Research paper Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors Jean-Ren e Pallandre a , Christophe Borg a , Didier Rognan b , Thibault Boibessot c , Vincent Luzet c , Semen Yesylevskyy d, e , Christophe Ramseyer d , Marc Pudlo c, * a Inserm 1098, EFS Bourgogne Franche Comt e, University of Franche-Comt e, IFR133, 8 rue du Dr Girod, 25020 Besançon, France b Laboratory for Therapeutical Innovation, UMR 7200 Universit e de Strasbourg/CNRS, MEDALIS Drug Discovery Center, F-67400 Illkirch, France c Nanomedicine Lab, EA e 4662, UFR SMP 19 rue Ambroise Par e Universit e de Franche Comt e, 25030 Besançon Cedex, France d Laboratoire Chrono Environnement UMR CNRS 6249, Facult e des Sciences et Techniques, La Bouloie, Universit e de Franche-Comt e, 25030, Besançon Cedex, France e Institute of Physics, National Academy of Sciences of Ukraine, Prospect Nauki, 46, Kyiv, 03039, Ukraine article info Article history: Received 2 July 2015 Received in revised form 25 August 2015 Accepted 27 August 2015 Available online 1 September 2015 Keywords: STAT3 Small-molecule inhibitor Aminotetrazole Anticancer drug Inflammation abstract The development of inhibitors blocking STAT3 transcriptional activity is a promising therapeutic approach against cancer and inflammatory diseases. In this context, the selectivity of inhibitors against the STAT1 transcription factor is crucial as STAT3 and STAT1 play opposite roles in the apoptosis of tumor cells and polarization of the immune response. A structure-based virtual screening followed by a luciferase-containing promoter assay on STAT3 and STAT1 signaling were used to identify a selective STAT3 inhibitor. An important role of the aminotetrazole group in modulating STAT3 and STAT1 inhib- itory activities has been established. Optimization of the hit compound leads to 23. This compound inhibits growth and survival of cells with STAT3 signaling pathway while displaying a minimal effect on STAT1 signaling. Moreover, it prevents lymphocyte T polarization into Th17 and Treg without affecting their differentiation into Th1 lymphocyte. © 2015 Elsevier Masson SAS. All rights reserved. 1. Introduction Signal Transducers and Activators of Transcription (STATs) are a family of seven cytoplasmic transcription factors (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6) with critical roles in the regulation of cell growth, cell differentiation, inflammation and immune response [1]. STAT proteins possess six structural regions including a N-domain, a coiled-coil domain, a DNA-binding domain, a linker domain, a Src Homology 2 (SH2) domain involved in proteineprotein interaction regulation and a tran- scription activation domain. STAT3 activation is mediated mainly by Src proteins or growth factor receptors. Activated Jak kinases phosphorylate tyrosine res- idues in the intracellular domains of these receptors [2]. STAT monomers bind with their SH2 domains to these receptor-docking sites allowing further interactions of STAT with Jak tyrosine kinases. Jak phosphorylates the tyrosine located at the C-terminal end of the STAT3 SH2 domain resulting in dimer formation through mutual SH2 domain-phosphotyrosine interactions. STAT dimers then translocate into the nucleus to mediate selective transcriptional activity [3]. Besides the transient activation in normal cells, constitutive or inappropriate activation of STAT3 has been found to directly contribute to oncogenesis in a wide variety of cancers [4]. In contrast, STAT1 is thought to play an opposite role via anti- proliferative and pro-apoptotic activities in tumor cells [5]. In addition, a transcriptional response to each STAT protein activation is highly specific and provides polarization of the immune response. An involvement of T helper 17 (Th17) lymphocyte Abbreviations: STAT, signal transducer and activation of transcription; SH2, Src Homoly 2; Th, T helper; OSM, oncostatin M; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide; NK, Natural Killer. * Corresponding author. E-mail addresses: [email protected] (J.-R. Pallandre), xtoph. [email protected] (C. Borg), [email protected] (D. Rognan), thibautboibessot@ hotmail.fr (T. Boibessot), [email protected] (V. Luzet), yesint4@gmail. com (S. Yesylevskyy), [email protected] (C. Ramseyer), marc. [email protected] (M. Pudlo). Contents lists available at ScienceDirect European Journal of Medicinal Chemistry journal homepage: http://www.elsevier.com/locate/ejmech http://dx.doi.org/10.1016/j.ejmech.2015.08.054 0223-5234/© 2015 Elsevier Masson SAS. All rights reserved. European Journal of Medicinal Chemistry 103 (2015) 163e174

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

lable at ScienceDirect
European Journal of Medicinal Chemistry 103 (2015) 163e174
Contents lists avai
European Journal of Medicinal Chemistry
journal homepage: http: / /www.elsevier .com/locate/ejmech
Research paper
Novel aminotetrazole derivatives as selective STAT3 non-peptideinhibitors
Jean-Ren�e Pallandre a, Christophe Borg a, Didier Rognan b, Thibault Boibessot c,Vincent Luzet c, Semen Yesylevskyy d, e, Christophe Ramseyer d, Marc Pudlo c, *
a Inserm 1098, EFS Bourgogne Franche Comt�e, University of Franche-Comt�e, IFR133, 8 rue du Dr Girod, 25020 Besançon, Franceb Laboratory for Therapeutical Innovation, UMR 7200 Universit�e de Strasbourg/CNRS, MEDALIS Drug Discovery Center, F-67400 Illkirch, Francec Nanomedicine Lab, EA e 4662, UFR SMP 19 rue Ambroise Par�e Universit�e de Franche Comt�e, 25030 Besançon Cedex, Franced Laboratoire Chrono Environnement UMR CNRS 6249, Facult�e des Sciences et Techniques, La Bouloie, Universit�e de Franche-Comt�e, 25030, Besançon Cedex,Francee Institute of Physics, National Academy of Sciences of Ukraine, Prospect Nauki, 46, Kyiv, 03039, Ukraine
a r t i c l e i n f o
Article history:Received 2 July 2015Received in revised form25 August 2015Accepted 27 August 2015Available online 1 September 2015
Keywords:STAT3Small-molecule inhibitorAminotetrazoleAnticancer drugInflammation
Abbreviations: STAT, signal transducer and activatHomoly 2; Th, T helper; OSM, oncostatin M; MTT, 3-(4diphenyltetrazolium bromide; NK, Natural Killer.* Corresponding author.
E-mail addresses: [email protected]@gmail.com (C. Borg), [email protected] (D.hotmail.fr (T. Boibessot), [email protected] (S. Yesylevskyy), christophe.ramseyer@[email protected] (M. Pudlo).
http://dx.doi.org/10.1016/j.ejmech.2015.08.0540223-5234/© 2015 Elsevier Masson SAS. All rights re
a b s t r a c t
The development of inhibitors blocking STAT3 transcriptional activity is a promising therapeuticapproach against cancer and inflammatory diseases. In this context, the selectivity of inhibitors againstthe STAT1 transcription factor is crucial as STAT3 and STAT1 play opposite roles in the apoptosis of tumorcells and polarization of the immune response. A structure-based virtual screening followed by aluciferase-containing promoter assay on STAT3 and STAT1 signaling were used to identify a selectiveSTAT3 inhibitor. An important role of the aminotetrazole group in modulating STAT3 and STAT1 inhib-itory activities has been established. Optimization of the hit compound leads to 23. This compoundinhibits growth and survival of cells with STAT3 signaling pathway while displaying a minimal effect onSTAT1 signaling. Moreover, it prevents lymphocyte T polarization into Th17 and Treg without affectingtheir differentiation into Th1 lymphocyte.
© 2015 Elsevier Masson SAS. All rights reserved.
1. Introduction
Signal Transducers and Activators of Transcription (STATs) are afamily of seven cytoplasmic transcription factors (STAT1, STAT2,STAT3, STAT4, STAT5a, STAT5b and STAT6) with critical roles in theregulation of cell growth, cell differentiation, inflammation andimmune response [1]. STAT proteins possess six structural regionsincluding a N-domain, a coiled-coil domain, a DNA-bindingdomain, a linker domain, a Src Homology 2 (SH2) domain
ion of transcription; SH2, Src,5-dimethylthiazol-2-yl)-2,5-
te.fr (J.-R. Pallandre), xtoph.Rognan), thibautboibessot@fr (V. Luzet), [email protected] (C. Ramseyer), marc.
served.
involved in proteineprotein interaction regulation and a tran-scription activation domain.
STAT3 activation is mediated mainly by Src proteins or growthfactor receptors. Activated Jak kinases phosphorylate tyrosine res-idues in the intracellular domains of these receptors [2]. STATmonomers bind with their SH2 domains to these receptor-dockingsites allowing further interactions of STATwith Jak tyrosine kinases.Jak phosphorylates the tyrosine located at the C-terminal end of theSTAT3 SH2 domain resulting in dimer formation through mutualSH2 domain-phosphotyrosine interactions. STAT dimers thentranslocate into the nucleus to mediate selective transcriptionalactivity [3].
Besides the transient activation in normal cells, constitutive orinappropriate activation of STAT3 has been found to directlycontribute to oncogenesis in a wide variety of cancers [4]. Incontrast, STAT1 is thought to play an opposite role via anti-proliferative and pro-apoptotic activities in tumor cells [5]. Inaddition, a transcriptional response to each STAT protein activationis highly specific and provides polarization of the immuneresponse. An involvement of T helper 17 (Th17) lymphocyte

J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174164
polarization contributes to the initiation of inflammatory diseasesas highlighted in colitis or psoriasis models. Here again, the balancebetween STAT3 and STAT1 was shown to be an important compo-nent in the tuning of the immune response as STAT3 activation isassociated with Th17 polarization while STAT1 signaling preventsTh17 polarization [6]. Therefore, the development of inhibitorsblocking STAT3 transcriptional activity is a relevant clinical issue [7]and the selectivity against STAT1 is crucial for the development ofnew STAT3 inhibitors with interesting pharmacodynamic profiles.
