EDITORIAL OFFICE London [email protected] The Macmillan Building, 4 Crinan Street, London N1 9XW, UK Tel: +44 (0)20 7843 3620; Fax: +44 (0)20 7843 3629 To subscribe and for more detailed information visit www.nature.com/reviews/drugdisc chief editor: Peter Kirkpatrick Senior editorS: Alexandra Flemming, Charlotte Harrison, Sarah Crunkhorn, Monica Hoyos Flight newS editor: Asher Mullard copy editor: Mariam Faruqi Senior copy editor (nrd): Man Tsuey Tse Senior copy editorS: Catriona Rodwell, Lucie Wootton copy editing Manager: Lewis Packwood art controLLer: Susanne Harris Senior art editorS: Vicky Summersby, Patrick Morgan, Kirsten Lee Managing production editor: Judith Shadwell Senior production editor: Simon Fenwick production controLLer: Natalie Smith Senior editoriaL aSSiStant: Laura Corns editoriaL aSSiStant: Ella Lines web production Manager: Deborah Anthony Marketing ManagerS: Tim Redding, Virginia Lee pubLiShing director: Peter Collins new york [email protected] Nature Publishing Group, 75 Varick Street, 9th floor, New York, NY 10013–1917, USA Tel: +1 212 726 9200; Fax: +1 212 696 9006 pubLiSher (biopharMa): Melanie Brazil cuStoMer ServiceS: [email protected] Copyright © 2012 Nature Publishing Group Research Highlight images courtesy of Getty Images unless otherwise credited. Printed in Wales by Cambrian Printers on acid-free paper. peter kirkpatrick aLexandra fLeMMing charLotte harriSon Monica hoyoS fLight aSher MuLLard Sarah crunkhorn EDITORS R ecent advances in RNA biology have accelerated the progress of a new generation of molecular therapies based on RNA, with several agents now in advanced clinical trials. In our first Review, Kole and colleagues compare and contrast the mechanisms of action and effects of three RNA-based therapeutic technologies — RNA interference, antisense oligonucleotides and steric-blocking oligonucleotides — and discuss their progress in the treatment of neuromuscular diseases, bacterial and viral infections, hypercholesterolaemia and cancer. A hallmark of tumour cells is an intrinsic or acquired resistance to apoptosis. This evasion of cell death is often aided by the abnormal expression of members of a family of anti-apoptotic proteins known as the inhibitor of apoptosis (IAP) proteins, which have been linked to tumour progression, treatment failure and poor prognosis in various cancers. In their Review, Fulda and Vucic provide an overview of IAP biology and discuss the therapeutic strategies that are being developed to target IAP proteins in human malignancies. Our final Review this month comprehensively discusses cognitive dysfunction in patients with psychiatric disorders, which is common and severely compromises the quality of life of patients but is largely not addressed by existing treatments that focus on emotional symptoms such as depression and anxiety. Millan and colleagues summarize the characteristics of cognitive dysfunction as well as the cerebral and cellular networks integrating and modulating cognition that are disrupted in psychiatric disorders. They also critically analyse current and emerging strategies for improving cognition in patients suffering from such diseases, and consider key challenges such as the development of more effective translational research approaches. Cognition in psychiatric disorders p141 RNA-based therapeutics p125 NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 87 IN THIS ISSUE © 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews Drug Discovery - February 2012
Oct 22, 2014
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

EDITORIAL OFFICE London [email protected] The Macmillan Building, 4 Crinan Street, London N1 9XW, UK Tel: +44 (0)20 7843 3620; Fax: +44 (0)20 7843 3629
To subscribe and for more detailed information visit www.nature.com/reviews/drugdisc
chief editor: Peter KirkpatrickSenior editorS: Alexandra Flemming, Charlotte Harrison, Sarah Crunkhorn, Monica Hoyos FlightnewS editor: Asher Mullardcopy editor: Mariam FaruqiSenior copy editor (nrd): Man Tsuey TseSenior copy editorS: Catriona Rodwell, Lucie Woottoncopy editing Manager: Lewis Packwoodart controLLer: Susanne HarrisSenior art editorS: Vicky Summersby, Patrick Morgan, Kirsten Lee
Managing production editor: Judith ShadwellSenior production editor: Simon Fenwickproduction controLLer: Natalie SmithSenior editoriaL aSSiStant: Laura CornseditoriaL aSSiStant: Ella Linesweb production Manager: Deborah AnthonyMarketing ManagerS: Tim Redding, Virginia LeepubLiShing director: Peter Collins
new york [email protected] Nature Publishing Group, 75 Varick Street, 9th floor, New York, NY 10013–1917, USA Tel: +1 212 726 9200; Fax: +1 212 696 9006
pubLiSher (biopharMa): Melanie Brazil
cuStoMer ServiceS: [email protected]
Copyright © 2012 Nature Publishing GroupResearch Highlight images courtesy of Getty Images unless otherwise credited.Printed in Wales by Cambrian Printers on acid-free paper.
peter kirkpatrick aLexandra fLeMMing charLotte harriSon
Monica hoyoS fLight aSher MuLLardSarah crunkhorn
EDITORS
Recent advances in RNA biology have accelerated the progress
of a new generation of molecular therapies based on RNA, with
several agents now in advanced clinical trials. In our first Review,
Kole and colleagues compare and contrast the mechanisms of
action and effects of three RNA-based therapeutic technologies — RNA
interference, antisense oligonucleotides and steric-blocking oligonucleotides
— and discuss their progress in the treatment of neuromuscular diseases,
bacterial and viral infections, hypercholesterolaemia and cancer.
A hallmark of tumour cells is an intrinsic or acquired resistance to apoptosis.
This evasion of cell death is often aided by the abnormal expression of
members of a family of anti-apoptotic proteins known as the inhibitor
of apoptosis (IAP) proteins, which have been linked to tumour progression,
treatment failure and poor prognosis in various cancers. In their Review,
Fulda and Vucic provide an overview of IAP biology and discuss the
therapeutic strategies that are being developed to target IAP proteins
in human malignancies. Our final Review this month comprehensively
discusses cognitive dysfunction in patients with psychiatric disorders,
which is common and severely compromises the quality of life of patients
but is largely not addressed by existing treatments that focus on emotional
symptoms such as depression and anxiety. Millan and colleagues summarize
the characteristics of cognitive dysfunction as well as the cerebral and
cellular networks integrating and modulating cognition that are disrupted
in psychiatric disorders. They also critically analyse current and emerging
strategies for improving cognition in patients suffering from such diseases,
and consider key challenges such as the development of more effective
translational research approaches.
Cognition in psychiatric disorders p141
RNA-based therapeutics p125
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 87
IN THIS ISSUE
© 2012 Macmillan Publishers Limited. All rights reserved

(Mis)treating the pharmacogenetic incidentalomeIsaac S. Kohane
Genome-wide screening is anticipated to accelerate the development of personalized medicine by identifying and exploiting new associations between genomic variants and drug responses. However, this goal could be undermined if care is not taken to minimize the impact of pharmacogenomic associations that turn out to have narrower implications than suggested by initial studies.
Isaac S. Kohane, M.D., Ph.D., is at the Harvard Medical School Center for Biomedical Informatics, 10 Shattuck Street, Boston, Massachusetts 02115, USA. e-mail: [email protected]:10.1038/nrd3659
Patients and health-care providers look forward to the era of precision medicine1, informed by molecular phenotypes, environmental modulators of physiology and a systems-orientated view of multiple pharmaco-logical interactions. Genome-wide screening technolo-gies are accelerating our advancement towards this era. However, at this early stage, the opportunities for being misled by multiple incidental genomics-based findings — the incidentalome2 — abound and are likely to grow. There are at least two ways in which the incidentalome can mislead and result in imprecise and potentially harmful medical practice: using genomic variants to identify a disease and using genomic variants to adjust drug dosage.
First, with regard to identifying the right disease, there is now a growing list of genomic variants that were once thought to have clinical significance and penetrance that would merit pharmacological or sur-gical intervention but have subsequently been found to have far less portent. Among the causes are incor-rect annotations and sequencing errors, both of which will probably be dramatically reduced by international efforts in the next few years. More problematic, how-ever, is the misleading nature of several mutations discovered within patient populations with a disease for which there are few studies in the general popu-lation. Failure to account for the genetic background and environmental modifiers that produced the origi-nal findings results in the mistaken impression that the probability of a patient having a specific mutation given a particular disease is of the same magnitude as the probability of the patient having the disease given that mutation.
This ascertainment error has already led to the over-turning of several hard-won beliefs about the highly penetrant nature of particular mutations. For exam-ple, the penetrance of hereditary haemochromatosis
protein (HFE) variants linked to clinically abnormal iron homeostasis was initially thought to be at least 80% but was found to be closer to 1% in the general population3. Other prominent examples include breast cancer 1 (BRCA1) and BRCA2 mutations associated with breast cancer risk whose documented penetrances have progressively decreased, and cystic fibrosis trans-membrane conductance regulator (CFTR) mutations that were previously thought to cause cystic fibrosis but have now been found (from new-born screening and follow-up studies) not to be associated with the disease4.
This genomic misidentification of disease is poten-tially harmful because risk-averse clinicians and patients may choose to proceed with therapies that are themselves morbid. For example, some women have undergone extensive surgery because they possessed variants in BRCA1 and BRCA2 that, over a decade after their identification, have still not been found to sig-nificantly increase the risk of breast cancer5. Similarly, long-term preventive pharmacotherapy — based on a genomic profile — with medications that are non-toxic in the short-term may present hazards after years of use. This may well result in clinical controversies analogous to those surrounding hormone replacement therapy, but scaled up by the multiplicity of individual-ized therapy regimens that are anticipated and increas-ingly seem likely.
Part of this problem could be solved in the future as more studies are conducted in the general popula-tion, accelerated by the use of electronic health record phenotyping6, to accurately establish the penetrance of genomic variants linked to disease. However, the prob-lem of multiple hypothesis testing is much thornier. Across the millions of variants measurable within a single human genome, even with 100% sensitivity and 99.9% specificity per test — which is an unrealistically
COMMENT
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 89
© 2012 Macmillan Publishers Limited. All rights reserved

optimistic estimate — most people will have at least one false positive test2. This challenge cannot be addressed by population studies alone, and will require the develop ment of a robust integrative systems-orientated medicine to accurately predict the physiological impli-cations of one or more variants.
The second main area in which the incidentalome could compromise personalized medicine is in the determination of the optimal dosage of drugs based on an analysis of genomic variants. Although most of the variance in the metabolism and clearance of some drugs, such as warfarin, can be captured by measuring a handful of genomic variants, this is not the case for many other drugs with narrow therapeutic windows. Nonetheless, there now appear to be thousands of vari-ants that can potentially further explain, and thereby allow the prediction of, the metabolism of specific drugs. For example, a recent review7 of copy number variants (CNVs) enumerated hundreds of CNVs across genes encoding proteins with known drug-metaboliz-ing activity, such as cytochrome P450-2D6 (CYP2D6), CYP2A6 and CYP11A1. In CYP2D6 alone, deletions ranged in prevalence from 2% to 34% in populations across the world, whereas the prevalence of duplica-tions ranged from 1% to 23%. Moreover, individuals who were empirically determined to be outlier metabo-lizers, such as poor metabolizers or ultra-metabolizers, were found to have a high rate of deletions and duplica-tions, respectively.
However, just as was the case for incidental findings of putative disease-causing variants, these findings do not necessarily mean that these CNVs will be predic-tive of metabolizer status. That is, the probability of a CNV in a metabolic gene in an individual who is an outlier metabolizer does not correspond to the prob-ability of an individual’s metabolizer status given that they possess that CNV. Many of the duplications may be non-functional, and other variants and/or regula-tory mechanisms may compensate for the CNV so that there is little net change in drug metabolism. Therefore using the variant alone to pick a dosage regimen may result in underdosing or toxic overdosing.
The potential solution to this problem is similar to that for the disease incidentalome, but more taxing: the effect of the mutation on drug metabolism and/or drug levels has to be empirically evaluated in the general population, and care taken to ensure that the observed differences are not related to confounders such as medi-cation compliance. Although robust systems-orientated medicine would be also helpful here, in the near term a reworking of post-marketing studies would be the most effective approach. That is, through information infrastructures such as the ‘Informed Cohort’8, patients would have the opportunity and ability to report on adverse events, share drug levels and genomic data from their health-care systems and receive expert-vetted rec-ommendations for dosage adjustment. This may well require the establishment of new mechanisms of col-laboration between the pharmaceutical industry and regulatory authorities. However, in the context of so many potentially useful therapies whose use could be seriously undermined by pharmacogenomic incidental findings, supporting timely mechanisms to gather the requisite data to determine the implications of genomic variants for drug dosage is essential for the attainment of precision medicine.
1. Committee on a Framework for Development of a New Taxonomy of Disease; National Research Council. Toward precision medicine: building a knowlege network for biomedical research and a new taxonomy of disease. (National Academies Press, Washington DC, 2011).
2. Kohane, I. S., Masys, D. R. & Altman, R. B. The incidentalome: a threat to genomic medicine. JAMA 296, 212–215 (2006).
3. Beutler, E. et al. Penetrance of 845G→A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 359, 211–218 (2002).
4. Thauvin-Robinet, C. et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J. Med. Genet. 46, 752–758 (2009).
5. Murray, M. L. et al. Follow-up of carriers of BRCA1 and BRCA2 variants of unknown significance: variant reclassification and surgical decisions. Genetics Med. 13, 998–1005 (2011).
6. Kohane, I. S. Using electronic health records to drive discovery in disease genomics. Nature Rev. Genet. 12, 417–428 (2011).
7. Johansson, I. & Ingelman-Sundberg, M. CNVs of human genes and their implication in pharmacogenetics. Cytogenetic Genome Res. 123, 195–204 (2008).
8. Kohane, I. S. et al. Medicine. Reestablishing the researcher-patient compact. Science 316, 836–837 (2007).
Competing interests statementThe author declares no competing financial interests.
90 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
C O M M E N T
© 2012 Macmillan Publishers Limited. All rights reserved

Globalization of clinical trials plateaus? p95
VEGF trap patent dispute p98
RuiPing Dong explains Merck’s Chinese R&D expansion p100
The neuropathic pain market p101
New drug approved for myelofibrosis p103
Asher Mullard
Last year the US Food and Drug Administration (FDA)’s Center for Drug Evaluation and Research (CDER) gave the green light to 24 new molecular entities and 6 new biologics. The approval of 30 new therapeutics is the most since 2004, which saw 36 products approved. The relative bumper crop, moreover, includes a substantial number of novel drugs that address major unmet medical needs, hit new targets and leverage the promise of genetic approaches to understanding disease.
“It is a really exciting list,” says Chris Milne, Associate Director
of the Tufts Center for the Study of Drug Development, in Boston, Massachusetts, USA. Andrew Jones, an analyst at Ernst & Young, agrees. “The thing to focus on is the level of innovation within the current crop of approvals,” he says. Among the stand-out statistics, he adds, is the approval of 11 first-in-class products.
Big winners among the companies involved included GlaxoSmithKline and Johnson & Johnson, which, with partners, both brought three new drugs to the market.
“In terms of approvals, I think the FDA did its job,” adds Eric Schmidt, an analyst at Cowen and Cowen. “The agency was engaged
in reviewing their drugs, in general they hit their timelines, and for the most part the decisions were not too surprising.” Nineteen of the approvals were granted to drugs in their first round of review.
Despite these positive signs for the industry, given the struggle over the past decade to get more new drugs to market, the usual caveats apply when looking for trends within the data set. “It is very difficult to read into 1 year’s numbers,” says Jones. Drug filings and approval decisions fluctuate from year to year because of financial concerns, unexpected clinical trial results and a host of other reasons. The long
2011 FDA drug approvalsThe US FDA approved 30 new therapeutics last year, including 11 first-in-class agents.
▶
Com
Stoc
k
NEWS & ANALYSIS
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 91
© 2012 Macmillan Publishers Limited. All rights reserved
Nature Reviews | Drug Discovery
Num
ber o
f dru
gs a
ppro
ved
60
50
40
30
20
10
01996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009
53
3
39
6
30
7
35
3
27
2
24
5
17
7
21
6
31
5
18
2
18
4
16
2
21
3
19
6
15
6
24
6
29
0
21
2
25
3
199519941993 2010 2011
New molecular entitiesBiologics license applications
development timelines and multifactorial review process also make it challenging, if not risky, to search for broader trends in research and development (R&D) productivity or pipeline strengths.
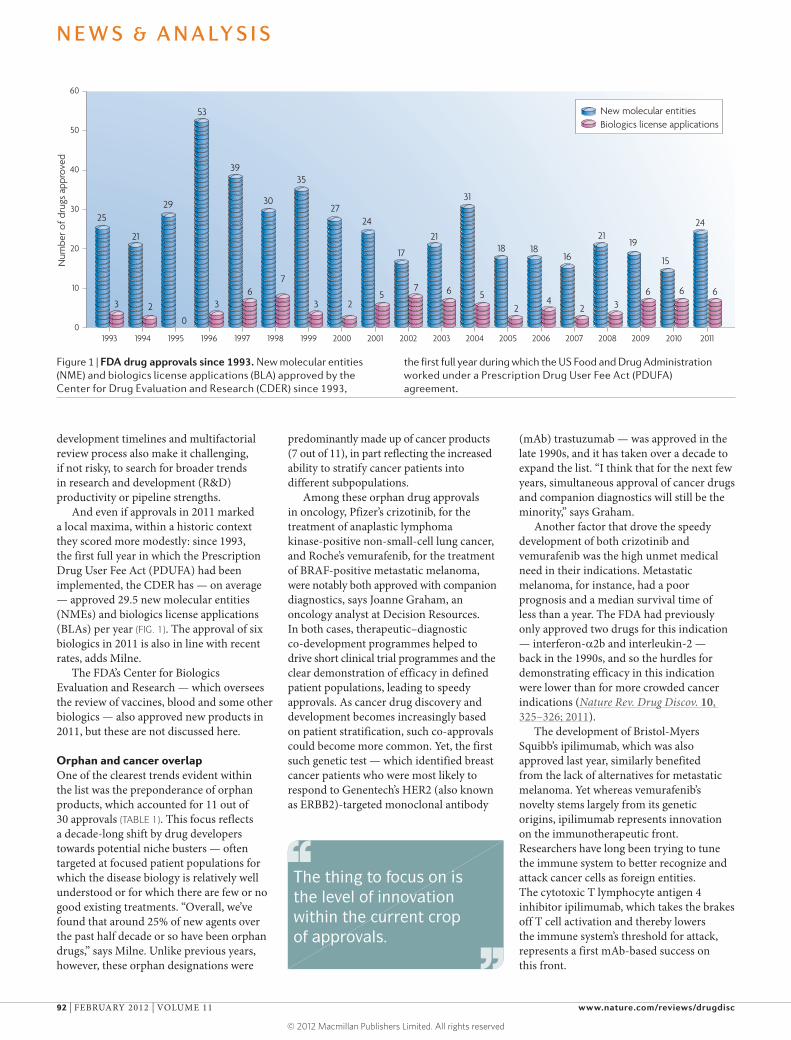
And even if approvals in 2011 marked a local maxima, within a historic context they scored more modestly: since 1993, the first full year in which the Prescription Drug User Fee Act (PDUFA) had been implemented, the CDER has — on average — approved 29.5 new molecular entities (NMEs) and biologics license applications (BLAs) per year (FIG. 1). The approval of six biologics in 2011 is also in line with recent rates, adds Milne.
The FDA’s Center for Biologics Evaluation and Research — which oversees the review of vaccines, blood and some other biologics — also approved new products in 2011, but these are not discussed here.
Orphan and cancer overlapOne of the clearest trends evident within the list was the preponderance of orphan products, which accounted for 11 out of 30 approvals (TABLE 1). This focus reflects a decade-long shift by drug developers towards potential niche busters — often targeted at focused patient populations for which the disease biology is relatively well understood or for which there are few or no good existing treatments. “Overall, we’ve found that around 25% of new agents over the past half decade or so have been orphan drugs,” says Milne. Unlike previous years, however, these orphan designations were
predominantly made up of cancer products (7 out of 11), in part reflecting the increased ability to stratify cancer patients into different subpopulations.
Among these orphan drug approvals in oncology, Pfizer’s crizotinib, for the treatment of anaplastic lymphoma kinase-positive non-small-cell lung cancer, and Roche’s vemurafenib, for the treatment of BRAF-positive metastatic melanoma, were notably both approved with companion diagnostics, says Joanne Graham, an oncology analyst at Decision Resources. In both cases, therapeutic–diagnostic co-development programmes helped to drive short clinical trial programmes and the clear demonstration of efficacy in defined patient populations, leading to speedy approvals. As cancer drug discovery and development becomes increasingly based on patient stratification, such co-approvals could become more common. Yet, the first such genetic test — which identified breast cancer patients who were most likely to respond to Genentech’s HER2 (also known as ERBB2)-targeted monoclonal antibody
(mAb) trastuzumab — was approved in the late 1990s, and it has taken over a decade to expand the list. “I think that for the next few years, simultaneous approval of cancer drugs and companion diagnostics will still be the minority,” says Graham.
Another factor that drove the speedy development of both crizotinib and vemurafenib was the high unmet medical need in their indications. Metastatic melanoma, for instance, had a poor prognosis and a median survival time of less than a year. The FDA had previously only approved two drugs for this indication — interferon-a2b and interleukin-2 — back in the 1990s, and so the hurdles for demonstrating efficacy in this indication were lower than for more crowded cancer indications (Nature Rev. Drug Discov. 10, 325–326; 2011).
The development of Bristol-Myers Squibb’s ipilimumab, which was also approved last year, similarly benefited from the lack of alternatives for metastatic melanoma. Yet whereas vemurafenib’s novelty stems largely from its genetic origins, ipilimumab represents innovation on the immunotherapeutic front. Researchers have long been trying to tune the immune system to better recognize and attack cancer cells as foreign entities. The cytotoxic T lymphocyte antigen 4 inhibitor ipilimumab , which takes the brakes off T cell activation and thereby lowers the immune system’s threshold for attack, represents a first mAb-based success on this front.
Figure 1 | FDA drug approvals since 1993. New molecular entities (NME) and biologics license applications (BLA) approved by the Center for Drug Evaluation and Research (CDER) since 1993,
the first full year during which the US Food and Drug Administration worked under a Prescription Drug User Fee Act (PDUFA) agreement.
The thing to focus on is the level of innovation within the current crop of approvals.
N E W S & A N A LY S I S
92 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
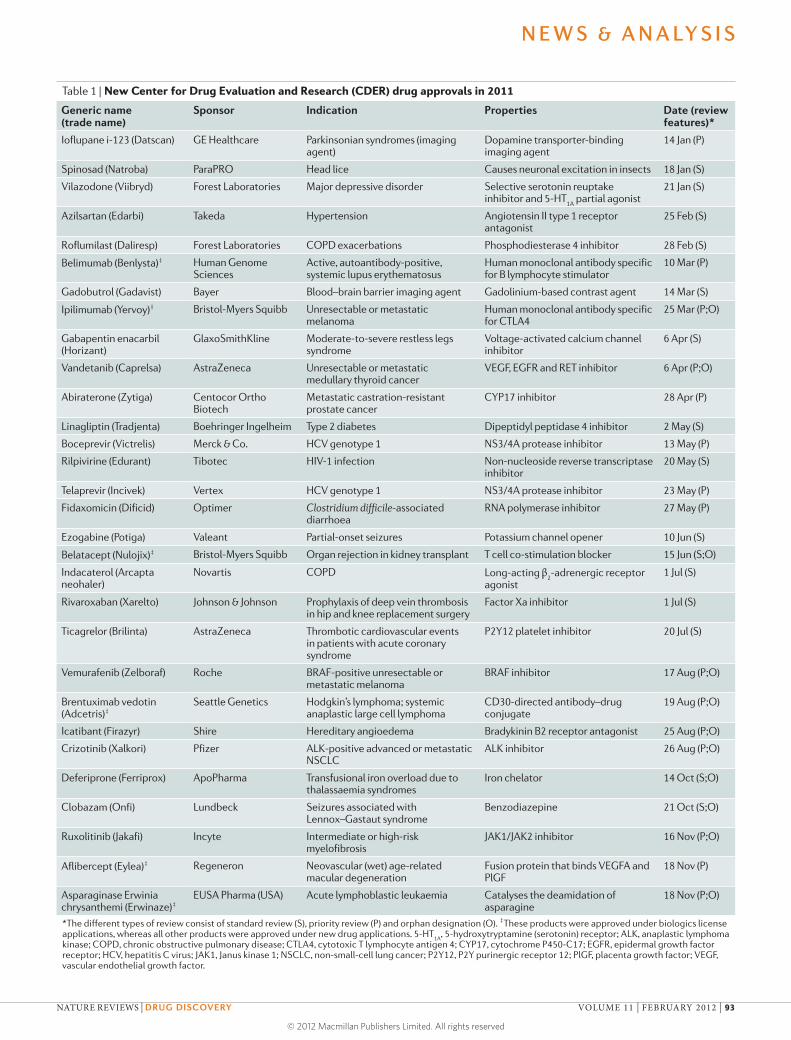
Table 1 | New Center for Drug Evaluation and Research (CDER) drug approvals in 2011
Generic name (trade name)
Sponsor Indication Properties Date (review features)*
Ioflupane i-123 (Datscan) GE Healthcare Parkinsonian syndromes (imaging agent)
Dopamine transporter-binding imaging agent
14 Jan (P)
Spinosad (Natroba) ParaPRO Head lice Causes neuronal excitation in insects 18 Jan (S)
Vilazodone (Viibryd) Forest Laboratories Major depressive disorder Selective serotonin reuptake inhibitor and 5-HT
1A partial agonist
21 Jan (S)
Azilsartan (Edarbi) Takeda Hypertension Angiotensin II type 1 receptor antagonist
25 Feb (S)
Roflumilast (Daliresp) Forest Laboratories COPD exacerbations Phosphodiesterase 4 inhibitor 28 Feb (S)
Belimumab (Benlysta)‡ Human Genome Sciences
Active, autoantibody-positive, systemic lupus erythematosus
Human monoclonal antibody specific for B lymphocyte stimulator
10 Mar (P)
Gadobutrol (Gadavist) Bayer Blood–brain barrier imaging agent Gadolinium-based contrast agent 14 Mar (S)
Ipilimumab (Yervoy)‡ Bristol-Myers Squibb Unresectable or metastatic melanoma
Human monoclonal antibody specific for CTLA4
25 Mar (P;O)
Gabapentin enacarbil (Horizant)
GlaxoSmithKline Moderate-to-severe restless legs syndrome
Voltage-activated calcium channel inhibitor
6 Apr (S)
Vandetanib (Caprelsa) AstraZeneca Unresectable or metastatic medullary thyroid cancer
VEGF, EGFR and RET inhibitor 6 Apr (P;O)
Abiraterone (Zytiga) Centocor Ortho Biotech
Metastatic castration-resistant prostate cancer
CYP17 inhibitor 28 Apr (P)
Linagliptin (Tradjenta) Boehringer Ingelheim Type 2 diabetes Dipeptidyl peptidase 4 inhibitor 2 May (S)
Boceprevir (Victrelis) Merck & Co. HCV genotype 1 NS3/4A protease inhibitor 13 May (P)
Rilpivirine (Edurant) Tibotec HIV-1 infection Non-nucleoside reverse transcriptase inhibitor
20 May (S)
Telaprevir (Incivek) Vertex HCV genotype 1 NS3/4A protease inhibitor 23 May (P)
Fidaxomicin (Dificid) Optimer Clostridium difficile-associated diarrhoea
RNA polymerase inhibitor 27 May (P)
Ezogabine (Potiga) Valeant Partial-onset seizures Potassium channel opener 10 Jun (S)
Belatacept (Nulojix)‡ Bristol-Myers Squibb Organ rejection in kidney transplant T cell co-stimulation blocker 15 Jun (S;O)
Indacaterol (Arcapta neohaler)
Novartis COPD Long-acting β2-adrenergic receptor
agonist1 Jul (S)
Rivaroxaban (Xarelto) Johnson & Johnson Prophylaxis of deep vein thrombosis in hip and knee replacement surgery
Factor Xa inhibitor 1 Jul (S)
Ticagrelor (Brilinta) AstraZeneca Thrombotic cardiovascular events in patients with acute coronary syndrome
P2Y12 platelet inhibitor 20 Jul (S)
Vemurafenib (Zelboraf) Roche BRAF-positive unresectable or metastatic melanoma
BRAF inhibitor 17 Aug (P;O)
Brentuximab vedotin (Adcetris)‡
Seattle Genetics Hodgkin’s lymphoma; systemic anaplastic large cell lymphoma
CD30-directed antibody–drug conjugate
19 Aug (P;O)
Icatibant (Firazyr) Shire Hereditary angioedema Bradykinin B2 receptor antagonist 25 Aug (P;O)
Crizotinib (Xalkori) Pfizer ALK-positive advanced or metastatic NSCLC
ALK inhibitor 26 Aug (P;O)
Deferiprone (Ferriprox) ApoPharma Transfusional iron overload due to thalassaemia syndromes
Iron chelator 14 Oct (S;O)
Clobazam (Onfi) Lundbeck Seizures associated with Lennox–Gastaut syndrome
Benzodiazepine 21 Oct (S;O)
Ruxolitinib (Jakafi) Incyte Intermediate or high-risk myelofibrosis
JAK1/JAK2 inhibitor 16 Nov (P;O)
Aflibercept (Eylea)‡ Regeneron Neovascular (wet) age-related macular degeneration
Fusion protein that binds VEGFA and PlGF
18 Nov (P)
Asparaginase Erwinia chrysanthemi (Erwinaze)‡
EUSA Pharma (USA) Acute lymphoblastic leukaemia Catalyses the deamidation of asparagine
18 Nov (P;O)
*The different types of review consist of standard review (S), priority review (P) and orphan designation (O). ‡These products were approved under biologics license applications, whereas all other products were approved under new drug applications. 5-HT
1A, 5-hydroxytryptamine (serotonin) receptor; ALK, anaplastic lymphoma
kinase; COPD, chronic obstructive pulmonary disease; CTLA4, cytotoxic T lymphocyte antigen 4; CYP17, cytochrome P450-C17; EGFR, epidermal growth factor receptor; HCV, hepatitis C virus; JAK1, Janus kinase 1; NSCLC, non-small-cell lung cancer; P2Y12, P2Y purinergic receptor 12; PlGF, placenta growth factor; VEGF, vascular endothelial growth factor.
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 93
© 2012 Macmillan Publishers Limited. All rights reserved

