
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


J. Erasmus and others
and clustering at sites of cell–cell contacts trigger dif-ferent signalling events that co-ordinate distinct as-pects of epithelial polarization (Braga and Yap, 2005).
Among the signalling pathways activated by E-cadherin-mediated adhesion are those triggered bythe members of the Rho family of small GTPases.The small GTPases RhoA, Rac1 and Cdc42 are thebest-studied members of this family and have beenshown to regulate adhesion mediated by different re-ceptors, such as integrins and nectins (Takai et al.,2008). Whether the modulation of cell–cell adhe-sion by Rho small GTPases derives from their abil-ity to reorganize the actin cytoskeleton in specificways has not been firmly established. For example,RhoA function is required for integrin adhesion andattachment to substratum induces stress fibres in dif-ferent cell types (Jaffe and Hall, 2005). AlthoughRhoA is necessary for stable cadherin-dependent ad-hesion (Braga, 2002), stress fibres are not triggeredby cadherin engagement at cell–cell contacts. Sim-ilarly, Rac1 has been shown to promote actin poly-merization and the branched array of actin filamentsduring lamellae protrusion, but in epithelia Rac par-ticipates in actin recruitment to clustered E-cadherincomplexes (Braga, 2002). Therefore during epithelialpolarization, functional analysis of the participationof Rho small GTPases in actin remodelling shouldnot be inferred from data obtained in different celltypes or from distinct receptors and stimuli.
It is important to understand how remodelling ofthe actin cytoskeleton by cadherin-mediated adhesionoccurs, as it provides a crucial process to strengthenand maintain cadherin complexes clustered at adhe-sion sites. Two models have been proposed of howjunctions are established between adjacent epithelialcells. In the first model, lamellae protrusions probeadjacent cells (McNeill et al., 1993; Adams et al.,1996; Krendel and Bonder, 1999; Ehrlichet al., 2002). Once transient cadherin interactions areestablished, a series of puncta (containing E-cadherinand F-actin) develops when the contacting membraneexpands as a straight line between approaching cells(McNeill et al., 1993; Adams et al., 1996; Adams andNelson, 1998). In the second model, transient cad-herin interactions are produced by ‘finger-like pro-trusions’ that resemble filopodia (Raich et al., 1999;Jacinto et al., 2000; Vasioukhin et al., 2000). Punctaform at the tip and/or sides of these structures andcadherin staining appears as a jagged pattern. Even-
tually, puncta coalesce as a straight line as adjacentcell membranes intermingle and ‘zipper up’ together.Finger-like protrusions containing F-actin are mostlyseen during cell–cell adhesion between fibroblastsor some types of epithelial cells when subconfluent(Yonemura et al., 1995; Gloushkankova et al., 1997;Vasioukhin et al., 2000).
The models above have in common two features:punctum as the unit of new cadherin-dependent con-tacts and cadherin localization as a straight line atstable epithelial junctions. However, the two mod-els differ on the cytoskeletal processes via which cellmembranes are placed close together during cell–celladhesion (lamellipodium versus ‘finger-like protru-sion’). These two actin processes have a very dis-tinct organization of actin filaments (i.e. branchedarray versus bundled filaments respectively) (Svitkinaand Borisy, 1999; Bailly et al., 2001; Svitkina et al.,2003). Yet, the structural organization of filament-ous actin in lamellipodium or filopodium (Pollardet al., 2000) is not consistent with the actin or-ganization observed in primary epithelial cells (i.e.a linear accumulation of F-actin coincident with cad-herin receptors and flanking peripheral thin bundles)(Zhang et al., 2005; Mege et al., 2006). Nevertheless,it is still feasible that lamellipodia and/or filopodia,which are required for the initial contact (Adams andNelson, 1998; Vasioukhin et al., 2000; Ehrlich et al.,2002), are transient and they quickly disassembleonce puncta are stable.
The above considerations may have implicationsfor the mechanisms by which cadherin complexesassociate indirectly with the cortical actin networkduring junction formation. Furthermore, they implythat the signalling pathways driven by early cadherin-mediated cell–cell contacts may differ substantially,depending on whether recognition of neighbouringcells involves lamellipodium or filopodium to initi-ate contacts. Regarding the first model (lamellipodia-mediated contacts), it is known that the small GTPaseRac is found at the lamellae when new cell–cell con-tacts are formed, but Rac comes off of the membranewhen contacts are stabilized (Ehrlich et al., 2002).In contrast, Rho localizes to the edges of expand-ing contacts, as tension develops during cell com-paction when early contacts become more stable (Ya-mada and Nelson, 2007). Inhibition of either RhoAor Rac1 efficiently destabilizes adhesion and removesE-cadherin from new or stable cell–cell contacts.
14 C© The Authors Journal compilation C© 2010 Portland Press Ltd

Newly formed E-cadherin contacts inactivate Cdc42 Research article
These results are consistent with the cadherin-dependent activation of Rac and Rho in differentepithelial cells (Braga, 2002).
The role of Cdc42 during E-cadherin-mediated ad-hesion has been more controversial. During dorsalclosure in Drosophila, Cdc42-driven filopodia havebeen shown to play a key role in the recognitionof specific segments in opposing sheets of epithelialcells and allow appropriate sealing of the membranes(Jacinto et al., 2000). However, as migration of epi-thelial sheets occurs during dorsal closure, it is feas-ible that the large number of filopodia observed maybe derived from the migratory process, rather thancell–cell adhesion itself. When cell–cell contacts areinitiated in mammalian cells, localization of Cdc42has not been demonstrated at filopodia. Furthermore,Cdc42 activation following E-cadherin junction as-sembly has not been determined in different epi-thelial cell types. To our knowledge, there is onlyone report in a breast tumour cell line that showsactive Cdc42 using expression of GFP–WASP-crib(green fluorescent protein–Wiskott–Aldrich syn-drome protein-Cdc42- and Rac-interacting bindingdomain), which specifically binds to activated Cdc42(Kim et al., 2000). In contrast, C-cadherin engage-ment inactivates Cdc42 (Noren et al., 2001).
The scarcity of information on whether Cdc42 isactivated by E-cadherin is paralleled by the con-tradictory data on the requirement of Cdc42 func-tion for cadherin adhesiveness. Some reports havedemonstrated that Cdc42 inhibition interferes withE-cadherin-mediated adhesion (Kodama et al., 1999;Du et al., 2008), whereas others show no effect(Bruewer et al., 2004). This is in contrast with thewell-established role of Rho and Rac in cadherin func-tion in different cell types (Braga, 2002; Braga andYap, 2005). Furthermore, the results available do notaddress whether (i) Cdc42 is specifically activated byE-cadherin clustering, (ii) Cdc42 is important for newassembly of cadherin cell–cell contacts or (iii) filopo-dia are necessary for de novo formation of junctionsunder conditions where migration is not involved.