Since indirect STAT3 blockade by Jak inhibitors is poorly selec-tive and could therefore cause severe adverse effects, several ap-proaches have been taken to directly inhibit STAT3 dimerization[8]. Several peptides and phosphopeptides were designed tomimic the amino acid sequence of STAT3, which interacts with theSH2 domain upon dimerization. However, strategies based on suchpeptides have shown several limitations including low cellpermeability and stability. Therefore, attention has been shiftedtowards small non-peptidic inhibitors [9] with better pharmaco-kinetic properties. Here, we aimed to design non-peptidic smallmolecules inhibiting STAT3 activity with enhanced selectivityagainst STAT1. For this purpose, a structure-based virtual screeningapproach was used to identify commercially available chemicalcompounds with the potential to bind STAT3 to the SH2 domain. Aluciferase-containing promoter assay was used to investigate theactivity of each candidate on STAT3 and STAT1 signaling. Thisapproach led to the identification of a prospective selective STAT3inhibitor (5). Then, an extensive structure activity relationshipsstudy was performed resulting in the molecule 23 and some otherderivatives. We demonstrate that an aminotetrazole groupincluded in the hydrophilic moiety impaired the ability of selectedchemical compounds to bind STAT1 while maintaining STAT3 in-hibition. The inhibitor 23 does not influence STAT1 signaling and iseffective in vitro in blocking STAT3-dependent physiological func-tions, such as cancer cell proliferation and Th17 polarization.
2. Results
2.1. Virtual screening for non-peptide inhibitors of STAT3 SH2domain
The potential interest to target the SH2 domain of STAT3 usingnon peptide low molecular-weight compounds prompted us toidentify prospective inhibitors by a docking-based virtual screeningof a commercially available compound library [10] targeting theavailable crystal structure of mouse STAT3 SH2 domain [11]. Thewell-known pTyr705-binding site (Lys591, Arg609, Ser611, Ser613)of the Stat3b homodimer was specifically selected for high-throughput docking. Virtual hit selection was based onknowledge-based scoring [12] aimed at identifying compounds,which share key interactions to the Stat3 pTyr705 binding site withthe co-crystallized phosphorylated peptide (Ala703-Pro704-pTyr705-Leu706-Lys707). A list of 9 out of 52 candidates was selected (Fig. 1A)based on commercial availability and the aqueous solubility ofcompounds. Compounds were then purchased and tested for theirability to inhibit STAT3 signaling in vitro. For this purpose a lucif-erase promoter assay including STAT3 response elements was usedas described previously [13]. Genetically modified HeLa cells withthe luciferase STAT3-promoter array were exposed to 50 mM ofselected candidate compounds and treated with Oncostatin M inorder to activate STAT3. Inhibitions of STAT3 luciferase activity of allstudied compounds are shown in Fig. 2. All candidates preventedthe induction of luciferase activity following Oncostatin M treat-ment. Compounds 2 and 6 were cytotoxic on Hela cells (data notshown) and were excluded from further analysis.
2.2. Influence of the selected STAT3 inhibitors on STAT1 activity
The selectivity of selected compounds for STAT3 vs. STAT1 in-hibition was tested as follows. HeLa cells containing a luciferasepromoter array with STAT1 response elements were used [14].STAT1 signaling was induced by interferon g. STAT3 luciferase as-says were performed in parallel to determine the selectivity of eachinhibitor. The well-known STAT3 SH2 domain inhibitor STA21 [15],was used as a control for STAT3 inhibition. The final concentrationof all candidates and STA21 was set to 50 mM. The candidate 5displayed selectivity close to that achieved with STA21 (Fig. 2). Thiscompound was therefore synthesized in our laboratory to confirminitial screening results and for further experiments. (Fig. 3)
2.3. Structure-activity relationships and development of inhibitor23
An extensive structure-activity relationships study was per-formed by synthetizing analogues of 5where each of three radicalsR1, R2, R3 (see Table 1) was substituted by different chemical groups.The stages of chemical synthesis of 5 and 23 are shown in Fig. 3whereas that of all other derivatives is shown in Figs. S1eS12.Each compound was assessed in STAT3 and STAT1-luciferase re-porter assays to evaluate its effect on STAT3 transcriptional activityand selectivity against STAT1. The cytotoxicity of each compoundwas tested after 24, 72 and 120 h of cell culture using a trypan blueexclusion assay or a viability test based on tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).
As a methyl on a phenyl moiety is quickly metabolized, a sig-nificant amount of work has been done to substitute this group (R1substituent, Table 1.1). Electron donating group (14) and hydro-philic groups (20, 21) reduced activity on STAT3 while small elec-tron withdrawing groups retained or increased activity andselectivity against STAT1 (11, 12, 16, 23). Among those, the nitrogroup at the para position (23) led to an effective STAT3 inhibitionand enhanced selectivity. The optimal position of the nitro sub-stituent has been confirmed to be the para position (24, 25). Anadditional nitro group did not improve the inhibitory activity (26),however an additional alkoxy substituent on the benzyl moiety(Table 1.2) leads to increase of activity depending on the size ofsubstituent. This last series of compounds led to the identificationof compound 30, which was endowed with high activity but lackedselectivity against STAT1. We also synthesized and tested a series ofcycles and heterocycles fused or linked to the benzyl moietywithout any convincing result (Table S1).
The methoxy group appeared to be the optimal R2 substituent,which cannot be removed or replaced (Tables 1.3 and 1.4). In theR1 ¼ 4-CH3 series, the activity on STAT3 was enhanced by modu-lating the hydrophobicity of the halogen (Table 1.5). Bromine at R3seems the best compromise between potency and selectivity butthis was not reproducible in the nitro series (Table 1.6).
2.4. Importance of the aminotetrazole group
Variations based on the partial or complete modification of theaminotetrazole group were set up to characterize the involvementof each parameter in inhibition and selectivity (Tables 1.7 and 1.8).The importance of the secondary amine was shown by the reducedactivity of the methylene derivative 44. Replacing the tetrazole of 5with a carboxylic acid in 45 significantly reduced both STAT3 andSTAT1 inhibition. Interestingly, non ionizable aldehyde and cyana-mide derivatives that share a planar structure displayed the samerange of inhibition as the tetrazole analogs. Among the R1 ¼ 4-CH3series, the cyanamide 43 and the aldehyde 42 were both able toinhibit STAT3 transcriptional activity. These experiments suggest

Fig. 1. Virtual screening identification of compound 5 as STAT3 inhibitor A) Structures of potential STAT3 inhibitors identified by virtual screening. asupplier code B) Docking pose ofcompound 5 in the SH2 domain of STAT3 (PDB code: 1BG1) [11]. The STAT3 backbone structure is displayed as a grey ribbon with a-helices as cylinders and b-strands as arrows.Important STAT3 side chains (cyan carbon atoms) for compound 5 (yellow carbon atoms) binding are indicated as sticks. Other heteroatoms (nitrogen, oxygen, chlorine) are coloredin blue, red and green, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174 165
that the planar structure of the hydrophilic part of inhibitors iscrucial for STAT3 binding. Indeed, the compounds 42 and 43 wereable to inhibit STAT1 activity whereas their tetrazole analog (5)exhibited reduced activity on STAT1 suggesting that a negativecharge on the tetrazole moiety is implied in selectivity. Thisexperiment has been reproduced in the R1 ¼ 4-NO2 series(Table 1.2) in which, all compounds except tetrazole analog 23showed STAT3 and STAT1 inhibitory functions, while 23 is the onlycompound with low activity on STAT1. We also tested a series ofcompounds with two acidic functions without convincing results(Table S2).
The role of the aminotetrazole group in binding to the STAT3SH2 domain was confirmed by in silico molecular modeling.Atomistic molecular dynamics simulations were performed to
sample the general dynamics of human STAT3 at a time scale of~60 ns. This provides a reliable basis for ensemble docking studiesof two series of inhibitors: R1 ¼ 4-NO2 series (23, 45, 46, 49) andR1 ¼ 4-CH3 series (5, 35, 42, 43). The distributions of the bestpredicted binding scores of these inhibitors to dimerization inter-face of STAT3 SH2 domain obtained for 200 representative proteinstructures from molecular dynamics simulations are shown inFig. 4.
