auf der Kieselgel-Oberfläche sollte mittels geeigneter spektroskopischer
Methoden wie der UV/vis-Spektroskopie, der 13C-CP-MAS-Festkörper-NMR-
Spektroskopie und der Röntgen-Photoelektronen-Spektroskopie (XPS) erfolgen. Die
Elementaranalyse, BET-Untersuchungen und elektrokinetische Messungen
(Zetapotential) sollten Hinweise zum Bedeckungsgrad und der Beladung an
funktionalisiertem PVAm auf Kieselgel geben.

2 Theoretische Grundlagen
17
2 Theoretische Grundlagen
2.1 Polyelektrolyte
Polyelektrolyte gehören zur Gruppe ionischer Polymere. Sie tragen ionische oder
dissoziierbare Gruppen als Bestandteil der Hauptkette oder seitenständig zu ihr. Die
Klassifizierung von ionischen Polymeren erfolgt je nach Art und Menge der
anionischen bzw. kationischen Gruppen wie sie in Schema 1 gezeigt ist.48
Schema 1: Klassifizierung ionischer Polymere nach Art und Menge an ionisch dissoziierbaren Gruppen.
Bei Polyelektrolyten ist die Anzahl der ionischen Gruppen pro Polymerkette so groß,
dass die Polymeren in der dissoziierbaren Form wasserlöslich sind. Polyelektrolyte
werden je nach Art der dissoziierbaren Gruppen in Polysäuren und Polybasen
unterteilt. Aus Polysäuren entstehen bei der Dissoziation unter Abspaltung von
Protonen Polyanionen. Als Polybasen werden jene Polyelektrolyte bezeichnet, die
als pro-ionische Gruppen solche enthalten, die u. a. in der Lage sind Protonen
aufzunehmen (z. B. durch Reaktion mit Säuren unter Salzbildung). Typische
Polybasen mit ketten- bzw. seitenständig dissoziierbaren Gruppen sind die Polymere
Polyvinylamin (PVAm), Polyethylenimin (PEI) und Polyvinylpyridin (PVPy), die zur
Gruppe der synthetischen Polelektrolyte gehören.48
hohe Anzahl ionischer Gruppen(eine pro Monomereinheit)
Polymere mit nur einer oder wenigen
ionischen Gruppen (Makroanionen, Makrokationen)
wenig polare Hauptkette und geringe Anzahl
ionischer Gruppen
definierte Anordnung von quartären Ammonium-
Gruppen innerhalb der Hauptkette
sowohl anionische als auch kationische Gruppen
Polyelektrolyte
Makroionen
Ionomere
Ionene
Polyampholyte
hohe Anzahl ionischer Gruppen(eine pro Monomereinheit)
Polymere mit nur einer oder wenigen
ionischen Gruppen (Makroanionen, Makrokationen)
wenig polare Hauptkette und geringe Anzahl
ionischer Gruppen
definierte Anordnung von quartären Ammonium-
Gruppen innerhalb der Hauptkette
sowohl anionische als auch kationische Gruppen
Polyelektrolyte
Makroionen
Ionomere
Ionene
Polyampholyte
Polyelektrolyte
Makroionen
Ionomere
Ionene
Polyampholyte

2 Theoretische Grundlagen
18
Nach ihrer Herkunft können Polyelektrolyte auch in Biopolymere wie Nucleinsäuren,
Polypeptide/Proteine und Alginsäuren sowie in chemisch modifizierte Biopolymere
wie Carboxymethylzellulose und Chitosan unterteilt werden.48
Es ist literaturbekannt, dass Polelektrolyte wie z. B. Polyvinylamin (PVAm) an feste
Oberflächen wie z. B. Kieselgel spontan adsorbieren.49-52 Die elektrostatische
Anziehung zwischen Polymermolekülen und einer entgegengesetzt geladenen Ober-
fläche wird u. a. dabei zum Aufbringen von Adsorptionsschichten ausgenutzt. Auch
kann eine Polymeradsorption an Metall- oder ungeladenen Festkörperoberflächen
erfolgen, wobei verschiedene thermodynamische Triebkräfte für die Adsorption
verantwortlich sind.53,54 Die Adsorption von Polymeren an einer festen Oberfläche
unterscheidet sich erheblich von der Adsorption kleiner Moleküle. Das Makromolekül
besitzt im adsorbierten Zustand noch Beweglichkeiten, so dass innerhalb der
Adsorptionsschicht Verformung und Faltung möglich ist. Daneben lässt die große
Atomanzahl des Makromoleküls mehrfache Bindungen an die Oberfläche zu.58
Bei der Adsorption von Polyelektrolyten an festen Oberflächen müssen daher
strukturelle und dynamische Aspekte berücksichtigt werden. Bei einem adsorbierten
Polymeren kann modellhaft zwischen drei Konformationen, die die adsorbierende
Polymerkette einnehmen kann, unterschieden werden. Die Segmente, die die
Oberfläche direkt berühren, werden dabei Zug (train) genannt. Teile die durch einen
Zug fixiert sind, aber ansonsten in die Flüssigkeit ragen, werden als Schwanz (tail)
bezeichnet. Segmente, die in die Flüssigkeit ragen, jedoch auf beiden Seiten von
Zügen begrenzt werden, werden Schlaufen (loops) genannt (s. Schema 2).1,59-61
Schema 2: Schematische Darstellung der Anordnung einer Polymerkette adsorbiert auf einer Substratoberfläche.
Substrat
loop
train
tail

2 Theoretische Grundlagen
19
Neben der Ladungsdichte des Polyelektrolyten spielen die Oberflächenladung des
Substrates und die Elektrolytkonzentration in der Lösung bei der Adsorption von
Polyelektrolyten auf festen Oberflächen eine wesentliche Rolle. Bei hohem
Elektrolytgehalt in der Lösung (Fremdsalzzugabe) und geringer Ladungsdichte
verhalten sich Polyelektrolyte weitgehend wie ungeladene Polymere.59,62 Ist die
Ionenstärke der Lösung gering, wird die Adsorption des Polymeren hauptsächlich
von den Ladungsverhältnissen zwischen Substrat und Polyelektrolyt beeinflusst.
Anhand von Schema 3 ist die Abhängigkeit der Polymerkonformation von der
Ladungsdichte am Beispiel der Adsorption eines kationischen Polyelektrolyten wie
z. B. PVAm an einer negativ geladenen Partikeloberfläche wie z. B. Kieselgel
dargestellt.62
Schema 3: Schematische Darstellung der Polymerkonformation am Beispiel der Adsorption an einer negativ geladenen Partikeloberfläche in Abhängigkeit von der kationischen Ladungsdichte des Polymeren.
Wie Schema 3 zeigt, ist bei Polymeren mit hohem Molekulargewicht und geringer
Ladungsdichte mit schwacher Adsorption von langen loops und tails der
Polymerkette zu rechnen. Mit zunehmender Ladungsdichte nimmt die Anzahl an
trains zu, während die Anzahl an loops und tails abnimmt. Für eine
Polymermodifizierung von Kieselgel mit kationischen Polyelektrolyten sollte die
Ladungsdichte des Polyelektrolyten im mittleren Bereich liegen, da hohe
Ladungsdichten zur Abstoßung der Polymerketten untereinander führen und damit
die Menge an adsorbiertem Polymeren (Polymerbeladung) wieder abnimmt.62
Nachfolgender Abschnitt 2.2 gibt einen kleinen Einblick der Möglichkeiten der
Herstellung, ausgewählter Eigenschaften und der Verwendung von Polyvinylamin.
+
+
++
++
+
+
+
+
++
++
++
++
+
+
+
+
++
++
++
++
++
++++
a) geringeLadungsdichte
b) mittlereLadungsdichte
c) hoheLadungsdichte

2 Theoretische Grundlagen
20
2.2 Polyvinylamin (PVAm)
Polyvinylamin (PVAm) ist ein wasserlösliches Polymer, bei dem die pro-ionischen
Aminogruppen wie in Schema 4 gezeigt in Abhängigkeit vom pH-Wert als freie
Basen oder protoniert vorliegen können. PVAm ist ein schwacher, kationischer
Polyelektrolyt, bei dem Ammoniumionen (–NH3⊕) als ionische Einheiten fungieren.
Die Gegenionen sind oft niedermolekular (z. B. Chlorid-Ionen).48
Schema 4: Schematische Darstellung des Säure-Base-Gleichgewichts von Polyvinylamin (PVAm) bei Variation des pH-Wertes durch z. B. Salzsäure.
Eine direkte Homopolymerisation von Vinylamin als Basismonomer zu PVAm ist nicht
möglich, da das tautomere Gleichgewicht weit auf der Seite von Ethylidenamin liegt
(siehe Schema 5).48
Schema 5: Schematische Darstellung des tautomeren Gleichgewichts zwischen Vinylamin und Ethylidenamin.
Schon in den 1940er Jahren wurde PVAm über die Polymerisation von
N-Vinylsuccinimid (s. Schema 6a) oder N-Vinylphtalimid (s. Schema 6b) mit
anschließender Hydrolyse des entstandenen Polymeren synthetisiert.63-65 Spätere
Synthesewege beinhalteten den von Polyacrylamid (s. Schema 6c) ausgehenden
Hofmann-Abbau.66 In den 1980er Jahren wurde PVAm über die Polymerisation von
N-Vinylamiden wie N-Vinylacetamid (s. Schema 6d) und anschließender Hydrolyse
des Polymeren synthetisiert.67-70
NH2x
HCl
yNH3
zNH2
Cl
C CH
NH2
H
HC C
H
HNH
H
H
Ethylidenamin

2 Theoretische Grundlagen
21
Schema 6: Schematische Darstellung von Synthesemöglichkeiten von PVAm über Hydrolyse von Poly-N-vinylsuccinimid (a) und Poly-N-vinylphtalimid (b), durch Hofmann-Abbau von Polyacrylamid (c) sowie über Hydrolyse von Poly-N-vinylacetamid (d).
Ein großer Nachteil der Hydrolysereaktionen von Poly-N-vinylamiden ist, dass
drastische Reaktionsbedingungen erforderlich sind. So muss im Sauren in
überschüssiger konzentrierter Salzsäure und im Basischen bei hoher Temperatur im
Autoklaven gearbeitet werden.67-70
Eine weitere Möglichkeit der Synthese von PVAm ist die Polymerisation von
N-Carbamat geschützten Vinylaminen wie N-Vinyl-t-butylcarbamat71,72 oder
N-Vinyl-benzylcarbamat73 mit anschließender Hydrolyse der entstandenen
Polymeren. Die Carbamat-Route zur Synthese von PVAm hat sich jedoch aufgrund
hoher Monomerkosten nicht durchgesetzt.
Durch die seit 1999 kommerzielle Verfügbarkeit von Vinylformamid (VFA) hat sich die
Herstellung von PVAm über saure oder basische Hydrolyse des wasserlöslichen
Polyvinylformamid (PVFA) als besonders günstig erwiesen. VFA wird durch
radikalische Polymerisation mit Hilfe geeigneter Initiatoren erhalten. Öl- und
wasserlösliche Azoverbindungen eignen sich als Starter am besten. Die
Formamidgruppe von PVFA kann unter moderaten Bedingungen sowohl sauer bis zu
80% als auch basisch quantitativ hydrolisiert werden, wobei die Ladungsdichte am
Polymerrückgrat durch die Wahl der Hydrolysebedingungen (Temperatur,
Reaktionszeit, Menge an Säure oder Base) beliebig eingestellt werden kann.2-7
NH2
n
N OO
nN OO
nC
O NH2
nNH
CO
CH3
n
a b c d
Hydrolyse HydrolyseHydrolyse Hofmann-Abbau

2 Theoretische Grundlagen
22
Die Möglichkeit der Teilverseifung zur gezielten Einstellung des Hydrolysegrades von
Polyvinylformamid (PVFA) ist in Schema 7 am Beispiel der basischen Hydrolyse zu
Poly(vinylformamid-co-vinylamin) gezeigt. Die abgespaltene Formylgruppe liegt dabei
als Natriumformiat im Reaktionssystem vor und kann über Dialyse entfernt werden.2
Der Einsatz mindestens äquimolarer Menge an Base bei der basischen Hydrolyse
von PVFA führt zu PVAm, was vollständig hydrolysiertem PVFA entspricht.
Schema 7: Schematische Darstellung der teilweisen Hydrolyse von Polyvinylformamid (PVFA) am Beispiel der basischen Hydrolyse mittels Natriumhydroxidlösung zu Poly(vinylformamid-co-vinylamin).
PVAm ist ein schwacher Polyelektrolyt, da der Dissoziationsgrad der ionischen
Gruppen und damit die Ladungsdichte entlang der Polymerkette eine Funktion des
pH-Wertes ist. PVAm weist eine sehr hohe kationische Ladungsdichte mit
~18 meq/g bei pH = 2 auf, was bei einem Molekulargewicht von Vinylamin mit
43 g·mol-1 einem Protonierungsgrad von etwa 78 % entspricht. Die Ladungsdichte
nimmt mit ~12.8 meq/g bei pH = 7 erwartungsgemäß mit steigendem pH-Wert ab,
wobei etwa 50 % der Aminogruppen protoniert vorliegen. Im Gegensatz zu anderen
Polymeren besitzt PVAm jedoch auch bei hohen pH-Werten (pH = 9 – 10) signifikant
hohe Ladungsdichten von ~6 meq/g, was einem Protonierungsgrad von etwa 25 %
entspricht.6,7
Die Änderung der Ladungsdichte in Abhängigkeit vom pH-Wert führt zu Änderungen
der Konformation der Polymerketten. Im stark Basischen liegen vor allem
undissoziierte Gruppen und damit hauptsächlich Polymerknäuel vor. Durch
Absenkung des pH-Wertes kommt es zu Knäuelaufweitungen, da die
Abstoßungskräfte gleichsinniger Ladungen zunehmen. Im stark Sauren liegen -
aufgrund der hohen Ladungsdichte - hauptsächlich lang gestreckte Polymerketten
vor.74,75
NH
CHO
x yNH2
zNH
CHO
NaOH / H2O
- HCOONa

2 Theoretische Grundlagen
23
Die vielseitig reaktionsfähigen Aminogruppen im PVAm bieten die Möglichkeit
polymeranalog mit strukturell verschiedenen Elektrophilen zu reagieren. Im
folgenden Schema 8 sind wichtige Derivatisierungsmöglichkeiten von PVAm
aufgeführt.8
Schema 8: Schematische Darstellung möglicher polymeranaloger Reaktionen von PVAm.
Auf Vinylformamid basierende Polymere erlauben die Synthese maßgeschneiderter
Polymere mit vielfältigsten Einsatzmöglichkeiten. So findet PVAm
Anwendungsmöglichkeiten z. B. in der Papierindustrie zur Verbesserung der
Bedruckbarkeit, Fixierung und Retention76-78 bei der Abwasserklärung als
Flockungsmittel79, bei der Erdölförderung80-82; als Schutzkolloid für
Polymerdispersionen83,84 und als Superabsorber85-88. Sie eignen sich aufgrund ihrer
hohen Ladungsdichte auch bei höheren pH-Werten zur Oberflächenmodifizierung
wie z. B. zur Hydrophilierung, zur Kationisierung, zum Korrosionsschutz sowie zur
Kompatibilitätsverbesserung.89,90
Eine weitere in dieser Arbeit genutzte, wichtige Funktionalisierungsmöglichkeit von
PVAm (in Schema 8 nicht gezeigt) ist die nucleophile aromatische Substitution von
Fluoraromaten, auf die im nächsten Abschnitt 2.3 kurz eingegangen wird.
RNCO
RHCO
Anhydrid
NH2
NHRHN
O
NHHN
R
Aminal
Harnstoff-Derivat
KationischesSalzOO
NH
OH
O
NHR
O Amid
M
Carbonat-salz
Metallionen-Komplex
H X
EZGN
H
OR
NH2
M
NH3X
CO32
NH3
NH3
H
NEZG EZG
EZG = Elektronen-ziehende Gruppe
Aminoplast mit Vernetzer XR
O
HN
Om
m = 1,2X = NH; O
HNOH
R
OR
PVAm
CO2

2 Theoretische Grundlagen
24
2.3 Nucleophile aromatische Substitution von Arylh alogeniden
Unter nucleophiler aromatischer Substitution werden im allgemeinen Reaktionen
verstanden, bei denen das am Aromaten angreifende, elektronenreiche Nucleophil
unter Einbeziehung eines Elektronenpaares eine neue Bindung zum Aromaten
knüpft, wobei ein anderer am aromatischen Ring befindlicher Substituent mit einem
Elektronenpaar das Substrat verlässt.
Dieser Austausch eines Substituenten kann entweder nach einem Additions-
Eliminierungs-Mechanismus oder einem Eliminierungs-Additions-Mechanismus
(„Arin-Mechanismus“) erfolgen.48 Viele nucleophile aromatische Substitutionen folgen
dem Additions-Eliminierungs-Mechanismus wie er in Schema 9 am Beispiel der
nucleophilen aromatischen Substitution von Sangers Reagenz und PVAm gezeigt
und im Rahmen der in dieser Arbeit untersuchten Substitutionsreaktionen zu
erwarten ist.20-22,91
Schema 9: Schematische Darstellung der nucleophilen aromatischen Substitution von Arylhalogeniden am Beispiel von Sangers Reagenz mit aliphatischen Aminen am Bsp. von PVAm nach bimolekularem Mechanismus (Addition-Eliminierung).
Das gebildete Carbanion als reaktive Zwischenstufe kann durch elektronenziehende
Gruppen wie R= –NO2, –CN, –X, die zur Delokalisierung der negativen Ladung
beitragen, stabilisiert werden. Dem entsprechend wird auch der Übergangszustand,
in dem die negative Ladung des Ringes generiert wird, durch Elektronenakzeptoren
stabilisiert. Aber auch die Natur des Halogens als austretende Gruppe ist von
Bedeutung, wobei dem Fluorsubstituenten eine auffallend hohe Reaktivität zukommt.
So zeigte u. a. Suhr 1964 durch kinetische Untersuchungen mittels UV/vis-
Spektroskopie, dass bei der nucleophilen aromatischen Substitution von
4-Nitroarylhalogeniden mit Piperidin in DMSO die Reaktionsgeschwindigkeit des
fluorsubstituierten Aromaten deutlich höher im Vergleich zu den entsprechenden
chlor- und jodsubstituierten Aromaten ist.12,92
k-1 k-2
R: Alkylrest der PVAm-Kette
O2N
NO2
F + + HFH2N-R O2N
NO2
NHRk1 k2
O2N
NO2
F
NH2R

2 Theoretische Grundlagen
25
Die höhere Reaktionsgeschwindigkeit bei der Umsetzung von Fluoraromaten wird
u. a. dem starken induktiven Effekt des Fluors zugeschrieben, da der elektrophile
Charakter des Kohlenstoffs am größten ist und die negative Ladung der
Zwischenstufe vom Fluorsubstituenten im induktiven Effekt am besten übernommen
werden kann. Auch tragen die Solvatationsenergien der gebildeten Fluoridionen zur
Gesamtenergie bei.20-22 Der Reaktion des gewünschten Nitroanilinderivates zurück
zur Zwischenstufe mit einer Reaktionsgeschwindigkeit k-2 (s. Schema 9) kommt
aufgrund der geringen Nucleophilie der Fluoridionen nur eine geringe Bedeutung zu.
Die Geschwindigkeit der Rückreaktion zur Ausgangsverbindung (k-1) wird durch
Substituenten, die am fluorsubstituierten Kohlenstoff der Zwischenstufe eine
Elektronenverarmung erzeugen (Stärkung der Kohlenstoff-Base-Bindung)
erniedrigt.12
Es ist bekannt, dass mit Zunahme der Anzahl elektronenziehender Gruppen wie z. B.
–NO2-Gruppen der Fluorsubstituent zur besseren Abgangsgruppe bei der
nucleophilen aromatischen Substitution mit aliphatischen Aminen im Vergleich zu
unsubstituiertem Fluorbenzen wird. Werden besser aktivierte Fluoraromaten für die
nucleophile aromatische Substitution eingesetzt, nimmt vor allem die Reaktions-
geschwindigkeit k1 des ersten Reaktionsschrittes (Anlagerung des Amins) zu. Die
Gesamtgeschwindigkeit wird dann jedoch nicht mehr vorwiegend durch die
Reaktionsgeschwindigkeit k1 (s. Schema 9), sondern auch von k2 der Zerfallsreaktion
der zwitterionischen Zwischenverbindungen unter Abspaltung von Fluoridionen
mitbestimmt. Wenn der erste Schritt der Gesamtreaktion der nucleophilen
aromatischen Substitution der geschwindigkeitsbestimmende ist, so sollte ein
Zusammenhang zwischen den beobachteten Reaktionsgeschwindigkeiten und der
Elektronenverteilung zu erwarten sein.12,20-22
Eigene Untersuchungen haben gezeigt, dass sehr stark aktivierte Fluoraromaten wie
Fluoranil bereits mit dem Lösungsmittel Wasser reagieren können.9 Um
Fluoraromaten zur Funktionalisierung von Polyvinylaminen in Wasser einsetzen zu
können, müssen diese einerseits durch Substituenten mit –M-Effekt und/oder
–I-Effekt für die nucleophile aromatische Substitution ausreichend aktiviert sein,
andererseits sollte die Aktivierung für einen nucleophilen Angriff auch nicht zu groß
sein, da sonst Nebenreaktionen mit dem Lösungsmittel zu erwarten sind.

2 Theoretische Grundlagen
26
Kinetische Untersuchungen der nucleophilen aromatischen Substitution von
2-Fluornitrobenzen (2-FNB) bzw. 4-Fluornitrobenzen (4-FNB) mit Natrium-Methanolat
in Methanol haben gezeigt, dass der ortho-Verbindung eine auffallend hohe
Reaktivität zukommt. So ist z. B. die Reaktionsgeschwindigkeit bei der Umsetzung
von Natrium-Methanolat mit 2-FNB fast dreimal höher im Vergleich zur Umsetzung
mit 4-FNB.20 Aufgrund der auffallend hohen Reaktivität von ortho-Nitroverbindungen
gegenüber entsprechender para-Nitroverbindungen wurden vor allem ortho-nitro-
substituierte Fluoraromaten für die nucleophile aromatische Substitution mit
Polyvinylaminen ausgewählt.20,91,93-95
Da die in dieser Arbeit eingesetzten aktivierten Fluoraromaten nicht oder nur
schwerlöslich in Wasser sind, müssen diese Verbindungen erst in eine
wasserlösliche Form überführt werden. Die in den letzten Jahren intensiv studierte
Einkapselung von hydrophoben Gastmolekülen in Cyclodextrinmoleküle ist dabei
eine elegante Möglichkeit wasserlösliche sog. Wirt-Gast-Komplexe zu erhalten.
Einen kurzen Einblick zu den Eigenschaften und Wirkprinzipien von Cyclodextrinen
soll nachfolgender Abschnitt 2.4 liefern.
2.4 Cyclodextrine
Cyclodextrine wurden bereits 1891 von Villiers et al. als Abbauprodukt von Stärke
isoliert.96 Im Jahre 1903 charakterisierte Schardinger sie als cyclische
Oligosaccharide, weshalb Cyclodextrine vor allem in älterer Literatur nach ihrem
Entdecker auch Schardinger-Dextrine genannt werden.97 Cyclodextrine werden auch
nach dem Stärkebestandteil Amylose Cycloamylosen, nach der Sammelbezeichnung
Glucane - für Polymere der Glucose - Cycloglucane genannt.48 Freudenberg et al.
beschrieb 1938 den Aufbau der Cyclodextrine aus 1,4-α-glycosidisch verbundenen
Glucoseeinheiten.98 Vor allem Cramer et al. untersuchte in den 1950´er Jahren die
Fähigkeit der Cyclodextrine zur Bildung von Einschlussverbindungen ohne
Ausbildung kovalenter chemischer Bindungen.99-102 Dies war zur damaligen Zeit eine
revolutionäre Idee, die auf die Ablehnung einiger prominenter Wissenschaftler traf.
Für lange Zeit galten Cyclodextrine eher als Kuriosität ohne technischen Nutzen. Seit
etwa 30 Jahren werden Cyclodextrine und ihre Anwendungen intensiv erforscht.

2 Theoretische Grundlagen
27
Der enzymatische Abbau von Stärke liefert ein Gemisch cyclischer und linearer
Maltooligosaccharide. Als Enzyme dienen verschiedene Cyclodextringlucosyl-
Transferasen (CGTasen) bakteriellen Ursprungs. Die bevorzugt gebildeten Ringe
enthalten 6, 7 oder 8 D-Glucoseeinheiten (α-, β-, γ-Cyclodextrin). Da die einzelnen
Cyclodextrine durch die CGTasen ineinander überführt werden können, wird durch
Zugabe selektiver Fällungsmittel nur das Cyclodextrinhomologe gebildet, welches
dem Reaktionsmedium laufend entzogen wird. Die einzelnen D-Glucoseeinheiten
sind wie bei den Stärkebestandteilen Amylopektin und Amylose 1,4-α-glycosidisch
miteinander verknüpft und in der Sesselkonformation (s. Schema 10) angeordnet.103
Schema 10: Schematische Darstellung der Strukturformeln von α-, β- und γ-Cyclodextrin.
Ein Cyclodextrinmolekül ähnelt einem hohlen Kegelstumpf (Torus), aus dessen
kleinerer Öffnung die primären OH-Gruppen (an C-6 gebunden) und aus dessen
größerer Öffnung die sekundären OH-Gruppen (an C-2 bzw. C-3 gebunden) ragen.
Aus sterischen Gründen ist die vollständige konformative Drehung eines
Glucosebausteines um die C-1-O-C-4´-Bindungen ausgeschlossen. Daher befinden
sich die Wasserstoffe H-3 und H-5 immer im Innenraum des Torus (Cavität) und die
Wasserstoffe H-1, H-2 und H-4 weisen nach außen.103
Schema 11 zeigt zur Veranschaulichung die molekulare und dreidimensionale
Anordnung am Beispiel von β-Cyclodextrin.
γ-Cyclodextrin β-Cyclodextrin α-Cyclodextrin

2 Theoretische Grundlagen
28
Schema 11: Schematische Darstellung der molekularen und dreidimensionalen Struktur von β-Cyclodextrin.
Während die Höhe des Kegelstumpfes bei α-, β- und γ-Cyclodextrin mit 7.9 − 8.0 Å
gleich groß ist, nimmt der Innendurchmesser der Cavität mit Zunahme der Anzahl
verknüpfter D-Glucoseeinheiten von α-Cyclodextrin zu γ-Cyclodextrin zu.103,104
Je nach Ringgröße binden Cyclodextrine im kristallinen Zustand 6 – 13 Gewichts%
Wasser. Kristallines β-Cyclodextrin z. B. enthält 12 Moleküle Kristallwasser pro
Makrocyclus. In Tab. 1 sind ausgewählte Eigenschaften von α-, β- und γ-Cyclodextrin
aufgeführt.104
Tabelle 1 : Ausgewählte Eigenschaften von α-, β- und γ-Cyclodextrin.
Cyclodextrin
Anzahl Glucose- einheiten
Molekular-gewicht
[g · mol-1]
Innenraum- Durchmesser
[Å]
Löslichkeit in Wasser (25 °C)
[g · l-1]
α-Cyclodextrin 6 972 4.7 − 5.2 145
β-Cyclodextrin 7 1135 6.0 − 6.4 18.5
γ-Cyclodextrin 8 1297 7.5 − 8.3 232
Die an C-2 gebundene OH-Gruppe der einen Glucoseeinheit eines Cyclodextrin-
moleküls kann mit einer an C-3 gebundenen OH-Gruppe einer benachbarten
Glucoseeinheit intramolekulare Wasserstoffbrückenbindungen ausbilden. Im
β-Cyclodextrin werden alle sechs möglichen Wasserstoffbrückenbindungen
ausgebildet, womit eine versteifte Struktur resultiert. Im Gegensatz dazu ist bei
α-Cyclodextrin eine Glucoseeinheit verdreht, wodurch nur vier intramolekulare
H-Brückenbindungen ausgebildet werden.105
oberer Rand: sekundäre OH-Gruppenunterer Rand: primäre OH-Gruppen
1,4-α-verknüpfte D-Glucose-Einheiten
C1HO
O
OH
C2HC4
C5
H
C6HOH
H
H
C3
O HHO
O
H
7
HO
HOOH
12
3
4 56
„hohler Kegelstumpf“
oberer Rand: sekundäre OH-Gruppenunterer Rand: primäre OH-Gruppen
1,4-α-verknüpfte D-Glucose-Einheiten
C1HO
O
OH
C2HC4
C5
H
C6HOH
H
H
C3
O HH
C1HO
O
OH
C2HC4
C5
H
C6HOH
H
H
C3
O HHO
O
H
7
HO
HOOH
12
3
4 56 O
O
H
7
HO
HOOH
O
O
H
7
HO
HOOH
12
3
4 56
„hohler Kegelstumpf“

2 Theoretische Grundlagen
29
Beim γ-Cyclodextrin wiederum sind aufgrund der erweiterten Ringgröße
(beweglichere Struktur) die intramolekularen Wasserstoffbrückenbindungen
geschwächt.105 Diese Unterschiede der Ausbildung intramolekularer Wasserstoff-
brückenbindungen bei α-, β- und γ-Cyclodextrin äußert sich u. a. in ihrer
Wasserlöslichkeit. So löst sich β-Cyclodextrin deutlich schlechter als γ-Cyclodextrin
und α-Cyclodextrin (s. Tab.1).104
Die besondere strukturelle Anordnung der 1,4-α-glycosidisch miteinander
verknüpften D-Glucoseeinheiten führt zu einem hydrophoben Innenraum des
Kegelstumpfes, während die Außenseite hydrophil ist. Auf diese Weise sind
Cyclodextrine leicht durch polare Lösungsmittel wie Wasser solvatisierbar und der
hydrophobe Innenraum gestattet über zwischenmolekulare Wechselwirkungen die
Einkapselung von hydrophoben Gastmolekülen (Wirt-Gast-Beziehung). Befindet sich
das Gastmolekül im Inneren der Cavität, werden die Addukte als Einschluss-
verbindungen bezeichnet, wobei der Gast vollständig oder partiell umschlossen sein
kann (s. Schema 12a+12b). Auch kann ein langes, dünnes Gastmolekül eine axiale
Einschlussverbindung und ein rundes, dickes Gastmolekül eine sandwichartige
Einschlussverbindung bilden (s. Schema 12c+12d). Befindet sich das Gastmolekül
außerhalb der Cavität, heißen die Addukte Anlagerungsverbindungen (Schema 12e).
Sind die Abmessungen des Gastmoleküls klein gegenüber der Cavität, können auch
mehrere Moleküle eingeschlossen sein (s. Schema 12f+12g).103
Schema 12: Topologie von Cyclodextrinaddukten: a) vollständiger, b) partieller, c) axialer, d) sandwichartiger Einschluss, e) deckelartige Anlagerungsverbindung f) 1:2-Einschlussverbindung und g) 2:2-Einschlussverbindung.
Für die Ausbildung einer 1:1-Einschlussverbindung muss das hydrophobe
Gastmolekül mit seiner Größe und Geometrie in die Cavität des
Cyclodextrinmoleküls passen. Die Stabilität der Wirt-Gast-Komplexe wird vom
Zusammenwirken der Wechselwirkungen der Gastmoleküle mit dem umgebenden
Cyclodextrin (hauptsächlich van-der-Waals-Kräfte und hydrophobe Wechsel-
wirkungen) bestimmt.104
e)b) c) d)a) f) g)e)e)b)b) c)c) d)d)a)a) f)f) g)g)

2 Theoretische Grundlagen
30
Aber auch Wasserstoffbrückenbindungen und sterische Effekte spielen für die
Komplexstabilität eine Rolle.104 Für eine Komplexbildung sind auch die im
hydrophoben Inneren des Cyclodextrins befindlichen Wassermoleküle verantwortlich.
Diese Wassermoleküle können, aufgrund energetisch ungünstigerer polar-apolarer
Wechselwirkungen, leicht durch weniger polare Gastmoleküle ausgetauscht
werden.106 Die freigewordenen Wassermoleküle tragen dann durch
Entropiezunahme − durch Aufnahme vom umgebenden Wasser − zur Stabilität der
Wirt-Gast-Komplexe bei.104 Für eine Komplexbildung in organischen Lösungsmitteln
ist daher zumindest eine geringe Menge an Wasser erforderlich.
In Lösung steht der gebildete Wirt-Gast-Komplex mit den nicht komplexierten
Gastmolekülen im thermodynamischen Gleichgewicht, wobei die Stabilität des Wirt-
Gast-Komplexes von der Lage dieses Gleichgewichtes abhängt. In Schema 13 ist
am Beispiel der Komplexierung von p-Xylol die Komplexbildung mit Cyclodextrin in
Wasser schematisch gezeigt, wobei das Gleichgewicht weit auf der Seite des Wirt-
Gast-Komplexes liegt.105,106
Schema 13: Schematische Darstellung der Komplexierung von p-Xylol mit Cyclodextrin.
Dabei spiegelt die Stabilitätskonstante die Lage des Gleichgewichts wider, die sich
bei einer 1:1-Einschlussverbindung nach dem Massenwirkungsgesetz aus dem
Quotienten der Konzentration des Cyclodextrin-Gast-Komplexes [CD-Gast] und dem
Produkt der Konzentrationen des Wirtes und des Gastes [CD] · [Gast] ergibt.105,106
CH3
CH3
+
CH3
CH3
CH3
CH3
CH3
CH3
+
CH3
CH3
CH3
CH3

2 Theoretische Grundlagen
31
Eine breite Palette von Anwendungsmöglichkeiten der Cyclodextrine ergibt sich
gerade aus ihrer Eigenschaft mit Gastmolekülen Wirt-Gast-Komplexe einzugehen.
Cyclodextrine werden generell als sicher bei oraler und nasaler Einnahme eingestuft.
Daher werden sie in der Nahrungsmittelindustrie u. a. zur Komplexierung von
Lebensmittelbestandteilen eingesetzt, um diese gegenüber Sauerstoff- und
Lichteinwirkung und vor thermischer Zersetzung zu schützen sowie Verluste durch
die Flüchtigkeit der Komponenten zu vermeiden. Zudem können Duft-, Aroma- und
Geschmacksstoffe maskiert (z. B. bei Knoblauchpillen) bzw. erhalten (z. B. bei
Kaugummis) werden. So kann z. B. bei Pflanzenölen die Autooxidation der
ungesättigten Fettsäuren mit Luftsauerstoff durch eine Komplexierung mit
Cyclodextrinen stark verzögert werden. Aus Cyclodextrinkomplexen mit Ölen,
Vitaminen und Aromastoffen können neben stabilen Komplexen zur Haltbarmachung
auch stabile Dispersionen in wässrigen Medien erhalten werden. Daher finden
Cyclodextrine auch in Kosmetika, vor allem in Hautcremes Verwendung.107,108
Im Pharmazeutischen Bereich haben Cyclodextrine inzwischen große Bedeutung
erlangt, da sich wasserunlösliche Wirkstoffe durch Komplexierung mit Cyclodextrinen
im Körper besser verteilen können. Auch im Haushalt finden Cyclodextrine bereits
Anwendung. So enthalten z. B. Waschmittel bestimmte Duftstoffe, die als
Cyclodextrin-Komplexe länger auf der Faser haften als die Parfümöle selbst. Auch
können auf Cyclodextrin basierende sog. Textilerfrischer zum Einsatz kommen
(Fébrèze®).107,108
Um die relativ schlechte Löslichkeit von vor allem β-Cyclodextrin in Wasser und in
organischen Lösungsmitteln zu verbessern, werden Cyclodextrine z. B. durch
O-Methylierung oder Hydroxypropylierung chemisch modifiziert. Das bedeutet, dass
bei einer Derivatisierung von β-Cyclodextrin neben sieben primären noch vierzehn
sekundäre OH-Gruppen verethert werden können.105,106 Soll als Zielprodukt z. B.
2,6-O-Dimethyl-β-cyclodextrin durch Modifizierung von β-Cyclodextrin synthetisiert
werden, müssen alle in C-3-Position befindlichen OH-Gruppen geschützt werden,
was synthetisch sehr aufwendig ist und die Produkte teuer macht. Da sich die in C-3-
Position befindlichen sekundären OH-Gruppen schlechter als die in C-2- und C-6-
Position befindlichen OH-Gruppen derivatisieren lassen, hat es sich in der groß-
technischen Anwendung durchgesetzt, auf die gezielte Modifikation zu verzichten.106

2 Theoretische Grundlagen
32
Beim statistisch methylierten β-Cyclodextrin liegen neben den primären C-6-OH-
Gruppen hauptsächlich die C-2-OH-Gruppen durch Methoxygruppen verethert vor.
Zu einem geringen Anteil sind auch die in C-2,C-3- oder C-3,C-6-Position
befindlichen OH-Gruppen verethert. So zeigt z. B. der vom Hersteller angegebene
Methylierungsgrad von Cavasol® W7 M (statistisch methyliertes β-Cyclodextrin) mit
1.6 – 1.9 pro Glucoseeinheit, dass auch Glucoseeinheiten existieren, die nicht (max.
0.2 %) oder nur monomethyliert vorliegen. Die statistische Methylierung hat den
Vorteil, dass - aufgrund der gestörten Ordnung von Wasserstoffbrückenbindungen -
die Löslichkeit in Wasser deutlich besser ist. Die Löslichkeit von Cavasol® W7 M in
Wasser ist mit 800 g · l-1 bei 25 °C deutlich größer im Vergleich zu 2,6- O-Dimethyl-
β-cyclodextrin (> 330 g · l-1) und β-Cyclodextrin (18.5 g · l-1).109,110
Aufgrund der besseren Wasserlöslichkeit wurde in der vorliegenden Arbeit - neben
der optimalen Größe der β-Cyclodextrin-Cavität und dem Preisvorteil gegenüber
reinem 2,6-O-Dimethyl-β-cyclodextrin - das statistisch methylierte Cyclodextrinderivat
Cavasol® W7 M eingesetzt.
2.5 Solvatochrome Eigenschaften von Nitroanilinen
Nitroaniline tragen mit der Nitrogruppe einen starken, elektronenziehenden
–M-Substituenten (Akzeptor) und mit der Aminogruppe einen starken,
elektronenliefernden +M-Substituenten (Donor) am Benzenring. Besteht die
Möglichkeit zur Resonanzstabilisierung führt dieser sog. push-pull-Effekt zu einer
charakteristischen Farbigkeit von Nitroanilin-Derivaten. Am Beispiel von 4-Nitroanilin
ist in Schema 14 die Resonanzstabilisierung über eine zwitterionische Struktur mit
p-chinoidem Charakter gezeigt.
Schema 14: Schematische Darstellung der Resonanzstabilisierung von 4-Nitroanilin (rechte Seite: Zwitterionische Resonanzstruktur mit p-chinoidem Charakter).
Resonanzstabilisierung über -NH 2- und NO 2-Gruppe
+M-Effektpush
-M-Effektpull
NH2NO
O
H2N NO
O

2 Theoretische Grundlagen
33
Die Anregung eines Elektrons aus dem freien Elektronenpaar des Stickstoffs (n→π*)
und die Konjugation von Doppelbindungen (π→π*) senkt die für Elektronen-
übergänge erforderlichen Energien bis hin in den sichtbaren Bereich. Die
langwelligste sichtbare Absorption steht in Verbindung mit einem intramolekularen
charge-transfer von der Aminogruppe (Donor) zur Nitrogruppe (Akzeptor).111
Es ist literaturbekannt, dass diese intramolekulare charge-transfer-Bande von
4-Nitroanilin-Derivaten mit Zunahme der Polarität des Lösungsmittels und mit
Zunahme der Elektronen-Donor-Stärke der Aminogruppe einer bathochromen
Verschiebung des UV/vis-Absorptionsmaximums (zu größerer Wellenlänge)
unterliegt.112,113
Die Verschiebung der Lage von UV/vis-Absorptionsbanden eines Chromophors in
Abhängigkeit der Natur des Lösungsmittels wird als Solvatochromie bezeichnet.
Positive Solvatochromie zeigen Chromophore wie z. B. 4-Nitroanilin, bei denen sich
das UV/vis-Absorptionsmaximum mit Zunahme der Lösungsmittelpolarität zu
größeren Wellenlängen verschiebt (bathochromer shift). Im Gegensatz dazu können
bestimmte solvatochrome Farbstoffe auch Verschiebungen des UV/vis-Absorptions-
maximums zu kleineren Wellenlängen (hypsochromer shift) mit Zunahme der
Lösungsmittelpolarität aufweisen, wobei dann von negativer Solvatochromie
gesprochen wird.111
Das Dipolmoment eines 4-Nitroanilin-Derivates ist im angeregten Zustand
(intramolekularer charge-transfer, chinoide Resonanzstruktur) größer als im
Grundzustand, so dass dieser von polaren Lösungsmitteln besser im Vergleich zum
Grundzustand stabilisiert wird, was in einer bathochromen Verschiebung des UV/vis-
Absorptionsmaximums resultiert.24,28-30,111
Das Dipolmoment von negativ solvatochromen Farbstoffen wiederum ist im
Grundzustand größer als im angeregten Zustand, so dass der dipolare Grundzustand
von polaren Lösungsmitteln besser stabilisiert wird. Dem entsprechend zeigen
negativ solvatochrome Farbstoffe eine stärkere Absenkung des Grundzustandes im
Vergleich zum angeregten Zustand mit Zunahme der Lösungsmittelpolarität, was
sich in einer hypsochromen Verschiebung der UV/vis-Absorptionsbande äußert.111
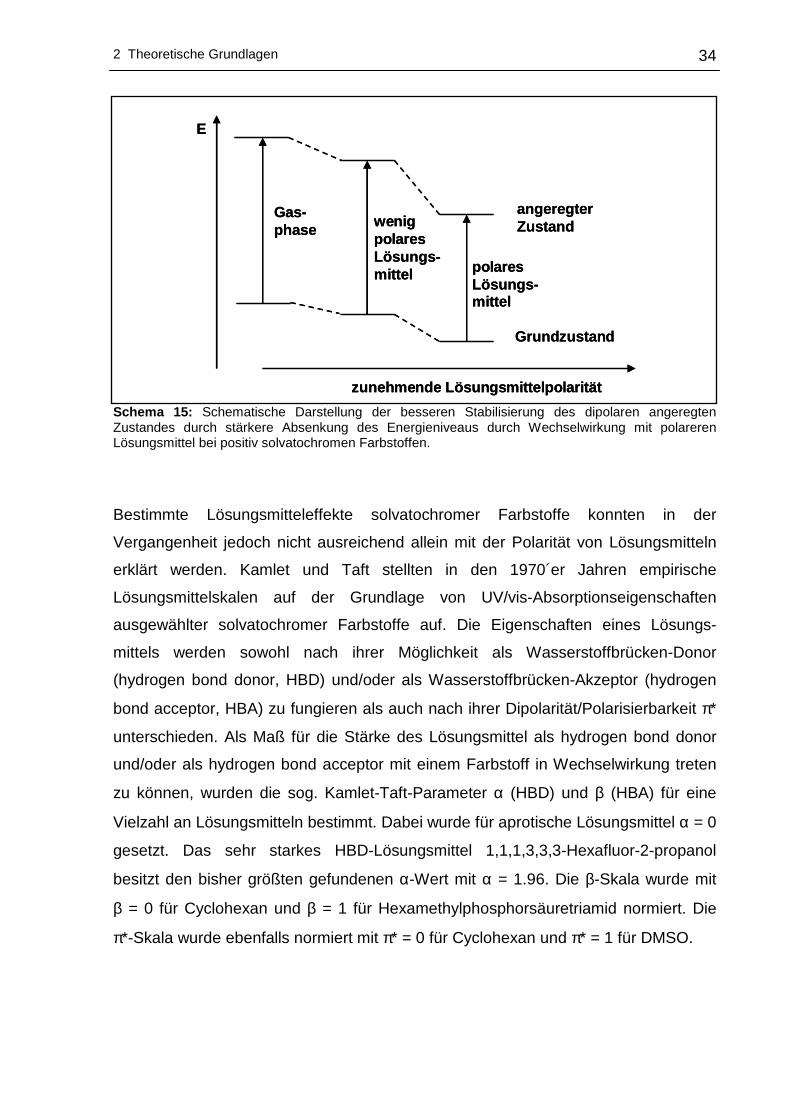
In Schema 15 ist gezeigt, wie der Grundzustand und der angeregte Zustand
(Energieniveaus für Elektronenübergänge) bei positiv solvatochromen Farbstoffen
bei der Solvatation durch Lösungsmittel unterschiedlicher Polarität unterschiedlich
gut stabilisiert werden.111
2 Theoretische Grundlagen
34
Schema 15: Schematische Darstellung der besseren Stabilisierung des dipolaren angeregten Zustandes durch stärkere Absenkung des Energieniveaus durch Wechselwirkung mit polareren Lösungsmittel bei positiv solvatochromen Farbstoffen.
Bestimmte Lösungsmitteleffekte solvatochromer Farbstoffe konnten in der
Vergangenheit jedoch nicht ausreichend allein mit der Polarität von Lösungsmitteln
erklärt werden. Kamlet und Taft stellten in den 1970´er Jahren empirische
Lösungsmittelskalen auf der Grundlage von UV/vis-Absorptionseigenschaften
ausgewählter solvatochromer Farbstoffe auf. Die Eigenschaften eines Lösungs-
mittels werden sowohl nach ihrer Möglichkeit als Wasserstoffbrücken-Donor
(hydrogen bond donor, HBD) und/oder als Wasserstoffbrücken-Akzeptor (hydrogen
bond acceptor, HBA) zu fungieren als auch nach ihrer Dipolarität/Polarisierbarkeit π*
unterschieden. Als Maß für die Stärke des Lösungsmittel als hydrogen bond donor
und/oder als hydrogen bond acceptor mit einem Farbstoff in Wechselwirkung treten
zu können, wurden die sog. Kamlet-Taft-Parameter α (HBD) und β (HBA) für eine
Vielzahl an Lösungsmitteln bestimmt. Dabei wurde für aprotische Lösungsmittel α = 0
gesetzt. Das sehr starkes HBD-Lösungsmittel 1,1,1,3,3,3-Hexafluor-2-propanol
besitzt den bisher größten gefundenen α-Wert mit α = 1.96. Die β-Skala wurde mit
β = 0 für Cyclohexan und β = 1 für Hexamethylphosphorsäuretriamid normiert. Die
π*-Skala wurde ebenfalls normiert mit π* = 0 für Cyclohexan und π* = 1 für DMSO.
E
zunehmende Lösungsmittelpolarität
wenigpolaresLösungs-mittel polares
Lösungs-mittel
Grundzustand
angeregterZustand
Gas-phase
E
zunehmende Lösungsmittelpolarität
wenigpolaresLösungs-mittel polares
Lösungs-mittel
Grundzustand
angeregterZustand
Gas-phase
2 Theoretische Grundlagen
35
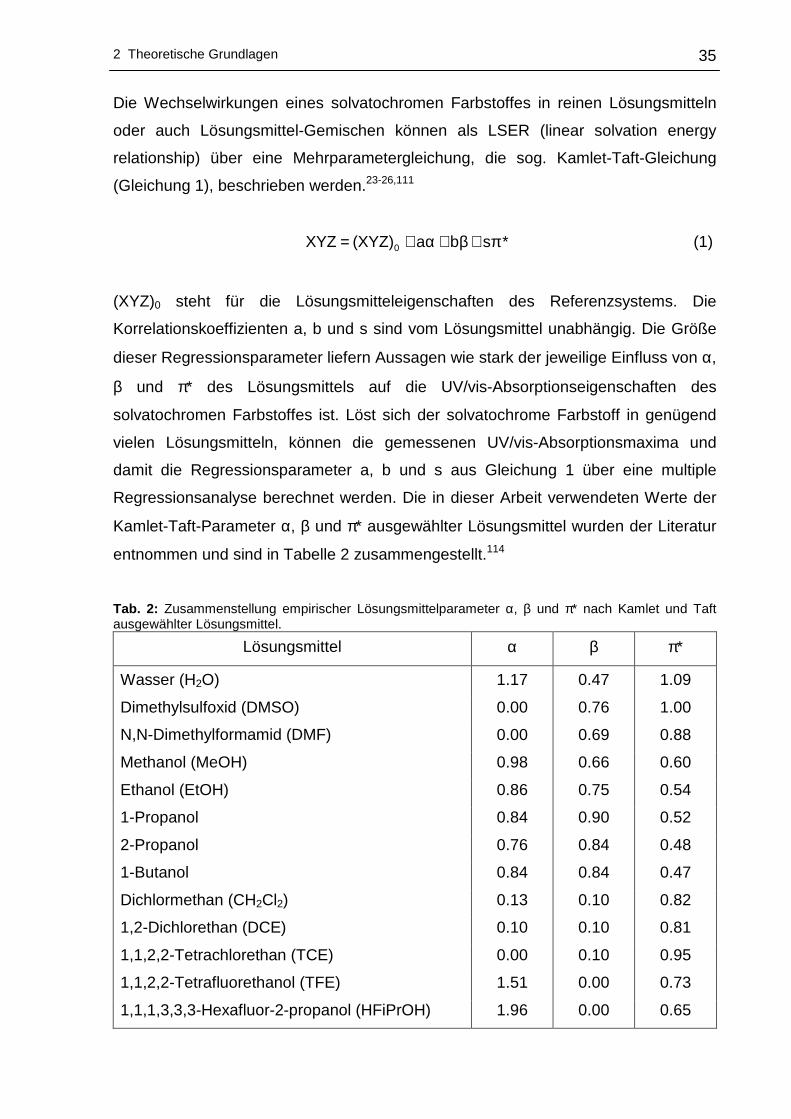
Die Wechselwirkungen eines solvatochromen Farbstoffes in reinen Lösungsmitteln
oder auch Lösungsmittel-Gemischen können als LSER (linear solvation energy
relationship) über eine Mehrparametergleichung, die sog. Kamlet-Taft-Gleichung
(Gleichung 1), beschrieben werden.23-26,111
*sba(XYZ)XYZ 0 π+β+α+= (1)
(XYZ)0 steht für die Lösungsmitteleigenschaften des Referenzsystems. Die
Korrelationskoeffizienten a, b und s sind vom Lösungsmittel unabhängig. Die Größe
dieser Regressionsparameter liefern Aussagen wie stark der jeweilige Einfluss von α,
β und π* des Lösungsmittels auf die UV/vis-Absorptionseigenschaften des
solvatochromen Farbstoffes ist. Löst sich der solvatochrome Farbstoff in genügend
vielen Lösungsmitteln, können die gemessenen UV/vis-Absorptionsmaxima und
damit die Regressionsparameter a, b und s aus Gleichung 1 über eine multiple
Regressionsanalyse berechnet werden. Die in dieser Arbeit verwendeten Werte der
Kamlet-Taft-Parameter α, β und π* ausgewählter Lösungsmittel wurden der Literatur
entnommen und sind in Tabelle 2 zusammengestellt.114
Tab. 2: Zusammenstellung empirischer Lösungsmittelparameter α, β und π* nach Kamlet und Taft ausgewählter Lösungsmittel.
Lösungsmittel α β π*
Wasser (H2O) 1.17 0.47 1.09
Dimethylsulfoxid (DMSO) 0.00 0.76 1.00
N,N-Dimethylformamid (DMF) 0.00 0.69 0.88
Methanol (MeOH) 0.98 0.66 0.60
Ethanol (EtOH) 0.86 0.75 0.54
1-Propanol 0.84 0.90 0.52
2-Propanol 0.76 0.84 0.48
1-Butanol 0.84 0.84 0.47
Dichlormethan (CH2Cl2) 0.13 0.10 0.82
1,2-Dichlorethan (DCE) 0.10 0.10 0.81
1,1,2,2-Tetrachlorethan (TCE) 0.00 0.10 0.95
1,1,2,2-Tetrafluorethanol (TFE) 1.51 0.00 0.73
1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH) 1.96 0.00 0.65

2 Theoretische Grundlagen
36
Am Beispiel von 4-Nitroanilin sind in Schema 16 zwei Möglichkeiten der Ausbildung
von Wasserstoffbrückenbindungen zum Lösungsmittel schematisch dargestellt.
4-Nitroanilin kann einerseits als HBD-Substrat über den Aminostickstoff in einem
HBA-Lösungsmittel (hydrogen bond acceptor, Typ B) und andererseits als HBA-
Substrat über den Sauerstoff der −NO2-Gruppe in einem HBD-Lösungsmittel
(hydrogen bond donor, Typ A) über Wasserstoffbrückenbindungen mit dem
Lösungsmittel in Wechselwirkung treten. Protische Lösungsmittel wie Alkohole und
Wasser können sowohl als HBA-Lösungsmittel als auch als HBD-Lösungsmittel
fungieren (s. Schema 16).23
Schema 16: Schematische Darstellung der Möglichkeit der Ausbildung von Wasserstoffbrücken-bindungen zwischen 4-Nitroanilin und protischen Lösungsmitteln (LM), die sowohl als hydrogen bond acceptor (HBA) als auch als hydrogen bond donor (HBD) fungieren können.
Beide Möglichkeiten der Ausbildung von Wasserstoffbrücken zum Lösungsmittel
(s. Schema 16) erniedrigen die Energie für Elektronenübergänge, da der dipolare
angeregte Zustand stärker als der Grundzustand stabilisiert wird.23
Ist die Aminogruppe z. B. durch zwei Ethylgruppen substituiert, kann dieses
4-Nitroanilin-Derivat nicht mehr als HBD-Substrat über den Aminostickstoff mit einem
HBA-Lösungsmittel (s. Typ B in Schema 16) in Wechselwirkung treten, da keine
Wasserstoffbrückenbindungen ausgebildet werden können. N,N-Diethyl-4-nitroanilin
kann lediglich über die −NO2-Gruppe als HBA-Substrat in einem HBD-Lösungsmittel
fungieren (s. Typ A in Schema 16).
Wasser ist deutlich polarer als Methanol (größerer π*-Wert) und aufgrund des
größeren α-Wertes ein etwas stärkeres HBD-Lösungsmittel. Methanol hingegen ist
ein stärkeres HBA-Lösungsmittel (größerer β-Wert) als Wasser. Somit könnten in
Wasser die H-Brückenbindungen überwiegend zur −NO2-Gruppe (Typ A in Schema
16) und in Methanol hingegen überwiegend zum Anilin-Wasserstoff (Typ B in
Schema 16) ausgebildet werden.
N NO
O
H
H
H OR
H OR
Typ B Typ A LM als HBA LM als HBD

3 Ergebnisse und Diskussion
37
3 Ergebnisse und Diskussion
3.1 Funktionalisierung von Polyvinylaminen in Wass er
3.1.1 Syntheseplanung und Charakterisierungsmöglic hkeiten
Die Funktionalisierung von Polyvinylaminen mit Hilfe aktivierter fluorsubstituierter
Aromaten wurde im Rahmen dieser Arbeit in Wasser als Lösungsmittel durchgeführt.
Wasser als Lösungsmittel bietet sich an, da es physiologisch und ökologisch
unbedenklich und im Vergleich zu den meisten organischen Lösungsmitteln
kostengünstiger ist. Um die nucleophile aromatische Substitution an hydrophoben
Fluoraromaten mit PVAm in Wasser durchführen zu können, kann sich
zweckmäßigerweise der Löslichkeitsvermittlung durch Cyclodextrine bedient werden.
Dabei wird die Eigenschaft der Cyclodextrine ausgenutzt hydrophobe Gastmoleküle
im Inneren aufzunehmen, wobei der gebildete Wirt-Gast-Komplex - bedingt durch die
hydrophile Außenseite der Cyclodextrine - wasserlöslich wird. Die Komplexierung der
hydrophoben Fluoraromaten erfolgte mit statistisch methyliertem 2,6-O-Dimethyl-β-
cyclodextrin (β-DMCD, Cavasol® W7 M) in Methanol. Zur besseren Übersichtlichkeit
wird in den nachfolgenden Abschnitten die in Schema 17 gezeigte Darstellung für
β-DMCD bzw. Cavasol® W7 M verwendet.
Schema 17: Schematische, vereinfachte Darstellung von 2,6-O-Dimethyl-β-cyclodextrin (β-DMCD).
Nach der Isolierung und der Trocknung des gebildeten Wirt-Gast-Komplexes kann
die nucleophile aromatische Substitution dieses Fluoraromat/β-DMCD-Komplexes mit
PVAm in Wasser zu den entsprechenden nitrophenyl-funktionalisierten Polyvinyl-
aminen vonstatten gehen. Ein Großteil des Lösungsmittels wird anschließend
abdestilliert, um das funktionalisierte PVAm in Aceton zur Fällung zu bringen.
O
O
H
7
H3CO
HOOCH3
12
3
4 5
6
7
8
O
O
H
7
H3CO
HOOCH3
12
3
4 5
6
7
8

3 Ergebnisse und Diskussion
38
Zur Veranschaulichung der einzelnen Syntheseschritte ist die Funktionalisierung von
PVAm in Wasser mittels statistisch methyliertem 2,6-O-Dimethyl-β-cyclodextrin
(β-DMCD) als Löslichkeitsvermittler im nachfolgenden Schema 18 kurz dargestellt.
Schema 18: Funktionalisierung von PVAm in Wasser mittels β-DMCD als Löslichkeitsvermittler.
Die nucleophile aromatische Substitution von aktivierten Fluoraromaten mit
Polyvinylaminen in Wasser erfolgte mit den in Schema 19 dargestellten acht
Fluoraromaten, die als Fluoraromat/β-DMCD-Komplexe eingesetzt wurden.
Schema 19: Eingesetzte aktivierte Fluoraromaten für die Funktionalisierung von Polyvinylaminen in Wasser über Löslichkeitsvermittlung mittels statistisch methyliertem 2,6-O-Dimethyl-β-cyclodextrin.
4-FNB
2-FNB FDNB
DFDNB 4-FNA 4-DMFNA
FDNstilben FNAzoB
F NO2F
O2N
F
O2N
NO2
F
O2N
NO2
F
F NH2
O2N
N
O2N
FCH3
CH3
F
O2N
NN
F
O2N
NO2
+
ββββ-DMCD-Komplex
CH3OHO
O
H
7CH3O
CH3O
HO
ββββ-DMCDLöslichkeitsvermittler
H2O 100°C
8 h
+
PVAm
FR
aktivierterFluoraromat
funktionalisiertes PVAm
R: elektronen-ziehende(r)Substituent(en)
R
NH2 NHx y
nNH2
FR
HF+
+
ββββ-DMCD-Komplex
CH3OHO
O
H
7CH3O
CH3O
HO
O
O
H
7CH3O
CH3O
HO
ββββ-DMCDLöslichkeitsvermittler
H2O 100°C
8 h
+
PVAm
FR
aktivierterFluoraromat
funktionalisiertes PVAm
R: elektronen-ziehende(r)Substituent(en)
R
NH2 NHx y
nNH2
FR
HF+

3 Ergebnisse und Diskussion
39
Die stattfindende Funktionalisierungsreaktion zeigt sich sofort nach Vereinigung
beider Reaktionskomponenten (Fluoraromat/β-DMCD-Komplex und Polyvinylamin)
durch einen Farbumschlag von farblos zu intensiv gelb bis purpurfarben je nach
eingesetztem Fluoraromat. Im UV/vis-Absorptionsspektrum funktionalisierter
Polyvinylamine zeigt sich die neue intramolekulare charge-transfer-Bande des
gebildeten π-Elektronen-push-pull-Systems (chromophore Polyvinylamin-Einheit).
Die Löslichkeit der funktionalisierten Polyvinylamine in Wasser und vor allem in
organischen Lösungsmitteln wie z. B. Methanol ist sehr gering, und erschwert die
Strukturaufklärung mit Hilfe 1H-Lösungs-NMR-spektroskopischer bzw. 13C-Lösungs-
NMR-spektroskopischer Untersuchungen. Daher wurden zur Identifizierung der
molekularen Struktur der jeweiligen chromophoren Einheit von funktionalisierten
Polyvinylaminen (mit Vergleich zu wohl definierten niedermolekularen
Modellverbindungen) und zur Ermittlung des Substitutionsgrades (Anteil
chromophorer Einheiten) hauptsächlich UV/vis-spektroskopische Untersuchungen
herangezogen.

3 Ergebnisse und Diskussion
40
3.1.2 Nitrophenyl-funktionalisierte Polyvinylamine mittels
4-Fluornitrobenzen (4-FNB)
Für die nucleophile aromatische Substitution von 4-Fluornitrobenzen (4-FNB) mit
Polyvinylamin (PVAm) in Wasser zu 4-nitrophenyl-funktionalisiertem PVAm
(PVAm 1) wurden vier Molverhältnisse (0.0763, 0.1526, 0.2289 und 0.3053) von
4-FNB/β-DMCD-Komplexen zu PVAm eingesetzt. Die Funktionalisierungsreaktion ist
in Schema 20 zur Veranschaulichung gezeigt. Es wurde sowohl PVAm mit einem
Molekulargewicht von 15,000 g·mol-1 als auch mit 400,000 g·mol-1 verwendet.
Schema 20: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 und 400,000 g·mol-1 mit 4-FNB/β-DMCD in Wasser und entsprechende Reaktionsbedingungen.
Die Funktionalisierung von PVAm zeigt sich durch einen Farbumschlag der
Reaktionslösung von farblos zu intensiv gelb, aufgrund der Bildung des push-pull-
Systems (chromophore Einheit). 4-Nitrophenyl-funktionalisiertes PVAm (PVAm 1)
löst sich sowohl gut in Wasser als auch gut in Methanol. In Abb. 1 sind UV/vis-
Absorptionsspektren von PVAm 1 mit einem Molekulargewicht von 15,000 g·mol-1 in
Wasser und in Methanol sowie eine Fotografie des festen Polymeren zur
Veranschaulichung gezeigt. 4-Nitrophenyl-funktionalisiertes PVAm (PVAm 1) zeigt
eine deutliche Verschiebung des UV/vis-Absorptionsmaximums zu größerer
Wellenlänge von 390 nm in Methanol zu 413 nm in Wasser. Es zeigt somit positive
Solvatochromie (bathochrome Verschiebung des UV/vis-Absorptionsmaximums mit
Zunahme der Lösungsmittelpolarität).
F NO2
NH2 NH
NO2
x y
+
NH2n
H2O
4-FNB/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
400,000 g · mol -1
100 °C8 h

3 Ergebnisse und Diskussion
41
Abb. 1: UV/vis-Absorptionsspektren von 4-nitrophenyl-funktionalisiertem PVAm (PVAm 1) in Wasser und Methanol sowie eine Fotografie des festen Polymeren.
Neben der Ausbildung von H-Brückenbindungen zum Lösungsmittel können auch
H-Brückenbindungen zwischen dem Anilinwasserstoff der chromophoren Einheit von
4-nitrophenyl-funktionalisiertem PVAm (PVAm 1) zu benachbarten NH2-Gruppen von
unsubstituierten PVAm-Einheiten eine Rolle spielen. UV/vis-spektroskopische
Untersuchungen von N-(4-Nitrophenyl)ethylendiamin zeigten, dass durch die
intramolekulare Wasserstoffbrückenbindung des Anilinwasserstoffs zur NH2-Gruppe
des aliphatischen Restes und durch die intermolekulare H-Brückenbindung zum
HBA-Lösungsmittel neben der Dipolarität/Polarisierbarkeit π* auch der β-Wert des
Lösungsmittels einen starken Einfluss auf die UV/vis-Absorptionseigenschaften
hat.115 Durch die Möglichkeit der Ausbildung von intramolekularen
H-Brücken zwischen den Anilinwasserstoffen zu NH2-Gruppen der unsubstituierten
PVAm-Einheiten verringert sich jedoch die Möglichkeit der Ausbildung von
intermolekularen H-Brücken zum Lösungsmittel über die Anilinwasserstoffe. Die
bathochrome Verschiebung des UV/vis-Absorptionsmaximums von 4-nitrophenyl-
funktionalisiertem PVAm in Methanol mit λmax = 390 nm zu λmax = 413 nm in Wasser
resultiert hauptsächlich aus der deutlich höheren Dipolarität/Polarisierbarkeit π* trotz
niedrigeren β-Wertes im Vergleich zu Methanol.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 350 400 450 500 550 600 650 700
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
NH2 NH
NO2
x y
15,000 g · mol -1
pH = 11
λλλλmax (H2O) = 413 nmλλλλmax (MeOH) = 390 nm
H2O
MeOH

3 Ergebnisse und Diskussion
42
2-Fluornitrobenzen (2-FNB)
Für die nucleophile aromatische Substitution von 2-Fluornitrobenzen (2-FNB) mit
PVAm (15,000 g·mol-1 und 400,000 g·mol-1) in Wasser zu 2-nitrophenyl-
funktionalisiertem PVAm (PVAm 2) wurden zur besseren Vergleichbarkeit die
gleichen Molverhältnisse (0.0763, 0.1526, 0.2289 und 0.3053) der 2-FNB/β-DMCD-
Komplexe zu PVAm eingesetzt, wie bei der Funktionalisierung mit 4-FNB.
Schema 21 zeigt anschaulich die Funktionalisierungsreaktion.
Schema 21: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 und 400,000 g·mol-1 mittels 2-FNB/β-DMCD in Wasser und entsprechende Reaktionsbedingungen.
Bei Zugabe der PVAm-Lösung zur wässrigen 2-FNB/β-DMCD-Komplex-Lösung ist
sofort ein Farbumschlag von farblos zu intensiv gelb aufgrund der Bildung des push-
pull-Systems (chromophore PVAm-Einheit) zu erkennen. Das ausgefällte und
getrocknete 2-nitrophenyl-funktionalisierte PVAm (PVAm 2) löst sich sowohl gut in
Wasser als auch gut in Methanol. Die mit 2-FNB/β-DMCD umgesetzten PVAm´s mit
einem Molekulargewicht von 400,000 g · mol-1 sind nicht mehr ganz so gut in Wasser
und in Methanol löslich. In Abb. 2 sind die UV/vis-Absorptionsspektren von PVAm 2
(mit einem eingesetztem Molverhältnis von 2-FNB zu PVAm von 0.3053) sowohl in
Wasser als auch in MeOH gezeigt. Das UV/vis-Absorptionsmaximum λmax in
Methanol liegt bei 432 nm. In Wasser ist - aufgrund der höheren
Lösungsmittelpolarität im Vergleich zu Methanol - eine bathochrome Verschiebung
des UV/vis-Absorptionsmaximums mit λmax = 450 nm zu erkennen.
Im Vergleich zu 4-nitrophenyl-funktionalisiertem PVAm (PVAm 1) mit
λmax(H2O) = 413 nm und λmax(MeOH) = 390 nm liegen die UV/vis-Absorptionsmaxima
von 2-nitrophenyl-funktionalisiertem PVAm (PVAm 2) bathochrom verschoben.
F
O2N NH2 NH
NO2
x y
+
NH2n
H2O
2-FNB/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
400,000 g · mol-1
100 °C8 h

3 Ergebnisse und Diskussion
43
Abb. 2: UV/vis-Absorptionsspektren von 2-nitrophenyl-funktionalisiertem PVAm (PVAm 2) in Wasser und in Methanol sowie eine Fotografie des festen Polymeren.
Bei PVAm 2 besteht die Möglichkeit der Ausbildung von intramolekularen
Wasserstoffbrückenbindungen der 1-Aminogruppe zur ortho-Nitrogruppe, die hilft die
Coplanarität der chinoiden Resonanzstruktur zu erhalten, die von polaren
Lösungsmitteln besser stabilisiert wird. Das äußert sich in einer weiteren
Energieabsenkung und damit in einer bathochromen Verschiebung des UV/vis-
Absorptionsmaximums im Vergleich zu PVAm 1.
Untersuchungen zu N-(2-Nitrophenyl)ethylendiamin zeigten, dass die intramolekulare
H-Brückenbindung der 1-Aminogruppe zur ortho-NO2-Gruppe sehr stabil ist und
keine intramolekulare H-Brücke zwischen dem Anilinwasserstoff und der NH2-Gruppe
des aliphatischen Restes ausgebildet wird. Es wurde postuliert, dass die HBA-
Fähigkeit β des Lösungsmittels keinen Einfluss auf die UV/vis-Absorptions-
eigenschaften hat.115,116
Es kann daher davon ausgegangen werden, dass die größere
Dipolarität/Polarisierbarkeit π* von Wasser im Vergleich zu Methanol den
entscheidenden Einfluss auf die UV/vis-Absorptionseigenschaften (positive
Solvatochromie) von 2-nitrophenyl-funktionalisiertem PVAm (PVAm 2) hat.
0
0.2
0.4
0.6
0.8
1
1.2
300 350 400 450 500 550 600 650 700 750 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
(5m
g in
50
ml)
[a.u
.]
NH2 NHNO2
x y
15,000 g · mol -1
pH = 11
λλλλmax (H2O) = 450 nmλλλλmax (MeOH) = 432 nm
MeOH
H2O

3 Ergebnisse und Diskussion
44
3.1.3 2-Nitro-4-aminobenzen-funktionalisierte Poly vinylamine mittels
4-Fluor-3-nitroanilin (4-FNA)
Für die Funktionalisierung von PVAm (15,000 g·mol-1) wurden die Molverhältnisse
von 4-FNA/β-DMCD zu PVAm mit 0.0763, 0.1526, 0.2289 und 0.3053 gewählt. Die
nucleophile aromatische Substitutionsreaktion von 4-FNA mit PVAm zu 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) mit den entsprechenden
Reaktionsbedingungen ist in Schema 22 gezeigt.
Schema 22: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 mittels 4-FNA/β-DMCD-Komplexen in Wasser und entsprechende Reaktionsbedingungen.
2-nitro-4-aminobenzen-funktionalisiertes PVAm (PVAm 3) löst sich gut in Wasser, in
Methanol und in 1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH). Weiterhin löst sich
PVAm 3 in geringen Mengen in Ethanol (EtOH), 1-Propanol, 2-Propanol,
1-Butanol, N,N-Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), Dichlormethan
(CH2Cl2) und in 2,2,2-Trifluorethanol (TFE). Dabei ist eine Änderung der Farbe des
gelösten Polymeren in Abhängigkeit vom Lösungsmittel zu erkennen.
Diese ausgeprägten solvatochromen Eigenschaften von 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) wurden mit Hilfe von UV/vis-spektroskopischen
Messungen untersucht. In Abb. 3 sind die UV/vis-Absorptionsspektren von PVAm 3
in Wasser, DMSO, EtOH, MeOH, CH2Cl2 und in HFiPrOH sowie eine Fotografie zur
Veranschaulichung gezeigt. Die ausgefällten, festen 2-nitro-4-aminobenzen-
funktionalisierten PVAm´s zeigen eine dunkelviolett bis schwarze Farbe.
F NH2
O2N NH2 NH
NO2
NH2
x y
+
NH2n
H2O
4-FNA/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
100 °C8 h
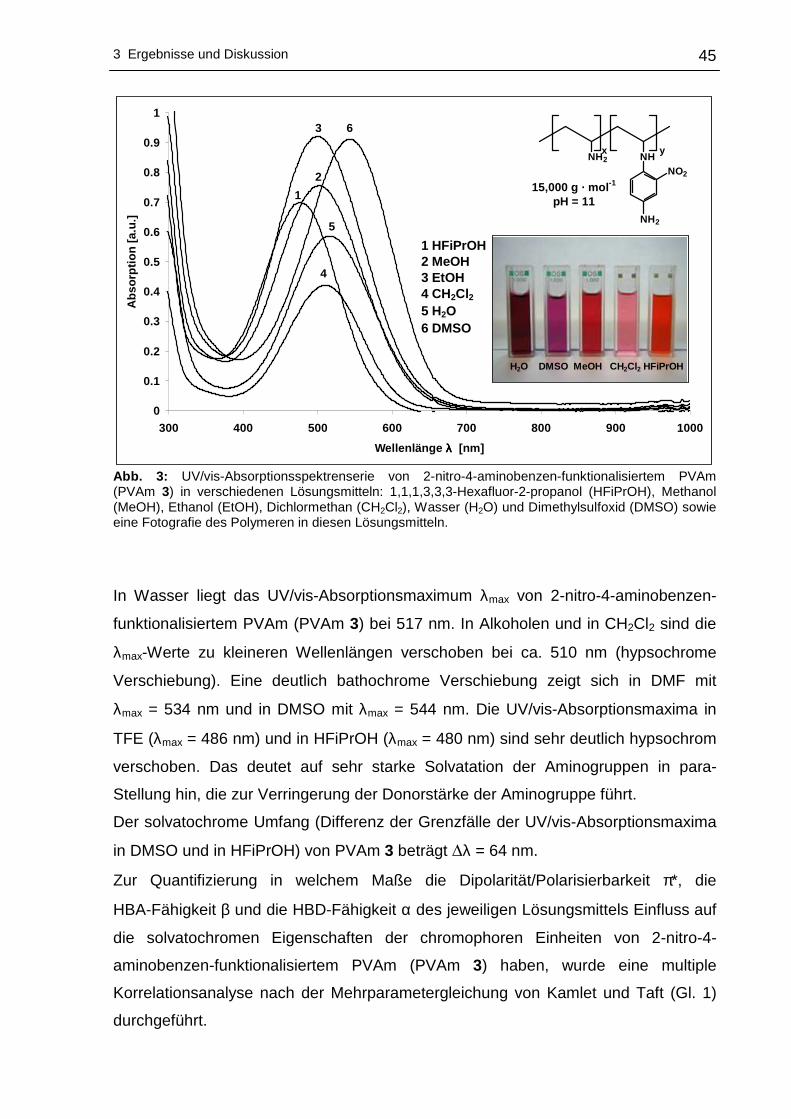
3 Ergebnisse und Diskussion
45
Abb. 3: UV/vis-Absorptionsspektrenserie von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) in verschiedenen Lösungsmitteln: 1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH), Methanol (MeOH), Ethanol (EtOH), Dichlormethan (CH2Cl2), Wasser (H2O) und Dimethylsulfoxid (DMSO) sowie eine Fotografie des Polymeren in diesen Lösungsmitteln.
In Wasser liegt das UV/vis-Absorptionsmaximum λmax von 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) bei 517 nm. In Alkoholen und in CH2Cl2 sind die
λmax-Werte zu kleineren Wellenlängen verschoben bei ca. 510 nm (hypsochrome
Verschiebung). Eine deutlich bathochrome Verschiebung zeigt sich in DMF mit
λmax = 534 nm und in DMSO mit λmax = 544 nm. Die UV/vis-Absorptionsmaxima in
TFE (λmax = 486 nm) und in HFiPrOH (λmax = 480 nm) sind sehr deutlich hypsochrom
verschoben. Das deutet auf sehr starke Solvatation der Aminogruppen in para-
Stellung hin, die zur Verringerung der Donorstärke der Aminogruppe führt.
Der solvatochrome Umfang (Differenz der Grenzfälle der UV/vis-Absorptionsmaxima
in DMSO und in HFiPrOH) von PVAm 3 beträgt ∆λ = 64 nm.
Zur Quantifizierung in welchem Maße die Dipolarität/Polarisierbarkeit π*, die
HBA-Fähigkeit β und die HBD-Fähigkeit α des jeweiligen Lösungsmittels Einfluss auf
die solvatochromen Eigenschaften der chromophoren Einheiten von 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) haben, wurde eine multiple
Korrelationsanalyse nach der Mehrparametergleichung von Kamlet und Taft (Gl. 1)
durchgeführt.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800 900 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.] 5
63
2
4
1
1 HFiPrOH2 MeOH3 EtOH4 CH2Cl25 H2O6 DMSO
NH2 NHNO2
NH2
x y
15,000 g · mol -1
pH = 11
H2O DMSO MeOH CH2Cl2 HFiPrOH

3 Ergebnisse und Diskussion
46
Die multiple Regressionsanalyse der aus den gemessenen Wellenlängen λmax
umgerechneten Wellenzahlen ṽmax über die α-, β- und π*-Werte der Lösungsmittel
(s. Tab. 2) ergibt für 2-nitro-4-aminobenzen-funktionalisiertes PVAm (PVAm 3) die
folgende Mehrparametergleichung (Gleichung 2).
*])[(~ πβαν 1.4781.1040.55720.77110cm PVAm 31max −−+=⋅ −−3 (2)
n = 11; r = 0.997; sd = 0.051; F<0.0001
Dabei bedeuten die einzelnen Parameter:
r…Korrelationskoeffizient bzw. Bestimmtheitsmaß,
n…Anzahl der Lösungsmittel,
sd…Standardabweichung und
F…Signifikanz der Mehrparametergleichung.
Werte für r gegen eins und F < 0.0001 stehen für die Güte der Korrelationsanalyse.
Die Vorzeichen der ermittelten lösungsmittelunabhängigen Korrelationskoeffizienten
a, b und s geben an, ob die Lösungsmittelparameter α, β und π* zu positiver
Solvatochromie (bathochrome Verschiebung) bzw. negativer Solvatochromie
(hypsochrome Verschiebung) der UV/vis-Absorptionsbande führen. Die Beträge von
a, b und s (siehe Gl. 2) zeigen, in welchem Maße der jeweilige
Lösungsmittelparameter Einfluss auf die Lage der UV/vis-Absorptionsbande besitzt.
Die Dipolarität/Polarisierbarkeit π* (s = -1.478) sowie die HBA-Fähigkeit β
(b = -1.104) weisen den größten Einfluss auf, die HBD-Fähigkeit α (a = 0.557)
hingegen zeigt den kleinsten Einfluss auf die solvatochromen Eigenschaften von
2-nitro-4-aminobenzen-funktionalisiertem PVAm 3 (Betrag von s/a = 2.65 bzw.
s/b = 1.34). Somit führen hohe π*-Werte und hohe β-Werte des Lösungsmittels zu
einer bathochromen Verschiebung der UV/vis-Absorptionsbande der chromophoren
Einheit von PVAm 3.
Die graphische Auftragung der nach der Mehrparametergleichung (Gl. 2)
berechneten Wellenzahlen ṽmax über die gemessenen Wellenzahlen ṽmax von 2-nitro-
4-aminobenzen-funktionalisiertem PVAm (PVAm 3) in 11 Lösungsmitteln ist in Abb. 4
gezeigt.

