The lowering of temperature to preserve tissues is not a new concept. Cooling organs that are to be used for transplantation is known to prolong their viability, and the discovery of well-preserved life forms in cold climates also points to the protective role of lowered temperatures. Although the phenomenon that profound temperature changes can protect the brain from various insults has been studied for quite some time, a report by Busto and colleagues 1 demonstrated that even small, clinically feasi- ble decreases in body temperature could prevent neuron death. This finding triggered renewed interest in cooling — or hypothermia — as a means of protecting the brain. Therapeutic hypothermia has been extensively studied in the laboratory. It is one of the most robust neuropro- tectants (that is, it protects neurons against apoptosis and necrosis) studied to date, and recent clinical studies have established a role for therapeutic cooling in neuroprotec- tion in some clinical conditions, including anoxic brain injury due to cardiac arrest 2,3 and hypoxic ischaemic neo- natal encephalopathy 4,5 . Furthermore, hypothermia has been widely shown in the laboratory to protect against experimental stroke, brain and spinal cord trauma, although this remains to be convincingly shown at the clinical level. Hypothermia is also an important part of the practice sometimes referred to as ‘suspended anima- tion’, which could be described as the slowing or cessation of essential metabolic processes without causing death 6 . Our increasing understanding of the metabolic and molecular events that occur following brain ischaemia and related acute neurological insults, such as hypoxia, trauma and brain haemorrhage (FIG. 1), has begun to shed light on the many facets of and pathways involved in ischaemic injury and how they are affected by thera- peutic hypothermia. Animal models of focal and global cerebral ischaemia are the most widely studied in terms of hypothermic neuroprotection. Focal cerebral ischae- mia models attempt to create brain injury that resem- bles stroke, whereas global cerebral ischaemia models attempt to imitate brain injury resulting from cardiac arrest. Focal cerebral ischaemia models can be further divided into models of permanent occlusion, where the parent vessel remains blocked (which could be said to represent most clinical strokes), and models of tem- porary occlusion, where the parent vessel is re-opened after a period of time. Hypothermia has also been stud- ied in models of brain trauma 7 and spinal cord trauma 8 , where it had beneficial results, but this area of research is beyond the scope of this Review. This Review focuses mostly on animal studies of brain ischaemia, including both focal and global cer- ebral ischaemia. These studies have collectively shown that hypothermia affects nearly every investigated cell death pathway, including pathways leading to excitotox- icity, apoptosis, inflammation and free radical produc- tion, and it is likely that no single factor can explain its underlying beneficial effect. We first discuss methods by which cooling is achieved in the laboratory. We then review findings from animal studies that have begun to reveal the effects of hypothermia during the acute, suba- cute and chronic phases of brain injury, and consider the future prospects of therapeutic hypothermia in terms of clinical translation and target identification. 1 Department of Neurology, University of California, San Francisco, California 94143‑0248, USA. 2 San Francisco Veterans Affairs Medical Center, San Francisco, California 94121, USA. 3 Department of Physiology, Kyungpook National University School of Medicine, Daegu, 700‑422, South Korea. Correspondence to M.A.Y. e‑mail: [email protected] doi:10.1038/nrn3174 Published online 22 February 2012 Apoptosis Innate, programmed cell death that is energy-dependent and leads to nuclear and cytoplasmic compaction with characteristic blebbing of the nucleus. It occurs during development but also in disease states. Necrosis Acute, uncontrolled cell death that leads to cell lysis. Neuroprotective mechanisms of hypothermia in brain ischaemia Midori A. Yenari 1,2 and Hyung Soo Han 3 Abstract | Cooling can reduce primary injury and prevent secondary injury to the brain after insults in certain clinical settings and in animal models of brain insult. The mechanisms that underlie the protective effects of cooling — also known as therapeutic hypothermia — are slowly beginning to be understood. Hypothermia influences multiple aspects of brain physiology in the acute, subacute and chronic stages of ischaemia. It affects pathways leading to excitotoxicity, apoptosis, inflammation and free radical production, as well as blood flow, metabolism and blood–brain barrier integrity. Hypothermia may also influence neurogenesis, gliogenesis and angiogenesis after injury. It is likely that no single factor can explain the neuroprotection provided by hypothermia, but understanding its myriad effects may shed light on important neuroprotective mechanisms. REVIEWS NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 267 © 2012 Macmillan Publishers Limited. All rights reserved

Neuroprotective Mechanisms of Hypothermia in Brain Ischaemia
Dec 15, 2015
buen articulo
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The lowering of temperature to preserve tissues is not a new concept. Cooling organs that are to be used for transplantation is known to prolong their viability, and the discovery of well-preserved life forms in cold climates also points to the protective role of lowered temperatures. Although the phenomenon that profound temperature changes can protect the brain from various insults has been studied for quite some time, a report by Busto and colleagues1 demonstrated that even small, clinically feasi-ble decreases in body temperature could prevent neuron death. This finding triggered renewed interest in cooling — or hypothermia — as a means of protecting the brain. Therapeutic hypothermia has been extensively studied in the laboratory. It is one of the most robust neuropro-tectants (that is, it protects neurons against apoptosis and necrosis) studied to date, and recent clinical studies have established a role for therapeutic cooling in neuroprotec-tion in some clinical conditions, including anoxic brain injury due to cardiac arrest2,3 and hypoxic ischaemic neo-natal encephalopathy4,5. Furthermore, hypothermia has been widely shown in the laboratory to protect against experimental stroke, brain and spinal cord trauma, although this remains to be convincingly shown at the clinical level. Hypothermia is also an important part of the practice sometimes referred to as ‘suspended anima-tion’ , which could be described as the slowing or cessation of essential metabolic processes without causing death6.
Our increasing understanding of the metabolic and molecular events that occur following brain ischaemia and related acute neurological insults, such as hypoxia, trauma and brain haemorrhage (FIG. 1), has begun to
shed light on the many facets of and pathways involved in ischaemic injury and how they are affected by thera-peutic hypothermia. Animal models of focal and global cerebral ischaemia are the most widely studied in terms of hypothermic neuroprotection. Focal cerebral ischae-mia models attempt to create brain injury that resem-bles stroke, whereas global cerebral ischaemia models attempt to imitate brain injury resulting from cardiac arrest. Focal cerebral ischaemia models can be further divided into models of permanent occlusion, where the parent vessel remains blocked (which could be said to represent most clinical strokes), and models of tem-porary occlusion, where the parent vessel is re-opened after a period of time. Hypothermia has also been stud-ied in models of brain trauma7 and spinal cord trauma8, where it had beneficial results, but this area of research is beyond the scope of this Review.
This Review focuses mostly on animal studies of brain ischaemia, including both focal and global cer-ebral ischaemia. These studies have collectively shown that hypothermia affects nearly every investigated cell death pathway, including pathways leading to excitotox-icity, apoptosis, inflammation and free radical produc-tion, and it is likely that no single factor can explain its underlying beneficial effect. We first discuss methods by which cooling is achieved in the laboratory. We then review findings from animal studies that have begun to reveal the effects of hypothermia during the acute, suba-cute and chronic phases of brain injury, and consider the future prospects of therapeutic hypothermia in terms of clinical translation and target identification.
1Department of Neurology, University of California, San Francisco, California 94143‑0248, USA.2San Francisco Veterans Affairs Medical Center, San Francisco, California 94121, USA. 3Department of Physiology, Kyungpook National University School of Medicine, Daegu, 700‑422, South Korea.Correspondence to M.A.Y. e‑mail: [email protected]:10.1038/nrn3174Published online 22 February 2012
ApoptosisInnate, programmed cell death that is energy-dependent and leads to nuclear and cytoplasmic compaction with characteristic blebbing of the nucleus. It occurs during development but also in disease states.
NecrosisAcute, uncontrolled cell death that leads to cell lysis.
Neuroprotective mechanisms of hypothermia in brain ischaemiaMidori A. Yenari1,2 and Hyung Soo Han3
Abstract | Cooling can reduce primary injury and prevent secondary injury to the brain after insults in certain clinical settings and in animal models of brain insult. The mechanisms that underlie the protective effects of cooling — also known as therapeutic hypothermia — are slowly beginning to be understood. Hypothermia influences multiple aspects of brain physiology in the acute, subacute and chronic stages of ischaemia. It affects pathways leading to excitotoxicity, apoptosis, inflammation and free radical production, as well as blood flow, metabolism and blood–brain barrier integrity. Hypothermia may also influence neurogenesis, gliogenesis and angiogenesis after injury. It is likely that no single factor can explain the neuroprotection provided by hypothermia, but understanding its myriad effects may shed light on important neuroprotective mechanisms.
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 267
© 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Neuroscience
Phases of ischaemic stroke
• Blood flow decrease
• Ionic homeostasis disturbance
• Apoptosis
• Inflammation by neutrophils, monocytes and microglia
• Necrotic debris removal
• Intracellular calcium increase • Cytokine production
• Stem cell proliferation, differentiation and maturation
• Glutamate release increase and excitotoxicity
• Cytotoxic oedema
• Membrane, mitochondrial and DNA damage
• Proteolytic enzyme activation
• Vasogenic oedema and intracranial pressure increase
• Angiogenesis
• Gliosis
• Reconnection of lost circuits
• Misfolding of proteins and enzyme dysfunction
• Reactive oxygen species production
• Stimulation of neurogenesis and angiogenesis
• Neurovascular remodelling and functional recovery
• Necrosis
Acute phase (minutes–hours) Subacute phase (hours–days) Chronic phase (weeks–months)
Methodological aspects of brain coolingMost laboratory studies of hypothermia use small rodents, although a few use larger mammals including non-human primates; in rodents, cooling is performed by applying a cooling blanket or by spraying water or alcohol on the anaesthetized animal’s fur. Cooling for longer durations (that is, for more than 1 day) in awake and freely moving animals can be accomplished by using automated misting systems and overhead fans9.
Studies aimed at determining the optimal parameters to achieve brain protection without causing additional adverse events have shown that relatively small decreases in brain temperature are as protective as lower tempera-tures10,11. Indeed, brain temperatures in the range of 30–34 °C (decreased from normal body temperatures of 36–38 °C) seem to provide protection that is as robust as temperatures below 25 °C. Timing and duration are also important factors for the effects of cooling, with early initiation increasing the likelihood of a good out-come. For example, in models of focal cerebral ischae-mia, protection is generally only observed if it is initiated within 1–2 h of ischaemia onset, although it can still be observed when cooling is delayed by more than 2 h, provided it is maintained for relatively long durations (24–48 h)12. The therapeutic time window seems wider in models of global cerebral ischaemia12. Importantly, only a few studies have demonstrated beneficial effects of cooling in terms of long-term benefit13,14 — many studies in animal models were carried out for relatively short time periods, with a relatively brief cooling period.
