No evidence of selection for mutational robustness during lethal mutagenesis of lymphocytic choriomeningitis virus Verónica Martín a,b , Ana Grande-Pérez c , Esteban Domingo a,b, ⁎ a Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), C/ Nicolás Cabrera, 1. Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain b Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Spain c Área de Genética, Facultad de Ciencias, Campus de Teatinos, Universidad de Málaga, 29071 Málaga, Spain abstract article info Article history: Received 9 April 2008 Returned to author for revision 5 May 2008 Accepted 13 May 2008 Available online 24 June 2008 Keywords: Quasispecies Error catastrophe Arenavirus Antiviral strategy Virus evolution Lethal mutagenesis is a transition towards virus extinction mediated by enhanced mutation rates during viral genome replication. Theoretical studies suggest that viruses can evolve towards regions of their fitness landscapes at which they display resistance to the deleterious effects of mutations. It has been suggested that such mutational robustness could jeopardize lethal mutagenesis. We have used the Arenavirus lymphocytic choriomeningitis virus (LCMV) to explore whether treatment with the mutagenic base analogue 5- fluorouracil (FU) selected for viral populations displaying resistance to lethal mutagenesis. Neither average LCMV populations with a history of FU mutagenesis, nor individual biological LCMV clones derived from those populations, displayed any resistance to lethal mutagenesis by FU. They were as sensitive to FU- induced extinction as LCMV populations and clones treated in parallel, but without a history of FU mutagenesis. Current evidence of the molecular events affecting quasispecies dynamics suggests that it is unlikely that a viral population can acquire mutational robustness under the increased mutation rates associated with mutagenic treatments. We consider mechanisms by which viruses could escape extinction by lethal mutagenesis, and provide evidence that mutational robustness is unlikely to be one of them. © 2008 Elsevier Inc. All rights reserved. Introduction RNA viruses replicate as dynamic distributions of closely related but non-identical genomes, termed viral quasispecies, in agreement with predictions of quasispecies theory, developed initially to explain self- organization and adaptability of early replicons as primitive life forms (Eigen, 1971; Eigen and Schuster, 1979; Eigen et al., 1988; Figlerowicz et al., 2003; Holland, 2006; Nowak, 2006; Biebricher and Domingo, 2007). The understanding of RNA viral populations as quasispecies has been instrumental to interpret virus adaptability and mechanisms of viral pathogenesis [reviews in Domingo et al., 2001, 2008; Holland, 2006]. One of the predictions of quasispecies theory is that there is a link between fidelity of viral replication and genome complexity, through an error threshold for maintenance of genetic information (Eigen and Schuster, 1979; Eigen et al., 1988; Biebricher and Eigen, 2005; Nowak, 2006). The error threshold defines a maximum average error rate compatible with maintenance of the instructions conveyed by an organized quasispecies. Persistent violation of the error thresh- old, so that no components of the mutant spectrum can survive to reconstruct new quasispecies, results in complete loss of information (Eigen and Biebricher, 1988; Eigen, 2002; Biebricher and Eigen, 2005). In the case of viruses, crossing such an error threshold by increasing the mutation rate during virus replication, should result in virus extinction. This prediction has been validated experimentally with several virus–host systems in cell culture and in vivo, and has led to the development of a new antiviral strategy termed lethal mutagenesis [Holland et al., 1990; Loeb et al., 1999; Harris et al., 2005; reviews in Graci and Cameron, 2002; Anderson et al., 2004; Domingo, 2005]. Alternative models that do not relate lethal mutagenesis to the quasispecies error catastrophe, and that emphasize a demographic component that unavoidably accompanies viral extinction, have also been proposed [reviewed in Bull et al., 2008]. Previous studies on the events underlying the extinction of lymphocytic choriomeningitis virus (LCMV) and foot-and-mouth disease virus (FMDV) by mutagenic nucleotide analogues have led to the following observations [summarized from Domingo et al., 2008]: (i) Extinction occurs with decreases of 10 2 - to 10 3 -fold in specific infectivity, and without modification of the consensus nucleotide sequence of the viral population (González-López et al., 2005; Grande- Pérez et al., 2005a). (ii) Low viral load and low viral fitness favor extinction (Sierra et al., 2000; Pariente et al., 2001). (iii) Mutagenic activity, and not only the inhibitory activity that is often associated with it, is necessary to achieve extinction (Pariente et al., 2003; Escarmís et al., 2008). (iv) Interfering interactions among components Virology 378 (2008) 185–192 ⁎ Corresponding author. Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), C/ Nicolás Cabrera, 1. Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain. Fax: +34 91 196 44 20. E-mail addresses: [email protected] (V. Martín), [email protected] (A. Grande-Pérez), [email protected] (E. Domingo). 0042-6822/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.virol.2008.05.016 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/yviro

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Virology 378 (2008) 185–192
Contents lists available at ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r.com/ locate /yv i ro
No evidence of selection for mutational robustness during lethal mutagenesis oflymphocytic choriomeningitis virus
Verónica Martín a,b, Ana Grande-Pérez c, Esteban Domingo a,b,⁎a Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), C/ Nicolás Cabrera, 1. Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spainb Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Spainc Área de Genética, Facultad de Ciencias, Campus de Teatinos, Universidad de Málaga, 29071 Málaga, Spain
⁎ Corresponding author. Centro de Biología MolecularNicolás Cabrera, 1. Universidad Autónoma de Madrid,Spain. Fax: +34 91 196 44 20.
E-mail addresses: [email protected] (V. Martín),(A. Grande-Pérez), [email protected] (E. Domingo)
0042-6822/$ – see front matter © 2008 Elsevier Inc. Aldoi:10.1016/j.virol.2008.05.016
a b s t r a c t
a r t i c l e i n f oArticle history:
Lethal mutagenesis is a tran Received 9 April 2008Returned to author for revision5 May 2008Accepted 13 May 2008Available online 24 June 2008Keywords:QuasispeciesError catastropheArenavirusAntiviral strategyVirus evolution
sition towards virus extinction mediated by enhanced mutation rates during viralgenome replication. Theoretical studies suggest that viruses can evolve towards regions of their fitnesslandscapes at which they display resistance to the deleterious effects of mutations. It has been suggested thatsuch mutational robustness could jeopardize lethal mutagenesis. We have used the Arenavirus lymphocyticchoriomeningitis virus (LCMV) to explore whether treatment with the mutagenic base analogue 5-fluorouracil (FU) selected for viral populations displaying resistance to lethal mutagenesis. Neither averageLCMV populations with a history of FU mutagenesis, nor individual biological LCMV clones derived fromthose populations, displayed any resistance to lethal mutagenesis by FU. They were as sensitive to FU-induced extinction as LCMV populations and clones treated in parallel, but without a history of FUmutagenesis. Current evidence of the molecular events affecting quasispecies dynamics suggests that it isunlikely that a viral population can acquire mutational robustness under the increased mutation ratesassociated with mutagenic treatments. We consider mechanisms by which viruses could escape extinction bylethal mutagenesis, and provide evidence that mutational robustness is unlikely to be one of them.
© 2008 Elsevier Inc. All rights reserved.