3 Ergebnisse und Diskussion
47
Abb. 4: Graphische Auftragung der berechneten über die gemessenen UV/vis-Absorptionsmaxima von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) in 11 Lösungsmitteln unter Berück-sichtigung der Lösungsmittelparameter α, β und π*.
Aus Abb. 4 wird deutlich, dass nur geringste Abweichungen von der Linearität
zwischen den gemessenen und berechneten Werten der UV/vis-Absorptionsmaxima
vorliegen (r = 0.997). Die solvatochromen Eigenschaften von 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) korrelieren sehr gut mit den empirischen
Lösungsmittelparametern α, β und π* nach Kamlet und Taft.
Eine ausführliche Diskussion der solvatochromen Eigenschaften von PVAm 3 mit
Vergleich zu denen anderer 2-nitro-4-aminobenzen-funktionalisierten Polyvinylamine
erfolgt später am Ende dieses Abschnittes 3.1.3.
18
18.5
19
19.5
20
20.5
21
21.5
18 18.5 19 19.5 20 20.5 21 21.5
ṽ max gemessen [cm -1· 10-3]
ṽm
ax b
erec
hnet
[cm
-1 ·
10-3
]
NH2 NHNO2
NH2
x y
3
DMSO
H2OCH2Cl2
EtOH
MeOH
HFiPrOH
1-Propanol 2-Propanol1-Butanol
DMF
TFE

3 Ergebnisse und Diskussion
48
N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA)
In Anlehnung an die Arbeiten von Giumanini et al. (N,N-Dimethylierung von
4-Fluoranilin) wurde N,N-Dimethyl-4-fluor-3-nitroanilin durch N,N-Dimethylierung von
4-Fluor-3-nitroanilin in schwefelsaurer Formaldehyd/THF-Lösung und Natrium-
borhydrid (NaBH4) als Reduktionsmittel synthetisiert.117 Eine ausführliche
Synthesevorschrift und die Charakterisierung des synthetisierten N,N-Dimethyl-4-
fluor-3-nitroanilin (4-DMFNA) sind in Abschnitt 5.6.1 (experimenteller Teil) aufgeführt.
Der von Giumanini postulierte Reaktionsmechanismus wurde für die in dieser Arbeit
durchgeführte N,N-Dimethylierung von 4-Fluor-3-nitroanilin übernommen (siehe
Schema 23).117
Schema 23: Schematische Darstellung des Mechanismus der N,N-Dimethylierung von 4-Fluor-3-nitroanilin zu N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA).
Der Vorteil dieser Syntheseroute besteht darin, dass das quartäre Ammoniumsalz
und das monomethylierte Anilin-Derivat als Nebenprodukte nicht entstehen.
Voraussetzung ist jedoch mindestens ein doppelter Überschuss an eingesetztem
Reduktionsmittel im Vergleich zum Anilinderivat. Giumanini et al. postulierten, dass
die Kationen-Spezies 7 viel leichter zu reduzieren ist als 4 (siehe Schema 23). Die
extrem geringe Konzentration an 7 (aufgrund des Gleichgewichts) verlangsamt die
zügige Methylierung des sekundären Amins 6 sobald es gebildet ist.117
H2C O H2C OH
F
O2N
NH 2
H
- H
F
O2N
NHH
CH2 OH
F
O2N
NH
CH2 OH2
H2O-
BH4 BH4
F
O2N
NH
CH2
F
O2N
NH
CH2F
O2N
NCH2 - H
OH2-
F
O2N
NCH3
CH3 BH4F
O2N
NCH2
CH3
F
O2N
NCH2
CH3
F
O2N
NCH2
CH3
OH2
F N
CH3
CH2
CH3
O2N
OH
F
O2N
NHCH2
CH3
OHF
O2N
NCH3
H
H2C OH
H2C OH
H+- H2O
F N
CH3
CH2
CH3
O2N
1
2
3
45
6
78
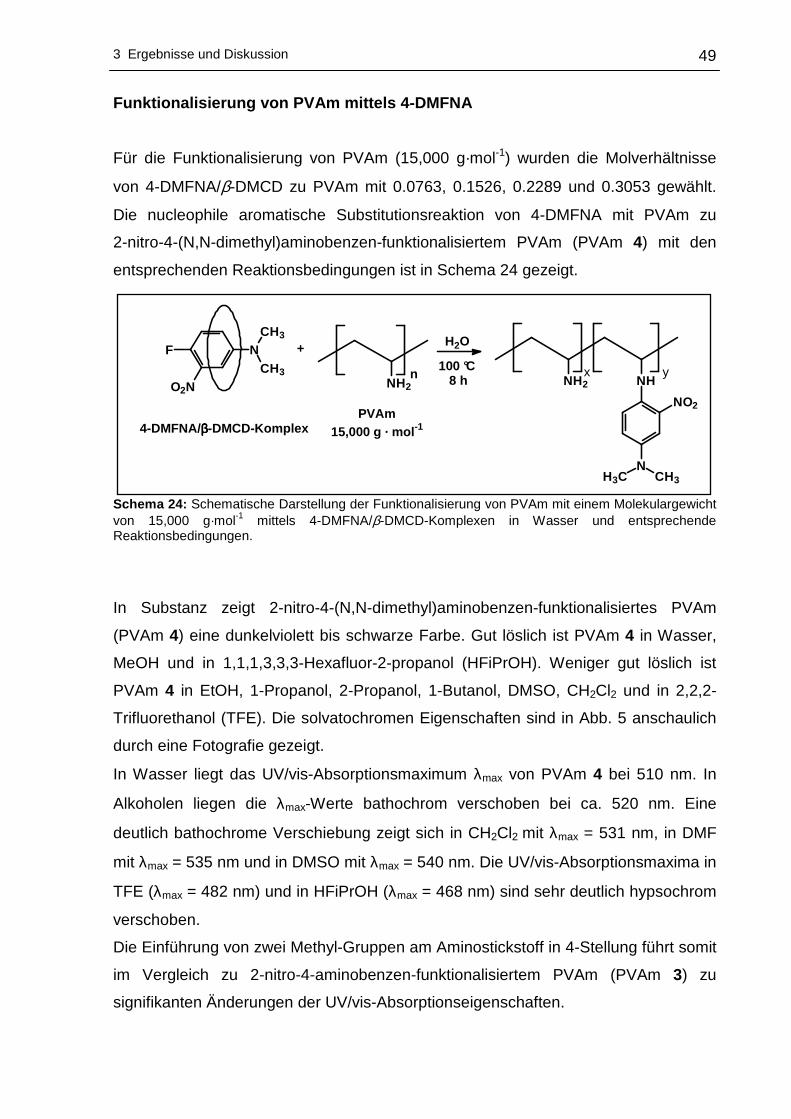
3 Ergebnisse und Diskussion
49
Funktionalisierung von PVAm mittels 4-DMFNA
Für die Funktionalisierung von PVAm (15,000 g·mol-1) wurden die Molverhältnisse
von 4-DMFNA/β-DMCD zu PVAm mit 0.0763, 0.1526, 0.2289 und 0.3053 gewählt.
Die nucleophile aromatische Substitutionsreaktion von 4-DMFNA mit PVAm zu
2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) mit den
entsprechenden Reaktionsbedingungen ist in Schema 24 gezeigt.
Schema 24: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 mittels 4-DMFNA/β-DMCD-Komplexen in Wasser und entsprechende Reaktionsbedingungen.
In Substanz zeigt 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PVAm
(PVAm 4) eine dunkelviolett bis schwarze Farbe. Gut löslich ist PVAm 4 in Wasser,
MeOH und in 1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH). Weniger gut löslich ist
PVAm 4 in EtOH, 1-Propanol, 2-Propanol, 1-Butanol, DMSO, CH2Cl2 und in 2,2,2-
Trifluorethanol (TFE). Die solvatochromen Eigenschaften sind in Abb. 5 anschaulich
durch eine Fotografie gezeigt.
In Wasser liegt das UV/vis-Absorptionsmaximum λmax von PVAm 4 bei 510 nm. In
Alkoholen liegen die λmax-Werte bathochrom verschoben bei ca. 520 nm. Eine
deutlich bathochrome Verschiebung zeigt sich in CH2Cl2 mit λmax = 531 nm, in DMF
mit λmax = 535 nm und in DMSO mit λmax = 540 nm. Die UV/vis-Absorptionsmaxima in
TFE (λmax = 482 nm) und in HFiPrOH (λmax = 468 nm) sind sehr deutlich hypsochrom
verschoben.
Die Einführung von zwei Methyl-Gruppen am Aminostickstoff in 4-Stellung führt somit
im Vergleich zu 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) zu
signifikanten Änderungen der UV/vis-Absorptionseigenschaften.
F N
O2N
CH3
CH3NH2 NH
NO2
NH3C CH3
x y
+
NH2n
H2O
4-DMFNA/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
100 °C8 h
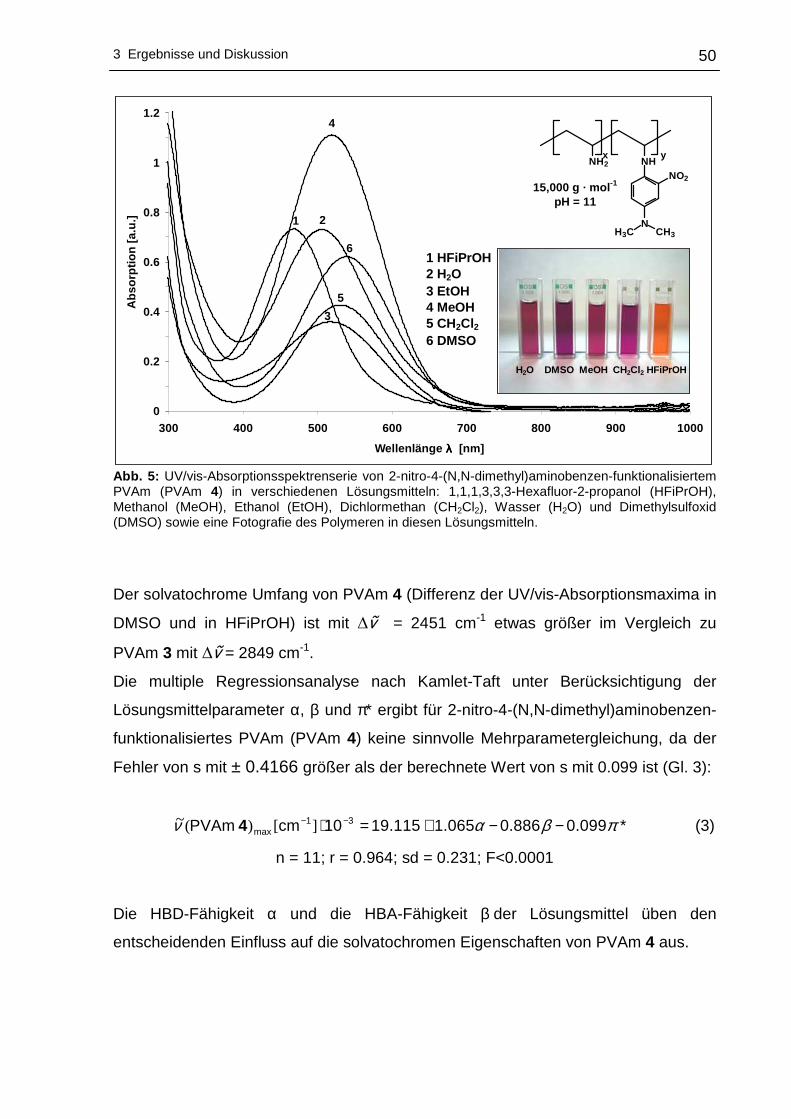
3 Ergebnisse und Diskussion
50
Abb. 5: UV/vis-Absorptionsspektrenserie von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) in verschiedenen Lösungsmitteln: 1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH), Methanol (MeOH), Ethanol (EtOH), Dichlormethan (CH2Cl2), Wasser (H2O) und Dimethylsulfoxid (DMSO) sowie eine Fotografie des Polymeren in diesen Lösungsmitteln.
Der solvatochrome Umfang von PVAm 4 (Differenz der UV/vis-Absorptionsmaxima in
DMSO und in HFiPrOH) ist mit ∆ṽ = 2451 cm-1 etwas größer im Vergleich zu
PVAm 3 mit ∆ṽ = 2849 cm-1.
Die multiple Regressionsanalyse nach Kamlet-Taft unter Berücksichtigung der
Lösungsmittelparameter α, β und π* ergibt für 2-nitro-4-(N,N-dimethyl)aminobenzen-
funktionalisiertes PVAm (PVAm 4) keine sinnvolle Mehrparametergleichung, da der
Fehler von s mit ± 0.4166 größer als der berechnete Wert von s mit 0.099 ist (Gl. 3):
*0.0990.8861.06519.11510cm PVAm 31max πβαν −−+=⋅ −− ][)(~ 4 (3)
n = 11; r = 0.964; sd = 0.231; F<0.0001
Die HBD-Fähigkeit α und die HBA-Fähigkeit β der Lösungsmittel üben den
entscheidenden Einfluss auf die solvatochromen Eigenschaften von PVAm 4 aus.
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700 800 900 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.] 2
6
4
3
5
1
1 HFiPrOH2 H2O3 EtOH4 MeOH5 CH2Cl26 DMSO
15,000 g · mol -1
pH = 11
NH2 NHNO2
NH3C CH3
x y
H2O DMSO MeOH CH2Cl2 HFiPrOH
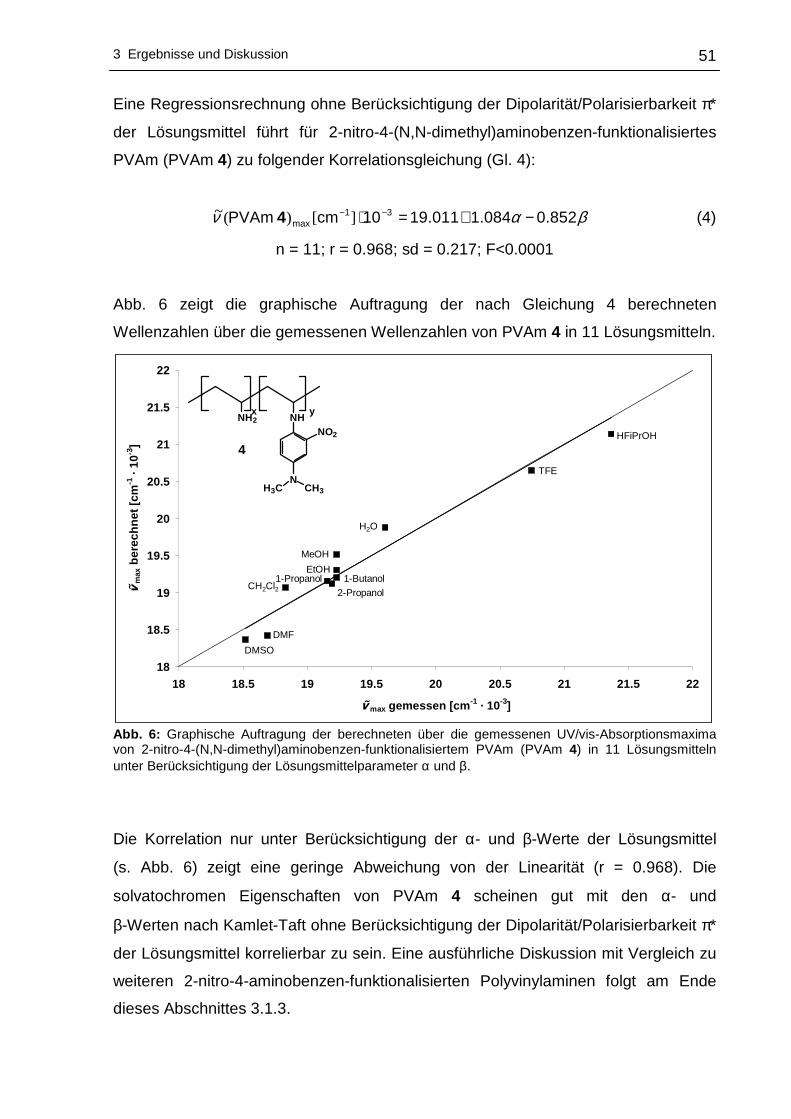
3 Ergebnisse und Diskussion
51
Eine Regressionsrechnung ohne Berücksichtigung der Dipolarität/Polarisierbarkeit π*
der Lösungsmittel führt für 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes
PVAm (PVAm 4) zu folgender Korrelationsgleichung (Gl. 4):
βαν 0.8521.08419.01110cm PVAm 31max −+=⋅ −− ][)(~ 4 (4)
n = 11; r = 0.968; sd = 0.217; F<0.0001
Abb. 6 zeigt die graphische Auftragung der nach Gleichung 4 berechneten
Wellenzahlen über die gemessenen Wellenzahlen von PVAm 4 in 11 Lösungsmitteln.
Abb. 6: Graphische Auftragung der berechneten über die gemessenen UV/vis-Absorptionsmaxima von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) in 11 Lösungsmitteln unter Berücksichtigung der Lösungsmittelparameter α und β.
Die Korrelation nur unter Berücksichtigung der α- und β-Werte der Lösungsmittel
(s. Abb. 6) zeigt eine geringe Abweichung von der Linearität (r = 0.968). Die
solvatochromen Eigenschaften von PVAm 4 scheinen gut mit den α- und
β-Werten nach Kamlet-Taft ohne Berücksichtigung der Dipolarität/Polarisierbarkeit π*
der Lösungsmittel korrelierbar zu sein. Eine ausführliche Diskussion mit Vergleich zu
weiteren 2-nitro-4-aminobenzen-funktionalisierten Polyvinylaminen folgt am Ende
dieses Abschnittes 3.1.3.
18
18.5
19
19.5
20
20.5
21
21.5
22
18 18.5 19 19.5 20 20.5 21 21.5 22
ṽ max gemessen [cm -1 · 10-3]
ṽm
ax b
erec
hnet
[cm
-1 ·
10-3
]
NH2 NHNO2
NH3C CH3
x y
DMSO
CH2Cl2
MeOH
EtOH
H2O
HFiPrOH
4
DMF
TFE
1-Propanol
2-Propanol
1-Butanol

3 Ergebnisse und Diskussion
52
Funktionalisierung von Poly- N-Methylvinylamin (PNMVAm) mittels 4-DMFNA
Es ist zu erwarten, dass die Einführung eines weiteren Substituenten am
Polymerrückrat, wie z. B. einer Methylgruppe am Aminostickstoff des PVAm zur
Erhöhung der Donorstärke führt, was die UV/vis-Absorptionseigenschaften des
entstehenden funktionalisierten Polymeren im Vergleich zu 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) und 2-nitro-4-(N,N-dimethyl)aminobenzen-
funktionalisiertem PVAm (PVAm 4) beeinflusst. Anstelle von PVAm wurde Poly-N-
Methylvinylamin (PNMVAm) zur Funktionalisierung mittels 4-DMFNA ausgewählt.
Poly-N-Methylvinylamin (PNMVAm) wurde durch saure Hydrolyse (2M HCl) aus
Poly-N-Methylvinylacetamid (196,000 g·mol-1) synthetisiert. Der pH-Wert wurde
mittels NaOH-Lösung auf pH = 11 eingestellt. Die ausführliche Reaktions-
durchführung und Charakterisierung ist in Abschnitt 5.2.1 (experimenteller Teil)
dargelegt. Zur Veranschaulichung ist die zusammengefasste Hydrolysereaktion
ausgehend von Poly-N-Methylvinylacetamid zum Poly-N-Methylvinylamin (PNMVAm)
in Schema 25 gezeigt.
Schema 25: Schematische Darstellung der Hydrolyse von Poly-N-Methylvinylacetamid zu Poly-N-Methylvinylamin (PNMVAm).
Für die Funktionalisierung von PNMVAm (196,000 g·mol-1) wurde ein Molverhältnis
von 4-DMFNA/β-DMCD zu PNMVAm mit 0.3053 gewählt. 60 ml der wässrigen Poly-
N-Methylvinylamin-Lösung (enthält 0.5 g des Polymeren) wurden für die nucleophile
aromatische Substitution von 4-DMFNA zu 2-nitro-4-(N,N-dimethyl)aminobenzen-
funktionalisiertem Poly-N-Methylvinylamin (PNMVAm 5) eingesetzt. Das ausgefällte
PNMVAm 5 zeigt als Feststoff eine dunkelviolett bis schwarze Farbe. 2-Nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertes PNMVAm (PNMVAm 5) löst sich mäßig in
Wasser, in Alkoholen, in DMF, in 1,2-Dichlorethan (DCE), in 1,1,2,2-Tetrachlorethan
(TCE) und in TFE. In DMSO, in CH2Cl2 und vor allem in HFiPrOH löst es sich gut.
NCH3H
nN
CH3O
CH3
n
2M HCl
10 d
NaOH + CH3COONa + NaCl + H2O

3 Ergebnisse und Diskussion
53
Abb. 7 zeigt UV/vis-Absorptionsspektren von PNMVAm 5 in Wasser, DMSO, EtOH,
MeOH, CH2Cl2 und in HFiPrOH sowie eine Fotografie zur Veranschaulichung.
Abb. 7: UV/vis-Absorptionsspektrenserie von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) in verschiedenen Lösungsmitteln: 1,1,1,3,3,3-Hexafluor-2-propanol (HFiPrOH), Methanol (MeOH), Ethanol (EtOH), Methylenchlorid (CH2Cl2), Wasser (H2O) und Dimethylsulfoxid (DMSO) sowie eine Fotografie des Polymeren in diesen Lösungsmitteln.
In Wasser liegt das UV/vis-Absorptionsmaximum λmax von 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) bei 510 nm. In
Alkoholen liegen die λmax-Werte hypsochrom verschoben bei ca. 500 nm. Eine
geringe bathochrome Verschiebung zeigt sich in TCE mit λmax = 515 nm, in DMF mit
λmax = 516 nm und in DMSO mit λmax = 520 nm.
In CH2Cl2 und DCE liegen die UV/vis-Absorptionsmaxima ähnlich wie in Wasser bei
510 nm. Die UV/vis-Absorptionsmaxima in TFE (λmax = 476 nm) und in HFiPrOH
(λmax = 462 nm) sind sehr deutlich hypsochrom verschoben.
Der solvatochrome Umfang von PNMVAm 5 (Differenz der UV/vis-Absorptions-
maxima in DMSO und in HFiPrOH) ist mit 2415 cm-1 etwas kleiner als bei PVAm 3
(∆ṽ = 2451 cm-1) und PVAm 4 (∆ṽ = 2849 cm-1).
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800 900 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
4
6
2
3
5
1
1 HFiPrOH2 EtOH3 MeOH4 H2O5 CH2Cl26 DMSO
196,000 g · mol -1
pH = 11
N NNO2
NH3C CH3
H3CH3C
H
x y
H2O DMSO MeOH CH2Cl2 HFiPrOH

3 Ergebnisse und Diskussion
54
Für 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PNMVAm (PNMVAm 5)
ergibt die multiple Regressionsanalyse nach Kamlet-Taft unter Berücksichtigung der
Lösungsmittelparameter α, β und π* folgende Mehrparametergleichung (Gl. 5):
*1.0650.7600.749 20.576 10cm PNMVAm 31max πβαν −−+=⋅ −− ][)(~ 5 (5)
n = 13; r = 0.947; sd = 0.218; F<0.0001
Aus Gleichung 5 wird deutlich, dass die HBD-Fähigkeit α mit a = 0.749 und die HBA-
Fähigkeit β mit b = -0.760 des Lösungsmittels nahezu gleichwertig einen geringeren
Einfluss auf die solvatochromen Eigenschaften der chromophoren Einheit von
PNMVAm 5 im Vergleich zur Dipolarität/Polarisierbarkeit π* mit s = 1.065 haben. Die
Beträge s/a = 1.42 und s/b = 1.40 sind annähernd gleich groß.
Die Auftragung der nach der Regressionsgeraden (Gl. 5) berechneten Wellenzahlen
ṽmax über die gemessenen Wellenzahlen ṽmax der chromophoren Einheit von 2-nitro-
4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) ist in Abb. 8
gezeigt.
Abb. 8: Graphische Auftragung der berechneten über die gemessenen UV/vis-Absorptionsmaxima von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) in 13 Lösungs-mitteln unter Berücksichtigung der Lösungsmittelparameter α, β und π*.
18.5
19
19.5
20
20.5
21
21.5
22
18.5 19 19.5 20 20.5 21 21.5 22
ṽ max gemessen [cm -1 · 10-3]
ṽm
ax b
erec
hnet
[cm
-1 ·
10-3
]
N NNO2
NH3C CH3
H3CH3C
H
x y
DMSO
CH2Cl2
MeOHEtOH
H2O
HFiPrOH
5
DMF
1-Propanol2-Propanol 1-Butanol
TFE
DCE
TCE

3 Ergebnisse und Diskussion
55
Alle Werte liegen gut in der Nähe der Geraden (r = 0.947), so dass die
solvatochromen Eigenschaften von 2-nitro-4-(N,N-dimethyl)aminobenzen-
funktionalisiertem PNMVAm (PNMVAm 5) mit den empirischen Lösungsmittel-
parametern α, β und π* nach Kamlet-Taft gut korrelierbar sind.
In der Tabelle 3 sind die Ergebnisse der UV/vis-spektroskopischen Untersuchungen
von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3), 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) und von 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) in verschiedenen
Lösungsmitteln und die entsprechenden Lösungsmittelparametern α, β und π* nach
Kamlet-Taft zusammengefasst.
Tab. 3: Zusammenfassung der Ergebnisse aus UV/vis-spektroskopischen Untersuchungen von 2-nitro-4-aminobenzen-funktionalisierten Polyvinylaminen PVAm 3, PVAm 4 und PNMVAm 5 mit Bezug zu den empirischen Lösungsmittelparametern α, β und π* nach Kamlet und Taft.
λmax [nm]
Lösungs-
mittel α β π*
NH2 NHNO2
NH2
x y
PVAm 3
NH2 NHNO2
NH3C CH3
x y
PVAm 4
x yN CH3
H
N CH3
NO2
NH3C CH3
PNMVAm 5
H2O 1.17 0.47 1.09 517 510 510
DMSO 0.00 0.76 1.00 544 540 520
DMF 0.00 0.69 0.88 534 535 516
MeOH 0.98 0.66 0.60 508 520 500
EtOH 0.86 0.75 0.54 509 520 500
1-Propanol 0.84 0.90 0.52 514 522 500
2-Propanol 0.76 0.84 0.48 510 521 501
1-Butanol 0.84 0.84 0.47 510 520 500
CH2Cl2 0.13 0.10 0.82 512 531 510
DCE 0.10 0.10 0.81 nicht löslich nicht löslich 510
TCE 0.00 0.10 0.95 nicht löslich nicht löslich 515
TFE 1.51 0.00 0.73 486 482 476
HFiPrOH 1.96 0.00 0.65 480 468 462

3 Ergebnisse und Diskussion
56
Die UV/vis-Absorptionsbanden der 2-nitro-4-aminobenzen-funktionalisierten
Polyvinylamine PVAm 3, PVAm 4 und PNMVAm 5 sind im Vergleich zu
entsprechenden niedermolekularen 2-Nitro-4-phenylendiamin-Derivaten bathochrom
verschoben.34,35 Das bedeutet, dass die Basizitäten der unsubstituierten Vinylamin-
Einheiten im Polymerrückrat möglicherweise Einfluss auf die Lage der UV/vis-
Absorptionsbanden der chromophoren Einheiten nehmen.
Durch die Möglichkeit der Ausbildung von intramolekularen H-Brückenbindungen bei
PVAm 3 und PVAm 4 (zwischen dem Anilinwasserstoff und der ortho-Nitrogruppe)
und der in 1-Stellung befindlichen Methylgruppe bei PNMVAm 5 ist davon
auszugehen, dass die solvatochromen Eigenschaften hauptsächlich von den
unterschiedlichen Möglichkeiten der Ausbildung von H-Brückenbindungen mit der in
para-Stellung befindlichen Aminogruppe bzw. para-N,N-Dimethylaminogruppe zum
Lösungsmittel herrühren.
Die Größe des α-Wertes (HBD Acidität) eines Lösungsmittels als Maß für die Stärke
als HBD-Lösungsmittel und die Größe des β-Wertes (HBA Basizität) als Maß für die
Stärke des Lösungsmittels als HBA-Lösungsmittel zu fungieren, haben
entscheidenden Einfluss auf die UV/vis-Absorptionseigenschaften von PVAm 3,
PVAm 4 und PNMVAm 5. Protische Lösungsmittel wie Wasser und Alkohole können
sowohl als HBA Lösungsmittel als auch als HBD Lösungsmittel wirken. So können
Wasserstoffbrückenbindungen zum einen über den para-Aminostickstoff bei
PVAm 3 in einem HBA-Lösungsmittel und zum anderen über die para-N,N-
Dimethylaminogruppe bei PVAm 4 und PNMVAm 5 in einem HBD-Lösungsmittel
ausgebildet werden.
In Wasser mit hohem α-Wert und hohem π*-Wert liegen die UV/vis-Absorptions-
maxima des PVAm 3 bei λmax = 517 nm und die der PVAm 4 und PNMVAm 5 bei
λmax = 510 nm. In DMSO als sehr polares, starkes HBA-Lösungsmittel mit hohem
π*- und einem höheren β-Wert als Wasser zeigen alle drei Polymeren die größte
bathochrome Verschiebung. In DMF mit einem etwas geringeren β- und π*-Wert als
DMSO sind die UV/vis-Absorptionsmaxima bathochrom verschoben im Vergleich zu
Wasser, jedoch etwas weniger als in DMSO. In Alkoholen liegen die UV/vis-
Absorptionsmaxima von PVAm 3 um 510 nm, bei PVAm 4 um 520 nm und bei
PNMVAm 5 um 500 nm.

3 Ergebnisse und Diskussion
57
In CH2Cl2 liegen die UV/vis-Absorptionsmaxima von allen drei Polymeren aufgrund
des höheren π*-Wertes im Vergleich zu Alkoholen bathochrom verschoben
(s. auch niedermolekulare 2-Nitro-4-phenylendiamin-Derivate).34,35
Alle drei 2-nitro-4-aminobenzen-funktionalisierten Polyvinylamine PVAm 3, PVAm 4
und PNMVAm 5 zeigen in HFiPrOH als sehr starkes HBD-Lösungsmittel (großer
α-Wert) eine deutlich hypsochrome Verschiebung der UV/vis-Absorptionsbanden.
Das deutet auf sehr starke Solvatation der Aminogruppen in 4-Stellung hin, die zur
starken Absenkung der Donorstärke der Aminogruppen führt. Die UV/vis-
Absorptionsmaxima liegen dann fast im Bereich von 2-nitrophenyl-funktionalisiertem
PVAm. In TFE sind die hypsochromen Verschiebungen der UV/vis-Absorptions-
maxima von PVAm 3, PVAm 4 und PNMVAm 5 aufgrund des etwas niedrigeren
α-Wertes und des etwas höheren π*-Wertes im Vergleich zu HFiPrOH etwas
geringer.
Die Einführung von zwei Methylgruppen in 4-Stellung (Erhöhung der Donorstärke der
4-Aminogruppe) bei PVAm 4 im Vergleich zu PVAm 3 äußert sich in Alkoholen und
CH2Cl2 in einer bathochromen Verschiebung des UV/vis-Absorptionsmaximums des
π-Elektronen-push-pull-Systems. Das zeigt sich auch bei niedermolekularen 2-Nitro-
4-phenylendiamin-Derivaten.34,35
Durch die Monoalkylsubstitution in 1-Stellung bei PVAm 3 und bei PVAm 4 können
über die –NH-Gruppen Wasserstoffbrückenbindungen zu den benachbarten ortho-
NO2-Gruppen ausgebildet werden, die helfen die molekulare Coplanarität zu
erhalten. Die Einführung einer weiteren Methylgruppe in 1-Stellung bei PNMVAm 5
erhöht zwar die Donorstärke des Aminostickstoffs im Vergleich zu PVAm 3 und
PVAm 4, doch durch die sterische Nähe zur ortho-Nitrogruppe wird die Coplanarität
gestört. Das führt zum Verlust an Resonanzstabilisierung, was sich bei PNMVAm 5
an der hypsochromen Verschiebung der UV/vis-Absorptionsbande im Vergleich zu
PVAm 4 in allen untersuchten Lösungsmitteln zeigt.118
In Tabelle 4 sind die Ergebnisse der Untersuchungen des solvatochromen
Verhaltens von PVAm 3, PVAm 4 und PNMVAm 5 in 11 bzw. 13 Lösungsmittel aus
Regressionsanalysen nach der Mehrparametergleichung von Kamlet und Taft (Gl. 1)
zusammengefasst. Der in Tab. 4 gezeigte solvatochrome Umfang (Differenz der
UV/vis-Absorptionsmaxima in DMSO und HFiPrOH) ist bei PVAm 4 mit
∆ṽ = 2849 cm-1 im Vergleich zu PVAm 3 mit ∆ṽ = 2451 cm-1 und PNMVAm 5 mit
∆ṽ = 2415 cm-1 am größten.
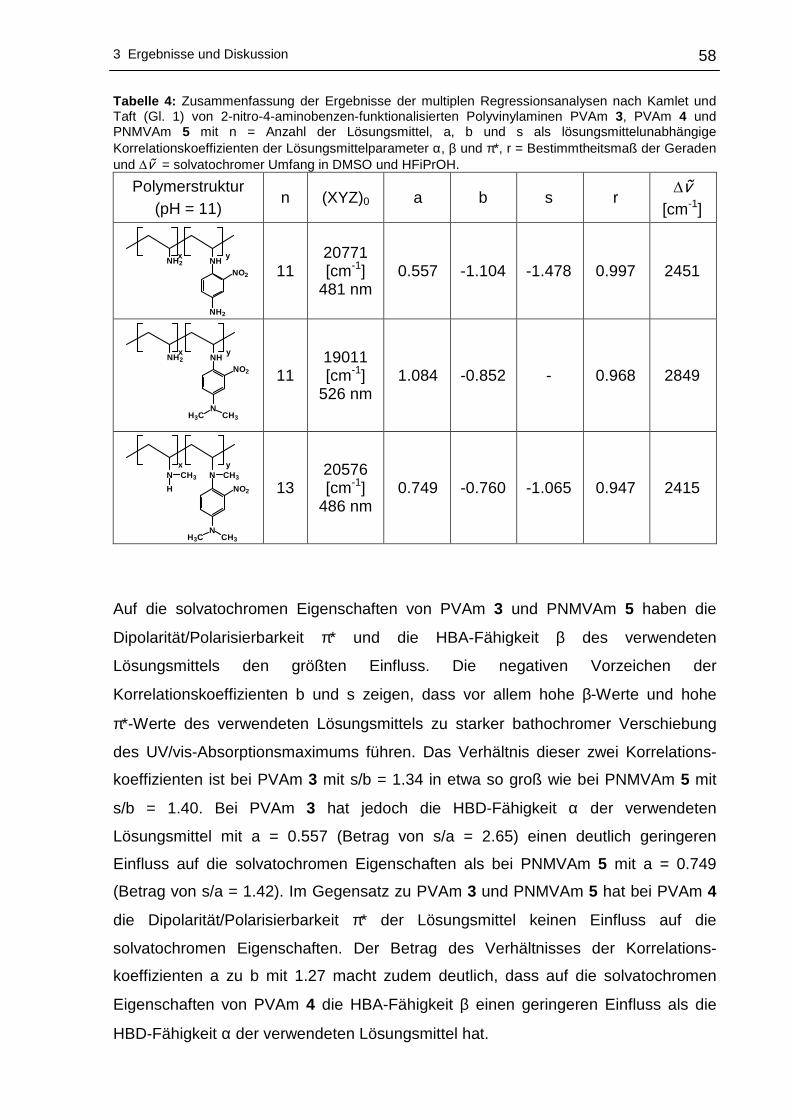
3 Ergebnisse und Diskussion
58
Tabelle 4: Zusammenfassung der Ergebnisse der multiplen Regressionsanalysen nach Kamlet und Taft (Gl. 1) von 2-nitro-4-aminobenzen-funktionalisierten Polyvinylaminen PVAm 3, PVAm 4 und PNMVAm 5 mit n = Anzahl der Lösungsmittel, a, b und s als lösungsmittelunabhängige Korrelationskoeffizienten der Lösungsmittelparameter α, β und π*, r = Bestimmtheitsmaß der Geraden und ∆ṽ = solvatochromer Umfang in DMSO und HFiPrOH.
Polymerstruktur (pH = 11)
n (XYZ)0 a b s r ∆ṽ
[cm-1]
NH2 NHNO2
NH2
x y
11 20771 [cm-1]
481 nm 0.557 -1.104 -1.478 0.997 2451
NH2 NHNO2
NH3C CH3
x y
11 19011 [cm-1]
526 nm 1.084 -0.852 - 0.968 2849
x yN CH3
H
N CH3
NO2
NH3C CH3
13 20576 [cm-1]
486 nm 0.749 -0.760 -1.065 0.947 2415
Auf die solvatochromen Eigenschaften von PVAm 3 und PNMVAm 5 haben die
Dipolarität/Polarisierbarkeit π* und die HBA-Fähigkeit β des verwendeten
Lösungsmittels den größten Einfluss. Die negativen Vorzeichen der
Korrelationskoeffizienten b und s zeigen, dass vor allem hohe β-Werte und hohe
π*-Werte des verwendeten Lösungsmittels zu starker bathochromer Verschiebung
des UV/vis-Absorptionsmaximums führen. Das Verhältnis dieser zwei Korrelations-
koeffizienten ist bei PVAm 3 mit s/b = 1.34 in etwa so groß wie bei PNMVAm 5 mit
s/b = 1.40. Bei PVAm 3 hat jedoch die HBD-Fähigkeit α der verwendeten
Lösungsmittel mit a = 0.557 (Betrag von s/a = 2.65) einen deutlich geringeren
Einfluss auf die solvatochromen Eigenschaften als bei PNMVAm 5 mit a = 0.749
(Betrag von s/a = 1.42). Im Gegensatz zu PVAm 3 und PNMVAm 5 hat bei PVAm 4
die Dipolarität/Polarisierbarkeit π* der Lösungsmittel keinen Einfluss auf die
solvatochromen Eigenschaften. Der Betrag des Verhältnisses der Korrelations-
koeffizienten a zu b mit 1.27 macht zudem deutlich, dass auf die solvatochromen
Eigenschaften von PVAm 4 die HBA-Fähigkeit β einen geringeren Einfluss als die
HBD-Fähigkeit α der verwendeten Lösungsmittel hat.
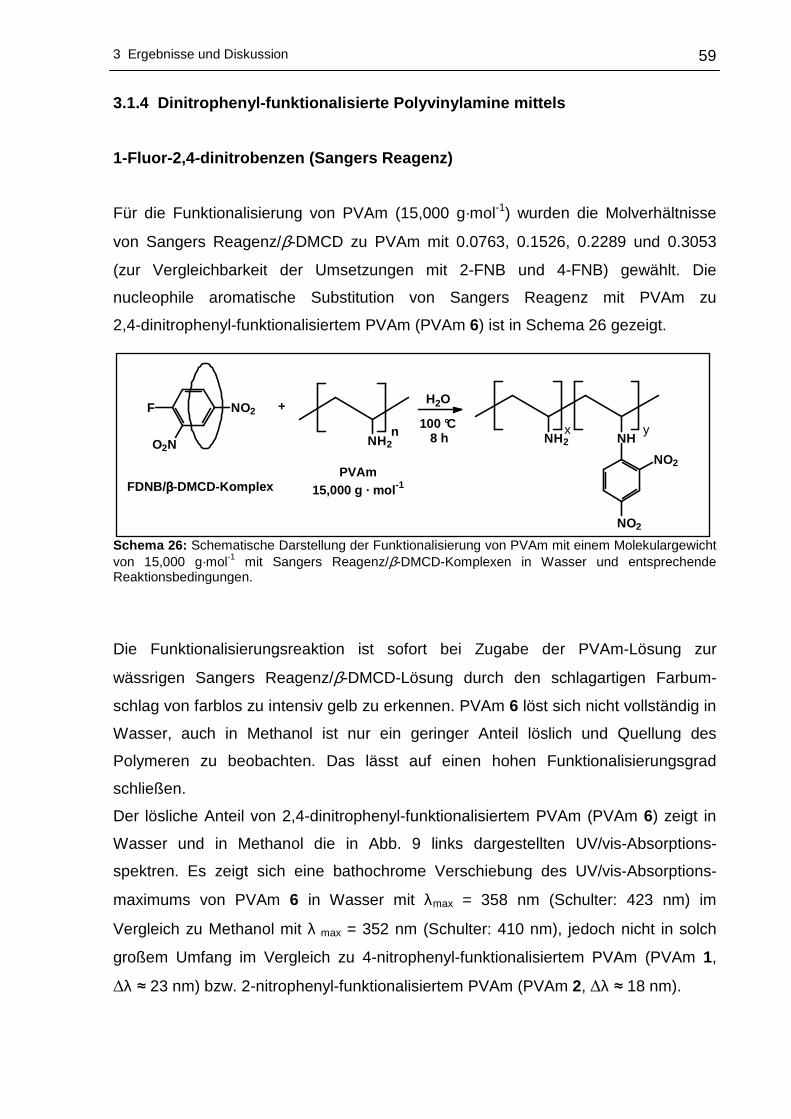
3 Ergebnisse und Diskussion
59
3.1.4 Dinitrophenyl-funktionalisierte Polyvinylami ne mittels
1-Fluor-2,4-dinitrobenzen (Sangers Reagenz)
Für die Funktionalisierung von PVAm (15,000 g·mol-1) wurden die Molverhältnisse
von Sangers Reagenz/β-DMCD zu PVAm mit 0.0763, 0.1526, 0.2289 und 0.3053
(zur Vergleichbarkeit der Umsetzungen mit 2-FNB und 4-FNB) gewählt. Die
nucleophile aromatische Substitution von Sangers Reagenz mit PVAm zu
2,4-dinitrophenyl-funktionalisiertem PVAm (PVAm 6) ist in Schema 26 gezeigt.
Schema 26: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 mit Sangers Reagenz/β-DMCD-Komplexen in Wasser und entsprechende Reaktionsbedingungen.
Die Funktionalisierungsreaktion ist sofort bei Zugabe der PVAm-Lösung zur
wässrigen Sangers Reagenz/β-DMCD-Lösung durch den schlagartigen Farbum-
schlag von farblos zu intensiv gelb zu erkennen. PVAm 6 löst sich nicht vollständig in
Wasser, auch in Methanol ist nur ein geringer Anteil löslich und Quellung des
Polymeren zu beobachten. Das lässt auf einen hohen Funktionalisierungsgrad
schließen.
Der lösliche Anteil von 2,4-dinitrophenyl-funktionalisiertem PVAm (PVAm 6) zeigt in
Wasser und in Methanol die in Abb. 9 links dargestellten UV/vis-Absorptions-
spektren. Es zeigt sich eine bathochrome Verschiebung des UV/vis-Absorptions-
maximums von PVAm 6 in Wasser mit λmax = 358 nm (Schulter: 423 nm) im
Vergleich zu Methanol mit λ max = 352 nm (Schulter: 410 nm), jedoch nicht in solch
großem Umfang im Vergleich zu 4-nitrophenyl-funktionalisiertem PVAm (PVAm 1,
∆λ ≈ 23 nm) bzw. 2-nitrophenyl-funktionalisiertem PVAm (PVAm 2, ∆λ ≈ 18 nm).
F NO2
O2N NH2 NH
NO2
NO2
x y
+
NH2n
H2O
FDNB/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
100 °C8 h

3 Ergebnisse und Diskussion
60
Abb. 9: Links: UV/vis-Absorptionsspektren des löslichen Anteils von 2,4-dinitrophenyl-funktionalisiertem PVAm (PVAm 6) in Wasser und in Methanol mit Fotografie des festen Polymeren und rechts: UV/vis-Absorptionsspektrum von 2,4-Dinitro-N-isopropylamin als Modellverbindung in Methanol.
Im festen Zustand hat PVAm 6 eine orange bis orange-braune Farbe
(s. Fotografie in Abb. 9). Die Resonanzstabilisierung der chromophoren Einheit von
PVAm 6 kann sowohl über die ortho-Nitrogruppe als auch über die para-Nitrogruppe
erfolgen.
Aufgrund der strukturellen Ähnlichkeit zur chromophoren Einheit des Polymeren
wurde 2,4-Dinitro-N-isopropylanilin als Modellverbindung ausgewählt. Die Synthese
erfolgte durch nucleophile aromatische Substitution von Sangers Reagenz mit
i-Propylamin in Toluol (s. Abschnitt 5.6. Experimenteller Teil).
Die Lage und Form der UV/vis-Absorptionsbande des löslichen Anteils von
2,4-dinitrophenyl-funktionalisiertem PVAm (PVAm 6, s. Abb. 9 links) weist mit
Vergleich zur Modellverbindung in Methanol (s. Abb. 9 rechts) auf die erfolgreiche
Bildung des π-Elektronen-push-pull-Systems - chromophore Einheit von PVAm 6 -
hin. Die schlechte Löslichkeit sowohl in Wasser als auch in Methanol deutet auf
hohen Umsatz an Sangers Reagenz und/oder Vernetzung der Polymerketten
untereinander hin, was in einem späteren Abschnitt näher diskutiert wird.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]NH2 NH
NO2
NO2
x y
410
15,000 g · mol -1
pH = 11
0.0
0.2
0.4
0.6
0.8
1.0
1.2
300 400 500 600 700
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
N
CH3
H3CH
NO2
NO2
349
410
2,4-Dinitro- N -isopropylanilin
352
MeOH
H2O
423
358

3 Ergebnisse und Diskussion
61
1,5-Difluor-2,4-dinitrobenzen (DFDNB)
Für die nucleophile aromatische Substitution von DFDNB mit PVAm (15,000 g·mol-1)
wurde ein Molverhältnis von DFDNB/β-DMCD zu PVAm mit 0.3053 gewählt. Die
Funktionalisierung von PVAm in Wasser mit DFDNB mittels β-DMCD als
Löslichkeitsvermittler zu 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm
(PVAm 7) ist in Schema 27 verdeutlicht.
Schema 27: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 mit DFDNB/β-DMCD-Komplexen in Wasser und entsprechende Reaktionsbedingungen.
PVAm 7 löst sich sowohl in Wasser als auch in Methanol nur sehr schlecht. Der
Vergleich der UV/vis-Absorptionsspektren des in Wasser und des in Methanol
löslichen Anteils von PVAm 7 mit der entsprechenden Modellverbindung 2,4-Dinitro-
N,N´-diisopropyl-1,5-phenylendiamin (Synthese und Charakterisierung siehe
Abschnitt 5.6) ist in Abb. 10 gezeigt. Als Feststoff hat PVAm 7 eine orange bis
orange-braune Farbe, was die Fotografie in Abb. 10 verdeutlichen soll.
Es ist erwartungsgemäß eine bathochrome Verschiebung der UV/vis-Absorptions-
doppelbande in Wasser (λ1 = 338 nm und λ2 = 423 nm) im Vergleich zu Methanol
(λ1 = 332 nm und λ2 = 414 nm) zu erkennen. Der solvatochrome Umfang ist jedoch
aufgrund der Kreuzkonjugation nicht so groß im Vergleich zum 2-nitrophenyl-
funktionalisiertem PVAm (PVAm 2) bzw. 4-nitrophenyl-funktionalisiertem PVAm
(PVAm 1).
Die entsprechende niedermolekulare Modellverbindung 2,4-Dinitro-N,N´-diisopropyl-
1,5-phenylendiamin zeigt ebenfalls eine UV/vis-Absorptionsdoppelbande mit
λ1 = 332 nm und λ2 = 417 nm (s. Abb. 10 rechts).
F
O2N
F
NO2
+
NH2n
H2O
DFDNB/ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
100 °C8 h NH2 NH
NO2
NO2
N
x y
H

3 Ergebnisse und Diskussion
62
Abb. 10: Links: UV/vis-Absorptionsspektren des löslichen Anteils von 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm (PVAm 7) in Wasser und in Methanol mit Fotografie des festen Polymeren und rechts: UV/vis-Absorptionsspektrum von 2,4-Dinitro-N,N´-diisopropyl-1,5-phenylendiamin als Modellverbindung in Methanol.
Die Lage und Form der UV/vis-Absorptionsbanden von 2,4-dinitro-1,5-
phenylendiamin-funktionalisiertenm PVAm (PVAm 7) deuten mit dem Vergleich des
UV/vis-Absorptionsspektrums der Modellverbindung auf Disubstitution von
1,5-Difluor-2,4-dinitrobenzen durch PVAm hin. Auch aufgrund der schlechten
Löslichkeit ist von teilweiser Disubstitution beider Fluorsubstituenten, und damit von
partieller Vernetzung der Polymerketten untereinander auszugehen, was später in
Abschnitt 3.2.3 näher diskutiert wird.
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
338
423
NH2 NH
NO2
NO2
N
x y
H
-0.1
0.1
0.3
0.5
0.7
0.9
1.1
1.3
1.5
1.7
300 400 500 600 700
Wellenlänge λλλλ [nm]A
bsor
ptio
n in
MeO
H [a
.u.]
332
417
NN
NO2O2N
HH
414
332
H2O
MeOH

3 Ergebnisse und Diskussion
63
3.1.5 2-Nitro-4-azobenzen-funktionalisiertes Polyv inylamin mittels
4-Fluor-3-nitroazobenzen (FNAzoB)
Für die Funktionalisierung von PVAm (15,000 g·mol-1) wurden die Molverhältnisse
von FNAzoB/β-DMCD zu PVAm mit 0.0763, 0.1526, 0.2289 und 0.3053 gewählt. Die
nucleophile aromatische Substitution von FNAzoB/β-DMCD mit PVAm zu 2-nitro-4-
azobenzen-funktionalisiertem PVAm (PVAm 8) ist in Schema 28 exemplarisch
gezeigt.
Schema 28: Schematische Darstellung der Funktionalisierung von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 mit FNAzoB/β-DMCD-Komplexen in Wasser und entsprechende Reaktionsbedingungen.
PVAm 8 löst sich in Wasser nur teilweise und in Methanol ist PVAm 8 sogar
unlöslich. Das könnte ein Indiz auf einen hohen Substitutionsgrad und damit einen
hohen Umsatz an 4-Fluor-3-nitroazobenzen sein.
Abb. 11 links zeigt ein UV/vis-Absorptionsspektrum des in Wasser löslichen Anteils
von 2-nitro-4-azobenzen-funktionalisiertem PVAm (PVAm 8). Der Vergleich der
UV/vis-Absorptionsspektren der entsprechenden Modellverbindung N-Isopropyl-4-
azobenzen-2-nitroanilin (Abb. 11 rechts), welche aus i-Propylamin und 4-Fluor-3-
nitroazobenzen synthetisiert wurde (Synthesedurchführung und Charakterisierung
siehe Abschnitt 5.6), zeigt in Methanol eine ähnliche Lage und Form der UV/vis-
Absorptionsbande wie der wasserlösliche Anteil von PVAm 8.
F N
O2N
N
NH2 NH
NO2
NN
x y+
NH2n
H2O
FNAzoB/ ββββ-DMCD-KomplexPVAm
15,000 g · mol -1
100 °C8 h
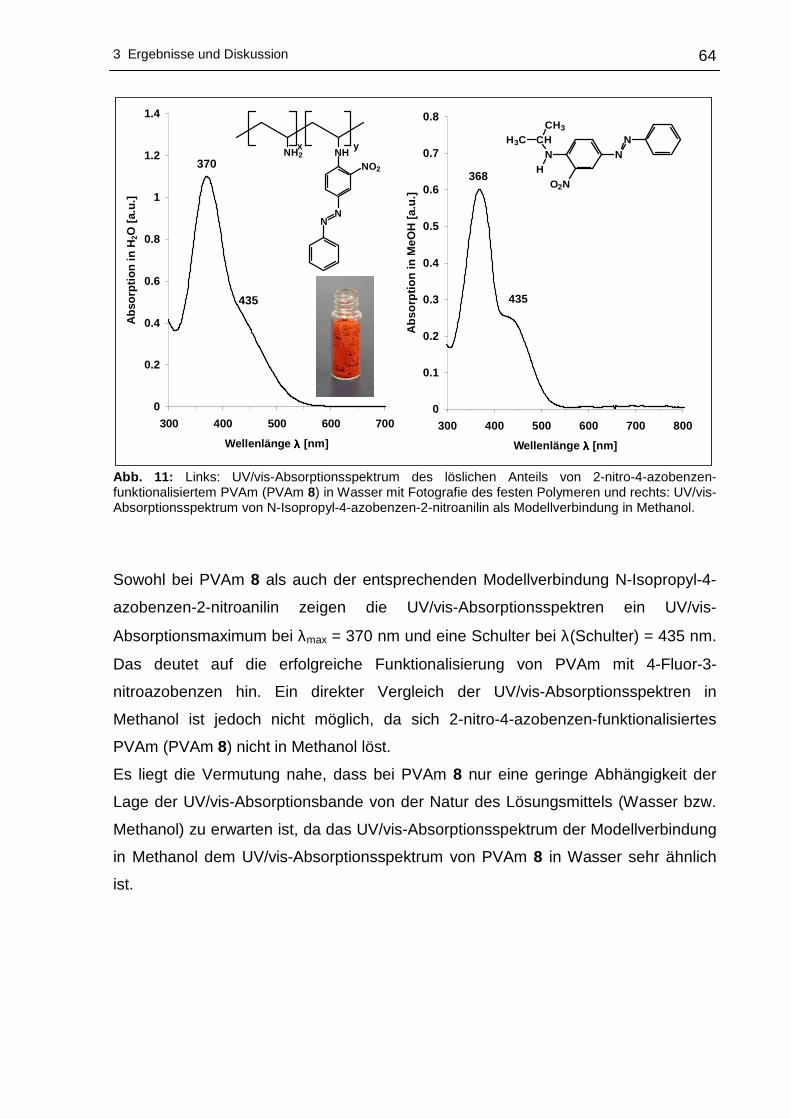
3 Ergebnisse und Diskussion
64
Abb. 11: Links: UV/vis-Absorptionsspektrum des löslichen Anteils von 2-nitro-4-azobenzen-funktionalisiertem PVAm (PVAm 8) in Wasser mit Fotografie des festen Polymeren und rechts: UV/vis-Absorptionsspektrum von N-Isopropyl-4-azobenzen-2-nitroanilin als Modellverbindung in Methanol.
Sowohl bei PVAm 8 als auch der entsprechenden Modellverbindung N-Isopropyl-4-
azobenzen-2-nitroanilin zeigen die UV/vis-Absorptionsspektren ein UV/vis-
Absorptionsmaximum bei λmax = 370 nm und eine Schulter bei λ(Schulter) = 435 nm.
Das deutet auf die erfolgreiche Funktionalisierung von PVAm mit 4-Fluor-3-
nitroazobenzen hin. Ein direkter Vergleich der UV/vis-Absorptionsspektren in
Methanol ist jedoch nicht möglich, da sich 2-nitro-4-azobenzen-funktionalisiertes
PVAm (PVAm 8) nicht in Methanol löst.
Es liegt die Vermutung nahe, dass bei PVAm 8 nur eine geringe Abhängigkeit der
Lage der UV/vis-Absorptionsbande von der Natur des Lösungsmittels (Wasser bzw.
Methanol) zu erwarten ist, da das UV/vis-Absorptionsspektrum der Modellverbindung
in Methanol dem UV/vis-Absorptionsspektrum von PVAm 8 in Wasser sehr ähnlich
ist.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
300 400 500 600 700
Wellenlänge λ λ λ λ [nm]
Abs
orpt
ion
in H
2O [a
.u.]
370NH2 NH
NO2
NN
x y
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
368
435
NCH
NN
O2NH
CH3
H3C
435

3 Ergebnisse und Diskussion
65
3.1.6 2,4´-Dinitrostilben-funktionalisiertes Polyv inylamin mittels
4-Fluor-3,4´-dinitrostilben (FDNstilben)
4-Fluor-3,4´-dinitrostilben (FDNstilben) wurde über eine Horner-Wadsworth-Emmons-
Reaktion119,120 aus kommerziell erhältlichen 4-Fluor-3-nitrobenzaldehyd und
4-Nitrobenzyl-diethylphosphonat mit Natriumethanolat (NaOEt) als Base in THF
synthetisiert. Schema 29 zeigt eine schematische Darstellung des
Reaktionsmechanismus einer Horner-Wadsworth-Emmons-Reaktion zum
gewünschten 4-Fluor-3,4´-dinitrostilben (FDNstilben).119,120
Schema 29: Schematische Darstellung des Mechanismus der Reaktion von 4-Fluor-3-nitrobenzaldehyd mit 4-Nitrobenzyldiethylphosphonat mittels Natriumethanolat als Base zu 4-Fluor-3,4´-dinitrostilben.
Die Synthesedurchführung und Charakterisierung des Fluoraromaten ist in Abschnitt
5.6.2 (Experimenteller Teil) ausführlich dargelegt. Ein Vorteil dieser Reaktion
gegenüber der Wittig-Reaktion ist die Wasserlöslichkeit des Phosphorsäureesters als
Nebenprodukt, der leichter abgetrennt werden kann als das bei der Wittig-Reaktion
gebildete Triphenylphospinoxid.48 Ein Nachteil ist die hier eingesetzte Base
Natriumethanolat, die durch Substitution des Fluorsubstituenten am 4-Fluor-3-
nitrobenzaldehyd zu 4-Ethoxy-3,4´-dinitrostilben als Nebenprodukt führen kann.
+
RCP
O H
HEtO
EtO
+
EtOH
Na EtO
Na
RCP
HOEtO
EtO
F
NO2
CO
H
Na
P
O
RC
H
CHO
F
NO2
EtO
EtO
Na
P
O
RC
H
CH
F
NO2
EtO
EtOOF
NO2
CC
H
H
O2NNa
P
OEtO
EtOO
NO2R:
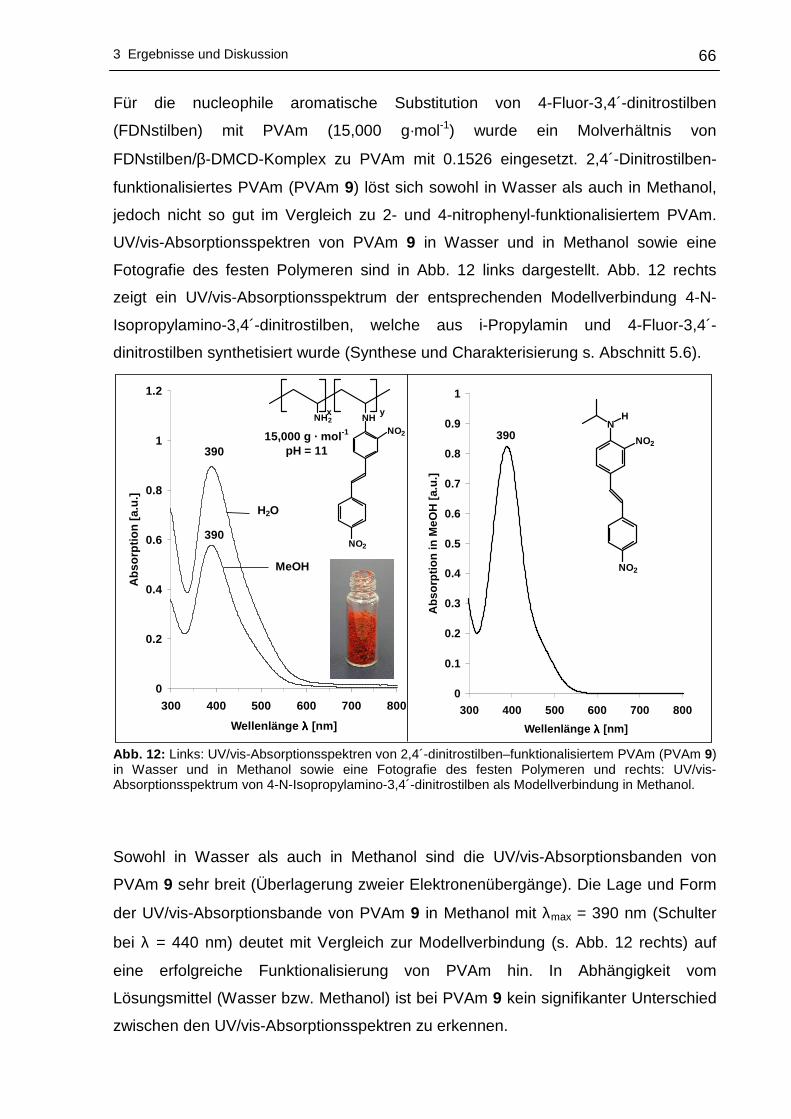
3 Ergebnisse und Diskussion
66
Für die nucleophile aromatische Substitution von 4-Fluor-3,4´-dinitrostilben
(FDNstilben) mit PVAm (15,000 g·mol-1) wurde ein Molverhältnis von
FDNstilben/β-DMCD-Komplex zu PVAm mit 0.1526 eingesetzt. 2,4´-Dinitrostilben-
funktionalisiertes PVAm (PVAm 9) löst sich sowohl in Wasser als auch in Methanol,
jedoch nicht so gut im Vergleich zu 2- und 4-nitrophenyl-funktionalisiertem PVAm.
UV/vis-Absorptionsspektren von PVAm 9 in Wasser und in Methanol sowie eine
Fotografie des festen Polymeren sind in Abb. 12 links dargestellt. Abb. 12 rechts
zeigt ein UV/vis-Absorptionsspektrum der entsprechenden Modellverbindung 4-N-
Isopropylamino-3,4´-dinitrostilben, welche aus i-Propylamin und 4-Fluor-3,4´-
dinitrostilben synthetisiert wurde (Synthese und Charakterisierung s. Abschnitt 5.6).
Abb. 12: Links: UV/vis-Absorptionsspektren von 2,4´-dinitrostilben–funktionalisiertem PVAm (PVAm 9) in Wasser und in Methanol sowie eine Fotografie des festen Polymeren und rechts: UV/vis-Absorptionsspektrum von 4-N-Isopropylamino-3,4´-dinitrostilben als Modellverbindung in Methanol.
Sowohl in Wasser als auch in Methanol sind die UV/vis-Absorptionsbanden von
PVAm 9 sehr breit (Überlagerung zweier Elektronenübergänge). Die Lage und Form
der UV/vis-Absorptionsbande von PVAm 9 in Methanol mit λmax = 390 nm (Schulter
bei λ = 440 nm) deutet mit Vergleich zur Modellverbindung (s. Abb. 12 rechts) auf
eine erfolgreiche Funktionalisierung von PVAm hin. In Abhängigkeit vom
Lösungsmittel (Wasser bzw. Methanol) ist bei PVAm 9 kein signifikanter Unterschied
zwischen den UV/vis-Absorptionsspektren zu erkennen.
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.] H2O
MeOH
390
390
NH2 NHNO2
NO2
x y
15,000 g · mol -1
pH = 11
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]390
N
NO2
NO2
H
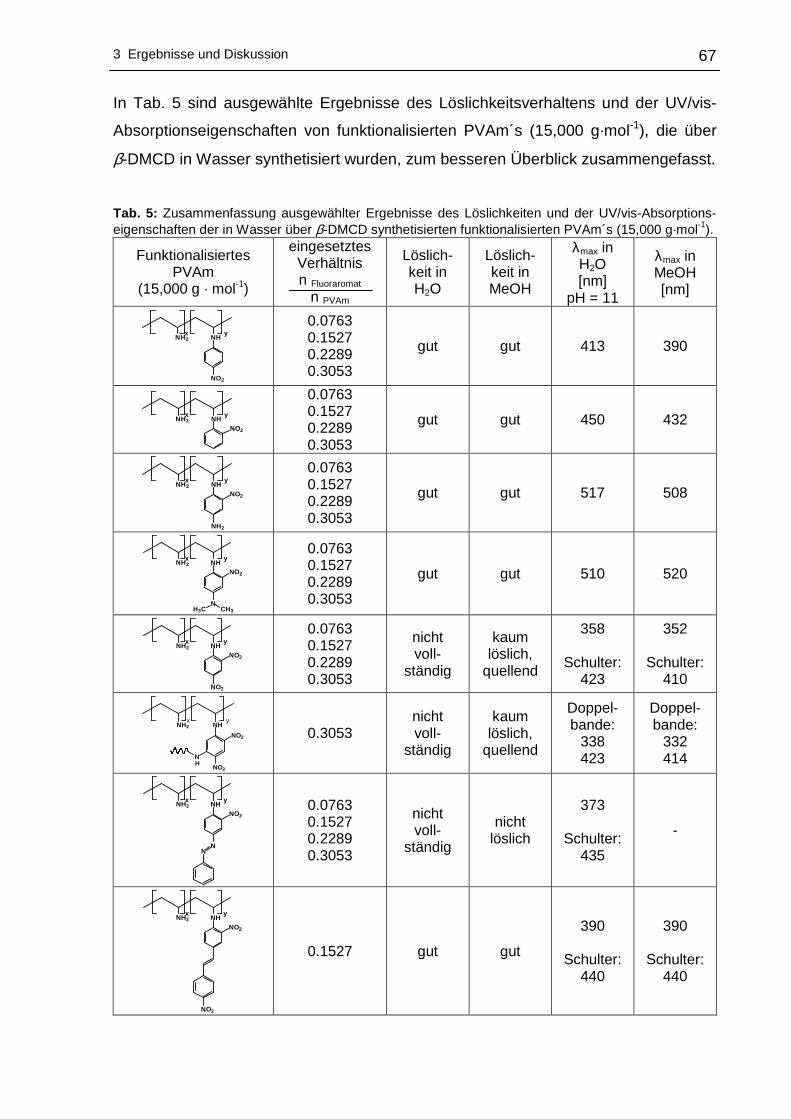
3 Ergebnisse und Diskussion
67
In Tab. 5 sind ausgewählte Ergebnisse des Löslichkeitsverhaltens und der UV/vis-
Absorptionseigenschaften von funktionalisierten PVAm´s (15,000 g·mol-1), die über
β-DMCD in Wasser synthetisiert wurden, zum besseren Überblick zusammengefasst.
Tab. 5: Zusammenfassung ausgewählter Ergebnisse des Löslichkeiten und der UV/vis-Absorptions-eigenschaften der in Wasser über β-DMCD synthetisierten funktionalisierten PVAm´s (15,000 g·mol-1).
Funktionalisiertes PVAm
(15,000 g · mol-1)
eingesetztes Verhältnis n Fluoraromat
n PVAm
Löslich-keit in H2O
Löslich-keit in MeOH
λmax in H2O [nm]
pH = 11
λmax in MeOH [nm]
0.0763 0.1527 0.2289 0.3053
gut gut 413 390
NH2 NHNO2
x y
0.0763 0.1527 0.2289 0.3053
gut gut 450 432
NH2 NHNO2
NH2
x y
0.0763 0.1527 0.2289 0.3053
gut gut 517 508
NH2 NHNO2
NH3C CH3
x y
0.0763 0.1527 0.2289 0.3053
gut gut 510 520
NH2 NHNO2
NO2
x y
0.0763 0.1527 0.2289 0.3053
nicht voll-
ständig
kaum löslich,
quellend
358
Schulter: 423
352
Schulter: 410
NH2 NH
NO2
NO2
N
x y
H
0.3053 nicht voll-
ständig
kaum löslich,
quellend
Doppel-bande:
338 423
Doppel-bande:
332 414
NH2 NHNO2
NN
x y
0.0763 0.1527 0.2289 0.3053
nicht voll-
ständig
nicht löslich
373
Schulter: 435
-
NH2 NHNO2
NO2
x y
0.1527 gut gut
390
Schulter: 440
390
Schulter: 440
NH2 NH
NO2
x y
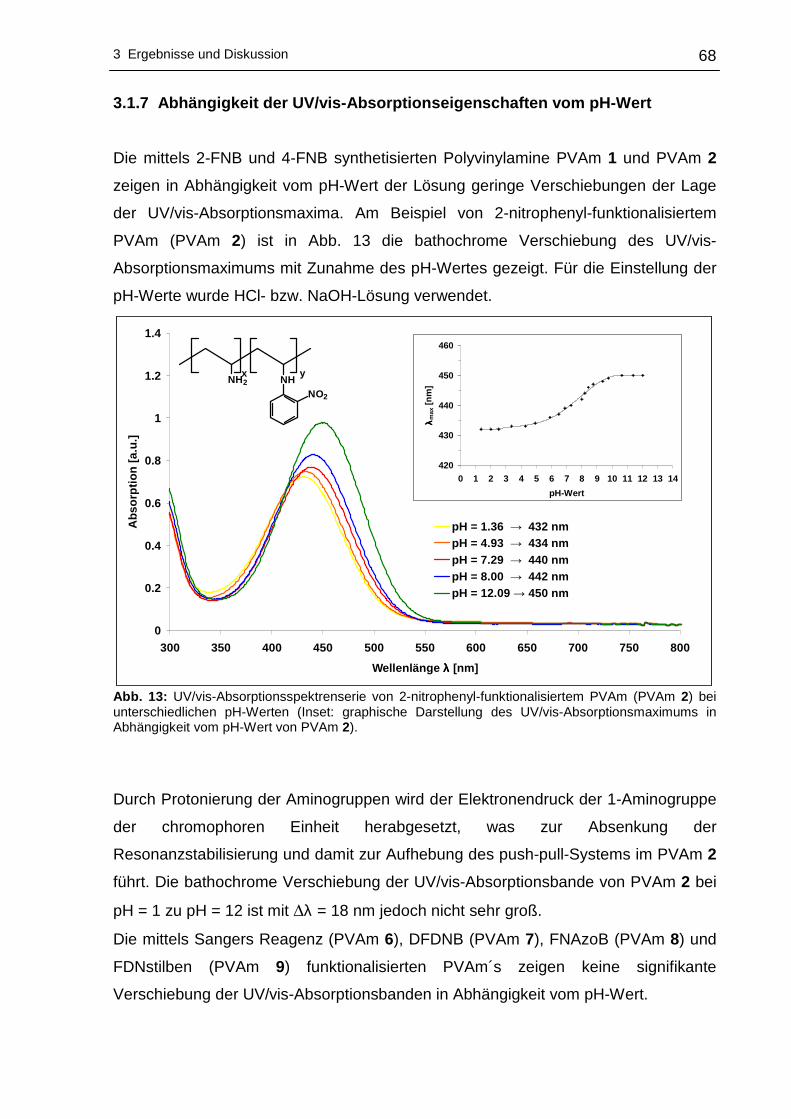
3 Ergebnisse und Diskussion
68
3.1.7 Abhängigkeit der UV/vis-Absorptionseigenscha ften vom pH-Wert
Die mittels 2-FNB und 4-FNB synthetisierten Polyvinylamine PVAm 1 und PVAm 2
zeigen in Abhängigkeit vom pH-Wert der Lösung geringe Verschiebungen der Lage
der UV/vis-Absorptionsmaxima. Am Beispiel von 2-nitrophenyl-funktionalisiertem
PVAm (PVAm 2) ist in Abb. 13 die bathochrome Verschiebung des UV/vis-
Absorptionsmaximums mit Zunahme des pH-Wertes gezeigt. Für die Einstellung der
pH-Werte wurde HCl- bzw. NaOH-Lösung verwendet.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
300 350 400 450 500 550 600 650 700 750 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
pH = 1.36 → 432 nmpH = 4.93 → 434 nmpH = 7.29 → 440 nmpH = 8.00 → 442 nmpH = 12.09 → 450 nm
NH2 NHNO2
x y
420
430
440
450
460
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
pH-Wert
λλ λλ max
[nm
]
Abb. 13: UV/vis-Absorptionsspektrenserie von 2-nitrophenyl-funktionalisiertem PVAm (PVAm 2) bei unterschiedlichen pH-Werten (Inset: graphische Darstellung des UV/vis-Absorptionsmaximums in Abhängigkeit vom pH-Wert von PVAm 2).
Durch Protonierung der Aminogruppen wird der Elektronendruck der 1-Aminogruppe
der chromophoren Einheit herabgesetzt, was zur Absenkung der
Resonanzstabilisierung und damit zur Aufhebung des push-pull-Systems im PVAm 2
führt. Die bathochrome Verschiebung der UV/vis-Absorptionsbande von PVAm 2 bei
pH = 1 zu pH = 12 ist mit ∆λ = 18 nm jedoch nicht sehr groß.
Die mittels Sangers Reagenz (PVAm 6), DFDNB (PVAm 7), FNAzoB (PVAm 8) und
FDNstilben (PVAm 9) funktionalisierten PVAm´s zeigen keine signifikante
Verschiebung der UV/vis-Absorptionsbanden in Abhängigkeit vom pH-Wert.

