Needleman-Wunsch with affine gaps Gap scores: (g)=-d-(g-1)e where d=2, e=1 Precedence: M, I x , I y M ( i , j )= max M ( i −1, j −1)+ s ( x i , y j ) I x ( i −1, j −1)+ s ( x i , y j ) I y ( i −1, j −1)+ s ( x i , y j ) ⎧ ⎨ ⎪ ⎩ ⎪ I x ( i , j )= max M ( i −1, j )− d I x ( i −1, j )− e ⎧ ⎨ ⎩ I y ( i , j )= max M ( i , j −1)− d I y ( i , j −1)− e ⎧ ⎨ ⎩ PAM 250 A C D A 2 -2 0 C 12 -5 D 4 Align the sequences: CA and DC

Needleman-Wunsch with affine gaps Gap scores: (g)=-d-(g-1)e where d=2, e=1 Precedence: M, I x, I y PAM 250 ACD A2-20 C12-5 D4 Align the sequences: CA.
Dec 22, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Needleman-Wunsch with affine gaps
Gap scores: (g)=-d-(g-1)e where d=2, e=1
Precedence: M, Ix, Iy
€
M(i, j) = max
M(i −1, j −1) + s(x i,y j )
Ix (i −1, j −1) + s(x i,y j )
Iy (i −1, j −1) + s(x i,y j )
⎧
⎨ ⎪
⎩ ⎪
Ix (i, j) = maxM(i −1, j) − d
Ix (i −1, j) − e
⎧ ⎨ ⎩
Iy (i, j) = maxM(i, j −1) − d
Iy (i, j −1) − e
⎧ ⎨ ⎩
PAM 250
A C D
A 2 -2 0
C 12 -5
D 4
Align the sequences: CA and DC

Multiple sequence alignment
Biology 162 Computational Genetics
Todd Vision2 Sep 2004

Preview• How to score a multiple alignment
– Sum of pairs scores– Weighting
• Generalizing pairwise alignment algorithms– Full dynamic programming– Carillo-Lipman
• Practical methods– Progressive– Iterative– Stochastic– Probabilistic
• Some final thoughts
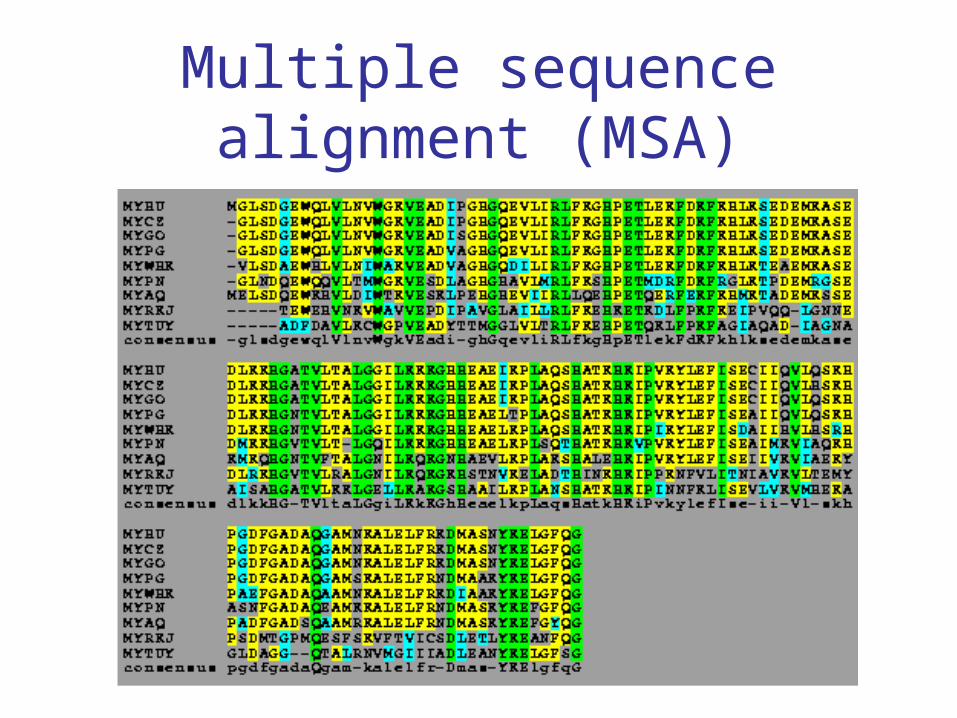
Multiple sequence alignment (MSA)