Another treatment modality advance came with the approval of Seattle Genetics’s brentuximab vedotin for the treatment of Hodgkin’s lymphoma and systemic anaplastic large cell lymphoma (Nature Rev. Drug Discov. 11, 19–20; 2012). Brentuximab vedotin is an antibody–drug conjugate in which a CD30-targeting mAb is attached to an antimitotic monomethyl auristatin E warhead, improving the specificity of the chemotherapeutic. The only other antibody–drug conjugate to ever get the green light in the United States is Wyeth’s (now Pfizer) gemtuzumab ozogamicin, which was approved in 2000 for acute myelogenous leukaemia but withdrawn in 2010 owing to lack of efficacy.
The only cancer product that was approved in 2011 without an orphan designation was Johnson & Johnson’s abiraterone, for prostate cancer (Nature Rev. Drug Discov. 10, 573–574; 2011). Given the high prevalence of this form of cancer, and the treatment duration, adds Graham, abiraterone is one of the few oncology drugs that “definitely has blockbuster potential”.
Despite the large cohort of cancer drugs last year, Milne notes that approvals in this therapeutic area seem disproportionally small when compared to its originating pipeline. “Cancer drugs tend to account for 35 –40% of the pipeline, and yet here we see they received under 30% of approvals last year,” he notes. “Cancer is looking a little tougher in terms of the metrics of success, time and money.”
Other major therapeutic areas also won a handful of approvals. In cardiology, the agency approved AstraZeneca’s platelet inhibitor ticagrelor for acute coronary syndromes, Johnson & Johnson’s factor Xa inhibitor rivaroxaban for prevention of deep vein thrombosis in adults undergoing hip and knee replacement surgery, and Takeda’s azilsartan as the eighth angiotensin II receptor blocker for hypertension. New respiratory products include Forest’s first-in-class phosphodiesterase 4 inhibitor roflumilast and Novartis’s long-acting β2-adrenergic receptor agonist indacaterol,
both for the treatment of chronic obstructive pulmonary disease.
Biotech bumpsMost of the sponsors submitting new drug applications for last year’s crop of approvals were large pharmaceutical companies, but biotech companies nevertheless succeeded in bringing some products to market in 2011 as well.
Vertex’s NS3/4A protease inhibitor telaprevir for the treatment of hepatitis C virus (HCV), for instance, was one of the most highly anticipated approvals in the history of biotech. It marked both the first major success for the company, founded in 1989. The drug — alongside Merck’s boceprevir, which was approved just a few days ahead of telaprevir and has the same mechanism of action — could also change the face of HCV therapy for million of patients who have previously avoided treatment because of low efficacy rates and high side-effect burdens.
“Those drugs are revolutionary and will change the course of HCV treatment forever,” says Schmidt. Both have been pegged as potential blockbusters.
Another biotech success, from a scientific perspective at least, was Human Genome Sciences’s B lymphocyte stimulator-targeting mAb belimumab, co-developed with GlaxoSmithKline. The immune-dampening mAb is the first new drug for the treatment of systemic lupus erythematosus in 50 years, and has cleared a clinical trial pathway for others to follow (Nature Rev. Drug Discov. 10, 243–245; 2011). It was also one of the early fruits of the genetic data deluge that started in the 1990s, highlighting the potential of genetics as a tool for drug discovery efforts.
Despite the therapeutic’s clinical and scientific novelty, however, its launch was a flop (sales repeatedly missed forecasts, and the company was forced to cut 150 jobs in January). Unfortunately, says Schmidt, poor launches have been the rule, rather than the exception, for biotech’s newcomers in 2011. Aside from telaprevir and products that were launched late in the year and so have not yet reported sales data, he says all the biotech launches have been “very poor”. “It may be that investors’ expectations were too high, that physicians don’t perceive the benefit the same way investors do, or just that it takes longer to get started with a drug launch than it used to.”
He holds out hope, however, that this won’t be the case for Incyte’s ruxolitinib, a first-in-class Janus kinase 1 (JAK1)/JAK2
inhibitor that was approved late in the year for myelofibrosis. “I do think that it is the one biotech drug from last year — along with telaprevir — that is truly differentiated, truly novel and will have a major impact on patients.”
In store for 2012“The number of new approvals is unlikely to be as high in 2012,” says Jones. The agency approved an unusually high percentage of NME and new-BLA candidates last year (>80%), he says, compared with recent precedents (around 50%). “In the absence of a significant upswing in the volume of drugs being reviewed, and assuming that the percentage of approvals reverts to recent norms, 2012 is likely to be a leaner year.”
Nevertheless, there are still some exciting potential stories.
Two obesity drugs — Arena’s lorcaserin and Vivus’s combination of phentermine and topiramate — will both have another attempt at approval, after closely followed rejections last year. Vertex’s ivacaftor, meanwhile, could become the first cystic fibrosis drug to target the underlying cause, rather than symptoms, of the disease (Nature Rev. Drug Discov. 10, 479–480; 2011).
On the cancer front, Graham highlights Genentech’s HER2-targeting pertuzumab as a possible contender. Genentech has submitted a BLA for the mAb — which binds to a different epitope compared to trastuzumab — for approval in the HER2-positive breast cancer setting, but hopes the therapeutic may eventually prove to be effective in HER2-negative patients as well. An FDA decision is expected in October 2012.
In the biotech space, Schmidt’s top picks for new 2012 approvals are Biogen Idec’s BG-12 and Medivation’s MDV3100. The small-molecule immunomodulator BG-12 is due to be filed for the treatment of relapsing–remitting multiple sclerosis in the first quarter of the year, ahead of a possible approval by the end of the year. A filing for Medivation’s oral androgen receptor signalling inhibitor MDV3100 in prostate cancer is also expected shortly, potentially presenting a competitor for abiraterone.
The number of new approvals is unlikely to be as high in 2012.
Cancer is looking a little tougher in terms of the metrics of success, time and money.
N E W S & A N A LY S I S
94 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
Nature Reviews | Drug Discovery
Loca
tion
of c
linic
al tr
ials
(%)
0
10
20
30
40
50
60
70
80
90
100
2000 2004 2008 2012 (projected)
North America Western Europe Rest of world
NEWS IN BRIEF
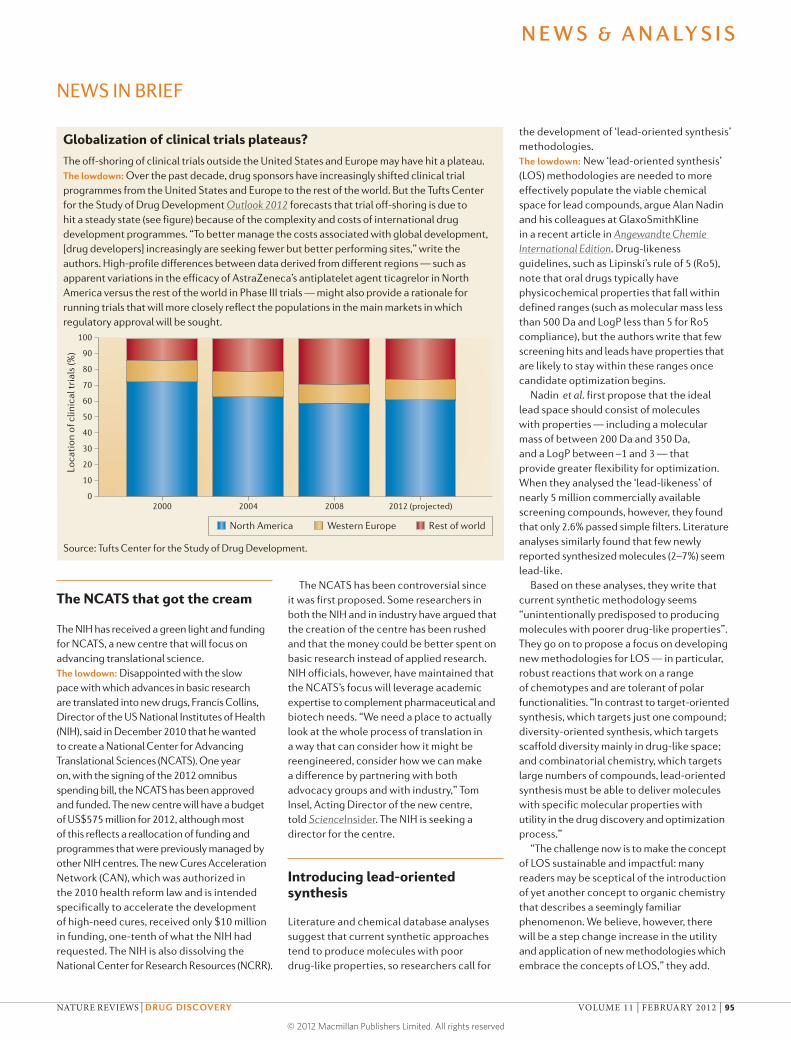
Globalization of clinical trials plateaus?The off-shoring of clinical trials outside the United States and Europe may have hit a plateau.The lowdown: Over the past decade, drug sponsors have increasingly shifted clinical trial programmes from the United States and Europe to the rest of the world. But the Tufts Center for the Study of Drug Development Outlook 2012 forecasts that trial off-shoring is due to hit a steady state (see figure) because of the complexity and costs of international drug development programmes. “To better manage the costs associated with global development, [drug developers] increasingly are seeking fewer but better performing sites,” write the authors. High-profile differences between data derived from different regions — such as apparent variations in the efficacy of AstraZeneca’s antiplatelet agent ticagrelor in North America versus the rest of the world in Phase III trials — might also provide a rationale for running trials that will more closely reflect the populations in the main markets in which regulatory approval will be sought.
Source: Tufts Center for the Study of Drug Development.
the development of ‘lead-oriented synthesis’ methodologies.The lowdown: New ‘lead-oriented synthesis’ (LOS) methodologies are needed to more effectively populate the viable chemical space for lead compounds, argue Alan Nadin and his colleagues at GlaxoSmithKline in a recent article in Angewandte Chemie International Edition. Drug-likeness guidelines, such as Lipinski’s rule of 5 (Ro5), note that oral drugs typically have physicochemical properties that fall within defined ranges (such as molecular mass less than 500 Da and LogP less than 5 for Ro5 compliance), but the authors write that few screening hits and leads have properties that are likely to stay within these ranges once candidate optimization begins.
Nadin et al. first propose that the ideal lead space should consist of molecules with properties — including a molecular mass of between 200 Da and 350 Da, and a LogP between –1 and 3 — that provide greater flexibility for optimization. When they analysed the ‘lead-likeness’ of nearly 5 million commercially available screening compounds, however, they found that only 2.6% passed simple filters. Literature analyses similarly found that few newly reported synthesized molecules (2–7%) seem lead-like.
Based on these analyses, they write that current synthetic methodology seems “unintentionally predisposed to producing molecules with poorer drug-like properties”. They go on to propose a focus on developing new methodologies for LOS — in particular, robust reactions that work on a range of chemotypes and are tolerant of polar functionalities. “In contrast to target-oriented synthesis, which targets just one compound; diversity-oriented synthesis, which targets scaffold diversity mainly in drug-like space; and combinatorial chemistry, which targets large numbers of compounds, lead-oriented synthesis must be able to deliver molecules with specific molecular properties with utility in the drug discovery and optimization process.”
“The challenge now is to make the concept of LOS sustainable and impactful: many readers may be sceptical of the introduction of yet another concept to organic chemistry that describes a seemingly familiar phenomenon. We believe, however, there will be a step change increase in the utility and application of new methodologies which embrace the concepts of LOS,” they add.
The NCATS has been controversial since it was first proposed. Some researchers in both the NIH and in industry have argued that the creation of the centre has been rushed and that the money could be better spent on basic research instead of applied research. NIH officials, however, have maintained that the NCATS’s focus will leverage academic expertise to complement pharmaceutical and biotech needs. “We need a place to actually look at the whole process of translation in a way that can consider how it might be reengineered, consider how we can make a difference by partnering with both advocacy groups and with industry,” Tom Insel, Acting Director of the new centre, told ScienceInsider. The NIH is seeking a director for the centre.
Introducing lead-oriented synthesis
Literature and chemical database analyses suggest that current synthetic approaches tend to produce molecules with poor drug-like properties, so researchers call for
The NCATS that got the cream
The NIH has received a green light and funding for NCATS, a new centre that will focus on advancing translational science.The lowdown: Disappointed with the slow pace with which advances in basic research are translated into new drugs, Francis Collins, Director of the US National Institutes of Health (NIH), said in December 2010 that he wanted to create a National Center for Advancing Translational Sciences (NCATS). One year on, with the signing of the 2012 omnibus spending bill, the NCATS has been approved and funded. The new centre will have a budget of US$575 million for 2012, although most of this reflects a reallocation of funding and programmes that were previously managed by other NIH centres. The new Cures Acceleration Network (CAN), which was authorized in the 2010 health reform law and is intended specifically to accelerate the development of high-need cures, received only $10 million in funding, one-tenth of what the NIH had requested. The NIH is also dissolving the National Center for Research Resources (NCRR).
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 95
© 2012 Macmillan Publishers Limited. All rights reserved

In one of the largest preclinical-stage deals ever, Abbott has agreed to pay US$400 million upfront to Reata Pharmaceuticals as part of an agreement to jointly develop and commercialize a series of second-generation oral antioxidant inflammation modulators (AIMs) with potential applications in cardiovascular disease, neurodegenerative disorders and immunology. Abbott and Reata, which signed a $450 million deal in September 2010 on Reata’s first-generation AIM bardoxolone methyl (now in Phase III trials), will share costs and profits on the preclinical AIMs in all newly licensed indications, except for selected autoimmune diseases such as rheumatoid arthritis. The first clinical study of an agent covered by the latest deal is expected to begin later this year.
Oxidative stress and inflammation are intimately interrelated and are common manifestations and mediators of many chronic disorders. Although strategies aimed at reducing oxidative stress to alleviate or prevent various diseases have been widely investigated, success so far has been elusive. “The past 40 years have been frustrating, as supplementation with direct antioxidants has failed, as has administration of specific individual antioxidant enzymes, or their mimetics, as drugs,” notes Professor Joe McCord, University of Colorado, USA.
Reata has pursued an alternative approach for reducing oxidative stress. Its AIMs act by potently activating NFE2-related factor 2 (NRF2), a ubiquitously expressed transcription factor that controls the expression of various genes involved in the oxidative stress response. “While NRF2 was originally thought to be primarily a regulator of the antioxidant enzymes, it is now known to participate in the regulation of many genes responsible for other stress-related processes such as inflammation and fibrosis, neurodegeneration and addictive behaviour, cancer chemoprevention, metastasis and drug resistance,” explains McCord. So, the activation of NRF2 may be beneficial in numerous disorders. “Because NRF2 production appears to decline with ageing, while free radical production increases, the regulation of NRF2 may be key to the management of the so-called ‘diseases of ageing’, which include cardiovascular disease, neurodegenerative diseases, cancer, type 2 diabetes and chronic failure of the kidneys and heart,” adds McCord.
Indeed, Reata’s bardoxolone methyl is showing promise in the treatment of advanced chronic kidney disease (CKD), a common disorder that is caused by conditions including high blood pressure and diabetes and for which current treatment options are limited. “Despite full adherence to the current standards of care, patients with severe CKD still progress to
end-stage renal disease (ESRD) and, especially in patients with diabetes, suffer increased risk for cardiovascular death and events,” notes Professor David Warnock, University of Alabama at Birmingham, USA, and Senior Medical Advisor and Consultant to Reata.
Bardoxolone methyl has successfully completed Phase II trials, including a study known as BEAM in which 227 patients with moderate to severe CKD and type 2 diabetes who were treated for 52 weeks with the AIM experienced a sustained improvement in kidney function (N. Engl. J. Med. 365, 327–336; 2011). Importantly, side effects were generally mild. However, “while the overall safety profile is encouraging at present, there is always the possibility that some unanticipated untoward effect may become evident when a large population of patients are exposed to this new class of agents”, cautions Warnock. The Phase III BEACON trial of bardoxolone methyl in patients with stage 4 CKD and type 2 diabetes is currently underway. “We should know within the next 2 years whether or not this new treatment approach makes an important contribution to reducing the occurrence of ESRD or cardiovascular death in this high-risk group of patients,” says Warnock.
The second-generation AIMs are anticipated to have several applications. However, as McCord notes: “It is unlikely that any NRF2-activating drug will achieve a ‘one size fits all’ status. A family of NRF2-activating drugs and dietary supplements will probably emerge to deal with the therapeutic and regulatory challenges of acute versus chronic versus preventative applications.”
Sarah Crunkhorn
D E A L WAT C H
Abbott boosts investment in NRF2 activators for reducing oxidative stress
N E W S & A N A LY S I S
BIOBUSINESS BRIEFS
96 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

BIOBUSINESS BRIEFS
Preliminary clinical trial data recently presented at the American Society of Hematology meeting showed that the Bruton’s tyrosine kinase (BTK) inhibitor PCI‑32765 was effective in treating in several types of B cell lymphoma. Furthermore, PCI‑32765 is at the centre of a deal — worth up to US$975 million — between the drug’s developer Pharmacyclics and Janssen Biotech.
“BTK is involved in signal transduction in the B cell receptor signalling pathway,” explains Simon Rule, a consultant haematologist at the Derriford Hospital, Plymouth, UK, “so this is a logical place to target B cells.”
As Louis Staudt, Chief of the lymphoid malignancies section at the Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, USA, further details, PCI‑32765 attacks a biological organizing principle of both chronic lymphocytic leukaemia and mantle cell lymphoma (both of which are types of B cell lymphomas). “A great deal of compelling — albeit circumstantial — evidence suggests that B cell receptor signalling is central to the pathogenesis of chronic lymphocytic leukaemia. The leukaemic cells often use highly related immunoglobulin variable regions in their B cell receptors, suggesting that they are reacting with either a self or foreign antigen. More recently, similar circumstantial evidence for the involvement
of B cell receptor signalling in mantle cell lymphoma has been reported.”
Interim analysis of a Phase II study in patients with relapsed or refractory mantle cell lymphoma showed that PCI‑32765 produced an objective response rate of 67% (16 out of 24 patients); in patients who had previously received the protease inhibitor bortezomib the objective response rate was 75%, compared with 58% in bortezomib‑naive patients.
According to Rule, who was an investigator on the trial, these results are very encouraging: “The response rates in the study of mantle cell lymphoma are better than with any other single‑agent drug yet described in the treatment of this disease. Many of these patients are heavily pretreated, so to have such a high response rate is stunning.”
In addition, in a Phase Ib/II follow‑up trial in patients with relapsed or refractory chronic lymphocytic leukaemia or small lymphocytic lymphoma, PCI‑32765 produced an objective response rate of 70%. Furthermore, in individuals with activated B cell‑like diffuse large B cell lymphoma — the most aggressive form of diffuse large B cell lymphoma — the drug induced tumour regression (2 complete responses and 1 partial response out of 9 relapsed or refractory patients).
Notably, PCI‑32765 binds irreversibly to its target, which could explain its positive effects. “The irreversible nature of PCI‑32765 endows the compound with
outstanding pharmacodyamics, leading to sustained inhibition of BTK during the course of treatment,” says Staudt, who was an investigator in one trial. “It also provides great specificity, because the cysteine residue with which the drug reacts is present in only 10 of the >500 kinases in the human genome.”
Other advantages of the drug are that it is given orally and has a modest side effect profile: “The paucity of side effects is extraordinary for a drug that is as active as this; indeed, the side effects seen to date in the Phase II study are what you might expect to see in the placebo arm,” enthuses Rule.
Under the terms of the agreement between Pharmacyclics and Janssen Biotech, the companies will collaborate on the development of PCI‑32765 for oncology and other indications, excluding inflammation and immune‑mediated conditions. Pharmacyclics will receive an upfront payment of $150 million and is eligible to receive an additional $825 million in development and regulatory milestone payments. There is one other inhibitor of BTK currently in clinical trials — AVL‑292 — which is in Phase I trials for B cell malignancies and autoimmune disease.
“Perhaps the most important take‑home message from these early clinical results is that B cell receptor signalling appears to be a pervasive feature of chronic lymphocytic leukaemia and mantle cell lymphoma that can be targeted therapeutically,” concludes Staudt. “It is likely that many other types of lymphoid malignancy will also depend on B cell receptor signalling — such as the activated B cell‑like subtype of diffuse large B cell lymphoma — and thus the potential utility of agents such as PCI‑32765 in these malignancies is tremendous.”
Charlotte Harrison
T R I A L WAT C H
BTK inhibitor shows positive results in B cell malignancies
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 1
© 2012 Macmillan Publishers Limited. All rights reserved

Regeneron and Genentech have settled their patent dispute over Eylea (aflibercept) — a treatment for wet age-related macular degeneration — which was launched onto the US market in November 2011. But the biotech companies are still disputing the same patents in relation to Zaltrap, a formulation of aflibercept that is in Phase III trials for colorectal and prostate cancer.
Aflibercept targets vascular endothelial growth factor (VEGF), which is involved in neovascularization and vascular permeability. The biologic is known as a VEGF trap; it acts as a soluble decoy receptor for two ligands that bind to VEGF receptors — VEGFA and placenta growth factor — therefore inhibiting ligand binding to VEGF receptor 1 and VEGF receptor 2.
Eylea and Zaltrap are developed by Regeneron (Zaltrap is developed in partnership with Sanofi) but both Regeneron and Genentech own patents related to VEGF traps; Genentech’s VEGF trap patents are collectively known as the Davis–Smyth patents.
In February 2011, on the same day that Regeneron applied for marketing approval of Eylea, it asked a US district court to determine that its activities related to the VEGF trap would not infringe on Genentech’s patents. Two months later Genentech sued Regeneron, alleging that Eylea infringed on the Davis–Smyth patents. This agreement settles the dispute.
Under the terms of the settlement, Regeneron will receive a non-exclusive licence to the Davies–Smyth patents and will make payments to Genentech based on US sales of Eylea until May 2016; the payments will be US$60 million on sales of Eylea reaching US$400 million, 4.75% on cumulative sales between $400 million and $3 billion, and 5.5% on sales over $3 billion. Regeneron anticipates that sales of Eylea will be between $140 million and $160 million in 2012.
However, the clash is not over yet; at the end of 2011 Genentech filed a suit against Regeneron and Sanofi, asserting that Zaltrap infringes on the Davis–Smyth patents.
Patent extensions for combination therapies
Two related rulings from the Court of Justice of the European Union (CJEU) have clarified how supplementary protection certificates (SPCs) — a form of patent extension — should be granted for combination therapies.
An SPC can grant up to a 5-year extension on market exclusivity after a patent has expired; its aim is to compensate for any
delay in marketing a product that is caused by the time taken to gain regulatory approval. SPCs are not a general extension to a patent; rather, they extend a patent with respect to a particular product. But there was uncertainty in the courts of some EU countries regarding the granting of SPCs for combination products, because these sometimes contain ingredients not listed in a corresponding patent or can include fewer ingredients than those listed in a patent.
Both cases centered on vaccines. Medeva (now part of UCB Pharma) owned a patent for a whooping cough vaccine that contained two ingredients: filamentous haemagglutinin and periactin. The company had tried to obtain SPCs to cover the use of these ingredients in vaccines for other diseases that included components not listed in the patent.
The second case involved several US universities, including Georgetown University after which the case took its name, that own patents related to vaccines for human papilloma virus (HPV). These patents are used to partly protect Gardasil and Cervarix; the universities tried to obtain an SPC for only a subset of the ingredients used in the HPV vaccines.
In deciding both cases together, the CJEU ruled that an SPC can be awarded for a subset of ingredients used in a combination therapy. However, an SPC can only be granted for those combination products that are specified
PATENT WATCH
Regeneron and Genentech’s VEGF trap dispute settles… and continues
in the patents; if a therapy includes additional ingredients, it cannot benefit from an SPC. The upshot of this? Patents now need to be written so that they describe as many product combinations as possible.Medeva and Georgetown rulings: http://curia.europa.eu/juris/document/document.jsf?docid=107305&pageIndex=0&doclang=en&mode=lst&dir=&occ=first&cid=1395548
WIPO launches patent licensing feature
The World Intellectual Property Organization (WIPO) — the body responsible for international patent applications — has announced a register that aims to promote the licensing of patents.
Under the scheme, which began on 1 January 2012, patent applicants who are interesting in licensing their inventions can indicate their wish to have information such as the licensing terms made available on the WIPO PatentScope database.WIPO licensing feature: http://www.wipo.int/pct/en/newslett/2011/12/article_0001.html
Charlotte Harrison
PATENT ADVISORS
Daniel M. Becker: Dechert, Mountain View, CA, USA.Luke Kempton: Wragge & Co., London, UK.Leslie Meyer-Leon: IP Legal Strategies, Boston, MA, USA.George W. Schlich: Schlich & Co., London, UK. John A. Tessensohn: Shusaku Yamamoto, Osaka, Japan.Philip Webber: Dehns, London, UK.
Com
Stoc
k
PHO
TOD
ISC
N E W S & A N A LY S I S
98 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
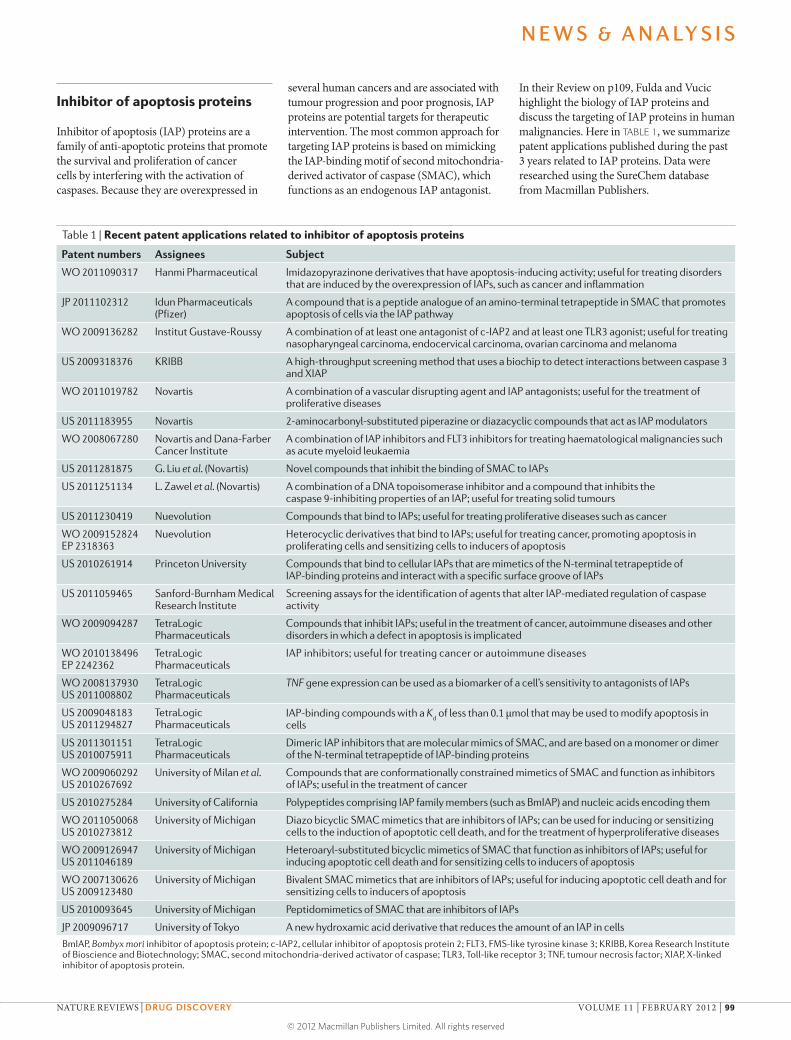
Table 1 | Recent patent applications related to inhibitor of apoptosis proteins
Patent numbers Assignees Subject
WO 2011090317 Hanmi Pharmaceutical Imidazopyrazinone derivatives that have apoptosis-inducing activity; useful for treating disorders that are induced by the overexpression of IAPs, such as cancer and inflammation
JP 2011102312 Idun Pharmaceuticals (Pfizer)
A compound that is a peptide analogue of an amino-terminal tetrapeptide in SMAC that promotes apoptosis of cells via the IAP pathway
WO 2009136282 Institut Gustave-Roussy A combination of at least one antagonist of c-IAP2 and at least one TLR3 agonist; useful for treating nasopharyngeal carcinoma, endocervical carcinoma, ovarian carcinoma and melanoma
US 2009318376 KRIBB A high-throughput screening method that uses a biochip to detect interactions between caspase 3 and XIAP
WO 2011019782 Novartis A combination of a vascular disrupting agent and IAP antagonists; useful for the treatment of proliferative diseases
US 2011183955 Novartis 2-aminocarbonyl-substituted piperazine or diazacyclic compounds that act as IAP modulators
WO 2008067280 Novartis and Dana-Farber Cancer Institute
A combination of IAP inhibitors and FLT3 inhibitors for treating haematological malignancies such as acute myeloid leukaemia
US 2011281875 G. Liu et al. (Novartis) Novel compounds that inhibit the binding of SMAC to IAPs
US 2011251134 L. Zawel et al. (Novartis) A combination of a DNA topoisomerase inhibitor and a compound that inhibits the caspase 9-inhibiting properties of an IAP; useful for treating solid tumours
US 2011230419 Nuevolution Compounds that bind to IAPs; useful for treating proliferative diseases such as cancer
WO 2009152824 EP 2318363
Nuevolution Heterocyclic derivatives that bind to IAPs; useful for treating cancer, promoting apoptosis in proliferating cells and sensitizing cells to inducers of apoptosis
US 2010261914 Princeton University Compounds that bind to cellular IAPs that are mimetics of the N-terminal tetrapeptide of IAP-binding proteins and interact with a specific surface groove of IAPs
US 2011059465 Sanford-Burnham Medical Research Institute
Screening assays for the identification of agents that alter IAP-mediated regulation of caspase activity
WO 2009094287 TetraLogic Pharmaceuticals
Compounds that inhibit IAPs; useful in the treatment of cancer, autoimmune diseases and other disorders in which a defect in apoptosis is implicated
WO 2010138496 EP 2242362
TetraLogic Pharmaceuticals
IAP inhibitors; useful for treating cancer or autoimmune diseases
WO 2008137930 US 2011008802
TetraLogic Pharmaceuticals
TNF gene expression can be used as a biomarker of a cell’s sensitivity to antagonists of IAPs
US 2009048183 US 2011294827
TetraLogic Pharmaceuticals
IAP-binding compounds with a Kd of less than 0.1 μmol that may be used to modify apoptosis in
cells
US 2011301151 US 2010075911
TetraLogic Pharmaceuticals
Dimeric IAP inhibitors that are molecular mimics of SMAC, and are based on a monomer or dimer of the N-terminal tetrapeptide of IAP-binding proteins
WO 2009060292 US 2010267692
University of Milan et al. Compounds that are conformationally constrained mimetics of SMAC and function as inhibitors of IAPs; useful in the treatment of cancer
US 2010275284 University of California Polypeptides comprising IAP family members (such as BmIAP) and nucleic acids encoding them
WO 2011050068 US 2010273812
University of Michigan Diazo bicyclic SMAC mimetics that are inhibitors of IAPs; can be used for inducing or sensitizing cells to the induction of apoptotic cell death, and for the treatment of hyperproliferative diseases
WO 2009126947 US 2011046189
University of Michigan Heteroaryl-substituted bicyclic mimetics of SMAC that function as inhibitors of IAPs; useful for inducing apoptotic cell death and for sensitizing cells to inducers of apoptosis
WO 2007130626 US 2009123480
University of Michigan Bivalent SMAC mimetics that are inhibitors of IAPs; useful for inducing apoptotic cell death and for sensitizing cells to inducers of apoptosis
US 2010093645 University of Michigan Peptidomimetics of SMAC that are inhibitors of IAPs
JP 2009096717 University of Tokyo A new hydroxamic acid derivative that reduces the amount of an IAP in cells
BmIAP, Bombyx mori inhibitor of apoptosis protein; c-IAP2, cellular inhibitor of apoptosis protein 2; FLT3, FMS-like tyrosine kinase 3; KRIBB, Korea Research Institute of Bioscience and Biotechnology; SMAC, second mitochondria-derived activator of caspase; TLR3, Toll-like receptor 3; TNF, tumour necrosis factor; XIAP, X-linked inhibitor of apoptosis protein.
Inhibitor of apoptosis proteins
Inhibitor of apoptosis (IAP) proteins are a family of anti-apoptotic proteins that promote the survival and proliferation of cancer cells by interfering with the acti vation of caspases. Because they are overexpressed in
several human cancers and are associated with tumour progression and poor prognosis, IAP proteins are potential targets for therapeutic intervention. The most common approach for targeting IAP proteins is based on mimicking the IAP-binding motif of second mitochondria-derived activator of caspase (SMAC), which functions as an endogenous IAP antagonist.
In their Review on p109, Fulda and Vucic highlight the biology of IAP proteins and discuss the targeting of IAP proteins in human malignancies. Here in TABLE 1, we summarize patent applications published during the past 3 years related to IAP proteins. Data were researched using the SureChem database from Macmillan Publishers.
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 99
© 2012 Macmillan Publishers Limited. All rights reserved