Although cooling can be easily achieved in small rodents, in humans it brings substantial challenges.
Because humans have a larger body mass and because of the co-morbidities such as diabetes and cardiovas-cular disease that typically occur in victims of cardiac arrest and stroke, cooling takes longer and may not be as well tolerated as in smaller animals12. In addition, early initiation is not always feasible in clinical settings. Furthermore, most stroke patients are not anaesthetized and are not generally comatose. Thus, shivering can occur during cooling, which not only causes discomfort but can also inhibit cooling efforts. These methodologi-cal aspects of therapeutic cooling raise questions as to whether the effects of hypothermia observed in the labo-ratory are durable, and whether findings from studies in young laboratory animals apply to older adult humans with various co-morbidities10,15.
Finally, the phenomenon of hibernation in some mammalian species also deserves some attention, as hibernation leads to marked reductions in body tem-perature and slowing of metabolism. The changes that occur in the brain during hibernation are similar to those that occur during therapeutic hypothermia. Further, an understanding of how animals enter and maintain hibernation may lead to important insights into how hypothermia should be applied in humans (BOX 1).
Acute effects of hypothermiaEffects on metabolism, blood flow and excitoxicity. The neuroprotective effects of cooling have largely been attrib-uted to the finding that lowered temperatures decrease the metabolic rate and reduce blood flow in the brain (see REF. 16 for a review). Temperature reductions decrease brain oxygen consumption and glucose metabolism
Figure 1 | The events involved in the pathogenesis of cerebral ischaemia are classified by their active time. The events associated with the acute phase initiate and manifest their actions within minutes to hours after the onset of stroke. These events include the loss of blood flow, loss of ion homeostasis, release of excitotoxic neurotransmitters, subcellular organelle damage, loss of normal protein structure and function, cell swelling followed by cell lysis, which gives rise to cytotoxic oedema, and necrosis. Damage to mitochondria can set the stage for the generation of reactive oxygen species when the occluded vessel is reperfused (reopened), because these mitochondria are no longer able to effectively neutralize reactive species. Necrotic debris can then give rise to many subacute events, which occur hours to days later (the subacute phase). Many of these processes are secondary to the initial ischaemic event, such as delayed cell death of cells (apoptosis) in the periphery of the infarct — an area that is exposed to less severe injury. In addition, necrotic debris can stimulate immune responses and activate proteases. The inflammatory response itself can lead to further reactive oxygen species generation. Although many subacute events could be considered damaging themselves, some of the factors generated may be important in setting the stage for processes of recovery and repair, such as neurogenesis and angiogenesis. In the chronic phase, which starts weeks to months later, many restorative processes occur, such as debris removal, cell genesis, synaptogenesis and remodelling.
R E V I E W S
268 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved

ReperfusionThe period of resumed blood flow to the tissue after arterial occlusion.
HyperaemiaHigher than normal blood flow.
TorporA prolonged state of energy conservation that allows heterothermic animals to tolerate limitations in resource availability that are encountered in extreme environments.
by about 5% per degree Celsius17. Hypothermia also preserves high-energy phosphate compounds, such as ATP, and maintains tissue pH. By preserving the brain’s metabolic stores, cooling can prevent the downstream consequences of increased lactate production (which is dependent on anaerobic metabolism) and the develop-ment of acidosis. The effect of cooling on cerebral blood flow is less straightforward. In the absence of injury, cool-ing decreases cerebral blood flow, whereas under injury conditions such as stroke, its effects are more complex. (BOX 2). During ischaemia, blood flow is markedly reduced as a result of vessel occlusion; however, when blood flow is restored (reperfusion), there is an overshoot of flow (hyper-aemia) followed by a gradual decline that occurs over a period of hours. Hypothermia actually blunts the imme-diate hyperaemia and prevents the gradual reduction in cerebral blood flow that follows17. In other types of brain injury, such as trauma, hypothermia has not always been shown to affect cerebral blood flow17.
Although reports have been inconsistent (see REF. 18 for a review), therapeutic cooling has also been shown to prevent the accumulation or release of excitotoxic amino acids such as glutamate. This may be attributable to the effect of cooling on metabolism, which preserves tissue ATP levels. ATP is needed to maintain ion gradients, and when these concentration gradients are disturbed, such as in the case of ischaemia, calcium influx occurs and leads to increased extracellular glutamate levels19. Hypothermia may also prevent the consequences of excitotoxicity by limiting calcium influx through AMPA channels. The glu-tamate receptor 2 (GluR2) subunit of the AMPA receptor is thought to limit calcium influx, and its downregulation by ischaemia20 may lead to the entry of excess calcium. Indeed, one study demonstrated that hypothermia atten-uates ischaemia-induced downregulation of GluR2 in a model of global cerebral ischaemia20.
However, several studies in rodents, particularly those examining mild and moderate hypothermia, have shown that these acute mechanisms do no fully explain the pro-tective effect of hypothermia. The changes in oxygen and
glucose consumption and in cerebral blood flow cannot explain the nonlinear protective effects of cooling21. That is, the extent of neuroprotection does not proportionately increase with temperature decreases. Further, the protec-tive effects of hypothermia cannot simply be explained by the prevention of ATP loss or reduction in extracellular glutamate as ATP loss and glutamate accumulation occur within minutes of ischaemia onset, whereas hypothermia can be effective even when cooling is initiated well after these events have occurred12. Furthermore, some stud-ies have shown that hypothermia merely delays many of these acute events15,22. The debate over whether hypo-thermia really provides permanent protection or merely delays the injury processes12 is important and still rel-evant today. In addition, the finding that cooling is also protective if it is initiated even hours after ischaemia onset suggests that hypothermia might have important effects on injurious processes in the subacute and even chronic phases of stroke and related insults, as discussed below (BOX 2).
Effects on early molecular events. Hypothermia has also been shown to affect other acute processes associated with ischaemia, including the induction of immediate early gene expression23 and the cellular stress response. The few studies that examined the effect of cooling on the stress response — specifically the expression of heat shock proteins, which are upregulated in response to various cellular stresses — are mixed. Some studies have shown that the expression of 70 kDa inducible heat shock protein (HSP70) is increased under hypothermic conditions24,25, and this might be consistent with its neu-roprotective properties26. However, other studies have shown that HSP70 expression is decreased under simi-lar conditions27, and others have shown no influence of cooling on its expression28. Thus, it is unclear whether hypothermic neuroprotection is mediated by altera-tions in the cellular stress response. The significance of the effect of hypothermia on immediate early gene expression is also unclear, as no subsequent studies have been carried out to systematically determine whether the alteration in gene expression underlies the protec-tive effect of hypothermia, or whether its suppression represents a global downregulation of transcription and contributes nothing to the observed protection.
MicroRNAs (miRNAs), a subset of non-coding RNAs, have been a topic of recent investigation in brain injury models, including stroke, where their expression increases as early as 2 h after ischaemia onset29. miRNAs are thought to play a part in silencing mRNAs, but they can also regulate a range of signalling pathways29. It is conceivable that they have an important role in stroke pathogenesis, and the roles of specific miRNAs are cur-rently under investigation. A recent report in a model of traumatic brain injury showed that cooling alters the expression of several miRNAs30. A few miRNAs, includ-ing miR-874 and miR-451, were most strongly affected: cooling decreased the expression of both miRNAs at 7 h, but miR-451 was increased by cooling at 24 h compared to normothermia. Further research is needed to better define their role in brain injury.
Box 1 | Hibernation
Hibernation is characterized by a prolonged state of energy conservation that animals such as arctic ground squirrels and bears routinely undergo in order to withstand extremely cold environments. In spite of the profound reduction of cerebral blood flow that ensues, hibernation causes no lasting brain injury and hibernating animals show relatively increased tolerance to ischaemic insults during and after the hibernation period119. Several studies have investigated the mechanisms underlying hibernation, and there seem to be many parallels between the brain changes that occur during hibernation and the adaptations that the brain undergoes in order to develop resistance to injury. These overlapping mechanisms include suppression of protein synthesis, excitotoxicity, inflammatory responses, oxidative stress and activation of cell death pathways120,121. Small hibernators such as arctic ground squirrels undergo regulated decreases in core body temperature to near or below freezing during torpor, whereas core body temperature in larger animals such as bears is far higher during hibernation. The metabolism of hibernating bears is reduced by 53% from the basal metabolic rate, even when core body temperature has returned to normothermic levels towards the end of hibernation122. As metabolic conditions of non-hibernating mammals under therapeutic hypothermia are similar to those of the hibernating bear, the therapeutic implications of understanding how animals achieve hibernation may provide useful insight into how to attain neuroprotection in humans.
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 269
© 2012 Macmillan Publishers Limited. All rights reserved

Subacute effects of hypothermiaThe subacute phase of stroke is considered to be the time during which secondary injury mechanisms occur, and this is generally considered to be anywhere from 1 to 7 days post-ischaemia. It is during this period that many reperfusion-related pathways are activated as a result of the increased generation of reactive oxygen species (ROS) by injured cells. In addition, inflammatory responses are activated during this period, along with other cell death pathways, including those leading to apoptosis. As a result, the extent of ischaemic injury can widen during the suba-cute period, and secondary injury such as blood–brain
barrier (BBB) disruption, oedema formation and haemor-rhage can occur. Several studies have now demonstrated that hypothermia affects multiple cell death and cell sur-vival pathways. Its beneficial effects of suppressing sev-eral cell death mechanisms and enhancing cell survival proteins might explain why hypothermia seems to be the most robust neuroprotectant studied to date.