Mind the gaps
GCGGCCCA TCAGGTAGTT GGTGGGCGGCCCA TCAGGTAGTT GGTGGGCGTTCCA TCAGCTGGTT GGTGGGCGTCCCA TCAGCTAGTT GGTGGGCGGCGCA TTAGCTAGTT GGTGA******** ********** *****
TTGACATG CCGGGG---A AACCGTTGACATG CCGGTG--GT AAGCCTTGACATG -CTAGG---A ACGCGTTGACATG -CTAGGGAAC ACGCGTTGACATC -CTCTG---A ACGCG******** ?????????? *****
Trivial
Difficult
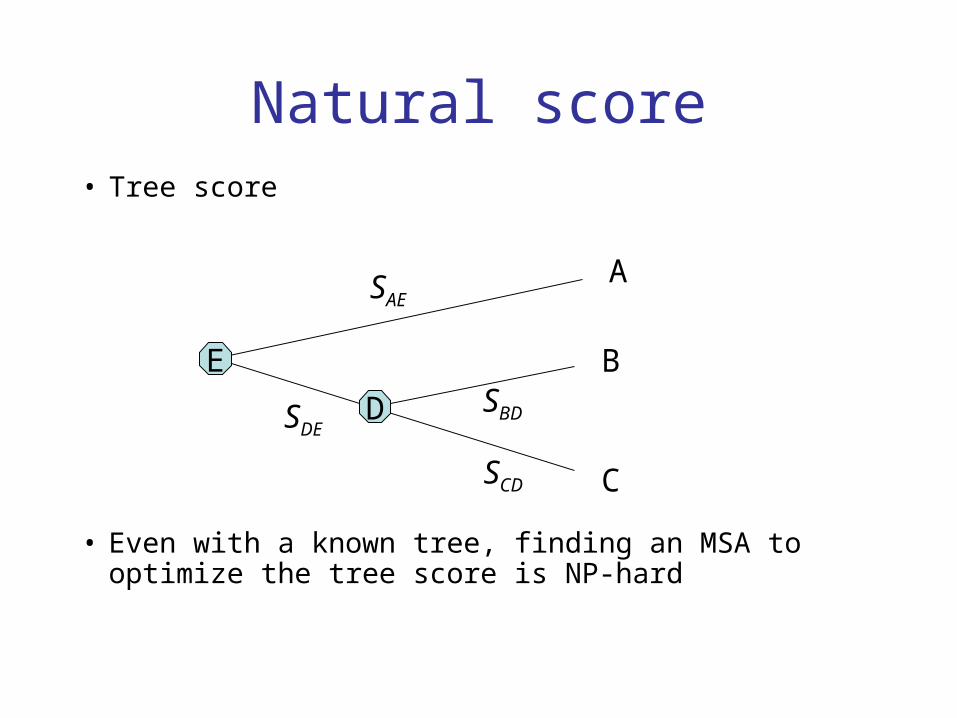
Natural score• Tree score
• Even with a known tree, finding an MSA to optimize the tree score is NP-hard
D
E
A
B
C
SAE
SDESBD
SCD
€
S=Sij
i,j are adjacent
∑

Star-tree scores
• Assume an unresolved phylogeny• Sum-of-pairs ()
• Entropy
• Consistency– Weighs agreement with external
evidence
€
S = s(ai,b j )i< j
∑
€
Scolumn = − qa ln2 qa( )a
∑

