2009;69:537-546. Cancer Res Brad N. Taylor, Rajeshwari R. Mehta, Tohru Yamada, et al. Enter Cancer Cells Noncationic Peptides Obtained From Azurin Preferentially Updated version http://cancerres.aacrjournals.org/content/69/2/537 Access the most recent version of this article at: Cited Articles http://cancerres.aacrjournals.org/content/69/2/537.full.html#ref-list-1 This article cites by 45 articles, 16 of which you can access for free at: Citing articles http://cancerres.aacrjournals.org/content/69/2/537.full.html#related-urls This article has been cited by 4 HighWire-hosted articles. Access the articles at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] Department at To order reprints of this article or to subscribe to the journal, contact the AACR Publications Permissions . [email protected] Department at To request permission to re-use all or part of this article, contact the AACR Publications Research. on November 21, 2013. © 2009 American Association for Cancer cancerres.aacrjournals.org Downloaded from Research. on November 21, 2013. © 2009 American Association for Cancer cancerres.aacrjournals.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

2009;69:537-546. Cancer Res Brad N. Taylor, Rajeshwari R. Mehta, Tohru Yamada, et al. Enter Cancer CellsNoncationic Peptides Obtained From Azurin Preferentially
Updated version
http://cancerres.aacrjournals.org/content/69/2/537
Access the most recent version of this article at:
Cited Articles
http://cancerres.aacrjournals.org/content/69/2/537.full.html#ref-list-1
This article cites by 45 articles, 16 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/69/2/537.full.html#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

Noncationic Peptides Obtained From Azurin Preferentially Enter
Cancer Cells
Brad N. Taylor,1Rajeshwari R. Mehta,
1Tohru Yamada,
1Fatima Lekmine,
1Konstantin Christov,
1
Ananda M. Chakrabarty,2Albert Green,
1Laura Bratescu,
1Anne Shilkaitis,
1
Craig W. Beattie,1and Tapas K. Das Gupta
1
Departments of 1Surgical Oncology and 2Microbiology and Immunology, University of Illinois College of Medicine, Chicago, Illinois
Abstract
Azurin, a member of the cupredoxin family of coppercontaining redox proteins, preferentially penetrates humancancer cells and exerts cytostatic and cytotoxic (apoptotic)effects with no apparent activity on normal cells. Amino acids50 to 77 (p28) of azurin seem responsible for cellularpenetration and at least part of the antiproliferative,proapoptotic activity of azurin against a number of solidtumor cell lines. We show by confocal microscopy andfluorescence-activated cell sorting that amino acids 50 to 67(p18) are a minimal motif (protein transduction domain)responsible for the preferential entry of azurin into humancancer cells. A combination of inhibitors that interfere withdiscrete steps of the endocytotic process and antibodies forcaveolae and Golgi-mediated transport revealed that theseamphipathic, A-helical peptides are unique. Unlike thecationic cell-penetrating peptides, A-helical antennapedia-like, or VP22 type peptides, p18 and p28 are not bound by cellmembrane glycosaminoglycans and preferentially penetratecancer cells via endocytotic, caveosome-directed, and caveo-some-independent pathways. Once internalized, p28, but notp18, inhibits cancer cell proliferation initially through acytostatic mechanism. These observations suggest the azurinfragments, p18 and p28, account for the preferential entry ofazurin into human cancer cells and a significant amount ofthe antiproliferative activity of azurin on human cancer cells,respectively. [Cancer Res 2009;69(2):537–46]
Introduction
Cell-penetrating peptides (CPP) are short amphipathic andcationic peptides and peptide derivatives, usually containingmultiple lysine and arginine residues (1). They form a class ofsmall molecules receiving significant attention as potentialtransport agents for a variety of cargoes including cytotoxic drugs(2, 3), antisense oligonucleotides (4), in gene therapy (5, 6), and asdecoy peptides (7). Small arginine-rich and other cationic peptidesgenerated from phage-displayed peptide libraries, initially charac-terized by the RGD (Arg-Gly-Asp) sequence that recognizes theintegrin family of cell surface receptors important to the invasionof tumor cells (8), are also potential carriers for imaging (9) andtherapeutic agents. These peptides enter cells in an energy-
dependent manner (9) and can distinguish between normal andmalignant vasculature (10, 11) and lymphatic tissue in murinesystems (9). The latter also induce apoptosis in tumor types, whichalso bind the peptide (9). However, even these recent additions tothe CPP armamentarium seem limited to binding to tumor-associated endothelial or lymphatic cells rather than directly andpreferentially penetrating a wide variety of malignant cells. Variousmechanisms for entry have been proposed; it now seems that themajority of CPPs enter cells via adsorptive-mediated endocytosisrather than direct penetration of the plasma membrane (1).Whatever the mechanisms of cell entry and intracellular disposi-tion, the overriding question regarding the potential pharmacologicapplication of CPPs is whether or not their intracellularconcentration in a target cell, in addition to that of any attendantcargo, is sufficient to elicit a pharmacologic response at levels thatare not toxic to nontarget cells (1). We have recently suggestedamino acids (aa) 50 to 77 of azurin, a cupredoxin secreted byPseudomonas aeruginosa , as a putative protein transductiondomain (PTD) responsible for the penetration of azurin intocancer cells (12), although we did not identify a route of cellularentry. The present study refines the PTD of azurin from aa 50 to 77(p28) to aa 50 to 67 (p18) and provides evidence for an endocytoticand nonendocytotic-mediated entry into normal and cancer cellsthat is not dependent on membrane bound glycosaminoglycans.Moreover, the COOH-terminal 12 aa of p28 accounts for asignificant amount of the antiproliferative activity of azurin onhuman cancer cells (13, 14).
Experimental Procedures
Cell culture and cell lines. Human cancer and noncancer(immortalized and nonimmortalized) cell lines were obtained fromAmerican Type Culture Collection [lung cancer (A549 and NCI-H23adenocarcinoma), normal lung (CCD-13Lu), prostate cancers(DU145 and LN-CAP), normal prostate (CRL11611), breast cancer(MCF-7), normal breast (MCF-10A), colon cancer (HCT116),normal colon (CCD33Co), fibrosarcoma (HT1080), and ovariancancer (SK-OV3 adenocarcinoma)]. Normal fibroblasts isolatedfrom skin were established in our laboratory. Normal ovarian cells(HOSE6-3) were a generous gift from Dr. S.W. Tsao (University ofHong Kong, Hong Kong, China). Melanoma lines (UISO-Mel-2, 23,29) were established and characterized in our laboratory (15). Allcells except UISO-Mel-2 (MEM-H) were cultured in MEM-E(Invitrogen) supplemented with 10% heat-inactivated fetal bovineserum (Atlanta Biological, Inc.), 100 units/mL penicillin, and 100Ag/mL streptomycin at 37jC in 5% CO2 or air.Proliferation assays/cell growth. Melanoma cells were seeded
(4 replicates) in flat-bottomed 24-well plates (Becton Dickinson) ata density of 12 � 103 cells per well. After 24 h, medium waschanged and fresh p18, p28, azurin, or a similar volume of medium
Note: Current address for B.N. Taylor: Caliper Life Sciences, 68 Elm Street,Hopkinton, MA 01748.
Requests for reprints: Tapas K. Das Gupta, 840 South Wood Street, Chicago, IL60612. Phone: 312-996-6134; Fax: 312-996-9365; E-mail: [email protected].
