1. Introduction 2. FDA-approved treatments for metastatic melanoma 3. Targeted therapy 4. Immunotherapy 5. Combination therapy 6. Expert opinion Review Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma Sara Tomei † , Ena Wang, Lucia Gemma Delogu, Francesco M Marincola & Davide Bedognetti † National Institutes of Health, Clinical Center and trans-NIH Center for Human Immunology (CHI), Department of Transfusion Medicine, Infectious Disease and Immunogenetics Section (IDIS), Bethesda, MD, USA Introduction: Melanoma is an aggressive disease characterized by a complex etiology. The discovery of key driving mutations (primarily BRAF mutations) led to the development of specific molecular inhibitors providing clinical benefit. Areas covered: Although BRAF-specific drugs have perhaps yielded the best results in melanoma-targeted therapy, there still remain several limitations, mostly due to the emergence of resistance and the lack of efficacy in patients without BRAF mutation. Novel drugs are currently being tested in clinical trials and showed encouraging results. Such drugs can specifically target molecular pathways aberrantly activated or repressed during melanoma development (targeted therapy) or act in a way to enhance the host immune system to fight cancer (immunotherapy). Here we provide a detailed overview of the current clinical strategies, which lay beyond BRAF-targeted therapy, spanning from molecular-targeted therapy to immunotherapy and to combination therapy. Expert opinion: Major advances in our understanding of the mechanisms behind melanoma development have led to the implementation of novel therapeutic drugs. Unfortunately, tools allowing prediction of responsiveness to a given treatment are not available yet. The increasing availability of high- throughput technologies will allow the elucidation of molecular mechanisms underlying responsiveness to cancer therapy and unveil an increased number of potential therapeutic targets. Keywords: adoptive T-cell transfer, combination therapy, immunotherapy, melanoma, targeted therapy Expert Opin. Biol. Ther. [Early Online] 1. Introduction Cutaneous melanoma is a highly heterogeneous disease characterized by a complex etiology. If diagnosed early, it can be potentially cured by surgical resection; however, once metastasized, it is highly refractory to conventional antineoplastic treatments, urging the identification of novel effective therapeutic solutions. High-throughput experimental strategies, such as gene mutation analysis and com- parative genomic hybridization, allowed the identification of crucial signaling pathways essential for melanoma development and progression [1]. The recognition of molecular aberrations occurring during melanoma development facilitated the application of new therapeutic strategies, which have improved the clinical management of melanoma patients [2,3]. The best results have been obtained after the introduction of BRAF inhibitors. BRAF is a serine-threonine kinase, member of the MAPK pathway; when activated, 10.1517/14712598.2014.890586 © 2014 Informa UK, Ltd. ISSN 1471-2598, e-ISSN 1744-7682 1 All rights reserved: reproduction in whole or in part not permitted Expert Opin. Biol. Ther. Downloaded from informahealthcare.com by Cornell University on 03/21/14 For personal use only.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1. Introduction
2. FDA-approved treatments for
metastatic melanoma
3. Targeted therapy
4. Immunotherapy
5. Combination therapy
6. Expert opinion
Review
Non-BRAF-targeted therapy,immunotherapy, and combinationtherapy for melanomaSara Tomei†, Ena Wang, Lucia Gemma Delogu, Francesco M Marincola &Davide Bedognetti†National Institutes of Health, Clinical Center and trans-NIH Center for Human Immunology
(CHI), Department of Transfusion Medicine, Infectious Disease and Immunogenetics Section
(IDIS), Bethesda, MD, USA
Introduction: Melanoma is an aggressive disease characterized by a complex
etiology. The discovery of key driving mutations (primarily BRAF mutations)
led to the development of specific molecular inhibitors providing clinical
benefit.
Areas covered: Although BRAF-specific drugs have perhaps yielded the best
results in melanoma-targeted therapy, there still remain several limitations,
mostly due to the emergence of resistance and the lack of efficacy in patients
without BRAF mutation. Novel drugs are currently being tested in clinical
trials and showed encouraging results. Such drugs can specifically target
molecular pathways aberrantly activated or repressed during melanoma
development (targeted therapy) or act in a way to enhance the host immune
system to fight cancer (immunotherapy). Here we provide a detailed overview
of the current clinical strategies, which lay beyond BRAF-targeted therapy,
spanning from molecular-targeted therapy to immunotherapy and to
combination therapy.
Expert opinion: Major advances in our understanding of the mechanisms
behind melanoma development have led to the implementation of novel
therapeutic drugs. Unfortunately, tools allowing prediction of responsiveness
to a given treatment are not available yet. The increasing availability of high-
throughput technologies will allow the elucidation of molecular mechanisms
underlying responsiveness to cancer therapy and unveil an increased number
of potential therapeutic targets.
Keywords: adoptive T-cell transfer, combination therapy, immunotherapy, melanoma,
targeted therapy
Expert Opin. Biol. Ther. [Early Online]
1. Introduction
Cutaneous melanoma is a highly heterogeneous disease characterized by a complexetiology. If diagnosed early, it can be potentially cured by surgical resection;however, once metastasized, it is highly refractory to conventional antineoplastictreatments, urging the identification of novel effective therapeutic solutions.High-throughput experimental strategies, such as gene mutation analysis and com-parative genomic hybridization, allowed the identification of crucial signalingpathways essential for melanoma development and progression [1]. The recognitionof molecular aberrations occurring during melanoma development facilitatedthe application of new therapeutic strategies, which have improved the clinicalmanagement of melanoma patients [2,3].
The best results have been obtained after the introduction of BRAF inhibitors.BRAF is a serine-threonine kinase, member of the MAPK pathway; when activated,
10.1517/14712598.2014.890586 © 2014 Informa UK, Ltd. ISSN 1471-2598, e-ISSN 1744-7682 1All rights reserved: reproduction in whole or in part not permitted
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

it transfers growth signals to the nucleus of the cells. The highsuccess of BRAF inhibitors is mainly due to the fact that mel-anoma development often relies on the activation of MAPKpathway. BRAF mutations occur in about 50 -- 60% ofmelanoma [4-7]. More than 90% of BRAF mutations resultin a single nucleotide mutation in the 600 codon(V600) [5,6]. Among them, about 90% of mutation are repre-sented by the valine to glutamic acid substitution(V600E) [5,6], associated with a 400-fold increased activity ofthe protein [8,9].Overall, approximately 50% of melanoma patients carry
the V600E mutation. Vemurafenib, a kinase inhibitor actingby blocking V600E mutated BRAF, have been extensivelyinvestigated in Phase II and III trials [4,10]. It has beenapproved by FDA in August 2011 for the treatment ofmetastatic or unresectable melanoma patients carrying theV600E mutation. In the interim analysis of the Phase III trial(BRIM-3), vemurafenib has been shown to induce an objec-tive response (OR) in 48% of the patients, significantly higherthan that obtained by dacarbazine (5%) [4]. Beyond tumorshrinkage, administration of vemurafenib resulted in a signif-icantly prolonged progression-free survival (PFS; 6.9 vs1.6 months; vemurafenib vs dacarbazine, respectively) and
overall survival (OS; 13.6 vs 9.7 months; vemurafenib vsdacarbazine), according to a recent update of the BRIM-3trial [11]. In the same setting, similar results in term of ORand PFS have been obtained by the BRAF inhibitor dabrafe-nib, which has been approved by FDA in May 2013. How-ever, the trial allowed crossover and was not poweredenough to detect differences in OS [12]. Even thoughPhase III trials focused on patients bearing V600E mutation,BRAF inhibitors may be similarly active in patients carryingother V600 mutations (i.e., V600K and V600D) [4,10,13,14].These observations led the European Medical Agency(EMA) to express favorable opinion on the use of vemurafe-nib and dabrafenib in metastatic or unresectable melanomapatients carrying any kind of BRAF V600 mutation. How-ever, despite BRAF inhibition has shown the most promisingresults in the field of melanoma targeted therapy, several chal-lenges remain. The first important obstacle is that half of mel-anoma patients do not carry V600 mutations and thereforecannot be treated with BRAF inhibitors. Moreover, a com-plete response (CR) only occurs in a very few cases(3 -- 6%), the duration of response rate is relatively short(5 -- 7 months) and all but few patients relapse within 1 or2 years through developing secondary resistance [4,10,12,13].Thus, much effort needs to be put into developing novel,more effective therapeutic approaches.
The purpose of this review is to discuss the most significantadvances in the treatment of metastatic melanoma, highlight-ing the novel therapeutic strategies (currently being tested inclinical trials [15]), which preclude BRAF treatment, encom-passing single molecular-targeted therapy, immunotherapy,and combinatorial therapeutic approaches.
2. FDA-approved treatments for metastaticmelanoma
To date there are four FDA-approved treatments for metastaticmelanoma besides the two aforementioned BRAF inhibitors:recombinant IL-2, ipilimumab (approved, respectively, in1998 and 2011; discussed in the following sections), dacarba-zine, and trametinib [4,16-18]. Dacarbazine is an alkylating agent,which was accepted as standard treatment for metastatic mela-noma in the 1970s [19,20], and it remains the only cytotoxic che-motherapy drug approved for the treatment of metastaticmelanoma. In preliminary studies, OR was reported in up to25% of patients [21]. However, in more rigorous randomizedtrials, dacarbazine has shown to induce an OR in about 10%of patients (range 5 -- 12%) [4,12,21,22]. CR is exceptionallyrare (1 -- 2%) [4,12,21,22], remission is in general transient [21],and advantage in OS has not been conclusively demonstratedin randomized trials. Although other cytotoxic agents (e.g.,fotemustine and temozolomide) have been shown to be at leastnot inferior to dacarbazine, none of them produces superiorbenefit to dacarbazine in terms of OS [21]. For this reason,dacarbazine is often chosen as control arm in randomized trialsassessing new therapeutics. Trametinib, a MAPK extracellular
Article highlights.
. A huge amount of novel therapeutic strategies arebeing developed for the treatment of metastaticmelanoma, including targeted therapies,immunotherapies, and combinatorial strategies.
. FDA recently approved two new drugs for metastaticmelanoma, namely: ipilimumab (anti-cytotoxicT-lymphocyte antigen 4 [anti-CTLA]-4), dabrafenib (BRAFinhibitor), and trametinib (MAPK extracellular signal-regulated kinase [MEK] inhibitor). Several additionaldrugs are being tested and Phase I/II clinical trials areproviding encouraging results.
. MAPK and Phosphatidylinositol 3-Kinase (PI3K) pathwaysare the most frequently altered pathways in melanoma.Pharmaceutical inhibitors, specifically blocking alteredoncogene proteins within these pathways, have beenproved to counteract melanoma development (e.g.,BRAF inhibitors, MEK inhibitors, AKT inhibitors).
. Steps forward have been made in the field of melanomaimmunotherapy, considering the exceptionalimmunogenicity of melanoma. These advances refer tohigh-dose IL-2, vaccination strategies, immunecheckpoint blockade, and adoptive T-cell transfer.
. Albeit targeted therapy and immunotherapy strategieshave revolutionized the clinical management ofmelanoma patients, it is becoming clear thatcombinatorial strategies are most likely to result in evenmore significant advances. Examples include BRAFinhibitors plus mTOR inhibitors, BRAF inhibitors plusMEK inhibitors, BRAF inhibitors plus PI3K inhibitor,combination of immune-checkpoint inhibitors and alsotargeted therapy plus immunotherapy.
This box summarizes key points contained in the article.
S. Tomei et al.
2 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

signal–regulated kinase (MEK) inhibitor (see following sec-tion), was approved by FDA on 29 May 2013 (concurrentlywith the approval of dabrafenib) for the treatment of patientswith unresectable or metastatic melanoma harboring BRAFV600E or V600K mutation [23].
Currently, several drugs are being tested and severalencouraging results have been recently published. Therapeuticstrategies for the treatment of melanoma can be systematicallydivided in three broad categories: targeted therapy, immuno-therapy, and combination therapy. Targeted treatments act bydirectly inhibiting molecules (e.g., proteins encoded by proto-ncogenes or growth factors), which are essential for the tumorgrowth. Immunotherapy treatments aim at stimulating theimmune system of the host to reject cancer cells. Combina-tion therapy refers to the use of multiple approaches toachieve a stronger anticancer response and overcome potentialmechanisms of resistance. Examples of the most effectivetreatments belonging to each of the above categories will begiven in the following sections of this review.
3. Targeted therapy
Biologic targeted therapy usually refers to a treatment that spe-cifically targets molecules involved in neoplastic process withless harm to normal cells. Targeted treatments act by blockingthe activity of specific molecules essential for cancer growthand development. The majority of targeted therapies includeeither small molecules or monoclonal antibodies. The testingand implementation of possible therapeutic drugs in mela-noma is growing up at an exponential rate as our understand-ing of the genetic heterogeneity of this disease improves.
An interesting recent study from Henary et al. [24] showedthat the administration of molecularly matched therapies pre-dicted better outcomes, emphasizing the relevance of targetedtherapy in the context of personalized medicine. Melanomacells can acquire a proliferative advantage or protectionagainst cell death by deregulating molecular pathways relevantto cell biology. The understanding of cell growth and molec-ular signaling pathways in melanoma is essential to developand implement new therapeutic drugs.
In the following, we provide an overview of the mostcommonly deregulated signaling pathways in melanomaresponsible for cell proliferation and cell survival, and themost relevant advance in clinical setting.
3.1 MAPK pathwayMAPK pathway is probably the best-studied signaling pathwayin cancer, most likely because of its essential role in melanomaonset and development [25]. A simplified representation ofMAPK pathway is given in Figure 1. Signaling through MAPKpathway occurs upon the binding of extracellular growth factorsto a variety of tyrosine kinase receptors (TKRs), including c-KITand c-MER, which in turn lead to the activation of RAS. RAS isa small GTP-binding protein, which can exist in three isoforms:HRAS (Harvey-RAS), KRAS (Kirsten-RAS), and NRAS
(neuroblastoma-RAS). NRAS is the most prevalent form inmelanoma being constitutively activated by point mutation inabout 15% of cases [26]. Once activated, NRAS binds BRAF,which further phosphorylates and activates MEK. MEK finallysignals to ERK, which translocates into the nucleus and enhan-ces the transcription of a bunch of transcription factors resultingin increased cell proliferation and increased cell survival.
Activating mutations within MAPK pathway account for90% of melanomas [27]. Studies have reported a constitutiveactivation of MAPK pathway through mutation and/or geneamplification of c-KIT. C-KIT gene encodes a cell-membranetyrosine kinase receptor, which plays an essential role in cellu-lar differentiation [28]. The role of c-KIT in chronic myeloge-nous leukemia (CML) and gastrointestinal stromal tumors(GIST) has been well established and the use of imatinib, anoral c-KIT inhibitor, has revolutionized the treatment of thesediseases [29].
The use of c-KIT inhibitors in melanoma represents anevolving matter of investigation. C-KIT mutations accountfor about 2% of cutaneous melanoma, albeit mutation and/oramplification of c-KIT are found in about 20 -- 25% in somemelanoma subtypes, such as mucosal melanoma, acral mela-noma, or melanoma arising from chronically sun-damagedskin [30,31]. Consistent with the essential role of c-KIT in mela-nocytes differentiation and migration, several melanomasexpress c-KIT. However, experimental evidence from studiesin thyroid and melanoma showed that the loss of this growthfactor receptor is strangely related to a worse malignantphenotype [32-34]. This may be explained by the acquisition ofa less-differentiated phenotype during tumor progression.A number of drugs targeting c-KIT have been developed andtheir effectiveness has been proved in GISTs. These drugsinclude sunitinib, nilotinib, sorafenib, dasatinib, and imatinib,and their therapeutic activity has been shown to be dependenton the c-KIT specific type of molecular alteration [35-37].
Results of c-KIT inhibitor trials inmetastatic melanomawereinitially disappointing. Three Phase II trials did not show clin-ical activity of imatinib in unselected melanoma patients[38-40]. The scenario substantially changed when the studiesfocused on melanoma patients carrying c-KIT mutations and/or amplifications, therefore highlighting the importance ofpatient selection in early targeted-therapy development.According to two Phase II trials, indeed, an OR is achieved inalmost 25% of patients, and temporary stabilization occurs inabout 50 -- 70% of subjects [31,41]. Although results in termsof OS seem encouraging (median OS from 11 to 14 months),the median PFS appears to be low, ranging from 3 to3.5 months [31,41]. Importantly, responses seem to be restrictedto patients carrying hot spot mutations either in exon 11, 13, ormultiple mutations [31,41]. There is a great interest for c-KITinhibition in mucosal and acral melanoma and other drugs,such as sunitinib, nilotinib, and dasatinib, are currently investi-gated in Phase II trials [42]. In view of the growing enthusiasm inthis field, the Society for Immunotherapy of Cancer (SITC)melanoma expert panels recommend to routine testing of
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 3
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