In the present study, we set out to address the abovequestions using normal human keratinocytes as ourmodel system for three reasons. First, keratinocyteshave been shown to contact neighbouring cells usingfilopodia in subconfluent cultures (Vasioukhin et al.,2000). Secondly, to rule out a potential contributionof migration to Cdc42 activation, we used only con-
fluent keratinocyte cultures grown in the absence ofcalcium ions (conditions in which cadherin receptorsare non-adhesive). When using confluent cultures,filopodia driven specifically from probing neighbour-ing cells can still occur, but migration-dependentfilopodial protrusions are prevented. Thirdly, freshmedium with serum activates small GTPases throughgrowth-factor stimulation and thus we induced cell–cell contacts by addition of calcium ions only.Under these conditions, we find that initiation of E-cadherin-mediated adhesion does not activate Cdc42.Instead, E-cadherin clustering is sufficient to inactiv-ate Cdc42 for up to 1 h following new junction form-ation. Our results are consistent with the absence offilopodia formation and the inability of Cdc42 inhib-ition to prevent assembly of new E-cadherin cell–celladhesion in confluent keratinocytes.
ResultsCadherin-mediated adhesion is necessary andsufficient to regulate Cdc42To determine whether Cdc42 is activated by junctionformation, we initially evaluated Cdc42 activationby pull-down assays using GST (glutathionetransferase)–PAK (p21-activated kinase)-crib, afusion protein containing the binding site of PAKfor Rac and Cdc42. Controls using keratinocytelysates loaded with GTP or GDP showed that,under our conditions, endogenous active Cdc42(Cdc42-GTP) can be specifically precipitated withGST–PAK-crib (Figure 1A). To specifically assessjunction-induced Cdc42 activation, two key differ-ences from previous reports were performed: (a) onlyconfluent keratinocyte cultures were used to excludethe contribution of cell spreading and migration forCdc42 activity levels; and (b) only calcium ions wereadded to the medium to avoid Cdc42 activationby fresh serum when the medium was replaced.Lysates were prepared from keratinocytes grown inthe absence of cell–cell contacts or at different timesof calcium-induced junction assembly. The levels ofactive Cdc42 (Cdc42-GTP) and Cdc42 protein in thelysates (total Cdc42) are shown in representative blots(Figure 1B), as well as the quantification (Figures 1Cand 1D; see the Materials and methods section fordetails). A marked decrease in the levels of activeCdc42 was observed as early as 15 min after cell–cellcontact formation (Figures 1C and 1D). Indeed, atime course using earlier time points showed that
www.biolcell.org | Volume 102 (1) | Pages 13–24 15

J. Erasmus and others
Figure 1 Cadherin-mediated adhesion is both necessary and sufficient to reduce Cdc42 activity levels during junctionassembly(A) Specificity of the pull-down assay. GST–PAK-crib precipitates active Cdc42 from keratinocyte lysates loaded with GTP[S] or
GDP. The amount of active (Cdc42-GTP) and total Cdc42 in the lysates were evaluated by Western blotting using anti-Cdc42
antibodies. (B) Cdc42 activity was evaluated during a time course of junction assembly (0–60 min). Cell–cell contacts were
induced by the addition of calcium ions for up to 60 min and lysates were incubated with GST–PAK-crib and probed with
anti-Cdc42 antibodies. Active Cdc42 (Cdc42-GTP) and total levels of Cdc42 protein in the lysates (total Cdc42) are shown in
representative blots. Cdc42 levels (active) were quantified and expressed relative to the starting point (no cell–cell contacts
values), arbitrarily set as 1, following a long time course (up to 60 min, C) or short time course (up to 15 min, D). (E) Cdc42
inactivation requires cadherin-mediated adhesion. Cadherin function was blocked by adding a cocktail of inhibitory antibodies
against E- and P-cadherin (cadh). Active Cdc42 levels were assessed at 15 and 30 min after the addition of calcium ions.
Controls were treated with non-immune IgG. Data are the mean +− S.E.M. of three experiments performed in duplicate. *P < 0.025;
**P < 0.004; ***P < 0.001; @P < 0.0005; $P < 0.00001.
Cdc42 inactivation is apparent after 5–10 min of newcell–cell contact assembly. The reduced level ofCdc42 activity was maintained for up to 1 h(Figure 1C).
We next asked whether Cdc42 inactivation was de-pendent on cadherin-mediated adhesion. A cocktailof inhibitory antibodies were used to block cadherin-dependent cell–cell adhesion (Braga et al., 1997),and calcium ions were added for 15 and 30 min(Figure 1E). Under these conditions, inactivation ofCdc42 was rescued, in particular after 30 min; levelsof active Cdc42 were comparable with those observed
in cultures without cell–cell contacts. In controls (ad-dition of non-immune IgG), active Cdc42 levels werereduced as seen earlier (Figures 1C and 1D). We con-clude that cadherin adhesive function does not activ-ate Cdc42. Instead, within the time frame investig-ated, cadherin clustering is necessary and sufficientfor Cdc42 inactivation.
