Tumor and Stem Cell Biology Novel Dyskerin-Mediated Mechanism of p53 Inactivation through Defective mRNA Translation Lorenzo Montanaro 1 , Maria Calienni 1 , Sara Bertoni 1 , Laura Rocchi 1 , Pasquale Sansone 2,5 , Gianluca Storci 1,5 , Donatella Santini 6 , Claudio Ceccarelli 3,5 , Mario Taffurelli 4 , Domenica Carnicelli 1 , Maurizio Brigotti 1 , Massimiliano Bonafè 1,5 , Davide Treré 1 , and Massimo Derenzini 3 Abstract In up to 60% of human cancers, p53 gene mutations are responsible for direct inactivation of the tumor suppressor function of p53. Alternative mechanisms of p53 inactivation described thus far mainly affect its posttranslational regulation. In X-linked dyskeratosis congenita, a multisystemic syndrome characterized by increased cancer susceptibility, mutations of the DKC1 gene encoding dyskerin cause a selective defect in the translation of a subgroup of internal ribosome entry site (IRES)–containing cellular mRNAs. In this study, we show that impairment of dyskerin function can cause p53 inactivation due to a defect in p53 mRNA transla- tion. siRNA-mediated reduction of dyskerin levels caused a decrease of p53 mRNA translation, protein levels, and functional activity, both in human breast cancer cells and in primary mammary epithelial progenitor cells. These effects seemed to be independent of the known role of dyskerin in telomerase function, and they were associated with a specific impairment of translation initiation mediated by IRES elements present in p53 mRNA. In a series of human primary breast cancers retaining wild-type p53, we found that low levels of dys- kerin expression were associated with reduced expression of p53-positive target genes. Our findings suggest that a dyskerin-mediated mechanism of p53 inactivation may occur in a subset of human tumors. Cancer Res; 70(11); 4767–77. ©2010 AACR. Introduction The tumor protein p53 plays a well-established role in pro- tecting against cancer by controlling the cellular response against various types of acute stress (1). The levels of p53 are generally influenced by its posttranslational degradation via the proteasome. In unstressed cells, p53 is indeed present in a latent state and is maintained at low levels through MDM2-dependent targeted degradation (2, 3). The exposure to different cellular stresses blocks p53 degradation, thus gi- ving rise to its accumulation (1). The effects of the cellular response to this p53 activation may be ascribed to the role of p53 as a transcriptional modulator that regulates the ex- pression of a wide and heterogeneous group of responsive genes. In general, p53 activation inhibits cell growth through cell cycle arrest or through induction of either proliferative senescence or apoptosis, thus preventing tumor development (4). In up to 60% of human cancers, p53 gene mutations di- rectly inactivate p53 function (5). Mechanisms leading to indirect p53 inactivation acting mainly on p53 posttransla- tional degradation have also been described in tumors (2, 3, 6–9). In addition to the block of its degradation, p53 induc- tion after stress also requires its neosynthesis, which entails the transcription (10) and the active translation of its mRNA (11). p53 mRNA translation seems to be regulated by proteins binding p53 mRNA (12–14) as well as by internal ribosome entry site (IRES) elements present in p53 mRNA (15), which are able to mediate translation initiation under stress. The impairment in proper translational control has been recently associated with cancer development and progres- sion (16). In the rare multisystemic syndrome X-linked dys- keratosis congenita (X-DC), mutations of the DKC1 gene encoding dyskerin, a pseudouridine synthase that modifies rRNA (17), cause a defect in the translation of a subgroup of IRES-containing cellular mRNAs (18) and are associated with increased cancer susceptibility (19, 20). The lack of dys- kerin function is associated with an increased susceptibility to breast carcinomas in the DKC1 hypomorphic mouse (21), and dyskerin expression and functions are also reduced in a subset of human breast carcinomas (22). In the present study, we assessed whether the lack of dyskerin may cause a p53 inactivation induced by a defect in IRES-mediated mRNA translation in human breast epithelial cells. Moreover, we investigated the relationship between DKC1 mRNA levels Authors' Affiliations: 1 Dipartimento di Patologia Sperimentale, 2 Dipartimento di Farmacologia e Tossicologia, 3 Dipartimento Clinico di Scienze Radiologiche e Istocitopatologiche, and 4 Dipartimento di Scienze Chirurgiche e Anestesiologiche, Università di Bologna; 5 Centro di Ricerca Biomedica Applicata (CRBA) and 6 Dipartimento di Ematologia, Oncologia e Medicina di Laboratorio, Policlinico S. Orsola- Malpighi, Bologna, Bologna, Italy Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). Corresponding Author: Lorenzo Montanaro, Dipartimento di Patologia Sperimentale, Università di Bologna, via San Giacomo 14, 40126 Bologna, Italy. Phone: 39-051-302874; Fax: 39-051-306861; E-mail: lorenzo. [email protected]. doi: 10.1158/0008-5472.CAN-09-4024 ©2010 American Association for Cancer Research. Cancer Research www.aacrjournals.org 4767

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Tumor and Stem Cell Biology
CancerResearch
Novel Dyskerin-Mediated Mechanism of p53 Inactivationthrough Defective mRNA Translation
Lorenzo Montanaro1, Maria Calienni1, Sara Bertoni1, Laura Rocchi1, Pasquale Sansone2,5, Gianluca Storci1,5,Donatella Santini6, Claudio Ceccarelli3,5, Mario Taffurelli4, Domenica Carnicelli1, Maurizio Brigotti1,Massimiliano Bonafè1,5, Davide Treré1, and Massimo Derenzini3
Abstract
Authors'2DipartimenScienze RScienze Chdi RicercaEmatologiaMalpighi, B
Note: SuppOnline (http
CorresponSperimentBologna, Itamontanaro@
doi: 10.115
©2010 Am
www.aacr
In up to 60% of human cancers, p53 gene mutations are responsible for direct inactivation of the tumorsuppressor function of p53. Alternative mechanisms of p53 inactivation described thus far mainly affect itsposttranslational regulation. In X-linked dyskeratosis congenita, a multisystemic syndrome characterized byincreased cancer susceptibility, mutations of the DKC1 gene encoding dyskerin cause a selective defect in thetranslation of a subgroup of internal ribosome entry site (IRES)–containing cellular mRNAs. In this study, weshow that impairment of dyskerin function can cause p53 inactivation due to a defect in p53 mRNA transla-tion. siRNA-mediated reduction of dyskerin levels caused a decrease of p53 mRNA translation, protein levels,and functional activity, both in human breast cancer cells and in primary mammary epithelial progenitor cells.These effects seemed to be independent of the known role of dyskerin in telomerase function, and they wereassociated with a specific impairment of translation initiation mediated by IRES elements present in p53mRNA. In a series of human primary breast cancers retaining wild-type p53, we found that low levels of dys-kerin expression were associated with reduced expression of p53-positive target genes. Our findings suggestthat a dyskerin-mediated mechanism of p53 inactivation may occur in a subset of human tumors. Cancer Res;70(11); 4767–77. ©2010 AACR.