3 Ergebnisse und Diskussion
69
Zusammenfassend sind in Tabelle 6 die Änderungen der UV/vis-Absorptions-
eigenschaften der chromophoren Einheiten von PVAm 1, PVAm 2, PVAm 6,
PVAm 7, PVAm 8 und PVAm 9 bei Änderung des pH-Wertes gezeigt.
Tab. 6: Zusammenfassung der UV/vis-Absorptionseigenschaften der mittels 4-FNB, 2-FNB, Sangers Reagenz, DFDNB, FNAzoB und FDNstilben funktionalisierten PVAm´s bei pH=1, pH=7 und pH=11.
λmax [nm] von funktionalisiertem PVAm eingesetzter
Fluoraromat pH = 1 pH = 7 pH = 11
F NO2
390 406 413
F
O2N 432 440 450
F
O2N
NO2
350
Schulter: 410
354
Schulter: 415
358
Schulter: 423
F
O2N
NO2
F
Doppelbande
335
416
Doppelbande
338
423
Doppelbande
338
423
F
O2N
NN
370
Schulter: 435
370
Schulter: 435
370
Schulter: 435
F
O2N
NO2
390
Schulter: 440
390
Schulter: 440
390
Schulter: 440
Sehr interessante UV/vis-Absorptionseigenschaften in Abhängigkeit vom
pH-Wert zeigen die mittels 4-FNA und 4-DMFNA funktionalisierten Polyvinylamine
PVAm 3, PVAm 4 und PNMVAm 5 auf die nachfolgend näher eingegangen wird.
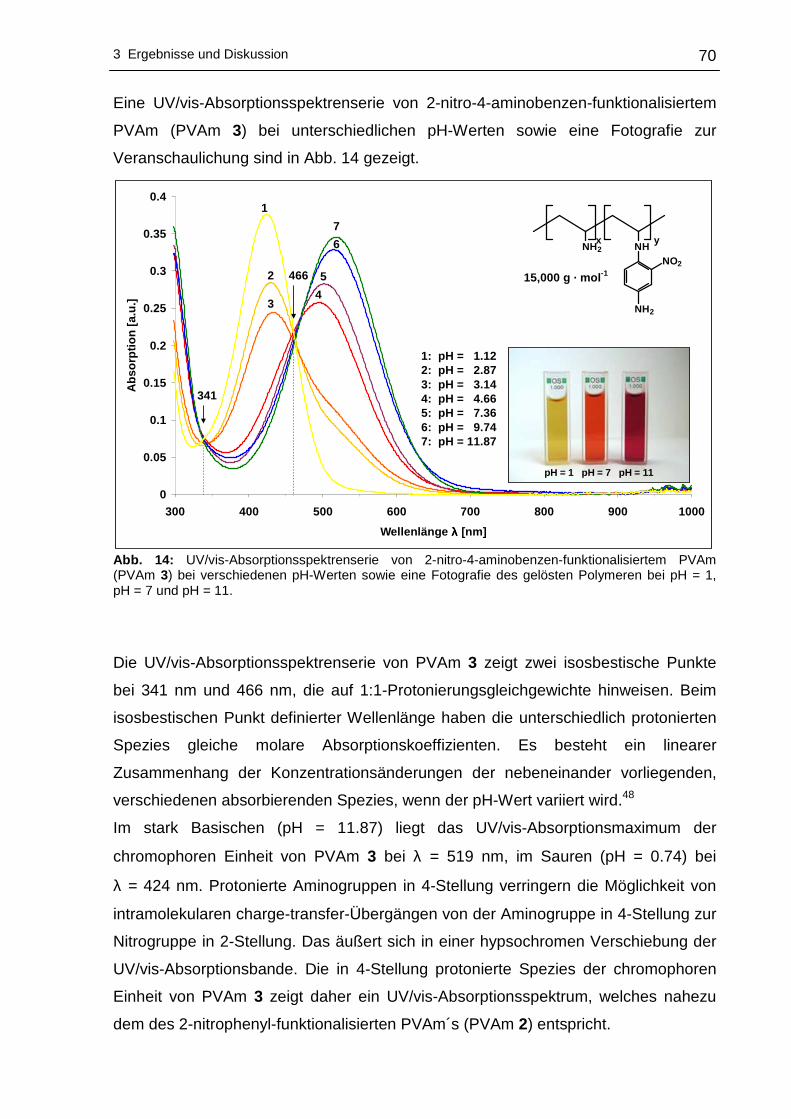
3 Ergebnisse und Diskussion
70
Eine UV/vis-Absorptionsspektrenserie von 2-nitro-4-aminobenzen-funktionalisiertem
PVAm (PVAm 3) bei unterschiedlichen pH-Werten sowie eine Fotografie zur
Veranschaulichung sind in Abb. 14 gezeigt.
Abb. 14: UV/vis-Absorptionsspektrenserie von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) bei verschiedenen pH-Werten sowie eine Fotografie des gelösten Polymeren bei pH = 1, pH = 7 und pH = 11.
Die UV/vis-Absorptionsspektrenserie von PVAm 3 zeigt zwei isosbestische Punkte
bei 341 nm und 466 nm, die auf 1:1-Protonierungsgleichgewichte hinweisen. Beim
isosbestischen Punkt definierter Wellenlänge haben die unterschiedlich protonierten
Spezies gleiche molare Absorptionskoeffizienten. Es besteht ein linearer
Zusammenhang der Konzentrationsänderungen der nebeneinander vorliegenden,
verschiedenen absorbierenden Spezies, wenn der pH-Wert variiert wird.48
Im stark Basischen (pH = 11.87) liegt das UV/vis-Absorptionsmaximum der
chromophoren Einheit von PVAm 3 bei λ = 519 nm, im Sauren (pH = 0.74) bei
λ = 424 nm. Protonierte Aminogruppen in 4-Stellung verringern die Möglichkeit von
intramolekularen charge-transfer-Übergängen von der Aminogruppe in 4-Stellung zur
Nitrogruppe in 2-Stellung. Das äußert sich in einer hypsochromen Verschiebung der
UV/vis-Absorptionsbande. Die in 4-Stellung protonierte Spezies der chromophoren
Einheit von PVAm 3 zeigt daher ein UV/vis-Absorptionsspektrum, welches nahezu
dem des 2-nitrophenyl-funktionalisierten PVAm´s (PVAm 2) entspricht.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
300 400 500 600 700 800 900 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
466
341
1: pH = 1.122: pH = 2.873: pH = 3.144: pH = 4.665: pH = 7.366: pH = 9.747: pH = 11.87
1
2
34
5
6
7
pH = 1 pH = 7 pH = 11
NH2 NHNO2
NH2
x y
15,000 g · mol -1
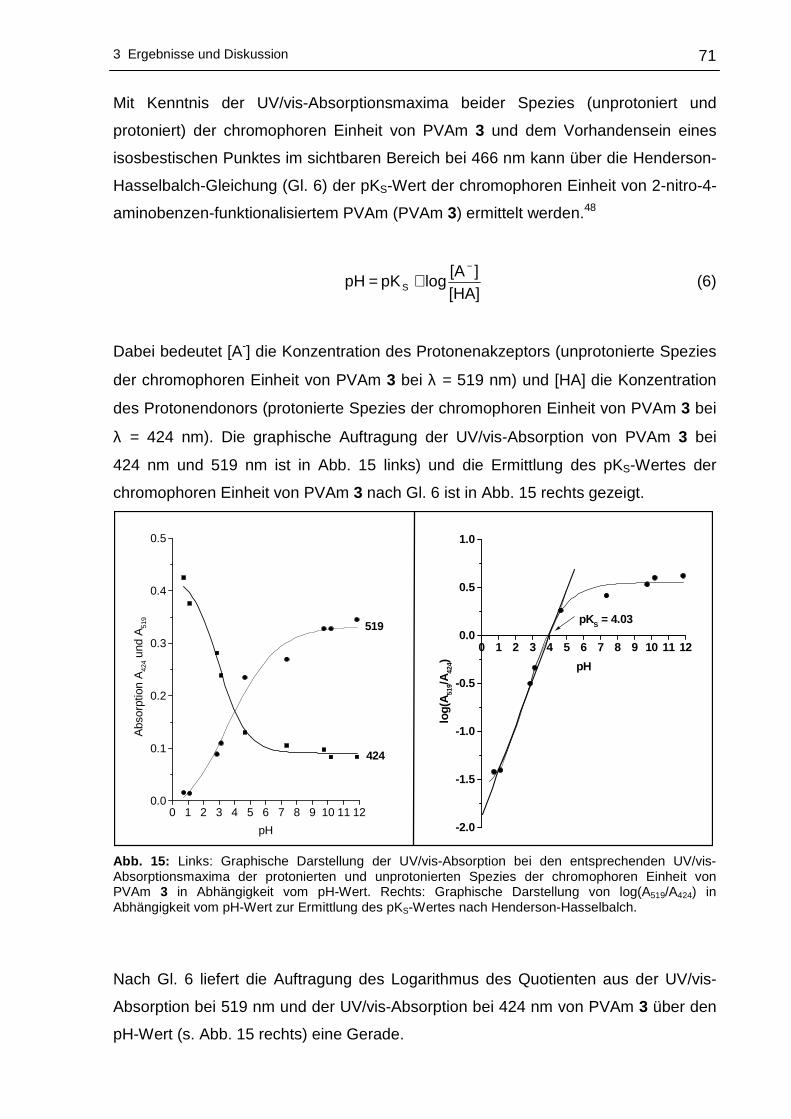
3 Ergebnisse und Diskussion
71
Mit Kenntnis der UV/vis-Absorptionsmaxima beider Spezies (unprotoniert und
protoniert) der chromophoren Einheit von PVAm 3 und dem Vorhandensein eines
isosbestischen Punktes im sichtbaren Bereich bei 466 nm kann über die Henderson-
Hasselbalch-Gleichung (Gl. 6) der pKS-Wert der chromophoren Einheit von 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) ermittelt werden.48
[HA]][A
logpKpH S
−
+= (6)
Dabei bedeutet [A-] die Konzentration des Protonenakzeptors (unprotonierte Spezies
der chromophoren Einheit von PVAm 3 bei λ = 519 nm) und [HA] die Konzentration
des Protonendonors (protonierte Spezies der chromophoren Einheit von PVAm 3 bei
λ = 424 nm). Die graphische Auftragung der UV/vis-Absorption von PVAm 3 bei
424 nm und 519 nm ist in Abb. 15 links) und die Ermittlung des pKS-Wertes der
chromophoren Einheit von PVAm 3 nach Gl. 6 ist in Abb. 15 rechts gezeigt.
Abb. 15: Links: Graphische Darstellung der UV/vis-Absorption bei den entsprechenden UV/vis-Absorptionsmaxima der protonierten und unprotonierten Spezies der chromophoren Einheit von PVAm 3 in Abhängigkeit vom pH-Wert. Rechts: Graphische Darstellung von log(A519/A424) in Abhängigkeit vom pH-Wert zur Ermittlung des pKS-Wertes nach Henderson-Hasselbalch.
Nach Gl. 6 liefert die Auftragung des Logarithmus des Quotienten aus der UV/vis-
Absorption bei 519 nm und der UV/vis-Absorption bei 424 nm von PVAm 3 über den
pH-Wert (s. Abb. 15 rechts) eine Gerade.
0 1 2 3 4 5 6 7 8 9 10 11 12
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
pKS = 4.03
log(
A51
9/A42
4) pH
0 1 2 3 4 5 6 7 8 9 10 11 120.0
0.1
0.2
0.3
0.4
0.5
424
519
Abs
orpt
ion
A42
4 und
A51
9
pH
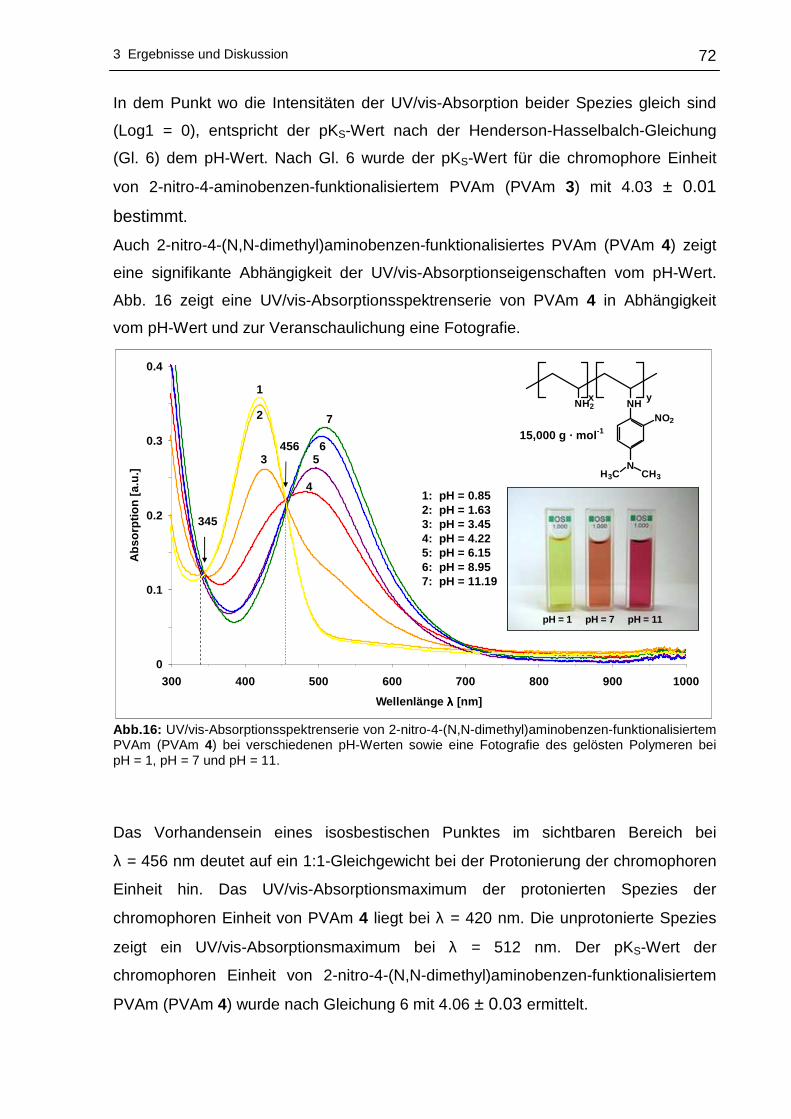
3 Ergebnisse und Diskussion
72
In dem Punkt wo die Intensitäten der UV/vis-Absorption beider Spezies gleich sind
(Log1 = 0), entspricht der pKS-Wert nach der Henderson-Hasselbalch-Gleichung
(Gl. 6) dem pH-Wert. Nach Gl. 6 wurde der pKS-Wert für die chromophore Einheit
von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) mit 4.03 ± 0.01
bestimmt.
Auch 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PVAm (PVAm 4) zeigt
eine signifikante Abhängigkeit der UV/vis-Absorptionseigenschaften vom pH-Wert.
Abb. 16 zeigt eine UV/vis-Absorptionsspektrenserie von PVAm 4 in Abhängigkeit
vom pH-Wert und zur Veranschaulichung eine Fotografie.
Abb.16: UV/vis-Absorptionsspektrenserie von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) bei verschiedenen pH-Werten sowie eine Fotografie des gelösten Polymeren bei pH = 1, pH = 7 und pH = 11.
Das Vorhandensein eines isosbestischen Punktes im sichtbaren Bereich bei
λ = 456 nm deutet auf ein 1:1-Gleichgewicht bei der Protonierung der chromophoren
Einheit hin. Das UV/vis-Absorptionsmaximum der protonierten Spezies der
chromophoren Einheit von PVAm 4 liegt bei λ = 420 nm. Die unprotonierte Spezies
zeigt ein UV/vis-Absorptionsmaximum bei λ = 512 nm. Der pKS-Wert der
chromophoren Einheit von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem
PVAm (PVAm 4) wurde nach Gleichung 6 mit 4.06 ± 0.03 ermittelt.
0
0.1
0.2
0.3
0.4
300 400 500 600 700 800 900 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
456
345
1: pH = 0.852: pH = 1.633: pH = 3.454: pH = 4.225: pH = 6.156: pH = 8.957: pH = 11.19
1
2
3
4
56
7NH2 NH
NO2
NH3C CH3
x y
15,000 g · mol -1
pH = 1 pH = 7 pH = 11
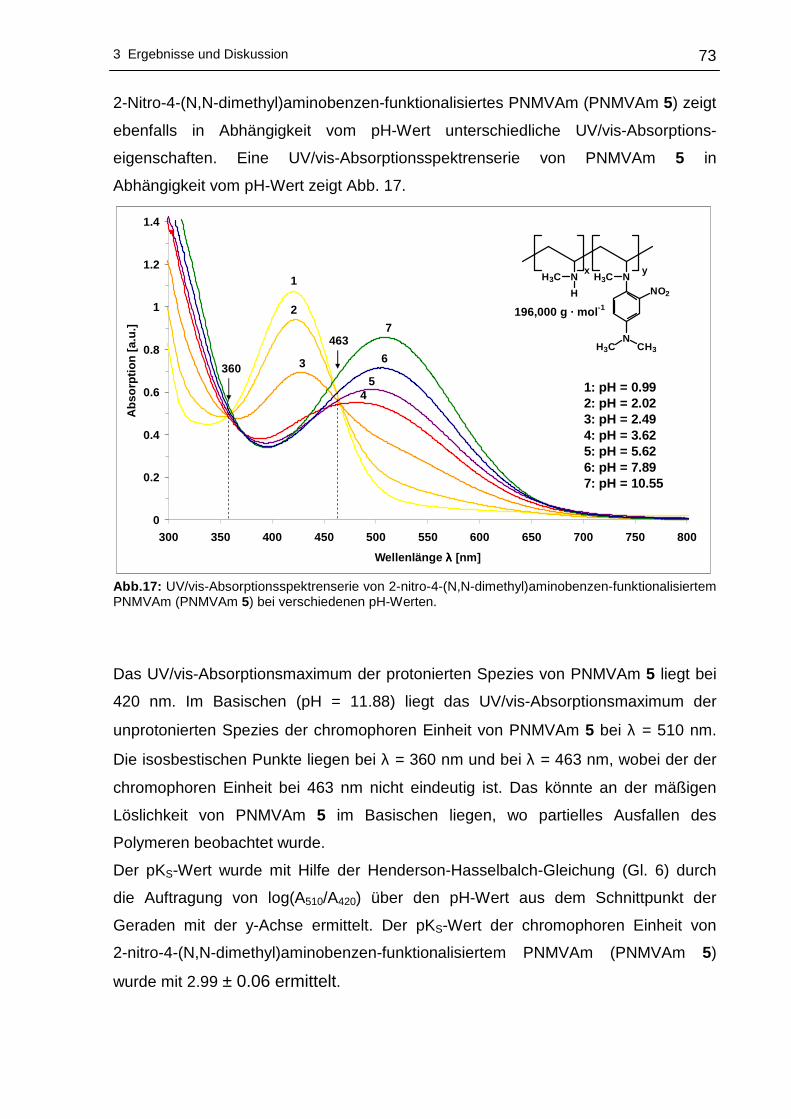
3 Ergebnisse und Diskussion
73
2-Nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PNMVAm (PNMVAm 5) zeigt
ebenfalls in Abhängigkeit vom pH-Wert unterschiedliche UV/vis-Absorptions-
eigenschaften. Eine UV/vis-Absorptionsspektrenserie von PNMVAm 5 in
Abhängigkeit vom pH-Wert zeigt Abb. 17.
Abb.17: UV/vis-Absorptionsspektrenserie von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5) bei verschiedenen pH-Werten.
Das UV/vis-Absorptionsmaximum der protonierten Spezies von PNMVAm 5 liegt bei
420 nm. Im Basischen (pH = 11.88) liegt das UV/vis-Absorptionsmaximum der
unprotonierten Spezies der chromophoren Einheit von PNMVAm 5 bei λ = 510 nm.
Die isosbestischen Punkte liegen bei λ = 360 nm und bei λ = 463 nm, wobei der der
chromophoren Einheit bei 463 nm nicht eindeutig ist. Das könnte an der mäßigen
Löslichkeit von PNMVAm 5 im Basischen liegen, wo partielles Ausfallen des
Polymeren beobachtet wurde.
Der pKS-Wert wurde mit Hilfe der Henderson-Hasselbalch-Gleichung (Gl. 6) durch
die Auftragung von log(A510/A420) über den pH-Wert aus dem Schnittpunkt der
Geraden mit der y-Achse ermittelt. Der pKS-Wert der chromophoren Einheit von
2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm (PNMVAm 5)
wurde mit 2.99 ± 0.06 ermittelt.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
300 350 400 450 500 550 600 650 700 750 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.] 463
360
1
2
3
45
6
7
1: pH = 0.992: pH = 2.023: pH = 2.494: pH = 3.625: pH = 5.626: pH = 7.897: pH = 10.55
N NNO2
NH3C CH3
H3CH3C
H
x y
196,000 g · mol -1

3 Ergebnisse und Diskussion
74
Die Ergebnisse der UV/vis-spektroskopischen Untersuchungen der 2-nitro-4-
aminobenzen-funktionalisierten Polyvinylamine PVAm 3, PVAm 4 und PNMVAm 5
bei unterschiedlichen pH-Werten sind zusammenfassend in Tabelle 7 dargestellt.
Tab. 7: Ergebnisse der UV/vis-spektroskopischen Untersuchungen zur pH-Abhängigkeit der UV/vis-Absorptionseigenschaften und die daraus ermittelten pKS-Werte der drei 2-nitro-4-aminobenzen-funktionalsierten Polyvinylamine PVAm 3, PVAm 4 und PNMVAm 5.
Polymerstruktur
(pH = 11)
chromophore Einheit
(pH = 11) pKS
NH2 NHNO2
NH2
x y
NH
O2N
NH
H
4.03 ± 0.01
NH2 NHNO2
NH3C CH3
x y
NH
O2N
NCH3
CH3
4.06 ± 0.03
x yN CH3
H
N CH3
NO2
NH3C CH3
N
O2N
NCH3
CH3
CH3
2.99 ± 0.06
Die pKS-Werte der chromophoren Einheiten von 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) und 2-nitro-4-(N,N-dimethyl)aminobenzen-
funktionalisiertem PVAm (PVAM 4) liegen im Bereich von Aniliniumionen in Wasser
(pKS = 4.6). Der pKS-Wert der chromophoren Einheit von PVAm 4 mit pKS = 4.06 ist
etwas größer als der von PVAm 3 mit pKS = 4.03. Die Einführung von zwei
Methylgruppen am 4-Aminostickstoff erhöht die Basizität, woraus eine etwas
schwächere Acidität resultiert. Der deutlich niedrigere pKS-Wert der chromophoren
Einheit von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PNMVAm
(PNMVAm 5) mit pKS = 2.99 beruht auf der in 1-Stellung eingeführten Methylgruppe,
die aus sterischen Gründen zum Herausdrehen der ortho-Nitrogruppe führt und
damit zu einem schwächeren +M-Effekt der 1-N-Methyl-Aminogruppe.118

3 Ergebnisse und Diskussion
75
Es ist literaturbekannt, dass die pKS-Werte von polysubstituierten Anilinen eine
lineare Abhängigkeit der Summe der Hammett-Konstanten (∑σ) zeigen (s. Gl. 7).121
0.08)4.45(0.16)2.88(pK S ±+⋅±−= ∑σ (7)
r2 = 0.955 n = 18, sd = 0.251
Dabei bedeuten die einzelnen Parameter der Regressionsstatistik:
r2…Korrelationskoeffizient, Bestimmtheitsmaß
n…Anzahl der polysubstituierten Aniline
sd…Standardabweichung.
Mit Hilfe von Gl. 7 konnten die σ -Hammett-Konstanten der ortho-Nitrogruppe von
2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) und 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) berechnet werden.
Ausgehend von den σpara-Werten der NH2-Gruppe (σpara = -0.66)122 und der N(CH3)2-
Gruppe (σpara = -0.63)122 wurden die σortho-Werte für die Nitrogruppe mit 0.806 für die
chromophore Einheit von PVAm 3 und mit 0.765 für die chromophore Einheit von
PVAm 4 bestimmt. Die berechneten Werte liegen in etwa im Bereich des
σpara-Wertes der Nitrogruppe (σpara = 0.78)122, so dass die ermittelten pKS-Werte der
chromophoren Einheiten von PVAm 3 und PVAm 4 daher sinnvoll erscheinen.
ESR-spektroskopische Untersuchungen von PVAm 3 und PVAm 4 zeigen sowohl als
Feststoff als auch in methanolischer Lösung einen möglicherweise partiellen Radikal-
Kationen-Charakter (s. Anhang S. 201). Die Signalmuster beider Polymere sind
gleich, was für eine gleiche zugrunde liegende Struktur der Radikale spricht. Die
Intensität der Signale ist jedoch nur sehr gering, so dass von nur wenigen Einheiten
dieser Spezies ausgegangen werden kann. In wässriger Lösung hingegen wurden
keine Radikal-Kationen festgestellt. Die Einführung einer –NO2-Gruppe in ortho-
Position zur Aminogruppe hat damit die Sensibilität der 1,4-Phenylendiamin-Derivate
gegenüber Luftsauerstoff herabgesetzt.31,34,35 Schon Michaelis zeigte 1939, dass bei
1,4-Phenylendiaminen bereits eine Methylgruppe in ortho-Stellung zu einer mono-
methylierten Aminogruppe ausreicht, um den Radikal-Kationen-Charakter zu
destabilisieren.40

3 Ergebnisse und Diskussion
76
3.1.8 Substitutionsgrad funktionalisierter Polyvin ylamine
Zur UV/vis-spektroskopischen Bestimmung der Substitutionsgrade der
funktionalisierten Polyvinylamine wurden niedermolekulare Modellverbindungen, die
strukturell in etwa der chromophoren Einheit entsprechen, synthetisiert. Dazu wurden
die Fluoraromaten mit i-Propylamin zu den entsprechenden Nitro-N-isopropylanilinen
umgesetzt (s. Abschnitt 5.6 Experimenteller Teil). Unter Zuhilfenahme der molaren
Absorptionskoeffizienten der Modellverbindungen und der spezifischen Absorptions-
koeffizienten der funktionalisierten PVAm´s kann über die UV/vis-Spektroskopie der
Substitutionsgrad s über eine literaturbekannte Gleichung (Gl. 8) ermittelt werden.123
SModell
eq
M
M
⋅′−⋅′
=εε
εs (8)
Dabei bedeuten die einzelnen Parameter:
s…Substitutionsgrad (Zahl der Substituenten pro Grundbaustein)
ε´ …spezifischer Absorptionskoeffizient des Polymeren [l·g-1·cm-1]
Meq…Molekulargewicht des Grundbausteines des Derivates (chromophore Einheit)
εModell…molarer Absorptionskoeffizient der Modellverbindung [l·mol-1·cm-1]
Ms…Molekulargewicht des eintretenden Substituenten.
Zur 1H-NMR-spektroskopischen Bestimmung des Substitutionsgrades von
funktionalisiertem PVAm wurde die Intensität der Aromaten-Signale ins Verhältnis zu
den CH- und CH2-Gruppen des Polymerrückrates gesetzt. Diese Methode wurde
zum Teil stichprobenartig zu Vergleichszwecken der UV/vis-spektroskopisch
ermittelten Substitutionsgrade verwendet. Aufgrund der schlechten Löslichkeit von
2,4-dinitro-phenyl-funktionalisiertem PVAm (PVAm 6 mittels Sangers Reagenz), von
2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm (PVAm 7 mittels DFDNB)
und von 2-nitro-4-azobenzen-funktionalisiertem PVAm (PVAm 8 mittels FNAzoB)
erweist sich sowohl die UV/vis-Spektroskopie als auch die 1H-NMR-Spektroskopie für
die Bestimmung der Substitutionsgrade als nicht geeignet. Die begrenzte Löslichkeit
von 2,4´-dinitrostilben-funktionalisiertem PVAm (PVAm 9 mittels FDNstilben) ist für
eine Substitutionsgradsbestimmung mit Hilfe der 1H-NMR-Spektroskopie nicht
ausreichend.
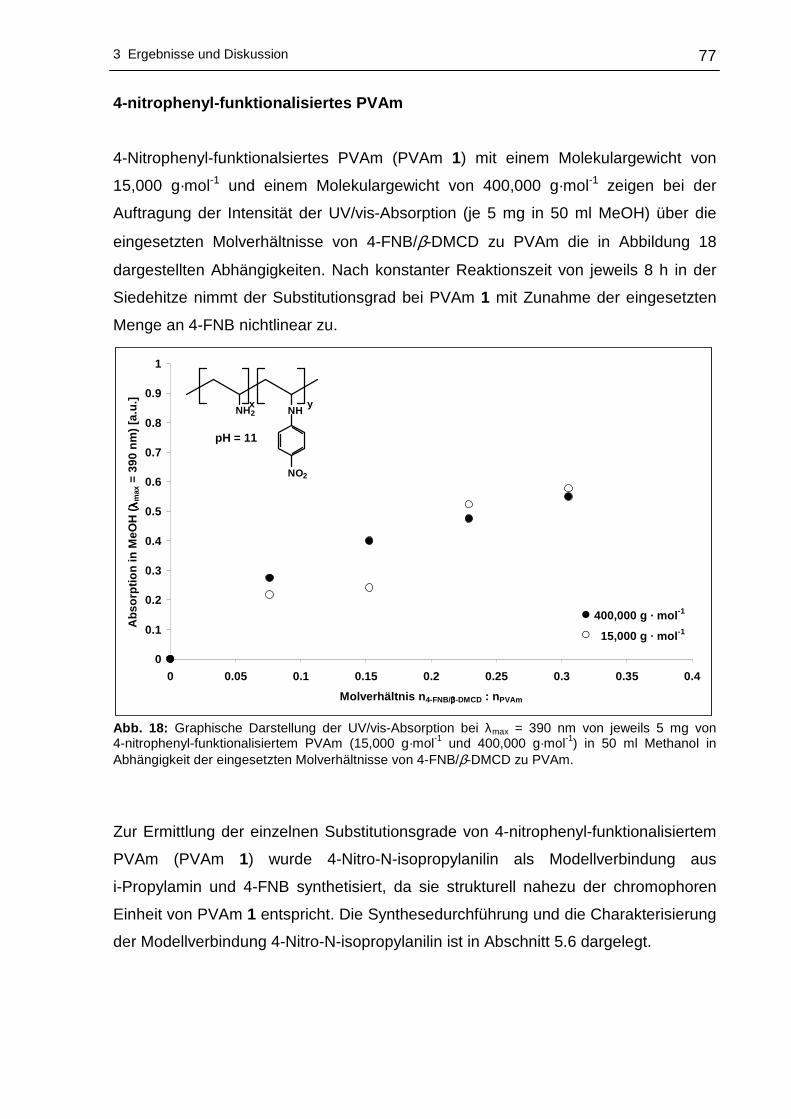
3 Ergebnisse und Diskussion
77
4-nitrophenyl-funktionalisiertes PVAm
4-Nitrophenyl-funktionalsiertes PVAm (PVAm 1) mit einem Molekulargewicht von
15,000 g·mol-1 und einem Molekulargewicht von 400,000 g·mol-1 zeigen bei der
Auftragung der Intensität der UV/vis-Absorption (je 5 mg in 50 ml MeOH) über die
eingesetzten Molverhältnisse von 4-FNB/β-DMCD zu PVAm die in Abbildung 18
dargestellten Abhängigkeiten. Nach konstanter Reaktionszeit von jeweils 8 h in der
Siedehitze nimmt der Substitutionsgrad bei PVAm 1 mit Zunahme der eingesetzten
Menge an 4-FNB nichtlinear zu.
Abb. 18: Graphische Darstellung der UV/vis-Absorption bei λmax = 390 nm von jeweils 5 mg von 4-nitrophenyl-funktionalisiertem PVAm (15,000 g·mol-1 und 400,000 g·mol-1) in 50 ml Methanol in Abhängigkeit der eingesetzten Molverhältnisse von 4-FNB/β-DMCD zu PVAm.
Zur Ermittlung der einzelnen Substitutionsgrade von 4-nitrophenyl-funktionalisiertem
PVAm (PVAm 1) wurde 4-Nitro-N-isopropylanilin als Modellverbindung aus
i-Propylamin und 4-FNB synthetisiert, da sie strukturell nahezu der chromophoren
Einheit von PVAm 1 entspricht. Die Synthesedurchführung und die Charakterisierung
der Modellverbindung 4-Nitro-N-isopropylanilin ist in Abschnitt 5.6 dargelegt.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4
Molverhältnis n 4-FNB/ββββ-DMCD : nPVAm
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
390
nm
) [a
.u.]
NH2 NH
NO2
x y
pH = 11
● 400,000 g · mol -1
○ 15,000 g · mol -1
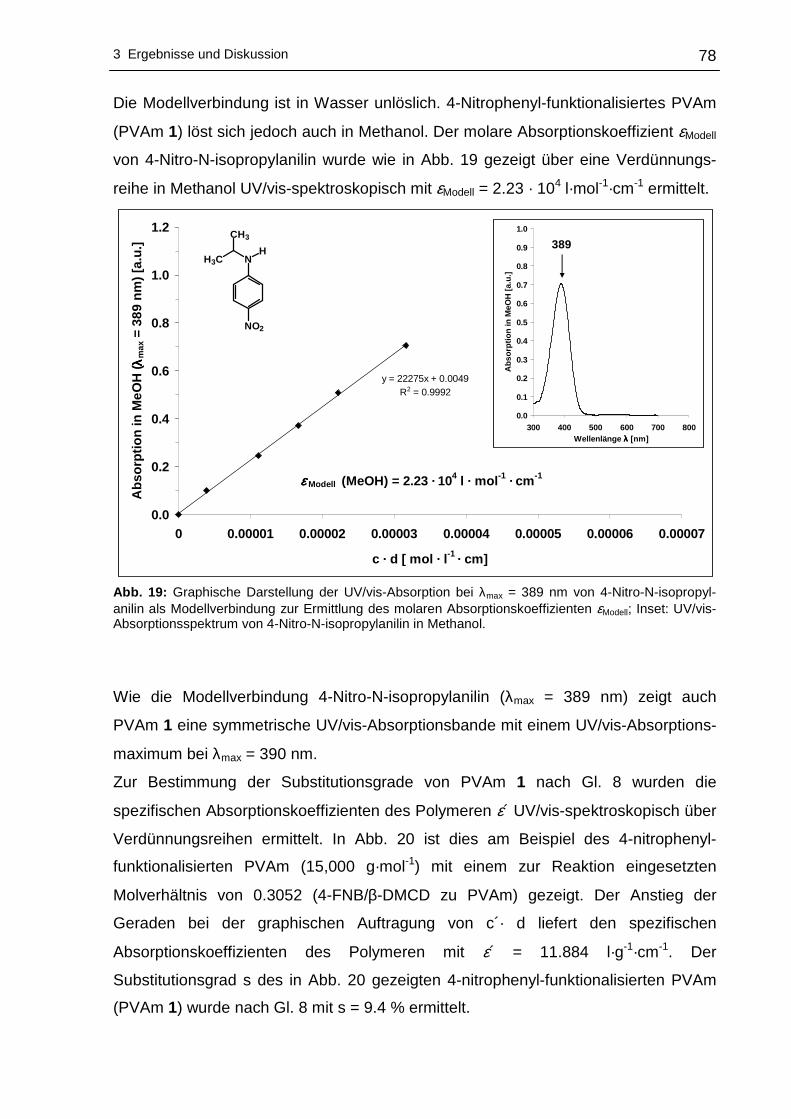
3 Ergebnisse und Diskussion
78
Die Modellverbindung ist in Wasser unlöslich. 4-Nitrophenyl-funktionalisiertes PVAm
(PVAm 1) löst sich jedoch auch in Methanol. Der molare Absorptionskoeffizient εModell
von 4-Nitro-N-isopropylanilin wurde wie in Abb. 19 gezeigt über eine Verdünnungs-
reihe in Methanol UV/vis-spektroskopisch mit εModell = 2.23 · 104 l·mol-1·cm-1 ermittelt.
Abb. 19: Graphische Darstellung der UV/vis-Absorption bei λmax = 389 nm von 4-Nitro-N-isopropyl-anilin als Modellverbindung zur Ermittlung des molaren Absorptionskoeffizienten εModell; Inset: UV/vis-Absorptionsspektrum von 4-Nitro-N-isopropylanilin in Methanol.
Wie die Modellverbindung 4-Nitro-N-isopropylanilin (λmax = 389 nm) zeigt auch
PVAm 1 eine symmetrische UV/vis-Absorptionsbande mit einem UV/vis-Absorptions-
maximum bei λmax = 390 nm.
Zur Bestimmung der Substitutionsgrade von PVAm 1 nach Gl. 8 wurden die
spezifischen Absorptionskoeffizienten des Polymeren ε´ UV/vis-spektroskopisch über
Verdünnungsreihen ermittelt. In Abb. 20 ist dies am Beispiel des 4-nitrophenyl-
funktionalisierten PVAm (15,000 g·mol-1) mit einem zur Reaktion eingesetzten
Molverhältnis von 0.3052 (4-FNB/β-DMCD zu PVAm) gezeigt. Der Anstieg der
Geraden bei der graphischen Auftragung von c´· d liefert den spezifischen
Absorptionskoeffizienten des Polymeren mit ε´ = 11.884 l·g-1·cm-1. Der
Substitutionsgrad s des in Abb. 20 gezeigten 4-nitrophenyl-funktionalisierten PVAm
(PVAm 1) wurde nach Gl. 8 mit s = 9.4 % ermittelt.
y = 22275x + 0.0049R2 = 0.9992
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 0.00001 0.00002 0.00003 0.00004 0.00005 0.00006 0.00007
c · d [ mol · l -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
389
nm
) [a
.u.]
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
300 400 500 600 700 800Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
389
εεεε Modell (MeOH) = 2.23 · 104 l · mol -1 · cm -1
N
CH3
H3CH
NO2

3 Ergebnisse und Diskussion
79
Abb. 20: Graphische Darstellung der UV/vis-Absorption von 4-nitrophenyl-funktionalisiertem PVAm (PVAm 1; 15,000 g·mol-1; eingesetztes Molverhältnis 4-FNB/β-DMCD zu PVAm: 0.3052) über c´ · d zur Ermittlung des spezifischen Absorptionskoeffizienten des Polymeren ε ´ in Methanol.
Zum stichprobenartigen Vergleich der UV/vis-spektroskopisch ermittelten
Substitutionsgrade wurde das 4-nitrophenyl-funktionalisierte PVAm (PVAm 1) mit
einem Molekulargewicht von 15,000 g·mol-1 und mit dem höchsten UV/vis-
spektroskopisch ermitteltem Substitutionsgrad (s = 9.4 %) in D2O gelöst und der
Substitutionsgrad mit Hilfe der 1H-NMR-Spektroskopie bestimmt. Mit dem Verhältnis
der Intensitäten der aromatischen Protonen zu denen der Polymerhauptkette ergibt
sich ein Substitutionsgrad von s = 10.0 %. Der 1H-NMR-spektroskopisch ermittelte
Substitutionsgrad von 4-nitrophenyl-funktionalisiertem PVAm stimmt mit dem über
die UV/vis-Spektroskopie ermitteltem Substitutionsgrad von 9.4 % im Fehlerbereich
sehr gut überein.
In Tabelle 8 sind die Ergebnisse der UV/vis-spektroskopischen Substitutions-
gradbestimmung aller synthetisierten 4-nitrophenyl-funktionalisierten PVAm´s
(15,000 g·mol-1 und 400,000 g·mol-1) zusammengefasst.
y = 11.884x + 0.0082R2 = 0.9989
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 0.02 0.04 0.06 0.08 0.1 0.12
c´ · d [g · mol -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
390
nm
) [a
.u.]
εεεε ´ = 11.884 l · g-1 · cm-1
Meq = 164.158 g · mol-1
MS = 122.112 g · mol-1
εεεε Modell = 2.23 · 104 l · mol -1 · cm-1
s = 9.35 %
SModell
eq
M
Ms
⋅′−⋅′
=εε
ε
NH2 NH
NO2
x y
15,000 g · mol -1
pH = 11
n4-FNB
nPVAm= 0.3052
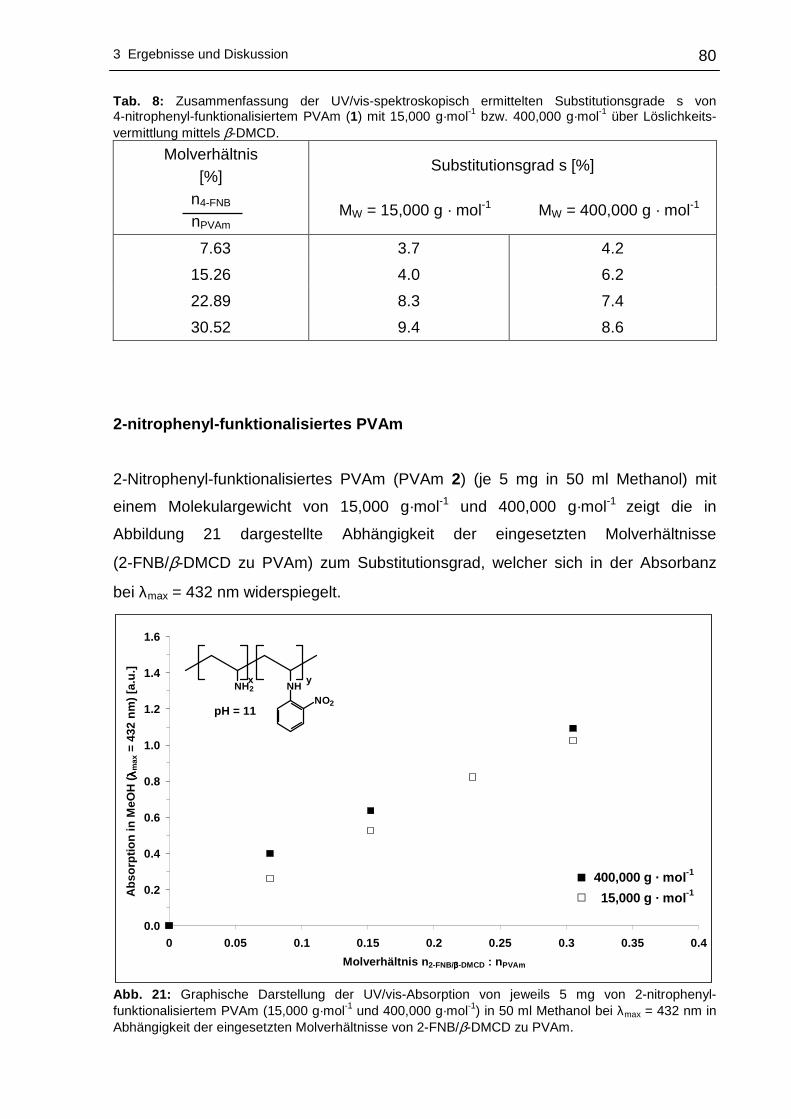
3 Ergebnisse und Diskussion
80
Tab. 8: Zusammenfassung der UV/vis-spektroskopisch ermittelten Substitutionsgrade s von 4-nitrophenyl-funktionalisiertem PVAm (1) mit 15,000 g·mol-1 bzw. 400,000 g·mol-1 über Löslichkeits-vermittlung mittels β-DMCD.
Molverhältnis [%]
n4-FNB
nPVAm
Substitutionsgrad s [%]
MW = 15,000 g · mol-1 MW = 400,000 g · mol-1
7.63 3.7 4.2
15.26 4.0 6.2
22.89 8.3 7.4
30.52 9.4 8.6
2-nitrophenyl-funktionalisiertes PVAm
2-Nitrophenyl-funktionalisiertes PVAm (PVAm 2) (je 5 mg in 50 ml Methanol) mit
einem Molekulargewicht von 15,000 g·mol-1 und 400,000 g·mol-1 zeigt die in
Abbildung 21 dargestellte Abhängigkeit der eingesetzten Molverhältnisse
(2-FNB/β-DMCD zu PVAm) zum Substitutionsgrad, welcher sich in der Absorbanz
bei λmax = 432 nm widerspiegelt.
Abb. 21: Graphische Darstellung der UV/vis-Absorption von jeweils 5 mg von 2-nitrophenyl-funktionalisiertem PVAm (15,000 g·mol-1 und 400,000 g·mol-1) in 50 ml Methanol bei λmax = 432 nm in Abhängigkeit der eingesetzten Molverhältnisse von 2-FNB/β-DMCD zu PVAm.
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4
Molverhältnis n 2-FNB/ββββ-DMCD : n PVAm
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
432
nm
) [a
.u.]
NH2 NHNO2
x y
pH = 11
■ 400,000 g · mol -1
□ 15,000 g · mol -1
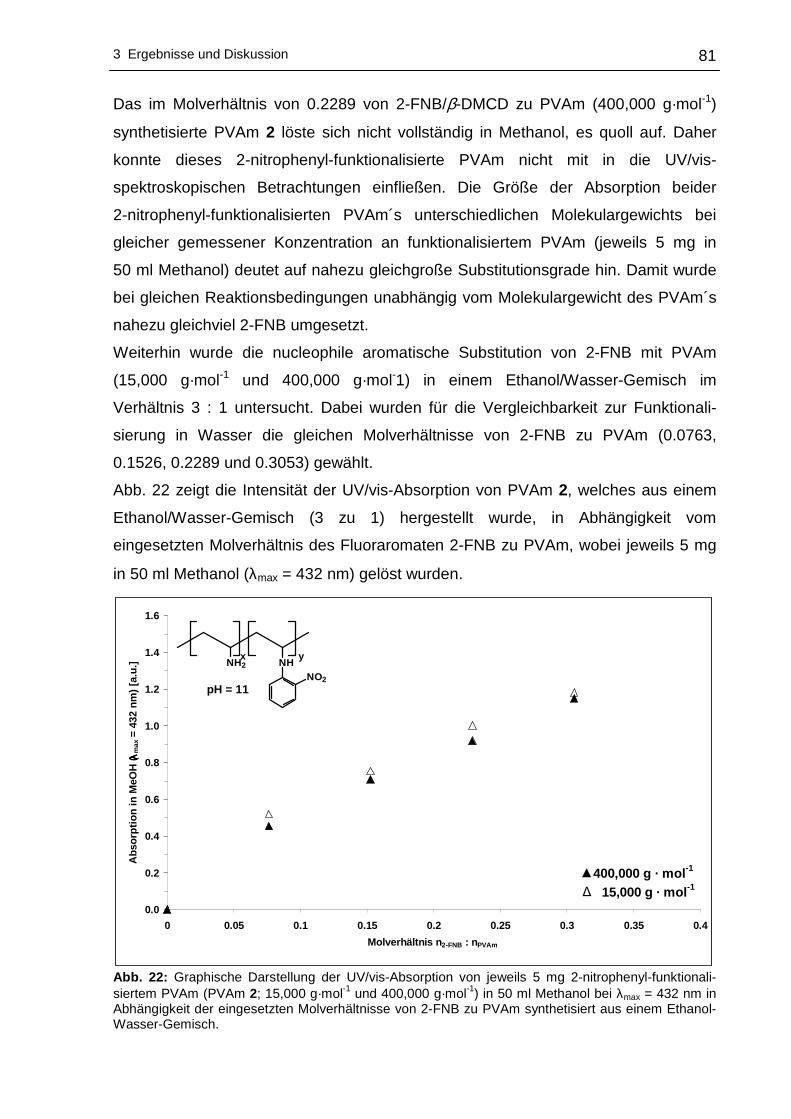
3 Ergebnisse und Diskussion
81
Das im Molverhältnis von 0.2289 von 2-FNB/β-DMCD zu PVAm (400,000 g·mol-1)
synthetisierte PVAm 2 löste sich nicht vollständig in Methanol, es quoll auf. Daher
konnte dieses 2-nitrophenyl-funktionalisierte PVAm nicht mit in die UV/vis-
spektroskopischen Betrachtungen einfließen. Die Größe der Absorption beider
2-nitrophenyl-funktionalisierten PVAm´s unterschiedlichen Molekulargewichts bei
gleicher gemessener Konzentration an funktionalisiertem PVAm (jeweils 5 mg in
50 ml Methanol) deutet auf nahezu gleichgroße Substitutionsgrade hin. Damit wurde
bei gleichen Reaktionsbedingungen unabhängig vom Molekulargewicht des PVAm´s
nahezu gleichviel 2-FNB umgesetzt.
Weiterhin wurde die nucleophile aromatische Substitution von 2-FNB mit PVAm
(15,000 g·mol-1 und 400,000 g·mol-1) in einem Ethanol/Wasser-Gemisch im
Verhältnis 3 : 1 untersucht. Dabei wurden für die Vergleichbarkeit zur Funktionali-
sierung in Wasser die gleichen Molverhältnisse von 2-FNB zu PVAm (0.0763,
0.1526, 0.2289 und 0.3053) gewählt.
Abb. 22 zeigt die Intensität der UV/vis-Absorption von PVAm 2, welches aus einem
Ethanol/Wasser-Gemisch (3 zu 1) hergestellt wurde, in Abhängigkeit vom
eingesetzten Molverhältnis des Fluoraromaten 2-FNB zu PVAm, wobei jeweils 5 mg
in 50 ml Methanol (λmax = 432 nm) gelöst wurden.
Abb. 22: Graphische Darstellung der UV/vis-Absorption von jeweils 5 mg 2-nitrophenyl-funktionali-siertem PVAm (PVAm 2; 15,000 g·mol-1 und 400,000 g·mol-1) in 50 ml Methanol bei λmax = 432 nm in Abhängigkeit der eingesetzten Molverhältnisse von 2-FNB zu PVAm synthetisiert aus einem Ethanol-Wasser-Gemisch.
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4
Molverhältnis n 2-FNB : nPVAm
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
432
nm
) [a
.u.] NH2 NH
NO2
x y
pH = 11
▲400,000 g · mol -1
∆ 15,000 g · mol -1
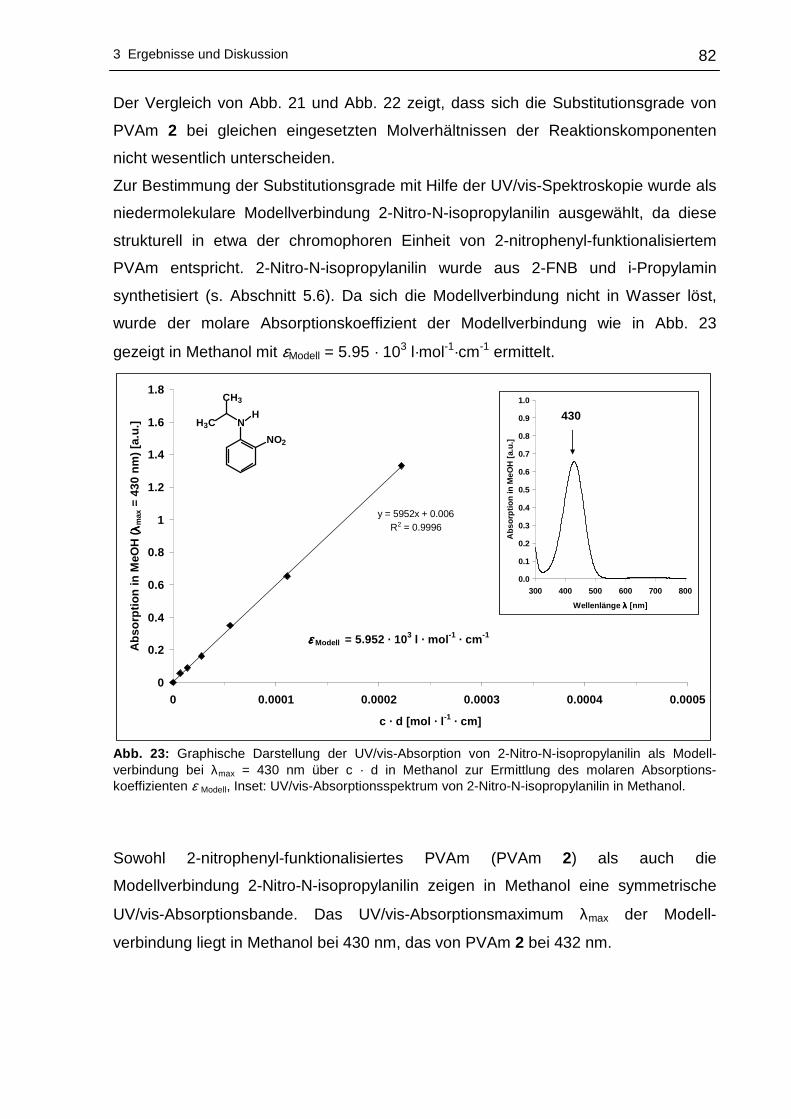
3 Ergebnisse und Diskussion
82
Der Vergleich von Abb. 21 und Abb. 22 zeigt, dass sich die Substitutionsgrade von
PVAm 2 bei gleichen eingesetzten Molverhältnissen der Reaktionskomponenten
nicht wesentlich unterscheiden.
Zur Bestimmung der Substitutionsgrade mit Hilfe der UV/vis-Spektroskopie wurde als
niedermolekulare Modellverbindung 2-Nitro-N-isopropylanilin ausgewählt, da diese
strukturell in etwa der chromophoren Einheit von 2-nitrophenyl-funktionalisiertem
PVAm entspricht. 2-Nitro-N-isopropylanilin wurde aus 2-FNB und i-Propylamin
synthetisiert (s. Abschnitt 5.6). Da sich die Modellverbindung nicht in Wasser löst,
wurde der molare Absorptionskoeffizient der Modellverbindung wie in Abb. 23
gezeigt in Methanol mit εModell = 5.95 · 103 l·mol-1·cm-1 ermittelt.
Abb. 23: Graphische Darstellung der UV/vis-Absorption von 2-Nitro-N-isopropylanilin als Modell-verbindung bei λmax = 430 nm über c · d in Methanol zur Ermittlung des molaren Absorptions-koeffizienten ε Modell, Inset: UV/vis-Absorptionsspektrum von 2-Nitro-N-isopropylanilin in Methanol.
Sowohl 2-nitrophenyl-funktionalisiertes PVAm (PVAm 2) als auch die
Modellverbindung 2-Nitro-N-isopropylanilin zeigen in Methanol eine symmetrische
UV/vis-Absorptionsbande. Das UV/vis-Absorptionsmaximum λmax der Modell-
verbindung liegt in Methanol bei 430 nm, das von PVAm 2 bei 432 nm.
y = 5952x + 0.006R2 = 0.9996
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 0.0001 0.0002 0.0003 0.0004 0.0005
c · d [mol · l -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
430
nm
) [a
.u.]
εεεε Modell = 5.952 · 103 l · mol -1 · cm-1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
430N
CH3
H3CH
NO2
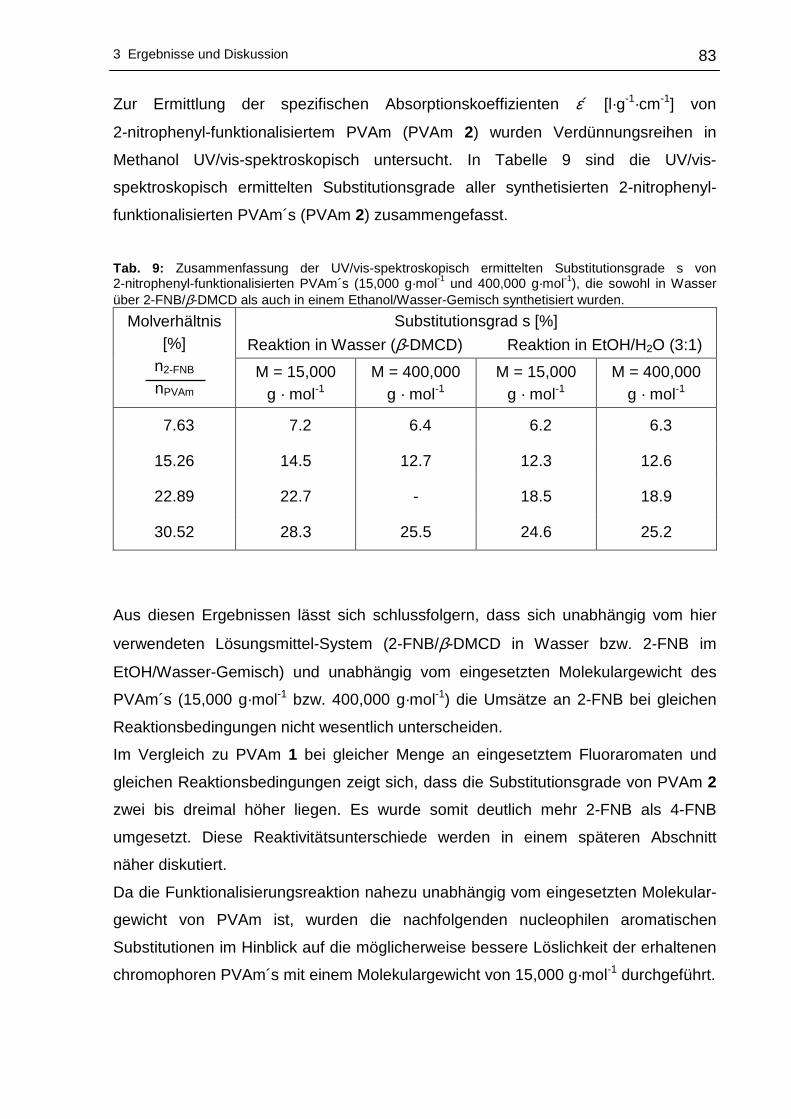
3 Ergebnisse und Diskussion
83
Zur Ermittlung der spezifischen Absorptionskoeffizienten ε´ [l·g-1·cm-1] von
2-nitrophenyl-funktionalisiertem PVAm (PVAm 2) wurden Verdünnungsreihen in
Methanol UV/vis-spektroskopisch untersucht. In Tabelle 9 sind die UV/vis-
spektroskopisch ermittelten Substitutionsgrade aller synthetisierten 2-nitrophenyl-
funktionalisierten PVAm´s (PVAm 2) zusammengefasst.
Tab. 9: Zusammenfassung der UV/vis-spektroskopisch ermittelten Substitutionsgrade s von 2-nitrophenyl-funktionalisierten PVAm´s (15,000 g·mol-1 und 400,000 g·mol-1), die sowohl in Wasser über 2-FNB/β-DMCD als auch in einem Ethanol/Wasser-Gemisch synthetisiert wurden.
Substitutionsgrad s [%]
Reaktion in Wasser (β-DMCD) Reaktion in EtOH/H2O (3:1)
Molverhältnis [%]
n2-FNB
nPVAm M = 15,000
g · mol-1 M = 400,000
g · mol-1 M = 15,000
g · mol-1 M = 400,000
g · mol-1
7.63 7.2 6.4 6.2 6.3
15.26 14.5 12.7 12.3 12.6
22.89 22.7 - 18.5 18.9
30.52 28.3 25.5 24.6 25.2
Aus diesen Ergebnissen lässt sich schlussfolgern, dass sich unabhängig vom hier
verwendeten Lösungsmittel-System (2-FNB/β-DMCD in Wasser bzw. 2-FNB im
EtOH/Wasser-Gemisch) und unabhängig vom eingesetzten Molekulargewicht des
PVAm´s (15,000 g·mol-1 bzw. 400,000 g·mol-1) die Umsätze an 2-FNB bei gleichen
Reaktionsbedingungen nicht wesentlich unterscheiden.
Im Vergleich zu PVAm 1 bei gleicher Menge an eingesetztem Fluoraromaten und
gleichen Reaktionsbedingungen zeigt sich, dass die Substitutionsgrade von PVAm 2
zwei bis dreimal höher liegen. Es wurde somit deutlich mehr 2-FNB als 4-FNB
umgesetzt. Diese Reaktivitätsunterschiede werden in einem späteren Abschnitt
näher diskutiert.
Da die Funktionalisierungsreaktion nahezu unabhängig vom eingesetzten Molekular-
gewicht von PVAm ist, wurden die nachfolgenden nucleophilen aromatischen
Substitutionen im Hinblick auf die möglicherweise bessere Löslichkeit der erhaltenen
chromophoren PVAm´s mit einem Molekulargewicht von 15,000 g·mol-1 durchgeführt.
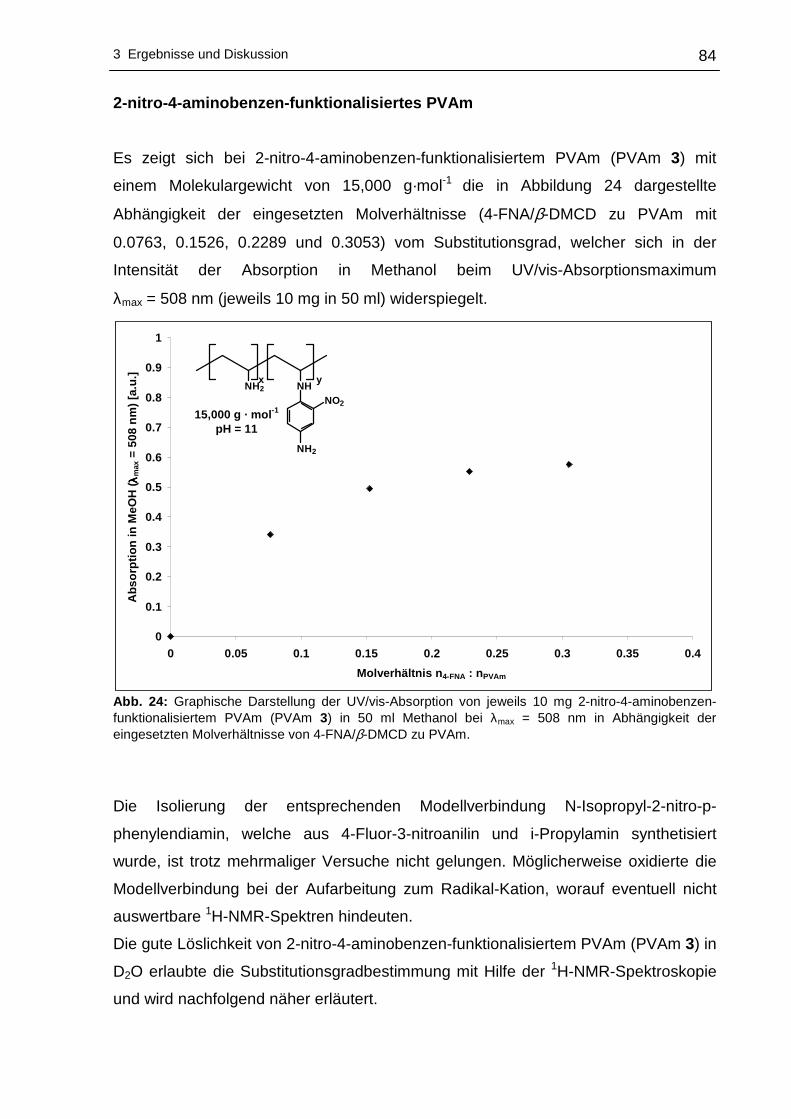
3 Ergebnisse und Diskussion
84
2-nitro-4-aminobenzen-funktionalisiertes PVAm
Es zeigt sich bei 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) mit
einem Molekulargewicht von 15,000 g·mol-1 die in Abbildung 24 dargestellte
Abhängigkeit der eingesetzten Molverhältnisse (4-FNA/β-DMCD zu PVAm mit
0.0763, 0.1526, 0.2289 und 0.3053) vom Substitutionsgrad, welcher sich in der
Intensität der Absorption in Methanol beim UV/vis-Absorptionsmaximum
λmax = 508 nm (jeweils 10 mg in 50 ml) widerspiegelt.
Abb. 24: Graphische Darstellung der UV/vis-Absorption von jeweils 10 mg 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) in 50 ml Methanol bei λmax = 508 nm in Abhängigkeit der eingesetzten Molverhältnisse von 4-FNA/β-DMCD zu PVAm.
Die Isolierung der entsprechenden Modellverbindung N-Isopropyl-2-nitro-p-
phenylendiamin, welche aus 4-Fluor-3-nitroanilin und i-Propylamin synthetisiert
wurde, ist trotz mehrmaliger Versuche nicht gelungen. Möglicherweise oxidierte die
Modellverbindung bei der Aufarbeitung zum Radikal-Kation, worauf eventuell nicht
auswertbare 1H-NMR-Spektren hindeuten.
Die gute Löslichkeit von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) in
D2O erlaubte die Substitutionsgradbestimmung mit Hilfe der 1H-NMR-Spektroskopie
und wird nachfolgend näher erläutert.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4
Molverhältnis n 4-FNA : n PVAm
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
508
nm
) [a
.u.]
NH2 NHNO2
NH2
x y
15,000 g · mol -1
pH = 11
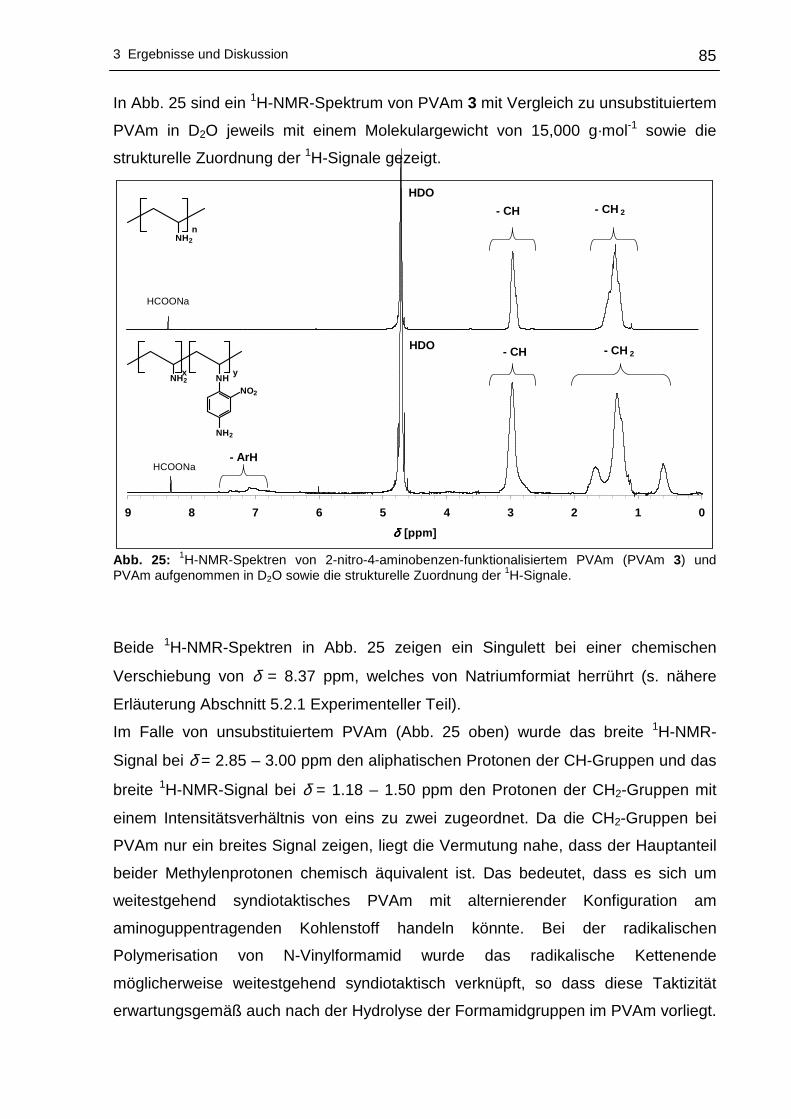
3 Ergebnisse und Diskussion
85
In Abb. 25 sind ein 1H-NMR-Spektrum von PVAm 3 mit Vergleich zu unsubstituiertem
PVAm in D2O jeweils mit einem Molekulargewicht von 15,000 g·mol-1 sowie die
strukturelle Zuordnung der 1H-Signale gezeigt.
Abb. 25: 1H-NMR-Spektren von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) und PVAm aufgenommen in D2O sowie die strukturelle Zuordnung der 1H-Signale.
Beide 1H-NMR-Spektren in Abb. 25 zeigen ein Singulett bei einer chemischen
Verschiebung von δ = 8.37 ppm, welches von Natriumformiat herrührt (s. nähere
Erläuterung Abschnitt 5.2.1 Experimenteller Teil).
Im Falle von unsubstituiertem PVAm (Abb. 25 oben) wurde das breite 1H-NMR-
Signal bei δ = 2.85 – 3.00 ppm den aliphatischen Protonen der CH-Gruppen und das
breite 1H-NMR-Signal bei δ = 1.18 – 1.50 ppm den Protonen der CH2-Gruppen mit
einem Intensitätsverhältnis von eins zu zwei zugeordnet. Da die CH2-Gruppen bei
PVAm nur ein breites Signal zeigen, liegt die Vermutung nahe, dass der Hauptanteil
beider Methylenprotonen chemisch äquivalent ist. Das bedeutet, dass es sich um
weitestgehend syndiotaktisches PVAm mit alternierender Konfiguration am
aminoguppentragenden Kohlenstoff handeln könnte. Bei der radikalischen
Polymerisation von N-Vinylformamid wurde das radikalische Kettenende
möglicherweise weitestgehend syndiotaktisch verknüpft, so dass diese Taktizität
erwartungsgemäß auch nach der Hydrolyse der Formamidgruppen im PVAm vorliegt.
0123456789
δδδδ [ppm]
NH2 NHNO2
NH2
x y
NH2
n
HCOONa
HDO
- CH - CH 2
- ArH
- CH 2- CH
HCOONa
HDO
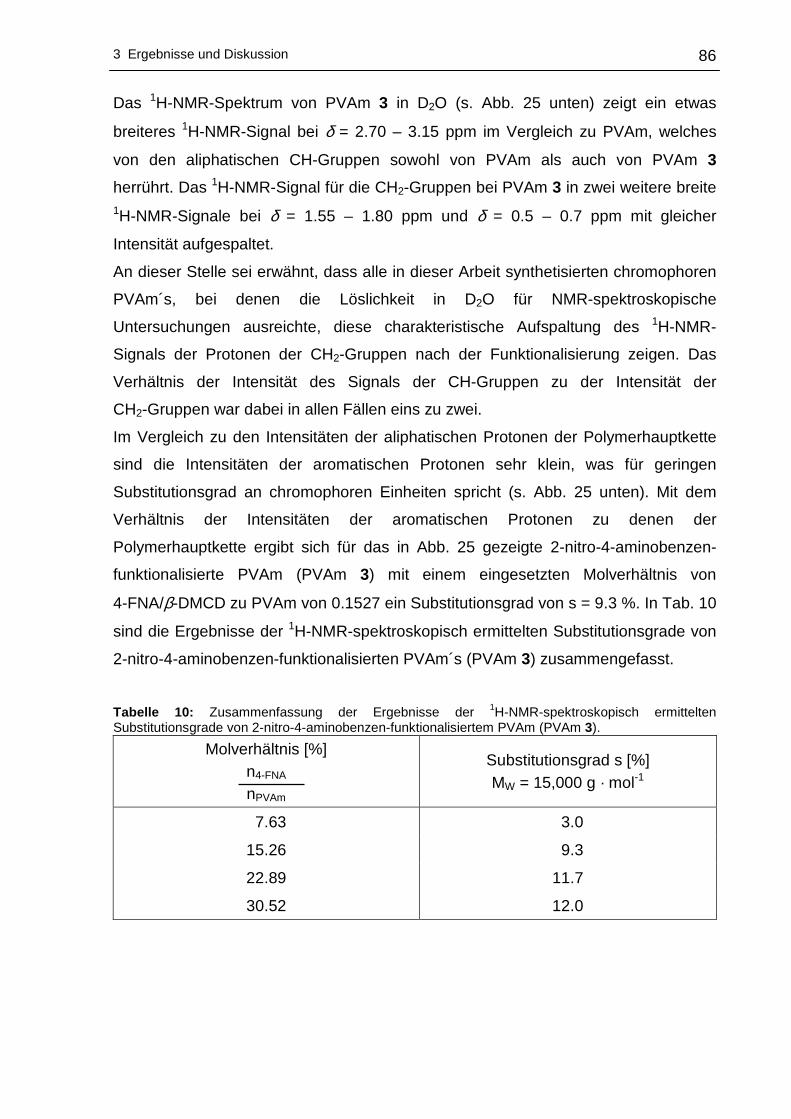
3 Ergebnisse und Diskussion
86
Das 1H-NMR-Spektrum von PVAm 3 in D2O (s. Abb. 25 unten) zeigt ein etwas
breiteres 1H-NMR-Signal bei δ = 2.70 – 3.15 ppm im Vergleich zu PVAm, welches
von den aliphatischen CH-Gruppen sowohl von PVAm als auch von PVAm 3
herrührt. Das 1H-NMR-Signal für die CH2-Gruppen bei PVAm 3 in zwei weitere breite 1H-NMR-Signale bei δ = 1.55 – 1.80 ppm und δ = 0.5 – 0.7 ppm mit gleicher
Intensität aufgespaltet.
An dieser Stelle sei erwähnt, dass alle in dieser Arbeit synthetisierten chromophoren
PVAm´s, bei denen die Löslichkeit in D2O für NMR-spektroskopische
Untersuchungen ausreichte, diese charakteristische Aufspaltung des 1H-NMR-
Signals der Protonen der CH2-Gruppen nach der Funktionalisierung zeigen. Das
Verhältnis der Intensität des Signals der CH-Gruppen zu der Intensität der
CH2-Gruppen war dabei in allen Fällen eins zu zwei.
Im Vergleich zu den Intensitäten der aliphatischen Protonen der Polymerhauptkette
sind die Intensitäten der aromatischen Protonen sehr klein, was für geringen
Substitutionsgrad an chromophoren Einheiten spricht (s. Abb. 25 unten). Mit dem
Verhältnis der Intensitäten der aromatischen Protonen zu denen der
Polymerhauptkette ergibt sich für das in Abb. 25 gezeigte 2-nitro-4-aminobenzen-
funktionalisierte PVAm (PVAm 3) mit einem eingesetzten Molverhältnis von
4-FNA/β-DMCD zu PVAm von 0.1527 ein Substitutionsgrad von s = 9.3 %. In Tab. 10
sind die Ergebnisse der 1H-NMR-spektroskopisch ermittelten Substitutionsgrade von
2-nitro-4-aminobenzen-funktionalisierten PVAm´s (PVAm 3) zusammengefasst.
Tabelle 10: Zusammenfassung der Ergebnisse der 1H-NMR-spektroskopisch ermittelten Substitutionsgrade von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3).
Molverhältnis [%] n4-FNA nPVAm
Substitutionsgrad s [%] MW = 15,000 g · mol-1
7.63 3.0
15.26 9.3
22.89 11.7
30.52 12.0

3 Ergebnisse und Diskussion
87
2-nitro-4-( N,N-dimethyl)aminobenzen-funktionalisiertes PVAm
2-Nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PVAm (PVAm 4) mit einem
Molekulargewicht von 15,000 g·mol-1 zeigt die in Abb. 26 dargestellte Abhängigkeit
des Substitutionsgrades vom eingesetzten Molverhältnis (4-DMFNA/β-DMCD zu
PVAm mit 0.0763, 0.1526, 0.2289 und 0.3053), wobei die Absorption in Methanol bei
λmax = 520 nm (je 10 mg PVAm 4 in 50 ml) über die jeweils eingesetzten
Molverhältnisse von 4-DMFNA/β-DMCD zu PVAm aufgetragen wurde.
Abb. 26: Graphische Darstellung der UV/vis-Absorption von jeweils 10 mg 2-nitro-4-(N,N-dimethyl)-aminobenzen-funktionalisiertem PVAm (PVAm 4) in 50 ml Methanol bei λmax = 520 nm in Abhängigkeit der eingesetzten Molverhältnisse von 4-DMFNA/β-DMCD zu PVAm.
Die Isolierung der entsprechenden Modellverbindung N1-Isopropyl-N4,N4-dimethyl-2-
nitro-p-phenylendiamin, welche aus N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA)
und i-Propylamin synthetisiert wurde, war nicht gelungen. Unumgesetztes 4-DMFNA
ließ sich nicht aus dem Reaktionsgemisch durch Umkristallisation in verschiedenen
Lösungsmitteln entfernen. Weder 4-DMFNA noch die Modellverbindung konnten
abgetrennt werden. Auch die Überführung des Phenylendiamin-Derivates zum HCl-
Salz und anschließende Deprotonierung der Aminogruppen brachte nicht den
gewünschten Erfolg.
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40
Molverhältnis n 4-DMFNA/nPVAm
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
520
nm
) [a
.u.]
NH2 NH2
NO2
NH3C CH3
x y
15,000 g · mol -1
pH = 11

3 Ergebnisse und Diskussion
88
Aufgrund der guten Löslichkeit von PVAm 4 in D2O wurden die Substitutionsgrade
daher ausschließlich mit Hilfe der 1H-NMR-Spektroskopie durch Vergleich der
Intensitäten der aromatischen Protonen zu denen der Polymerhauptkette ermittelt. In
Tabelle 11 sind die 1H-NMR-spektroskopisch ermittelten Substitutionsgrade s der vier
synthetisierten 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisierten PVAm´s
(PVAm 4) in Bezug auf die eingesetzten Molverhältnisse der Reaktionskomponenten
4-DMFNA/β-DMCD zu PVAm zusammengefasst.
Tabelle 11: Zusammenfassung der 1H-NMR-spektroskopisch ermittelten Substitutionsgrade s von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) mit einem Molekulargewicht von 15,000 g·mol-1 in Bezug auf die eingesetzten Molverhältnisse von 4-DMFNA/β-DMCD zu PVAm.
Molverhältnis [%] n4-DMFNA
nPVAm
Substitutionsgrad s [%] M = 15,000 g · mol-1
7.63 3.0
15.26 6.0
22.89 8.7
30.52 11.3
Die Substitutionsgrade von PVAm 4 sind im Vergleich zu 2-nitro-4-aminobenzen-
funktionalisiertem PVAm (PVAm 3) bei gleichem eingesetzten Molverhältnis von
Fluoraromat/β-DMCD zu PVAm etwas geringer (vgl. Tabelle 10).
Die Erhöhung der Donorstärke der in 4-Stellung befindlichen Aminogruppe durch
N,N-Dimethylierung bei PVAm 4 im Vergleich zu PVAm 3 wirkt dem Elektronenzug
der aktivierenden Nitrogruppe in 2-Stellung entgegen. Damit ist der fluorsubstituierte
Kohlenstoff weniger für einen nucleophilen Angriff von Aminogruppen des PVAm´s
positiviert, die Substitutionsreaktion verläuft somit etwas langsamer. Auf die
Reaktivität von 4-DMFNA im Vergleich zu 4-FNA bei der Funktionalisierung von
PVAm wird in einem späteren Abschnitt näher eingegangen.

3 Ergebnisse und Diskussion
89
2-nitro-4-( N,N-dimethyl)aminobenzen-funktionalisiertes PNMVAm
2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes PNMVAm (PNMVAm 5) mit
einem Molekulargewicht von 196,000 g·mol-1 und einem eingesetzten Molverhältnis
von 4-DMFNA/β-DMCD zu PNMVAm mit 0.3053 löst sich sowohl in Wasser als auch
in Methanol. Jedoch ist die Löslichkeit nicht so gut im Vergleich zu 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) und 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) und nicht ausreichend für
eine Substitutionsgradbestimmung mit Hilfe der 1H-NMR-Spektroskopie.
Die Bestimmung des Substitutionsgrades s von PNMVAm 5 erfolgte daher
ausschließlich UV/vis-spektroskopisch. Die der chromophoren Einheit in etwa
strukturell entsprechende Modellverbindung N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-
p-phenylendiamin wurde aus 4-DMFNA und N-Methyl-i-Propylamin synthetisiert. Eine
ausführliche Synthesebeschreibung und Charakterisierung von N1-Isopropyl-
N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin ist in Abschnitt 5.6 (Experimenteller Teil)
dargelegt.
Da die Modellverbindung in Wasser schwer löslich, PNMVAm 5 jedoch in Methanol
gut löslich ist, wurde der molare Absorptionskoeffizient der Modellverbindung εModell
über eine Verdünnungsreihe in Methanol UV/vis-spektroskopisch ermittelt. In Abb. 27
ist die graphische Auftragung des Produktes aus der Konzentration c und der
Schichtdicke der Küvette über die Absorption von N1-Isopropyl-N1,N4,N4-trimethyl-2-
nitro-p-phenylendiamin bei λmax in Methanol gezeigt. Weiterhin ist in Abb. 27 ein
UV/vis-Absorptionsspektrum der Modellverbindung in Methanol abgebildet. Das
UV/vis-Absorptionsmaximum der relativ breiten UV/vis-Absorptionsbande liegt bei
λmax = 440 nm. Der molare Absorptionskoeffizient der Modellverbindung
N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin (rotes Öl) ist in Methanol
mit εModell = 8.375 · 102 l·mol-1·cm-1 relativ klein.
Es ist literaturbekannt, dass strukturell ähnliche 2-Nitro-p-phenylendiamin-Derivate
ebenfalls niedrige molare Absorptionskoeffizienten aufweisen. So zeigt im Vergleich
N1,N1,N4,N4-tetramethyl-2-nitro-p-phenylendiamin (dunkelrotes Öl) in Ethanol ein
UV/vis-Absorptionsmaximum bei λmax = 445 nm und einen molaren Absorptions-
koeffizienten von 1.2 · 103 l·mol-1·cm-1 in Cyclohexan.35
3 Ergebnisse und Diskussion
90
Abb. 27: Graphische Darstellung der UV/vis-Absorption von N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin in Methanol über c · d bei λmax = 440 nm zur Ermittlung des molaren Absorptions-koeffizienten der Modellverbindung ε Modell, Inset: UV/vis-Absorptionsspektrum von N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin in Methanol.
Mit Kenntnis des spezifischen Absorptionskoeffizienten von PNMVAm 5 mit
ε´ = 0.8655 l·g-1·cm-1 (33 mg in 50 ml) und des molaren Absorptionskoeffizienten der
Modellverbindung N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin in
Methanol wurde der Substitutionsgrad nach Gl. 8 von PNMVAm 5 mit s = 27.1 %
bestimmt.
Der Substitutionsgrad liegt bei einem eingesetzten Molverhältnis von
4-DMFNA/β-DMCD zu PVAm mit 0.3053 im Vergleich zu 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) mit s = 11.3 % deutlich
höher. Das könnte an der längeren Reaktionszeit von 12 h in der Siedehitze während
der Funktionalisierungsreaktion im Vergleich zu 8 h bei der Synthese von PVAm 4
liegen. Der höhere Substitutionsgrad beim Einsatz von sekundärem Polyamin im
Vergleich zu primärem Polyamin (PVAm) könnte auch von der größeren Donorstärke
der monomethylierten 1-Aminogruppe herrühren. Aus sterischen Gründen sollte
jedoch die Bildung der zwitterionischen Zwischenstufe mit sekundärem Polyamin bei
der nucleophilen aromatischen Substitution erschwert sein.
y = 837.46x + 0.0046R2 = 0.9996
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 0.0002 0.0004 0.0006 0.0008 0.001 0.0012 0.0014 0.0016 0.0018 0.002
c · d [mol · l -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
440
nm
) [a
.u.]
N
CH3
H3CCH3
NO2
NCH3H3C
εεεε Modell = 837.5 l · mol -1 · cm-1
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
λλλλmax = 440 nm

3 Ergebnisse und Diskussion
91
2,4´-dinitrostilben-funktionalisiertes PVAm
2,4´-dinitrostilben-funktionalisiertes PVAm (PVAm 9) löst sich sowohl in Wasser als
auch in Methanol, jedoch nicht so gut im Vergleich zu 2- bzw. 4-nitrophenyl-
funktionalisiertem PVAm (PVAm 2 bzw. PVAm 1). Für die Synthese der der
chromophoren Einheit in etwa strukturell entsprechenden Modellverbindung wurde
4-Fluor-3,4´-dinitrostilben (FDNstilben) mit i-Propylamin zur Reaktion gebracht
(s. Abschnitt 5.6 Experimenteller Teil).
Da die Modellverbindung in Wasser unlöslich, das 2,4´-dinitrostilben-funktionalisierte
PVAm (PVAm 9) in Methanol löslich ist, wurde der molare Absorptionskoeffizient
εModell über eine Verdünnungsreihe in Methanol wie in Abb. 28 gezeigt mit
εModell = 2.67 · 104 l mol-1 cm-1 bestimmt.
Abb. 28: Graphische Darstellung der UV/vis-Absorption bei λmax = 390 nm von 4-N-Isopropylamino-
3,4´-dinitrostilben in Methanol über c · d zur Ermittlung des molaren Absorptionskoeffizienten ε Modell, Inset: UV/vis-Absorptionsspektrum von 4-N-Isopropylamino-3,4´-dinitrostilben in Methanol.
Das UV/vis-Absorptionsmaximum λmax der Modellverbindung 4-N-Isopropylamino-
3,4´-dinitrostilben liegt in Methanol wie das von 2,4´-dinitrostilben-funktionalisiertem
PVAm (PVAm 9) bei 390 nm.
y = 26721x + 0.0095R2 = 0.9994
0
0.2
0.4
0.6
0.8
1
1.2
0 0.00001 0.00002 0.00003 0.00004 0.00005 0.00006
c · d [mol · l -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
390
nm
) [a
.u.]
εεεε Modell = 2.67 · 104 l · mol -1 · cm-1
NCH
O2NH
CH3
H3C NO2
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
λmax = 390 nm
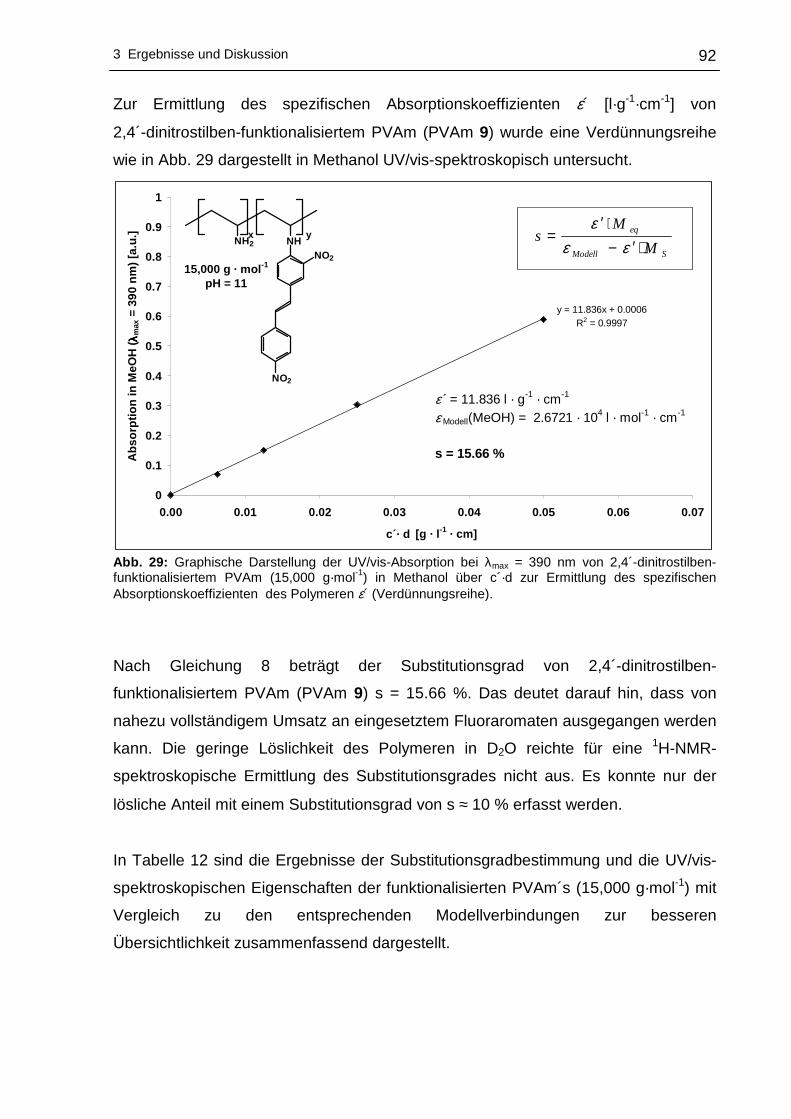
3 Ergebnisse und Diskussion
92
Zur Ermittlung des spezifischen Absorptionskoeffizienten ε´ [l·g-1·cm-1] von
2,4´-dinitrostilben-funktionalisiertem PVAm (PVAm 9) wurde eine Verdünnungsreihe
wie in Abb. 29 dargestellt in Methanol UV/vis-spektroskopisch untersucht.
Abb. 29: Graphische Darstellung der UV/vis-Absorption bei λmax = 390 nm von 2,4´-dinitrostilben-funktionalisiertem PVAm (15,000 g·mol-1) in Methanol über c´·d zur Ermittlung des spezifischen Absorptionskoeffizienten des Polymeren ε´ (Verdünnungsreihe).
Nach Gleichung 8 beträgt der Substitutionsgrad von 2,4´-dinitrostilben-
funktionalisiertem PVAm (PVAm 9) s = 15.66 %. Das deutet darauf hin, dass von
nahezu vollständigem Umsatz an eingesetztem Fluoraromaten ausgegangen werden
kann. Die geringe Löslichkeit des Polymeren in D2O reichte für eine 1H-NMR-
spektroskopische Ermittlung des Substitutionsgrades nicht aus. Es konnte nur der
lösliche Anteil mit einem Substitutionsgrad von s ≈ 10 % erfasst werden.
In Tabelle 12 sind die Ergebnisse der Substitutionsgradbestimmung und die UV/vis-
spektroskopischen Eigenschaften der funktionalisierten PVAm´s (15,000 g·mol-1) mit
Vergleich zu den entsprechenden Modellverbindungen zur besseren
Übersichtlichkeit zusammenfassend dargestellt.
y = 11.836x + 0.0006R2 = 0.9997
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07
c´· d [g · l -1 · cm]
Abs
orpt
ion
in M
eOH
(λλ λλ m
ax =
390
nm
) [a
.u.]
ε ´ = 11.836 l · g-1 · cm-1
ε Modell(MeOH) = 2.6721 · 104 l · mol-1 · cm-1
s = 15.66 %
SModell
eq
M
Ms
⋅′−⋅′
=εε
εNH2 NH
NO2
NO2
x y
15,000 g · mol -1
pH = 11
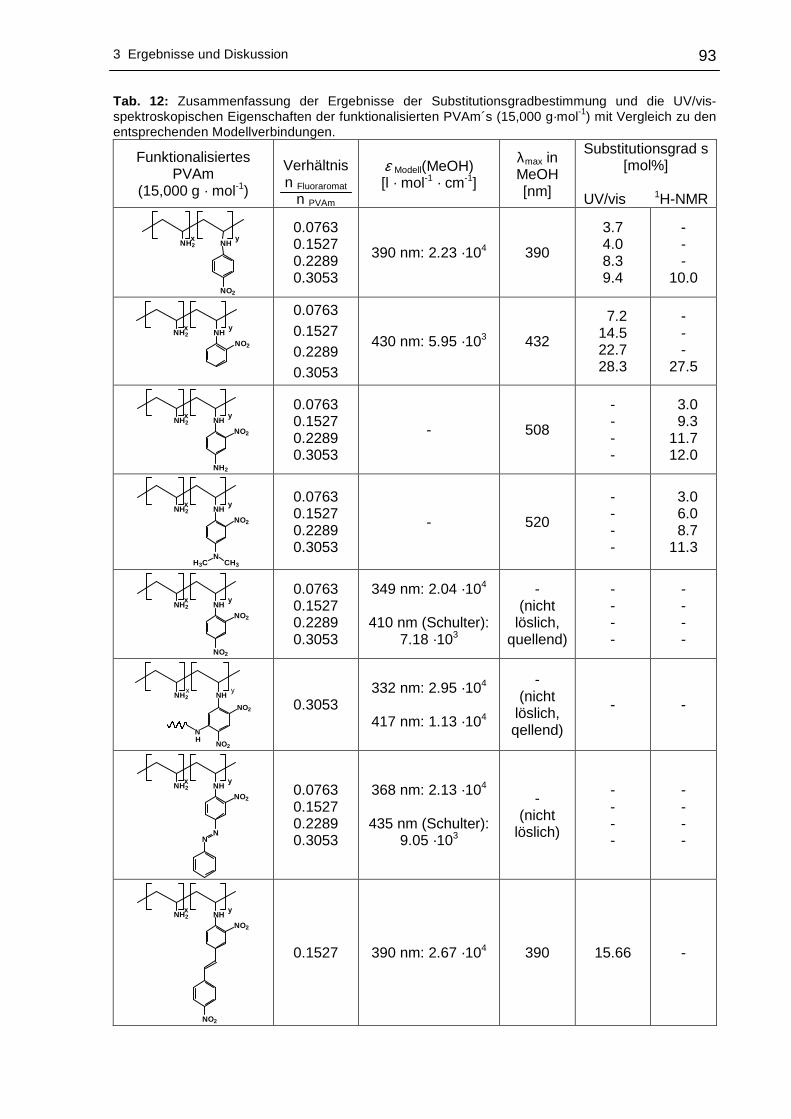
3 Ergebnisse und Diskussion
93
Tab. 12: Zusammenfassung der Ergebnisse der Substitutionsgradbestimmung und die UV/vis-spektroskopischen Eigenschaften der funktionalisierten PVAm´s (15,000 g·mol-1) mit Vergleich zu den entsprechenden Modellverbindungen.
Funktionalisiertes PVAm
(15,000 g · mol-1)
Verhältnis n Fluoraromat
n PVAm
ε Modell(MeOH) [l · mol-1 · cm-1]
λmax in MeOH [nm]
Substitutionsgrad s [mol%]
UV/vis 1H-NMR
NH2 NH
NO2
x y
0.0763 0.1527 0.2289 0.3053
390 nm: 2.23 ·104 390
3.7 4.0 8.3 9.4
- - -
10.0
NH2 NHNO2
x y
0.0763 0.1527 0.2289 0.3053
430 nm: 5.95 ·103 432
7.2 14.5 22.7 28.3
- - -
27.5
NH2 NHNO2
NH2
x y
0.0763 0.1527 0.2289 0.3053
- 508
- - - -
3.0 9.3 11.7 12.0
NH2 NHNO2
NH3C CH3
x y
0.0763 0.1527 0.2289 0.3053
- 520
- - - -
3.0 6.0 8.7 11.3
NH2 NHNO2
NO2
x y
0.0763 0.1527 0.2289 0.3053
349 nm: 2.04 ·104
410 nm (Schulter):
7.18 ·103
- (nicht
löslich, quellend)
- - - -
- - - -
NH2 NH
NO2
NO2
N
x y
H
0.3053 332 nm: 2.95 ·104
417 nm: 1.13 ·104
- (nicht
löslich, qellend)
- -
NH2 NHNO2
NN
x y
0.0763 0.1527 0.2289 0.3053
368 nm: 2.13 ·104
435 nm (Schulter):
9.05 ·103
- (nicht
löslich)
- - - -
- - - -
NH2 NHNO2
NO2
x y
0.1527 390 nm: 2.67 ·104 390 15.66 -

3 Ergebnisse und Diskussion
94
3.1.9 Kinetische Untersuchungen mittels UV/vis-Spe ktroskopie
3.1.9.1 Meßprinzip und Auswertung
Die sich während der nucleophilen aromatischen Substitution von aktivierten
Fluoraromaten mit PVAm bildenden nitrophenyl-funktionalisierten PVAm´s zeichnen
sich durch charakteristische UV/vis-Absorptionsspektren im Bereich von
400 − 520 nm aus. Diese Funktionalisierungsreaktionen können gut in Abhängigkeit
von der Reaktionszeit UV/vis-spektroskopisch verfolgt werden.
Zur Ermittlung von Aktivierungsparametern wurden zeitabhängige UV/vis-
Absorptionsspektren während der Funktionalisierung von PVAm, welches in großem
Überschuss eingesetzt wurde, aufgenommen. Beim entsprechenden UV/vis-
Absorptionsmaximum der neu gebildeten UV/vis-Absorptionsbande(n) je nach
eingesetztem Fluoraromat wurde die UV/vis-Absorption in Abhängigkeit von der
Reaktionszeit graphisch aufgetragen. Aus den charakteristisch gekrümmten
Messkurven wurden mittels nichtlinearer Regression nach dem Modell für
Reaktionen pseudo-monomolekularer Ordnung (Gl. 9) die Geschwindigkeits-
konstanten k der Bildung des jeweiligen nitrophenyl-funktionalisierten PVAm´s bei
verschiedenen Reaktionstemperaturen bestimmt.13
( )tk10 eAAy ⋅−⋅+= (9)
Dabei bedeuten die Regressionskoeffizienten:
A0 = erreichte UV/vis-Endabsorption bei t = ∞
A1 = Gesamtabsorptionsänderung zwischen t = 0 und t = ∞
k = Geschwindigkeitskonstante in s-1.
Voraussetzung für die Auswertung nach Gl. 9 für Reaktionen pseudo-erster Ordnung
bei zwei Reaktionspartnern ist der Einsatz einer der beiden Reaktionskomponenten
in großem Überschuss, damit dessen Konzentration während der Reaktion als
nahezu konstant angesehen werden kann. Die wahre Geschwindigkeitskonstante
zweiter Ordnung ergibt sich aus dem Quotienten der Geschwindigkeitskonstanten k
und der Konzentration der im Überschuss vorliegenden Reaktionskomponenten, in
diesem Falle PVAm.