I2009 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-08-2932
www.aacrjournals.org 537 Cancer Res 2009; 69: (2). January 15, 2009
Research Article
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

without peptide (8 replicates) added daily for 72 h. Cells were thencounted in a Beckman Coulter (Z1 coulter particle counter). Valuesrepresent the mean F SD of four replicates.3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bro-
mide assay. Melanoma cells were seeded at a density of 2,000cells per well and allowed to attach for 24 h. Freshly preparedpeptide (10 AL) or culture medium was then added to each well.After 24 h, medium was changed and p18, p28, or azurin wereadded daily. After 72 h of incubation, 10 AL of 3-(4,5-dimethylth-iazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent(Trevigen) was added to each well, the samples incubated for3 h at room temperature, 100 AL of detergent added to each well,and incubated for an additional 3 h at 37jC. Absorbance wasmeasured with a SpectraMax 340 plate reader (Molecular DevicesCorporation) and percent change in the absorbance at 570 nm intreated cells relative to untreated controls determined. Valuesrepresent the meanF SD. Significance between control and treatedgroups was determined by Student’s t test.Peptide synthesis. All azurin-derived peptides including p18,
Leu50-Gly67 LSTAADMQGVVTDGMASG, p28 Leu50-Asp77
LSTAADMQGVVTDGMASGLDKDYLKPDD, p18b Val60-Asp77
VTDGMASGLDKDYLKPDD, p12 Gly66-Asp77 SGLDKDYLKPDD,mitogen-activated protein, Mastoparan-7, and poly arginine(Arg8) were synthesized by CS Bio, Inc., as >95% purity and massbalance.Predictive modeling for azurin peptides. We used GENETYX
software (ver. 6.1) to generate Robson structure models for azurinderived peptides (16). The MAPAS software was used to identifystrong membrane contacts and define regions of the proteinsurface that form such contacts (17). If a protein, i.e., azurin, has amembranephilic residue score (MRS) of >3, membranephilic areascore (MAS) of >60%, and coefficient of membranephilic asymme-try (Kmpha) of >2.5, there is a high probability that the protein has atrue membrane-contacting region.Peptide/protein labeling. Peptides were dissolved in 1 mL PBS
mixed with Alexa Fluor 568 dye (Molecular Probes) at a 1:2 protein/dye ratio, 100 AL sodium bicarbonate were added, and the mixturewere incubated overnight at 4jC with continuous stirring. Labeledpeptide was dialyzed against cold-PBS using Slide-A-Lyzer DialysisCassettes 1000 MWCO for p12 and 2000 MWCO for others (PierceBiotechnology).Cell penetration/confocal analysis. Cells were seeded over-
night on glass coverslips at 37jC under 5% CO2, rinsed with freshmedium, and incubated at 37jC for 2 h in prewarmed mediumcontaining Alexa Fluor 568–labeled azurin peptides (20 Amol/L),Arg8 (5 Amol/L), or medium alone. After incubation, coverslipswere rinsed 3� with PBS, fixed in 2.5% formalin for 5 min, andwashed 2� in PBS, once in d.i.H2O, and mounted in mediumcontaining 1.5 Ag/mL 4¶,6-diamidino-2-phenylindole (DAPI) tocounter stain nuclei (VECTASHIELD; Vector Laboratories).Cellular uptake and distribution were photographed under aninverted confocal laser scanning microscope (Model LC510; CarlZeiss Inc.).Peptide colocalization with lysosomes or mitochondria was
determined by incubating cells growing on a glass coverslip for2 h at 37jC with Alexa Fluor 568–labeled azurin or peptides.Mitrotracker (MitroTracker Green FM) or lysotracker (LysoTrackerGreen DND-26; Invitrogen Corporation) was added ( final concen-tration, 1 Amol/L) for the last 30 min of incubation. Cells wererinsed 3� with PBS, fixed in 2.5% formalin for 5 min, washed2� with PBS, and incubated in 0.1% Triton-X100 in PBS for 15 min.
Cells were then incubated with 1 Ag/mL rabbit anti-human golgin97 or anti-human caveolin 1 (Abcam) in PBS with 1% bovine serumalbumin. After 1 h of incubation at 4jC, coverslips were washedonce with PBS, incubated 10 min in PBS containing Alexa Fluor468–conjugated goat anti-rabbit antibody, washed 2� in PBS, andonce in d.i.H2O. Coverslips were mounted in medium containing1.5 Ag/mL DAPI. Colocalization (yellow) of Alexa Fluor 568 (red)and Alexa Fluor 468 (green) was analyzed and photographed.UISO-Mel-2 cells on coverslips were preincubated in MEM-H
containing 100 Ag/mL heparin sulfate (HS; Sigma-Aldrich) for30 min and p18, p28, or Arg8 were added to bring the finalconcentration to 20 Amol/L. After 1 h, coverslips were washed,fixed, and analyzed as described.Cell penetration by fluorescence-activated cell sorting. Cells
(1.0 � 106/500 AL PBS) were incubated for 2 h at 37jC with AlexaFluor–568 labeled p18 or p28 (20 Amol/L), Arg8 (5 Amol/L), ormedium alone, washed 3� in PBS, fixed in 2.5% formalin for 5 min,washed twice in PBS, resuspended in 200 AL PBS, passed through ascreen to obtain a single-cell suspension, and analyzed with aMoFlo Cell Sorter (Dako) Eex 568 nm and Eem 603 nm. The foldincrease of the mean fluorescence intensity (MFI) over backgroundlevels represents mean fluorescence of three separate experiments.Entry inhibitors. UISO-Mel-2 cells (3 � 105 per 300 AL)
maintained in phenol red–free, serum-free MEM-H at 37jC, werepretreated with the following inhibitors (Sigma-Aldrich): chlor-promazine (CPZ), amiloride, nystatin, filipin, methyl-h-cyclodextrin(MhCD), brefeldin A (BFA), Taxol, sodium azide, cytochalasin D,staurosporine, ouabain, wortmannin, monensin, nocodazole,benzyl-2-acetamido-2-deoxy-a-D-galactopyranoside, (BnGalNac),tunicamycin, and neuraminidase. Final concentrations werederived from the dose response curves of individual inhibitors.Alexa Fluor 568–labeled p18 or p28 (20 Amol/L) was then added,incubated for 1 h, and the cells washed, fixed, and prepared for flowcytometric analysis.Cell membrane toxicity assays/lactate dehydrogenase leak-
age assay. A lactate dehydrogenase (LDH) leakage assay wasperformed according to the manufacturer’s instructions (CytoTox-One; Promega) with 100 AL of UISO-Mel-2 cells (5 � 103).Cells without peptides/proteins were used as a negative control.Experiments were carried out in triplicate (data representmean F SE).Hemolysis assay. Human whole blood samples (2–3 mL) were
centrifuged for 10 min at 1,000 � g , the pellets washed once withPBS, once with HKR buffer (pH7.4; ref. 18), resuspended in HKRbuffer to 4% erythrocytes, and 50 AL transferred to a 1.5-mL tubewith 950 AL of peptides, azurin (5, 50, and 100 Amol/L) or 0.1%Triton X-100 in HRK buffer to disrupt the RBC membrane. MAPand Mastoparan7 (Bachem California, Inc.) were used as positivecontrols. After 30 min at 37jC with rotation, tubes were centrifugedfor 2 min at 1,000 � g , 300 AL of supernatants transferred to a96-well plate, and absorbance recorded at 540 nm.Kinetics of entry. UISO-Mel-2 cells (5 � 105 cells) were
suspended in MEM-H without phenol red. Reactions were startedby adding either Alexa Fluor 568-conjugated p18 at 0, 10, 20, 50,100, 150, and 200 Amol/L for 5, 10, 15, and 20 s, or Alexa Fluor568–conjugated p28 at 1, 10, 25, 50, 100, 150, and 200 Amol/L for30, 60, 90, and 120 s on ice. After incubation, 1 mL of cold-PBS wasadded to the 250 AL reaction in mixture. Cells were centrifugedtwice at 600 � g for 2 min at 4jC. At least 10,000 fixed cells wereanalyzed by flow cytometry in each reaction, and their backgroundand relative fluorescence were calculated.