Localized activation of Cdc42 during junctionassemblyThe measurement of global levels of Cdc42 activationby pull-down assays may, however, not be sensitive
16 C© The Authors Journal compilation C© 2010 Portland Press Ltd
Newly formed E-cadherin contacts inactivate Cdc42 Research article
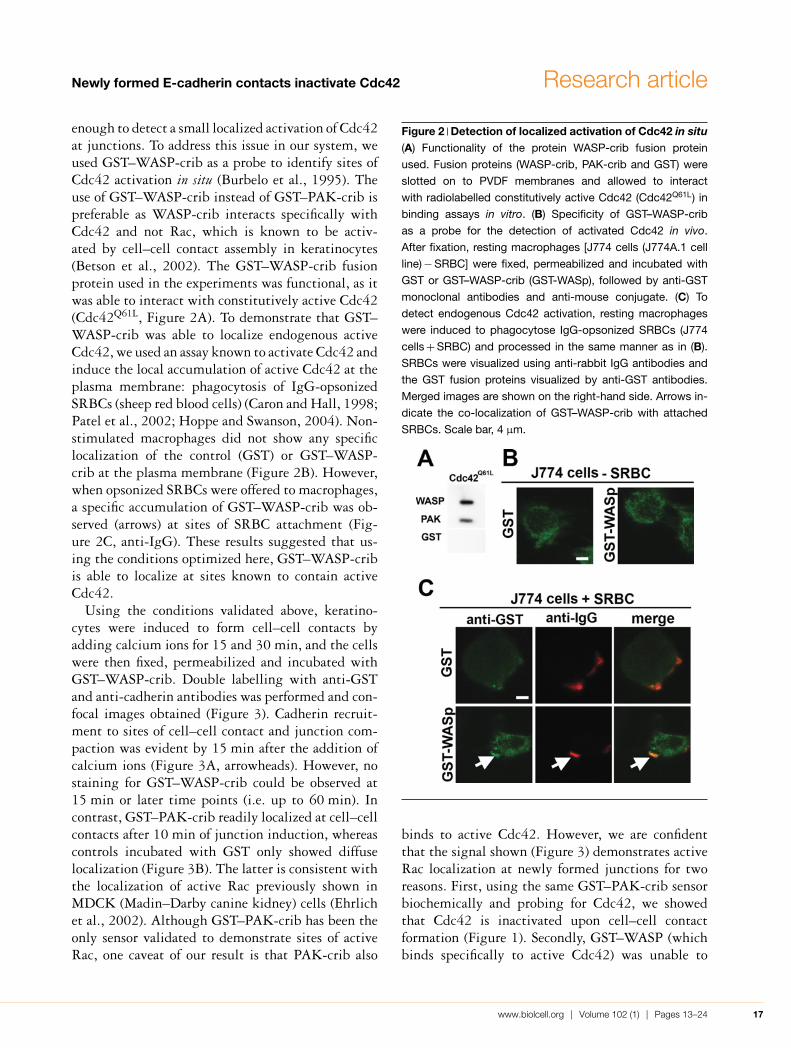
enough to detect a small localized activation of Cdc42at junctions. To address this issue in our system, weused GST–WASP-crib as a probe to identify sites ofCdc42 activation in situ (Burbelo et al., 1995). Theuse of GST–WASP-crib instead of GST–PAK-crib ispreferable as WASP-crib interacts specifically withCdc42 and not Rac, which is known to be activ-ated by cell–cell contact assembly in keratinocytes(Betson et al., 2002). The GST–WASP-crib fusionprotein used in the experiments was functional, as itwas able to interact with constitutively active Cdc42(Cdc42Q61L, Figure 2A). To demonstrate that GST–WASP-crib was able to localize endogenous activeCdc42, we used an assay known to activate Cdc42 andinduce the local accumulation of active Cdc42 at theplasma membrane: phagocytosis of IgG-opsonizedSRBCs (sheep red blood cells) (Caron and Hall, 1998;Patel et al., 2002; Hoppe and Swanson, 2004). Non-stimulated macrophages did not show any specificlocalization of the control (GST) or GST–WASP-crib at the plasma membrane (Figure 2B). However,when opsonized SRBCs were offered to macrophages,a specific accumulation of GST–WASP-crib was ob-served (arrows) at sites of SRBC attachment (Fig-ure 2C, anti-IgG). These results suggested that us-ing the conditions optimized here, GST–WASP-cribis able to localize at sites known to contain activeCdc42.
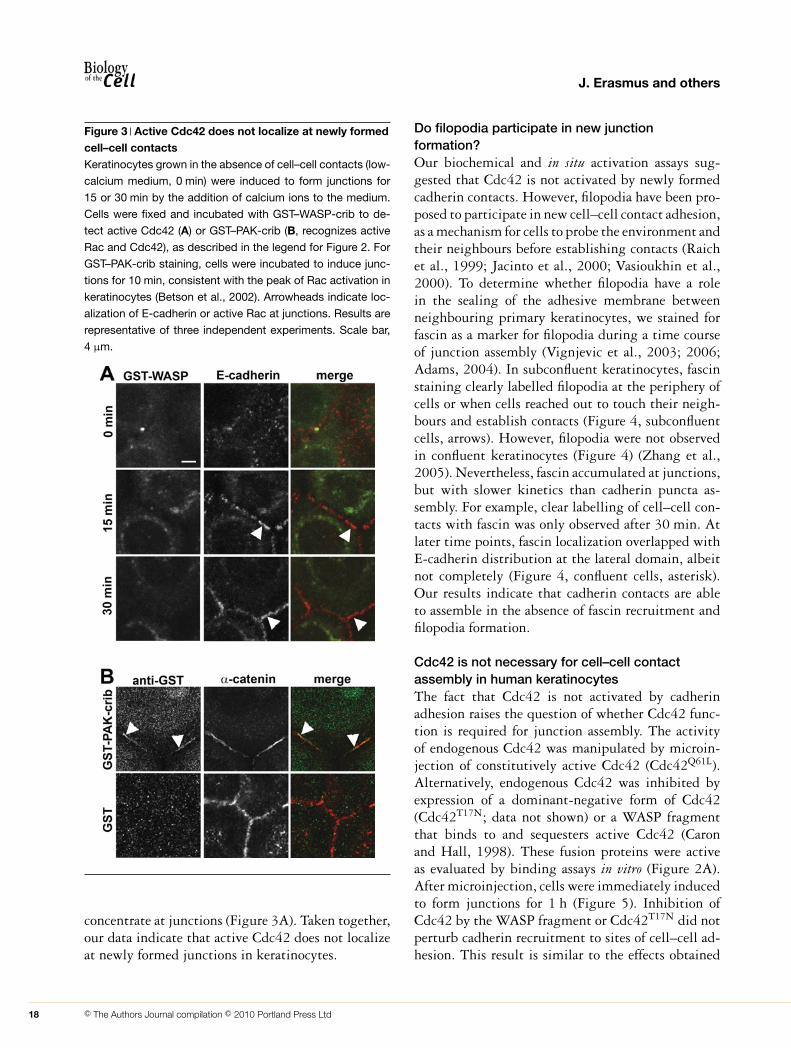
Using the conditions validated above, keratino-cytes were induced to form cell–cell contacts byadding calcium ions for 15 and 30 min, and the cellswere then fixed, permeabilized and incubated withGST–WASP-crib. Double labelling with anti-GSTand anti-cadherin antibodies was performed and con-focal images obtained (Figure 3). Cadherin recruit-ment to sites of cell–cell contact and junction com-paction was evident by 15 min after the addition ofcalcium ions (Figure 3A, arrowheads). However, nostaining for GST–WASP-crib could be observed at15 min or later time points (i.e. up to 60 min). Incontrast, GST–PAK-crib readily localized at cell–cellcontacts after 10 min of junction induction, whereascontrols incubated with GST only showed diffuselocalization (Figure 3B). The latter is consistent withthe localization of active Rac previously shown inMDCK (Madin–Darby canine kidney) cells (Ehrlichet al., 2002). Although GST–PAK-crib has been theonly sensor validated to demonstrate sites of activeRac, one caveat of our result is that PAK-crib also
Figure 2 Detection of localized activation of Cdc42 in situ(A) Functionality of the protein WASP-crib fusion protein
used. Fusion proteins (WASP-crib, PAK-crib and GST) were
slotted on to PVDF membranes and allowed to interact
with radiolabelled constitutively active Cdc42 (Cdc42Q61L) in
binding assays in vitro. (B) Specificity of GST–WASP-crib
as a probe for the detection of activated Cdc42 in vivo.
After fixation, resting macrophages [J774 cells (J774A.1 cell
line) − SRBC] were fixed, permeabilized and incubated with
GST or GST–WASP-crib (GST-WASp), followed by anti-GST
monoclonal antibodies and anti-mouse conjugate. (C) To
detect endogenous Cdc42 activation, resting macrophages
were induced to phagocytose IgG-opsonized SRBCs (J774
cells + SRBC) and processed in the same manner as in (B).