Introduction
RNAviruses replicate as dynamic distributions of closely relatedbutnon-identical genomes, termed viral quasispecies, in agreement withpredictions of quasispecies theory, developed initially to explain self-organization and adaptability of early replicons as primitive life forms(Eigen, 1971; Eigen and Schuster, 1979; Eigen et al., 1988; Figlerowiczet al., 2003; Holland, 2006; Nowak, 2006; Biebricher and Domingo,2007). The understanding of RNAviral populations as quasispecies hasbeen instrumental to interpret virus adaptability and mechanisms ofviral pathogenesis [reviews in Domingo et al., 2001, 2008; Holland,2006]. One of the predictions of quasispecies theory is that there is alink between fidelity of viral replication and genome complexity,through an error threshold for maintenance of genetic information(Eigen and Schuster, 1979; Eigen et al., 1988; Biebricher and Eigen,2005; Nowak, 2006). The error threshold defines a maximum averageerror rate compatible with maintenance of the instructions conveyedby an organized quasispecies. Persistent violation of the error thresh-old, so that no components of the mutant spectrum can survive to
“Severo Ochoa” (CSIC-UAM), C/Cantoblanco, 28049, Madrid,
l rights reserved.
reconstruct new quasispecies, results in complete loss of information(Eigen and Biebricher, 1988; Eigen, 2002; Biebricher and Eigen, 2005).In the case of viruses, crossing such an error threshold by increasingthe mutation rate during virus replication, should result in virusextinction. This prediction has been validated experimentally withseveral virus–host systems in cell culture and in vivo, and has led to thedevelopment of a new antiviral strategy termed lethal mutagenesis[Holland et al., 1990; Loeb et al., 1999; Harris et al., 2005; reviews inGraci and Cameron, 2002; Anderson et al., 2004; Domingo, 2005].Alternative models that do not relate lethal mutagenesis to thequasispecies error catastrophe, and that emphasize a demographiccomponent that unavoidably accompanies viral extinction, have alsobeen proposed [reviewed in Bull et al., 2008].
Previous studies on the events underlying the extinction oflymphocytic choriomeningitis virus (LCMV) and foot-and-mouthdisease virus (FMDV) by mutagenic nucleotide analogues have led tothe following observations [summarized from Domingo et al., 2008]:(i) Extinction occurs with decreases of 102- to 103-fold in specificinfectivity, and without modification of the consensus nucleotidesequence of the viral population (González-López et al., 2005; Grande-Pérez et al., 2005a). (ii) Low viral load and low viral fitness favorextinction (Sierra et al., 2000; Pariente et al., 2001). (iii) Mutagenicactivity, and not only the inhibitory activity that is often associatedwith it, is necessary to achieve extinction (Pariente et al., 2003;Escarmís et al., 2008). (iv) Interfering interactions among components

186 V. Martín et al. / Virology 378 (2008) 185–192
of the mutant spectra play a key role in virus extinction (González-López et al., 2004; Grande-Pérez et al., 2005a,b; Perales et al., 2007).Participation of a class of interfering genomes, termed defectors, inviral extinction was supported by the observation that loss ofinfectious LCMV preceded loss of replicating LCMV RNA, and by insilico simulations of the fate of infectious genomes in the course of viralreplication in the presence or absence of interfering genomes (Grande-Pérez et al., 2005b).These results led to the proposal of the lethal defec-tionmodel of virus extinction. Interferencewithin quasispecies has beenobserved also with pre-extinction FMDV populations (González-Lópezet al., 2004), with specific capsid and polymerase mutants of FMDV(Perales et al., 2007), and with several proteins and the cre replicationRNA element of poliovirus (PV) (Crowder and Kirkegaard, 2005). At lowmutagenic intensities, virusmaygo extinct by the action of defectors, andat high mutagenic intensities, the system may collapse as a result ofincreasing interfering interactions, and also of lethal mutations thateliminate viable genomes (González-López et al., 2004; Arias et al., 2005;Grande-Pérez et al., 2005a,b; Takeuchi and Hogeweg, 2007).
A critical issue regarding lethal mutagenesis as an antiviral strategyis whether virus mutants resistant to the mutagenic activities can beselected, and whether they can jeopardize extinction. Mutant RNAviruses with decreased sensitivity to inhibitors of viral replication arealmost universally selected in cell culture and in vivo, due to thecombined effects of high mutation rates during viral replication andthe requirement of one or very few mutations to decrease virussensitivity to an antiviral inhibitor (Domingo, 2003; Figlerowicz et al.,2003). PV, hepatitis C virus (HCV) and FMDV that show resistanceto the nucleoside analogue ribavirin (1-β-D-ribofuranosyl-1, 2, 3-triazole-3-carboxamide) –which is mutagenic for these viruses–, havebeen selected and the relevant mutations were located in thecorresponding viral polymerases (Crotty et al., 2000, 2001; Pfeifferand Kirkegaard, 2003; Vo et al., 2003; Young et al., 2003; Freistadtet al., 2004; Castro et al., 2005; Pfeiffer and Kirkegaard, 2005b,a;Vignuzzi et al., 2006; Sierra et al., 2007). The administration of FU tomice can prevent the establishment of a persistent infection in vivo.This experiment represented the first evidence of the feasibility of alethal mutagenesis approach in vivo (Ruiz-Jarabo et al., 2003).Ribavirin acted as a potent inhibitor of LCMV replication, and nomutagenic activity could be documented (Ruiz-Jarabo et al., 2003).The effects on the levels of viral replication and particle production oftreatments with ribavirin and FU suggest different mechanisms ofaction (Ruiz-Jarabo et al., 2003). Although mutations that decreasesensitivity to mutagenic nucleotide analogues are likely to diminishthe effectiveness of lethal mutagenesis treatments, the effect ofmutations that confer resistance to ribavirin on virus extinction hasnot been quantified. It should be also considered that resistancemutations often entail a fitness cost and that low fitness virus in vivocould be easier to clear by the host immune responses.
An alternative mechanism that has been proposed to act againstextinction by lethal mutagenesis is robustness (Sanjuan et al., 2007).Robustness has been variously defined as the invariance of phenotypesin the face of perturbation (de Visser et al., 2003) or, more specificallyfor the case of viruses, as phenotypic constancy in the face ofmutationalchanges in the genome (Montville et al., 2005). Two populations ofvesicular stomatitis virus (VSV) manifested different sensitivity to 5-fluorouracil (FU)mutagenesis, attributed to oneof thepopulations lyingon a flat fitness surface that rendered the virus less sensitive to thedeleterious effects of mutations (Sanjuan et al., 2007). Fitness surfacerefers in this case to the shape that values of replicative capacity willfollow when genetic variations of the virus or environmental changesoccur. It follows the general adaptive landscape concept initiallyproposed by (Wright, 1931). The possible acquisition of increasedresistance to the deleterious effects of mutations by virtue of virusesfinding a flat fitness surface was predicted by theoretical studies(Schuster and Swetina, 1988; Wilke et al., 2001), and it has beensupported by some experimental results. A VSV mutant behaved as a
neutral mutant relative to the wild type virus under standardreplication conditions, but displayed a selective disadvantage whenenvironmental perturbations intervened during competitive replica-tion (Quer et al., 1996). In particular, FU mutagenesis prompted thedominance of the wild type over the mutant VSV. The neutral mutantincluded a number of mutations relative to the wild type that mighthave rendered it vulnerable to additional mutations (Quer et al., 2001).Evidence of robustness has been also obtained with viroids replicatingin UV-irradiated plants (Codoner et al., 2006).