Entropy as used in a sequence logo

SP scores: pros and cons
• Pros– Easy, intuitive, work OK
• Cons– Substitution scores based on pairs of residues
– Inconsistent behavior with k• One mismatch matters more when k is large than
when k is small
– Gap penalties undefined for s(-,-)€
log(pab /qaqb ) + log(pac /qaqc ) + log(pbc /qbqc )
log(pabc /qaqbqc )

Natural gap penalties
• Gap costs in multiple alignment should be equal to sum of gap costs in induced pairwise alignments
• Computationally prohibitive to compute for most algorithms
• Instead, quasi-natural gap costs are computed– They are almost always identical

Weighted SP scores
• Scores are not independent due to (unaccounted for) shared ancestry
• To correct this, sum-of-pairs scores from related sequences can be down-weighted
• Variety of weighting schemes exist• Tree-based weighting is simplest
– Assign weights proportional to sum of branch lengths on a phylogenetic tree
– Obviously requires a tree (but we have an approximate tree in some algorithms)

Full dynamic programming
• We have k sequences of length n– Recursion equations are similar to pairwise case– We can use a simple extension of pairwise
scoring– As before, we can guarantee an optimal
alignment
• The problem is we must fill out a k-dimensional hypercube– Time and space grow exponentially in k– At least O(k22knk)– Computationally prohibitive even for a moderate
number of short sequences

Carillo-Lipman algorithm• Reduce volume of hypercube that is searched• Upper bound on score
– Score of optimal MSA is less than or equal to sum of scores of optimal pairwise alignments
• Lower bound on score– Score of optimal MSA must be greater or equal to
score of heuristic MSA
• Projections in each dimension defined by optimal pairwise alignments and induced heuristic alignments
• Optimum path is bounded by projections in all dimensions

Carillo-Lipman algorithm

Carillo-Lipman algorithm
• Only works for SP scoring function• Implemented in MSA software
– Can still only tackle small cases (eg 15 sequences of length 300)

Practical global alignment methods
• Progressive– Uses a guide tree to reduce the problem to
multiple pairwise alignments
• Iterative– Initialized with a fast multiple alignment– Sequences are randomly partitioned and
pairwise aligned until convergence
• Stochastic– Genetic algorithms as an example
• Probabilistic– Hidden Markov models

Progressive Alignment• Fast, but no guarantee of finding the
optimum • Implementations: Feng-Doolittle,
ClustalW, Pileup• Steps
– Compute all k(k-1)/2 pairwise alignments– Use alignment scores to construct guide tree– Perform pairwise alignments beginning at the
leaves of the guide tree and working toward the root

Pairwise score matrix
Sequence 1
Sequence 2
Sequence 3
Sequence 4
Sequence 5
Sequence 1
S12 S13 S14 S15
Sequence 2
S23 S24 S25
Sequence 3
S34 S35
Sequence 4
S45
Sequence 5

Sequence 2
Sequence 3
Sequence 4
Sequence 5
Sequence 1
Guide Tree
2
4
31

New Problem
• How to align a sequence to an alignment?• Or two alignments to each other?
• Feng-Doolittle solution– Choose highest scoring pair of sequences
between the two groups to guide the alignment
• ClustalW solution– Profile alignment: compute generalized sum
of pairs score

Profiles
Profile I
1 2 3 4 ---------- a w w w wpos c w w w w g w w w w t w w w w 1 1 1 1
Profile II
1 2 3 4 ---------- a w w w wpos c w w w w g w w w w t w w w w 1 1 1 1

ClustalW- ad hoc improvements
• Variable substitution matrix• Encourage gaps preferentially in structural loops
– Residue-specific gap penalties– Reduced penalties in hydrophilic regions
• Reduced gap penalties in positions already containing gaps
• Increased gap opening penalties in flanking sequence of gap