SRBCs were visualized using anti-rabbit IgG antibodies and
the GST fusion proteins visualized by anti-GST antibodies.
Merged images are shown on the right-hand side. Arrows in-
dicate the co-localization of GST–WASP-crib with attached
SRBCs. Scale bar, 4 μm.
binds to active Cdc42. However, we are confidentthat the signal shown (Figure 3) demonstrates activeRac localization at newly formed junctions for tworeasons. First, using the same GST–PAK-crib sensorbiochemically and probing for Cdc42, we showedthat Cdc42 is inactivated upon cell–cell contactformation (Figure 1). Secondly, GST–WASP (whichbinds specifically to active Cdc42) was unable to
www.biolcell.org | Volume 102 (1) | Pages 13–24 17
J. Erasmus and others
Figure 3 Active Cdc42 does not localize at newly formedcell–cell contactsKeratinocytes grown in the absence of cell–cell contacts (low-
calcium medium, 0 min) were induced to form junctions for
15 or 30 min by the addition of calcium ions to the medium.
Cells were fixed and incubated with GST–WASP-crib to de-
tect active Cdc42 (A) or GST–PAK-crib (B, recognizes active
Rac and Cdc42), as described in the legend for Figure 2. For
GST–PAK-crib staining, cells were incubated to induce junc-
tions for 10 min, consistent with the peak of Rac activation in
keratinocytes (Betson et al., 2002). Arrowheads indicate loc-
alization of E-cadherin or active Rac at junctions. Results are
representative of three independent experiments. Scale bar,
4 μm.
concentrate at junctions (Figure 3A). Taken together,our data indicate that active Cdc42 does not localizeat newly formed junctions in keratinocytes.
Do filopodia participate in new junctionformation?Our biochemical and in situ activation assays sug-gested that Cdc42 is not activated by newly formedcadherin contacts. However, filopodia have been pro-posed to participate in new cell–cell contact adhesion,as a mechanism for cells to probe the environment andtheir neighbours before establishing contacts (Raichet al., 1999; Jacinto et al., 2000; Vasioukhin et al.,2000). To determine whether filopodia have a rolein the sealing of the adhesive membrane betweenneighbouring primary keratinocytes, we stained forfascin as a marker for filopodia during a time courseof junction assembly (Vignjevic et al., 2003; 2006;Adams, 2004). In subconfluent keratinocytes, fascinstaining clearly labelled filopodia at the periphery ofcells or when cells reached out to touch their neigh-bours and establish contacts (Figure 4, subconfluentcells, arrows). However, filopodia were not observedin confluent keratinocytes (Figure 4) (Zhang et al.,2005). Nevertheless, fascin accumulated at junctions,but with slower kinetics than cadherin puncta as-sembly. For example, clear labelling of cell–cell con-tacts with fascin was only observed after 30 min. Atlater time points, fascin localization overlapped withE-cadherin distribution at the lateral domain, albeitnot completely (Figure 4, confluent cells, asterisk).Our results indicate that cadherin contacts are ableto assemble in the absence of fascin recruitment andfilopodia formation.
Cdc42 is not necessary for cell–cell contactassembly in human keratinocytesThe fact that Cdc42 is not activated by cadherinadhesion raises the question of whether Cdc42 func-tion is required for junction assembly. The activityof endogenous Cdc42 was manipulated by microin-jection of constitutively active Cdc42 (Cdc42Q61L).Alternatively, endogenous Cdc42 was inhibited byexpression of a dominant-negative form of Cdc42(Cdc42T17N; data not shown) or a WASP fragmentthat binds to and sequesters active Cdc42 (Caronand Hall, 1998). These fusion proteins were activeas evaluated by binding assays in vitro (Figure 2A).After microinjection, cells were immediately inducedto form junctions for 1 h (Figure 5). Inhibition ofCdc42 by the WASP fragment or Cdc42T17N did notperturb cadherin recruitment to sites of cell–cell ad-hesion. This result is similar to the effects obtained
18 C© The Authors Journal compilation C© 2010 Portland Press Ltd

Newly formed E-cadherin contacts inactivate Cdc42 Research article
Figure 4 Localization of fascin, a filopodial marker,during induction of cell–cell contactsSubconfluent or confluent keratinocytes were induced to form
junctions for different time points (0–30 min), fixed and la-
belled with antibodies against E-cadherin (green) and fascin
(red). Arrows indicate the concentration of fascin at filopodia;
arrowheads point to the recruitment of E-cadherin at sites of
cell–cell contacts; asterisks indicate the concentration of fas-
cin at cell–cell contacts at later time points (30 min). Results
are representative of three independent experiments. Scale
bar, 8 μm.
by microinjection of control GST (Figure 5). In con-trast, activated Cdc42 partially inhibited junctionformation within the time frame investigated. Theseresults are in agreement with the idea that, in or-der for stable cadherin junctions to be formed in
Figure 5 Cdc42 is not required for new cell–cell contactformation in keratinocytesConfluent keratinocytes, grown in low-calcium medium,
were microinjected with activated Cdc42 (Cdc42Q61L),
GST–WASP-crib or dominant-negative Cdc42 (Cdc42T17N) to
inhibit Cdc42 function, and junctions were induced to form
with the addition of calcium for 1 h. As a control, GST was
also microinjected. Cells were fixed and stained for E-cad-
herin; injected cells were visualized by co-injection of dex-
tran–Cy3 (indocarbocyanine) (injected). Arrows indicate the
absence of E-cadherin from cell–cell contacts; arrowheads
indicate E-cadherin at junctions. Results are representative of
three independent experiments. Scale bar, 40 μm.
keratinocytes, Cdc42 activation and function are notnecessary.
DiscussionIn the present study, we demonstrate for the first timethat Cdc42 is not activated following new assemblyof E-cadherin-dependent adhesion. On the contrary,Cdc42 is specifically inactivated by E-cadherin clus-tering and its activity levels remain low for up to 1 h.
www.biolcell.org | Volume 102 (1) | Pages 13–24 19

J. Erasmus and others
However, it is feasible that Cdc42 activity levels mayincrease later on, a prediction that is supported by themany important functions of Cdc42 in epithelia (seebelow). Consistent with our data in keratinocytes, inMDCK cells expressing C-cadherin, direct cadherinengagement does not activate Cdc42, but activatesRac1 instead (Noren et al., 2001). It is also possiblethat there are cell-type specific signalling events withrespect to Cdc42 activation and that Cdc42 may beactive following stimulation of other cell–cell adhe-sion receptors, such as nectin (Takai et al., 2008).