In view of this previous evidence, and due to the relevance of apossiblemutational robustness to the detrimental effects of mutationsin a lethal mutagenesis approach, we have examined whetherenhanced mutagenesis resulted in selection of variants of theprototypic LCMV that displayed mutational robustness, and therebyjeopardize the use of lethal mutagenesis as an antiviral strategy tocombat arenavirus infections. Notably, several arenaviruses, chieflyLassa virus, are a group of highly significant human pathogens forwhich no effective vaccines, and only limited possibilities forpharmacological intervention, are currently available [reviewed inOldstone, 2002]. Furthermore, evidence indicates that LCMV is aneglected humanpathogen (Fischer et al., 2006; Jamieson et al., 2006).The LCMV genome consists of two segments of single stranded RNA(small or S, of 3376 nucleotides; large or L, of 7228 nucleotides) ofnegative polarity. Each segment uses an ambisense coding strategy todirect the synthesis of two viral polypeptides. The S segment encodesthe virus nucleoprotein (NP) and glycoprotein precursor GPC, which isposttranslationally processed into mature GP1 and GP2 proteins viaproteolytic cleavage by the cellular protease S1P. The L segmentencodes the virus RNA-dependent RNA polymerase (L polymerase)and the small RING finger protein Z that is the counterpart of thematrix (M) protein found in many other enveloped negative strandRNA viruses (Buchmeier et al., 2007). LCMV is highly sensitive to FU-mediated extinction (Grande-Pérez et al., 2002; Ruiz-Jarabo et al.,2003), and contributed the first proof of principle of the feasibility of alethal mutagenesis approach in vivo, by documenting that FUprevented the establishment of a persistent LCMV infection in mice(Ruiz-Jarabo et al., 2003). Here we provide evidence against evolutionof LCMV towards mutational robustness in the course of mutagenesistreatments that eventually lead to virus extinction.
Results
Sensitivity to 5-fluorouracil mutagenesis of LCMV populations with orwithout a history of 5-fluorouracil mutagenesis
LCMV (Arm 53b) can be extinguished by treatment with FU in adose-dependent manner (Grande-Pérez et al., 2002; Ruiz-Jarabo et al.,2003). In serial, low multiplicity of infection (m.o.i.) passages in BHK-21 cells (as detailed inMaterials andmethods), concentrations of FU of50 to 100 μg/ml resulted in virus extinction in 2 to 3 passages, while FUconcentrations of 10 to 20 μg/ml resulted in reduction of virus titer butno viral extinction for at least 13 passages (Grande-Pérez et al., 2002;Martín et al., unpublished results).We chose four LCMV populations totest their sensitivity to FUmutagenesis: Cp5 and Cp9, that correspondto LCMV serially passaged in BHK-21 cells 5 and 9 times, respectively,in the absence of FU; and FU-20p5 and FU-20p9 that correspond toLCMV serially passaged in BHK-21 cells 5 and 9 times, respectively, inthe presence of 20 μg/ml FU (Fig. 1). Duplicate samples of the fourpopulations were subjected to three additional serial passages eitherin the absence or the presence of FU (100 μg/ml). In all cases, ex-tinction occurred in the first or second passage in the presence of FU,and not in its absence (Fig. 2). Extinction was ascertained by absenceof infectivity and absence of specific RT-PCR amplifiable, LCMV-specific sequences, after three blind passages of the viral population inthe absence of mutagens, as detailed in Materials and methods. Thus,the results confirm the sensitivity to FU-induced lethal mutagenesis of
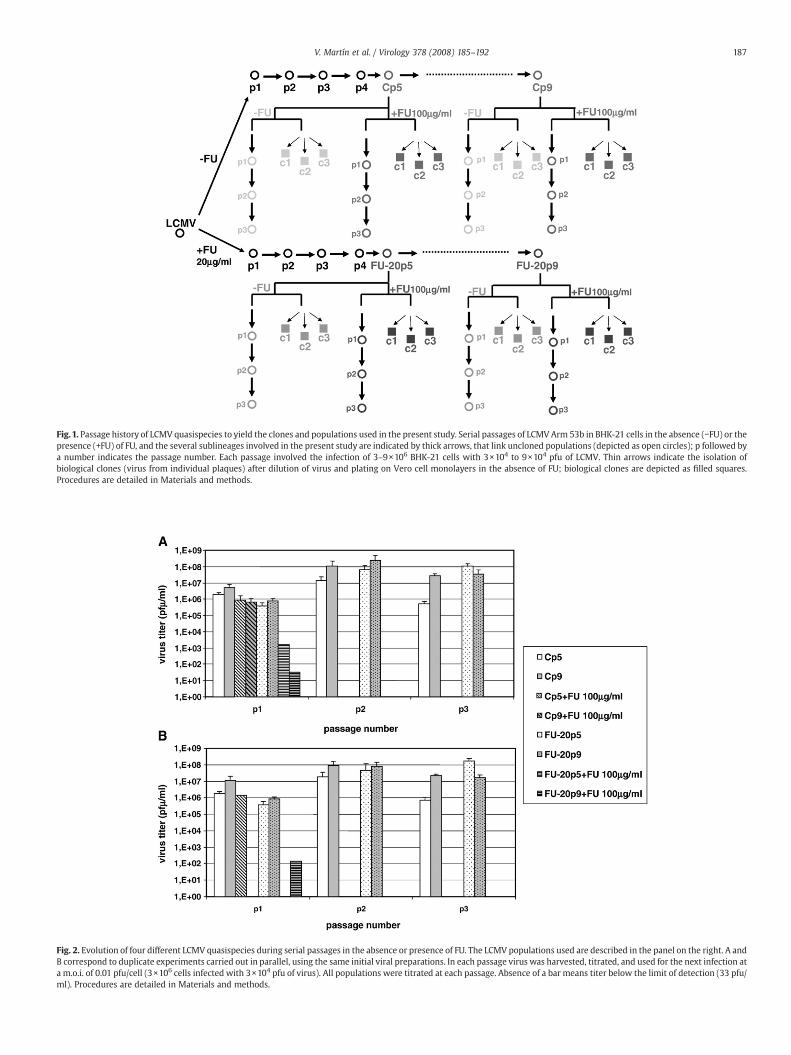
Fig. 2. Evolution of four different LCMV quasispecies during serial passages in the absence or presence of FU. The LCMV populations used are described in the panel on the right. A andB correspond to duplicate experiments carried out in parallel, using the same initial viral preparations. In each passage virus was harvested, titrated, and used for the next infection ata m.o.i. of 0.01 pfu/cell (3×106 cells infected with 3×104 pfu of virus). All populations were titrated at each passage. Absence of a bar means titer below the limit of detection (33 pfu/ml). Procedures are detailed in Materials and methods.