c-KITmutations in clinical practice [43]. Whether acquisition ofresistances to imatinib in melanoma is due to the acquisition ofadditional c-KIT mutations, as demonstrated in GIST [44],remains to be elucidated [42].Similarly to c-KIT, another important RTK in melanoma
is c-MER tyrosine kinase (MERTK); c-MER is a member ofthe TAM (TYRO, AXL, MERTK) family of RTKs. A recentstudy from Schlegel and colleagues [45] has pointed c-MER asa potential therapeutic target in melanoma. In this work, theywere able to show that treatment of melanoma cell lines withUNC1062, a novel c-MER inhibitor, induced apoptosis andinhibited invasion of melanoma cells, encouraging a deepertesting of such drug.In addition to the deregulation of RTKs, several MAPK
downstream molecules are often altered in melanoma. Thefirst oncogene to be discovered was NRAS [46]. As mentionedearlier, NRAS is a member of the rat sarcoma (RAS) proteinfamily, together with HRAS and KRAS. RAS are GTP-binding proteins, which, once activated by RTKs, can signala complex network of downstream molecular pathways.Although evidence shows KRAS and HRAS to be implicatedin melanoma [47,48], NRAS is the most active form. Mostcommon mutations in NRAS gene have been found locatedat codon 61 [49], causing the impairment of NRAS enzymaticactivity to switch GTP to GDP. Albeit several attempts havebeen made to directly target NRAS, no clinical success hasbeen reached yet. One probable explanation is the low
concentration range (picomolar) required for the affinity ofGTP to bind NRAS, which made the development of GTP-specific antagonists impossible so far [50]. Nevertheless, signif-icant effort has been spent to indirectly inhibit NRAS by tar-geting key post-translational modifications, such asfarnesylation. Farnesylation consists in a lipid modificationnecessary for NRAS membrane association. The inhibitionof farnesylation process results in the blockage of NRAS trans-location to the plasma membrane. A study from Niessner andcolleagues [51] has shown that the farnesyltransferase inhibitor(FTI) lonafarnib was also able to inhibit mTOR signalingpathway enhancing apoptosis induced by sorafenib inmelanoma cell lines. However, testing FTI tipifarnib (alsocalled R115777) in a Phase II clinical trial failed to obtainencouraging results [52], most likely due to aspecific responsesas farnesylation occurs in the maturation process of severalproteins other than RAS. Although in this trial patients wereunselected for NRAS mutation, the intrinsic difficulty todevelop NRAS inhibitors and the scarce activity of thisapproach in other tumors have shifted the efforts to inhibitMAPK pathway toward more effective targets.
Another important protein kinase belonging to MAPKpathway is MEK, and its inhibition is currently under investi-gation. Considering that MEK activation has been shown tobe a resistance mechanism by which melanoma treated withBRAF inhibitors relapse, MEK inhibition was tested in com-bination with BRAF inhibitors (see combination therapy
c-KIT
NRAS
BRAF
MEK1/2
ERK1/2
PI3K PTEN
Cytosol
AKT
mTOR
p53
MDM2
PDK1
GSK3!
!-catenin
IKK NF"BBAD
BCL2
BAX
GPCR
GNAQ/GNA11 PKC
ELK MITF JUN
Proliferation, differentiation
PIP2
PIP3
Figure 1. Simplified representation of MAPK and PI3K pathways. Red bolts indicate targets of molecular alterationsin melanoma.AKT: V-Akt murine thymoma Viral Oncogene Homolog; BAD: BCL2-associated agonist of cell death; BAX: BCL2-associated X protein; BCL2: B cell lymphoma-2;
BRAF: V-raf murine sarcoma viral oncogene homolog B; c-KIT: V-Kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; ELK: ETS-domain protein; ERK1/
2: Extracellular signal--regulated kinase 1 and 2; GPCR: G protein--coupled receptor; GSK3b: Glycogen synthase kinase 3 b; IKK: IkB kinase; JUN: V-Jun avian sar-
coma virus 17 oncogene homolog; MDM2: Mouse double minute-2; MEK1/2: MAPK extracellular signal–regulated kinase 1 and 2; MITF: Microphthalmia transcrip-
tion factor; mTOR: Mammalian target of rapamycin; NFkB: nuclear factor-kB; NRAS: Neuroblastoma RAS; p53: Tumor protein 53; PDK1: Phosphoinositide-
dependent kinase-1; PI3K: Phosphoinositide-3 kinase; PIP: Phosphatidylinositol phosphate; PKC: Protein kinase C; PTEN: Phosphatase and tensin homolog.
S. Tomei et al.
4 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

section subsequently), yielding extremely promising results.MEK proteins include MEK1 and MEK2, which are theonly substrate of BRAF. Mutations on MEK genes are veryrare, although they have been reported in a few cases [49].However, the acquisition of MEK1 mutations P124L andQ56P have been demonstrated to confer resistance to BRAFand MEK inhibitors [53]. A recent review by Salama andKim [54] lists a broad variety of MEK inhibitors currentlyunder investigation. Worthy of note, selumetinib is an oralpotent inhibitor of MEK1 and MEK2 kinases. The efficacyof this drug was evaluated in a large Phase II clinical trial inadvanced melanoma patients. Unfortunately, the study failedto show a difference of the OR rate and PFS between thetreatment arm and the control group (temozolomide) [55].Interestingly, five out of six responders were BRAF mutated.Combination of selumetinib and dacarbazine was reportedto be clinically active in a Phase II randomized trial (PFS5.6 vs 3.0 months, selumetinb plus dacarbazine vs dacarbazinealone), although nonsignificant benefit in terms of OS wasobserved [56].
Several clinical trials are still testing MEK inhibitors inmelanoma, given the fact that MAPK reactivation is oftendriven by MEK signaling. Among the MEK inhibitors tested,trametinib has shown so far the most promising results. In aPhase III study, 322 patients carrying V600E or V600K muta-tion were randomized to receive either trametinib or chemo-therapy (either decarbazine or paclitaxel) [23]. A statisticalsignificant prolongation of PFS was shown for patients treatedwith trametinib compared to the control group (4.8 vs1.5 months). OR rate was also higher in trametinib arm thanin chemotherapy arm (22 vs 8%). Despite the crossover to tra-metinib at progression, the 6 months OS was significantlyhigher in the trametinib arm (81 vs 67%) [23]. On the basisof these results, FDA approved trametinib for the treatmentof metastatic melanoma harboring V600E or V600K muta-tions. Importantly, tremetinib is not active if administered topatients progressing on a BRAF inhibitor, stressing the needto better identify the window of efficacy of trametinib inBRAF mutant melanoma [57].
However, patients carrying NRAS mutations cannot bene-fit from such treatment, urging the need to find other addi-tional treatment options. Recently, Ascierto and colleagues[58] in an open-label, nonrandomized, Phase II trial assessedthe efficacy of MEK162, a potent MEK inhibitor, in patientscarrying NRAS or V600E BRAF mutations. In addition to anOR rate of 20% in BRAF-mutated patients, they were able toshow that MEK162 has activity in NRAS mutant patients(20% of OR rate), although no CR was observed. However,all patients relapsed within 9 months. A similar OR rate(18%) in patients bearing NRAS mutation has been observedwith the use of pimasertib [59], another MEK inhibitor underclinical development.
These proof-of-principle studies pinpoint MEK inhibitionas potential new approach for NRAS mutant melanoma,which till now has few treatment options. Once again, the
main limitation of MAPK-targeted therapy is the rapid devel-opment of resistance. However, while melanoma can take dif-ferent roads to counteract to MAPK-targeted inhibition, thereactivation of the extracellular-signal-regulated kinases(ERK) appears to be a common feature [60,61]. Two isoformsof ERK, ERK1 and ERK2, transmit proliferative signalsonce phosphorylated by the upstream MEK proteins. Quiteinterestingly, Morris and colleagues [62] recently identified apotent ATP-competitive ERK inhibitor by screening a libraryof 5 million compounds. Such inhibitor, called SCH772984,showed promising clinically attractive results as it was able toinhibit ERK phosphorylation in a dose-dependent manner.Testing this compound in vivo is warranted to prove itsefficacy and its potential relevance in clinical settings.
3.2 Phosphatidylinositol 3-kinase pathwayPhosphatidylinositol 3-kinase (PI3K) pathway is the secondmost frequently deregulated pathway in melanoma afterMAPK pathway (Figure 1). Similarly to MAPK, PI3K cascadeis triggered by RTKs and RAS protein. Once activated byNRAS, PI3K can catalyze the phosphorylation of phosphati-dylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol3,4,5-trisphosphate (PIP3). PIP3 functions as a second mes-senger, and its production is antagonized by the tumorsuppressor phosphatase and tensin homolog (PTEN).PIP3 can activate AKT (through the phosphatidylinositol-dependent kinase 1, PDK1), which, in turn, phosphorylatesmTOR and multiple additional targets determining increasedoncogenic transformation, survival, proliferation, and cell-cycle regulation [63]. The deregulation of PI3K pathway inmelanoma occurs through activation of PI3K (encoded byPI3KCA gene), PTEN loss, and AKT activation. The inde-pendent therapeutic relevance of inhibiting PI3K pathwayitself has not been well established yet, however, preclinicalevidence suggests that PI3K pathway inhibition may be arelevant adjunct to MAPK-targeted therapy. Several smallmolecule inhibitors targeting AKT and mTOR activity havebeen developed and tested in melanoma cell lines and xeno-grafted animal models [64]. However, the number of clinicalstudies currently evaluating their clinical efficacy is very low.The AKT inhibitors wortmannin and LY294002 have beenshown to block AKT activity in a variety of functional studies,but their application in clinical settings is limited by their off-target activities [63,65]. Several derivatives of rapamycin, suchas temsirolimus, have also been used as mTOR inhibitorsand have demonstrated clinical activity in other malignanciessuch as renal cell carcinoma. Nevertheless, the administrationof temsirolimus (either used alone or in combination with themultiple kinase inhibitor sorafenib) failed to show significantclinical activity in Phase II trials [66,67].
Even though several additional AKT inhibitors, such asAPI-2, SR13668, BI-69A11, GSK690693, and MEK-2206are under investigation, it is widely believed that PI3K path-way inhibition works better when combined to other targetedtherapies. An interesting study by Werzowa and colleagues
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 5
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

assessed the potential efficacy of PI3K pathway vertical inhibi-tion in melanoma cell lines and xenograft models [68]. Theconcurrent treatment of rapamycin and the mTOR inhibitorPI-103 had a potent antitumor activity both in vitro andin vivo. Although in general PI3K and mTOR inhibitionseems to be a promising strategy against cancer developmentand some drugs have been already approved for the treatmentof renal cancer [69], such drugs did not obtain the same successin melanoma. The reason behind this phenomenon is notclear yet albeit it has been hypothesized that the ability ofrapamycin derivatives to phosphorylate AKT may impedetheir clinical success [70]. Nevertheless, MEK and PI3Kcombination seems promising. An interesting in vitro studyby Greger et al. showed that combinations of BRAF, MEK,and PI3K/mTOR inhibitors overcome acquired resistance todabrafenib, mediated by NRAS or MEK mutations [71].Another study showed that combined MEK and PI3K/mTOR pathway inhibition induces regression of NRAS-mutant melanoma cell lines [72]. An increasing number ofclinical trials combining PI3K and MAPK inhibitions inmelanoma are currently undergoing or in development.As a note, inhibition of Bcl-2, a key anti-apoptotic protein
expressed in cancer cells, through the antisense oligonucleo-tide oblimersen seemed to increase OS in a subset of patients(i.e., those with normal or low level of LDH) [73], but thesefindings were not confirmed in a recent randomizedPhase III trial [74].In light of recent in vitro or in vivo studies, modulation cell-
cycle checkpoints (which are de-regulated in melanoma) andinhibition of G protein-coupled receptor signaling (found tobe activated in up to 80% of uveal melanoma) have recentlygained the attention of clinical investigators [42,72]. Remark-ably, two very recent investigations have found that > 70% ofmelanoma bears somatic mutation of telomerase reverse tran-scriptase (TERT) promoter, therefore highlighting anotherimportant oncogenic mechanism [75,76].
3.3 AngiogenesisBecause melanoma is a highly vascularized cancer, severalattempts to therapeutically target angiogenic mechanisms inmelanoma have been explored. Anti-angiogenic drugs havebeen developed and demonstrated to be useful in differenttypes of cancer. Still, their clinical benefit in melanoma isnot clear. Among the anti-angiogenic drugs, bevacizumab, ahumanized monoclonal antibody targeting the VEGF A(VEGF-A), has been approved for use in certain malignancies,such as colon (in combination with chemotherapy), kidney(in combination with IFN-a), lung (in combination withchemotherapy), and brain cancers.In metastatic melanoma, the use of bevacizumab as single
agent, or in combination with IFN-a failed to show signifi-cant clinical activity [77]. According to a large randomizedPhase II trial (BEAM) enrolling 314 treatment naıve patients,bevacizumab, in combination with chemotherapy enhances,though not significantly, PFS and OR (4.2 vs 5.6 month;
16 vs 26%; PFS and OR, bevacizumab vs bevacizumab pluscarboplatin and paclitaxel). OS advantage was initially signif-icant (8.6 vs 12.3 months; p = 0.04), but it was reduced in asubsequent analysis with longer follow-up, (9.2 vs12.3 months; p = 0.19) [78]. Although it is not clear if thiscombination will move forward, the BEAM study, which isoften considered a positive Phase II trial, still represents oneof the best evidence of the activity of anti-angiogenic therapyin advanced melanoma [79].
Interestingly, Del Vecchio and co-workers [80] evaluated theclinical and biological activity of the use of bevacizumab incombination with fotemustine, a chemotherapeutic drugwidely used in Europe. The authors were able to show thatsuch regimen had important clinical activity as first-line treat-ment of metastatic melanoma, obtaining an encouraging OSof 20.5 months (PFS = 8.8 months, OR 15%), althoughgrade 3 -- 4 hematological toxicity was quite high.
Attempts to use bevacizumab in clinical trials employingother combinatorial setting are currently underway and resultsare awaited. Recently, a preliminary analysis of 20 patientsenrolled in a Phase I study assessing combination of the cyto-toxic T-lymphocyte antigen 4 (CTLA-4) mAb ipilimumaband bevacizumab showed an intriguing OR of 38%; all theresponses lasted > 6 months [81].
Similarly to VEGF-A mAbs, other drugs targeting theVEGF receptors (VEGFRs) have been tested in melanoma.Among them, sorafenib, which inhibits tyrosine kinases asso-ciated with VEGF and plateled-derived growth factor, failedto demonstrate significant clinical activity in combinationwith chemotherapy in Phase III trials [82,83]. The more prom-ising ones as single agents are axitinib and lenvatinib and theirinvestigations alone or in combination are moving forward. Ina multicenter, Phase II study, axitinib monotherapy resultedin a 19% OR rate [84]. Similarly, in a Phase I evaluation ofpatients with solid malignancies, including melanoma, lenva-tinib (also called E7080) demonstrated an OR of 21% [85].
Based on this evidence, it is likely that anti-angiogenictherapies might be helpful in treating melanoma especiallywhen combined with other drugs and much effort should bespent in identifying the more beneficial anti-angiogenic com-bination therapies. Moreover, other novel molecules, as thosemimicking natural anti-angiogenic agent inhibitors (e.g.,endostatin), inhibitors of placental growth factor, and agentsblocking HIFa, a key molecule in hypoxia signaling, arecurrently under development [86].
4. Immunotherapy
Cancer immunotherapy is meant as the treatment aiming atannealing tumors by inducing, enhancing, or suppressing spe-cific immune responses. Aside from the molecular alterationswithin proliferative pathways described earlier, severalimmune alterations have been reported in melanoma, includ-ing decreased number of peripheral B and T cells, increasednumber of regulatory T cells, impaired activation of natural
S. Tomei et al.
6 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