In confluent keratinocytes, we find that Cdc42 in-hibition does not prevent formation of new cadherincell–cell contacts. Similar results were obtained inmature junctions in MDCK cells (Takaishi et al.,1997; Bruewer et al., 2004), indicating that Cdc42inactivation does not grossly impair the mainten-ance of cell–cell contacts in this cell type. However,in other studies long-term Cdc42 inactivation leadsto reduced cadherin localization in mature contacts inMDCK cells (Kodama et al., 1999) or Cdc42 knock-out immortalized keratinocytes (Du et al., 2008).The latter long-term effect of Cdc42 inhibitionmay occur most likely via regulation of E-cadherinturnover and/or polarized distribution. Cdc42 local-izes primarily at the Golgi and has a clear role inintracellular trafficking of different surface and intra-cellular proteins (Jaffe and Hall, 2005). Cdc42 hasbeen shown to play a role in recycling/transport of E-cadherin and other basolateral proteins (Musch et al.,2001; Rojas et al., 2001; Bruewer et al., 2004; Izumiet al., 2004; Wu et al., 2007), and thus can indir-ectly affect adhesion (Kodama et al., 1999; Wu et al.,2007; Du et al., 2008). It is possible that inhibitionof Cdc42 may indirectly interfere with cadherin ad-hesion by down-regulating the amount of receptorsavailable at the surface. The net effect is perturbedcell–cell adhesion, but, as overall protein traffickingmay be perturbed, these effects are far from specificto cadherin receptors.
However, our data does not imply that appropri-ate cell–cell adhesion maturation occurs in the ab-sence of Cdc42 function. Four lines of evidence sug-gest that Cdc42 is important for epithelial adhesivesystems. First, in different epithelial models, Cdc42has been implicated in epithelial polarization andthe appropriate assembly and functionality of tightjunctions (Rojas et al., 2001; Braga, 2002; Brueweret al., 2004). Secondly, by regulating the localiza-
tion and activity of the Par complex, Cdc42 modu-lates the polarized distribution of different proteinsinto apical and basolateral domains (Jaffe and Hall,2005). Thirdly, there is also evidence that Cdc42may regulate junction morphology. Constitutive ac-tivation of Cdc42 results in tightly compacted junc-tions between adjacent contacting cells (Stoffler et al.,1998; Kodama et al., 1999). Interestingly, upon de-pletion of the exchange factor Tuba, an upstreamregulator of Cdc42, junctions become undulated in-stead of a straight line in-between neighbouring cells(Otani et al., 2006). Finally, the confluence of epi-thelial cells also appears to regulate the activation ofRac and Cdc42 (Noren et al., 2001).
Our results suggest that, as opposed to Rho andRac, there is no clear evidence that Cdc42 has adirect effect on the cadherin-dependent cytoskeletalremodelling that is important to stabilize new cell–cell contacts. We show that Cdc42-dependent filopo-dial protrusions are not strictly required for cell–celladhesion to be established. If lamellae or filopodiaare involved in the initial events of cell–cell con-tact assembly, these actin structures must be verytransient and labile. Indeed, in human keratino-cytes or MDCK cells, the pattern of labelled actinincorporation at junctions does not resemble actinlabelling of either lamellipodia or filopodia (Theriotand Mitchison, 1991; McNeill et al., 1993;Mallavarapu and Mitchison, 1999; Zhang et al.,2005). Furthermore, electron microscopy studies inepithelial cells reveal a distinct compact organiza-tion of bundles in a circumferential ring (Yonemuraet al., 1995; Mege et al., 2006; Yamada and Nelson,2007), as opposed to the actin meshwork found inlamellae or tight bundles of filopodia (Pollard et al.,2000). These differences in F-actin organization haveimportant implications for identification of the regu-latory mechanisms involved in junction assembly andstabilization.
Filopodia are clearly visible in subconfluent kerat-inocyte cultures (Vasioukhin et al., 2000), as assessedby the presence of finger-like protrusions enriched infascin (the present study). Although Cdc42 is inac-tivated upon cadherin clustering, it is feasible thatother small GTPase family members could inducefilopodia during induction of cell–cell contacts (Jaffeand Hall, 2005). In contrast, although E-cadherin isdetected at cell–cell contacts as early as 5 min aftercalcium addition to confluent keratinocytes (Zhang
20 C© The Authors Journal compilation C© 2010 Portland Press Ltd

Newly formed E-cadherin contacts inactivate Cdc42 Research article
et al., 2005), fascin is recruited to junctions at amuch later time point (30 min). This result indicatesthat protrusion of fascin-labelled filopodia does notcorrelate temporally with assembly of cadherin con-tacts. Instead, fascin co-localization with junctionalactin (Zhang et al., 2005) at later time points maysuggest its potential role in the stabilization of thisactin population. In conclusion, there is no evidenceof the requirement of filopodia or Cdc42 activationduring de novo assembly of cell–cell contacts in kerat-inocytes. Thus it is important to revisit the issue ofCdc42 function in E-cadherin adhesion in other celltypes under conditions that do not involve migration-triggered filopodia or the addition of fresh mediumto induce cell–cell contacts (as serum activates Cdc42via growth factor receptors).
It remains to be established whether a potentialactivation of Cdc42 at later time points is cadherin-dependent or -independent. However, there are otheradhesion receptors that become localized at junctionsand can potentially activate Cdc42. For example,nectins are calcium-independent adhesive receptorsthat, when engaged at cell–cell contacts, are knownto activate Cdc42 (Takai et al., 2008). Another pos-sibility is integrins, which are cell-matrix adhesivereceptors that also localize at cell–cell contacts in epi-thelial cells. Although the specific function of integ-rins at junctions is unclear, it is feasible that integrinsmay activate Cdc42 signalling pathways similarly towhat shown when Cdc42 is localized at focal adhe-sions (Jaffe and Hall, 2005).
Nevertheless, Cdc42 has essential functions in epi-thelia which extend beyond the assembly of junc-tional complexes. In the skin, ablation of Cdc42 doesnot interfere with cell–cell contacts, but rather thepolarized secretion of extracellular matrix compon-ents and maintenance of the basement membrane inthe skin (Wu et al., 2006). At least three main cel-lular processes are regulated by Cdc42 in epithelialmorphogenesis. Cdc42 has been shown to be import-ant for epithelial lumen formation in vitro in MDCKcells (Jaffe and Hall, 2005; Martin-Belmonte andMostov, 2008) and in the regulation of spindle ori-entation during cell division to maintain epithelialpolarization (Jaffe and Hall, 2005). In other celltypes, Cdc42 participates in the re-positioning of thenucleus and MTOC (microtubule-organizing centre)following different stimuli (Etienne-Manneville andHall, 2001; Tzima et al., 2003; Gomes et al., 2005;
Desai et al., 2009). Thus it is conceivable that sim-ilar changes may be regulated by Cdc42 in epithelialcells.