Institute), with WuXi PharmaTech and with BeiGene. We hope that through the new R&D Asia Centre we will be able to collaborate with more academic institutes in China.
Many industry watchers argue that key drivers behind the whole industry’s investment in China are the need to forge ties with government to ensure market access and its supply of cheap labour. Are these fair points?No matter where you go, you need to have good communications with local authorities to make sure that you understand local needs and that your development plan will be accepted. If you have an R&D centre that increases your presence in China, it will certainly give you more opportunities to discuss programmes with them.
With regard to cheap labour, it is one of the reasons why a lot of the companies came to China and India. But for our industry it is innovation that matters. And if you really only wanted cheap labour, there are other countries that you could go to that are even cheaper. We didn’t come to China for cheap labour, we came here for its talent and ability to innovate.
Can you point to evidence of China’s innovative potential?One example we can see is from publications in first-class journals like Nature, Science and Cell. If you look over the past 10 years, you see a significant increase in the number of papers in these journals in which the first author is from China. These are not Chinese researchers studying in another country, but Chinese researchers who are working in China. This increase in publications demonstrates that there is high-quality science in China.
Given the costs of bringing a new drug to market, can novel medicines be affordable in China?We do have affordability and market access issues in China and in other emerging markets. Because of this, we will work to complement our innovative products with branded generics and innovative branded generics, which might have more convenient dosing or a superior pharmacokinetic profile. But, due to economic growth, we will see in the next few years a growing middle class that will demand, and be able to pay for, innovative drugs.
What lessons have you learnt from earlier expansions into Asia in terms of fostering successful innovative R&D in China?If you look at Japan 30 years ago, every major US and European pharmaceutical company had a research facility there. But if you look now, there is little basic research anymore. They only have development facilities there. An important lesson I have learnt from this is that it is not enough to just copy what you do in the Western countries and move the same programmes to Asia. You have to ask the question: why do you believe you can do better in China than in the United States and in Europe?
For certain diseases, the answer is that you find more doctors, patients, specialists and resources in China. Not many people do basic research on gastric cancer in the United States or the United Kingdom, for example, because there is not a big unmet need there. But in China it is a top killer and so there is a lot of basic and clinical research.
A second thing that we have learnt is that we need to partner with local companies. In China, for example, we have partnered with BGI (formerly Beijing Genomics
What are your current R&D capabilities in China, and how do you plan to expand these?People have the perception that Merck does not yet have R&D in China, but we actually do. Since 2005, we have had one of our three global data management centres in China. We also have our global biostatistician centre in China, and a China development group that includes regulatory, clinical, operation and pharmacovigilance groups. Overall, we have about 300 R&D staff in China.
By 2014, we plan to have 600 R&D staff. They will work in two key areas. A first focus will be on developing drugs in China for China. Historically, we have had a huge gap between when a drug launches in the United States and Europe versus when it launches in China, and so we need to accelerate our Chinese launches. For this, we will expand our regulatory, clinical research operations, project management and pharmacovigilance teams.
Second, because the environment for innovation has significantly improved in China, we wanted to grow R&D in China so that it can contribute to our global pipeline. Firstly, our researchers will focus on customizing our portfolio for Chinese patients, who have some common diseases in China that are not common in the Western countries. We want to focus on developing drugs for hepatocellular carcinoma and oesophageal cancer, for example.
What other areas do you foresee as potential growth areas for China in terms of innovative R&D?GlaxoSmithKline has moved neuroscience to China, Lilly has moved diabetes to China, and Pfizer has moved infectious diseases to China. These three areas, as well as cardiovascular disease and vaccines, are forecast to grow in China.
AN AUDIENCE WITH…
RuiPing Dong Late last year Merck & Co. announced plans to spend US$1.5 billion to bolster research and development (R&D) in China, one of the industry’s largest investments in the country to date. The firm’s initial plans include, by 2014, the construction of an Asian R&D headquarters in Beijing and a doubling of their Chinese research staff count to 600 employees. Overseeing the expansion is RuiPing Dong, head of emerging markets R&D. Prior to joining Merck in 2010, Dong headed up Bristol-Myers Squibb’s R&D efforts in Asia-Pacific and the emerging markets, and supervised AstraZeneca’s oncology programmes. Speaking with Asher Mullard, he explains the strategy behind the Chinese R&D expansion.
N E W S & A N A LY S I S
100 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
2010 2011 2012 2013 2014 2015 2016 2017 2018 2019 2020
ARC-4558RalfinamideAVP-923AmiKetEladurHorizantNucynta EROther*GenericQutenzaGraliseLidodermCymbaltaLyrica
Billi
ons
of U
S$
FROM THE ANALYST’S COUCH
The neuropathic pain marketSarah Nightingale Munro sofa by Donna Wilson, courtesy of www.scp.co.uk
Neuropathic pain is a chronic pain condition caused by a primary lesion or dysfunction in the nervous system. It can be a consequence of many different insults, such as trauma, neuronal injury or infection. The most commonly studied neuropathic pain subtypes include diabetic neuropathic pain (DNP), postherpetic neuralgia (PHN) and HIV-related neuropathic pain. Collectively, these three conditions were estimated to affect over 6 million people across the seven major pharmaceutical markets (the United States, Japan, France, Germany, Italy, Spain and the United Kingdom) in 2010 (REF. 1). However, the total affected population is considerably larger owing to the number of additional neuropathic pain conditions, such as neuropathic lower back pain, cancer-related neuropathic pain, complex regional pain syndrome and postoperative neuropathic pain.
Current therapeutic optionsOwing to the limited understanding of the precise aetiology of many neuropathic pain conditions, current pharmacological treatments encompass an array of drug classes including anticonvulsants, antidepressants, narcotic analgesics and topical anaesthetics. However, with just seven drugs approved for neuropathic pain conditions across the seven
major markets (late 2011), and because of the limited efficacy of these drugs, off-label prescribing is widespread.
The current standard treatment is the anticonvulsant pregabalin (Lyrica; Pfizer). It alters neuronal activity through modulation of the calcium channels and agonism of the GABA (γ-aminobutyric acid) α2δ subunit. This dual mechanism of action leads to efficacy in many neuropathic pain conditions. Lyrica has the broadest neuropathic pain label of all marketed agents but its analgesic efficacy profile leaves much room for improvement; it is associated with several central nervous system (CNS)-related side effects including sedation and dizziness.
Another leading product is the anti - depressant duloxetine (Cymbalta; Eli Lilly). It is a serotonin and noradrenaline reuptake inhibitor indicated for the treatment of DNP. However, Cymbalta is expected to face fierce competition from generic duloxetine in the European and US markets following its patent expiry in December 2012 and June 2013, respectively.
In recent years there has been a shift towards the use of transdermal therapies for the management of neuropathic pain. Since the 1990s, Lidoderm (5% lidocaine patch; Endo/Grünenthal/Teikoku) has dominated the topical neuropathic pain market. In 2010
however, Qutenza (8% capsaicin patch; NeurogesX/Astellas Pharma) entered the US and European markets for the treatment of PHN. Qutenza offers an application frequency of once every 3 months, which is a substantial improvement on Lidoderm’s once-daily dosing frequency. However, Qutenza’s painful and cumbersome application process is expected to limit future uptake.
The newest compounds to enter the neuropathic pain market are Gralise (extended release gabapentin; Depomed) and Nucynta ER (extended release tapentadol; Johnson & Johnson). Gralise is a once-daily formulation of the anticonvulsant gabapentin and was approved for the management of PHN in January 2011, making it the first oral treatment to obtain approval from the US Food and Drug Administration (FDA) for neuropathic pain in over 6 years. Despite its improved dosing regime, Gralise offers limited clinical differentiation from other established treatment options and this is likely to hinder the product’s success. By contrast, Nucynta ER, which obtained FDA approval for the treatment of chronic pain in August 2011, is likely to achieve blockbuster sales in the neuropathic pain market. It possesses a dual mode of action — agonism of the μ-opioid receptor and inhibition of noradrenaline reuptake — and has demonstrated efficacy in both neuropathic and nociceptive pain conditions. As a result, it is expected to be prescribed for conditions involving both pain types, such as chronic lower back pain. Its opioid-sparing effects and reduced potential for abuse will be key clinical advantages for Nucynta ER in neuropathic pain conditions requiring long-term management.
Unmet needsIn the absence of disease-modifying or curative agents for the management of neuropathic pain, improved analgesic efficacy remains a primary unmet need. It is estimated that only one in four patients with neuropathic pain experiences over 50% pain relief. The pharmaceutical industry has so far struggled to improve on current therapeutic options, owing to the complexity of identifying the most appropriate targets to investigate.
Currently available drugs also produce several significant side effects such as ▶
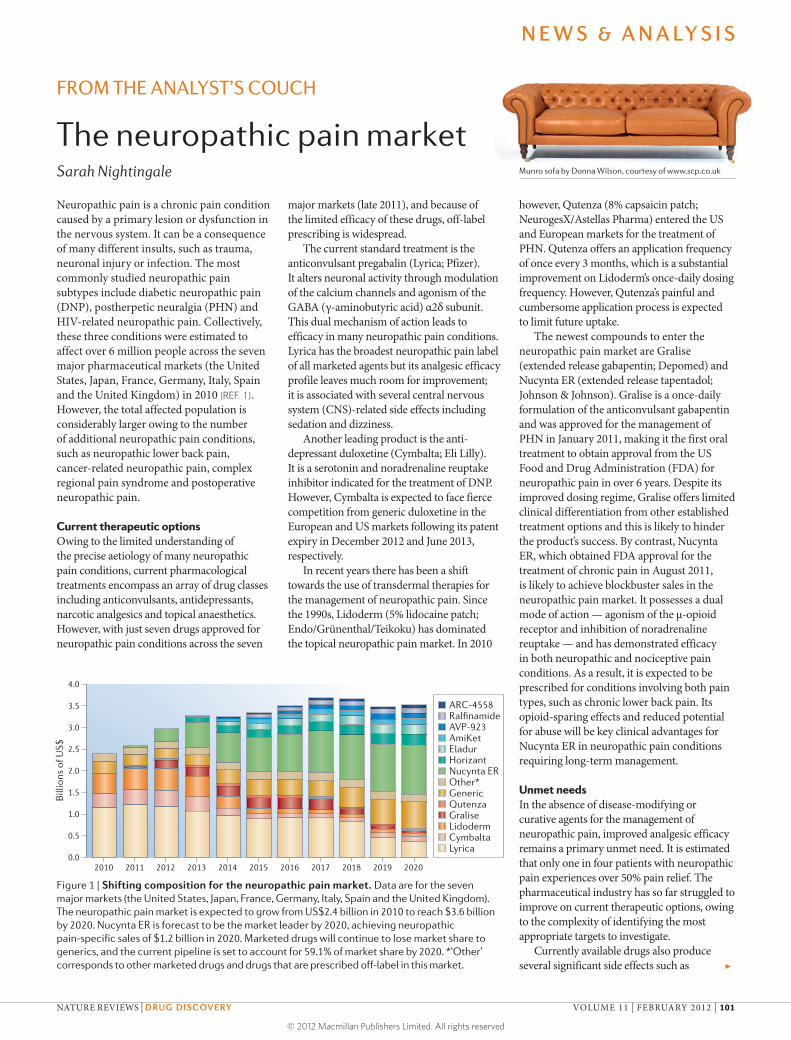
Figure 1 | Shifting composition for the neuropathic pain market. Data are for the seven major markets (the United States, Japan, France, Germany, Italy, Spain and the United Kingdom). The neuropathic pain market is expected to grow from US$2.4 billion in 2010 to reach $3.6 billion by 2020. Nucynta ER is forecast to be the market leader by 2020, achieving neuropathic pain-specific sales of $1.2 billion in 2020. Marketed drugs will continue to lose market share to generics, and the current pipeline is set to account for 59.1% of market share by 2020. *‘Other’ corresponds to other marketed drugs and drugs that are prescribed off-label in this market.
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 101
© 2012 Macmillan Publishers Limited. All rights reserved
NEUROPATHIC PAIN | MARKET INDICATORS
▶ drowsiness, dizziness and somnolence, which negatively affect patients’ quality of life. Furthermore, there is also increasing pressure on cost-effectiveness. Drug developers not only have to demonstrate efficacy to physicians but are also facing the increasing challenge of demonstrating a cost–benefit to regulatory bodies and insurers.
Key drugs in developmentThe neuropathic pain pipeline is one of the most extensive in the CNS arena, with 79 candidates known to be under clinical investigation in 2011 (REF. 2). A summary of key late-stage compounds can be found in TABLE 1.
The pipeline contains an array of novel candidates and agents offering improved dosing convenience. The most advanced pipeline candidate is Horizant (gabapentin enacarbil; XenoPort/GlaxoSmithKline), which offers once- or twice-daily dosing and is currently pending approval in the United States for the treatment of PHN. However, it will face competition from other branded and generic gabapentin compounds in the marketplace, in particular from the once-daily extended release gabapentin, Gralise. Other compounds aiming to reduce the dosing frequency include: the controlled release pregabalin, which is in Phase III development for the once-daily treatment of neuropathic pain conditions; and Eladur (bupivacaine
patch; Durect/King/Pfizer), which is in Phase II development for the treatment of PHN and requires one patch application every 3 days. Although Eladur improves on Lidoderm’s once-daily application, it falls short of Qutenza’s 3-monthly dosing frequency. However, the absence of severe application-site side effects such as those associated with Qutenza will be a clinical advantage for Eladur.
AmiKet (ketamine and amitriptyline; EpiCept) and ARC-4558 (clonidine; Arcion) are two topical ointment formulations that are set to offer a more versatile application method than the patch formulation. This is of particular importance for the management of peripheral neuropathic pain types that are often localized to the hands and feet, where patch application can be challenging. Although AmiKet is expected to become the first of the topical ointments in the pipeline to enter the market, both agents will offer novel mechanisms of action in the neuropathic pain market. Drugs with novel mechanisms of action represent useful additions to the neuropathic pain market given the high percentage of patients who do not respond to current therapy options. Other novel candidates under clinical investigation include the cough suppressant dextromethorphan (AVP-923; IriSys/Avanir) and the selective sodium channel blocker ralfinamide (Newron).
Market outlookIn addition to the expansion of existing brands into new geographical regions, the uptake of new neuropathic pain products is expected to offset generic erosion, expanding the seven major market neuropathic pain sales from US$2.4 billion in 2010 to peak sales of $3.6 billion by 2020. Seven pipeline candidates are expected to enter the neuropathic pain market across the seven major markets by 2020 (FIG. 1). Over this forecast period, sales of currently marketed products are projected to decrease threefold as leading brands such as Cymbalta and Lyrica experience generic erosion. Nevertheless, the anticipated uptake of Nucynta ER in the neuropathic lower back pain market is expected to drive market growth as there are currently no treatments indicated for this prevalent condition.
Sarah Nightingale is an associate analyst at Datamonitor, 119 Farringdon Road,
London EC1R 3DA, UK. e‑mail: [email protected]
doi:10.1038/nrd3624
1. Datamonitor. Epidemiology: Peripheral Neuropathic pain — A common co-morbidity for HIV, diabetes, and herpes zoster. HC00221–001 (October 2011)
2. Datamonitor. R&D Trends: Neuropathic pain — Extensive and diverse pipeline driven by high unmet need. HC00064–006 (July 2011).
Competing interests statementThe author declares no competing financial interests.
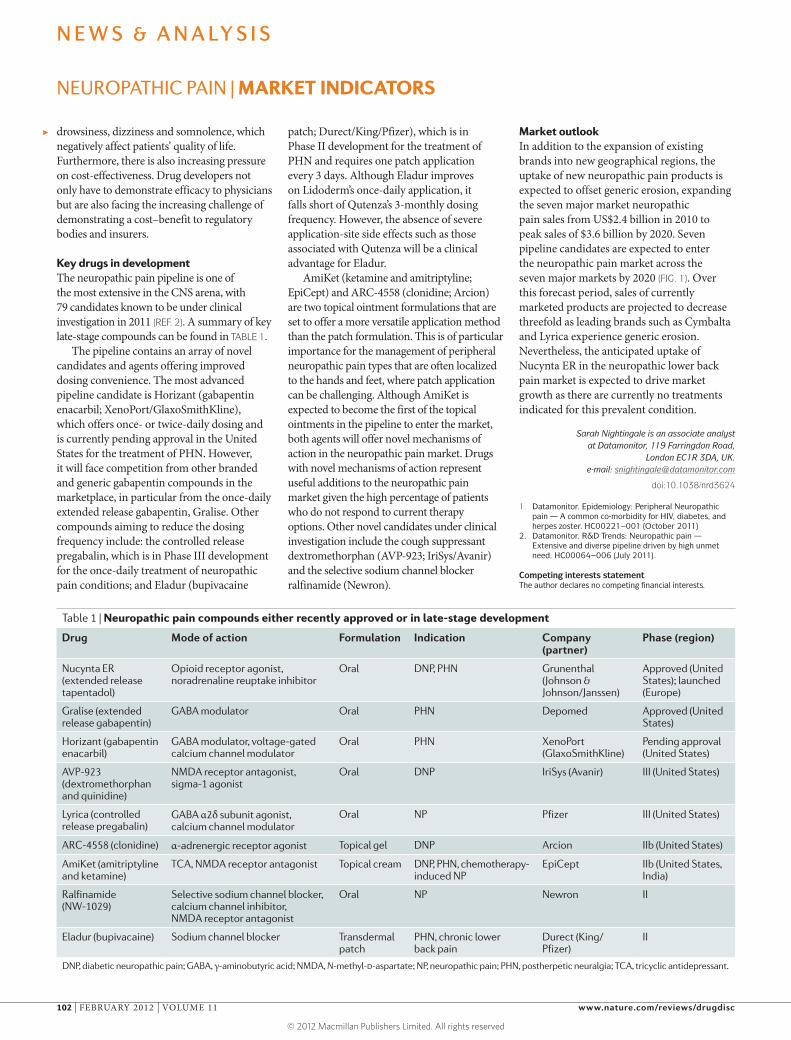
Table 1 | Neuropathic pain compounds either recently approved or in late-stage development
Drug Mode of action Formulation Indication Company (partner)
Phase (region)
Nucynta ER (extended release tapentadol)
Opioid receptor agonist, noradrenaline reuptake inhibitor
Oral DNP, PHN Grunenthal (Johnson & Johnson/Janssen)
Approved (United States); launched (Europe)
Gralise (extended release gabapentin)
GABA modulator Oral PHN Depomed Approved (United States)
Horizant (gabapentin enacarbil)
GABA modulator, voltage-gated calcium channel modulator
Oral PHN XenoPort (GlaxoSmithKline)
Pending approval (United States)
AVP-923 (dextromethorphan and quinidine)
NMDA receptor antagonist, sigma-1 agonist
Oral DNP IriSys (Avanir) III (United States)
Lyrica (controlled release pregabalin)
GABA α2δ subunit agonist, calcium channel modulator
Oral NP Pfizer III (United States)
ARC-4558 (clonidine) α-adrenergic receptor agonist Topical gel DNP Arcion IIb (United States)
AmiKet (amitriptyline and ketamine)
TCA, NMDA receptor antagonist Topical cream DNP, PHN, chemotherapy- induced NP
EpiCept IIb (United States, India)
Ralfinamide (NW-1029)
Selective sodium channel blocker, calcium channel inhibitor, NMDA receptor antagonist
Oral NP Newron II
Eladur (bupivacaine) Sodium channel blocker Transdermal patch
PHN, chronic lower back pain
Durect (King/Pfizer)
II
DNP, diabetic neuropathic pain; GABA, γ-aminobutyric acid; NMDA, N-methyl-d-aspartate; NP, neuropathic pain; PHN, postherpetic neuralgia; TCA, tricyclic antidepressant.
N E W S & A N A LY S I S
102 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
N
N
NH
N
•H3PO4
N
CNH
(R)-3-(4-(7H-pyrrolo[2,3d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate; C17H18N6
•H3PO4; Mr = 404.36
Ruxolitinib phosphate
FRESH FROM THE PIPELINE
RuxolitinibRuben A. Mesa, Uma Yasothan and Peter Kirkpatrick
In November 2011, ruxolitinib (Jakafi; Incyte/Novartis), a small-molecule inhibitor of Janus kinases, was approved by the US Food and Drug Administration for the treatment of patients with intermediate or high-risk myelofibrosis, including primary myelofibrosis, post-polycythaemia vera myelofibrosis and post-essential thrombocythaemia myelofibrosis.
Primary myelofibrosis, polycythaemia vera (PV) and essential thrombocythaemia (ET) are a group of myeloproliferative neoplasms (MPNs). They have clinical characteristics that in advanced cases include progressive myelofibrosis, anaemia and enlargement of the spleen, and symptoms that include fatigue and bone pain1,2. Standard treatments for these MPNs, such as myelosuppressive therapy with hydroxyurea, do not significantly affect the natural history of the disease, and are also not very effective in managing disease symptoms1,2.
Basis of discoveryJanus kinases (JAKs) are a family of four cytoplasmic tyrosine kinases (JAK1, JAK2, JAK3 and non-receptor tyrosine kinase 2
(TYK2)) that mediate signals from the receptors for various cytokines and growth factors that have a key role in haematopoiesis and immune function2. In 2005, several groups reported the presence of an activating mutation in the JAK2 gene, which results in a valine to phenyalanine substitution at codon 617 (JAK2V617F), in a substantial proportion of patients with various MPNs2,3. Ectopic expression of JAK2V617F in mice results in MPN-like phenotypes, supporting an important role for this mutation, and JAK2 in general, in human MPNs2,3.
Drug propertiesRuxolitinib (FIG. 1) was discovered as an inhibitor of JAK1 and JAK2 (REF. 3). In a mouse model of JAK2V617F-positive MPNs, oral administration of ruxolitinib markedly reduced splenomegaly and levels of circulating inflammatory cytokines, preferentially eliminated JAK2V617F-mutant cells and increased survival3,4. It showed promising effects in a Phase I/II trial involving 153 patients with myelofibrosis5, supporting its further clinical evaluation.
Clinical dataThe efficacy and safety of ruxolitinib were studied in two Phase III trials in patients with myelofibrosis (primary myelofibrosis, post-PV myelofibrosis or post-ET myelofibrosis)4. In both studies, patients had palpable splenomegaly at least 5 cm below the costal margin and a risk category of intermediate-2 or high based on the International Working Group Consensus Criteria4. The starting dose of ruxolitinib was based on platelet count; doses were then individualized based on tolerability and efficacy, with a maximum dose in any group of 20 mg orally twice daily4.
Study 1 was a double-blind, randomized placebo-controlled trial involving 309 patients who were either refractory to or were not candidates for available therapy; patients were randomized to ruxolitinib or placebo in a 1:1 ratio4. Patients had a median spleen volume of 2,595 cm3, measured by magnetic resonance imaging (MRI) or computed tomography (CT) (the upper limit of normal volume is ~300 cm3)4. The primary efficacy end point was the proportion of patients
achieving a ≥35% reduction from the baseline in spleen volume at week 24 as measured by MRI or CT4. Secondary end points included the proportion of patients with a ≥50% reduction in total symptom score from the baseline to week 24, as measured by the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary4. This daily diary score captures the six core symptoms of myelofibrosis (abdominal discomfort, pain under left ribs, night sweats, itching, bone and/or muscle pain and early satiety), with scores for each symptom ranging from 0 (absent) to 10 (worst imaginable), which are added to create a total score with a maximum of 60. At baseline, the mean total symptom score was 18.0 in the ruxolitinib group and 16.5 in the placebo group4.
Study 2 was an open-label trial involving 219 patients who were randomized in a 2:1 ratio to ruxolitinib or the best available therapy (such as hydroxyurea and glucocorticoids), selected by the investigator on a patient-by-patient basis4. Patients had a median spleen volume of 2,381 cm3 as measured by MRI or CT4. The primary efficacy end point was the proportion of patients achieving ≥35% reduction from the baseline in spleen volume at week 48 as measured by MRI or CT4.
In both studies, a significantly larger proportion of patients in the groups that received ruxolitinib achieved the primary efficacy end point than the patients in the comparator groups: 42% of patients in study 1 compared with 1% in the placebo group; and 29% of patients in study 2 compared with 0% in the group receiving the best available therapy4. In addition, in study 1 46% of patients had a ≥50% reduction in total symptom score by week 24, compared with 5% of patients in the placebo group4. All six of the symptoms contributed to the higher total symptom score response rate in the group treated with ruxolitinib4.
IndicationsRuxolitinib is approved by the US Food and Drug Administration (FDA) for the treatment of patients with intermediate or high-risk myelofibrosis, including primary myelofibrosis, post-PV myelofibrosis and post-ET myelofibrosis4. ▶
Figure 1 | Ruxolitinib. Ruxolitinib (previously known as INCB018424) was discovered as a selective inhibitor of Janus kinases (JAKs). IC
50
values for inhibition of JAK1, JAK2 and JAK3 in enzyme assays were 3.3 nM, 2.8 nM and 428 nM, respectively3.
N E W S & A N A LY S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 103
© 2012 Macmillan Publishers Limited. All rights reserved

ANALYSIS | MYELOFIBROSIS
▶
Box 1 | Market for myelofibrosis
Analysing the market for myelofibrosis is Uma Yasothan, IMS Health, London, UK.
It is estimated that ~100,000 patients are afflicted with primary myelofibrosis, polycythaemia vera (PV) and essential thrombocythaemia (ET) in the United States alone2. Ruxolitinib (Jakafi; Incyte/Novartis), an oral Janus kinase inhibitor, is the first drug to be approved specifically to treat patients with intermediate or high-risk myelofibrosis, including primary myelofibrosis, post-PV myelofibrosis and post-ET myelofibrosis. It is launching with a fairly broad label into a disease area with a high unmet need and with plenty of opportunity to grow. Initial perceptions of physicians on efficacy and/or safety are also likely to change with the release of additional long-term safety data and survival data, which — if favourable — could contribute to increased uptake. Potential barriers to its adoption include its high cost; Incyte states that ruxolitinib will cost US$7,000 a month, or $84,000 per year, for insured patients.
Novartis holds the rights to ruxolitinib outside the United States, and its experience with imatinib (Gleevec) for chronic myeloid leukaemia is expected to be valuable in establishing a launch platform. Additional Phase II studies are currently underway with ruxolitinib for PV and ET, and other potential indications include rheumatoid arthritis. Analysts’ projections for ruxolitinib sales reflect its promising clinical profile; sales estimates range from $56 million to $67 million in 2012, with projected worldwide peak sales of more than $1 billion by 2015 (Wei, T et al. Jefferies Equity Research Report. 7 Nov 2011; Schmidt, E. & Bishop, N. Cowen & Company Biotechnology Report. 8 Dec 2011).
Analysing issues in the treatment of myelofibrosis is Ruben A. Mesa, M.D., Chair, Division of Hematology and Medical Oncology and Professor of Medicine, Mayo Clinic, Arizona, USA.
Treatment strategies for myelofibrosis (primary, post-PV or post-ET) can be stratified according to three goals: first, alleviation of the disease burden, whether that is improvement of cytopaenias, constitutional symptoms or splenomegaly; second, the potential to delay disease progression; and third, cure through allogeneic stem cell transplantation (ASCT).
The recent approval of the JAK1/JAK2 inhibitor ruxolitinib is likely to substantially alter front-line therapy of patients with myelofibrosis, with strategies being based on estimates of disease prognosis and symptom burden. First, asymptomatic low-risk patients, who have a median survival of 185 months, are best managed by observation (Supplementary information S1 (figure)). Interferon-based therapy might delay disease progression in early-stage myelofibrosis but this is currently experimental6. Low-risk patients with symptoms that are not included in prognostic scores (such as fatigue and pruritus) might benefit from JAK inhibition.
Patients with myelofibrosis classified as ‘intermediate-1 risk’ have a median survival of 78 months, and with this shortened survival, decision-making becomes more complex. Front-line therapy for patients who are primarily afflicted with splenomegaly and related symptoms should include
JAK2 inhibition. Patients primarily with anaemia (and/or thrombocytopaenia) may be considered for an anaemia-based therapy, such as an immunomodulatory drug (for example, lenalidomide)7 or androgens8. In the future, a JAK2 inhibitor that could improve anaemia, such as CYT387, which is currently in Phase I/II trials (ClinicalTrials.gov identifier: NCT00935987), might also be an option. A case could also be made to consider ASCT in such patients if they are exceptional candidates for transplantation.
Patients with myelofibrosis classified as ‘intermediate-2’ or high-risk have a median survival of 16–35 months, and ASCT should be front-line therapy for good candidates. For individuals who elect not to proceed with ASCT, or who are suboptimal candidates, front-line therapy would again involve JAK2 inhibition with ruxolitinib for patients whose burden of illness is primarily splenomegaly and related symptoms. Patients who primarily have anaemia may benefit from an immunomodulatory drug, androgens, a JAK2 inhibitor or entry into an alternative clinical trial.
Finally, individuals who present with blast-phase MPN, who have a median survival of less than 3 months, should undergo rapid induction chemotherapy if they are candidates for ASCT, and if this returns their disease to the chronic phase they should proceed to an allotransplant. If they are not initially candidates for ASCT because of age or co-morbidities, options include: hypomethylation therapy for high-risk acute myeloid leukaemias, such as
with 5-azacitidine9; an appropriate clinical trial for elderly patients or for high-risk acute leukaemia; or supportive care if none of these options is appropriate.
The rapid progress from the discovery of the JAK2V617F mutation in 2005 to the approval of ruxolitinib in 2011 has involved a period of unprecedented effort in evaluating the pathogenesis and developing new treatment options for patients with myelofibrosis and, subsequently, patients with earlier-stage MPNs. Further advances in treatment are anticipated to come through combination strategies and new pathway inhibitors. Trials are currently being planned with combinations to enhance ASCT outcomes (JAK2 inhibitor prior to ASCT), improve anaemia responses (JAK2 inhibitor plus either immunomodulatory drugs or androgens), enhance anti-stem-cell effects (JAK2 inhibitors plus hypomethylation therapies or interferon) and/or enhance a regression of intramedullary fibrosis (JAK2 inhibitors with Hedgehog pathway inhibitors or antibodies against pro-fibrotic cytokines).
Ruben A. Mesa is at the Division of Hematology & Medical Oncology, Mayo Clinic, Scottsdale,
Arizona 85259, USA.
Uma Yasothan is at IMS Health, 7 Harewood Avenue, London NW1 6JB, UK.
Peter Kirkpatrick is at Nature Reviews Drug Discovery.
e-mails: [email protected]; [email protected]; [email protected]
doi:10.1038/nrd3652
1. Vannucchi, A. M. et al. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J. Clin. 59, 171–191 (2009).
2. Quintás-Cardama, A. et al. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nature Rev. Drug Discov. 10, 127–140 (2011).
3. Quintás-Cardama, A. et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115, 3109–3117 (2010).
4. US Food and Drug Administration. FDA labeling information — Jakafi. FDA website [online], http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf (2011).
5. Verstovsek, S. et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 363, 1117–1127 (2010).
6. Silver, R. T. et al. Recombinant interferon-alpha may retard progression of early primary myelofibrosis: a preliminary report. Blood 117, 6669–6672 (2011).
7. Mesa, R. A. et al. Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) Phase 2 trial E4903. Blood 116, 4436–4438 (2010).
8. Cervantes, F. et al. Efficacy and tolerability of danazol as a treatment for the anaemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patients. Br. J. Haematol. 129, 771–775 (2005).
9. Thepot, S. et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 116, 3735–3742 (2010).
Competing financial interestsThe authors declare no competing financial interests.
N E W S & A N A LY S I S
104 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