Fig.1. Passage history of LCMV quasispecies to yield the clones and populations used in the present study. Serial passages of LCMV Arm 53b in BHK-21 cells in the absence (−FU) or thepresence (+FU) of FU, and the several sublineages involved in the present study are indicated by thick arrows, that link uncloned populations (depicted as open circles); p followed bya number indicates the passage number. Each passage involved the infection of 3–9×106 BHK-21 cells with 3×104 to 9×104 pfu of LCMV. Thin arrows indicate the isolation ofbiological clones (virus from individual plaques) after dilution of virus and plating on Vero cell monolayers in the absence of FU; biological clones are depicted as filled squares.Procedures are detailed in Materials and methods.
187V. Martín et al. / Virology 378 (2008) 185–192
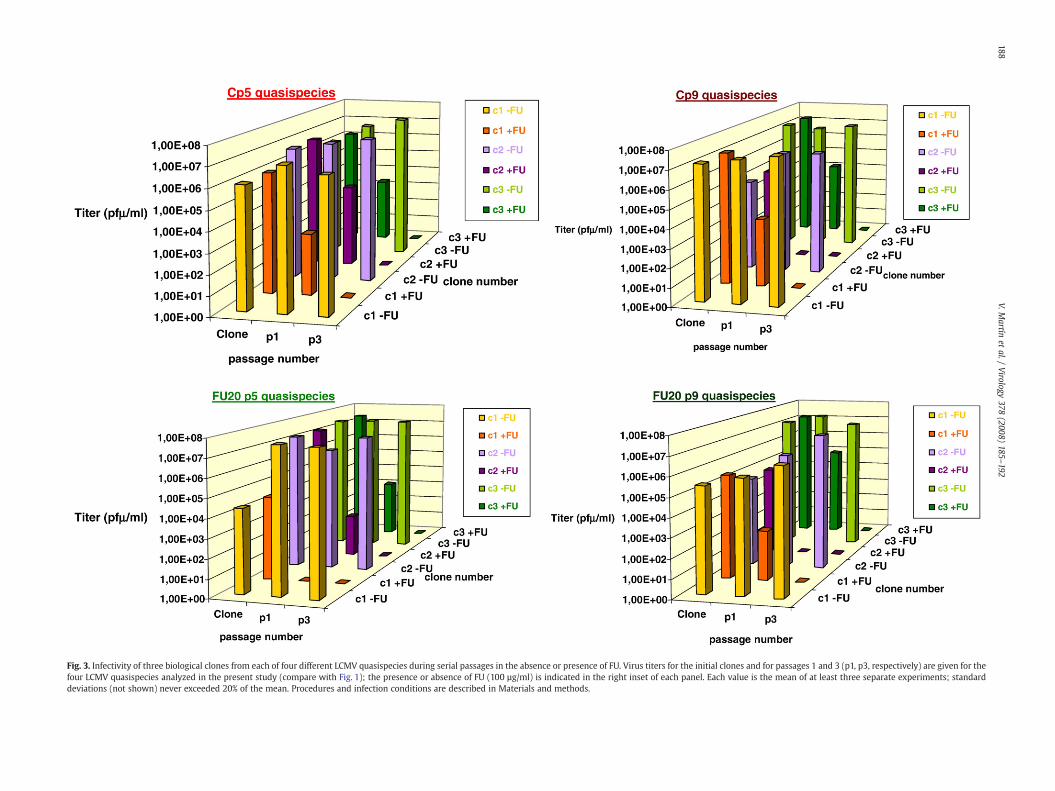
Fig. 3. Infectivity of three biological clones from each of four different LCMV quasispecies during serial passages in the absence or presence of FU. Virus titers for the initial clones and for passages 1 and 3 (p1, p3, respectively) are given for thefour LCMV quasispecies analyzed in the present study (compare with Fig. 1); the presence or absence of FU (100 µg/ml) is indicated in the right inset of each panel. Each value is the mean of at least three separate experiments; standarddeviations (not shown) never exceeded 20% of the mean. Procedures and infection conditions are described in Materials and methods.
188V.M
artínet
al./Virology
378(2008)
185–192

189V. Martín et al. / Virology 378 (2008) 185–192
LCMV, irrespective of a prior history of replication in the presence ofFU, and provide no evidence of acquisition of mutational robustness ofLCMV in the course of mutagenesis.
Sensitivity to 5-fluorouracil mutagenesis of biological clones of LCMVisolated from populations with or without a history of 5-fluorouracilmutagenesis
Mutagenized LCMV populations contain defective genomes thatcan interfere with the replication of infectious LCMV, and maycontribute to virus extinction, in agreement with the lethal defectionmodel of virus extinction (Grande-Pérez et al., 2005b) (see Introduc-tion). Therefore, it could be argued that the sensitivity of unclonedLCMV populations to FU was favored by defector genomes present inthe population, but that biological clones from the populations treatedwith FU had acquired mutational robustness. To test this possibility, 3biological clones from each of the LCMV populations Cp5, Cp9, FU-20p5 and FU-20p9 were isolated (as described in Materials andmethods), and subjected to 3 serial passages either in the absence orpresence of FU (100 μg/ml). Extinction was observed at passage 2 to 3for all clones passaged in the presence of FU, and not in its absence(Fig. 3), irrespective of the prior history of FU-induced mutagenesis ofthe populations fromwhich the clones were isolated. No difference inthe time (number of passages) required to reach extinction wasobserved between clones isolated from populations without a historyof FU treatment (Cp5, Cp9) and those with a history of FU treatment(FU-20p5, FU-20p9) (Fig. 3). Thus both, uncloned LCMV populations,and biological clones derived from those populations, did not showany resistance to FU-induced extinction, despite a prior history of FUmutagenesis.
Discussion
Lethal mutagenesis is currently under intense investigation as apotential new antiviral strategy, with an ongoing phase 2 clinical trialwith AIDS patients under treatment with an antiviral mutagenicnucleoside analogue (Harris et al., 2005). New nucleotide analoguesare currently under study that could find an application in lethalmutagenesis of viruses (Harki et al., 2006; Graci et al., 2007; Harkiet al., 2007; Graci et al., 2008; Moriyama et al., 2008). A possibleclinical application of lethal mutagenesis to persistent ribovirus andretrovirus infections is encouraged by the documented extinction ofmany viruses as a result of their replication in the presence ofmutagenic agents [reviewed in Graci and Cameron, 2002; Andersonet al., 2004; Domingo, 2005]. Decreases in viral load as a result ofenhanced mutagenesis of the virus may contribute to virus clearanceby the host immune response. Therefore, it is of utmost importance toidentify those mechanisms that may lead to selection of viral mutants(or viral populations) resistant to the mutagenic treatment, and thatmay permit viruses to evade extinction. The question is particularlypertinent given the widespread occurrence of viruses resistant to (oreven dependent on) antiviral inhibitors (Domingo, 2003; Figlerowiczet al., 2003; Baldwin and Berkhout, 2007). At least two extinction-escape mechanisms can be envisaged: (i) selection of virus mutantsthat, because of substitutions in the viral polymerase, are deficient inthe incorporation of one or several mutagenic nucleotide analogues,and (ii) acquisition of mutational robustness (insensitivity to thedeleterious effects ofmutations). Mechanism (i) has been documentedwith the isolation of PV, HCV and FMDV polymerase mutants withdecreased sensitivity to ribavirin (Pfeiffer and Kirkegaard, 2003,2005b; Vo et al., 2003; Pfeiffer and Kirkegaard, 2005b; Vignuzziet al., 2006; Sierra et al., 2007). Mechanism (ii) has been proposedon the basis of the comparison of two VSV populations, but themolecular basis underlying the increased mutational robustnessattributed to one of the VSV populations was not identified (Sanjuanet al., 2007).