Taken together, our data strongly indicate thatCdc42 is not activated or required for the assembly ofnew E-cadherin cell–cell contacts. Given the essentialfunctions of Cdc42 for epithelial morphogenesis re-ported above, an important question to be addressedin the future is the identification of the receptor thatis responsible for Cdc42 activation during epithelialpolarization and the molecular mechanisms that par-ticipate in different Cdc42-dependent morphogen-etic events.
Materials and methodsCell cultureNormal human keratinocytes (strains Sf, Kf and AEK, passage2–5) were co-cultured with a fibroblast feeder layer (J2), asdescribed previously (Rheinwald, 1989). Keratinocytes grownwithout cell–cell adhesion were cultured essentially as reportedpreviously [low-calcium medium (Hodivala and Watt, 1994)].Only confluent cultures maintained in low-calcium mediumwere used in all experiments. Cell–cell adhesion was inducedby the addition of calcium ions (1.8 mM) or by transferring cellsinto standard-calcium medium (calcium switch).
To assess the levels of Cdc42 activity during induction ofcell–cell adhesion using biochemical methods, confluent kerat-inocytes grown in low-calcium medium were used (one 56 cm2
plate per reaction tube). Cell–cell adhesion was induced for dif-ferent periods of time by the addition of calcium ions to a finalconcentration of 1.8 mM (to avoid addition of fresh serum andactivation of small GTPases). For localized Cdc42 activation,keratinocytes grown to confluence in low-calcium medium wereinduced to assemble junctions by adding calcium ions to 1.8 mMand fixed at different time points.
Cells from the murine macrophage J774A.1 cell line weremaintained in complete medium [DMEM (Dulbecco’s mod-ified Eagle’s medium; Invitrogen)] supplemented with 10%heat-inactivated FCS (foetal calf serum; PAA Laboratories) and100 units/ml penicillin and 100 μg/ml streptomycin (Invitro-gen), as described previously (Caron and Hall, 1998; Patel et al.,2002). For testing of localized Cdc42 activation during phago-cytosis, J774A.1 cells were challenged with IgG-opsonized SR-BCs (TCS), as described previously (Caron and Hall, 1998; Patelet al., 2002). Briefly, SRBCs (0.1 μl per 13 mm coverslip) wereopsonized with rabbit anti-SRBC IgG (Cappel) during 30 minin 1 ml of GVB (gelatin veronal buffer; Sigma Chemical) andwashed once with GVB. IgG–SRBCs were allowed to adhere for15 min at 4◦C and synchronized phagocytosis was inducedfor 15 min at 37◦C. After phagocytic challenge, cells werewashed once with PBS and fixed in cold 4% (w/v) paraform-aldehyde for 15 min at 4◦C.
Antibodies and immunofluorescenceThe following monoclonal antibodies were used: anti-E-cadherin[ECCD-2 (rat; Hirai et al., 1989) or HECD-1 (mouse;Shimoyama et al., 1989)], anti-Cdc42 (clone 44; Transduction
www.biolcell.org | Volume 102 (1) | Pages 13–24 21

J. Erasmus and others
Laboratories), anti-GST (clone GST-2; Sigma) and anti-fascin(clone 55K-2; Dako). Secondary antibodies were bought fromJackson ImmunoResearch Laboratories. FITC-conjugated phal-loidin was from Sigma and anti-(mouse horseradish peroxidase)antibody was from Pierce.
Unless otherwise specified, staining was performed as reportedpreviously (Braga et al., 1997). Cells were fixed in 3% paraform-aldehyde for 10 min at room temperature and permeabilized in0.1% Triton X-100 in PBS/10% FCS. For fascin staining, cellswere fixed in cold methanol for 3 min.
Production of fusion proteinsProduction of GST–PAK-crib protein was performed asdescribed previously (Betson et al., 2002). For the GST–WASP-crib fusion protein, bacteria were cultured overnight and in-duced with 0.5 mM IPTG (isopropyl β-D-thiogalactoside; Cal-biochem) for 3 h at 30◦C. The bacterial pellet was resuspen-ded in PBS buffer containing 1 mM EGTA, 1 mM MgCl2,5 μg/ml leupeptin, 5 μg/ml Pefabloc (Sigma), 5 μg/ml pep-statin and 50 mM PMSF. Following sonication and centrifu-gation (21350 g, 20 min, 4◦C), the supernatant was transferredinto a fresh tube, DTT (dithiothreitol; Sigma) was added to a con-centration of 5 mM and pre-equilibrated glutathione–Sepharose4B (Pharmacia) beads were added. After 30 min rotation at 4◦C,beads were washed four times in the same buffer as above and fu-sion proteins were eluted in PBS containing 10 mM glutathione(Sigma). Samples were dialysed (15 mM Tris, pH 7.5, 150 mMNaCl, 5 mM MgCl2 and 0.1 mM DTT), concentrated, aliquotedand snap frozen in liquid nitrogen. For experiments, each ali-quot was used once to ensure that the activity of the fusion pro-teins was preserved.
Cdc42 activation biochemical assaysActivation assays were performed using GST–PAK-crib, as de-scribed previously (Betson et al., 2002), but using anti-Cdc42antibody to assess active and total Cdc42. Specific bands weredetected by chemiluminescence (Pierce). Quantification was per-formed by scanning different film exposures to ensure a linearrange of the signal obtained (Arcus 1200 AGFA scanner) andusing TotalLab software (Nonlinear Dynamics). Values of activeCdc42 were corrected for the total level of Cdc42 protein presentin the lysates, and the amount obtained in low-calcium culturesarbitrarily set as 1. Inhibition of cadherin function during thecalcium switch was performed by pre-incubation with a cocktailof antibodies against E-cadherin and P-cadherin followed by theaddition of calcium ions for 15 min (Braga et al., 1997; Betsonet al., 2002).
Localization of active Cdc42 and RacLocalized Cdc42 activation was performed by staining withGST–WASP-crib fusion protein. After fixation in 3% para-formaldehyde in buffer A (50 mM Tris/HCl, pH 7.5, 5 mMMgCl2 and 100 mM NaCl), keratinocytes or J774A.1 macro-phages stimulated as described above were washed three times inbuffer A. Thereafter, cells were incubated with GST–WASP-crib(titrated to 25 nM) diluted in buffer A with 10% FCS at roomtemperature for 1 h. After three washes in buffer A, cells wereincubated with anti-(mouse GST) antibody, followed by anti-[mouse Cy3 (indocarbocyanine)] antibody conjugate diluted in
buffer A with 10% FCS. Double labelling was performed by in-cubation with anti-E-cadherin antibody (rat monoclonal, kerat-inocytes) or anti-IgG (anti-rabbit polyclonal antibody, J774A.1).Staining using GST–PAK-crib was performed essentially as de-scribed above, but the optimal concentration of GST–PAK-cribwas titrated for each batch of fusion protein.