Introduction
The tumor protein p53 plays a well-established role in pro-tecting against cancer by controlling the cellular responseagainst various types of acute stress (1). The levels of p53are generally influenced by its posttranslational degradationvia the proteasome. In unstressed cells, p53 is indeed presentin a latent state and is maintained at low levels throughMDM2-dependent targeted degradation (2, 3). The exposureto different cellular stresses blocks p53 degradation, thus gi-ving rise to its accumulation (1). The effects of the cellularresponse to this p53 activation may be ascribed to the roleof p53 as a transcriptional modulator that regulates the ex-pression of a wide and heterogeneous group of responsivegenes. In general, p53 activation inhibits cell growth through
Affiliations: 1Dipartimento di Patologia Sperimentale,to di Farmacologia e Tossicologia, 3Dipartimento Clinico diadiologiche e Istocitopatologiche, and 4Dipartimento diirurgiche e Anestesiologiche, Università di Bologna; 5CentroBiomedica Applicata (CRBA) and 6Dipartimento di
, Oncologia e Medicina di Laboratorio, Policlinico S. Orsola-ologna, Bologna, Italy
lementary data for this article are available at Cancer Research://cancerres.aacrjournals.org/).
ding Author: Lorenzo Montanaro, Dipartimento di Patologiaale, Università di Bologna, via San Giacomo 14, 40126ly. Phone: 39-051-302874; Fax: 39-051-306861; E-mail: lorenzo.unibo.it.
8/0008-5472.CAN-09-4024
erican Association for Cancer Research.
journals.org
cell cycle arrest or through induction of either proliferativesenescence or apoptosis, thus preventing tumor development(4). In up to 60% of human cancers, p53 gene mutations di-rectly inactivate p53 function (5). Mechanisms leading toindirect p53 inactivation acting mainly on p53 posttransla-tional degradation have also been described in tumors (2,3, 6–9). In addition to the block of its degradation, p53 induc-tion after stress also requires its neosynthesis, which entailsthe transcription (10) and the active translation of its mRNA(11). p53 mRNA translation seems to be regulated by proteinsbinding p53 mRNA (12–14) as well as by internal ribosomeentry site (IRES) elements present in p53 mRNA (15), whichare able to mediate translation initiation under stress.The impairment in proper translational control has been
recently associated with cancer development and progres-sion (16). In the rare multisystemic syndrome X-linked dys-keratosis congenita (X-DC), mutations of the DKC1 geneencoding dyskerin, a pseudouridine synthase that modifiesrRNA (17), cause a defect in the translation of a subgroupof IRES-containing cellular mRNAs (18) and are associatedwith increased cancer susceptibility (19, 20). The lack of dys-kerin function is associated with an increased susceptibilityto breast carcinomas in the DKC1 hypomorphic mouse (21),and dyskerin expression and functions are also reduced in asubset of human breast carcinomas (22). In the presentstudy, we assessed whether the lack of dyskerin may causea p53 inactivation induced by a defect in IRES-mediatedmRNA translation in human breast epithelial cells. Moreover,we investigated the relationship between DKC1 mRNA levels
4767

Montanaro et al.
4768
and the expression of p53 target genes in primary humanbreast carcinomas. Our results show that the decrease of dys-kerin levels reduces p53 levels and activity through a defectin IRES-dependent p53 mRNA translation, and that in prima-ry tumors, low levels of dyskerin expression are associatedwith reduced p53 activity, thus suggesting the existence ofa translational mechanism involving dyskerin for the inacti-vation of p53 in human tumors.
Materials and Methods
Cells and reagentsCells were obtained from the American Type Culture
Collection and IZSLER. During the study, cell line identitywas verified by DNA fingerprinting and day-by-day mor-phology checking. Subconfluent cells were treated withMG115 and MG132 (10 nmol/L; Calbiochem), ionizing ra-diation (6 Gy), doxorubicin (0.5, 1, and 4 μmol/L), and de-feroxamine (100 μmol/L; Sigma-Aldrich). For survivalassays, cells were either trypsinized and counted aftertrypan blue staining or formalin fixed and stained with0.05% crystal violet for 10 minutes.Mammospheres (MS) were obtained as previously de-
scribed, downscaling the method for small tissue quantities(300–900 mg; ref. 23). Fresh surgical specimens, obtainedfrom patients with ductal breast carcinoma who underwentquadrantectomy or mastectomy, were collected to generateMS. Normal and tumor samples were histologically charac-terized to ensure the proper classification of normal andtumor tissue. Particular care was paid to generate MSfrom specimens in which only normal or tumor tissueswere detectable at histologic examination. Briefly, tissueswere placed in sterile Epicult (StemCell Technologies),minced, and incubated for 6 to 12 hours in the presenceof 1,000 units of collagenase/hyaluronidase enzyme mix(StemCell Technologies). Samples were centrifuged at80 × g for 2 minutes. The pellet was digested with dispaseand DNase for 3 minutes (StemCell Technologies) andthen pelleted at 450 × g for 5 minutes. Pellets were resus-pended, filtered through a 40-μm nylon mesh (BD),and plated into low attachment wells (Corning), filledwith mammary epithelial growth medium (MEGM) andsupplemented with 10 ng/mL epidermal growth factor,10 ng/mL basic fibroblast growth factor, 10 μg/mL insu-lin, 1 μmol/L hydrocortisone, and aliquots of gentamycinand amphotericine (Cambrex). Primary MS began to formafter 4 to 6 days and were processed at day 10. Experi-mental procedures were performed on secondary MS,generated by incubating primary MS in 1× trypsin-EDTAsolution (Cambrex) for 3 minutes, followed by two washesin complete MEGM and filtration through a 40-μm nylonmesh. When indicated, MS were treated with 5 μmol/Ldoxorubicin for 24 hours.