Effects on cell death pathways. Hypothermia has been shown to affect several aspects of apoptotic cell death. There are two main pathways that lead to apoptosis (FIG. 2). The intrinsic pathway is thought to stem from within
Box2 | Phase-specific effects of hypothermia in cerebral ischaemia
In the acute phase, cooling has beneficial effects on the brain as it preserves metabolic stores and maintains tissue homeostasis, decreases excitatory amino acid accumulation and alters immediate early gene expression. During the subacute phase, hypothermia alters apoptotic cell death pathways in such a way as to favour cell survival. It also prevents blood–brain barrier (BBB) disruption and its downstream effects, such as oedema and brain haemorrhage. Hypothermia also inhibits many pro-inflammatory responses, and enhances the expression of trophic factors. In the chronic phase, the beneficial effects of hypothermia can still be observed, even when cooling was applied during only the acute and early subacute phase. Weeks to months after cooling (that is, in the chronic phase), the brain seems to have increased precursor cell generation and differentiation, increased synaptogenesis and improved functional recovery compared to normothermic brains.
Phase Effect
Acute phase Differential alteration of cerebral blood flow
Preservation of energy stores
Reduction of excitatory amino acids
Decrease of lactate production and cellular acidosis
Improvement of brain glucose metabolism
Alteration of immediate early gene expression
Alteration of the cellular stress response
Subacute phase Prevention of apoptotic death:
• decreased BAX expression (pro-apoptotic) and increased BCL-2 expression (anti-apoptotic)
• inhibition of caspase-dependent pathway (cytochrome c, APAF1) and non-dependent pathway (AIF)
• inhibition of extrinsic apoptosis pathway
• blocking of pro-apoptotic PKCδ activation and anti-apoptotic PKCε degradation
• preservation of anti-apoptotic AKT activity and attenuation of pro-apoptotic PTEN action
Inhibition of inflammation:
• decreased inflammatory cell infiltration
• decreased activation of immune transcription factors
• reduced production of free radicals
Reduction of BBB disruption:
• preservation of vascular endothelial function
• reduction of extracellular protease expression and activity
• reduction of brain haemorrhage
Increase of neurotrophins and their receptors
Chronic phase Enhanced differentiation of precursor cells
Enhanced angiogenesis
Increase of neural and oligodendrocyte precursor cells
Induced neurite outgrowth and increased neuronal connectivity
AIF, apoptosis inducing factor; PKC, protein kinase C; PTEN, phosphatase and tensin homologue.
R E V I E W S
270 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved

Procaspase 8
Caspase 8
FAS
Caspase 3 Caspase 9
Caspase 3
FADD
Activated caspases
Apoptosis
APAF1
BCL-2
Cytochrome c
BAXBID
tBID AIF
Intracellular apoptotic signals
PKCδ
PKCε
+
Intrinsic pathway
Extrinsic pathway
Caspase-independentpathway
FASL
+
+
Cooling
Cooling
Cooling
Cooling
Nature Reviews | Neuroscience
the cell at the level of the mitochondria31, whereas the extrinsic pathway is triggered via a cell surface receptor32. Hypothermia can affect both pathways, but whether cooling has any effect on neuron survival depends on whether apoptosis is occurring in a given model or para-digm. Models of moderate ischaemic injury — such as models of global cerebral ischaemia or mild focal cerebral ischaemia (30 minutes or shorter durations of middle cerebral artery occlusion (MCAO)) — lead to predomi-nantly apoptotic cell death, whereas more severe insults result in predominantly necrotic cell death.
Cooling can interfere with the intrinsic (or mito-chondrial) pathway by changing the expression of BCL-2 family members, reducing cytochrome c release and decreasing caspase activation33. In models of global cerebral ischaemia, hypothermia leads to reduc-tions in pro-apoptotic BCL-2 family members such as BCL-2-associated X (BAX) and increases in the anti-apoptotic member BCL-2. Acting downstream of BCL-2 family proteins, protein kinase Cδ (PKCδ) is a PKC iso-form that has been shown to contribute to ischaemic injury34, and caspase 3 leads to translocation of PKCδ from the cytosol to the mitochondria and to the nucleus to induce apoptosis35. By contrast, a different isoform, PKCε is anti-apoptotic, and is degraded by caspases. Interestingly, hypothermia does not seem to alter over-all levels of PKCδ36, but it blocks its translocation to the mitochondria and the nucleus and stimulates the action of PKCε after ischaemia37.
The extrinsic apoptotic pathway also seems to be acti-vated in ischaemic brain injury. The most widely studied apoptosis-inducing receptor and ligand in this pathway are FAS and FASL, respectively. Genetic or pharmacologi-cal disruption of this pathway has been shown to improve outcome from experimental stroke38,39, and hypothermia seems to suppress the expression of both proteins in models in which neuroprotection is observed40. In order for FAS to initiate apoptosis via caspase 8 activation, its ligand FASL must bind FAS. How FASL binds FAS is somewhat unclear: many reports indicate that FASL must be present on the cell’s surface in order to engage FAS41, but other reports indicate that FASL must first be cleaved from the surface by activated matrix metalloproteinases (MMPs) and solubilized42. Hypothermia seems to pre-vent this cleavage, as levels of soluble FASL are decreased in cooled rodent brains, as are levels of several MMPs43–
46. The decreased level of soluble FASL was also associ-ated with decreased caspase 8 activation, which occurs downstream of FAS activation40.
In models of more severe stroke (MCAO of 2 h or longer), hypothermia does not seem to affect BCL-2 family members or caspase activation but prevents cytochrome c release47. These observations might be explained by a third, caspase-independent apop-totic pathway that involves direct cell killing through the release of apoptosis-inducing factor (AIF) from mitochondria48. Indeed, in a more severe model of MCAO, hypothermia reduced apoptotic cell death and suppressed AIF translocation49.
Other, more recently studied molecules involved in apoptosis are affected by hypothermia as well. Phosphatase
Figure 2 | Apoptotic pathways. Apoptosis occurs through three main pathways. The extrinsic pathway begins outside the cell, with the activation of specific death receptors on the cell surface. FAS is one of many such death receptors that are activated by death ligands. The ligand for FAS is FASL. Signalling from death receptors activates a signalling complex through a death-inducing signalling complex (DISC). Activation of DISC, a complex of a death ligand, death receptor and adaptor proteins, such as FAS- associated death domain protein (FADD), leads to caspase 8 activation. Activated caspase 8 initiates the caspase cascade, leading ultimately to cleavage and activation of effector caspases such as caspase 3. Cleaved caspases eventually cause apoptosis. The intrinsic pathway is initiated from within the cell in response to ‘death signals’ such as DNA damage, a defective cell cycle, detachment from the extracellular matrix, hypoxia, loss of cell survival factors or other types of severe cellular stress. The intrinsic apoptotic pathway hinges on the balance of activity between the pro-apoptotic (BCL-2 antagonist/killer (BAK), BCL-2-associated X protein (BAX), BCL-2-related ovarian killer protein (BOK), BH3-interacting domain death agonist (BID), and so on) and anti-apoptotic (BCL-2, BCL-XL, and so on) members of the BCL-2 superfamily of proteins, which are thought to influence mitochondrial signals. Mitochondrial apoptotic signalling leads to the release of cytochrome c from the mitochondrial intermembrane space to the cytosol, where it forms an apoptosome with apoptotic pro-tease-activating factor 1 (APAF1). The apoptosome activates caspase 9 and proceeds down the same common pathway as the extrinsic pathway, leading to caspase 3 activation and, eventually, apoptosis. Protein kinase C (PKC) family members can be either pro-apoptotic (PKCδ) or anti-apoptotic (PKCε). PKCδ translocates from the cytosol to the mitochondria to initiate apoptosis, whereas PKCε prevents apoptosis. There is also crosstalk between the intrinsic and extrinsic pathways, as death receptor-activated caspase 8 can cleave the pro-apoptotic BCL-2 family member BID to a truncated form (tBID), and then can also initiate the intrinsic pathway through the mitochondria. A third apoptotic pathway has also been described, where apoptosis- inducing factor (AIF) is released from the mitochondria and directly leads to apoptosis without activating caspases. Cooling affects several aspects of apoptotic cell death pathways (white boxes).
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 271
© 2012 Macmillan Publishers Limited. All rights reserved

and tensin homologue (PTEN) is a tumour suppressor molecule with pro-apoptotic functions. PTEN deletion has been shown to prevent ischaemic brain injury50; how-ever, PTEN phosphorylation, which leads to its deactiva-tion, is normally decreased in brain ischaemia18. Under conditions of hypothermia in which neuroprotection was observed, phosphorylated PTEN levels were preserved, but not under hypothermic conditions that did not result in neuroprotection36. Thus, the deactivated form of this pro-apoptotic protein seems to be associated with hypo-thermic neuroprotection. The mechanisms underlying this association require further investigation.
Effects on survival pathways. Several neurotrophic factors in the brain have been studied with regard to their therapeutic potential in various acute neuro-logical insults. These proteins control synaptic func-tion and plasticity and sustain neuronal cell survival, morphol ogy and differentiation. In animal models of brain insult, exogenous administration of one or more of these factors improved functional neuro logical out-come without necessarily affecting lesion size. In studies in which hypothermia had neuroprotective effects fol-lowing ischaemic brain insult, levels of brain-derived neu rotrophic factor (BDNF)51,52, glial-derived neu-rotrophic factor (GDNF)53 and neurotrophin54 were increased in the brain. Further, hypothermia increased extracellular signal-regulated kinase (ERK) phospho-rylation, a downstream element of BDNF signalling52,55. However, ERK signalling itself does not seem to account for the protective effect of hypothermia, as pharmaco-logic inhibition of ERK by U0126 failed to prevent the benefit of hypothermia56.
Hypothermia also upregulates other survival factors. As described above, hypothermia upregulates the anti-apoptotic protein BCL-2 (REFS 57,58) and also promotes activation of AKT59, a serine/threonine protein kinase that has multiple roles in glucose metabolism, cell proliferation,
apoptosis, transcription and cell migration. After phospho-rylation by phosphoinositide 3-kinase (PI3K), activated AKT phosphorylates (that is, inactivates) pro-apoptotic proteins such as glycogen synthase 3β (GSK3β) and BCL-2 antagonist of cell death (BAD). In a model of ischaemic brain injury, hypothermia conferred neuro protection by maintaining AKT activity, and this protection was lost when hypothermia was applied in combination with an AKT inhibitor59. Thus, although hypothermia has gener-ally been documented to suppress or decrease metabo-lism and protein expression, it can also upregulate or maintain proteins involved in cell survival and growth. The family of cold-shock proteins, which are upregulated at lower temperatures, has been studied in systems other than the CNS, but might also be relevant in hypothermia-induced brain protection (BOX 3).