Inhibition of Cdc42 functionMicroinjection of fusion proteins was performed as reported pre-viously (Braga et al., 1997). The following fusion proteins wereused: GST (pGEX-2T), activated Cdc42 (pGEX-2T-Cdc42Q61L),dominant-negative Cdc42 (pGEX-2T-Cdc42T17N) or pGEX-KG-WASP. The functionality of the proteins was checked bybinding assays in vitro. Keratinocytes grown in low-calcium me-dium were microinjected with the fusion proteins and cell–cellcontacts induced immediately for 1 h, fixed and stained for E-cadherin.
FundingWork in the Braga laboratory was supported byCancer Research UK [grant number C1282/A5960/G18481 (to J.E.)]; the Wellcome Trust [grant num-ber 081357/Z/06/Z (to S.N.)]; and the MedicalResearch Council (non-Clinical Senior Fellowship toV.M.M.B.). S.A. was supported by a PRAXIS XXIFellowship from the Portuguese Ministry of Scienceand Technology. Work in the Caron laboratory wassupported by the Medical Research Council [grantnumber G0700154]; and the Biotechnological andBiological Sciences Research Council (BBSRC) viathe Centre for Integrative Systems Biology at Imper-ial College (CISBIC) [grant number BB/C519670/1].
ReferencesAdams, J.C. (2004) Roles of fascin in cell adhesion and motility. Curr.
Opin. Cell Biol. 16, 590–596Adams, C.L. and Nelson, W.J. (1998) Cytomechanics of
cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10,572–577
Adams, C.L., Nelson, W.J. and Smith, S.J. (1996) Quantitativeanalysis of cadherin–catenin–actin reorganization duringdevelopment of cell–cell adhesion. J. Cell Biol. 135, 1899–1911
Bailly, M., Ichetovkin, I., Grant, W., Zebda, N., Machesky, L.M.,Segall, J.E. and Condeelis, J. (2001) The F-actin side bindingactivity of the Arp2/3 complex is essential for actin nucleation andlamellipod extension. Curr. Biol. 11, 620–625
Betson, M., Lozano, E., Zhang, J. and Braga, V.M.M. (2002) Racactivation upon cell–cell contact formation is dependent onsignaling from the epidermal growth factor receptor. J. Biol. Chem.277, 36962–36969
Braga, V.M.M. (2002) Cell–cell adhesion and signalling. Curr. Opin.Cell Biol. 14, 546–556
Braga, V.M. and Yap, A.S. (2005) The challenges of abundance:epithelial junctions and small GTPase signalling. Curr. Opin. CellBiol. 17, 466–474
22 C© The Authors Journal compilation C© 2010 Portland Press Ltd

Newly formed E-cadherin contacts inactivate Cdc42 Research article
Braga, V.M.M., Machesky, L.M., Hall, A. and Hotchin, N.A. (1997) Thesmall GTPases Rho and Rac are required for the establishment ofcadherin-dependent cell–cell contacts. J. Cell Biol. 137,1421–1431
Bruewer, M., Hopkins, A.M., Hobert, M.E., Nusrat, A. and Madara,J.L. (2004) RhoA, Rac1, and Cdc42 exert distinct effects onepithelial barrier via selective structural and biochemicalmodulation of junctional proteins and F-actin. Am. J. Physiol. CellPhysiol. 287, C327–C335
Burbelo, P.D., Drechsel, D. and Hall, A. (1995) A conserved bindingmotif defines numerous candidate target proteins for both Cdc42and Rac GTPases. J. Biol. Chem. 270, 29071–29074
Caron, E. and Hall, A. (1998) Identification of two distinctmechanisms of phagocytosis controlled by different Rho GTPases.Science 282, 1717–1721
Desai, R.A., Gao, L., Raghavan, S., Liu, W.F. and Chen, C.S. (2009)Cell polarity triggered by cell–cell adhesion via E-cadherin. J. CellSci. 122, 905–911
Du, D., Pedersen, E., Wang, Z., Karlsson, R., Chen, Z., Wu, X. andBrakebusch, C. (2008) Cdc42 is crucial for the maturation ofprimordial cell junctions in keratinocytes independent of Rac1.Exp. Cell Res. 315, 1480–1489
Ehrlich, J.S., Hansen, M.D. and Nelson, W.J. (2002) Spatio-temporalregulation of Rac1 localization and lamellipodia dynamics duringepithelial cell–cell adhesion. Dev. Cell 3, 259–270
Etienne-Manneville, S. and Hall, A. (2001) Integrin-mediatedactivation of Cdc42 controls cell polarity in migrating astrocytesthrough PKCζ. Cell 106, 489–496
Gloushkankova, N.A., Alieva, N.A., Krendel, M.F., Bonder, E.M.,Feder, H.H., Vasiliev, J.M. and Gelfand, I.M. (1997) Cell–cellcontact changes the dynamics of lamellar activity innon-transformed epitheliocytes but not in their ras-transformeddescendants. Proc. Natl. Acad. Sci. U.S.A. 94, 879–883
Gomes, E.R., Jani, S. and Gundersen, G.G. (2005) Nuclear movementregulated by Cdc42, MRCK, myosin, and actin flow establishesMTOC polarization in migrating cells. Cell 121, 451–463
Hirai, Y., Nose, A., Kobayashi, S. and Takeichi, M. (1989) Expressionand role of E- and P-cadherin adhesion molecules in embryonichistogenesis. I. Lung epithelial morphogenesis. Development 105,263–270
Hodivala, K.J. and Watt, F.M. (1994) Evidence that cadherins play arole in the downregulation of integrin expression that occurs duringkeratinocyte terminal differentiation. J. Cell Biol. 124, 589–600
Hoppe, A.D. and Swanson, J.A. (2004) Cdc42, Rac1, and Rac2display distinct patterns of activation during phagocytosis.Mol. Biol. Cell 15, 3509–3519
Izumi, G., Sakisaka, T., Baba, T., Tanaka, S., Morimoto, K. and Takai,Y. (2004) Endocytosis of E-cadherin regulated by Rac and Cdc42small G proteins through IQGAP1 and actin filaments. J. Cell Biol.166, 237–248
Jacinto, A., Wood, W., Balayo, T., Turmaine, M., Martinez-Arias, A.and Martin, P. (2000) Dynamic actin-based epithelial adhesion andcell matching during Drosophila dorsal closure. Curr. Biol. 10,1420–1426
Jaffe, A.B. and Hall, A. (2005) Rho GTPases: biochemistry andbiology. Ann. Rev. Cell Dev. Biol. 21, 247–269
Kim, S.H., Zhigang, L. and Sacks, D.B. (2000) E-cadherin-mediatedcell–cell attachment activates Cdc42. J. Biol. Chem. 275,36999–37005
Kodama, A., Takaishi, K., Nakano, K., Nishioka, H. and Takai, Y.(1999) Involvement of Cdc42 small G protein in cell–cell adhesion,migration and morphology of MDCK cells. Oncogene 18,3996–4006
