Nerve Growth Factor Restores p53 Function in Pituitary Tumor Cell Lines via trkA-Mediated Activation of Phosphatidylinositol 3-Kinase MARCO FACCHETTI, DANIELA UBERTI, MAURIZIO MEMO, AND CRISTINA MISSALE Division of Pharmacology, Department of Biomedical Sciences and Biotechnology and Centre of Excellence on Diagnostic and Therapeutic Innovation, University of Brescia, 25123 Brescia, Italy Two groups of prolactinomas were identified, one slowly proliferating and responsive to bromocrip- tine and one fast proliferating and bromocriptine resistant. Nerve growth factor (NGF) inhibits pro- liferation of bromocriptine-resistant cells by mech- anisms that are still unclear. The tumor suppressor p53 is one of the key reg- ulators of cell proliferation and in most tumors, but not pituitary adenomas, it is inactivated by genomic mutations. Here we investigated whether in prolactinoma cell lines NGF influences cell cycle-related pathways involving p53. By using conformation-specific antibodies and immunocy- tochemistry we found that in bromocriptine-resis- tant cells p53 adopts a mutant conformation that precludes its nuclear translocation and transcrip- tional activity. NGF administration to these cells refolds p53 into wild-type tertiary structure, pro- motes its nuclear translocation, and restores its DNA-binding activity as demonstrated by the tran- scriptional activation of p21 Cip1/WAF1 and the re- sulting down-regulation of different cyclins and cy- clin-dependent kinase 2. Inactivation of trkA, but not of p75 NTR , and wortmannin prevented NGF- induced p53 nuclear translocation. Thus, in pro- lactinoma cells p53 is inactivated by conforma- tional mutation and cytoplasmic segregation. This defect is reversible because NGF reconstitutes ac- tive p53 in these cells. This effect of NGF is exclu- sively mediated by trkA through activation of phos- phatidylinositol-3-kinase and may be related to its growth-inhibitory action. (Molecular Endocrinology 18: 162–172, 2004) P ROLACTINOMAS ARE THE most frequently oc- curring adenomas in the human pituitary. In pre- vious studies we have developed and characterized two phenotypically different groups of human prolacti- noma cell lines (1, 2). Those derived from tumors re- fractory to the dopaminomimetic therapy (NR) not only lack D 2 receptors, but also have a high proliferation rate in vitro and high tumorigenic potential in vivo, whereas those derived from the dopamine-sensitive prolactinomas (R) proliferate slowly and lack tumori- genicity (1). Proliferation of R cells, as well as the expression of D 2 receptors, is under the control of an autocrine loop mediated by nerve growth factor (NGF) that is disrupted in NR cells (2). NGF administration to NR not only induces D 2 receptor expression, but also remarkably inhibits the proliferation rate and abro- gates the anchorage-independent clonal growth in soft agar and the tumorigenic potential in vivo of these cells (1, 3). The molecular mechanisms underlying the latter effects are still elusive. NGF interacts with trkA and p75 NTR receptors (4). The tyrosine kinase trkA signals through a Ras-depen- dent pathway ending with activation of the mitogen- activated phosphokinases (Erk) (5) and through other enzymes, such as phosphatidylinositol 3-kinase (PI3K) (6) and phospholipase C (PLC) (7), whereas p75 NTR stimulates ceramide production and activates both nuclear factor-B (NF-B) and c-Jun N-terminal kinase (8). How these receptors act, individually or together, to regulate specific cell responses to NGF and the nature of the intracellular signals activated are key questions in NGF signal transduction. We have re- cently reported that, in prolactinoma cells, D 2 receptor expression is controlled by NGF via binding to p75 NTR and activation of NF-B, in a trkA-independent way (9). However, the receptor subtypes and intracellular pathways involved in NGF-mediated growth arrest of these cells have not been identified so far. The role of trkA and p75 NTR in cell growth control is still contro- versial. In particular, trkA has been shown to promote proliferation in some tumor cell lines (10, 11) but to induce cell growth inhibition in others (12–14). Simi- larly, it has been reported that p75 NTR is a tumor suppressor in certain tumors (15, 16) but is mitogenic in others (17). Therefore, identifying the mechanisms involved in NGF-mediated cell growth inhibition in pro- lactinoma cells may help to clarify this issue. Cell proliferation is regulated at specific check points in the cell cycle (18). In particular, an important Abbreviations: cdk, Cyclin-dependent kinase; HRP, horse- radish peroxidase; NF-B, nuclear factor B; NGF, nerve growth factor; NP-40, Nonidet P-40; PI3K, phosphatidylino- sitol 3-kinase; PLC, phospholipases C; WB, Western blot. Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community. 0888-8809/04/$15.00/0 Molecular Endocrinology 18(1):162–172 Printed in U.S.A. Copyright © 2004 by The Endocrine Society doi: 10.1210/me.2003-0190 162

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Nerve Growth Factor Restores p53 Function inPituitary Tumor Cell Lines via trkA-MediatedActivation of Phosphatidylinositol 3-Kinase
MARCO FACCHETTI, DANIELA UBERTI, MAURIZIO MEMO, AND CRISTINA MISSALE
Division of Pharmacology, Department of Biomedical Sciences and Biotechnology and Centre ofExcellence on Diagnostic and Therapeutic Innovation, University of Brescia, 25123 Brescia, Italy
Two groups of prolactinomas were identified, oneslowly proliferating and responsive to bromocrip-tine and one fast proliferating and bromocriptineresistant. Nerve growth factor (NGF) inhibits pro-liferation of bromocriptine-resistant cells by mech-anisms that are still unclear.
The tumor suppressor p53 is one of the key reg-ulators of cell proliferation and in most tumors,but not pituitary adenomas, it is inactivated bygenomic mutations. Here we investigated whetherin prolactinoma cell lines NGF influences cellcycle-related pathways involving p53. By usingconformation-specific antibodies and immunocy-tochemistry we found that in bromocriptine-resis-tant cells p53 adopts a mutant conformation thatprecludes its nuclear translocation and transcrip-tional activity. NGF administration to these cells
refolds p53 into wild-type tertiary structure, pro-motes its nuclear translocation, and restores itsDNA-binding activity as demonstrated by the tran-scriptional activation of p21Cip1/WAF1 and the re-sulting down-regulation of different cyclins and cy-clin-dependent kinase 2. Inactivation of trkA, butnot of p75NTR, and wortmannin prevented NGF-induced p53 nuclear translocation. Thus, in pro-lactinoma cells p53 is inactivated by conforma-tional mutation and cytoplasmic segregation. Thisdefect is reversible because NGF reconstitutes ac-tive p53 in these cells. This effect of NGF is exclu-sively mediated by trkA through activation of phos-phatidylinositol-3-kinase and may be related to itsgrowth-inhibitory action. (Molecular Endocrinology18: 162–172, 2004)
PROLACTINOMAS ARE THE most frequently oc-curring adenomas in the human pituitary. In pre-
vious studies we have developed and characterizedtwo phenotypically different groups of human prolacti-noma cell lines (1, 2). Those derived from tumors re-fractory to the dopaminomimetic therapy (NR) not onlylack D2 receptors, but also have a high proliferationrate in vitro and high tumorigenic potential in vivo,whereas those derived from the dopamine-sensitiveprolactinomas (R) proliferate slowly and lack tumori-genicity (1). Proliferation of R cells, as well as theexpression of D2 receptors, is under the control of anautocrine loop mediated by nerve growth factor (NGF)that is disrupted in NR cells (2). NGF administration toNR not only induces D2 receptor expression, but alsoremarkably inhibits the proliferation rate and abro-gates the anchorage-independent clonal growth insoft agar and the tumorigenic potential in vivo of thesecells (1, 3). The molecular mechanisms underlying thelatter effects are still elusive.
NGF interacts with trkA and p75NTR receptors (4).The tyrosine kinase trkA signals through a Ras-depen-dent pathway ending with activation of the mitogen-activated phosphokinases (Erk) (5) and through otherenzymes, such as phosphatidylinositol 3-kinase (PI3K)(6) and phospholipase C� (PLC�) (7), whereas p75NTR
stimulates ceramide production and activates bothnuclear factor-�B (NF-�B) and c-Jun N-terminal kinase(8). How these receptors act, individually or together,to regulate specific cell responses to NGF and thenature of the intracellular signals activated are keyquestions in NGF signal transduction. We have re-cently reported that, in prolactinoma cells, D2 receptorexpression is controlled by NGF via binding to p75NTR
and activation of NF-�B, in a trkA-independent way(9). However, the receptor subtypes and intracellularpathways involved in NGF-mediated growth arrest ofthese cells have not been identified so far. The role oftrkA and p75NTR in cell growth control is still contro-versial. In particular, trkA has been shown to promoteproliferation in some tumor cell lines (10, 11) but toinduce cell growth inhibition in others (12–14). Simi-larly, it has been reported that p75NTR is a tumorsuppressor in certain tumors (15, 16) but is mitogenicin others (17). Therefore, identifying the mechanismsinvolved in NGF-mediated cell growth inhibition in pro-lactinoma cells may help to clarify this issue.