The maintenance of protein homeo-stasis (proteostasis) is vital for cell function and involves a tightly regu-lated network of pathways control-ling the synthesis, folding, transport and degradation of proteins. Loss of proteostatic control can lead to the accumulation and aggregation of misfolded proteins — features that are associated with numerous conditions including metabolic diseases, cancer and neurodegen-erative disorders. Now, Calamini and colleagues describe a series of novel small-molecule proteostasis regulators that activate the heat shock response (HSR) and restore protein homeostasis in various models of protein conformational diseases.
Crucial to proteostasis are the cytosolic HSR and the unfolded protein response (UPR), which are induced under conditions of cellular
stress and act to restore protein balance in the cytoplasm and endo-plasmic reticulum, respectively. The HSR is governed by a family of heat shock factors (HSFs) — primarily HSF1, which promotes the expres-sion of genes encoding molecular chaperones, such as the heat shock proteins (HSPs), that guide the con-formation of proteins during biogen-esis and prevent their misfolding and aggregation. Enhancing the activity of HSF1 and the concentrations of molecular chaperones has previously been shown to restore proteostasis in several disease models. With this in mind, Calamini and colleagues set out to identify novel small-molecule proteostasis regulators.
To do this, the authors developed a human cell-based high-throughput screen to identify activators of the HSR. Using two independent primary screens encompassing a total of 900,000 compounds, they obtained and confirmed 263 positive hits. Clustering active compounds by structure resulted in 233 hits grouped into seven clusters, and identified three common scaffolds across the two primary screens. Fourteen com-pounds were selected as represen-tative small-molecule ‘proteostasis regulators’ for subsequent studies.
Further in vitro experiments confirmed that the newly identi-fied molecules activated the HSR; they induced mRNA and protein expression of several HSPs in an HSF1-dependent manner. Analysis of the gene signatures of the proteo-stasis regulators revealed that some molecules activated additional stress-responsive proteostasis network pathways, including the UPR and the antioxidant stress response. Of the five compounds that were shown to
most strongly activate HSF1, three significantly protected cells from death induced by severe heat shock and from apoptosis induced by oxidative stress.
Next, the authors assessed whether the proteostasis regulators could restore proteostasis in two cellular models of conformational diseases — Huntington’s disease and cystic fibrosis. Indeed, the com-pounds reduced aggregate formation of a mutant huntingtin protein con-taining a polyglutamine repeat, and rescued trafficking and cell surface expression of a mutant cystic fibrosis transmembrane conductance regula-tor in the respective disease models. Furthermore, in a Caenorhabditis elegans model of polyglutamine diseases, the proteostasis regulators reduced the aggregation of expanded polyglutamines and ameliorated aggregation-associated toxicity, restoring motility to close to that of control animals.
In contrast to current known small-molecule activators of the HSR, these compounds did not act by causing protein misfolding, protea-some inhibition or HSP90 inhibition.
These findings support further investigation of small-molecule pro-teostasis regulators for the potential treatment of multiple proteopathies, and the novel molecules identified in this study represent promising first-generation tool compounds.
Sarah Crunkhorn
ORIGINAL RESEARCH PAPERS Calamini, B. et al. Small-molecule proteostasis regulators for protein conformational diseases. Nature Chem. Biol. 25 Dec 2011 (doi:10.1038/nchembio.763)FURTHER READING Neef, D. et al. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nature Rev. Drug Discov. 10, 930–944 (2011)
P R OT E I N C O N F O R M AT I O N A L D I S E A S E S
Rescuing protein homeostasis
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
Nature Reviews Drug Discovery | AOP, published online 20 January 2012; doi:10.1038/nrd3662
© 2012 Macmillan Publishers Limited. All rights reserved

Although preclinical studies suggest that fibroblast growth factor 21 (FGF21) might have disease-modifying properties in obesity and type 2 diabetes, recom-binant FGF21 protein has poor pharmacokinetics, which precludes its clinical use. Now, writing in Science Translational Medicine, a group from Genentech describes the identification and characteriza-tion of an agonistic antibody that is specific for FGF receptor 1 (FGFR1) and has long-lasting anti diabetic and lipid-lowering properties.
The authors used phage display technology to generate two mono clonal antibodies that had sub nanomolar affinity for FGFR1b and FGFR1c but did not bind to any other FGFR isoforms or to the FGF co-receptor β-klotho. One antibody — R1MAb1 — was chosen for further studies.
In a mouse model of diabetes (db/db mice), a single injection of R1MAb1 normalized blood glucose levels for more than 1 week, and levels remained lower than in control mice (immunolgobulin G-treated mice) for more than 30 days, including at time points when serum concentra-tions of R1MAb1 could not be detected. Administration of R1MAb1 also lowered fasting glucose levels (but did not cause hypoglycaemia) and lowered fasting serum insulin levels, suggesting that R1MAb1 had insulin-sensitizing effects.
In addition, the authors determined that R1MAb1 exerts
its agonistic effects by promoting homodimerization and activation of FGFR1, and this action mediates the antidiabetic effects of the antibody.
Next, the authors investigated the mechanisms responsible for the antidiabetic activity of R1MAb1 and compared it to FGF21. Both FGF21 and R1MAb1 had similar (but not identical) in vivo effects, such as reducing serum insulin and lipid levels as well as lowering blood glucose levels, but although FGF21 increased serum levels of β-hydroxybutyrate (a ketone body mostly produced in the liver) in lean mice, R1MAb1 did not. In addition, both FGF21 and the antibody activated common signalling pathways, such as phosphorylation of extracellular signal-regulated kinase (ERK) and MAPK/ERK kinase (MEK) in adipose tissue, but unlike FGF21 the antibody did not induce phosphorylation in the liver. These findings suggested that R1MAb1 acts on adipose tissue, whereas FGF21 acts on the liver as well as adipose tissue. Indeed, intraperitoneal injec-tion of R1MAb1 had no effect on blood glucose levels, insulin resistance or glucose tolerance in mice with compromised adipose tissue.
Further studies showed that, like FGF21, in adipose tissue R1MAb1 induces expression of the nuclear receptor transcriptional co-activator PPARγ co-activator 1α and downstream genes that are linked to oxidative metabolism.
Further work is needed to fully determine the properties of R1MAb1
and to determine any potential side effects before it could be used in humans. Nevertheless, this study shows that R1MAb1 has superior pharmacokinetic properties to recombinant FGF21 — which has a half-life of about 30 minutes in rodents and about 2 hours in mon-keys — and its administration leads to sustained antidiabetic effects in a mouse model.
Charlotte Harrison
ORIGINAL RESEARCH PAPER Wu, A. L. et al. Amelioration of type 2 diabetes by antibody-mediated activation of fibroblast growth factor receptor 1. Sci. Transl. Med. 3, 113ra126 (2011)FURTHER READING Beenken, A. & Mohammadi, M. The FGF family: biology, pathophysiology and therapy. Nature Rev. Drug Discov. 8, 235–253 (2009)
O B E S I T Y A N D D I A B E T E S
An FGFR antibody with long-lasting effects
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
Nature Reviews Drug Discovery | AOP, published online 20 January 2012; doi:10.1038/nrd3660
© 2012 Macmillan Publishers Limited. All rights reserved

The recurrence and mortality rate of patients with glioblastoma multi forme (GBM) is close to 100%. At present, therapy consists of surgical debulking of the tumour followed by radiation therapy and chemo therapy. Preclinical studies have shown that therapeutically engineered stem cells may be well suited to treat GBM but these studies have highlighted the difficulty of delivering and retaining the cells in the target area. In a study published in Nature Neuroscience, Khalid Shah and colleagues describe a mouse resection model of GBM in which stem cells encapsulated in a bio degradable synthetic
extracellular matrix (sECM) are able to home to the tumour, delay tumour regrowth and increase survival.
Most in vivo models of GBM focus on targeting the intact tumour, so to mimic the clinical scenario the authors first developed a mouse resection model using fluorescent human U87 GBM cells that can be visualized over time. They showed that resecting established tumours, which were generated by implanting low or high numbers of GBM cells, significantly prolonged survival.
Next, they assessed the survival of mouse neural stem cells (NSCs) in an sECM, as well as their ability to prolif-erate and secrete proteins, in vitro and in vivo. Interestingly, their viability in the fluorescent resection model was greater than non-encapsulated mouse NSCs, and over 4 days the cells were found to migrate out of the capsule and specifically home to the tumours. In this study, the cells were engineered to express and secrete TNF-related apoptosis-inducing ligand (TRAIL), a cytotoxic agent that induces apoptosis in approximately 50% of GBM cells. In culture, the encapsulated mouse NSC-TRAIL cells significantly reduced the viability of TRAIL-sensitive human GBM cells by activating caspase 3 and caspase 7 as well as caspase 8. Three days after implanting the cells in the resection cavity of the mouse model, an 80% decrease in residual tumour cells was observed, along with a marked increase in caspase 3 and caspase 7
activity. This suppression of tumour growth was maintained for over 40 days, and all of the treated mice were alive 42 days after resection. By contrast, the median survival of mice treated with encapsulated mouse NSCs that did not express TRAIL was 14.5 days following resection.
Finally, the authors carried out a similar assessment of the therapeutic potential of encapsulated TRAIL-expressing human bone marrow-derived mesenchymal stem cells (MSCs) on the TRAIL-sensitive pri-mary human invasive glioma cell line GBM8. Similarly to the mouse NSCs, the human MSCs induced apoptosis of the malignant cells in a time- and caspase-dependent manner, and decreased tumour volume in vivo.
Together, these findings highlight the benefits of encapsulation for stem cell therapies. In the brain, transplanting sECM-encapsulated stem cells increased their retention time in the resection cavity, thus allowing them to exert their therapeutic action more effectively. Encapsulation of stem cells engineered with other antitumour agents or therapeutic proteins might be useful for the treatment of other pathologies besides GBM.
Monica Hoyos Flight
ORIGINAL RESEARCH PAPER Kauer, T. M. et al. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nature Neurosci. 25 Dec 2011 (doi:10.1038/nn.3019)
D R U G D E L I V E RY
Encapsulation improves therapeutic stem cell action
PHOTODISC
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
Nature Reviews Drug Discovery | AOP, published online 20 January 2012; doi:10.1038/nrd3661
© 2012 Macmillan Publishers Limited. All rights reserved

The concept of drug-likeness — which is based on the observation that physicochemical properties of drugs, such as molecular mass and lipophilicity, tend to fall within a relatively narrow range of the possible values — is widely used to prioritize compounds in early-stage drug discovery. Hopkins and col-leagues, writing in Nature Chemistry, now present a novel approach for assessing drug-likeness that could overcome some important limita-tions of established approaches and might also better reflect the intuitive assessments of experienced medicinal chemists about the attractiveness of candidate compounds.
Standard approaches for assessing drug-likeness — which are widely used to filter large numbers of com-pounds to select those for inclusion in screening collections — typically apply several simple pass/fail cut-offs for individual physicochemical properties. For example, a compound
would fail the classic Lipinski ‘rule of 5’ guidelines (Ro5) for oral bioavail-ability if two or more of the following criteria are met: molecular mass >500 Da; calculated octanol–water partition coefficient (cLogP; a measure of lipophilicity) >5, number of hydrogen-bond donors >5; number of hydrogen-bond acceptors >10.
However, the lack of discrimina-tion of such approaches beyond simply passing or failing on particular properties means that compounds for which all properties are close to the cut-offs are considered to be equal to those that in fact may be much more likely to provide the starting point for a successful drug. Indeed, given that some properties such as lipophilicity may be more important than others, a compound that fails the Ro5 owing to a molecular mass slightly above the cut-off (for example, 502 Da) but has a favourable lipophilicity (for example, cLogP = 2) could be more drug-like than one that passes with properties just inside the limits (for example, molecular mass 499 Da, cLogP = 4.9).
To address this limitation, Hopkins and colleagues developed a measure named quantitative estimate of drug-likeness (QED), whose value ranges between 0 and 1, with 1 being the most drug-like. First, the numeri-cal or categorical physicochemical descriptors of interest (measured on different scales) are described by a desirability function that reflects the extent to which the criterion in question is favourable. These are then integrated into the single dimension-less QED score, and — importantly — the weight that each descriptor has in
the integrated value can be altered to reflect the relative importance of the descriptor to drug-likeness.
To assess how well QED could distinguish drugs from non-drugs, the authors compared its performance with other drug-likeness measures by using a set of 771 oral drugs from the DrugBank database as a positive group and small-molecule ligands from the Protein Data Bank as the negative set. QED outperformed the Ro5, as well as all other commonly used measures tested. An advantage of QED is that it provides a way to rank compounds whether they fail the Ro5 or not, and interestingly it was found that oral drugs that fail the Ro5 have QED values over a broad range, from nearly 0 to 0.8.
Additional research provided further support for the use of QED as a transparent and straightforward measure to allow compounds to be more effectively ranked by their relative merit. For example, a survey of medicinal chemists found that QED scores reflected their views on whether members of a large compound set were attractive or not for further progression if they were screening hits. Finally, the authors also suggest that this measure could be used to efficiently prioritize drug targets based on the potential to iden-tify drug-like molecules (assessed by QED) that bind to a site on the target.
Peter KirkpatrickORIGINAL RESEARCH PAPER Bickerton, G. R. et al. Quantifying the chemical beauty of drugs. Nature Chem. 24 Jan 2012 (doi: 10.1038/nchem.1243)FURTHER READING Gleeson, M. P. et al. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nature Rev. Drug Discov. 10, 197–208 (2011)
M E D I C I N A L C H E M I S T RY
Shades of chemical beauty
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
© 2012 Macmillan Publishers Limited. All rights reserved

Inhibiting the oncogenic kinase BCR–ABL1, which causes chronic myeloid leukaemia (CML), is a para-digm for clinically successful targeted therapy. However, drug-resistant mutations frequently emerge dur-ing clinical treatment. A new study shows that attempting to inhibit drug-resistant BCR–ABL1 mutants can result in a counterproductive activation of oncogenic signalling, and suggests a synergistic strategy to overcome this resistance.
Richard Marais and colleagues studied the effects of various protein kinase inhibitors on human mela-noma cells that express oncogenic NRASQ61L. They noticed that imatinib, nilotinib and dasatininb, which are all inhibitors of BCR–ABL1, paradoxically hyperactivated oncogenic signalling through the RAF–MEK–ERK kinase pathway. This also occurred in cells with activating KRAS mutations.
One possible mechanism of hyperactivating RAF–MEK–ERK signalling in RAS-mutant cells is through the partial inhibition of BRAF and CRAF: these kinases become active as both homodimers and heterodimers; when one RAF monomer is inhibited it can bind and activate a non-drug-bound RAF monomer. The authors confirmed that all three BCR–ABL1 inhibitors induced homodimerization and het-erodimerization of BRAF and CRAF, and that the activation of signalling was dependent on a physical interac-tion between these RAF proteins and an activated RAS protein.
Are these potential off-target effects of BCR–ABL1 inhibitors on RAF proteins also seen in more relevant cell types expressing BCR–ABL1? In Ba/F3 mouse pro-B
cells and human CML cells that both expressed BCR–ABL1, treat-ment with imatinib, nilotinib or dasatinib blocked RAF–MEK–ERK oncogenic signalling. However, in equivalent cells that expressed BCR–ABL1T315I (a clinically observed BCR–ABL1 mutant that is resistant to all of these inhibitors), treatment caused BRAF–CRAF heterodimerization and the hyper-activation of RAF–MEK–ERK signalling. Overall, these results suggest a model in which activation of RAS proteins (through mutation or through BCR–ABL1 activity) primes cells for RAF–MEK–ERK pathway hyperactivation through the off-target effects of BCR–ABL1 inhibitors on RAF proteins.
To test whether the RAF–MEK–ERK pathway hyperactivation could be therapeutically counteracted, the authors tested nilotinib in combina-tion with a MEK inhibitor in Ba/F3 cells and in human CML cell lines that both expressed BCR–ABL1T315I: these agents inhibited cell growth and induced apoptosis only in combination. Moreover, nilotinib
synergized with a MEK inhibitor in primary, patient-derived BCR–ABL1T315I CML cells ex vivo, and also in Ba/F3 BCR–ABL1T315I mouse allo-grafts in vivo when treatment started concurrently with the injection of cells. This synergy was also seen in a human CML cell line in which the resistance to BCR–ABL1 inhibitors was mediated by the overexpression of the tyrosine kinase LYN rather than by secondary BCR–ABL1 mutations.
It will be interesting to determine the effectiveness of this combination, relative to other therapeutic strate-gies, for treating established, drug-resistant CML in mice, and hopefully in patients with CML.
Darren J. Burgess Assistant Editor, Nature Reviews Cancer
and Nature Reviews Genetics
This article originally appeared in Nature Rev. Cancer (doi:10.1038/nrc3211).
ORIGINAL RESEARCH PAPER Packer, L. M. et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell 20, 715–727 (2011)
A N T I C A N C E R D R U G S
Keeping one step ahead
GET
TY
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
© 2012 Macmillan Publishers Limited. All rights reserved

L U N G D I S E A S E
Blocking TGFβ improves emphysemaSignalling mediated by transforming growth factor-β (TGFβ) is dysregulated in lung disorders such as emphysema. This study showed that blockade of TGFβ improved disease symptoms in a mouse model of cigarette smoke-induced emphysema. Administration of a TGFβ-specific neutralizing antibody prevented alveolar cell death, and improved lung architecture and lung mechanics. The clinically used angiotensin receptor type 1 antagonist losartan — which blocks TGFβ signalling — additionally normalized oxidative stress, inflammation, metalloproteinase activation and elastin remodelling, suggesting that TGFβ-targeted therapies could have potential in lung disease.ORIGINAL RESEARCH PAPER Podowski, M. et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J. Clin. Invest. 122, 229–240 (2012)
D R U G S A F E T Y
Predicting adverse drug reactionsCami et al. describe a computational network-based method for predicting adverse drug reactions (ADRs). They collected drug safety data from 2005 and used it to construct a network of known drug–ADR associations. This was used to train a logistic regression model to predict unknown side effects of drugs in the network. The performance of the model was evaluated by comparing these predictions with the new drug–ADR associations that were reported between 2006 and 2010. The model was able to predict seven out of eight drugs that were deemed to be associated with ADRs, highlighting that predictive network methods can be used to predict ADRs.ORIGINAL RESEARCH PAPER Cami, A. et al. Predicting adverse drug events using pharmacological network models. Sci. Transl. Med. 3, 114ra127 (2011)
N E U R O D E G E N E R AT I V E D I S O R D E R S
A neuroprotective role for sirtuin 1Cellular metabolism has a key role in the pathogenesis of Huntington’s disease (HD), which is caused by the accumulation of mutant huntingtin protein (HTT). These two studies show that sirtuin 1 (SIRT1), an NAD-dependent protein deacetylase involved in the control of cellular metabolism, has neuroprotective effects in mouse models of HD. Jiang et al. showed that overexpression of SIRT1 in mice with HD (N171-82Q mice) improved motor function, decreased brain atrophy, and attenuated the metabolic abnormalities and decline in brain-derived neurotrophic factor (BDNF) concentration induced by mutant HTT. Furthermore, they showed that mutant HTT interacts with SIRT1 to inhibit its deacetylase activity, which prevents its pro-survival function. In agreement with these findings, Jeong et al. showed that brain-specific SIRT1 overexpression improved the survival, neuropathology and expression of BDNF in another mouse model of HD (R6/2 mice), and that the deacetylase activity of the enzyme is required for its neuroprotective effects. They also showed that mutant HTT disrupts the interaction between cAMP-responsive element binding protein (CREB) and CREB-regulated transcription co-activator 1 (TORC1; indentified as a new SIRT1 substrate), which suppresses the transcription of BDNF; overexpression of SIRT1 restores this interaction by deacetylating and activating TORC1. Together, these findings suggest that modulation of SIRT1 could be beneficial in HD.ORIGINAL RESEARCH PAPERS Jiang, M. et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nature Med. 18 Dec 2011 (doi:10.1038/nm.2558) | Jeong, H. et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nature Med. 18 Dec 2011 (doi:10.1038/nm.2559)
IN BRIEF
R E S E A R C H H I G H L I G H T S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012
© 2012 Macmillan Publishers Limited. All rights reserved

Evasion of cell death is a characteristic feature of human cancers1 and represents a key cause of resistance to cur-rent treatment approaches2. Therefore, reactivation of cell death programmes in cancer cells is a promising strategy to overcome resistance to treatment, which is one of the major unsolved problems in clinical oncology. Although apoptosis is currently the best characterized form of pro-grammed cell death, additional modes of programmed cell death exist — for example, necroptosis is a regulated form of necrosis3. In principle, cell death pathways can be blocked by the abnormal expression of anti-apoptotic molecules in cancer cells. Inhibitor of apoptosis (IAP) proteins comprise a family of anti-apoptotic proteins that promote pro-survival signalling pathways and prevent the activa-tion of the effector phase of apoptosis by interfering with the activation of caspases. As overexpression of IAP pro-teins frequently occurs in various human cancers and has been linked to tumour progression, treatment failure and poor prognosis, IAP proteins are considered as promis-ing targets for therapeutic intervention. Thus, there has been much effort over the past decade to develop drugs targeting this group of proteins. Drugs that are designed to neutralize the pro-oncogenic functions of IAP proteins may be more effective at either directly eliciting cell death or lowering the threshold for cell death induction when they are combined with additional anticancer therapeutics.
IAP protein familyInhibition of apoptosis is crucial for the survival of cancer cells as well as neuronal cells, and is also crucial for the efficient replication of many viruses. The first members of the IAP protein family, encoded by the iap genes of the baculoviruses Orgyia pseudotsugata and Cydia pomo-nella, were discovered in an elegant genetic screen aimed at identifying anti-apoptotic genes that are instrumental for viral replication4,5. Subsequently, through functional screens and, more commonly, homology-searching bio-informatics screens, IAP proteins were discovered in all metazoans. There are eight IAP proteins in humans: neu-ronal apoptosis inhibitory protein (also known as BIRC1), cellular IAP1 (c-IAP1; also known as BIRC2), cellular IAP2 (c-IAP2; also known as BIRC3), X chromosome-linked IAP (XIAP; also known as BIRC4), survivin (also known as BIRC5), ubiquitin-conjugating BIR domain enzyme apollon (also known as BIRC6), melanoma IAP (ML-IAP; also known as BIRC7), and IAP-like protein 2 (ILP2; also known as BIRC8) (reviewed in REFS 6,7) (BOX 1).
All IAP proteins contain the signature baculoviral IAP repeat (BIR) domain, and some IAP proteins (such as c-IAP1, c-IAP2, XIAP and ML-IAP) also have a carboxy-terminal RING domain. These RING domain-containing IAP proteins act as E3 ubiquitin ligases — a function
1Institute for Experimental Cancer Research in Pediatrics, Goethe University Frankfurt, Komturstr. 3a, 60528 Frankfurt, Germany.2Department of Early Discovery Biochemistry, Genentech Inc., 1 DNA Way, South San Francisco, California 94080, USA.e‑mails: [email protected]; [email protected]:10.1038/nrd3627
Inhibitor of apoptosis(IAP). An anti-apoptotic protein family that encompasses the caspase inhibitor X chromosome-linked IAP and the regulators of nuclear factor-κB signalling — the cellular IAPs.
Baculoviral IAP repeat(BIR). A signature zinc ion-coordinating domain that is crucial for caspase inhibition and many protein–protein interactions.
Targeting IAP proteins for therapeutic intervention in cancerSimone Fulda1 and Domagoj Vucic2
Abstract | Evasion of apoptosis is one of the crucial acquired capabilities used by cancer cells to fend off anticancer therapies. Inhibitor of apoptosis (IAP) proteins exert a range of biological activities that promote cancer cell survival and proliferation. X chromosome-linked IAP is a direct inhibitor of caspases — pro-apoptotic executioner proteases — whereas cellular IAP proteins block the assembly of pro-apoptotic protein signalling complexes and mediate the expression of anti-apoptotic molecules. Furthermore, mutations, amplifications and chromosomal translocations of IAP genes are associated with various malignancies. Among the therapeutic strategies that have been designed to target IAP proteins, the most widely used approach is based on mimicking the IAP-binding motif of second mitochondria-derived activator of caspase (SMAC), which functions as an endogenous IAP antagonist. Alternative strategies include transcriptional repression and the use of antisense oligo nucleotides. This Review provides an update on IAP protein biology as well as current and future perspectives on targeting IAP proteins for therapeutic intervention in human malignancies.
REVIEWS
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 109
© 2012 Macmillan Publishers Limited. All rights reserved