RNA interferenceDKC1- and p53-specific double-stranded siRNAs and ap-
propriate controls were obtained from Invitrogen. DKC1RNA interference (RNAi) was done with either single or three
Cancer Res; 70(11) June 1, 2010
pooled siRNA oligonucleotides (Invitrogen). p53 RNAi wasdone using the specific Stealth validated RNAi DuoPak.siRNAs were transfected in adherent cells using Lipofectamine2000 (Invitrogen) and in MS using in vitro JET-PEI reagent(Polyplus Transfection).
Western blotSDS-PAGE and immunoblotting were carried out accord-
ing to standard procedures. Antibodies were anti-dyskerin(Santa Cruz Biotechnology), anti-p53 (clone BP-53-12, Novo-castra), anti-p21 (clone SX118, Dako), and anti–β-actin(Sigma-Aldrich).
Real-time and semiquantitative PCRTotal RNA was extracted from frozen samples using Trizol
reagent (Invitrogen) and reverse transcribed using the High-Capacity cDNA Archive Kit (Applied Biosystems). Real-timePCR analysis was done in a Gene Amp 7000 Sequence Detec-tion System (Applied Biosystems) using the TaqMan ap-proach. For each sample, three replicates were analyzed.Sets of primers and fluorogenic probes specific for DKC1,p53, p21, PUMA, and MDM2 mRNAs were purchased fromApplied Biosystems, whereas hTR-specific primers and probewere synthesized as described (22). The relative amounts ofthe studied target genes were calculated using the expressionof human β-glucuronidase (Applied Biosystems) and 18SRNA (for polysomal analysis) as an endogenous control.The final results were determined as follows: N target =2−(ΔCt sample − ΔCt calibrator), where ΔCt values of the sampleand calibrator were determined by subtracting the Ct valueof the endogenous control gene from the Ct value of eachtarget gene. Telomerase activity was measured by real-timePCR using the Quantitative Telomerase Detection Kit (AlliedBiotech, Inc.). Each evaluation was done in triplicate using1 μg of protein extract. For the other mRNAs studied bysemiquantitative reverse transcription-PCR (RT-PCR), thefollowing primers and annealing temperatures were used,respectively: p21 mRNA, 5′-TGGGGATGTCCGTCAGAACC,5′ TGGAGTGGTAGAAATCTCTCATGCT, 59°C; BNIP3,5′-CGTTCCAGCCTCGGTTTCTATTTA, 5-CGCCTTCCA-ATATAGATCCCCAAT, 51°C; PUMA, 5′-CAGACTGT-GAATCCTGTGCT, 5′-ACAGTATCTTACAGGCTGCC, 62°C.Cycling conditions were predenaturation step at 95°C for2 minutes; 28 cycles of denaturation at 95°C for 1 minute,annealing at the appropriate temperature for 1 minute,extension at 72°C for 1 minute; final extension at 72°C for7 minutes.
Analysis of protein synthesis in cellsProtein synthesis was measured as the rate of incorpora-
tion of labeled leucine during a 30-minute incubation of thesubconfluent cell monolayers in complete medium contain-ing 50 mg/L leucine and trace amounts of [3H]leucine as pre-viously described (24).
Isolation of polyribosomal mRNASubconfluent cells were washed in PBS at 4°C. The
cellular pellet was lysed in 2 volumes of 10 mmol/L
Cancer Research

Lack of Dyskerin Inactivates p53
Tris-HCl (pH 7.4), 10 mmol/L NaCl, 3 mmol/L MgCl20.5% NP40 for 10 minutes at 4°C. The lysates were thencentrifuged at 14,000 × g for 10 minutes at 4°C and thesupernatant was used for the isolation of polyribosomes.Lysates were stratified onto a 15% to 50% sucrose gradientin 30 mmol/L HEPES/KOH (pH 7.5), 80 mmol/L KCl, 1.8mmol/L Mg-acetate, and centrifuged at 4°C for 15 hours at40,000 × g. From gradients, 1-mL fractions were collectedand their absorbance was read at 260 nm. Polyribosomalfractions were pooled and centrifuged at 100,000 × g for15 hours at 4°C. RNA was extracted from pellets using Trizolreagent.
DNA and mRNA transfectionFor the evaluation of p53 transcriptional activity, cells
were transfected with 300 ng of DNA per sample ofthe PathDetect p53 cis-reporter gene plasmid (Stratagene).As transfection efficiency control, 100 ng of pRL-CMV(Promega) per sample were cotransfected in each case.After 24 hours, cells were harvested for luciferase assay.For the RNA transfection, capped mRNA was transcribedfrom linearized pR-CrPV-IRES-F (gift from Dr. DavideRuggero, University of California, San Francisco, SanFrancisco, CA; ref. 18) and pR-p53-IRES-F plasmids (giftfrom Dr. Barsanjit Mazumder, Cleveland State University,Cleveland, OH; ref. 25) using the mMessage mMachineT7 kit (Ambion) and cells were transfected with 1 μg ofRNA per sample using DMRIEC (Invitrogen). After 8-hourtransfection, cells were harvested and analyzed with dual-luciferase kit (Promega).
Patient materialsOne hundred nineteen breast carcinomas were selected
from a series of consecutive patients who had undergonesurgical resection for primary breast carcinoma at theSurgical Department of the University of Bologna be-tween 1994 and 2006, on the sole basis of frozen tissueavailability. Nuclear immunostaining of p53 (clone BP53-12.1, Biogenex) and p21 (clone EA10, Oncogene Science)was assessed by image cytometry and expressed as the per-centage of labeled nuclear area over the total neoplasticnuclear area (labeling index). For assessing p53 status,we considered that the p53 gene was mutated when morethan 10% of the tumor cell nuclei were stained and p21 la-beling index was less than 10% (26). In a subset of cases (34of 119), p53 status was also assessed by direct sequencing.Tumors were grouped (high, medium, and low expression)according to DKC1 mRNA levels measured by real-timeRT-PCR using the previously described survival-associatedcutoffs (22).