Mutational robustness has been suggested as a mechanism forviruses to reach a mutation-selection balance in the face of highmutation rates during RNA genome replication and retrotranscription(Drake and Holland, 1999; Holland, 2006; Domingo, 2007). Robust-ness offers a mechanism by which viruses can avoid (or attenuate) thedeleterious effects of mutations. The proposal assumes that virusescan evolve to increase the fraction of genomic residues that are neutralor quasi-neutral [reviewed in Wilke and Adami, 2003], but themolecular events underlying such evolution are not known. Since ithas been proposed that high mutation rates favor evolution towardsmutational robustness (Montville et al., 2005; Elena et al., 2007), timewas ripe to suggest that mutational robustness could be a mechanismto escape lethal mutagenesis (Sanjuan et al., 2007).
In the present study, we have found no evidence of even minimalalterations of sensitivity to extinction, either of uncloned LCMVpopulations or of biological clones, after treatment with FU. Not even atrend towards anyminimal increased resistance to extinctionwas notedin LCMV populations or clones that had been subjected to 9 passages inthe presence of FU, when compared either with their parentalpopulations or with passaged populations not subjected to FUmutagenesis. It could be argued that many more replication roundsmight be needed for LCMV to evolve towards mutational robustness.However, nine passages are more than double the number of passagesneeded for LCMVextinction under conditions of lethalmutagenesis, andhere we have tested robustness as a possible impairment of virusextinction. The number of passages allowed for LCMV to acquirerobustness was commensurate with the number of passages requiredfor extinction (Grande-Pérez et al., 2002, 2005a,b; Ruiz-Jarabo et al.,2003; Grande-Pérez et al., 2005a,b). Therefore, mutational robustnesssuggested as a resistance mechanism against lethal mutagenesis(Sanjuan et al., 2007) does not operate in LCMV under our experimentalconditions, and, therefore, robustness cannot be regarded as a generalmechanism of resistance to lethal mutagenesis. Contrary to detection ofribavirin-resistant mutants of FMDV (Sierra et al., 2007), we have failedto obtain FMDV or LCMV populations that show decreased sensitivity toFU. Althoughwe cannot exclude that suchmutants might be eventuallyobtained, and that viral polymerases with decreased capacity toincorporate FU might exist, our current evidence suggests that theyareunlikely tobe readily selectedduringa lethalmutagenesis treatment.
Basic observations with RNA virus genomes and their evolutionrender unlikely mutational robustness as a mechanism to escape lethalmutagenesis. One is that in RNA viruses both the genome itself andencoded proteins (most of them multifunctional) determine the viralphenotype. Therefore, the neutral space of genomic nucleotides mustbe extremely reduced in RNA viruses. Despite theoretical and experi-mental evidence of RNA genome populations displaying differentsensitivity to mutations (Quer et al., 1996, 2001; Wilke et al., 2001;Codoner et al., 2006; Schuster and Stadler, 2008) it is unlikely that apathway towards increased robustness can be found under theenhanced mutagenesis conditions and within the time span inherentto a lethal mutagenesis treatment. A revealing comparison has beenprovided by biological FMDV clones that accumulated mutations (thatreached 1.5×10−2 substitutions per genomic nucleotide), following 409plaque-to-plaque transfers, without extinction (Escarmís et al., 2008).Experimental and in silico evidence suggests that the occurrence at lowfrequency of compensatory mutations allowed viral subpopulations tobe refounded for virus survival regardless of extinction of many clonespresent in the population (Lázaro et al., 2002, 2003; Manrubia et al.,2005; Escarmís et al., 2008). Despite FMDV harbouring a potential forsurvival of minority subpopulations, no such a survival has beenobserved when the same FMDV clones were subjected to lethalmutagenesis (Pariente et al., 2005; Escarmís et al., 2008). Nocompensatory mutations could be selected in the face of heavymutagenesis, and no slow-replicating subpopulations, if they werepresent, could find their way towards dominance in the populations.Modulation of replication rate is likely to require multiple coordinated

190 V. Martín et al. / Virology 378 (2008) 185–192
mutations at both the polymerase and other accompanying proteins.Such requirements are unlikely to be achieved under enhancedmutagenesis, unless slowly-replicating, mutagenesis-refractory sub-populations preexisted in substantial proportions in the evolving RNAviral quasispecies. Experimental evidence to support this possibility islacking.
Evidence suggests that real viruses do not move within smoothfitness landscapes (Wright, 1931, 1982) but, rather, that they movethrough rough landscapes. It is extremely unlikely than in a short time(limited number of replication rounds) a virus will encounter the flatfitness surface to render it robust to mutations, andmore so under theinstability conditions enforced by enhanced mutagenesis. Evidencepoints towards long-termevolution as themore realistic conditions forviruses to increase mutational robustness. The case of the VSV mutantthat lost its neutrality under FUmutagenesis (and other environmentalperturbations) (Quer et al., 1996, 2001) involved a relatively enhancedresistance to mutagenesis of the wild type VSV relative to the mutant,but certainlywild typeVSVwasnot located in afitness surface of such anature as to render it insensitive to enhanced mutagenesis. This wasextensively documented in the pioneer studies of John Holland andcolleagues on error catastrophe of wild type VSV (Holland et al., 1990;Lee et al., 1997). We conclude that mutational robustness of LCMV isunlikely to play any significant role in preventing or delaying lethalmutagenesis of LCMV during replication in BHK-21 cells.
Materials and methods
Cells and virus
Virus infections and maintenance of baby hamster kidney (BHK-21) and Vero cells were carried out as described (Sierra et al., 2000;Grande-Pérez et al., 2002; Meyer et al., 2002). LCMV strain Armstrong(Arm) 53b is a triple plaque-purified clone from LCMV Arm CApassaged four times in BHK-21 cells. Virus infectivity in samples fromthe supernatant of infected cultures was determined by plaque assayon Vero cell monolayers, because the virus does not form lytic plaqueson BHK-21 cells. Vero cell monolayers (1×106 cells in six-well dishes)were infected in duplicate with serial dilutions of the virus prepara-tions. After 90 min of absorption at 37 °C, 7% CO2, the virus wasremoved and cells were overlaid with 4 ml of DMEM supplementedwith 0.7% FCS, 2% L-glutamine/50 μg/ml, gentamicin, and 0.3% agar.After 7 days, cells were stained with 2% crystal violet in 2%formaldehyde, and viral plaques were counted.
History of the viral quasispecies used
Four different LCMV quasispecies were chosen from previous work(Grande-Pérez et al., 2002). Cp5 and Cp9 are LCMV Arm 53b passagedin BHK-21 cells five and nine times, respectively, in absence of FU. Eachpassage involved infection of 3–9×106 cells with 3–9×104 104 pfu ofLCMV. FU-20p5 and FU-20p9 are LCMV Arm 53b passaged in BHK-21cells five and nine times, respectively, in presence of 20 μg/ml of FU.This concentration of FU was not sufficient to lead to LCMV extinction(Grande-Pérez et al., 2002). Each passage involved infection of 3–9×106 cells with 3–9×104 104 pfu of LCMV (Grande-Pérez et al., 2002).