Cell proliferation is regulated at specific checkpoints in the cell cycle (18). In particular, an important
Abbreviations: cdk, Cyclin-dependent kinase; HRP, horse-radish peroxidase; NF-�B, nuclear factor �B; NGF, nervegrowth factor; NP-40, Nonidet P-40; PI3K, phosphatidylino-sitol 3-kinase; PLC, phospholipases C; WB, Western blot.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/04/$15.00/0 Molecular Endocrinology 18(1):162–172Printed in U.S.A. Copyright © 2004 by The Endocrine Society
doi: 10.1210/me.2003-0190
162

restriction point in the G1 phase, which is controlled bythe tumor suppressor protein p53 and its effectorp21Cip1/WAF1, has been identified (18). The importanceof this mechanism is demonstrated by the fact thatinactivation of this pathway is related to tumorigene-sis. More than 50% of all human cancers show inac-tivating mutations of the p53 gene (19) and in cells thatretain wild-type p53, other defects in this pathwayhave been identified. In particular, changes in the ter-tiary structure from a wild-type to a mutant conforma-tion (20, 21) and aberrant cytoplasmic sequestrationare emerging as important mechanisms of p53 inac-tivation (22–26). Studies on pituitary tumors, in con-trast to most of solid tumors, have failed to identifymutations in the p53 gene (27–29). These findings,together with the observation that p53 is implicated inNGF-induced growth arrest of neuronal cells (30, 31),prompted us to test the hypothesis that p53 mightmediate the antiproliferative action of NGF in prolacti-noma cells.
We found that in the more transformed NR cells, butnot in the more differentiated R phenotype, p53adopts a mutant conformation that precludes its nu-clear localization and transcriptional activity. This de-fect is reversible because NGF administration to thesecells refolds p53 into the wild-type conformation, pro-motes its nuclear translocation, and restores its tran-scriptional activity. This effect, resulting in the expres-sion of p21Cip1/WAF1 and subsequent down-regulationof different cyclins and cyclin-dependent kinases(cdks), is mediated by trkA-induced activation of thePI3K-Akt pathway in a p75NTR-independent way.
RESULTS
Expression, Conformation, and CellularLocalization of p53 in Prolactinoma Cells
The expression of p53 was evaluated by both RT-PCRand Western blot (WB) in two prolactinoma cell lines,one R, characterized by low proliferation rate and NGFproduction, and one NR, which has high proliferationrate and lacks NGF production. RT-PCR analysis re-vealed that p53 mRNA was present in both R and NRcell lines at a similar expression level (Fig. 1A, lanes 1and 2) and that exposure of NR cells to 100 ng/ml NGFfor 5 d did not substantially modify p53 mRNA expres-sion (Fig. 1A, lane 3). Direct amplification of the RNA,i.e. omitting the reverse transcription reaction, did notproduce any band (data not shown), confirming thespecificity of the PCR. Similar results were obtained byWB with the specific anti-p53 antibody that recognizesboth wild-type and mutated p53 forms. A representa-tive WB is reported in Fig. 1B and the densitometricanalysis of three independent blots, with p53 signalsnormalized to the corresponding �-tubulin staining, isreported in Fig. 1C. R and NR cells exhibited com-parable p53 levels, and treatment of NR cells withNGF did not modify p53 concentration.
There is now increasing evidence that one of theintrinsic properties of p53 is conformational flexibilitythat causes it to adopt at least two tertiary structuresin vivo, a wild-type functionally active and a mutantfunctionally inactive conformation (32). The conforma-tional status of p53 in R and NR prolactinoma cell lineswas evaluated by immunoprecipitation with conforma-tion-specific monoclonal antibodies that allow dis-criminating folded vs. unfolded p53. In particular, thewild-type form is reactive with the PAb1620 antibody,which binds to a denaturation-sensitive epitope withinthe DNA-binding domain, whereas the so-called “mu-tant” form reacts with PAb240 antibody recognizing aprimary epitope that is cryptic in the wild-type confor-mation (32, 33). Immunoprecipitated p53 was de-tected by WB with the polyclonal antibody CM-1. Asshown in the representative immunoprecipitation re-ported in Fig. 2, in R cells p53 had high reactivity withPAb1620 (lane 3), but not with PAb240 (lane 2), sug-gesting that in this cell line p53 adopts its wild-type
Fig. 1. Expression of p53 in Prolactinoma CellsA, p53 mRNA levels in R and untreated or NGF-treated NR
cells. The cDNA was PCR amplified (28 cycles) with specificprimers as described in Materials and Methods. The amountof cDNA in each sample was determined by PCR amplifica-tion of �-actin. B, Representative WB analysis of p53 proteinin R, NR, and NGF-treated NR cells. Aliquots of cell proteins(15 �g/lane) were resolved by 10% SDS-PAGE and immu-noreacted with the anti-p53 antibody as described in Mate-rials and Methods. The amount of proteins in each lane wasevaluated by immunoreaction with the �-tubulin antibody. C,Densitometric analysis of p53 normalized to the correspond-ing �-tubulin levels. Bars represent the mean � SE of threeexperiments.
Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas Mol Endocrinol, January 2004, 18(1):162–172 163
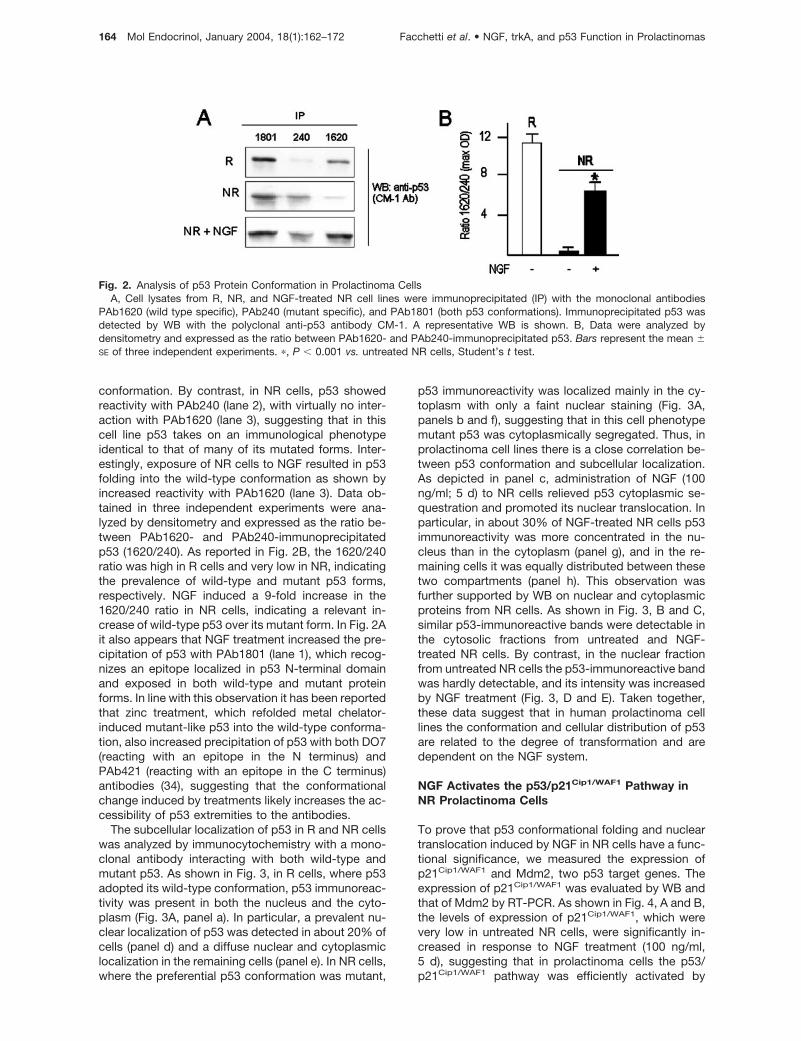
conformation. By contrast, in NR cells, p53 showedreactivity with PAb240 (lane 2), with virtually no inter-action with PAb1620 (lane 3), suggesting that in thiscell line p53 takes on an immunological phenotypeidentical to that of many of its mutated forms. Inter-estingly, exposure of NR cells to NGF resulted in p53folding into the wild-type conformation as shown byincreased reactivity with PAb1620 (lane 3). Data ob-tained in three independent experiments were ana-lyzed by densitometry and expressed as the ratio be-tween PAb1620- and PAb240-immunoprecipitatedp53 (1620/240). As reported in Fig. 2B, the 1620/240ratio was high in R cells and very low in NR, indicatingthe prevalence of wild-type and mutant p53 forms,respectively. NGF induced a 9-fold increase in the1620/240 ratio in NR cells, indicating a relevant in-crease of wild-type p53 over its mutant form. In Fig. 2Ait also appears that NGF treatment increased the pre-cipitation of p53 with PAb1801 (lane 1), which recog-nizes an epitope localized in p53 N-terminal domainand exposed in both wild-type and mutant proteinforms. In line with this observation it has been reportedthat zinc treatment, which refolded metal chelator-induced mutant-like p53 into the wild-type conforma-tion, also increased precipitation of p53 with both DO7(reacting with an epitope in the N terminus) andPAb421 (reacting with an epitope in the C terminus)antibodies (34), suggesting that the conformationalchange induced by treatments likely increases the ac-cessibility of p53 extremities to the antibodies.