Cancer Research
Cancer Res 2009; 69: (2). January 15, 2009 538 www.aacrjournals.org
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
I125 labeling of azurin and competition assays. Peptidebinding and entry was determined using a whole cell assay withUISO-Mel-2 cells in HEPES solution (50,000 cells/mL), wereincubated for 30 min at 37jC with increasing concentrations(0–175 nmol/L) of radiolabeled azurin in the presence/absence of
1,000-fold excess of unlabeled p18, p28, or azurin, then washedthrice with ice cold PBS, and radioactivity remaining in the cellpellet counted. Radioactivity in cells incubated with 125I azurinalone was considered total binding; radioactivity in the presence ofunlabeled azurin, p18, or p28 was considered nonspecific binding.
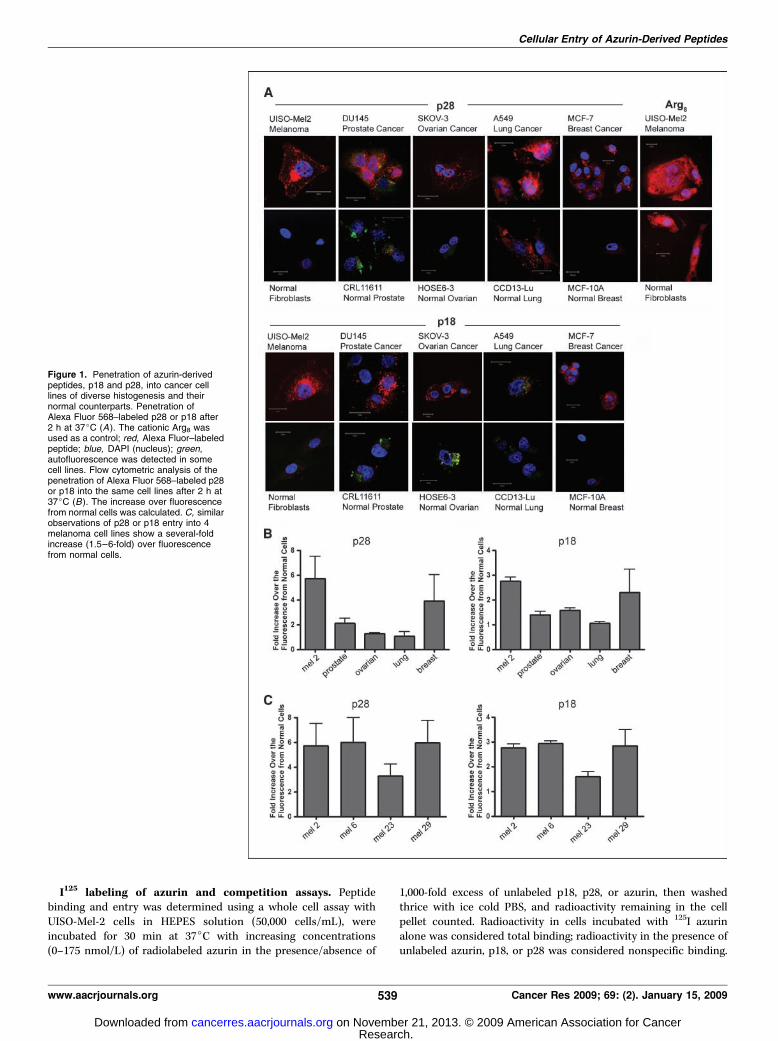
Figure 1. Penetration of azurin-derivedpeptides, p18 and p28, into cancer celllines of diverse histogenesis and theirnormal counterparts. Penetration ofAlexa Fluor 568–labeled p28 or p18 after2 h at 37jC (A). The cationic Arg8 wasused as a control; red, Alexa Fluor–labeledpeptide; blue, DAPI (nucleus); green,autofluorescence was detected in somecell lines. Flow cytometric analysis of thepenetration of Alexa Fluor 568–labeled p28or p18 into the same cell lines after 2 h at37jC (B). The increase over fluorescencefrom normal cells was calculated. C, similarobservations of p28 or p18 entry into 4melanoma cell lines show a several-foldincrease (1.5–6-fold) over fluorescencefrom normal cells.
Cellular Entry of Azurin-Derived Peptides
www.aacrjournals.org 539 Cancer Res 2009; 69: (2). January 15, 2009
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

Specific binding was determined by subtracting nonspecificbinding from total binding and Scatchard plots generated.
Results
The NH2-terminal domain of p28 is responsible forpreferential entry into cancer cells. We initially defined aa50 to 77 of azurin as a putative PTD for cell penetration (12), whichfits well with structural evidence for an a-helical regionencompassing residues 54 to 67 of azurin stabilizing the azurinmolecule (19). Confocal analyses initially suggested that p28 andp18 of p28/azurin (Fig. 1A) penetrated human cancer cells withsimilar efficiency but did not penetrate histologically matchednormal cell lines to the same degree (Fig. 1A ; ref. 12). A singularexception was CCD13-Lu, a cell line derived from lung fibroblasts.The cationic Arg8 was rapidly and efficiently taken up intofibroblasts (Fig. 1A) and all other normal cell lines tested (data notshown). These observations were confirmed by a more sensitivefluorescence-activated cell sorting (FACS) analyses (Fig. 1B) wherep28 fluorescence was f0.5 to 6 and p18 f0.5- to 3-fold higherthan the corresponding normal cell line, with the exception of lungcancer. A similar pattern in intracellular fluorescence intensity wasobserved within a melanomas, where the relative intensity of p18was f50% of that observed with p28 (Fig. 1C). Fluorescenceintensity over background was also consistently lower in cell pairsexposed to p18 than p28 (data not shown), again suggesting lessp18 entered individual cells. In all cases, the degree of entry of p18
and p28 into either cancer or normal cells was less than thatobserved with Arg8, where no preference for entry was observed(Fig. 1A). The predicted Robson structure (data not shown) of p18suggests that the COOH-terminal aas form a partial h-sheet. Thisand the shorter length of p18, which lacks the hydrophilic COOH-terminal 10 aas (aa 68–77) of p28, suggests that p18, as a putativePTD for azurin, may have a more rapid entry into cancer andnormal cells via a nonendocytotic over an endocytotic ormembrane receptor mediated process. MAPAS data (MRS, 3.74;MAS, 87.1; Kmpha, 2.37) predict that aas 69, 70, 75, 76, and 85 ofazurin provide the best opportunity for membrane contact,suggesting the COOH-terminal region of p28, not present on p18(aa, 50–67), is most likely to contact specific residues on the cellmembrane, irrespective of the status of a cell.The preferential penetration of p18 and p28 was confirmed by
exposing the same cell lines to azurin 60 to 77 (p18b), or aa 66 to 77(p12), the COOH-terminal 12 aa of p28 (Fig. 2A and B). Here, thepreferential penetration observed with p18 and p28 was completelyabolished. p18b (theoretical pI 4.13) has a short a-helix and partialh-sheet (16), and is extremely hydrophilic, which together maynegate preferential entry. p12 (theoretical pI 4.33) lacks asecondary a-helical structure but is also hydrophilic, suggestingoverall hydrophilcity may be a major contributor to the decrease inselectivity of cell penetration.Cell penetration is not a result of membrane disruption. Cell
penetration by azurin, p28, and p18 does not result frommembrane disruption. An LDH assay (Fig. 3A) suggested that
Figure 2. Entry of azu 60 to 77 (p18b)and azu 66 to 77 (p12) into cancer andnormal cells. Cells were incubated withAlexa Fluor 568–labeled p18b (A ) or p12(B) at 37jC for 2 h and images recordedby confocal microscopy. Red, AlexaFluor–labeled peptide; blue, API (nucleus).Green or yellow, Autofluorescence innormal cell lines (green+red).