killer (NK) cells (due to downregulation of activatingligands), acquisition of tolerance by CD8+ T cells (due toincreased expression of inhibitory receptors), increased levelof pro-tumorigenic cytokines such as TGF-b, TNF-a, IL-6,IL-8, and IL-1, and altered expression of immune-relatedgenes [87,88].
Immunotherapy strategies for melanoma treatment carry ahigh clinical relevance considering the exceptional immuno-genicity of such disease. The immunogenicity of melanomahas been established based on the high number oftumor-infiltrating lymphocytes (TIL) found in resected mela-noma samples, and several strategies have been proposed toconquer melanoma taking advantage of this peculiar feature.A diverse set of melanoma antigens have been identified astarget of cellular immune response, which can be groupedinto three broad categories: tumor differentiation antigens(TDA), including melanoma antigen recognized by T cells 1(MART-1), glycoprotein 100 (gp100), tyrosinase (TYR),tyrosinase-related protein 1 (TYRP1), and 2 (TYRP2), whichare found expressed in normal melanocytes and are overex-pressed in melanoma; tumor-specific antigen (TSA), includ-ing the melanoma antigen (MAGE), the B melanomaantigen (BAGE), and G antigen (GAGE) gene families, theNY-ESO-1 (also known as CTAG1), which are found in nor-mal gametic cells in the testes and in cancers in addition tomelanoma but are not expressed by normal melanocytes;and mutated antigens, including b-catenin, which can befound in different tissues. Additionally, several melanomaantigens have been identified as targets of humoral immuneresponses.
The expression of melanoma antigens is of great impor-tance as it allows the innate and adaptive immune systemto track and destroy nascent tumor cells, through a mech-anism known as ‘immune surveillance’. However, cancercells can escape immune surveillance by becoming poorlyimmunogenic. This phenomenon has been commonlyreferred to as ‘immune editing’ [89]. Several mechanismsof immune editing have been described, including thedownregulation of antigen presentation MHC moleculesby the tumor cells themselves, the secretion of soluble fac-tors and cytokines, such as TGF-b and IL-10, whichinhibit effective immune response, the creation of animmune suppressive environment by recruiting inhibitoryimmune cells, such as T-reg, NKs, and macrophages, andthe expression of negative regulators of the immune system,including programmed death ligand 1 (PD-L1) and theCTLA-4 [90].
We report subsequently the best-characterized immuno-therapy strategies for the treatment of melanoma.
4.1 Nonspecific immunotherapy: IL-2IL-2 is a pro-inflammatory cytokine regulating T cells andNK cells homeostasis. Studies in animal models and humanshave shown that IL-2 has strong antitumor activity [91,92]
and high-dose IL-2 (HD-IL-2) was approved by FDA in1998 for the treatment of metastatic melanoma.
HD-IL-2 was initially shown to have dose-dependent anti-cancer effect by the group of Steven Rosenberg at theNational Cancer Institute [93]. FDA approved HD-IL-2 in1998 based on the observation that it can induce long-termremissions. In large retrospective analyses, administration ofHD-IL-2 induce an OR in about 15% of patients (range13 -- 19%) of patients, and CR in 4 -- 6% [92,94-96]. However,in a recent randomized Phase III trial, these percentagesdropped from 10 to 6% for OR and from 2 (two patients)to 1% (one patient) for CR when evaluation was assessed bycentral review. Even though the real rate of OR needs to bedetermined, complete-remission under HD-IL-2 appear tobe extremely durable (median CR duration > 14 years,according to a long follow-up analysis of a retrospectivecasuistic) [95].
However, HD-IL-2-related adverse events are severe (e.g.,hemodynamic shock, respiratory insufficiency, and neurotox-icity) and require intensive inpatient care [21]. The opinions ofEuropean and US specialists about the risk--benefit ratio ofthis treatment largely diverge. In light of the potentiallycurative role of HD-IL-2, the US National ComprehensiveCancer Network US guidelines still list HD-IL-2 as treatmentoption in a subset of (clinically fit) patients, if the center has aconsiderable experience in the management of this regimen.Even more, the (US) melanoma panelists of the Society forImmunotherapy of Cancer (SITC) recommend the adminis-tration of HD-IL-2 as first-line treatment in good perfor-mance status patients without central nervous systemmetastases, even in presence of BRAF mutation [43]. Con-versely, the European Society of Medical Oncology (ESMO)melanoma panelists do not encourage its routine use inclinical practice, in view of the high toxicity and the lack ofrandomized trials demonstrating survival advantage in theoverall population [97].
Considering the relatively low benefit--risk ratio, one of themain limitations of HD-IL-2 is the lack of pretreatment toolsto predict patients who are likely to respond to the treatment.Recently, in a proteomic study, we showed that low levels offibronectin and VEGF, an angiogenetic factor with immuno-suppressive functions, predict response to HD-IL-2 [98] inmetastatic melanoma patients. Although these findings needto be prospectively validated, Hodi and co-workers recentlyreported that low levels of VEGF also predict OS and clinicalresponse to ipilimumab in metastatic melanoma [99] Notewor-thy, an interesting study from Joseph and colleagues hasrecently shown that patients harboring NRAS mutation carrya higher likelihood to respond to therapy [96], linking, for thefirst time, alterations of the MAPK pathway to the responsive-ness to HD-IL-2.
Although IL-2 has been used in oncology since > 20 years, itsmechanism of action is still not completely understood. Onlyfew studies have attempted to comprehensively understandthe mechanisms behind the cancer rejection mediated by IL-2.
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 7
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

Our group performed a series of proteomic and transcrip-tomic analyses of peripheral mononuclear cells (PBMCs)and pre- and post-melanoma metastasis using fine-needleaspiration biopsies (FNA) obtained from patients undergoingsystemic IL-2 administration [98,100-103]. We observed thatadministration of IL-2 induces a cytokine storm already afterthe first administration, where most of the cytokines investi-gated (e.g., IFN-g , IL-10, and TNF-a) increased significantlycompared with the baseline value. CXCR3 ligands (i.e.,CXCL9, 10 and 11) and CCR5 ligands (i.e., CCL3 andCCL4) were found among the chemokines that significantlyincreased early after treatment [100,101].Even when investigated by whole genome gene-expression
analysis, the effects of HD-IL-2 were dramatic withinPBMCs, whereas effects within the tumor microenvironmentwere lesion-specific and limited [103]. Altogether these findingssuggest that the main effect of IL-2 in the tumor is the activa-tion of innate mechanisms driven by monocytes and eventu-ally sustained by NK, and the tumor infiltrating lymphocytes.These cells in concert can initiate an inflammatory chemo-
tactic cascade, orchestrated by the activation of the IRF-1/IFN-g pathway and inducing release of related specificchemotactic ligands (i.e., CXCR3 and CCR5 ligands) withconsequent polarized (Th1) recruitment of other immunecells later on [104]. Importantly, the degree of inflammationinduced by HD-IL-2 correlates with the development oftumor rejection [102,103].
4.2 Vaccination strategiesCancer vaccines aim at treating cancer by stimulating thehost’s immune reaction against cancer cells. The implementa-tion of therapeutic vaccines to treat melanoma remains achallenge [105-107].Broadly, melanoma vaccines are classified into the follow-
ing main categories: whole-cell vaccines (either autologousor allogeneic, prepared using melanoma cells or subcellularfraction); dendritic-cell (DC) vaccines (which take advantageof the highly specialization of DCs to present antigens andinduce primary cellular immune responses); peptide vaccines(which use melanoma antigens to stimulate cytotoxicT lymphocytes response against tumor cells); ganglioside vac-cines (which take advantage of the high expression of ganglio-sides on the surface of melanoma cells to generate an immuneresponse against them); DNA vaccines (composed of nakedDNA expression plasmids that include a gene encoding thetarget antigens from tumor cells); viral vector vaccines (inwhich a virus is used as vector of melanoma antigens); andoncolytic vaccine (in which an oncolytic virus replicating incancer cells is used to mediate tumor cell lysis) [107-110].The best-characterized vaccines for melanoma treatment
are the peptide-based ones directed to activate T-cell response[111]. The recognition of the critical role of T cells in theimmune-mediated treatment of melanoma dates back to1988, when Rosenberg and colleagues first showed the regres-sion of metastatic melanoma through the autologous transfer
of tumor infiltrating lymphocytes combined with IL-2 [112].In 1991, van der Bruggen et al. described for the first time agene encoding an antigen recognized by T cells (MAGE-1)[113]. This study, followed by the description of the firstcancer-specific epitope HLA-A1 restricted [114], gave molecu-lar accuracy to this rather disregarded field and providedthe opportunity to investigate with scientific precision thephenomenon of immune-mediated cancer rejection. Thisled, in turn, to the characterization of a myriad of tumor anti-gens and their immunodominant peptides, with consequentimplementation of active immunization strategies [115].
The study of the tumor-antigen-specific immunizationrestricted to an individual epitopic determinant offered theopportunity to study the dynamic of the immune responsein humans by reducing the algorithm of tumor--host cross-talk to a specific HLA/epitope interaction with the comple-mentary T-cell receptor [116]. These studies have shown that,in order to develop clinical active immunotherapies, factorsbeyond T-cell/HLA/epitope interactions must be considered.In fact, the large majority of the studies failed to report anassociation between specific response against the administeredantigens and clinical benefit [104]. Humoral and/or cellularimmune response against vaccine antigens are necessary forthe development of an effective antitumor response but theyare certainly not sufficient [117]. Other factors, as the presenceof co-stimulatory or inhibitory stimuli in tumor microenvi-ronment, genetic polymorphisms of the host, genetic of thetumors, and environmental factors (i.e., intestinal micro-biota) [118], can certainly modify the algorithm of immuneresponse, making the evaluation of a single parameter unliketo predict the clinical benefit [119-121].
Besides local toxicity, vaccines are in general extremely welltolerated. However, in melanoma, as well as in many othersolid tumors, vaccination alone has failed to induce significanttumor regression, the OR rate being around 3 -- 4% [122].Nevertheless, recent randomized Phase III trials in othermalignancies (i.e., prostate cancer) have shown that vaccineadministration can prolong survival even in absence of detect-able tumor shrinkage [123]. The convincing detection of aremarkable survival benefit dissociated with measurablechanges in the tumor size forced us to reconsider the mecha-nism of actions of vaccinations. An interesting interpretationof these paradigmatic results comes from the analysis of tumorkinetic following different treatments. By applying a rathernovel mathematical model to Phase II trials in prostate cancerpatients receiving chemotherapy or PROSTVAC vaccine(consisting in recombinant poxviral vectors containing trans-genes for prostate specific antigen (PSA) and the costimula-tory molecules B7), Fojo and Schlom showed thatchemotherapy determines an initial reduction of tumor bur-den followed, at relapse, by a tumor-growth rate similar tothat before chemotherapy [124,125]. Vaccination, however,modifies clinical outcome (OS) by reducing tumor-growthrate. The same mechanisms likely explain differences betweensurvival curves in BRAF and ipilimumab trials (Figure 2).
S. Tomei et al.
8 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.
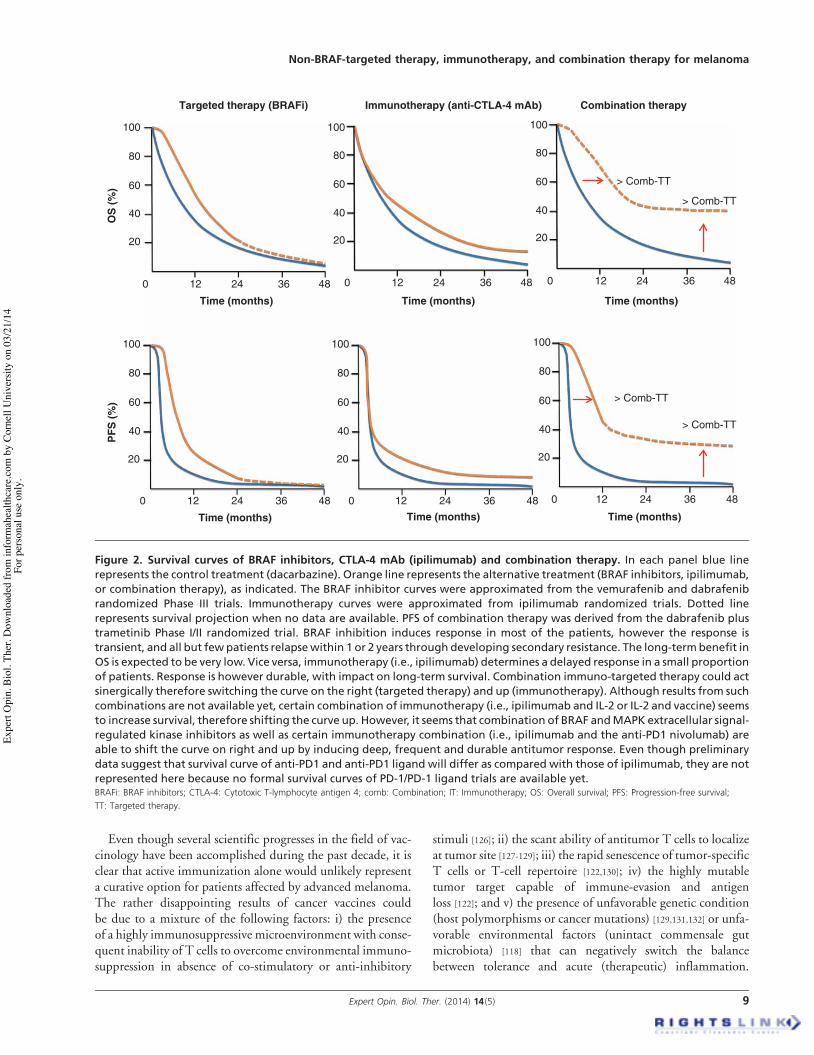
Even though several scientific progresses in the field of vac-cinology have been accomplished during the past decade, it isclear that active immunization alone would unlikely representa curative option for patients affected by advanced melanoma.The rather disappointing results of cancer vaccines couldbe due to a mixture of the following factors: i) the presenceof a highly immunosuppressive microenvironment with conse-quent inability of T cells to overcome environmental immuno-suppression in absence of co-stimulatory or anti-inhibitory
stimuli [126]; ii) the scant ability of antitumor T cells to localizeat tumor site [127-129]; iii) the rapid senescence of tumor-specificT cells or T-cell repertoire [122,130]; iv) the highly mutabletumor target capable of immune-evasion and antigenloss [122]; and v) the presence of unfavorable genetic condition(host polymorphisms or cancer mutations) [129,131,132] or unfa-vorable environmental factors (unintact commensale gutmicrobiota) [118] that can negatively switch the balancebetween tolerance and acute (therapeutic) inflammation.
OS
(%)
PFS
(%)
Time (months) Time (months) Time (months)
Time (months)Time (months)Time (months)
> Comb-TT
> Comb-TT
> Comb-TT
> Comb-TT
Targeted therapy (BRAFi)
100
80
60
40
20
100
80
60
40
20
100
80
60
40
20
0 12 24 36 48 0 12 24 36 48 120 24 36 48
Immunotherapy (anti-CTLA-4 mAb) Combination therapy
100
80
60
40
20
100
80
60
40
20
100
80
60
40
20
0 12 24 36 48 0 12 24 36 48 0 12 24 36 48
Figure 2. Survival curves of BRAF inhibitors, CTLA-4 mAb (ipilimumab) and combination therapy. In each panel blue linerepresents the control treatment (dacarbazine). Orange line represents the alternative treatment (BRAF inhibitors, ipilimumab,or combination therapy), as indicated. The BRAF inhibitor curves were approximated from the vemurafenib and dabrafenibrandomized Phase III trials. Immunotherapy curves were approximated from ipilimumab randomized trials. Dotted linerepresents survival projection when no data are available. PFS of combination therapy was derived from the dabrafenib plustrametinib Phase I/II randomized trial. BRAF inhibition induces response in most of the patients, however the response istransient, and all but few patients relapsewithin 1 or 2 years through developing secondary resistance. The long-termbenefit inOS is expected to be very low. Vice versa, immunotherapy (i.e., ipilimumab) determines a delayed response in a small proportionof patients. Response is however durable, with impact on long-term survival. Combination immuno-targeted therapy could actsinergically therefore switching the curve on the right (targeted therapy) and up (immunotherapy). Although results from suchcombinations are not available yet, certain combination of immunotherapy (i.e., ipilimumab and IL-2 or IL-2 and vaccine) seemsto increase survival, therefore shifting the curve up. However, it seems that combination of BRAF andMAPK extracellular signal-regulated kinase inhibitors as well as certain immunotherapy combination (i.e., ipilimumab and the anti-PD1 nivolumab) areable to shift the curve on right and up by inducing deep, frequent and durable antitumor response. Even though preliminarydata suggest that survival curve of anti-PD1 and anti-PD1 ligand will differ as compared with those of ipilimumab, they are notrepresented here because no formal survival curves of PD-1/PD-1 ligand trials are available yet.BRAFi: BRAF inhibitors; CTLA-4: Cytotoxic T-lymphocyte antigen 4; comb: Combination; IT: Immunotherapy; OS: Overall survival; PFS: Progression-free survival;
TT: Targeted therapy.
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 9
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