It is clearly seen that the inhibitors containing the tetrazolegroup (5, 23, and 35) show much stronger binding energies thanthe inhibitors, which lack this group. The peaks of energy distri-butions for 5, 23 and 35 are similar and located near�6 kcal/mol. Incontrast, the peaks for other inhibitors are between �5.5and �5 kcal/mol. This result clearly shows that the tetrazole group
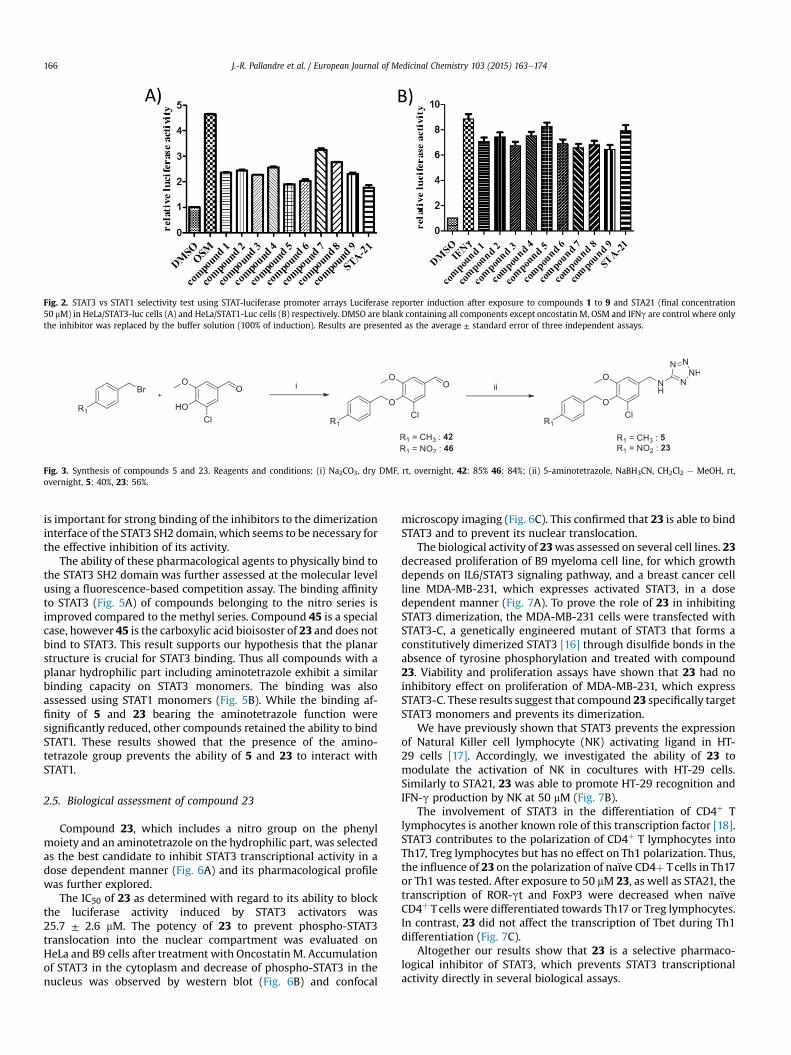
Fig. 2. STAT3 vs STAT1 selectivity test using STAT-luciferase promoter arrays Luciferase reporter induction after exposure to compounds 1 to 9 and STA21 (final concentration50 mM) in HeLa/STAT3-luc cells (A) and HeLa/STAT1-Luc cells (B) respectively. DMSO are blank containing all components except oncostatin M, OSM and IFNg are control where onlythe inhibitor was replaced by the buffer solution (100% of induction). Results are presented as the average ± standard error of three independent assays.
Fig. 3. Synthesis of compounds 5 and 23. Reagents and conditions: (i) Na2CO3, dry DMF, rt, overnight, 42: 85% 46: 84%; (ii) 5-aminotetrazole, NaBH3CN, CH2Cl2 e MeOH, rt,overnight, 5: 40%, 23: 56%.
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174166
is important for strong binding of the inhibitors to the dimerizationinterface of the STAT3 SH2 domain, which seems to be necessary forthe effective inhibition of its activity.
The ability of these pharmacological agents to physically bind tothe STAT3 SH2 domain was further assessed at the molecular levelusing a fluorescence-based competition assay. The binding affinityto STAT3 (Fig. 5A) of compounds belonging to the nitro series isimproved compared to the methyl series. Compound 45 is a specialcase, however 45 is the carboxylic acid bioisoster of 23 and does notbind to STAT3. This result supports our hypothesis that the planarstructure is crucial for STAT3 binding. Thus all compounds with aplanar hydrophilic part including aminotetrazole exhibit a similarbinding capacity on STAT3 monomers. The binding was alsoassessed using STAT1 monomers (Fig. 5B). While the binding af-finity of 5 and 23 bearing the aminotetrazole function weresignificantly reduced, other compounds retained the ability to bindSTAT1. These results showed that the presence of the amino-tetrazole group prevents the ability of 5 and 23 to interact withSTAT1.
2.5. Biological assessment of compound 23
Compound 23, which includes a nitro group on the phenylmoiety and an aminotetrazole on the hydrophilic part, was selectedas the best candidate to inhibit STAT3 transcriptional activity in adose dependent manner (Fig. 6A) and its pharmacological profilewas further explored.
The IC50 of 23 as determined with regard to its ability to blockthe luciferase activity induced by STAT3 activators was25.7 ± 2.6 mM. The potency of 23 to prevent phospho-STAT3translocation into the nuclear compartment was evaluated onHeLa and B9 cells after treatment with Oncostatin M. Accumulationof STAT3 in the cytoplasm and decrease of phospho-STAT3 in thenucleus was observed by western blot (Fig. 6B) and confocal
microscopy imaging (Fig. 6C). This confirmed that 23 is able to bindSTAT3 and to prevent its nuclear translocation.
The biological activity of 23was assessed on several cell lines. 23decreased proliferation of B9 myeloma cell line, for which growthdepends on IL6/STAT3 signaling pathway, and a breast cancer cellline MDA-MB-231, which expresses activated STAT3, in a dosedependent manner (Fig. 7A). To prove the role of 23 in inhibitingSTAT3 dimerization, the MDA-MB-231 cells were transfected withSTAT3-C, a genetically engineered mutant of STAT3 that forms aconstitutively dimerized STAT3 [16] through disulfide bonds in theabsence of tyrosine phosphorylation and treated with compound23. Viability and proliferation assays have shown that 23 had noinhibitory effect on proliferation of MDA-MB-231, which expressSTAT3-C. These results suggest that compound 23 specifically targetSTAT3 monomers and prevents its dimerization.
We have previously shown that STAT3 prevents the expressionof Natural Killer cell lymphocyte (NK) activating ligand in HT-29 cells [17]. Accordingly, we investigated the ability of 23 tomodulate the activation of NK in cocultures with HT-29 cells.Similarly to STA21, 23 was able to promote HT-29 recognition andIFN-g production by NK at 50 mM (Fig. 7B).
The involvement of STAT3 in the differentiation of CD4þ Tlymphocytes is another known role of this transcription factor [18].STAT3 contributes to the polarization of CD4þ T lymphocytes intoTh17, Treg lymphocytes but has no effect on Th1 polarization. Thus,the influence of 23 on the polarization of naïve CD4þ T cells in Th17or Th1 was tested. After exposure to 50 mM 23, as well as STA21, thetranscription of ROR-gt and FoxP3 were decreased when naïveCD4þ T cells were differentiated towards Th17 or Treg lymphocytes.In contrast, 23 did not affect the transcription of Tbet during Th1differentiation (Fig. 7C).
Altogether our results show that 23 is a selective pharmaco-logical inhibitor of STAT3, which prevents STAT3 transcriptionalactivity directly in several biological assays.

Table 1Structure Activity Relationships (SAR) of compound 5 derivativesa.
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174 167

Fig. 4. Probability density distribution of docking binding scores on human STAT3homology model. Binding scores resulted from docking on 200 frames extracted frommolecular dynamics trajectory of human STAT3 homology model for compounds 23(black), 5 (red) and 35 (black) which are aminotetrazole derivatives* and 42, 46(green), 45 (red) and 43, 49 (blue) which are aldehyde, carboxylic acid and cyanamidederivatives respectively. (For interpretation of the references to colour in this figurelegend, the reader is referred to the web version of this article.)
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174168
3. Discussion
A structure-based virtual screening approach followed by me-dicinal chemistry optimization has been used to identify a novelseries of Stat3 inhibitors. First, virtual screening was utilized to
Fig. 5. Binding affinity to STAT3 and STAT1 of compounds 5, 43, 42, 23, 45, 46, 49. Specificwith A) 5-FAMeG(pTyr)LPQTV-CONH2 a STAT3-bound fluorescently labeled peptide and B)
select non-peptide commercially available compounds able tomimic the phosphotyrosine peptide co-crystallized with the Stat3bSH2 domain (Fig. 1).
Their evaluation by a Stat3-dependent luciferase reporter(Fig. 2A) then led to the identification of compound 5 as the mostpromising candidate. An extensive set of derivatives was synthe-sized (Fig. 3 and Table 1) and subjected to an in vitro luciferase genereporter assay. This led to identification of 23, a new pharmaco-logical agent devoid of inhibitory activity on STAT1 but blockingSTAT3 transcriptional activity with an IC50 of 25.7 mM.
Compound 23 Stat3 inhibitory activity is similar to that describedfor STA21 [15], a natural inhibitor of complex chemical structure,whose chemical modification is cumbersome [19]. Niclosamide, aknown anthelmintic [20], is actually one of the best small moleculeinhibitors of STAT3. It inhibits STAT3-mediated luciferase reporteractivity at submicromolar concentrations but no structure activityrelationshipshaveeverbeenestablished for thiscompound.Thesameapplies to HJC0123 [21], which displays inhibitory properties at 5 mM.Stattic [22] displays avery high affinity in afluorescence polarization-basedbindingassaybut seemstobind irreversibly toSTAT3.Celecoxib[23], SI31eV3-32 [24], STX-0119 [25] and isophtalate derivatives [26]seem to be in the same range of activity as 23, while LY5 [27], phe-nylquinoline derivatives [28], benzothiazole analogues [29] and 4-aminophenol derivatives [30] displayed better results than 23. Thedevelopment of STAT3 inhibitor S3I-201 [31] opened the studies ofsalicylic derivatives [32] that bind to the STAT3 SH2 domain. Furtherdevelopment led to BP-1-102 [33] exhibiting 30e40% inhibition at aconcentration of 15 mM in a STAT3 luciferase reporter assay.