Cancer Research
Cancer Res 2009; 69: (2). January 15, 2009 540 www.aacrjournals.org
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
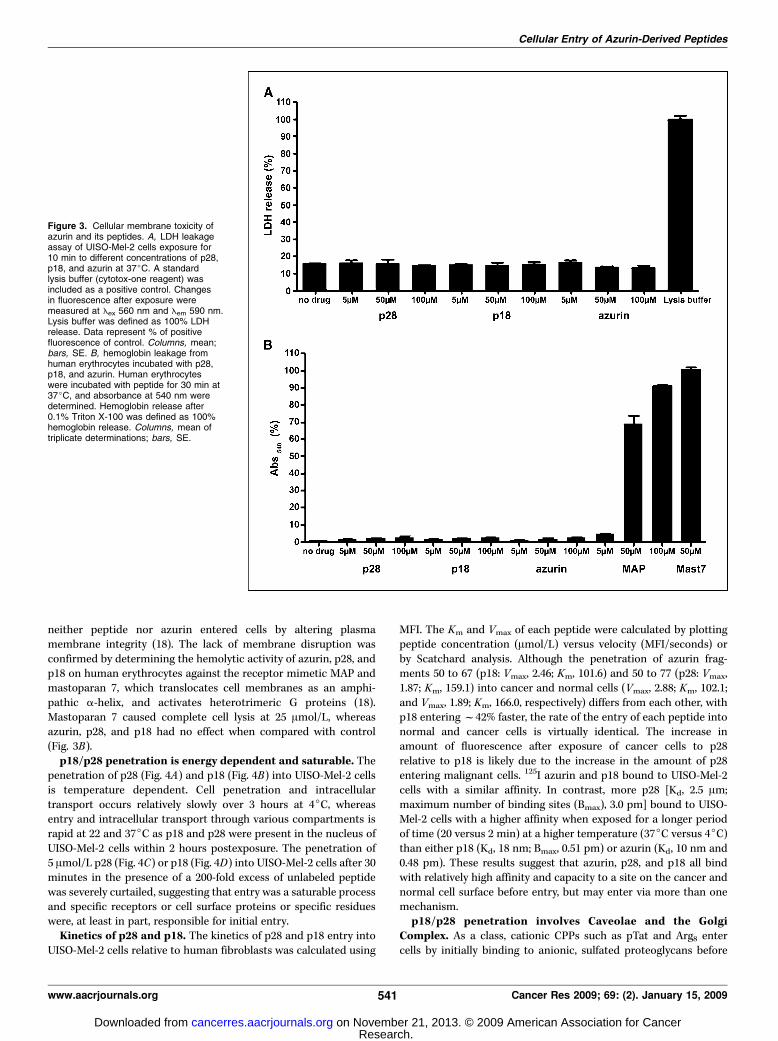
neither peptide nor azurin entered cells by altering plasmamembrane integrity (18). The lack of membrane disruption wasconfirmed by determining the hemolytic activity of azurin, p28, andp18 on human erythrocytes against the receptor mimetic MAP andmastoparan 7, which translocates cell membranes as an amphi-pathic a-helix, and activates heterotrimeric G proteins (18).Mastoparan 7 caused complete cell lysis at 25 Amol/L, whereasazurin, p28, and p18 had no effect when compared with control(Fig. 3B).p18/p28 penetration is energy dependent and saturable. The
penetration of p28 (Fig. 4A) and p18 (Fig. 4B) into UISO-Mel-2 cellsis temperature dependent. Cell penetration and intracellulartransport occurs relatively slowly over 3 hours at 4jC, whereasentry and intracellular transport through various compartments israpid at 22 and 37jC as p18 and p28 were present in the nucleus ofUISO-Mel-2 cells within 2 hours postexposure. The penetration of5 Amol/L p28 (Fig. 4C) or p18 (Fig. 4D) into UISO-Mel-2 cells after 30minutes in the presence of a 200-fold excess of unlabeled peptidewas severely curtailed, suggesting that entry was a saturable processand specific receptors or cell surface proteins or specific residueswere, at least in part, responsible for initial entry.Kinetics of p28 and p18. The kinetics of p28 and p18 entry into
UISO-Mel-2 cells relative to human fibroblasts was calculated using
MFI. The Km and Vmax of each peptide were calculated by plottingpeptide concentration (Amol/L) versus velocity (MFI/seconds) orby Scatchard analysis. Although the penetration of azurin frag-ments 50 to 67 (p18: Vmax, 2.46; Km, 101.6) and 50 to 77 (p28: Vmax,1.87; Km, 159.1) into cancer and normal cells (Vmax, 2.88; Km, 102.1;and Vmax, 1.89; Km, 166.0, respectively) differs from each other, withp18 enteringf42% faster, the rate of the entry of each peptide intonormal and cancer cells is virtually identical. The increase inamount of fluorescence after exposure of cancer cells to p28relative to p18 is likely due to the increase in the amount of p28entering malignant cells. 125I azurin and p18 bound to UISO-Mel-2cells with a similar affinity. In contrast, more p28 [Kd, 2.5 Am;maximum number of binding sites (Bmax), 3.0 pm] bound to UISO-Mel-2 cells with a higher affinity when exposed for a longer periodof time (20 versus 2 min) at a higher temperature (37jC versus 4jC)than either p18 (Kd, 18 nm; Bmax, 0.51 pm) or azurin (Kd, 10 nm and0.48 pm). These results suggest that azurin, p28, and p18 all bindwith relatively high affinity and capacity to a site on the cancer andnormal cell surface before entry, but may enter via more than onemechanism.p18/p28 penetration involves Caveolae and the Golgi
Complex. As a class, cationic CPPs such as pTat and Arg8 entercells by initially binding to anionic, sulfated proteoglycans before
Figure 3. Cellular membrane toxicity ofazurin and its peptides. A, LDH leakageassay of UISO-Mel-2 cells exposure for10 min to different concentrations of p28,p18, and azurin at 37jC. A standardlysis buffer (cytotox-one reagent) wasincluded as a positive control. Changesin fluorescence after exposure weremeasured at Eex 560 nm and Eem 590 nm.Lysis buffer was defined as 100% LDHrelease. Data represent % of positivefluorescence of control. Columns, mean;bars, SE. B, hemoglobin leakage fromhuman erythrocytes incubated with p28,p18, and azurin. Human erythrocyteswere incubated with peptide for 30 min at37jC, and absorbance at 540 nm weredetermined. Hemoglobin release after0.1% Triton X-100 was defined as 100%hemoglobin release. Columns, mean oftriplicate determinations; bars, SE.