Therefore, novel generation of vaccines [133] could be effectivewhen combined with other therapies (e.g., inflammatory cyto-kines, immune-checkpoint inhibitors, chemotherapy, or radia-tion therapy, see combination therapy section), with the goalto overcome the barriers listed earlier. Vaccination could beeven more successful when used in patients with low tumorburden (i.e., adjuvant setting), or in selected patients particu-larly sensitive to immune-manipulation (i.e., those bearingspecific polymorphisms).Only very few vaccination strategies have shown promising
results in Phase II trials and are currently being tested inPhase III randomized trials (in addition to gp100 vaccineplus IL-2 [134], see combination therapy). In a Phase II trialon stage III/IV M1a melanoma, MAGE-A3 vaccine com-bined with AS15 immunostimulant (a mixture ofQS21 saponin, TLR4, and TLR9 agonist) induced an ORin 11% of patients (including 2% of CR) and a median OSof 33 months [135]. The advantage of vaccination was evenhigher in patients bearing an inflammatory gene-signaturecentered on IRF-1/IFN-g-related transcripts [117,136]. Thisadjuvant--vaccine combination was selected for the Phase IIItrial. Unfortunately, it has been recently announced that thestudy did not meet its first co-primary end point as it didnot significantly extend disease-free survival (DFS) whencompared to placebo [137]. The result of the other co-primaryend point (DFS in patients bearing the inflammatory genesignature identified in Phase II trial) is expected in 2015. Inthe OPTIM Phase III trials by Kaufman et al. T-VEC, anoncolytic immunotherapy derived from herpes simplex virustype-1 designed to selectively replicate within tumors and toproduce GM-CSF to enhance systemic antitumor immuneresponses, induced OR in 26% of patients, including 11%of CR. According to the interim analysis, there was also atrend in OS favoring vaccination (HR: 0.79; vaccination vsGM-CSF alone) [138,139]. Allovectin-7 (a plasmid--lipid com-plex containing the DNA sequences encoding HLA-B7 andb2 microglobulin) in combination with dacarbazine [140],and M-VAX (an autologous hapten-modified vaccine), aloneor in combination with IL-2 (NCT00477906), represent theother two vaccination strategies that are currently being eval-uated in Phase III trials [141]. Notably, there is an increasinginterest for testing nanoparticle as vaccine adjuvant [142].Functionalizing carbon nanotubes are particularly promisingin view of their theranostic (i.e., both diagnostic and thera-peutic) potential. In fact, functionalized carbon nanotubescan stimulate monocytes to release pro-inflammatory cyto-kines and chemokines associated with T helper 1 polarizationand immune-mediated tumor rejection [143]. Moreover,because of their high echogenicity these nanotubes could bepotentially used as ultrasound contrast agents [144].An emerging and promising approach to generate tumor-
specific T cells is represented by chimeric antigen receptor(CAR) or T-cell receptor (TCR) engineered T cells. Studiesin B-cell hematological malignancies have shown that engi-neered T cells against CD19+ can mediate deep tumor
remission [145]. In melanoma, the administration of modifiedT cells expressing high avidity anti-gp100 or anti-MART-1TCR had induced a considerable OR rate (19 -- 30%), withfew complete remissions (one patient in the MART-1 trial)[146]. However, patients exhibited destruction of melanocytesin skin, eye, and ear, which also express, although at lowerlevel, these antigens. Similarly, administration of engineeredT cells against MAGE-A3 is associated with high incidenceof neurotoxicity because of cross reactivity with MAGE pro-teins expressed by brain [146]. Therefore, it is imperative toselect antigens that are not expressed by normal cells and togenerate T cells that do not cross-react with epitope withinself-antigens [146]. The most promising results have beenobtained by targeting NY-ESO (OR in 5/10 melanomapatients, including 2 CR) [146].
4.3 Immune checkpoint blockadeAn extremely promising approach used in clinical settings tocounteract the immune escape mechanisms mounted bycancer cells is the inhibition of the CTLA-4 (Figure 3) [147].CTLA-4 is a key player in establishing immune toleranceand one of the main regulators of T-cell-mediated antitumorimmune responses. The major function of CTLA-4 is tomodulate T cells at the time of their initial response to anti-gen [148]. In fact, although CTLA-4 is expressed by activatedCD8+ effector T cells, its key role relies in the regulation ofT CD4+ populations through down modulation of helperT-cell activity and enhancement of regulatory T-cell immu-nosuppressive function [147,148].
The possible role of CTLA-4 blockage in clinical settingwas proposed in 1995 by Allison and Krummel [149,150]. It isnow clear that antibodies binding the extracellular domainof CTLA-4 can, therefore, block its inhibitory signals andenhance the immune system to fight against cancer cells.Two anti-CTLA-4 monoclonal antibodies, that is, ipilimu-mab and tremelimumab, are currently being applied in meta-static melanoma setting. The former has been approved byFDA in March 2011.
Ipilimumab is a fully human IgG1 monoclonal antibody.Results of ipilimumab trials have boldly increased the enthu-siasm in the field of cancer immunotherapy and deservedetailed description. Two Phase III trials have conclusivelydemonstrated its efficacy in previously treated (ipilimumabmonotherapy, MDX010-020 trial) [17], or naive (ipilimu-mab+dacrabazine, CA184-024 trial) [22] advanced melanomapatients. In the MDX010-20, ipilimumab showed to increaseOS, as compared with gp100 vaccine alone (median OS:10.1 vs 6.4 months, ipilimumab vs gp100 vaccine, p =0.003) [17]. The differences in terms of OS increased withthe time. The 1- and 2-year OS were 46 and 24% in ipilimu-mab arm, and 25 and 14% in the vaccine arm, respectively [17].In the CA184-024 study median OS was 11.1 months in thedacarbazine-ipilimumab arm, and 9.1 months in dacarbazinealone arm [22]. A recent follow-up analysis of this latter trialhighlights the progressive benefit in OS associated with
S. Tomei et al.
10 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

ipilimumab. The 1-, 2-, 3- and 4-year OS were, respectively,48, 29, 21, and 19% in the ipilimumab-dacrabazine arm and36, 18, 12 and 10% in the dacarbazine arm [151]. Therefore,ipilimumab almost doubles the 4-year survival rate. Althoughresponses were few in number, they were more likely to occurin patients treated with ipilimumab. As for the MDX010-20-the OR rate was 11% (including 1.5% of CR) in the ipilimu-mab arm, and 1.5% in the gp100 arm. No CRs were observedin the gp100 alone arm [17]. As for CA184-024, the OR ratewas 15% (including 1.5% of CR) in the ipilimumab-dacarbazine arm and 10% (including 1% of CR) in the dacar-bazine alone arm [22]. Even though 15% of OR rate is notparticularly high, the responses were more durable in patientstreated with ipilimumab (OR duration: 19 months vs8 months, dacarbazine plus ipilimumab vs dacarbazine) [22].Similar (although less pronounced) differences were observedin term of PFS in the two studies. The continuous follow-upof patients enrolled in three Phase II trials also confirm thelong-term benefit of ipilimumab, with a 5-year OS rangingfrom 12 to 28% and 38 to 50% for previously treated andnaıve patients [152,153]. These results should, however, be takenwith caution, being derived from early Phase trials. However,the US ipilimumab expanded access program, which prospec-tively surveyed the activity and safety of ipilimumabin > 800 (pre-treated) patients, showed a 2- and 3-year OSrate of 22 and 17%, respectively [154]. Remarkably, theseresults are very close to those obtained in Phase III trials.According to Phase II trials, the combinations of ipilimumabplus fotemustine (NIBIT-M1 trial) [155], and ipilimumab plustemozolomide [156] seem also to be promising. To evaluate theresponse to treatment, the NIBIT-M1 trial was the first toemploy the immune-related criteria, instead than the classicWHO or RECIST criteria for solid tumors. In fact, the onsetof clinical response is often delayed, leading to problems inapplying classic criteria of response. Response can be achievedafter stabilization or even after progression, and CR can occuryears after an initial remission. These observations led to definenew criteria (immune-related response criteria; irRC) forthe definition of response in immunotherapeutic trials [157].
Tremelimumab is a fully human IgG2monoclonal antibody.The G2 isotype was chosen to diminish complement activationand cytokine storm. A recent Phase III trials failed to show a sig-nificant advantage in terms of OS when compared standardchemotherapy (dacarbazine or temozolamide) in treatmentnaıve advanced melanoma patients (median OS: 12.6 vs10.7 months, p = 0.13) [158]. Response rate was also similar inthe two arms (11 vs 10%, tremelimumab vs chemotherapy,respectively) although duration was significantly longer aftertremelimumab (35.8 vs 13.7 months, p = 0.001). However,tremelimumab survival curves are similar to those obtained inthe Phase III ipilimumab trials in the same patient setting.The 2- and 3-year OS were 26 and 21% in patients treatedwith tremelimumab and 23 and 17% in patients treated withchemotherapy. The failure to detect significant differencescould be due, at least in part, to the enrichment in patients
more responsive to chemotherapy or to study contaminations.Moreover, although the trial did not allow crossover, a propor-tion of patients randomized to receive chemotherapy likelyreceived ipilimumab at progression. During trial conduct, infact, ipilimumab became widely available for patients random-ized in the control arm [158]. Dosage and schedules could alsohave influenced the results. Nevertheless, promising results areobtained in Phase I/II trial, in pretreated melanoma [159,160]
and mesothelioma patients [161]. Overall, we believe there is stillroom for this drug in metastatic melanoma setting. One of themain limitations of the CTLA-4 blockades is that immuneresponse takes time to be effective and about 70% of patientsprogress within the first 6 months [17,22]. Common immune-related adverse event include colitis, hepatitis, and endochrino-paties, and can be life threatening in a small but considerableproportion of patients. In view of the toxic profile, anotherlimitation of this approach is that so far there are no toolsallowing to define with confidence the patients who will benefitfrom this approach.
Another critical immune checkpoint is represented byPD-1. Four anti-PD1 and 3 anti-PD1 ligands mAbs arecurrently in clinical development (Figure 3, Table 1).
PD-1 is induced once T cells become activated. Themajor function of PD1 is to repress the activity of effectorT cells in peripheral tissues to limit autoimmunity or collat-eral damages in response to infections [148]. PD1 has twoknown ligands, PD-L1 (B7-H1) and PDL-2 (B7-DC)[162,163], which are expressed by tumor cells to avoid T-cellmediated lysis.
Results of large Phase I/II trials of two anti-PD1 mAbs(nivolumab/BMS-936558 and lambrolizumab/MK-3475) andanti-PD-1 ligand mAbs (BMS-936559 and MPDL3280A)are extremely encouraging. If paradigmatic pattern of responseof CTLA-4 (delayed and slow effect on tumor shrinkageand prolonged OS) in a small proportion of patients haveforced us to define (or redefine) mechanism of action ofimmunotherapy, recent results of Phase I/II anti-PD-1 andanti-PD-1 ligand trials have shown once again that pattern ofresponse among different immune manipulations could beextremely different. Surprisingly, anti-PD-1 or anti-PD-1mAb induce tumor shrinkage in most patients with advancedmelanoma, lung, and kidney cancers [164-166]. OR rate inmelanoma cohorts were 31, 38, 17, and 29% for nivolumab[165,167], lambrolizumab [166], BMS-936559 [164], andMPDL3280A [168], respectively. Tumor shrinkage was alreadyevident at the time of the first assessment (at 6 or 12 weeks),and was extremely durable (1 year or beyond in most cases).Importantly, toxicity (most immunomediated) was muchlower as compared with that observed in ipilimumab trials.Although the magnitude of the clinical benefit needs to beassessed through (undergoing) randomized trials and longerfollow-up, the pattern of response and the (favorable) safetyprofile of anti-PD1/PD-1 ligands appear to be quite unique.Importantly, PD-1/PD-1 blockage was effective in patientsalready previously treated with ipilimumab, therefore,
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 11
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

confirming that CTLA-4 and PD-1 immune-suppressive path-ways are not redundant.Besides the co-inhibitory interactions described earlier, co-
stimulatory signaling between antigen presenting cells (APC)and T cells could also be targeted to enhance antitumorimmunity. A schematic representation of the clinical develop-ment of immune-checkpoint inhibitors and co-stimulatorymolecules in melanoma is provided in Figure 3. The statusof the corresponding clinical trials (updated as of January2014) is provided in Table 1.Agonist antibodies aimed at enhancing T and NK func-
tions (e.g., CD40, CD137 (4-1BB), anti-CD134 (OX40),and glucocorticoid-induced TNF receptor (GITR)) are beingevaluated in early phase trials [169]. Perhaps, the most clini-cally relevant examples in this regard come from the
interactions between OX40 and OX40L and betweenCD40 and CD40L.
OX40 (also known as CD134) is a TNF-receptor familymember, which is transiently expressed by T cells upon thebinding to the TCR. The engagement of OX40 promotesa vast spectrum of T-cell responses, including increasedproliferation, cytokine production, and increased differentiationand survival. The interaction between OX40 and its ligand,OX40L, occurs in 48 h after antigen recognition by T cells[170]. Targeting of OX40/OX40L has been proposed as a rele-vant approach for autoimmunity and cancer [171]. Preclinicalstudies have shown that ligation of OX40 via agonist anti-OX40 mAB or OX40L-Ig fusion proteins can induce robustT-cell-mediated antitumor immunity [172,173]. In a recentPhase I trial [174], Curti and colleagues showed that the
IpilimumabTremelimumab
T cell
OX40 Anti-OX40
CTLA-4CD80/CD86
Dendritic cell
Tumor
OX40L
CD40L
Clinical development status
Stimulatory signalInhibitory signal
FDA approvedPhase III trialsPhase II trialsPhase I trials
R Recruiting trials
Macrophage
MHC-II LAG-3BMS-986016IMP-321
GITRL GITR
?B7-H3
PD-L1/PD-L2 PD-1NivolumabLambrolizumabMPDL3280ACT-011
CD137L CD137 Urelumab
TRX518
MGA271
R RRR R
R
BMS-936559AMP-224MEDI4736RR
RR R
R
R
R
R RRR
CP-870,893DacetuzumabChi Lob 7/4Lucatumumab
CD40
Figure 3. Immune-checkpoints, co-stimulatory signaling, and clinical development of immune-targeted therapy. Stimulatorysignaling is in green, inhibitory signaling is in red. The clinical development status is represented by colored rectangles andrefers to trials that have recruited (or are currently recruiting) patients with advanced or metastatic melanoma. The symbol ‘R’highlights trials currently recruiting melanoma patients. Name of the drug is next to the corresponding targeted molecule(either receptor or ligand). Status of clinical trials is update as of January 2014 (source: clinicaltrials.gov; more detailsin Table 1). Represented expression of ligands and receptors should not be considered exhaustive: for example, PD1L-PD2L arealso expressed by antigen presenting cells as macrophages and dendritic cells (not shown in the figure); MHC-II is alsoexpressed by dendritic cells and can be expressed by melanoma cells as well.CTLA-4: Cytotoxic T-lymphocyte antigen 4.
S. Tomei et al.
12 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

Table 1. Agents targeting immune-checkpoints and co-stimulatory signaling*.
Agent Clinical trials
BMS-986016 (anti-LAG3) Phase I trial in solid tumors as monotherapy and in combination with nivolumabrecruiting (NCT01968109)
IMP321 (anti-LAG3) Phase I/II trial in combination with vaccine in melanoma recruiting (NCT01308294)IPILIMUMAB (anti-CTLA-4) FDA approved for melanoma; two positive Phase III trials (published) in previously
treated melanoma patients as monotherapy versus combination with gp100 vaccineversus gp100 vaccine (NCT00094653) and in naıve patients in combination withdacarbazine versus dacarbazine completed (NCT00324155); Phase III trials in melanomaas monotherapy versus combination with nivolumab versus nivolumab (NCT01844505)or versus lambrolizumab (NCT01866319) recruiting; Phase III trials in small-cell lungcancer (NCT0145076) and non-small-cell lung cancer (NCT01285609) in combinationwith chemotherapy versus chemotherapy alone recruiting; enrollment completed in aPhase III trial in prostate cancer versus placebo (NCT01057810); > 60 Phase I/II trialsevaluating combinations with targeted therapy, radiotherapy, or variousimmunotherapies in melanoma and in other cancers are currently recruiting
TREMELIMUMAB (anti-CTLA-4) Negative Phase III trial in melanoma versus chemotherapy published (NCT00257205);Phase I trial in melanoma in combination with the anti-CD40 mAbCP-870,893 (NCT01103635) recruiting; Phase II trial in mesothelioma recruiting(NCT01843374); Phase I trial in non-small-cell lung cancer in combination with gefitinib(NCT02040064) or the anti-PD-L1 mAb MEDI4736 (NCT02000947) and in solid tumorsin combination with MEDI4736 (NCT01975831) recruiting; Phase I trial in combinationwith chemoembolization or ablation in liver cancer recruiting (NCT01853618)
Anti-OX40 (OX40 agonist) Encouraging results from a Phase I trial in multiple cancers recently published(NCT01644968); Phase II trial in melanoma as monotherapy (NCT01416844) withdrawnprior to enrollment; Phase I/II in combination with ipilimumab (NCT01689870) recentlysuspended; Phase I/II trials in breast cancer patients with lung metastases incombination with radiotherapy (NCT01862900) and in prostate cancer in combinationwith radiotherapy and cyclophosphamide recruiting (NCT01303705)
CP-870,893 (CD40 agonist) Phase I trial in melanoma in combination with tremelimumab, recruiting(NCT01103635); Phase I trial in melanoma in combination with vaccination ongoing butnot recruiting (NCT01008527); Phase I trial in pancreatic cancer in combination withchemotherapy recruiting (NCT01456585); two Phase I trials in solid tumors(NCT00607048) and in pancreatic cancer (NCT00711191) in combination withchemotherapy completed and listed as ‘having results’
Dacetuzumab (SGN-40 or huS2C6;anti-CD-40)
Phase I/II trial completed in leukemia patients (NCT00283101); 6 Phase I and 2 Phase IItrials completed in multiple myeloma and non-Hodgkin’s lymphoma
Chi Lob 7/4 (anti-CD40) A Phase I trial in multiple cancers ongoing but not recruiting (NCT01561911)Lucatumumab (HCD122;anti-CD40) Phase I trial for follicular lymphoma patients in combination with bendamustine
completed (NCT01275209)Urelumab (BMS-663513; anti-CD137) Phase II trial in melanoma completed (NCT00612664), Phase I trial in melanoma in
combination with ipilimumab withdrawn prior to enrollment (NCT00803374); Phase Itrials in non-Hodgkin’s lymphoma recruiting (NCT01471210; NCT01775631); Phase Itrials in advanced tumors (NCT00351325, NCT00309023) and non-small-cell lungcancer (NCT00461110) terminated
TRX518 (anti-GITR) Phase I trial in melanoma and other solid tumors recruiting (NCT01239134)MGA271 (anti-BT-H3) Phase I trials in melanoma (NCT01918930) and in melanoma and other solid tumors
(NCT01391143) recruitingNivolumab (BMS-936558/MDX-1106;Anti-PD1)
Positive results of a Phase I trials in patients with melanoma and other solid tumors(NCT00730639) or in melanoma in combination with ipilimumab (NCT01024231)published; Phase I trials in melanoma in combination with vaccination recruiting(NCT01176474, NCT01176461); Phase III trial in melanoma as monotherapy versusdacarbazine (NCT01721772) or versus combination with ipilimumab versus ipilimumab(NCT01844505) recruiting; Phase II trials in melanoma as monotherapy versuscombination with ipilimumab versus ipilimumab (NCT01927419) or given sequentiallywith ipilimumab recruiting (NCT01783938); Phase I trial in solid tumors in combinationwith anti-KIR (NCT01714739), IL-21 (NCT01629758), and the anti-LAG1 mAbBMS-986016 (NCT01968109) recruiting; Phase I trials assessing combination withipilimumab in melanoma (NCT01024231) and other combinations in other solid orliquid tumors recruiting
*Updated as of January 2014 (source: clinicaltrial.gov).
CTLA-4: Cytotoxic T-lymphocyte antigen 4; LAG3: Lymphocyte activation genes 3; PD-L1: Programmed death ligand 1.
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 13
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