3 Ergebnisse und Diskussion
95
Welches Verhältnis beider Reaktionskomponenten zueinander eingesetzt wird, hängt
von der zu erwartenden UV/vis-Endabsorption des Chromophoren ab, die einen Wert
über 1.8 nicht überschreiten sollte (Bandenstauchung und Verbreiterung). Ist die
Bildung des Chromophoren sehr schnell und/oder die Verdünnung zu gering,
womöglich sind die Zeitabstände zwischen den einzelnen Messungen zu groß, so
äußert sich das in schlecht oder nicht auswertbaren Kurven bei der zeitlichen
Auftragung der UV/vis-Absorption. Ein zu zeitiges Abbrechen der UV/vis-
spektroskopischen Verfolgung der Reaktion liefert eine Gerade, die nur den Beginn
der Reaktion anzeigt. Die Regressionsstatistik nach Gl. 9 ist dann mit einem großen
Fehler behaftet. Daher muss für jedes Reaktionssystem vorher ausprobiert werden,
welche Konzentrationen und welches Zeitfenster günstig für die Messungen sind.
Mit Kenntnis der Geschwindigkeitskonstanten k bei unterschiedlichen Temperaturen
kann über die Arrhenius-Gleichung (Gl. 10) durch graphisches Auftragen von lnk
über T-1 aus dem Anstieg der Geraden die Aktivierungsenergie EA bestimmt
werden.48 Dabei ist zu beachten, dass mit dieser Methode die Reaktionsgeschwindig-
keitskonstante k der Bildung von nitrophenyl-funktionalisiertem PVAm bei der
nucleophilen aromatischen Substitution von Fluoraromaten mit PVAm experimentell
erfasst wird und nicht die Geschwindigkeitskonstanten k1 bzw. k2 der Teilschritte.
−⋅= RT
EA
eAk (10)
Die Aktivierungsenthalpie ∆H‡ wird durch graphisches Auftragen von ln(k · T-1) über
T-1 aus dem Anstieg der Geraden und die Aktivierungsentropie ∆S‡ aus dem
Schnittpunkt mit der y-Achse mit Hilfe der Eyring-Gleichung (Gl. 11) bestimmt.48
−
≠≠
⋅⋅=RT∆H
R∆S
B eehTk
k (11)
Die Freie Aktivierungsenthalpie ∆G‡ wurde über die Gibbs-Helmholtz-Gleichung
(Gl. 12) berechnet.48
≠≠≠ −= ST∆∆H∆G (12)

3 Ergebnisse und Diskussion
96
3.1.9.2 Funktionalisierung von Polyvinylamin (PVAm ) mittels
4-Fluornitrobenzen (4-FNB)
Eine charakteristische UV/vis-Absorptionsspektrenserie aufgenommen während der
nucleophilen aromatischen Substitution von 4-FNB/β-DMCD mit PVAm (327-facher
Überschuss, 0.3375 mol · l-1)) in Abhängigkeit von der Reaktionszeit bei 60°C i n
Wasser bei pH = 11 zeigt Abb. 30.
Abb. 30: UV/vis-Absorptionsspektrenserie aufgenommen während der nucleophilen aromatischen Substitution von 4-FNB/β-DMCD mit PVAm in Wasser (Zunahme der Intensität der neu gebildeten UV/vis-Absorptionsbande bei λmax = 413 nm von 4-nitrophenyl-funktionalisiertem PVAm).
Die neu auftretende UV/vis-Absorptionsbande bei λmax = 413 nm spiegelt die Bildung
von 4-nitrophenyl-funktionalisiertem PVAm wider. Es ist literaturbekannt, dass die
Komplexbildung von niedermolekularen 4-Nitroanilinderivaten mit z. B. Cyclodextrin
zu einer bathochromen Verschiebung des UV/vis-Absorptionsmaximums führt.124,125
Die Lage des UV/vis-Absorptionsmaximum zeigt jedoch, dass β-DMCD keinen
Einfluss auf die UV/vis-Absorptionsbande hat und somit keine Wechselwirkungen mit
der neu gebildeten chromophoren Gruppe eingeht.
-0.1
0.1
0.3
0.5
0.7
0.9
1.1
1.3
300 350 400 450 500 550 600 650 700
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
NH2 NH
NO2
x y
60°C
41316 h14 h12 h10 h
8 h6 h4 h3 h2 h1 h0 h
15,000 g · mol -1
pH = 11

3 Ergebnisse und Diskussion
97
Die graphische Auftragung der UV/vis-Absorption bei entsprechendem UV/vis-
Absorptionsmaximum λmax = 413 nm über die Reaktionszeit t ergibt gekrümmte
Kurven für Reaktionen pseudo-monomolekularer Ordnung. Abb. 31 zeigt die UV/vis-
Absorption der Reaktion von 4-FNB/β-DMCD mit PVAm in Wasser in Abhängigkeit
von der Reaktionszeit bei 50°C, 60°C, 70°C and 80°C . Mittels nichtlinearer
Regression nach Gleichung 9 wurden die Geschwindigkeitskonstanten k, welche
erwartungsgemäß mit Zunahme der Reaktionstemperatur zunehmen, bestimmt.
Abb. 31: Graphische Darstellung der UV/vis-Absorption bei λmax = 413 nm über der Reaktionszeit (gekrümmte Kurven für Reaktionen pseudo-monomolekularer Ordnung) bei 50°C, 60°C, 70°C und 80°C zur Ermittlung der Geschwindigkeitskonstanten k der Funktionalisierung von PVAm mittels 4-FNB.
Die Aktivierungsenergie EA der nucleophilen aromatischen Substitution von
4-FNB/β-DMCD mit PVAm wurde nach Arrhenius aus dem Anstieg der Geraden
durch graphische Auftragung von lnk über T-1 nach Gleichung 10 bestimmt
(vgl. Abb. 35). Die Aktivierungsenergie EA für die nucleophile aromatische
Substitution von 4-FNB/β-DMCD mit PVAm in Wasser wurde mit 67.5 ± 1.04 kJ·mol-1
ermittelt. Der Fehlerbereich der Aktivierungsenergie wurde dabei aus dem
Bestimmtheitsmaß r2 der Geraden berechnet.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 2 4 6 8 10 12 14 16 18 20 22 24
Zeit [h]
Abs
ortio
n (
λλ λλ max
= 4
13 n
m)
[a.u
.] 60°C
70°C
k50°C = 1.13 · 10-6 s-1
k60°C = 3.11 · 10-6 s-1
k70°C = 5.33 · 10-6 s-1
k80°C = 1.00 · 10-5 s-1
50°C
80°C
NH2 NH
NO2
x y
15,000 g · mol-1pH = 11
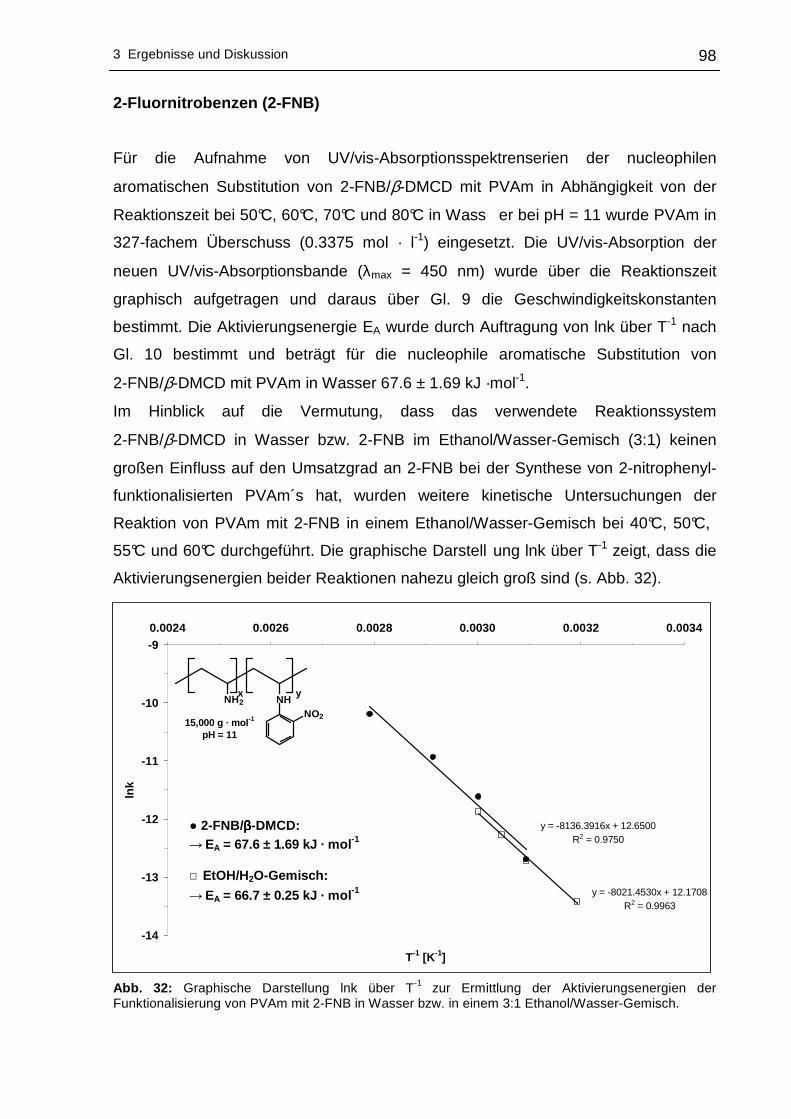
3 Ergebnisse und Diskussion
98
2-Fluornitrobenzen (2-FNB)
Für die Aufnahme von UV/vis-Absorptionsspektrenserien der nucleophilen
aromatischen Substitution von 2-FNB/β-DMCD mit PVAm in Abhängigkeit von der
Reaktionszeit bei 50°C, 60°C, 70°C und 80°C in Wass er bei pH = 11 wurde PVAm in
327-fachem Überschuss (0.3375 mol · l-1) eingesetzt. Die UV/vis-Absorption der
neuen UV/vis-Absorptionsbande (λmax = 450 nm) wurde über die Reaktionszeit
graphisch aufgetragen und daraus über Gl. 9 die Geschwindigkeitskonstanten
bestimmt. Die Aktivierungsenergie EA wurde durch Auftragung von lnk über T-1 nach
Gl. 10 bestimmt und beträgt für die nucleophile aromatische Substitution von
2-FNB/β-DMCD mit PVAm in Wasser 67.6 ± 1.69 kJ ·mol-1.
Im Hinblick auf die Vermutung, dass das verwendete Reaktionssystem
2-FNB/β-DMCD in Wasser bzw. 2-FNB im Ethanol/Wasser-Gemisch (3:1) keinen
großen Einfluss auf den Umsatzgrad an 2-FNB bei der Synthese von 2-nitrophenyl-
funktionalisierten PVAm´s hat, wurden weitere kinetische Untersuchungen der
Reaktion von PVAm mit 2-FNB in einem Ethanol/Wasser-Gemisch bei 40°C, 50°C,
55°C und 60°C durchgeführt. Die graphische Darstell ung lnk über T-1 zeigt, dass die
Aktivierungsenergien beider Reaktionen nahezu gleich groß sind (s. Abb. 32).
Abb. 32: Graphische Darstellung lnk über T-1 zur Ermittlung der Aktivierungsenergien der Funktionalisierung von PVAm mit 2-FNB in Wasser bzw. in einem 3:1 Ethanol/Wasser-Gemisch.
y = -8136.3916x + 12.6500R2 = 0.9750
y = -8021.4530x + 12.1708R2 = 0.9963
-14
-13
-12
-11
-10
-90.0024 0.0026 0.0028 0.0030 0.0032 0.0034
T-1 [K -1]
lnk
● 2-FNB/ββββ-DMCD:→ EA = 67.6 ± 1.69 kJ · mol -1
□ EtOH/H2O-Gemisch:
→ EA = 66.7 ± 0.25 kJ · mol -1
NH2 NHNO2
x y
15,000 g · mol -1
pH = 11

3 Ergebnisse und Diskussion
99
Durch graphisches Auftragen von ln(k·T-1) über T-1 wurden die Aktivierungs-
enthalpien ∆H‡ aus den Anstiegen der Geraden und die Aktivierungsentropien ∆S‡
aus den Schnittpunkten mit der y-Achse mit Hilfe der Eyring-Gleichung (Gl. 11)
bestimmt.
In Tabelle 13 sind die Ergebnisse der kinetischen Untersuchungen
(Geschwindigkeitskonstante k, Aktivierungsenergie EA, Aktivierungsenthalpie ∆H‡
und Aktivierungsentropie ∆S‡) der Funktionalisierung von PVAm mittels 2-FNB zum
einen in Wasser über Löslichkeitsvermittlung mittels β-DMCD und zum anderen in
einem Ethanol/Wasser -Lösungsmittelgemisch (3:1) zusammengefasst.
Tab. 13: Zusammenfassung der Ergebnisse der kinetischen Untersuchungen zur Ermittlung der Aktivierungsparameter: Geschwindigkeitskonstante k, Aktivierungsenergie EA, Aktivierungsenthalpie ∆H‡ und Aktivierungsentropie ∆S‡) der Funktionalisierung von PVAm mit 2-FNB in Wasser über β-DMCD und in einem Ethanol/Wasser-Gemisch.
Fluoraromat
(FA)
nPVAm
nFA
Τ
[°C]
k
[s-1]
pH = 11
EA
[kJ·mol-1]
Arrhenius
∆H‡
[kJ·mol-1]
Eyring
∆S‡
[J·K-1·mol-1]
Eyring
F
O2N
β-DMCD/H2O
327
50
60
70
85
3.08 · 10-6
9.06 · 10-6
1.79 · 10-5
3.74 · 10-5
67.6 ± 1.69 64.82 -149.16
F
O2N
EtOH/H2O
327
40
50
55
60
1.49 · 10-6
3.03 · 10-6
4.66 · 10-6
6.95 · 10-6
66.7 ± 0.25 64.01 -152.69
Die Unterschiede der Aktivierungsparameter sind sehr gering. Daher ist es nicht
verwunderlich, dass sich − bei gleichem eingesetzten Molverhältnis von 2-FNB zu
PVAm im EtOH/Wasser-Lösungsmittelgemisch und 2-FNB/β-DMCD zu PVAm in
Wasser − die Substitutionsgrade von 2-nitrophenyl-funktionalisiertem PVAm kaum
unterscheiden. Der Umsatzgrad an 2-FNB ist nahezu unabhängig von den in dieser
Arbeit verwendeten Lösungsmittelsystemen (s. Abschnitt 3.1.8 S. 83).
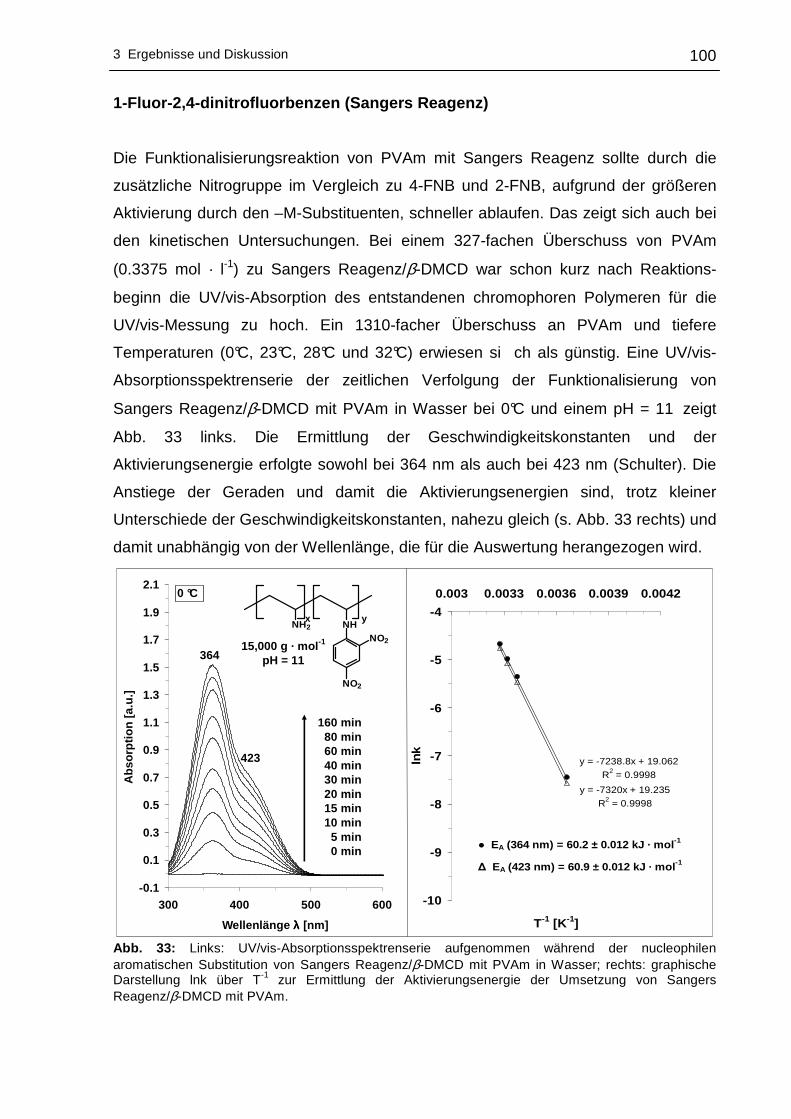
3 Ergebnisse und Diskussion
100
1-Fluor-2,4-dinitrofluorbenzen (Sangers Reagenz)
Die Funktionalisierungsreaktion von PVAm mit Sangers Reagenz sollte durch die
zusätzliche Nitrogruppe im Vergleich zu 4-FNB und 2-FNB, aufgrund der größeren
Aktivierung durch den –M-Substituenten, schneller ablaufen. Das zeigt sich auch bei
den kinetischen Untersuchungen. Bei einem 327-fachen Überschuss von PVAm
(0.3375 mol · l-1) zu Sangers Reagenz/β-DMCD war schon kurz nach Reaktions-
beginn die UV/vis-Absorption des entstandenen chromophoren Polymeren für die
UV/vis-Messung zu hoch. Ein 1310-facher Überschuss an PVAm und tiefere
Temperaturen (0°C, 23°C, 28°C und 32°C) erwiesen si ch als günstig. Eine UV/vis-
Absorptionsspektrenserie der zeitlichen Verfolgung der Funktionalisierung von
Sangers Reagenz/β-DMCD mit PVAm in Wasser bei 0°C und einem pH = 11 zeigt
Abb. 33 links. Die Ermittlung der Geschwindigkeitskonstanten und der
Aktivierungsenergie erfolgte sowohl bei 364 nm als auch bei 423 nm (Schulter). Die
Anstiege der Geraden und damit die Aktivierungsenergien sind, trotz kleiner
Unterschiede der Geschwindigkeitskonstanten, nahezu gleich (s. Abb. 33 rechts) und
damit unabhängig von der Wellenlänge, die für die Auswertung herangezogen wird.
Abb. 33: Links: UV/vis-Absorptionsspektrenserie aufgenommen während der nucleophilen aromatischen Substitution von Sangers Reagenz/β-DMCD mit PVAm in Wasser; rechts: graphische Darstellung lnk über T-1 zur Ermittlung der Aktivierungsenergie der Umsetzung von Sangers Reagenz/β-DMCD mit PVAm.
-0.1
0.1
0.3
0.5
0.7
0.9
1.1
1.3
1.5
1.7
1.9
2.1
300 400 500 600
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
0 °C
160 min80 min60 min40 min30 min20 min15 min10 min
5 min0 min
NH2 NHNO2
NO2
x y
364
423
15,000 g · mol -1
pH = 11
y = -7238.8x + 19.062
R2 = 0.9998
y = -7320x + 19.235
R2 = 0.9998
-10
-9
-8
-7
-6
-5
-4
0.003 0.0033 0.0036 0.0039 0.0042
T-1 [K -1]
lnk
● EA (364 nm) = 60.2 ± 0.012 kJ · mol -1
∆ EA (423 nm) = 60.9 ± 0.012 kJ · mol -1
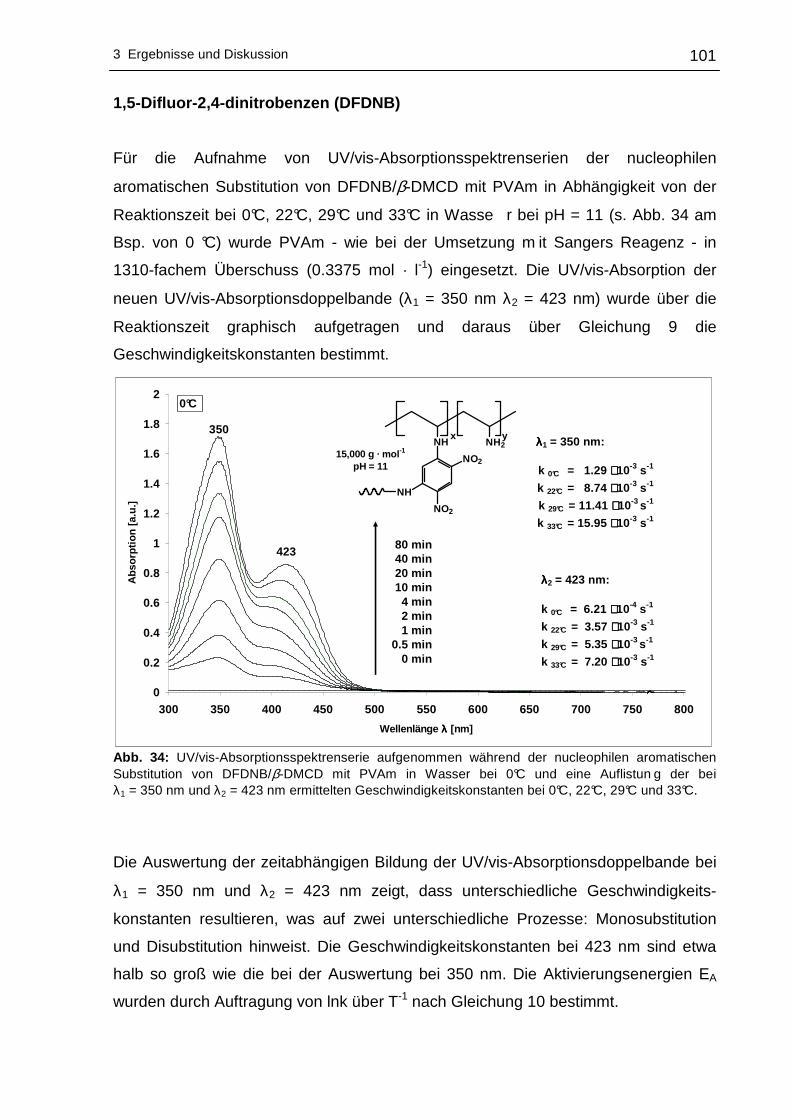
3 Ergebnisse und Diskussion
101
1,5-Difluor-2,4-dinitrobenzen (DFDNB)
Für die Aufnahme von UV/vis-Absorptionsspektrenserien der nucleophilen
aromatischen Substitution von DFDNB/β-DMCD mit PVAm in Abhängigkeit von der
Reaktionszeit bei 0°C, 22°C, 29°C und 33°C in Wasse r bei pH = 11 (s. Abb. 34 am
Bsp. von 0 °C) wurde PVAm - wie bei der Umsetzung m it Sangers Reagenz - in
1310-fachem Überschuss (0.3375 mol · l-1) eingesetzt. Die UV/vis-Absorption der
neuen UV/vis-Absorptionsdoppelbande (λ1 = 350 nm λ2 = 423 nm) wurde über die
Reaktionszeit graphisch aufgetragen und daraus über Gleichung 9 die
Geschwindigkeitskonstanten bestimmt.
Abb. 34: UV/vis-Absorptionsspektrenserie aufgenommen während der nucleophilen aromatischen Substitution von DFDNB/β-DMCD mit PVAm in Wasser bei 0°C und eine Auflistun g der bei λ1 = 350 nm und λ2 = 423 nm ermittelten Geschwindigkeitskonstanten bei 0°C, 22°C, 29°C und 33°C.
Die Auswertung der zeitabhängigen Bildung der UV/vis-Absorptionsdoppelbande bei
λ1 = 350 nm und λ2 = 423 nm zeigt, dass unterschiedliche Geschwindigkeits-
konstanten resultieren, was auf zwei unterschiedliche Prozesse: Monosubstitution
und Disubstitution hinweist. Die Geschwindigkeitskonstanten bei 423 nm sind etwa
halb so groß wie die bei der Auswertung bei 350 nm. Die Aktivierungsenergien EA
wurden durch Auftragung von lnk über T-1 nach Gleichung 10 bestimmt.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
300 350 400 450 500 550 600 650 700 750 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
80 min40 min20 min10 min
4 min2 min1 min
0.5 min0 min
0°C
x yNH NH2
NO2
NO2
NH
423
350
k 0°C = 1.29 ⋅⋅⋅⋅ 10-3 s-1
k 22°C = 8.74 ⋅⋅⋅⋅ 10-3 s-1
k 29°C = 11.41 ⋅⋅⋅⋅ 10-3 s-1
k 33°C = 15.95 ⋅⋅⋅⋅ 10-3 s-1
k 0°C = 6.21 ⋅⋅⋅⋅ 10-4 s-1
k 22°C = 3.57 ⋅⋅⋅⋅ 10-3 s-1
k 29°C = 5.35 ⋅⋅⋅⋅ 10-3 s-1
k 33°C = 7.20 ⋅⋅⋅⋅ 10-3 s-1
λλλλ1 = 350 nm:
λλλλ2 = 423 nm:
15,000 g · mol -1
pH = 11
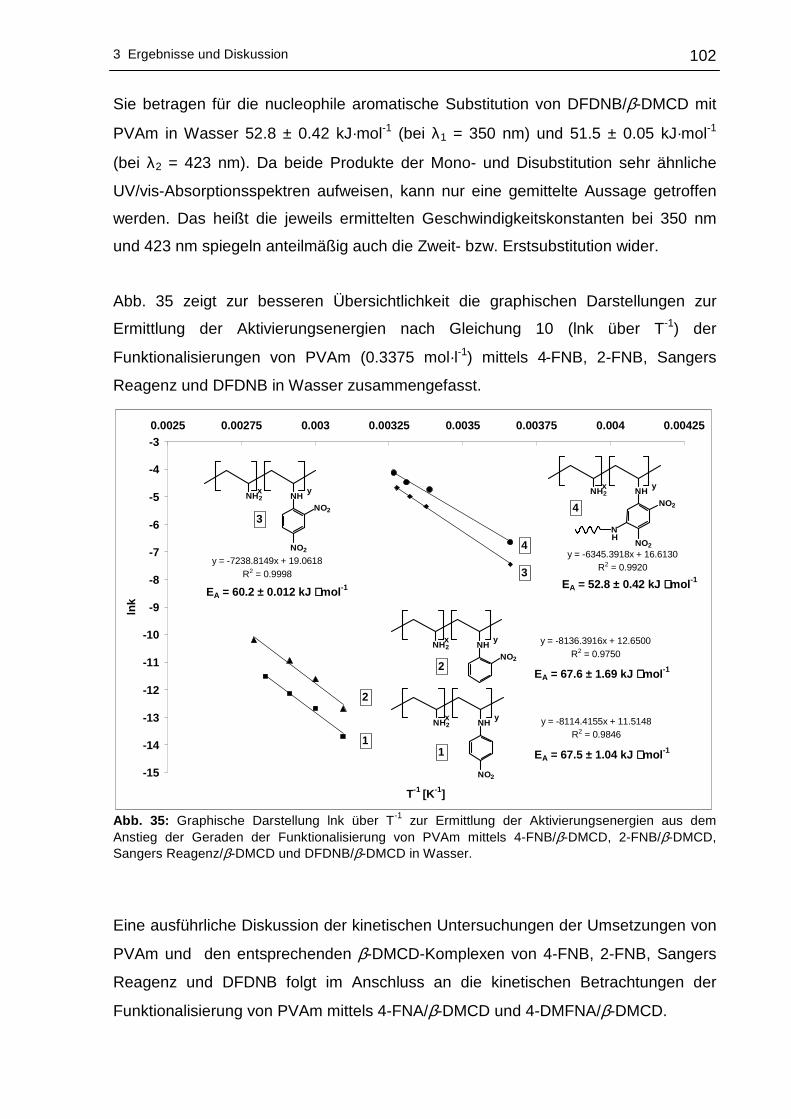
3 Ergebnisse und Diskussion
102
Sie betragen für die nucleophile aromatische Substitution von DFDNB/β-DMCD mit
PVAm in Wasser 52.8 ± 0.42 kJ·mol-1 (bei λ1 = 350 nm) und 51.5 ± 0.05 kJ·mol-1
(bei λ2 = 423 nm). Da beide Produkte der Mono- und Disubstitution sehr ähnliche
UV/vis-Absorptionsspektren aufweisen, kann nur eine gemittelte Aussage getroffen
werden. Das heißt die jeweils ermittelten Geschwindigkeitskonstanten bei 350 nm
und 423 nm spiegeln anteilmäßig auch die Zweit- bzw. Erstsubstitution wider.
Abb. 35 zeigt zur besseren Übersichtlichkeit die graphischen Darstellungen zur
Ermittlung der Aktivierungsenergien nach Gleichung 10 (lnk über T-1) der
Funktionalisierungen von PVAm (0.3375 mol·l-1) mittels 4-FNB, 2-FNB, Sangers
Reagenz und DFDNB in Wasser zusammengefasst.
Abb. 35: Graphische Darstellung lnk über T-1 zur Ermittlung der Aktivierungsenergien aus dem Anstieg der Geraden der Funktionalisierung von PVAm mittels 4-FNB/β-DMCD, 2-FNB/β-DMCD, Sangers Reagenz/β-DMCD und DFDNB/β-DMCD in Wasser.
Eine ausführliche Diskussion der kinetischen Untersuchungen der Umsetzungen von
PVAm und den entsprechenden β-DMCD-Komplexen von 4-FNB, 2-FNB, Sangers
Reagenz und DFDNB folgt im Anschluss an die kinetischen Betrachtungen der
Funktionalisierung von PVAm mittels 4-FNA/β-DMCD und 4-DMFNA/β-DMCD.
y = -8114.4155x + 11.5148R2 = 0.9846
y = -8136.3916x + 12.6500R2 = 0.9750
y = -6345.3918x + 16.6130R2 = 0.9920
y = -7238.8149x + 19.0618R2 = 0.9998
-15
-14
-13
-12
-11
-10
-9
-8
-7
-6
-5
-4
-30.0025 0.00275 0.003 0.00325 0.0035 0.00375 0.004 0.00425
T-1 [K -1]
lnk
1EA = 67.5 ± 1.04 kJ ⋅ ⋅ ⋅ ⋅ mol -11
2
EA = 67.6 ± 1.69 kJ ⋅ ⋅ ⋅ ⋅ mol -1 2
EA = 60.2 ± 0.012 kJ ⋅⋅⋅⋅ mol -1
3
3
4
EA = 52.8 ± 0.42 kJ ⋅⋅⋅⋅ mol -1
4
x yNH2 NH
NO2
NO2
NH
NH2 NHNO2
NO2
x y
NH2 NHNO2
x y
NH2 NH
NO2
x y

3 Ergebnisse und Diskussion
103
4-Fluor-3-nitroanilin (4-FNA)
In Abb. 36 (links) ist eine charakteristische UV/vis-Absorptionsspektrenserie
aufgenommen während der nucleophilen aromatischen Substitution von
4-FNA/β-DMCD mit PVAm in Abhängigkeit von der Reaktionszeit bei 60°C in Wasser
bei pH = 11 gezeigt. PVAm (0.3375 mol·l-1) wurde dabei in 164-fachem Überschuss
eingesetzt, da ein 327-facher Überschuss wie bei der Funktionalisierung von PVAm
mittels 2-FNB und 4-FNB zu kleinen UV/vis-Endabsorptionen (< 0.5) und sehr langen
Messzeiten führte.
Abb. 36: Links: UV/vis-Absorptionsspektrenserie aufgenommen während der nucleophilen aromatischen Substitution von 4-FNA/β-DMCD mit PVAm in Wasser; rechts: graphische Darstellung lnk über T-1 zur Ermittlung der Aktivierungsenergie der Umsetzung von 4-FNA/β-DMCD mit PVAm.
Die neu auftretende UV/vis-Absorptionsbande bei λmax = 519 nm zeigt die Bildung
von 2-nitro-4-aminobenzen-funktionalisiertem PVAm. Die Funktionalisierung von
PVAm mittels 4-FNA/β-DMCD wurde weiterhin bei 50 °C und 80 °C UV/vis-
spektroskopisch verfolgt. Die UV/vis-Absorption bei λmax = 519 wurde über die
Reaktionszeit bei 50°C, 60°C und 80°C graphisch auf getragen und daraus über Gl. 9
die Geschwindigkeitskonstanten bestimmt. Aus der Auftragung von lnk über T-1 nach
Gl. 10 wurde die Aktivierungsenergie mit 67.4 ± 0.16 kJ·mol-1 ermittelt (s. Abb. 36
rechts).
0
0.2
0.4
0.6
0.8
1
1.2
350 450 550 650 750 850
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
519
60°C
42 h36 h30 h25 h20 h15 h10 h 5 h 0 h
NH2 NHNO2
NH2
x y
15,000 g · mol -1
pH = 11y = -8102.1593x + 10.6763
R2 = 0.9977
-16
-15
-14
-13
-12
-110.0027 0.0028 0.0029 0.003 0.0031 0.0032
T-1 [K -1]
lnk
EA = 67.4 ± 0.16 kJ · mol -1

3 Ergebnisse und Diskussion
104
N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA)
Für die kinetischen Untersuchungen der Funktionalisierung von PVAm mittels
4-DMFNA in Wasser bei pH = 11 wurde PVAm (0.3375 mol·l-1) in 64-fachem
Überschuss zum 4-DMFNA/β-DMCD-Komplex eingesetzt. Die Konzentration von
4-DMFNA wurde aufgrund der zu geringen UV/vis-Endabsorption und sehr langer
Messzeiten im Vergleich zu 4-FNA verdoppelt.
Die Geschwindigkeitskonstanten wurden über Gl. 9 durch die graphische Auftragung
der UV/vis-Absorption bei λmax über der Reaktionszeit bestimmt (s. Abb. 37 links). Mit
Hilfe von Gl. 10 wurde die Aktivierungsenergie aus den Geschwindigkeitskonstanten
bei 32°C, 60°C und 70°C durch graphisches Auftragen von lnk über T-1 (s. Abb. 37
rechts) aus dem Anstieg der Geraden mit 76.5 kJ·mol-1 ermittelt.
Abb. 37: Links: graphische Darstellung der UV/vis-Absorption bei λmax über die Reaktionszeit der Funktionalisierung von PVAm mittels 4-DMFNA/β-DMCD in Wasser; rechts: graphische Darstellung lnk über T-1 zur Ermittlung der Aktivierungsenergie der Umsetzung von 4-DMFNA/β-DMCD mit PVAm.
Eine ausführliche Diskussion der ermittelten Aktivierungsparameter der einzelnen
Funktionalisierungsreaktionen von im Überschuss eingesetztem PVAm
(0.3375 mol·l-1) folgt im Anschluss an Tabelle 14, in der übersichtlich die Ergebnisse
der kinetischen Untersuchungen nach pseudo-erster Ordnung mit Hilfe der UV/vis-
Spektroskopie zusammengefasst sind.
0
0.5
1
1.5
2
2.5
0 20 40 60 80 100
Zeit [h]
Abs
orpt
ion
bei
λλ λλ max
[a.u
.]
k32°C = 3.0464 · 10-7 s-1
k60°C = 3.7770 · 10-6 s-1
k70°C = 8.6297 · 10-6 s-1
NH2 NHNO2
NH3C CH3
x y
15,000 g · mol -1
pH = 11
32 °C
60 °C
70 °Cy = -9196.51x + 15.13
R2 = 1.00
-16
-15
-14
-13
-12
-11
-100.0028 0.003 0.0032 0.0034
T-1 [K -1]
lnk
EA = 76.5 ± 0.00 kJ · mol -1
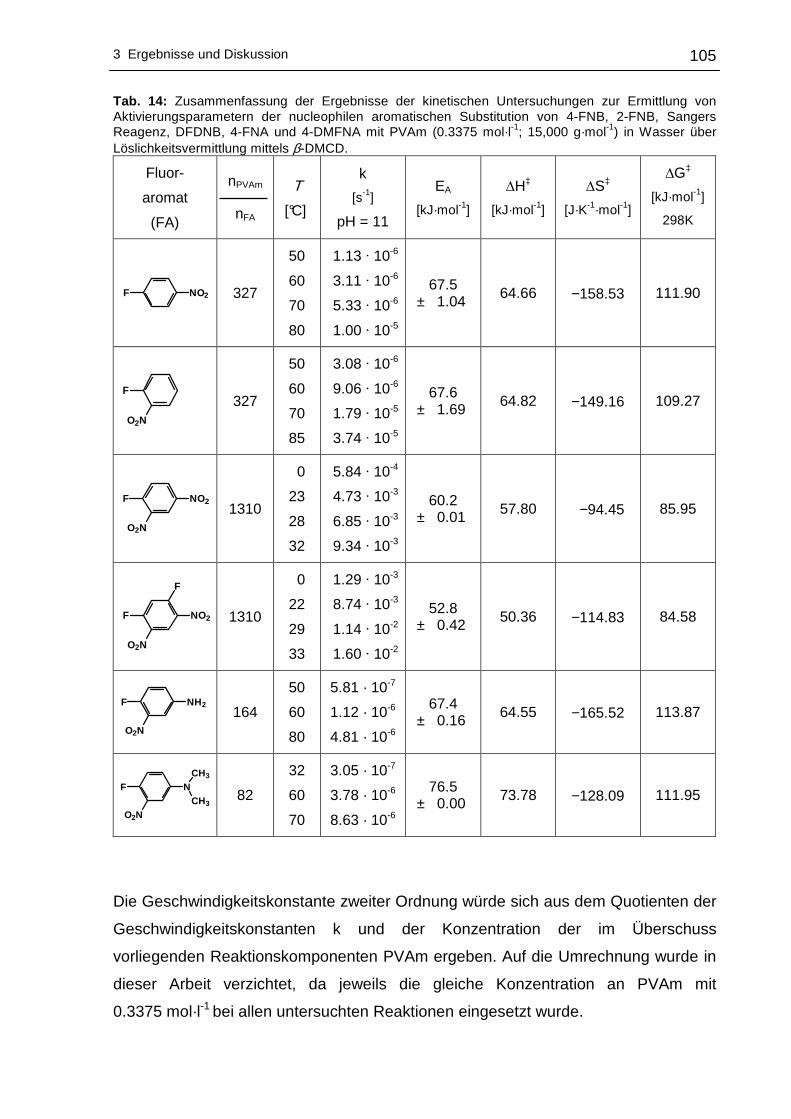
3 Ergebnisse und Diskussion
105
Tab. 14: Zusammenfassung der Ergebnisse der kinetischen Untersuchungen zur Ermittlung von Aktivierungsparametern der nucleophilen aromatischen Substitution von 4-FNB, 2-FNB, Sangers Reagenz, DFDNB, 4-FNA und 4-DMFNA mit PVAm (0.3375 mol·l-1; 15,000 g·mol-1) in Wasser über Löslichkeitsvermittlung mittels β-DMCD.
Fluor-
aromat
(FA)
nPVAm
nFA
Τ
[°C]
k
[s-1]
pH = 11
EA
[kJ·mol-1]
∆H‡
[kJ·mol-1]
∆S‡
[J·K-1·mol-1]
∆G‡
[kJ·mol-1]
298K
F NO2
327
50
60
70
80
1.13 · 10-6
3.11 · 10-6
5.33 · 10-6
1.00 · 10-5
67.5 ± 1.04 64.66 −158.53 111.90
F
O2N
327
50
60
70
85
3.08 · 10-6
9.06 · 10-6
1.79 · 10-5
3.74 · 10-5
67.6 ± 1.69 64.82 −149.16 109.27
F
O2N
NO2
1310
0
23
28
32
5.84 · 10-4
4.73 · 10-3
6.85 · 10-3
9.34 · 10-3
60.2 ± 0.01 57.80 −94.45 85.95
F
O2N
NO2
F
1310
0
22
29
33
1.29 · 10-3
8.74 · 10-3
1.14 · 10-2
1.60 · 10-2
52.8 ± 0.42 50.36 −114.83 84.58
F NH2
O2N
164
50
60
80
5.81 · 10-7
1.12 · 10-6
4.81 · 10-6
67.4 ± 0.16 64.55 −165.52 113.87
N
O2N
FCH3
CH3
82
32
60
70
3.05 · 10-7
3.78 · 10-6
8.63 · 10-6
76.5 ± 0.00 73.78 −128.09 111.95
Die Geschwindigkeitskonstante zweiter Ordnung würde sich aus dem Quotienten der
Geschwindigkeitskonstanten k und der Konzentration der im Überschuss
vorliegenden Reaktionskomponenten PVAm ergeben. Auf die Umrechnung wurde in
dieser Arbeit verzichtet, da jeweils die gleiche Konzentration an PVAm mit
0.3375 mol·l-1 bei allen untersuchten Reaktionen eingesetzt wurde.

3 Ergebnisse und Diskussion
106
Die Aktivierungsentropien ∆S‡ sind bei allen untersuchten Funktionalisierungs-
reaktionen negativ, wie es die Vereinigung zweier neutraler Moleküle zu einer
zwitterionischen Zwischenstufe erwarten lässt.
Aufgrund gleicher Reaktionsbedingungen (50°C, 60°C und 70°C) und gleicher
Konzentrationsverhältnisse der eingesetzten Reaktionspartner (PVAm im 327-fachen
Überschuss, 0.3375 mol·l-1) können die Geschwindigkeitskonstanten bei der
Umsetzung von 4-FNB/β-DMCD und 2-FNB/β-DMCD mit PVAm direkt miteinander
verglichen werden. Die Geschwindigkeitskonstanten bei der nucleophilen
Substitution von 2-FNB/β-DMCD mit PVAm bei 50°C, 60°C und 70°C sind etwa
dreimal so groß wie die der Umsetzung von 4-FNB/β-DMCD mit PVAm. Das deckt
sich mit dem etwa zwei bis dreimal höheren Substitutionsgrad von 2-nitrophenyl-
funktionalisiertem PVAm im Vergleich zu 4-nitrophenyl-funktionalisiertem PVAm bei
gleichen Reaktionsbedingungen (s. Abschnitt 3.1.4). Die Aktivierungsenergien für die
nucleophile aromatische Substitution von 4-FNB/β-DMCD und 2-FNB/β-DMCD sind
mit 67.6 kJ·mol-1 und 67.5 kJ·mol-1 nahezu gleich. Der messbare Reaktivitäts-
unterschied rührt einerseits von der höheren zu überwindenden Aktivierungsentropie
her (ortho–NO2-Gruppe erschwert sterisch die Anlagerung des Amins) und anderer-
seits setzt die Wechselwirkung der ortho–NO2-Gruppe mit dem Anilinwasserstoff der
chromophoren Einheit die Energie des Übergangszustandes herab, was wiederum
zur Begünstigung der Reaktion führt. Die Beschleunigung der Reaktion durch solche
Nachbargruppeneffekte der ortho–NO2-Gruppe sind literaturbekannt.20,91,93-95
Sangers Reagenz ist durch einen weiteren –M-Substituenten erwartungsgemäß für
eine nucleophile aromatische Substitution mit PVAm besser aktiviert als 2-FNB und
4-FNB. Demzufolge nimmt die Aktivierungsenergie mit 60.2 kJ·mol-1 für die
nucleophile aromatische Substitution signifikant ab.
Die Einführung eines weiteren Fluorsubstituenten bei DFDNB führt zu einer weiteren
Absenkung der Aktivierungsenergie auf 52.8 kJ·mol-1, was auf den –I Effekt des
zweiten Fluorsubstituenten zurückzuführen ist. Allerdings wird der Effekt auf die
Aktivierungsentropie im Vergleich zu Sangers Reagenz etwas kompensiert.
In guter Näherung können die Geschwindigkeitskonstanten der Umsetzung von
Sangers Reagenz/β-DMCD und DFDNB/β-DMCD miteinander verglichen werden.
Die Geschwindigkeitskonstanten bei der Reaktion von DFDNB/β-DMCD sind etwa
doppelt so groß wie die der Reaktion von Sangers Reagenz/β-DMCD mit PVAm.

3 Ergebnisse und Diskussion
107
Der direkte Vergleich der Geschwindigkeitskonstanten der Umsetzung von
4-FNA/β-DMCD und 2-FNB/β-DMCD ist aufgrund der unterschiedlich eingesetzten
Konzentrationsverhältnisse nicht möglich. Die nahezu gleich großen Aktivierungs-
energien zeigen, dass die Aminogruppe in 1-Stellung bei 4-Fluor-3-nitroanilin
(4-FNA) kaum Einfluss auf die Aktivierung (durch Elektronenzug der ortho-
Nitrogruppe) zu haben scheint. Im Gegensatz dazu hat die Einführung von zwei
Methylgruppen am 1-Aminostickstoff bei N,N-Dimethyl-4-fluor-3-nitroanilin
(4-DMFNA) im Vergleich zu 4-FNA einen großen Einfluss auf die Aktivierungs-
energie. Der größere +M-Effekt der Aminogruppe im Vergleich zu 4-FNA verringert
die Reaktivität für die nucleophile aromatische Substitution mit PVAm entscheidend.
Der Vergleich zur Aktivierungsentropie der Reaktion von i-Propylamin mit 4-FNB in
DMSO mit −∆S‡ = 38.9 J·K-1·mol-1 zeigt, dass bei der Funktionalisierung von PVAm
mit 4-FNB eine höhere Aktivierungsentropie (−∆S‡ = 158.5 J·K-1·mol-1) überwunden
werden muss.13,126 Die Polymerkette, die bei pH = 11 der wässrigen Lösung als
statistisches Knäuel vorliegt, muss daher einen signifikanten Einfluss auf die
Reaktivität haben. Es ist bekannt, dass die Nucleophilie primärer Alkylamine bei der
nucleophilen aromatischen Substitution von Fluoraromaten im Wesentlichen durch
sterische Faktoren bestimmt wird, so dass verzweigte primäre Amine deutlich
langsamer reagieren als unverzweigte.126,127 Die Basizität des Amins hat auf die
Reaktivität keinen großen Einfluss, so dass der räumliche Bau des Amins in vielen
Fällen ausschlaggebend für die Geschwindigkeit der Substitutionsreaktion ist.126,127
Der geschwindigkeitsbestimmende Schritt der nucleophilen aromatischen
Substitution (Additions-Eliminierungs-Mechanismus) ist jedoch auch abhängig von
der Wahl des Lösungsmittels. Je nachdem wie gut das Lösungsmittel in der Lage ist
die zwitterionische Zwischenstufe zu stabilisieren, kann der erste oder der zweite
Teilschritt der Substitutionsreaktion geschwindigkeitsbestimmend sein. So ist z. B.
bei der Umsetzung von 4-Fluornitrobenzen mit i-Propylamin in DMSO der erste
Schritt der geschwindigkeitsbestimmende, während in Toluol der zweite Schritt der
Substitutionsreaktion geschwindigkeitsbestimmend ist.130,131
Am Beispiel der nucleophilen aromatischen Substitution von 1-Chlor-2,4-dinitro-
benzen mit Piperidin wurden die Reaktionsgeschwindigkeiten sowohl in 12
aprotischen als auch in 10 protischen Lösungsmitteln untersucht. In allen Fällen war
dabei die Addition von Piperidin zur Bildung der Zwischenstufe der geschwindigkeits-
bestimmende Schritt.91

3 Ergebnisse und Diskussion
108
Die Reaktionsgeschwindigkeit bei der nucleophilen aromatischen Substitution von
1-Chlor-2,4-dinitrobenzen mit Piperidin nimmt in aprotischen Lösungsmitteln mit
Zunahme der Polarität des Lösungsmittels zu, da die Bildung der zwitterionischen
Zwischenstufe von polaren Lösungsmitteln besser stabilisiert werden kann. Diese
Korrelation zwischen der Reaktivität und der Polarität in aprotischen HBA-Lösungs-
mitteln (hydrogen bond acceptor) lässt den Schluss zu, dass starke intramolekulare
Wasserstoffbrückenbindungen zwischen dem Ammonium-Wasserstoff und der ortho-
Nitrogruppe verantwortlich für die Stabilisierung der Zwischenstufe und des
korrespondierenden angeregten Zustandes sind. Die Reaktivität in protischen
Lösungsmitteln ist etwas geringer im Vergleich zu den meisten aprotischen
Lösungsmitteln. Die Reaktivität des eingesetzten Aromaten ist invers proportional zur
Fähigkeit des Lösungsmittels als HBD-Lösungsmittel (hydrogen bond donor) zu
fungieren, welche sich im Lösungsmittelparameter α nach Kamlet und Taft
widerspiegelt. Die relativ geringen Reaktionsgeschwindigkeiten bei der nucleophilen
aromatischen Substitution in protischen Lösungsmitteln sind daher das Ergebnis
starker Solvatation der Aminogruppe der eingesetzten Base.91
In Wasser als gutes HBD-Lösungsmittel mit hohem α-Wert (α = 1.17) werden somit
die Aminogruppen von PVAm stark solvatisiert. Zudem wird bei der nucleophilen
aromatischen Substitution von aktivierten Fluoraromaten die zwitterionische
Zwischenstufe und der korrespondierende angeregte Zustand aufgrund der hohen
Dipolarität/Polarisierbarkeit π* von Wasser (π* = 1.09) stabilisiert. Es kann daher
davon ausgegangen werden, dass der erste Schritt der nucleophilen aromatischen
Substitution als langsamster der geschwindigkeitsbestimmende Schritt ist. Das
zeigen auch die Ergebnisse der kinetischen Untersuchungen, da die Reaktions-
geschwindigkeit mit der Elektronenverteilung am eingesetzten Fluoraromaten und mit
der zu überwindenden Aktivierungsentropie zur Bildung der Zwischenstufe direkt
zusammenhängt.
Die bisher angestellten kinetischen Untersuchungen der Reaktionen von Fluor-
aromaten mit PVAm wurden bei pH = 11 durchgeführt, um für die Funktionali-
sierungsreaktion von PVAm einen großen Anteil an unprotonierten Aminogruppen
vorliegen zu haben. Eine Erniedrigung des pH-Wertes der Reaktionslösung
(Erhöhung des Protonierungsgrades von PVAm) sollte daher auf die Reaktions-
geschwindigkeit der nucleophilen aromatischen Substitution von aktivierten
Fluoraromaten einen Einfluss haben.

3 Ergebnisse und Diskussion
109
3.1.9.3 Einfluss des pH-Wertes
Im Basischen liegen die Polymerketten von kationischen Polyelektrolyten wie PVAm
hauptsächlich als statistische Knäuel vor. Durch die Protonierung von kationischen
Polyelektrolyten (z. B. PVAm: −NH2 + H⊕ → −NH3⊕) wird die Ladungsdichte erhöht
und durch die Abstoßungskräfte gleich geladener Ionen kommt es zur
Knäuelaufweitung. Im stark Sauren liegen daher vor allem lang gestreckte
Polymerketten vor.74,75
PVAm weist die höchste kationische Ladungsdichte eines Polymeren auf, die bisher
bekannt ist. Im Gegensatz zu anderen Polymeren besitzt PVAm jedoch auch bei
hohen pH-Werten (pH = 9−10) eine signifikante Ladungsdichte von konstant
6 meq/g, was einem etwa 25 %igem Protonierungsgrad entspricht. Bei pH = 7
(12.8 meq/g) ist von 55 %igem und bei pH = 4 von etwa 75 %igem Protonierungs-
grad auszugehen. Die angegebenen Ladungsdichten beziehen sich auf ein PVAm
mit einem Molekulargewicht von 340,000 g·mol-1 (Lupamin® 9095).7
Die Ladungsdichten von Lupamin® 9095 wurden dabei über Polyelektrolyttitration
nach Horn mit Kaliumpolyvinylsulfat (anionischer Polyelektrolyt) und zur Endpunkt-
erkennung mittels eines metachromatischen Farbstoffes bestimmt.7,128
Mit abnehmendem pH-Wert einer PVAm-lösung nimmt also die Ladungsdichte und
durch Knäuelaufweitung der Polymerketten die Viskosität der Lösung zu. Eine
charakteristische Kenngröße der Viskosität einer Polymerlösung ist die Grenz-
viskositätszahl [η] oder auch Staudinger-Index genannt. Der Staudinger-Index steht
mit dem Molekulargewicht des Polymeren über die Kuhn-Mark-Houwink-Sakurada-
Gleichung (Gleichung 13) in empirischem Zusammenhang.74
[ ] αη MK ⋅= (13)
Der sog. K-Wert ist eine von der gelösten Substanz und vom Lösungsmittel
abhängige Konstante. Der Exponent α ist eine Größe, welche für die Konformation
der Polymerketten steht und für die Grenzfälle des idealen Knäuels mit α = 0.5 und
für starre, lineare Ketten mit α = 2 festgelegt ist.74 Je größer das Molekulargewicht M
und je gestreckter die Polymerketten sind, desto größer die Viskosität der
Polymerlösung.
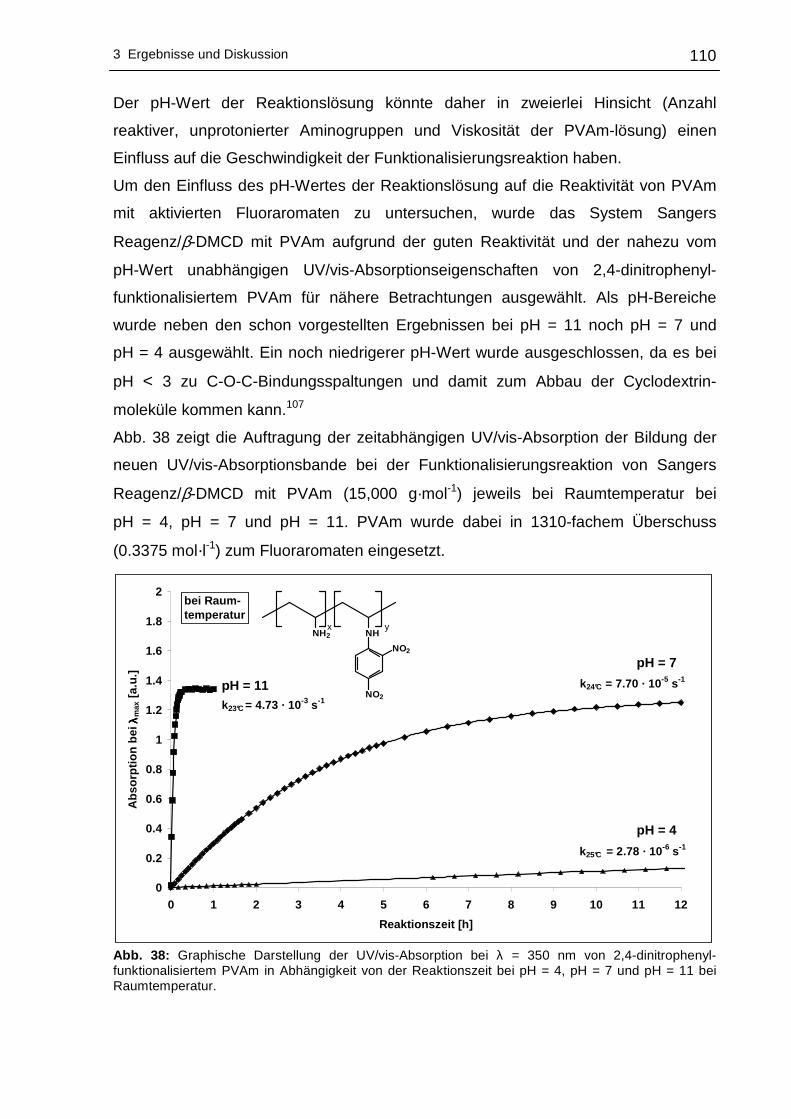
3 Ergebnisse und Diskussion
110
Der pH-Wert der Reaktionslösung könnte daher in zweierlei Hinsicht (Anzahl
reaktiver, unprotonierter Aminogruppen und Viskosität der PVAm-lösung) einen
Einfluss auf die Geschwindigkeit der Funktionalisierungsreaktion haben.
Um den Einfluss des pH-Wertes der Reaktionslösung auf die Reaktivität von PVAm
mit aktivierten Fluoraromaten zu untersuchen, wurde das System Sangers
Reagenz/β-DMCD mit PVAm aufgrund der guten Reaktivität und der nahezu vom
pH-Wert unabhängigen UV/vis-Absorptionseigenschaften von 2,4-dinitrophenyl-
funktionalisiertem PVAm für nähere Betrachtungen ausgewählt. Als pH-Bereiche
wurde neben den schon vorgestellten Ergebnissen bei pH = 11 noch pH = 7 und
pH = 4 ausgewählt. Ein noch niedrigerer pH-Wert wurde ausgeschlossen, da es bei
pH < 3 zu C-O-C-Bindungsspaltungen und damit zum Abbau der Cyclodextrin-
moleküle kommen kann.107
Abb. 38 zeigt die Auftragung der zeitabhängigen UV/vis-Absorption der Bildung der
neuen UV/vis-Absorptionsbande bei der Funktionalisierungsreaktion von Sangers
Reagenz/β-DMCD mit PVAm (15,000 g·mol-1) jeweils bei Raumtemperatur bei
pH = 4, pH = 7 und pH = 11. PVAm wurde dabei in 1310-fachem Überschuss
(0.3375 mol·l-1) zum Fluoraromaten eingesetzt.
Abb. 38: Graphische Darstellung der UV/vis-Absorption bei λ = 350 nm von 2,4-dinitrophenyl-funktionalisiertem PVAm in Abhängigkeit von der Reaktionszeit bei pH = 4, pH = 7 und pH = 11 bei Raumtemperatur.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
0 1 2 3 4 5 6 7 8 9 10 11 12
Reaktionszeit [h]
Abs
orpt
ion
bei
λλ λλ max
[a.u
.]
pH = 11
pH = 7
bei Raum-temperatur
pH = 4
NH2 NH
NO2
NO2
x y
k23°C = 4.73 · 10-3 s-1
k24°C = 7.70 · 10-5 s-1
k25°C = 2.78 · 10-6 s-1
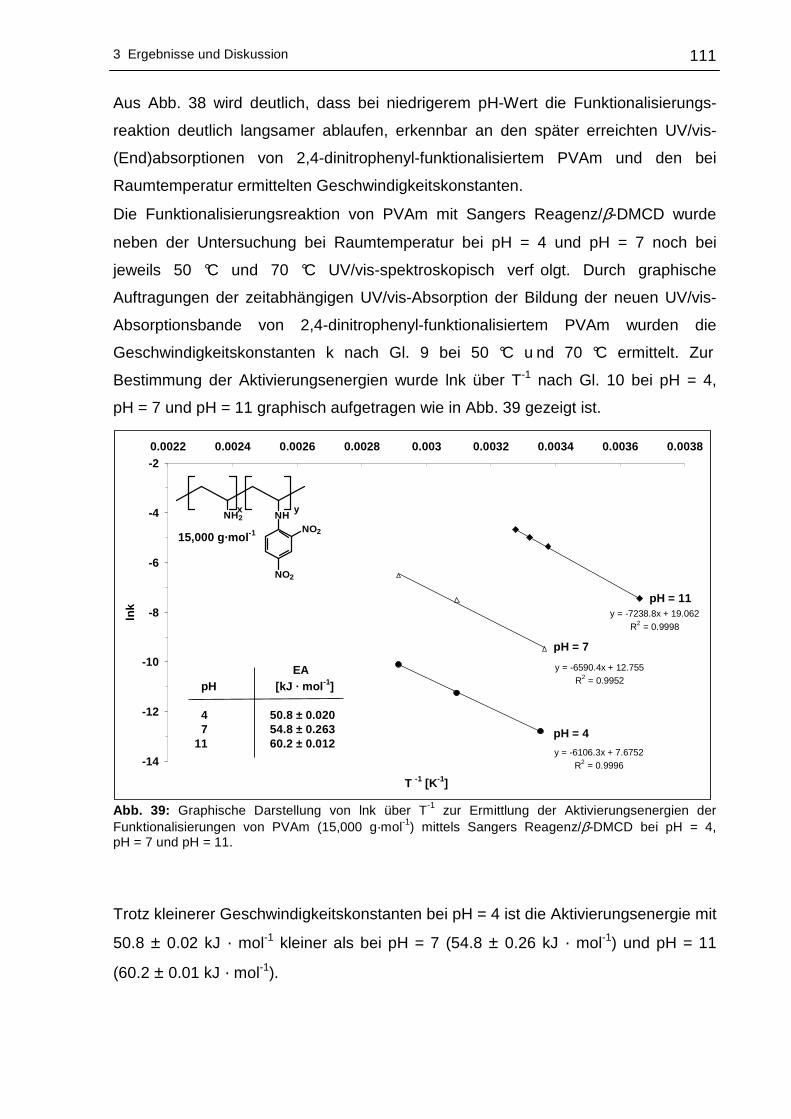
3 Ergebnisse und Diskussion
111
Aus Abb. 38 wird deutlich, dass bei niedrigerem pH-Wert die Funktionalisierungs-
reaktion deutlich langsamer ablaufen, erkennbar an den später erreichten UV/vis-
(End)absorptionen von 2,4-dinitrophenyl-funktionalisiertem PVAm und den bei
Raumtemperatur ermittelten Geschwindigkeitskonstanten.
Die Funktionalisierungsreaktion von PVAm mit Sangers Reagenz/β-DMCD wurde
neben der Untersuchung bei Raumtemperatur bei pH = 4 und pH = 7 noch bei
jeweils 50 °C und 70 °C UV/vis-spektroskopisch verf olgt. Durch graphische
Auftragungen der zeitabhängigen UV/vis-Absorption der Bildung der neuen UV/vis-
Absorptionsbande von 2,4-dinitrophenyl-funktionalisiertem PVAm wurden die
Geschwindigkeitskonstanten k nach Gl. 9 bei 50 °C u nd 70 °C ermittelt. Zur
Bestimmung der Aktivierungsenergien wurde lnk über T-1 nach Gl. 10 bei pH = 4,
pH = 7 und pH = 11 graphisch aufgetragen wie in Abb. 39 gezeigt ist.
Abb. 39: Graphische Darstellung von lnk über T-1 zur Ermittlung der Aktivierungsenergien der Funktionalisierungen von PVAm (15,000 g·mol-1) mittels Sangers Reagenz/β-DMCD bei pH = 4, pH = 7 und pH = 11.
Trotz kleinerer Geschwindigkeitskonstanten bei pH = 4 ist die Aktivierungsenergie mit
50.8 ± 0.02 kJ · mol-1 kleiner als bei pH = 7 (54.8 ± 0.26 kJ · mol-1) und pH = 11
(60.2 ± 0.01 kJ · mol-1).
y = -7238.8x + 19.062R2 = 0.9998
y = -6590.4x + 12.755R2 = 0.9952
y = -6106.3x + 7.6752R2 = 0.9996-14
-12
-10
-8
-6
-4
-20.0022 0.0024 0.0026 0.0028 0.003 0.0032 0.0034 0.0036 0.0038
T -1 [K -1]
lnk
NH2 NHNO2
NO2
x y
pH = 4
pH = 7
pH = 11
EA pH [kJ · mol -1] 4 50.8 ± 0.020 7 54.8 ± 0.263 11 60.2 ± 0.012
15,000 g·mol -1
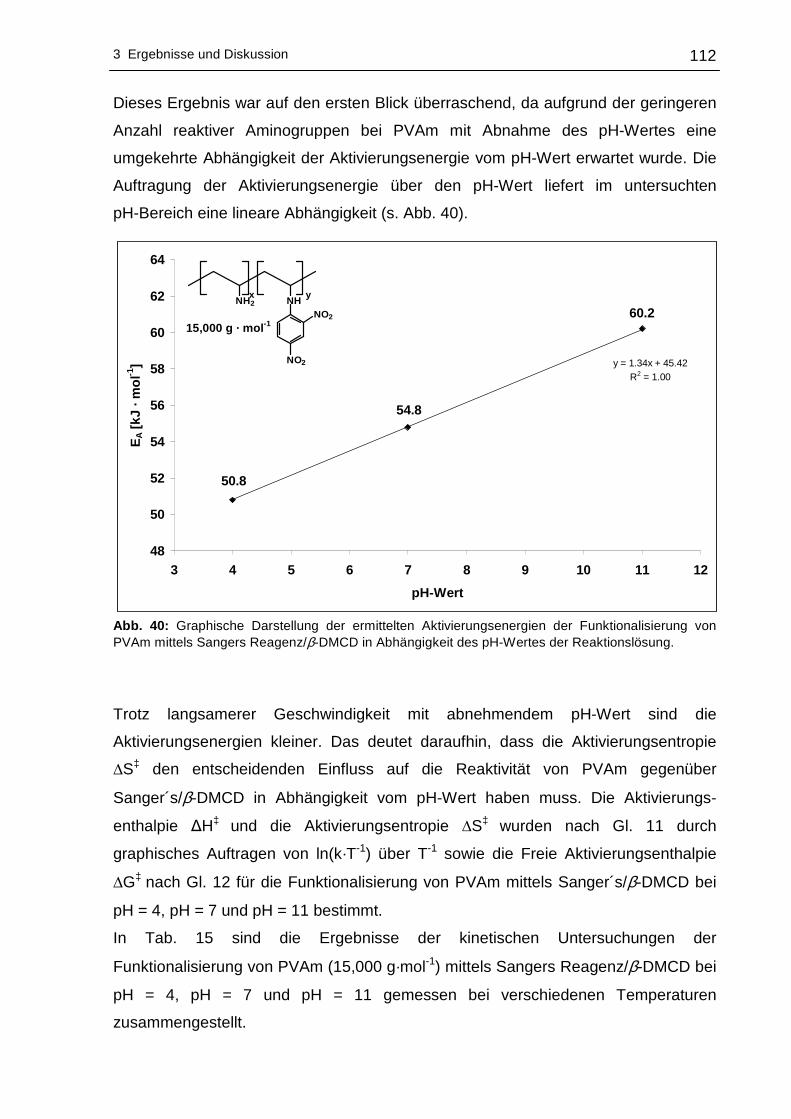
3 Ergebnisse und Diskussion
112
Dieses Ergebnis war auf den ersten Blick überraschend, da aufgrund der geringeren
Anzahl reaktiver Aminogruppen bei PVAm mit Abnahme des pH-Wertes eine
umgekehrte Abhängigkeit der Aktivierungsenergie vom pH-Wert erwartet wurde. Die
Auftragung der Aktivierungsenergie über den pH-Wert liefert im untersuchten
pH-Bereich eine lineare Abhängigkeit (s. Abb. 40).
Abb. 40: Graphische Darstellung der ermittelten Aktivierungsenergien der Funktionalisierung von PVAm mittels Sangers Reagenz/β-DMCD in Abhängigkeit des pH-Wertes der Reaktionslösung.
Trotz langsamerer Geschwindigkeit mit abnehmendem pH-Wert sind die
Aktivierungsenergien kleiner. Das deutet daraufhin, dass die Aktivierungsentropie
∆S‡ den entscheidenden Einfluss auf die Reaktivität von PVAm gegenüber
Sanger´s/β-DMCD in Abhängigkeit vom pH-Wert haben muss. Die Aktivierungs-
enthalpie ∆H‡ und die Aktivierungsentropie ∆S‡ wurden nach Gl. 11 durch
graphisches Auftragen von ln(k·T-1) über T-1 sowie die Freie Aktivierungsenthalpie
∆G‡ nach Gl. 12 für die Funktionalisierung von PVAm mittels Sanger´s/β-DMCD bei
pH = 4, pH = 7 und pH = 11 bestimmt.
In Tab. 15 sind die Ergebnisse der kinetischen Untersuchungen der
Funktionalisierung von PVAm (15,000 g·mol-1) mittels Sangers Reagenz/β-DMCD bei
pH = 4, pH = 7 und pH = 11 gemessen bei verschiedenen Temperaturen
zusammengestellt.
y = 1.34x + 45.42R2 = 1.00
48
50
52
54
56
58
60
62
64
3 4 5 6 7 8 9 10 11 12
pH-Wert
EA [k
J ·
mol
-1]
50.8
54.8
60.2NH2 NH
NO2
NO2
x y
15,000 g · mol -1
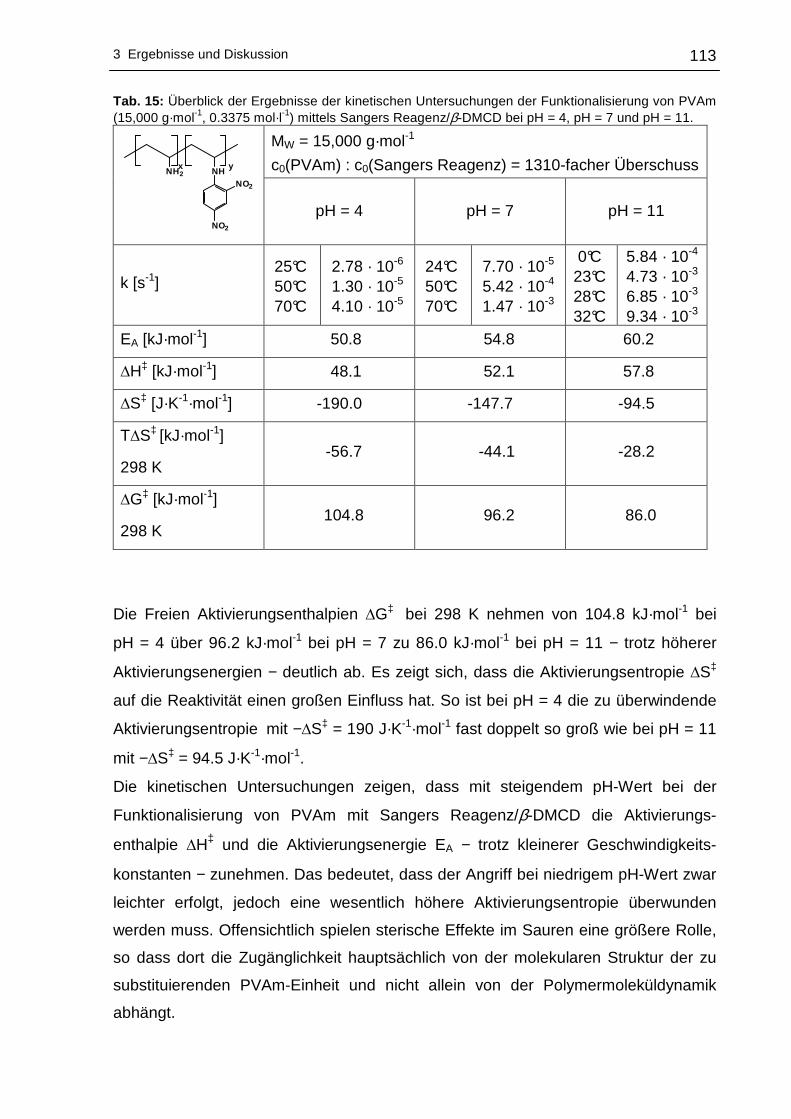
3 Ergebnisse und Diskussion
113
Tab. 15: Überblick der Ergebnisse der kinetischen Untersuchungen der Funktionalisierung von PVAm (15,000 g·mol-1, 0.3375 mol·l-1) mittels Sangers Reagenz/β-DMCD bei pH = 4, pH = 7 und pH = 11.
MW = 15,000 g·mol-1
c0(PVAm) : c0(Sangers Reagenz) = 1310-facher Überschuss
NH2 NHNO2
NO2
x y
pH = 4 pH = 7 pH = 11
k [s-1] 25°C 50°C 70°C
2.78 · 10-6 1.30 · 10-5 4.10 · 10-5
24°C 50°C 70°C
7.70 · 10-5 5.42 · 10-4 1.47 · 10-3
0°C 23°C 28°C 32°C
5.84 · 10-4
4.73 · 10-3
6.85 · 10-3
9.34 · 10-3 EA [kJ·mol-1] 50.8 54.8 60.2
∆H‡ [kJ·mol-1] 48.1 52.1 57.8
∆S‡ [J·K-1·mol-1] -190.0 -147.7 -94.5
T∆S‡ [kJ·mol-1]
298 K -56.7 -44.1 -28.2
∆G‡ [kJ·mol-1]
298 K 104.8 96.2 86.0
Die Freien Aktivierungsenthalpien ∆G‡ bei 298 K nehmen von 104.8 kJ·mol-1 bei
pH = 4 über 96.2 kJ·mol-1 bei pH = 7 zu 86.0 kJ·mol-1 bei pH = 11 − trotz höherer
Aktivierungsenergien − deutlich ab. Es zeigt sich, dass die Aktivierungsentropie ∆S‡
auf die Reaktivität einen großen Einfluss hat. So ist bei pH = 4 die zu überwindende
Aktivierungsentropie mit −∆S‡ = 190 J·K-1·mol-1 fast doppelt so groß wie bei pH = 11
mit −∆S‡ = 94.5 J·K-1·mol-1.
Die kinetischen Untersuchungen zeigen, dass mit steigendem pH-Wert bei der
Funktionalisierung von PVAm mit Sangers Reagenz/β-DMCD die Aktivierungs-
enthalpie ∆H‡ und die Aktivierungsenergie EA − trotz kleinerer Geschwindigkeits-
konstanten − zunehmen. Das bedeutet, dass der Angriff bei niedrigem pH-Wert zwar
leichter erfolgt, jedoch eine wesentlich höhere Aktivierungsentropie überwunden
werden muss. Offensichtlich spielen sterische Effekte im Sauren eine größere Rolle,
so dass dort die Zugänglichkeit hauptsächlich von der molekularen Struktur der zu
substituierenden PVAm-Einheit und nicht allein von der Polymermoleküldynamik
abhängt.