Compound 23 shows a moderate activity in comparison to someof the above-mentioned compounds but it benefits from extensivestructure function studies, which identified an important role ofthe aminotetrazole group in modulating relative STAT1 and STAT3inhibitory activities. Tetrazole substitution by aldehyde, cyanamideor carboxylic acid in the parent 5 structure (Table 1.7) hampered theselectivity against STAT1. Similar results were achieved using 23series (Table 1.8). The influence of the tetrazole moiety was alsoconfirmed in numerical simulations involving molecular dynamicand ensemble docking studies (Fig. 4). Finally, inhibition of bindingof fluorescein-labeled phosphopeptides, which bind to the SH2domains of STAT3 (Fig. 5) suggests that selectivity appears to be at amolecular level.
binding on SH2 domain are determined in fluorescence-polarization assay respectively5-FAM-G(pTyr)YIKTE-CONH2 a STAT1-bound fluorescently labeled peptide.

Fig. 6. Effect of 23 at various doses on the STAT3 activity and translocation into nucleus. A) STAT3 and STAT1 luciferase activities at various doses; B) phospho-STAT3 of (1) HeLatotal extract, (2) B9 total extract, (3) HeLa cytoplasm fraction, (4) HeLa nuclear fraction, (5) B9 cytoplasm fraction, (6) B9 nuclear fraction, after exposure to Oncostatin M observed bywestern blot; C) Confocal microscopy on HeLa cells without and after exposure to 23 at 100 mM and revealed by anti-p-Tyr-STAT3 (pTyr705) (3E2) and anti-STAT3 (79D7). DAPI wasused for nuclear staining (Molecular Probes).
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174 169
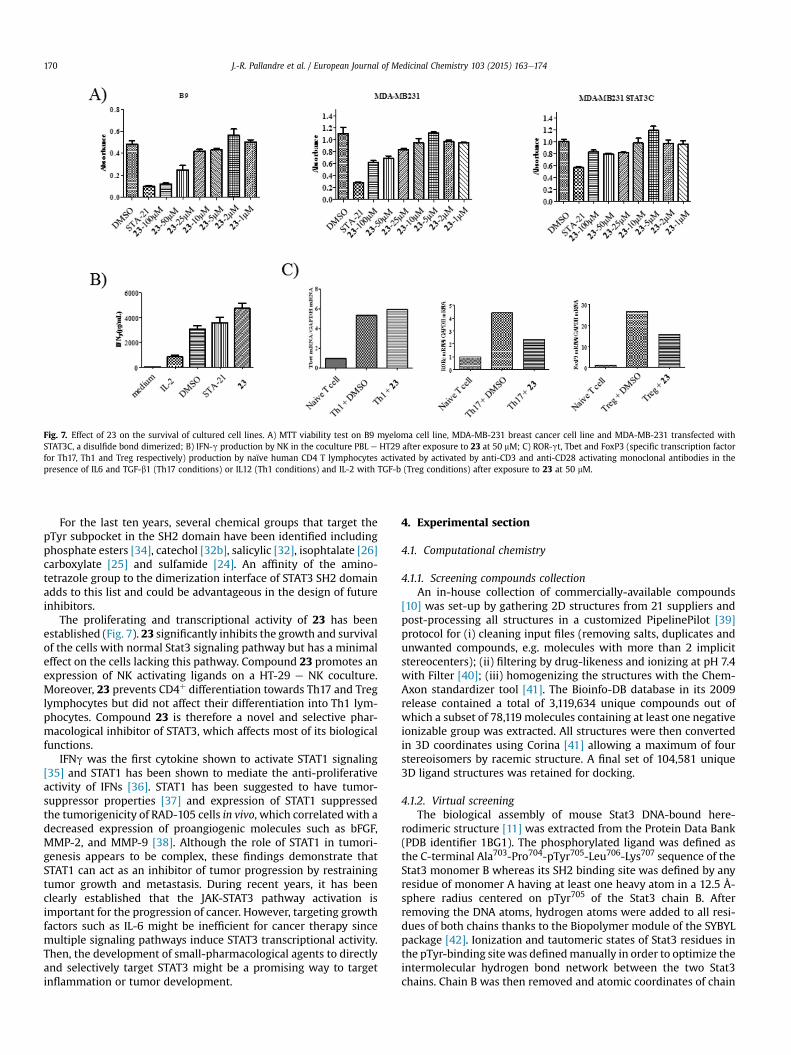
Fig. 7. Effect of 23 on the survival of cultured cell lines. A) MTT viability test on B9 myeloma cell line, MDA-MB-231 breast cancer cell line and MDA-MB-231 transfected withSTAT3C, a disulfide bond dimerized; B) IFN-g production by NK in the coculture PBL e HT29 after exposure to 23 at 50 mM; C) ROR-gt, Tbet and FoxP3 (specific transcription factorfor Th17, Th1 and Treg respectively) production by naïve human CD4 T lymphocytes activated by activated by anti-CD3 and anti-CD28 activating monoclonal antibodies in thepresence of IL6 and TGF-b1 (Th17 conditions) or IL12 (Th1 conditions) and IL-2 with TGF-b (Treg conditions) after exposure to 23 at 50 mM.
J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174170
For the last ten years, several chemical groups that target thepTyr subpocket in the SH2 domain have been identified includingphosphate esters [34], catechol [32b], salicylic [32], isophtalate [26]carboxylate [25] and sulfamide [24]. An affinity of the amino-tetrazole group to the dimerization interface of STAT3 SH2 domainadds to this list and could be advantageous in the design of futureinhibitors.
The proliferating and transcriptional activity of 23 has beenestablished (Fig. 7). 23 significantly inhibits the growth and survivalof the cells with normal Stat3 signaling pathway but has a minimaleffect on the cells lacking this pathway. Compound 23 promotes anexpression of NK activating ligands on a HT-29 e NK coculture.Moreover, 23 prevents CD4þ differentiation towards Th17 and Treglymphocytes but did not affect their differentiation into Th1 lym-phocytes. Compound 23 is therefore a novel and selective phar-macological inhibitor of STAT3, which affects most of its biologicalfunctions.
IFNg was the first cytokine shown to activate STAT1 signaling[35] and STAT1 has been shown to mediate the anti-proliferativeactivity of IFNs [36]. STAT1 has been suggested to have tumor-suppressor properties [37] and expression of STAT1 suppressedthe tumorigenicity of RAD-105 cells in vivo, which correlated with adecreased expression of proangiogenic molecules such as bFGF,MMP-2, and MMP-9 [38]. Although the role of STAT1 in tumori-genesis appears to be complex, these findings demonstrate thatSTAT1 can act as an inhibitor of tumor progression by restrainingtumor growth and metastasis. During recent years, it has beenclearly established that the JAK-STAT3 pathway activation isimportant for the progression of cancer. However, targeting growthfactors such as IL-6 might be inefficient for cancer therapy sincemultiple signaling pathways induce STAT3 transcriptional activity.Then, the development of small-pharmacological agents to directlyand selectively target STAT3 might be a promising way to targetinflammation or tumor development.
4. Experimental section
4.1. Computational chemistry
4.1.1. Screening compounds collectionAn in-house collection of commercially-available compounds
[10] was set-up by gathering 2D structures from 21 suppliers andpost-processing all structures in a customized PipelinePilot [39]protocol for (i) cleaning input files (removing salts, duplicates andunwanted compounds, e.g. molecules with more than 2 implicitstereocenters); (ii) filtering by drug-likeness and ionizing at pH 7.4with Filter [40]; (iii) homogenizing the structures with the Chem-Axon standardizer tool [41]. The Bioinfo-DB database in its 2009release contained a total of 3,119,634 unique compounds out ofwhich a subset of 78,119 molecules containing at least one negativeionizable group was extracted. All structures were then convertedin 3D coordinates using Corina [41] allowing a maximum of fourstereoisomers by racemic structure. A final set of 104,581 unique3D ligand structures was retained for docking.
4.1.2. Virtual screeningThe biological assembly of mouse Stat3 DNA-bound here-
rodimeric structure [11] was extracted from the Protein Data Bank(PDB identifier 1BG1). The phosphorylated ligand was defined asthe C-terminal Ala703-Pro704-pTyr705-Leu706-Lys707 sequence of theStat3 monomer B whereas its SH2 binding site was defined by anyresidue of monomer A having at least one heavy atom in a 12.5 Å-sphere radius centered on pTyr705 of the Stat3 chain B. Afterremoving the DNA atoms, hydrogen atoms were added to all resi-dues of both chains thanks to the Biopolymer module of the SYBYLpackage [42]. Ionization and tautomeric states of Stat3 residues inthe pTyr-binding sitewas definedmanually in order to optimize theintermolecular hydrogen bond network between the two Stat3chains. Chain B was then removed and atomic coordinates of chain

J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174 171
A saved for docking. Docking of the filtered set of the BioinfoDBdatabase was done using standard “speed1 settings” of the PLANTSdocking program [43] and the Chemplp scoring function.
Hit selection was based on a two-step knowledge-based pro-tocol aimed at identifying compounds that share phosphorylatedpeptide key interactions with key residues of Stat3. For this pur-pose, all docking poses were converted into 1D protein-ligandinteraction fingerprints (IFP) using the in-house developed IFPprogram [12]. A first reference IFP (IFP1: 4 bits/residue) was definedbetween pTyr705 (Stat3 B chain) and its anchoring residues(Lys591, Arg609) in chain A, considering polar interactions only(hydrogen bonds and salt bridges). A second IFP (IFP2: 7 bits/res-idue) was then defined between the phosphorylated peptideAla703-Pro704-pTyr705-Leu706-Lys707 of chain B and its full bindingsite (Lys591, Arg595, Arg609, Ser611, Ser613, Ser614, Thr620,Phe621, Trp623, Lys626, Ile634, Gln635, Ser636, Val637, Glu638,Pro639, Tyr640, Tyr657, Ile659, Cys712, Val713, Thr714, Pro715,Phe716) in chain A considering all interactions (hydrophobic, aro-matic, hydrogen bonds and salt bridges) [12]. Tanimoto coefficientswere then computed between the interaction fingerprints of allBioinfoDB docking poses and each of the two reference IFPs. Poseswith a Tanimoto coefficient of 1.0 (IFP1) and higher than 0.5 (IFP2)were selected next, leaving a final set of 52 non-redundant hits.