RING domainA ubiquitin ligase domain defined by a catalytic zinc finger-like module that coordinates two zinc ions.
IAP antagonistsMolecules that bind to inhibitor of apoptosis (IAP) proteins and block IAP-mediated inhibition of cell death.
Extrinsic (death receptor) pathway of apoptosisAn apoptotic pathway initiated at the plasma membrane by specific transmembrane receptors of the tumour necrosis factor receptor superfamily. Ligation of these receptors results in the formation of a death-inducing signalling complex that drives caspase 8 activation.
that appears to be increasingly important for their role in survival and signalling pathways8,9. In addition to the BIR and RING domains, IAP proteins have several other domains that are mostly involved in protein–pro-tein interactions10–16 (BOX 1). In this Review we focus on the classical anti-apoptotic IAP proteins: XIAP, c-IAP1, c-IAP2 and ML-IAP. The biological functions of other IAP proteins have been reviewed in REFS 17–19.
Functions of IAP proteinsRegulation of programmed cell death by IAP proteins. The execution of programmed cell death can proceed via distinct pathways, including apoptosis, necroptosis and autophagic cell death3. As IAP antagonists do not appear to be involved in the regulation of autophagy2,19, this form of programmed cell death is not discussed here. Although apoptosis, in most circumstances, involves the activa-tion of caspases as effector molecules, caspase activity negatively controls necroptosis3. There are two major apoptosis signalling pathways: the extrinsic (death recep-tor) pathway of apoptosis and the intrinsic (mitochondrial) pathway of apoptosis20 (FIG. 1).
The extrinsic (death receptor) pathway of apop-tosis is characteristically initiated by the ligation of
death receptors on the cell surface via their respective ligands21. Death receptors belong to the tumour necrosis factor receptor (TNFR) family and comprise CD95 (also known as FAS), TNF-related apoptosis-inducing ligand (TRAIL; also known as APO2L) receptors and TNFR, among other receptors21. The binding of death receptor ligands such as CD95 ligand, TRAIL or TNF to their respective receptors stimulates the recruitment of sig-nalling molecules to the activated death receptor and the formation of the so-called death-inducing signalling complex (DISC)21. The DISC contains FAS-associated death domain protein (FADD) as an adaptor molecule, the initiator caspases caspase 8 and caspase 10 as well as — depending on the cell line — cellular FLICE-like inhibitory protein (also known as CFLAR)21. Following its recruitment to the DISC, caspase 8 becomes acti-vated via oligomerization21. Activated caspase 8 can then either directly cleave downstream effector caspases such as caspase 3 or it can indirectly promote caspase 3 activation by initiating mitochondrial perturbations via cleavage of BID (BH3-interacting domain death agonist)21. Following its processing into the active form — truncated BID — by caspase 8, truncated BID is translocated to the mitochondria to stimulate mito-chondrial outer membrane permeabilization21.
The intrinsic (mitochondrial) pathway of apoptosis is mediated by the release of apoptogenic proteins, includ-ing cytochrome c and second mitochondria-derived activa-tor of caspase (SMAC; also known as DIABLO), from the mitochondrial intermembrane space into the cytosol22. In the cytosol, cytochrome c promotes the assembly of the apoptosome complex, which contains cytochrome c, caspase 9 and apoptotic protease-activating factor 1 (REF. 22). This apoptosome complex drives the activa-tion of caspase 9 and subsequently of caspase 3 (REF. 22). SMAC promotes caspase activation and apoptosis by binding to several IAP proteins, including XIAP, c-IAP1 and c-IAP2, via its amino-terminal amino acid residues (Ala-Val-Pro-Ile) that are highly conserved through evo-lution23–25. The mitochondrial pathway of apoptosis is tightly controlled by pro- and anti-apoptotic proteins of the BCL-2 family26.
Although IAP proteins were initially thought to block apoptosis by directly binding to and inhibiting caspases, it was later shown that only XIAP is a potent direct inhibitor of caspase 3, caspase 7 and caspase 9 (REF. 27) (FIG. 1). Structural studies have shown that inhibition of caspase 9 by XIAP involves the interaction of the pep-tide-binding groove of the BIR3 domain of XIAP with the N terminus of the small subunit p12 of processed caspase 9; this region contains a four-amino-acid IAP-binding sequence that is homologous to a sequence that is also present in SMAC28,29. In addition, the interaction of the BIR3 domain of XIAP with caspase 9 prevents the homodimerization of caspase 9 and its subsequent activation28,29.
Inhibition of caspase 3 and caspase 7 involves the interaction of the linker region between the BIR1 and BIR2 domains of XIAP with the substrate-binding site of the activated caspase30–33. The peptide-binding groove on the surface of the BIR2 domain of XIAP also makes
Box 1 | Structural organization of human IAP proteins
Eight inhibitor of apoptosis (IAP) proteins are found in humans: neuronal apoptosis inhibitory protein (NAIP; also known as BIRC1); cellular IAP1 (c-IAP1; also known as BIRC2); cellular IAP2 (c-IAP2; also known as BIRC3); X chromosome-linked IAP (XIAP; also known as BIRC4); survivin (also known as BIRC5); BIR-containing ubiquitin-conjugating BIR domain enzyme apollon (also known as BIRC6); melanoma IAP (ML-IAP; also known as Livin or BIRC7); and IAP-like protein 2 (ILP2; also known as BIRC8) (reviewed in REFS 6,7). The unifying feature of all IAP proteins is the baculoviral IAP repeat (BIR) domain. Up to three copies of this conserved 70–80 amino-acid zinc-binding domain are present in IAP proteins232,233 (for example, ML-IAP and survivin contain one copy, whereas XIAP and c-IAP proteins contain three copies). BIR domains are required for the majority of the IAP-mediated protein–protein interactions and for the inhibition of apoptosis. Several IAP proteins (such as c-IAP1, c-IAP2, XIAP and ML-IAP) contain a carboxy-terminal RING domain, which provides these IAP proteins with a E3 ubiquitin ligase activity8,9. Besides BIR and RING domains, c-IAPs, XIAP and ILP2 harbour a centrally located ubiquitin-asso-ciated (UBA) domain10,11. The UBA domains of IAP proteins can bind to monoubiquitin as well as polyubiquitin chains composed of various linkages10,11. Some IAP proteins also have additional protein domains such as the caspase recruitment domain (CARD) of c-IAP proteins, the ubiquitin-conjugating (UBC) domain of apollon, the coiled-coil region of survivin, and the nucleotide-binding and oligomerization domain as well as the leucine-rich repeat (LRR) domains of NAIP12–16.
NACHT, domain present in NAIP, MHC class II transactivator (CIITA), 20-hydroxyeicosatetraenoic acid synthase (HET-E) and transition protein 1 (TP1). Nature Reviews | Drug Discovery
NAIP
cIAP1
cIAP2
XIAP
MLIAP
ILP2
Survivin
Apollon
BIR RING Coiled-coilNACHT CARDUBA UBCLRR
R E V I E W S
110 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Intrinsic (mitochondrial) pathway of apoptosisAn apoptotic pathway that is triggered by cellular stresses (such as DNA damage) or intracellular stimuli (such as the production of reactive oxygen species or calcium overload).
Tumour necrosis factor(TNF). A cytokine that is involved in systemic inflammation and is necessary for the single-agent pro-apoptotic activity of inhibitor of apoptosis protein antagonists.
Second mitochondria-derived activator of caspase(SMAC). A pro-apoptotic protein that leaves mitochondria and enters the cytoplasm following apoptotic insult; in the cytoplasm, it binds to and neutralizes inhibitor of apoptosis proteins.
Nuclear factor-κB(NF-kB). A protein complex that regulates the expression of several cytokines and anti-apoptotic proteins. NF-kB signalling proceeds via the canonical or non-canonical pathways.
Canonical NF-κB pathwayA signalling pathway mediated by nuclear factor-κB (NF-κB) that predominantly results in the activation of transcription factor RELA (also known as p65)–p50 heterodimers.
important interactions with the N termini of the small subunits of partially processed caspase 3 and caspase 7 (REF. 34). Other IAP proteins, such as c-IAP proteins and ML-IAP, are not potent inhibitors of caspases27,35. Instead, they bind to SMAC with high affinities and prevent it from blocking XIAP-mediated inhibition of caspases35. In addition, the E3 ubiquitin ligase activity of IAP proteins allows them to regulate the stability of many cell death modulators and effectors — including caspases and IAP proteins themselves — with direct consequences on cellular survival (reviewed in REF. 36).
Besides SMAC, additional IAP-binding proteins have been identified, including high temperature requirement protein A2 (HTRA2, also known as OMI), which is a mammalian homologue of the bacterial heat-inducible serine protease HtrA37–40, apoptosis-related protein in the TGFb signalling pathway (ARTS; also known as SEPT4)235 and XIAP-associated factor 1 (XAF1), which binds to several IAP proteins includ-ing XIAP, c-IAP1, c-IAP2, ML-IAP, IAP-like protein 2 and neuronal apoptosis inhibitory protein41,42.
Role of IAP proteins in signalling pathways. In addi-tion to controlling cellular survival by inhibiting cell death, several IAP proteins — particularly c-IAP1 and c-IAP2 — are crucial regulators of pro-survival nuclear factor-κB (NF-κB) signalling pathways (FIG. 2). The NF-κB transcription factor family has five members, and the homodimers and heterodimers of these proteins regulate the transcription of NF-κB-responsive genes including cIAP2, TNF, BCL2 and many other modula-tors of apoptosis (reviewed in REF. 43). The expression of NF-κB-regulated genes can be achieved through canonical (classical) and non-canonical (alternative) signalling pathways.
In the canonical NF-κB pathway, the inhibitor of NF-κB (IκB) protein binds to NF-κB proteins to keep them in the cytoplasm of unstimulated cells. The binding of TNF to TNFR1 triggers the formation of a signalling com-plex, which recruits TNFR1-associated death domain protein, receptor-interacting protein 1 (RIP1) and TNFR-associated factor 2 (TRAF2), as well as c-IAP1 and c-IAP2, leading to c-IAP-dependent RIP1 ubiqui-tylation44–48. Ubiquitylated RIP1 serves as a platform for the recruitment of the kinase complex formed by TGFβ-activated kinase 1 and TAK1-binding protein (the TAK1–TAB kinase complex), the scaffold protein NF-κB essential modulator (NEMO), and the IκB kinase (IKK) complex, and the linear ubiquitin chain assembly complex. This leads to the activation of IKKβ, which in turn phosphorylates IκBα, prompting its polyubiquityla-tion and subsequent degradation (reviewed in REF. 43).
The ubiquitin ligase activity of c-IAP proteins is instrumental for the activation of the canonical NF-κB signalling pathway by TNF as well as by other ligands of the TNF family, such as TNF ligand superfamily mem-ber 15 (also known as TL1A), TNF-related weak inducer of apoptosis (TWEAK; also known as TNFSF12), LIGHT, CD27 (also known as TNFRSF7) and CD40 ligand as well as transmembrane activator and CAML interactor (TACI; also known as TNFRSF13B)36,49. The
other E3 ubiquitin ligase that participates in these signal-ling pathways is HOIL1-interacting protein (HOIP)48,50. In coordination with haem-oxidized IRP2 ubiquitin ligase 1 (HOIL1; also known as RBCK1) and Sharpin, HOIP promotes the assembly of linear ubiquitin chains on NEMO and RIP1, and stabilizes the signalling complexes51–53.
In contrast to their positive role in the regulation of canonical NF-κB signalling, c-IAP proteins negatively regulate non-canonical NF-κB signalling by mediating degradative ubiquitylation of NF-κB-inducing kinase (NIK; also known as MAP3K14)59 (FIG. 2). NIK protein levels are extremely low in unstimulated cells owing to constitutive ubiquitin-dependent proteasomal degrada-tion of NIK within a cytoplasmic complex comprising c-IAP proteins, TRAF2, TRAF3 and NIK54–60. Following the binding of their ligands, several members of the TNFR family — including the TWEAK receptor FN14 (fibroblast growth factor-inducible 14; also known as TNFRSF12A), the CD40 ligand receptor (also known as TNFRSF5) and the lymphotoxin-β receptor (LT-βR) — engage TRAF2, TRAF3 and the c-IAP proteins into their signalling complexes, which causes c-IAP-depend-ent ubiquitylation and degradation of the c-IAP proteins themselves, in addition to the degradation of TRAF2 and TRAF3 (REFS 36,49,61). The depletion of c-IAP pro-teins, TRAF2 and TRAF3 leads to NIK stabilization and accumulation as well as the subsequent phosphorylation and activation of IKKα62,63. Activated IKKα phosphoryl-ates the NF-κB precursor protein p100 (also known as NF-κB2), causing its partial proteasomal processing to generate the p52 NF-κB subunit (reviewed in REF. 64). Thus, c-IAP proteins are crucial regulators of NF-κB activation, and their antagonism could have a consider-able impact on these signalling pathways.
IAP proteins in tumoursThe expression and/or function of IAP proteins are deregulated in many human cancers because of genetic aberrations, an increase in their mRNA or protein expression or the loss of endogenous inhibitors such as SMAC. The expression levels of IAP proteins and their antagonists have been correlated with clinical param-eters and cancer prognosis in several retrospective tri-als, as discussed below. However, these results should be interpreted with caution because of the low numbers of samples studied in some reports and the limitations of currently available reagents for analysing the expression of IAP proteins in tissue specimens. Therefore, addi-tional studies are required to evaluate the prognostic value of IAP proteins in human malignancies.
XIAP and c-IAP proteins in cancer. Genetic evidence indicates that cIAP1 and cIAP2 are proto-oncogenes that are affected by chromosomal aberrations in can-cers. The 11q21-q23 amplification, which harbours both cIAP1 and cIAP2, has been identified in vari-ous types of tumours (Supplementary information S1 (table)), including esophageal carcinoma65, hepatocel-lular carcinoma, cervical cancer66, liver cancer67, medul-loblastoma68, glioblastoma69, non-small-cell lung cancer
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 111
© 2012 Macmillan Publishers Limited. All rights reserved
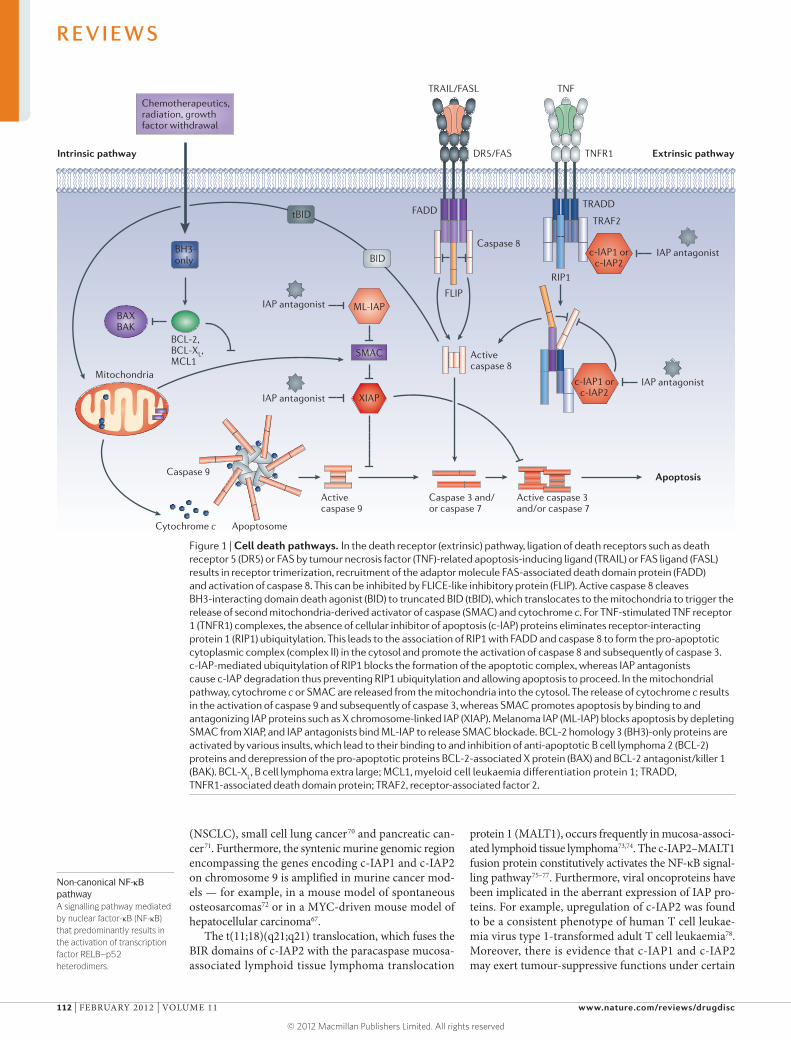
Non-canonical NF-κB pathwayA signalling pathway mediated by nuclear factor-κB (NF-κB) that predominantly results in the activation of transcription factor RELB–p52 heterodimers.
(NSCLC), small cell lung cancer70 and pancreatic can-cer71. Furthermore, the syntenic murine genomic region encompassing the genes encoding c-IAP1 and c-IAP2 on chromosome 9 is amplified in murine cancer mod-els — for example, in a mouse model of spontaneous osteosarcomas72 or in a MYC-driven mouse model of hepatocellular carcinoma67.
The t(11;18)(q21;q21) translocation, which fuses the BIR domains of c-IAP2 with the paracaspase mucosa-associated lymphoid tissue lymphoma translocation
protein 1 (MALT1), occurs frequently in mucosa-associ-ated lymphoid tissue lymphoma73,74. The c-IAP2–MALT1 fusion protein constitutively activates the NF-κB signal-ling pathway75–77. Furthermore, viral oncoproteins have been implicated in the aberrant expression of IAP pro-teins. For example, upregulation of c-IAP2 was found to be a consistent phenotype of human T cell leukae-mia virus type 1-transformed adult T cell leukaemia78. Moreover, there is evidence that c-IAP1 and c-IAP2 may exert tumour-suppressive functions under certain
Figure 1 | Cell death pathways. In the death receptor (extrinsic) pathway, ligation of death receptors such as death receptor 5 (DR5) or FAS by tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) or FAS ligand (FASL)results in receptor trimerization, recruitment of the adaptor molecule FAS-associated death domain protein (FADD) and activation of caspase 8. This can be inhibited by FLICE-like inhibitory protein (FLIP). Active caspase 8 cleaves BH3-interacting domain death agonist (BID) to truncated BID (tBID), which translocates to the mitochondria to trigger the release of second mitochondria-derived activator of caspase (SMAC) and cytochrome c. For TNF-stimulated TNF receptor 1 (TNFR1) complexes, the absence of cellular inhibitor of apoptosis (c-IAP) proteins eliminates receptor-interacting protein 1 (RIP1) ubiquitylation. This leads to the association of RIP1 with FADD and caspase 8 to form the pro-apoptotic cytoplasmic complex (complex II) in the cytosol and promote the activation of caspase 8 and subsequently of caspase 3. c-IAP-mediated ubiquitylation of RIP1 blocks the formation of the apoptotic complex, whereas IAP antagonists cause c-IAP degradation thus preventing RIP1 ubiquitylation and allowing apoptosis to proceed. In the mitochondrial pathway, cytochrome c or SMAC are released from the mitochondria into the cytosol. The release of cytochrome c results in the activation of caspase 9 and subsequently of caspase 3, whereas SMAC promotes apoptosis by binding to and antagonizing IAP proteins such as X chromosome-linked IAP (XIAP). Melanoma IAP (ML-IAP) blocks apoptosis by depleting SMAC from XIAP, and IAP antagonists bind ML-IAP to release SMAC blockade. BCL-2 homology 3 (BH3)-only proteins are activated by various insults, which lead to their binding to and inhibition of anti-apoptotic B cell lymphoma 2 (BCL-2) proteins and derepression of the pro-apoptotic proteins BCL-2-associated X protein (BAX) and BCL-2 antagonist/killer 1 (BAK). BCL-X
L, B cell lymphoma extra large; MCL1, myeloid cell leukaemia differentiation protein 1; TRADD,
TNFR1-associated death domain protein; TRAF2, receptor-associated factor 2.
Nature Reviews | Drug Discovery
ML-IAP
XIAP
SMAC
Activecaspase 9
Caspase 9
ApoptosomeCytochrome c
Caspase 3 and/or caspase 7
Active caspase 3 and/or caspase 7
Apoptosis
BH3-only
Mitochondria
BCL-2,BCL-XL,MCL1
BAXBAK
FLIP
RIP1
Caspase 8
Activecaspase 8
DR5/FAS
TRAIL/FASL
TNFR1
TRADD
TRAF2
TNF
FADD
IAP antagonist
Intrinsic pathway Extrinsic pathway
Chemotherapeutics, radiation, growth factor withdrawal
IAP antagonist
IAP antagonist
IAP antagonist
c-IAP1 orc-IAP2
c-IAP1 orc-IAP2
tBID
BID
R E V I E W S
112 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Duke’s stagingClassification system for colorectal cancer based on pathology.
circumstances. In multiple myeloma, deletions of c-IAP1 and c-IAP2 were found to cause constitutive aberrant activation of the non-canonical NF-κB pathway via stabi-lization of NIK54,55.
There are diverging reports on the prognostic rel-evance of XIAP in acute myeloid leukaemia (AML). Although two studies reported that low levels of XIAP expression correlated with better overall survival and favourable cytogenetics in AML79,80, another study found no prognostic impact of XIAP expression in this malig-nancy81. Interestingly, a three-gene expression signature including c-IAP2 as one of the three highly expressed genes was identified to accurately predict poor overall survival in newly diagnosed patients with AML82. In childhood de novo AML, high levels of XIAP mRNA and XIAP protein constitute an independent poor prognos-tic factor83,84, whereas in paediatric T cell acute lympho-blastic leukaemia (ALL), high levels of XIAP expression correlated with poor response to prednisone85. Patients with chronic lymphocytic leukaemia (CLL) who expe-rienced progressive disease were reported to express higher levels of several IAP proteins, but lower levels of SMAC compared to patients with stable disease86. In addition, upregulation of c-IAP1 was observed in CLL cells that were resistant to DNA damage-induced apo-ptosis87; however, in another study the levels of c-IAP1, c-IAP2 and XIAP expression in patients with CLL did not correlate with prognostic factors or sensitivity to the chemotherapeutic drug fludarabine88. Overexpression of XIAP, which occurred in 55% of patients with diffuse large B cell lymphoma, significantly correlated with poor clinical outcome89.
In clear-cell renal cell carcinoma (RCC), XIAP expression levels were reported to significantly increase with advancing tumour stage, tumour dedifferentia-tion and aggressive tumour growth90,91. In addition, the XIAP/SMAC ratio steadily increased, as SMAC expres-sion remained largely unaltered91. Furthermore, XIAP expression was identified as an independent prognostic parameter in RCC where it correlates with a lower 5-year survival rate90,92. In bladder cancer, high levels of XIAP expression correlated with tumour dedifferentiation and significantly lower recurrence-free survival rates93, and independently predicted the recurrence of non-muscular invasive bladder cancer in a multivariate analysis93. In addition, high levels of nuclear c-IAP1 expression signifi-cantly correlated with tumour stage and tumour grade as well as decreased overall and recurrence-free survival94.
Notably, higher levels of XIAP expression correlated with venous invasion, Duke’s staging, tumour dediffer-entiation and lower survival rates in colorectal cancer, and proved to be an independent prognostic factor by multivariate analysis95. In addition, high levels of c-IAP2 expression correlated with a significantly lower survival rate in colorectal cancer96. Furthermore, c-IAP2 expres-sion in pancreatic ductal adenocarcinomas was higher than in normal tissues, which correlated with a lower survival rate97. In hepatocellular carcinoma, a high XIAP/XAF1 ratio in cirrhotic tissues or elevated XIAP expression were identified as indicators of poor sur-vival98,99. An elevated XIAP/XAF1 ratio also correlated
with poorer survival in gastric adenocarcinoma100. Unexpectedly, high XIAP levels correlated with a lower rather than higher probability of tumour recurrence in prostate cancer, and XIAP was identified as an inde-pendent predictor of tumour recurrence in a multivari-ate analysis101. Similarly, in an independent study higher levels of XIAP expression were associated with longer relapse-free survival102.
In breast carcinoma, nuclear — but not cytoplas-mic — expression of XIAP correlated with shortened overall survival103. In addition, high levels of nuclear c-IAP1 were predictive of poor overall survival and local recurrence-free survival in cervical squamous cell car-cinomas, and were shown to be independent prognostic factors by multivariate analysis66; XIAP or c-IAP2 levels, however, were not correlated with disease stage or sur-vival in this type of cancer104.
The prognostic relevance of individual IAP proteins in NSCLC is not yet clear, as one study reported that overall survival was significantly increased in patients with high levels of XIAP105, but another study reported that the expression of c-IAP1, c-IAP2 and XIAP was not predictive of the response to chemotherapy in patients with advanced NSCLC106. In melanoma, higher levels of XIAP expression correlated with progressive disease stage107. In addition, c-IAP1 expression was reported to correlate with nodal metastasis in squamous cell carci-noma of the tongue108. Nuclear c-IAP1 expression corre-lated with lymph node metastasis and advanced disease stage in head and neck squamous cell carcinomas, but its association with poor prognosis was not found to be statistically significant109.
ML-IAP in cancer. ML-IAP is not expressed in most normal human tissues but it is expressed in various cancers including melanoma and renal cancer 110–112 (Supplementary information S1 (table)). Interestingly, elevated levels of ML-IAP were associated with a favour-able rather than poor prognosis in some studies, possi-bly because of the pro-apoptotic function of its cleaved form113. The expression profile of ML-IAP in melanomas demonstrates a strong bias for primary and especially metastatic melanomas, with marginal to no expression in melanocytes or naevi110,112. This preference for ML-IAP expression in melanomas is attributed to the regulation of ML-IAP by the lineage survival oncogene microphthal-mia-associated transcription factor (MITF)114.
In RCCs, ML-IAP mRNA and ML-IAP protein expression was detected in about 40% of tumour speci-mens, whereas the adjacent normal renal tissues were negative for ML-IAP115. By comparison, transcripts of both ML-IAP splice variants — the α- and β-isoforms — were found to be expressed in the tumour tissue as well as in the corresponding normal renal tissue without showing any obvious qualitative differences111. Although a high level of nuclear ML-IAP expression was identi-fied as an independent favourable predictor of progres-sion-free survival and cancer-specific survival in RCC by a multivariate analysis116, in another study ML-IAP expression did not correlate with pathological or clinical parameters or with patient outcome115.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 113
© 2012 Macmillan Publishers Limited. All rights reserved
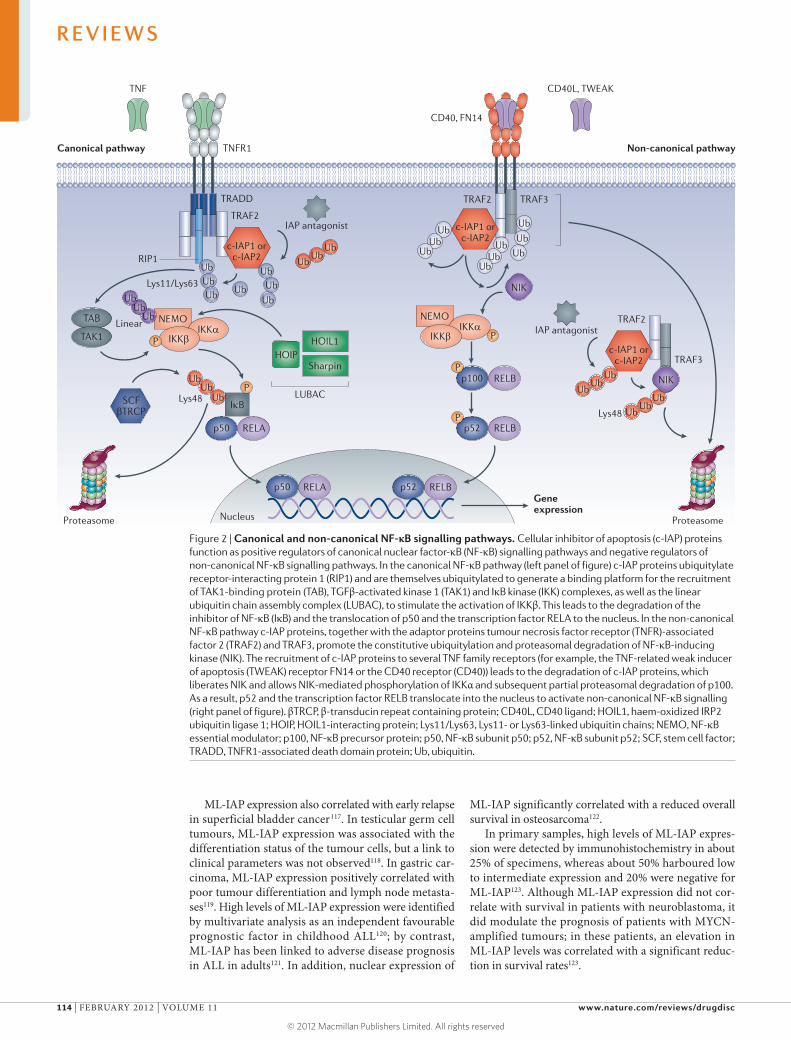
ML-IAP expression also correlated with early relapse in superficial bladder cancer117. In testicular germ cell tumours, ML-IAP expression was associated with the differentiation status of the tumour cells, but a link to clinical parameters was not observed118. In gastric car-cinoma, ML-IAP expression positively correlated with poor tumour differentiation and lymph node metasta-ses119. High levels of ML-IAP expression were identified by multivariate analysis as an independent favourable prognostic factor in childhood ALL120; by contrast, ML-IAP has been linked to adverse disease prognosis in ALL in adults121. In addition, nuclear expression of
ML-IAP significantly correlated with a reduced overall survival in osteosarcoma122.
In primary samples, high levels of ML-IAP expres-sion were detected by immunohistochemistry in about 25% of specimens, whereas about 50% harboured low to intermediate expression and 20% were negative for ML-IAP123. Although ML-IAP expression did not cor-relate with survival in patients with neuroblastoma, it did modulate the prognosis of patients with MYCN-amplified tumours; in these patients, an elevation in ML-IAP levels was correlated with a significant reduc-tion in survival rates123.
Figure 2 | Canonical and non-canonical NF-κB signalling pathways. Cellular inhibitor of apoptosis (c-IAP) proteins function as positive regulators of canonical nuclear factor-κB (NF-κB) signalling pathways and negative regulators of non-canonical NF-κB signalling pathways. In the canonical NF-κB pathway (left panel of figure) c-IAP proteins ubiquitylate receptor-interacting protein 1 (RIP1) and are themselves ubiquitylated to generate a binding platform for the recruitment of TAK1-binding protein (TAB), TGFβ-activated kinase 1 (TAK1) and IκB kinase (IKK) complexes, as well as the linear ubiquitin chain assembly complex (LUBAC), to stimulate the activation of IKKβ. This leads to the degradation of the inhibitor of NF-κB (IκB) and the translocation of p50 and the transcription factor RELA to the nucleus. In the non-canonical NF-κB pathway c-IAP proteins, together with the adaptor proteins tumour necrosis factor receptor (TNFR)-associated factor 2 (TRAF2) and TRAF3, promote the constitutive ubiquitylation and proteasomal degradation of NF-κB-inducing kinase (NIK). The recruitment of c-IAP proteins to several TNF family receptors (for example, the TNF-related weak inducer of apoptosis (TWEAK) receptor FN14 or the CD40 receptor (CD40)) leads to the degradation of c-IAP proteins, which liberates NIK and allows NIK-mediated phosphorylation of IKKα and subsequent partial proteasomal degradation of p100. As a result, p52 and the transcription factor RELB translocate into the nucleus to activate non-canonical NF-κB signalling (right panel of figure). βTRCP, β-transducin repeat containing protein; CD40L, CD40 ligand; HOIL1, haem-oxidized IRP2 ubiquitin ligase 1; HOIP, HOIL1-interacting protein; Lys11/Lys63, Lys11- or Lys63-linked ubiquitin chains; NEMO, NF-κB essential modulator; p100, NF-κB precursor protein; p50, NF-κB subunit p50; p52, NF-κB subunit p52; SCF, stem cell factor; TRADD, TNFR1-associated death domain protein; Ub, ubiquitin.
Nature Reviews | Drug Discovery
HOIP
p52
TNFR1Canonical pathway Non-canonical pathway
TRADD
TRAF2
RIP1
Lys11/Lys63
LUBAC
Linear
TNF
UbUbUbUb
Ub
UbUb
Ub
UbUb
Ub
UbUb
Ub
UbUb
CD40, FN14
TRAF3
TRAF3
TRAF2
TRAF2
CD40L, TWEAK
SCFβTRCP
UbUb
Ub
UbUb
Ub
Ub
Ub
Ub
NIK
NIK
Lys48
Proteasome Proteasome
Pp50 RELA
P
p100
TAB
TAK1
P
NEMO
HOIL1
Sharpin
IKKαIKKβ
NEMOIKKα
IKKβP
UbUb
Ub
IκB
UbUb
UbLys48P
p52p50 RELA RELB
RELB
RELB
Nucleus
Gene expression
IAP antagonist
IAP antagonist
c-IAP1 orc-IAP2
c-IAP1 orc-IAP2
c-IAP1 orc-IAP2
R E V I E W S
114 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