3 Ergebnisse und Diskussion
114
3.1.9.4 Einfluss des Molekulargewichtes von PVAm
Da literaturbekannt ist, dass Copolymere aus Vinylamin und Acrylamid
unterschiedlicher Anteile an Vinylamin-Einheiten (55−78%) bei pH = 4 ein Maximum
der reduzierten Viskosität [η] durchlaufen129, galt es zu untersuchen, ob die
Reaktivität von PVAm bei der nucleophilen aromatischen Substitution von Sangers
Reagenz allein von der Anzahl der reaktiv vorliegenden primären Aminogruppen
und/oder auch von der damit verbundenen Änderung der Viskosität abhängig ist.
Daher wurde die Funktionalisierung von PVAm zum einen mit einem höheren
Molekulargewicht und zum anderen durch Zugabe von KCl-Fremdsalz kinetisch mit
Hilfe der UV/vis-Spektroskpie untersucht.
Die Funktionalisierungsreaktion wurde am System Sangers Reagenz/β-DMCD und
PVAm mit einem Molekulargewicht von 400,000 g·mol-1 bei pH = 4, pH = 7 und
pH = 11 UV/vis-spektroskopisch untersucht. Dabei wurde PVAm zur Vergleichbarkeit
vorhergehender Untersuchungen (MW = 15,000 g·mol-1) in einem 1310-fachem
Überschuss (0.3375 mol·l-1) zu Sangers Reagenz eingesetzt. Für die kinetischen
Untersuchungen wurden zeitabhängige UV/vis-Absorptionsspektren aufgenommen
und aus der graphischen Auftragung der UV/vis-Absorption (beim entsprechenden
UV/vis-Absorptionsmaximum) in Abhängigkeit von der Reaktionszeit die
Geschwindigkeitskonstanten nach Gl. 9 bestimmt. Bei der Funktionalisierung von
PVAm (400,000 g·mol-1) mittels Sangers Reagenz/β-DMCD wurden die
nachfolgenden Geschwindigkeitskonstanten ermittelt: k30°C (pH = 11) = 4.65 · 10-3 s-1,
k25°C (pH = 7) = 6.00 · 10-5 s-1 und k30°C (pH = 4) = 2.31 · 10-6 s-1. Auch bei einem
höheren Molekulargewicht von PVAm zeigt sich, dass mit Abnahme des pH-Wertes
die Geschwindigkeitskonstanten bei gleicher Reaktionstemperatur kleiner sind, somit
die Reaktionen langsamer verlaufen.
Die Funktionalisierungsreaktion wurde weiterhin bei pH = 4 bei 50 °C und 75 °C und
bei pH = 7 bei 50 °C und 70 °C in Abhängigkeit von der Reaktionszeit UV/vis-
spektroskopisch verfolgt. Bei pH = 11 wurden neben 30 °C geringere Temperaturen
gewählt (0 °C und 20 °C), da die Funktionalisierung sreaktion von PVAm mittels
Sangers Reagenz/β-DMCD bei höheren Temperaturen sehr schnell verläuft und
daher zeitlich schlecht UV/vis-spektroskopisch verfolgbar ist.
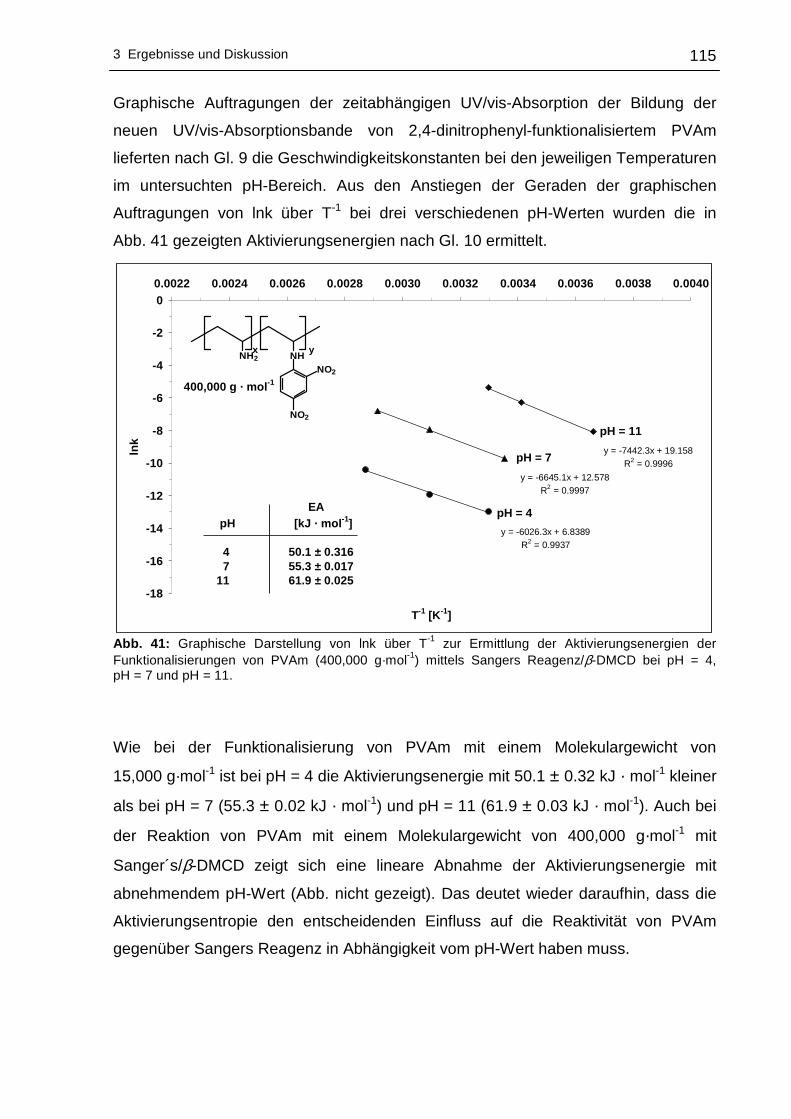
3 Ergebnisse und Diskussion
115
Graphische Auftragungen der zeitabhängigen UV/vis-Absorption der Bildung der
neuen UV/vis-Absorptionsbande von 2,4-dinitrophenyl-funktionalisiertem PVAm
lieferten nach Gl. 9 die Geschwindigkeitskonstanten bei den jeweiligen Temperaturen
im untersuchten pH-Bereich. Aus den Anstiegen der Geraden der graphischen
Auftragungen von lnk über T-1 bei drei verschiedenen pH-Werten wurden die in
Abb. 41 gezeigten Aktivierungsenergien nach Gl. 10 ermittelt.
Abb. 41: Graphische Darstellung von lnk über T-1 zur Ermittlung der Aktivierungsenergien der Funktionalisierungen von PVAm (400,000 g·mol-1) mittels Sangers Reagenz/β-DMCD bei pH = 4, pH = 7 und pH = 11.
Wie bei der Funktionalisierung von PVAm mit einem Molekulargewicht von
15,000 g·mol-1 ist bei pH = 4 die Aktivierungsenergie mit 50.1 ± 0.32 kJ · mol-1 kleiner
als bei pH = 7 (55.3 ± 0.02 kJ · mol-1) und pH = 11 (61.9 ± 0.03 kJ · mol-1). Auch bei
der Reaktion von PVAm mit einem Molekulargewicht von 400,000 g·mol-1 mit
Sanger´s/β-DMCD zeigt sich eine lineare Abnahme der Aktivierungsenergie mit
abnehmendem pH-Wert (Abb. nicht gezeigt). Das deutet wieder daraufhin, dass die
Aktivierungsentropie den entscheidenden Einfluss auf die Reaktivität von PVAm
gegenüber Sangers Reagenz in Abhängigkeit vom pH-Wert haben muss.
y = -6026.3x + 6.8389R2 = 0.9937
y = -6645.1x + 12.578R2 = 0.9997
y = -7442.3x + 19.158R2 = 0.9996
-18
-16
-14
-12
-10
-8
-6
-4
-2
00.0022 0.0024 0.0026 0.0028 0.0030 0.0032 0.0034 0.0036 0.0038 0.0040
T-1 [K -1]
lnk
NH2 NHNO2
NO2
x y
400,000 g · mol -1
pH = 4
pH = 7
pH = 11
EA pH [kJ · mol -1] 4 50.1 ± 0.316 7 55.3 ± 0.017 11 61.9 ± 0.025

3 Ergebnisse und Diskussion
116
Die Aktivierungsenthalpien ∆H‡, die Aktivierungsentropien ∆S‡ nach Gl. 10 sowie die
Freien Aktivierungsenthalpien ∆G‡ nach Gl. 7 wurden bestimmt und sind für
pH = 4, pH = 7 und pH = 11 mit den entsprechend ermittelten Geschwindigkeits-
konstanten k zusammenfassend in Tabelle 16 aufgeführt.
Tab. 16: Überblick der Ergebnisse der kinetischen Untersuchungen der Funktionalisierung von PVAm (400,000 g·mol-1) mittels Sangers Reagenz/β-DMCD bei pH = 4, pH = 7 und pH = 11.
MW = 400,000 g·mol-1
c0(PVAm) : c0(Sangers Reagenz) = 1310-facher Überschuss
NH2 NHNO2
NO2
x y
pH = 4 pH = 7 pH = 11
k [s-1]
30°C
50°C
75°C
2.31 · 10-6
6.61 · 10-6
3.00 · 10-5
25°C
50°C
70°C
6.00 · 10-5
3.50 · 10-4
1.11 · 10-3
0°C
20°C
30°C
3.10 · 10-4
1.91 · 10-3
4.65 · 10-3
EA [kJ·mol-1] 50.1 55.3 61.9
∆H‡ [kJ·mol-1] 42.2 52.6 59.5
∆S‡ [J·K-1·mol-1] -197.1 -149.2 -93.6
T∆S‡ [kJ·mol-1]
298 K -58.8 -44.5 -27.9
∆G‡ [kJ·mol-1]
298 K 101.0 97.1 87.4
Die Aktivierungsparameter sind im Vergleich zu denen der Umsetzung von Sangers
Reagenz/β-DMCD mit PVAm mit einem Molekulargewicht von 15,000 g·mol-1
(vgl. Tab. 15) nahezu identisch.
Das Molekulargewicht des verwendeten PVAm´s und damit die Viskosität der Lösung
hat für die Funktionalisierungsreaktion mittels Sangers Reagenz in über
1000-fachem Unterschuss zu PVAm offensichtlich keinen Einfluss auf die Reaktivität.
Es liegt die Vermutung nahe, dass bei solch großem Überschuss an PVAm
gegenüber Sangers Reagenz die Kettenbeweglichkeit des Polymeren nicht erfasst
wird, da möglicherweise nur rein zufällige Aminogruppen des Polymerrückgrates von
PVAm substituiert werden.
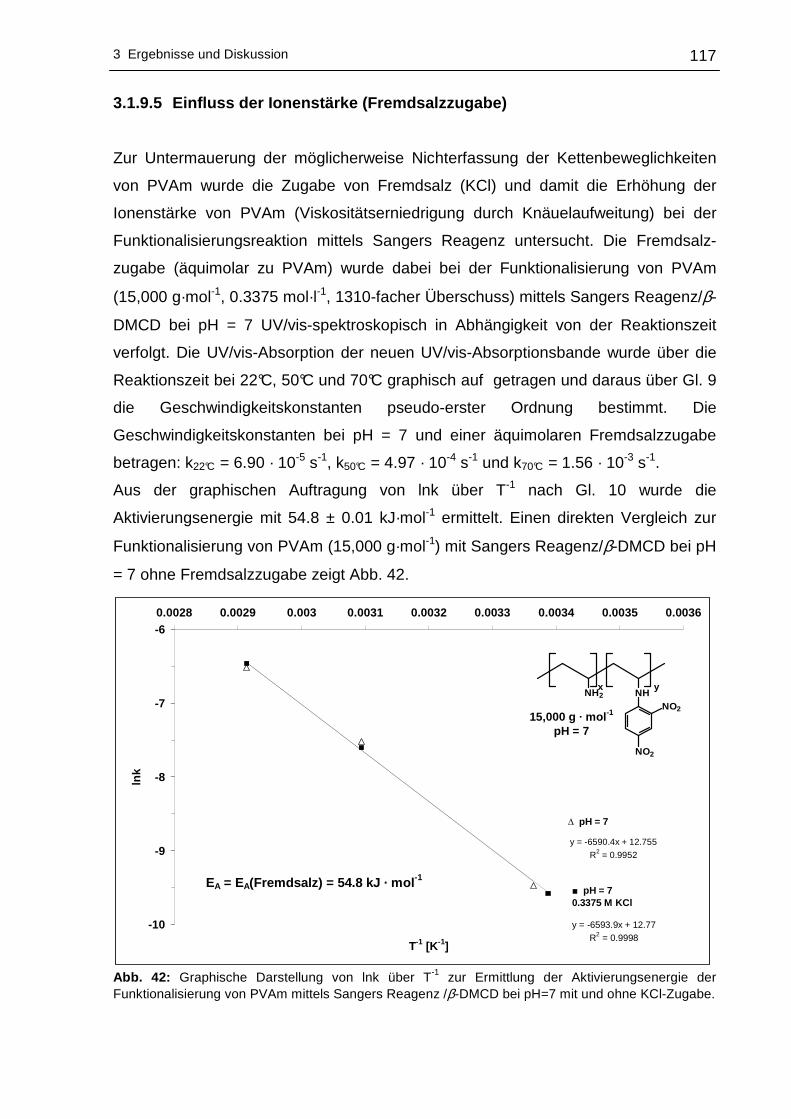
3 Ergebnisse und Diskussion
117
3.1.9.5 Einfluss der Ionenstärke (Fremdsalzzugabe)
Zur Untermauerung der möglicherweise Nichterfassung der Kettenbeweglichkeiten
von PVAm wurde die Zugabe von Fremdsalz (KCl) und damit die Erhöhung der
Ionenstärke von PVAm (Viskositätserniedrigung durch Knäuelaufweitung) bei der
Funktionalisierungsreaktion mittels Sangers Reagenz untersucht. Die Fremdsalz-
zugabe (äquimolar zu PVAm) wurde dabei bei der Funktionalisierung von PVAm
(15,000 g·mol-1, 0.3375 mol·l-1, 1310-facher Überschuss) mittels Sangers Reagenz/β-
DMCD bei pH = 7 UV/vis-spektroskopisch in Abhängigkeit von der Reaktionszeit
verfolgt. Die UV/vis-Absorption der neuen UV/vis-Absorptionsbande wurde über die
Reaktionszeit bei 22°C, 50°C und 70°C graphisch auf getragen und daraus über Gl. 9
die Geschwindigkeitskonstanten pseudo-erster Ordnung bestimmt. Die
Geschwindigkeitskonstanten bei pH = 7 und einer äquimolaren Fremdsalzzugabe
betragen: k22°C = 6.90 · 10-5 s-1, k50°C = 4.97 · 10-4 s-1 und k70°C = 1.56 · 10-3 s-1.
Aus der graphischen Auftragung von lnk über T-1 nach Gl. 10 wurde die
Aktivierungsenergie mit 54.8 ± 0.01 kJ·mol-1 ermittelt. Einen direkten Vergleich zur
Funktionalisierung von PVAm (15,000 g·mol-1) mit Sangers Reagenz/β-DMCD bei pH
= 7 ohne Fremdsalzzugabe zeigt Abb. 42.
Abb. 42: Graphische Darstellung von lnk über T-1 zur Ermittlung der Aktivierungsenergie der Funktionalisierung von PVAm mittels Sangers Reagenz /β-DMCD bei pH=7 mit und ohne KCl-Zugabe.
y = -6590.4x + 12.755
R2 = 0.9952
y = -6593.9x + 12.77R2 = 0.9998
-10
-9
-8
-7
-60.0028 0.0029 0.003 0.0031 0.0032 0.0033 0.0034 0.0035 0.0036
T-1 [K -1]
lnk
NH2 NHNO2
NO2
x y
∆ pH = 7
■ pH = 70.3375 M KCl
15,000 g · mol -1
pH = 7
EA = EA(Fremdsalz) = 54.8 kJ · mol -1
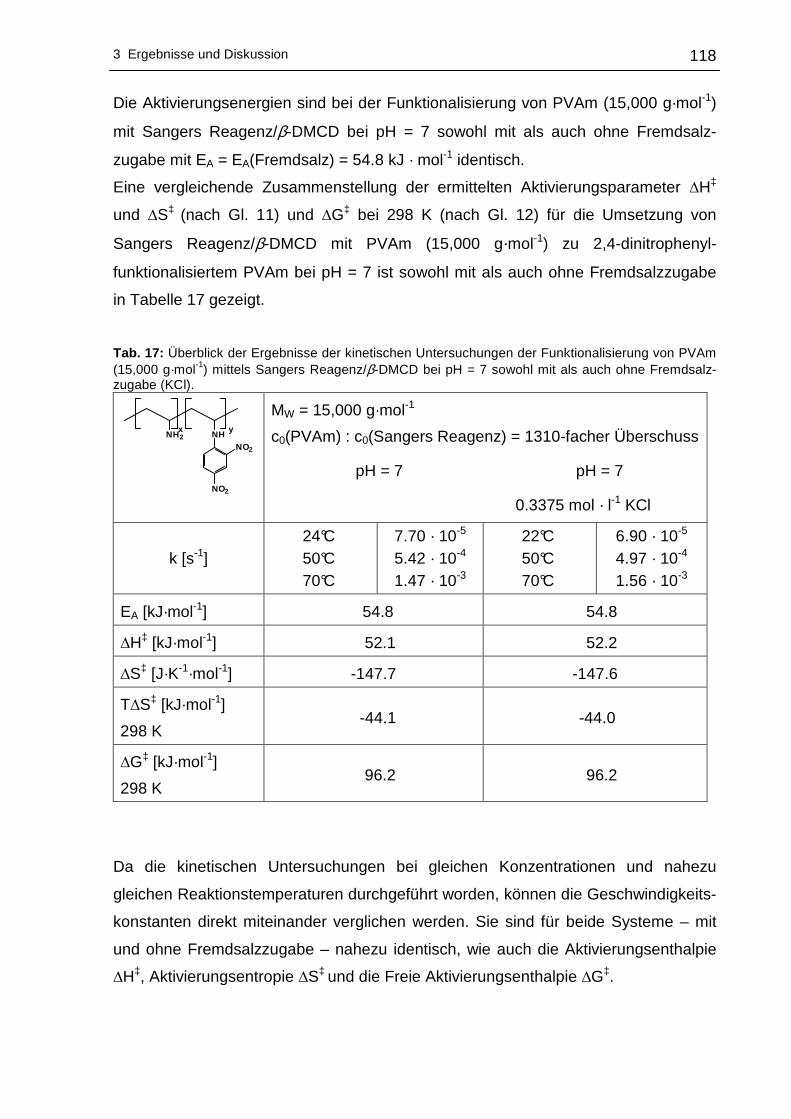
3 Ergebnisse und Diskussion
118
Die Aktivierungsenergien sind bei der Funktionalisierung von PVAm (15,000 g·mol-1)
mit Sangers Reagenz/β-DMCD bei pH = 7 sowohl mit als auch ohne Fremdsalz-
zugabe mit EA = EA(Fremdsalz) = 54.8 kJ · mol-1 identisch.
Eine vergleichende Zusammenstellung der ermittelten Aktivierungsparameter ∆H‡
und ∆S‡ (nach Gl. 11) und ∆G‡ bei 298 K (nach Gl. 12) für die Umsetzung von
Sangers Reagenz/β-DMCD mit PVAm (15,000 g·mol-1) zu 2,4-dinitrophenyl-
funktionalisiertem PVAm bei pH = 7 ist sowohl mit als auch ohne Fremdsalzzugabe
in Tabelle 17 gezeigt.
Tab. 17: Überblick der Ergebnisse der kinetischen Untersuchungen der Funktionalisierung von PVAm (15,000 g·mol-1) mittels Sangers Reagenz/β-DMCD bei pH = 7 sowohl mit als auch ohne Fremdsalz-zugabe (KCl).
NH2 NHNO2
NO2
x y
MW = 15,000 g·mol-1
c0(PVAm) : c0(Sangers Reagenz) = 1310-facher Überschuss
pH = 7 pH = 7
0.3375 mol · l-1 KCl
k [s-1] 24°C 50°C 70°C
7.70 · 10-5 5.42 · 10-4 1.47 · 10-3
22°C 50°C 70°C
6.90 · 10-5 4.97 · 10-4 1.56 · 10-3
EA [kJ·mol-1] 54.8 54.8
∆H‡ [kJ·mol-1] 52.1 52.2
∆S‡ [J·K-1·mol-1] -147.7 -147.6
T∆S‡ [kJ·mol-1]
298 K -44.1 -44.0
∆G‡ [kJ·mol-1]
298 K 96.2 96.2
Da die kinetischen Untersuchungen bei gleichen Konzentrationen und nahezu
gleichen Reaktionstemperaturen durchgeführt worden, können die Geschwindigkeits-
konstanten direkt miteinander verglichen werden. Sie sind für beide Systeme – mit
und ohne Fremdsalzzugabe – nahezu identisch, wie auch die Aktivierungsenthalpie
∆H‡, Aktivierungsentropie ∆S‡ und die Freie Aktivierungsenthalpie ∆G‡.

3 Ergebnisse und Diskussion
119
Eine Fremdsalzzugabe und die damit verbundene Viskositätserniedrigung hat bei der
Funktionalisierung mittels Sangers Reagenz bei in sehr großem Überschuss
eingesetztem PVAm (1310-fach) keinen Einfluss auf die Reaktivität. Die
Substitutionsreaktion am PVAm-Polymerrückgrat erfolgt bei sehr geringer
Konzentration des eingesetzten Fluoraromaten für Reaktionen pseudo-mono-
molekularer Ordnung (1310-facher Überschuss an PVAm) eher zufällig. Die
Viskosität der Lösung und damit die Zugänglichkeit der PVAm-Ketten
(Polymermoleküldynamik) haben bei nucleophilen aromatischen Substitutionen von
in großem Unterschuss eingesetzten Fluoraromaten mit PVAm keinen Einfluss auf
die Aktivierungsparameter.
Um den Einfluss der Kettenbeweglichkeit von PVAm-Ketten auf die Reaktivität bei
nucleophilen aromatischen Substitutionen von aktivierten Fluoraromaten erfassen zu
können, müsste der Fluoraromat in deutlich höheren Konzentrationen zu PVAm
eingesetzt werden. Dann müsste jedoch ein anderer kinetischer Ansatz (keine
Reaktion pseudo-erster Ordnung) zur Auswertung zeitabhängig gemessener UV/vis-
Absorptionsspektren herangezogen werden. UV/vis-spektroskopische Messungen
von Funktionalisierungsreaktionen bei denen der Fluoraromat aufgrund seiner
großen Konzentration schnell zu großer Intensität der UV/vis-Absorption der neu
gebildeten, chromophoren PVAm-Einheiten führt, ist durch den Detektor des
genutzten UV/vis-Gerätes auf eine bis zu 1.8 messbare UV/Vis-Absorption limitiert.
Aufgrund der relativ hohen molaren Absorptionskoeffizienten nitrophenyl-
funktionalisierter PVAm´s stößt die in dieser Arbeit genutzte Messmethode schnell an
ihre Grenzen.
Um Funktionalisierungsreaktionen von PVAm mit z. B. Sangers Reagenz in z. B.
äquimolarem Verhältnis UV/vis-spektroskopisch verfolgen zu können, müsste in sehr
großer Verdünnung durch das Lösungsmittel gearbeitet werden. Aufgrund dieser
Verdünnung müssten jedoch für spürbare Viskositätsunterschiede sehr hohe
Molekulargewichte von PVAm für Untersuchungen des Einflusses von Ketten-
beweglichkeiten eingesetzt werden. Neben hohen Molekulargewichten von PVAm
könnte die Zugänglichkeit der Polymerketten zusätzlich auch durch Fremdsalz-
zugaben beeinflusst werden.

3 Ergebnisse und Diskussion
120
3.2 Polymermodifizierung von Kieselgel H (KGH)
3.2.1 Syntheseplanung und Charakterisierungsmöglic hkeiten
Zur Funktionalisierung von anorganischen Trägermaterialien wie z. B. Kieselgel
stehen prinzipiell drei Möglichkeiten der Polymermodifizierung zur Verfügung. Es
können Kieselgelpartikel zuerst mit PVAm beladen und anschließend mit
Cyclodextrin/Fluoraromat-Komplexen umgesetzt oder in einer bereits nitrophenyl-
funktionalisierten PVAm-Lösung suspendiert werden, wobei in beiden Fällen für die
Modifizierung im wässrigen Milieu Cyclodextrin als Löslichkeitsvermittler des
Fluoraromaten notwendig wäre.
Schema 30 zeigt die in dieser Arbeit genutzte dritte Möglichkeit der
Polymermodifizierung durch Umsetzung von auf Kieselgel adsorbierten
Fluoraromaten mit PVAm in Wasser am Beispiel von 4-Fluornitrobenzen (4-FNB), mit
dem Vorteil, dass die nucleophile aromatische Substitution an der Grenzfläche
Kieselgel/Wasser ohne Einsatz von Cyclodextrin vonstatten gehen kann.
Schema 30: Modifizierung von Kieselgel H durch Adsorption der entsprechenden Fluoraromaten und Umsetzung mit PVAm in Wasser und weitere Verfahrensweise der Charakterisierung der erhaltenen chromophoren Partikel und überstehender chromophorer Polymerlösung.
Kiesel - gel H 400,000 g/mol Adsorption
aus EtOH
4-FNB beladene Partikel
NH2n
PVAm F NO2
4-nitrophenyl -funktionalisiertes PVAm auf Kieselgel
NO2N
H
NO2N
H
• Soxhlet-Extraktion der Partikelerst mit Wasser dann mit Aceton
• Trocknung unter Vakuum
• Charakterisierung mittels UV-vis,EA, BET, XPS, Zetapotential,…
• Ausfällen des Polymeren in Aceton• Trocknung unter Vakuum
Bestimmung der Funktionalisierungs-grade mittels UV-vis- bzw. 1H-NMR-Spektroskopie
Filtration der modifizierten Feststoff-Partikel
ModifiziertePartikel
ÜberstehendePolymerlösung
• Soxhlet-Extraktion der Partikelerst mit Wasser dann mit Aceton
• Trocknung unter Vakuum
• Charakterisierung mittels UV-vis,EA, BET, XPS, Zetapotential,…
• Ausfällen des Polymeren in Aceton• Trocknung unter Vakuum
Bestimmung der Funktionalisierungs-grade mittels UV-vis- bzw. 1H-NMR-Spektroskopie
Filtration der modifizierten Feststoff-Partikel
ModifiziertePartikel
ÜberstehendePolymerlösung

3 Ergebnisse und Diskussion
121
Für die Funktionalisierung an Kieselgel wurde ausschließlich PVAm mit einem
Molekulargewicht von 400,000 g·mol-1 eingesetzt, da Erfahrungen aus der Literatur
zeigten, dass bei geringen Molekulargewichten weniger an Polymer adsorbiert
werden konnte.50 Zur besseren Vergleichbarkeit der einzelnen Umsetzungen wurden
die Komponenten immer im gleichen Molverhältnis von Kieselgel zu PVAm zu
Fluoraromat mit 10 : 1 : 0.3 eingesetzt. Die Reaktionszeit betrug jeweils 12 Stunden
in der Siedehitze. Die in Schema 31 gezeigten aktivierten Fluoraromaten wurden für
die Funktionalisierung von PVAm auf Kieselgel H ausgewählt.
Schema 31: Eingesetzte aktivierte Fluoraromaten für die Funktionalisierung von PVAm (400,000 g·mol-1) auf Kieselgel H.
Die erhaltenen polymermodifizerten Kieselgele wurden abgetrennt und gründlich
über Soxhlet-Extraktionen von möglicherweise unumgesetztem Fluoraromat und
überschüssigem, nicht adsorbiertem funktionalisiertem PVAm erst mit Wasser und
anschließend mit Aceton oder Ethanol gewaschen.
4-FNB
2-FNB FDNB
DFDNB 4-FNA 4-DMFNA
FDNstilben FNAzoB
F NO2F
O2N
F
O2N
NO2
F
O2N
NO2
F
F NH2
O2N
N
O2N
FCH3
CH3
F
O2N
NN
F
O2N
NO2
N
HO2N
NO2
F NO2
FNAzoB-2N-DNP-FNA
HN N
N
F NO2
O2N
N

3 Ergebnisse und Diskussion
122
Um Aussagen bezüglich des Substitutionsgrades der funktionalisierten PVAm´s
treffen zu können, wurde die überbleibende farbige, wässrige Polymerlösung sofort
nach der Abtrennung der Feststoffpartikel in Aceton ausgefällt und abgetrennt, um
eine weitere Umsetzung des nicht umgesetzten Fluoraromaten mit PVAm zu
unterbinden. So kann die direkte Vergleichbarkeit beider Polymeren auf Kieselgel
und in der Lösung gewährleistet werden. Eine ausführliche Synthesedurchführung
zur Modifizierung von Kieselgel H mit PVAm und der Funktionalisierung mittels
entsprechender Fluoraromaten ist in Abschnitt 5.5 (Experimenteller Teil) dargelegt.
Die erfolgreiche Fixierung von funktionalisiertem PVAm auf Kieselgel zeigt die
Farbigkeit der synthetisierten Pulver, die trotz gründlichen Waschens über Soxhlet-
Extraktionen (Wasser, Aceton bzw. Ethanol) nicht verloren ging.
UV/vis-spektroskopische Untersuchungen der modifizierten Kieselgelpartikel wurden
zur Identifizierung der molekularen Struktur der chromophoren Einheiten von PVAm
mit Vergleich zu wohl definierten niedermolekularen Modellverbindungen
herangezogen, die aus den entsprechenden Fluoraromaten und i-Propylamin
synthetisiert wurden. Als Referenz diente dabei reines Kieselgel H.
Weiterhin wurden zur Aufklärung der chemischen Struktur der chromophoren PVAm-
Einheiten Röntgen-photoelektronenspektroskopische Untersuchungen (XPS)
herangezogen. Für elektrokinetische Untersuchungen (Zetapotential) wurden Mikro-
elektrophorese-Experimente der modifizierten Kieselgele zur Untersuchung des
Ladungsverhaltens in wässriger Lösung als Funktion des pH-Wertes durchgeführt.
Verschiebungen der isoelektrischen Punkte (IEP) - im Vergleich zu unmodifiziertem
KGH - geben Hinweise auf die Anzahl der substituierten Aminogruppen von PVAm. 13C-CP-MAS-Festkörper-NMR-spektroskopische Untersuchungen, BET-Messungen
und Elementaranalysen komplettieren die Charakterisierung der chromophoren,
polymermodifizierten Kieselgele.
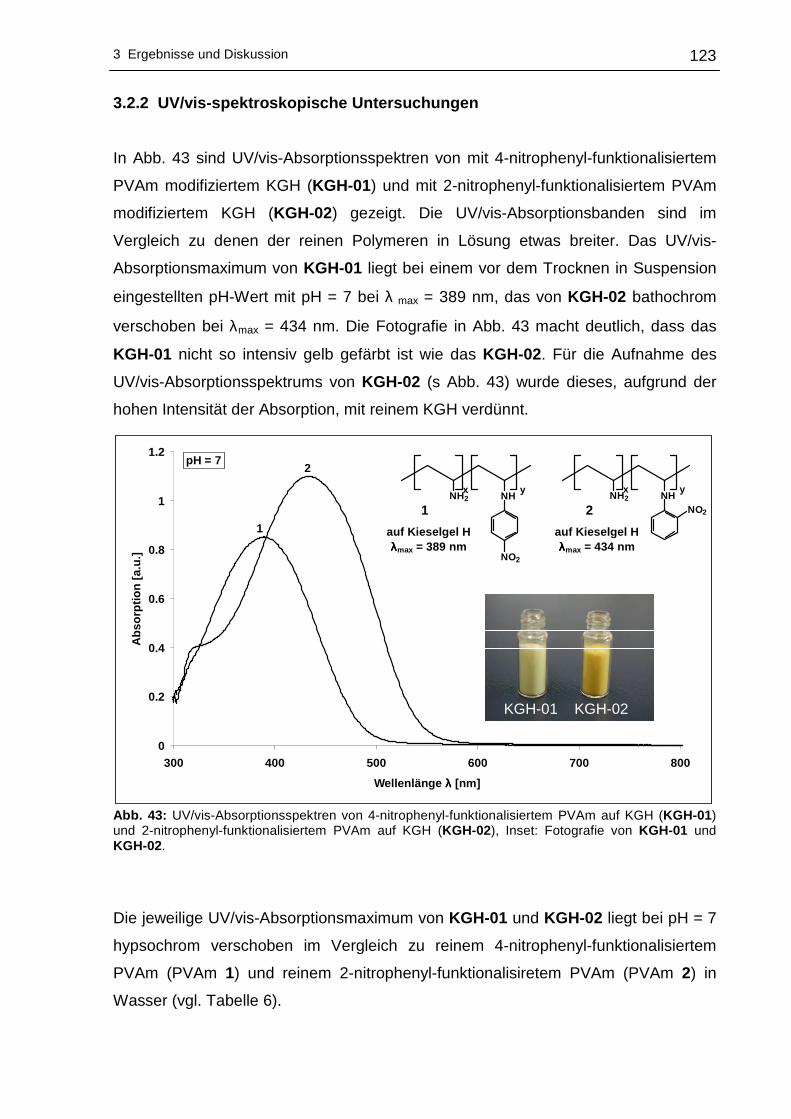
3 Ergebnisse und Diskussion
123
3.2.2 UV/vis-spektroskopische Untersuchungen
In Abb. 43 sind UV/vis-Absorptionsspektren von mit 4-nitrophenyl-funktionalisiertem
PVAm modifiziertem KGH (KGH-01) und mit 2-nitrophenyl-funktionalisiertem PVAm
modifiziertem KGH (KGH-02) gezeigt. Die UV/vis-Absorptionsbanden sind im
Vergleich zu denen der reinen Polymeren in Lösung etwas breiter. Das UV/vis-
Absorptionsmaximum von KGH-01 liegt bei einem vor dem Trocknen in Suspension
eingestellten pH-Wert mit pH = 7 bei λ max = 389 nm, das von KGH-02 bathochrom
verschoben bei λmax = 434 nm. Die Fotografie in Abb. 43 macht deutlich, dass das
KGH-01 nicht so intensiv gelb gefärbt ist wie das KGH-02. Für die Aufnahme des
UV/vis-Absorptionsspektrums von KGH-02 (s Abb. 43) wurde dieses, aufgrund der
hohen Intensität der Absorption, mit reinem KGH verdünnt.
Abb. 43: UV/vis-Absorptionsspektren von 4-nitrophenyl-funktionalisiertem PVAm auf KGH (KGH-01) und 2-nitrophenyl-funktionalisiertem PVAm auf KGH (KGH-02), Inset: Fotografie von KGH-01 und KGH-02.
Die jeweilige UV/vis-Absorptionsmaximum von KGH-01 und KGH-02 liegt bei pH = 7
hypsochrom verschoben im Vergleich zu reinem 4-nitrophenyl-funktionalisiertem
PVAm (PVAm 1) und reinem 2-nitrophenyl-funktionalisiretem PVAm (PVAm 2) in
Wasser (vgl. Tabelle 6).
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
1
2
NH2 NH
NO2
x y
auf Kieselgel Hλλλλmax = 389 nm
1NH2 NH
NO2
x y
auf Kieselgel Hλλλλmax = 434 nm
2
KGH-01 KGH-02
pH = 7

3 Ergebnisse und Diskussion
124
Die funktionalisierten PVAm´s sind daher möglicherweise neben den primären
Aminogruppen der PVAm-Einheiten auch über den Aminostickstoff der
chromophoren Einheit über Wasserstoffbrückenbindungen an die Silanolgruppen der
Kieselgel-Oberfläche gebunden (s. Schema 32), was zu weniger Resonanz-
stabilisierungsenergie (Schwächung des push-Charakters der Aminogruppe der
chromophoren Einheit) und damit zu einer hypsochromen Verschiebung der UV/vis-
Absorptionsbande im Vergleich zur Lage der UV/vis-Absorptionsbande in wässriger
Lösung führt.
Schema 32: Schematische Darstellung der möglichen Anbindung von 4-nitrophenyl-funktionalisiertem PVAm an eine Kieselgel-Oberfläche.
Der Substitutionsgrad von 4-nitrophenyl- und 2-nitrophenyl-funktionalisiertem PVAm
der überstehenden Lösung der KGH-Suspension wurde mit Hilfe der entsprechenden
Modellverbindungen (4-Nitro-N-isopropylanilin bzw. 2-Nitro-N-isopropylanilin) über
Verdünnungsreihen in Methanol UV/vis-spektroskopisch nach Gleichung 8 bestimmt.
Der Substitutionsgrad von 4-nitrophenyl-funktionalisiertem PVAm auf KGH-01 ist mit
7.0 % im Vergleich zu 2-nitrophenyl-funktionalisiertem PVAm auf KGH-02 mit 34.5 %
deutlich geringer. Für die Umsetzung von 2-FNB mit PVAm an der Kieselgel-
oberfläche ist von nahezu vollständigem Umsatz an 2-FNB auszugehen.
N NH
NO2
H HNH2 NH3
O
H
Si
O
H
Si
O
H
Si
O
Si
Kieselgel-Oberfläche
4-nitrophenyl-funktionalisiertes
PVAmN N
H
NO2
H HNH2 NH3
O
H
Si
O
H
Si
O
H
Si
O
Si
Kieselgel-Oberfläche
4-nitrophenyl-funktionalisiertes
PVAm
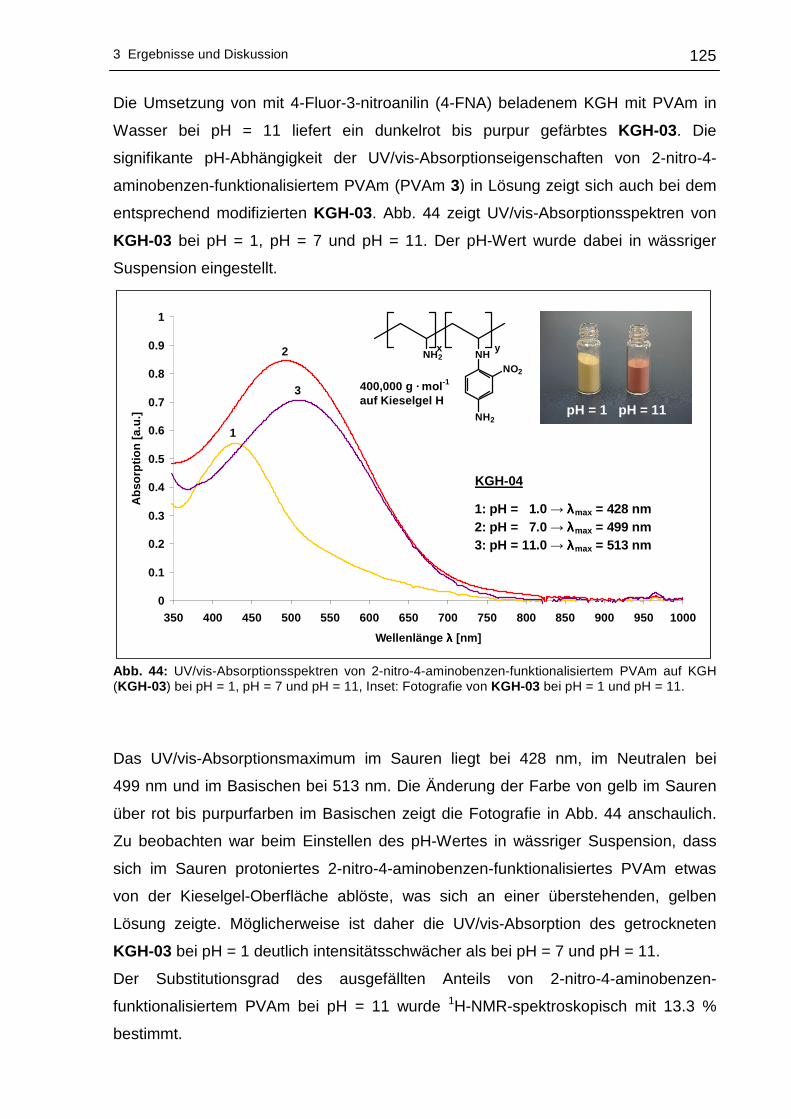
3 Ergebnisse und Diskussion
125
Die Umsetzung von mit 4-Fluor-3-nitroanilin (4-FNA) beladenem KGH mit PVAm in
Wasser bei pH = 11 liefert ein dunkelrot bis purpur gefärbtes KGH-03. Die
signifikante pH-Abhängigkeit der UV/vis-Absorptionseigenschaften von 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) in Lösung zeigt sich auch bei dem
entsprechend modifizierten KGH-03. Abb. 44 zeigt UV/vis-Absorptionsspektren von
KGH-03 bei pH = 1, pH = 7 und pH = 11. Der pH-Wert wurde dabei in wässriger
Suspension eingestellt.
Abb. 44: UV/vis-Absorptionsspektren von 2-nitro-4-aminobenzen-funktionalisiertem PVAm auf KGH (KGH-03) bei pH = 1, pH = 7 und pH = 11, Inset: Fotografie von KGH-03 bei pH = 1 und pH = 11.
Das UV/vis-Absorptionsmaximum im Sauren liegt bei 428 nm, im Neutralen bei
499 nm und im Basischen bei 513 nm. Die Änderung der Farbe von gelb im Sauren
über rot bis purpurfarben im Basischen zeigt die Fotografie in Abb. 44 anschaulich.
Zu beobachten war beim Einstellen des pH-Wertes in wässriger Suspension, dass
sich im Sauren protoniertes 2-nitro-4-aminobenzen-funktionalisiertes PVAm etwas
von der Kieselgel-Oberfläche ablöste, was sich an einer überstehenden, gelben
Lösung zeigte. Möglicherweise ist daher die UV/vis-Absorption des getrockneten
KGH-03 bei pH = 1 deutlich intensitätsschwächer als bei pH = 7 und pH = 11.
Der Substitutionsgrad des ausgefällten Anteils von 2-nitro-4-aminobenzen-
funktionalisiertem PVAm bei pH = 11 wurde 1H-NMR-spektroskopisch mit 13.3 %
bestimmt.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
350 400 450 500 550 600 650 700 750 800 850 900 950 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
1: pH = 1.0 → λλλλmax = 428 nm2: pH = 7.0 → λλλλmax = 499 nm3: pH = 11.0 → λλλλmax = 513 nm
NH2 NHNO2
NH2
x y
400,000 g · mol -1
auf Kieselgel H
1
2
3
pH = 1 pH = 11pH = 1 pH = 11
KGH-04
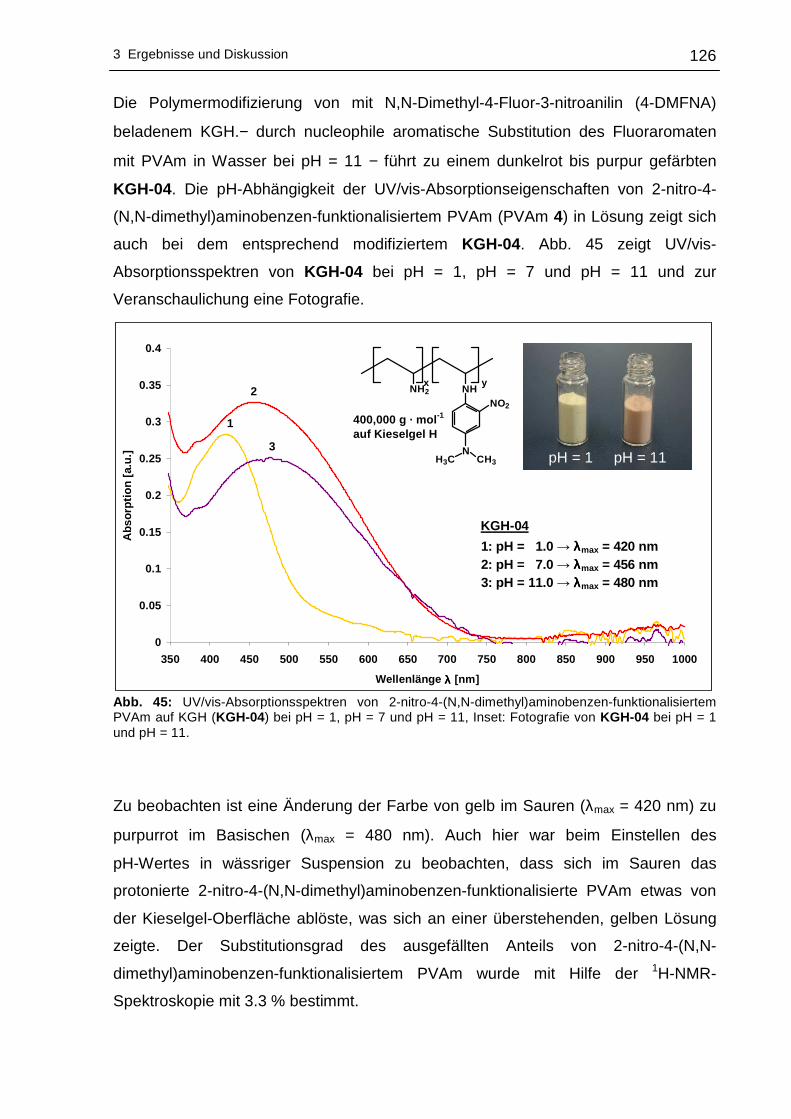
3 Ergebnisse und Diskussion
126
Die Polymermodifizierung von mit N,N-Dimethyl-4-Fluor-3-nitroanilin (4-DMFNA)
beladenem KGH.− durch nucleophile aromatische Substitution des Fluoraromaten
mit PVAm in Wasser bei pH = 11 − führt zu einem dunkelrot bis purpur gefärbten
KGH-04. Die pH-Abhängigkeit der UV/vis-Absorptionseigenschaften von 2-nitro-4-
(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) in Lösung zeigt sich
auch bei dem entsprechend modifiziertem KGH-04. Abb. 45 zeigt UV/vis-
Absorptionsspektren von KGH-04 bei pH = 1, pH = 7 und pH = 11 und zur
Veranschaulichung eine Fotografie.
Abb. 45: UV/vis-Absorptionsspektren von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm auf KGH (KGH-04) bei pH = 1, pH = 7 und pH = 11, Inset: Fotografie von KGH-04 bei pH = 1 und pH = 11.
Zu beobachten ist eine Änderung der Farbe von gelb im Sauren (λmax = 420 nm) zu
purpurrot im Basischen (λmax = 480 nm). Auch hier war beim Einstellen des
pH-Wertes in wässriger Suspension zu beobachten, dass sich im Sauren das
protonierte 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisierte PVAm etwas von
der Kieselgel-Oberfläche ablöste, was sich an einer überstehenden, gelben Lösung
zeigte. Der Substitutionsgrad des ausgefällten Anteils von 2-nitro-4-(N,N-
dimethyl)aminobenzen-funktionalisiertem PVAm wurde mit Hilfe der 1H-NMR-
Spektroskopie mit 3.3 % bestimmt.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
350 400 450 500 550 600 650 700 750 800 850 900 950 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
1: pH = 1.0 → λλλλmax = 420 nm2: pH = 7.0 → λλλλmax = 456 nm3: pH = 11.0 → λλλλmax = 480 nm
NH2 NHNO2
NH3C CH3
x y
400,000 g · mol -1
auf Kieselgel H1
2
3pH = 1 pH = 11
KGH-04

3 Ergebnisse und Diskussion
127
Die Umsetzung von mit Sangers Reagenz und DFDNB beladenen KGH´s mit PVAm
in Wasser bei pH = 11 liefern intensiv gelb gefärbte Kieselgele KGH-05 und KGH-06.
Beide Kieselgele wurden für die Aufnahme von UV/vis-Absorptionsspektren mit
reinem KGH verdünnt. Abb. 46 zeigt UV/vis-Absorptionsspektren und eine Fotografie
von mit 2,4-dinitrophenyl-funktionalisiertem PVAm modifiziertem KGH (KGH-05) und
mit 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm modifiziertem KGH
(KGH-06).
Abb. 46: UV/vis-Absorptionsspektren von 2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (KGH-05) und 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm auf KGH (KGH-06), Inset: Fotografie von KGH-05 und KGH-06.
Die UV/vis-Absorptionsbanden der Pulverproben sind sehr breit. Das UV/vis-
Absorptionsmaximum von KGH-05 liegt bei 424 nm, das von KGH-06 bei 420 nm. Im
UV/vis-Absorptionsspektrum von KGH-05 zeigt sich zusätzlich eine Schulter um
575 nm. Das könnte ein Hinweis auf Vernetzung des Polymeren durch Substitution
der para-Nitrogruppe von 2,4-dinitrophenyl-funktionalisiertem PVAm durch eine
Aminogruppe des PVAm´s sein.
Die schlechte Löslichkeit des ausgefällten, nicht adsorbierten Anteils der
funktionalisierten PVAm´s auf KGH-05 und auf KGH-06 reichten weder für eine 1H-NMR-spektroskopische Bestimmung noch für eine UV/vis-spektroskopische
Bestimmung des Substitutionsgrades aus.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
250 300 350 400 450 500 550 600 650 700 750 800
Wellenlänge λλλλ [nm]
Abs
ortio
n [a
.u.]
5
6
auf Kieselgel Hλλλλmax = 424 nm
NH2 NHNO2
NO2
x y
5
auf Kieselgel Hλλλλmax = 420 nm
6
KGH-05 KGH-06
NH2 NH
NO2
NO2
N
x y
H
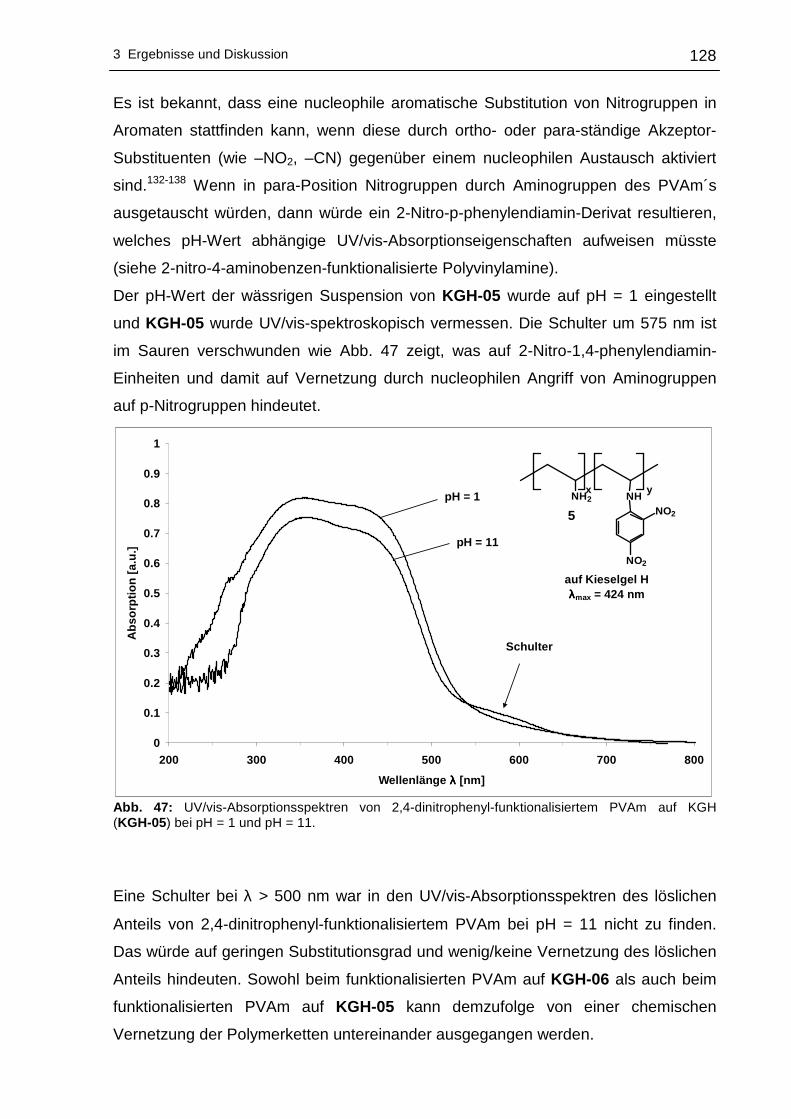
3 Ergebnisse und Diskussion
128
Es ist bekannt, dass eine nucleophile aromatische Substitution von Nitrogruppen in
Aromaten stattfinden kann, wenn diese durch ortho- oder para-ständige Akzeptor-
Substituenten (wie –NO2, –CN) gegenüber einem nucleophilen Austausch aktiviert
sind.132-138 Wenn in para-Position Nitrogruppen durch Aminogruppen des PVAm´s
ausgetauscht würden, dann würde ein 2-Nitro-p-phenylendiamin-Derivat resultieren,
welches pH-Wert abhängige UV/vis-Absorptionseigenschaften aufweisen müsste
(siehe 2-nitro-4-aminobenzen-funktionalisierte Polyvinylamine).
Der pH-Wert der wässrigen Suspension von KGH-05 wurde auf pH = 1 eingestellt
und KGH-05 wurde UV/vis-spektroskopisch vermessen. Die Schulter um 575 nm ist
im Sauren verschwunden wie Abb. 47 zeigt, was auf 2-Nitro-1,4-phenylendiamin-
Einheiten und damit auf Vernetzung durch nucleophilen Angriff von Aminogruppen
auf p-Nitrogruppen hindeutet.
Abb. 47: UV/vis-Absorptionsspektren von 2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (KGH-05) bei pH = 1 und pH = 11.
Eine Schulter bei λ > 500 nm war in den UV/vis-Absorptionsspektren des löslichen
Anteils von 2,4-dinitrophenyl-funktionalisiertem PVAm bei pH = 11 nicht zu finden.
Das würde auf geringen Substitutionsgrad und wenig/keine Vernetzung des löslichen
Anteils hindeuten. Sowohl beim funktionalisierten PVAm auf KGH-06 als auch beim
funktionalisierten PVAm auf KGH-05 kann demzufolge von einer chemischen
Vernetzung der Polymerketten untereinander ausgegangen werden.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
200 300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
auf Kieselgel Hλλλλmax = 424 nm
NH2 NHNO2
NO2
x y
5
pH = 11
pH = 1
Schulter

3 Ergebnisse und Diskussion
129
Die Polymermodifizierung von mit 4-Fluor-3-nitroazobenzen (FNAzoB) und 4-Fluor-
3,4´-dinitrostilben (FDNstilben) beladenen KGH´s durch nucleophile aromatische
Substitution der Fluoraromaten mit PVAm in Wasser bei pH = 11 liefern intensiv
orangefarbene Kieselgele KGH-07 und KGH-10. Abb. 48 zeigt UV/vis-Absorptions-
spektren und eine Fotografie von mit 2-nitro-4-azobenzen-funktionalisiertem PVAm
modifiziertem KGH (KGH-07) und mit 2,4´-dinitrostilben-funktionalisiertem PVAm
modifiziertem KGH (KGH-10), die aufgrund der Farbintensität mit reinem KGH
verdünnt werden mussten.
Abb. 48: UV/vis-Absorptionsspektren von 2-nitro-4-azobenzen-funktionalisiertem PVAm auf KGH (KGH-07) und 2,4´-dinitrostilben-funktionalisiertem PVAm auf KGH (KGH-10), Inset: Fotografie von KGH-07 und KGH-10.
Die UV/vis-Absorptionsbanden der Pulverproben sind sehr breit. Das UV/vis-
Absorptionsmaximum von KGH-07 liegt bei 455 nm, das von KGH-10 bei 400 nm.
Aufgrund der Unlöslichkeit von 2-nitro-4-azobenzen-funktionalisiertem PVAm auf
KGH-07 in Methanol konnte der Substitutionsgrad UV/vis-spektroskopisch nicht
bestimmt werden. Die Löslichkeit in Wasser war für die Bestimmung des
Substitutionsgrades mittels 1H-NMR-Spektroskopie zu gering.
Das 2,4´-dinitrostilben-funktionalisiertes PVAm der überstehenden Lösung (KGH-10)
löst sich in Methanol, so dass der Substitutionsgrad UV/vis-spektroskopisch nach
Gl. 3 mit s = 17.6 % bestimmt wurde.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
250 300 350 400 450 500 550 600 650 700 750 800 850 900 950 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
7
10
auf Kieselgel Hλλλλmax = 455 nm
NH2 NHNO2
NN
x yNH2 NH
NO2
NO2
x y
auf Kieselgel Hλλλλmax = 400 nm
7 10
KGH-07 KGH-10
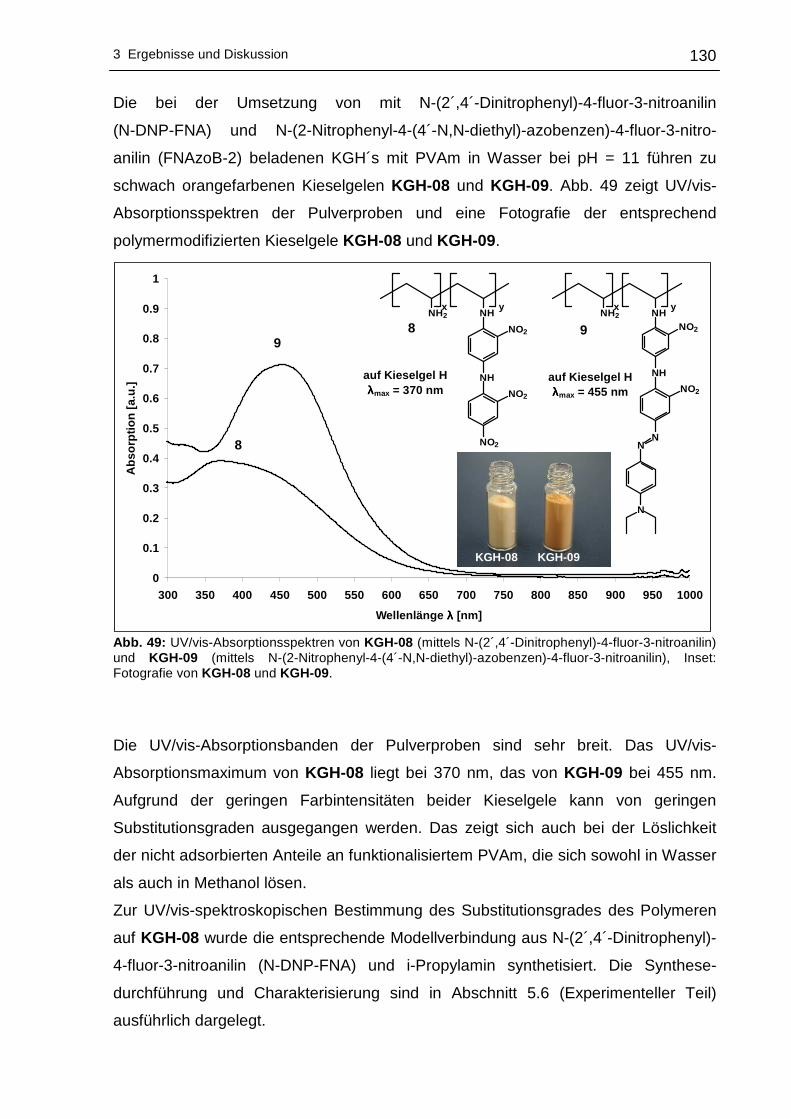
3 Ergebnisse und Diskussion
130
Die bei der Umsetzung von mit N-(2´,4´-Dinitrophenyl)-4-fluor-3-nitroanilin
(N-DNP-FNA) und N-(2-Nitrophenyl-4-(4´-N,N-diethyl)-azobenzen)-4-fluor-3-nitro-
anilin (FNAzoB-2) beladenen KGH´s mit PVAm in Wasser bei pH = 11 führen zu
schwach orangefarbenen Kieselgelen KGH-08 und KGH-09. Abb. 49 zeigt UV/vis-
Absorptionsspektren der Pulverproben und eine Fotografie der entsprechend
polymermodifizierten Kieselgele KGH-08 und KGH-09.
Abb. 49: UV/vis-Absorptionsspektren von KGH-08 (mittels N-(2´,4´-Dinitrophenyl)-4-fluor-3-nitroanilin) und KGH-09 (mittels N-(2-Nitrophenyl-4-(4´-N,N-diethyl)-azobenzen)-4-fluor-3-nitroanilin), Inset: Fotografie von KGH-08 und KGH-09.
Die UV/vis-Absorptionsbanden der Pulverproben sind sehr breit. Das UV/vis-
Absorptionsmaximum von KGH-08 liegt bei 370 nm, das von KGH-09 bei 455 nm.
Aufgrund der geringen Farbintensitäten beider Kieselgele kann von geringen
Substitutionsgraden ausgegangen werden. Das zeigt sich auch bei der Löslichkeit
der nicht adsorbierten Anteile an funktionalisiertem PVAm, die sich sowohl in Wasser
als auch in Methanol lösen.
Zur UV/vis-spektroskopischen Bestimmung des Substitutionsgrades des Polymeren
auf KGH-08 wurde die entsprechende Modellverbindung aus N-(2´,4´-Dinitrophenyl)-
4-fluor-3-nitroanilin (N-DNP-FNA) und i-Propylamin synthetisiert. Die Synthese-
durchführung und Charakterisierung sind in Abschnitt 5.6 (Experimenteller Teil)
ausführlich dargelegt.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 350 400 450 500 550 600 650 700 750 800 850 900 950 1000
Wellenlänge λλλλ [nm]
Abs
orpt
ion
[a.u
.]
auf Kieselgel Hλλλλmax = 455 nm
x yNH2 NH
NO2
NH
NO2
NN
N
9
8
9
auf Kieselgel Hλλλλmax = 370 nm
x yNH2 NH
NO2
NH
NO2
NO2
8
KGH-08 KGH-09
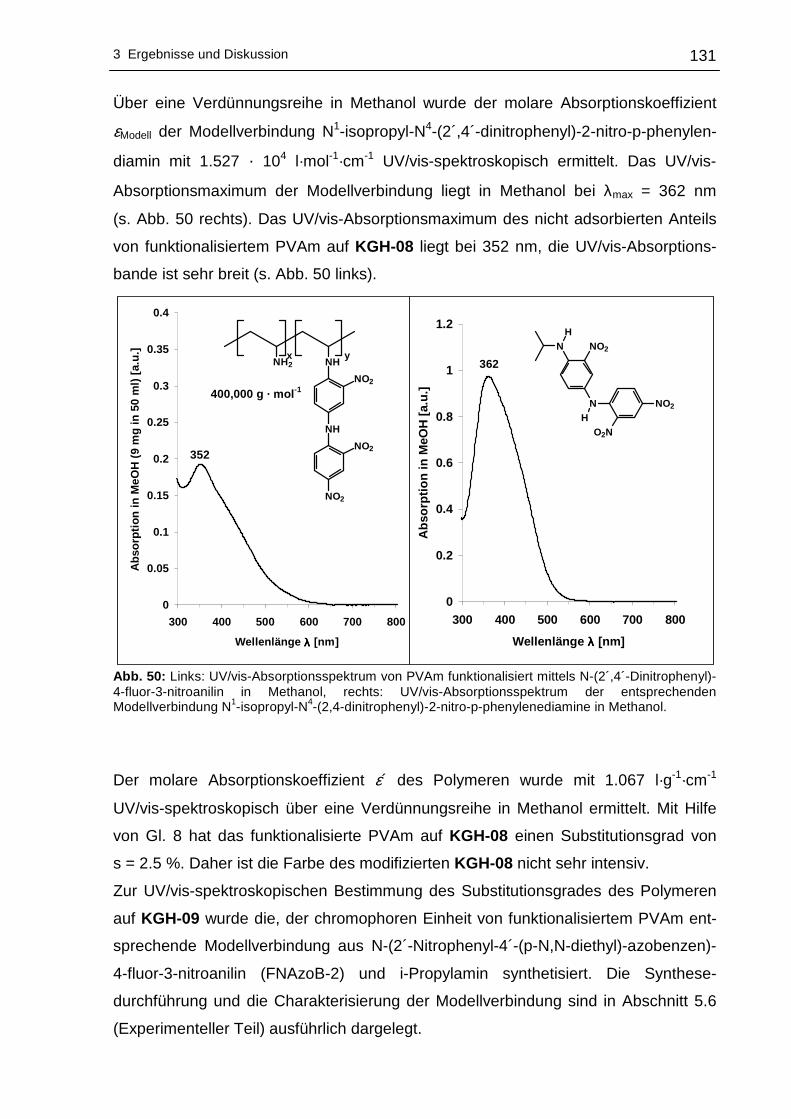
3 Ergebnisse und Diskussion
131
Über eine Verdünnungsreihe in Methanol wurde der molare Absorptionskoeffizient
εModell der Modellverbindung N1-isopropyl-N4-(2´,4´-dinitrophenyl)-2-nitro-p-phenylen-
diamin mit 1.527 · 104 l·mol-1·cm-1 UV/vis-spektroskopisch ermittelt. Das UV/vis-
Absorptionsmaximum der Modellverbindung liegt in Methanol bei λmax = 362 nm
(s. Abb. 50 rechts). Das UV/vis-Absorptionsmaximum des nicht adsorbierten Anteils
von funktionalisiertem PVAm auf KGH-08 liegt bei 352 nm, die UV/vis-Absorptions-
bande ist sehr breit (s. Abb. 50 links).
Abb. 50: Links: UV/vis-Absorptionsspektrum von PVAm funktionalisiert mittels N-(2´,4´-Dinitrophenyl)-4-fluor-3-nitroanilin in Methanol, rechts: UV/vis-Absorptionsspektrum der entsprechenden Modellverbindung N1-isopropyl-N4-(2,4-dinitrophenyl)-2-nitro-p-phenylenediamine in Methanol.
Der molare Absorptionskoeffizient ε´ des Polymeren wurde mit 1.067 l·g-1·cm-1
UV/vis-spektroskopisch über eine Verdünnungsreihe in Methanol ermittelt. Mit Hilfe
von Gl. 8 hat das funktionalisierte PVAm auf KGH-08 einen Substitutionsgrad von
s = 2.5 %. Daher ist die Farbe des modifizierten KGH-08 nicht sehr intensiv.
Zur UV/vis-spektroskopischen Bestimmung des Substitutionsgrades des Polymeren
auf KGH-09 wurde die, der chromophoren Einheit von funktionalisiertem PVAm ent-
sprechende Modellverbindung aus N-(2´-Nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-
4-fluor-3-nitroanilin (FNAzoB-2) und i-Propylamin synthetisiert. Die Synthese-
durchführung und die Charakterisierung der Modellverbindung sind in Abschnitt 5.6
(Experimenteller Teil) ausführlich dargelegt.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
(9
mg
in 5
0 m
l) [a
.u.]
x yNH2 NH
NO2
NH
NO2
NO2
352
400,000 g · mol -1
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
N
NO2
O2N
N
H
H
NO2
362
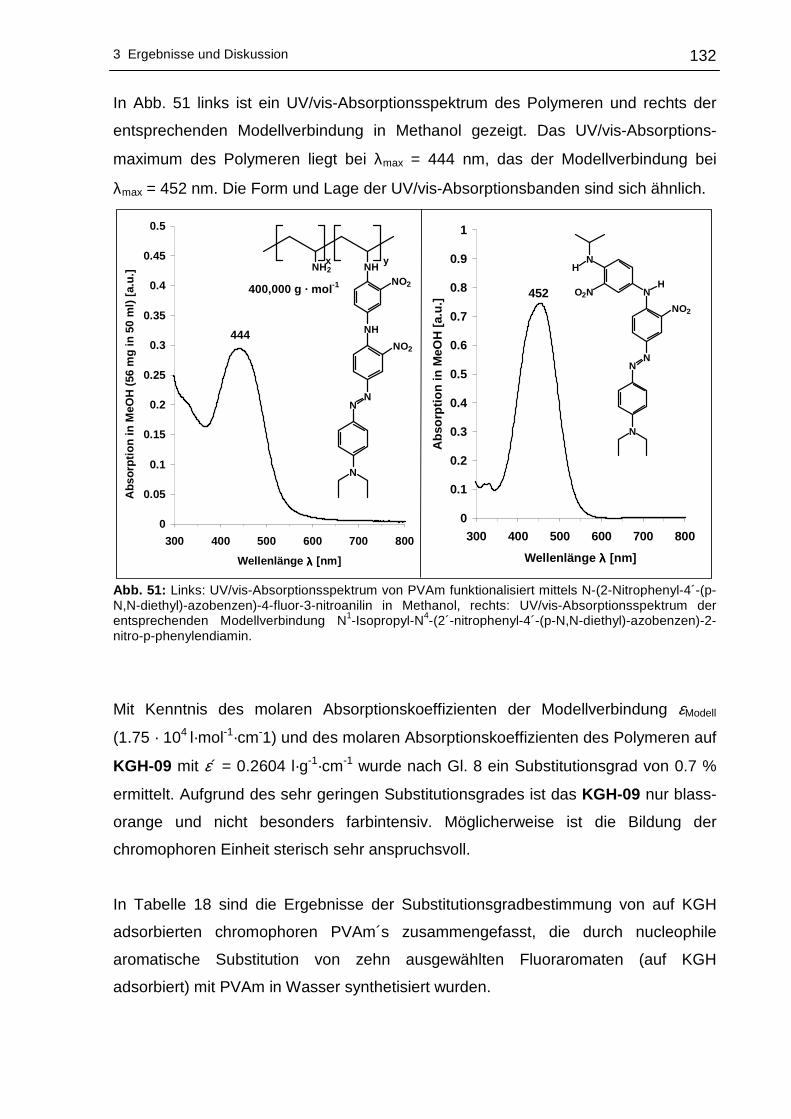
3 Ergebnisse und Diskussion
132
In Abb. 51 links ist ein UV/vis-Absorptionsspektrum des Polymeren und rechts der
entsprechenden Modellverbindung in Methanol gezeigt. Das UV/vis-Absorptions-
maximum des Polymeren liegt bei λmax = 444 nm, das der Modellverbindung bei
λmax = 452 nm. Die Form und Lage der UV/vis-Absorptionsbanden sind sich ähnlich.
Abb. 51: Links: UV/vis-Absorptionsspektrum von PVAm funktionalisiert mittels N-(2-Nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-4-fluor-3-nitroanilin in Methanol, rechts: UV/vis-Absorptionsspektrum der entsprechenden Modellverbindung N1-Isopropyl-N4-(2´-nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-2-nitro-p-phenylendiamin.
Mit Kenntnis des molaren Absorptionskoeffizienten der Modellverbindung εModell
(1.75 · 104 l·mol-1·cm-1) und des molaren Absorptionskoeffizienten des Polymeren auf
KGH-09 mit ε´ = 0.2604 l·g-1·cm-1 wurde nach Gl. 8 ein Substitutionsgrad von 0.7 %
ermittelt. Aufgrund des sehr geringen Substitutionsgrades ist das KGH-09 nur blass-
orange und nicht besonders farbintensiv. Möglicherweise ist die Bildung der
chromophoren Einheit sterisch sehr anspruchsvoll.
In Tabelle 18 sind die Ergebnisse der Substitutionsgradbestimmung von auf KGH
adsorbierten chromophoren PVAm´s zusammengefasst, die durch nucleophile
aromatische Substitution von zehn ausgewählten Fluoraromaten (auf KGH
adsorbiert) mit PVAm in Wasser synthetisiert wurden.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0.5
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
(56
mg
in 5
0 m
l) [a
.u.]
x yNH2 NH
NO2
NH
NO2
NN
N
400,000 g · mol -1
444
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
300 400 500 600 700 800
Wellenlänge λλλλ [nm]
Abs
orpt
ion
in M
eOH
[a.u
.]
452
NH
NH
NO2
N
O2N
N
N
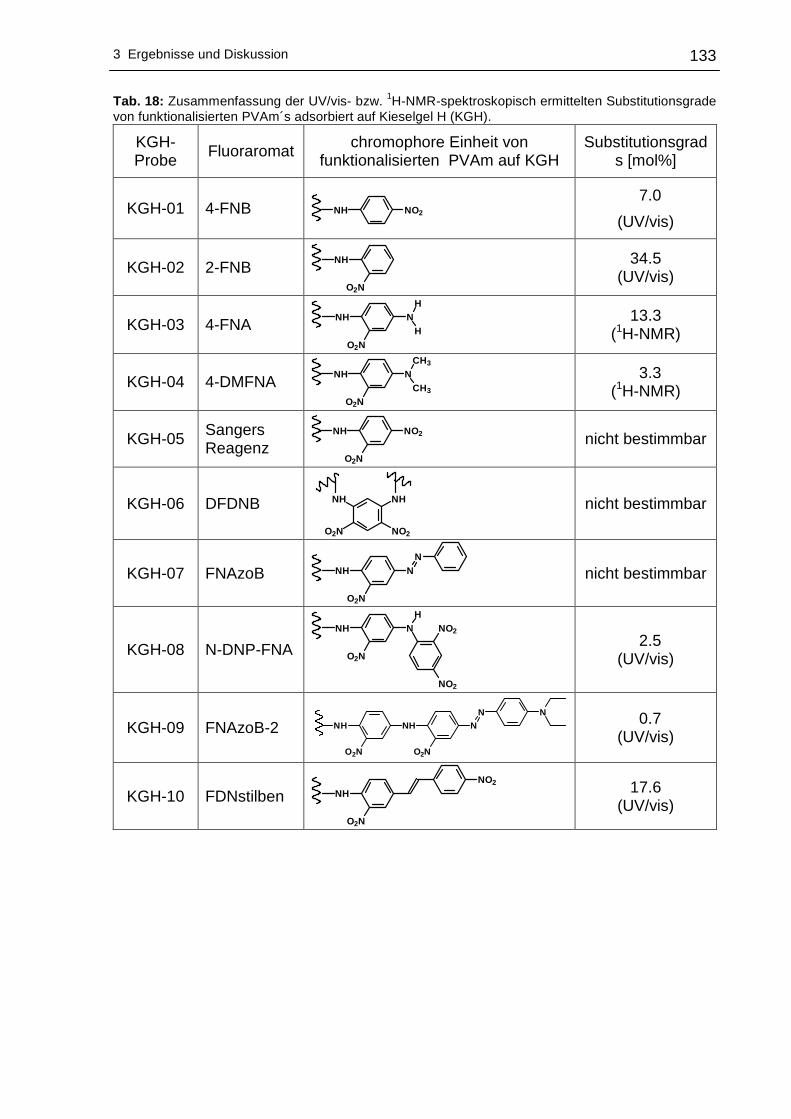
3 Ergebnisse und Diskussion
133
Tab. 18: Zusammenfassung der UV/vis- bzw. 1H-NMR-spektroskopisch ermittelten Substitutionsgrade von funktionalisierten PVAm´s adsorbiert auf Kieselgel H (KGH).
KGH-Probe
Fluoraromat chromophore Einheit von funktionalisierten PVAm auf KGH
Substitutionsgrad s [mol%]
KGH-01 4-FNB NO2NH
7.0
(UV/vis)
KGH-02 2-FNB NH
O2N
34.5 (UV/vis)
KGH-03 4-FNA NH
O2N
NH
H
13.3 (1H-NMR)
KGH-04 4-DMFNA NH
O2N
NCH3
CH3
3.3 (1H-NMR)
KGH-05 Sangers Reagenz
NH
O2N
NO2
nicht bestimmbar
KGH-06 DFDNB
NO2
NH
O2N
NH
nicht bestimmbar
KGH-07 FNAzoB NH
O2N
NN
nicht bestimmbar
KGH-08 N-DNP-FNA
NH
O2N
NH
NO2
NO2
2.5 (UV/vis)
KGH-09 FNAzoB-2
NH
O2N
NN
NH
O2N
N
0.7 (UV/vis)
KGH-10 FDNstilben NH
O2N
NO2
17.6 (UV/vis)