4.1.3. Molecular dynamics simulations and ensemble dockingThe structure of human STAT3 monomer was constructed by
homology modeling using the full-length sequence and the humanSTAT1 crystal structure 1BF5 as a template. The homology modelingwas performed with the SWISS- MODEL server [44]. The modelobtained was subject to all-atommolecular dynamics simulations inwater in the NPT ensemble at a temperature of 300 K and a pressureof 1 atm. GROMACS 4.6.5 software suit with GROMOS 54A7forcefield was used. The protein was energy minimized and subject to20 ns simulationwith restrained backbone followed by unrestrained60 ns production simulation. The equilibrationwasmonitored by theRMSD of Ca atoms. The last 10 ns of the trajectory were used toextract 200 frames at 5 ps intervals. These frames were used as therepresentative structures in ensemble docking simulations.
The docking procedurewas performed as follows. The structuresof selected inhibitors were optimized using Gaussian09 at theB3LYP/6e31þþG(d) level of theory and their ESP partial chargedwere computed. The ligands and the corresponding protein struc-tures were prepared for docking using the MGLTools-1.5.6. Thedocking was performed with the Autodock Vina [45]. Analysis ofthe docking results was performed with custom software based onthe Python binding of the Pteros molecular modeling library [46].
4.2. STAT inhibitors synthesis
Starting material reagents and analytical grade solvents werepurchased from SigmaeAldrich and Acros. All reactions wereroutinely checked by TLC using Merck Kieselgel 60 F254 aluminumplates and visualized by UV light. IR spectra were performed on aSpectrum65 perkin Elmer with UATR and principal absorptions aregiven in cm-1. 1H and 13C NMR spectra were recorded in thespecified deuterated solvent at 300 MHz and 75 MHz on a BruckerAC 300 instrument. Chemical shifts are expressed in parts permillion (d) relative to the solvent signal and the coupling constants Jare given in Hertz (Hz). ESI e MS analyses were carried out at theService Commun d'Analyse, ICMR e UMR CNRS 6229e51,100Reims.
4.2.1. 3-chloro-5-methoxy-4-(4-methylbenzyloxy)benzaldehyde 42The 4-methylbenzyl chloride (540mg, 3.84 mmol) was added to
a suspension of the 5-chlorovanilline (596 mg, 3.20 mmol), sodium
iodide (1.153 g, 7.69 10�3 mol) and sodium carbonate (815 mg,8.3 mmol) in dry acetone. The reaction mixture was stirred toreflux. Overnight and then it was diluted with water and extractedthree times with ethylacetate. The combined organic layer waswashed with brine, dried over sodium sulfate, filtered and evapo-rated under vacuum. The product was obtained by flash chroma-tography (cyclohexane e dichloromethane: 4e6) as a white solid(788 mg, 85%). IR (ATR): 3051, 3026, 2920, 2838, 2741, 1691, 1589,1572 cm�1. 1H NMR (300 MHz, CDCl3) ppm 9.83 (s, 1H), 7.48 (s, 1H),7.39 (d, J¼ 7.7 Hz, 2H), 7.35 (s, 1H), 7.17 (d, J¼ 7.7 Hz, 2H), 5.13 (s,2H), 3.93 (s, 3H), 2.35 (s, 3H). 13C NMR (75 MHz, CDCl3) ppm 189.9,154.5,149.6,138.1,133.5,132.4,129.3,129.0,128.6,125.8,109.4, 75.0,56.2, 21.2.
4.2.2. N-(3-chloro-5-methoxy-4-(4-methylbenzyloxy)benzyl)-2H-tetrazole-5-amine 5
A solution of 42 (154 mg, 0.53 mmol) and 5-aminotetrazole(90 mg, 0.11 mmol) in a mixture of methanol and dichloro-methane (a suspension was prepared in methanol and dichloro-methane was added until dissolution) was stirred at rt for 2 h. Thensodium cyanoborohydride (40 mg, 0.64 mmol) was added and thereaction mixture was further stirred overnight. The reaction wasquenched with water. The product was extracted with ethylacetate.The combined organic layers were washed with brine and driedover sodium sulfate. The product was obtained by column chro-matography (dichloromethane e methanol: 95-5) as a white solid(76 mg, 40%). IR (ATR): 3253, 3146, 3051, 2923, 2880, 1624, 1578,1537, 1518 cm�1. 1H NMR (300 MHz, DMSO D6) ppm 14.65 (s, 1H),7.53 (t, J¼ 6.2 Hz, 1H), 7.34 (d, J¼ 7.8 Hz, 2H), 7.18 (d, J¼ 7.8 Hz, 2H),7.05 (d, J¼ 1.1 Hz, 1H), 6.98 (d, J¼ 1.1 Hz, 1H), 4.90 (s, 2H), 4.34 (d,J¼ 6.2 Hz, 2H), 3.84 (s, 3H), 2.31 (s, 3H). 1H NMR (300 MHz, DMSOD6) ppm 153.4, 152.1, 142.2, 137.1, 136.1, 133.9, 128.6, 128.2, 126.7,119.7, 110.9, 73.9, 56.0, 46.1, 20.7. HRMS (ESI) calcd forC17H18ClN5O2Na 382.1047 [(M þ Na)þ]; found 382.1053.
4.2.3. 3-chloro-5-methoxy-4-(4-nitrobenzyloxy)benzaldehyde 46The 4-nitrobenzyl bromide (1.19 g, 5.51 mmol) was added to a
suspension of the 5-chlorovanilline (1.03 g, 5.51mmol), and sodiumcarbonate (876 mg, 8.3 mmol) in dry DMF. The reaction mixturewas stirred at rt overnight and then it was diluted with water andextracted three times with ethylacetate. The combined organiclayer was washed with brine, dried over with sodium sulfate,filtered and evaporated under vacuum. The product was obtainedby recrystallization (cyclohexane e dichloromethane: 4e6) as awhite fluffy solid (1.5 g, 84%). IR (ATR): 3107, 3085, 3008, 2950,2868, 1697, 1606, 1593, 1574, 1517 cm�1. 1H NMR (300 MHz, DMSOD6) d ppm 9.91 (s, 1H), 8.27 (d, J¼ 7.2 Hz, 2H), 7.78 (d, J¼ 7.2 Hz, 2H),7.68 (s, 1H), 7.56 (s, 1H), 5.30 (s, 2H), 3.95 (s, 3H). 13C NMR (75MHz,DMSO D6) 191.0, 153.8, 148.0, 147.1, 144.3, 132.7, 128.7, 127.6, 124.0,123.4, 111.0, 73.0, 56.4.
4.2.4. N-(3-chloro-5-methoxy-4-(4-nitrobenzyloxy)benzyl)-2H-tetrazole-5-amine 23
A solution of 46 (1.03 g, 3.20 mmol) and 5-aminotetrazole(545 mg, 6.41 mmol) in a mixture of methanol and dichloro-methane (a suspension was prepared in methanol and dichloro-methane was added until dissolution) was stirred at r.t. for 2 h.Then sodium cyanoborohydride (242 mg, 3.84 mmol) was addedand the reaction mixture was further stirred overnight. The reac-tion was quenched with water. The product was extracted withethylacetate. The combined organic layers were washed with brineand dried over with sodium sulfate. The solvent was removed invacuum and the product was obtained by recrystallization (cyclo-hexane e dichloromethane:4e6) as a white solid (700 mg, 56%). IR(ATR): 3274, 3161, 3076, 2940, 2885, 2850, 2779, 2694, 1643, 1602,

J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174172
1578, 1523 cm�1. 1H NMR (300 MHz, DMSO D6) d ppm 14.69 (s, 1H),8.26 (d, J¼ 8.5 Hz, 2H), 7.76 (d, J¼ 8.5 Hz, 2H), 7.58 (s, 1H), 7.09 (s,1H), 7.01 (s, 1H), 5.11 (s, 2H), 4.35 (d, J¼ 4.3 Hz, 2H), 3.83 (s, 3H). 13CNMR (75 MHz, DMSO D6) d ppm 153.3, 148.9, 147.0, 144.8, 142.0,136.6, 128.6, 126.6, 123.4, 119.7, 111.0, 72.8, 56.1, 46.1. HRMS (ESI)calcd for C16H16ClN6O4 391.0922 [(M þ H)þ]; found 391.0918.
4.3. Biological assays
4.3.1. Cells and reagentsHuman breast cancer (MDA-MB-231) and colorectal adenocar-
cinoma cell line (HT-29) were obtained from ATCC (the AmericanType Culture Collection, Manassas, VA, USA).
HeLa/STAT3-luc cell line, containing a promoter with STAT3element responses controlling the luciferase gene, was obtainedfrom Panomics (Redwood city, CA, USA). HeLa/STAT3-Luc cell linewas cultured with hygromycin selection (500 mg/mL). Expositionsof this cell line to Oncostatin M enabled the induction of luciferaseactivity.
Cells were grown in Dulbeccos's Modified Eagles Medium(Gibco-BRL, France), supplemented with 50 U/ml penicillin, 50 mg/ml streptomycin, 2mM L-glutamine,1mMNa pyruvate (Gibco-BRL,France) and 10% decomplemented fetal calf serum (Seromed,France).