Effects on inflammatory mediators. Although the inflammation that accompanies many acute neurologi-cal conditions is thought to contribute to tissue recovery and repair, studies have shown that it can also exacerbate acute brain injury60. The injured brain stimulates innate immune responses leading to activation of microglia and circulating leukocytes, and these immune cells can then release various molecules, including ROS, proteases and pro-inflammatory cytokines. These molecules can acti-vate more inflammatory cells, leading to a vicious cycle of cell death and immune activation.
Several animal studies have shown that inhibiting various aspects of this immune response by brain cool-ing can have beneficial effects on neurological out-come following brain ischaemia and injury61 (FIG. 2). Hypothermia indeed affects many aspects of this immune response. It lowers numbers of neutrophils and activated microglia in the ischaemic area and reduces levels of many inflammatory mediators including ROS62 and reactive nitrogen species63, adhesion molecules64, pro-inflammatory cytokines (such as interleukin-1β (IL-1β), tumour necrosis factor-α (TNFα) and IL-6)59,60 and the chemokines CC-chemokine ligand 2 (CCL2; also known as MCP1) and C-C motif chemokine 20 (also known as MIP3α)65,66. However, anti-inflamma-tory cytokines such as IL-10 and transforming growth factor-β (TGFβ) are reduced by hypothermia as well67,68, indicating that hypothermia does not have a purely anti-inflammatory effect.
Hypothermia also suppresses the activation of nuclear factor-κB (NF-κB)69, a major transcription factor that can activate many inflammation-related genes. The mechanism by which hypothermia suppresses NF-κB activation — in inflammatory cells such as microglia and also in neurons and astrocytes — depends on the type of cerebral insult. In models of focal brain ischaemia, hypothermia prevented nuclear NF-κB translocation and DNA binding by inhibiting the activity of inhibitor of NF-κB kinase (IKK). IKK is required for the phos-phorylation and degradation of NF-κB inhibitor (IκB), thereby allowing NF-κB to enter the nucleus, where it can upregulate target genes69,70. In models of global cer-ebral ischaemia, hypothermia also decreased nuclear translocation of NF-κB, but its regulatory proteins IκB
Box3 | Cold shock proteins
Mammalian cells generally respond to cold temperatures by arresting the cell cycle and inhibiting protein translation and gene transcription. Work in prokaryotic cells has revealed a class of cold-inducible (or ‘cold shock’) proteins that enable bacteria to withstand decreased temperatures. Two of these genes, cold-inducible RNA-binding protein (CIRBP) and RNA binding motif protein 3 (RBM3), are specifically induced by mild hypothermia123. They belong to a highly conserved glycine-rich RNA-binding protein family and are known to regulate translation by acting as RNA chaperones124. A few studies have begun to explore their significance in the brain. CIRBP mRNA in the cortex and hippocampus was found to increase after brain ischaemia in rats, with even higher increases in ischaemic brains of rats that had been exposed to hypothermia125. CIRBP levels were also increased in neural stem cell lines cultured under a moderately low temperature (32 ºC) compared with neural stem cell lines cultured under normal temperature (37 ºC)98. Even though the capacity of cold shock proteins to increase the survival of cells (by inhibiting apoptosis) and enhance activation of the extracellular signal-related kinase signalling pathway has been reported in mammalian cell lines126, this has not been directly studied in brain injury models. Knockdown of CIRBP by small interfering RNA in fibroblasts prevented the anti-apoptotic effect of cooling117, indicating a crucial role for CIRBP in the neuroprotective effect of hypothermia. Future research on the effects and mechanisms of cold shock proteins in the brain will contribute to a broader understanding of the effects of therapeutic cooling and lead to the exploration of cold shock proteins as a therapeutic target.
R E V I E W S
272 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved

and IKK were unaffected71. Notably, NF-κB also regu-lates genes involved in cell survival and growth72; thus, the net effect of hypothermia-induced suppression of NF-κB activity is difficult to predict.
Hypothermia also affects the mitogen-activated protein kinase (MAPK) pathway, another important enzyme system that regulates inflammation, in a cell-type dependent manner. In stimulated cultured micro-glia, hypothermia suppresses ERK signalling73, but in experimental stroke, hypothermia actually activates ERK in brain endothelial cells. This activation leads to decreased levels of intercellular adhesion molecule 1 (ICAM1), which is one of many inflammatory factors regulated by the MAPK signalling pathway and a type of adhesion molecule involved in attracting circulating inflammatory cells to the CNS74.
In summary, hypothermia has a largely suppressive effect on inflammation, and this anti-inflammatory property might serve as a major protective mechanism in ischaemic conditions. In a similar way, mild hypother-mia reduced inflammatory responses in a model of brain inflammation where cell death does not occur, and this suggests that inflammatory responses are temperature sensitive64.
Effects on the blood–brain barrier. The restoration of blood flow after ischaemia can lead to secondary injuries including oedema and haemorrhage, which are consequences of BBB disruption. BBB disruption after stroke or other brain injuries is caused by struc-tural and functional impairment of components of the neurovascular unit, including tight-junction proteins, transport proteins, basement membrane, endothelial cells, astrocytes, pericytes and neurons. Models of brain ischaemia, trauma and intracerebral haemorrhage have shown that mild to moderate hypothermia protects the BBB and prevents oedema formation44,75–79. Specifically, hypothermia prevents the activation of proteases respon-sible for degrading the extracellular matrix, such as the MMPs43–46,80,81. Activated MMPs have been shown to degrade several tight-junction proteins that make up the BBB, leading to oedema formation and brain haem-orrhage. Hypothermia reduces the proteolytic activity of MMPs and the consequent degradation of vascular basement membrane proteins82 and the extracellular matrix proteins agrin and laminin82. In addition to sup-pressing MMPs, hypothermia increases the expression of endogenous MMP inhibitors, such as metalloproteinase inhibitor 2 (also known as TIMP2).
In a rat focal cerebral ischaemia model, preservation of BBB integrity by hypothermia was documented even 5 days after the cessation of hypothermia80, demonstrat-ing the potential long-term benefits of hypothermia. Hypothermia also preserves vascular morphology, as the distortion of endothelial cells and their separation from the basement membrane that is associated with ischae-mia was attenuated by cooling in a model of experimen-tal stroke82. In addition, it seems to affect other cells that make up the BBB, such as pericytes, which are increas-ingly recognized to have an important regulatory role in BBB integrity. In a model of MCAO, pericytes were
found to migrate away from the vessel wall, leading to basement membrane disorganization, and moderate hypothermia prevented this83.
Hypothermia also attenuates oedema formation by preserving the brain’s water balance. Aquaporins are a family of water channel proteins that control the move-ment of water across cell membranes. Aquaporin 4 is the predominant type of aquaporin in the microvasculature of the CNS and has been observed in the end-foot mem-brane of astrocytes. Aquaporin 4 expression is increased in reactive astrocytes in cerebral ischaemic lesions57, and deleting Aqp4 reduces brain oedema following MCAO84. Mild hypothermia reduced brain oedema formation by suppressing aquaporin 4 expression in models of intra-cerebral haemorrhage85 and cardiac arrest86. In sum-mary, by preserving the structural proteins and cells that constitute the BBB, and by inhibiting the activation of damaging proteases and preventing the opening of water channels, hypothermia prevents secondary brain injury from brain oedema and haemorrhage.
In addition to water channel regulation, hypother-mia modulates molecular transport across the BBB. One study demonstrated that hypothermia decreased multi-drug resistance protein 1 (MDR1)-mediated transport without affecting passive diffusion and paracellular transport87. MDR1 is a type of transport protein that mobilizes drugs and drug metabolites through transcel-lular pathways. The finding that hypothermia can affect drug transport highlights the need for pharmacokinetic studies: drugs that may be co-administered during cool-ing should be studied for any temperature-dependent changes in pharmacokinetics.
By protecting BBB integrity, hypothermia can limit brain oedema and increases in intracranial pressure. However, hypothermia may not always lead to BBB protection and subsequent neuroprotection. Studies in models of protease-induced brain haemorrhage lead-ing to BBB degradation have shown that hypother-mia has inconsistent effects on functional outcome in these models. Compared to brain ischaemia models, in which several laboratories have consistently shown hypothermia-induced neuroprotection, in brain haem-orrhage models hypothermia leads to neurological improvement in some laboratories, whereas others report no improvement or even a slight worsening of neurological outcome76,77,88,89. It should be noted that hypothermia can also inhibit coagulation by decreas-ing fibrinogen generation and compromising thrombin generation90. The net result of cooling in this situation might increase the chance of bleeding91. Thus, as cooling could lead to expansion of bleeding, hypothermia may be a less effective neuroprotectant in brain haemorrhage.
Chronic effects of hypothermiaChronic effects of hypothermia have been observed weeks to months after injury and well after cooling has ceased. Recent work has focused on whether the acute hypothermic treatment has lasting effects, and whether hypothermia might affect recovery and repair mecha-nisms that occur in the brain long after the acute and subacute phases of injury.
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 273
© 2012 Macmillan Publishers Limited. All rights reserved

NestinAn intermediate filament protein used as a marker for CNS stem cells.
Effects on neurogenesis. Recent studies have investi-gated the brain’s regenerative capacity following focal cerebral ischaemia and traumatic brain injury92. The influence of therapeutic hypothermia on neurogenesis and the brain’s ability to reorganize synaptic connectiv-ity has been studied by a few groups, but is currently far from clear. After brain insults such as stroke, neurons in the ischaemic area lose synaptic connectivity and undergo cell death93. However, it is becoming increas-ingly recognized that endogenous restorative processes are also activated after ischaemia, leading to neurogen-esis and synaptogenesis92. However, these restorative processes are incomplete, as evidenced by the persis-tent disability seen in most stroke and brain trauma vic-tims. Clearly, being able to enhance these endogenous properties would have clinical benefits. Even though rodent studies have shown that acute brain insults ini-tiate the proliferation of neural stem cells in the subven-tricular zone and the hippocampal subgranular zone93, neurogenesis in the uninjured aged brain is markedly reduced94. This is relevant as these types of brain insult occur commonly in old age, whereas gliogenesis is not affected by age95. Spontaneous recovery through neuro-genesis is limited in brain injury, and there is an obvious need to develop strategies to improve regenerative pro-cesses such as proliferation of neuronal precursor cells, migration of precursor cells to the injury area, differen-tiation into mature neurons and reconnection between neurons. Here, we review the influence of hypothermia on neurological recovery and repair (FIG. 3).