The subcellular localization of p53 in R and NR cellswas analyzed by immunocytochemistry with a mono-clonal antibody interacting with both wild-type andmutant p53. As shown in Fig. 3, in R cells, where p53adopted its wild-type conformation, p53 immunoreac-tivity was present in both the nucleus and the cyto-plasm (Fig. 3A, panel a). In particular, a prevalent nu-clear localization of p53 was detected in about 20% ofcells (panel d) and a diffuse nuclear and cytoplasmiclocalization in the remaining cells (panel e). In NR cells,where the preferential p53 conformation was mutant,
p53 immunoreactivity was localized mainly in the cy-toplasm with only a faint nuclear staining (Fig. 3A,panels b and f), suggesting that in this cell phenotypemutant p53 was cytoplasmically segregated. Thus, inprolactinoma cell lines there is a close correlation be-tween p53 conformation and subcellular localization.As depicted in panel c, administration of NGF (100ng/ml; 5 d) to NR cells relieved p53 cytoplasmic se-questration and promoted its nuclear translocation. Inparticular, in about 30% of NGF-treated NR cells p53immunoreactivity was more concentrated in the nu-cleus than in the cytoplasm (panel g), and in the re-maining cells it was equally distributed between thesetwo compartments (panel h). This observation wasfurther supported by WB on nuclear and cytoplasmicproteins from NR cells. As shown in Fig. 3, B and C,similar p53-immunoreactive bands were detectable inthe cytosolic fractions from untreated and NGF-treated NR cells. By contrast, in the nuclear fractionfrom untreated NR cells the p53-immunoreactive bandwas hardly detectable, and its intensity was increasedby NGF treatment (Fig. 3, D and E). Taken together,these data suggest that in human prolactinoma celllines the conformation and cellular distribution of p53are related to the degree of transformation and aredependent on the NGF system.
NGF Activates the p53/p21Cip1/WAF1 Pathway inNR Prolactinoma Cells
To prove that p53 conformational folding and nucleartranslocation induced by NGF in NR cells have a func-tional significance, we measured the expression ofp21Cip1/WAF1 and Mdm2, two p53 target genes. Theexpression of p21Cip1/WAF1 was evaluated by WB andthat of Mdm2 by RT-PCR. As shown in Fig. 4, A and B,the levels of expression of p21Cip1/WAF1, which werevery low in untreated NR cells, were significantly in-creased in response to NGF treatment (100 ng/ml,5 d), suggesting that in prolactinoma cells the p53/p21Cip1/WAF1 pathway was efficiently activated by
Fig. 2. Analysis of p53 Protein Conformation in Prolactinoma CellsA, Cell lysates from R, NR, and NGF-treated NR cell lines were immunoprecipitated (IP) with the monoclonal antibodies
PAb1620 (wild type specific), PAb240 (mutant specific), and PAb1801 (both p53 conformations). Immunoprecipitated p53 wasdetected by WB with the polyclonal anti-p53 antibody CM-1. A representative WB is shown. B, Data were analyzed bydensitometry and expressed as the ratio between PAb1620- and PAb240-immunoprecipitated p53. Bars represent the mean �SE of three independent experiments. �, P � 0.001 vs. untreated NR cells, Student’s t test.
164 Mol Endocrinol, January 2004, 18(1):162–172 Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas

NGF. Similarly, NGF administration to NR cells in-duced a significant increase in the expression ofMdm2 mRNA (Fig. 4C).
Because p21Cip1/WAF1 acts as an inhibitor of differ-ent cyclins and cdks to initiate G1 arrest, we measuredthe expression of cell cycle-related proteins in un-treated and NGF-treated NR cells. The expression ofcyclin E and cdk2 was evaluated by WB, whereascyclin A, which was hardly detectable by WB (data notshown), was measured by RT-PCR. A representativeWB is reported in Fig. 4E. Densitometric analysis ofthree experiments with cyclin E and cdk2 normalizedto the corresponding values of �-tubulin is shown inFig. 4, F and G. Exposure of NR cells to NGF resultedin a 42% � 1.7% decrease of cyclin E expression (Fig.4F) and a 62% � 1.4% decrease of cdk2 expression(Fig. 4G). Similarly, as a result of NGF administration,cyclin A mRNA was significantly decreased (Fig. 4D).Thus, according to its antiproliferative effects and as aresult of activation of the p53/p21Cip1/WAF1 pathway, inNR cells NGF impairs the formation of cyclin A-cdk2and cyclin E-cdk2 complexes that are essential for cellcycle progression at the G1/S transition (18).
NGF-Induced p53 Nuclear Translocation IsMediated by trkA-Dependent Activation of PI3K
To evaluate the contribution of trkA and p75NTR re-ceptors to NGF-induced p53 nuclear translocation,each receptor was individually inactivated, and p53subcellular localization was investigated by immuno-cytochemistry. On the basis of previous studies (9),p75NTR-mediated responses were inhibited using aspecific anti-p75NTR antibody at the concentration of100 ng/ml, and trkA was inactivated by inhibiting itsintrinsic tyrosine kinase activity with 1 �M genistein.As illustrated in Fig. 5B, genistein prevented NGF-induced trkA phosphorylation at Tyr490. The ratioptrkA (max OD)/�-tubulin (max OD) was 0.13 � 0.01(mean � SE; n � 3) in NGF-treated cells and 0.052 �0.02 (mean � SE; n � 3) in cells exposed to NGF in thepresence of genistein. Genistein also abolished theeffects of NGF on p53 trafficking. A representativeimmunocytochemistry is depicted in Fig. 5A. As inuntreated NR cells (Fig. 5A, panels a and e), also in NRcells exposed to NGF in the presence of 1 �M
genistein, p53 immunoreactivity was confined to thecytoplasm (panels d and h). By contrast, p53 nucleartranslocation induced by NGF in NR cells was notmodified by inhibition of p75NTR signaling, as shownby the specific p53 immunostaining that, as in cellsexposed to NGF (panels b and f), also in cells exposedto NGF in the presence of the anti-p75NTR antibody,was mainly localized into the nucleus (panels c and g).In these conditions the anti-p75NTR antibody com-pletely prevented NGF-induced NF-�B activation (Fig.5C), indicating that it efficiently blocked p75NTR-mediated effects. To further demonstrate the role ofNGF-mediated trkA activation in the regulation of p53trafficking, NGF-secreting R cells were either deprived
Fig. 3. Subcellular Distribution of p53 in Prolactinoma CellsA, R, NR, and NGF-treated NR cells were fixed with meth-
anol at �20 C and processed for p53 immunostaining asdescribed in Materials and Methods. Panel a, Distribution ofp53 immunoreactivity in R cells detected at �40 magnifica-tion; panels d and e, representative R cells detected at �100magnification showing that in some cells p53 staining isconcentrated into the nucleus (d) and in others it is equallydistributed between the nucleus and the cytoplasm (e); panelb, distribution of p53 immunoreactivity in NR cells detected at�40 magnification; panel f, representative NR cells detectedat �100 magnification showing that p53 immunostaining issegregated into the cytoplasm; panel c, distribution of p53immunoreactivity in NGF-treated NR cells (�40 magnifica-tion); panels g and h, representative NGF-treated NR cellsdetected at �100 magnification showing that in some cellsp53 immunostaining is preferentially localized into the nu-cleus (g), whereas in others (h) it is equally distributed be-tween the nucleus and the cytoplasm. Scale bar, 100 �m. B,Representative WB analysis of p53 in cytoplasmic extractsfrom untreated and NGF-treated NR cells. Aliquots of pro-teins (15 �g/lane) were analyzed for p53 and �-tubulin levelsas described in Materials and Methods. C, Densitometricanalysis of cytoplasmic p53 normalized to the corresponding�-tubulin levels. Bars represent the mean � SE of three ex-periments. D, Representative WB analysis of p53 in nuclearextracts from untreated and NGF-treated NR cells. Aliquotsof proteins (30 �g/lane) were analyzed for p53 and �-actinlevels as described in Materials and Methods. E, Densitomet-ric analysis of nuclear p53 normalized to the corresponding�-actin levels. Bars represent the mean � SE of three exper-iments. �, P � 0.001 vs. untreated cells, Student’s t test.
Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas Mol Endocrinol, January 2004, 18(1):162–172 165

of secreted NGF with an anti-NGF monoclonal anti-body, as previously described (2), or exposed togenistein or to the anti-p75NTR antibody. A represen-tative p53 immunostaining is illustrated in Fig. 6. p53immunoreactivity, which in untreated R cells was dis-tributed in both the nucleus and cytoplasm with highnuclear staining (panel A), underwent cytoplasmic re-tention both during NGF deprivation (panel B) andgenistein treatment (panel C), but not during inactiva-tion of p75NTR (panel D). These data suggest thatNGF-dependent p53 nuclear localization in humanprolactinomas is mediated by the trkA receptor in ap75NTR-independent way.
With the aim of identifying the intracellular effectorsinvolved in trkA-mediated p53 nuclear translocation,Erk activation was blocked by PD 98059 (35), and PI3Kwas inhibited by wortmannin (36). The representativeresults reported in Fig. 7A show that, as in NGF-treated NR cells (panels a and c), also in cells exposedto NGF in the presence of 50 �M PD98059, p53 im-munostaining was concentrated into the nucleus (pan-els b and d), suggesting that this compound did not
interfere with NGF action. Control experiments, per-formed to evaluate the efficiency of PD 98059 treat-ment, showed that Erk phosphorylation induced byNGF in NR cells was abolished by coincubation withthis compound (Fig. 7, B and C). Figure 8A reports arepresentative immunocytochemistry demonstratingthat 500 nM wortmannin completely blocked p53 nu-clear translocation induced by NGF in NR cells.Whereas in NGF-treated NR cells, p53 immunoreac-tivity was concentrated into the nucleus (panels b ande), in cells exposed to NGF in the presence of wort-mannin, as in untreated cells (panels a and d), p53immunoreactivity was in fact segregated into the cy-toplasm (panels c and f). In line with these obser-vations, wortmannin also abolished the stimulatoryeffect of NGF on p21Cip1/WAF1 expression (Fig. 8, Band C). The effect of wortmannin on NGF-inducedp21Cip1/WAF1 expression was dose dependent over therange of 5 nM to 0.5 �M Fig. 8D). Taken together, thesedata rule out the possibility that the Ras-Erk pathwaymay be involved in the control of p53 trafficking elic-ited by NGF and strongly suggest a crucial involve-
Fig. 4. NGF Induces the Expression of p21Cip1/WAF1 and Down-Regulates Different Cyclins and cdk2 in NR Prolactinoma CellsA, Effect of NGF on p21Cip1/WAF1 expression. Proteins (30 �g/lane) from untreated and NGF-treated NR cells were analyzed by
WB with the anti-p21Cip1/WAF1 antibody. To ensure equal protein loading, membranes were stripped and reprobed with theanti-�-tubulin antibody. B, Densitometric analysis of p21Cip1/WAF1 normalized to the corresponding �-tubulin levels. Barsrepresent the mean � SE of three experiments. �, P � 0.001 vs. untreated cells, Student’s t test. C, Effect of NGF treatment onMdm2 mRNA levels. The amount of cDNA in each sample was checked by PCR amplification with �-actin primers. Data arerepresentative of three experiments. D, RT-PCR analysis of cyclin A mRNA levels in untreated and NGF-treated NR cells. Dataare representative of three experiments. E, Representative WB showing the effect of NGF on cyclin E and cdk2 expression. F andG, Densitometric analysis of cyclin E (F) and cdk2 (G) normalized to the corresponding �-tubulin levels. Bars represent the mean �SE of three experiments. �, P � 0.001 vs. untreated cells, Student’s t test.
166 Mol Endocrinol, January 2004, 18(1):162–172 Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas

ment of PI3K. To strengthen this finding, we evaluatedwhether NGF activates PI3K in NR cells by measuringthe phosphorylation of its effector Akt (37) in WB ex-periments with a specific antiphospho(Ser473)-Akt(pAkt) antibody. As reported in Fig. 9, exposure of NRcells to NGF resulted in a sustained Akt phosphoryla-tion. Phospho-Akt was hardly detectable in untreatedcells (Fig. 9A, lane 1) and in cells treated with NGF for24 h (lane 2). By contrast, pAkt was 6-fold increasedafter a 2-d NGF treatment (lane 3) and was still highafter 5 d (lane 4). Densitometric analysis of three ex-periments with pAkt normalized to the corresponding
values of �-tubulin is shown in Fig. 9B. Because a 5-dadministration of the potent PLC� inhibitor U-73122was prevented by its toxicity, we investigated whetherNGF might activate this enzyme in prolactinoma cells.As reported in Fig. 10, a 5-d NGF treatment did notmodify the levels of PLC� measured by WB with aspecific antibody (lanes 1 and 2). To evaluate the effectof NGF on PLC� phosphorylation, protein extractsobtained from untreated or NGF-treated NR cells wereimmunoprecipitated with an antiphosphotyrosine (p-Tyr) antibody, and the resulting proteins were immu-noblotted with a monoclonal anti-PLC� antibody.Constitutively phosphorylated PLC� was detectable inNR cells maintained in culture in the presence of se-rum (lanes 3 and 5). Exposure of these cells to 100ng/ml NGF for either 5 min or 5 d did not change thelevels of phosphorylated PLC� (lanes 4 and 6). Similarresults were obtained in serum-starved cells treatedwith NGF for 5 min. No specific signals were detect-able in either untreated or NGF-treated cells (lanes 7and 8). Thus PLC� apparently does not contribute tothe effects of NGF on prolactinoma cells.
DISCUSSION
Disruption of p53 function is frequently correlated withtumorigenesis. Mutations occurring within the DNA-
Fig. 5. Inactivation of trkA Prevents NGF-Mediated p53 Nu-clear Translocation in NR Cells
A, Cells were fixed in methanol at �20 C and processed forp53 immunostaining. Panel a, subcellular localization of p53in untreated NR cells detected at �40 magnification; panel e,representative NR cells detected at �100 magnificationshowing that p53 immunostaining is segregated into the cy-toplasm; panel b, distribution of p53 immunoreactivity inNGF-treated NR cells detected at �40 magnification; panel f,representative NGF-treated NR cells detected at �100 mag-nification showing that p53 immunostaining is prevalent intothe nucleus; panel c, subcellular distribution of p53 in cellstreated with NGF in the presence of the anti- p75NTR antibody(�40 magnification); panel g, �100 magnification of repre-sentative NR cells exposed to both NGF and anti-p75NTR
antibody showing that p53 immunostaining is more concen-trated in the nucleus than in the cytoplasm; panel d, distri-bution of p53 immunoreactivity in cells treated with NGF inthe presence of 1 �g/ml genistein detected at �40 magnifi-cation; panel h, �100 magnification of representative NRcells exposed to both NGF and genistein showing that p53immunostaining is cytoplasmically segregated. Scale bar,100 �m. B, Effect of genistein on NGF-induced phosphory-lation of trkA at Tyr490. A representative WB obtained withthe Tyr490-phospho-trkA-specific antibody is shown. Lane 1,NGF-treated cells; lane 2, NGF and genistein-treated cells. C,The efficiency of the anti-p75NTR antibody as an inhibitor ofp75NTR-mediated responses was evaluated by EMSA with aspecific NF-�B sequence (9). Lane 1, NGF-treated cells; lane2, NGF- and p75NTR antibody-treated cells. Data are repre-sentative of three experiments.