administration of an anti-OX40 mouse mAB in patients withadvanced cancer had an acceptable toxicity profile and wasable to induce regression of at least one metastatic lesion in12 out of 30 patients. The therapy also increased CD8 T-cellIFN-g production in two of three patients with melanoma.Moreover, in another recent study, Redmond and col-leagues [175] demonstrated that the targeting of OX40 in combi-nation with IL2 therapy increased tumor regression inpreclinical tumor model and restored the function of anergictumor-reactive CD8 T cells in mice, leading to enhancedsurvival.CD40 is a cell-surface molecule, member of the TNF-
receptor family, expressed by APC, such as dendritic cells,B cells, and monocytes, but also by many nonimmune cellsand a wide range of tumors [169]. The natural ligand ofCD40 is CD145, which is expressed on T cells. It hasbeen shown that in tumor-bearing hosts, CD40 agonist trig-ger effective immune response against tumor-associated anti-gens [176-178]. Four CD40 agonist mABs have beeninvestigated in clinical trials so far, namely: CP-870,893,dacetuzumab, Chi Lob 7/4, and lucatumumab [169,179].However, these treatments showed wide heterogeneity rang-ing from strong agonism (CP-870,893) to antagonism (luca-tumumab), suggesting that their mechanism of action mightbe different. A study employing CP-870,893 as CD40 ago-nist demonstrated that a single intravenous infusion ofCP-870,893 was well tolerated and an objective PR wasobserved in four patients with metastatic melanoma (14%
of all patients and 27% of melanoma patients) [180]. Addi-tional clinical trials employing CD40 agonists are ongoingand results are strongly awaited.
Blockage of other inhibitory pathways as the immunosup-pressive enzyme indoleamine 2,3-dioxygenas (IDO) seemspromising in preclinical study [181], and clinical studies areongoing (NCT01961115; NCT00739609). Drugs blockingother immune-checkpoints such as T-cell membrane protein3 (TIM3; inhibitor receptor) and B7-H4 (inhibitory ligand),TGF-b, and KIR inhibitory receptors are under develop-ment [148,169]. Notably, trials evaluating blockage of the inhib-itory receptor lymphocyte activation genes 3 (LAG3), theinhibitory ligand B7-H3 are ongoing [148,182,183] .
Finally, the recent discovery that themanipulation ofBACH2gene can shift the balance between effector and regulatory T-cellresponse also represents a novel starting point for the develop-ment of next-generation immunotherapeutic approaches [184].
4.4 Adoptive cell therapyAdoptive cell therapy (ACT) transfer is a form of passiveimmunotherapy, which consists in the ex vivo expansion ofautologous antitumor reactive lymphocytes and their reinfu-sion into lymphodepleted patients, accompanied by the exog-enous administration of IL-2 to further boost their antitumoractivity. Lymphodepletion, in fact, seems to favor the antitu-mor activity of ACT by suppressing regulatory T cells andlimiting host tumor immunosuppressive factors. ACT wasdeveloped by the group of Steven Rosenberg at the National
Table 1. Agents targeting immune-checkpoints and co-stimulatory signaling* (continued).
Agent Clinical trials
Lambrolizumab (MK-3475 anti-PD1) Phase III trial in melanoma as monotherapy versus ipilimumab (NCT01866319)recruiting; Phase II trial in melanoma ongoing but not recruiting (NCT01704287);Phase I trials in melanoma, solid tumors or non-small-cell lung cancer recruiting(NCT01295827, NCT01848834, NCT01840579); Phase II/III trials in non-small-cell lungcancer versus docetaxel recruiting (NCT01905657); Phase II trial in tumor withmicrosatellite instability recruiting (NCT01876511); positive results of the melanomacohort of the NCT01295827 study recently published
MPDL3280A (anti-PD1) Phase I trial in melanoma or hematologic malignancies (NCT01375842), melanoma(combination with vemurafenib, NCT01656642), or in solid tumors in combination withbevacizumab or chemotherapy (NCT01633970) recruiting; several Phase I-II trials andone Phase III trial in lung cancer recruiting (NCT02008227)
CT-011 (anti-PD1) Phase II trial in melanoma completed (NCT01435369); Phase II trials in prostate, renal,pancreatic cancer and hematological malignancies in combination with chemotherapyor vaccine recruiting; other Phase I and Phase II trials in gastrointestinal tumors andhematological malignancies recently concluded
BMS-936559 (MDX-1105; anti-PD-L1) Phase I trials in melanoma (NCT01455103) and hematological malignancies(NCT01452334) withdrawn before enrollment; a Phase I trial in solid cancer is ongoingbut not recruiting (NCT00729664)
AMP-224 (anti-PD-L2) Phase I trial in solid cancers ongoing but not recruiting (NCT01352884)MEDI4736 (anti-PD-L1) Phase I/II trial in melanoma in combination with dabrafenib or dabrafenib and
trametinib recruiting (NCT02027961); Phase I trial in japanese patients with solid tumorsrecruiting (NCT01938612); Phase I trial in solid tumors recruiting (NCT01693562);Phase I trial in combination with tremelimumab in solid tumors (NCT01975831) or innon-small-lung cancer recruiting (NCT02000947)
*Updated as of January 2014 (source: clinicaltrial.gov).
CTLA-4: Cytotoxic T-lymphocyte antigen 4; LAG3: Lymphocyte activation genes 3; PD-L1: Programmed death ligand 1.
S. Tomei et al.
14 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

Cancer Institute and has shown encouraging results inPhase II clinical trials. In a recent analysis evaluating93 heavily pretreated metastatic melanoma patients enrolledin three Phase II trials, ACT was able to induce OR in 56%of patients. Complete remission was obtained in 20 patients(21.5%), with 19 patients potentially cured as they had ongo-ing regression beyond 3 years [185]. A similar OR rate (48%,including 6.5% of CR), measured using immune-relatedresponse criteria (irRC), had been obtained by Radvanyiet al. in a Phase II trial enrolling 31 patients [186].
Beyond the toxicity related to HD-IL-2 administration, themain limitation associated to ACT is that TIL expansionrequires highly specialized personnel. Using the traditionalapproach, TILs are generally successfully grown from only50 to 60% of the patients and require a long period of time(about 5 weeks). Simplified protocols using short-term culturesderive TILs from up to 90% of patients in a shorter period oftime (10 -- 18 days) [187]. Rosenberg’s group, however, recentlyreported a global OR of 28% (CR rate 7%), in randomizedPhase II trials comparing two different TIL protocols (youngTILs vs CD8 enriched TILs) [188]. By using young TILsBesser et al., in a Phase II trial enrolling 70 patients, reportedan encouragingOR of 40% in the evaluated population, includ-ing 5 CR [187]. In all the ACT trials responses were durable andpatients in CR very rarely relapsed. The results of these Phase IItrials need to be taken with caution, and the true response rateneeds to be evaluated in randomized Phase III trials. Neverthe-less, ACT seems to represent a potential curative therapy forpatients withmetastatic melanoma [189]. Randomizedmulticen-ter trials aimed at evaluating the true response rate and OSadvantage of ACT represent the reasonable next step.
Despite these encouraging results, the dynamic of TILmigration at tumor site is still not completely clear.
Studies where TILs were labeled with Indium 111 showedthat TILs migration in the tumor site does not follow alinear kinetic. After re-infusion, which is followed by IL-2administration, TIL localize in lung, spleen, and liver butnot at tumor sites [190,191]. The migration of TILs at tumorsite is detectable only after 24 to 48 h. It is possible that thisearly compartmentalization is mediated by the release(following HD-IL-2 administration) of specific chemokines(i.e., CXCR3 and CCR5 ligands) from resident immunecells and stromal cell in peripheral oragans (e.g., spleen,lung, and liver) [129,192]. Intriguingly, by analyzing a largecohort of patients treated with adoptive therapy and HD-IL-2, we recently showed that the downregulation ofCXCR3 and CCR5 receptors in TILs correlates with thefrequency and the degree of response [129]. This receptordown-modulation is due to the transcript downregulationand/or to the presence of common polymorphism (CCR5D32, which codify for a nonfunctioning receptor) [129]. Itis tempting to hypothesize that a reduced, but not absent,expression of these receptors might prevent the earliersequestration of TIL by extra-tumoral sites and paradoxi-cally allow their subsequent localization to the tumor
when the cytokine storm has subsided and the tumor repre-sents the only tissue maintaining chemokine secretion [129].
5. Combination therapy
Combination therapy is intended as the use of multiple ther-apies to treat a single disease with the aim to overcome thehurdles of mono-therapeutic strategies [193]. Combining che-motherapy agents represents a pillar of the medical treatmentof virtually all the major liquid and solid malignancies, withexception of melanoma and renal cell carcinoma. In thesetwo tumors combination of cytotoxic agents failed to showincrease efficacy as compared with monotherapy. Inmelanoma, > 40 randomized trials have evaluated combina-tion of chemotherapy +/- cytokines (IFN-a and/or IL-2)[21]. It is now clear that although polychemotherapy +/-IFN-a and/or IL-2 in general enhance the OR rate, they donot increase the OS [21]. The failure of polychemotherapy isin general attributed to the intrinsic chemo-resistance ofmelanoma. As for first generation immunochemotherapy, itshould be noted that, because of safety issues, IFN-a and orIL-2 were administered at low dose, while their maximalantitumor effect is in general achieved using high dosages.Nevertheless, prospective trials have shown that combinationof HD-IL-2 and IFN-a does not increase OS, as comparedwith HD-IL-2 alone [194,195].
This depressing scenario dramatically changed after adventof targeted therapy immune checkpoint inhibitors. Althoughthe mono-targeted therapy and immunotherapy strategieshave revolutionized the clinical management of melanomapatients, it is becoming clear that combination strategiesare most likely to result in even more significant advances.Building upon the early success of mono-therapy agents inmelanoma, trials involving new combination therapy areunderway. It has been thought that combining vaccineswith anti-CTLA-4 could produce a synergic effect. Whenipilimumab was administered together with peptide vaccina-tion (gp100), the combined treatment was more effective(both in term of PFS and OS) than gp100 alone [17]. How-ever, there were no significant differences in both PFS andOS between ipilimumab alone and ipilimumab combinedwith vaccine [30]. Conversely, gp100 vaccine and IL-2 canact in a synergistic way. In a Phase III randomized trials inmetastatic melanoma patients, Schwartzentruber et al.showed that the addition of the gp100 vaccine to HD-IL-2 can significantly increase the number of responses(CR: 7 vs 1%, and OR: 16 vs 6%, IL-2 + gp100 vs IL-2,respectively, p = 0.03) and lengthen PFS survival (medianPFS 2.2 vs 1.6 months, p = 0.008), with similar trend inOS [134]. Two recent studies showed that efficacy ofCTLA-4 can be enhanced by the concomitant administrationof immune-stimulant. In a recent randomized Phase II trialHodi et al. showed that the addition of GM-CSF to ipilimu-mab increase OS (1 year OS: 68 vs 51%, GM-CSF plus ipi-limumab vs ipilimumab alone, p = 0.03) and was also
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 15
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

associate with less toxicity [196]. Remarkably, long-term fol-low-up Phase I/II trials in metastatic melanoma patientshave suggested that the addition of IL-2 to ipilimumab coulddramatically improve the rate of durable CR (CR rate: 17 vs7%, ipilimumab + IL-2 vs ipilimumab +/- gp 100 vaccine)and extend five overall survival up to 25% [197].Overall, these apparently clashing results underline once
again the need to understand the mechanisms of action ofsimultaneous administration by designing clinical or transla-tion trial aimed at evaluating the effect of immunotherapyin vivo in humans [198]. However, in view of the huge numberof potential combinations, novel generations of clinical trialsusing factorial design are needed [199] to select the morepromising combinations.Another possible combination approach is the use of tar-
geted therapy together with immunotherapy [200]. The ratio-nale supporting this type of combination approach relies onthe possibility to achieve a deeper and long lasting antitumoreffect (Figure 2C), as pointed out by Ribas et al. [201].BRAF inhibitors, for example, induce almost immediate
tumor shrinkage in most patients (median time to response1.5 months). Responses are, however, transient, and the effectof long-term survival is predicted to be very limited(Figure 2A). Conversely, CTLA-4 blockades induce less num-ber of clinical responses (usually delayed), but they are muchmore durable (Figure 2B).A number of observations support the target-immunother-
apy combination. Several cytotoxic agents have been shown toinduce antitumor immune responses by promoting cell deathwith consequent release of tumor-associated antigens [202]. Inmelanoma, this could be achieved through targeted therapy [203],rather than with conventional chemotherapy. Moreover, con-stitutive activation of oncogenic pathways, as the BRAF-MAPK pathway, can induce several immunosuppressive mech-anisms [204,205], including promotion of stromal cell-mediatedimmunosuppression via induction of IL-1 [7,206]. In addition,BRAF inhibition can enhance melanoma antigens expressionand create a more favorable immune environment, facilitatingthe effectiveness of immunotherapy [89,207-209].A study evaluating combination of ipilimumab and vemura-
fenib was object of a Phase I study by Ribas et al. [210]. Althoughthis combination is supported by a strong rationale, investiga-tors noticed an unacceptable hepatotoxicity, which promptedthem to close prematurely the study. This example reinforcesthe need to carefully evaluate the safety of combinationapproach in appropriate early trials [210].Very recently, two additional prospective studies in
metastatic melanoma have highlighted the potentiality of com-bining highly effective agents. In a first Phase I/II randomizedtrial, combination of BRAF inhibitor dabrafenib andtrametinib resulted superior to dabrafenib alone [23]. The twodrugs combined at full monotherapy dose resulted in 76% ofOR (including 9% of CR), while 54% of patients achievedOR in the dabrafenib group (including 4% of CR; p = 0.03).Progression-free survival in the combination was 9.4 months,
as compared with 5.8 months in the monotherapy group(p < 0.001). Responses were also more durable with the combi-nation (10.2 vs 5.6). Importantly, > 40% of patients were aliveand progression-free at 1 year in the combination arm, as com-pared with 10% in the monotherapy arm. Combination wasalso feasible in term of safety [211]. This landmark study demon-strated that the inhibition of the same pathways through highlyeffective targeted therapy agents (vertical inhibition) coulddramatically impact on the course of this aggressive disease,overcoming (at least in part) mechanisms of resistance occur-ring during monotherapy. Several trials also employing thera-pies targeting different pathways are underway; examplesinclude vemurafenib (BRAF inhibitor) plus everolimus or tem-sirolmus (mTOR inhibitors, identifier NCT01596140),MEK162 plus AMG479 (insulin-like growth factor1 receptorinhibitor, identifier NCT015 62899), vemurafenib plusBKM120 (a PI3K inhibitor, identifier NCT01512251).
In a second Phase I trial, combination of PD-1 andCTLA-4 blockers (nivolumab and ipilimumab) resulted, atthe maximum tolerated dose, in 53% of OR rate, with tumorregression of 80% or more in every patient who had response[212]. The profound effect of concurrent combination wasalready evident at the first imaging assessment (3 months aftertreatment initiation). Responses were extremely durable andongoing in 90% patients who had a response (duration rang-ing from 6 to 72 weeks at the time of data analysis). Thispattern of response (rapid, deep, and durable) appeared tobe distinct of that observed in previous immune-checkpointblockade trials. Importantly, toxicity was qualitatively similarto that of monotherapy [212].
6. Expert opinion
Although major advances have been made for the treatment ofmetastatic melanoma, with combination therapies paving theroad toward clinical success, much effort needs to be spent inidentifying patients who are more likely to respond to a giventreatment.
It is becoming now clear that patients who are most likelyto reject tumors following immunotherapy are the ones whodisplay a constitutive activation of the IFN-g/signal trans-ducers and activators of transcription (STAT)-1/IRF-1 axis(Th1 phenotype) [81,117,121] with IRF-1 playing a central rolein this regard [1,117,121,192,213]. Gene signatures predictive ofresponse to immunotherapy (i.e., IL-2-based therapy, vac-cines) invariably included genes belonging to the followingfunctional modules:
1) IFN-g/T helper 1 module (STAT-1/IRF-1/IFN-g-stimulated genes pathway);
2) The specific cytotoxic recruitment module (CXCR3/CXCR3 ligand and CCR5/CCR5 ligand pathway);
3) The immuno-effector function module (Granzyme/Perforin/Granulisin/TIA-1 pathway).
S. Tomei et al.
16 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