Progressive alignment: major weakness
• Errors introduced in the alignment of subgroups are propagated through all subsequent steps
• There is no provision for correcting such errors once they happen
• Local optimum versus global optimum

Iterative alignment
• Again capitalizes on the ease of pairwise alignment between groups of sequences
• Allows for gaps to be removed and positions to be shifted in each iteration
• Some algorithms guarantee convergence given long enough
• Can be several orders of magnitude slower than progressive methods
• Most successful implementation: PRRN
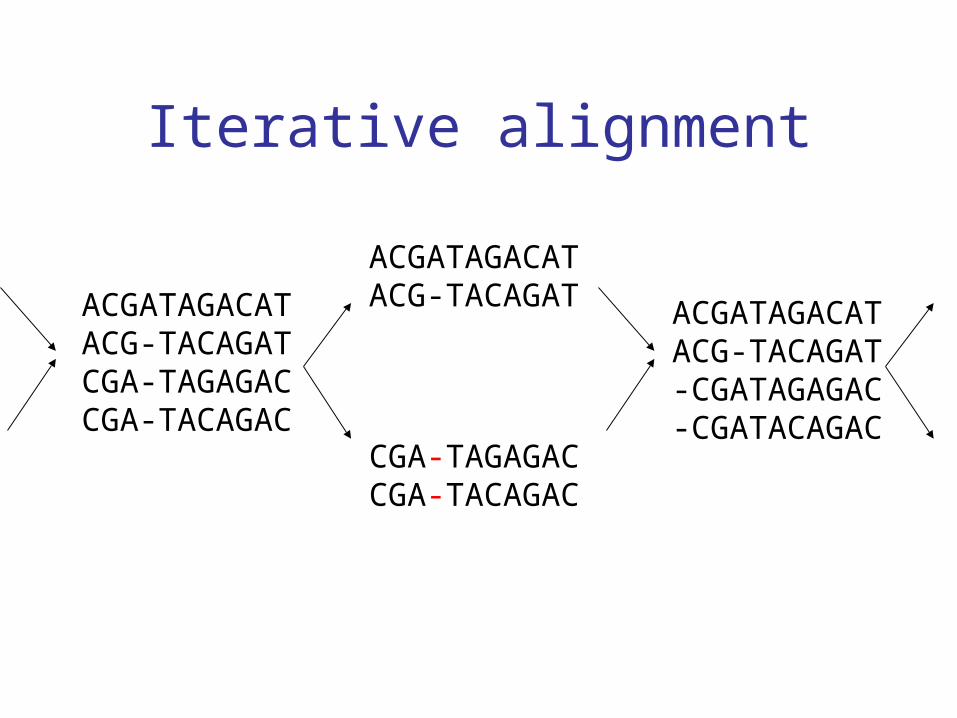
Iterative alignment
CGA-TAGAGACCGA-TACAGAC
ACGATAGACATACG-TACAGATACGATAGACAT
ACG-TACAGATCGA-TAGAGACCGA-TACAGAC
ACGATAGACATACG-TACAGAT-CGATAGAGAC-CGATACAGAC

T-COFFEE• Uses consistency as an objective
function– Evaluates consistency with pairs of residues
found in optimal local alignments and heuristic global alignment
• The consistency function can also incorporate extraneous information (such as structural constraints)
• Among the most successful of approaches when % identity is moderate to good

Dialign
• A multiple local alignment algorithm• Informally, it works by chaining
together ungapped segments from dotplots
• Does not explicitly score gaps at all• May contain unaligned regions
flanked by aligned regions

Stochastic methods
• Genetic algorithms (eg SAGA)– Initalize with population of heuristic alignments– Evaluate ‘fitness’ of individual alignments
• Can employ computationally intensive scoring functions
– Create new generation of alignments• Select parents according to fitness• ‘Cross-over’ attributes of parents• Apply mutation to perturb progeny alignments
– Return to ‘evaluate fitness’ step– Stopping rule