3 Ergebnisse und Diskussion
134
3.2.3 13C-CP-MAS-Festkörper-NMR-spektroskopische Untersuchu ngen
Hochaufgelöste NMR-Spektren von Festkörpern können mit der normalen PFT-
Technik (Puls-Fourier-Transform-Technik) nicht aufgenommen werden. Dipol-Dipol-
und Quadrupol-Feldgradienten-Wechselwirkungen, die in Lösung herausgemittelt
werden, führen zu starker Linienverbreiterung. Die Anisotropie der chemischen
Verschiebung bewirkt breite Linienformen und die Spin-Gitter-Relaxationszeiten von
Kernen wie 13C sind in Festkörpern sehr lang. Hochaufgelöste Festkörper-NMR-
spektren können erhalten werden, wenn die Probe mit hoher Geschwindigkeit um
eine Achse rotiert, die den sog. „magischen Winkel“ von 54.73° einschließt. Durch
dieses Magic Angle Spinning (MAS) entspricht das Pulverspektrum prinzipiell dem
Lösungsspektrum.139
Die direkte Detektion von 13C Kernen ist jedoch aufgrund der geringen Häufigkeit, der
niedrigen Spinpolarisation und niedriger Signalintensitäten schwierig. Um diese
Schwierigkeiten zu umgehen, wird daher eine Festkörper-NMR-Methode benötigt,
die 1H-NMR-typisch hohe Polarisationen und kurze Relaxationszeiten mit der
hochauflösenden Detektion von 13C-Kernen kombiniert. Eine Verstärkung der Signale
„seltener“ Kerne wie 13C erreicht man durch den Transfer der Polarisation „häufiger“
Kerne (üblicherweise 1H-Kerne) bei der sog. Kreuzpolarisation (Cross Polarization).
Die Methode beruht darauf, dass die Magnetisierung leicht von stark polarisierten
Kernen auf schwach polarisierte Kerne übergeht, sobald die Kerne passend
zueinander in Kontakt gebracht werden. Die Kombination von CP und MAS bei der
protonenverstärkten NMR-Spektroskopie eröffnet einen routinemäßigen Zugang zu
hochempfindlichen, hochaufgelösten 13C-NMR-Spektren von Festkörpern.140
Durch 13C-CP-MAS-Festkörper-NMR-spektroskopische Untersuchungen von mit
funktionalisiertem PVAm beladenen Kieselgel-Partikeln konnte die molekulare
Struktur der gebildeten chromophoren Einheiten am Polymerrückgrat neben den
unsubstituierten PVAm-Einheiten charakterisiert werden. Für die Zuordnung der
einzelnen 13C-Signale der polymermodifizierten Kieselgele wurden 13C-Lösungs-
NMR-spektren von niedermolekularen Modellverbindungen (N-Isopropyl-2-nitroanilin-
Derivate) herangezogen. An zwei ausgewählten Beispielen − mit hohen
Substitutionsgraden an chromophoren PVAm-Einheiten − ist nachfolgend gezeigt,
wie die molekulare Struktur von funktionalisiertem PVAm auf KGH mit Hilfe der 13C- NMR-Spektroskopie charakterisiert wurde.
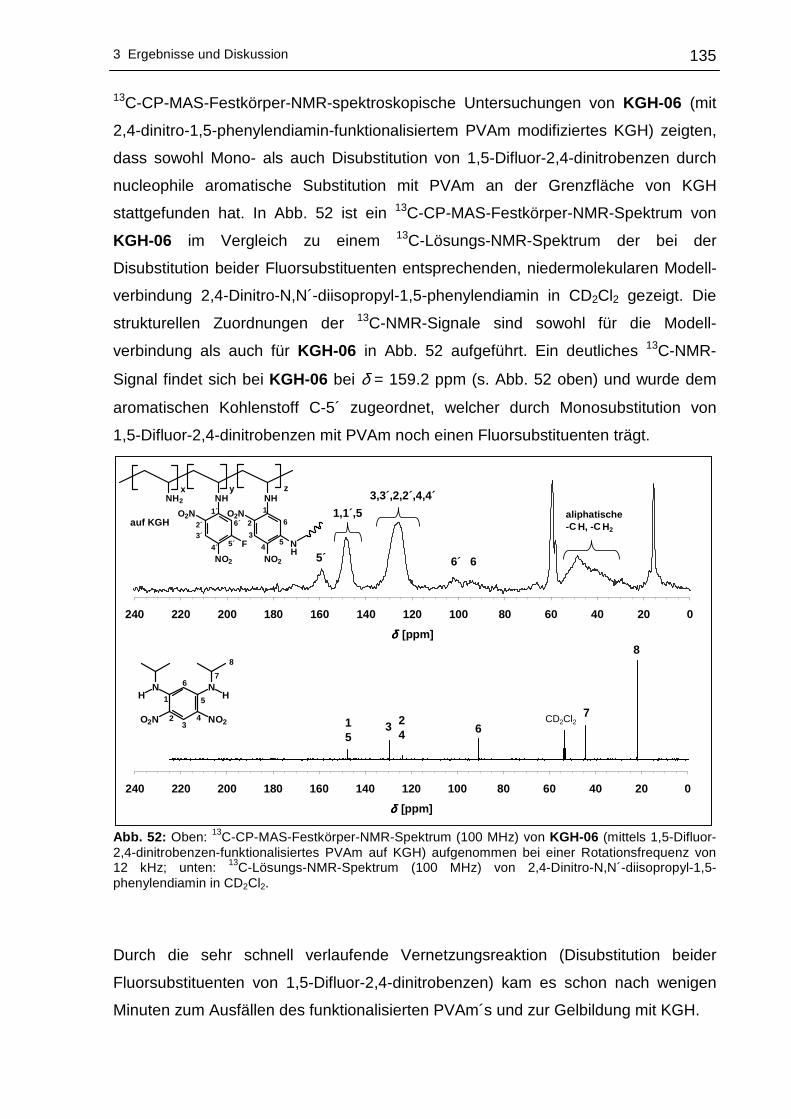
3 Ergebnisse und Diskussion
135
13C-CP-MAS-Festkörper-NMR-spektroskopische Untersuchungen von KGH-06 (mit
2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm modifiziertes KGH) zeigten,
dass sowohl Mono- als auch Disubstitution von 1,5-Difluor-2,4-dinitrobenzen durch
nucleophile aromatische Substitution mit PVAm an der Grenzfläche von KGH
stattgefunden hat. In Abb. 52 ist ein 13C-CP-MAS-Festkörper-NMR-Spektrum von
KGH-06 im Vergleich zu einem 13C-Lösungs-NMR-Spektrum der bei der
Disubstitution beider Fluorsubstituenten entsprechenden, niedermolekularen Modell-
verbindung 2,4-Dinitro-N,N´-diisopropyl-1,5-phenylendiamin in CD2Cl2 gezeigt. Die
strukturellen Zuordnungen der 13C-NMR-Signale sind sowohl für die Modell-
verbindung als auch für KGH-06 in Abb. 52 aufgeführt. Ein deutliches 13C-NMR-
Signal findet sich bei KGH-06 bei δ = 159.2 ppm (s. Abb. 52 oben) und wurde dem
aromatischen Kohlenstoff C-5´ zugeordnet, welcher durch Monosubstitution von
1,5-Difluor-2,4-dinitrobenzen mit PVAm noch einen Fluorsubstituenten trägt.
Abb. 52: Oben: 13C-CP-MAS-Festkörper-NMR-Spektrum (100 MHz) von KGH-06 (mittels 1,5-Difluor-2,4-dinitrobenzen-funktionalisiertes PVAm auf KGH) aufgenommen bei einer Rotationsfrequenz von 12 kHz; unten: 13C-Lösungs-NMR-Spektrum (100 MHz) von 2,4-Dinitro-N,N´-diisopropyl-1,5-phenylendiamin in CD2Cl2.
Durch die sehr schnell verlaufende Vernetzungsreaktion (Disubstitution beider
Fluorsubstituenten von 1,5-Difluor-2,4-dinitrobenzen) kam es schon nach wenigen
Minuten zum Ausfällen des funktionalisierten PVAm´s und zur Gelbildung mit KGH.
020406080100120140160180200220240
δδδδ [ppm]
15
24
3 67
8
CD2Cl2
NN
NO2O2N
HH 1
23
4
5
67
8
020406080100120140160180200220240
δδδδ [ppm]
5´
1,1´,5
3,3´,2,2´,4,4´
6´ 6
aliphatische-C H, -C H2
1
2
3
45
61´
6´
5´4´
3´2´
x y z
H
NH2 NH
O2N
NO2
F
NH
O2N
NO2
N
auf KGH
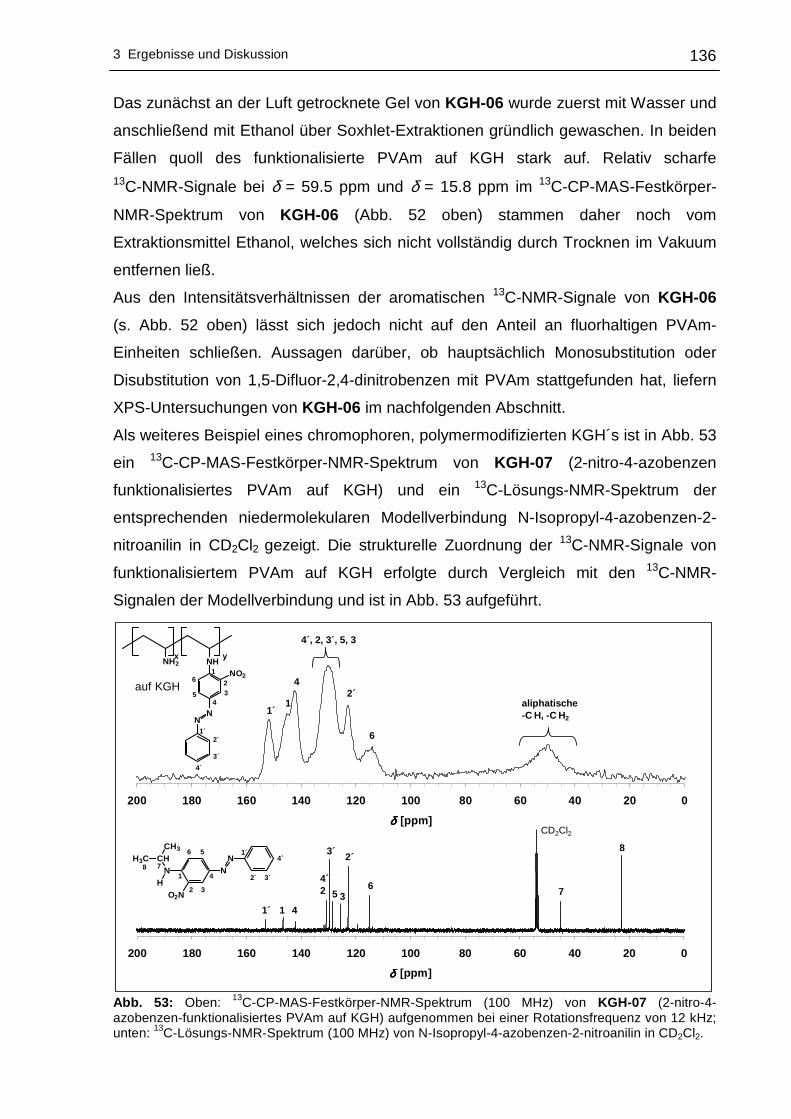
3 Ergebnisse und Diskussion
136
Das zunächst an der Luft getrocknete Gel von KGH-06 wurde zuerst mit Wasser und
anschließend mit Ethanol über Soxhlet-Extraktionen gründlich gewaschen. In beiden
Fällen quoll des funktionalisierte PVAm auf KGH stark auf. Relativ scharfe 13C-NMR-Signale bei δ = 59.5 ppm und δ = 15.8 ppm im 13C-CP-MAS-Festkörper-
NMR-Spektrum von KGH-06 (Abb. 52 oben) stammen daher noch vom
Extraktionsmittel Ethanol, welches sich nicht vollständig durch Trocknen im Vakuum
entfernen ließ.
Aus den Intensitätsverhältnissen der aromatischen 13C-NMR-Signale von KGH-06
(s. Abb. 52 oben) lässt sich jedoch nicht auf den Anteil an fluorhaltigen PVAm-
Einheiten schließen. Aussagen darüber, ob hauptsächlich Monosubstitution oder
Disubstitution von 1,5-Difluor-2,4-dinitrobenzen mit PVAm stattgefunden hat, liefern
XPS-Untersuchungen von KGH-06 im nachfolgenden Abschnitt.
Als weiteres Beispiel eines chromophoren, polymermodifizierten KGH´s ist in Abb. 53
ein 13C-CP-MAS-Festkörper-NMR-Spektrum von KGH-07 (2-nitro-4-azobenzen
funktionalisiertes PVAm auf KGH) und ein 13C-Lösungs-NMR-Spektrum der
entsprechenden niedermolekularen Modellverbindung N-Isopropyl-4-azobenzen-2-
nitroanilin in CD2Cl2 gezeigt. Die strukturelle Zuordnung der 13C-NMR-Signale von
funktionalisiertem PVAm auf KGH erfolgte durch Vergleich mit den 13C-NMR-
Signalen der Modellverbindung und ist in Abb. 53 aufgeführt.
Abb. 53: Oben: 13C-CP-MAS-Festkörper-NMR-Spektrum (100 MHz) von KGH-07 (2-nitro-4-azobenzen-funktionalisiertes PVAm auf KGH) aufgenommen bei einer Rotationsfrequenz von 12 kHz; unten: 13C-Lösungs-NMR-Spektrum (100 MHz) von N-Isopropyl-4-azobenzen-2-nitroanilin in CD2Cl2.
020406080100120140160180200
δδδδ [ppm]
020406080100120140160180200
δδδδ [ppm]
NCH
NN
O2NH
CH3
H3C
1
2 3
4
56
78
1´
2´ 3´
4´
NH2 NHNO2
NN
x y
1
23
45
6
1´2´
3´
4´
auf KGH
1´1
4
4´, 2, 3´, 5, 3
2´
6
aliphatische-C H, -C H2
1´ 1 4
3´
3
2´
6 7
8
4´2 5
CD2Cl2

3 Ergebnisse und Diskussion
137
3.2.4 Röntgen-Photoelektronenspektroskopische Unte rsuchungen (XPS)
Die XPS ist eine Methode der Elektronenspektroskopie, mit der Bindungszustände
analysiert werden können. Wird ein Atomkern von weicher Röntgenstrahlung
getroffen, so wird ein Elektron aus einer inneren Bahn (z. B. K-Niveau) zum
Verlassen der Bahn gezwungen. Aus der in Elektronenvolt gemessenen kinetischen
Energie dieses Photoelektrons lässt sich bei Kenntnis der Röntgenanregungsenergie
die Bindungsenergie des durch den Photoeffekt emittierten Elektrons bestimmen. Die
XPS findet insbesondere Anwendung zur Analyse von Probenoberflächen.48
XPS-Spektren wurden von zehn chromophoren PVAm-modifizierten KGH´s
aufgenommen. Als Röntgenstrahlungsquelle diente Mono-Al Kα-Strahlung mit einer
Aufnahmeleistung der Röntgenröhre von 300 W bei 20 mA.
In den Übersichtsspektren aller modifizierten KGH´s wurden folgende Elemente
nachgewiesen:
Kohlenstoff: als C 1s Peak,
Stickstoff: als N 1s und N KLL Peaks,
Sauerstoff: als O 1s, O 2s, O KL1 = O KL23L23, O KL2 = O KL1L23 und O KL3 = O
KL1L1 Peaks,
Fluor: als F 1s, F KL1 = F KL23L23, F KL2 = F KL1L23 und F KL3 = F KL1L1
Peaks,
Silicium: als Si 2p3/2, Si 2p1/2 und Si 2s Peaks.
Bei allen Proben erfolgte eine Überkompensation der Aufladung, die durch das
Verschieben des C 1s Peaks auf die Bindungsenergie (BE) gesättigter Kohlen-
wasserstoffe CxHy mit einer BE = 285.00 eV korrigiert wurde.141
In Abbildung 54 (links) sind Übersichts-XPS-Spektren von reinem KGH (a),
PVAm/KGH (b), 2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (c, KGH-05)
und 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm auf KGH (d, KGH-06)
mit der Zuordnung der entsprechend erhaltenen Signale gezeigt.
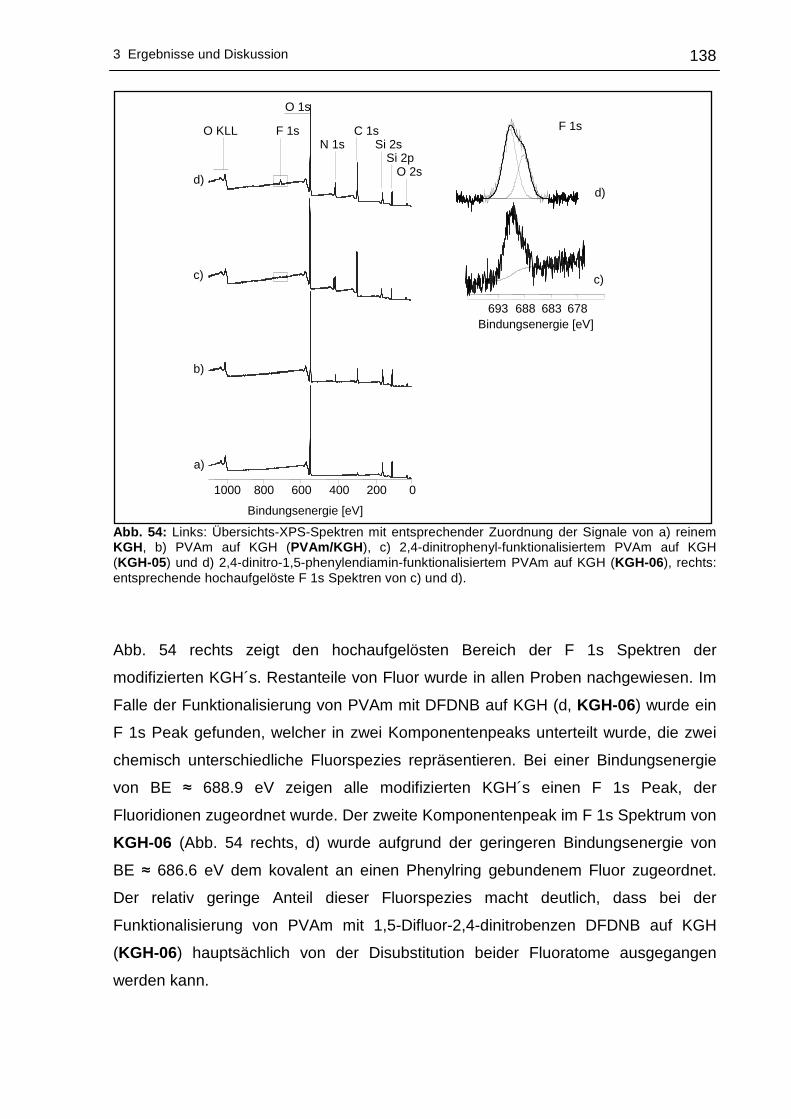
3 Ergebnisse und Diskussion
138
Abb. 54: Links: Übersichts-XPS-Spektren mit entsprechender Zuordnung der Signale von a) reinem KGH, b) PVAm auf KGH (PVAm/KGH ), c) 2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (KGH-05) und d) 2,4-dinitro-1,5-phenylendiamin-funktionalisiertem PVAm auf KGH (KGH-06), rechts: entsprechende hochaufgelöste F 1s Spektren von c) und d).
Abb. 54 rechts zeigt den hochaufgelösten Bereich der F 1s Spektren der
modifizierten KGH´s. Restanteile von Fluor wurde in allen Proben nachgewiesen. Im
Falle der Funktionalisierung von PVAm mit DFDNB auf KGH (d, KGH-06) wurde ein
F 1s Peak gefunden, welcher in zwei Komponentenpeaks unterteilt wurde, die zwei
chemisch unterschiedliche Fluorspezies repräsentieren. Bei einer Bindungsenergie
von BE ≈ 688.9 eV zeigen alle modifizierten KGH´s einen F 1s Peak, der
Fluoridionen zugeordnet wurde. Der zweite Komponentenpeak im F 1s Spektrum von
KGH-06 (Abb. 54 rechts, d) wurde aufgrund der geringeren Bindungsenergie von
BE ≈ 686.6 eV dem kovalent an einen Phenylring gebundenem Fluor zugeordnet.
Der relativ geringe Anteil dieser Fluorspezies macht deutlich, dass bei der
Funktionalisierung von PVAm mit 1,5-Difluor-2,4-dinitrobenzen DFDNB auf KGH
(KGH-06) hauptsächlich von der Disubstitution beider Fluoratome ausgegangen
werden kann.
1000 800 600 400 200 0
a)
b)
c)
O 1s
O KLL C 1sN 1s Si 2s
Si 2p
F 1s
d)
693 683688 678
F 1s
c)
O 2s
d)
Bindungsenergie [eV]
Bindungsenergie [eV]
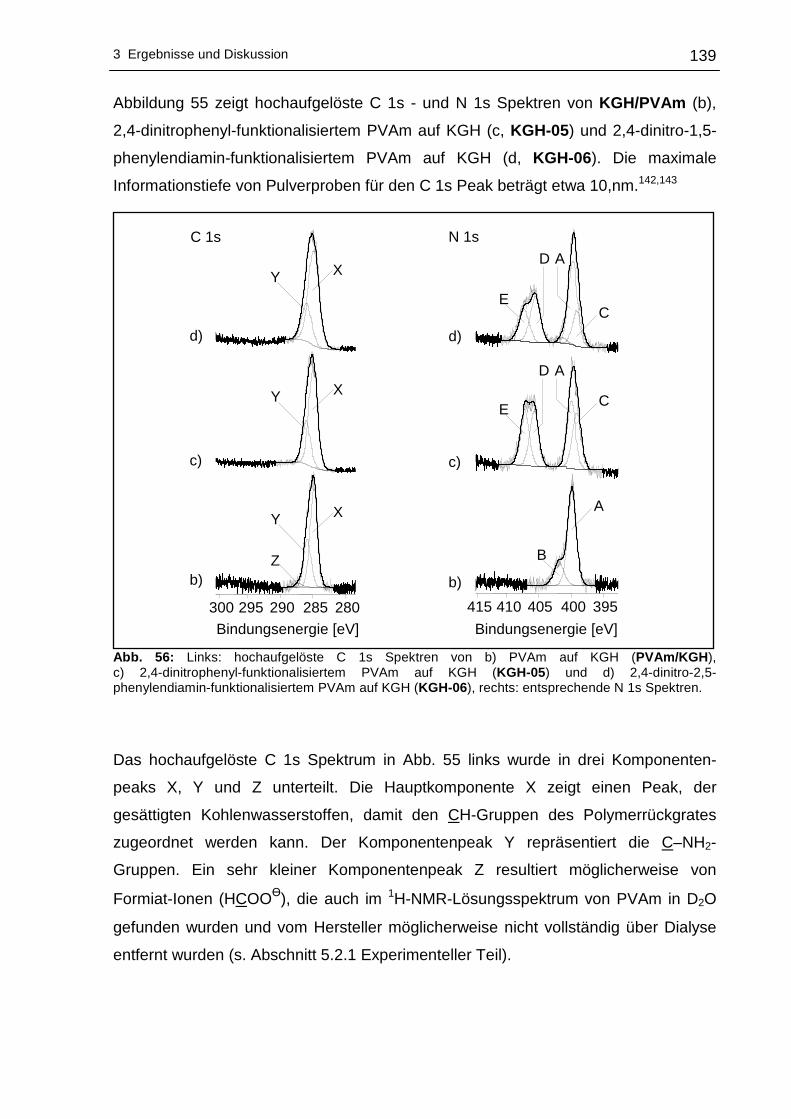
3 Ergebnisse und Diskussion
139
Abbildung 55 zeigt hochaufgelöste C 1s - und N 1s Spektren von KGH/PVAm (b),
2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (c, KGH-05) und 2,4-dinitro-1,5-
phenylendiamin-funktionalisiertem PVAm auf KGH (d, KGH-06). Die maximale
Informationstiefe von Pulverproben für den C 1s Peak beträgt etwa 10,nm.142,143
Abb. 56: Links: hochaufgelöste C 1s Spektren von b) PVAm auf KGH (PVAm/KGH ), c) 2,4-dinitrophenyl-funktionalisiertem PVAm auf KGH (KGH-05) und d) 2,4-dinitro-2,5-phenylendiamin-funktionalisiertem PVAm auf KGH (KGH-06), rechts: entsprechende N 1s Spektren.
Das hochaufgelöste C 1s Spektrum in Abb. 55 links wurde in drei Komponenten-
peaks X, Y und Z unterteilt. Die Hauptkomponente X zeigt einen Peak, der
gesättigten Kohlenwasserstoffen, damit den CH-Gruppen des Polymerrückgrates
zugeordnet werden kann. Der Komponentenpeak Y repräsentiert die C–NH2-
Gruppen. Ein sehr kleiner Komponentenpeak Z resultiert möglicherweise von
Formiat-Ionen (HCOOӨ), die auch im 1H-NMR-Lösungsspektrum von PVAm in D2O
gefunden wurden und vom Hersteller möglicherweise nicht vollständig über Dialyse
entfernt wurden (s. Abschnitt 5.2.1 Experimenteller Teil).
A
B
415 405 395400410
b)
C
AD
c)
C
AD
E
d)
N 1s
XY
C 1s
300 290 280285295
b)
c)
d)
XY
E
XY
Z
Bindungsenergie [eV] Bindungsenergie [eV]

3 Ergebnisse und Diskussion
140
Die durch die Umsetzung von PVAm mit den entsprechenden Fluoraromaten
gebildeten sekundären Aminogruppen und die damit enthaltenen C–NO2-Gruppen
können im C 1s Spektrum nicht vom Komponentenpeak Y unterschieden werden.
Das hochaufgelöste N 1s Spektrum in Abb. 55 rechts (b) zeigt, dass primäre
Aminogruppen (C–NH2, Komponentenpeak A) mit protonierten Aminogruppen
(C–NH3⊕, Komponentenpeak B) im Gleichgewicht stehen. Weiterhin wird deutlich,
dass das N 1s Spektrum von KGH-05 (b) und KGH-06 (c) neben dem N 1s Peak bei
400 eV noch einen Peak bei 406 eV zeigt. Das Signal um 406 eV repräsentiert die an
einen Phenylring gebundenen –NO2-Gruppen.144 Es ist noch einmal aufgespaltet in
zwei Signale (Komponentenpeak D und E). Diese Aufspaltung des C–NO2 Peaks in
zwei Komponentenpeaks D und E spricht für intramolekulare Wasserstoffbrücken-
bindungen zwischen Aminogruppen und ortho-Nitrogruppen.
Das Signal um 406 eV für an Phenylringe gebundene –NO2-Gruppen zeigen nur die
Kieselgele KGH-02, -05, -06, -07 und -10, wo ein hoher Anteil an chromophoren
PVAm-Einheiten im funktionalisierten PVAm erreicht wurde. Bei KGH-01, -03, -04,
-08 und -09 wurde um 406 eV kein Signal gefunden. Die maximale Informationstiefe
für N 1s Photoelektronen erzeugt mit Al Kα-Röntgenstrahlung ist nicht größer als
8 nm.141,142 Möglicherweise können die –NO2-Gruppe in der Tiefe der Kieselgel-
partikel nicht detektiert werden. Das ist ein Hinweis darauf, dass die Substitutions-
reaktion in Abhängigkeit der Reaktivität des Fluoraromaten in unterschiedlicher
Weise vonstatten geht. Das bedeutet, dass die weniger reaktiven Fluoraromaten in
den Poren mit PVAm reagiert haben könnten und damit die chromophoren Gruppen
mehr in Oberflächennähe des Kieselgels lokalisiert sind. Wohingegen die reaktiveren
Fluoraromaten schon vor Erreichen der Poren in der Peripherie der Kieselgelpartikel
mit PVAm reagiert haben könnten.
Zur Quantifizierung wurden aus den Peakflächen der Übersichtsspektren unter
Berücksichtigung der relativen Empfindlichkeitsfaktoren und der energieabhängigen
Spektrometer-Transmissionsfunktion die Elementarverhältnisse (Atomverhältnisse)
ermittelt, wobei das Hintergrundspektrum in Anlehnung an Shirley subtrahiert
wurde.145 Die auf diese Weise ermittelten Elementarverhältnisse verschiedener
Proben sind miteinander vergleichbar. Tabelle 19 zeigt eine Zusammenfassung der
Elementarverhältnisse aus den XPS-Untersuchungen aller modifizierten Kieselgele.

3 Ergebnisse und Diskussion
141
Tab. 19: Zusammenfassung der quantitativen Analyse der Elementarverhältnisse von mit funktionalisierten PVAm´s modifizierten KGH´s aus XPS-Untersuchungen.
Fluoraromat KGH-Probe
[N]:[C] [O]:[C] [F]:[C] [Si]:[C] [Si]:[O]
- KGH rein - 14.811 - 5.240 0.354
4-FNB KGH-01 0.293 2.752 0.013 0.999 0.383
2-FNB KGH-02 0.305 2.233 0.013 0.830 0.372
4-FNA KGH-03 0.186 0.165 0.007 0.584 0.354
4-DMFNA KGH-04 0.236 2.077 0.015 0.759 0.366
Sangers Reagenz KGH-05 0.410 0.922 0.005 0.215 0.232
DFDNB KGH-06 0.361 0.989 0.030 0.267 0.270
FNAzoB KGH-07 0.298 0.573 0.010 0.181 0.318
N-DNP-FNA KGH-08 0.279 2.537 0.013 0.912 0.360
FNAzoB-2 KGH-09 0.258 2.027 0.016 0.754 0.372
FDNstilben KGH-10 0.243 1.306 0.007 0.422 0.323
Für SiO2 sollte das theoretische Atomverhältnis [Si]:[O] = 0.5 betragen. Der höhere
Sauerstoffanteil kann auf das Vorhandensein einer hohen Anzahl an Silanolgruppen
(Si–OH-Gruppen) und/oder auf adsorptiv gebundenes Wasser zurückgeführt werden.
Das [F]:[C]-Atomverhältnis ist bei allen KGH-Proben sehr gering. Das [Si]:[C]-
Atomverhältnis der Umsetzung von PVAm mit für die nucleophile aromatische
Substitution besser aktivierten Fluoraromaten auf KGH ist deutlich kleiner als die der
Umsetzung mit weniger reaktiven Fluoraromaten (siehe Tab. 19 [Si]:[C]-Atom-
verhältnisse von KGH-05, -06 und -07 im Vergleich zu KGH-01, -08 und -09). Da die
Informationstiefe bei der XPS etwa 10 nm beträgt, weist der geringe Siliziumanteil
darauf hin, dass sich auf der KGH-Oberfläche möglicherweise eine dickere Schicht
an funktionalisiertem PVAm befindet, wenn die Funktionalisierung von PVAm mit
besser aktivierten Fluoraromaten wie Sangers Reagenz, DFDNB und FNAzoB
erfolgt.
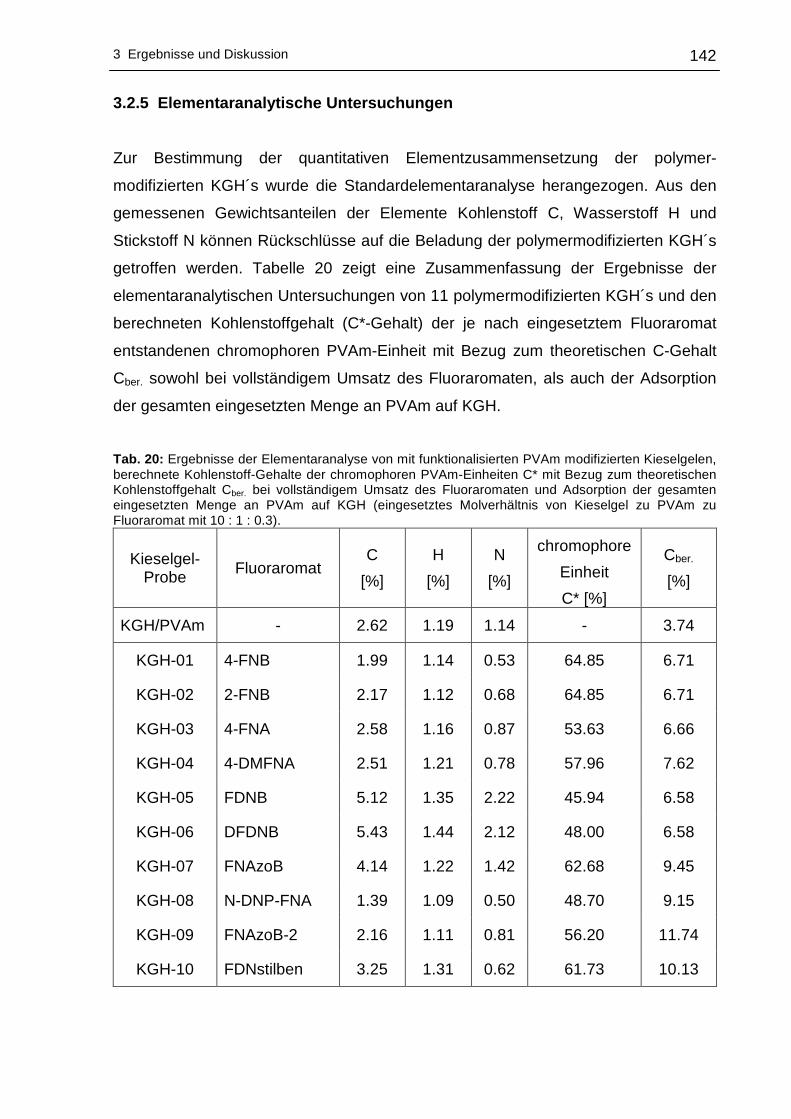
3 Ergebnisse und Diskussion
142
3.2.5 Elementaranalytische Untersuchungen
Zur Bestimmung der quantitativen Elementzusammensetzung der polymer-
modifizierten KGH´s wurde die Standardelementaranalyse herangezogen. Aus den
gemessenen Gewichtsanteilen der Elemente Kohlenstoff C, Wasserstoff H und
Stickstoff N können Rückschlüsse auf die Beladung der polymermodifizierten KGH´s
getroffen werden. Tabelle 20 zeigt eine Zusammenfassung der Ergebnisse der
elementaranalytischen Untersuchungen von 11 polymermodifizierten KGH´s und den
berechneten Kohlenstoffgehalt (C*-Gehalt) der je nach eingesetztem Fluoraromat
entstandenen chromophoren PVAm-Einheit mit Bezug zum theoretischen C-Gehalt
Cber. sowohl bei vollständigem Umsatz des Fluoraromaten, als auch der Adsorption
der gesamten eingesetzten Menge an PVAm auf KGH.
Tab. 20: Ergebnisse der Elementaranalyse von mit funktionalisierten PVAm modifizierten Kieselgelen, berechnete Kohlenstoff-Gehalte der chromophoren PVAm-Einheiten C* mit Bezug zum theoretischen Kohlenstoffgehalt Cber. bei vollständigem Umsatz des Fluoraromaten und Adsorption der gesamten eingesetzten Menge an PVAm auf KGH (eingesetztes Molverhältnis von Kieselgel zu PVAm zu Fluoraromat mit 10 : 1 : 0.3).
Kieselgel-Probe Fluoraromat
C
[%]
H
[%]
N
[%]
chromophore
Einheit
C* [%]
Cber.
[%]
KGH/PVAm - 2.62 1.19 1.14 - 3.74
KGH-01 4-FNB 1.99 1.14 0.53 64.85 6.71
KGH-02 2-FNB 2.17 1.12 0.68 64.85 6.71
KGH-03 4-FNA 2.58 1.16 0.87 53.63 6.66
KGH-04 4-DMFNA 2.51 1.21 0.78 57.96 7.62
KGH-05 FDNB 5.12 1.35 2.22 45.94 6.58
KGH-06 DFDNB 5.43 1.44 2.12 48.00 6.58
KGH-07 FNAzoB 4.14 1.22 1.42 62.68 9.45
KGH-08 N-DNP-FNA 1.39 1.09 0.50 48.70 9.15
KGH-09 FNAzoB-2 2.16 1.11 0.81 56.20 11.74
KGH-10 FDNstilben 3.25 1.31 0.62 61.73 10.13

3 Ergebnisse und Diskussion
143
Die Grundeinheit im unfunktionalisierten PVAm weist einen Kohlenstoffgehalt von
55.78 % und einen Stickstoffgehalt von 32.52 % auf. Bei vollständiger Adsorption des
eingesetzten PVAm´s auf KGH (0.3589 g Polymer auf 5 g KGH) würde für
KGH/PVAm ein C-Gehalt von 3.74 % resultieren. Das Verhältnis von gemessenem
Kohlenstoffgehalt von KGH/PVAm mit 2.62 % zu berechnetem Kohlenstoffgehalt
ergibt, dass etwa 70 % von eingesetztem PVAm auf der KGH-Oberfläche adsorbiert
bleiben. Im Mittel befinden sich bei KGH/PVAm daher 0.05 g Polymer auf 1 g KGH.
Ist der berechnete C-Gehalt der chromophoren PVAm-Einheit höher gegenüber der
unfunktionalisierten PVAm-Einheit, so führt die Funktionalisierung von PVAm bei
angenommen gleicher adsorbierter Menge an Polymer zu einem höheren C-Gehalt
des Hybridmaterials im Vergleich zu KGH/PVAm .
Daher wäre für KGH-01, -02, -04, -07 und -10 ein höherer C-Gehalt der Hybrid-
materialien zu erwarten, wenn ebenfalls 0.05 g an funktionalisiertem PVAm auf 1 g
KGH adsorptiv gebunden wären. Die Kieselgele KGH-01 und KGH-02 besitzen
jedoch einen deutlich geringeren C-Gehalt als KGH/PVAm . Es scheint, dass bei
KGH-01 (Substitutionsgrad s = 7.0 mol%) und KGH-02 (s = 34.5 mol%) weniger
Polymer auf der Kieselgeloberfläche im Vergleich zu KGH/PVAm gebunden ist.
Da die Anzahl substituierter PVAm-Einheiten bei KGH-04 mit s = 3.3 mol% gering
und der berechnete C-Gehalt der chromophoren Einheit mit 57.96 % nur etwas höher
als der von PVAm ist, spricht der etwas niedrigere C-Gehalt des Hybridmaterials für
ähnliche oder etwas geringere Polymerbeladung im Vergleich zu KGH/PVAm .
Die chromophoren Einheiten von funktionalisiertem PVAm bei KGH-07 und KGH-10
(s = 17.6 mol%) haben einen miteinander vergleichbaren, höheren C-Gehalt als die
PVAm-Einheit, wodurch ein deutlich höherer C-Gehalt des Hybridmaterials mit
4.14 % bzw. 3.25 % resultiert. Da der Substitutionsgrad von dem auf KGH-07
adsorbierten Polymeren nicht bestimmt werden konnte, sind keine Rückschlüsse auf
die Schichtdicke im Vergleich zu KGH/PVAm möglich. Es scheint jedoch bei KGH-07
mehr an Fluoraromat als bei KGH-10 umgesetzt worden zu sein, da dass Verhältnis
vom gemessenen C-Gehalt zum berechneten C-Gehalt Cber. (bei vollständigem
Umsatz und Adsorption) bei KGH-07 mit ca. 49 % höher ist als bei KGH-10 mit 32 %.
Allerdings könnte auch die Polymerbeladung bei KGH-07 größer sein als bei
KGH-10, worauf vor allem die [Si]:[C]-Atomverhältnisse aus den XPS-
Untersuchungen (vgl. Tab. 19) von KGH-07 mit [Si]:[C] = 0.181 und von KGH-10 mit
[Si]:[C] = 0.422 hindeuten.

3 Ergebnisse und Diskussion
144
Die berechneten C-Gehalte der chromophoren Einheiten von funktionalisiertem
PVAm auf KGH-05, -06 und -08 sind niedriger als der berechnete C-Gehalt der
PVAm-Einheit. Bei gleicher adsorbierter Menge an funktionalisiertem Polymer (0.05 g
Polymer auf 1 g KGH) müsste ein niedrigerer C-Gehalt im Vergleich zu KGH/PVAm
resultieren.
Der mehr als doppelt so große gemessene C-Gehalt von KGH-05 und KGH-06
deutet jedoch daraufhin, dass eine sehr viel dickere Schicht an Polymer auf der
Kieselgeloberfläche im Vergleich zu KGH/PVAm adsorptiv gebunden vorliegt. Für
diesen Befund könnten Vernetzungsreaktionen der Polymerketten untereinander
verantwortlich sein, die zu größerer Beladung der Kieselgel-Partikel und
Agglomeration führen. Auch die XPS-Untersuchungen deuteten bei KGH-05 und
KGH-06 aufgrund der geringen [Si]:[C]-Atomverhältnisse auf größere Beladung der
Kieselgelpartikel hin.
Im Gegensatz dazu ist bei KGH-08 aufgrund des sehr geringen Substitutionsgrades
von s = 2.5 mol% und des niedrigen C-Gehaltes von 1.38 % von einer wesentlich
niedrigeren Polymerbelegung im Vergleich zu KGH/PVAm auszugehen.
Bei KGH-03 (s = 13.3 mol%) scheint in etwa die gleiche Menge an Polymer wie bei
KGH/PVAm adsorbiert zu sein, da der gemessene C-Gehalt in etwa dem von
KGH/PVAm entspricht, wobei sich die C-Gehalte der chromophoren Einheit und
PVAm nicht wesentlich unterscheiden. Der berechnete C-Gehalt der chromophoren
Einheit des auf KGH-09 adsorbierten funktionalisierten PVAm´s entspricht in etwa
dem berechneten C-Gehalt von PVAm. Da der Substitutionsgrad bei KGH-09 mit nur
s = 0.7 mol% sehr gering ist, der gemessene C-Gehalt des Hybridmaterials jedoch
niedriger als bei KGH/PVAm ist, scheint weniger Polymer im Vergleich zu
KGH/PVAm auf der Oberfläche von KGH adsorptiv gebunden zu sein.
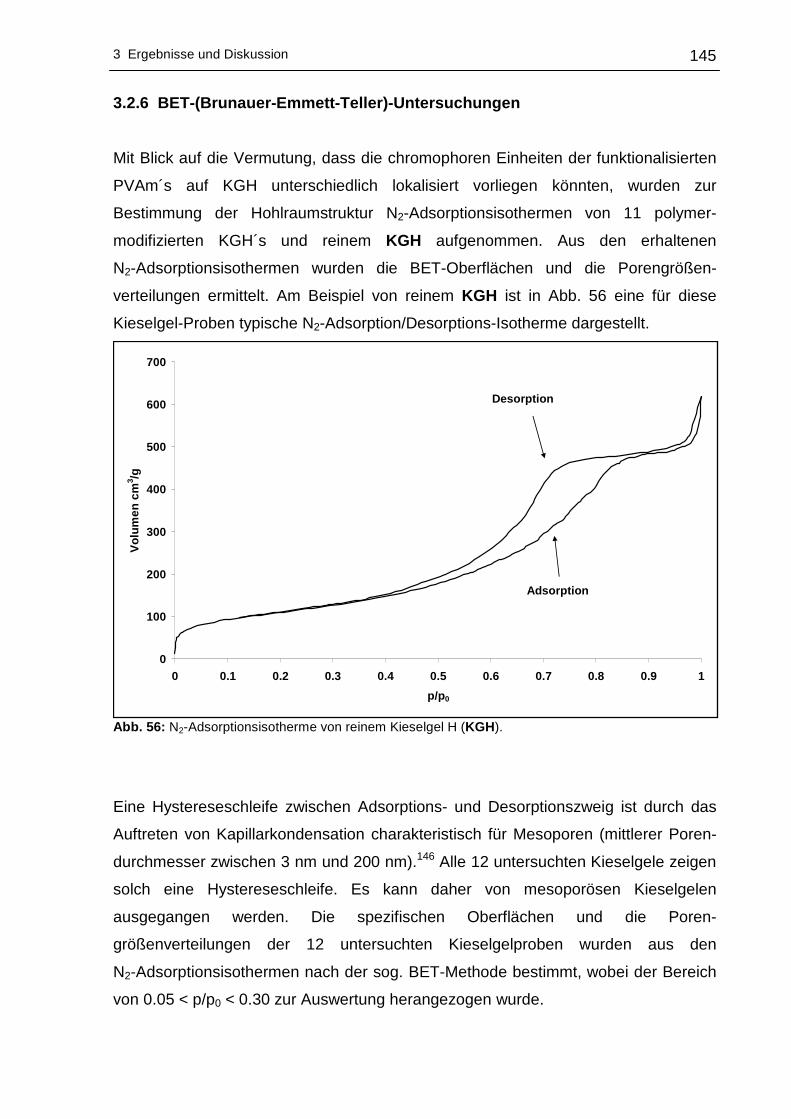
3 Ergebnisse und Diskussion
145
3.2.6 BET-(Brunauer-Emmett-Teller)-Untersuchungen
Mit Blick auf die Vermutung, dass die chromophoren Einheiten der funktionalisierten
PVAm´s auf KGH unterschiedlich lokalisiert vorliegen könnten, wurden zur
Bestimmung der Hohlraumstruktur N2-Adsorptionsisothermen von 11 polymer-
modifizierten KGH´s und reinem KGH aufgenommen. Aus den erhaltenen
N2-Adsorptionsisothermen wurden die BET-Oberflächen und die Porengrößen-
verteilungen ermittelt. Am Beispiel von reinem KGH ist in Abb. 56 eine für diese
Kieselgel-Proben typische N2-Adsorption/Desorptions-Isotherme dargestellt.
Abb. 56: N2-Adsorptionsisotherme von reinem Kieselgel H (KGH).
Eine Hystereseschleife zwischen Adsorptions- und Desorptionszweig ist durch das
Auftreten von Kapillarkondensation charakteristisch für Mesoporen (mittlerer Poren-
durchmesser zwischen 3 nm und 200 nm).146 Alle 12 untersuchten Kieselgele zeigen
solch eine Hystereseschleife. Es kann daher von mesoporösen Kieselgelen
ausgegangen werden. Die spezifischen Oberflächen und die Poren-
größenverteilungen der 12 untersuchten Kieselgelproben wurden aus den
N2-Adsorptionsisothermen nach der sog. BET-Methode bestimmt, wobei der Bereich
von 0.05 < p/p0 < 0.30 zur Auswertung herangezogen wurde.
0
100
200
300
400
500
600
700
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
p/p0
Vol
umen
cm
3 /g
Adsorption
Desorption

3 Ergebnisse und Diskussion
146
Für reines KGH wurde eine spezifische BET-Oberfläche von 395 m2·g-1 und eine
enge Porenradienverteilung ermittelt, die in Abb. 57 links (grau unterlegt) gezeigt ist.
Abb. 57: Porenradienverteilungen von links: reinem KGH (grau) und mit unfunktionalisiertem PVAm modifiziertem KGH (KGH/PVAm )und rechts: von KGH-05 (grau) und KGH-01.
Abb. 57 zeigt einerseits, dass eine Polymermodifizierung von reinem KGH zur
Verbreiterung der Porengrößenverteilung führt und das andererseits das KGH/PVAm
eine Porenradienverteilung mit einem Hauptanteil bei 5 – 10 nm im Gegensatz zu
reinem KGH mit 2 – 5 nm aufweist. Das mittels Sangers Reagenz funktionalisierte
PVAm auf KGH (KGH-05) zeigt wiederum eine Porenradienverteilung mit einem
Hauptanteil von 2 – 5 nm, das mittels 4-FNB funktionalisierte PVAm auf KGH
(KGH-01) einen Hauptanteil bei 5 – 10 nm.
In Tabelle 21 sind die Ergebnisse der BET-Untersuchungen von zehn mit
funktionalisiertem PVAm modifizierten KGH´s, reinem KGH und mit PVAm
beladenem KGH − mit Bezug zur Polymerbeladung aus den elementaranalytischen
Untersuchungen (EA) − zusammengefasst.
1 5 10 1000
20
40
60
80
100
KGH-05
KGH-01
Vol
umen
[%]
Porenradius [nm]
1 5 10 1000
20
40
60
80
100
KGH/PVAm
KGH
Vol
umen
[%]
Porenradius [nm]
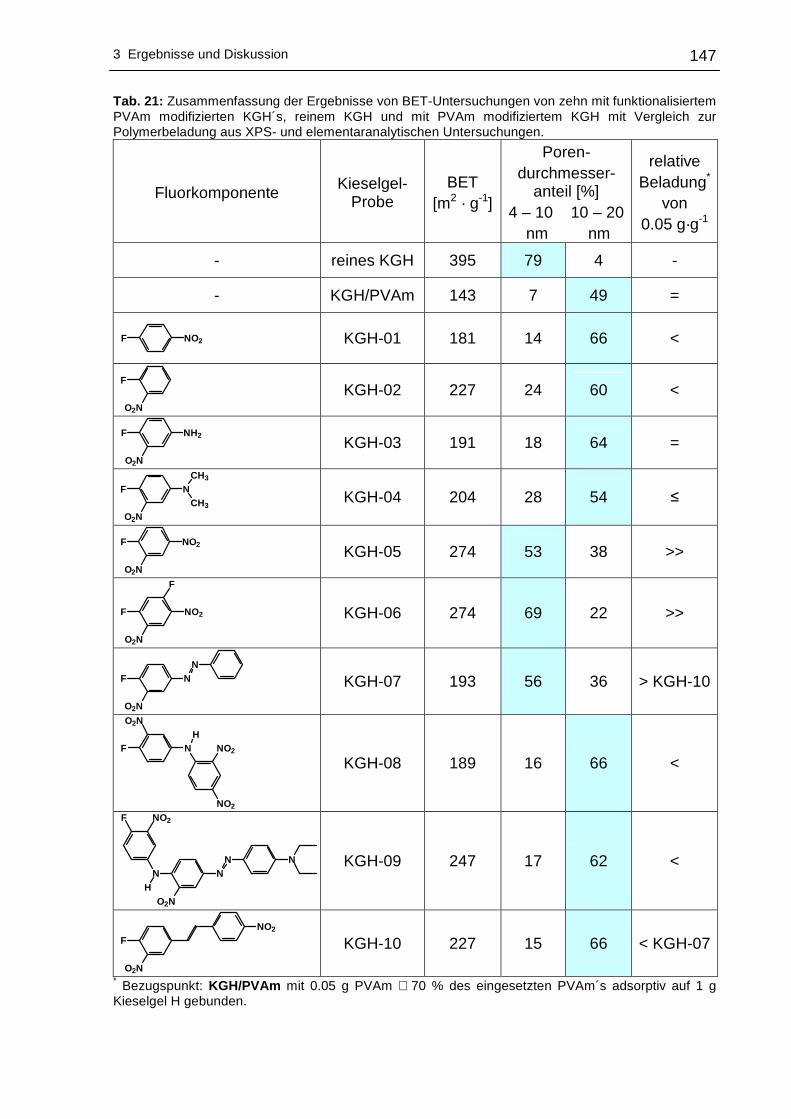
3 Ergebnisse und Diskussion
147
Tab. 21: Zusammenfassung der Ergebnisse von BET-Untersuchungen von zehn mit funktionalisiertem PVAm modifizierten KGH´s, reinem KGH und mit PVAm modifiziertem KGH mit Vergleich zur Polymerbeladung aus XPS- und elementaranalytischen Untersuchungen.
Fluorkomponente Kieselgel-
Probe BET
[m2 · g-1]
Poren- durchmesser-
anteil [%] 4 – 10 10 – 20 nm nm
relative Beladung*
von 0.05 g·g-1
- reines KGH 395 79 4 -
- KGH/PVAm 143 7 49 =
F NO2
KGH-01 181 14 66 <
F
O2N KGH-02 227 24 60 <
F NH2
O2N KGH-03 191 18 64 =
N
O2N
FCH3
CH3
KGH-04 204 28 54 ≤
F
O2N
NO2
KGH-05 274 53 38 >>
F
O2N
NO2
F
KGH-06 274 69 22 >>
F
O2N
NN
KGH-07 193 56 36 > KGH-10
NH
F NO2
NO2
O2N
KGH-08 189 16 66 <
N
O2N
NN
H
F NO2
N
KGH-09 247 17 62 <
F
O2N
NO2
KGH-10 227 15 66 < KGH-07
* Bezugspunkt: KGH/PVAm mit 0.05 g PVAm ≅ 70 % des eingesetzten PVAm´s adsorptiv auf 1 g Kieselgel H gebunden.

3 Ergebnisse und Diskussion
148
Ausgenommen der Kieselgele KGH-05, -06 und -07 liegt der Hauptanteil der
Porendurchmesser (50 – 60 %) wie der von KGH/PVAm im Bereich von 10 – 20 nm.
Die Kieselgele KGH-05, -06 und -07 hingegen zeigen einen Hauptanteil der
Porendurchmesser wie die von reinem KGH mit einer Größe von 4 – 10 nm, was ein
Indiz dafür ist, dass der Hauptanteil an Poren vom PVAm wahrscheinlich nicht
aufgefüllt, sondern erhalten geblieben ist.
Die spezifische Oberfläche der Kieselgelpartikel sinkt durch die Beladung mit
funktionalisiertem PVAm drastisch ab. Die Modifizierung von KGH mit reinem PVAm
zu KGH/PVAm bewirkt ein Absinken der spezifischen BET-Oberfläche zu 143 m2·g-1
im Vergleich zu reinem KGH mit 395 m2·g-1. Eine visuell beobachtbare schnelle
Gelbildung zwischen Polymeren und Kieselgel-Partikeln zeigte sich bei den
Synthesen zu KGH-02, -05, -06, und -07. Das spiegelt sich außer bei KGH-07 in den
größeren spezifischen Oberflächen bis 274 m2·g-1 im Vergleich KGH/PVAm zu wider,
da die Poren möglicherweise nicht aufgefüllt sind.
Weniger reaktive Fluoraromaten könnten hauptsächlich in den Poren mit PVAm
reagiert haben, was dann zu einer kleineren spezifischen Oberfläche im Vergleich zu
den mit besser aktivierten Fluoraromaten synthetisierten KGH´s führen würde. Das
zeigt sich bei KGH-01, -03, -04 und -08 mit einer spezifischen Oberfläche von
189 bis 204 m2·g-1.
KGH-07 hat trotz eines Hauptanteils der Porengröße wie KGH (4 – 10 nm) eine
relativ kleine spezifische Oberfläche von 193 m2·g-1. KGH-09 wiederum besitzt mit
247 m2·g-1 eine deutlich größere spezifische Oberfläche als KGH/PVAm , wo
aufgrund des geringen Substitutionsgrades (s = 0.7 mol%) eher mit einer zu
KGH/PVAm vergleichbar großen Oberfläche gerechnet wurde.
Da KGH-10 eine ähnliche Porengrößenverteilung (> 60% mit 10 – 20 nm) aufweist
wie KGH-02 und beide KGH´s die gleiche spezifische Oberfläche von 227 m2·g-1
besitzen, könnte sich auch bei KGH-10 weniger Polymer auf der Oberfläche befinden
im Vergleich zu KGH/PVAm (0.05 g Polymer auf 1g KGH).
Auch die BET-Untersuchungen weisen neben den XPS-Untersuchungen darauf hin,
dass die weniger reaktiven Fluoraromaten hauptsächlich in den Poren, die
reaktiveren Fluoraromaten jedoch schnell – schon vor Erreichen der Poren in der
Peripherie der Partikel – mit PVAm reagiert haben könnten.

3 Ergebnisse und Diskussion
149
3.2.7 Elektrokinetische Untersuchungen (Zetapotent ial)
Durch elektrokinetische Messungen können Ladungsverteilungen an Grenzflächen
untersucht werden, die vorliegen, wenn sich feste Partikel im thermodynamischen
Gleichgewicht mit einer Flüssigkeit befinden. Die Verwendung von Wasser als
flüssige Phase bietet die Möglichkeit den die Partikel umgebenden pH-Wert zu
variieren und damit die Adsorption von H⊕ -Ionen und OHӨ-Ionen auf der Oberfläche
zu kontrollieren. Um bei Änderung des pH-Wertes die Ionenstärke nahezu konstant
zu halten, wird ein Inertsalz wie z. B. KCl zugesetzt.
Befindet sich eine Festkörperoberfläche im chemischen Gleichgewicht mit einer
Elektrolytlösung, so liegt bei freier und unabhängiger Beweglichkeit der
Ladungsträger (Ionen) an der Phasengrenze eine andere Ladungsverteilung als im
Inneren der jeweiligen Phase vor. Der Überschuss einer Ionensorte und die relative
Verarmung deren Gegenionen an der Phasengrenze wird als elektrochemische
Doppelschicht bezeichnet. Die Ursachen für die Anreicherung einer Ionensorte an
der Phasengrenze können in der Dissoziation saurer Molekülgruppen oder der
bevorzugten Adsorption von Anionen oder Kationen liegen. Aufgrund der
unterschiedlichen Anzahl von Ladungen an der Phasengrenze und den
Volumenphasen bildet sich ein elektrisches Potential aus.147
Die modellhafte Beschreibung der elektrochemischen Doppelschicht eines
Festkörpers, der mit einer verdünnten Elektrolytlösung im Gleichgewicht steht,
basiert auf Arbeiten von Gouy, Chapman, Stern und Graham. Dieses GCSG-Modell
unterteilt die elektrochemische Doppelschicht in eine starre/immobile und in eine
diffuse/mobile Schicht. Wird ein äußeres Kraftfeld so angelegt, dass es zur
Relativbewegung zwischen fester und flüssiger Phase kommt, verbleibt der starre
Teil der elektrochemischen Doppelschicht an der Festkörperoberfläche, während der
mobile Teil sich mit der Volumenphase der Flüssigkeit bewegt. Aufgrund der
Relativbewegung muss eine Ebene existieren, in der ein Abscheren des mobilen
Teils der elektrochemischen Doppelschicht erfolgt. Das Potential in dieser
Scherebene wird als elektrokinetisches Potential bzw. Zetapotential ζ bezeichnet.
Das Zetapotential ist daher ein Indikator für den Aufbau der elektrochemischen
Doppelschicht. Mit Änderung der Zusammensetzung der flüssigen Phase (z. B. die
Änderung des pH-Wertes) ändert sich auch die elektrochemische Doppelschicht, die
sich in einer Änderung des Zetapotentials zeigt.147
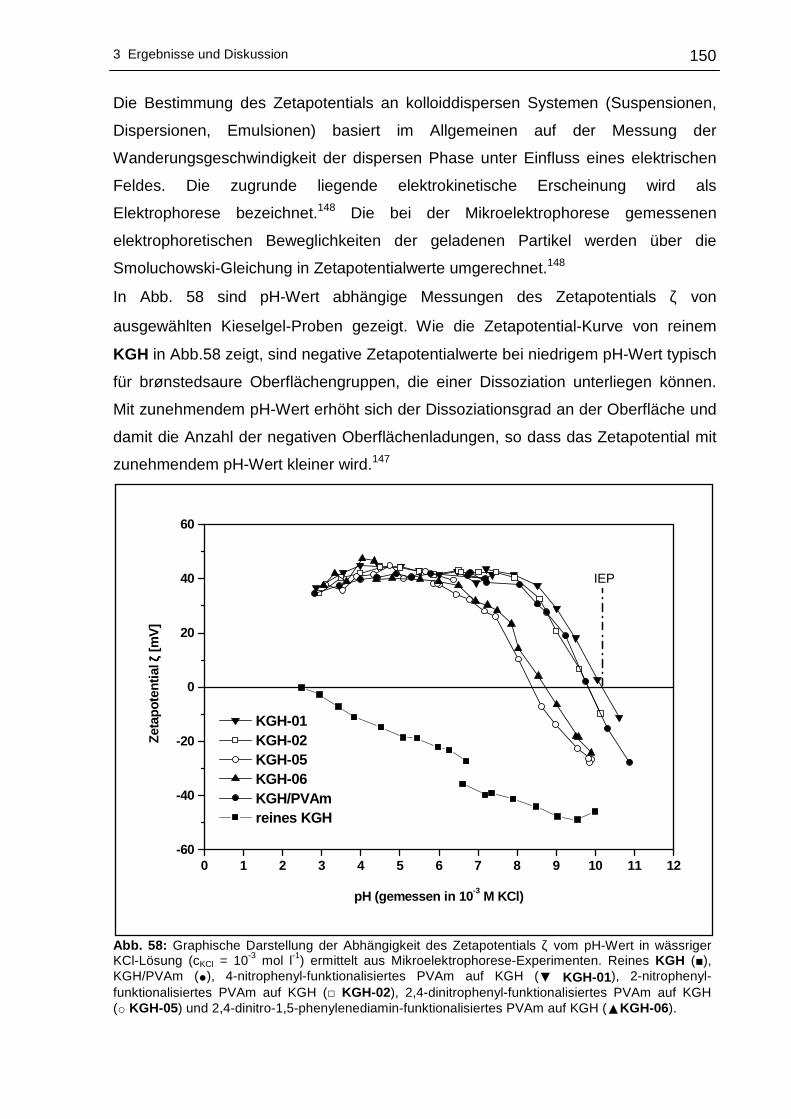
3 Ergebnisse und Diskussion
150
Die Bestimmung des Zetapotentials an kolloiddispersen Systemen (Suspensionen,
Dispersionen, Emulsionen) basiert im Allgemeinen auf der Messung der
Wanderungsgeschwindigkeit der dispersen Phase unter Einfluss eines elektrischen
Feldes. Die zugrunde liegende elektrokinetische Erscheinung wird als
Elektrophorese bezeichnet.148 Die bei der Mikroelektrophorese gemessenen
elektrophoretischen Beweglichkeiten der geladenen Partikel werden über die
Smoluchowski-Gleichung in Zetapotentialwerte umgerechnet.148
In Abb. 58 sind pH-Wert abhängige Messungen des Zetapotentials ζ von
ausgewählten Kieselgel-Proben gezeigt. Wie die Zetapotential-Kurve von reinem
KGH in Abb.58 zeigt, sind negative Zetapotentialwerte bei niedrigem pH-Wert typisch
für brønstedsaure Oberflächengruppen, die einer Dissoziation unterliegen können.
Mit zunehmendem pH-Wert erhöht sich der Dissoziationsgrad an der Oberfläche und
damit die Anzahl der negativen Oberflächenladungen, so dass das Zetapotential mit
zunehmendem pH-Wert kleiner wird.147
Abb. 58: Graphische Darstellung der Abhängigkeit des Zetapotentials ζ vom pH-Wert in wässriger KCl-Lösung (cKCl = 10-3 mol l-1) ermittelt aus Mikroelektrophorese-Experimenten. Reines KGH (■), KGH/PVAm (●), 4-nitrophenyl-funktionalisiertes PVAm auf KGH (▼ KGH-01), 2-nitrophenyl- funktionalisiertes PVAm auf KGH (□ KGH-02), 2,4-dinitrophenyl-funktionalisiertes PVAm auf KGH (○ KGH-05) und 2,4-dinitro-1,5-phenylenediamin-funktionalisiertes PVAm auf KGH (▲KGH-06).
0 1 2 3 4 5 6 7 8 9 10 11 12-60
-40
-20
0
20
40
60
KGH-01 KGH-02 KGH-05 KGH-06 KGH/PVAm reines KGH
IEP
Zeta
pote
ntia
l ζζ ζζ
[mV
]
pH (gemessen in 10 -3 M KCl)
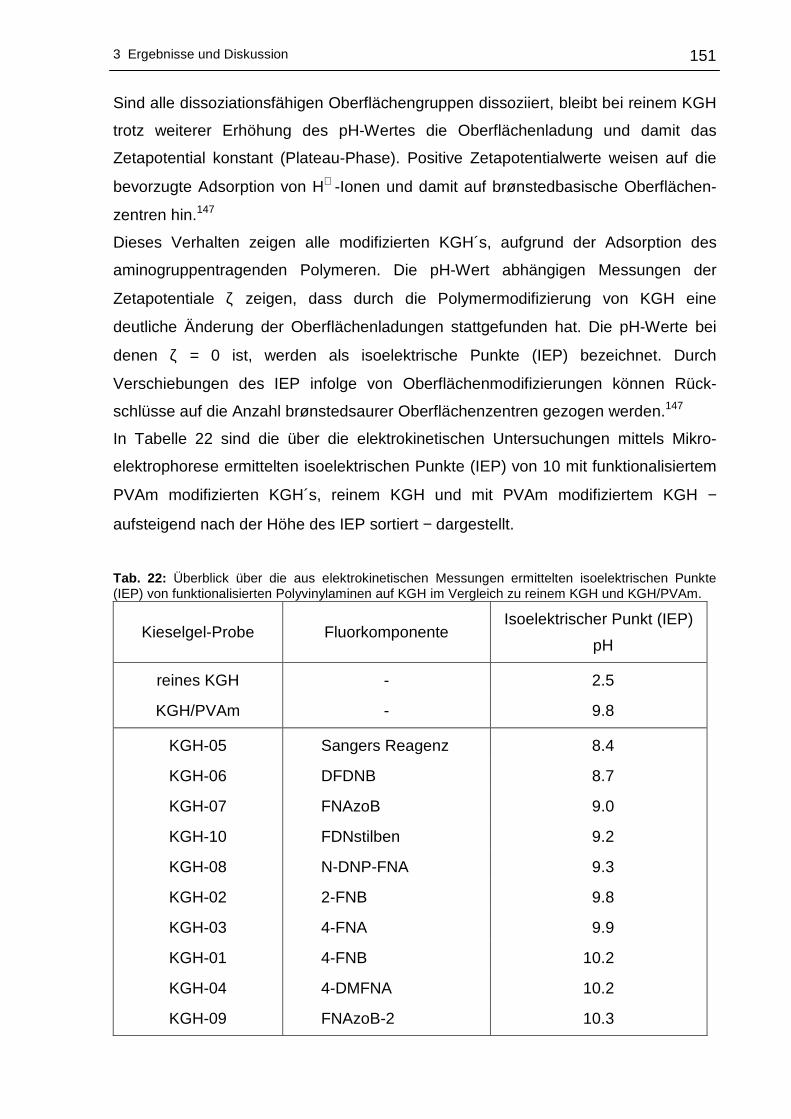
3 Ergebnisse und Diskussion
151
Sind alle dissoziationsfähigen Oberflächengruppen dissoziiert, bleibt bei reinem KGH
trotz weiterer Erhöhung des pH-Wertes die Oberflächenladung und damit das
Zetapotential konstant (Plateau-Phase). Positive Zetapotentialwerte weisen auf die
bevorzugte Adsorption von H⊕ -Ionen und damit auf brønstedbasische Oberflächen-
zentren hin.147
Dieses Verhalten zeigen alle modifizierten KGH´s, aufgrund der Adsorption des
aminogruppentragenden Polymeren. Die pH-Wert abhängigen Messungen der
Zetapotentiale ζ zeigen, dass durch die Polymermodifizierung von KGH eine
deutliche Änderung der Oberflächenladungen stattgefunden hat. Die pH-Werte bei
denen ζ = 0 ist, werden als isoelektrische Punkte (IEP) bezeichnet. Durch
Verschiebungen des IEP infolge von Oberflächenmodifizierungen können Rück-
schlüsse auf die Anzahl brønstedsaurer Oberflächenzentren gezogen werden.147
In Tabelle 22 sind die über die elektrokinetischen Untersuchungen mittels Mikro-
elektrophorese ermittelten isoelektrischen Punkte (IEP) von 10 mit funktionalisiertem
PVAm modifizierten KGH´s, reinem KGH und mit PVAm modifiziertem KGH −
aufsteigend nach der Höhe des IEP sortiert − dargestellt.
Tab. 22: Überblick über die aus elektrokinetischen Messungen ermittelten isoelektrischen Punkte (IEP) von funktionalisierten Polyvinylaminen auf KGH im Vergleich zu reinem KGH und KGH/PVAm.
Kieselgel-Probe Fluorkomponente Isoelektrischer Punkt (IEP)
pH
reines KGH - 2.5
KGH/PVAm - 9.8
KGH-05 Sangers Reagenz 8.4
KGH-06 DFDNB 8.7
KGH-07 FNAzoB 9.0
KGH-10 FDNstilben 9.2
KGH-08 N-DNP-FNA 9.3
KGH-02 2-FNB 9.8
KGH-03 4-FNA 9.9
KGH-01 4-FNB 10.2
KGH-04 4-DMFNA 10.2
KGH-09 FNAzoB-2 10.3

3 Ergebnisse und Diskussion
152
Die Anzahl brønstedsaurer Oberflächenzentren von reinem KGH nimmt durch die
Polymermodifizierung drastisch ab. Durch die Adsorption an der Kieselgeloberfläche
neutralisiert das aminogruppentragende Polymer zum einen die sauren Oberflächen-
zentren von reinem KGH und liefert zusätzlich brønstedbasische Aminogruppen. Da
die Verschiebung des IEP´s von pH = 2.5 (reines KGH) zu pH > 9 (modifizierte
KGH´s) sehr deutlich ist, scheint die Oberflächenbedeckung der polymermodifizierten
KGH´s recht dicht zu sein. Wäre sehr wenig Polymer adsorbiert, so dass noch viele
brønstedsaure Oberflächenzentren von Kieselgel H neben wenigen brønsted-
basischen Oberflächenzentren von PVAm vorliegen, würde sich das in einer
geringeren Verschiebung des IEP bis zu pH = 7 äußern.
Die Verschiebung des IEP zu pH = 9.8 KGH/PVAm soll für vergleichende
Betrachtungen als Richtwert dienen. So zeigt sich, dass in Abhängigkeit der
Reaktivität des eingesetzten Fluoraromaten unterschiedliche Werte des IEP´s zu
finden sind. Die Umsetzung von PVAm mit weniger aktivierten Fluoraromaten zu den
Kieselgelen KGH-01, -03, -04 und -09 führen zu einem IEP bei pH ≈ 10, was für
etwas bessere Bedeckung der Kieselgeloberfläche im Vergleich zu KGH/PVAm
spricht. Wohingegen die durch die Umsetzung von PVAm mit besser aktivierten
Fluoraromaten modifizierten Kieselgele KGH-05, -06, -07 und -10 kleinere IEP´s als
KGH/PVAm zeigen, was auf weniger Abschirmung/Bedeckung der brønstedsauren
Oberflächenzentren von Kieselgel hindeutet.
Die Unterschiede der erhaltenen IEP´s der polymermodifizierten KGH´s sind
wiederum ein Hinweis darauf, dass die für die nucleophile aromatische Substitution
stark aktivierten Fluoraromaten mit PVAm sehr schnell schon vor Erreichen der
Adsorption in den Poren reagieren, wobei diese teilweise vom Polymeren
unausgefüllt bleiben. Freie – von den Aminogruppen des PVAm´s nicht neutralisierte
Silanolgruppen des KGH´s – resultieren in einer weniger starken Verschiebung des
IEP´s. Weniger reaktive Fluoraromaten reagieren in Oberflächennähe (in den Poren)
und die nahezu komplette Oberflächenbeschichtung bewirkt eine Verschiebung des
IEP´s zu größeren pH-Werten, da die –Si–OH-Gruppen von den –NH2-Gruppen des
PVAm´s auch in den Poren neutralisiert werden.
In Tabelle 23 sind zur besseren Übersichtlichkeit ausgewählte Ergebnisse der BET-,
XPS- und elektrokinetischen Untersuchungen von 10 mit funktionalisiertem PVAm
beladenen KGH´s, von reinem KGH und von mit PVAm modifiziertem KGH
zusammengefasst.
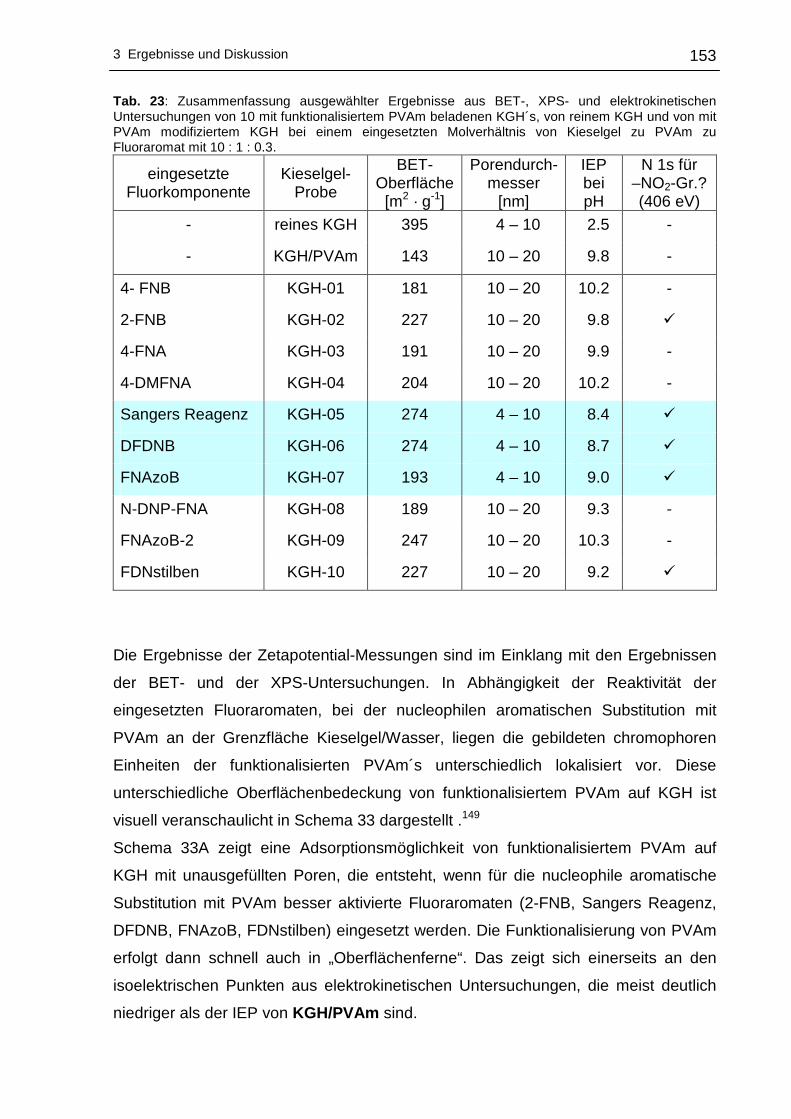
3 Ergebnisse und Diskussion
153
Tab. 23: Zusammenfassung ausgewählter Ergebnisse aus BET-, XPS- und elektrokinetischen Untersuchungen von 10 mit funktionalisiertem PVAm beladenen KGH´s, von reinem KGH und von mit PVAm modifiziertem KGH bei einem eingesetzten Molverhältnis von Kieselgel zu PVAm zu Fluoraromat mit 10 : 1 : 0.3.
eingesetzte Fluorkomponente
Kieselgel-Probe
BET- Oberfläche
[m2 · g-1]
Porendurch-messer
[nm]
IEP bei pH
N 1s für –NO2-Gr.? (406 eV)
- reines KGH 395 4 – 10 2.5 -
- KGH/PVAm 143 10 – 20 9.8 -
4- FNB KGH-01 181 10 – 20 10.2 -
2-FNB KGH-02 227 10 – 20 9.8 �
4-FNA KGH-03 191 10 – 20 9.9 -
4-DMFNA KGH-04 204 10 – 20 10.2 -
Sangers Reagenz KGH-05 274 4 – 10 8.4 �
DFDNB KGH-06 274 4 – 10 8.7 �
FNAzoB KGH-07 193 4 – 10 9.0 �
N-DNP-FNA KGH-08 189 10 – 20 9.3 -
FNAzoB-2 KGH-09 247 10 – 20 10.3 -
FDNstilben KGH-10 227 10 – 20 9.2 �
Die Ergebnisse der Zetapotential-Messungen sind im Einklang mit den Ergebnissen
der BET- und der XPS-Untersuchungen. In Abhängigkeit der Reaktivität der
eingesetzten Fluoraromaten, bei der nucleophilen aromatischen Substitution mit
PVAm an der Grenzfläche Kieselgel/Wasser, liegen die gebildeten chromophoren
Einheiten der funktionalisierten PVAm´s unterschiedlich lokalisiert vor. Diese
unterschiedliche Oberflächenbedeckung von funktionalisiertem PVAm auf KGH ist
visuell veranschaulicht in Schema 33 dargestellt .149
Schema 33A zeigt eine Adsorptionsmöglichkeit von funktionalisiertem PVAm auf
KGH mit unausgefüllten Poren, die entsteht, wenn für die nucleophile aromatische
Substitution mit PVAm besser aktivierte Fluoraromaten (2-FNB, Sangers Reagenz,
DFDNB, FNAzoB, FDNstilben) eingesetzt werden. Die Funktionalisierung von PVAm
erfolgt dann schnell auch in „Oberflächenferne“. Das zeigt sich einerseits an den
isoelektrischen Punkten aus elektrokinetischen Untersuchungen, die meist deutlich
niedriger als der IEP von KGH/PVAm sind.

3 Ergebnisse und Diskussion
154
Andererseits sind die im Vergleich zu KGH/PVAm meist deutlich höheren
spezifischen BET-Oberflächen und die im Vergleich zu reinem KGH gleichen
Porengrößen ein Indiz für unausgefüllte Poren (s. Schema 33A).
Schema 33: Schematische Darstellung der Adsorption von funktionalisiertem PVAm auf KGH durch Substitution von A: höher reaktiven Fluoraromaten und B: weniger reaktiven Fluoraromaten mit PVAm.
Im Falle von Sangers Reagenz und DFDNB können zudem auch Vernetzungs-
reaktionen von funktionalisierten PVAm-Ketten untereinander stattfinden und die
Poren von Kieselgel H werden nicht vollständig ausgefüllt. Da bei den XPS-Unter-
suchungen nur in den N 1s Spektren von KGH-02, -05, -06, -07 und -10 ein Signal
um 406 eV (für an Phenylringe gebundene –NO2-Gruppen) gefunden wurde, deutet
alles darauf hin, dass besser aktivierte Fluoraromaten mit PVAm nach dem in
Schema 33A dargestellten Adsorptionsmodell reagieren könnten.149
Bei den Kieselgelen KGH-01, -03, -04, -08 und -09 konnte bei den XPS-
Untersuchungen kein N 1s Signal bei 406 eV detektiert werden. Zum einen könnte
das an den geringen Substitutionsgraden des funktionalisierten PVAm´s auf KGH
(bei KGH-03, -08 und -09) und zum anderen an der begrenzten Informationstiefe für
N 1s Photoelektronen liegen, die für eine Detektierung der sich in der Tiefe/Poren
befindlichen –NO2-Gruppen möglicherweise nicht ausreicht. Daher könnten weniger
aktivierte Fluoraromaten hauptsächlich, wie in Schema 33B gezeigt, in den Poren
des Kieselgels mit PVAm reagiert haben. Dafür sprechen neben den XPS-
Untersuchungen auch die aus den elektrokinetischen Messungen ermittelten IEP´s,
die meist größer sind (dichtere Oberflächenbedeckung von KGH) als der IEP von
KGH/PVAm und die meist kleineren spezifischen BET-Oberflächen.149
NH2NH2
NH2
NH2
NH2
NH2
NH
NH
A
NH2NH2
NH2
NH2
NH2
NH2
NH
NH
NH2NH2
NH2
NH2
NH2
NH2
NH
NH
A B
NH2
NH2
NH2
NH2
NH2
H2N
NH2
BB
NH2
NH2
NH2
NH2
NH2
H2N
NH2
NH2
NH2
NH2
NH2
NH2
H2N
NH2

4 Zusammenfassung und Ausblick
155
4 Zusammenfassung und Ausblick
Der zentrale Gegenstand dieser Arbeit war die eingehende Untersuchung der
nucleophilen aromatischen Substitution von aktivierten Fluoraromaten mit Polyvinyl-
aminen in Wasser, um neue chromophore wasserlösliche Polyelektrolyte zu
synthetisieren. Es galt aufbauend auf eigenen, orientierenden Voruntersuchungen
das synthetische Potential weiter auszubauen und auszuloten.9,10 Als aminogruppen-
tragende Polymere wurden Polyvinylamine mit zwei unterschiedlichen
Molekulargewichten (Mw = 15,000 g·mol-1 bzw. 400,000 g·mol-1) von der BASF AG
sowie ein Poly-N-Methylvinylamin (PNMVAm, 196,000 g·mol-1) eingesetzt. Die
nucleophile aromatische Substitution ausgewählter Fluoraromaten mit Polyvinyl-
aminen in Wasser gelingt über die Komplexierung der Fluorkomponenten mittels
Cyclodextrin zur Löslichkeitsvermittlung. Für die Funktionalisierungsreaktionen
wurden die verwendeten Fluoraromaten mittels statistisch methyliertem
2,6-O-Dimethyl-β-cyclodextrin (CAVASOL®, β-DMCD) von Wacker im äquimolaren
Verhältnis komplexiert und als 1:1-Komplexe für die Reaktionen mit Polyvinylaminen
eingesetzt.
Es erwies sich als notwendig, dass die verwendeten Fluoraromaten für die
nucleophile aromatische Substitution durch Substituenten mit –M-Effekt (wie –NO2-
Gruppen) in Konjugation zum Fluorsubstituenten aktiviert sein müssen, da die
Reaktionstemperatur der siedenden wässrigen PVAm-Lösung für weniger aktivierte
Fluoraromaten nicht ausreicht.9,10 Sehr gut zur Funktionalisierung von PVAm in
Wasser eigneten sich folgende Fluoraromaten: 2-Fluornitrobenzen (2-FNB),
2,4-Dinitrofluorbenzen (Sangers Reagenz), 1,5-Difluor-2,4-dinitrobenzen (DFDNB),
4-Fluor-3-nitroazobenzen (FNAzoB) und 4-Fluor-3,4´-dinitrostilben (FDNstilben).
Bei der Funktionalisierung von Polyvinylaminen mit weniger aktivierten Fluor-
aromaten wie N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA), N-(2´,4´-Dinitrophenyl)-
4-fluor-3-nitroanilin (N-DNP-FNA) und N-(2´-Nitrophenyl-4´-(p-N,N-diethyl)-
azobenzen)-4-fluor-3-nitroanilin (FNAzoB-2) konnten nur geringe Substitutionsgrade
an chromophoren Einheiten bei gleichen Reaktionsbedingungen erhalten werden.
Eine Erhöhung der eingesetzten Menge an Fluoraromat führte bei gleichen
Reaktionsbedingungen zur Erhöhung des Substitutionsgrades.