Fig. 6. Disruption of NGF-Mediated Autocrine Mechanismsin R Cells Results in p53 Nuclear Exclusion
A, p53 immunoreactivity in representative R cells (�100magnification) showing either nuclear or both cytoplasmicand nuclear localization. B, p53 immunoreactivity in repre-sentative R cells exposed to the anti-NGF antibody (�100magnification) showing a preferential cytoplasmic localiza-tion. C, p53 immunoreactivity in representative R cells ex-posed to genistein (�100 magnification) showing a preferen-tial cytoplasmic localization. D, p53 immunoreactivity inrepresentative R cells exposed to the anti-p75NTR antibody(�100 magnification) showing both nuclear and cytoplasmiclocalization. Scale bar, 100 �m.
Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas Mol Endocrinol, January 2004, 18(1):162–172 167

binding domain are the prevalent mechanism of p53inactivation in half of all human tumors (19). Othermechanisms, however, such as interaction with cellu-lar proteins enhancing p53 degradation (38, 39) or withcytoplasmic anchor proteins that prevent p53 nuclearlocalization (26, 40–43), can also nullify p53 activity.Although sequence analysis showed that p53 is wildtype in the overwhelming majority of pituitary adeno-mas (27–29), accumulation of this protein has beendetected in a proportion of these tumors (44–46), es-pecially the invasive type. The existence of functional
defects in p53-dependent pathways in pituitary ade-nomas, however, has never been clearly demon-strated. In this study we report, for the first time, thatthe phenotype of human prolactinoma cell lines isrelated to p53 conformation, cellular localization, andtranscriptional activity and that NGF is crucial to main-tain p53 in its active form.
A critical feature for tumor suppression is a “wild-type”conformation of p53. Mutations in the p53 DNA-bindingdomain frequently result in the expression of a proteinwith an inactive mutant conformation (19). Yet wild-typep53 is characterized by intrinsic flexibility that causes it totransiently adopt either wild-type or mutant conforma-tions in a cell cycle-dependent manner (32, 47). In par-ticular, the wild-type conformation correlates with
Fig. 7. The Erk Inhibitor PD 98059 Does Not Affect NGF-Induced p53 Nuclear Translocation in NR Cells
A, p53 immunostaining in NR cells treated with NGF in theabsence (panel a) and in the presence (panel b) of 50 �M PD98059; panels c and d, �100 magnification of representativecells exposed to either NGF (c) or NGF and PD 98059 (d)showing that in both conditions p53 immunoreactivity waspreferentially localized into the nucleus. Scale bar, 100 �m. B,50 �M PD 98059 prevented NGF-induced Erk1/2 phosphor-ylation in NR cells. Cells were treated with NGF in the ab-sence or in the presence of PD 98059 for different times.Erk1/2 phosphorylation was evaluated by WB with a specificantibody that selectively recognizes the phosphorylatedforms of Erk1/2. C, Densitometric analysis of pErk1/2 nor-malized to the corresponding �-tubulin levels. Bars representthe mean � SE of three experiments. �, P � 0.001 vs. NGF-treated cells, Student’s t test.
Fig. 8. Inhibition of PI3K Abolishes NGF-Induced p53 Nu-clear Translocation and p21Cip1/WAF1 Expression in NR Cells
Cells were treated with 100 ng/ml NGF for 5 d in theabsence or in the presence of the PI3K inhibitor wortmannin(0.5 �M), and p53 cellular localization was evaluated by im-munocytochemistry (A). In untreated cells p53 immunoreac-tivity was segregated into the cytoplasm (panels a and d),whereas in NGF-treated cells it was preferentially localizedinto the nucleus (panels b and e). In cells exposed to bothNGF and wortmannin p53 was sequestered into the cyto-plasm (panels c and f). Scale bar, 100 �m. B, RepresentativeWB showing the expression of p21Cip1/WAF1 in untreated cellsand in cells exposed to NGF in the absence or in the presenceof wortmannin. Lane 1, Untreated cells; lane 2, NGF-treatedcells; lane 3, NGF- and wortmannin-treated cells. C, Densi-tometric analysis of p21Cip1/WAF1 normalized to the corre-sponding �-tubulin levels. Bars represent the mean � SE ofthree experiments. �, P � 0.001 vs. untreated cells, Student’st test. D, Dose-response curve of wortmannin effect on NGF-induced p21Cip1/WAF1 expression.
168 Mol Endocrinol, January 2004, 18(1):162–172 Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas

growth suppression, whereas the mutant tertiary struc-ture promotes proliferation in cells expressing wild-typep53 (47). In this study we report that p53 adopts differentconformations in R and NR prolactinoma cell lines, dueto the presence or the absence of secreted NGF in theculture media. In particular, in the more differentiated,slowly proliferating, NGF-secreting R cells, p53 adoptsthe wild-type conformation, whereas in the fast prolifer-ating NR cells, which do not produce NGF, p53 takes onthe mutant conformation. Moreover, according to previ-ous data obtained in fibroblast and neuroblastoma celllines (21), in prolactinoma cells a close correlation wasalso found between p53 conformation and its cellularlocalization. The wild-type p53 form was present, in fact,in both the nucleus and the cytoplasm in R cells, whereasthe mutant p53 form was cytoplasmically segregated inNR. Thus, in the slowly proliferating R cells p53 exhibitsfunctional conformation and localization, but in the fast-proliferating NR cell line, p53 is inactivated by conforma-tional mutation and cytoplasmic segregation. Along thisline, aberrant cytoplasmic sequestration of p53 has beendetected in many human tumors (22, 48–51). Interest-ingly, p53 structural alteration in prolactinomas is revers-ible and depends on NGF. Inactivation of secreted NGFresulted, in fact, in p53 nuclear exclusion in R cells,whereas NGF administration to NR cells refolded p53into the wild-type conformation, promoted its nucleartranslocation, and restored its specific DNA-binding ac-tivity as demonstrated by the transcriptional activation ofp21Cip1/WAF1 and Mdm2. Moreover, as a result of
p21Cip1/WAF1 expression, cyclin A, cyclin E, and cdk2were remarkably down-regulated in NGF-treated cells,an effect related to cell cycle arrest at the G1 to S tran-sition (18) that may represent the mechanism of thereported antiproliferative effect of NGF on these cell lines(1). Along this line, NGF-induced inhibition of cell pro-liferation in PC12 cells (12) and in NIH-3T3 cells (13) hasbeen reported to be mediated by activation of p21Cip1/
WAF1. Taken together these data suggest that escape ofNR cells from NGF control results in a reversible stabili-zation of p53 in the mutant conformation likely leading totheir fast-proliferating phenotype. It is worth noting thatthe effects of NGF on p53 conformation, cellular local-ization, and transcriptional activity were detectable aftera long-term treatment. Although this could be related tothe doubling time of NR cells (�2 d), the possibility can-not be excluded that other factors induced by NGF,rather than NGF itself, might regulate p53 conformation.
NGF transduces its effects by interacting with twotypes of surface receptors. From a functional point ofview, signaling by trkA and p75NTR, which are oftenpresent on the same cell, may be synergistic, indepen-dent, or antagonistic. We have previously shown that thep75NTR receptor plays a critical, trkA-independent role inthe control of D2 receptor expression in prolactinoma celllines (9). In this paper we demonstrate that trkA is thereceptor that, in a p75NTR-independent way, mediatesthe effect of NGF on p53 conformation and nuclear lo-calization. Taken together, these data thus suggest thatin prolactinoma cell lines, even if trkA and p75NTR arecoexpressed (1, 2), they mediate individual and indepen-dent functions of NGF. Whether p75NTR and trkA interactto mediate other effects of NGF on these cells remains tobe established. The role of trkA in the control of tumorgrowth is still controversial. TrkA operates, in fact, as atypical mitogenic growth factor receptor in some tumorcell lines (10, 11) but induces differentiation and inhibitionof cell growth in others (12–14). These observations,together with our present data, suggest that the func-tional effects of trkA are related to the cellular back-ground in which it is expressed, rather than to its intrinsicsignaling properties.