Probabilistic methods
• Hidden Markov Models– Models that generate MSAs– Many parameters to fit
• Probability of each residue in each column• Probability of entering gap states between columns
– Perform poorly on unaligned sequences– But are commonly used in signature databases
• Perform well for finding matches to already aligned sequences
• Efficient algorithms exist for aligning sequences to HMMs
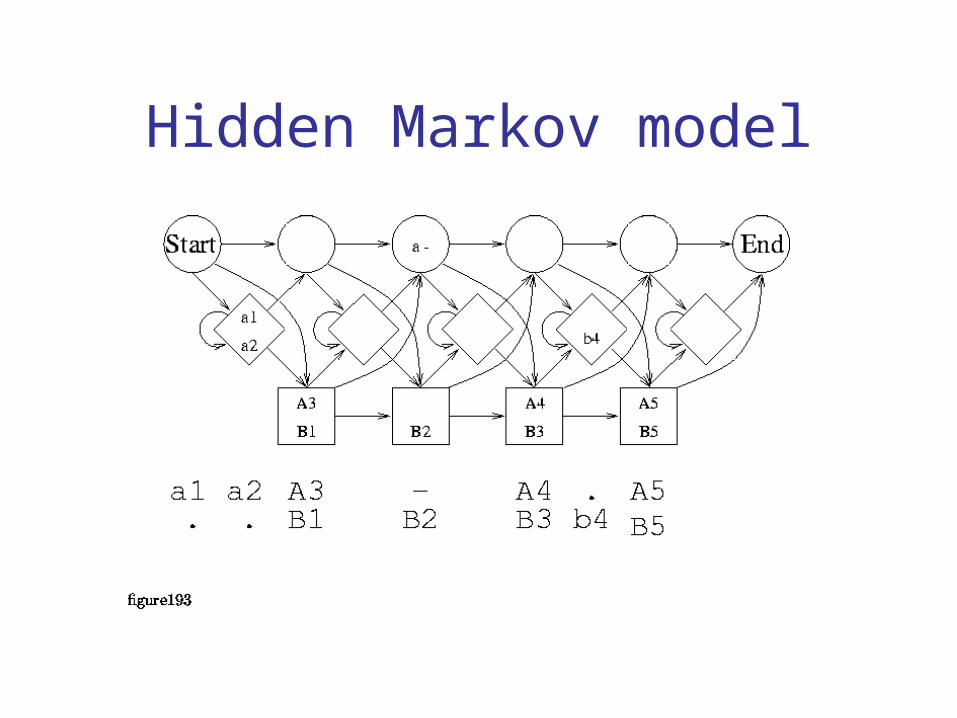
Hidden Markov model

How do you know when you’ve got the right
answer?• Short answer: you don’t.• Structural superposition typically
used to evaluate methodologies• BAliBASE: database of curated
reference alignments

Comparison of test and reference alignments
• Modified SP score– Frequency with which pairs of
residues aligned in test are aligned in reference
• Column score– Frequency with which entire columns
of residues are aligned in both test and reference

Be skeptical!
• MSA is a hard problem– Computationally– Biologically
• There is no ‘one size fits all’ algorithm
• No two algorithms need agree

The future of MSA
• Chances are your new sequence matches something already in the database
• It may soon be a rarity to generate an MSA from scratch– Signature databases currently allow
local alignment of a query to a pre-existing local multiple alignment (eg InterProScan)

Summary
• Challenges in MSA– Even bounded dynamic programming is
impractical – Appropriate scoring is not obvious
• How MSA is achieved in practice– Fastest
• Progressive pairwise alignment
– Slower• Iterative alignment• Stochastic alignment
• Automated MSAs require manual scrutiny

Reading Assignment
• Pertsemlidis A, Fondon JW (2002) Having a BLAST with bioinformatics (and avoiding BLASTphemy), 10 pgs.http://genomebiology.com/2001/2/10/reviews/2002.1
Related Documents