Cooling has been shown to differentially affect neu-rogenesis in uninjured animals. In one study that exam-ined neurogenesis in the developing brain, reduction of brain temperature to 30 °C for 21 h decreased the number of proliferating cells in the subgranular zone of the hippocampus, but not the periventricular zone96. However, under conditions of hypoxia–ischaemia in the developing brain, hypothermia to 33 °C enhanced the maturation of neural progenitor cells in the stria-tum and inhibited apoptosis of proliferating neural stem cells that were already increased by ischaemic stimuli97. The mechanism of reduced apoptosis seems to be linked to the cooling-induced upregulation of BCL-2 (REF. 97). In a study of cultured neural stem cells, mild hypother-mia also inhibited apoptosis, increased the number of nestin positive cells and inhibited stem cell differen-tiation into astrocytes98. Adult rodents exposed to fore-brain ischaemia and subjected to mild hypothermia had increased numbers of newborn neurons in the dentate gyrus compared to animals exposed to ischaemia with-out cooling99. By contrast, another study in adult rats with forebrain ischaemia showed that hypothermia had no effect on neurogenesis100; however, the duration of hypothermia in this study was rather short (33 °C for 45 min) and occurred relatively early, either during the ischaemic period or during the immediate reperfusion phase. Thus, it is possible that hypothermia may not have any effect on neurogenesis if it is not applied dur-ing a critical time window (or windows) that has yet to be clearly defined. More research in this area is needed; in particular to determine the optimal conditions under
which cooling might be expected to positively influ-ence neurogenesis and whether cooling may improve neurogenesis in aged brains exposed to ischaemia and related insults.
Effects on gliogenesis and angiogenesis. Oligo-dendrocytes, although less studied than neurons, suc-cumb to brain insults and undergo cell death with a susceptibility that is similar to neurons, and hypo-thermia attenuates trauma-induced oligodendrocyte cell death, demyelination and circuit dysfunction101,102. Hypothermia (32 °C) increased the number of oligoden-drocyte precursor cells in a primary culture taken from embryonic mouse brains103. As a result, greater num-bers of oligodendrocyte precursor cells that undergo cell cycle progression were maintained in a less well- differentiated state. However, an in vivo study using a hypoxia model in preterm fetal sheep demonstrated that hypothermia (30 °C) was associated with an overall reduction in hypoxia-induced loss of immature oligo-dendrocytes, although it did not prevent the hypoxia-induced reduced proliferation of oligodendrocytes within the periventricular white matter104.
Reports of the effects of hypothermia on endo-genous cell genesis in the injured and uninjured brains are somewhat conflicting. Some reports96,104 indicate that hypothermia suppresses stem cell proliferation, whereas many reports indicate the opposite98,99,103, and some even suggest that cooling promotes progenitor cell differentiation towards neurogenesis over gliogenesis97,98. Hypothermia to temperatures lower than 30 °C seems to suppress cell proliferation and phase-specific and nonspecific cell cycle arrest as a result of reduced energy supply96,105. However, small temperature decreases seem to protect against progenitor cell death98,104. Thus, we speculate that mild hypothermia enables the differen-tiation of precursor cells while preventing apoptosis, and that cooling to lower temperatures seems detrimental to cells and blocks their proliferation.
Astrogliogenesis and angiogenesis are thought to con-tribute to brain regeneration following brain injury93,106, but this has not been studied intensively. Astrocytes comprise the largest population of cells in the ischaemic core during the subacute to chronic period after stroke107, and reactive astrocytes are the main component of the glial scar. However, glial scar formation in the brain can obstruct neurite outgrowth and regeneration108,109, and blocking astrocyte activation and related reactions can exacerbate inflammation and increase injury responses109. Thus, enhancement of gliogenesis may do some harm. How hypothermia affects gliogenesis has not yet been studied in any depth. Mild hypothermia has been shown to enhance angiogenesis in focal cerebral ischaemia110, spinal cord injury111 and traumatic brain injury models112. Although these angiogenic effects by hypothermia are presumably beneficial for repair processes, their signifi-cance is still uncertain. In fact, a few studies suggest that angiogenesis may actually be detrimental to brain repair. For example, one study of acute stroke patients showed that an early dominance of pro-angiogenic factors (including platelet-derived growth factors (PDGFs),
R E V I E W S
274 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved
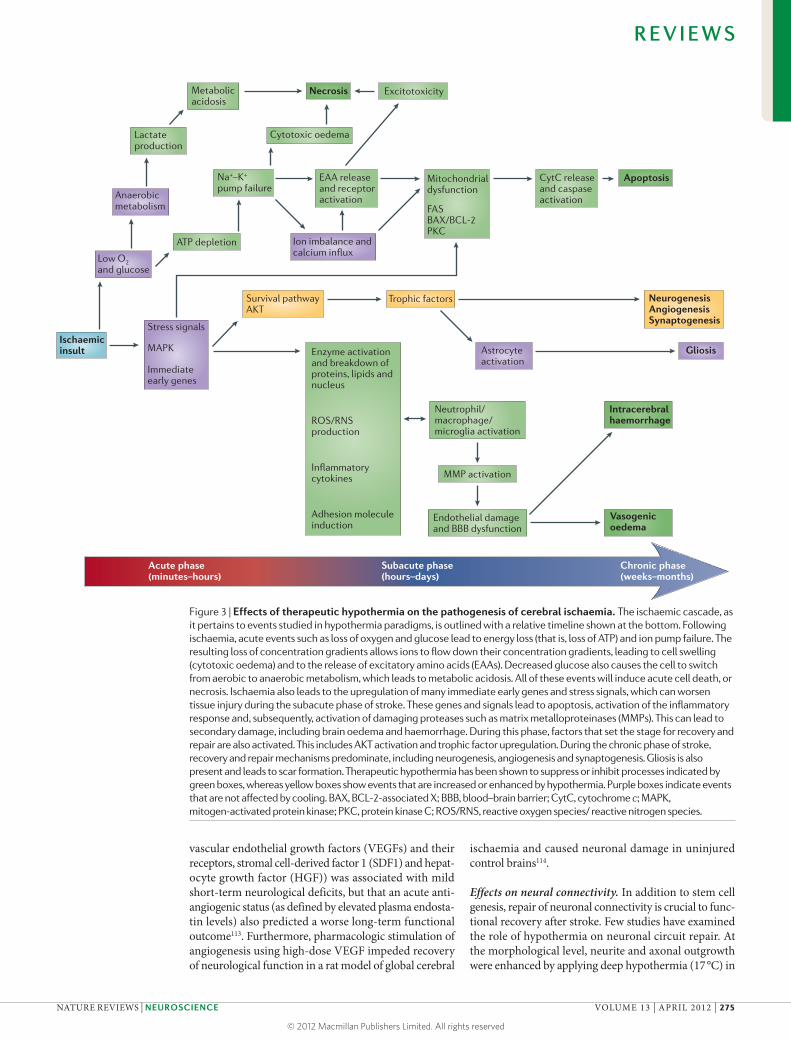
Nature Reviews | Neuroscience
Cytotoxic oedemaLactate production
Excitotoxicity
Anaerobicmetabolism
Na+–K+ pump failure
Metabolicacidosis
Necrosis
Apoptosis
Low O2and glucose
Ischaemic insult
ATP depletion
EAA releaseand receptoractivation
CytC releaseand caspaseactivation
Mitochondrialdysfunction
FASBAX/BCL-2PKC
Ion imbalance and calcium influx
Survival pathwayAKT
Trophic factors
Stress signals
Immediateearly genes
Enzyme activationand breakdown of proteins, lipids and nucleus
ROS/RNSproduction
Inflammatorycytokines
Adhesion moleculeinduction
NeurogenesisAngiogenesisSynaptogenesis
GliosisAstrocyteactivation
Neutrophil/macrophage/microglia activation
MMP activation
Intracerebralhaemorrhage
Vasogenicoedema
Endothelial damageand BBB dysfunction
Acute phase(minutes–hours)
Subacute phase(hours–days)
Chronic phase(weeks–months)
MAPK
vascular endothelial growth factors (VEGFs) and their receptors, stromal cell-derived factor 1 (SDF1) and hepat-ocyte growth factor (HGF)) was associated with mild short-term neurological deficits, but that an acute anti-angiogenic status (as defined by elevated plasma endosta-tin levels) also predicted a worse long-term functional outcome113. Furthermore, pharmacologic stimulation of angiogenesis using high-dose VEGF impeded recovery of neurological function in a rat model of global cerebral
ischaemia and caused neuronal damage in uninjured control brains114.
Effects on neural connectivity. In addition to stem cell genesis, repair of neuronal connectivity is crucial to func-tional recovery after stroke. Few studies have examined the role of hypothermia on neuronal circuit repair. At the morphological level, neurite and axonal outgrowth were enhanced by applying deep hypothermia (17 °C) in
Figure 3 | Effects of therapeutic hypothermia on the pathogenesis of cerebral ischaemia. The ischaemic cascade, as it pertains to events studied in hypothermia paradigms, is outlined with a relative timeline shown at the bottom. Following ischaemia, acute events such as loss of oxygen and glucose lead to energy loss (that is, loss of ATP) and ion pump failure. The resulting loss of concentration gradients allows ions to flow down their concentration gradients, leading to cell swelling (cytotoxic oedema) and to the release of excitatory amino acids (EAAs). Decreased glucose also causes the cell to switch from aerobic to anaerobic metabolism, which leads to metabolic acidosis. All of these events will induce acute cell death, or necrosis. Ischaemia also leads to the upregulation of many immediate early genes and stress signals, which can worsen tissue injury during the subacute phase of stroke. These genes and signals lead to apoptosis, activation of the inflammatory response and, subsequently, activation of damaging proteases such as matrix metalloproteinases (MMPs). This can lead to secondary damage, including brain oedema and haemorrhage. During this phase, factors that set the stage for recovery and repair are also activated. This includes AKT activation and trophic factor upregulation. During the chronic phase of stroke, recovery and repair mechanisms predominate, including neurogenesis, angiogenesis and synaptogenesis. Gliosis is also present and leads to scar formation. Therapeutic hypothermia has been shown to suppress or inhibit processes indicated by green boxes, whereas yellow boxes show events that are increased or enhanced by hypothermia. Purple boxes indicate events that are not affected by cooling. BAX, BCL-2-associated X; BBB, blood–brain barrier; CytC, cytochrome c; MAPK, mitogen-activated protein kinase; PKC, protein kinase C; ROS/RNS, reactive oxygen species/ reactive nitrogen species.