Virus infections
Semiconfluent monolayers of BHK-21 (3×106 cells in 100-mmdiameter dishes) were infected at a m.o.i. of 0.01 pfu per cell, in 10 mlof DMEM supplemented with 10% FCS, 2% L-glutamine, 0.52% glucoseand 50 μg/ml gentamicin, either in the absence or the presence of FU(200 μg/ml), following described procedures (Grande-Pérez et al.,2002). Supernatants from infected cells (and from mock-infectedcells) were harvested 48 h postinfection, clarified by centrifugation at2500 rpm for 30min at 4 °C, and stored at −80 °C. Virus infectivity was
determined by plaque assay on Vero cell monolayers as described(Ahmedet al.,1988). Infectivity ranged from3.6×105 to 3.4×108 pfu/mlin the absence of FU, and 1.3×106 to b33 pfu/ml in the presence of FU.The absence of plaques in supernatants from mock-infected culturesascertained the absence of contaminationwith virus. Values shown arethe mean of at least three determinations.
Selection of individual biological clones from LCMV quasispecies
Vero cells monolayers (1×106 cells in six-well dishes) wereinfected with serial dilutions of LCMV, and after 90 minutes ofadsorption, the virus was removed and cells were overlaid with 4 mlof DMEM (without phenol red), and supplemented with 0.7% FCS, 2%L-glutamine/50 μg/ml, gentamicin, 0.3% agar. No FU was included inthe medium. After 4–5 days, virus from individual plaques werevisualized under visible light (facilitated by absence of phenol red),and the contents of individual plaques were resuspended in 500 μl ofDMEM without FCS, and titrated. Titer of resuspended LCMV bio-logical clones and passaged once in M-24 well ranged from 104 to107 pfu/ml.
Drug treatment
Preparation of FU stock solutions, determination of BHK-21 cellviability, and procedures for infections in the presence of thismutagenic base analogue were performed as described (Grande-Pérez et al., 2002). Al least 80% cell survival was scored after the FUtreatments performed in the present study, in agreement withprevious determination (Sierra et al., 2000). LCMV was consideredextinguished when no reverse transcriptase-PCR (RT-PCR)-amplifi-able material, and no infectivity (b33 pfu/ml, limit of detection) couldbe observed after three blind passages of the undiluted viralpopulation in BHK-21 cells in standard culture medium, in theabsence of mutagens (Sierra et al., 2000; Pariente et al., 2001).
RNA extraction and reverse transcription PCR
RNA was extracted with Trizol (Invitrogen) from supernatants ofinfected cultures, following the manufacturer's protocol. RNAs wereamplified by RT-PCR by using ThermoScript reverse transcriptase(Invitrogen) and a reverse (antisense) primer at 60 °C for 45 min,followed by PCR with Pfu DNA polymerase (Promega). cDNAs wereanalyzed by agarose gel electrophoresis.
Acknowledgments
We are indebted to J.C. de la Torre and J. J. Holland for valuableadvice throughout this project, and for the critical reading of themanuscript. We thank M. Dávila and A.I. de Ávila for expert technicalassistance. Work supported by grant BFU2005-00863 and FundaciónRamón Areces. CIBERehd is funded by Instituto de Salud Carlos III. A.Grande-Perez was supported by a contract Ramón y Cajal from MEC.
References
Ahmed, R., Simon, R.S., Matloubian, M., Kolhekar, S.R., Southern, P.J., Freedman, D.M.,1988. Genetic analysis of in vivo-selected viral variants causing chronic infection:importance of mutation in the L RNA segment of lymphocytic choriomeningitisvirus. J. Virol. 62 (9), 3301–3308.
Anderson, J.P., Daifuku, R., Loeb, L.A., 2004. Viral error catastrophe by mutagenicnucleosides. Annu. Rev., Microbiol. 58, 183–205.
Arias, A., Agudo, R., Ferrer-Orta, C., Perez-Luque, R., Airaksinen, A., Brocchi, E., Domingo, E.,Verdaguer, N., Escarmis, C., 2005. Mutant viral polymerase in the transition of virus toerror catastrophe identifies a critical site forRNAbinding. J.Mol. Biol. 353 (5),1021–1032.
Baldwin, C., Berkhout, B., 2007. HIV-1 drug-resistance and drug-dependence. Retro-virology 4, 78.
Biebricher, C.K., Eigen, M., 2005. The error threshold. Virus Res. 107 (2), 117–127.Biebricher, C.K., Domingo, E., 2007. The advantage of the high genetic diversity in RNA
viruses. Future Virol. 2 (1), 35–38.

191V. Martín et al. / Virology 378 (2008) 185–192
Buchmeier, M.J., d.l.T., J.C., Peters, C.J., 2007. Arenaviridae: the viruses and theirreplication, In: Knipe, D.M., Howley, P.M., et al. (Eds.), Fields Virology, 5th ed.Lappincott Williams & Wilkins, Phyladelphia, pp. 1791–1827.
Bull, J.J., Sanjuan, R., Wilke, C.O., 2008. Lethal Mutagenesis, In: Domingo, E., Parrish, C.,Holland, J.J., et al. (Eds.), Origin and evolution of viruses, 2nd edition. Elsevier,Amsterdam (The Netherlands), pp. 207–218.
Castro, C., Arnold, J.J., Cameron, C.E., 2005. Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective.Virus Res. 107 (2), 141–149.
Codoner, F.M., Daros, J.A., Sole, R.V., Elena, S.F., 2006. The fittest versus the flattest:experimental confirmation of the quasispecies effect with subviral pathogens. PLoSPathog. 2 (12), e136.
Crotty, S., Maag, D., Arnold, J.J., Zhong, W., Lau, J.Y.N., Hong, Z., Andino, R., Cameron, C.E.,2000. The broad-spectrum antiviral ribonucleotide, ribavirin, is an RNA virusmutagen. Nat. Med 6, 1375–1379.
Crotty, S., Cameron, C.E., Andino, R., 2001. RNA virus error catastrophe: direct moleculartest by using ribavirin. Proc. Natl. Acad. Sci. U. S. A. 98 (12), 6895–6900.
Crowder, S., Kirkegaard, K., 2005. Trans-dominant inhibition of RNAviral replication canslow growth of drug-resistant viruses. Nat. Genet. 37 (7), 701–709.
de Visser, J.A., Hermisson, J., Wagner, G.P., Ancel Meyers, L., Bagheri-Chaichian, H.,Blanchard, J.L., Chao, L., Cheverud, J.M., Elena, S.F., Fontana,W., Gibson, G., Hansen, T.F.,Krakauer, D., Lewontin, R.C., Ofria, C., Rice, S.H., von Dassow, G., Wagner, A., Whitlock,M.C., 2003. Perspective: evolution and detection of genetic robustness. Evolut. Int. J.Org. Evolut. 57 (9), 1959–1972.