Statistical analysisThe χ2 or Mann-Whitney U test, when appropriate, was
used for the comparisons among groups. Agreement be-tween scores was assessed by the κ statistics. All statis-tics were obtained using the SPSS statistical softwarepackage (SPSS, Inc.). P values <0.05 were regarded as sta-tistically significant.
www.aacrjournals.org
Results
Lack of dyskerin hinders p53 mRNA translation inhuman breast cancer cellsWe selectively reduced DKC1 mRNA levels by transient
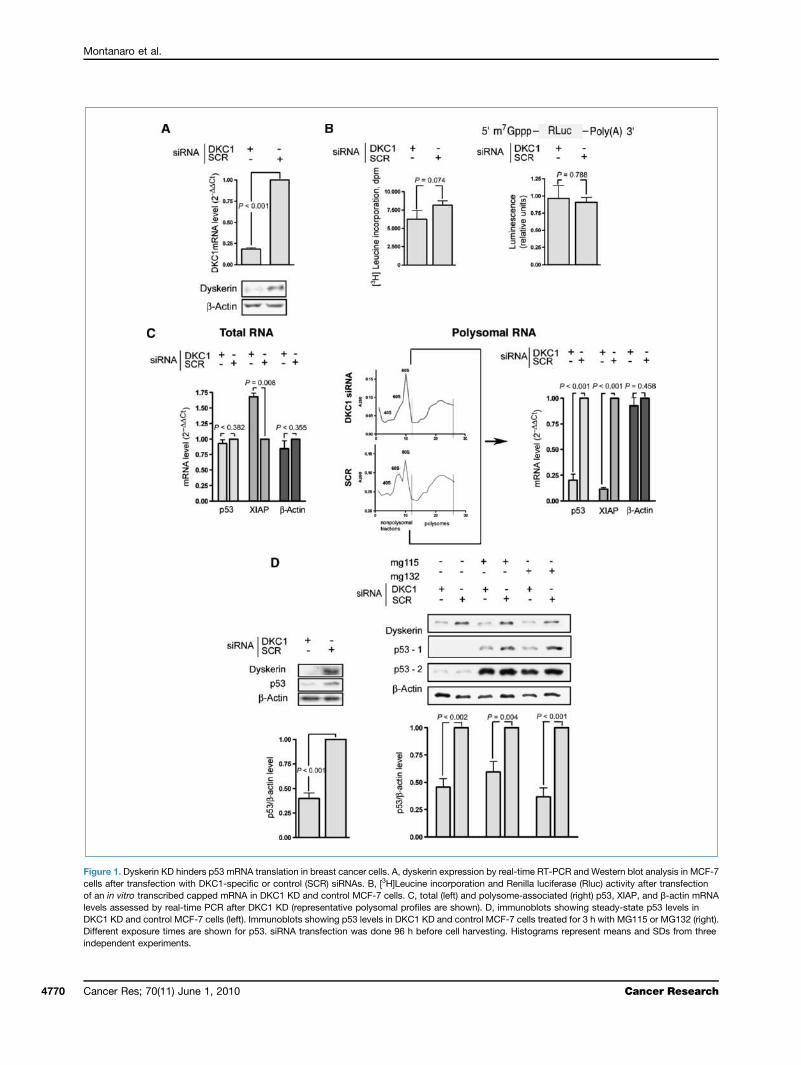
RNAi-mediated knockdown (KD) in p53 wild-type MCF-7breast cancer cells. This approach led to a strong reductionin DKC1 mRNA and protein lasting for at least 96 hours(Fig. 1A). The reduction of dyskerin levels did not cause aglobal decrease in protein synthesis, as evaluated by [3H]leu-cine incorporation, as well as in mRNA translation, evaluatedby measuring the activity of firefly luciferase after transfec-tion of an in vitro transcribed capped mRNA (Fig. 1B).Whereas DKC1 KD had no significant effects on global p53mRNA levels (Fig. 1C, left), it strongly decreased the recruit-ment of p53 mRNA to polysomal fractions (Fig. 1C, right). Aspreviously described for DKC1 mutations (17), DKC1 KD alsoinfluenced the translation of the IRES-containing XIAPmRNA, whereas it did not affect the translation of the house-keeping β-actin mRNA (Fig. 1C). XIAP mRNA levels areknown to be constant and XIAP regulation seems to occurspecifically at the translational level (27). Surprisingly, ourresults show that after DKC1 KD, there is a 1.7-fold increasein XIAP mRNA. We may hypothesize that, because DKC1 KDstrongly decreases XIAP translation, a feedback networkmight increase XIAP mRNA transcription or stability.We then investigated whether the observed translational
defect of p53 mRNA affects p53 level. Our results indicatethat DKC1 KD strongly downregulated the p53 levels at thesteady state (Fig. 1D, left). We confirmed these results byusing three different siRNAs for DKC1 KD also (Supplemen-tary Fig. S1). To evaluate if the decrease of dyskerin affectsp53 levels independently from its proteasome-mediateddegradation, we compared p53 levels in control and DKC1KD MCF-7 cells, exposed to the specific proteasomechemical inhibitors MG115 and MG132. Also under theseconditions, DKC1 KD strongly reduced p53 levels, showingthat the reduction of dyskerin levels downregulates p53le-vels independently of proteasome-mediated degradation(Fig. 1D, right).
Dyskerin reduction impairs p53 function in humanbreast cancer cellsTo evaluate the functional consequences on p53 of the
translational defect observed, first we assessed if DKC1 KDleads to a reduction in p53 transcriptional activity. To thispurpose, a reporter assay in which firefly luciferase is placedunder the control of a repeated p53-specific response ele-ment was used. In DKC1 KD cells, we observed a strong re-duction of p53 transcriptional activity, similar to thatobtained through the transfection of p53-specific siRNAs(Fig. 2A). Next, to acquire more information on the effectof dyskerin reduction and p53 function, we exposed DKC1KD MCF-7 cells to different stresses activating the p53-de-pendent response. After exposure to ionizing radiations,DKC1 KD reduced the p53 accumulation and the expressionof the p53 target gene product p21 (ref. 28; Fig. 2B). DKC1 KDalso induced a strong reduction of p53 accumulation after
Cancer Res; 70(11) June 1, 2010 4769
Montanaro et al.