NGF binding to trkA results in the activation of dif-ferent signaling effectors. Our analysis with specificinhibitors and by measuring Akt phosphorylationstrongly pointed to PI3K as the most plausible intra-cellular messenger mediating the effects of NGF onthe p53/p21Cip1/WAF1 pathway. PI3K has been impli-cated in disparate cell responses including protectionfrom apoptosis, stimulation of cell proliferation, andtumor formation (37). Nevertheless, there is evidencethat in different tumor cell lines PI3K is required forp53-dependent induction of p21Cip1/WAF1 in responseto antitumor agents (53–55). Our data, showing that inprolactinoma cell lines the trkA-PI3K pathway is in-volved in the activation of p21Cip1/WAF1 in response toNGF, are in line with these observations.
In conclusion, our data, showing that the tertiary struc-ture and cellular localization of wild-type p53 are relatedto the degree of transformation of human prolactinoma
Fig. 9. NGF Promotes Akt Phosphorylation in NR CellsCells were treated with NGF for different times, and Akt
phosphorylation was evaluated by WB with a specific an-tiphospho-Akt (Ser473). A, Representative WB showing thelevels of pAkt. Lane 1, Untreated cells; lane 2, 24-h NGFtreatment; lane 3, 2-d NGF treatment; lane 4, 5-d NGF treat-ment. B, Densitometric analysis of pAkt normalized to thecorresponding �-tubulin levels. Bars represent the means �SE of three experiments. �, P � 0.001 vs. untreated cells.
Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas Mol Endocrinol, January 2004, 18(1):162–172 169

cell lines, suggest that disruption of p53 function bymechanisms different from genomic mutation mightcontribute to the development of these tumors. More-over, the finding that p53 conformation may be shiftedfrom mutant-like to wild type by NGF suggests that thisstructural alteration is reversible, an observation thatmight open new therapeutic perspectives, if such a de-fect is found also in primary pituitary adenomas.
MATERIALS AND METHODS
Prolactinoma Cell Cultures and Treatments
R and NR prolactinoma cells were grown in Ham’s F10 me-dium supplemented with 2.5% fetal bovine serum, 15%horse serum (Euroclone, Celbio, Milano, Italy) 4 mM glu-tamine, and 100 U of penicillin-streptomycin at 37 C and 5%CO2. NR cells were treated with 100 ng/ml NGF (2.5S,mouse, Alomone Labs, Jerusalem, Israel) for 5 d in the ab-sence or in the presence of either mouse anti-p75NTR mono-clonal antibody (100 ng/ml; Chemicon International, Te-mecula, CA), or PD98059 (50 �M; Calbiochem, San Diego,CA) or genistein (1 �g/ml; BIOMOL Research Laboratories,Inc., Plymouth, PA) or wortmannin (0.5 �M, Calbiochem). Rcells were exposed either to the anti-NGF antibody (50 ng/ml;Chemicon) or to 1 �g/ml genistein or to the anti-p75NTR
antibody (100 ng/ml). NGF and genistein were added once atthe beginning of treatment, whereas the anti-p75NTR anti-body, the anti-NGF antibody, PD 98059, and wortmanninwere added to the cultures every day.
Immunoprecipitation and WB
Cells were lysed in 50 mM Tris-HCl (pH 7.6) containing 150mM NaCl, 0.5% sodium deoxycholate, 0.5% Nonidet P-40(NP-40), 1 mM NaF, 1 mM Na3VO4, and a mixture of proteaseinhibitors (Complete Mini Protease Inhibitors, Roche Molec-ular Biochemicals, Basel, Switzerland) and centrifuged at18,000 � g for 20 min at 4 C. The resulting supernatantscontaining cell proteins were stored at �20 C. Aliquots ofproteins (10–30 �g) were resolved by SDS-PAGE and trans-ferred onto nitrocellulose membranes. After blotting for 1 h atroom temperature in Tris-buffered saline (TBS) containing0.1% Tween 20 and 5% low-fat dry milk, membranes wereincubated overnight at 4 C using the following antibodies:anti-p53 monoclonal antibody PAb421 (1.5 �g/ml) (Onco-gene Science, Inc., Boston, MA); anti-p21Cip1/WAF1 monoclo-nal antibody (1 �g/ml) (PharMingen International, San Diego,CA); anti-phospho-p44/42 Erk (Thr202/Tyr204) polyclonalantibody (1:1,000 dilution) (Cell Signaling, Beverly MA);anti-cdk2 monoclonal antibody (1:200 dilution) (Santa CruzBiotechnology, Heidelberg, Germany); anti-phospho-trkA
(Tyr490) polyclonal antibody (1:1,000 dilution in Tris-bufferedsaline containing BSA 5% and Tween 20 0.1%) (Cell Signal-ing); antiphospho-Akt (Ser473) antibody (Cell Signaling); anti-cyclin E monoclonal antibody (Upstate Biotechnology, Inc.,Lake Placid, NY). To ensure equal protein loading, mem-branes were stripped with a WB recycling kit (ChemiconInternational) and reprobed with anti-�-tubulin antibody (1:1,500 dilution) (Neo-Markers, Fremont, CA).
To isolate nuclear and cytoplasmic proteins, cells wereresuspended in ice-cold 10 mM HEPES (pH 7.9) containing1.5 mM MgCl2, 10 mM KCl, and the protease inhibitors, incu-bated on ice for 5 min, added with 10% NP-40, mixed byvortex for 10 sec, and centrifuged at 5,000 � g for 5 min. Thesupernatants containing the cytoplasmic proteins werestored at �20 C, and the pellets were resuspended in ice-cold 20 mM HEPES (pH 7.9) containing 25% glycerol, 420 mM
NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and the protease inhib-itors, shaken for 15 min at 4 C, and centrifuged at 18,000 �g for 20 min at 4 C. The supernatants containing the nuclearproteins were stored at �20 C. Aliquots of cytoplasmic (15�g/lane) and nuclear proteins (30 �g/lane) were resolved bySDS-PAGE and processed as described above to detectp53. To ensure equal protein loading, membranes werestripped and reprobed with �-actin antibody (1:500 dilution)(Sigma, St. Louis, MO) for the nuclear fraction and with �-tubulin antibody (1:1,500 dilution) for the cytoplasmic frac-tion. For detection, an enhanced chemiluminescence system(Amersham International, Cardiff, UK) was used with horse-radish peroxidase (HRP)-conjugated secondary antibodies(antirabbit, 1:2,000 dilution, Santa Cruz Biotechnology, Inc.;and antimouse, 1:1,500 dilution, DAKO, Copenhagen, Den-mark). To detect Tyr490-phosphorylated trkA, an enhancedchemiluminescent system allowing visualization of proteins inthe low femtogram range (Supersignal West, Pierce Chemi-cal, Milano, Italy) with a HRP-conjugated secondary antibody(Pierce Chemical) was used. The blots obtained from at leastthree independent experiments were analyzed by densitom-etry, and the specific signals were normalized to the corre-sponding �-tubulin or �-actin staining.
To detect tyrosine-phosphorylated PLC�, aliquots of thesupernatant containing the cytoplasmic proteins (100 �g)were immunoreacted overnight at 4 C with 2 �g of a mono-clonal antiphosphotyrosine (pTyr) antibody (TransductionLaboratories, Inc., Lexington, KY). The resulting immuno-complexes were precipitated by incubation with protein A-Sepharose (Santa Cruz Biotechnology, Inc.) for 2 h at roomtemperature. The pellets were resuspended in 50 mM Tris-HCl, pH 7.6, containing 150 mM NaCl, 0.5% sodium deoxy-cholate, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, and theprotease inhibitors, centrifuged at 18,000 � g at 4 C, resus-pended in 20 �l of sample buffer and resolved by SDS-PAGE.Membranes were incubated overnight at 4 C with the mono-clonal anti-PLC� antibody (1:250 dilution, Transduction Lab-oratories, Inc.). The immunoreaction was detected by en-hanced chemiluminescence as previously described.
Fig. 10. NGF Does Not Activate PLC� in NR Prolactinoma CellsTo evaluate PLC� expression, cytoplasmic proteins were analyzed by WB. To evaluate PLC� phosphorylation, aliquots of
cytoplasmic proteins were immunoprecipitated (IP) with the antiphosphotyrosine antibody, and the resulting immunocomplexeswere detected by WB with the anti-PLC� antibody. Lanes 1, 3, and 5, Untreated NR cells; lanes 2 and 4, 5-d NGF treatment; lane6, 5-min NGF treatment; lane 7, serum-starved untreated NR cells; lane 8, NGF treatment of serum-starved NR cells. Data arerepresentative of three experiments.