This phenotype is also characterized by the counter-activation of suppressive mechanisms. Therefore, it is not sur-prising that high expression of IDO and PD-1 ligands, whichare both IFN-g inducible genes, have been found to be asso-ciated with responsiveness to ipilimumab [214] and anti-PD1treatment [165]. The determinants of this responsive immunephenotype are object of investigations.
Coordinate enhancement of these modules following treat-ment is associated with responsiveness to IL-2 [103], ipilimu-mab [215], and other immunotherapies, including vaccines[216] and Toll-like receptor agonists [217]. These observationssuggest that, although distinct immunotherapies clearly actin very different ways, their final therapeutic relies on the acti-vation of similar and redundant downstream mechanisms, asrecently reviewed elsewhere [121].
It is currently becoming accepted that immune-mediatedtumor rejection is mediated by both the genetic backgroundof the host and the genetic make-up of individual cancer cellscarrying their own specific mutations [119,120,218]. Yet, the rel-ative weight played by each of those factors is not completelyunderstood as well as their contribution to mediate respon-siveness to a given cancer therapy. The relevance of the geneticbackground of the host in mediating tumor rejection has beenrecently witnessed by two studies from our group. In the firstone, we hypothesized that polymorphisms of IRF5 gene,known to be implicated in autoimmune diseases and in par-ticular to systemic lupus erythematosus, might play a role inthe immune-mediated rejection of the tumor. Concordantto our hypothesis, we were able to show that the IRF5 geneticvariants protecting against the development of autoimmunitywere highly predictive of nonresponsiveness to adoptive ther-apy in melanoma [131]. These results support the link betweenautoimmunity and immune responsiveness. In the secondstudy, we assessed the prognostic relevance of genetic varia-tions of CTLA-4 and HLA polymorphisms in melanomapatients who received adjuvant IFN-a [132]. Multivariate anal-ysis revealed that a five-marker genotyping signature based onHLA-B38, HLA-C15, HLA-C3 DRB1*15, and CTLA-4CT60* polymorphisms, was predictive of OS in 284 mela-noma patients. The findings of these two studies strengthenthe relevance of inherited genetic variations in mediatingtumor rejection induced by immunotherapy regimens.
Aside from inherited genetic variations, somatic mutationsacquired during tumor development are likely to influencetumor rejection. The most evident proof that the geneticbackground of individual cancer cells plays a role in mediatingcancer rejection comes from the observation of mixedresponses, a rare phenomenon where multiple metastasesfrom the same patient respond differently to a given therapy.By comparing mixed metastases from two melanoma patientswho received immunotherapy, we were able to show thatmetastases who responded to the treatment were the oneswho had upregulated genes associated with antigen presenta-tion and IFN-mediated rejection [216]. These findings demon-strate that tumors sharing the same host’s genetic backgroundcan behave differently depending on the differential expres-sion of genes involved in triggering an IFN-mediated immunesignaling. It should be also noted that cancer cells share thesame genetic background of the host, thus, tumor behavioris concurrently influenced by inherited variations other thansomatic mutations.
Environmental factors also play a role in this regard, andevidence accumulated in the recent years show that it is acombination of all the three categories that affect tumorrejection rather than each single one alone [218]. Understand-ing the mechanisms underlying tumor rejection is essentialto implement personalized therapies.
We believe that this goal is reachable only by understand-ing the complex interplay between the genetic backgroundof the host, the genetic make-up of individual cancer cells,and environmental factors. The availability of high-throughput technologies allowed the investigators to startthe race toward cancer-personalized medicine. We believethat, in the future, the integration of the ‘omic’ researchapproaches will allow a more potent understanding of thealgorithm governing tumor rejection and responsiveness toa given therapy and lead to improved clinical managementof melanoma patients.
Declaration of interest
The authors have no competing interests to declare and havereceived no funding in preparation of the manuscript.
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 17
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

BibliographyPapers of special note have been highlighted as
either of interest (!) or of considerable interest(!!) to readers.
1. Spivey TL, De Giorgi V, Zhao Y, et al.
The stable traits of melanoma genetics:
an alternate approach to target discovery.
BMC Genomics 2012;13:156
2. Ibrahim N, Haluska FG. Molecular
pathogenesis of cutaneous melanocytic
neoplasms. Annu Rev Pathol
2009;4:551-79
3. Palmieri G, Capone M, Ascierto ML,
et al. Main roads to melanoma.
J Transl Med 2009;7:86
4. Chapman PB, Hauschild A, Robert C,
et al. Improved survival with
vemurafenib in melanoma with BRAF
V600E mutation. N Engl J Med
2011;364:2507-16
5. Davies H, Bignell GR, Cox C, et al.
Mutations of the BRAF gene in human
cancer. Nature 2002;417:949-54
6. Colombino M, Lissia A, Capone M,
et al. Heterogeneous distribution of
BRAF/NRAS mutations among Italian
patients with advanced melanoma.
J Transl Med 2013;11:202
7. Tomei S, Civini S, Bedognetti D, et al.
The immune-related role of BRAF
mutation in melanoma [abstract p788].
J Immuno Therapy of Cancer 2012;23:9
8. Pollock PM, Harper UL, Hansen KS,
et al. High frequency of BRAF mutations
in nevi. Nat Genet 2003;33:19-20
9. Kumar R, Angelini S, Snellman E,
Hemminki K. BRAF mutations are
common somatic events in melanocytic
nevi. J Invest Dermatol 2004;122:342-8
10. Sosman JA, Kim KB, Schuchter L, et al.
Survival in BRAF V600-mutant
advanced melanoma treated with
vemurafenib. N Engl J Med
2012;366:707-14
11. Chapman PB, Hauschild A, Robert C,
et al. Updated overall survival (OS)
results for BRIM-3, a phase III
randomized, open-label, multicenter trial
comparing BRAF inhibitor vemurafenib
(vem) with dacarbazine (DTIC) in
previously untreated patients with
BRAFV600E-mutated melanoma.
J Clin Oncol 2012:30(Suppl):
abstract 8502
12. Hauschild A, Grob JJ, Demidov LV,
et al. Dabrafenib in BRAF-mutated
metastatic melanoma: a multicentre,
open-label, phase 3 randomised
controlled trial. Lancet 2012;380:358-65
13. Ascierto PA, Minor D, Ribas A, et al.
Phase II trial (BREAK-2) of the BRAF
inhibitor dabrafenib (GSK2118436) in
patients with metastatic melanoma.
J Clin Oncol 2013;31:3205-11
14. Ascierto PA, Kirkwood JM, Grob JJ,
et al. The role of BRAF V600 mutation
in melanoma. J Transl Med 2012;10:85
15. Clinical Trials. Avaialable from http://
clinicaltrial.gov/)
16. Tsao H, Atkins MB, Sober AJ.
Management of cutaneous melanoma.
N Engl J Med 2004;351:998-1012
17. Hodi FS, O’Day SJ, McDermott DF,
et al. Improved survival with ipilimumab
in patients with metastatic melanoma.
N Engl J Med 2010;363:711-23.. First Phase III study showing an
advantage of overall survival in
metastatic melanoma
using immunotherapy.
18. Eigentler TK, Meier F, Garbe C. Protein
kinase inhibitors in melanoma.
Expert Opin Pharmacother
2013;14(16):2195-201
19. Benjamin RS. Chemotherapy of
malignant melanoma. World J Surg
1979;3:321-8
20. Luce JK. Chemotherapy of malignant
melanoma. Cancer 1972;30:1604-15
21. Garbe C, Eigentler TK, Keilholz U,
et al. Systematic review of medical
treatment in melanoma: current status
and future prospects. Oncologist
2011;16:5-24
22. Robert C, Thomas L, Bondarenko I,
et al. Ipilimumab plus dacarbazine for
previously untreated metastatic
melanoma. N Engl J Med
2011;364:2517-26
23. Flaherty KT, Robert C, Hersey P, et al.
Improved survival with MEK inhibition
in BRAF-mutated melanoma. N Engl
J Med 2012;367:107-14.. First Phase III study showing an
advantage of overall survival in
metastatic melanoma using non-BRAf
target therapy MEK inhibitor.
24. Henary H, Hong DS, Falchook GS,
et al. Melanoma patients in a phase I
clinic: molecular aberrations, targeted
therapy and outcomes. Ann Oncol
2013;24(8):2158-65
25. Smalley KS. A pivotal role for ERK in
the oncogenic behaviour of malignant
melanoma? Int J Cancer
2003;104:527-32
26. Colombino M, Capone M, Lissia A,
et al. BRAF/NRAS mutation frequencies
among primary tumors and metastases in
patients with melanoma. J Clin Oncol
2012;30:2522-9
27. Nikolaou VA, Stratigos AJ, Flaherty KT,
Tsao H. Melanoma: new insights and
new therapies. J Invest Dermatol
2012;132:854-63
28. Yoshida H, Kunisada T, Grimm T, et al.
Review: melanocyte migration and
survival controlled by SCF/c-kit
expression. J Investig Dermatol
Symp Proc 2001;6:1-5
29. Hochhaus A. Imatinib mesylate (Glivec,
Gleevec) in the treatment of chronic
myelogenous leukemia (CML) and
gastrointestinal stromal tumors (GIST).
Ann Hematol 2004;83(Suppl 1):S65-6
30. Beadling C, Jacobson-Dunlop E,
Hodi FS, et al. KIT gene mutations and
copy number in melanoma subtypes.
Clin Cancer Res 2008;14:6821-8
31. Carvajal RD, Antonescu CR,
Wolchok JD, et al. KIT as a therapeutic
target in metastatic melanoma. JAMA
2011;305:2327-34. First study showing that imatinib
mesylate is active in a subset of
metastatic melanoma patients carrying
c-KIT mutations and amplification.
32. Natali PG, Nicotra MR, Winkler AB,
et al. Progression of human cutaneous
melanoma is associated with loss of
expression of c-kit proto-oncogene
receptor. Int J Cancer 1992;52:197-201
33. Shen SS, Zhang PS, Eton O, Prieto VG.
Analysis of protein tyrosine kinase
expression in melanocytic lesions by
tissue array. J Cutan Pathol
2003;30:539-47
34. Tomei S, Mazzanti C, Marchetti I, et al.
c-KIT receptor expression is strictly
associated with the biological behaviour
of thyroid nodules. J Transl Med
2012;10:7
35. Guo T, Agaram NP, Wong GC, et al.
Sorafenib inhibits the imatinib-resistant
KITT670I gatekeeper mutation in
S. Tomei et al.
18 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

gastrointestinal stromal tumor.
Clin Cancer Res 2007;13:4874-81
36. Prenen H, Cools J, Mentens N, et al.
Efficacy of the kinase inhibitor
SU11248 against gastrointestinal stromal
tumor mutants refractory to imatinib
mesylate. Clin Cancer Res
2006;12:2622-7
37. Schittenhelm MM, Shiraga S,
Schroeder A, et al. Dasatinib
(BMS-354825), a dual SRC/ABL kinase
inhibitor, inhibits the kinase activity of
wild-type, juxtamembrane, and activation
loop mutant KIT isoforms associated
with human malignancies. Cancer Res
2006;66:473-81
38. Ugurel S, Hildenbrand R, Zimpfer A,
et al. Lack of clinical efficacy of imatinib
in metastatic melanoma. Br J Cancer
2005;92:1398-405
39. Kim KB, Eton O, Davis DW, et al.
Phase II trial of imatinib mesylate in
patients with metastatic melanoma.
Br J Cancer 2008;99:734-40
40. Wyman K, Atkins MB, Prieto V, et al.
Multicenter Phase II trial of high-dose
imatinib mesylate in metastatic
melanoma: significant toxicity with no
clinical efficacy. Cancer
2006;106:2005-11
41. Guo J, Si L, Kong Y, et al. Phase II,
open-label, single-arm trial of imatinib
mesylate in patients with metastatic
melanoma harboring c-Kit mutation or
amplification. J Clin Oncol
2011;29:2904-9
42. Flaherty KT, Hodi FS, Fisher DE. From
genes to drugs: targeted strategies for
melanoma. Nat Rev Cancer
2012;12:349-61
43. Kaufman HL, Kirkwood JM, Hodi FS,
et al. The Society for Immunotherapy of
Cancer consensus statement on tumour
immunotherapy for the treatment of
cutaneous melanoma. Nat Rev
Clin Oncol 2013;10:588-98.. This manuscript provides the first
guidelines on the use of
immunotherapy in melanoma patients.
44. Antonescu CR. The GIST paradigm:
lessons for other kinase-driven cancers.
J Pathol 2011;223:251-61
45. Schlegel J, Sambade MJ, Sather S, et al.
MERTK receptor tyrosine kinase is a
therapeutic target in melanoma.
J Clin Invest 2013;123:2257-67
46. Albino AP, Le Strange R, Oliff AI, et al.
Transforming ras genes from human
melanoma: a manifestation of tumour
heterogeneity? Nature 1984;308:69-72
47. Milagre C, Dhomen N, Geyer FC, et al.
A mouse model of melanoma driven by
oncogenic KRAS. Cancer Res
2010;70:5549-57
48. Tomei S, Adams S, Uccellini L, et al.
Association between HRAS rs12628 and
rs112587690 polymorphisms with the
risk of melanoma in the North American
population. Med Oncol
2012;29:3456-61
49. COSMIC (Catalogue Of Somatic
Mutations In Cancer. Available from:
http://cancer.sanger.ac.uk/cancergenome/
projects/cosmic
50. Posch C, Ortiz-Urda S. NRAS mutant
melanoma--undrugable? Oncotarget
2013;4:494-5
51. Niessner H, Beck D, Sinnberg T, et al.
The farnesyl transferase inhibitor
lonafarnib inhibits mTOR signaling and
enforces sorafenib-induced apoptosis in
melanoma cells. J Invest Dermatol
2011;131:468-79
52. Gajewski TF, Salama AK,
Niedzwiecki D, et al. Phase II study of
the farnesyltransferase inhibitor
R115777 in advanced melanoma
(CALGB 500104). J Transl Med
2012;10:246
53. Emery CM, Vijayendran KG,
Zipser MC, et al. MEK1 mutations
confer resistance to MEK and B-RAF
inhibition. Proc Natl Acad Sci USA
2009;106:20411-16
54. Salama AK, Kim KB. MEK Inhibition in
the treatment of advanced melanoma.
Curr Oncol Rep 2013;15:473-82
55. Kirkwood JM, Bastholt L, Robert C,
et al. Phase II, open-label, randomized
trial of the MEK1/2 inhibitor
selumetinib as monotherapy versus
temozolomide in patients with advanced
melanoma. Clin Cancer Res
2012;18:555-67
56. Robert C, Dummer R, Gutzmer R, et al.
Selumetinib plus dacarbazine versus
placebo plus dacarbazine as first-line
treatment for BRAF-mutant metastatic
melanoma: a phase 2 double-blind
randomised study. Lancet Oncol
2013;14:733-40
57. Kim KB, Kefford R, Pavlick AC, et al.
Phase II study of the MEK1/
MEK2 inhibitor Trametinib in patients
with metastatic BRAF-mutant cutaneous
melanoma previously treated with or
without a BRAF inhibitor. J Clin Oncol
2013;31:482-9
58. Ascierto PA, Schadendorf D, Berking C,
et al. MEK162 for patients with
advanced melanoma harbouring NRAS
or Val600 BRAF mutations: a non-
randomised, open-label phase 2 study.
Lancet Oncol 2013;14:249-56
59. Lebbe C, Lesimple T, Raymond E, et al.
Pimasertib (MSC1936369B/AS703026),
a selective oral MEK1/2 inhibitor, shows
clinical activity in cutaneous and uveal
metastatic melanoma in the phase I
program [abstract C07]. EADO 2012
Annual Meeting; 2012
60. Basile KJ, Abel EV, Dadpey N, et al.
In vivo MAPK reporting reveals the
heterogeneity in tumoral selection of
resistance to RAF inhibitors. Cancer Res
2013;73(23):7101-10
61. Nazarian R, Shi H, Wang Q, et al.
Melanomas acquire resistance to B-RAF
(V600E) inhibition by RTK or N-RAS
upregulation. Nature 2010;468:973-7
62. Morris EJ, Jha S, Restaino CR, et al.
Discovery of a novel ERK inhibitor with
activity in models of acquired resistance
to BRAF and MEK inhibitors.
Cancer Discov 2013;3:742-50
63. Wangari-Talbot J, Chen S. Genetics of
melanoma. Front Genet 2012;3:330
64. Madhunapantula SV, Robertson GP.
Therapeutic Implications of Targeting
AKT Signaling in Melanoma.
Enzyme Res 2011;2011:327923
65. Knight ZA, Shokat KM. Chemically
targeting the PI3K family.
Biochem Soc Trans 2007;35:245-9
66. Margolin K, Longmate J, Baratta T,
et al. CCI-779 in metastatic melanoma:
a phase II trial of the California Cancer
Consortium. Cancer 2005;104:1045-8
67. Margolin KA, Moon J, Flaherty LE,
et al. Randomized phase II trial of
sorafenib with temsirolimus or tipifarnib
in untreated metastatic melanoma
(S0438). Clin Cancer Res
2012;18:1129-37
68. Werzowa J, Koehrer S, Strommer S,
et al. Vertical inhibition of the
mTORC1/mTORC2/PI3K pathway
shows synergistic effects against
melanoma in vitro and in vivo.
J Invest Dermatol 2011;131:495-503
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 19
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