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 275
© 2012 Macmillan Publishers Limited. All rights reserved

CoagulopathiesAbnormal conditions of blood clotting or blood clot lysis.
AutophagyThe breakdown of a cell’s own components by the lysosome.
AnoikisA form of programmed cell death that is activated when cells detach from the extracellular matrix
organotypic brain slices115,116. A genomic analysis study in a rat model of traumatic brain injury demonstrated that mild hypothermia had a significant effect on gene expres-sion. An analysis of hippocampal gene expression profiles from rats exposed to hypothermia following traumatic brain injury revealed statistically significant differences in 133 transcripts compared to injured normothermic rats. Of these, 57 transcripts were upregulated and 76 were downregulated after injury. Those genes belonging to synapse organization and biogenesis were especially upregulated in hypothermic animals compared to nor-mothermic animals117. Although the scientific literature is still scant, current data suggest that overall, hypothermia supports regenerative processes by enhancing synapse formation and reorganization.
To summarize, although the full effect of therapeutic hypothermia in brain repair is unclear, under specific con-ditions it seems to have a beneficial role in protecting stem cells, promoting their proliferation and differentiation and possibly encouraging recovery of neural circuitry.
Conclusions and future directionsHypothermia is a remarkable phenomenon that pre-serves tissues and limits injury. It is perhaps the most robust neuroprotectant studied in the laboratory and has also been shown to have efficacy in humans. It affects nearly every metabolic, molecular and cellular event in cell death to promote tissue preservation (FIG. 3). More recent studies have shown that hypothermia can also favourably modulate endogenous regenerative and restorative properties. This multi-faceted aspect of therapeutic hypothermia may suggest that the goal of neuroprotection requires multi-target approaches.
It may also be possible to combine hypothermia with other therapeutic modalities, such as neuroprotect-ants or thrombolytic agents, to extend the therapeutic window of the drugs or of hypothermia itself. The use of hypothermia in combination may lead to the re- examination of the many neuroprotectant drugs that failed at the clinical level, as many drugs may not have been studied in the most optimal manner.
In spite of the robust protective effects of therapeu-tic hypothermia demonstrated in the laboratory, there are several obstacles that hamper the application of this
method in the clinic, including how to achieve effective cooling in humans, how to prevent untoward effects (including systemic complications118 and potential harmful effects of hypothermia, as discussed above) and how to identify patient populations that are most likely to benefit from therapeutic hypothermia. These issues highlight the need to develop more sophisticated translational research tools in the laboratory. Animal models of ischaemic injury and the methods of cooling used in the laboratory are quite different from those used in the clinic. Thus, an effort to more precisely sim-ulate the clinical condition and treatment conditions — for example, therapeutic cooling of patients after cardiac arrest usually lasts 12–24 h as opposed to the 2 h often used in animal studies — might provide solu-tions for better and wider application of therapeutic hypothermia in the clinic. In addition, investigations to overcome complications of systemic hypothermia, such as shivering, infection and coagulopathies, are lacking. These complications are serious at the clinical level, but are largely ignored in the laboratory. Approaches to pharmacologically induced hypothermia are on the horizon12 and are attractive because they are less invasive and would not require the use of specialized equipment.
From a research standpoint, hypothermia may also be used as a model of neuroprotection in which potential therapeutic targets could be identified and investigated, but this requires more studies on how hypothermia influences signalling pathways. In particular, studies investigating the effects of hypothermia on cell death pathways require an updated view on these pathways, as the traditional concepts of necrosis versus apoptosis are not sufficient to explain many of the injury mecha-nisms in the brain. Other types of cell death, such as autophagy and anoikis, have yet to be studied in the context of therapeutic cooling.
In summary, therapeutic hypothermia has its place in the laboratory, translational and clinical realms as a model of neuroprotection and now as a treatment for some ischaemic brain injuries. However, it is clear that more research is still needed to understand its biological significance as well as how it can be effectively applied in additional clinical conditions.
1. Busto, R. et al. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J. Cereb. Blood Flow Metab. 7, 729–738 (1987).This study was the first to show that relatively small decreases in temperature resulted in remarkable neuroprotection and led to a resurgence of interest in the field of therapeutic hypothermia.
2. Bernard, S. A. et al. Treatment of comatose survivors of out‑of‑hospital cardiac arrest with induced hypothermia. N. Engl. J. Med. 346, 557–563 (2002).
3. The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 346, 549–556 (2002).References 2 and 3 are prospective clinical studies that showed that mild hypothermia improved neurological outcomes in victims of cardiac arrest.
4. Gluckman, P. D. et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365, 663–670 (2005).
A clinical trial of selective head cooling that showed improvement in neurological outcomes in some neonates with hypoxic ischaemic encephalopathy.
5. Shankaran, S. et al. Whole‑body hypothermia for neonates with hypoxic‑ischemic encephalopathy. N. Engl. J. Med. 353, 1574–1584 (2005).A clinical trial of whole body cooling that showed improvement in neurological outcomes in some neonates with hypoxic ischaemic encephalopathy.
6. Nozari, A. et al. Suspended animation can allow survival without brain damage after traumatic exsanguination cardiac arrest of 60 minutes in dogs. J. Trauma 57, 1266–1275 (2004).
7. Dietrich, W. D. & Bramlett, H. M. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics 7, 43–50 (2010).
8. Dietrich, W. D., Atkins, C. M. & Bramlett, H. M. Protection in animal models of brain and spinal cord injury with mild to moderate hypothermia. J. Neurotrauma 26, 301–312 (2009).
9. Colbourne, F., Sutherland, G. R. & Auer, R. N. An automated system for regulating brain temperature in
awake and freely moving rodents. J. Neurosci. Methods 67, 185–190 (1996).
10. van der Worp, H. B., Sena, E. S., Donnan, G. A., Howells, D. W. & Macleod, M. R. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta‑analysis. Brain 130, 3063–3074 (2007).This paper provided a systematic review and meta-analysis of the evidence for efficacy of hypothermia in animal models of ischaemic stroke based on more than 100 publications.
11. Krieger, D. W. & Yenari, M. A. Therapeutic hypothermia for acute ischemic stroke: what do laboratory studies teach us? Stroke 35, 1482–1489 (2004).
12. Yenari, M., Kitagawa, K., Lyden, P. & Perez‑Pinzon, M. Metabolic downregulation: a key to successful neuroprotection? Stroke 39, 2910–2917 (2008).This paper reviews how therapeutic hypothermia and ischaemic preconditioning, which are both widely studied models of neuroprotection in the laboratory, have moved from basic science studies to clinical studies.
R E V I E W S
276 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved

13. Colbourne, F., Li, H. & Buchan, A. M. Indefatigable CA1 sector neuroprotection with mild hypothermia induced 6 hours after severe forebrain ischemia in rats. J. Cereb. Blood Flow Metab. 19, 742–749 (1999).This study demonstrated remarkable and durable neuroprotection by therapeutic cooling in a global cerebral ischaemia model, even when cooling was started as late as 6 h from ischaemia onset.
14. Maier, C. M., Sun, G. H., Kunis, D., Yenari, M. A. & Steinberg, G. K. Delayed induction and long‑term effects of mild hypothermia in a focal model of transient cerebral ischemia: neurological outcome and infarct size. J. Neurosurg. 94, 90–96 (2001).
15. Meloni, B. P., Mastaglia, F. L. & Knuckey, N. W. Therapeutic applications of hypothermia in cerebral ischaemia. Ther. Adv. Neurol. Disord. 1, 12–35 (2008).
16. Erecinska, M., Thoresen, M. & Silver, I. A. Effects of hypothermia on energy metabolism in mammalian central nervous system. J. Cereb. Blood Flow Metab. 23, 513–530 (2003).
17. Yenari, M., Wijman, C. & Steinberg, G. in Hypothermia in Neurocritical Care (eds Mayer, S. & Sessler, D.) 141–178 (Marcel Dekker, New York, 2004).
18. Zhao, H., Steinberg, G. K. & Sapolsky, R. M. General versus specific actions of mild‑moderate hypothermia in attenuating cerebral ischemic damage. J. Cereb. Blood Flow Metab. 27, 1879–1894 (2007).This paper provides a comprehensive description of the protective mechanisms of hypothermia from laboratory data to facilitate clinical translation of therapeutic hypothermia.
19. Lee, J. M., Zipfel, G. J. & Choi, D. W. The changing landscape of ischaemic brain injury mechanisms. Nature 399, A7–A14 (1999).
20. Colbourne, F., Grooms, S. Y., Zukin, R. S., Buchan, A. M. & Bennett, M. V. Hypothermia rescues hippocampal CA1 neurons and attenuates down‑regulation of the AMPA receptor GluR2 subunit after forebrain ischemia. Proc. Natl Acad. Sci. USA 100, 2906–2910 (2003).
21. Ginsberg, M. D., Sternau, L. L., Globus, M. Y., Dietrich, W. D. & Busto, R. Therapeutic modulation of brain temperature: relevance to ischemic brain injury. Cerebrovasc. Brain Metab. Rev. 4, 189–225 (1992).
22. Dietrich, W. D., Busto, R., Alonso, O., Globus, M. Y. & Ginsberg, M. D. Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J. Cereb. Blood Flow Metab. 13, 541–549 (1993).
23. Kamme, F., Campbell, K. & Wieloch, T. Biphasic expression of the fos and jun families of transcription factors following transient forebrain ischaemia in the rat. Effect of hypothermia. Eur. J. Neurosci. 7, 2007–2016 (1995).
24. Terao, Y. et al. Hypothermia enhances heat‑shock protein 70 production in ischemic brains. Neuroreport 20, 745–749 (2009).
25. Cullen, K. E. & Sarge, K. D. Characterization of hypothermia‑induced cellular stress response in mouse tissues. J. Biol. Chem. 272, 1742–1746 (1997).