170 Mol Endocrinol, January 2004, 18(1):162–172 Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas

Conformation-Specific Immunoprecipitation of p53
Conformation-specific immunoprecipitation of p53 was per-formed according to Verhaegh (33). Cells were lysed in theimmunoprecipitation nondenaturing buffer (10 mM Tris, pH 7.6,containing 140 mM NaCl and 0.5% NP-40 and protease inhib-itors) for 20 min on ice and cleared from cell debris by centrif-ugation. To prevent nonspecific binding, the supernatant wasprecleared with 10% protein A-Sepharose (Santa Cruz Biotech-nology, Inc.) for 20 min on ice, followed by centrifugation. Forimmunoprecipitation of p53, 1 �g of the monoclonal antibodiesPAb1620 (wild type specific, Oncogene Science, Inc.), PAb240(mutant specific, Neo-Markers), or PAb1801 (recognizing bothwild-type and mutant p53, Oncogene Science, Inc.) was addedto the precleared supernatant for 2 h at 4 C. Immune complexeswere collected with protein A-Sepharose (Santa Cruz Biotech-nology, Inc.) and washed five times with immunoprecipitationbuffer. Immunoprecipitated p53 was detected by WB using theanti-p53 polyclonal antibody CM-1 (1:1200 dilution) (Novocas-tra, Newcastle, UK), which recognizes both mutant and wild-type p53 and a HRP-conjugated secondary antibody. Data ob-tained from three independent experiments were analyzed bydensitometry and expressed as the ratio between PAb1620-and PAb240-immunoprecipitated p53.
Immunocytochemistry
Cells were plated at low density on poly-L-lysine-coated glasscover slips, fixed with methanol at �20 C for 5 min, and per-meabilized for 20 min at room temperature in PBS containing0.2% Triton X-100 and 10% normal rabbit serum (DAKO Corp.).Cells were incubated overnight at 4 C with the monoclonalanti-p53 antibody PAb421 (20 �g/ml in PBS containing 0.2%Triton X-100 and 1% normal rabbit serum) and then with thebiotinylated rabbit antimouse secondary antibody (1:400 dilu-tion in PBS containing 0.2% Triton X-100 and 1% normal rabbitserum) (DAKO Corp.) for 1 h at room temperature. After threerinses with PBS, cells were incubated with the avidin-biotincomplex (ABC, DAKO Corp.) for 45 min at room temperature.Peroxidase staining was obtained by incubation in 0.06% 3,3-diaminobenzidine and 0.01% H2O2 in PBS buffer. Cells werevisualized at both �40 and �100 magnification.
RNA Isolation and RT-PCR
Total RNA was isolated using the Nucleospin-RNA II Kit(CLONTECH, Palo Alto, CA). Two micrograms of each sam-ple were transcribed into cDNA by using the murine Moloneyleukemia virus reverse transcriptase (Promega Corp., Madi-son, WI) and oligo(dT)18 as a primer. p53 was amplified withthe oligonucleotides 5�-CTGAGGTTGGCTCTGACTGTAC-CACCATCC-3� and 5�-CTCATTCAGCTCTCGGA ACATCTC-GAAGCG-3�, which generate a 371-bp fragment. The reac-tion was performed for 28 cycles (94 C, 1 min; 66 C, 30 sec;72 C, 1 min). Mdm2 was amplified for 28 cycles (94 C, 1 min;52 C, 30 sec; 72 C, 1 min) with the oligonucleotides 5�-GAAAGAGGTTCTTTTTTATCTTGG-3� and 5�-ATTTTCTTCT-GTCTCACTAATTGC-3�, which generate a 364-bp fragment.To amplify cyclin A, the oligonucleotides 5�-TCCTTGGAAAG-CAAACAGTAAA-3� and 5�-AACCCACTTTAGGTTTACATTT-3�, generating a 332-bp fragment, were used, and the reac-tion was performed for 35 cycles (94 C, 1 min; 50 C, 30 sec;72 C, 4 min). Amplification with 5�-TAAAGACCTCTAT-GCCAACACAGT-3� and 5�-CACGATGGAGGGCCGGACT-CATC-3� primers encoding a fragment of human �-actin (94C, 1 min; 60 C, 30 sec; 72 C, 1 min; 25 cycles) was performedas a control of the amount of cDNA in each sample. Allreactions were performed within the linear range of amplifi-cation. Omission of the reverse transcription was performedas a control of the PCR specificity. The PCR products wereanalyzed on 1% agarose gels stained with ethidium bromide.
Acknowledgments
Received May 23, 2003. Accepted September 26, 2003.Address all correspondence and requests for reprints to:
Cristina Missale, Ph.D., Department of Biomedical Sciencesand Biotechnology, University of Brescia, Viale Europa 11,25123 Brescia, Italy. E-mail: [email protected].
This work was supported by grants from Ministero Universitae Ricerca (MIUR 9906153187 and MIUR 2002067251) and byFondazione Italiana per la Ricerca sul Cancro (FIRC) to C.M.
REFERENCES
1. Missale C, Boroni F, Losa M, Giovanelli M, Zanellato A,Dal Toso R, Balsari A, Spano PF 1993 Nerve growthfactor suppressed the transforming phenotype of humanprolactinomas. Proc Natl Acad Sci USA 90:5781–5785
2. Missale C, Losa M, Sigala S, Balsari A, Giovanelli M, SpanoPF 1996 Nerve growth factor controls proliferation and pro-gression of human prolactinoma cell lines through an auto-crine mechanism. Mol Endocrinol 10:272–285
3. Missale C, Losa M, Boroni F, Giovanelli M, Balsari A,Spano PF 1995 Nerve growth factor and bromocriptine:a sequential therapy for human bromocriptine-resistantprolactinomas. Br J Cancer 72:1397–1399
4. Chao MV, Hempstead BL 1995 p75 and trk: a two re-ceptor system. Trends Neurosci 18:321–326
5. Kaplan DR, Stephens RM 1994 Neurotrophin signal trans-duction by the trk receptor. J Neurobiol 25:1404–1417
6. Soltoff SP, Rabin SL, Cantley LC, Kaplan DR 1992 Nervegrowth factor promotes the activation of phosphatidyl-inositol 3-kinase and its association with the trk tyrosinekinase. J Biol Chem 267:17472–17477
7. Choi DY, Toledo-Aral JJ, Segal R, Halegoua S 2001Sustained signalling by phospholipase C-� mediatesnerve growth factor-triggered gene expression. Mol CellBiol 21:2695–2705
8. Carter BD, Lewin GR 1997 Neurotrophins live or let die:does p75 decide? Neuron 18:187–190
9. Fiorentini C, Guerra N, Facchetti M, Finardi A, Tiberio L,Schiaffonati L, Spano PF, Missale C 2002 Nerve growthfactor regulates dopamine D2 receptor expression in pro-lactinoma cell lines via p75NGFR-mediated activation ofnuclear factor-�B. Mol Endocrinol 16:353–366
10. Descamps S, Toillon RA, Adriaenssens E, Pawlowski W,Cool SM, Nurcombe V, Le Bourhis X, Boilly B, Peyrat JP,Hondermarck H 2001 Nerve growth factor stimulatesproliferation and survival of human breast cancer cellsthrough two distinct signalling pathways. J Biol Chem276:17864–17870
11. Sortino MA, Condorelli F, Vancheri C, Chiarenza A, Ber-nardini R, Consoli U, Canonico PL 2000 Mitogenic effectof nerve growth factor (NGF) in LNCaP prostate adeno-carcinoma cells: role of the high- and low-affinity NGFreceptors. Mol Endocrinol 14:124–136
12. Hughes AL, Gollapudi L, Sladek TL, Neet KE 2000 Me-diation of nerve growth factor-driven cell cycle arrest inPC12 cells by p53. J Biol Chem 275:37829–37837
13. Decker SJ 1995 Nerve growth factor-induced growtharrest and induction of p21Cip1/WAF1 in NIH-3T3 cellsexpressing TrkA. J Biol Chem 270:30841–30844
14. Matsushima H, Bogenmann E 1993 Expression of trkAcDNA in neuroblastomas mediates differentiation in vitroand in vivo. Mol Cell Biol 13:7447–7456
15. Krygier S, Djakiew D 2002 Neurotrophin receptorp75(NTR) suppresses growth and nerve growth factor-mediated metastasis of human prostate cancer cells. IntJ Cancer 98:1–7