4 Zusammenfassung und Ausblick
156
Der Substitutionsgrad von funktionalisiertem Polyvinylamin wurde insbesondere mit
Hilfe der UV/vis-Spektroskopie durch Vergleich der Intensität der UV/vis-Absorption
von funktionalisiertem Polyvinylamin mit der Intensität der UV/vis-Absorption einer
niedermolekularen Modellverbindung in Methanol bestimmt. Die Modellverbindungen
wurden nach strukturellen Gesichtspunkten der chromophoren Einheit des jeweils
funktionalisierten Polyvinylamins entsprechend synthetisiert. Ließ es die Löslichkeit
der funktionalisierten Polyvinylamine in D2O zu, wurde auch die 1H-NMR-
Spektroskopie zur Bestimmung der jeweiligen Substitutionsgrade herangezogen,
indem die Intensitäten der 1H-NMR-Signale der aromatischen Protonen mit den 1H-NMR-Signalen der Protonen der Polymerhauptkette ins Verhältnis gesetzt
wurden.
Die Polymere, die durch die nucleophile aromatische Substitution von stärker
aktivierten Fluoraromaten wie 2,4-Dinitrofluorbenzen (Sangers Reagenz),
1,5-Difluor-2,4-dinitrobenzen (DFDNB) und 4-Fluor-3-nitroazobenzen (FNAzoB) mit
PVAm erhalten wurden, waren sehr schlecht bzw. unlöslich in Wasser und/oder
Methanol, so dass eine Substitutionsgradbestimmung nicht möglich war.
Um nicht nur aus den erhaltenen Substitutionsgraden der jeweils funktionalisierten
Polyvinylamine Rückschlüsse auf die Reaktivität der jeweils eingesetzten
Fluoraromaten zu ziehen, wurden kinetische Untersuchungen der Funktionalisierung
von PVAm mittels aktivierter Fluoraromaten in Wasser mit Hilfe der UV/vis-
Spektroskopie durchgeführt. Dazu wurde die nucleophile aromatische Substitution
von ausgewählten Fluoraromaten mit PVAm durch Variation der Temperatur, des
pH-Wertes und des Molekulargewichts in Abhängigkeit von der Reaktionszeit UV/vis-
spektroskopisch verfolgt. Die Geschwindigkeit der Funktionalisierungsreaktion stieg
erwartungsgemäß mit der Temperatur an.
Die Reaktionsgeschwindigkeit bei der nucleophilen Substitution von 2-Fluornitro-
benzen (2-FNB) mit PVAm in Wasser ist etwa dreimal größer als die der Umsetzung
von 4-Fluornitrobenzen (4-FNB). Das erklärt den etwa zwei bis dreimal höheren
Substitutionsgrad von 2-nitrophenyl-funktionalisiertem PVAm im Vergleich zu
4-nitrophenyl-funktionalisiertem PVAm bei gleichen Reaktionsbedingungen. Die
Aktivierungsenergien EA für die nucleophile aromatische Substitution von 4-FNB bzw.
2-FNB mit PVAm in Wasser über Löslichkeitsvermittlung mittels β-DMCD sind jedoch
mit 67.6 kJ · mol-1 bzw. 67.5 kJ · mol-1 nahezu gleich groß.

4 Zusammenfassung und Ausblick
157
Der Reaktivitätsunterschied rührt von der unterschiedlichen Aktivierungsentropie ∆S‡
her, die bei der Substitution von 2-FNB mit PVAm bei gleichen Reaktions-
bedingungen um etwa 10 J·K-1·mol-1 kleiner ist im Vergleich zur Umsetzung mit
4-FNB. Der ortho-Effekt der –NO2-Gruppe bei 2-FNB führt möglicherweise zu
Nachbargruppeneffekten, welche in der Literatur oft diskutiert werden.20,91,93-95
Sangers Reagenz ist durch zwei –M-Substituenten erwartungsgemäß für eine
nucleophile aromatische Substitution mit PVAm besser aktiviert als 2-FNB und
4-FNB. Demzufolge nimmt die Aktivierungsenergie mit 60.2 kJ·mol-1 für die
nucleophile aromatische Substitution deutlich ab. Die Einführung eines weiteren
Fluorsubstituenten bei DFDNB führt zu einer weiteren Absenkung der Aktivierungs-
energie mit EA = 52.8 kJ·mol-1, was auf den –I-Effekt des zweiten Fluorsubstituenten
zurückzuführen ist. Die nucleophile aromatische Substitution von DFDNB/β-DMCD
verläuft fast doppelt so schnell wie die der Reaktion von Sangers Reagenz/β-DMCD
mit PVAm in Wasser.
Die Aminogruppe bei 4-Fluor-3-nitroanilin (4-FNA) scheint trotz des +M-Effektes auf
die Aktivierung bei der Funktionalisierungsreaktion kaum Einfluss zu haben. Die
Aktivierungsenergie ist nahezu gleich der Aktivierungsenergie der nucleophilen
aromatischen Substitution von 2-FNB mit PVAm. Die Aktivierungsenergie der
nucleophilen aromatischen Substitution von N,N-Dimethyl-4-fluor-3-nitroanilin
(4-DMFNA) mit PVAm in Wasser hingegen ist mit 76.5 kJ·mol-1 deutlich höher im
Vergleich zur Umsetzung mit 4-FNA. Der größere +M-Effekt der in 1-Stellung
dimethylierten Aminogruppe verringert die Reaktivität für die nucleophile aromatische
Substitution doch erwartungsgemäß entscheidend.
Die kinetischen Untersuchungen mit Hilfe der UV/vis-Spektroskopie zeigten
weiterhin, dass die Polymerkette selbst einen Einfluss auf die Reaktivität bei der
nucleophilen aromatischen Substitution von Fluoraromaten hat. Bei der Umsetzung
von PVAm mit 4-FNB/β-DMCD in Wasser (pH = 11) wurde im Vergleich zur Reaktion
von i-Propylamin mit 4-FNB in DMSO (−∆S‡ = 38.9 J·K-1·mol-1) eine deutlich höhere
zu überwindende Aktivierungsentropie mit −∆S‡ = 158.5 J·K-1·mol-1 gefunden.126,127
Die geringere Reaktivität von PVAm im Vergleich zu i-Propylamin könnte auf
sterische Faktoren zurückzuführen sein.

4 Zusammenfassung und Ausblick
158
Ein höherer pH-Wert der wässrigen PVAm-Lösungen ist für die Funktionalisierung
von PVAm mittels Fluoraromaten sinnvoll, um den Protonierungsgrad (Anzahl von
Ammoniumgruppen –NH3⊕) zu Gunsten von primären Aminogruppen (–NH2) gering
zu halten, was zur Beschleunigung der Funktionalisierungsreaktion führt.
Die Aktivierungsenthalpie und die Aktivierungsenergie nahmen bei der
Funktionalisierung von PVAm mit Sangers Reagenz – trotz größerer Geschwindig-
keitskonstanten – mit steigendem pH-Wert ab. Bei der Funktionalisierung von PVAm
mit Sangers Reagenz war die Aktivierungsentropie bei pH = 4 fast doppelt so groß im
Vergleich zur Funktionalisierung bei pH = 11. Das deutet darauf hin, dass der Angriff
bei der nucleophilen aromatischen Substitution von Fluoraromaten mit PVAm bei
niedrigem pH-Wert zwar leichter erfolgt, jedoch eine wesentlich höhere
Aktivierungsentropie überwunden werden muss. Offensichtlich spielen sterische
Effekte im Sauren eine größere Rolle, so dass dort die Zugänglichkeit hauptsächlich
von der molekularen Struktur der zu substituierenden PVAm-Einheit und weniger von
der gesamten Polymermoleküldynamik abhängt.
Die Ergebnisse der kinetischen Untersuchungen lassen den Schluss zu, dass wie bei
den meisten nucleophilen aromatischen Substitutionen von aktivierten Fluoraromaten
die Anlagerung des Aminostickstoffs der primären Aminogruppe von PVAm an den
aromatischen Ring zur Bildung der zwitterionischen Zwischenstufe der geschwindig-
keitsbestimmende Schritt ist.
Die mittels 4-FNB und 2-FNB synthetisierten 4-nitrophenyl-funktionalisierten PVAm´s
(PVAm 1) und 2-nitrophenyl-funktionalisierten PVAm´s (PVAm 2) zeigen in
Abhängigkeit vom pH-Wert der Lösung nur geringe Verschiebungen der Lage des
UV/vis-Absorptionsmaximums. Das UV/vis-Absorptionsmaximum von PVAm 1 liegt
bei pH = 1 bei 390 nm, bei pH = 7 bei 406 nm und bei pH = 11 bei 413 nm. Das
UV/vis-Absorptionsmaximum von PVAm 2 liegt bei pH = 1 bei 432 nm, bei pH = 7 bei
440 nm und bei pH = 11 bei 450 nm. Die mittels 1-Fluor-2,4-dinitrobenzen (Sangers
Reagenz), 1,5-Difluor-2,4-dinitrobenzen (DFDNB), 4-Fluor-3-nitroazobenzen
(FNAzoB) und 4-Fluor-3,4´-dinitrostilben (FDNstilben) funktionalisierten PVAm´s
weisen in Abhängigkeit vom pH-Wert keine signifikante Verschiebung der Lage der
UV/vis-Absorptionsbanden auf. Sehr interessant sind die optischen Eigenschaften
von 2-nitro-4-aminobenzen-funktionalisierten Polyvinylaminen, die sowohl in
Abhängigkeit vom pH-Wert als auch in Abhängigkeit von der Natur des Lösungs-
mittels signifikante Verschiebungen der UV/Vis-Absorptionsbanden aufweisen.

4 Zusammenfassung und Ausblick
159
Das UV/vis-Absorptionsmaximum der chromophoren Einheit von 2-nitro-4-
aminobenzen-funktionalisiertem PVAm (PVAm 3) liegt im stark Basischen bei
519 nm, im stark Sauren bei 424 nm. Das Vorhandensein eines isosbestischen
Punktes bei 466 nm erlaubte mit Hilfe der Henderson-Hasselbalch-Gleichung die
Ermittlung des pKs-Wertes der chromophoren Einheit mit pKS = 4.06.
Die pH-Wert abhängigen UV/vis-spektroskopischen Untersuchungen von 2-nitro-4-
(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) und von 2-nitro-4-
(N,N-dimethyl)aminobenzen-funktionalisiertem Poly-N-Methylvinylamin (PNMVAm 5)
zeigten signifikante Unterschiede der UV/vis-Absorptionseigenschaften in
Abhängigkeit vom pH-Wert. Die pKS-Werte der chromophoren Einheiten konnten mit
pKS = 4.03 für PVAm 4 und mit pKS = 2.99 für PNMVAm 5 ermittelt werden. Die
Einführung von zwei Methylgruppen am 4-Aminostickstoff bei PVAm 4 erhöht die
Basizität im Vergleich zu PVAm 3. Der im Vergleich zu PVAm 3 und PVAm 4 deutlich
niedrigere pKS-Wert der chromophoren Einheit von PNMVAm 5 beruht auf der in
1-Stellung eingeführten Methylgruppe, die zum Herausdrehen der ortho-Nitrogruppe
führt und damit zu einem schwächeren +M-Effekt der 1-N-Methyl-Aminogruppe.
Alle untersuchten funktionalisierten Polyvinylamine zeigen mehr oder weniger
ausgeprägte solvatochrome Eigenschaften. In Abhängigkeit von der Natur des
Lösungsmittels wurde das jeweilige UV/vis-Absorptionsmaximum der chromophoren
Polyvinylamin-Einheit mit Zunahme der Lösungsmittelpolarität bathochrom – zu
größeren Wellenlängen – verschoben (positive Solvatochromie).
Aufgrund der guten Löslichkeit von 2-nitro-4-aminobenzen-funktionalisierten
Polyvinylaminen (PVAm 3, PVAm 4 und PNMVAm 5) in einer Reihe an
Lösungsmitteln konnten die solvatochromen Eigenschaften mit Hilfe der Kamlet-Taft-
Gleichung quantifiziert werden. Hohe α-Werte der Lösungsmittel und damit die
Fähigkeit als HBD-Lösungsmittel (hydrogen bond donor) mit der chromophoren
Einheit in Wechselwirkung zu treten, führen bei allen drei 2-nitro-4-aminobenzen-
funktionalisierten Polyvinylaminen zu einer hypsochromen Verschiebung der UV/vis-
Absorptionsbande (zu kleineren Wellenlängen). Auf die solvatochromen
Eigenschaften von PVAm 3 weisen vor allem die Dipolarität/Polarisierbarkeit π* und
die Fähigkeit des Lösungsmittels als HBA-Lösungsmittel (hydrogen bond acceptor)
zu fungieren den größten Einfluss auf. Somit führt bei PVAm 3 ein hoher
π*-Wert und/oder ein hoher β-Wert des Lösungsmittels zu einer bathochromen
Verschiebung der UV/vis-Absorptionsbande der chromophoren Einheit.

4 Zusammenfassung und Ausblick
160
Die solvatochromen Eigenschaften von PVAm 4 sind unabhängig von der
Dipolarität/Polarisierbarkeit π* des verwendeten Lösungsmittels. Die Fähigkeit des
Lösungsmittels als HBA-Lösungsmittel (β-Wert) zu wirken, hat auf die solvato-
chromen Eigenschaften von PVAm 4 einen etwas geringeren Einfluss als die
Fähigkeit als HBD-Lösungsmittel (hydrogen bond donor, α-Wert) mit der
chromophoren Einheit in Wechselwirkung zu treten.
Die solvatochromen Eigenschaften von PNMVAm 5 werden hauptsächlich von der
Dipolarität/Polarisierbarkeit π* des Lösungsmittels bestimmt. Die Fähigkeit des
Lösungsmittels als HBD-Lösungsmittel (α-Wert) und/oder HBA-Lösungsmittel
(β-Wert) zu wirken, spielt in gleichem Maße eine geringere Rolle als die
Dipolarität/Polarisierbarkeit π*. Somit führen hohe π*-Werte und/oder hohe β-Werte
zu bathochromer Verschiebung der UV/vis-Absorptionsbande von PNMVAm 5.
Die nucleophile aromatische Substitution von Fluoraromaten mittels Polyvinylaminen
ist somit eine elegante Methode zur Darstellung von chromophoren wasserlöslichen
Polymeren. Durch die Funktionalisierung von PVAm besteht die Möglichkeit die
bisherigen Anwendungsmöglichkeiten von PVAm zu erweitern.
In zukünftigen Arbeiten könnte eine direkte Funktionalisierung von Polyvinylamin mit
Stilben- und Azoverbindungen weiter studiert werden, da diese als optische,
reversible Schalter, zur Nanostrukturierung von Oberflächen und für NLO-
Anwendungen von sehr großer Bedeutung sind.41-47
Es konnte weiterhin gezeigt werden, dass die nucleophile aromatische Substitution
von Fluoraromaten mit PVAm auch an der Phasengrenze Feststoff/Wasser zur
Oberflächenmodifizierung genutzt werden kann. Durch Umsetzung von wässriger
PVAm-Lösung mit auf Kieselgel H adsorbiertem 4-FNB, 2-FNB, 4-FNA, 4-DMFNA,
Sangers Reagenz, DFDNB, FNAzoB, N-(2´,4´-Dinitro-phenyl)-4-fluor-3-nitroanilin (N-
DNP-FNA), N-(2´-Nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-4-fluor-3-nitroanilin
(FNAzoB-2) und FDNstilben wurden farbige Kieselgele erhalten. Das jeweils
funktionalisierte PVAm blieb irreversibel auf der Festkörperoberfläche fixiert. In
Abhängigkeit der Reaktivität des Fluoraromaten liegen die chromophoren Einheiten
der funktionalisierten PVAm´s auf Kieselgel H unterschiedlich lokalisiert vor. Die für
die nucleophile aromatische Substitution mit PVAm weniger aktivierten
Fluoraromaten 4-FNB, 4-FNA, 4-DMFNA, N-DNP-FNA und FNAzoB-2 haben
möglicherweise hauptsächlich in Oberflächennähe – in den Poren – mit den
Aminogruppen von PVAm reagiert.

4 Zusammenfassung und Ausblick
161
Die für die nucleophile aromatische Substitution mit PVAm hingegen besser
aktivierten Fluoraromaten 2-FNB, Sangers Reagenz, DFDNB, FNAzoB und
FDNstilben haben möglicherweise hauptsächlich schon vor Erreichen der Poren in
der Peripherie der Partikel mit PVAm reagiert.
Über die nucleophile aromatische Substitution von an Festkörperoberflächen
gebundenen Fluoraromaten mit Polyvinylaminen ist es somit möglich zu
chromophoren PVAm-Kieselgel-Hybridmaterialien zu gelangen. Die Eigenschaften
der funktionalisierten Polyvinylamine lassen sich dabei mit denen des anorganischen
Trägers im Hybridmaterial kombinieren. So zeigte 2-nitro-4-aminobenzen-
funktionalisiertes PVAm und 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertes
PVAm jeweils adsorbiert auf KGH eine jeweils dem funktionalisierten Polymeren in
Lösung entsprechende Änderung der Lage der UV/vis-Absorptionsbande in
Abhängigkeit vom pH-Wert. Somit könnten neben den 2-nitro-4-aminobenzen-
funktionalisierten Polyvinylaminen PVAm 3, PVAm 4 und PNMVAm 5 auch die
entsprechend polymermodifizierten Kieselgele als pH-Sensoren fungieren.
Da Polyvinylamin aus Wasser in sehr definierten, nanometerdicken Schichten auf
Glas- und Siliconsubstraten adsorbiert49-51, könnte die in dieser Arbeit entwickelte
einfache Methode ein eleganter Weg sein, um Stilben- und Azofarbstoffeinheiten
direkt an Oberflächen zu binden. Diese einfach zu präparierenden Schichten
nachfolgend photochemisch schaltend zu gestalten, wäre eine sehr elegante
Synthese und demzufolge eine große Herausforderung.

5 Experimenteller Teil
162
5 Experimenteller Teil
5.1 Analysenmethoden und verwendete Geräte
UV/vis-Spektroskopie
Für die UV/vis-spektroskopischen Untersuchungen wurde ein MCS 400
Spektrometer der Firma Carl Zeiss Jena GmbH verwendet. Die Aufnahme von
UV/vis-Absorptionsspektren der funktionalisierten Polyvinylamine erfolgte in Küvetten
mit einer optischen Weglänge von 1.0 cm und für kinetische Untersuchungen in
Lösung mit Hilfe einer Tauchküvette TSM 5 mittels Glasfaseroptik. Die modifizierten
Kieselgele wurden mit einem speziellen Messkopf aufgenommen, indem die
Pulverprobe auf einer Quarzglasplatte über der Glasfaseroptik positioniert wurde.
NMR-Spektroskopie
Flüssig-NMR-Spektren wurden mit einem Gemini 2000 der Firma VARIAN bei
300 MHz bzw. einem UNITY INOVA 400 der Firma VARIAN bei 400 MHz an der
TU Chemnitz (Professur für Organische Chemie) aufgenommen. Die Aufnahme der 1H-NMR-Spektren erfolgte bei 300 MHz bzw. 400 MHz, die der 13C-NMR-Spektren
bei 75 MHz bzw. 100 MHz. Als interner Standard diente das jeweilige
Lösungsmittelsignal von D2O, CDCl3, d6-Aceton bzw. CD2Cl2. Die Multiplizität eines
Signals wird durch folgende Abkürzungen oder auch Kombinationen dieser
angegeben durch: br. = breit, s = Singulett, d = Dublett, t = Triplett, q = Quartett und
m = Multiplett. 13C-CP-MAS-Festkörper-NMR-Spektren modifizierter Kieselgele wurden mit einem
Bruker Avance 400 NMR-Spektrometer bei 100.62 MHz aufgenommen. Die
Festkörperproben rotierten jeweils mit 15 kHz für mehrere Tage im Probenkopf. Als
interner Standard diente Adamantan.
Elementaranalyse
Elementaranalysen wurden an der TU Chemnitz (Professur für Organische Chemie)
mit einem VARIO EL CHN-Analysator und an der Friedrich-Schiller-Universität Jena
mit einem VARIO EL III der Firma Elementaranalysensysteme GmbH Hanau
durchgeführt. An der Martin-Luther-Universität Halle-Wittenberg wurde mit einem
CHNS 932 Elementaranalysator der Firma Leco gemessen.

5 Experimenteller Teil
163
BET-Untersuchungen
Für die Ermittlung der BET-Oberflächen und Porenvolumina von reinem Kieselgel H
und von polymermodifizierten Kieselgelen kam das Instrument Sorptomatik 1900 der
Firma Fisons an der Professur Technische Chemie der TU Chemnitz zum Einsatz,
wobei die Proben vorher im Vakuum ausgeheizt wurden.
Röntgen-Photoelektronen-Spektroskopie (XPS)
Die XPS-Messungen von reinem Kieselgel H und polymermodifizierten Kieselgelen
wurden am Leibniz-Institut für Polymerforschung e. V. in Dresden mit einem AXIS
ULTRA der Firma Kratos Analytical (UK) durchgeführt. Als Röntgenquelle diente
Mono-Al Kα-Strahlung. Die Aufnahmeleistung der Röntgenröhre war 300 W bei
20 mA. Die Übergangsenergie des Analysators betrug für Übersichtsspektren 160 eV
bzw. für aufgelöste Spektren 20 eV. Bei allen Proben erfolgte eine
Überkompensation der Aufladung, die durch das Verschieben des C 1s Peaks auf
die Bindungsenergie (BE) gesättigter Kohlenwasserstoffe CxHy mit einer
BE = 285.00 eV korrigiert wurde.141
elektrokinetische Messungen (Zetapotential)
Die elektrokinetischen Messungen zur Ermittlung des Zetapotentials von reinem
Kieselgel H und polymermodifizierten Kieselgelen in Abhängigkeit vom pH-Wert
wurden am Leibniz-Institut für Polymerforschung e. V. in Dresden mit einem
ZetaSizer 3 der Firma Malvern (UK) durchgeführt. Als Inertsalz wurde KCl verwendet
(0.001 M KCl). Die Partikelgeschwindigkeit im elektrischen Feld wurde mittels Laser
DOPPLER Anemometrie unter Verwendung eines He-Ne-Lasers bestimmt. Die bei
der Mikroelektrophorese gemessenen elektrophoretischen Beweglichkeiten der
geladenen Partikel wurden anschließend über die sog. Smoluchowski-Gleichung in
Zetapotentialwerte umgerechnet.148
ESR-Spektroskopie
Die ESR-Untersuchungen wurden mit einem ESP 300 E Spektrometer der Firma
Bruker an der Friedrich-Schiller-Universität in Jena durchgeführt. Die Modulations-
frequenz betrug 100 kHz und die Amplitude 5 G. Die Spinkonzentration der
Polymerprobe wurde durch Vergleich der Peakfläche des Signals der Probe mit dem
Signal einer Standardprobe bestimmt (CuSO4·5H2O).
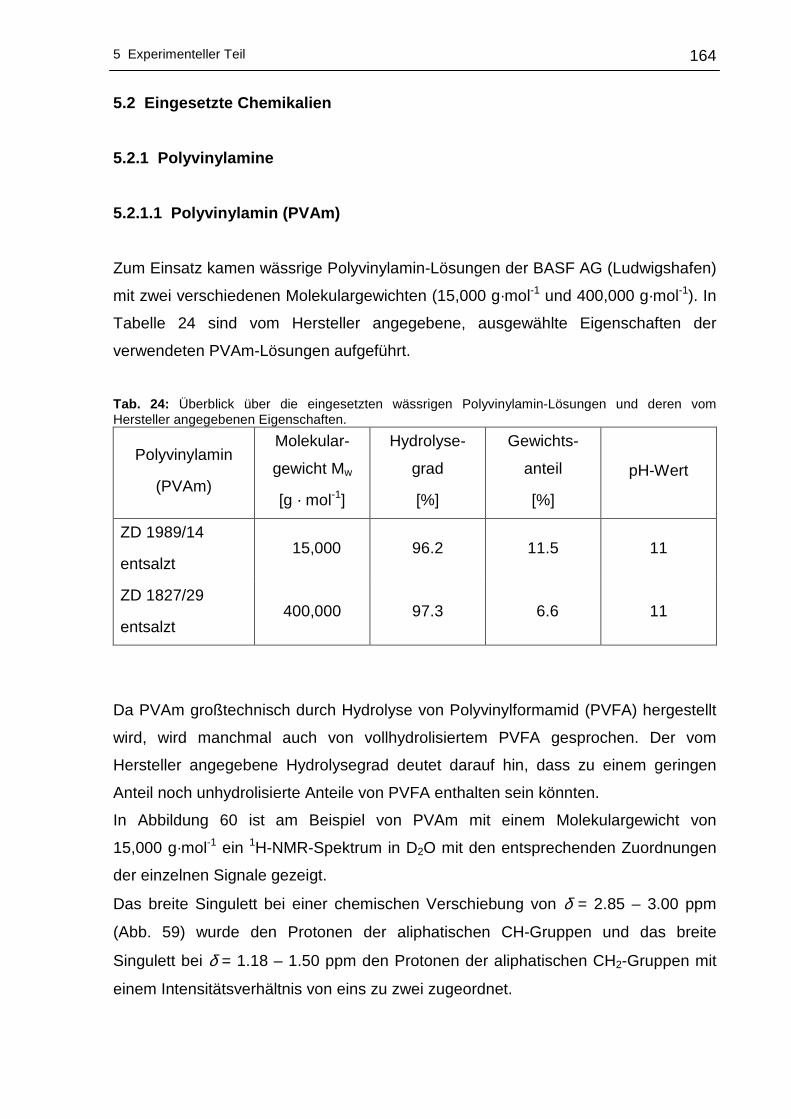
5 Experimenteller Teil
164
5.2 Eingesetzte Chemikalien
5.2.1 Polyvinylamine
5.2.1.1 Polyvinylamin (PVAm)
Zum Einsatz kamen wässrige Polyvinylamin-Lösungen der BASF AG (Ludwigshafen)
mit zwei verschiedenen Molekulargewichten (15,000 g·mol-1 und 400,000 g·mol-1). In
Tabelle 24 sind vom Hersteller angegebene, ausgewählte Eigenschaften der
verwendeten PVAm-Lösungen aufgeführt.
Tab. 24: Überblick über die eingesetzten wässrigen Polyvinylamin-Lösungen und deren vom Hersteller angegebenen Eigenschaften.
Polyvinylamin
(PVAm)
Molekular-
gewicht Mw
[g · mol-1]
Hydrolyse-
grad
[%]
Gewichts-
anteil
[%]
pH-Wert
ZD 1989/14
entsalzt 15,000 96.2 11.5 11
ZD 1827/29
entsalzt 400,000 97.3 6.6 11
Da PVAm großtechnisch durch Hydrolyse von Polyvinylformamid (PVFA) hergestellt
wird, wird manchmal auch von vollhydrolisiertem PVFA gesprochen. Der vom
Hersteller angegebene Hydrolysegrad deutet darauf hin, dass zu einem geringen
Anteil noch unhydrolisierte Anteile von PVFA enthalten sein könnten.
In Abbildung 60 ist am Beispiel von PVAm mit einem Molekulargewicht von
15,000 g·mol-1 ein 1H-NMR-Spektrum in D2O mit den entsprechenden Zuordnungen
der einzelnen Signale gezeigt.
Das breite Singulett bei einer chemischen Verschiebung von δ = 2.85 – 3.00 ppm
(Abb. 59) wurde den Protonen der aliphatischen CH-Gruppen und das breite
Singulett bei δ = 1.18 – 1.50 ppm den Protonen der aliphatischen CH2-Gruppen mit
einem Intensitätsverhältnis von eins zu zwei zugeordnet.
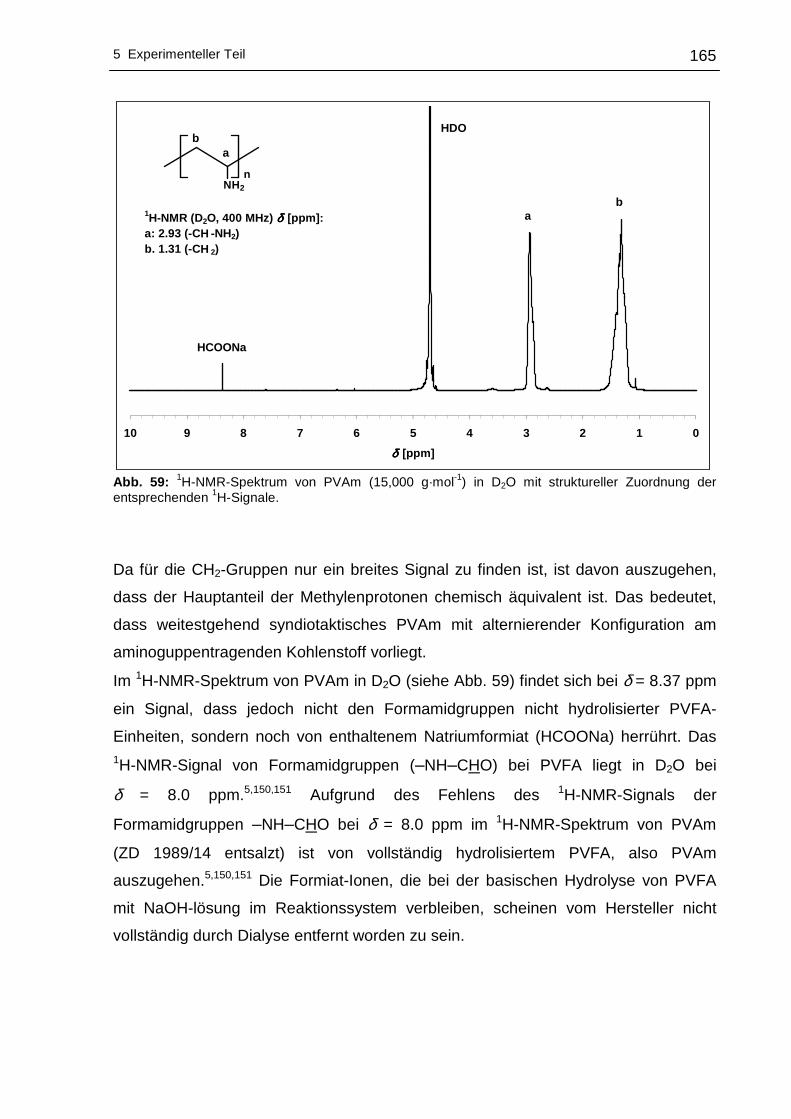
5 Experimenteller Teil
165
Abb. 59: 1H-NMR-Spektrum von PVAm (15,000 g·mol-1) in D2O mit struktureller Zuordnung der entsprechenden 1H-Signale.
Da für die CH2-Gruppen nur ein breites Signal zu finden ist, ist davon auszugehen,
dass der Hauptanteil der Methylenprotonen chemisch äquivalent ist. Das bedeutet,
dass weitestgehend syndiotaktisches PVAm mit alternierender Konfiguration am
aminoguppentragenden Kohlenstoff vorliegt.
Im 1H-NMR-Spektrum von PVAm in D2O (siehe Abb. 59) findet sich bei δ = 8.37 ppm
ein Signal, dass jedoch nicht den Formamidgruppen nicht hydrolisierter PVFA-
Einheiten, sondern noch von enthaltenem Natriumformiat (HCOONa) herrührt. Das 1H-NMR-Signal von Formamidgruppen (–NH–CHO) bei PVFA liegt in D2O bei
δ = 8.0 ppm.5,150,151 Aufgrund des Fehlens des 1H-NMR-Signals der
Formamidgruppen –NH–CHO bei δ = 8.0 ppm im 1H-NMR-Spektrum von PVAm
(ZD 1989/14 entsalzt) ist von vollständig hydrolisiertem PVFA, also PVAm
auszugehen.5,150,151 Die Formiat-Ionen, die bei der basischen Hydrolyse von PVFA
mit NaOH-lösung im Reaktionssystem verbleiben, scheinen vom Hersteller nicht
vollständig durch Dialyse entfernt worden zu sein.
012345678910
δδδδ [ppm]
HCOONa
ab
HDO
1H-NMR (D2O, 400 MHz) δδδδ [ppm]:a: 2.93 (-CH -NH2) b. 1.31 (-CH 2)
NH2
n
ab
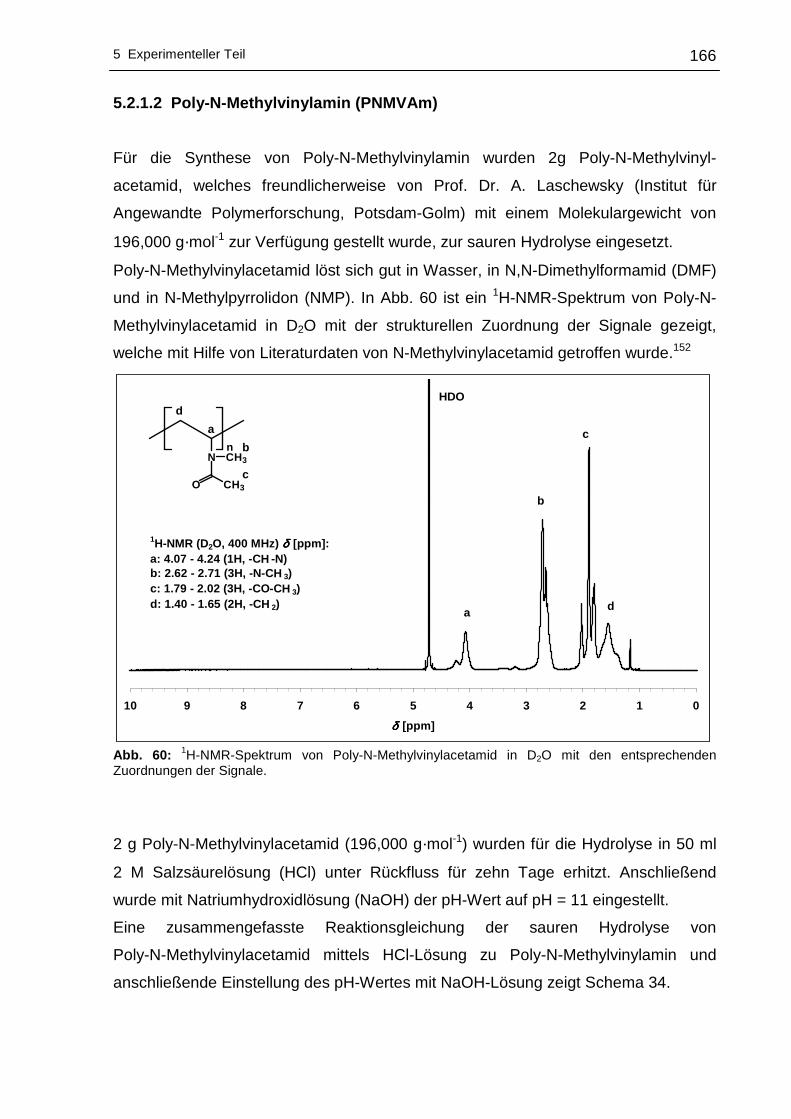
5 Experimenteller Teil
166
5.2.1.2 Poly- N-Methylvinylamin (PNMVAm)
Für die Synthese von Poly-N-Methylvinylamin wurden 2g Poly-N-Methylvinyl-
acetamid, welches freundlicherweise von Prof. Dr. A. Laschewsky (Institut für
Angewandte Polymerforschung, Potsdam-Golm) mit einem Molekulargewicht von
196,000 g·mol-1 zur Verfügung gestellt wurde, zur sauren Hydrolyse eingesetzt.
Poly-N-Methylvinylacetamid löst sich gut in Wasser, in N,N-Dimethylformamid (DMF)
und in N-Methylpyrrolidon (NMP). In Abb. 60 ist ein 1H-NMR-Spektrum von Poly-N-
Methylvinylacetamid in D2O mit der strukturellen Zuordnung der Signale gezeigt,
welche mit Hilfe von Literaturdaten von N-Methylvinylacetamid getroffen wurde.152
Abb. 60: 1H-NMR-Spektrum von Poly-N-Methylvinylacetamid in D2O mit den entsprechenden Zuordnungen der Signale.
2 g Poly-N-Methylvinylacetamid (196,000 g·mol-1) wurden für die Hydrolyse in 50 ml
2 M Salzsäurelösung (HCl) unter Rückfluss für zehn Tage erhitzt. Anschließend
wurde mit Natriumhydroxidlösung (NaOH) der pH-Wert auf pH = 11 eingestellt.
Eine zusammengefasste Reaktionsgleichung der sauren Hydrolyse von
Poly-N-Methylvinylacetamid mittels HCl-Lösung zu Poly-N-Methylvinylamin und
anschließende Einstellung des pH-Wertes mit NaOH-Lösung zeigt Schema 34.
012345678910
δδδδ [ppm]
a
b
c
d
HDO
N CH3
CH3O
n
a
b
c
d
1H-NMR (D2O, 400 MHz) δδδδ [ppm]:a: 4.07 - 4.24 (1H, -CH -N)b: 2.62 - 2.71 (3H, -N-CH 3)c: 1.79 - 2.02 (3H, -CO-CH 3)d: 1.40 - 1.65 (2H, -CH 2)

5 Experimenteller Teil
167
Schema 34: Schematische Darstellung der Hydrolysereaktion von Poly-N-Methylvinylacetamid zu Poly-N-Methylvinylamin (PNMVAm).
Ein kleiner Teil der wässrigen Poly-N-Methylvinylamin-Lösung wurde in Aceton
ausgefällt. Abb. 61 zeigt ein 1H-NMR-Spektrum von Poly-N-Methylvinylamin
(PNMVAm) in D2O mit den entsprechenden Zuordnungen der einzelnen Signale.
Abb. 61: 1H-NMR-Spektrum von Poly-N-Methylvinylamin in D2O und Zuordnung der Signale.
Das 1H-NMR-Spektrum in Abb. 61 deutet mit Vergleich zu Abb. 60 auf vollständige
Hydrolyse von Poly-N-Methylvinylacetamid zu Poly-N-Methylvinylamin (PNMVAm)
hin. Bei einer chemischen Verschiebung von δ = 1.85 ppm ist ein Signal erkennbar,
welches dem noch enthaltenen Fällungsmittel Aceton zugeordnet wurde.139
NCH3H
nN
CH3O
CH3
n
2M HCl
10 d
NaOH + CH3COONa + NaCl + H2O
012345678910
δδδδ [ppm]
HDO
Aceton
a
b
c
1H-NMR (D2O, 400 MHz) δδδδ [ppm]:a: 2.59 - 2.70 (1H, -CH -N)b: 2.30 (3H, -NH-CH 3)c: 1.38 - 1.69 (2H, -CH 2)
NH
CH3
n
a
b
c
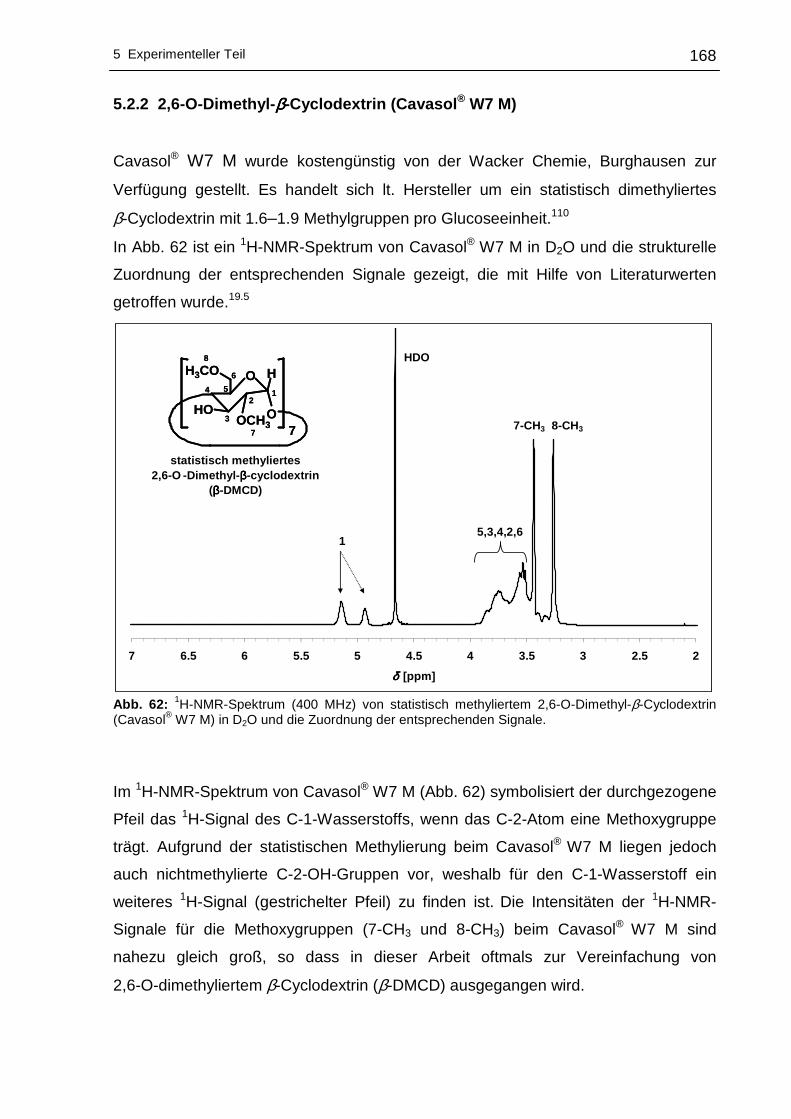
5 Experimenteller Teil
168
5.2.2 2,6-O-Dimethyl- ββββ-Cyclodextrin (Cavasol ® W7 M)
Cavasol® W7 M wurde kostengünstig von der Wacker Chemie, Burghausen zur
Verfügung gestellt. Es handelt sich lt. Hersteller um ein statistisch dimethyliertes
β-Cyclodextrin mit 1.6–1.9 Methylgruppen pro Glucoseeinheit.110
In Abb. 62 ist ein 1H-NMR-Spektrum von Cavasol® W7 M in D2O und die strukturelle
Zuordnung der entsprechenden Signale gezeigt, die mit Hilfe von Literaturwerten
getroffen wurde.19.5
Abb. 62: 1H-NMR-Spektrum (400 MHz) von statistisch methyliertem 2,6-O-Dimethyl-β-Cyclodextrin (Cavasol® W7 M) in D2O und die Zuordnung der entsprechenden Signale.
Im 1H-NMR-Spektrum von Cavasol® W7 M (Abb. 62) symbolisiert der durchgezogene
Pfeil das 1H-Signal des C-1-Wasserstoffs, wenn das C-2-Atom eine Methoxygruppe
trägt. Aufgrund der statistischen Methylierung beim Cavasol® W7 M liegen jedoch
auch nichtmethylierte C-2-OH-Gruppen vor, weshalb für den C-1-Wasserstoff ein
weiteres 1H-Signal (gestrichelter Pfeil) zu finden ist. Die Intensitäten der 1H-NMR-
Signale für die Methoxygruppen (7-CH3 und 8-CH3) beim Cavasol® W7 M sind
nahezu gleich groß, so dass in dieser Arbeit oftmals zur Vereinfachung von
2,6-O-dimethyliertem β-Cyclodextrin (β-DMCD) ausgegangen wird.
22.533.544.555.566.57
δδδδ [ppm]
O
O
H
7
H3CO
HOOCH3
12
3
4 5
6
7
8
O
O
H
7
H3CO
HOOCH3
12
3
4 5
6
7
8
5,3,4,2,61
8-CH37-CH3
HDO
statistisch methyliertes2,6-O -Dimethyl- ββββ-cyclodextrin
(ββββ-DMCD)

5 Experimenteller Teil
169
5.2.3 Fluoraromaten
Für die Funktionalisierung von Polyvinylaminen kamen die in Schema 35 gezeigten
10 Fluoraromaten zum Einsatz. 4-Fluornitrobenzen (4-FNB), 2-Fluornitrobenzen
(2-FNB) und 1-Fluor-2,4-dinitrobenzen (FDNB, Sangers Reagenz) von Sigma-
Aldrich, 1,5-Difluor-2,4-dinitrobenzen (DFDNB) von Merck und 4-Fluor-3-nitroanilin
(4-FNA) von ABCR waren kommerziell erhältlich. 4-FNB, 2-FNB and Sangers
Reagenz wurden zur Reinigung unter Vakuum destilliert. Weiße Kristalle von DFDNB
wurden ohne weitere Reinigung eingesetzt. Braunes Pulver von 4-FNA wurde aus
Ethanol umkristallisiert.
Schema 35: Schematische Darstellung der eingesetzten aktivierten Fluoraromaten für die nucleophile aromatische Substitution mit Polyvinylaminen.
4-FNB
2-FNB FDNB
DFDNB 4-FNA 4-DMFNA
FDNstilben FNAzoB
F NO2F
O2N
F
O2N
NO2
F
O2N
NO2
F
F NH2
O2N
N
O2N
FCH3
CH3
F
O2N
NN
F
O2N
NO2
N
HO2N
NO2
F NO2
FNAzoB-2N-DNP-FNA
HN N
N
F NO2
O2N
N

5 Experimenteller Teil
170
Die Fluoraromaten 4-Fluor-3-nitroazobenzen (FNAzoB), N-(2´,4´-Dinitrophenyl)-4-
fluor-3-nitroanilin (N-DNP-FNA) und N-(2´-Nitrophenyl-4´-(p-N,N-diethyl)-azobenzen)-
4-fluor-3-nitroanilin (FNAzoB-2) wurden freundlicherweise von A. Seifert (Professur
Polymerchemie, TU Chemnitz) zur Verfügung gestellt.
Die Synthesedurchführung zur Darstellung von N,N-Dimethyl-4-fluor-3-nitroanilin
(4-DMFNA) und 4-Fluor-3,4´-dinitrostilben (FDNstilben) und deren Charakterisierung
sind im Abschnitt 5.6 ausführlich dargelegt.
5.2.4 Kieselgel H (KGH)
Kieselgel H wurde von Fluka (Sigma-Aldrich) käuflich erworben. Die Partikelgröße
von 5 − 40 µm wurde vor allem für die elektrokinetischen Untersuchungen
(Zetapotential) ausgewählt. Weitere Angaben zu den Partikel-Eigenschaften wurden
vom Hersteller nicht getroffen. Es wurden rasterelektronenmikroskopische
Aufnahmen von reinem Kieselgel H (s. Abb. 63) mit einem SEM 515 der Firma
Philips an der Professur Analytik an Festkörperoberflächen (Institut für Physik) an der
TU Chemnitz bei einer angelegten Spannung von 25 kV aufgenommen.
Abb. 63: Rasterelektronenmikroskopische Aufnahmen von reinem Kieselgel H (KGH).
Aus Abb. 63 wird deutlich, dass kleinste kugelförmige Partikel zu größeren
Aggregaten zusammengelagert sind. Es handelt sich um ein mesoporöses Material
mit einer spezifischen BET-Oberfläche von 395 m2·g-1 wie BET-Untersuchungen
ergaben (s. Abschnitt 3.2.6).
10 µm10 µm2 µm2 µm

5 Experimenteller Teil
171
5.3 Funktionalisierung von Polyvinylaminen mittels Fluoraromaten
Komplexierung von Fluoraromaten mit 2,6-O-Dimethyl-β-cyclodextrin (β-DMCD):
Unterschiedliche Mengen des Fluoraromaten (1.77 mmol, 3.54 mmol, 5.31 mmol,
7.09 mmol) wurden jeweils mit der äquimolaren Menge an (β-DMCD) in 30 ml
Methanol gelöst und über Nacht gerührt. Der gebildete β-DMCD-Komplex wurde vom
Lösungsmittel befreit (Rotationsverdampfer) und im Vakuum getrocknet. Ob die Zeit
zur Komplexierung (über Nacht) für eine vollständige Komplexierung des
Fluoraromaten ausreichte, wurde mittels Dünnschichtchromatographie überprüft.
in Wasser über Löslichkeitsvermittlung mit 2,6-O-Dimethyl-β-cyclodextrin:
Zur klaren wässrigen Lösung des β-DMCD-Komplexes (100 ml) wurden jeweils 1.0 g
PVAm enthaltene wässrige Lösungen (15,000 g·mol-1, 8.7 ml, 11.5 Gew%) bzw.
15.2 ml (400,000 g·mol-1, 6.6 Gew%) zugegeben. Für die Funktionalisierung von
Poly-N-Methylvinylamin (PNMVAm) wurde eine 0.5 g Polymer enthaltene wässrige
Lösung (60 ml) zugegeben. Die Lösung wurde unter Rückfluss zum Sieden erhitzt.
Noch vor Erreichen der Siedetemperatur erkannte man den Beginn der Reaktion an
der Änderung der Farbe der Reaktionslösung. Um das funktionalisierte Polymere in
Aceton zur Fällung zu bringen, wurde der Großteil des Wassers abdestilliert, so dass
die Gesamtzeit der Reaktion 8 h im Falle von PNMVAm 12 h betrug. Das gefällte
Polymere wurde abzentrifugiert (Teflonzentrifugengefäß) und mit Aceton solange
gewaschen bis die überstehende Lösung farblos blieb. Der Feststoff wurde im
Teflonzentrifugengefäß im Vakuum über Kieselgel getrocknet. Teflongefäße waren
notwendig, da sowohl PVAm als auch die funktionalisierten PVAm´s sehr gut an
Glasoberflächen haften.
im Ethanol/Wasser-Gemisch ohne β-DMCD:
Unterschiedliche Mengen des Fluoraromaten 2-Fluornitrobenzen (0.25 g, 0.5 g,
0.75 g, 1.0 g) wurden in 30 ml Ethanol gelöst und 8.7 ml (1.0 g; 15,000 g·mol-1) der
wässrigen PVAm-Lösung zugegeben. Die Reaktionsmischung wurde unter Rückfluss
zum Sieden erhitzt. Der Großteil des Ethanols wurde so abdestilliert, dass die
Reaktionszeit insgesamt 8 h betrug. Das Polymere wurde dann in Aceton ausgefällt
und mit Aceton gewaschen. Die Trocknung erfolgte in Teflonzentrifugengefäßen im
Vakuum.

5 Experimenteller Teil
172
5.4 Kinetische Untersuchungen der Funktionalisieru ng von PVAm
in Wasser bei pH = 11 mit MW = 15,000 g·mol-1:
Für die kinetischen Untersuchungen mit Hilfe der UV/vis-Spektroskopie wurden
jeweils 4.4 ml (0.5 g festes Polymer) der PVAm-Lösung mit einem Molekulargewicht
von 15,000 g·mol-1 in 25 ml destilliertem Wasser bei der gewünschten Temperatur
bei pH = 11 temperiert. Die Fluoraromat/β-DMCD-Komplexe von 4-Fluornitrobenzen
(4-FNB), 2-Fluornitro-benzen (2-FNB), 2,4-Dinitrofluorbenzen (Sangers Reagenz),
1,5-Difluor-2,4-dinitrobenzen (DFDNB), 4-Fluor-3-nitroanilin (4-FNA) und
N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA) wurden jeweils in 5 ml destilliertem
Wasser gelöst und mittels Einmalspritze zur PVAm-Lösung gegeben.
in Ethanol/Wasser bei pH = 11 mit MW = 15,000 g·mol-1:
Jeweils 4.4 ml (0.5 g festes Polymer) der PVAm-Lösung mit einem Molekulargewicht
von 15,000 g·mol-1 wurden in 25 ml Ethanol bei der gewünschten Temperatur
temperiert. Die 2-FNB/β-DMCD-Komplexe wurden jeweils in 5 ml Ethanol gelöst und
mittels Einmalspritze zur PVAm-Lösung gegeben.
In Tabelle 25 sind die eingesetzten Fluoraromaten mit den entsprechenden
Molverhältnissen zu PVAm und die Reaktionsbedingungen für die Funktionalisierung
von PVAm mit einem Molekulargewicht von 15,000 g·mol-1 in Wasser über
Löslichkeitsvermittlung mittels β-DMCD bei pH = 11 sowie ohne Löslichkeits-
vermittlung am Beispiel der Funktionalisierung mit 2-FNB in einem Ethanol/Wasser-
Gemisch zusammengestellt.

5 Experimenteller Teil
173
Tab. 25: Überblick der eingesetzten Fluoraromaten mit den entsprechenden Molverhältnissen zu PVAm (15,000 g·mol-1) und die Reaktionsbedingungen für die Funktionalisierungsreaktion bei pH = 11 in Wasser über Löslichkeitsvermittlung mittels β-DMCD sowie ohne Löslichkeitsvermittlung in einem EtOH/H2O-Gemisch am Beispiel der Funktionalisierung von PVAm mit 2-Fluornitrobenzen.
Fluoraromat (FA)
c0(PVAm) [mol·l-1]
c0(FA) [mol·l-1]
nPVAm
nFA
Τ [°C]
Zeit [h]
F NO2
0.3375 1.03 ·10-3 327
50 60 70 80
19 17 5
3.5 F
O2N
0.3375 1.03 ·10-3
327
50 60 70 85
20 16 7 3
F
O2N EtOH/H2O
0.3375 1.03 ·10-3 327
40 50 55 60
24 24 24 24
F
O2N
NO2
0.3375 2.58 ·10-4 1310
0 23 28 32
3 1
0.5 0.5
F
O2N
NO2
F
0.3375 2.58 ·10-4 1310
0 22 29 33
3 1
0.5 0.5
F NH2
O2N
0.3375 2.06 ·10-3 164 50 60 80
67 42 19
N
O2N
FCH3
CH3
0.3375 4.12 ·10-3 82 32 60 70
67 50 25
mit Sangers Reagenz bei Variation des pH-Wertes und Fremdsalzzugabe (KCl):
Es wurden 15 ml der PVAm-Stammlösung (11.5 Gew%; 15,000 g·mol-1) bzw. 30.4 ml
(6.6 Gew%; 400,000 g·mol-1) mit verdünnter Salzsäure auf pH = 4 und pH = 7 bzw.
mit verdünnter Natronlauge auf pH = 11 so eingestellt, dass das Gesamtvolumen pro
Lösung jeweils 100 ml betrug. Aus den PVAm-Lösungen mit einem Molekulargewicht
von 15,000 g·mol-1 wurden jeweils 29.4 ml (0.5 g festes Polymer), aus den PVAm-
Lösungen mit einem Molekulargewicht von 400,000 g·mol-1 wurden jeweils 32.6 ml
bei der gewünschten Temperatur temperiert.

5 Experimenteller Teil
174
Für die Fremdsalzzugabe wurde die äquimolare Menge an Kaliumchlorid (0.8655 g)
zur PVAm-lösung (15,000 g·mol-1) ins Temperiergefäß gegeben. PVAm wurde zum
Vergleich der Funktionalisierung bei pH = 11 in 1310-fachem Überschuss zu Sangers
Reagenz eingesetzt. Dafür wurde aus einer Stammlösung von Sangers Reagenz in
Methanol (33 mg in 100 ml) jeweils 5 ml (8.866 mmol) mit der äquimolaren Menge an
β-DMCD in 10 ml Methanol komplexiert. Die Sangers Reagenz/β-DMCD-Komplexe
wurden in jeweils 5 ml destilliertem Wasser gelöst und mittels Einmalspritze zur
PVAm-Lösung gegeben.
In Tabelle 26 sind die Reaktionsbedingungen der UV/vis-spektroskopischen
Verfolgung der Funktionalisierung von PVAm (15,000 g·mol-1 und 400,000 g·mol-1)
mittels Sangers Reagenz/β-DMCD in Wasser bei pH = 4, pH = 7 und pH = 11 sowie
bei pH = 7 unter Zugabe von Fremdsalz (KCl) bei verschiedenen Temperaturen
zusammengestellt.
Tab. 26: Zusammenstellung der Reaktionsbedingungen der UV/vis-spektroskopischen Verfolgung der Funktionalisierung von PVAm (15,000 g·mol-1 und 400,000 g·mol-1) mittels Sangers Reagenz/β-DMCD in Wasser bei pH = 4, pH = 7 und pH = 11 sowie bei pH = 7 unter Zugabe von Fremdsalz (KCl) bei verschiedenen Temperaturen.
NH2 NHNO2
NO2
x y
pH = 4
T [°C] Zeit [h]
pH = 7
T [°C] Zeit [h]
pH = 11
T [°C] Zeit [h]
Mn = 15,000 g · mol-1
c0(PVAm) = 0.3375 mol·l-1 c0(Sangers Reagenz) = 2.577 · 10-4 mol · l-1
25°C 50°C 70°C
18 18 6
24°C 50°C 70°C
20 3 1
siehe Tab. 25
siehe Tab. 25
Mn = 400,000 g · mol-1
c0(PVAm) = 0.3088 mol·l-1 c0(Sangers Reagenz) = 2.358 · 10-4 mol · l-1
30°C 50°C 75°C
17 15.5
6
25°C 50°C 70°C
15 3
1.25
0 20 30
2 1.5 1
Mn = 15,000 g · mol-1
c0(KCl) = 0.3375 mol·l-1 c0(PVAm) = 0.3375 mol·l-1 c0(Sangers Reagenz) = 2.577 · 10-4 mol · l-1
- - 22°C 50°C 70°C
17 4 1
- -

5 Experimenteller Teil
175
5.5 Polymermodifizierung von Kieselgel H (KGH)
Für die Polymermodifizierung von Kieselgel H wurden jeweils 5 g KGH (83.33 mmol)
mit jeweils 2.5 mmol der in Schema 35 gezeigten 10 Fluoraromaten über Nacht in
Ethanol bzw. je nach Löslichkeit auch in Aceton suspendiert. Das Lösungsmittel
wurde am Rotationsverdampfer vollständig entfernt. Das mit Fluoraromat beladene
Kieselgel H wurde in 50 ml Wasser suspendiert. Je 5.4 ml (8.33 mmol, 0.3589 g) der
PVAm-lösung mit einem Molekulargewicht von 400,000 g·mol-1 und einem pH = 11
wurden zugegeben und die Reaktionsmischung wurde für 12 h zum Sieden erhitzt.
Teilweise zeigte sich schon nach wenigen Minuten eine Gelbildung, so dass zur
besseren Durchmischung der gallertartigen Masse die Menge des Suspensions-
mittels Wasser erhöht wurde. Nach Beendigung der Reaktionszeit wurden die
chromophoren Kieselgele in Teflon-Zentrifugengefäßen von der wässrigen Phase
abgetrennt. Die wässrige, chromophore Phase des funktionalisierten PVAm´s wurde
sofort in eiskaltem Aceton ausgefällt und nach mehrmaligem Waschen mit Aceton im
Vakuum getrocknet. Die Menge der zur Fällung gebrachten, funktionalisierten
PVAm´s war teilweise aufgrund der starken Verdünnung der wässrigen PVAm-
Lösung gering. Die farbigen Kieselgelpartikel wurden über Soxhlet-Extraktionen erst
mit Wasser und anschließend mit Aceton oder Ethanol gewaschen. Die Trocknung
der chromophoren Kieselgele erfolgte im Vakuum. In Tabelle 27 sind die
eingesetzten Fluoraromaten zur Modifizierung von KGH, die verwendeten
Lösungsmittel für die Adsorption der Fluoraromaten an KGH und die jeweiligen
Beobachtungen während der Modifizierung zusammenfassend dargestellt.
Tabelle 27: Zusammenstellung der eingesetzten Fluoraromaten für die Modifizierung von Kieselgel H (KGH) und Beobachtungen während der Modifizierung.
KGH-Probe Fluoraromat (FA)
Lösungsmittel für Adsorption
des FA
Gelbildung in 50 ml Wasser?
zusätzliche Menge an Wasser
KGH-01 4-FNB Ethanol nein - KGH-02 2-FNB Ethanol ja 200 ml KGH-03 4-FNA Ethanol nein - KGH-04 4-DMFNA Ethanol nein - KGH-05 Sangers Reagenz Ethanol ja 200 ml KGH-06 DFDNB Ethanol ja 200 ml KGH-07 FNAzoB Aceton ja 200 ml KGH-08 N-DNP-FNA Aceton nein - KGH-09 FNAzoB-2 Aceton nein - KGH-10 FDNstilben Ethanol nein -

5 Experimenteller Teil
176
5.6 Synthesevorschriften und Charakterisierungen
5.6.1 N,N-Dimethyl-4-fluor-3-nitroanilin (4-DMFNA)
15 ml einer 3M Schwefelsäure werden in 50 ml THF in einem 500 ml Zweihalskolben
unter Eiskühlung vorgelegt. Anschließend werden 9.5 ml (0.128 mol) Formaldehyd-
lösung zugegeben. Separat zur Reaktionsmischung werden in einem Becherglas 5 g
(0.032 mol) 4-Fluor-3-nitroanilin (4-FNA) in 200 ml THF gelöst und 5.81 g
(0.1536 mol) gemörsertes Natriumborhydrid (NaBH4) zugegeben. Die Hälfte dieser
Suspension wird über einen Tropftrichter langsam zur Formaldehyd/THF-Lösung
gegeben. Es bildet sich ein weißer Feststoff in orange-roter Reaktionslösung. Nach
Zugabe von weiteren 15 ml 3M H2SO4 wird die andere Hälfte der Suspension
zugetropft. Anschließend werden 50 ml destilliertes Wasser unter Gasentwicklung
zugegeben und solange gerührt bis kein im Überschuss eingesetztes NaBH4 mehr
zersetzt wird. Durch Zugabe von KOH-Plätzchen wird die stark saure Lösung basisch
gemacht, wobei ein Farbumschlag von orange-rot zu dunkelrot und eine Phasen-
trennung zwischen Wasser und THF erkennbar ist. Das Lösungsmittelgemisch wird
zur Abtrennung des weißen Feststoffes in einen 500 ml Scheidetrichter filtriert. Die
wässrige Phase wird zweimal mit je 50 ml Diethylether (Et2O) ausgeschüttelt. Die
organischen Phasen werden vereinigt und mit 20 ml gesättigter NaCl-Lösung
gewaschen. Die organische Phase (THF/Et2O) wird mit Na2SO4 getrocknet. Nach
Filtration wird der Großteil des Lösungsmittelgemisches abdestilliert. Im Gefrier-
schrank kristallisiert das Produkt aus der orange-roten Lösung nach und nach aus.
rote Kristalle
Schmelzbereich: 37−41 °C
Ausbeute: 94.9 % (bezogen auf 4-FNA)
Elementaranalyse (kalkuliert): C: 51.70 (52.17), H: 5.23 (4.93), N: 14.78 (15.21).
1H-NMR (CD2Cl2; 300 MHz) δ [ppm]: 7.21 (dd, 1H, H-2, 4J[a,19F] = 5.49 Hz, 4J[a,c] = 3.30 Hz); 7.12 (dd, 1H, H-5, 3J[b,c] = 9.34 Hz, 3J[b,19F] = 10.44 Hz);
6.90 (ddd, 1H, H-6, 3J[b,c] = 9.34 Hz, 4J[c,19F] = 3.85 Hz, 4J[a,c] = 3.30 Hz);
2.97 (s, 6H, H-7). In Abb. 64 ist ein 1H-NMR-Spektrum von 4-DMFNA in CD2Cl2 mit
der strukturellen Zuordnung der 1H-Signale gezeigt.
N
O2N
FCH3
CH31
23
4
5 6
7

5 Experimenteller Teil
177
Abb. 64: 1H-NMR-Spektrum von 4-DMFNA in CD2Cl2 und strukturelle Zuordnung der 1H-Signale.
13C-NMR (CD2Cl2; 75 MHz) δ [ppm]: 148.71 (C-1); 147.36 (C-4); 146.20 (C-3); 118.76
(C-6); 118.61 (C-5); 107.60 (C-2); 40.69 (C-7). 13C-NMR-Spektrum von 4-DMFNA in
CD2Cl2 und strukturelle Zuordnung der 13C-Signale siehe Abb. 65.
Abb. 65: 13C-NMR-Spektrum von 4-DMFNA in CD2Cl2 und strukturelle Zuordnung der 13C-Signale.
00.511.522.533.544.555.566.577.5
δδδδ [ppm]
6.86.977.17.27.3
δδδδ [ppm]
H-7
CD2Cl2
H-2
H-5
H-6
Wasserim CD2Cl2
N
O2N
FCH3
CH31
23
4
5 6
7
020406080100120140160180200
δδδδ [ppm]
C-1
C-6
C-2
C-4
C-3
C-7
CD2Cl2
C-5
N
O2N
FCH3
CH31
23
4
5 6
7

5 Experimenteller Teil
178
5.6.2 4-Fluor-3,4´-dinitrostilben (FDNstilben)
4-Fluor-3,4´-dinitrostilben (FDNstilben) wurde über eine Horner-Wadsworth-Emmons-
Reaktion119,120 aus 4-Fluor-3-nitrobenzaldehyd (Aldrich) und 4-Nitrobenzyl-
diethylphosphonat (Acros) mit Natriumethanolat (NaOEt) als Base in Tetrahydrofuran
(THF) synthetisiert (s. Schema 36).
Schema 36: Schematische Darstellung der Reaktion von 4-Fluor-3-nitrobenzaldehyd mit 4-Nitrobenzyldiethylphosphonat über eine Horner-Wadsworth-Emmons-Reaktion in Tetrahydrofuran zu 4-Fluor-3,4´-dinitrostilben (FDNstilben).
In einem 250ml-Dreihalskolben mit Schutzgaseinleitung werden 3.23 g
4-Nitrobenzyldiethylphosphonat (0.0118mol) in 50 ml trockenem THF gelöst und auf
0°C (Eiskühlung) abgekühlt. Anschließend werden aus einem 50ml-Tropftrichter mit
Druckausgleich 0.8048 g (0.0118 mol, M = 68.05 g·mol-1) Natriumethanolat (NaOEt)
in 50 ml THF zur Phosphonatlösung langsam zugetropft (ca. 20 min). Dabei färbt
sich die Reaktionslösung intensiv rot. Dann werden 2.00 g (0.0118 mol) 4-Fluor-3-
nitrobenzaldehyd gelöst in 50 ml THF ebenfalls langsam über einen 50ml-
Tropftrichter zugegeben (ca. 30min). Die intensiv rot gefärbte Reaktionslösung wird
über Nacht bei Raumtemperatur gerührt. Die Reaktionslösung gibt man dann in ca.
400 ml destilliertes Wasser, wobei sofort ein käsiger, gelber Niederschlag ausfällt. Mit
verdünnter HCl-Lösung wird neutralisiert, wobei noch etwas mehr Reaktionsprodukt
ausfällt. Der gelbe Niederschlag wird über eine Fritte abgesaugt und zuerst mit
Ethanol gewaschen und anschließend aus einem THF/Wasser-Gemisch
(Kühlschrank) umkristallisiert.
Das gewünschte Reaktionsprodukt 4-Fluor-3,4´-dinitrostilben wird mit einem
Schmelzpunkt von 212−216 °C und einer Ausbeute von 44 % (bezogen auf 4-F luor-
3-nitrobenzaldehyd) erhalten. Als Nebenprodukt konnte 4-Ethoxy-3,4´-dinitrostilben 1H-NMR-spektroskopisch identifiziert werden.
F
O2N
CHO + CH2P
O
EtO
OEt
NO2 F
O2N
NO2
M = 169,11 g/mol M = 273,22 g/mol M = 288,23 g/mol
NaOEt
THF
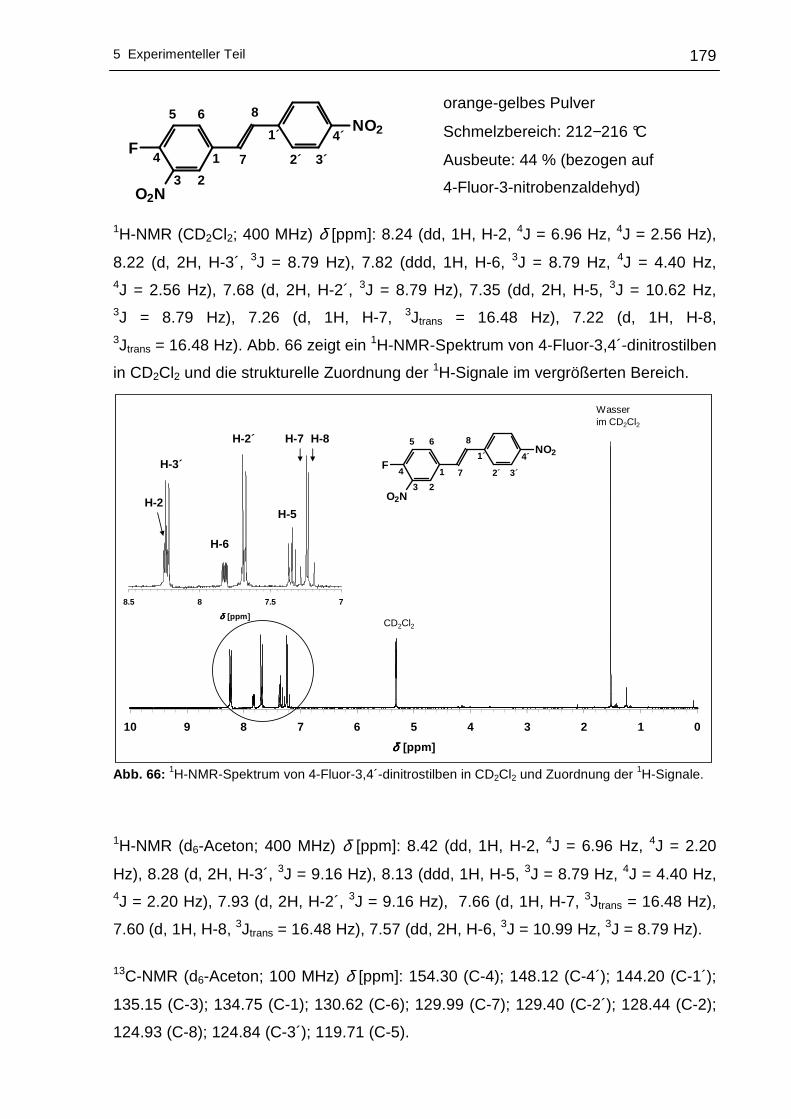
5 Experimenteller Teil
179
orange-gelbes Pulver
Schmelzbereich: 212−216 °C
Ausbeute: 44 % (bezogen auf
4-Fluor-3-nitrobenzaldehyd)
1H-NMR (CD2Cl2; 400 MHz) δ [ppm]: 8.24 (dd, 1H, H-2, 4J = 6.96 Hz, 4J = 2.56 Hz),
8.22 (d, 2H, H-3´, 3J = 8.79 Hz), 7.82 (ddd, 1H, H-6, 3J = 8.79 Hz, 4J = 4.40 Hz, 4J = 2.56 Hz), 7.68 (d, 2H, H-2´, 3J = 8.79 Hz), 7.35 (dd, 2H, H-5, 3J = 10.62 Hz, 3J = 8.79 Hz), 7.26 (d, 1H, H-7, 3Jtrans = 16.48 Hz), 7.22 (d, 1H, H-8, 3Jtrans = 16.48 Hz). Abb. 66 zeigt ein 1H-NMR-Spektrum von 4-Fluor-3,4´-dinitrostilben
in CD2Cl2 und die strukturelle Zuordnung der 1H-Signale im vergrößerten Bereich.
Abb. 66: 1H-NMR-Spektrum von 4-Fluor-3,4´-dinitrostilben in CD2Cl2 und Zuordnung der 1H-Signale.
1H-NMR (d6-Aceton; 400 MHz) δ [ppm]: 8.42 (dd, 1H, H-2, 4J = 6.96 Hz, 4J = 2.20
Hz), 8.28 (d, 2H, H-3´, 3J = 9.16 Hz), 8.13 (ddd, 1H, H-5, 3J = 8.79 Hz, 4J = 4.40 Hz, 4J = 2.20 Hz), 7.93 (d, 2H, H-2´, 3J = 9.16 Hz), 7.66 (d, 1H, H-7, 3Jtrans = 16.48 Hz),
7.60 (d, 1H, H-8, 3Jtrans = 16.48 Hz), 7.57 (dd, 2H, H-6, 3J = 10.99 Hz, 3J = 8.79 Hz).
13C-NMR (d6-Aceton; 100 MHz) δ [ppm]: 154.30 (C-4); 148.12 (C-4´); 144.20 (C-1´);
135.15 (C-3); 134.75 (C-1); 130.62 (C-6); 129.99 (C-7); 129.40 (C-2´); 128.44 (C-2);
124.93 (C-8); 124.84 (C-3´); 119.71 (C-5).
F
O2N
NO2
1
23
4
5 6
7
8
1´
2´ 3´
4´
012345678910
δδδδ [ppm]
CD2Cl2
Wasser im CD2Cl2
F
O2N
NO2
1
23
4
5 6
7
8
1´
2´ 3´
4´
77.588.5
δδδδ [ppm]
H-2
H-3´
H-6
H-2´
H-5
H-7 H-8

5 Experimenteller Teil
180
5.6.3 Modellverbindungen
In Anlehnung an die Arbeiten von Nudelman et al. (Umsetzung von
4-Fluornitrobenzen bzw. 2-Fluornitrobenzen mit Isopropylamin) wurden die Modell-
verbindungen aus den entsprechenden Fluoraromaten und Isopropylamin
synthetisiert.130 Eine allgemeingültige Reaktionsgleichung der nucleophilen
aromatischen Substitution von Isopropylamin mit aktivierten Fluoraromaten zeigt
Schema 37.
Schema 37: Schematische Darstellung der Reaktion von aktivierten Fluoraromaten mit Isopropylamin im Überschuss in Toluen bei 80°C zu den entsprechen den N-Isopropylanilin-Derivaten.
Allgemeine Synthesevorschrift nach Nudelman et al.:130
Der Fluoraromat wird in 50 − 70 ml Toluol gelöst. Isopropylamin wird in 5-fachem
Überschuss (bei Disubstitution in 10-fachem Überschuss) zugegeben, um das durch
die nucleophile aromatische Substitution des Fluoraromaten entstehende HF als
Isopropylammoniumfluorid abzufangen. Die Reaktionslösung wird bei 80 °C für 20 h
unter Rückfluss erhitzt. Während der Reaktion und besonders beim Abkühlen der
gelben Lösung fällt nach und nach mehr HF-Salz aus, welches abfiltriert und mit
Toluen gewaschen wird. Das Filtrat wird über eine Destillation vom Lösungsmittel
und von unumgesetztem Isopropylamin befreit. Der gelb bis orangefarbene
Rückstand wird aus Methanol umkristallisiert.
4-Nitro- N-isopropylanilin
Die Modellverbindung wurde bereits im Rahmen meiner Diplomarbeit synthetisiert
und ist nur zur Vollständigkeit hier noch einmal aufgeführt.9
intensiv gelbe Kristalle
Schmelzpunkt: 83–85 °C
Ausbeute: 74 % (bezogen auf 4-Fluornitrobenzen).
NH
CH3
H3CNO2
H3C
H3CNH2+
im Überschuss
F
R
R: elektronen-ziehende(r) Substituent(en)
F- (CH3)2CHNH3
Toluen / 80°C
N
CH3
H3C
H R