Domingo, E., 2003. Quasispecies and the development of new antiviral strategies. Prog.Drug Res. 60, 133–158.
Domingo, E., 2005. Virus entry into error catastrophe as a new antiviral strategy. VirusRes. 107, 115–228.
Domingo, E., 2007. Virus evolution. In: Knipe, D.M., Howley, P.M., et al. (Eds.), FieldsVirology. Lappincott Williams & Wilkins, Philadelphia, pp. 389–421.
Domingo, E., Biebricher, C., Eigen, M., Holland, J.J., 2001. Quasispecies and RNA VirusEvolution: Principles and Consequences. Landes Bioscience, Austin.
Domingo, E., Escarmis, C., Menéndez-Arias, L., Perales, C., Herrera, M., Novella, I.,Holland, J., 2008. Viral quasiespecies:dynamics, interactions and pathogenesis, In:Domingo, E., Parrish, C., Holland, J.J., et al. (Eds.), Origin and Evolution of viruses,2nd edition. Elsevier, Amsterdam (The Netherlands), pp. 87–118.
Drake, J.W., Holland, J.J., 1999. Mutation rates among RNAviruses. Proc. Natl. Acad. Sci.U. S. A. 96, 13910–13913.
Eigen, M., 1971. Selforganization of matter and the evolution of biological macro-molecules. Naturwissenschaften 58 (10), 465–523.
Eigen, M., 2002. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. U. S. A.99 (21), 13374–13376.
Eigen, M., Schuster, P., 1979. The Hypercycle. A Principle of Natural Self-organization.Springer, Berlin.
Eigen, M., Biebricher, C.K., 1988. Sequence space and quasispecies distribution. In:Domingo, E., Ahlquist, P., Holland, J.J., et al. (Eds.), RNA Genetics. 3CRC Press, BocaRaton, FL., pp. 211–245.
Eigen, M., McCaskill, J., Schuster, P., 1988. Molecular quasi-species. J. Phys. Chem. 92,6881–6891.
Elena, S.F., Wilke, C.O., Ofria, C., Lenski, R.E., 2007. Effects of population size andmutation rate on the evolution of mutational robustness. Evolut. Int. J. Org. Evolut.61 (3), 666–674.
Escarmís, C., Lazaro, E., Arias, A., Domingo, E., 2008. Repeated bottleneck transfers canlead to non-cytocidal forms of a cytopathic virus: implications for viral extinction.J. Mol. Biol. 376 (2), 367–379.
Figlerowicz, M., Alejska, M., Kurzynska-Kokorniak, A., Figlerowicz, M., 2003. Geneticvariability: the key problem in the prevention and therapy of RNA-based virusinfections. Med Res. Rev. 23 (4), 488–518.
Fischer, S.A., Graham,M.B., Kuehnert, M.J., Kotton, C.N., Srinivasan, A.,Marty, F.M., Comer,J.A., Guarner, J., Paddock, C.D., DeMeo, D.L., Shieh, W.J., Erickson, B.R., Bandy, U.,DeMaria Jr., A., Davis, J.P., Delmonico, F.L., Pavlin, B., Likos, A., Vincent,M.J., Sealy, T.K.,Goldsmith, C.S., Jernigan, D.B., Rollin, P.E., Packard, M.M., Patel, M., Rowland, C.,Helfand, R.F., Nichol, S.T., Fishman, J.A., Ksiazek, T., Zaki, S.R., 2006. Transmission oflymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354(21), 2235–2249.
Freistadt, M.S., Meades, G.D., Cameron, C.E., 2004. Lethal mutagens: broad-spectrumantivirals with limited potential for development of resistance? Drug Resist. Updat.7 (1), 19–24.
González-López, C., Gómez-Mariano, G., Escarmís, C., Domingo, E., 2005. Invariantaphthovirus consensus nucleotide sequence in the transition to error catastrophe.Infect. Genet. Evolut. 5, 366–374.
González-López, C., Arias, A., Pariente, N., Gómez-Mariano, G., Domingo, E., 2004.Preextinction viral RNA can interfere with infectivity. J. Virol. 78 (7), 3319–3324.
Graci, J.D., Cameron, C.E., 2002. Quasispecies, error catastrophe, and the antiviralactivity of ribavirin. Virology 298 (2), 175–180.
Graci, J.D., Harki, D.A., Korneeva, V.S., Edathil, J.P., Too, K., Franco, D., Smidansky, E.D.,Paul, A.V., Peterson, B.R., Brown, D.M., Loakes, D., Cameron, C.E., 2007. Lethalmutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J. Virol.81 (20), 11256–11266.
Graci, J.D., Too, K., Smidansky, E.D., Edathil, J.P., Barr, E.W., Harki, D.A., Galarraga, J.E.,Bollinger Jr., J.M., Peterson, B.R., Loakes, D., Brown, D.M., Cameron, C.E., 2008. Lethalmutagenesis of picornaviruses with N-6-modified purine nucleoside analogues.Antimicrob. Agents Chemother. 52 (3), 971–979.
Grande-Pérez, A., Sierra, S., Castro, M.G., Domingo, E., Lowenstein, P.R., 2002. Molecularindetermination in the transition to error catastrophe: systematic elimination oflymphocytic choriomeningitis virus through mutagenesis does not correlate
linearly with large increases in mutant spectrum complexity. Proc. Natl. Acad. Sci.U. S. A. 99 (20), 12938–12943.
Grande-Pérez, A., Gómez-Mariano, G., Lowenstein, P.R., Domingo, E., 2005a. Mutagen-esis-induced, large fitness variations with an invariant arenavirus consensusgenomic nucleotide sequence. J. Virol. 79 (16), 10451–10459.
Grande-Pérez, A., Lazaro, E., Lowenstein, P., Domingo, E., Manrubia, S.C., 2005b.Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. U. S. A.102 (12), 4448–4452.
Harki,D.A.,Graci, J.D., Galarraga, J.E., Chain,W.J.,Cameron, C.E., Peterson,B.R., 2006. Synthesisand antiviral activity of 5-substituted cytidine analogues: identification of a potentinhibitor of viral RNA-dependent RNA polymerases. J. Med. Chem. 49 (21), 6166–6169.
Harki, D.A., Graci, J.D., Edathil, J.P., Castro, C., Cameron, C.E., Peterson, B.R., 2007.Synthesis of a universal 5-nitroindole ribonucleotide and incorporation into RNA bya viral RNA-dependent RNA polymerase. Chembiochem 8 (12), 1359–1362.
Harris, K.S., Brabant,W., Styrchak, S., Gall, A., Daifuku, R., 2005. KP-1212/1461, a nucleosidedesigned for the treatment of HIV by viral mutagenesis. Antiviral Res. 67 (1), 1–9.
Holland, J., 2006. Transitions in understanding of RNA viruses: an historical perspective.Curr. Top. Microbiol. Immunol. 299, 371–401.
Holland, J.J., Domingo, E., de la Torre, J.C., Steinhauer, D.A., 1990. Mutation frequencies atdefined single codon sites in vesicular stomatitis virus and poliovirus can beincreased only slightly by chemical mutagenesis. J. Virol. 64, 3960–3962.