4770
Figure 1. Dyskerin KD hinders p53 mRNA translation in breast cancer cells. A, dyskerin expression by real-time RT-PCR and Western blot analysis in MCF-7cells after transfection with DKC1-specific or control (SCR) siRNAs. B, [3H]Leucine incorporation and Renilla luciferase (Rluc) activity after transfectionof an in vitro transcribed capped mRNA in DKC1 KD and control MCF-7 cells. C, total (left) and polysome-associated (right) p53, XIAP, and β-actin mRNAlevels assessed by real-time PCR after DKC1 KD (representative polysomal profiles are shown). D, immunoblots showing steady-state p53 levels inDKC1 KD and control MCF-7 cells (left). Immunoblots showing p53 levels in DKC1 KD and control MCF-7 cells treated for 3 h with MG115 or MG132 (right).Different exposure times are shown for p53. siRNA transfection was done 96 h before cell harvesting. Histograms represent means and SDs from threeindependent experiments.
Cancer Res; 70(11) June 1, 2010 Cancer Research

Lack of Dyskerin Inactivates p53
Figure 2. Reduction of dyskerin levels downregulates p53-mediated response in breast cancer cells. A, p53 transcriptional activity after DKC1, p53, andcontrol (SCR) siRNA transfection, assessed by reporter assay. B, immunoblots showing p53 and p21 levels in DKC1 KD and control cells 8 h afterionizing radiation exposure. C, immunoblots showing p53 levels in DKC1 KD and control cells after doxorubicin treatment (top). Cell death (by trypan blueexclusion test; bottom left) and growth (after crystal violet staining; bottom right) analyses under doxorubicin treatment are also shown. D, immunoblotsand RT-PCR analysis showing p53 and CA-IX mRNA levels, respectively, in DKC1 KD and control cells for 48 h with deferoxamine. Cell death analysisunder DFX treatment is also shown. Experiments were done in MCF-7 cells. siRNA transfection was done 96 h before cell harvesting. Histograms represent meansand SDs from three independent experiments.
Cancer Res; 70(11) June 1, 2010www.aacrjournals.org 4771

Montanaro et al.
4772
treatment with the genotoxic agent doxorubicin (Fig. 2C).This effect was associated with an increased cellular resis-tance to p53-dependent doxorubicin-induced death and witha growth advantage under doxorubicin treatment (Fig. 2C).Moreover, DKC1 KD strongly inhibited the p53 accumulationinduced by the hypoxia-mimetic agent deferoxamine (Fig. 2D).In these conditions, the expression of the hypoxia-induced, p53negatively regulated carbonic anhydrase IX gene (29) wasstrongly upregulated, indicating a reduction in p53-mediatedtranscriptional repression. After DKC1 KD, cells were alsomore resistant to deferoxamine-mediated cell death, an obser-vation consistent with the known prosurvival effect of p53reduction and carbonic anhydrase IX overexpression underhypoxia-like conditions (refs. 30, 31; Fig. 2D). Significantly,we confirmed that the reduction of dyskerin levels also affectsp53 accumulation in an additional p53 wild-type breast cancercell line such as ZR-75-1 (ref. 32; Supplementary Fig. S2).
Dyskerin reduction impairs p53 function in primarymammary gland progenitorsTo verify if the observed impairment of p53 function may
also occur in human nonneoplastic primary breast epithelialcells, we evaluated the effect of DKC1 KD incultures of pro-genitor cells derived from normal mammary gland tissue
Cancer Res; 70(11) June 1, 2010
expanded as MS (23, 33). siRNA sequences were effectivelydelivered in MS, producing a strong DKC1 KD (Fig. 3A). Al-though the material obtained from this source was too scarcefor assessing p53 levels by Western blot analysis, we wereable to document that DKC1 KD reduced the expression ofdirect p53 targets such as p21 (28), PUMA (34), and BNIP3(35) both at the steady state and after exposure to doxorubi-cin (Fig. 3B), thus indicating a decrease of p53 activity. More-over, dyskerin reduction led to an increase in the size of theMS, as it occurs in consequence of p53 KD (Fig. 3C). Also in asimilar experiment performed in MS obtained from a humanprimary breast carcinoma (36), DKC1 KD lowered the levelsof p21 and BNIP3 mRNAs (Fig. 3D). Besides providing infor-mation on primary breast epithelial cells of human origin,these results on MS are particularly relevant in light of theobservation that carcinogenesis in the mammary glandmight result from transformation of progenitor cells by thederegulation of self-renewal pathways in which p53 is largelyinvolved (37, 38).
Dyskerin reduction acts on p53 independently of itsrole in telomeraseOne well-characterized result of dyskerin dysfunction is
the impairment of telomerase complex functionality due
Figure 3. The reduction of dyskerin levels downregulates p53 activity in human primary mammary gland progenitors. A, DKC1 mRNA levels by real-timeRT-PCR in human normal mammary gland tissue after 96 h in DKC1 KD and control (SCR) cells. B, semiquantitative RT-PCR analysis showing expression ofp21, PUMA, and BNIP3 in primary human MS from normal tissue after DKC1 KD at the basal level and after doxorubicin treatment. C, size of MS fromhuman normal mammary gland tissue 96 h after DKC1 and p53 KD. Phase-contrast microscopy micrographs and MS size distribution are shown.Bar, 100 μm. D, DKC1 mRNA levels by real-time RT-PCR and expression of p21, PUMA, and BNIP3 by semiquantitative RT-PCR analysis in primary humanMS from tumoral tissue 96 h after DKC1 KD.