The mouse B cell hybridoma B9, known to proliferate in ainterleukin 6 (IL-6) dependent manner, was maintained in RPMI1640 complemented with 2 mM Glutamine, 50 mM 2-Mercaptoethanol (2 ME), 50 pg/ml recombinant human IL-6(Peprotech, France).
4.3.2. Generation of reporter cell linesThe STAT1 reporter 8x-GAS-Luc plasmid was obtained by Dr C.
K. Glass, (Department of Medicine, University of California, SanDiego, CA, U.S.A.) The plasmid DNA was transfected into cervicalepithelial HeLa cells, and stable puromycin-resistant (1 mg/ml)clones were selected. Transfections were carried out with Lip-ofectAmine 2000 (Invitrogen, Carlsbad, CA), according to themanufacturer's protocol.
STAT1 signaling was induced by exposing HeLa/STAT1-Luc cellsto IFN-g and monitored using luciferase activity.
As previously described in the work of Bedel R. et al. [17]transfection of Stat3C expression vector or mock vector intoMDA-MB-231 cells was carried out using lipofectamine 2000, fol-lowed by selection in medium supplemented with 1 mg/ml puro-mycin (Sigma, France).
4.3.3. STAT promoter assaysTo identify STAT3 inhibitor, biochemical compounds were
assessed for their ability to interfere with STAT3 and/or STAT1signaling using the STAT3-luc or STAT1-luc HeLa cells treated withoncostatin M or IFN-g respectively. For that purpose, cells wereseeded in a 24-well plate and incubated overnight in humidifiedincubator at 37 �C with 5% CO2. Cells were treated with vehicle orinhibitors at 50 mM for 1 h 100 ng/ml of Oncostatin M (Peprotech,France) or 20 ng/mL of IFN-g (Peprotech, France) were added inculture medium for 6 h. Then, medium was removed by aspirationand 150 mL of lysis buffer (Promega, Madison, USA) was added toeach well before a further incubation at room temperature andwith constant shaking. Finally, 50 mL of each lysatewere transferredinto a new white 96-well plate for luciferase assay. To induceluciferase activity, 50 mL of luciferase substrate (Promega) wasadded. The plate was immediately read using a luminometer(Delphia Perkin Elmer, Waltham, MA, USA).
4.3.4. Fluorescence polarization assayFluorescence polarization (FP) assay was conducted based on
fluorescence signal differences between free and STAT3-boundfluorescently labeled peptide as described by Schust and Berg[22]. Briefly, all reactions contained 10 nM of the fluorescent pep-tide 5-FAM eG(pTyr) LPQTV-CONH2 (Genscript, Piscataway, NJ,USA) and 100 nM GST-tagged, full-length human STAT3 protein(SignalChem, Richmond, BC,Canada) in 96-well black plates (PerkinElmer). For STAT1, we used the fluorescent peptide 5-FAM-G(pTyr)YIKTE- CONH2 (Genscript, Piscataway, NJ, USA)and the GST-taggedhuman STAT1b protein (SignalChem, Richmond, BC,Canada).
For evaluating compounds, STAT3 or STAT1 protein was incu-bated with various concentrations of 23 or vehicle at room tem-perature for 60 min in the assay buffer (50 mM NaCl, 10 mMHepes,pH 7.5, 1 mM EDTA, 0.01% Triton-X100, 2 mM dithiothreitol).Fluorescent peptide was added at a final concentration of 10 nMand incubated for 30 min at room temperature following which theFP measurements were examined by Envision 2102 MultilabelReader (Perkin Elmer, Waltham, MA, USA) using FITC FP Dualmodule with excitation filter of FITC FP 480 and emission filter ofFITC FP P-pol535 and S-pol535.
4.3.5. Western blottingCells were lysed in RIPA buffer with antiproteases cocktail
(complete Mini EDTA-free; Roche, France), then sonicated in ice.After BCA protein assay quantification, cells extracts were fractionedby SDS-PAGE. The proteins were then transferred on PVDF mem-branes that were blocked with 5% non-fat milk in Tris Buffer Salinewith 0.1% Tween-20 (TBST) buffer for 90 min at room temperature.Membranes were incubated overnight at 4 �C with primary anti-bodies at dilution 1:1000 human anti-p-Tyr-STAT3 (pTyr705) (3E2)and anti-STAT3 (79D7), anti-p-Tyr-STAT1 (pTyr701) (58D6), anti-STAT1 (9H2), lamin B1, histone H1 (#9138, #4904, #9167, #9176,Cell signaling, Massachusetts, USA). b-actin, 1: 106 was used as aninternal standard for loading. Immunodetection was carried out byusing horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG as a secondary antibody and enhanced chem-iluminescence detection reagents. Chemiluminescencewas detectedusing a camera and Bio-1D software (WilbereLourmat, France).
4.3.6. Confocal microscopyHeLa cells were cultured on poly-L-lysine coated glass slides
(Sigma) and incubated overnight at 37 �C. Vehicle or inhibitorswere incubated during 2 h at 37 �C. Cells were fixed in 3.7% form-aldehyde and permeabilized with 0.1% Triton X- 100 for 15 min andfurther washed 3 times with PBS. After 20 min of blocking in 20%foetal bovine serum (FBS) and washing, cells were stained with theappropriate mAbs or an isotype-matched control antibody inphosphate-buffered saline (PBS) containing 1% bovine serum al-bumin (BSA) for 2 h at room temperature. Afterwards, slides werewashed and incubated with the appropriate secondary antibodiesfor 1 h at room temperature. Cells were then washed with PBS andlabeled with DAPI and covered with cover slides with Dako fluo-rescent mounting medium (Dako, France). Confocal images werecollected with an Olympus FV1000 laser scanning confocal micro-scope (Olympus, Center Valley, PA, USA).
Antibodies used were anti-p-Tyr-STAT3 (pTyr705) (3E2) andanti-STAT3 (79D7). DAPI was used for nuclear staining (MolecularProbes).
4.3.7. In vitro differentiation of T cellsNaïve CD4 T cells (CD4þ CD62Lþ) were obtained from spleens of
C57BL/6 mice by magnetic selection (CD4þ CD62L þ T Cell Isola-tion Kit II, mouse Miltenyi Biotec) according to the manufacturer'srecommendations.

J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174 173
Isolated naïve CD4þ T cell were stimulated with plate-boundanti-CD3 (2 mg/mL; 145-2C11; BioXcell) and anti-CD28 (2 mg/mL;PV-1; BioXcell) and were polarized into effector CD4þ T lympho-cytes subsets without cytokines (Th0 cells) or with IL-12 (10 ng/mL), anti-IL-4 (10 mg/mL) (Th1 cells) or with TGF-b1 (2 ng/mL), IL-6(30 ng/mL) antieIFNeg (10 mg/mL), anti-IL-4 (10 mg/mL) (Th17) orwith TGF-b1 (5 ng/mL), IL-2 (20 ng/mL) antieIFNeg (10 mg/mL)(Treg).
After 2 days of polarization, the cells were collected for real-timequantitative PCR analysis.
4.3.8. Real-time quantitative PCRTotal RNA from T cells was extracted with RNeasy mini-kit
(Qiagen). Reverse-transcription was performed using High Capac-ity RNA-to-cDNA polymerase (Life Technologies) and was analyzedby the Taqman method (Applied Biosystems) with the CFX-96 real-time PCR system, according to the manufacturer's instructions(Biorad). Expression of target RNA was normalized to mouseGAPDH. Primers used were: RORc (Mm00441139_m1), Foxp3(Mm00475156_m1) and T-bet (Mm00450960_m1).
4.3.9. Cell line proliferation assaysMTTassay was performed to determine the effects of compound
23 on cell proliferation. Briefly, MDA-MB-231 and MDA-MB-231STAT3C or B9 cell line were plated in a 96 well tissue cultureplate (10,000 cells per well) and incubated for 24 h. After incuba-tion the cells were treated with vehicle or 23 (50 mM final con-centration) for 72 h. After incubation, freshly prepared MTT (5 mg/ml) in PBS was added to each well and incubated for 3 h and theplate was read at 570 nm by Envision 2102 Multilabel Reader.
4.3.10. Assessment of cytokine productionFor this purpose, human peripheral blood lymphocytes were
cultured during 1 day with IL-2 (1000UI/mL), or with the tumor cellline (HT-29 andMDA-MB-231). After 24 h, cell culture supernatantswere assayed by ELISA for human IFN-g (Diaclone, France) ac-cording to the manufacturer's protocol.
Acknowledgment
We thank Dr C. K. Glass (Department of Medicine, University ofCalifornia, San Diego, CA, U.S.A.) for the STAT1 reporter 8x-GAS-Lucplasmid. The authors are thankful to the conseil r�egional de FrancheComt�e support grant 2012-0167, and to the Supercomputing Centerof the IN2P3-CNRS (Villeurbanne, France) for the allocation ofcomputing time.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2015.08.054.
References
[1] a) J.E. Darnell, Transcription factors as targets for cancer therapy, Nat. Rev.Cancer 2 (2002) 740e749, http://dx.doi.org/10.1038/nrc906;b) J.E. Darnell, STATs and gene regulation, Science 277 (1997) 1630e1635,http://dx.doi.org/10.1126/science.277.5332.1630.
[2] a) A.C. Greenlund, M.A. Farrar, B.L. Viviano, R.D. Schreiber, Ligand-induced IFNgamma receptor tyrosine phosphorylation couples the receptor to its signaltransduction system (p91), EMBO J. 13 (7) (1994) 1591e1600;b) K. Shuai, G.R. Stark, I.M. Kerr, J.E. Darnell Jr., A single phosphotyrosineresidue of Stat91 required for gene activation by interferon-gamma, Science261 (1993) 1744e1746, http://dx.doi.org/10.1126/science.7690989.