Jamieson, D.J., Ellis, J.E., Jernigan, D.B., Treadwell, T.A., 2006. Emerging infectious diseaseoutbreaks: old lessons and new challenges for obstetrician–gynecologists. Am. J.Obstet. Gynecol. 194 (6), 1546–1555.
Lázaro, E., Escarmís, C., Domingo, E., Manrubia, S.C., 2002. Modeling viral genomefitness evolution associated with serial bottleneck events: evidence of stationarystates of fitness. J. Virol. 76 (17), 8675–8681.
Lazaro, E., Escarmis, C., Perez-Mercader, J., Manrubia, S.C., Domingo, E., 2003. Resistanceof virus to extinction on bottleneck passages: study of a decaying and fluctuatingpattern of fitness loss. Proc. Natl. Acad. Sci. U. S. A. 100 (19), 10830–10835.
Lee, C.H., Gilbertson, D.L., Novella, I.S., Huerta, R., Domingo, E., Holland, J.J., 1997.Negative effects of chemical mutagenesis on the adaptive behavior of vesicularstomatitis virus. J. Virol. 71 (5), 3636–3640.
Loeb, L.A., Essigmann, J.M., Kazazi, F., Zhang, J., Rose, K.D., Mullins, J.I., 1999. Lethalmutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U. S. A.96, 1492–1497.
Manrubia, S.C., Escarmis, C., Domingo, E., Lazaro, E., 2005. High mutation rates,bottlenecks, and robustness of RNA viral quasispecies. Gene 347 (2), 273–282.
Meyer, B.J., de La Torre, J.C., Southern, P.J., 2002. Arenaviruses: genomic RNAstranscription, and replication. In: Oldstone, M.B.A., et al. (Eds.), Curr. Top. Microbiol.Immunol., 262. Springer, Berlin, pp. 139–157.
Montville, R., Froissart, R., Remold, S.K., Tenaillon, O., Turner, P.E., 2005. Evolution ofmutational robustness in an RNA virus. PLoS Biol. 3 (11), e381.
Moriyama, K., Suzuki, T., Negishi, K., Graci, J.D., Thompson, C.N., Cameron, C.E.,Watanabe, M., 2008. Effects of introduction of hydrophobic group on ribavirin baseon mutation induction and anti-RNA viral activity. J. Med. Chem. 51 (1), 159–166.
Nowak, M.A., 2006. Evolutionary Dynamics. The Belknap Press of Harvard UniversityPress, Cambridge.
Oldstone, M.B.A. (Ed.), 2002. Arenaviruses I and II. Current Topics in Microbiol. andImmunol. Springer, Berlin. 262 and 263.
Pariente, N., Sierra, S., Lowenstein, P.R., Domingo, E., 2001. Efficient virus extinction bycombinations of a mutagen and antiviral inhibitors. J. Virol. 75 (20), 9723–9730.
Pariente, N., Airaksinen, A., Domingo, E., 2003. Mutagenesis versus inhibition in theefficiency of extinction of foot-and-mouth disease virus. J. Virol. 77 (12), 7131–7138.
Pariente, N., Sierra, S., Airaksinen, A., 2005. Action of mutagenic agents and antiviralinhibitors on foot-and-mouth disease virus. Virus Res. 107 (2), 183–193.
Perales, C., Mateo, R., Mateu, M.G., Domingo, E., 2007. Insights into RNA virus mutantspectrum and lethal mutagenesis events: replicative interference and complemen-tation by multiple point mutants. J. Mol. Biol. 369 (4), 985–1000.
Pfeiffer, J.K., Kirkegaard, K., 2003. A single mutation in poliovirus RNA-dependent RNApolymerase confers resistance to mutagenic nucleotide analogs via increasedfidelity. Proc. Natl. Acad. Sci. U. S. A. 100 (12), 7289–7294.
Pfeiffer, J.K., Kirkegaard, K., 2005a. Increased fidelity reduces poliovirus fitness underselective pressure in mice. PLoS Pathogens 1, 102–110.
Pfeiffer, J.K., Kirkegaard, K., 2005b. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in thereplicon RNA. J. Virol. 79 (4), 2346–2355.
Quer, J., Huerta, R., Novella, I.S., Tsimring, L., Domingo, E., Holland, J.J., 1996.Reproducible nonlinear population dynamics and critical points during replicativecompetitions of RNA virus quasispecies. J. Mol. Biol. 264 (3), 465–471.
Quer, J., Hershey, C.L., Domingo, E., Holland, J.J., Novella, I.S., 2001. Contingent neutralityin competing viral populations. J. Virol. 75 (16), 7315–7320.
Ruiz-Jarabo, C.M., Ly, C., Domingo, E., de la Torre, J.C., 2003. Lethal mutagenesis of theprototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 308 (1),37–47.
Sanjuan, R., Cuevas, J.M., Furio, V., Holmes, E.C., Moya, A., 2007. Selection for robustnessin mutagenized RNA viruses. PLoS Genet. 3 (6), e93.
Schuster, P., Swetina, J., 1988. Stationary mutant distributions and evolutionaryoptimization. Bull. Math. Biol. 50 (6), 635–660.
Schuster, P., Stadler, P., 2008. Early replicons: origin and evolution, In: Domingo, E.,Parrish, C., Holland, J.J., et al. (Eds.), Origin and evolution of viruses, 2nd edition.Elsevier, Amsterdam (The Netherlands), pp. 1–42.
Sierra, S., Dávila, M., Lowenstein, P.R., Domingo, E., 2000. Response of foot-and-mouthdisease virus to increased mutagenesis. Influence of viral load and fitness in loss ofinfectivity. J. Virol. 74, 8316–8323.
Sierra, M., Airaksinen, A., González-López, C., Agudo, R., Arias, A., Domingo, E., 2007.

192 V. Martín et al. / Virology 378 (2008) 185–192
Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin:implications for error catastrophe. J. Virol. 81, 2012–2024.
Takeuchi, N., Hogeweg, P., 2007. Error-threshold exists in fitness landscapes with lethalmutants. BMC Evol. Biol. 7, 15 author reply 15.
Vignuzzi, M., Stone, J.K., Arnold, J.J., Cameron, C.E., Andino, R., 2006. Quasispeciesdiversity determines pathogenesis through cooperative interactions in a viralpopulation. Nature 439, 344–348.
Vo, N.V., Young, K.C., Lai, M.M.C., 2003. Mutagenic and inhibitory effects of ribavirin onhepatitis C virus RNA polymerase. Biochemistry 42 (35), 10462–10471.
Wilke, C.O., Adami, C., 2003. Evolution ofmutational robustness.Mutat. Res. 522 (1–2), 3–11.Wilke, C.O., Ronnewinkel, C., Martinetz, T., 2001. Dynamic fitness landscapes in
molecular evolution. Phys. Rep. 349, 395–446.Wright, S., 1931. Evolution in Mendelian populations. Genetics 16, 97–159.Wright, S., 1982. Character change, speciation, and the higher taxa. Evolution 36,
427–443.Young, K.C., Lindsay, K.L., Lee, K.J., Liu, W.C., He, J.W., Milstein, S.L., Lai, M.M., 2003.
Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus duringribavirin monotherapy. Hepatology 38 (4), 869–878.
Related Documents