SMAC in cancer. Low-level SMAC expression cor-related with advanced tumour stage, high grade and unfavourable prognosis with reduced recurrence-free and tumour-specific survival in RCC124,125. In blad-der carcinoma, SMAC expression inversely correlated with the stage and grade of bladder cancer, and 5-year recurrence-free or disease-specific survival rates were higher in patients expressing higher levels of SMAC126. Lack of SMAC expression in colorectal carcinoma cor-related with a higher incidence of metastasis and shorter survival, and was an independent poor prognostic indi-cator127. Similarly, high levels of SMAC expression in rectal cancer tissues before treatment were identified as an independent prognostic factor for significantly improved 5-year disease-free survival rates128.
In breast carcinoma, SMAC expression was found to inversely correlate with tumour stage and infiltra-tive growth129. Higher levels of SMAC expression in endometrial cancers significantly correlated with lower tumour grade and longer disease-specific survival130. High SMAC expression was associated with early local recurrence rate in cervical cancer131, whereas low lev-els of SMAC were linked with progressive tumour stage and poorer prognosis in lung cancer132. In addition, high levels of SMAC expression in AML were reported to be an independent prognostic factor associated with longer overall survival, higher complete remission rate and a favourable karyotype133.
Targeting IAP proteins for cancer therapyGiven their elevated expression in many tumour types, their functional importance for the survival of tumour tissues and their resistance to anticancer therapies, it is not surprising that IAP proteins are considered to be attractive targets for therapeutic intervention in human malignancies. Several targeting strategies have been explored thus far and, among them, small-molecule IAP antagonists and antisense oligonucleotides have garnered the most attention7,134. FIGURE 3 shows a selec-tion of compounds currently in preclinical or clinical development.
SMAC-derived peptides. Almost 15 years ago it was dis-covered that the Drosophila melanogaster pro-apoptotic proteins Reaper, Head involution defective (HID; also known as Wrinkled) and the cell death protein Grim can bind to and inhibit the anti-apoptotic activity of D. mel-anogaster and baculoviral IAP proteins; this suggested that IAP proteins could be effectively antagonized135,136. Furthermore, these studies demonstrated that several amino acid residues at the N terminus of endogenous D. melanogaster antagonists are responsible for the bind-ing and antagonism of IAP proteins. The subsequent identification of the human IAP antagonist SMAC and structural studies on the BIR domains of IAP as well as the SMAC protein and SMAC-derived peptides defined the precise region that is involved in binding to selected BIR domains of IAP23,137,138.
The revelation that the IAP-binding motif contains only four amino acids (Ala-Val-Pro-Ile), coupled with a detailed understanding of the topology of the SMAC
peptide binding site on the BIR domains of IAP, spurred considerable interest as it presented a potential binding pocket for small-molecule therapeutic antagonists24,25. Indeed, peptides derived from the N terminus of mature active SMAC were shown to mimic the activity of the SMAC protein and were used to validate the targeting of IAP proteins by small-molecule antagonists134,139. Ensuing explorations of possible permutations of IAP BIR domain-binding peptides revealed that the naturally occurring alanine residue in the N-terminal position is highly preferred for binding and that the Ala-Val-Pro-Phe sequence found in D. melanogaster HID was among the best binding sequences136,140,141.
SMAC peptides could effectively block IAP–caspase interactions and sensitize a variety of cancers to pro-apoptotic stimuli142, indicating a wide applicability of this approach. Even more importantly, a crucial study demonstrated the in vivo antitumour efficacy of SMAC peptides in combination with TRAIL in a malignant glioma xenograft model143. However, although SMAC-based peptides showed efficacy in cell culture experi-ments, and even in xenograft studies, they do not possess good pharmacological properties. Therefore, they could not be considered for development as therapeutic agents. Nevertheless, these and other early studies provided sci-entific and technological validation for the development of small-molecule IAP antagonists for the treatment of human cancers142–145.
SMAC-mimicking IAP antagonists. The development of SMAC-based peptidomimetics identified novel struc-tures with improved pharmacological properties and improved IAP-binding affinities146–149. An important step in the development of these peptidomimetics was the introduction of increased rigidity into the central scaffold. For instance, one study reported a compound with a rigid bicyclic scaffold and N-methylalanine in the P1 position that effectively bound to the BIR domains of IAP and promoted the death of treated cancer cells as a single agent150. Parallel and subsequent efforts allowed the identification of several different monovalent IAP antagonists that were capable of inducing apoptosis and sensitizing cancer cells to a broad spectrum of death-acti-vating stimuli151–154. A second category of SMAC mimet-ics are bivalent or dimeric compounds, which consist of two SMAC mimetics connected with a chemical linker.
Interestingly, Lipinski’s rules (also known as the rule-of-five guidelines), a set of parameters that evalu-ate the drug-likeness of a chemical compound155, do not predict the potential utility of bivalent IAP antagonists. However, a serendipitous discovery of a bivalent SMAC mimetic compound has led to the development of a new class of IAP antagonists and considerably expanded the repertoire of IAP-targeting agents156. Bivalent or dimeric IAP antagonist compounds are composed of two mon-ovalent units that are connected through a chemical linker7 (FIG. 3). Bivalent antagonists generally display higher binding affinities than monovalent compounds for IAP constructs; they promote the dimerization of BIR2–BIR3 constructs of c-IAP1, and BIR3 constructs of XIAP59,157.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 115
© 2012 Macmillan Publishers Limited. All rights reserved
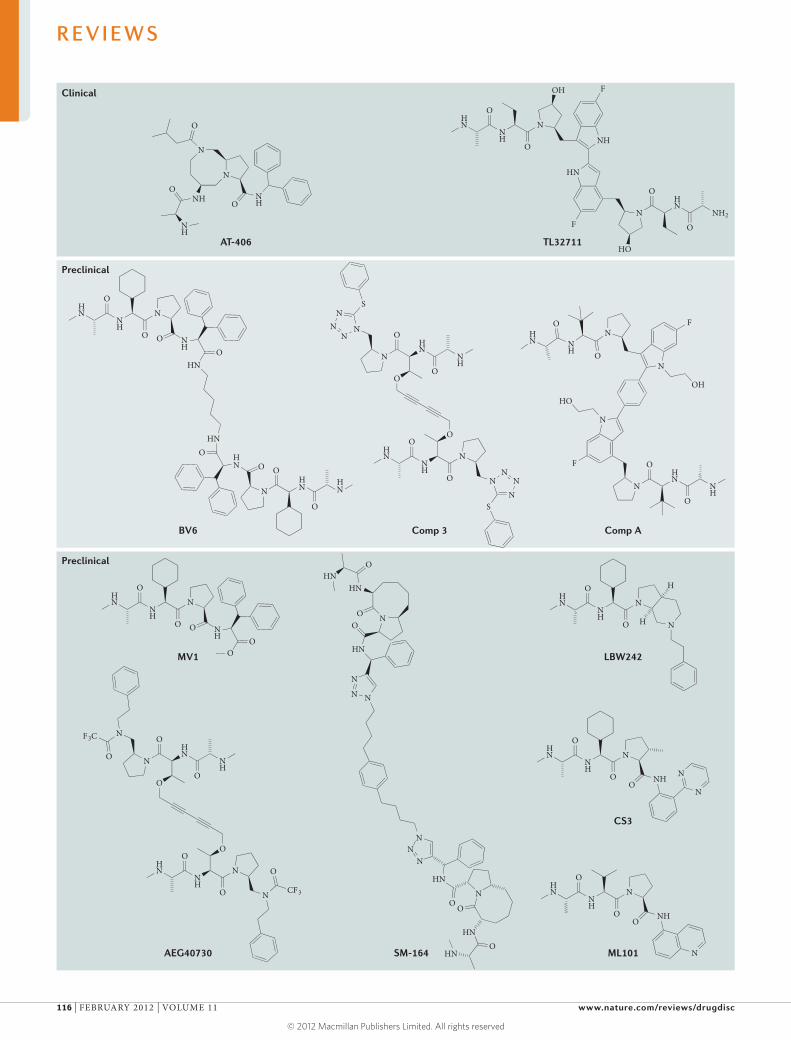
Nature Reviews | Drug Discovery
HN
NH
OO
O
HN
NH
O
O
N
N
O
N
N
O
F3C
O
CF3HN
NH
N
O
OO
NH
N
HN
NH
N
O
O NH O
O
O
HN
NH
N
O
O N
H
H
HN
NH
N
O
OO
NHN
N
F
N
OH
N
ONH
OHN
N
HO
F
N
HN
NH
O
O
HN
FN
HO
HN
NH2
O
O
NH
F
NNH
HN
OH
O
O
HN
NH
OO
O
HN
NH
O
O
N
N
NN
N
S
O
N
N
N
NN
S
HN
HN
HN
NH
N
O
O NH
NH
NNH
NH
O
O
O O
O
O
N
N
NHO
NH
O
NHO
AT-406
Comp 3
TL32711
CS3
BV6
AEG40730 ML101
Comp A
LBW242MV1
SM-164
HN
OHN
ONO
N
N
N
HN
NN
N
HN
N
O
O
O
HN
HN
Clinical
Preclinical
Preclinical
R E V I E W S
116 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

However, the most striking feature of bivalent antag-onists is their high potency in cell death assays and tumour growth inhibition assays44,59,60,158. It is difficult to provide a clear explanation for this increased potency, as the activity of bivalent compounds is often much higher (10-fold to 100-fold) than the activity observed when two monovalent compounds are combined. It has been shown that bivalent antagonists can effectively promote c-IAP dimerization and the consequent ubiquitin-mediated proteasomal degradation of c-IAP proteins59,60. However, the ability of bivalent compounds to simulta-neously engage the BIR2 and BIR3 domains of XIAP, which results in prominent caspase 3 and caspase 7 acti-vation, is probably more relevant159,160. In cells in which XIAP represents a major resistance factor, efficient neu-tralization of this unique endogenous inhibitor of cas-pases is particularly important for the combined use of IAP antagonists with other death-activating stimuli160,161.
Mechanism of action of IAP antagonists. IAP antago-nists bind to XIAP, c-IAP1, c-IAP2 and ML-IAP, and prevent the association of these proteins with caspases and SMAC; they also prevent XIAP-mediated inhibi-tion of caspase 3, caspase 7 and caspase 9. However, IAP antagonists have the most profound effect on c-IAP lev-els. Monovalent and bivalent IAP antagonists stimulate rapid autoubiquitylation and proteasomal degradation of c-IAP1 and c-IAP2 (REFS 59,60) by inducing confor-mational changes in c-IAP proteins that stimulate their E3 ubiquitin ligase activity, which leads to the degrada-tion of c-IAP proteins by the proteasome.
Until recently, it was not clear how the binding of IAP antagonists to the BIR3 domain of c-IAP proteins affects the activity of the E3 ligase RING domain, which is sepa-rated from the BIR3 domain by a few hundred amino acids and several protein–protein interaction domains. Recent publications have shown that dimerization of the RING domain is essential for the E3 ligase activ-ity of c-IAP proteins and that IAP antagonists promote dimer formation and E3 ubiquitin ligase activation162,163. Furthermore, biochemical and structural studies have revealed that the unliganded, multidomain c-IAP1 sequesters the RING domain within a compact, mono-meric structure that blocks its dimerization164 (FIG. 4). When antagonists bind to the BIR3 domain of c-IAP1 they block crucial BIR3–RING domain interactions, causing conformational rearrangements that enable the dimerization of the RING domain and the formation of the active E3 ligase164 (FIG. 4).
Elevated E3 ligase activity of c-IAP proteins promotes the ubiquitylation of several proteins that reside in the same receptor-associated or cytoplasmic protein com-plexes, such as RIP1 (REFS 44,45). Ubiquitylation of RIP1 leads to the activation of the canonical NF-κB signalling pathway and the expression of NF-κB-responsive genes. However, as the c-IAP proteins are themselves targeted for degradation, this burst of E3 ubiquitin ligase activ-ity is relatively short. Without c-IAP proteins, NIK can accumulate and stimulate the activation of the non-canonical NF-κB pathway59,60. Both the canonical and non-canonical NF-κB pathways are likely to be impor-tant for IAP antagonist-stimulated expression of several cytokines and anti-apoptotic proteins, and spatiotem-poral differences in the activation of these signalling pathways could determine their relative contributions to gene expression.
TNF that is produced in an autocrine or paracrine manner triggers TNFR1-mediated signalling59,60,151,158 that would normally activate NF-κB and mitogen-activated protein kinase (MAPK) signalling pathways9. However, in the absence of c-IAP proteins RIP1 cannot be ubiqui-tylated, which allows it to bind to FADD and caspase 8, and consequently activate apoptosis44,45. Therefore, IAP antagonist-mediated activation of the E3 ligase activity of c-IAP proteins and the consequent elimination of these proteins acts like a switch to transform pro-survival TNF signalling into a pro-apoptotic pathway. These mechanis-tic findings constitute a foundation for the further devel-opment of IAP antagonists and for the identification of diagnostic and pharmacodynamic biomarkers.
Development of c-IAP- and XIAP-selective antagonists. IAP antagonists were initially designed to have a broad specificity and target different IAP proteins (for exam-ple, XIAP, ML-IAP, c-IAP1 and c-IAP2). However, recent findings have defined biological roles of individual IAP proteins, which — coupled with the structural informa-tion on the selected BIR domains of these proteins — incited the design of IAP antagonists that are specific for a particular IAP protein or a group of IAP proteins. As a result of these efforts, an IAP antagonist has been synthesized that is selective for c-IAP1 and c-IAP2 (the c-IAP-selective antagonist CS3)165 (FIG. 3). CS3 can induce the death of sensitive cancer cells, promote the degradation of c-IAP1 and c-IAP2, and activate canoni-cal and non-canonical NF-κB signalling pathways165. Although it is capable of inducing apoptosis, CS3 was a much weaker inducer of cell death in short-term and long-term viability assays in comparison with a chemi-cally similar pan-selective IAP antagonist, PS1 (REF. 165). Therefore, although the single-agent pro-apoptotic activity and activation of NF-κB signalling pathways, as well as cytokine production, rely on the antagonism of c-IAP proteins, both c-IAP and XIAP should be targeted simultaneously for efficient stimulation of cell death.
Much more effort has been devoted to targeting XIAP to prevent it from binding to and inhibiting caspase 3 (REF. 166). A high-throughput screening approach iden-tified TWX-024, which is a compound that can disrupt the XIAP–caspase 3 interaction and sensitize otherwise
▶
Figure 3 | Structure and development status of selected IAP antagonists. The following inhibitor of apoptosis (IAP) antagonists are currently at the preclinical stage: MV1 (Genentech)59; BV6 (Genentech)59; Comp 3 (Joyant Pharmaceuticals)156; Comp A (TetraLogic Pharmaceuticals)60; AEG40730 (Aegera Therapeutics)44; SM-164 (University of Michigan)157; LBW242 (Novartis)151; CS3 (Genentech)165; and ML101 (Sanford-Burnham Medical Research Institute)169. IAP antagonists that are currently in Phase I clinical trials are pan-selective agents that broadly target all IAP proteins228. These antagonists include AT-406 (Ascenta Therapeutics/University of Michigan)234 and TL32711 (Tetralogic Pharmaceuticals)229. Not shown are the monovalent agents GDC-0917 (Genentech) (ClinicalTrials.gov identifier: NCT01226277) and LCL161 (Novartis)228, and the bivalent agent IAP antagonist AEG35156 (Aegera Therapeutic )230, which are also in Phase I clinical trials.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 117
© 2012 Macmillan Publishers Limited. All rights reserved

resistant cancer cells to TRAIL treatment167. A different screen of a combinatorial chemistry library identified a structurally unrelated compound — TPI-1396-34, a molecule from the polyphenylurea class of compounds — that reversed XIAP-mediated blockade of caspase 3 activity and displayed antitumour activity in vivo168. More recent efforts using rational design identified a series of tripeptides that displayed reasonable selectivity for the BIR2 domain of XIAP and were capable of sensi-tizing cancer cells to TRAIL treatment169. Unfortunately, none of these compounds has been further optimized to yield highly selective and efficacious XIAP antagonists, despite the urgent need to target this unique inhibi-tor of caspases. The ideal XIAP-targeting agent should enable simultaneous antagonism of the BIR2 and BIR3 domains, thus allowing more efficient activation of cas-pase 3 and caspase 7 as well as caspase 9.
XIAP antisense oligonucleotides. Single-stranded anti-sense oligonucleotides are short patches of synthetic DNA that usually contain 12–30 oligonucleotides. The complementarity of this stretch of DNA to a correspond-ing mRNA strand provides the basis for the target gene-specific interference with the translational machinery, which leads to the degradation of the target mRNA and downregulation of target protein expression170.
In preclinical cancer models, low nanomolar concen-trations of the antisense oligonucleotide AEG35156 were reported to efficiently decrease XIAP mRNA and XIAP protein levels171. As a single agent, AEG35156 displayed potent antitumour activity in vivo in xenograft mouse models of prostate, colon, ovarian and lung cancer, and caused complete tumour regression in combination with taxanes171,172. Importantly, the in vivo antitumour effects of AEG35156 correlated with suppression of XIAP lev-els in the tumour171. In addition, suppression of XIAP protein levels resulted in enhanced apoptosis following in vitro treatment with various chemotherapeutic com-pounds, the death receptor ligand TRAIL or after radia-tion therapy171,173–177.
The first-in-human study with AEG35156 in patients with advanced refractory cancers demonstrated that the
compound was well tolerated and showed predictable pharmacokinetic properties, dose-dependent changes in circulating biomarkers of cell death as well as some clinical evidence of antitumour activity178.
In a non-randomized Phase I/II trial evaluating the XIAP antisense oligonucleotide AEG35156 in combi-nation with re-induction chemotherapy (that is, high-dose cytarabine and idarubicin), AEG35156 proved to be well tolerated in all 56 patients, with the excep-tion of two who experienced peripheral neuropathy179. Pharmacodynamic studies showed a dose-dependent knockdown of XIAP mRNA179. Similarly, another report confirmed that AEG35156 treatment resulted in a dose-dependent reduction in XIAP mRNA lev-els in this Phase I/II trial in patients with AML180. This reduction in XIAP expression was associated with the induction of apoptosis in one out of four patients within the Phase I trial and four out of five patients within the Phase II trial, with preferential induction of apoptosis in CD34+38– AML stem cells180. Furthermore, high doses of AEG35156 were associated with higher overall response rates179. Importantly, 10 out of 11 patients (91%) receiving high-dose AEG35156 in combination with re-induction chemotherapy achieved complete remission after the fail-ure of a prior single induction regimen179.
In contrast to these results obtained in non-randomized Phase I/II trials, a subsequent randomized Phase II study in patients with primary refractory AML showed no improvement in remission rates following the addition of the AEG35156 XIAP antisense oligonucleotide to the re-induction chemotherapy protocol181. However, as phar-macodynamic studies were not conducted in this trial as previously done in the non-randomized Phase I/II trials, it remains unknown whether efficient target knockdown of XIAP mRNA was achieved.
Combination therapiesIAP antagonists have been extensively studied in combination protocols together with other cytotoxic agents including anticancer drugs, small-molecule sig-nal transduction inhibitors, proteasome inhibitors and death receptor ligands as well as with radiation therapy.
Nature Reviews | Drug Discovery
RINGBIR3 UBA CARD Ub
Antagonistbinding
DegradationDimerizationand activation
Closed monomer Open monomer Open and active dimer Degradation, apoptosis
Figure 4 | Model for the induction of c-IAP protein ubiquitin ligase activity by IAP antagonists. Unstimulated cellular inhibitor of apoptosis (c-IAP) proteins adopt a closed and inactive monomeric conformation. The binding of IAP antagonists to the baculoviral IAP repeat 3 (BIR3) domain disrupts this state by blocking crucial BIR3 interactions with the RING domain and prompting a change in conformation, which exposes the RING domain and allows it to dimerize and form an active ubiquitin ligase. Ensuing autoubiquitylation leads to proteasomal degradation of c-IAP proteins and initiation of apoptosis. CARD, caspase recruitment domain; UBA, ubiquitin-associated domain; Ub, ubiquitin.
R E V I E W S
118 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Combination therapies with chemotherapeutics. IAP antagonists have been shown to sensitize various can-cers towards chemotherapy, including pancreatic, lung, colon, prostate, breast and skin cancer as well as acute leukaemia, in combinations with different classes of cytotoxic drugs, such as gemcitabine, doxorubicin, etoposide, cisplatin, 5-fluorouracil, vinorelbine, irinote-can and cytarabine143,182–195 (Supplementary informa-tion S2 (table)). Mechanistic studies have recently shown that IAP antagonists that simultaneously neu-tralize c-IAP1 or c-IAP2, in addition to XIAP, initiate a RIP1-mediated cell death pathway in a TNF-dependent or TNF-independent manner that promotes chemo-therapy-induced apoptosis184,187. Elevated levels of the TNF protein were also detected in the tumour tissue of xenografted or transgenic mice in vivo following the administration of IAP antagonists and cytotoxic drugs, which correlated with the antitumour response in vivo194.
In line with the notion that IAP antagonists can initi-ate NF-κB activation as well as autocrine and/or paracrine TNF signalling, the inhibition of NF-κB has been shown to attenuate the SMAC mimetic-mediated chemosensi-tization in pancreatic cancer, pointing to a pro-apoptotic role of NF-κB in this context182. In lung carcinoma the XIAP antagonist compound XAC 1396-11 was reported to act in concert with cisplatin and vinorelbine in cells186, whereas the natural product XIAP inhibitor embelin exerted cytotoxicity against cisplatin-resistant lung car-cinoma cells183. Similarly, downregulation or antagonism of ML-IAP sensitizes several cancer cell lines to cell death stimulated by chemotherapeutics142,197. There is evidence that the combination of IAP antagonists with chemo-therapy exerts some tumour selectivity, as little toxicity was reported for IAP antagonists and anticancer drugs on non-malignant human cells such as peripheral blood lymphocytes184.
Combination therapies with death receptor agonists. In addition to chemotherapeutics, IAP antagonists have been extensively studied in combination with death receptor ligands — particularly TRAIL receptor ago-nists — in various cancers including breast, prostate, colon and bladder cancer, cholangiocarcinoma, pan-creatic carcinoma, lung cancer, melanoma, leukaemia, bone tumours, glioblastoma, neuroblastoma, head and neck squamous cell carcinoma as well as multiple myeloma143,156,188–191,193,195,196,198–212 (Supplementary infor-mation S2 (table)). In pancreatic cancer — one of the most lethal types of cancer — IAP antagonists were shown to synergize with soluble agonistic antibodies for TRAIL or the TRAIL receptor to induce apoptosis and suppress tumour growth in vivo143,204,211. Furthermore, SMAC mimetics have been reported to decrease TRAIL-stimulated invasion and metastasis even in cancers such as cholangiocarcinoma that do not respond to the SMAC mimetic-mediated sensitization to pro-apoptotic death receptor signalling196.
Moreover, IAP antagonists and TRAIL were found to cooperatively induce apoptosis in CLL, including in patients with unfavourable prognostic features such the chromosome 17p deletion, cellular tumour antigen
p53 mutation, unmutated variable heavy-chain (VH) genes or a chemoresistant form of the disease203,210. This pro-apoptotic activity of IAP antagonists and TRAIL in CLL was confirmed by an independent study203. Besides TRAIL, IAP antagonists were reported to act in concert with a hexameric form of the CD95 ligand or agonis-tic CD95-specific antibodies to trigger apoptotic cell death207,212,213 and bypass the need for mitochondrial amplification of death receptor-mediated apoptosis160,161. Although antagonism of XIAP is crucial for the syner-gistic activity of IAP antagonists and death receptors, neutralization of c-IAP proteins and FLICE-like inhibi-tory protein is also important for this pro-apoptotic combination160,200,208.
In addition, IAP antagonists have been reported to switch the cell’s response to TNF — from survival to cell death — via their ability to stimulate the autoubiquity-lation and degradation of c-IAP1 and c-IAP2, thereby favouring the formation of the TNFR1-stimulated pro-apoptotic cytoplasmic complex (complex II) that engages FADD and caspase 8 (REFS 59,60,156,158,214). In apoptosis-resistant cells pharmacological or genetic depletion of c-IAP1 and c-IAP2 can potentiate TNF-induced necroptosis by enhancing the formation of the necrosome complex — a cytosolic complex containing RIP1, RIP3 and FADD215,216. Thus, IAP antagonists can act in combination with death receptor ligands to trigger apoptosis as well as non-apoptotic forms of cell death.
Combination therapies with radiation. Small-molecule IAP antagonists have been reported to enhance radio-sensitivity in several types of cancer, such as pancreatic cancer, prostate cancer and glioblastoma (including glioblastoma-initiating cancer stem cells)192,217–221. In an orthotopic glioblastoma model in nude mice, the admin-istration of the SMAC mimetic LBW242 enhanced the antitumour effect of radiation and temozolomide — the standard-of-care treatment for glioblastoma — and sig-nificantly prolonged the survival of mice192. In addition, LBW242 enhanced the inhibitory effect of radiotherapy on endothelial cells192. Although the SMAC mimetic BV6 was shown to stimulate NF-κB activation, which was required for the SMAC mimetic-conferred radio-sensitization of glioblastoma cells221, the IAP antago-nist SH-130 was reported to inhibit radiation-induced NF-κB activation in prostate cancer cells220. SH-130 enhanced the antitumour effect of X-ray radiation in a mouse xenograft model of prostate cancer without increasing systemic toxicity220. Furthermore, the natural IAP antagonist embelin was shown to enhance radia-tion-induced cell death in prostate carcinoma219.
Combination therapies with kinase inhibitors. Intriguingly, IAP antagonists have been reported to potentiate the effects of several kinase inhibitors, includ-ing inhibitors of FMS-like tyrosine kinase 3 (FLT3) or ERBB as well as inhibitors of platelet-derived growth factor receptor, insulin-like growth factor receptor or epidermal growth factor receptor (EGFR, also known as ERBB1)222–225. For example, embelin was reported to overcome the acquired resistance to EGFR and ERBB2
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 119
© 2012 Macmillan Publishers Limited. All rights reserved

(also known as HER2) inhibitors in breast carcinoma, which has been linked to the upregulation of XIAP226. Similarly, SMAC mimetics enhanced apoptosis follow-ing treatment with ERBB inhibitors such as trastuzumab (Herceptin; Roche/Genentech), lapatinib (Tykerb; GlaxoSmithKline) or gefitinib (Iressa; AstraZeneca/Teva) in HER2-overexpressing breast cancer cells224.
Furthermore, the SMAC mimetics LCL161 and LBW242 were shown to augment the effects of the break-point cluster region (BCR)–ABL inhibitor nilotinib (Tasigna; Novartis) or the FLT3 inhibitor PKC412 in acute leukaemia models in vitro as well as in vivo222,227. In glio-blastoma, LBW242 acted in concert with several receptor tyrosine kinase inhibitors, including imatinib (Gleevec; Novartis), nilotinib, NVP-AWW541 and PKI166, to pro-mote downstream caspase activation and apoptosis223. In multiple myeloma, LBW242 was reported to induce addi-tive and/or synergistic antitumour activity in combination with proteasome inhibitors such as bortezomib (Velcade; Millennium Pharmaceuticals) and NPI-0052 (REF. 190). Similarly, a combination of SMAC mimetics and the pro-teasome inhibitor bortezomib was shown to cooperate in triggering apoptosis in melanoma cells198.
Clinical development of IAP antagonistsCurrently, there are at least five clinical trials that are testing the applicability of small-molecule IAP antago-nists as anticancer treatments (FIG. 3). Ongoing Phase I trials conducted by Genentech, Novartis and Ascenta Therapeutics are examining the safety and pharma-cological properties of monovalent IAP antagonists, and Phase I trials run by Aegera Therapeutics/Human Genome Sciences and TetraLogic Pharmaceuticals are studying bivalent antagonists (FIG. 3). Monovalent antag-onists could be orally administered to patients, which may allow more flexibility in dosing regimens, especially for combinations with other standard-of-care agents that stimulate cell death. Conversely, bivalent IAP antagonists have been shown to be more efficacious in preclinical studies44,59. This higher potency should probably prompt a very careful dosing schedule in clinical studies. In addi-tion, owing to their size, bivalent antagonists have to be administered intravenously, probably no more than once a week. Thus, it remains to be determined whether the pharmacodynamic or pharmacokinetic profiles of mono- or bivalent IAP antagonists are more suitable for clinical application.
Novartis reported the results of a Phase I study of LCL161, an orally bioavailable IAP antagonist, and indicated that this antagonist was well tolerated in patients with cancer — no dose-limiting toxicities were reported228. LCL161 also demonstrated target antagonism by causing c-IAP1 protein degradation and upregulation of circulating chemokines such as interleukin-8 and monocyte chemoattractant protein 1 (REF. 228). Similar observations were reported by Aegera Therapeutics/Human Genome Sciences and TetraLogic Pharmaceuticals for HGS1029 and TL32711, respec-tively229,230. The bivalent IAP antagonists HGS1029 and TL32711 both displayed proportional pharmacokinet-ics across their dose ranges and were well tolerated,
with grade 2 transient lymphopaenia and neutrophilia reported in some patients229,230. As expected, in the high-est dose cohorts both of these reagents stimulated the downmodulation of c-IAP1 and the elevation of mono-cyte chemoattractant protein 1 levels as well as serum levels of processed caspase 3 and caspase 7.
Conclusions and future perspectivesFor the successful development of IAP antagonists as therapeutic agents, it will be important to identify pre-dictive biomarkers to select the optimal patient popu-lation (or populations) for this therapeutic approach. As the loss of c-IAP1 can be expected in non-tumour tissues, as well as in responsive and non-responsive malignancies229,230, c-IAP1 degradation could potentially serve as an indicator of IAP antagonist activity but not as a predictive biomarker of treatment outcome. The increase in apoptotic markers such as the processing of caspase 3 and caspase 7 should be a much better indica-tor of expected efficacy.
However, as there is evidence that IAP antagonists can also regulate TNF-induced necroptosis in addition to apoptosis215,216, caspase activation might not always be an indicator of IAP antagonist activity. Thus far, TNF has emerged as a crucial factor for the single-agent activity of IAP antagonists in cancer cells. However, it remains to be determined whether or not TNF levels (or its production) may represent suitable markers to identify patients who will be likely to respond to IAP antagonists. Furthermore, it is still not clear which tissue (or tissues) or cells in the organism are the primary source of TNF production in vivo. In addition, do other cytokines that are produced as a result of IAP antagonist treatment con-tribute to the antitumour effect, and may these prove to be better biomarkers of treatment response?
Another important question is which IAP protein is a crucial target for combination treatments. XIAP seems to be an instrumental target for the combination of IAP antagonists with death receptor-agonistic antibodies or ligands. However, the efficacy of the combination of IAP antagonists with different chemotherapeutics, radiation therapy or kinase inhibitors is less clear. Targeting c-IAP proteins would potentially generate single-agent activ-ity but neutralizing XIAP-mediated caspase inhibition is undoubtedly important as well. If TNF signalling has an important role for a particular treatment combination with IAP antagonists, one could postulate that targeting of c-IAP proteins might be needed. If the contribution of IAP antagonists to effective cell killing requires the lowering of the apoptotic threshold through caspase activation, XIAP might be a crucial target.
As IAP proteins are important regulators of various physiological and pathophysiological processes — for example, the regulation of the immune response — therapeutic targeting of IAP proteins may also repre-sent a strategy in other human diseases besides cancer. Furthermore, one could envision using IAP antago-nists as immunotherapeutics to enhance dendritic cell maturation and to promote the development of tumour-specific T cell immunity, as demonstrated in the proof-of-concept study by Dougan et al.231.
R E V I E W S
120 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

In summary, their elevated expression in many tumour types, potent anti-apoptotic activity and their contribu-tion to pro-survival signalling pathways make IAP pro-teins promising targets for drug development in cancer. Preclinical studies have indicated that the therapeutic potential of IAP antagonists might best be exploited in combination protocols, including conventional
chemotherapeutic drugs, signal transduction modula-tors, death receptor agonists or radiation therapy. We hope that continued research efforts will generate even more insight into the functions of IAP proteins in apoptotic and non-apoptotic cell death, signal transduction pathways, cell division and protein turnover, and thereby open addi-tional facets for therapeutic intervention.
1. Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
2. Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 124, 511–515 (2009).
3. Galluzzi, L. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 (2012).
4. Crook, N. E., Clem, R. J. & Miller, L. K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 67, 2168–2174 (1993).This study identified the first viral iap gene from the Cydia pomonella granulovirus baculovirus.
5. Birnbaum, M. J., Clem, R. J. & Miller, L. K. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J. Virol. 68, 2521–2528 (1994).
6. Salvesen, G. S. & Duckett, C. S. IAP proteins: blocking the road to death’s door. Nature Rev. Mol. Cell Biol. 3, 401–410 (2002).
7. Ndubaku, C., Cohen, F., Varfolomeev, E. & Vucic, D. Targeting inhibitor of apoptosis (IAP) proteins for therapeutic intervention. Future Med. Chem. 1, 1509–1525 (2009).
8. Vaux, D. L. & Silke, J. IAPs, RINGs and ubiquitylation. Nature Rev. Mol. Cell Biol. 6, 287–297 (2005).
9. Varfolomeev, E. & Vucic, D. (Un)expected roles of c-IAPs in apoptotic and NF-κB signaling pathways. Cell Cycle 7, 1511–1521 (2008).
10. Blankenship, J. W. et al. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2. Biochem. J. 417, 149–160 (2009).
11. Gyrd-Hansen, M. et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-κB as well as cell survival and oncogenesis. Nature Cell Biol. 10, 1309–1317 (2008).
12. Hauser, H. P., Bardroff, M., Pyrowolakis, G. & Jentsch, S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 141, 1415–1422 (1998).
13. Chen, Z. et al. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun. 264, 847–854 (1999).
14. Jeyaprakash, A. A. et al. Structure of a survivin–borealin–INCENP core complex reveals how chromosomal passengers travel together. Cell 131, 271–285 (2007).
15. Liston, P. et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379, 349–353 (1996).
16. Wilmanski, J. M., Petnicki-Ocwieja, T. & Kobayashi, K. S. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J. Leukoc. Biol. 83, 13–30 (2008).
17. Altieri, D. C. Validating survivin as a cancer therapeutic target. Nature Rev. Cancer 3, 46–54 (2003).
18. Hunter, A. M., LaCasse, E. C. & Korneluk, R. G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 12, 1543–1568 (2007).
19. Geddes, K., Magalhaes, J. G. & Girardin, S. E. Unleashing the therapeutic potential of NOD-like receptors. Nature Rev. Drug Discov. 8, 465–479 (2009).
20. Fulda, S. & Debatin, K. M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 (2006).
21. Ashkenazi, A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 19, 325–331 (2008).
22. Fulda, S., Galluzzi, L. & Kroemer, G. Targeting mitochondria for cancer therapy. Nature Rev. Drug Discov. 9, 447–464 (2010).
23. Chai, J. et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862 (2000).
24. Liu, Z. et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 408, 1004–1008 (2000).
25. Wu, G. et al. Structural basis of IAP recognition by Smac/DIABLO. Nature 408, 1008–1012 (2000).
26. Adams, J. M. & Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 (2007).
27. Eckelman, B. P., Salvesen, G. S. & Scott, F. L. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 7, 988–994 (2006).
28. Srinivasula, S. M. et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410, 112–116 (2001).
29. Shiozaki, E. N. et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 11, 519–527 (2003).
30. Huang, Y. et al. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104, 781–790 (2001).
31. Chai, J. et al. Structural basis of caspase-7 inhibition by XIAP. Cell 104, 769–780 (2001).
32. Riedl, S. J. et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell 104, 791–800 (2001).
33. Silke, J. et al. Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J. 20, 3114–3123 (2001).
34. Scott, F. L. et al. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 24, 645–655 (2005).
35. Vucic, D. et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem. J. 385, 11–20 (2005).
36. Vucic, D., Dixit, V. M. & Wertz, I. E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nature Rev. Mol. Cell Biol. 12, 439–452 (2011).
37. Hegde, R. et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277, 432–438 (2002).
38. Verhagen, A. M. et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 277, 445–454 (2002).
39. Martins, L. M. et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 277, 439–444 (2002).
40. Suzuki, Y. et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8, 613–621 (2001).
41. Liston, P. et al. Identification of XAF1 as an antagonist of XIAP anti-caspase activity. Nature Cell Biol. 3, 128–133 (2001).
42. Arora, V. et al. Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)–XAF1 complex. J. Biol. Chem. 282, 26202–26209 (2007).
43. Scheidereit, C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene 25, 6685–6705 (2006).
44. Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
45. Varfolomeev, E. et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 (2008).
46. Mahoney, D. J. et al. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl Acad. Sci. USA 105, 11778–11783 (2008).
47. Dynek, J. N. et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 (2010).
48. Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
49. Varfolomeev, E. et al. Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by TNF family receptors. Sci. Signal. (in the press).
50. Tokunaga, F. et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nature Cell Biol. 11, 123–132 (2009).
51. Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
52. Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471, 637–641 (2011).
53. Tokunaga, F. et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).The studies in REFS 50–53 identified Sharpin as a novel component of the linear ubiquitin chain assembly complex.
54. Keats, J. J. et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 12, 131–144 (2007).
55. Annunziata, C. M. et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130 (2007).The studies in REFS 54 and 55 identified genetic abnormalities in several genes, including c-IAP proteins, of the canonical and non-canonical NF-κB pathways in multiple myeloma.
56. Grech, A. P. et al. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-κB activation in mature B cells. Immunity 21, 629–642 (2004).
57. He, J. Q. et al. Rescue of TRAF3-null mice by p100 NF-κ B deficiency. J. Exp. Med. 203, 2413–2418 (2006).
58. Demchenko, Y. N. et al. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood 115, 3541–3552 (2010).
59. Varfolomeev, E. et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 (2007).This paper demonstrated that IAP antagonists induce apoptosis by triggering c-IAP1 degradation, NF-κB activation and autocrine TNF production, and identified c-IAP proteins as E3 ligases for NIK.
60. Vince, J. E. et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 (2007).This study demonstrated that IAP antagonists induce apoptosis by triggering c-IAP1 degradation, NF-κB activation and autocrine TNF production.
61. Vince, J. E. et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1–TRAF2 complex to sensitize tumor cells to TNFα. J. Cell Biol. 182, 171–184 (2008).
62. Xiao, G., Harhaj, E. W. & Sun, S. C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 7, 401–409 (2001).
63. Senftleben, U. et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 293, 1495–1499 (2001).
64. Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem. Pharmacol. 72, 1161–1179 (2006).
65. Imoto, I. et al. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 61, 6629–6634 (2001).
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 121
© 2012 Macmillan Publishers Limited. All rights reserved