Cancer Research

Lack of Dyskerin Inactivates p53
Figure 4. Reduction of dyskerin levels acts on p53 independently of its effects on telomerase activity. A, DKC1 mRNA expression, TERC levels measured byreal-time PCR, and quantitative telomerase activity by a telomere repeat amplification protocol measured by real-time PCR in pBabe-TER MCF-7 cellsand in control pBabe empty vector MCF-7 cells after DKC1 and control (SCR) siRNA transfection. B, representative immunoblots showing dyskerin and p53levels in TERC-overexpressing pBabe-TER and in pBabe empty vector MCF-7 control cells after DKC1 and control (SCR) siRNA transfection at thesteady state and after doxorubicin treatment. C, expression of p21 and PUMA mRNAs by real-time RT-PCR in pBabe-TER and in control pBabe emptyvector MCF-7 cells after DKC1 and control (SCR) siRNA transfection at the steady state and after doxorubicin treatment. siRNA transfections were done96 h before cell harvesting. All histograms show fold changes relative to the values observed in the control pBabe empty vector MCF-7 cells. Histogramsrepresent means and SDs from three independent experiments.
Cancer Res; 70(11) June 1, 2010www.aacrjournals.org 4773

Montanaro et al.
4774
to telomerase RNA component (TERC) destabilization(39). This was observed also in MCF-7 cells after DKC1KD (Fig. 4A). In cells derived from X-DC patients, theoverexpression of TERC is sufficient to rescue the telo-merase defect associated with DKC1 gene mutation (40).To rule out the possibility that the effect of DKC1 KD onp53 may be in any way linked to the impairment of telo-merase function, we generated, by retroviral transduction,MCF-7 cells stably overexpressing TERC (MCF-7 pBabe-TER; ref. 41). In these cells, due to the high rate of TERCtranscription, DKC1 KD did not cause a significant impair-ment in telomerase activity (Fig. 4A) but strongly loweredp53 levels both at the steady state and after doxorubicintreatment, similarly to what occurred in the empty vector–infected control cells (MCF-7 pBabe empty vector; Fig. 4B).Also, DKC1 KD reduced the expression levels of p53 targetgenes such as p21 and PUMA after doxorubicin treatment
Cancer Res; 70(11) June 1, 2010
both in MCF-7 pBabe empty vector cells and in MCF-7pBabe-TER, showing that p53 activity is not influencedby the effect of dyskerin reduction on telomerase activity(Fig. 4C). These results indicate that the reduction in dyskerinlevels affects p53 levels and function independently of its rolein telomerase activity.
Dyskerin reduction induces defectivep53-IRES–mediated translationWe then investigated the mechanism leading to the alter-
ation in p53 translation. The translation of a group of IRES-containing mRNAs is defective in cells bearing dysfunctionaldyskerin (18). We evaluated how IRES-mediated translation isregulated in our model by measuring the activity of a fireflyluciferase encoded by a downstream cistron under the controlof an IRES sequence in a bicistronic mRNA. As occurs in X-DC(18), DKC1 KD in MCF-7 cells induced a defective translation
Figure 5. Reduction of dyskerin levels impairs p53-IRES–mediated translation. A, IRES-mediated translation assessed by measuring the FLuc and RLucactivity in MCF-7 cells after DKC1 and control (SCR) siRNA transfection and 8 h after transfection with a bicistronic mRNA transcribed from pRLCrPV-IRES. B, p53-IRES–mediated translation after DKC1 KD and 8 h after transfection with a bicistronic mRNA transcribed from pRL p53-IRES inMCF-7 cells with or without 72-h deferoxamine treatment. siRNA transfection was done 96 h before cell harvesting. Histograms represent means and SDsfrom three independent experiments.
Cancer Research

Lack of Dyskerin Inactivates p53
mediated by viral IRES sequences, such as the cricket paraly-sis virus (CrPV)-IRES (Fig. 5A). Using a similar bicistronicmRNA approach, we then evaluated whether the translationmediated by an IRES sequence described in p53 mRNA can beinfluenced by dyskerin levels. DKC1 KD strongly impaired thep53-IRES–mediated translation both at the steady state andunder deferoxamine (Fig. 5B). Remarkably, such hypoxia-likeconditions elicited a strong reduction in the global levels ofprotein synthesis and cap-dependent translation (Fig. 5Band Supplementary Fig. S3A), similarly to what occurs inthe presence of low oxygen tension (42). On the other hand,p53 accumulation required active mRNA translation (Supple-mentary Fig. S3B). According to this, p53-IRES–mediatedtranslation substantially increased under deferoxamine treat-ment (Fig. 5B). In principle, such an increase should stronglycontribute to maintaining p53 synthesis when cap-dependenttranslation is repressed. The observed impairment of p53-IRES–mediated translation should then be considered particularly rel-evant for p53 function under stress conditions. The obtainedresults were confirmed in MCF-7 cells using three differentsiRNA sequences and in other breast cancer–derived cell linessuch as ZR-75-1 and MDA-MB-468 (Supplementary Fig. S4).
Dyskerin expression and p53 function inhuman breast cancerWe next aimed at investigating whether a similar mecha-
nism could be present also in primary breast tumors. To thisend, we measured DKC1 mRNA expression in a series of
www.aacrjournals.org
primary breast carcinomas. Supplementary Table S1 sum-marizes the biopathologic features of the studied series. Totest the relationship between dyskerin expression and p53activity in this series, we have first defined the p53 statusby immunohistochemical analysis. The putatively p53-mutatedtumors were identified by both the presence of p53 and theabsence of p21 in the same specimen (26). We also assessedthe agreement of the immunohistochemical method of p53mutation detection with direct sequencing in a subgroupof cases using the κ statistics. A κ value of 1.00 indicates per-fect agreement, and 0 indicates a level of agreement bychance only. The obtained κ value was 0.72, indicating a goodconcordance. Then, in the whole subgroup of immunohisto-chemically defined p53 wild-type tumors, we measured byreal-time RT-PCR the expression levels of p21 and MDM2,two p53 target genes. The expression of these two genes re-sulted to be significantly associated with DKC1 mRNA levels(Fig. 6A), indicating a reduction of p53 activity in tumors ex-pressing low dyskerin mRNA levels, grouped according to thepreviously described cutoff values of DKC1 mRNA expression(22). In the same series, we also studied the relationship be-tween p53 status and dyskerin expression. We found thattumors with low dyskerin expression were more frequentlycharacterized by a p53 wild-type status than tumors withhigh dyskerin expression (Fig. 6B), thus supporting thehypothesis that decreased dyskerin expression may representan alternative mechanism of p53 inactivation, which, in somecases, replaces the p53 mutation mechanism.