[3] a) J.N. Ihle, STATs: signal transducers and activators of transcription, Cell 84(1996) 331e334;b) M.H. Heim, The Jak-Stat pathway: cytokine signaling from the receptor tothe nucleus, J. Recept. Signal. Transduct. Res. 19 (1999) 75e120, http://
dx.doi.org/10.3109/10799899909036638.[4] H. Yu, R. Jove, The STATs of cancer - new molecular targets come of age, Nat.
Rev. Cancer 4 (2004) 97e105, http://dx.doi.org/10.1038/nrc1275.[5] S. Pensa, G. Regis, D. Boselli, F. Novelli, V. Poli, STAT1 and STAT3 in tumori-
genesis: two sides of the same coin?, in: A. Stephanou (Ed.), JAK-STATPathway in Disease, Landes Bioscience, Austin, 2008, pp. 100e121.
[6] A. Amadi-Obi, C.R. Yu, X. Liu, R.M. Mahdi, G.L. Clarke, R.B. Nussenblatt, I. Gery,Y.S. Lee, C.E. Egwuagu, TH17 cells contribute to uveitis and scleritis and areexpanded by IL-2 and inhibited by IL-27/STAT1, Nat. Med. 13 (6) (2007)711e718, http://dx.doi.org/10.1038/nm1585.
[7] L. Costantino, D. Barlocco, STAT3 as a target for cancer drug discovery, Curr.Med. Chem. 15 (2008) 834e843, http://dx.doi.org/10.2174/092986708783955464.
[8] a) A.K. Mankan, F.R. Greten, Inhibiting signal transducer and activator oftranscription 3: rationality and rationale design of inhibitors, Expert Opin.Invest. Drugs 20 (2011) 1263e1275, http://dx.doi.org/10.1517/13543784.2011.601739;b) A. Lavecchia, C. Di Giovanni, E. Novellino, STAT-3 Inhibitors: state of the artand new horizons for cancer treatment, Curr. Med. Chem. 18 (2011)2359e2375, http://dx.doi.org/10.2174/092986711795843218;c) B.D. Page, D.P. Ball, P.T. Gunning, Signal transducer and activator of tran-scription 3 inhibitors: a patent review, Expert Opin. Ther. Pat. 21 (1) (2011)65e83, http://dx.doi.org/10.1517/13543776.2011.539205.
[9] B. Debnath, S. Xu, N. Neamati, Small molecule inhibitors of signal transducerand activator of transcription 3 (Stat3) protein, J. Med. Chem. 55 (15) (2012)6645e6668, http://dx.doi.org/10.1021/jm300207s.
[10] http://bioinfo-pharma.u-strasbg.fr/bioinfo.[11] S. Becker, B. Groner, C.W. Muller, Three-dimensional structure of the Stat3-
beta homodimer bound to DNA, Nature 394 (1998) 145e151, http://dx.doi.org/10.1038/28101.
[12] G. Marcou, D. Rognan, Optimizing fragment and scaffold docking by use ofmolmecular interaction fingeprints, J. Chem. Inf. Model 47 (2007) 195e207,http://dx.doi.org/10.1021/ci600342e.
[13] R. Buettner, R. Corzano, R. Rashid, J. Lin, M. Senthil, M. Hedvat, A. Schroeder,A. Mao, A. Herrmann, J. Yim, H. Li, Y.C. Yuan, K. Yakushijin, F. Yakushijin,N. Vaidehi, R. Moore, G. Gugiu, T.D. Lee, R. Yip, Y. Chen, R. Jove, D. Horne,J.C. Williams, Alkylation of cysteine 468 in Stat3 defines a novel site fortherapeutic development, ACS Chem. Biol. 6 (5) (2011) 432e443, http://dx.doi.org/10.1021/cb100253e.
[14] L. Klampfer, J. Huang, L.A. Swaby, L. Augenlicht, Requirement of histonedeacetylase activity for signaling by STAT1, J. Biol. Chem. 279 (29) (2004)30358e30368, http://dx.doi.org/10.1074/jbc.M401359200.
[15] H. Song, R. Wang, S. Wang, J. Lin, A low-molecular-weight compounddiscovered through virtual database screening inhibits Stat3 function inbreast cancer cells, Proc. Natl. Acad. Sci. U. S. A. 102 (13) (2005) 4700e4705,http://dx.doi.org/10.1073/pnas.0409894102.
[16] G. Niu, K.L. Wright, M. Huang, L. Song, E. Haura, J. Turkson, S. Zhang, T. Wang,D. Sinibaldi, D. Coppola, R. Heller, L.M. Ellis, J. Karras, J. Bromberg, D. Pardoll,R. Jove, H. Yu, Constitutive Stat3 activity up-regulates VEGF expression andtumor angiogenesis, Oncogene 21 (13) (2002) 2000e2008, http://dx.doi.org/10.1038/sj/onc/1205260.
[17] R. Bedel, A. Thiery-Vuillemin, C. Grandclement, J. Balland, J.P. Remy-Martin,B. Kantelip, J.R. Pallandre, X. Pivot, C. Ferrand, P. Tiberghien, C. Borg, Novel rolefor STAT3 in transcriptional regulation of NK immune cell targeting receptorMICA on cancer cells, Cancer Res. 71 (5) (2011) 1615e1626, http://dx.doi.org/10.1158/0008-5472.
[18] C.E. Egwuagu, STAT3 in CD4þ T helper cell differentiation and inflammatorydiseases, Cytokine 47 (3) (2009) 149e156, http://dx.doi.org/10.1016/j.cyto.2009.07.003.
[19] D. Bhasin, K. Cisek, T. Pandharkar, N. Regan, C. Li, B. Pandit, J. Lin, P.-K. Li,Design, synthesis, and studies of small molecule STAT3 inhibitors, Bioorg.Med. Chem. Lett. 18 (2008) 391e395, http://dx.doi.org/10.1016/j.bmcl.2007.10.031.
[20] X. Ren, L. Duan, Q. He, Z. Zhang, Y. Zhou, D. Wu, J. Pan, D. Pei, K. Ding, Iden-tification of niclosamide as a new small-molecule inhibitor of the STAT3signaling pathway, ACS Med. Chem. Lett. 1 (2010) 454e459, http://dx.doi.org/10.1021/ml100146z.
[21] H. Chen, Z. Yang, C. Ding, L. Chu, Y. Zhang, K. Terry, H. Liu, Q. Shen, J. Zhou,Fragment-based drug design and identification of HJC0123, a novel orallybioavailable STAT3 inhibitor for cancer therapy, Eur. J. Med. Chem. 62 (2013)498e507, http://dx.doi.org/10.1016/j.ejmech.2013.01.023.
[22] J. Schust, B. Sperl, A. Hollis, T.U. Mayer, T. Berg, Stattic: a small-molecule in-hibitor of STAT3 activation and dimerization, Chem. Biol. 13 (2006)1235e1242, http://dx.doi.org/10.1016/j.chembiol.2006.09.018.
[23] H. Li, A. Liu, Z. Zhao, Y. Xu, J. Lin, D. Jou, C. Li, Fragment-based drug design anddrug repositioning using multiple ligand simultaneous docking (MLSD):identifying celecoxib and template compounds as novel inhibitors of signaltransducer and activator of transcription 3 (STAT3), J. Med. Chem. 54 (2011)5592e5596, http://dx.doi.org/10.1021/jm101330h.
[24] V.M. Shahani, P. Yue, S. Haftchenary, W. Zhao, J.L. Lukkarila, X. Zhang, D. Ball,C. Nona, P.T. Gunning, J. Turkson, Identification of purine-scaffold small-molecule inhibitors of Stat3 activation by QSAR Studies, ACS Med. Chem. Lett.2 (2011) 79e84, http://dx.doi.org/10.1021/ml100224d.
[25] K. Matsuno, Y. Masuda, Y. Uehara, H. Sato, A. Muroya, O. Takahashi,T. Yokotagawa, T. Furuya, T. Okawara, M. Otsuka, N. Ogo, T. Ashizawa,

J.-R. Pallandre et al. / European Journal of Medicinal Chemistry 103 (2015) 163e174174
C. Oshita, S. Tai, H. Ishii, Y. Akiyama, A. Asai, Identification of a new series ofSTAT3 inhibitors by virtual screening, ACS Med. Chem. Lett. 1 (2010)371e375, http://dx.doi.org/10.1021/ml1000273.
[26] C. Saturnino, C. Palladino, M. Napoli, M.S. Sinicropi, A. Botta, M. Sala,A. Carcereri de Prati, E. Novellino, H. Suzuki, Synthesis and biological evalu-ation of new N-alkylcarbazole derivatives as STAT3 inhibitors: preliminarystudy, Eur. J. Med. Chem. 60 (2013) 112e119, http://dx.doi.org/10.1016/j.ejmech.2012.11.004.
[27] W. Yu, H. Xiao, J. Lin, C. Li, Discovery of novel STAT3 small molecule inhibitorsvia in silico site-directed fragment-based drug design, J. Med. Chem. 56 (2013)4402e4412, http://dx.doi.org/10.1021/jm400080c.
[28] J. Xu, D.C. Cole, C.-P.B. Chang, R. Ayyad, M. Asselin, W. Hao, J. Gibbons,S.A. Jelinsky, K.A. Saraf, K. Park, Inhibition of the signal transducer and acti-vator of transcription-3 (STAT3) signaling pathway by 4-oxo-1-phenyl-1,4-dihydroquinoline-3-carboxylic acid esters, J. Med. Chem. 51 (2008)4115e4121, http://dx.doi.org/10.1021/jm701271y.