69. Hudes G, Carducci M, Tomczak P,
et al. Temsirolimus, interferon alfa, or
both for advanced renal-cell carcinoma.
N Engl J Med 2007;356:2271-81
70. Hay N. The Akt-mTOR tango and its
relevance to cancer. Cancer Cell
2005;8:179-83
71. Greger JG, Eastman SD, Zhang V, et al.
Combinations of BRAF, MEK, and
PI3K/mTOR inhibitors overcome
acquired resistance to the BRAF inhibitor
GSK2118436 dabrafenib, mediated by
NRAS or MEK mutations.
Mol Cancer Ther 2012;11:909-20
72. Posch C, Moslehi H, Feeney L, et al.
Combined targeting of MEK and PI3K/
mTOR effector pathways is necessary to
effectively inhibit NRAS mutant
melanoma in vitro and in vivo.
Proc Natl Acad Sci USA
2013;110:4015-20
73. Bedikian AY, Millward M,
Pehamberger H, et al. Bcl-2 antisense
(oblimersen sodium) plus dacarbazine in
patients with advanced melanoma: the
Oblimersen Melanoma Study Group.
J Clin Oncol 2006;24:4738-45
74. Bedikian AY, Lebbe C, Robert C, et al.
Survival in a phase III, randomized,
double-blind study of dacarbazine with
or without oblimersen (Bcl-2 antisense)
in patients with advanced melanoma and
low-normal serum lactate dehydrogenase
(LDH; AGENDA). J Clin Oncol
2011;29(Suppl):abstract 8531
75. Horn S, Figl A, Rachakonda PS, et al.
TERT promoter mutations in familial
and sporadic melanoma. Science
2013;339:959-61
76. Huang FW, Hodis E, Xu MJ, et al.
Highly recurrent TERT promoter
mutations in human melanoma. Science
2013;339:957-9
77. Varker KA, Biber JE, Kefauver C, et al.
A randomized phase 2 trial of
bevacizumab with or without daily low-
dose interferon alfa-2b in metastatic
malignant melanoma. Ann Surg Oncol
2007;14:2367-76
78. Kim KB, Sosman JA, Fruehauf JP, et al.
BEAM: a randomized phase II study
evaluating the activity of bevacizumab in
combination with carboplatin plus
paclitaxel in patients with previously
untreated advanced melanoma.
J Clin Oncol 2012;30:34-41
79. Minor DR. Bevacizumab advanced
melanoma (BEAM) was a positive trial.
J Clin Oncol 2012;30:2023;
author reply 2023-2024. This is an interesting comment on the
BEAM trial, describing why inhibiting
angiogenesis represents an approach
that should not be dismissed.
80. Del Vecchio M, Mortarini R, Canova S,
et al. Bevacizumab plus fotemustine as
first-line treatment in metastatic
melanoma patients: clinical activity and
modulation of angiogenesis and
lymphangiogenesis factors.
Clin Cancer Res 2010;16:5862-72.. An interesting Phase II study
evaluating combination of
bevacizumab and chemotherapy and
assessing biological correlates.
81. Hodi FS, Friedlander PA, Atkins MB,
et al. A phase I trial of ipilimumab plus
bevacizumab in patients with
unresectable stage III or stage IV
melanoma. J Clin Oncol
2011;29(Suppl):abstract 8511
82. Flaherty KT, Lee SJ, Zhao F, et al.
Phase III trial of carboplatin and
paclitaxel with or without sorafenib in
metastatic melanoma. J Clin Oncol
2013;31:373-9
83. Hauschild A, Agarwala SS, Trefzer U,
et al. Results of a phase III, randomized,
placebo-controlled study of sorafenib in
combination with carboplatin and
paclitaxel as second-line treatment in
patients with unresectable stage III or
stage IV melanoma. J Clin Oncol
2009;27:2823-30
84. Fruehauf J, Lutzky J, McDermott D,
et al. Multicenter, phase II study of
axitinib, a selective second-generation
inhibitor of vascular endothelial growth
factor receptors 1, 2, and 3, in patients
with metastatic melanoma.
Clin Cancer Res 2011;17:7462-9
85. Hong DS, Koetz BS, Kurzrock R, et al.
Phase I dose-escalation study of E7080, a
selective tyrosine kinase inhibitor,
administered orally to patients with solid
tumors. J Clin Oncol
2010;28:15s(Suppl):abstract 2540
86. Corrie PG, Basu B, Zaki KA. Targeting
angiogenesis in melanoma: prospects for
the future. Ther Adv Med Oncol
2010;2:367-80
87. Ascierto ML, De Giorgi V, Liu Q, et al.
An immunologic portrait of cancer.
J Transl Med 2011;9:146. A comprehensive review proposing an
immunological interpretation
of cancer.
88. Shimanovsky A, Jethava A, Dasanu CA.
Immune alterations in malignant
melanoma and current immunotherapy
concepts. Expert Opin Biol Ther
2013;13:1413-27
89. Wilmott JS, Long GV, Howle JR, et al.
Selective BRAF inhibitors induce marked
T-cell infiltration into human metastatic
melanoma. Clin Cancer Res
2012;18:1386-94
90. Gajewski TF, Meng Y, Harlin H.
Immune Suppression in the tumor
microenvironment. J Immunother
2006;29:233-40
91. Krieg C, Letourneau S, Pantaleo G,
Boyman O. Improved IL-2
immunotherapy by selective stimulation
of IL-2 receptors on lymphocytes and
endothelial cells. Proc Natl Acad
Sci USA 2010;107:11906-11
92. Atkins MB, Lotze MT, Dutcher JP, et al.
High-dose recombinant interleukin
2 therapy for patients with metastatic
melanoma: analysis of 270 patients
treated between 1985 and 1993.
J Clin Oncol 1999;17:2105-16
93. Rosenberg SA, Mule JJ, Spiess PJ, et al.
Regression of established pulmonary
metastases and subcutaneous tumor
mediated by the systemic administration
of high-dose recombinant interleukin 2.
J Exp Med 1985;161:1169-88
94. Phan GQ, Attia P, Steinberg SM, et al.
Factors associated with response to
high-dose interleukin-2 in patients with
metastatic melanoma. J Clin Oncol
2001;19:3477-82
95. Smith FO, Downey SG, Klapper JA,
et al. Treatment of metastatic melanoma
using interleukin-2 alone or in
conjunction with vaccines.
Clin Cancer Res 2008;14:5610-18
96. Joseph RW, Sullivan RJ, Harrell R, et al.
Correlation of NRAS mutations with
clinical response to high-dose IL-2 in
patients with advanced melanoma.
J Immunother 2012;35:66-72
97. Dummer R, Hauschild A,
Guggenheim M, et al. Melanoma:
ESMO Clinical Practice Guidelines for
S. Tomei et al.
20 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

diagnosis, treatment and follow-up.
Ann Oncol 2010;21(Suppl 5):v194-7
98. Sabatino M, Kim-Schulze S, Panelli MC,
et al. Serum vascular endothelial growth
factor and fibronectin predict clinical
response to high-dose interleukin-
2 therapy. J Clin Oncol
2009;27:2645-52
99. Yuan J, Zhou J, Dong Z, et al.
Pre-treatment serum vascular endothelial
growth factor is associated with clinical
response and overall survival in advanced
melanoma patients treated with
ipilimumab. J Immuno Ther Cancer
2013;1(Suppl 1):P247
100. Panelli MC, Wang E, Phan G, et al.
Gene-expression profiling of the response
of peripheral blood mononuclear cells
and melanoma metastases to systemic
IL-2 administration. Genome Biol
2002;3:RESEARCH0035
101. Panelli MC, White R, Foster M, et al.
Forecasting the cytokine storm following
systemic interleukin (IL)-
2 administration. J Transl Med
2004;2:17
102. Wang E, Miller LD, Ohnmacht GA,
et al. Prospective molecular profiling of
melanoma metastases suggests classifiers
of immune responsiveness. Cancer Res
2002;62:3581-6
103. Weiss GR, Grosh WW,
Chianese-Bullock KA, et al. Molecular
insights on the peripheral and
intratumoral effects of systemic high-dose
rIL-2 (aldesleukin) administration for the
treatment of metastatic melanoma.
Clin Cancer Res 2011;17:7440-50. An interesting prospective study on
gene expression in tumor and PBMC
following IL-2 administration.
104. Bedognetti D, Wang E, Sertoli MR,
Marincola FM. Gene-expression profiling
in vaccine therapy and immunotherapy
for cancer. Expert Rev Vaccines
2010;9:555-65
105. Blanchard T, Srivastava PK, Duan F.
Vaccines against advanced melanoma.
Clin Dermatol 2013;31:179-90
106. Mocellin S, Rossi CR, Nitti D, et al.
Dissecting tumor responsiveness to
immunotherapy: the experience of
peptide-based melanoma vaccines.
Biochim Biophys Acta 2003;1653:61-71
107. Lens M. The role of vaccine therapy in
the treatment of melanoma. Expert Opin
Biol Ther 2008;8:315-23
108. Terando AM, Faries MB, Morton DL.
Vaccine therapy for melanoma: current
status and future directions. Vaccine
2007;25(Suppl 2):B4-16
109. Ascierto ML, Worschech A, Yu Z, et al.
Permissivity of the NCI-60 cancer cell
lines to oncolytic Vaccinia Virus
GLV-1h68. BMC Cancer 2011;11:451
110. Reinboth J, Ascierto ML, Chen NG,
et al. Correlates between host and viral
transcriptional program associated with
different oncolytic vaccinia virus isolates.
Hum Gene Ther Methods
2012;23:285-96
111. Wang E, Phan GQ, Marincola FM.
T-cell-directed cancer vaccines: the
melanoma model. Expert Opin
Biol Ther 2001;1:277-90
112. Rosenberg SA, Packard BS,
Aebersold PM, et al. Use of tumor-
infiltrating lymphocytes and interleukin-2
in the immunotherapy of patients with
metastatic melanoma. A preliminary
report. N Engl J Med 1988;319:1676-80
113. van der Bruggen P, Traversari C,
Chomez P, et al. A gene encoding an
antigen recognized by cytolytic T
lymphocytes on a human melanoma.
J Immunol 2007;178:2617-21
114. Traversari C, van der Bruggen P,
Luescher IF, et al. A nonapeptide
encoded by human gene MAGE-1 is
recognized on HLA-A1 by cytolytic
T lymphocytes directed against tumor
antigen MZ2-E. J Exp Med
1992;176:1453-7
115. Alexandrescu DT, Ichim TE,
Riordan NH, et al. Immunotherapy for
melanoma: current status and
perspectives. J Immunother
2010;33:570-90
116. Wang E, Selleri S, Sabatino M, et al.
Spontaneous and treatment-induced
cancer rejection in humans. Expert Opin
Biol Ther 2008;8:337-49
117. Wang E, Bedognetti D, Marincola FM.
Prediction of response to anticancer
immunotherapy using gene signatures.
J Clin Oncol 2013;31:2369-71. An interesting mini-review on the role
of gene-expression in predicting
outcome to immunotherapy.
118. Iida N, Dzutsev A, Stewart CA, et al.
Commensal bacteria control cancer
response to therapy by modulating the
tumor microenvironment. Science
2013;342:967-70. An interesting study showing the role of
gut microbiota in modulating the tumor
microenvironment and thereby
influencing response to immunotherapy.
119. Wang E, Bedognetti D, Tomei S,
Marincola FM. Common pathways to
tumor rejection. Ann NY Acad Sci
2013;1284:75-9
120. Wang E, Tomei S, Marincola FM.
Reflections upon human cancer immune
responsiveness to T cell-based therapy.
Cancer Immunol Immunother
2012;61:761-70. This manuscript reflects on the
mechanisms behind patients’
individual tumor rejection and
immune responsiveness.
121. Galon J, Angell HK, Bedognetti D,
Marincola FM. The continuum of cancer
immunosurveillance: prognostic,
predictive, and mechanistic signatures.
Immunity 2013;39:11-26.. An updated review summarizing and
explaining striking analogies between
prognostic and predictive
gene-expression signatures.
122. Klebanoff CA, Acquavella N, Yu Z,
Restifo NP. Therapeutic cancer vaccines:
are we there yet? Immunol Rev
2011;239:27-44
123. Kantoff PW, Higano CS, Shore ND,
et al. Sipuleucel-T immunotherapy for
castration-resistant prostate cancer.
N Engl J Med 2010;363:411-22
124. Stein WD, Gulley JL, Schlom J, et al.
Tumor regression and growth rates
determined in five intramural NCI
prostate cancer trials: the growth rate
constant as an indicator of therapeutic
efficacy. Clin Cancer Res
2011;17:907-17
125. Schlom J. Therapeutic cancer vaccines:
current status and moving forward.
J Natl Cancer Inst 2012;104:599-613
126. Motz GT, Coukos G. Deciphering and
reversing tumor immune Suppression.
Immunity 2013;39:61-73
127. Salerno EP, Shea SM, Olson WC, et al.
Activation, dysfunction and retention of
T cells in vaccine sites after injection of
incomplete Freund’s adjuvant, with or
without peptide.
Cancer Immunol Immunother
2013;62:1149-59
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 21
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