Cellular Entry of Azurin-Derived Peptides
www.aacrjournals.org 541 Cancer Res 2009; 69: (2). January 15, 2009
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

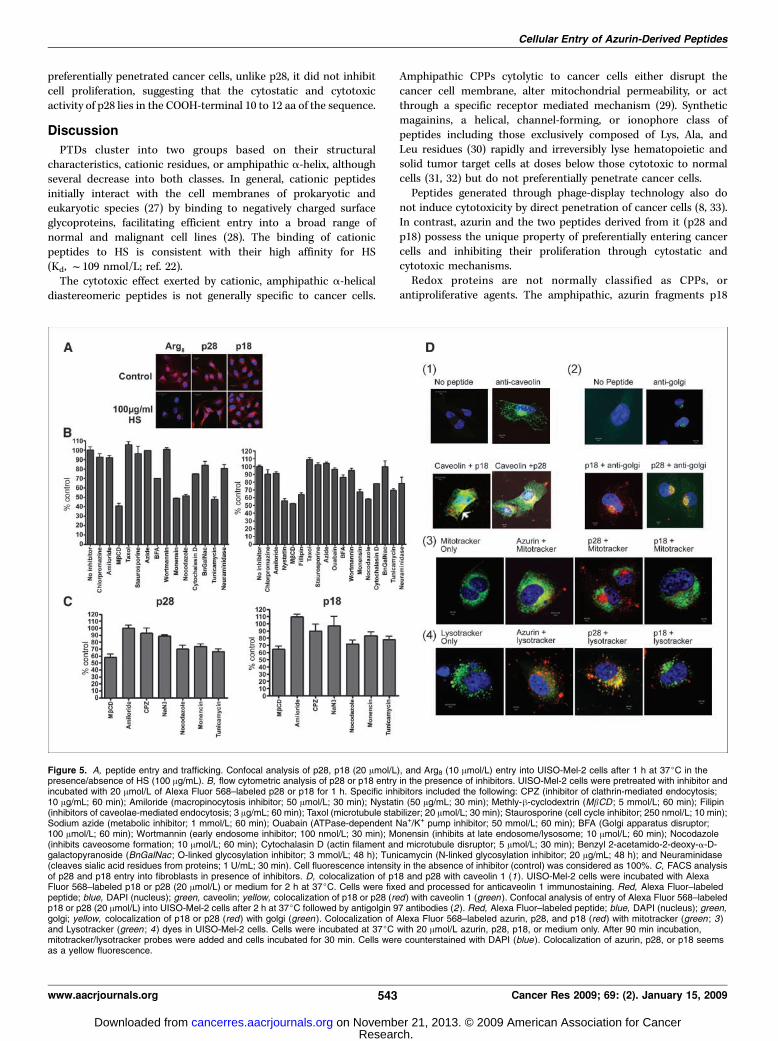
endocytosis (1). Incubation of p28 and p18 and Arg8 with UISO-Mel-2 cells under serum-free conditions in the presence/absenceof (HS) reduced the amount of intracellular Arg8 but did not alterthe entry of either p28 or p18 (Fig. 5A). The penetration of p18,and p28 (Fig. 5B) into UISO-Mel-2 cells in the presence or absenceof a specific inhibitor of O-linked glycosylation, BnGalNac, andneuraminidase, which cleaves sialic acid residues, was alsoinhibited. However, tunicamycin, an inhibitor of N-linked glycosyl-ation, significantly reduced the penetration of p18 and p28 acrossthe cell membrane (P < 0.05).Inhibitors of energy-dependent transport mechanisms, i.e., ATP.
Sodium azide (Fig. 5B) and ouabain (Na+K+ ATPase pump) did notinhibit the penetration of either peptide, suggesting nonendocy-totic pathways might also be involved in the penetration of thesepeptides. CPZ, which inhibits clathrin mediated endocytosis, alsohad no effect on penetration, nor did the macropinocytosisinhibitor amiloride (Fig. 5B). Stabilization of microtubules withTaxol had no effect on penetration, but disruption of actinfilaments and macropinocytosis with Cytochalasin D produced asmall (f20%), reproducible inhibition of p18 and p28 penetration.The lack of effect of amiloride suggests that the inhibitory activityof Cytochalasin D is probably through its effect on actin filaments.Inhibition of the cell cycle with staurosporine did not blockpenetration, suggesting that penetration was not cell cycle specific.The lack of effect of staurosporine on p18 and p28 penetration ofthe cancer cell plasma membrane also suggests that a Src kinase/tyrosine kinase–dependent pathway was not involved in penetra-tion (20), was dynamin independent, and hence independent ofcaveolae budding (21). In contrast, nocodazole, which disruptscaveolae transport and inhibitors of cholesterol mobilization and,hence, caveolae-mediated endocytosis, inhibited penetration 50%to 65%.Wortmannin, an inhibitor of early endosome formation, did not
block the intracellular accumulation of either p18 or p28 suggestingthat, unlike cholera toxin (22), a caveolae to early endosomepathway is not involved in the intracellular trafficking of p18 andp28. The lack of early endosome involvement in the intracellular
trafficking of p18 and p28 also suggests that clathrin-mediatedendocytosis is not involved in internalization of these peptides(22, 23).Monensin, which inhibits late endosome/lysosome, and BFA, a
disruptor of the Golgi apparatus, reduced the intracellularaccumulation of both peptides with a greater inhibitory effect onp28 (f30%) than p18 (f10%; Fig. 5B).The penetration of p28 and p18 into fibroblasts was also
inhibited by MhCD, nocodazole, monensin, and tunicamycin, butnot by amiloride, sodium azide, and CPZ (Fig. 5C). This suggeststhat at least one mechanism of entry into cancer and normal cellsmay be similar, but additional preferential accumulation intocancer cells may be a function of the number of commonmembrane receptors or structures, i.e., caveolae (Fig. 5B, 1 and 2).p18 and p28 colocalized with caveolin-1 and golgin 97 antibodies(Fig. 5D, 1 and 2), confirming these organelles are involved in theintracellular trafficking of p18 and p28. Interestingly, azurin, butneither p18 nor p28, colocalized with mitochondrial specificfluorescence (Fig. 5D, 3). In contrast, p28 and azurin, but notp18, colocalized with lysosomes, (Fig. 5D, 4).Functional analysis of p28 and p18. Azurin inhibits the growth
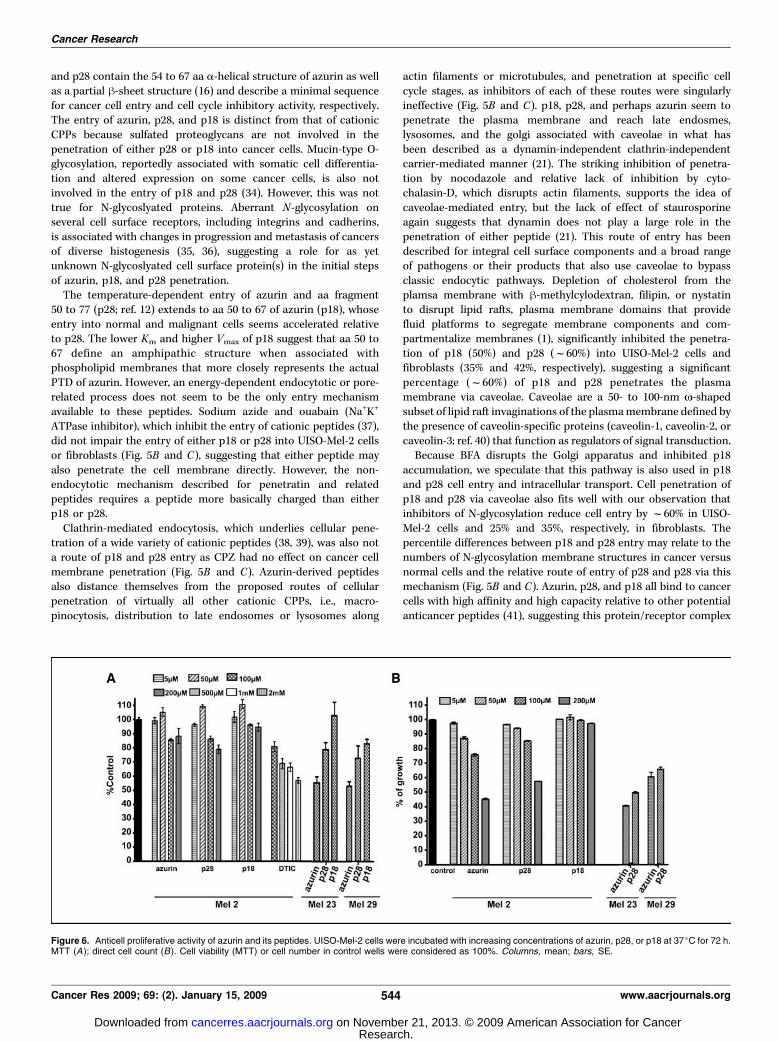
of several human cancer cell lines in vitro (14, 24–26) and in vivo(13). Figure 6A and B illustrate the effect of p18 and p28 relative toazurin and dacarbazine (DTIC) on UISO-Mel-2 cells as determinedby MTT and cell count. Azurin decreased (P < 0.05) cell survivalat 100 and 200 Amol/L f15% (Fig. 6A). p28 inhibited cell survival14% and 22% (P < 0.05) at 100 and 200 Amol/L, respectively. Incontrast, p18 had no effect, whereas DTIC produced a significantdose-related decrease on UISO-Mel-2 survival. Azurin and p28(200 Amol/L) also significantly decreased the survival of UISO-Mel-23 and 29 cells, whereas p18 had no significant effect. In contrast,azurin and p28 produced a dose-related decrease in cell number(Fig. 6B). p18 had no effect on UISO-Mel-2 cell proliferation. Theapparent increase (f30–35%; UISO-Mel-2) in p28 and azurininhibition of melanoma cell proliferation, measured directly,suggests that the inhibitory effect may reside primarily at the levelof cell cycle with apoptosis subsequent to any delay. Although p18
Figure 4. Temperature-dependent and competitive internalization of p28 and p18 into UISO-Mel-2 cells. Penetration of Alexa Fluor 568–labeled p28 (A) orp18 (B) at 20 Amol/L was evaluated by confocal microscopy at different temperatures. Red, Alexa Fluor–labeled peptide; blue, DAPI (nucleus). C and D, confocalanalysis of entry of Alexa Fluor 568–labeled p28 (C) or p18 (D) at 5 Amol/L into UISO-Mel-2 cells after 30 min at 37jC in the presence/absence of unlabeledpeptide (200-fold excess). RT, room temperature.