66. Imoto, I. et al. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 62, 4860–4866 (2002).
67. Zender, L. et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267 (2006).
68. Reardon, D. A. et al. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 57, 4042–4047 (1997).
69. Weber, R. G., Sommer, C., Albert, F. K., Kiessling, M. & Cremer, T. Clinically distinct subgroups of glioblastoma multiforme studied by comparative genomic hybridization. Lab. Invest. 74, 108–119 (1996).
70. Dai, Z. et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 12, 791–801 (2003).
71. Bashyam, M. D. et al. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 7, 556–562 (2005).
72. Ma, O. et al. MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res. 69, 2559–2567 (2009).
73. Dierlamm, J. et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood 93, 3601–3609 (1999).
74. Akagi, T. et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene 18, 5785–5794 (1999).
75. Zhou, H., Du, M. Q. & Dixit, V. M. Constitutive NF-kB activation by the t(11;18)(q21;q21) product in MALT lymphoma is linked to deregulated ubiquitin ligase activity. Cancer Cell 7, 425–431 (2005).
76. Morgan, J. A. et al. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. 59, 6205–6213 (1999).The studies in REFS 74, 75 and 77 identified the t(11;18)(q21;q21) translocation and the corresponding c-IAP2–MALT1 fusion protein in MALT lymphoma.
77. Varfolomeev, E., Wayson, S. M., Dixit, V. M., Fairbrother, W. J. & Vucic, D. The inhibitor of apoptosis protein fusion c‑IAP2•MALT1 stimulates NF-κB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 281, 29022–29029 (2006).
78. Waldele, K. et al. Requirement of the human T-cell leukemia virus (HTLV-1) tax-stimulated HIAP‑1 gene for the survival of transformed lymphocytes. Blood 107, 4491–4499 (2006).
79. Tamm, I. et al. XIAP expression correlates with monocytic differentiation in adult de novo AML: impact on prognosis. Hematol. J. 5, 489–495 (2004).
80. Tamm, I. et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 6, 1796–1803 (2000).
81. Carter, B. Z. et al. Caspase-independent cell death in AML: caspase inhibition in vitro with pan-caspase inhibitors or in vivo by XIAP or survivin does not affect cell survival or prognosis. Blood 102, 4179–4186 (2003).
82. Hess, C. J. et al. Activated intrinsic apoptosis pathway is a key related prognostic parameter in acute myeloid leukemia. J. Clin. Oncol. 25, 1209–1215 (2007).
83. Tamm, I. et al. High expression levels of X-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 10, 3737–3744 (2004).
84. Sung, K. W. et al. Overexpression of X-linked inhibitor of apoptosis protein (XIAP) is an independent unfavorable prognostic factor in childhood de novo acute myeloid leukemia. J. Korean Med. Sci. 24, 605–613 (2009).
85. Hundsdoerfer, P., Dietrich, I., Schmelz, K., Eckert, C. & Henze, G. XIAP expression is post-transcriptionally upregulated in childhood ALL and is associated with glucocorticoid response in T-cell ALL. Pediatr. Blood Cancer 55, 260–266 (2010).
86. Grzybowska-Izydorczyk, O., Cebula, B., Robak, T. & Smolewski, P. Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur. J. Cancer 46, 800–810 (2010).
87. Vallat, L. et al. The resistance of B-CLL cells to DNA damage-induced apoptosis defined by DNA microarrays. Blood 101, 4598–4606 (2003).
88. Silva, K. L. et al. Apoptotic effect of fludarabine is independent of expression of IAPs in B-cell chronic lymphocytic leukemia. Apoptosis 11, 277–285 (2006).
89. Hussain, A. R. et al. Prognostic significance of XIAP expression in DLBCL and effect of its inhibition on AKT signalling. J. Pathol. 222, 180–190 (2010).
90. Ramp, U. et al. XIAP expression is an independent prognostic marker in clear-cell renal carcinomas. Hum. Pathol. 35, 1022–1028 (2004).
91. Yan, Y. et al. Disturbed balance of expression between XIAP and Smac/DIABLO during tumour progression in renal cell carcinomas. Br. J. Cancer 91, 1349–1357 (2004).
92. Mizutani, Y. et al. Overexpression of XIAP expression in renal cell carcinoma predicts a worse prognosis. Int. J. Oncol. 30, 919–925 (2007).
93. Li, M., Song, T., Yin, Z. F. & Na, Y. Q. XIAP as a prognostic marker of early recurrence of nonmuscular invasive bladder cancer. Chin. Med. J. 120, 469–473 (2007).
94. Che, X. et al. Nuclear cIAP1 overexpression is a tumor stage- and grade-independent predictor of poor prognosis in human bladder cancer patients. Urol. Oncol. 25 Jul 2011 (doi:10.1016/j.urolonc.2010.12.016).
95. Xiang, G., Wen, X., Wang, H., Chen, K. & Liu, H. Expression of X-linked inhibitor of apoptosis protein in human colorectal cancer and its correlation with prognosis. J. Surg. Oncol. 100, 708–712 (2009).
96. Krajewska, M. et al. Analysis of apoptosis protein expression in early-stage colorectal cancer suggests opportunities for new prognostic biomarkers. Clin. Cancer Res. 11, 5451–5461 (2005).
97. Esposito, I. et al. Overexpression of cellular inhibitor of apoptosis protein 2 is an early event in the progression of pancreatic cancer. J. Clin. Pathol. 60, 885–895 (2007).
98. Augello, C. et al. Inhibitors of apoptosis proteins (IAPs) expression and their prognostic significance in hepatocellular carcinoma. BMC Cancer 9, 125 (2009).
99. Shi, Y. H. et al. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology 48, 497–507 (2008).
100. Shibata, T. et al. Disturbed expression of the apoptosis regulators XIAP, XAF1, and Smac/DIABLO in gastric adenocarcinomas. Diagn. Mol. Pathol. 16, 1–8 (2007).
101. Seligson, D. B. et al. Expression of X-linked inhibitor of apoptosis protein is a strong predictor of human prostate cancer recurrence. Clin. Cancer Res. 13, 6056–6063 (2007).
102. Krajewska, M. et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin. Cancer Res. 9, 4914–4925 (2003).
103. Zhang, Y. et al. X-linked inhibitor of apoptosis positive nuclear labeling: a new independent prognostic biomarker of breast invasive ductal carcinoma. Diagn. Pathol. 6, 49 (2011).
104. Liu, S. S. et al. Anti-apoptotic proteins, apoptotic and proliferative parameters and their prognostic significance in cervical carcinoma. Eur. J. Cancer 37, 1104–1110 (2001).
105. Ferreira, C. G. et al. Expression of X-linked inhibitor of apoptosis as a novel prognostic marker in radically resected non-small cell lung cancer patients. Clin. Cancer Res. 7, 2468–2474 (2001).
106. Ferreira, C. G. et al. Assessment of IAP (inhibitor of apoptosis) proteins as predictors of response to chemotherapy in advanced non-small-cell lung cancer patients. Ann. Oncol. 12, 799–805 (2001).
107. Hiscutt, E. L. et al. Targeting X-linked inhibitor of apoptosis protein to increase the efficacy of endoplasmic reticulum stress-induced apoptosis for melanoma therapy. J. Invest. Dermatol. 130, 2250–2258 (2010).
108. Qi, S. et al. Expression of cIAP-1 correlates with nodal metastasis in squamous cell carcinoma of the tongue. Int. J. Oral Maxillofac. Surg. 37, 1047–1053 (2008).
109. Tanimoto, T. et al. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 224, 141–151 (2005).
110. Vucic, D., Stennicke, H. R., Pisabarro, M. T., Salvesen, G. S. & Dixit, V. M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 10, 1359–1366 (2000).
111. Wagener, N. et al. Expression of inhibitor of apoptosis protein livin in renal cell carcinoma and non-tumorous adult kidney. Br. J. Cancer 97, 1271–1276 (2007).
112. Gong, J. et al. Melanoma inhibitor of apoptosis protein is expressed differentially in melanoma and melanocytic naevus, but similarly in primary and metastatic melanomas. J. Clin. Pathol. 58, 1081–1085 (2005).
113. Nachmias, B. et al. Caspase-mediated cleavage converts livin from an antiapoptotic to a proapoptotic factor: implications for drug-resistant melanoma. Cancer Res. 63, 6340–6349 (2003).
114. Dynek, J. N. et al. Microphthalmia-associated transcription factor is a critical transcriptional regulator of melanoma inhibitor of apoptosis in melanomas. Cancer Res. 68, 3124–3132 (2008).
115. Kempkensteffen, C. et al. Expression of the apoptosis inhibitor livin in renal cell carcinomas: correlations with pathology and outcome. Tumour Biol. 28, 132–138 (2007).
116. Haferkamp, A. et al. High nuclear livin expression is a favourable prognostic indicator in renal cell carcinoma. BJU Int. 102, 1700–1706 (2008).
117. Gazzaniga, P. et al. Expression and prognostic significance of LIVIN, SURVIVIN and other apoptosis-related genes in the progression of superficial bladder cancer. Ann. Oncol. 14, 85–90 (2003).
118. Kempkensteffen, C. et al. Expression of splicing variants of the inhibitor of apoptosis livin in testicular germ cell tumors. Tumour Biol. 29, 76–82 (2008).
119. Wang, T. S. et al. Expression of livin in gastric cancer and induction of apoptosis in SGC-7901 cells by shRNA-mediated silencing of livin gene. Biomed. Pharmacother. 64, 333–338 (2010).
120. Choi, J. et al. Expression of livin, an antiapoptotic protein, is an independent favorable prognostic factor in childhood acute lymphoblastic leukemia. Blood 109, 471–477 (2007).
121. El-Mesallamy, H. O., Hegab, H. M. & Kamal, A. M. Expression of inhibitor of apoptosis protein (IAP) livin/BIRC7 in acute leukemia in adults: correlation with prognostic factors and outcome. Leuk. Res. 35, 1616–1622 (2011).
122. Nedelcu, T. et al. Livin and Bcl-2 expression in high-grade osteosarcoma. J. Cancer Res. Clin. Oncol. 134, 237–244 (2008).
123. Kim, D. K. et al. Expression of inhibitor-of-apoptosis protein (IAP) livin by neuroblastoma cells: correlation with prognostic factors and outcome. Pediatr. Dev. Pathol. 8, 621–629 (2005).
124. Mizutani, Y. et al. Downregulation of Smac/DIABLO expression in renal cell carcinoma and its prognostic significance. J. Clin. Oncol. 23, 448–454 (2005).
125. Kempkensteffen, C. et al. Expression levels of the mitochondrial IAP antagonists Smac/DIABLO and Omi/HtrA2 in clear-cell renal cell carcinomas and their prognostic value. J. Cancer Res. Clin. Oncol. 134, 543–550 (2008).
126. Mizutani, Y., Katsuoka, Y. & Bonavida, B. Prognostic significance of second mitochondria-derived activator of caspase (Smac/DIABLO) expression in bladder cancer and target for therapy. Int. J. Oncol. 37, 503–508 (2010).
127. Endo, K. et al. Clinical significance of Smac/DIABLO expression in colorectal cancer. Oncol. Rep. 21, 351–355 (2009).
128. Yan, H. et al. Prognostic value of Smac expression in rectal cancer patients treated with neoadjuvant therapy. Med. Oncol. 25 Jan 2011 (doi:10.1007/s12032-011-9819-x).
129. Pluta, P. et al. Correlation of Smac/DIABLO protein expression with the clinico-pathological features of breast cancer patients. Neoplasma 58, 430–435 (2011).
130. Dobrzycka, B. et al. Prognostic significance of smac/DIABLO in endometrioid endometrial cancer. Folia Histochem. Cytobiol. 48, 678–681 (2011).
131. Arellano-Llamas, A. et al. High Smac/DIABLO expression is associated with early local recurrence of cervical cancer. BMC Cancer 6, 256 (2006).
R E V I E W S
122 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

132. Sekimura, A. et al. Expression of Smac/DIABLO is a novel prognostic marker in lung cancer. Oncol. Rep. 11, 797–802 (2004).
133. Pluta, A. et al. Influence of high expression of Smac/DIABLO protein on the clinical outcome in acute myeloid leukemia patients. Leuk. Res. 34, 1308–1313 (2010).
134. Vucic, D. & Fairbrother, W. J. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin. Cancer Res. 13, 5995–6000 (2007).
135. Vucic, D., Kaiser, W. J., Harvey, A. J. & Miller, L. K. Inhibition of Reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs). Proc. Natl Acad. Sci. USA 94, 10183–10188 (1997).
136. Vucic, D., Kaiser, W. J. & Miller, L. K. Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol. Cell Biol. 18, 3300–3309 (1998).
137. Du, C., Fang, M., Li, Y., Li, L. & Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 (2000).
138. Verhagen, A. M. et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53 (2000).The studies in REFS 138 and 139 identified the mitochondrial protein SMAC as a mammalian IAP antagonist that promotes caspase activation by neutralizing IAP proteins.
139. LaCasse, E. C. et al. IAP-targeted therapies for cancer. Oncogene 27, 6252–6275 (2008).
140. Franklin, M. C. et al. Structure and function analysis of peptide antagonists of melanoma inhibitor of apoptosis (ML-IAP). Biochemistry 42, 8223–8231 (2003).
141. Grether, M. E., Abrams, J. M., Agapite, J., White, K. & Steller, H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 9, 1694–1708 (1995).
142. Vucic, D. et al. SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP). J. Biol. Chem. 277, 12275–12279 (2002).This was the first evidence that SMAC-mimicking peptides can sensitize cancer cells to chemotherapeutic agents.
143. Fulda, S., Wick, W., Weller, M. & Debatin, K. M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nature Med. 8, 808–815 (2002).This was the first in vivo proof-of-concept study to show that antagonizing IAP proteins by a SMAC mimetic represents a promising strategy for sensitizing cancer cells to TRAIL-induced apoptosis.
144. Arnt, C. R., Chiorean, M. V., Heldebrant, M. P., Gores, G. J. & Kaufmann, S. H. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J. Biol. Chem. 277, 44236–44243 (2002).
145. Yang, L. et al. Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 63, 831–837 (2003).
146. Oost, T. K. et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 47, 4417–4426 (2004).
147. Sharma, S. K., Straub, C. & Zawel, L. Development of peptidomimetics targeting IAPs. Int. J. Pept. Res. Ther. 12, 21–32 (2006).
148. Kipp, R. A. et al. Molecular targeting of inhibitor of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry 41, 7344–7349 (2002).
149. Flygare, J. A. & Fairbrother, W. J. Small-molecule pan-IAP antagonists: a patent review. Expert Opin. Ther. Pat. 20, 251–267 (2010).
150. Zobel, K. et al. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 1, 525–533 (2006).
151. Gaither, A. et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-α signaling. Cancer Res. 67, 11493–11498 (2007).
152. Sun, H. et al. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc. Chem. Res. 41, 1264–1277 (2008).
153. Cohen, F. et al. Orally bioavailable antagonists of inhibitor of apoptosis proteins based on an azabicyclooctane scaffold. J. Med. Chem. 52, 1723–1730 (2009).
154. Sun, H. et al. Structure-based design, synthesis, and evaluation of conformationally constrained mimetics of the second mitochondria-derived activator of caspase that target the X-linked inhibitor of apoptosis protein/caspase-9 interaction site. J. Med. Chem. 47, 4147–4150 (2004).
155. Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
156. Li, L. et al. A small molecule Smac mimic potentiates TRAIL- and TNFα-mediated cell death. Science 305, 1471–1474 (2004).
157. Sun, H. et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J. Am. Chem. Soc. 129, 15279–15294 (2007).
158. Petersen, S. L. et al. Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic- induced apoptosis. Cancer Cell 12, 445–456 (2007).
159. Gao, Z. et al. A dimeric Smac/diablo peptide directly relieves caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of XIAP by Smac/Diablo. J. Biol. Chem. 282, 30718–30727 (2007).
160. Varfolomeev, E. et al. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J. Biol. Chem. 284, 34553–34560 (2009).
161. Jost, P. J. et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460, 1035–1039 (2009).
162. Feltham, R. et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 286, 17015–17028 (2011).
163. Mace, P. D. et al. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 283, 31633–31640 (2008).
164. Dueber, E. C. et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334, 376–380 (2011).
165. Ndubaku, C. et al. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem. Biol. 4, 557–566 (2009).
166. Schimmer, A. D., Dalili, S., Batey, R. A. & Riedl, S. J. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 13, 179–188 (2006).
167. Wu, T. Y., Wagner, K. W., Bursulaya, B., Schultz, P. G. & Deveraux, Q. L. Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem. Biol. 10, 759–767 (2003).
168. Schimmer, A. D. et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 5, 25–35 (2004).
169. Gonzalez-Lopez, M. et al. Design, synthesis and evaluation of monovalent Smac mimetics that bind to the BIR2 domain of the anti-apoptotic protein XIAP. Bioorg. Med. Chem. Lett. 21, 4332–4336 (2011).
170. Jansen, B. & Zangemeister-Wittke, U. Antisense therapy for cancer — the time of truth. Lancet Oncol. 3, 672–683 (2002).
171. LaCasse, E. C. et al. Preclinical characterization of AEG35156/GEM 640, a second-generation antisense oligonucleotide targeting X-linked inhibitor of apoptosis. Clin. Cancer Res. 12, 5231–5241 (2006).
172. Shaw, T. J., Lacasse, E. C., Durkin, J. P. & Vanderhyden, B. C. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int. J. Cancer 122, 1430–1434 (2008).
173. Cao, C., Mu, Y., Hallahan, D. E. & Lu, B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene 23, 7047–7052 (2004).
174. McManus, D. C. et al. Loss of XIAP protein expression by RNAi and antisense approaches sensitizes cancer cells to functionally diverse chemotherapeutics. Oncogene 23, 8105–8117 (2004).
175. Holt, S. V., Brookes, K. E., Dive, C. & Makin, G. W. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol. Rep. 25, 1177–1181 (2011).
176. Hu, Y. et al. Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin. Cancer Res. 9, 2826–2836 (2003).
177. Amantana, A., London, C. A., Iversen, P. L. & Devi, G. R. X-linked inhibitor of apoptosis protein inhibition induces apoptosis and enhances chemotherapy sensitivity in human prostate cancer cells. Mol. Cancer Ther. 3, 699–707 (2004).
178. Dean, E. et al. Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion in patients with advanced refractory cancer. J. Clin. Oncol. 27, 1660–1666 (2009).
179. Schimmer, A. D. et al. PhaseI/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J. Clin. Oncol. 27, 4741–4746 (2009).
180. Carter, B. Z. et al. XIAP antisense oligonucleotide (AEG35156) achieves target knockdown and induces apoptosis preferentially in CD34+38– cells in a Phase 1/2 study of patients with relapsed/refractory AML. Apoptosis 16, 67–74 (2011).
181. Schimmer, A. D. et al. Addition of AEG35156 XIAP antisense oligonucleotide in reinduction chemotherapy does not improve remission rates in patients with primary refractory acute myeloid leukemia in a randomized Phase II study. Clin. Lymphoma Myeloma Leuk. 11, 433–438 (2011).
182. Stadel, D. et al. Requirement of NF-κB for Smac mimetic-mediated sensitization of pancreatic carcinoma cells for gemcitabine-induced apoptosis. Neoplasia (in the press).
183. Cheng, Y. J. et al. XIAP-mediated protection of H460 lung cancer cells against cisplatin. Eur. J. Pharmacol. 627, 75–84 (2010).
184. Loeder, S. et al. RIP is required for IAP inhibitor-mediated sensitization of childhood acute leukemia cells to chemotherapy-induced apoptosis. Leukemia (in the press).
185. Carter, B. Z. et al. Small-molecule XIAP inhibitors derepress downstream effector caspases and induce apoptosis of acute myeloid leukemia cells. Blood 105, 4043–4050 (2005).
186. Dean, E. J. et al. A small molecule inhibitor of XIAP induces apoptosis and synergises with vinorelbine and cisplatin in NSCLC. Br. J. Cancer 102, 97–103 (2010).
187. Probst, B. L. et al. Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-α-dependent manner. Cell Death Differ. 17, 1645–1654 (2010).
188. Bockbrader, K. M., Tan, M. & Sun, Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene 24, 7381–7388 (2005).
189. Metwalli, A. R. et al. Smac mimetic reverses resistance to TRAIL and chemotherapy in human urothelial cancer cells. Cancer Biol. Ther. 10, 885–892 (2010).
190. Chauhan, D. et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM). Blood 109, 1220–1227 (2007).
191. Guo, F. et al. Ectopic overexpression of second mitochondria-derived activator of caspases (Smac/diablo) or cotreatment with N-terminus of Smac/diablo peptide potentiates epothilone B derivative-(BMS 247550) and Apo-2L/TRAIL-induced apoptosis. Blood 99, 3419–3426 (2002).
192. Ziegler, D. S. et al. A small-molecule IAP inhibitor overcomes resistance to cytotoxic therapies in malignant gliomas in vitro and in vivo. Neuro Oncol. 13, 820–829 (2011).
193. Servida, F. et al. Novel second mitochondria-derived activator of caspases (Smac) mimetic compounds sensitize human leukemic cell lines to conventional chemotherapeutic drug-induced and death receptor-mediated apoptosis. Invest. New Drugs 29, 1264–1275 (2011).
194. Dineen, S. P. et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 70, 2852–2861 (2010).
195. Fandy, T. E., Shankar, S. & Srivastava, R. K. Smac/DIABLO enhances the therapeutic potential of chemotherapeutic drugs and irradiation, and sensitizes TRAIL-resistant breast cancer cells. Mol. Cancer 7, 60 (2008).
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 123
© 2012 Macmillan Publishers Limited. All rights reserved

196. Fingas, C. D. et al. A Smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology 52, 550–561 (2010).
197. Crnkovic-Mertens, I., Hoppe-Seyler, F. & Butz, K. Induction of apoptosis in tumor cells by siRNA-mediated silencing of the livin/ML-IAP/KIAP gene. Oncogene 22, 8330–8336 (2003).
198. Lecis, D. et al. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and bortezomib. Br. J. Cancer 102, 1707–1716 (2010).
199. Dai, Y. et al. A Smac-mimetic sensitizes prostate cancer cells to TRAIL-induced apoptosis via modulating both IAPs and NF-κB. BMC Cancer 9, 392 (2009).
200. Cheung, H. H., Mahoney, D. J., Lacasse, E. C. & Korneluk, R. G. Down-regulation of c-FLIP enhances death of cancer cells by Smac mimetic compound. Cancer Res. 69, 7729–7738 (2009).
201. Ren, X., Xu, Z., Myers, J. N. & Wu, X. Bypass NFκB-mediated survival pathways by TRAIL and Smac. Cancer Biol. Ther. 6, 1031–1035 (2007).
202. Petrucci, E. et al. A small molecule Smac mimic potentiates TRAIL-mediated cell death of ovarian cancer cells. Gynecol. Oncol. 105, 481–492 (2007).
203. Frenzel, L. P. et al. Novel X-linked inhibitor of apoptosis inhibiting compound as sensitizer for TRAIL-mediated apoptosis in chronic lymphocytic leukaemia with poor prognosis. Br. J. Haematol. 152, 191–200 (2011).
204. Stadel, D. et al. TRAIL-induced apoptosis is preferentially mediated via TRAIL receptor 1 in pancreatic carcinoma cells and profoundly enhanced by XIAP inhibitors. Clin. Cancer Res. 16, 5734–5749 (2010).
205. Siegelin, M. D., Gaiser, T. & Siegelin, Y. The XIAP inhibitor embelin enhances TRAIL-mediated apoptosis in malignant glioma cells by down-regulation of the short isoform of FLIP. Neurochem. Int. 55, 423–430 (2009).
206. Haag, C. et al. Identification of c-FLIPL and c-FLIPS as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 60, 225–237 (2011).
207. Kater, A. P. et al. Inhibitors of XIAP sensitize CD40-activated chronic lymphocytic leukemia cells to CD95-mediated apoptosis. Blood 106, 1742–1748 (2005).
208. Lu, J. et al. Therapeutic potential and molecular mechanism of a novel, potent, nonpeptide, Smac mimetic SM-164 in combination with TRAIL for cancer treatment. Mol. Cancer Ther. 10, 902–914 (2011).
209. Mori, T. et al. Effect of the XIAP inhibitor embelin on TRAIL-induced apoptosis of pancreatic cancer cells. J. Surg. Res. 142, 281–286 (2007).
210. Loeder, S. et al. A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res. 69, 8977–8986 (2009).
211. Vogler, M. et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 69, 2425–2434 (2009).
212. Loeder, S., Drensek, A., Jeremias, I., Debatin, K. M. & Fulda, S. Small molecule XIAP inhibitors sensitize childhood acute leukemia cells for CD95-induced apoptosis. Int. J. Cancer 126, 2216–2228 (2010).
213. Geserick, P. et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054 (2009).
214. Wang, L., Du, F. & Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
215. Laukens, B. et al. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 13, 971–979 (2011).
216. Vanlangenakker, N. et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 18, 656–665 (2011).
217. Vellanki, S. H. et al. Small-molecule XIAP inhibitors enhance γ-irradiation-induced apoptosis in glioblastoma. Neoplasia 11, 743–752 (2009).
218. Giagkousiklidis, S., Vellanki, S. H., Debatin, K. M. & Fulda, S. Sensitization of pancreatic carcinoma cells for γ-irradiation-induced apoptosis by XIAP inhibition. Oncogene 26, 7006–7016 (2007).
219. Dai, Y. et al. Natural IAP inhibitor embelin enhances therapeutic efficacy of ionizing radiation in prostate cancer. Am. J. Cancer Res. 1, 128–143 (2011).
220. Dai, Y. et al. Molecularly targeted radiosensitization of human prostate cancer by modulating inhibitor of apoptosis. Clin. Cancer Res. 14, 7701–7710 (2008).
221. Berger, R. et al. NF-κB is required for Smac mimetic-mediated sensitization of glioblastoma cells for γ-irradiation-induced apoptosis. Mol. Cancer Ther. 10, 1867–1875 (2011).
222. Weisberg, E. et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: effects on mutant FLT3-expressing cells. Mol. Cancer Ther. 6, 1951–1961 (2007).
223. Ziegler, D. S. et al. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J. Clin. Invest. 118, 3109–3122 (2008).
224. Foster, F. M. et al. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 11, R41 (2009).
225. Weisberg, E. et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia 24, 2100–2109 (2010).
226. Aird, K. M., Ghanayem, R. B., Peplinski, S., Lyerly, H. K. & Devi, G. R. X-linked inhibitor of apoptosis protein inhibits apoptosis in inflammatory breast cancer cells with acquired resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol. Cancer Ther. 9, 1432–1442 (2010).
227. Weisberg, E. et al. Beneficial effects of combining a type II ATP competitive inhibitor with an allosteric competitive inhibitor of BCR-ABL for the treatment of imatinib-sensitive and imatinib-resistant CML. Leukemia 24, 1375–1378 (2010).
228. Infante, J. R. et al. A Phase I study of LCL-161, an oral IAP inhibitor, in patients with advanced cancer. in:
Proceedings of the 101st Annual Meeting of the American Association for Cancer Research (17–21 Apr 2010; Washington DC, USA; Abstract 2775).
229. Amaravadi, R. K. et al. Phase 1 study of the Smac mimetic TL32711 in adult subjects with advanced solid tumors and lymphoma to evaluate safety, pharmacokinetics, pharmacodynamics and antitumor activity. in: Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research (2–6 Apr 2011; Orlando, Florida; Abstract LB-406).
230. Sikic, B. I. et al. Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of HGS1029, an inhibitor of apoptosis protein (IAP) inhibitor, in patients (Pts) with advanced solid tumors: Results of a phase I study. J. Clin. Oncol. (Meeting Abstracts) 29, 3008 (2011).
231. Dougan, M. et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J. Exp. Med. 207, 2195–2206 (2010).
232. Hinds, M. G., Norton, R. S., Vaux, D. L. & Day, C. L. Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nature Struct. Biol. 6, 648–651 (1999).
233. Sun, C. et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 401, 818–822 (1999).
234. Cai, Q. et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J. Med. Chem. 54, 2714–2726 (2011).
235. Larisch, S. et al. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nature Cell Biol. 2, 915–921 (2000).
AcknowledgementsWe thank W. Fairbrother, K. Deshayes, E. Dueber and other researchers at Genentech who provided help with comments, figures and suggestions; we also thank C. Hugenberg for expert secretarial assistance. This work has been partially supported by grants from the German Research Foundation (Deutsche Forschungsgemeinschaft), the Federal Ministry of Education and Research (BMBF), the José Carreras Foundation, the European Commission and IAP6/18 (to S.F.).
Competing interests statementThe authors declare competing financial interests: see Web version for details.
FURTHER INFORMATIONSimone Fulda’s homepage: http://www.kinderkrebsstiftung-frankfurt.de/institut/index.html Domagoj Vucic’s homepage: http://www.gene.com/gene/research/postdoctoral/mentors/cellbio/vucic/index.htmlClinicalTrials.gov website (A Study Evaluating the Safety, Tolerability and Pharmacokinetics of GDC-0917 Administered to Patients With Refractory Solid Tumors or Lymphoma): http://clinicaltrials.gov/ct2/show/NCT01226277
SUPPLEMENTARY INFORMATIONSee online article: S1 (table) | S2 (table)
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
R E V I E W S
124 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Most currently marketed drugs are small molecules that target proteins such as enzymes and receptors, which represent a considerably small subset of total cellular pro-teins. By contrast, oligonucleotides are macro molecules that target pre-mRNA and mRNA — the carriers of genetic information before it is translated into proteins. Because mRNAs encode all cellular proteins, oligonucleo-tides targeting mRNA could prove to be effective for targets and diseases that are not treatable by current drugs. For example, genetic diseases — in which the defect in the gene can be best repaired by manipulating DNA or RNA rather than the protein — such as Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) are two such diseases (discussed below). This Review covers the following three approaches that exploit oligonucleotides: RNA interference (RNAi), antisense oligonucleotides and steric-blocking oligonucleotides. These three approaches involve the binding of comple-mentary oligonucleotides to target RNA through base pairing, and therefore all three are — in essence — operating by an antisense mechanism. However, they differ substantially in their downstream mechanisms of action and the functional outcomes they produce.
RNAi and antisense oligonucleotides — which will be discussed only briefly as they have been extensively reviewed in the literature (see REFS 1,2) — modulate
gene expression by inducing enzymatic degradation of targeted mRNA and the removal of the disease-causing gene product, such as an oncogene or a pro-inflammatory cytokine. As cellular enzymes need to recognize these antisense compounds, they can only be chemically modified to a limited degree, which limits our ability to enhance their pharmacological qualities. Antisense compounds that modulate RNA function by blocking the access of cellular machinery to RNA, and so do not lead to degradation of the target RNA, are the main focus of this Review. This different mode of action leads to outcomes such as the repair of a defective RNA or the generation of a novel protein, which cannot be achieved using RNAi or antisense oligonucleotides. Furthermore, because RNA-blocking oligonucleotides do not need to exploit cellular enzymes for their activity, they can be subjected to more extensive chemical modifications that improve their drug-like qualities.
The poor intracellular uptake of RNAi, antisense oligo nucleotides and steric-blocking oligonucleotides is a major impediment to their use as therapeutics. This is the main reason why the antiviral drug fomivirsen3 is currently the only approved antisense drug (although the drug was discontinued in 2004 as the market for the drug diminished). Recent advances in chemistries that improve the intracellular delivery of antisense oligonucleotides, as
1AVI BioPharma, 3450 Monte Villa Parkway, Bothell, Washington 98021, USA.2Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, New York 11724, USA.3Department of Molecular, Cellular and Developmental Biology, 219 Prospect Street, Yale University, New Haven, Connecticut 06520, USA.Correspondence to R.K. e‑mail: [email protected]:10.1038/nrd3625 Published online 20 January 2012
RNA interference(RNAi). A form of post- transcriptional gene silencing in which the expression or transfection of double- stranded RNA induces degradation by nucleases of the homologous endogenous transcripts, resulting in the reduction or loss of gene activity.
RNA therapeutics: beyond RNA interference and antisense oligonucleotidesRyszard Kole1, Adrian R. Krainer2 and Sidney Altman3
Abstract | Here, we discuss three RNA-based therapeutic technologies exploiting various oligonucleotides that bind to RNA by base pairing in a sequence-specific manner yet have different mechanisms of action and effects. RNA interference and antisense oligonucleotides downregulate gene expression by inducing enzyme-dependent degradation of targeted mRNA. Steric-blocking oligonucleotides block the access of cellular machinery to pre-mRNA and mRNA without degrading the RNA. Through this mechanism, steric-blocking oligonucleotides can redirect alternative splicing, repair defective RNA, restore protein production or downregulate gene expression. Moreover, they can be extensively chemically modified to acquire more drug-like properties. The ability of RNA-blocking oligonucleotides to restore gene function makes them best suited for the treatment of genetic disorders. Positive results from clinical trials for the treatment of Duchenne muscular dystrophy show that this technology is close to achieving its clinical potential.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 125
© 2012 Macmillan Publishers Limited. All rights reserved