[29] M. Zhang, W. Zhu, Y. Li, Discovery of novel inhibitors of signal transducer andactivator of transcription 3 (STAT3) signaling pathway by virtual screening,Eur. J. Med. Chem. 62 (2013) 301e310, http://dx.doi.org/10.1016/j.ejmech.2013.01.009.
[30] M. Zhang, W. Zhu, N. Ding, W. Zhang, Y. Li, Identification and characterizationof small molecule inhibitors of signal transducer and activator of transcription3 (STAT3) signaling pathway by virtual screening, Bioorg. Med. Chem. Lett. 23(2013) 2225e2229, http://dx.doi.org/10.1016/j.bmcl.2013.01.056.
[31] K. Siddiquee, S. Zhang, W.C. Guida, M.A. Blaskovich, B. Greedy, H.R. Lawrence,M.L.R. Yip, R. Jove, M.M. McLaughlin, N.J. Lawrence, S.M. Sebti, J. Turkson,Selective chemical probe inhibitor of Stat3, identified through structure-basedvirtual screening, induces antitumor activity, Proc. Natl. Acad. Sci. U. S. A. 104(18) (2007) 7391e7396, http://dx.doi.org/10.1073/pnas.0609757104.
[32] a) S. Fletcher, B.D.G. Page, X. Zhang, P. Yue, Z.H. Li, S. Sharmeen, J. Singh,W. Zhao, A.D. Schimmer, S. Trudel, J. Turkson, P.T. Gunning, Antagonism of theStat3eStat3 protein dimer with salicylic acid based small molecules, Chem-MedChem 6 (2011) 1459e1470, http://dx.doi.org/10.1002/cmdc.201100194;b) B.D.G. Page, S. Fletcher, P. Yue, Z. Li, X. Zhang, S. Sharmeen, A. Datti,J.L. Wrana, S. Trudel, A.D. Schimmer, J. Turkson, P.T. Gunning, Identification ofa non-phosphorylated, cell permeable, small molecule ligand for the Stat3SH2 domain, Bioorg. Med. Chem. Lett. 21 (2011) 5605e5609, http://dx.doi.org/10.1016/j.bmcl.2011.06.056;c) X. Zhang, P. Yue, S. Fletcher, W. Zhao, P.T. Gunning, J. Turkson, A novelsmall-molecule disrupts Stat3 SH2 domainphosphotyrosine interactions andStat3-dependent tumor processes, Biochem. Pharmacol. 79 (2010)1398e1409, http://dx.doi.org/10.1016/j.bcp.2010.01.001;d) S. Fletcher, J. Singh, X. Zhang, P. Yue, B.D.G. Page, S. Sharmeen,V.M. Shahani, W. Zhao, A.D. Schimmer, J. Turkson, P.T. Gunning, Disruption oftranscriptionally active stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities, Chem-BioChem 10 (2009) 1959e1964, http://dx.doi.org/10.1002/cbic.200900172;e) W. Hao, Y. Hu, C. Niu, X. Huang, C.-P.B. Chang, J. Gibbons, J. Xu, Discovery ofthe catechol structural moiety as a Stat3 SH2 domain inhibitor by virtualscreening, Bioorg. Med. Chem. Lett. 18 (2008) 4988e4992, http://dx.doi.org/10.1016/j.bmcl.2008.08.032.
[33] a) X. Zhang, P. Yue, B.D.G. Page, T. Li, W. Zhao, A.T. Namanja, D. Paladino,J. Zhao, Y. Chen, P.T. Gunning, J. Turkson, Orally bioavailable small-moleculeinhibitor of transcription factor Stat3 regresses human breast and lung can-cer xenografts, Proc. Natl. Acad. Sci. U. S. A. 109 (24) (2012) 9623e9628,http://dx.doi.org/10.1073/pnas.1121606109;b) B.D.G. Page, D.C. Croucher, Z.H. Li, S. Haftchenary, V.H. Jimenez-Zepeda,J. Atkinson, P.A. Spagnuolo, Y.L. Wong, R. Colaguori, A.M. Lewis,A.D. Schimmer, S. Trudel, P.T. Gunning, Inhibiting aberrant signal transducerand activator of transcription protein activation with tetrapodal, smallmolecule Src homology 2 domain binders: promising agents against multiplemyeloma, J. Med. Chem. 56 (2013) 7190e7200, http://dx.doi.org/10.1021/jm3017255.
[34] a) P.T. Gunning, M.P. Glenn, K.A. Siddiquee, W.P. Katt, E. Masson, S.M. Sebti,
J. Turkson, A.D. Hamilton, Targeting protein-protein interactions: suppressionof Stat3 dimerization with rationally designed small-molecule, nonpeptidicSH2 domain binders, ChemBioChem 9 (2008) 2800e2803, http://dx.doi.org/10.1002/cbic.200800291;b) K.A.Z. Siddiquee, P.T. Gunning, M. Glen, W.P. Katt, S. Zhang, C. Schroeck,S.M. Sebti, R. Jove, A.D. Hamilton, J. Turkson, An oxazole-based small-moleculeStat3 inhibitor modulates Stat3 stability and processing and induces anti-tumor cell effects, ACS Chem. Biol. 2 (2007) 787e798, http://dx.doi.org/10.1021/cb7001973;c) P.K. Mandai, W.S. Liaeo, J.S. McMurray, Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3, Org.Lett. 11 (2009) 3394e3397, http://dx.doi.org/10.1021/ol9012662;d) D.R.I.V. Coleman, Z. Ren, P.K. Mandal, A.G. Cameron, G.A. Dyer, S. Muranjan,M. Campbell, X. Chen, J.S. McMurray, Investigation of the binding de-terminants of phosphopeptides targeted to the Src homology 2 domain of thesignal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor, J. Med. Chem. 48 (2005) 6661e6670, http://dx.doi.org/10.1021/jm050513m;e) V.M. Shahani, P. Yue, S. Fletcher, S. Sharmeen, M.A. Sukhai, D.P. Luu,X. Zhang, H. Sun, W. Zhao, A.D. Schimmer, J. Turkson, P.T. Gunning, Design,synthesis, and in vitro characterization of novel hybrid peptidomimetic in-hibitors of STAT3 protein, Bioorg. Med. Chem. 19 (2011) 1823e1838, http://dx.doi.org/10.1016/j.bmc.2010.12.010;f) P.T. Gunning, W.P. Katt, M. Glenn, K. Siddique, J.S. Kim, R. Jove, S.M. Sebti,J. Turkson, A.D. Hamilton, Isoform selective inhibition of STAT1 or STAT3homo-dimerization via peptidomimetic probes: structural recognition of STATSH2 domains, Bioorg. Med. Chem. Lett. 17 (2007) 1875e1878, http://dx.doi.org/10.1016/j.bmcl.2007.01.077.
[35] a) K. Shuai, C. Schindler, V.R. Prezioso, J.E. Darnell Jr., Activation of tran-scription by IFN-gamma:tyrosine phosphorylation of a91-kD DNA bindingprotein, Science 258 (1992) 1808e1812, http://dx.doi.org/10.1126/sci-ence.1281555;b) C. Schindler, K. Shuai, V.R. Prezioso, J.E. Darnell Jr., Interferon-depend-enttyrosine phosphorylationofalatentcytoplasmictranscriptionfactor, Science257 (1992) 809e813, http://dx.doi.org/10.1126/science.1496401.
[36] J.F. Bromberg, C.M. Horvath, Z. Wen, R.D. Schreiber, J.E. Darnell Jr., Tran-scriptionally active Stat1 is required for the antiproliferative effects of bothinter- feron alpha and interferon gamma, Proc. Natl. Acad. Sci. U. S. A. 93(1996) 7673e7678.
[37] G.P. Dunn, C.M. Koebel, R.D. Schreiber, Interferons, immunity and cancerimmunoediting, Nat. Rev. Immunol. 6 (2006) 836e848, http://dx.doi.org/10.1038/nri1961.
[38] S. Huang, C.D. Bucana, M. Van Arsdall, I.J. Fidler, STAT1 negatively regulatesangiogenesis, tumorigenicity and metastasis of tumor cells, Oncogene 21(2002) 2504e2512, http://dx.doi.org/10.1038/sj/onc/1205341.
[39] Pipeline Pilot, version 7.5, Accelrys, San Diego, CA 92121, USA.[40] Filter, version 2., OpenEye Scientific Software, Santa Fe, NM 87508, USA.[41] Chemaxon Kft, 1031 Budapest, Hungary.[42] SYBYL, version 8.0, Tripos, St.Louis, MO 63101, USA.[43] O. Korb, T. Stützle, T.E. Exner, Empirical scoring functions for advanced
protein-ligand docking with PLANTS, J. Chem. Inf. Model. 49 (2009) 84e96,http://dx.doi.org/10.1021/ci800298z.
[44] M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer,T.G. Cassarino, M. Bertoni, L. Bordoli, T. Schwede, SWISS-MODEL: modellingprotein tertiary and quaternary structure using evolutionary information,Nucleic Acids Res. 42 (2014) 252e258, http://dx.doi.org/10.1093/nar/gku340(Online).
[45] O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy ofdocking with a new scoring function, efficient optimization and multi-threading, J. Comput. Chem. 31 (2010) 455e461, http://dx.doi.org/10.1002/jcc.21334.
[46] S.O. Yesylevskyy, Pteros: fast and easy to use open-source Cþþ library formolecular analysis, J. Comput. Chem. 33 (2012) 1632e1636, http://dx.doi.org/10.1002/jcc.22989.
Related Documents