26. Yenari, M. A. et al. Antiapoptotic and anti‑inflammatory mechanisms of heat‑shock protein protection. Ann. NY Acad. Sci. 1053, 74–83 (2005).
27. Kumar, K., Wu, X., Evans, A. T. & Marcoux, F. The effect of hypothermia on induction of heat shock protein (HSP)‑72 in ischemic brain. Metab. Brain Dis. 10, 283–291 (1995).
28. Xu, L., Yenari, M. A., Steinberg, G. K. & Giffard, R. G. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J. Cereb. Blood Flow Metab. 22, 21–28 (2002).
29. Vemuganti, R. The microRNAs and stroke: no need to be coded to be counted. Transl. Stroke Res. 1, 158–160 (2010).
30. Truettner, J. S., Alonso, O. F., Bramlett, H. M. & Dietrich, W. D. Therapeutic hypothermia alters microRNA responses to traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 31, 1897–1907 (2011).This paper demonstrated that several miRNAs are also extremely temperature sensitive (in addition to many well-known temperature-sensitive mRNAs).
31. Green, D. R. & Reed, J. C. Mitochondria and apoptosis. Science 281, 1309–1312 (1998).
32. Ashkenazi, A. & Dixit, V. M. Death receptors: signaling and modulation. Science 281, 1305–1308 (1998).
33. Liu, L. & Yenari, M. A. Therapeutic hypothermia: neuroprotective mechanisms. Front. Biosci. 12, 816–825 (2007).
34. Bright, R. et al. Protein kinase C δ mediates cerebral reperfusion injury in vivo. J. Neurosci. 24, 6880–6888 (2004).
35. Raval, A. P. et al. Protein kinase C δ cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation. J. Cereb. Blood Flow Metab. 25, 730–741 (2005).
36. Lee, S. M., Zhao, H., Maier, C. M. & Steinberg, G. K. The protective effect of early hypothermia on PTEN phosphorylation correlates with free radical inhibition in rat stroke. J. Cereb. Blood Flow Metab. 29, 1589–1600 (2009).
37. Shimohata, T., Zhao, H. & Steinberg, G. K. εPKC may contribute to the protective effect of hypothermia in a rat focal cerebral ischemia model. Stroke 38, 375–380 (2007).
38. Martin‑Villalba, A. et al. CD95 ligand (Fas‑L/APO‑1L) and tumor necrosis factor‑related apoptosis‑inducing ligand mediate ischemia‑induced apoptosis in neurons. J. Neurosci. 19, 3809–3817 (1999).
39. Rosenbaum, D. M. et al. Fas (CD95/APO‑1) plays a role in the pathophysiology of focal cerebral ischemia. J. Neurosci. Res. 61, 686–692 (2000).
40. Liu, L. et al. FasL shedding is reduced by hypothermia in experimental stroke. J. Neurochem. 106, 541–550 (2008).
41. Cunningham, L. A., Wetzel, M. & Rosenberg, G. A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 50, 329–339 (2005).
42. Powell, W. C., Fingleton, B., Wilson, C. L., Boothby, M. & Matrisian, L. M. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr. Biol. 9, 1441–1447 (1999).
43. Hamann, G. F. et al. Mild to moderate hypothermia prevents microvascular basal lamina antigen loss in experimental focal cerebral ischemia. Stroke 35, 764–769 (2004).
44. Lee, J. E., Yoon, Y. J., Moseley, M. E. & Yenari, M. A. Reduction in levels of matrix metalloproteinases and increased expression of tissue inhibitor of metalloproteinase‑2 in response to mild hypothermia therapy in experimental stroke. J. Neurosurg. 103, 289–297 (2005).
45. Truettner, J. S., Alonso, O. F. & Dalton Dietrich, W. Influence of therapeutic hypothermia on matrix metalloproteinase activity after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 25, 1505–1516 (2005).
46. Wagner, S. et al. Topographically graded postischemic presence of metalloproteinases is inhibited by hypothermia. Brain Res. 984, 63–75 (2003).
47. Yenari, M. A. et al. Mild hypothermia attenuates cytochrome c release but does not alter Bcl‑2 expression or caspase activation after experimental stroke. J. Cereb. Blood Flow Metab. 22, 29–38 (2002).
48. Susin, S. A. et al. Molecular characterization of mitochondrial apoptosis‑inducing factor. Nature 397, 441–446 (1999).
49. Zhao, H. et al. Conditions of protection by hypothermia and effects on apoptotic pathways in a rat model of permanent middle cerebral artery occlusion. J. Neurosurg. 107, 636–641 (2007).
50. Shi, G. D. et al. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem. Biophys. Res. Commun. 404, 941–945 (2011).
51. Vosler, P. S., Logue, E. S., Repine, M. J. & Callaway, C. W. Delayed hypothermia preferentially increases expression of brain‑derived neurotrophic factor exon III in rat hippocampus after asphyxial cardiac arrest. Brain Res. Mol. Brain Res. 135, 21–29 (2005).
52. D’Cruz, B. J. et al. Hypothermic reperfusion after cardiac arrest augments brain‑derived neurotrophic factor activation. J. Cereb. Blood Flow Metab. 22, 843–851 (2002).
53. Schmidt, K. M., Repine, M. J., Hicks, S. D., DeFranco, D. B. & Callaway, C. W. Regional changes in glial cell line‑derived neurotrophic factor after cardiac arrest and hypothermia in rats. Neurosci. Lett. 368, 135–139 (2004).
54. Boris‑Moller, F., Kamme, F. & Wieloch, T. The effect of hypothermia on the expression of neurotrophin mRNA in the hippocampus following transient cerebral ischemia in the rat. Brain Res. Mol. Brain Res. 63, 163–173 (1998).
55. Hicks, S. D., Parmele, K. T., DeFranco, D. B., Klann, E. & Callaway, C. W. Hypothermia differentially increases extracellular signal‑regulated kinase and stress‑activated protein kinase/c‑Jun terminal kinase activation in the hippocampus during reperfusion after asphyxial cardiac arrest. Neuroscience 98, 677–685 (2000).
56. D’Cruz, B. J., Logue, E. S., Falke, E., DeFranco, D. B. & Callaway, C. W. Hypothermia and ERK activation after cardiac arrest. Brain Res. 1064, 108–118 (2005).
57. Slikker, W., Desai, V. G., Duhart, H., Feuers, R. & Imam, S. Z. Hypothermia enhances bcl‑2 expression and protects against oxidative stress‑induced cell death in Chinese hamster ovary cells. Free Radic. Biol. Med. 31, 405–411 (2001).
58. Zhang, Z., Sobel, R. A., Cheng, D., Steinberg, G. K. & Yenari, M. A. Mild hypothermia increases Bcl‑2 protein expression following global cerebral ischemia. Brain Res. Mol. Brain Res. 95, 75–85 (2001).
59. Zhao, H. et al. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J. Neurosci. 25, 9794–9806 (2005).
60. Wang, Q., Tang, X. N. & Yenari, M. A. The inflammatory response in stroke. J. Neuroimmunol. 184, 53–68 (2007).
61. Ceulemans, A. G. et al. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J. Neuroinflammation 7, 74 (2010).
62. Perrone, S. et al. Whole body hypothermia and oxidative stress in babies with hypoxic‑ischemic brain injury. Pediatr. Neurol. 43, 236–240 (2010).
63. Han, H. S., Qiao, Y., Karabiyikoglu, M., Giffard, R. G. & Yenari, M. A. Influence of mild hypothermia on inducible nitric oxide synthase expression and reactive nitrogen production in experimental stroke and inflammation. J. Neurosci. 22, 3921–3928 (2002).
64. Deng, H., Han, H. S., Cheng, D., Sun, G. H. & Yenari, M. A. Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke 34, 2495–2501 (2003).
65. Meybohm, P. et al. Mild hypothermia alone or in combination with anesthetic post‑conditioning reduces expression of inflammatory cytokines in the cerebral cortex of pigs after cardiopulmonary resuscitation. Crit. Care 14, R21 (2010).
66. Terao, Y. et al. Macrophage inflammatory protein‑3α plays a key role in the inflammatory cascade in rat focal cerebral ischemia. Neurosci. Res. 64, 75–82 (2009).
67. Matsui, T. & Kakeda, T. IL‑10 production is reduced by hypothermia but augmented by hyperthermia in rat microglia. J. Neurotrauma 25, 709–715 (2008).
68. Truettner, J. S., Suzuki, T. & Dietrich, W. D. The effect of therapeutic hypothermia on the expression of inflammatory response genes following moderate traumatic brain injury in the rat. Brain Res. Mol. Brain Res. 138, 124–134 (2005).
69. Yenari, M. A. & Han, H. S. Influence of hypothermia on post‑ischemic inflammation: role of nuclear factor kappa B (NFκB). Neurochem. Int. 49, 164–169 (2006).
70. Han, H. S., Karabiyikoglu, M., Kelly, S., Sobel, R. A. & Yenari, M. A. Mild hypothermia inhibits nuclear factor‑κB translocation in experimental stroke. J. Cereb. Blood Flow Metab. 23, 589–598 (2003).
71. Webster, C. M. et al. Inflammation and NFκB activation is decreased by hypothermia following global cerebral ischemia. Neurobiol. Dis. 33, 301–312 (2009).
72. Mattson, M. P., Culmsee, C., Yu, Z. & Camandola, S. Roles of nuclear factor κB in neuronal survival and plasticity. J. Neurochem. 74, 443–456 (2000).
73. Schmitt, K. R. et al. Hypothermia suppresses inflammation via ERK signaling pathway in stimulated microglial cells. J. Neuroimmunol. 189, 7–16 (2007).
74. Choi, J. S. et al. Mild hypothermia attenuates intercellular adhesion molecule‑1 induction via activation of extracellular signal‑regulated kinase‑1/2 in a focal cerebral ischemia model. Stroke Res. Treat. 2011, 846716 (2011).
75. Dietrich, W. D., Busto, R., Halley, M. & Valdes, I. The importance of brain temperature in alterations of the blood‑brain barrier following cerebral ischemia. J. Neuropathol. Exp. Neurol. 49, 486–497 (1990).
76. Kawanishi, M. et al. Effect of delayed mild brain hypothermia on edema formation after intracerebral hemorrhage in rats. J. Stroke Cerebrovasc. Dis. 17, 187–195 (2008).