Cancer Research
Cancer Res 2009; 69: (2). January 15, 2009 542 www.aacrjournals.org
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
preferentially penetrated cancer cells, unlike p28, it did not inhibitcell proliferation, suggesting that the cytostatic and cytotoxicactivity of p28 lies in the COOH-terminal 10 to 12 aa of the sequence.
Discussion
PTDs cluster into two groups based on their structuralcharacteristics, cationic residues, or amphipathic a-helix, althoughseveral decrease into both classes. In general, cationic peptidesinitially interact with the cell membranes of prokaryotic andeukaryotic species (27) by binding to negatively charged surfaceglycoproteins, facilitating efficient entry into a broad range ofnormal and malignant cell lines (28). The binding of cationicpeptides to HS is consistent with their high affinity for HS(Kd, f109 nmol/L; ref. 22).The cytotoxic effect exerted by cationic, amphipathic a-helical
diastereomeric peptides is not generally specific to cancer cells.
Amphipathic CPPs cytolytic to cancer cells either disrupt thecancer cell membrane, alter mitochondrial permeability, or actthrough a specific receptor mediated mechanism (29). Syntheticmagainins, a helical, channel-forming, or ionophore class ofpeptides including those exclusively composed of Lys, Ala, andLeu residues (30) rapidly and irreversibly lyse hematopoietic andsolid tumor target cells at doses below those cytotoxic to normalcells (31, 32) but do not preferentially penetrate cancer cells.Peptides generated through phage-display technology also do
not induce cytotoxicity by direct penetration of cancer cells (8, 33).In contrast, azurin and the two peptides derived from it (p28 andp18) possess the unique property of preferentially entering cancercells and inhibiting their proliferation through cytostatic andcytotoxic mechanisms.Redox proteins are not normally classified as CPPs, or
antiproliferative agents. The amphipathic, azurin fragments p18
Figure 5. A, peptide entry and trafficking. Confocal analysis of p28, p18 (20 Amol/L), and Arg8 (10 Amol/L) entry into UISO-Mel-2 cells after 1 h at 37jC in thepresence/absence of HS (100 Ag/mL). B, flow cytometric analysis of p28 or p18 entry in the presence of inhibitors. UISO-Mel-2 cells were pretreated with inhibitor andincubated with 20 Amol/L of Alexa Fluor 568–labeled p28 or p18 for 1 h. Specific inhibitors included the following: CPZ (inhibitor of clathrin-mediated endocytosis;10 Ag/mL; 60 min); Amiloride (macropinocytosis inhibitor; 50 Amol/L; 30 min); Nystatin (50 Ag/mL; 30 min); Methly-h-cyclodextrin (MbCD ; 5 mmol/L; 60 min); Filipin(inhibitors of caveolae-mediated endocytosis; 3 Ag/mL; 60 min); Taxol (microtubule stabilizer; 20 Amol/L; 30 min); Staurosporine (cell cycle inhibitor; 250 nmol/L; 10 min);Sodium azide (metabolic inhibitor; 1 mmol/L; 60 min); Ouabain (ATPase-dependent Na+/K+ pump inhibitor; 50 mmol/L; 60 min); BFA (Golgi apparatus disruptor;100 Amol/L; 60 min); Wortmannin (early endosome inhibitor; 100 nmol/L; 30 min); Monensin (inhibits at late endosome/lysosome; 10 Amol/L; 60 min); Nocodazole(inhibits caveosome formation; 10 Amol/L; 60 min); Cytochalasin D (actin filament and microtubule disruptor; 5 Amol/L; 30 min); Benzyl 2-acetamido-2-deoxy-a-D-galactopyranoside (BnGalNac ; O-linked glycosylation inhibitor; 3 mmol/L; 48 h); Tunicamycin (N-linked glycosylation inhibitor; 20 Ag/mL; 48 h); and Neuraminidase(cleaves sialic acid residues from proteins; 1 U/mL; 30 min). Cell fluorescence intensity in the absence of inhibitor (control) was considered as 100%. C, FACS analysisof p28 and p18 entry into fibroblasts in presence of inhibitors. D, colocalization of p18 and p28 with caveolin 1 (1). UISO-Mel-2 cells were incubated with AlexaFluor 568–labeled p18 or p28 (20 Amol/L) or medium for 2 h at 37jC. Cells were fixed and processed for anticaveolin 1 immunostaining. Red, Alexa Fluor–labeledpeptide; blue, DAPI (nucleus); green, caveolin; yellow, colocalization of p18 or p28 (red ) with caveolin 1 (green ). Confocal analysis of entry of Alexa Fluor 568–labeledp18 or p28 (20 Amol/L) into UISO-Mel-2 cells after 2 h at 37jC followed by antigolgin 97 antibodies (2). Red, Alexa Fluor–labeled peptide; blue, DAPI (nucleus); green,golgi; yellow, colocalization of p18 or p28 (red) with golgi (green ). Colocalization of Alexa Fluor 568–labeled azurin, p28, and p18 (red) with mitotracker (green ; 3)and Lysotracker (green ; 4 ) dyes in UISO-Mel-2 cells. Cells were incubated at 37jC with 20 Amol/L azurin, p28, p18, or medium only. After 90 min incubation,mitotracker/lysotracker probes were added and cells incubated for 30 min. Cells were counterstained with DAPI (blue ). Colocalization of azurin, p28, or p18 seemsas a yellow fluorescence.