16. Khwaja F, Djakiew D 2003 Inhbition of cell cycle effectorsof proliferation in bladder tumor epithelial cells by thep75NTR tumor suppressor. Mol Carcinog 36:153–160
Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas Mol Endocrinol, January 2004, 18(1):162–172 171

17. Weis C, Wiesenhofer B, Humpel C 2002 Nerve growthfactor plays a divergent role in mediating growth of rat C6glioma cells via binding to the p75 neurotrophin receptor.J Neurooncol 56:59–67
18. Johnson DG, Walker CL 1999 Cyclins and cell cyclecheckpoints. Annu Rev Pharmacol Toxicol 39:295–312
19. Bullock AN, Fresht AR 2001 Rescuing the function ofmutant p53. Nat Rev Cancer 1:68–76
20. Webley KM, Shorthouse AJ, Royds JA 2000 Effect ofmutation and conformation on the function of p53 incolorectal cancer. J Pathol 191:361–367
21. Gaitonde SV, Riley JR, Qiao D, Martinez JD 2000 Con-formational phenotype of p53 is linked to nuclear trans-location. Oncogene 19:4042–4049
22. Moll UM, Riou G, Levine AJ 1992 Two distinct mecha-nisms alter p53 in breast cancer: mutation and nuclearexclusion. Proc Natl Acad Sci USA 89:7262–7266
23. Moll UM, Ostermeyer AG, Haladay AG, Windfield B, Fra-zier M, Zambetti 1996 Cytoplasmic sequestration of wildtype p53 protein impairs the G1 checkpoint after DNAdamage. Mol Cell Biol 16:1126–1137
24. Schlamp CL, Poulsen GL, Nork TM, Nickells RW 1997Nuclear exclusion of wild-type p53 in immortalized humanretinoblastoma cells. J Natl Cancer Inst 89:1530–1536
25. Ueda H, Ullrich SJ, Gangemi JD, Kappel CA, Ngo L, Fei-telson MA, Jay G 1995 Functional inactivation but not struc-tural mutation of p53 causes liver cancer. Nat Genet9:41–47
26. Nikolaev AY, Li M, Puskas N, Qin J, Gu W 2003 Parc: acytoplasmic anchor for p53. Cell 112:20–40
27. Herman V, Drazin NZ, Gonsky R, Melmed S 1993 Molecularscreening of pituitary adenomas for gene mutations andrearrangements. J Clin Endocrinol Metab 77:50–55
28. Levy A, Hall L, Yendall WA, Lightman SL 1994 p53 genemutations in pituitary adenomas: rare events. Clin Endo-crinol (Oxf) 41:809–814
29. Pei L, Melmed S, Scheithauer BW, Kovacs K, Prager D1994 H-ras mutations in human pituitary carcinoma me-tastasis. J Clin Endocrinol Metab 78:842–846
30. Eizenberg O, Faber-Elman A, Gottlieb E, Oren M, RotterV, Schwartz M 1996 p53 plays a regulatory role in differ-entiation and apoptosis of central nervous system-asso-ciated cells. Mol Cell Biol 16:5178–5185
31. Gollapudi L, Neet KE 1997 Different mechanisms forinhibition of cell proliferation via cell cycle proteins inPC12 cells by nerve growth factor and staurosporine.J Neurosci Res 49:461–474
32. Meplan C, Richard MJ, Hainaut P 2000 Redox signallingand transition metals in the control of the p53 pathway.Biochem Pharmacol 59:25–33
33. Verhaegh GW, Richard MJ, Hainaut P 1997 Regulation ofp53 by metal ions and by antioxidants: dithiocarbamatedown-regulates p53 DNA-binding activity by increasing theintracellular level of copper. Mol Cell Biol 17:5699–5706
34. Meplan C, Richard MJ, Hainaut P 2000 Metalloregulationof the tumor suppressor protein p53: zinc mediates therenaturation of p53 after exposure to metal chelators invitro and in intact cells. Oncogene 19:5227–5236
35. Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR 1995A synthetic inhibitor of the mitogen-activated protein kinasecascade. Proc Natl Acad Sci USA 92:7686–7689
36. Woscholski R, Kodaki T, Mc Kinnon M, Waterfield MD,Parker PS 1994 A comparison of demethoxyviridinand wortmannin as inhibitors of phosphatidylinositol3-kinase. FEBS Lett 342:109–114
37. Vivanco I, Sawyers CL 2002 The phosphatidylinositol3-kinase-Akt pathway in human cancer. Nat Rev Cancer2:489–501
38. Brown JP, Pagano M 1997 Mechanism of p53 degrada-tion. Biochim Biophys Acta 1332:O1–O6
39. Haupt Y, Maya R, Kazaz A, Oren M 1997 Mdm2 pro-motes the rapid degradation of p53. Nature 387:296–299
40. Fontoura BM, Atienza CA, Sorokina EA, Moromoto T,Carroll RB 1997 Cytoplasmic p53 polypeptide is associ-ated with ribosomes. Mol Cell Biol 17:3146–3154
41. Giannakakou P, Sackett DL, Ward Y, Webster KR, Bla-gosklonny MV, Fojo T 2000 p53 is associated with cel-lular microtubules and is transported to the nucleus bydynein. Nat Cell Biol 2:709–717
42. Klotzsche O, Etzrodt D, Hohenberg H, Bohn W, DeppertW 1998 Cytoplasmic retention of mutant tsp53 is depen-dent on an intermediate filament (vimentin) scaffold. On-cogene 16:3423–3434
43. Metcalfe S, Weeds A, Okorokov AL, Milner J, CockmanM, Pope B 1999 Wild-type p53 protein shows calcium-dependent binding to F-actin. Oncogene 18:2351–2355
44. Thapar K, Scheithauer BW, Kovacs K, Pernicone P, LawsER 1996 P53 expression in pituitary adenomas andcarcinomas: correlation with invasiveness and tumorgrowth fractions. Neurosurgery 38:765–771
45. Green VL, White MC, Hipkin LJ, Jeffreys TV, Foy PM,Atkin SL 1997 Apoptosis and p53 tumor suppressorgene protein expression in human anterior pituitary ad-enomas. Eur J Endocrinol 136:382–387
46. Suliman M, Royds J, Cullen D, Timperley W, Powell T,Battersby R, Jones TH 2001 Mdm2 and the p53 pathwayin human pituitary adenomas. Clin Endocrinol (Oxf) 54:317–325
47. Milner J 1995 Flexibility: the key to p53 function? TrendsBiochem Sci 20:49–51
48. Moll UM, LaQuaglia M, Bernard M, Riou G 1995 Wild-type p53 protein undergoes cytoplasmic sequestration inundifferentiated neuroblastomas but not in differentiatedtumors. Proc Natl Acad Sci USA 92:4407–4411
49. Isaacs JS, Hardman R, Carman TA, Barret JC, WeissmanBE 1998 Differential subcellular p53 localization andfunction in N- and S-type neuroblastoma cell lines. CellGrowth Differ 9:545–555
50. Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, LeeAKC 1995 p53 gene mutations, p53 protein accumula-tion and compartmentalization in colorectal adenocarci-noma. Am J Pathol 147:790–798
51. Weiss J, Heine M, Corner B, Pilch H, Jung EG 1995Expression of p53 protein in malignant melanoma: clin-icopathological and prognostic implications. Br J Derma-tol 133:23–31
52. Deleted in proof53. Mitsuuchi Y, Johnson SW, Selvakumaran M, Williams SJ,
Hamilton TC, Testa JR 2000 The phosphatidylinositol3-kinase/AKT signal transduction pathway plays a criticalrole in the expression of p21WAF1/CIP1/SDI1 induced bycisplatin and paclitaxel. Cancer Res 60:5390–5394
54. Fukuchi K, Watanabe H, Tomoyasu S, Ichimura S, Tat-sumi K, Gomi K 2000 Phosphatidylinositol 3-kinase in-hibitors, wortmannin or LY294002, inhibited accumula-tion of p21 protein after �-irradiation by stabilization ofthe protein. Biochim Biophys Acta 1496:207–220
55. Okaichi K, Suzuki K, Morita N, Ikeda M, Takhashi H, Mat-suda N, Watanabe M, Okumura Y 2002 Low dose of wort-mannin reduces radiosensitivity of human glioblastomacells through the p53 pathway. Oncol Rep 9:859–862
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
172 Mol Endocrinol, January 2004, 18(1):162–172 Facchetti et al. • NGF, trkA, and p53 Function in Prolactinomas
Related Documents