Krendel, M.F. and Bonder, E.M. (1999) Analysis of actin filamentbundle dynamics during contact formation in live epithelial cells.Cell Motil. Cytoskeleton 43, 296–309
Mallavarapu, A. and Mitchison, T. (1999) Regulated actincytoskeleton assembly at filopodium tips controls their extensionand retraction. J. Cell Biol. 146, 1097–1106
Martin-Belmonte, F. and Mostov, K. (2008) Regulation of cell polarityduring epithelial morphogenesis. Curr. Opin. Cell Biol. 20,227–234
McNeill, H., Ryan, T.A., Smith, S.J. and Nelson, J.W. (1993) Spatialand temporal dissection of immediate and early events followingcadherin-mediated epithelial cell adhesion. J. Cell Biol. 120,1217–1226
Mege, R.M., Gavard, J. and Lambert, M. (2006) Regulation ofcell–cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18,541–548
Musch, A., Cohen, D., Kreitzer, G. and Rodriguez-Boulan, E. (2001)Cdc42 regulates the exit of apical and basolateral proteins fromthe trans-Golgi network. EMBO J. 20, 2171–2179
Noren, N.K., Niessen, C.M., Gumbiner, B.M. and Burridge, K. (2001)Cadherin engagement regulates Rho family GTPases.J. Biol. Chem. 276, 33305–33308
Otani, T., Ichii, T., Aono, S. and Takeichi, M. (2006) Cdc42 GEF Tubaregulates the junctional configuration of simple epithelial cells. J.Cell Biol. 175, 135–146
Patel, J.C., Hall, A. and Caron, E. (2002) Vav regulates activation ofRac but not Cdc42 during FcγR-mediated phagocytosis. Mol. Biol.Cell 13, 1215–1226
Pollard, T.D., Blanchoin, L. and Mullins, R.D. (2000) Molecularmechanisms controlling actin filament dynamics in non musclecells. Annu. Rev. Biophys. Biomol. Struct. 29, 545–576
Raich, W.B., Agbunag, C. and Hardin, J. (1999) Rapid epithelial-sheetsealing in the Caenorhabditis elegans embryo requirescadherin-dependent filopodial priming. Curr. Biol. 9,1139–1146
Rheinwald, J.G. (1989) Methods for clonal growth and serialcultivation of normal human epidermal keratinocytes andmesothelial cells, In Cell Growth and Division: A PracticalApproach (Baserga, R., ed.) pp. 81–94, IRL Press, Oxford
Rojas, R., Ruiz, W.G., Leung, S.-M., Jou, T.-S. and Apodaca, G.(2001) Cdc42-dependent modulation of tight junctions andmembrane protein traffic in polarized Madin–Darby canine kidneycells. Mol. Biol. Cell 12, 2257–2274
Shimoyama, Y., Yoshida, T., Terada, M., Shimosato, Y., Abe, O. andHirohashi, S. (1989) Molecular cloning of human Ca2+-dependentcell–cell adhesion molecule homologous to mouse placentalcadherin: its low expression in human placental tissues. J. CellBiol. 109, 1787–1794
Stoffler, H.E., Honnert, U., Bauer, C.A., Hofer, D., Schwarz, H., Muller,R.T., Drenckhahn, D. and Bahler, M. (1998) Targeting of themyosin-I myr 3 to intercellular adherens type junctions induced bydominant active Cdc42 in HeLa cells. J. Cell Sci. 111,2779–2788
Svitkina, T.M. and Borisy, G.G. (1999) Arp2/3 complex and actindepolymerizing factor/cofilin in dendritic organization andtreadmilling of actin filament array lamellipodia. J. Cell Biol. 145,1009–1026
Svitkina, T.M., Bulanova, E.A., Chaga, O.Y., Vignjevic, D.M., Kojima,S., Vasiliev, J.M. and Borisy, G.G. (2003) Mechanism of filopodiainitiation by reorganization of a dendritic network. J. Cell Biol. 160,409–421
Takai, Y., Miyoshi, J., Ikeda, W. and Ogita, H. (2008) Nectins andnectin-like molecules: roles in contact inhibition of cell movementand proliferation. Nat. Rev. Mol. Cell Biol. 9, 603–615
Takaishi, K., Sasaki, T., Kotani, H., Nishioka, H. and Takai, Y. (1997)Regulation of cell–cell adhesion by Rac and Rho small G proteinsin MDCK cells. J. Cell Biol. 139, 1047–1059
Theriot, J.A. and Mitchison, T.J. (1991) Actin microfilament dynamicsin locomoting cells. Nature 352, 126–131
www.biolcell.org | Volume 102 (1) | Pages 13–24 23

J. Erasmus and others
Tzima, E., Kiosses, W.B., del Pozo, M.A. and Schwartz, M.A. (2003)Localized Cdc42 activation, detected using a novel assay,mediates microtubule organizing center positioning in endothelialcells in response to fluid shear stress. J. Biol. Chem. 278,31020–31023
Vasioukhin, V., Bauer, C., Yin, M. and Fuchs, E. (2000) Directed actinpolymerization is the driving force for epithelial cell–cell adhesion.Cell 100, 209–219
Vignjevic, D., Yarar, D., Welch, M.D., Peloquin, J., Svitkina, T. andBorisy, G.G. (2003) Formation of filopodia-like bundles in vitro froma dendritic network. J. Cell Biol. 160, 951–962
Vignjevic, D., Kojima, S., Aratyn, Y., Danciu, O., Svitkina, T. andBorisy, G.G. (2006) Role of fascin in filopodial protrusion. J. CellBiol. 174, 863–875
Wu, X., Quondamatteo, F. and Brakebusch, C. (2006) Cdc42expression in keratinocytes is required for the maintenance of thebasement membrane in skin. Matrix Biol. 25, 466–474
Wu, X., Li, S., Chrostek-Grashoff, A., Czuchra, A., Meyer, H.,Yurchenco, P.D. and Brakebusch, C. (2007) Cdc42 is crucialfor the establishment of epithelial polarity duringearly mammalian development. Dev. Dyn. 236,2767–2778
Yamada, S. and Nelson, W.J. (2007) Localized zones of Rho and Racactivities drive initiation and expansion of epithelial cell-celladhesion. J. Cell Biol. 178, 517–527
Yonemura, S., Itoh, M., Nagafuchi, A. and Tsukita, S. (1995)Cell-to-cell adherens junction formation and actin filamentorganization: similarities and differences between non-polarizedfibroblasts and polarized epithelial cells. J. Cell Sci. 108,127–142
Zhang, J., Betson, M., Erasmus, J., Zeikos, K., Bailly, M., Cramer, L.and Braga, V.M.M. (2005) Actin at cell–cell junctions is composedof two dynamic and functional populations. J. Cell Sci. 118,5549–5562
Received 18 March 2009/25 June 2009; accepted 7 July 2009
Published as Immediate Publication 7 July 2009, doi:10.1042/BC20090048
24 C© The Authors Journal compilation C© 2010 Portland Press Ltd
Related Documents