Cellular Entry of Azurin-Derived Peptides
www.aacrjournals.org 543 Cancer Res 2009; 69: (2). January 15, 2009
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
and p28 contain the 54 to 67 aa a-helical structure of azurin as wellas a partial h-sheet structure (16) and describe a minimal sequencefor cancer cell entry and cell cycle inhibitory activity, respectively.The entry of azurin, p28, and p18 is distinct from that of cationicCPPs because sulfated proteoglycans are not involved in thepenetration of either p28 or p18 into cancer cells. Mucin-type O-glycosylation, reportedly associated with somatic cell differentia-tion and altered expression on some cancer cells, is also notinvolved in the entry of p18 and p28 (34). However, this was nottrue for N-glycoslyated proteins. Aberrant N-glycosylation onseveral cell surface receptors, including integrins and cadherins,is associated with changes in progression and metastasis of cancersof diverse histogenesis (35, 36), suggesting a role for as yetunknown N-glycoslyated cell surface protein(s) in the initial stepsof azurin, p18, and p28 penetration.The temperature-dependent entry of azurin and aa fragment
50 to 77 (p28; ref. 12) extends to aa 50 to 67 of azurin (p18), whoseentry into normal and malignant cells seems accelerated relativeto p28. The lower Km and higher Vmax of p18 suggest that aa 50 to67 define an amphipathic structure when associated withphospholipid membranes that more closely represents the actualPTD of azurin. However, an energy-dependent endocytotic or pore-related process does not seem to be the only entry mechanismavailable to these peptides. Sodium azide and ouabain (Na+K+
ATPase inhibitor), which inhibit the entry of cationic peptides (37),did not impair the entry of either p18 or p28 into UISO-Mel-2 cellsor fibroblasts (Fig. 5B and C), suggesting that either peptide mayalso penetrate the cell membrane directly. However, the non-endocytotic mechanism described for penetratin and relatedpeptides requires a peptide more basically charged than eitherp18 or p28.Clathrin-mediated endocytosis, which underlies cellular pene-
tration of a wide variety of cationic peptides (38, 39), was also nota route of p18 and p28 entry as CPZ had no effect on cancer cellmembrane penetration (Fig. 5B and C). Azurin-derived peptidesalso distance themselves from the proposed routes of cellularpenetration of virtually all other cationic CPPs, i.e., macro-pinocytosis, distribution to late endosomes or lysosomes along
actin filaments or microtubules, and penetration at specific cellcycle stages, as inhibitors of each of these routes were singularlyineffective (Fig. 5B and C). p18, p28, and perhaps azurin seem topenetrate the plasma membrane and reach late endosmes,lysosomes, and the golgi associated with caveolae in what hasbeen described as a dynamin-independent clathrin-independentcarrier-mediated manner (21). The striking inhibition of penetra-tion by nocodazole and relative lack of inhibition by cyto-chalasin-D, which disrupts actin filaments, supports the idea ofcaveolae-mediated entry, but the lack of effect of staurosporineagain suggests that dynamin does not play a large role in thepenetration of either peptide (21). This route of entry has beendescribed for integral cell surface components and a broad rangeof pathogens or their products that also use caveolae to bypassclassic endocytic pathways. Depletion of cholesterol from theplamsa membrane with h-methylcylodextran, filipin, or nystatinto disrupt lipid rafts, plasma membrane domains that providefluid platforms to segregate membrane components and com-partmentalize membranes (1), significantly inhibited the penetra-tion of p18 (50%) and p28 (f60%) into UISO-Mel-2 cells andfibroblasts (35% and 42%, respectively), suggesting a significantpercentage (f60%) of p18 and p28 penetrates the plasmamembrane via caveolae. Caveolae are a 50- to 100-nm N-shapedsubset of lipid raft invaginations of the plasmamembrane defined bythe presence of caveolin-specific proteins (caveolin-1, caveolin-2, orcaveolin-3; ref. 40) that function as regulators of signal transduction.Because BFA disrupts the Golgi apparatus and inhibited p18
accumulation, we speculate that this pathway is also used in p18and p28 cell entry and intracellular transport. Cell penetration ofp18 and p28 via caveolae also fits well with our observation thatinhibitors of N-glycosylation reduce cell entry by f60% in UISO-Mel-2 cells and 25% and 35%, respectively, in fibroblasts. Thepercentile differences between p18 and p28 entry may relate to thenumbers of N-glycosylation membrane structures in cancer versusnormal cells and the relative route of entry of p28 and p28 via thismechanism (Fig. 5B and C). Azurin, p28, and p18 all bind to cancercells with high affinity and high capacity relative to other potentialanticancer peptides (41), suggesting this protein/receptor complex
Figure 6. Anticell proliferative activity of azurin and its peptides. UISO-Mel-2 cells were incubated with increasing concentrations of azurin, p28, or p18 at 37jC for 72 h.MTT (A); direct cell count (B). Cell viability (MTT) or cell number in control wells were considered as 100%. Columns, mean; bars, SE.
Cancer Research
Cancer Res 2009; 69: (2). January 15, 2009 544 www.aacrjournals.org
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

localizes in caveolae and is internalized, eventually moving(via caveosomes) to the golgi, endoplasmic reticulum, and nucleus.Macropinocytosis does not seem to be involved nor does any entrymechanism that includes peptide transport to early endosomes. Inaddition to caveolar-mediated entry, our kinetic analysis alsosuggests that p28 and p18 penetrate the plasma membrane via anonclathrin-caveolae–mediated process. A clathrin- and caveolin-independent pathway can exist as a constitutive internalizationmechanism, such as for the interleukin 2 receptor (42) andfor certain glycosyl-phosphatidylinositol–anchored proteins (43).Clathrin- and caveolin-independent endocytosis is also used bypathogens to invade cells, either exclusively, as for the murinepolyoma virus (44), or in combination with a conventional pathway,as is the case for the influenza virus (45). An increase in caveolin-1expression in cancer cells over normal cells is not likely to be thesole basis for the preferential entry of azurin, p28, and p18 intocancer cells. Fibroblasts and a number of other normal cells alsohave significant numbers of caveolae on their surface (23).However, it provides an avenue for additional investigation intothe preferential penetration of these peptides into cancer cells.Our results suggest that the cellular penetration of aa 50 to 67
and 50 to 77 of azurin is unique relative to all current CPPs in its
preference for cancer cells, and that the COOH-terminal 10 to 12aas of p28, aa 50 to 77 of azurin, contain the domain responsible forcell cycle inhibition and apoptotic activity. As such, these novelpeptides could potentially serve as either a novel chemotherapeuticagent, adjunct to extant drugs or as CPPs to transport cargosincluding imaging agents into a broad range of cancer cells.
Disclosure of Potential Conflicts of Interest
This work was supported through a sponsored research agreement between CDGTherapeutics, Inc. and UIC. CDG Therapeutics, Inc. has an exclusive licensingagreement for the development and commercialization of cupredoxin-derivedpeptides. A.M. Chakrabarty and T.K. Das Gupta are cofounders of and shareholdersin CDG Therapeutics, Inc. T. Yamada is one of the inventors and potentially willreceive a share of the royalty received by UIC through the licensing of the technology.C.W. Beattie is CSO of CDG Therapeutics, Inc. All terms of this arrangement aremanaged by UIC in accordance with its conflict of interest policies.
Acknowledgments
Received 8/1/2008; revised 9/24/2008; accepted 10/23/2008.Grant support: A sponsored research agreement between CDG Therapeutics, Inc.,
and UIC.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
Cellular Entry of Azurin-Derived Peptides
www.aacrjournals.org 545 Cancer Res 2009; 69: (2). January 15, 2009
References
1. Fischer PM. Cellular uptake mechanisms and potentialtherapeutic utility of peptidic cell delivery vectors:Progress 2001–2006. Med Res Rev 2007;27:755–95.
2. Gusarova GA, Wang IC, Major ML, et al. A cell-penetrating ARF peptide inhibitor of FoxM1 in mousehepatocellular carcinoma treatment. J Clin Invest 2007;117:99–111.
3. Melnick A. Targeting aggressive B-cell lymphomaswith cell-penetrating peptides. Biochem Soc Trans 2007;35:802–6.
4. Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR,Juliano RL. Conjugates of antisense oligonucleotideswith the Tat and antennapedia cell-penetrating pep-tides: effects on cellular uptake, binding to targetsequences, and biologic actions. Pharm Res 2002;19:744–54.
5. El-Andaloussi S, Johansson HJ, Lundberg P, Langel U.Induction of splice correction by cell-penetratingpeptide nucleic acids. J Gene Med 2006;8:1262–73.
6. Cashman SM, Sadowski SL, Morris DJ, Frederick J,Kumar-Singh R. Intercellular trafficking of adenovirus-delivered HSV VP22 from the retinal pigment epitheli-um to the photoreceptors–implications for gene ther-apy. Mol Ther 2002;6:813–23.
7. Fisher L, Soomets U, Cortes Toro V, et al. Cellulardelivery of a double-stranded oligonucleotide NFnBdecoy by hybridization to complementary PNA linked toa cell-penetrating peptide. Gene Ther 2004;11:1264–72.
8. Pasqualini R, Koivunen E, Ruoslahti E. a v integrins asreceptors for tumor targeting by circulating ligands. NatBiotechnol 1997;15:542–6.
9. Laakkonen P, Akerman ME, Biliran H, et al. Antitumoractivity of a homing peptide that targets tumorlymphatics and tumor cells. Proc Natl Acad Sci U S A2004;101:9381–6.