128. Hailemichael Y, Dai Z, Jaffarzad N,
et al. Persistent antigen at vaccination
sites induces tumor-specific CD8(+) T
cell sequestration, dysfunction and
deletion. Nat Med 2013;19:465-72
129. Bedognetti D, Spivey TL, Zhao Y, et al.
CXCR3/CCR5 pathways in metastatic
melanoma patients treated with adoptive
therapy and interleukin-2. Br J Cancer
2013;109:2412-23. An interesting study evaluating a
combinatorial approach to biomarker
discovery in cancer immunotherapy.
130. Gattinoni L, Lugli E, Ji Y, et al.
A human memory T cell subset with
stem cell-like properties. Nat Med
2011;17:1290-7
131. Uccellini L, De Giorgi V, Zhao Y, et al.
IRF5 gene polymorphisms in melanoma.
J Transl Med 2012;10:170
132. Wang E, Zhao Y, Monaco A, et al.
A multi-factorial genetic model for
prognostic assessment of high risk
melanoma patients receiving adjuvant
interferon. PLoS One 2012;7:e40805
133. Parmiani G, Cimminiello C, Maccalli C.
Increasing immunogenicity of cancer
vaccines to improve their clinical
outcome. Expert Rev Vaccines
2013;12:1111-13
134. Schwartzentruber DJ, Lawson DH,
Richards JM, et al. gp100 peptide
vaccine and interleukin-2 in patients with
advanced melanoma. N Engl J Med
2011;364:2119-27.. The first randomized trial showing
that vaccination could be effective in
metastatic melanoma when combined
to other modalities.
135. Kruit WH, Suciu S, Dreno B, et al.
Selection of immunostimulant AS15 for
active immunization with MAGE-A3
protein: results of a randomized phase II
study of the European Organisation for
Research and Treatment of Cancer
Melanoma Group in Metastatic
Melanoma. J Clin Oncol
2013;31:2413-20
136. Ulloa-Montoya F, Louahed J, Dizier B,
et al. Predictive gene signature in
MAGE-A3 antigen-specific cancer
immunotherapy. J Clin Oncol
2013;31:2388-95
137. Brichard VG. Selection of patients for
cancer immunotherapy. Society for
immunotherapy of cancer
2013 Annual Meeting, oral presentation
138. Kaufman HL. Vaccines for melanoma
and renal cell carcinoma. Semin Oncol
2012;39:263-75
139. Andtbacka R, Collichio FA, Amatruda T,
et al. OPTiM: a randomized phase III
trial of talimogene laherparepvec
(T-VEC) versus subcutaneous (SC)
granulocyte-macrophage colony-
stimulating factor (GM-CSF) for the
treatment (tx) of unresected stage IIIB/C
and IV melanoma. J Clin Oncol
2013;31(Suppl):abstract LBA9008
140. Chowdhery R, Gonzalez R. Immunologic
therapy targeting metastatic melanoma:
allovectin-7. Immunotherapy
2011;3:17-21
141. Berd D, Sato T, Maguire HC Jr, et al.
Immunopharmacologic analysis of an
autologous, hapten-modified human
melanoma vaccine. J Clin Oncol
2004;22:403-15
142. Gottardi R, Douradinha B. Carbon
nanotubes as a novel tool for vaccination
against infectious diseases and cancer.
J Nanobiotechnology 2013;11:30
143. Pescatori M, Bedognetti D, Venturelli E,
et al. Functionalized carbon nanotubes as
immunomodulator systems. Biomaterials
2013;34:4395-403
144. Delogu LG, Vidili G, Venturelli E, et al.
Functionalized multiwalled carbon
nanotubes as ultrasound contrast agents.
Proc Natl Acad Sci USA
2012;109:16612-17
145. Restifo NP, Dudley ME, Rosenberg SA.
Adoptive immunotherapy for cancer:
harnessing the T cell response.
Nat Rev Immunol 2012;12:269-81.. A comprehensive review on the
advances in adoptive therapy.
146. Hinrichs CS, Restifo NP. Reassessing
target antigens for adoptive T-cell
therapy. Nat Biotechnol
2013;31:999-1008
147. Wang XY, Zuo D, Sarkar D, Fisher PB.
Blockade of cytotoxic T-lymphocyte
antigen-4 as a new therapeutic approach
for advanced melanoma.
Expert Opin Pharmacother
2011;12:2695-706
148. Pardoll DM. The blockade of immune
checkpoints in cancer immunotherapy.
Nat Rev Cancer 2012;12:252-64
149. Allison JP, Krummel MF. The Yin and
Yang of T cell costimulation. Science
1995;270:932-3
150. Krummel MF, Allison JP. CD28 and
CTLA-4 have opposing effects on the
response of T cells to stimulation.
J Exp Med 1995;182:459-65
151. Maio M, Bondarenko I, Robert C, et al.
Four-year survival update for metastatic
melanoma (MM) patients (pts) treated
with ipilimumab (IPI) + dacarbazine
(DTIC) on Phase 3 study CA184-024.
Ann Oncol 23(9s Suppl):
abstract 1127P
152. Wolchok JD, Weber JS, Maio M, et al.
Four-year survival rates for patients with
metastatic melanoma who received
ipilimumab in phase II clinical trials.
Ann Oncol 2013;24:2174-80
153. Maio M, Di Giacomo AM, Robert C,
Eggermont AM. Update on the role of
ipilimumab in melanoma and first data
on new combination therapies.
Curr Opin Oncol 2013;25:166-72
154. Hamid O, Hwu W, Richards J, et al.
Ipilimumab (Ipi) expanded access
program (EAP) for patients (pts) with
stage III/IV melanoma: 10 mg/kg cohort
interim results. J Clin Oncol
2012;30(Suppl):abstract 8508
155. Di Giacomo AM, Ascierto PA, Pilla L,
et al. Ipilimumab and fotemustine in
patients with advanced melanoma
(NIBIT-M1): an open-label, single-arm
phase 2 trial. Lancet Oncol
2012;13:879-86
156. Patel SP, Hwu W, Kim KB, et al. Phase
II study of the frontline combination of
ipilimumab and temozolomide in
patients with metastatic melanoma.
J Clin Oncol 2012;30(Suppl):
abstract 8514
157. Wolchok JD, Hoos A, O’Day S, et al.
Guidelines for the evaluation of immune
therapy activity in solid tumors:
immune-related response criteria.
Clin Cancer Res 2009;15:7412-20
158. Ribas A, Kefford R, Marshall MA, et al.
Phase III randomized clinical trial
comparing tremelimumab with
standard-of-care chemotherapy in
patients with advanced melanoma.
J Clin Oncol 2013;31:616-22
159. Ribas A. Clinical development of the
anti-CTLA-4 antibody tremelimumab.
Semin Oncol 2010;37:450-4
160. Tarhini AA, Cherian J, Moschos SJ,
et al. Safety and efficacy of combination
immunotherapy with interferon alfa-2b
and tremelimumab in patients with stage
S. Tomei et al.
22 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

IV melanoma. J Clin Oncol
2012;30:322-8
161. Calabro L, Morra A, Fonsatti E, et al.
Tremelimumab for patients with
chemotherapy-resistant advanced
malignant mesothelioma: an open-label,
single-arm, phase 2 trial. Lancet Oncol
2013;14:1104-11
162. Freeman GJ, Long AJ, Iwai Y, et al.
Engagement of the PD-1
immunoinhibitory receptor by a novel
B7 family member leads to negative
regulation of lymphocyte activation.
J Exp Med 2000;192:1027-34
163. Latchman Y, Wood CR, Chernova T,
et al. PD-L2 is a second ligand for PD-1
and inhibits T cell activation.
Nat Immunol 2001;2:261-8
164. Brahmer JR, Tykodi SS, Chow LQ,
et al. Safety and activity of
anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med
2012;366:2455-65
165. Topalian SL, Hodi FS, Brahmer JR,
et al. Safety, activity, and immune
correlates of anti-PD-1 antibody in
cancer. N Engl J Med 2012;366:2443-54
166. Hamid O, Robert C, Daud A, et al.
Safety and tumor responses with
lambrolizumab (anti-PD-1) in
melanoma. N Engl J Med
2013;369:134-44
167. Sznol M, Kluger H, Hodi FS, et al.
Survival and long-term follow-up of
safety and response in patients (pts) with
advanced melanoma (MEL) in a phase I
trial of nivolumab (anti-PD-1; BMS-
936558; ONO-4538). J Clin Oncol
2013;31(Suppl):abstract CRA9006
168. Hamid O, Sosman J, Lawrence D, et al.
Clinical activity, safety, and biomarkers of
MPDL3280A, an engineered PD-L1
antibody in patients with locally advanced
or metastatic melanoma (mM).
J Clin Oncol 2013;31(Suppl):
abstract 9010
169. Melero I, Grimaldi AM, Perez-Gracia JL,
Ascierto PA. Clinical development of
immunostimulatory monoclonal
antibodies and opportunities for
combination. Clin Cancer Res
2013;19:997-1008
170. Sugamura K, Ishii N, Weinberg AD.
Therapeutic targeting of the effector
T-cell co-stimulatory molecule OX40.
Nat Rev Immunol 2004;4:420-31
171. Redmond WL, Weinberg AD. Targeting
OX40 and OX40L for the treatment of
autoimmunity and cancer.
Crit Rev Immunol 2007;27:415-36
172. Watts TH. TNF/TNFR family members
in costimulation of T cell responses.
Annu Rev Immunol 2005;23:23-68
173. Croft M. Control of immunity by the
TNFR-related molecule OX40 (CD134).
Annu Rev Immunol 2010;28:57-78
174. Curti BD, Kovacsovics-Bankowski M,
Morris N, et al. OX40 Is a Potent
Immune-Stimulating Target in Late-
Stage Cancer Patients. Cancer Res
2013;73:7189-98
175. Redmond WL, Triplett T, Floyd K,
Weinberg AD. Dual anti-OX40/IL-
2 therapy augments tumor
immunotherapy via IL-2R-mediated
regulation of OX40 expression.
PLoS One 2012;7:e34467
176. French RR, Chan HT, Tutt AL,
Glennie MJ. CD40 antibody evokes a
cytotoxic T-cell response that eradicates
lymphoma and bypasses T-cell help.
Nat Med 1999;5:548-53
177. Diehl L, den Boer AT, Schoenberger SP,
et al. CD40 activation in vivo overcomes
peptide-induced peripheral cytotoxic
T-lymphocyte tolerance and augments
anti-tumor vaccine efficacy. Nat Med
1999;5:774-9
178. Sotomayor EM, Borrello I, Tubb E,
et al. Conversion of tumor-specific
CD4+ T-cell tolerance to T-cell priming
through in vivo ligation of CD40.
Nat Med 1999;5:780-7
179. Vonderheide RH, Glennie MJ. Agonistic
CD40 antibodies and cancer therapy.
Clin Cancer Res 2013;19:1035-43
180. Vonderheide RH, Flaherty KT,
Khalil M, et al. Clinical activity and
immune modulation in cancer patients
treated with CP-870,893, a novel
CD40 agonist monoclonal antibody.
J Clin Oncol 2007;25:876-83
181. Spranger S, Gajewski T. Therapeutic
efficacy of combined blockade of CTLA-
4 +/- PD-L1 +/- IDO is associated with
re-activation of T cells directly within the
tumor microenvironment. J Immuno
Ther Cancer 2013;1(Suppl 1):O8
182. Brignone C, Gutierrez M, Mefti F, et al.
First-line chemoimmunotherapy in
metastatic breast carcinoma: combination
of paclitaxel and IMP321 (LAG-3Ig)
enhances immune responses and
antitumor activity. J Transl Med
2010;8:71
183. Poirier N, Haudebourg T, Brignone C,
et al. Antibody-mediated depletion of
lymphocyte-activation gene-3 (LAG-3(+))
-activated T lymphocytes prevents
delayed-type hypersensitivity in non-
human primates. Clin Exp Immunol
2011;164:265-74
184. Roychoudhuri R, Hirahara K,
Mousavi K, et al. BACH2 represses
effector programs to stabilize T(reg)-
mediated immune homeostasis. Nature
2013;498:506-10
185. Rosenberg SA, Yang JC, Sherry RM,
et al. Durable complete responses in
heavily pretreated patients with
metastatic melanoma using T-cell transfer
immunotherapy. Clin Cancer Res
2011;17:4550-7
186. Radvanyi LG, Bernatchez C, Zhang M,
et al. Specific lymphocyte subsets predict
response to adoptive cell therapy using
expanded autologous tumor-infiltrating
lymphocytes in metastatic melanoma
patients. Clin Cancer Res
2012;18:6758-70
187. Besser MJ, Shapira-Frommer R,
Itzhaki O, et al. Adoptive transfer of
tumor-infiltrating lymphocytes in
patients with metastatic melanoma:
intent-to-treat analysis and efficacy after
failure to prior immunotherapies.
Clin Cancer Res 2013;19:4792-800
188. Dudley ME, Gross CA, Somerville RP,
et al. Randomized selection design trial
evaluating CD8+-enriched versus
unselected tumor-infiltrating lymphocytes
for adoptive cell therapy for patients with
melanoma. J Clin Oncol 2013;31:2152-9
189. Phan GQ, Rosenberg SA. Adoptive cell
transfer for patients with metastatic
melanoma: the potential and promise of
cancer immunotherapy. Cancer Control
2013;20:289-97
190. Bedognetti D, Wang E,
Sato-Matsushita M, et al. Molecular
profiling of immunotherapeutic
resistance. In: Prendergsat GC,
Jaffee EM, editors, Cancer
immunotherapy, immune suppression
and tumor growth. 2nd edition. Elsevier,
2013.
191. Fisher B, Packard BS, Read EJ, et al.
Tumor localization of adoptively
transferred indium-111 labeled tumor
infiltrating lymphocytes in patients with
Non-BRAF-targeted therapy, immunotherapy, and combination therapy for melanoma
Expert Opin. Biol. Ther. (2014) 14 (5) 23
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.

metastatic melanoma. J Clin Oncol
1989;7:250-61
192. Spivey TL, Uccellini L, Ascierto ML,
et al. Gene expression profiling in acute
allograft rejection: challenging the
immunologic constant of rejection
hypothesis. J Transl Med 2011;9:174
193. Ascierto PA, Marincola FM.
Combination therapy: the next
opportunity and challenge of medicine.
J Transl Med 2011;9:115
194. Marincola FM, White DE, Wise AP,
Rosenberg SA. Combination therapy
with interferon alfa-2a and interleukin-2
for the treatment of metastatic cancer.
J Clin Oncol 1995;13:1110-22
195. Daponte A, Signoriello S, Maiorino L,
et al. Phase III randomized study of
fotemustine and dacarbazine versus
dacarbazine with or without interferon-
alpha in advanced malignant melanoma.
J Transl Med 2013;11:38
196. Hodi FS, Lee SJ, McDermott DF, et al.
Multicenter, randomized phase II trial of
GM-CSF plus ipilimumab versus ipi
alone in metastatic melanoma.
J Clin Oncol 2013;31(Suppl):
abstract CRA9007
197. Prieto PA, Yang JC, Sherry RM, et al.
CTLA-4 blockade with ipilimumab:
long-term follow-up of 177 patients with
metastatic melanoma. Clin Cancer Res
2012;18:2039-47
198. Marincola FM. The trouble with
translational medicine. J Intern Med
2011;270:123-7
199. Simon R. Novel clinical trial designs
for development of immunotherapy
combinations. SITC Cancer
Immunotherapy Clinical Trials:
Concepts and Challenges Meeting
2013, oral presentation
200. Yang Y, Lizee G, Hwu P. Strong
emerging rationale for combining
oncogene-targeted agents with
immunotherapy. Oncoimmunology
2013;2:e22730
201. Ribas A, Hersey P, Middleton MR, et al.
New challenges in endpoints for drug
development in advanced melanoma.
Clin Cancer Res 2012;18:336-41
202. Zitvogel L, Galluzzi L, Smyth MJ,
Kroemer G. Mechanism of action of
conventional and targeted anticancer
therapies: reinstating immunosurveillance.
Immunity 2013;39:74-88
203. Ribas A, Wolchok JD. Combining
cancer immunotherapy and targeted
therapy. Curr Opin Immunol
2013;25:291-6
204. Castelli C, Sensi M, Lupetti R, et al.
Expression of interleukin 1 alpha,
interleukin 6, and tumor necrosis factor
alpha genes in human melanoma clones
is associated with that of mutated N-RAS
oncogene. Cancer Res 1994;54:4785-90
205. Sumimoto H, Imabayashi F, Iwata T,
Kawakami Y. The BRAF-MAPK
signaling pathway is essential for cancer-
immune evasion in human melanoma
cells. J Exp Med 2006;203:1651-6
206. Khalili JS, Liu S, Rodriguez-Cruz TG,
et al. Oncogenic BRAF(V600E)
promotes stromal cell-mediated
immunosuppression via induction of
interleukin-1 in melanoma.
Clin Cancer Res 2012;18:5329-40
207. Frederick DT, Piris A, Cogdill AP, et al.
BRAF inhibition is associated with
enhanced melanoma antigen expression
and a more favorable tumor
microenvironment in patients with
metastatic melanoma. Clin Cancer Res
2013;19:1225-31
208. Liu C, Peng W, Xu C, et al. BRAF
inhibition increases tumor infiltration by
T cells and enhances the antitumor
activity of adoptive immunotherapy in
mice. Clin Cancer Res 2013;19:393-403
209. Boni A, Cogdill AP, Dang P, et al.
Selective BRAFV600E inhibition
enhances T-cell recognition of melanoma
without affecting lymphocyte function.
Cancer Res 2010;70:5213-19
210. Ribas A, Hodi FS, Callahan M, et al.
Hepatotoxicity with combination of
vemurafenib and ipilimumab. N Engl
J Med 2013;368:1365-6
211. Flaherty KT, Infante JR, Daud A, et al.
Combined BRAF and MEK inhibition in
melanoma with BRAF V600 mutations.
N Engl J Med 2012;367:1694-703
212. Wolchok JD, Kluger H, Callahan MK,
et al. Nivolumab plus ipilimumab in
advanced melanoma. N Engl J Med
2013;369:122-33
213. Murtas D, Maric D, De Giorgi V, et al.
IRF-1 responsiveness to IFN-gamma
predicts different cancer immune
phenotypes. Br J Cancer 2013;109:76-82
214. Hamid O, Schmidt H, Nissan A, et al.
A prospective phase II trial exploring the
association between tumor
microenvironment biomarkers and
clinical activity of ipilimumab in
advanced melanoma. J Transl Med
2011;9:204
215. Ji RR, Chasalow SD, Wang L, et al. An
immune-active tumor microenvironment
favors clinical response to ipilimumab.
Cancer Immunol Immunother
2012;61:1019-31
216. Carretero R, Wang E, Rodriguez AI,
et al. Regression of melanoma metastases
after immunotherapy is associated with
activation of antigen presentation and
interferon-mediated rejection genes.
Int J Cancer 2012;131:387-95
217. Panelli MC, Stashower ME, Slade HB,
et al. Sequential gene profiling of basal
cell carcinomas treated with imiquimod
in a placebo-controlled study defines the
requirements for tissue rejection.
Genome Biol 2007;8:R8
218. Wang E, Uccellini L, Marincola FM.
A genetic inference on cancer immune
responsiveness. Oncoimmunology
2012;1:520-5
AffiliationSara Tomei†1,2, Ena Wang1,3,
Lucia Gemma Delogu4,
Francesco M Marincola1,3 &
Davide Bedognetti*1
†,*Authors for correspondence1National Institutes of Health, Clinical Center
and trans-NIH Center for Human Immunology
(CHI), Department of Transfusion Medicine,
Infectious Disease and Immunogenetics Section
(IDIS), Bethesda, MD 20892, USA
E-mail: [email protected] Cornell Medical College in Qatar,
Department of Genetic Medicine,
P.O. Box 24144, Doha, Qatar
E-mail: [email protected] Medical and Research Center,
Research Branch, P.O. Box 26999, Doha, Qatar4Universita degli Studi di Sassari,
Dipartimento di Chimica e Farmacia,
07100 Sassari, Italy
S. Tomei et al.
24 Expert Opin. Biol. Ther. (2014) 14(5)
Expe
rt O
pin.
Bio
l. Th
er. D
ownl
oade
d fro
m in
form
ahea
lthca
re.c
om b
y Co
rnel
l Uni
vers
ity o
n 03
/21/
14Fo
r per
sona
l use
onl
y.
Related Documents