77. MacLellan, C. L., Davies, L. M., Fingas, M. S. & Colbourne, F. The influence of hypothermia on outcome after intracerebral hemorrhage in rats. Stroke 37, 1266–1270 (2006).
78. Oda, Y., Gao, G., Wei, E. P. & Povlishock, J. T. Combinational therapy using hypothermia and the immunophilin ligand FK506 to target altered pial arteriolar reactivity, axonal damage, and blood‑brain barrier dysfunction after traumatic brain injury in rat. J. Cereb. Blood Flow Metab. 31, 1143–1154 (2011).
R E V I E W S
NATURE REVIEWS | NEUROSCIENCE VOLUME 13 | APRIL 2012 | 277
© 2012 Macmillan Publishers Limited. All rights reserved

79. Preston, E. & Webster, J. A two‑hour window for hypothermic modulation of early events that impact delayed opening of the rat blood‑brain barrier after ischemia. Acta Neuropathol. 108, 406–412 (2004).
80. Nagel, S. et al. Minocycline and hypothermia for reperfusion injury after focal cerebral ischemia in the rat: effects on BBB breakdown and MMP expression in the acute and subacute phase. Brain Res. 1188, 198–206 (2008).
81. Lee, K. M., Jang, J. H., Park, J. S., Kim, D. S. & Han, H. S. Effect of mild hypothermia on blood brain barrier disruption induced by oleic acid in rats. Genes Genomics 31, 89–98 (2009).
82. Baumann, E., Preston, E., Slinn, J. & Stanimirovic, D. Post‑ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and SPARC, and the blood‑brain barrier disruption after global cerebral ischemia. Brain Res. 1269, 185–197 (2009).
83. Duz, B., Oztas, E., Erginay, T., Erdogan, E. & Gonul, E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: an ultrastructural study. Cryobiology 55, 279–284 (2007).
84. Manley, G. T. et al. Aquaporin‑4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nature Med. 6, 159–163 (2000).
85. Dai, D. W. et al. Effect of local mild hypothermia on expression of aquaporin‑4 following intracerebral hemorrhage in rats. Zhonghua Yi Xue Za Zhi 86, 906–910 (2006) (in Chinese).
86. Xiao, F. et al. Cerebral cortical aquaporin‑4 expression in brain edema following cardiac arrest in rats. Acad. Emerg. Med. 11, 1001–1007 (2004).
87. Jin, J. S. et al. Effect of therapeutic moderate hypothermia on multi‑drug resistance protein 1‑mediated transepithelial transport of drugs. Neurol. Med. Chir. 46, 321–327 (2006).
88. Fingas, M., Penner, M., Silasi, G. & Colbourne, F. Treatment of intracerebral hemorrhage in rats with 12 h, 3 days and 6 days of selective brain hypothermia. Exp. Neurol. 219, 156–162 (2009).This is a comprehensive study of therapeutic hypothermia in a brain haemorrhage model.
89. Kollmar, R. et al. Hypothermia reduces perihemorrhagic edema after intracerebral hemorrhage. Stroke 41, 1684–1689 (2010).
90. Martini, W. Z. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J. Trauma 67, 202–209 (2009).
91. Martini, W. Z. The effects of hypothermia on fibrinogen metabolism and coagulation function in swine. Metabolism 56, 214–221 (2007).
92. Kernie, S. G. & Parent, J. M. Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiol. Dis. 37, 267–274 (2010).
93. Font, M. A., Arboix, A. & Krupinski, J. Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr. Cardiol. Rev. 6, 238–244 (2010).
94. Shruster, A., Melamed, E. & Offen, D. Neurogenesis in the aged and neurodegenerative brain. Apoptosis 15, 1415–1421 (2010).
95. Jinno, S. Decline in adult neurogenesis during aging follows a topographic pattern in the mouse hippocampus. J. Comp. Neurol. 519, 451–466 (2011).
96. Kanagawa, T. et al. A decrease of cell proliferation by hypothermia in the hippocampus of the neonatal rat. Brain Res. 1111, 36–40 (2006).
97. Xiong, M. et al. Post‑ischemic hypothermia promotes generation of neural cells and reduces apoptosis by Bcl‑2 in the striatum of neonatal rat brain. Neurochem. Int. 58, 625–633 (2011).
98. Saito, K. et al. Moderate low temperature preserves the stemness of neural stem cells and suppresses apoptosis of the cells via activation of the cold‑inducible RNA binding protein. Brain Res. 1358, 20–29 (2010).
99. Silasi, G. & Colbourne, F. Therapeutic hypothermia influences cell genesis and survival in the rat hippocampus following global ischemia. J. Cereb. Blood Flow Metab. 31, 1725–1735 (2011).This paper showed that prolonged hypothermia positively interacts with post-ischaemic repair processes, such as neurogenesis, resulting in improved functional outcome.
100. Lasarzik, I. et al. Mild hypothermia has no long‑term impact on postischemic neurogenesis in rats. Anesth. Analg. 109, 1632–1639 (2009).
101. Lotocki, G. et al. Oligodendrocyte vulnerability following traumatic brain injury in rats. Neurosci. Lett. 499, 143–148 (2011).
102. Lotocki, G. et al. Oligodendrocyte vulnerability following traumatic brain injury in rats: effect of moderate hypothermia. Therapeutic Hypothermia and Temperature Management 1, 43–51 (2011).
103. Imada, S. et al. Hypothermia‑induced increase of oligodendrocyte precursor cells: possible involvement of plasmalemmal voltage‑dependent anion channel 1. J. Neurosci. Res. 88, 3457–3466 (2010).
104. Bennet, L. et al. The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J. Physiol. 578, 491–506 (2007).
105. Matijasevic, Z., Snyder, J. E. & Ludlum, D. B. Hypothermia causes a reversible, p53‑mediated cell cycle arrest in cultured fibroblasts. Oncol. Res. 10, 605–610 (1998).
106. Gopurappilly, R. et al. Stem cells in stroke repair: current success & future prospects. CNS Neurol. Disord. Drug Targets 10, 741–756 (2011).
107. Li, L. et al. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia 58, 1610–1619 (2010).
108. Hawthorne, A. L. et al. The unusual response of serotonergic neurons after CNS injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. J. Neurosci. 31, 5605–5616 (2011).
109. Trendelenburg, G. & Dirnagl, U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia 50, 307–320 (2005).
110. Xie, Y. C., Li, C. Y., Li, T., Nie, D. Y. & Ye, F. Effect of mild hypothermia on angiogenesis in rats with focal cerebral ischemia. Neurosci. Lett. 422, 87–90 (2007).
111. Kao, C. H., Chio, C. C., Lin, M. T. & Yeh, C. H. Body cooling ameliorating spinal cord injury may be neurogenesis‑, anti‑inflammation‑ and angiogenesis‑associated in rats. J. Trauma 70, 885–893 (2011).
112. Kuo, J. R. et al. Brain cooling‑stimulated angiogenesis and neurogenesis attenuated traumatic brain injury in rats. J. Trauma 69, 1467–1472 (2010).
113. Navarro‑Sobrino, M. et al. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis 216, 205–211 (2011).
114. Manoonkitiwongsa, P. S., Schultz, R. L., McCreery, D. B., Whitter, E. F. & Lyden, P. D. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. J. Cereb. Blood Flow Metab. 24, 693–702 (2004).
115. Schmitt, K. R. et al. Hypothermia‑induced neurite outgrowth is mediated by tumor necrosis factor‑α. Brain Pathol. 20, 771–779 (2010).
116. Schmitt, K. R. et al. S100B modulates IL‑6 release and cytotoxicity from hypothermic brain cells and inhibits hypothermia‑induced axonal outgrowth. Neurosci. Res. 59, 168–173 (2007).
117. Feng, J. F. et al. Effect of therapeutic mild hypothermia on the genomics of the hippocampus after moderate traumatic brain injury in rats. Neurosurgery 67, 730–742 (2010).
118. Sessler, D. I. Complications and treatment of mild hypothermia. Anesthesiology 95, 531–543 (2001).
119. Frerichs, K. U. & Hallenbeck, J. M. Hibernation in ground squirrels induces state and species‑specific tolerance to hypoxia and aglycemia: an in vitro study in hippocampal slices. J. Cereb. Blood Flow Metab. 18, 168–175 (1998).
120. Dave, K. R. et al. Protein kinase C epsilon activation delays neuronal depolarization during cardiac arrest in the euthermic arctic ground squirrel. J. Neurochem. 110, 1170–1179 (2009).
121. Drew, K. L. et al. Hypoxia tolerance in mammalian heterotherms. J. Exp. Biol. 207, 3155–3162 (2004).
122. Toien, O. et al. Hibernation in black bears: independence of metabolic suppression from body temperature. Science 331, 906–909 (2011).
123. Han, H. S. & Yenari, M. A. Effect of gene expression by therapeutic hypothermia in cerebral ischemia. Future Neurol. 2, 435–440 (2007).This review covers the spectrum of gene expression changes resulting from hypothermia, including temperature-sensitive RNAs.
124. Al‑Fageeh, M. B. & Smales, C. M. Cold‑inducible RNA binding protein (CIRP) expression is modulated by alternative mRNAs. RNA 15, 1164–1176 (2009).
125. Liu, A., Zhang, Z., Li, A. & Xue, J. Effects of hypothermia and cerebral ischemia on cold‑inducible RNA‑binding protein mRNA expression in rat brain. Brain Res. 1347, 104–110 (2010).
126. Sakurai, T. et al. Cirp protects against tumor necrosis factor‑α‑induced apoptosis via activation of extracellular signal‑regulated kinase. Biochim. Biophys. Acta 1763, 290–295 (2006).
AcknowledgementsThis work was supported by grants from the US National Institutes of Health (NS40516 to M.Y.), the Veteran’s Merit Award (M.Y.), the Korea Healthcare technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A100870 to H.S.H.) and the Industrial Strategic technology development program (10035197 to H.S.H.), which is funded by the Ministry of Knowledge Economy (MKE), Korea. Grants to M.Y. were administered by the Northern California Institute for Research and Education, and supported by resources of the Veterans Affairs Medical Center, San Francisco, California.
Competing interests statementThe authors declare no competing financial interests.
R E V I E W S
278 | APRIL 2012 | VOLUME 13 www.nature.com/reviews/neuro
© 2012 Macmillan Publishers Limited. All rights reserved
Related Documents