10. Joyce JA, Laakkonen P, Bernasconi M, Bergers G,Ruoslahti E, Hanahan D. Stage-specific vascularmarkers revealed by phage display in a mouse modelof pancreatic islet tumorigenesis. Cancer Cell 2003;4:393–403.
11. Enback J, Laakkonen P. Tumour-homing peptides:tools for targeting, imaging and destruction. BiochemSoc Trans 2007;35:780–3.
12. Yamada T, Fialho AM, Punj V, Bratescu L, Gupta TK,Chakrabarty AM. Internalization of bacterial redox
protein azurin in mammalian cells: entry domain andspecificity. Cell Microbiol 2005;7:1418–31.
13. Punj V, Bhattacharyya S, Saint-Dic D, et al. Bacterialcupredoxin azurin as an inducer of apoptosis andregression in human breast cancer. Oncogene 2004;23:2367–78.
14. Yang DS, Miao XD, Ye ZM, et al. Bacterial redoxprotein azurin induce apoptosis in human osteosarco-ma U2OS cells. Pharmacol Res 2005;52:413–21.
15. Rauth S, Kichina J, Green A, Bratescu L, Das GuptaTK. Establishment of a human melanoma cell linelacking p53 expression and spontaneously metastasiz-ing in nude mice. Anticancer Res 1994;14:2457–63.
16. Garnier J, Osguthorpe DJ, Robson B. Analysis of theaccuracy and implications of simple methods forpredicting the secondary structure of globular proteins.J Mol Biol 1978;120:97–120.
17. Sharikov Y, Walker RC, Greenberg J, et al. MAPAS: atool for predicting membrane-contacting protein surfa-ces. Nat Methods 2008;5:119.
18. Saar K, Lindgren M, Hansen M, et al. Cell-penetratingpeptides: a comparative membrane toxicity study. AnalBiochem 2005;345:55–65.
19. Manetto GD, Grasso DM, Milardi D, et al. Therole played by the a-helix in the unfolding pathwayand stability of azurin: switching between hierarchicand nonhierarchic folding. Chembiochem 2007;8:1941–9.
20. Pelkmans L, Puntener D, Helenius A. Local actinpolymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002;296:535–9.
21. Kirkham M, Parton RG. Clathrin-independentendocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta2005;1746:349–63.
22. Tran D, Carpentier JL, Sawano F, Gorden P, Orci L.Ligands internalized through coated or noncoatedinvaginations follow a common intracellular pathway.Proc Natl Acad Sci U S A 1987;84:7957–61.
23. Parton RG, Richards AA. Lipid rafts and caveolae asportals for endocytosis: new insights and commonmechanisms. Traffic 2003;4:724–38.
24. Yamada T, Hiraoka Y, Ikehata M, et al. Apoptosis orgrowth arrest: Modulation of tumor suppressor p53’sspecificity by bacterial redox protein azurin. Proc NatlAcad Sci U S A 2004;101:4770–5.
25. Chaudhari A, Mahfouz M, Fialho AM, et al.Cupredoxin-cancer interrelationship: azurin bindingwith EphB2, interference in EphB2 tyrosine phosphor-ylation, and inhibition of cancer growth. Biochemistry2007;46:1799–810.
26. Ye Z, Peng H, Fang Y, Feng J, Yang DS. Theconstruction of the eukaryotic expression plasmidpcDNA3.1/azurin and the increased apoptosis ofU2OS cells transfected with it. Cell Mol Biol Lett2007;12:407–21.
27. Kondejewski LH, Lee DL, Jelokhani-Niaraki M,Farmer SW, Hancock RE, Hodges RS. Optimization ofmicrobial specificity in cyclic peptides by modulation ofhydrophobicity within a defined structural framework.J Biol Chem 2002;277:67–74.
28. Fuchs SM, Raines RT. Pathway for polyarginine entryinto mammalian cells. Biochemistry 2004;43:2438–44.
29. Leuschner C, Hansel W. Membrane disrupting lyticpeptides for cancer treatments. Curr Pharm Des 2004;10:2299–310.
30. Javadpour MM, Juban MM, Lo WC, et al. De novoantimicrobial peptides with low mammalian celltoxicity. J Med Chem 1996;39:3107–13.
31. Cruciani RA, Barker JL, Zasloff M, Chen HC,Colamonici O. Antibiotic magainins exert cytolyticactivity against transformed cell lines through chan-nel formation. Proc Natl Acad Sci U S A 1991;88:3792–6.
32. Papo N, Shai Y. New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumorcells compared to normal cells. Biochemistry 2003;42:9346–54.
33. Gehlsen KR, Argraves WS, Pierschbacher MD,Ruoslahti E. Inhibition of in vitro tumor cell invasionby Arg-Gly-Asp-containing synthetic peptides. J Cell Biol1988;106:925–30.
34. Brooks SA, Carter TM, Bennett EP, Clausen H,Mandel U. Immunolocalisation of members of thepolypeptide N-acetylgalactosaminyl transferase (ppGal-NAc-T) family is consistent with biologically relevantaltered cell surface glycosylation in breast cancer. ActaHistochem 2007;109:273–84.
35. Partridge EA, Le Roy C, Di Guglielmo GM, et al.Regulation of cytokine receptors by Golgi N-glycanprocessing and endocytosis. Science 2004;306:120–4.
36. Seales EC, Jurado GA, Brunson BA, Wakefield JK,Frost AR, Bellis SL. Hypersialylation of h1 integrins,
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from

Cancer Research
Cancer Res 2009; 69: (2). January 15, 2009 546 www.aacrjournals.org
observed in colon adenocarcinoma, may contribute tocancer progression by up-regulating cell motility.Cancer Res 2005;65:4645–52.
37. Sandvig K, Olsnes S. Entry of the toxic proteins abrin,modeccin, ricin, and diphtheria toxin into cells. II. Effectof pH, metabolic inhibitors, and ionophores andevidence for toxin penetration from endocytoticvesicles. J Biol Chem 1982;257:7504–13.
38. Richard JP, Melikov K, Vives E, et al. Cell-penetratingpeptides. A reevaluation of the mechanism of cellularuptake. J Biol Chem 2003;278:585–90.
39. Potocky TB, Menon AK, Gellman SH. Cytoplasmicand nuclear delivery of a TAT-derived peptide and a h-
peptide after endocytic uptake into HeLa cells. J BiolChem 2003;278:50188–94.
40. Scherer PE, Lewis RY, Volonte D, et al. Cell-type andtissue-specific expression of caveolin-2. Caveolins 1 and2 co-localize and form a stable hetero-oligomericcomplex in vivo . J Biol Chem 1997;272:29337–46.
41. Hohla F, Schally AV, Kanashiro CA, et al. Growthinhibition of non-small-cell lung carcinoma by BN/GRPantagonist is linked with suppression of K-Ras, COX-2,and pAkt. Proc Natl Acad Sci U S A 2007;104:18671–6.
42. Lamaze C, Dujeancourt A, Baba T, Lo CG, BenmerahA, Dautry-Varsat A. Interleukin 2 receptors anddetergent-resistant membrane domains define a cla-
thrin-independent endocytic pathway. Mol Cell 2001;7:661–71.
43. Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomesvia a distinct cdc42-regulated, clathrin-independentpinocytic pathway. Dev Cell 2002;2:411–23.
44. Ewers H, Smith AE, Sbalzarini IF, Lilie H, Koumout-sakos P, Helenius A. Single-particle tracking of murinepolyoma virus-like particles on live cells and artificialmembranes. Proc Natl Acad Sci U S A 2005;102:15110–5.
45. Sieczkarski SB, Whittaker GR. Influenza virus canenter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 2002;76:10455–64.
Research. on November 21, 2013. © 2009 American Association for Cancercancerres.aacrjournals.org Downloaded from
Related Documents