Figure 6. DKC1 mRNA expressionlevels and p53 function and status inhuman breast carcinomas. A, p21 andMDM2 mRNA expression by real-timePCR in p53 wild-type breast carcinomasgrouped according to dyskerin mRNAexpression levels. Gray box plots showmRNA values within the three DKC1mRNA groups. The median value isdepicted by horizontal columns, theinterquartile range by boxes, and theminimum and maximum values byvertical columns. The expression inp53-mutated tumors is also shown forreference (white box plots). B,frequencies of p53-mutated primarybreast carcinomas grouped according toDKC1 mRNA.
Cancer Res; 70(11) June 1, 2010 4775

Montanaro et al.
4776
Discussion
Our results show a novel dyskerin-dependent mechanismof p53 inactivation acting on p53 mRNA translation. The re-duction of dyskerin levels impairs p53 function indepen-dently of its well-known role in telomerase activity andcauses a strong defect in p53-IRES–mediated translation ini-tiation. Data obtained on primary breast cancer specimenssuggest that the described dyskerin-dependent mechanismof p53 inactivation may be active in a subset of humanbreast carcinomas.It has been previously shown that the efficient translation
of p53 mRNA is necessary for proper p53-mediated response(11). In this study, for the first time, we report that the lack ofdyskerin impinges p53 mRNA translation and efficient p53-mediated response. This result provides a rationale for ex-plaining the increased tumor susceptibility in X-DC. In fact,this phenotype can result from the contribution of thealtered translation of p53 mRNA as well as of other IRES-containing mRNAs that code for tumor suppressors, suchas p27 mRNA, as already described (18). In a recent report,an enhanced p53 response was identified in cells from micebearing a specific truncating dyskerin mutation also found inthe X-DC family (43). Our findings in the breast epithelial cel-lular types studied are not in line with such an observation.We may attempt to explain this discrepancy with the differ-ent approaches used to affect dyskerin function. In fact, inthe previous study, the particular properties of the DKC1 mu-tation used may affect dyskerin function differently than theKD approach used in the present report. Our results alsoindicate that the defect in p53 translation and function isindependent of the relevant role played by dyskerin in telo-merase function. Given the well-established role of dyskerinin the site-specific rRNA uridine modification (44), this find-ing supports the proposed model in which an intrinsic ribo-somal defect might affect the translation of specific cellularmRNAs (45). Our results also indicate that the reduction ofdyskerin levels induced a strong defect in p53-IRES–mediatedtranslation. Studies using different experimental approachesindicate that several cellular mRNAs coding for tumor sup-pressors contain an IRES element (15, 46, 47) and that theloss of proper IRES-mediated translation can contribute to
Cancer Res; 70(11) June 1, 2010
tumorigenesis (18, 48). The observation that the decreaseof IRES translation is associated with a strong reduction inthe activity of p53 supports the involvement of defectiveIRES-mediated translation in tumorigenesis.In the DKC1 hypomorphic mice, the defective uridine
modification in rRNA has been associated with a high sus-ceptibility to tumors, including breast carcinomas (21). Wepreviously showed that dyskerin expression is extremely var-iable in human sporadic tumors of various histologic origins(22). In particular, in breast carcinomas, tumors character-ized by very low dyskerin levels also exhibit defective rRNApseudouridylation (22). In the present study, the observationsobtained in human primary breast carcinomas indicate thatp53 function is also reduced in the presence of low dyskerinlevels. It is also worth recalling that mutation of the p53 geneoccurs in only about 20% of breast carcinomas, whereas inother solid tumors the mutation rates are about 70% (49, 50).Because the proper function of dyskerin is necessary forefficient p53 mRNA translation, it is tempting to speculatethat, in breast carcinomas not bearing direct p53 gene muta-tions, the alteration in dyskerin function could be one of themechanisms resulting in a decrease of p53 activity, thus con-tributing to the neoplastic phenotype.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Kathleen Collins (University of California at Berkeley,Berkeley, CA), Dr. B. Mazumder, and D. Ruggero for reagents, andDr. P. Scaglioni for helpful suggestions.
Grant Support
Pallotti Legacy for Cancer Research and Association for International CancerResearch grant 09-0083 (L. Montanaro).
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received 11/02/2009; revised 03/29/2010; accepted 03/31/2010; publishedOnlineFirst 05/25/2010.
References
1. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol CellBiol 2007;8:275–83.2. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid deg-
radation of p53. Nature 1997;387:296–9.3. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by
Mdm2. Nature 1997;387:299–303.4. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev
Cancer 2002;2:594–604.5. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in
human cancers. Science 1991;253:49–53.6. Linares LK, Hengstermann A, Ciechanover A, Müller S, Scheffner M.
HdmX stimulates Hdm2-mediated ubiquitination and degradation ofp53. Proc Natl Acad Sci U S A 2003;100:12009–14.
7. Danovi D, Meulmeester E, Pasini D, et al. Amplification of Mdmx
(or Mdm4) directly contributes to tumor formation by inhibiting p53tumor suppressor activity. Mol Cell Biol 2004;24:5835–43.
8. Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev1998;12:2984–91.
9. Colaluca IN, Tosoni D, Nuciforo P, et al. NUMB controls p53 tumoursuppressor activity. Nature 2008;451:76–80.
10. Hollstein M, Hainaut P. Massively regulated genes: the example ofTP53. J Pathol 2010;220:164–73.
11. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Par-ticipation of p53 protein in the cellular response to DNA damage.Cancer Res 1991;51:6304–11.
12. Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, DeppertW. Negative feedback regulation of wild-type p53 biosynthesis.EMBO J 1995;14:4442–9.
Cancer Research

Lack of Dyskerin Inactivates p53
13. Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53translation and induction after DNA damage by ribosomal proteinL26 and nucleolin. Cell 2005;123:49–63.
14. Grover R, Raym PS, Das S. Polypyrimidine tract binding proteinregulates IRES-mediated translation of p53 isoforms. Cell Cycle2008;7:2189–98.