5 Experimenteller Teil
181
2-Nitro- N-isopropylanilin
orangefarbenes Öl
Ausbeute: 97 % (bezogen auf 2-Fluornitrobenzen)
Elementaranalyse (kalkuliert): C: 60.032 (59.99), H: 6.661 (6.71), N: 15.329 (15.55).
1H-NMR (CDCl3; 300 MHz) δ [ppm]: 8.13 (d, 1H, H-3, 3J = 8.79 Hz), 7.99 (br. s, 1H,
-NH), 7.39 (dd, 1H, H-5, 3J = 7.69 Hz, 3J = 8.79 Hz),
6.84 (d, 1H, H-6, 3J = 8.79 Hz), 6.57 (dd, 1H, H-4, 3J = 7.69 H, 3J = 8.79 Hz),
3.82 (m, 1H, H-7, -CH-(CH3)2, 3J = 6.60 Hz), 1.31 (d, 6H, H-8, -CH-(CH3)2,
3J = 6.60 Hz).
Ein 1H-NMR-Spektrum von 2-Nitro-N-isopropylanilin in CDCl3 und die strukturelle
Zuordnung der 1H-Signale ist Abb. 67 mit Vergleich zu Literaturwerten von 2-Nitro-N-
isopropylanilin in CDCl3 gezeigt.130
Abb. 67: 1H-NMR-Spektrum von 2-Nitro-N-isopropylanilin in CDCl3 mit entsprechender Zuordnung der 1H-NMR-Signale mit Vergleich zu Literaturwerten.
NH
CH3
H3C
O2N
1
2 3
4
56
7
8
00.511.522.533.544.555.566.577.588.59
δ δ δ δ [ppm]
H-7
1H-NMR (CDCl3, 300 MHz) δδδδ [ppm]: Lit.: 130
H-3: 8.13 (d; 1H) 8.11 -NH: 7.99 (-NH; 1H) 8.00H-5: 7.39 (dd; 1H) 7.39 H-6: 6.84 (d;1H) 6.82 H-4: 6.57 (dd; 1H) 6.57H-7: 3.82 (m; 1H) 3.78H-8: 1.31 (d; 6H) 1.32
NH
CH3
H3C
O2N
1
2 3
4
56
7
8
H-8
H-3 H-4H-5H-6
-NH
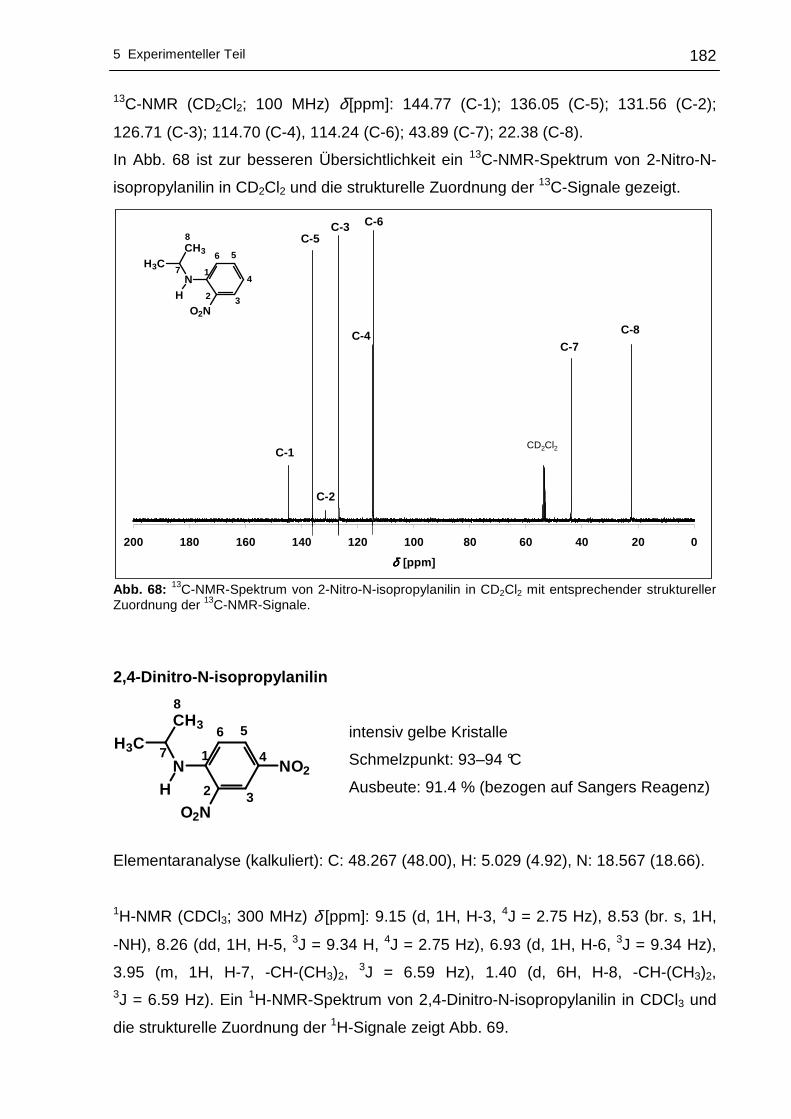
5 Experimenteller Teil
182
13C-NMR (CD2Cl2; 100 MHz) δ [ppm]: 144.77 (C-1); 136.05 (C-5); 131.56 (C-2);
126.71 (C-3); 114.70 (C-4), 114.24 (C-6); 43.89 (C-7); 22.38 (C-8).
In Abb. 68 ist zur besseren Übersichtlichkeit ein 13C-NMR-Spektrum von 2-Nitro-N-
isopropylanilin in CD2Cl2 und die strukturelle Zuordnung der 13C-Signale gezeigt.
Abb. 68: 13C-NMR-Spektrum von 2-Nitro-N-isopropylanilin in CD2Cl2 mit entsprechender struktureller Zuordnung der 13C-NMR-Signale.
2,4-Dinitro- N-isopropylanilin
intensiv gelbe Kristalle
Schmelzpunkt: 93–94 °C
Ausbeute: 91.4 % (bezogen auf Sangers Reagenz)
Elementaranalyse (kalkuliert): C: 48.267 (48.00), H: 5.029 (4.92), N: 18.567 (18.66).
1H-NMR (CDCl3; 300 MHz) δ [ppm]: 9.15 (d, 1H, H-3, 4J = 2.75 Hz), 8.53 (br. s, 1H,
-NH), 8.26 (dd, 1H, H-5, 3J = 9.34 H, 4J = 2.75 Hz), 6.93 (d, 1H, H-6, 3J = 9.34 Hz),
3.95 (m, 1H, H-7, -CH-(CH3)2, 3J = 6.59 Hz), 1.40 (d, 6H, H-8, -CH-(CH3)2,
3J = 6.59 Hz). Ein 1H-NMR-Spektrum von 2,4-Dinitro-N-isopropylanilin in CDCl3 und
die strukturelle Zuordnung der 1H-Signale zeigt Abb. 69.
NH
CH3
H3C
O2N
NO21
2 3
4
56
7
8
020406080100120140160180200
δδδδ [ppm]
NH
CH3
H3C
O2N
1
2 3
4
56
7
8
C-1
C-5
C-2
C-3
C-4
C-6
C-7
C-8
CD2Cl2
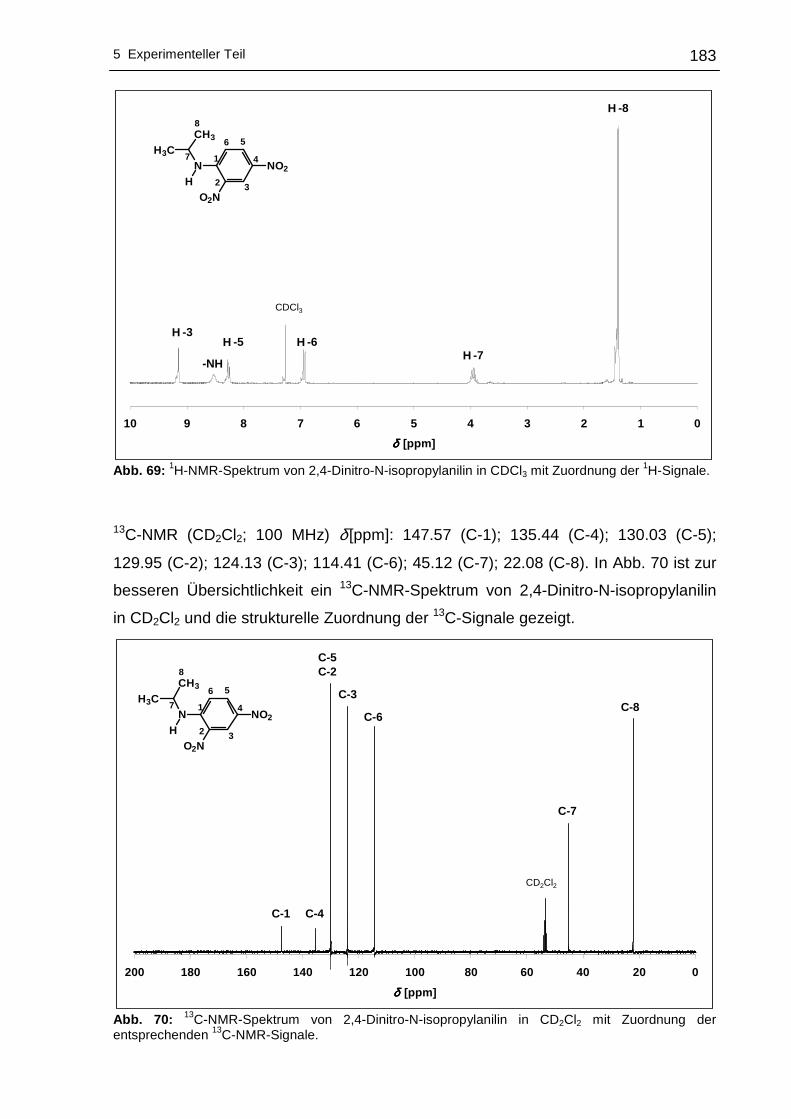
5 Experimenteller Teil
183
Abb. 69: 1H-NMR-Spektrum von 2,4-Dinitro-N-isopropylanilin in CDCl3 mit Zuordnung der 1H-Signale.
13C-NMR (CD2Cl2; 100 MHz) δ [ppm]: 147.57 (C-1); 135.44 (C-4); 130.03 (C-5);
129.95 (C-2); 124.13 (C-3); 114.41 (C-6); 45.12 (C-7); 22.08 (C-8). In Abb. 70 ist zur
besseren Übersichtlichkeit ein 13C-NMR-Spektrum von 2,4-Dinitro-N-isopropylanilin
in CD2Cl2 und die strukturelle Zuordnung der 13C-Signale gezeigt.
Abb. 70: 13C-NMR-Spektrum von 2,4-Dinitro-N-isopropylanilin in CD2Cl2 mit Zuordnung der entsprechenden 13C-NMR-Signale.
012345678910
δδδδ [ppm]
H -3
H -8
H -7H -6H -5
-NH
CDCl3
NH
CH3
H3C
O2N
NO21
2 3
4
56
7
8
020406080100120140160180200
δδδδ [ppm]
NH
CH3
H3C
O2N
NO21
2 3
4
56
7
8
C-1
C-5C-2
C-3
C-6
C-7
C-8
C-4
CD2Cl2
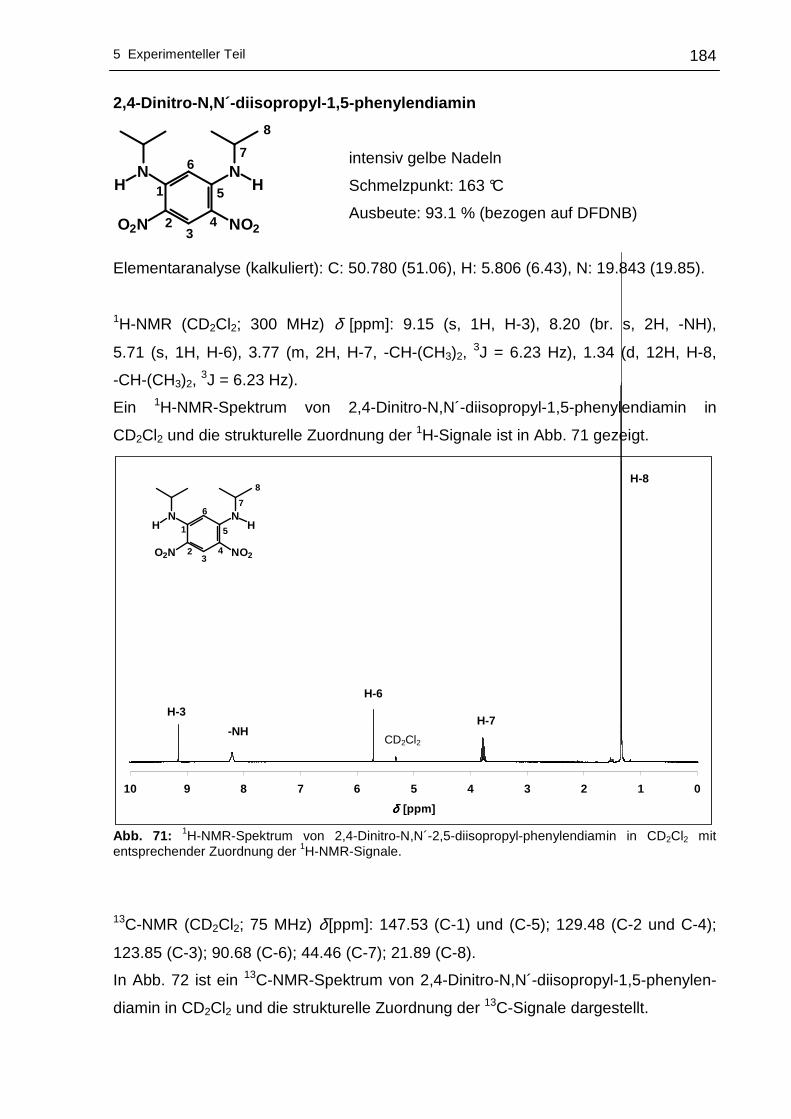
5 Experimenteller Teil
184
2,4-Dinitro- N,N´-diisopropyl-1,5-phenylendiamin
intensiv gelbe Nadeln
Schmelzpunkt: 163 °C
Ausbeute: 93.1 % (bezogen auf DFDNB)
Elementaranalyse (kalkuliert): C: 50.780 (51.06), H: 5.806 (6.43), N: 19.843 (19.85).
1H-NMR (CD2Cl2; 300 MHz) δ [ppm]: 9.15 (s, 1H, H-3), 8.20 (br. s, 2H, -NH),
5.71 (s, 1H, H-6), 3.77 (m, 2H, H-7, -CH-(CH3)2, 3J = 6.23 Hz), 1.34 (d, 12H, H-8,
-CH-(CH3)2, 3J = 6.23 Hz).
Ein 1H-NMR-Spektrum von 2,4-Dinitro-N,N´-diisopropyl-1,5-phenylendiamin in
CD2Cl2 und die strukturelle Zuordnung der 1H-Signale ist in Abb. 71 gezeigt.
Abb. 71: 1H-NMR-Spektrum von 2,4-Dinitro-N,N´-2,5-diisopropyl-phenylendiamin in CD2Cl2 mit entsprechender Zuordnung der 1H-NMR-Signale.
13C-NMR (CD2Cl2; 75 MHz) δ [ppm]: 147.53 (C-1) und (C-5); 129.48 (C-2 und C-4);
123.85 (C-3); 90.68 (C-6); 44.46 (C-7); 21.89 (C-8).
In Abb. 72 ist ein 13C-NMR-Spektrum von 2,4-Dinitro-N,N´-diisopropyl-1,5-phenylen-
diamin in CD2Cl2 und die strukturelle Zuordnung der 13C-Signale dargestellt.
NN
NO2O2N
HH 1
23
4
5
67
8
012345678910
δδδδ [ppm]
H-3
-NH
H-6
H-7
H-8
CD2Cl2
NN
NO2O2N
HH 1
23
4
5
67
8
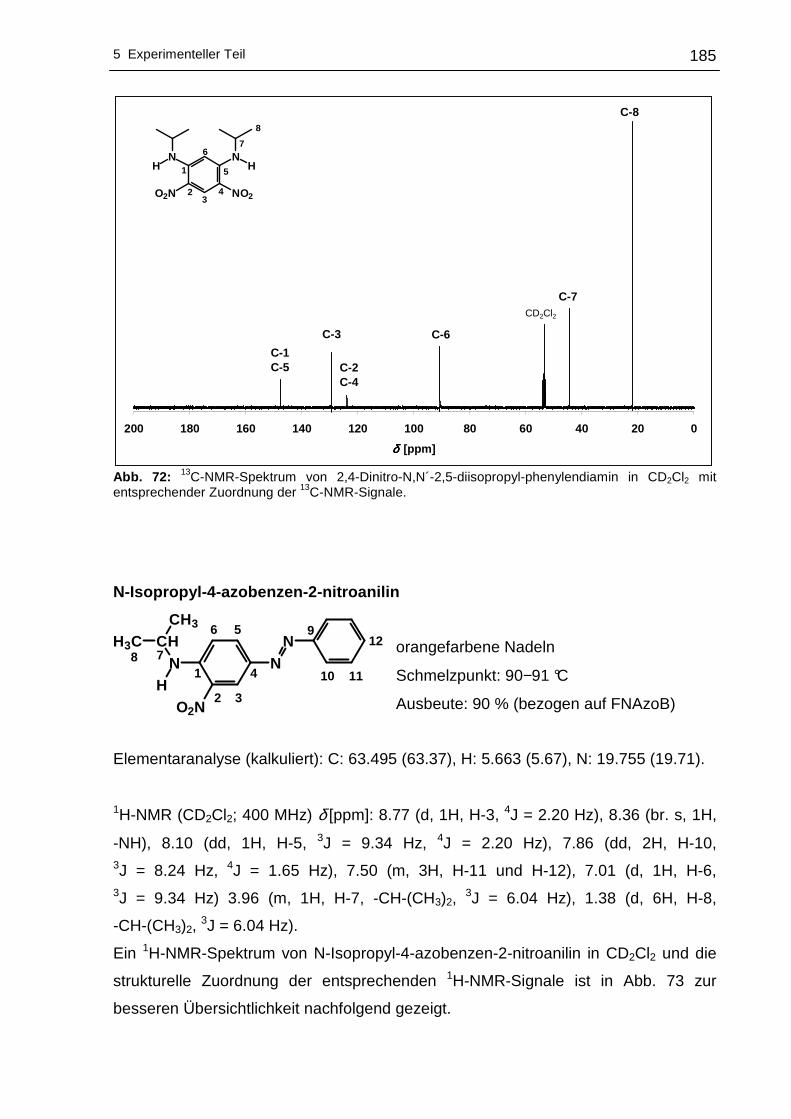
5 Experimenteller Teil
185
Abb. 72: 13C-NMR-Spektrum von 2,4-Dinitro-N,N´-2,5-diisopropyl-phenylendiamin in CD2Cl2 mit entsprechender Zuordnung der 13C-NMR-Signale.
N-Isopropyl-4-azobenzen-2-nitroanilin
orangefarbene Nadeln
Schmelzpunkt: 90−91 °C
Ausbeute: 90 % (bezogen auf FNAzoB)
Elementaranalyse (kalkuliert): C: 63.495 (63.37), H: 5.663 (5.67), N: 19.755 (19.71).
1H-NMR (CD2Cl2; 400 MHz) δ [ppm]: 8.77 (d, 1H, H-3, 4J = 2.20 Hz), 8.36 (br. s, 1H,
-NH), 8.10 (dd, 1H, H-5, 3J = 9.34 Hz, 4J = 2.20 Hz), 7.86 (dd, 2H, H-10, 3J = 8.24 Hz, 4J = 1.65 Hz), 7.50 (m, 3H, H-11 und H-12), 7.01 (d, 1H, H-6, 3J = 9.34 Hz) 3.96 (m, 1H, H-7, -CH-(CH3)2,
3J = 6.04 Hz), 1.38 (d, 6H, H-8,
-CH-(CH3)2, 3J = 6.04 Hz).
Ein 1H-NMR-Spektrum von N-Isopropyl-4-azobenzen-2-nitroanilin in CD2Cl2 und die
strukturelle Zuordnung der entsprechenden 1H-NMR-Signale ist in Abb. 73 zur
besseren Übersichtlichkeit nachfolgend gezeigt.
NCH
NN
O2NH
CH3
H3C
1
2 3
4
56
78
9
10 11
12
020406080100120140160180200
δδδδ [ppm]
C-1C-5 C-2
C-4
C-3 C-6
C-7
C-8
CD2Cl2
NN
NO2O2N
HH 1
23
4
5
67
8
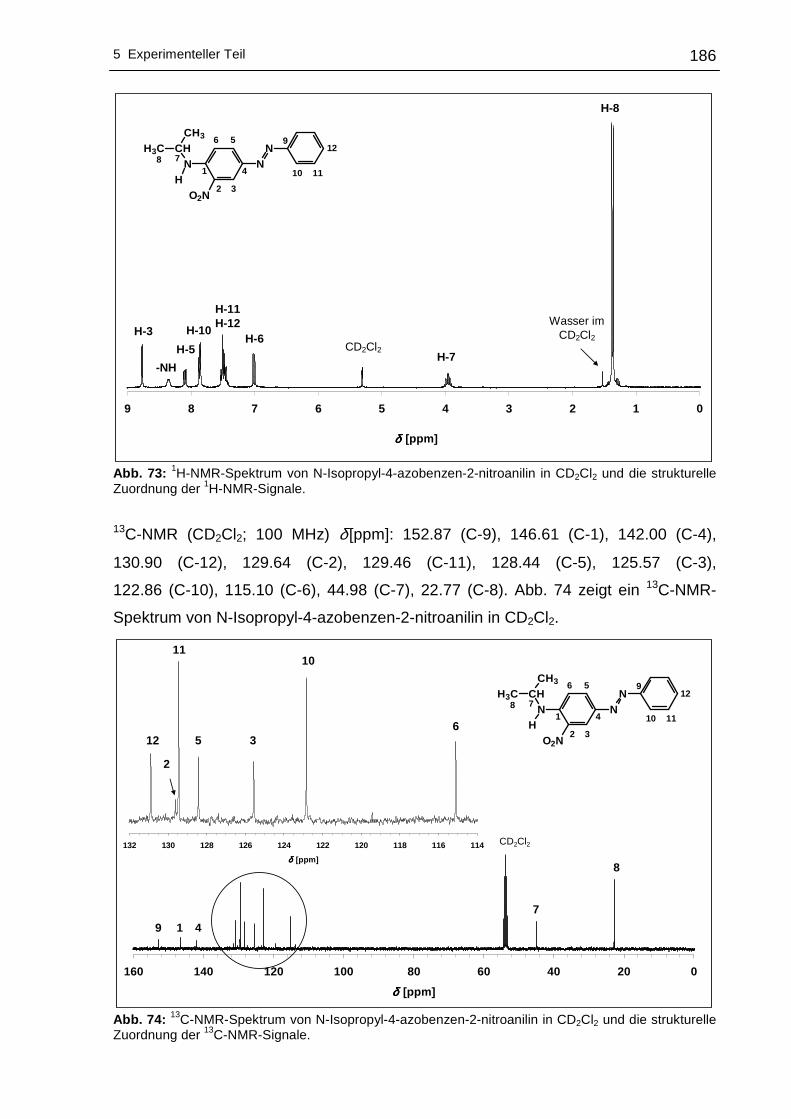
5 Experimenteller Teil
186
Abb. 73: 1H-NMR-Spektrum von N-Isopropyl-4-azobenzen-2-nitroanilin in CD2Cl2 und die strukturelle Zuordnung der 1H-NMR-Signale. 13C-NMR (CD2Cl2; 100 MHz) δ [ppm]: 152.87 (C-9), 146.61 (C-1), 142.00 (C-4),
130.90 (C-12), 129.64 (C-2), 129.46 (C-11), 128.44 (C-5), 125.57 (C-3),
122.86 (C-10), 115.10 (C-6), 44.98 (C-7), 22.77 (C-8). Abb. 74 zeigt ein 13C-NMR-
Spektrum von N-Isopropyl-4-azobenzen-2-nitroanilin in CD2Cl2.
Abb. 74: 13C-NMR-Spektrum von N-Isopropyl-4-azobenzen-2-nitroanilin in CD2Cl2 und die strukturelle Zuordnung der 13C-NMR-Signale.
0123456789
δδδδ [ppm]
H-3
-NH
H-5
H-10
H-11H-12
H-6
H-7
H-8
CD2Cl2
Wasser imCD2Cl2
NCH
NN
O2NH
CH3
H3C
1
2 3
4
56
78
9
10 11
12
020406080100120140160
δδδδ [ppm]
114116118120122124126128130132
δδδδ [ppm]
9 1 4
12
2
11
5 3
10
6
7
8
CD2Cl2
NCH
NN
O2NH
CH3
H3C
1
2 3
4
56
78
9
10 11
12

5 Experimenteller Teil
187
4-N-Isopropylamino-3,4´-dinitrostilben orangerote Kristalle
Schmelzpunkt: 167−169 °C
Ausbeute: 47 % (bezogen auf
FDNstilben)
Elementaranalyse (kalkuliert): C: 62.10 (62.38), H: 4.77 (5.23), N: 12.56 (12.84).
1H-NMR (d6-Aceton; 400 MHz) δ [ppm]: 8.35 (d, 1H, H-2, 4J = 2.20 Hz), 8.23 (dd, 1H,
H-3´, 3J = 9.16 Hz, 4J = 2.20 Hz), 8.14 (br. s, 1H, -NH), 7.95 (dd, 1H, H-6, 3J = 9.16 Hz, 4J = 2.20 Hz), 7.85 dd, 2H, H-2´, 3J = 9.16 Hz, 4J = 2.20 Hz),
7.52 (d, 1H, H-10, 3Jtrans = 16. 48 Hz), 7.31 (d, 1H, H-9, 3Jtrans = 16. 48 Hz),
7.19 (d, 1H, H-5, 3J = 9.16 Hz), 4.05 (m, 1H, H-7, -CH-(CH3)2, 3J = 6.23 Hz),
1.37 (d, 6H, H-8, -CH-(CH3)2, 3J = 6.23 Hz).
In Abb. 75 ist ein 1H-NMR-Spektrum von 4-N-Isopropylamino-3,4´-dinitrostilben in
d6-Aceton mit der strukturellen Zuordnung der 1H-NMR-Signale dargestellt.
Abb. 75: 1H-NMR-Spektrum von 4-N-Isopropylamino-3,4´-dinitrostilben in d6-Aceton und die strukturelle Zuordnung der 1H-NMR-Signale.
0123456789
δδδδ [ppm]
7.07.27.47.67.88.08.28.48.6
δδδδ [ppm]
d6-Aceton
Wasser imd6-Aceton
H-8
H-7
H-10 H-9H-5
H-2´H-3´
H-2
H-6
-NH
NCH
O2NH
CH3
H3C NO2
1
23
4
5 6
789
10
1´
2´ 3´
4´
NCH
O2NH
CH3
H3C NO2
1
23
4
5 6
789
10
1´
2´ 3´
4´
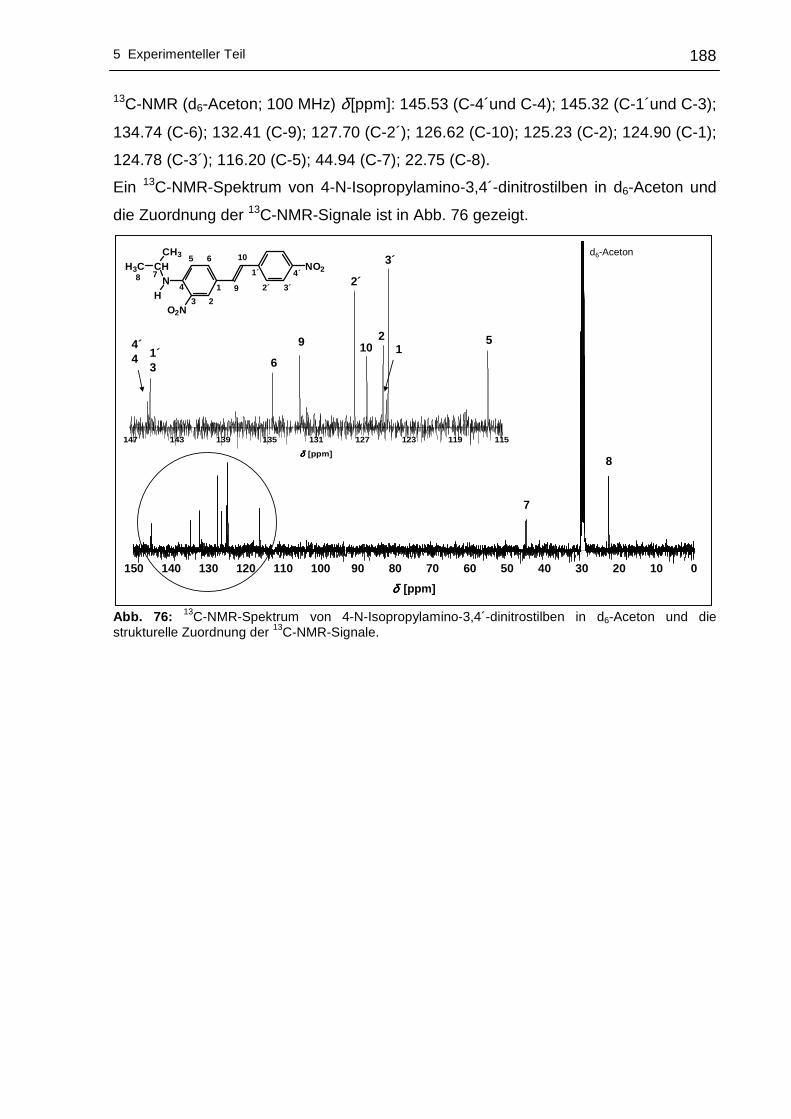
5 Experimenteller Teil
188
13C-NMR (d6-Aceton; 100 MHz) δ [ppm]: 145.53 (C-4´und C-4); 145.32 (C-1´und C-3);
134.74 (C-6); 132.41 (C-9); 127.70 (C-2´); 126.62 (C-10); 125.23 (C-2); 124.90 (C-1);
124.78 (C-3´); 116.20 (C-5); 44.94 (C-7); 22.75 (C-8).
Ein 13C-NMR-Spektrum von 4-N-Isopropylamino-3,4´-dinitrostilben in d6-Aceton und
die Zuordnung der 13C-NMR-Signale ist in Abb. 76 gezeigt.
Abb. 76: 13C-NMR-Spektrum von 4-N-Isopropylamino-3,4´-dinitrostilben in d6-Aceton und die strukturelle Zuordnung der 13C-NMR-Signale.
0102030405060708090100110120130140150
δδδδ [ppm]
NCH
O2NH
CH3
H3C NO2
1
23
4
5 6
789
10
1´
2´ 3´
4´
4´4 1´
3 6
9
2´
3´
102
15
7
8
d6-Aceton
115119123127131135139143147
δδδδ [ppm]
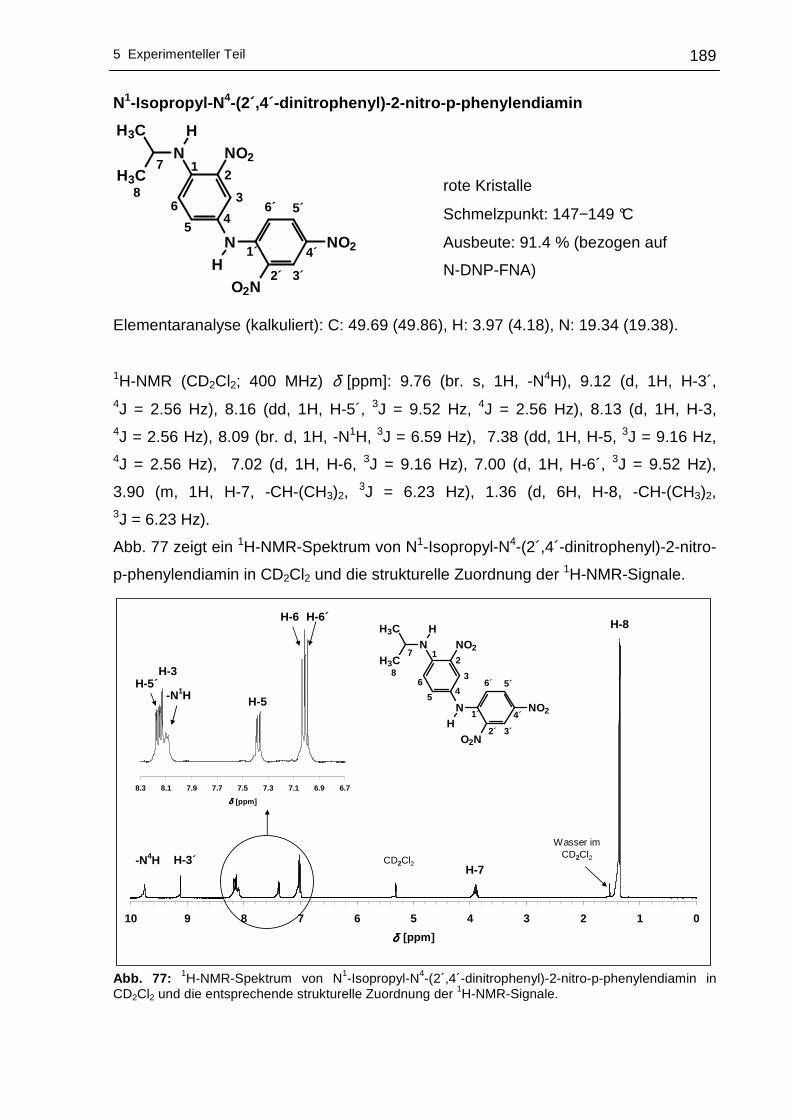
5 Experimenteller Teil
189
N1-Isopropyl- N4-(2´,4´-dinitrophenyl)-2-nitro- p-phenylendiamin
rote Kristalle
Schmelzpunkt: 147−149 °C
Ausbeute: 91.4 % (bezogen auf
N-DNP-FNA)
Elementaranalyse (kalkuliert): C: 49.69 (49.86), H: 3.97 (4.18), N: 19.34 (19.38).
1H-NMR (CD2Cl2; 400 MHz) δ [ppm]: 9.76 (br. s, 1H, -N4H), 9.12 (d, 1H, H-3´, 4J = 2.56 Hz), 8.16 (dd, 1H, H-5´, 3J = 9.52 Hz, 4J = 2.56 Hz), 8.13 (d, 1H, H-3, 4J = 2.56 Hz), 8.09 (br. d, 1H, -N1H, 3J = 6.59 Hz), 7.38 (dd, 1H, H-5, 3J = 9.16 Hz, 4J = 2.56 Hz), 7.02 (d, 1H, H-6, 3J = 9.16 Hz), 7.00 (d, 1H, H-6´, 3J = 9.52 Hz),
3.90 (m, 1H, H-7, -CH-(CH3)2, 3J = 6.23 Hz), 1.36 (d, 6H, H-8, -CH-(CH3)2,
3J = 6.23 Hz).
Abb. 77 zeigt ein 1H-NMR-Spektrum von N1-Isopropyl-N4-(2´,4´-dinitrophenyl)-2-nitro-
p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 1H-NMR-Signale.
Abb. 77: 1H-NMR-Spektrum von N1-Isopropyl-N4-(2´,4´-dinitrophenyl)-2-nitro-p-phenylendiamin in CD2Cl2 und die entsprechende strukturelle Zuordnung der 1H-NMR-Signale.
012345678910
δδδδ [ppm]
6.76.97.17.37.57.77.98.18.3
δδδδ [ppm]
-N4H H-3´H-7
CD2Cl2
Wasser imCD2Cl2
N
NO2
O2N
N
H3C
H3C
H
H
NO21
2
3
45
6
1´
2´ 3´
4´
5´6´
7
8
H-8
H-5´H-3
-N1H H-5
H-6 H-6´
N
NO2
O2N
N
H3C
H3C
H
H
NO21
2
3
45
6
1´
2´ 3´
4´
5´6´
7
8
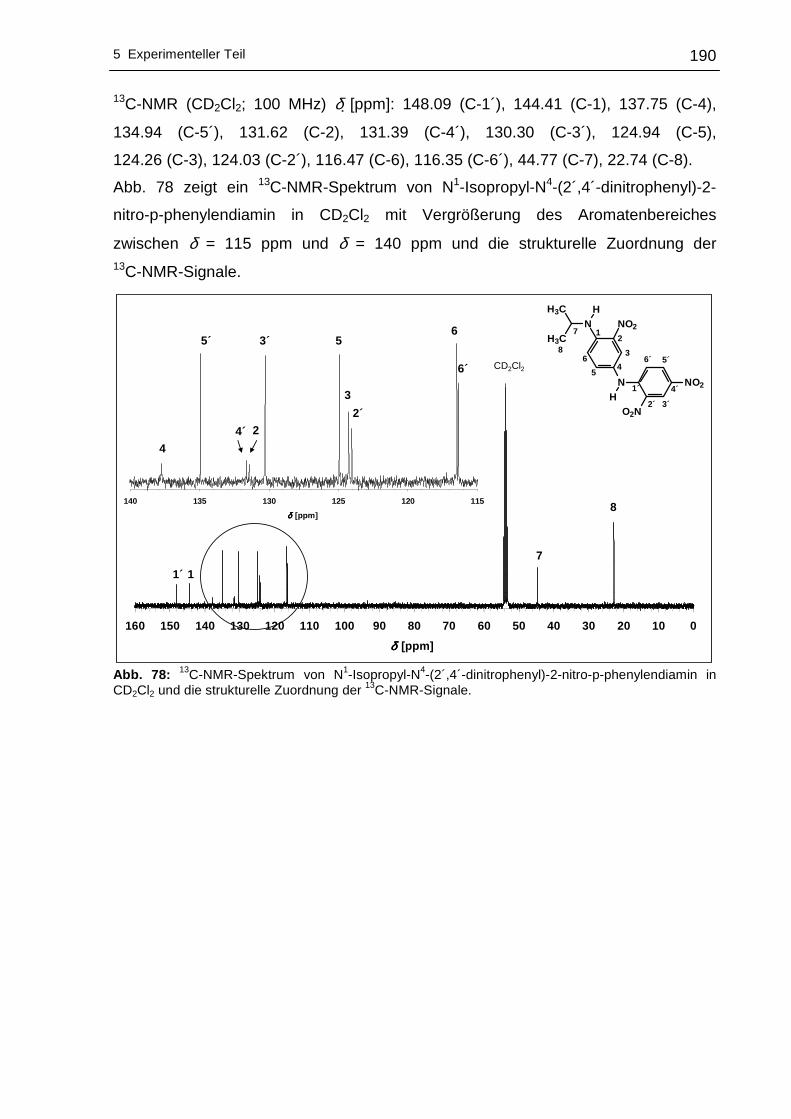
5 Experimenteller Teil
190
13C-NMR (CD2Cl2; 100 MHz) δ [ppm]: 148.09 (C-1´), 144.41 (C-1), 137.75 (C-4),
134.94 (C-5´), 131.62 (C-2), 131.39 (C-4´), 130.30 (C-3´), 124.94 (C-5),
124.26 (C-3), 124.03 (C-2´), 116.47 (C-6), 116.35 (C-6´), 44.77 (C-7), 22.74 (C-8).
Abb. 78 zeigt ein 13C-NMR-Spektrum von N1-Isopropyl-N4-(2´,4´-dinitrophenyl)-2-
nitro-p-phenylendiamin in CD2Cl2 mit Vergrößerung des Aromatenbereiches
zwischen δ = 115 ppm und δ = 140 ppm und die strukturelle Zuordnung der 13C-NMR-Signale.
Abb. 78: 13C-NMR-Spektrum von N1-Isopropyl-N4-(2´,4´-dinitrophenyl)-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 13C-NMR-Signale.
0102030405060708090100110120130140150160
δδδδ [ppm]
N
NO2
O2N
N
H3C
H3C
H
H
NO21
2
3
45
6
1´
2´ 3´
4´
5´6´
7
8
115120125130135140
δδδδ [ppm]
7
8
CD2Cl2
1´ 1
4
5´ 3´ 5
3
6
6´
2´
4´ 2

5 Experimenteller Teil
191
N1-Isopropyl- N4-(2´-nitrophenyl-4´-( p-N,N-diethyl)-azobenzen)-2-nitro- p-
phenylendiamin
dunkelrot bis violette Kristalle
Schmelzpunkt: 220−222 °C
Ausbeute: 87.1 % (bezogen auf
FNAzoB-2)
Elementaranalyse (kalkuliert): C: 60.90 (61.09), H: 5.34 (5.95), N: 19.86 (19.95).
1H-NMR (CD2Cl2; 400 MHz) δ [ppm]: 9.05 (br. s, 1H, -N4H, -NH), 8.64 (d, 1H, H-3´, 4J = 2.56 Hz), 8.13 (d, 1H, H-3, 4J = 2.56 Hz), 8.06 (br. d, 1H, -N1H, 3J = 7.33 Hz),
7.95 (dd, 1H, H-5´, 3J = 9.16 Hz, 4J = 2.56 Hz), 7.78 (d, 2H, H-10, 3J = 9.16 Hz),
7.42 (d, 2H, H-5, 3J = 9.16 Hz), 7.06 (d, 1H, H-6´, 3J = 9.16 Hz), 6.99 (d, 1H, H-6, 3J = 9.16 Hz), 6.72 (d, 2H, H-11, 3J = 9.16 Hz), 3.89 (m, 1H, H-7, -CH-(CH3)2, 3J = 6.59 Hz), 3.44 (qu, 4H, H-13, -CH2-CH3,
3J = 6.96 Hz), 1.35 (d, 6H, H-8,
-CH-(CH3)2, 3J = 6.59 Hz), 1.21 (tr, 6H, H-14, -CH2-CH3,
3J = 6.96 Hz).
Abb. 79 zeigt ein 1H-NMR-Spektrum von N1-Isopropyl-N4-(2´-nitrophenyl-4´-(N,N-
diethyl)-azobenzen)-2-nitro-p-phenylendiamin in CD2Cl2.
Abb. 79: 1H-NMR-Spektrum von N1-Isopropyl-N4-(2´-nitrophenyl-4´-(N,N-diethyl)-azobenzen)-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 1H-NMR-Signale.
NO2NH
NH
NN N
O2N
12
3
45
6
7
8
9
10 11
12
13 14
1´
2´ 3´
4´
5´6´
012345678910
δδδδ [ppm]
-N4H H-3´H-7
H-13
H-8
H-14
CD2Cl2
Wasser imCD2Cl2
6.46.66.877.27.47.67.888.28.4
δδδδ [ppm]
H-3 -N1H
H-5´
H-10
H-5
H-6´
H-6
H-11NH
NH
NO2
N
O2N
N
N
1
23
4
56
7
8
910
1112
1´
2´
3´4´
5´
6´
13
14
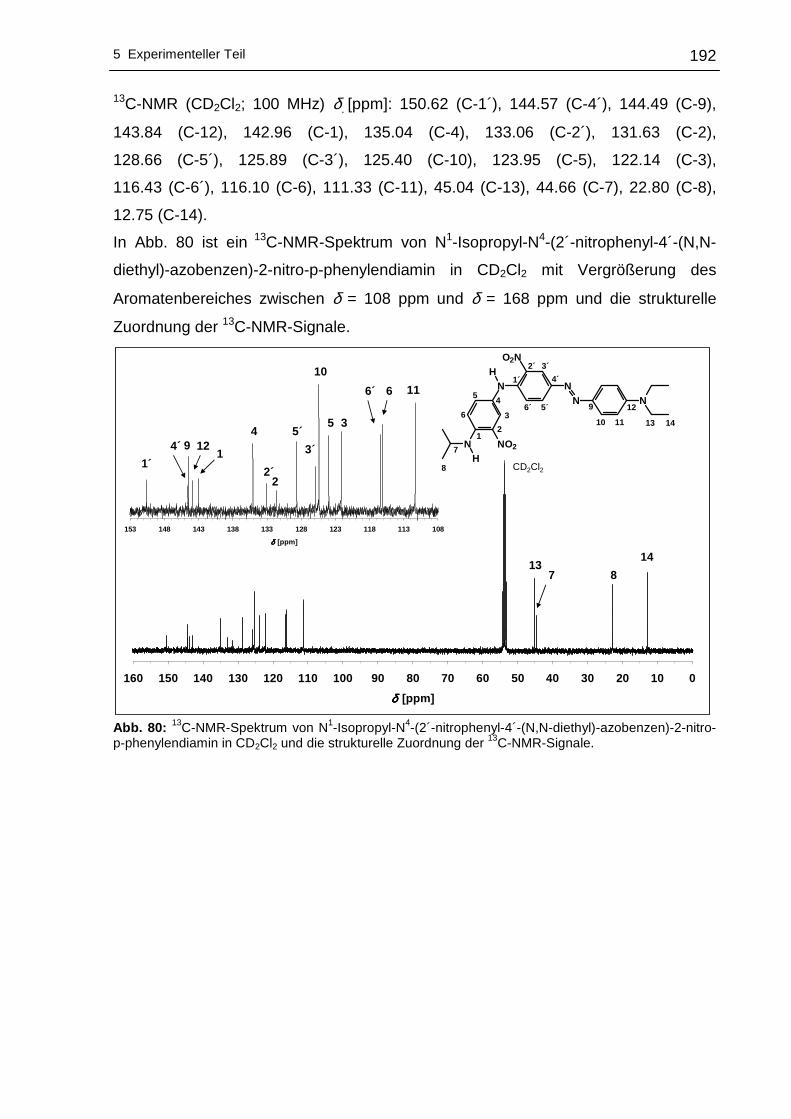
5 Experimenteller Teil
192
13C-NMR (CD2Cl2; 100 MHz) δ �[ppm]: 150.62 (C-1´), 144.57 (C-4´), 144.49 (C-9),
143.84 (C-12), 142.96 (C-1), 135.04 (C-4), 133.06 (C-2´), 131.63 (C-2),
128.66 (C-5´), 125.89 (C-3´), 125.40 (C-10), 123.95 (C-5), 122.14 (C-3),
116.43 (C-6´), 116.10 (C-6), 111.33 (C-11), 45.04 (C-13), 44.66 (C-7), 22.80 (C-8),
12.75 (C-14).
In Abb. 80 ist ein 13C-NMR-Spektrum von N1-Isopropyl-N4-(2´-nitrophenyl-4´-(N,N-
diethyl)-azobenzen)-2-nitro-p-phenylendiamin in CD2Cl2 mit Vergrößerung des
Aromatenbereiches zwischen δ = 108 ppm und δ = 168 ppm und die strukturelle
Zuordnung der 13C-NMR-Signale.
Abb. 80: 13C-NMR-Spektrum von N1-Isopropyl-N4-(2´-nitrophenyl-4´-(N,N-diethyl)-azobenzen)-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 13C-NMR-Signale.
0102030405060708090100110120130140150160
δδδδ [ppm]
108113118123128133138143148153
δδδδ [ppm]
NO2NH
NH
NN N
O2N
12
3
45
6
7
8
9
10 11
12
13 14
1´
2´ 3´
4´
5´6´
1´
9 121
4´
2´
4
2
5´
3´
10
5 3
66´ 11
137 8
14
CD2Cl2

5 Experimenteller Teil
193
N1-Isopropyl- N1,N4,N4-trimethyl-2-nitro- p-phenylendiamin
In 30 ml Toluol werden 0.5 g (2.715 mmol) N,N-Dimethyl-4-fluor-3-nitroanilin
(4-DMFNA) gelöst. Die orangefarbene Lösung wird auf 60 °C erhitzt und 1.4 ml
von N-Isopropylmethylamin (Fluka) − 5-facher Überschuss zu 4-DMFNA − werden
unter Schutzgas in die Lösung eingespritzt. Die Reaktionslösung wird für 14 h bei
60 °C gerührt. Dabei färbt sich die Lösung nach und nach intensiver orange-rot. Ein
Ausfallen von N-Isopropylmethylamin-HF-Salz ist nicht zu beobachten. Anschließend
wird der Großteil an Toluen und möglicherweise unumgesetztes Amin über eine
Destillation entfernt. Der rote, ölige Rückstand wird zur Entfernung von
möglicherweise enthaltenem HF-Salz in Wasser aufgenommen und das Produkt
über einen Scheidetrichter mit Diethylether abgetrennt. Die rote Etherphase wird am
Rotationsverdampfer vom Lösungsmittel befreit und ein rotes Öl bleibt zurück.
rotes Öl
Ausbeute: 67.8 % (bezogen auf 4-DMFNA)
1H-NMR (CD2Cl2; 400 MHz) δ [ppm]: 7.14 (d, 1H, H-6, 3J = 8.59 Hz), 6.86 (dd, 1H,
H-5, 3J = 8.59 Hz, 4J = 3.13 Hz), 6.84 (d, 1H, H-3, 4J = 3.13 Hz), 3.22 (m, 1H, H-7,
-CH-(CH3)2, 3J = 6.25 Hz), 2.91 (s, 6H, H-10, N4-(CH3)2), 2.60 (s, 3H, H-9, N1-CH3),
1.37 (d, 6H, H-8, -CH-(CH3)2, 3J = 6.25 Hz).
13C-NMR (CD2Cl2; 100 MHz) δ �[ppm]: 147.44 (C-1); 146.78 (C-4); 135.52 (C-2);
125.24 (C-5); 116. 77 (C-6); 107.19 (C-3); 55.00 (C-9); 40.70 (C-10); 35.89 (C-7);
19.70 (C-8).
Ein 1H-NMR-Spektrum von N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin
in CD2Cl2 ist mit Vergrößerung des Aromatenbereiches zwischen δ = 6.8 ppm und
δ = 7.2 ppm und der strukturellen Zuordnung der 1H-NMR-Signale in Abb. 81
dargestellt. Abb. 82 zeigt ein 13C-NMR-Spektrum von N1-Isopropyl-N1,N4,N4-
trimethyl-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der
entsprechenden 13C-NMR-Signale.
N
O2N
CH3
CH3
NH3C
CH3
H3C1
2 3
4
56
7
8
9 10

5 Experimenteller Teil
194
Abb. 81: 1H-NMR-Spektrum von N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 1H-NMR-Signale.
Abb. 82: 13C-NMR-Spektrum von N1-Isopropyl-N1,N4,N4-trimethyl-2-nitro-p-phenylendiamin in CD2Cl2 und die strukturelle Zuordnung der 13C-NMR-Signale.
00.511.522.533.544.555.566.577.58
δδδδ [ppm]
6.86.856.96.9577.057.17.157.2
δδδδ [ppm]
CD2Cl2
N
O2N
CH3
CH3
NH3C
CH3
H3C1
2 3
4
56
7
8
9 10
H-7
H-10
H-9
H-8
RSB
H-6H-3
H-5
0102030405060708090100110120130140150160
δδδδ [ppm]
N
O2N
CH3
CH3
NH3C
CH3
H3C1
2 3
4
56
7
8
9 10
41 2
5
6
3
7
10
9
8
CD2Cl2

Literaturverzeichnis
195
6 Literaturverzeichnis
[1] Dautzenberg, H.; Jäger, W.; Kötz, J.; Phillip, J.; Seidel, C.; Stscherbina, D. Polyelectrolytes: Formation, Characterization and Application; Carl Hanser Verlag, München, 1994. [2] Technisches Merkblatt N-Vinylformamid, Mitsubishi Kasai, Japan, 1997. [3] Pinschmidt Jr., R. K.; Sagl, D. J. Polymeric Materials Encyclopedia; ed.: Salamone, J. C.; CRC Press New York, 1996, S. 7095 ff. [4] Gu, L.; Zhu, S.; Hrymak, A. N.; Pelton, R. H. Polymer 2001, 42, 3077–3086. [5] Gu, L.; Zhu, S.; Hrymak J. Appl. Polym. Sci. 2002, 86, 3412–3419. [6] Kröner, M.; Dupuis, J., Winter, M. J. Prakt. Chem. 2000, 342, 115–131. [7] Produkt-Informationsbroschüre zu Lupaminen®, BASF AG, Ludwigshafen,
2000. [8] Pinschmidt, R. K.; Renz, W. L.; Caroll, W. E.; Yacoub, K.; Drescher, J.; Nordquist, A. F.; Chen, N. J. Macromol. Sci.-Pure Appl. Chem. 1997, A34, 1885–1905. [9] Roth, I. Diplomarbeit 2001, TU Chemnitz, Fakultät für Naturwissenschaften, Institut für Chemie, Professur Polymerchemie (Prof. S. Spange). [10] Roth, I.; Spange, S. Macromol. Rapid Commun. 2001, 22, 1288–1291. [11] Meier, H. Angew. Chem. 2005, 117, 2536–2561. [12] Suhr, H. Chem. Ber. 1964, 97, 3277–3283. [13] Roth, I.; Spange, S. Macromolecules 2005, 38, 10034–10041. [14] Suhr, H. Tetrahedron Letters 1966, 47, 5871–5874. [15] Bellamy, A. J.; King, D. S.; Golding, P. Propellants, Explosives, Pyrotechnics 2004, 29, 166–177. [16] Broxton, T. J.; Christie, J. R.; Theodoridis, D. J. Phys. Org. Chem. 1993, 6, 535–538. [17] Correa, N. M.; Durantini, E. N.; Silber, J. J. J. Org. Chem. 2000, 65, 6427–
6433. [18] Zingaretti, L.; Boscatto, L.; Chiacchiera, S. M.; Silber, J. J. ARCIVOC 2003, 189–200. [19] 1) Jeromin, J., Noll, O., Ritter, H. Macromol. Chem. Phys. 1998, 199, 2641– 2645.
2) Jeromin, J., Ritter, H. Macromolecules 1999, 32, 5236–5239. 3) Glöckner, P., H. Ritter, H. Macromol. Rapid Commun. 1999, 20, 602–605. 4) Fischer, M., Ritter, H. Macromol. Rapid Commun. 2000, 21, 142–145. 5) Storsberg, J., Ritter, H. Macromol. Rapid Commun. 2000, 21, 236–241. 6) Glöckner, P., Metz, N., Ritter, H. Macromolecules 2000, 33, 4288–4290. 7) Casper, P. Glöckner, P., Ritter, H. Macromolecules 2000, 33, 4361–4364. 8) Reihmann, M. H., Ritter, H. Macromol. Chem. Phys. 2000, 201, 798–804. 9) Glöckner, P.; Ritter, H. Macromol. Chem. Phys. 2000, 201, 2455–2457.
10) Alupei, V., Ritter, H. Macromol. Rapid Commun. 2001, 22, 1349–1353. 11) Bernhardt, S., Glöckner, P., Theis, A.; Ritter, H. Macromolecules 2001, 34, 1647–1649. 12) Storsberg, J.; Glöckner, P.; Eigner, M.; Schnoller, U.; Ritter, H.; Voit, B.; Nuyken, O. Designed Monomers and Polymers 2001, 4, 9–17.

Literaturverzeichnis
196
13) Ritter, H., Tabatabai, M. Prog. Polym. Sci. 2002, 27, 1713–1720. 14) Storsberg, J., Ritter, H. Macromol. Chem. Phys. 2002, 203, 812–818. 15) Heinenberg, M.; Ritter, H. Macromol. Chem. Phys. 2002, 203, 1804–1810. 16) Alupei, I. C.; Alupei, V.; Ritter, H. Macromol. Rapid Commun. 2002, 23, 55– 58. 17) Ritter, H.; Schmelzer, M.; Tabatabai, M. e-Polymers 2002, no. 003. 18) Tonami, H.; Uyama, H.; Kobayashi, S.; Reihmann, S.; Ritter, H. e-Polymers 2002, no. 039. 19) Theis, A.; Ritter, H. Macromol. Chem. Phys. 2003, 204, 1297–1304. 20) Choi, S. W.; Kretschmann, O.; Ritter, H.; Ragnoli, M.; Galli, G. Macromol. Chem. Phys. 2003, 204, 1475–1479. 21) Storsberg, J.; van Aert, H.; van Roost, C.; Ritter, H. Macromolecules 2003, 36, 50–53. 22) Ritter, H.; Schwarz-Barac, S.; Stein, P. Macromolecules 2003, 36, 318–322. 23) Pang, Y. J.; Ritter, H.; Tabatabai, M. Macromolecules 2003, 36, 7090–7093. 24) Mejias, L.; Schollmeyer, D.; Sepulveda-Boza, S.; Ritter, H. Macromolecular Bioscience 2003, 3, 395–399. 25) Schwarz-Barac, S.; Ritter, H. J. Macromol. Sci.-Pure Appl. Chem. 2003, A40, 437–448. 26) Alupei, I. C.; Alupei, V.; Ritter, H. Macromol. Rapid Commun. 2003, 24, 527– 531. 27) Choi, S. W.; Ritter, H. Macromol. Rapid Commun. 2004, 25, 716–719. 28) Sarvothaman, M. K.; Ritter, H. Macromol. Rapid Commun. 2004, 25, 1948– 1952. 29) Choi, S.; Ritter, H. J. Macromol. Sci.-Pure Appl. Chem. 2005, A42, 321– 325. 30) Ritter, H.; Steffens, C.; Storsberg, J. e-Polymers 2005, no. 034. 31) Cinar, H.; Kretschmann, O.; Ritter, H. Macromolecules 2005, 38, 5078– 5082. 32) Choi, S. W.; Frank, W.; Ritter, H. Reactive & Functional Polymers 2006, 66, 149–156. 33) Steffens, C.; Choi, S. W.; Ritter, H. Macromol. Rapid Commun. 2006, 27, 542–547. 34) Köllisch, H.; Barner-Kowollik, C.; Ritter, H. Macromol. Rapid Commun. 2006, 27, 848–853.
[20] Sauer, J.; Huisgen, R. Angew. Chem. 1960, 9, 294–315. [21] Schmid, G. H. Organic Chemistry, 1. Aufl. , Mosby-Verlag, St. Louis, 1996. [22] Morrison, R. T.; Boyd, R. N.; Lehrbuch der Organischen Chemie, 3.Aufl., VCH Weinheim, 1986. [23] Kamlet, M. J; Taft, R. W. J. Am. Chem. Soc. 1976, 98, 377–383. [24] Kamlet, M. J; Abboud, J.-L.; Taft, R. W. J. Am. Chem. Soc. 1977, 99, 6027–
6038. [25] Kamlet, M. J., Jones, M. E.; Abboud, J.-L.; Taft, R. W. J. Chem. Soc. Perkin Trans ΙΙ 1979, 342–348. [26] Kamlet, M. J.; Abboud, J.-L. M.; Abraham, M. H.; Taft, R. W. J. Org. Chem. 1983, 48, 2877–2887.

Literaturverzeichnis
197
[27] Laurence, C.; Nicolet, P.; Tawfik-Dalati, M.; Abboud, J.-L. M.; Notario, R. J. Phys. Chem. 1994, 98, 5807–5816. [28] Helburn, R.; Ullah, N.; Mansour, G.; Maxka, J. J. Phys. Org. Chem. 1997, 10, 42–48. [29] Helburn, R.; Dijiba, Y.; Mansour, G.; Maxka, J. Langmuir 1998, 14, 7147–
7154. [30] Mansour, G.; Creedon, W.; Dorrestein, P. C.; Maxka, J.; MacDonald, J. C.; Helburn, R. J. Org. Chem. 2001, 66, 4050–4054. [31] Corbett, J. F. Spectrochim. Acta 1967, 23A, 2315–2332. [32] Corbett, J. F. “The Chemistry of Synthetic Dyes”, Vol. 5, ed. by K. Venkataraman, Academic Press, New York, 1971, S. 508 ff.. [33] Griffiths, J.; Lockwood, M.; Roozpeikar, B. J. Chem. Soc. Perkin Trans ΙΙ 1977, 1608–1610. [34] Chu, K.-Y.; Griffiths, J. J. Chem. Soc. Perkin. Trans. Ι, 1978, 406–408. [35] Chu, K.-Y.; Griffiths, J. J. Chem. Soc. Perkin. Trans. Ι, 1978, 1194–1198. [36] Wurster, C.; Sendtner, R. Ber. Dtsch. Chem. Ges. 1879, 12, 522–528. [37] Wurster, C.; Sendtner, R. Ber. Dtsch. Chem. Ges. 1879, 12, 1803–1807. [38] Willstätter, R.; Piccard, Ber. Dtsch. Chem. Ges. 1908, 41, 1458–1475. [39] Michaelis, L. J. Am. Chem. Soc. 1931, 53, 2953–2962. [40] Michaelis, L.; Schubert, M. P.; Granick, S. J. Am. Chem. Soc. 1939, 61, 1981–
1992. [41] Lehn, J.-M. Supramolecular Chemistry, VCH: Weinheim, 1995. [42] Balzani, V.; Scandola, F. Supramolecular Photochemistry, Ellis Horwood: New York, 1991. [43] Kawata, S.; Kawata, Y. Chem. Rev. 2000, 100, 1777–1788. [44] Delaire, J. A.; Nakatani, K. Chem. Rev. 2000, 100, 1817–1845. [45] Moerner, W. E.; Silence, S. M. Chem. Rev. 1994, 94, 127–155. [46] Matsui, M.; Tsuge, M.; Funabiki, K.; Shibata, K.; Muramatsu, H.; Hirota, K.; Hosoda, M.; Tai, K.; Shiozaki, H.; Kim, M.; Nakatsu, K. J. Fluorine Chem. 1999, 97, 207–212. [47] Luo, L.; Hua, J.; Qui, J.; Cheng, J.; Shen, Y.; Lu, Z.; Wang, P.; Ye, C. Chem. Commun. 2001, 2, 171–172. [48] CD Römpp Chemielexikon, Version 1.0, Georg Thieme Verlag, Stuttgart/New York, 1995. [49] Spange, S.; Meyer, T.; Voigt, I.; Eschner, M.; Estel, E.; Pleul, D.; Simon, F. Adv. Polym. Sci. 2004, 165, 43–78. [50] Voigt, I.; Simon, F.; Komber, H.; Jacobasch, H.-J.; Spange, S. Colloid Polym. Sci. 2000, 278, 48–56. [51] Voigt, I.; Simon, F.; Estel, E.; Spange, S. Langmuir 2001, 17, 3080–3086. [52] Bucatariu, F.; Simon, F.; Spange, S.; Schwarz, S.; Dragan, S. Macromol. Symp. 2004, 210, 219–228. [53] Spange, S.; Wolf, S.; Simon, F. Progr. Colloid Polym. Sci. 2006, 132, 110–116. [54] Haupt, B. J.; Ennis, J.; Sevick, E. M. Langmuir 1999, 15, 3886–3892. [55] Poptoshev, E.; Rutland, M. W.; Claesson, P. M. Langmuir 1999, 15, 7789–
7794. [56] Poptoshev, E.; Rutland, M. W.; Claesson, P. M. Langmuir 2000, 16, 1987–
1992.

Literaturverzeichnis
198
[57] Filho, N. L. D.; do Cormo, D. R. in Encyclopedia of Surface and Colloid Science Marcel Dekker, New York, 2004, S. 1–20. [58] Patat, F.; Killmann, E.; Schliebener, C. Fortschr. Hochpolym.-Forsch.1964, 3, 332–393. [59] Radeva, Tsetska in Encyclopedia of Surface and Colloid Science, Marcel Dekker, New York, 2002, S. 547–557. [60] Smith-Palmer, T, Pelton, R. in Encyclopedia of Surface and Colloid Science, Marcel Dekker, New York, 2002, S. 2207–2224. [61] Arys, X.; Jonas, A. M., Laschewsky, A.; Legras, R. in Supramolecular Polymers, ed.: Ciferri, A., Marcel Dekker, New York, 2000, S. 505–555. [62] Eklund, D.; Lindström, T. Paper chemistry - an introduction, DT Paperscience puplications, Finnland, 1991, S. 145–179. [63] Hallensleben, M. L. in Ullmann´s Encyclopedia of Polymer Science and Engineering, 5th ed., 1992, ed.: B. Elvers, S. Hawkins, G. Schulz, A21, 752ff. [64] Hanford, W. E.; Stevenson, H. B. US Pat. 2,365,340 1944. [65] Reynolds, D. D.; Kenyon, W. O. J. Am. Chem. Soc. 1947, 69, 911–915. [66] Tanaka, H. J. J. Polym. Sci. Part B 1978, 16, 87-89. [67] Dawson, D. J.; Gless, R. D.; Wingard Jr., R. E. J. Am. Chem. Soc. 1976, 98, 5996–6000. [68] Stackman, R. W.; Summerville, R. H. Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 242–246. [69] Overberger, C. G.; Kikiyotani, S. J. Polym. Sci. Chem. Ed. 1983, 21, 525–540. [70] Dawson, D. J.; Brock, P. J. US Pat. 4,393,174 1983. [71] Hart, R. Makromol. Chem. 1959, 32, 51–56. [72] Uyama, H.; Kato, H.; Kobayashi, S. Polym. J. 1994, 26, 858–863. [73] Hart, R. J. Polym. Sci. 1958, 29, 629–636. [74] Elias, H.-G. Makromoleküle: Struktur-Eigenschaften-Synthesen Stoffe, 2. Aufl., 1972, Hüthig & Wepf Verlag, Basel, Heidelberg, S. 271–275. [75] Kirwan, L. J.; Papastavrou, G.; Borkovec, M. Nano Lett. 2004, 4, 149–152. [76] Linhart, F.; Auhorn, W. Das Papier 1992, 10A, V38–V45. [77] Riebeling, U.; De Clercq, A.; Stange, A.; Sendhoff, N.; Nilz, C.; Kröner, M. EP 553 135 1990. [78] Wang, F.; Tanaka, H. J. Appl. Polym. Sci. 2000, 78, 1805–1810. [79] Brunnmüller, F.; Schneider, R.; Kröner, M.; Müller, H.; Linhart, F.; Burkert, H.; Beyer, K.-H. EP 071 050 1981. [80] Pinschmidt, R. K. US 4 931 194 1990. [81] Pinschmidt, R. K.; Vijayendran, B. R.; Lai, Ta-W. US 5 085 787 1992. [82] Shu, P. US 5 134 176 1992. [83] Lenney, W. E.; Sagl, D. US 5 470 903 1995. [84] Cabrera, I. EP 894 809 1997. [85] Beihoffer, T.; Mitchell, M.; Darlington, J. W.; Anderson, M. WO 99/25745 1997. [86] Beihoffer, T.; Mitchell, M. US 6 159 591 2000. [87] Beihoffer, T.; Mitchell, M.; Trzupek, L. T.; Darlington, J. W.; Anderson, M. US 6 194 631 2001. [88] Beihoffer, T.; Mitchell, M. US 6 235 965 2001. [89] Keller, H.; Hoffmann, G.; Denzinger, W.; Fässler, R. EP 672 467 1994. [90] Schrell, A.; Huber, B. EP 692 559 1994.

Literaturverzeichnis
199
[91] Nudelman, N. S. in Supplement F2: The Chemistry of Amino, Nitroso, Nitro and Related Groups, ed.: Patai, S., John Wiley & Sons, Ltd.; 1996, 1215–
1300. [92] Suhr, H. Chem. Ber. 1964, 97, 3268–3276. [93] Bunnett, J. F.; Morath, R. J. J. Amer. Chem. Soc. 1955, 77, 5051–5055. [94] Bamkole, T. O.; Hirst, J.; Udoessien, E. I. J. Chem. Soc. Perkin ΙΙ 1973, 110–
114. [95] Bamkole, T. O.; Hirst, J.; Udoessien, E. I. J. Chem. Soc. Perkin ΙΙ 1973, 2114–
2119. [96] Villiers, A. Compt. Rend. Acad. Sci. Paris 1891, 112, 536–538. [97] Schardinger, F Z. Untersuch. Nahr. Genußm. 1903, 6, 865–880. [98] Freudenberg, K.; Meyer-Delius, M. Ber. Dtsch. Chem. Ges. 1938, 71, 1596–
1600. [99] Cramer, F. Chem. Ber. 1951, 84, 851–854. [100] v. Dietrich, H.; Cramer, F. Chem. Ber. 1954, 87, 806–817. [101] Cramer, F.; Henglein, F. M. Chem. Ber. 1957, 90, 2561–2571. [102] Cramer, F.; Henglein, F. M. Chem. Ber. 1957, 90, 2572–2575. [103] Wenz, G. Angew. Chem. 1994, 106, 851–870. [104] Saenger, W. Angew. Chem. 1980, 92, 343–361. [105] Szejtli, J. Chem. Rev. 1998, 98, 1743–1753. [106] Szejtli, J. J. Mater. Chem. 1997, 7, 575–587. [107] Gröger, M.; Kretzer, E. K.; Woyke, A. Science Forum der Universität Siegen 2001, www.science-forum.de/download/cyclodex.pdf. [108] Bourchard-Tuch, C. Chem. Unserer Zeit 2005, 39, 137–139. [109] Technisches Merkblatt und Sicherheitsdatenblatt Heptakis(2,6-O-dimethyl)-β-
cyclodextrin. [110] Technisches Merkblatt und EU-Sicherheitsdatenblatt CAVASOL W7 M (91/155/EWG); Wacker Chemie, Burghausen, 2006. [111] Reichardt, C. Solvents and Solvents Effects in Organic Chemistry, 3. ed., Wiley-VCH, Weinheim, 2003, S. 329ff. [112] Nagakura, S.; Kojima, M.; Murayama, Y. J. Mol. Spectrosc. 1964, 13, 174–
192. [113] Minesinger, R. R.; Kamlet, M. J. J. Phys. Chem. 1974, 78, 494–497. [114] Marcus, Y. Chem. Soc. Rev. 1993, 409–419. [115] Giacomelli, L.; Cattana, R.; Anunziata, J.; Silber, J. J., Hedrera, M.; Salerno, A.; Perillo, I. J. Phys. Org. Chem. 1994, 7, 162–168. [116] Cattana, R.; Silber, J. J.; Anunziata, J. Can. J. Chem. 1992, 70, 2677–2682. [117] Giumanini, A.G.; Chiavari, G.; Musiani, M. M.; Rossi, P. Synthesis 1980, 9, 743–746. [118] Roth, I.; Jbarah, A. A.; Holze, R.; Friedrich, M.; Spange, S. Macromol. Rapid Commun. 2006, 27, 193–199. [119] Wadsworth, W. S.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. [120] Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. [121] Gross, R. C.; Seybold, P. G.; Peralta-Inga, Z.; Murray, J. S.; Politzer, P. J. Org. Chem. 2001, 66, 6919–6925. [122] Brown, H. C. in Determination of Organic Structures by Physical Methods, Eds.: Braude, E. A. and Nachod, F. C., Academic Press, New York, 1955.

Literaturverzeichnis
200
[123] Schurz, J. Physikalische Chemie der Hochpolymeren, Springer Verlag,1974, 116–119. [124] Lo Meo, P.; D´Anna, F.; Gruttadauria, M.; Riela, S.; Noto, R. Org. Biomol. Chem. 2003, 1, 1584–1590. [125] Lo Meo, P.; D´Anna, F.; Gruttadauria, M.; Riela, S.; Noto, R. Tetrahedron 2004, 60, 9099–9111. [126] Suhr, H. Liebigs Ann. Chem. 1965, 687, 175–182. [127] Suhr, H. Liebigs Ann. Chem. 1965, 689, 109–117. [128] Horn, D.; Progr. Colloid & Polymer Sci. 1978, 65, 251–264. [129] Hu, Z.; Zhang, S.; Yang, J.; Chen, Y. J. Appl. Polym. Chem. 2003, 89, 3889–
3893. [130] Nudelman, N. S.; Cerdeira, S. J. Chem. Soc. Perkin Trans II 1986, 695–698. [131] Nudelman, N. S.; Mancini, P. M. E.; Martinez, R. D.; Vottero, L. R. J. Chem. Soc. Perkin Trans II 1987, 951–954. [132] Bunnett, J. F.; Zahler, R. E. Chem. Rev. 1951, 49, 273–389. [133] Parker, R. E.; Read, T. O. J. Chem. Soc. 1962, 3149–3153. [134] Bartoli, G.; Todesco, P. E. Acc. Chem. Res. 1977, 10, 125–132. [135] Beck, J. R. Tetrahedron 1978, 34, 2057–2068. [136] Effenberger, F.; Streicher, W. Chem. Ber. 1991, 124, 157–162. [137] Effenberger, F.; Koch, M.; Streicher, W. Chem. Ber. 1991, 124, 163–173. [138] Ono, N. The Nitro Group in Organic Synthesis, Wiley-VCH, 2001, S. 182–230. [139] Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der organischen Chemie, 5. Aufl., GeorgThieme Verlag, Stuttgart, N. Y., 1995. [140] Laws, D. D.; Bitter, H.-L. L.; Jerschow, A. Angew. Chem. 2002, 114, 3224–
3259. [141] Beamson, G.; Briggs, D. High resolution of organic polymers, The Sienta ESCA 300 Database, J. Wiley & Sons, Chichester, New York, Brisbane, Toronto, Singapur, 1992. [142] Seah, M. P.; Dench, W. A. Surface Interface Analysis 1979, 1, 2. [143] Briggs, D. Characterization of surfaces, Pergamon, Oxford, 1989. [144] Liu, Yi-Chun; McCreery, R. L. J. Am. Chem. Soc. 1995, 117, 11254–11259. [145] Shirley, D.A. Phys. Rev. B 1972, 5, 4709–4714. [146] Unger, K. Angew. Chem. 1972, 8, 331–343. [147] Jacobasch, H.-J.; Simon, F.; Bellmann, C. tm - Technisches Messen: Sensoren, Geräte, Systeme 1996,63, 439–446. [148] Nitzsche, R.; Simon, F. tm - Technisches Messen: Sensoren, Geräte, Systeme 1997, 64, 106–113. [149] Roth, I.; Simon, F.; Bellmann, C.; Seifert, A.; Spange, S. Chem. Mater. 2006, 18, 4730–4739. [150] Madl, A.; Spange, S.; Waldbach, T.; Anders, E.; Mahr, N. Macromol. Chem. Phys. 1999, 200, 1495–1505. [151] Madl, A.; Spange, S. Macromol. Symp. 2000, 161, 149–157. [152] 1H-NMR-Spektrum von N-Methylvinylacetamid in CDCl3 bei 399.65 MHz unter www.aist.go.jp/RIODB/SDBS/cgi-bin/direct_frame_top.cgi?lang=eng
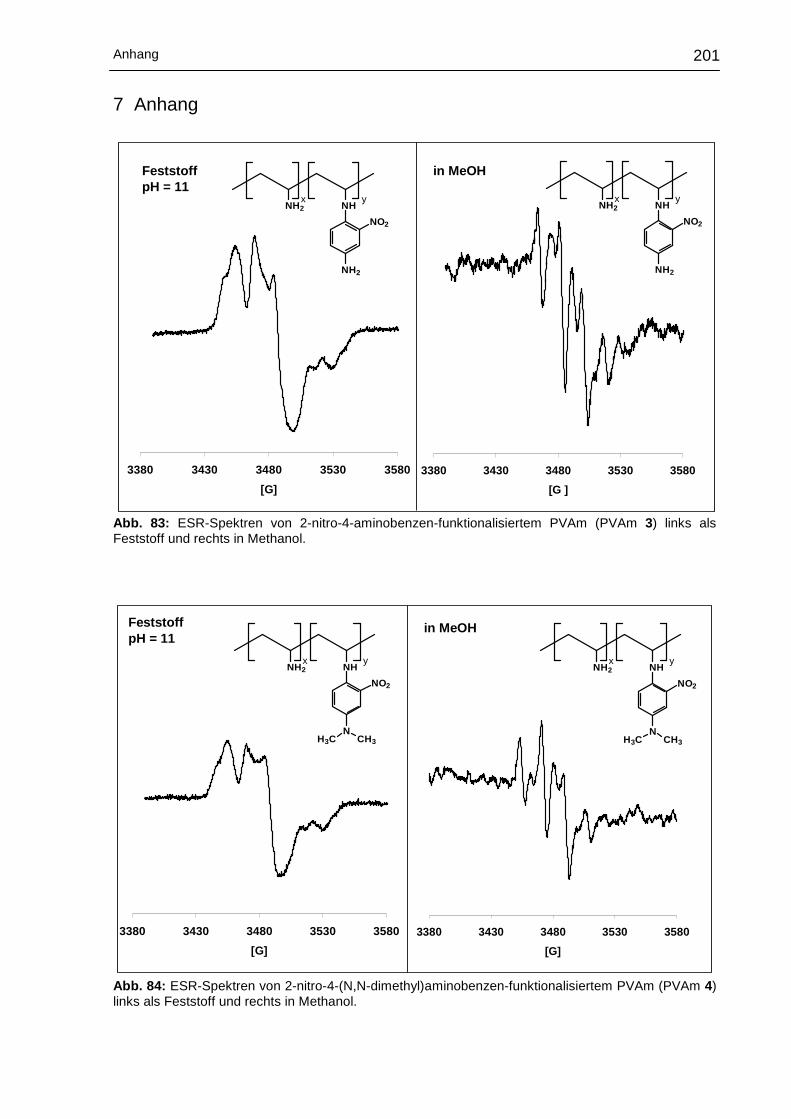
Anhang
201
7 Anhang
Abb. 83: ESR-Spektren von 2-nitro-4-aminobenzen-funktionalisiertem PVAm (PVAm 3) links als Feststoff und rechts in Methanol.
Abb. 84: ESR-Spektren von 2-nitro-4-(N,N-dimethyl)aminobenzen-funktionalisiertem PVAm (PVAm 4) links als Feststoff und rechts in Methanol.
3380 3430 3480 3530 3580
[G]
NH2 NH
NH2
NO2
x y
FeststoffpH = 11
3380 3430 3480 3530 3580
[G ]
NH2 NH
NH2
NO2
x y
in MeOH
3380 3430 3480 3530 3580
[G]
NH2 NH
N
NO2
H3C CH3
x y
FeststoffpH = 11
3380 3430 3480 3530 3580
[G]
NH2 NH
N
NO2
H3C CH3
x y
in MeOH

Selbständigkeitserklärung
202
Selbständigkeitserklärung
Hiermit erkläre ich, dass die vorliegende Arbeit selbständig und nur unter
Verwendung der angegebenen Quellen und Hilfsmittel angefertigt wurde.
Chemnitz, den 19.12.2006 Isabelle Roth

Lebenslauf/wissenschaftlicher Werdegang
203
Lebenslauf
Name: Isabelle Roth
geb. am: 06.04.1976 in Plauen i. V.
Familienstand: ledig
Schulbildung: 1994 Abitur in Reichenbach i. V.
Studium: 10/1994 − 03/1996 BWL-Studium an der TU Chemnitz
Studiengangwechsel
04/1996 − 08/2001 Chemie-Studium an der TU Chemnitz
Schwerpunktfach: Physikalische Chemie
Wahlpflichtfach: Polymerchemie
Diplomarbeit: „Nucleophile Substitutionsreaktionen fluorsubstituierter
Aromaten mit Polyvinylamin“
TU Chemnitz, Fakultät für Naturwissenschaften, Institut für
Chemie, Professur Polymerchemie (Prof. S. Spange)
Abschluss: Diplom
Promotion: 09/2001 − 07/2006
wissenschaftliche Mitarbeiterin an der TU Chemnitz,
Fakultät für Naturwissenschaften, Institut für Chemie,
Professur Polymerchemie (Prof. S. Spange)
seit 08/2006
wissenschaftliche Mitarbeiterin an der TU Chemnitz,
Fakultät für Maschinenbau, Institut für Allgemeinen
Maschinenbau und Kunststofftechnik,
Professur Strukturleichtbau und Kunststoffverarbeitung
(Prof. L. Kroll)

Lebenslauf/wissenschaftlicher Werdegang
204
Wissenschaftliche Leistungen
Beiträge zu Tagungen, Seminaren und Workshops:
• "Synthesis and Structure of Novel Chromophoric Ormosils" (Poster)
A. Seifert, I. Roth, H. Müller, S. Hesse, C. Jäger, S. Spange
16. − 21.09.2001 Sol-Gel-Tagung “11th International Workshop on Glasses,
Ceramics, Hybrids and Nanocomposites from Gels” in Abano Terme, Italien.
• "Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylamin und Hybridmaterialien" (Poster)
I. Roth, A. Seifert, S. Spange
18. − 19.03.2002 GDCh-Fachgruppentagung Makromolekulare Chemie
„Funktionspolymere für Systemlösungen“ in Darmstadt.
• "Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylamin und Hybridmaterialien" (Vortrag)
S. Spange, N. Moszner, A. Seifert, I. Roth
27.02. − 01.03.2003 Makromolekulares Kolloquium in Freiburg.
• "Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylamin und Hybridmaterialien" (Vortrag)
I. Roth, S. Spange
Doktorandenseminar des Institutes für Chemie der TU Chemnitz, Oktober 2003.
• "Synthese chromophorer Polymere mit dipolaren Funktionalitäten" (Poster)
S. Anders, I. Roth, S. Spange
15. − 16.03.2004 GDCh-Fachgruppentagung Makromolekulare Chemie
„Fortschritte bei der Synthese und Charakterisierung von Polymeren“ in
Düsseldorf.

Lebenslauf/wissenschaftlicher Werdegang
205
• "Surface Functionalization of Inorganic Materials from aqueous Solution using
Polyvinylformamide-co-polyvinylamines" (Poster)
I. Roth, S. Wolf, F. Simon, C. Bellmann, S. Spange
18. − 19.11.2004 zum 6th IPF Colloquium “Advanced Heterogeneous Polymer
Materials” in Dresden.
• "Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylamin und Hybridmaterialien" (Poster)
I. Roth, A. Seifert, S. Spange
05. − 06.12.2004 Workshop „Struktur und Dynamik von Polymeren in
eingeschränkten Geometrien unterschiedlicher Topologie“ in Großbothen.
Veröffentlichungen:
• "Nucleophilic Substitution of 4-Fluoronitrobenzene with Polyvinylamine in Water
Mediated by Cyclodextrins."
I. Roth, S. Spange, Macromol. Rapid Commun. 2001, 22, 1288−1291.
Anmerkung: cover picture
• "Solvatochromic Azamethine Dyes for Probing the Polarity of Gold-Cluster-
Functionalized Silica Particles."
S. Spange, D. Kunzmann, R. Sens, A. Seifert, I. Roth, W. Thiel, Chem. Eur. J.
2003, 9, 4161−4167.
• "A solvatochromic dye for probing significantly the dipolarity/polarizability of
HBD (hydrogen bond donating) environments."
S. Spange, R. Sens, Y. Zimmermann, A. Seifert, I. Roth, S. Anders,
K. Hofmann, New J. Chem. 2003, 27, 520−524.

Lebenslauf/wissenschaftlicher Werdegang
206
• "Nucleophile aromatische Substitution als Werkzeug zur Funktionalisierung von
Polyvinylamin und Hybridmaterialien."
A. Seifert, I. Roth, S. Spange, N. Moszner, Macromol. Chem. Phys. 2003, 204,
F30−F31, Sonderauflage für die Teilnehmer des Makromolekularen
Kolloquiums vom 27.02. − 01.03.2003 in Freiburg.
ISSN: 1521-3935 (online); ISSN: 1022-1352 (print)
• "Kinetic Studies on the Nucleophilic Aromatic Substitution of Fluoronitrobenzene
Derivatives with Polyvinylamine in Water mediated by 2,6-O-Dimethyl-β-
cyclodextrin."
I. Roth, S. Spange, Macromolecules 2005, 38, 10034−10041.
• "2-Nitro-1,4-diaminobenzene-functionalized Polyvinylamines as water soluble
UV-vis-sensitive pH-sensors."
I. Roth, A. A. Jbarah, R. Holze, M. Friedrich, S. Spange, Macromol. Rapid
Commun. 2006, 27, 193−199.
• "Fabrication of Silica Particles functionalized with Chromophores and Amino
groups Using Synergism of Polyvinylamine Adsorption and Nucleophilic
Aromatic Substitution with Fluoroaromatics."
I. Roth, F. Simon, C. Bellmann, A. Seifert, S. Spange, Chem. Mater. 2006, 18,
4730−4739.
Related Documents