15. Yang DQ, Halaby MJ, Zhan Y. The identification of an internal ribo-somal entry site in the 5′-untranslated region of p53 mRNA providesa novel mechanism for the regulation of its translation following DNAdamage. Oncogene 2006;25:4613–9.
16. Schneider RJ, Sonenberg N. Translational control in cancer develop-ment and progression. In: Translational control in biology andmedicine. In: MathewsMB, Sonenberg N, Hershey JWB, editors. ColdSpring Harbor (NY): Cold Spring Harbor Laboratory Press; 2007.
17. Meier UT. The many facets of H/ACA ribonucleoproteins. Chromoso-ma 2005;114:1–14.
18. Yoon A, Peng G, Brandenburger Y, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science2006;312:902–6.
19. Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosiscongenita is caused by mutations in a highly conserved gene withputative nucleolar functions. Nat Genet 1998;19:32–8.
20. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 2000;110:768–79.
21. Ruggero D, Grisendi S, Piazza F, et al. Dyskeratosis congenita andcancer in mice deficient in ribosomal RNA modification. Science2003;299:259–62.
22. Montanaro L, Brigotti M, Clohessy J, et al. Dyskerin expression influ-ences the level ofribosomal RNA pseudo-uridylation and telomeraseRNA component in human breast cancer. J Pathol 2006;210:10–8.
23. Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation andtranscriptional profiling of human mammary stem/progenitor cells.Genes Dev 2003;17:1253–70.
24. Petronini PG, Tramacere M, Mazzini A, Piedimonte G, Silvotti L,Borghetti AF. Hyperosmolarity-induced stress proteins in chickembryo fibroblasts. Exp Cell Res 1987;172:450–62.
25. Chaudhuri S, Vyas K, Kapasi P, et al. Human ribosomal protein L13ais dispensable for canonical ribosome function but indispensable forefficient rRNA methylation. RNA 2007;13:2224–37.
26. Nenutil R, Smardova J, Pavlova S, et al. Discriminating functionaland non-functional p53 in human tumours by p53 and MDM2 immu-nohistochemistry. J Pathol 2005;207:251–9.
27. Holcik M. Translational upregulation of the X-linked inhibitor of apo-ptosis. Ann N Y Acad Sci 2003;1010:249–58.
28. El-Deiry WS, Harper JW, O'Connor PM, et al. WAF1/CIP1 is inducedin p53-mediated G1 arrest and apoptosis. Cancer Res 1994;54:1169–74.
29. Kaluzová M, Kaluz S, Lerman MI, Stanbridge EJ. DNA damage is aprerequisite for p53-mediated proteasomal degradation of HIF-1α inhypoxic cells and downregulation of the hypoxia marker carbonicanhydrase IX. Mol Cell Biol 2004;24:5757–66.
30. Hammond EM, Giaccia AJ. The role of p53 in hypoxia-inducedapoptosis. Biochem Biophys Res Commun 2005;331:718–25.
31. Sansone P, Storci G, Tavolari S, et al. IL-6 triggers malignant features
www.aacrjournals.org
in mammospheres from human ductal breast carcinoma and normalmammary gland. J Clin Invest 2007;117:3988–4002.
32. Wasielewski M, Elstrodt F, Klijn JG, Berns EM, Schutte M. Thirteennew p53 gene mutants identified among 41 human breast cancer celllines. Breast Cancer Res Treat 2006;99:97–101.
33. Mani SA, GuoW, LiaoMJ, et al. The epithelial-mesenchymal transitiongenerates cells with properties of stem cells. Cell 2008;133:704–15.
34. Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediatesthe apoptotic response to p53 in colorectal cancer cells. Proc NatlAcad Sci U S A 2003;100:1931–6.
35. Yan J, Yun H, Yang Y, Jing B, Feng C, Song-bin F. Upregulation ofBNIP3 promotes apoptosis of lung cancer cells that were induced byp53. Biochem Biophys Res Commun 2006;346:501–7.
36. Storci G, Sansone P, Trere D, et al. The basal-like breast carcinomaphenotype is regulated by SLUG gene expression. J Pathol 2008;214:25–38.
37. Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal path-ways, and carcinogenesis. Breast Cancer Res 2005;7:86–95.
38. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: anew concept in cancer therapeutics. BioDrugs 2007;21:299–310.
39. Mitchell JR, Wood E, Collins K. A telomerase component is defec-tive in the human disease dyskeratosis congenita. Nature 1999;402:551–5.
40. Wong JM, Collins K. Telomerase RNA level limits telomere main-tenance in X-linked dyskeratosis congenita. Genes Dev 2006;20:2848–58.
41. Montanaro L, Calienni M, Ceccarelli C, et al. Relationship betweendyskerin expression and telomerase activity in human breast cancer.Cell Oncol 2008;30:483–90.
42. Braunstein S, Karpisheva K, Pola C, et al. A hypoxia-controlledcap-dependent to cap-independent translation switch in breastcancer. Mol Cell 2007;28:501–12.
43. Gu BW, Bessler M, Mason PJ. A pathogenic dyskerin mutationimpairs proliferation and activates a DNA damage response inde-pendent of telomere length in mice. Proc Natl Acad Sci U S A2008;105:10173–8.
44. Wang C, Query CC, Meyer UT. Immunopurified small nucleolarribonucleoprotein particles pseudouridylate rRNA independently oftheir association with phosphorylated Nopp140. Mol Cell Biol2002;22:8457–66.
45. Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, andcancer. Am J Pathol 2008;173:301–10.
46. Kullmann M, Göpfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibitp27 translation via an IRES element in the p27 5′UTR. Genes Dev2002;16:3087–99.
47. Wilker EW, van Vugt MA, Artim SA, et al. 14-3-3σ controls mitotictranslation to facilitate cytokinesis. Nature 2007;446:329–32.
48. Barna M, Pusic A, Zollo O, et al. Suppression of Myc oncogenicactivity by ribosomal protein haploinsufficiency. Nature 2008;456:971–5.
49. Gasco M, Shami S, Crook T. The p53 pathway in breast cancer.Breast Cancer Res 2002;4:70–6.
50. Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update.Endocr Relat Cancer 2006;13:293–325.
Cancer Res; 70(11) June 1, 2010 4777
Related Documents