Antisense oligonucleotidesOligonucleotides that bind to complementary mRNA by base pairing and induce cleavage of targeted mRNA by ribonuclease H, an enzyme that degrades RNA in RNA–DNA duplexes.
Small interfering RNAs(siRNAs). Synthetic, short, 21–22-nucleotide-long double-stranded RNAs with chemical modifications designed to increase their stability and cellular uptake. One strand of siRNA hybridizes to targeted mRNA and allows mRNA degradation.
RNA-induced silencing complex(RISC). A multiprotein complex that, when combined with small interfering RNA (siRNA), affects mRNA degradation. A key component of RISC is an endonuclease, argonaute 2, which cleaves the targeted mRNA within the siRNA–mRNA duplex.
well as differences that characterize the three technologies, are highlighted in this Review. Aptamers, which are more structurally complex than oligonucleotide RNA-based drugs, and interact directly with proteins rather than complementary RNA, are not covered in this article4.
Oligonucleotides that degrade target mRNARNAi. RNAi was first demonstrated in a nematode5 (Caenorhabditis elegans), in which the delivery of exo-genous, long, double-stranded RNA (dsRNA) effectively silenced the expression of a gene (encoding a myofilament protein) by inducing the degradation of a homo logous host mRNA. The mechanism that mediated gene silencing involved degradation of dsRNA into small interfering RNAs (siRNAs) — double-stranded RNA fragments that are 21–22 nucleotides long and interact with a multiprotein RNA-induced silencing complex (RISC). Within the RISC, the siRNA is unwound, the sense strand is discarded, and the antisense or guide strand binds to mRNA. When siRNA is fully complementary to its target, the endo nuclease argonaute 2 — a component of the RISC — cleaves the mRNA 10 and 11 nucleotides downstream from the 5′ end of the antisense strand6 (FIG. 1a).
Although RNAi could not initially be detected in mammalian cells, later studies showed that these cells lacked the ability to cleave dsRNA into siRNA. The discovery that synthetic siRNA can enter the RISC and degrade targeted mRNA when delivered to cultured human cells7 led to a rapid increase in the amount of research related to siRNA. In 2002, the first full year after the discovery of siRNA, a query for siRNA in PubMed resulted in 234 citations; for the 2002–2010 period it lists 33,009 citations, of which 7,241 were from 2010. Notably, a first successful in vivo study was carried out as early as 2003, using naked siRNA to knockdown FAS mRNA in a mouse model of fulminant hepatitis8. The increase in the amount of RNAi research also resulted in the founding of companies — such as Alnylam Pharmaceuticals and Sirna Therapeutics — focused on the development of siRNA as a promising therapeutic platform.
Systemically delivered unmodified siRNA is rapidly degraded by nucleases circulating in the bloodstream; it has a half-life in plasma of approximately a few minutes, and its uptake into target organs and cells is generally poor but it has shown some success in liver delivery8. Chemical modifications to promote metabolic stability and improve target cell penetration have been introduced to overcome such problems. However, it has become apparent that the interaction of siRNAs with the cellular RISC machinery presents a challenge for their use as therapeutics, because only limited chemical modifi-cations can be introduced into siRNA for it to remain functional within the RISC. In the antisense strand, phosphorothioate internucleotide linkages at the 3′ end, and 2′-O-methyl (2′-OMe) nucleotide substitutions (FIG. 2) in one or two internal nucleotides, are tolerated and improve the resistance of the siRNA to nucleases. The sense strand can be modified more heavily (that is, more internal nucleotides can carry 2′-OMe nucleotide substitutions) without substantially reducing efficacy (reviewed in REF. 9).
Because of the difficulty in achieving intracellular delivery of siRNAs, they were administered locally in the majority of initial clinical trials. These included: intra-vitreal injection for the treatment of macular degener-ation (ClinicalTrials.gov identifier: NCT00363714); intranasal delivery for respiratory syncytial virus (ClinicalTrials.gov identifier: NCT00658086); and direct injections in skin lesions for pachyonychia congenita, a rare genetic skin disorder (ClinicalTrials.gov identifier: NCT00716014). It was noted that intranasal delivery in humans results in only minimal systemic distribution of the drug10, indicating that in the absence of delivery agents unmodified siRNAs have a limited bioavailability in humans.
Two clinical trials that are currently underway aim to tackle the problems associated with systemic siRNA delivery by combining siRNA with delivery-enhancing agents. In one trial, a cocktail of two siRNAs, ALN-VSP02, has been formulated with lipid particles and targeted to mRNAs encoding kinesin spindle protein and vascular endothelial growth factor, which are essen-tial for tumour proliferation and tumour-supporting angiogenesis, respectively (see the 4 June 2011 press release on the Alnylam Pharmaceuticals website; ClinicalTrials.gov identifier: NCT00882180). In the second trial (of CALAA-01), siRNA has been formu-lated in a cyclodextrin–adamantane polyethylene glycol particle that includes a targeting component, human transferrin protein, which targets the siRNA to ribo-nucleotide reductase mRNA (ClinicalTrials.gov identi-fier: NCT00689065). These delivery-enhancing moieties should improve the cellular uptake of siRNA; if successful, these clinical studies will be of substantial interest.
A series of lipid-based, liver-directed siRNA carrier particles was recently tested in a mouse model of haemo-philia and in healthy cynomolgus monkeys. Systemic delivery of the formulated siRNAs reduced the levels of factor VII mRNA and glyceraldehyde-3-phosphate dehydrogenase mRNA at remarkably low doses of 0.01 mg per kg in mice and 0.1 mg per kg in monkeys, respectively11. A single high dose of the preparation was well tolerated in both species. As the effects of siRNAs were examined only in the liver, it is not known whether they were taken up by other tissues. Nevertheless, these results represent dramatic improvements in siRNA delivery to specific target tissues — in this case the liver.
Another problem associated with RNAi technology — which stems from mechanisms of RNAi — is the off-target effects of siRNAs. Short dsRNAs, termed micro-RNAs (miRNAs), are produced in mammalian cells and as a class they control the efficiency of the translation of mRNAs. Synthetic siRNAs may interfere with this pro-cess, thus mimicking the effects of miRNAs. Specifically, siRNAs can enter the RISC and bind with certain base pairs mismatched to untargeted mRNAs, thus acting like endogenous miRNAs and leading to off-target gene silencing12. In addition, siRNAs can activate an innate immune response via activation of Toll-like receptors, leading to undesirable side effects such as the induction of pro-inflammatory cytokines or interferon-α6,12. Taken together, these developments indicate that systemic
R E V I E W S
126 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
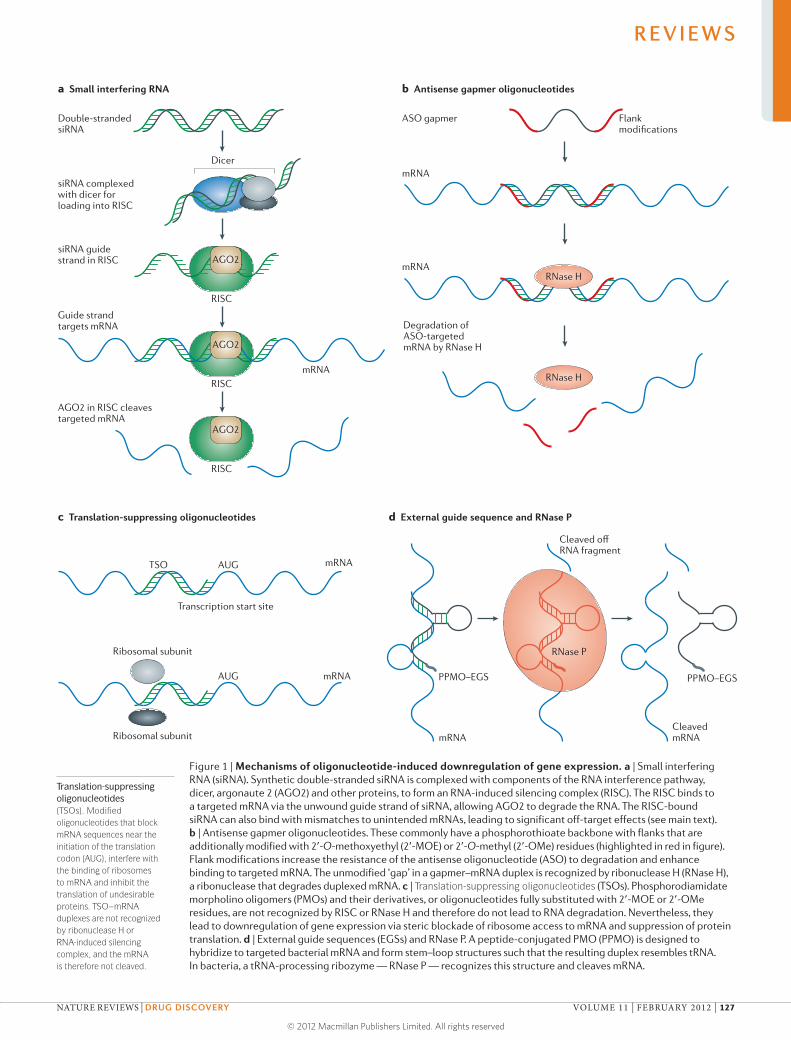
Nature Reviews | Drug Discovery
Dicer
RISC
AGO2
RISCmRNA
mRNA
AGO2
RISC
AGO2
a Small interfering RNA
Double-stranded siRNA
b Antisense gapmer oligonucleotides
ASO gapmer Flankmodifications
siRNA complexed with dicer for loading into RISC
siRNA guidestrand in RISC
Guide strand targets mRNA
AGO2 in RISC cleavestargeted mRNA
Transcription start site
mRNARNase H
Degradation ofASO-targetedmRNA by RNase H
RNase H
c Translation-suppressing oligonucleotides d External guide sequence and RNase P
RNase P
TSO AUG mRNA
AUG mRNA
Ribosomal subunit
Ribosomal subunit
mRNA
PPMO–EGS
CleavedmRNA
Cleaved offRNA fragment
PPMO–EGS
Translation-suppressing oligonucleotides(TSOs). Modified oligonucleotides that block mRNA sequences near the initiation of the translation codon (AUG), interfere with the binding of ribosomes to mRNA and inhibit the translation of undesirable proteins. TSO–mRNA duplexes are not recognized by ribonuclease H or RNA-induced silencing complex, and the mRNA is therefore not cleaved.
Figure 1 | Mechanisms of oligonucleotide-induced downregulation of gene expression. a | Small interfering RNA (siRNA). Synthetic double-stranded siRNA is complexed with components of the RNA interference pathway, dicer, argonaute 2 (AGO2) and other proteins, to form an RNA-induced silencing complex (RISC). The RISC binds to a targeted mRNA via the unwound guide strand of siRNA, allowing AGO2 to degrade the RNA. The RISC-bound siRNA can also bind with mismatches to unintended mRNAs, leading to significant off-target effects (see main text). b | Antisense gapmer oligonucleotides. These commonly have a phosphorothioate backbone with flanks that are additionally modified with 2′-O-methoxyethyl (2′-MOE) or 2′-O-methyl (2′-OMe) residues (highlighted in red in figure). Flank modifications increase the resistance of the antisense oligonucleotide (ASO) to degradation and enhance binding to targeted mRNA. The unmodified ‘gap’ in a gapmer–mRNA duplex is recognized by ribonuclease H (RNase H), a ribonuclease that degrades duplexed mRNA. c | Translation-suppressing oligonucleotides (TSOs). Phosphorodiamidate morpholino oligomers (PMOs) and their derivatives, or oligonucleotides fully substituted with 2′-MOE or 2′-OMe residues, are not recognized by RISC or RNase H and therefore do not lead to RNA degradation. Nevertheless, they lead to downregulation of gene expression via steric blockade of ribosome access to mRNA and suppression of protein translation. d | External guide sequences (EGSs) and RNase P. A peptide-conjugated PMO (PPMO) is designed to hybridize to targeted bacterial mRNA and form stem–loop structures such that the resulting duplex resembles tRNA. In bacteria, a tRNA-processing ribozyme — RNase P — recognizes this structure and cleaves mRNA.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 127
© 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
N
O
N
O
O
P ON
Base
BaseO
N
O
N
O
O
P O
Base
Base
O
NNH
H
N
O
N
OO
P ON
Base
BaseO
ONH
RXRRXRRXRRXRXAcetylated
O
OP
O
S O
O
O
Base
Base
O
PO
S O
OCH3
OOCH3
O
OP
O
O O
O
O
Base
Base
PO
O O
OH
OH
O
OP
O
S O
O
O
Base
Base
OCH3
PO
S O
OCH3
O
OP
O
S O
O
O
Base
Base
PO
S O
O
PO
S O
O
Base
Base
O
PO
S O
OO
O
DNA (PS)
PMO PPMO PMOplus
2′-MOE 2′-OMe LNA RNA
delivery of chemically modified siRNA is not very effective and that more work will be needed to achieve sufficient therapeutic activity and specificity in the absence of delivery agents.
It has now been over a decade since the discoveries of RNAi and siRNAs but despite some promising clin-ical trial results the industry remains cautious about the therapeutic potential of RNAi-based drugs. For example, although Merck acquired Sirna Therapeutics for US$1.1 billion, it was quoted in 2009 as remaining sceptical about the development of siRNA-based drugs13; meanwhile, Roche, Novartis and Pfizer14 decided to dramatically reduce or eliminate their RNAi research programmes, which raised the question “Is RNAi dead?” in a 2011 editorial15 (see the article on the GenomeWeb website, and the article on the Proactive Investors website). This is reminiscent of a previous question — “Does antisense exist?” — that was raised in a commentary in 1995 (REF. 16). The history of anti-sense oligonucleotides (BOX 1), which now seem to be well on their way to becoming drugs, suggests that
RNAi will eventually succeed as a therapeutically viable technology. As a possible harbinger of further progress, an upcoming clinical trial will test chemically modified siRNA for the treatment of diabetic macular oedema (ClinicalTrials.gov identifier: NCT01445899).
Antisense oligonucleotides. Since the first application of a short fragment of unmodified DNA in cell cul-ture as an antisense oligonucleotide, by Zamecnik and Stephenson17 in 1978, remarkable progress has been made in oligonucleotide drug development. Numerous chemical modifications that improve the drug-like prop-erties of DNA have been introduced (BOX 1), an antisense drug has been marketed and successful clinical trials are underway, as described below. Currently, a typical antisense oligonucleotide drug candidate is about 20 nucleotides long and has a phosphorothioate linkage (FIG. 2) between the nucleosides that form the backbone. In addition, five nucleotides at each flank are further modified (FIG. 2) to protect the antisense oligonucleotide from exonucleases, thus increasing its stability in vivo.
Figure 2 | Oligonucleotide chemistries. All oligonucleotides are negatively charged. Phosphorothioate (PS) backbones, as well as 2′-O-methoxyethyl (2′-MOE) and 2′-O-methyl (2′-OMe) substituents, increase resistance to degradation and promote protein binding to target RNA. Locked nucleic acid (LNA) modification markedly increases the binding of the oligonucleotide to the targeted mRNA (see top panel). In phosphorodiamidate morpholino oligomers (PMOs), ribose (RNA) or deoxyribose (DNA) is replaced with morpholine rings, and the phosphorothioate or phosphodiester (RNA) groups are replaced with uncharged phosphorodiamidate groups, resulting in a compound that is neutral and very resistant to degradation (see bottom panel). Positively charged piperazine residues in positively charged PMOs (PMOplus), or positively charged arginine-rich peptides in peptide-conjugated PMOs (PPMOs), dramatically improve the intracellular uptake of the oligomers.
R E V I E W S
128 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

This design leaves a central 10-nucleotide phosphoro-thioate gap (hence the term ‘gapmers’) that allows the cleavage of targeted mRNA by ribonuclease H (RNase H)18 (FIG. 1b). The modified flanks also improve the binding of the antisense oligonucleotide to mRNA and reduce the side effects that are associated with the presence of phosphorothioate residues (BOX 1).
In contrast to siRNA, which tolerates only limited modifications to remain RISC-compatible, more extensive chemical modifications in gapmers do not abrogate RNase H activity. One such modification, 2′-O-methoxyethyl (2′-MOE) ribonucleoside (FIG. 2), is present in two anti-sense oligonucleotides that have recently been successful in clinical trials: mipomersen (also known as ISIS 301012) and custirsen (also known as OGX-111)19,20.
Mipomersen is a 2′-MOE phosphorothioate anti-sense oligonucleotide gapmer that targets mRNA encoding apolipoprotein B100 (APOB100) expressed in the liver; APOB100 is a protein that is involved in the production of low-density lipoprotein cholesterol
(LDL-C; also known as ‘bad’ cholesterol). Mipomersen reduces the levels of APOB100 secreted from liver cells into the bloodstream. In a recent Phase III trial, mipomersen lowered LDL-C levels by ~25% in patients with homozygous familial hypercholesterolaemia who had very high LDL-C levels (up to 300 mg per dl); by comparison, placebo treatment lowered LDL-C levels by ~3%. Twenty-six out of thirty-four treated patients incurred injection-site reactions, and four had a signifi-cant increase in alanine aminotransferase levels, which is indicative of liver stress19. These signals need to be carefully monitored, especially in a chronic disease.
Custirsen is also a phosphorothioate antisense oligo-nucleotide gapmer with 2′-MOE flanks, and it targets the mRNA encoding clusterin, an anti-apoptotic chaper-one protein that is upregulated in cancer cells. Significant declines in serum levels of clusterin were seen in the Phase II trial of custirsen in patients with advanced meta-static prostate cancer; the trial included 41 patients in each arm, and patients were treated with a combination
Box 1 | The ups and downs of antisense oligonucleotides
In 1978 Zamecnik and Stephenson17,96 found that a 13‑nucleotide‑long oligodeoxynucleotide that was complementary to a target sequence in Rous sarcoma virus RNA inhibited viral replication and protein translation in vitro; the field of antisense oligonucleotides was born. Remarkably, in this pioneering work the authors even introduced chemical modifications at the 3′ and 5′ ends of the oligonucleotide to reduce its degradation by cellular nucleases, which improved its activity, and they demonstrated that the same antisense oligonucleotide used against another avian virus was less effective, presumably because the three mismatches in the target sequence weakened its binding. Thus, two key themes — improvements in antisense oligonucleotide efficacy via chemical modifications, and the need for specificity controls — were established at the very birth of the field.
It took almost 10 years before the next major advancement in the field. The most consequential was the introduction of phosphorothioate internucleotide linkages97 (FIG. 2), followed by the addition of 2′‑O‑methyl‑modified nucleotides at the 3′ and 5′ ends, which protected the antisense oligonucleotides from degradation by nucleases98. These modifications dramatically increased the stability of antisense oligonucleotides in cell culture and in vivo, while still allowing ribonuclease H (RNase H)-mediated cleavage of RNA in phosphorothioate antisense oligonucleotide–RNA duplexes (reviewed in REF. 18). Several phosphorothioate antisense oligonucleotide drug candidates progressed through various stages of drug development but only one — fomivirsen — was registered (in 1998) as a treatment for cytomegalovirus‑induced retinitis in immunocompromised patients with AIDS. The drug, a 21‑mer oligonucleotide, was delivered via intraocular injection. This choice of indication and delivery contributed to the success of the drug. Intravitreal distribution of the drug dramatically limited the necessary dose (330 μg per 0.05 ml) and eliminated systemic exposure of the patient to the drug, thus avoiding any potential side effects. Consequently, 20 years after Zamecnik’s discovery, the first antisense drug reached the market3.
Although off‑target effects have not been a serious issue for phosphorothioate antisense oligonucleotides, this backbone imparts a significant, hybridization‑independent toxicity profile that varies with different sequences. The effects include increased coagulation time, pro‑inflammatory effects and activation of the complement pathway. At higher concentrations, phosphorothioate antisense oligonucleotides lead to renal tubule changes and thrombocytopaenia99. In addition, phosphorothioate antisense oligonucleotides that contain certain sequences induce a strong immunostimulatory response through their interactions with Toll‑like receptors100 or they bind directly to proteins, leading to unexpected spurious effects. These results led to an article entitled: “Does antisense exist?”16. Some companies continued the development of phosphorothioate antisense oligonucleotides until very recently.
Oblimersen, which was developed by Genta as a potential anticancer drug, is one such example. Oblimersen targets the mRNA of the gene encoding B cell lymphoma 2 (BCL-2), an anti-apoptotic protein that is overexpressed in numerous cancers. It is noteworthy how difficult the attempts have been to bring oblimersen to market. Searching the ClinicalTrials.gov database for oblimersen shows that since 1999, this drug has been tested in 45 clinical trials. Unfortunately, recent trials for the treatment of advanced melanoma and myeloma, which enrolled almost 1,000 patients, demonstrated either very modest effects of the drug (for the treatment of melanoma) or no effect of the drug101,102 (for the treatment of myeloma). These efforts were certainly costly. In April 2002 Genta and Aventis (now Sanofi) entered into a development agreement for oblimersen that was valued at the time at US$480 million (See the article on the Forbes website). Today, Genta’s stock is no longer listed on a regular NASDAQ stock exchange board, and the oblimersen programme was terminated after additional trials missed the expected primary targets103.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 129
© 2012 Macmillan Publishers Limited. All rights reserved

External guide sequence(EGS). A short RNA sequence designed to bind to targeted mRNA and form a structure that is recognized by a tRNA-processing ribozyme, ribonuclease P. Ribonuclease P cleaves mRNA and thereby downregulates the function of a targeted gene.
Splice-switching oligonucleotides(SSOs). Chemically modified oligonucleotides that block sequences in pre-mRNA that are involved in pre-mRNA splicing, and redirect mRNA splicing pathways. SSO–pre-mRNA duplexes are not recognized by ribonuclease H or the RNA-induced silencing complex, and the pre-mRNA is thus not cleaved.
Alternative splicingSplicing of pre-mRNA to yield more than one kind of mRNA — that is, different splice variants — by frequently including or excluding an exon.
Translational reading frameArrangement of mRNA nucleotides into triplets (codons) that, when read by the ribosome, are translated into one amino acid per codon. The reading frame usually starts with a translation initiation codon, AUG — for example, AUG UUU ACA GCA. Deletion of a nucleotide, such as the third uridylic in the second codon, changes the reading frame to AUG ‘UUA CAG CA’, thus preventing the translation of the desired protein.
of custirsen and docetaxel or docetaxel alone20. Custirsen was well tolerated, although fever was observed in some patients. The most encouraging outcome was the increase in median overall survival in patients treated with custirsen, which was ~24 months compared with ~17 months for patients treated with docetaxel alone. Differences in other measures were not statistically sig-nificant. Nevertheless, on the strength of these results two Phase III trials were initiated (see the 30 September 2010 press release on the OncoGenex website). Custirsen was not effective in a Phase II study of women with meta-static breast cancer21, suggesting that the efficacy of the drug may be cancer-specific.
The first-generation antisense phosphorothioate drug fomivirsen was approved 20 years after the discovery of the first antisense oligonucleotide; if custirsen and mipomersen are approved in the near future, another 10 years will have elapsed from the approval of fomivirsen to the approval of these new, second-generation, gapmer-type antisense drugs. This timescale is similar to that for the development of monoclonal antibodies — another class of macromolecular drugs (BOX 1). See TABLE 1 for a comparison of the RNA therapeutics discussed in this Review.
Steric-blocking oligonucleotidesIn parallel to the development of gapmer antisense oligo-nucleotides, oligonucleotides were developed that do not induce RNase H-mediated cleavage of mRNA but instead act by blocking targeted RNA without inducing its degradation. Early examples of these oligonucleotides included oligonucleoside methylphosphonates22 and phosphorodiamidate morpholino oligomers23 (PMOs) (FIG. 2). The outcomes of this approach depend on the nucleotide sequence elements in mRNA or pre-mRNA that are targeted. These outcomes can include modula-tion of splicing when pre-mRNA is targeted, blockade of mRNA translation or RNA folding, and external guide sequence (EGS)-directed mRNA degradation by a tRNA-processing ribozyme, RNase P. They can also be used to block toxic RNAs that would otherwise sequester protein factors at their expanded triplet repeats (FIGS 1,3). These applications are highlighted below.
Splice-switching oligonucleotides. Oligonucleotide-induced modulation of splicing leads to several out-comes in cell culture and in vivo that have potential therapeutic value. These outcomes are not achievable with siRNA or classic gapmer antisense oligonucleo-tides, which only downregulate gene expression. Splice-switching oligo nucleotides (SSOs), oligonucleotides that modulate pre-mRNA splicing, can repair defective RNA and restore the production of essential proteins; they can also generate novel proteins with desirable properties and regulate the presence of disease-related splice variant proteins. The latter outcome is achieved by modulation of alternative splicing of pre-mRNA. As over 95% of all human genes produce splice variant proteins by alternative splicing, modulation of alter-native splicing may be applicable to a multitude of diseases24.
To modulate pre-mRNA splicing, oligonucleotides must block RNA sequences that are essential for splicing and prevent the interaction of splicing factors — such as RNA-binding proteins, small nuclear RNAs and other components of the spliceosome — with the pre-mRNA (BOX 2). The chemistries that have been shown to work in animal models include peptide nucleic acids (PNAs), alternating locked nucleic acids (LNAs) and deoxynucleo tide oligonucleotides, fully modified (non-gapmer) 2′-substituted oligonucleotides and PMO-based oligomers25–28 (FIG. 2). The latter two SSO chemistries have been used in clinical trials that tested splicing modulation as a treatment for DMD29–32 (FIG. 3a).
Duchenne muscular dystrophyDMD is a severe genetic disorder that affects one in 3,500 newborn males. It causes muscle wasting that leads to loss of mobility by 10–12 years of age, and death in the mid-20s resulting from failure of respiratory and car-diac functions. The genetic defects that underlie DMD are mostly DNA deletions within the gene that encodes dystrophin, a protein that connects intracellular actin filaments to the sarcolemma membrane and as such is essential for maintaining the integrity of that membrane. In its absence, muscle fibres disintegrate at a rate that outpaces muscle regeneration mechanisms.
In most cases of DMD, the deletions disrupt the translational reading frame, thus abrogating expression of the dystrophin protein. By contrast, in a milder form of the disease, Becker muscular dystrophy (BMD), the deletions mostly preserve the reading frame, allowing the production of truncated but partially functional dys-trophin. Remarkably, even though the mutant dystrophin produced in BMD may have large internal deletions, and is produced at levels below normal, its activity is largely sufficient to retain muscle function; in some affected indi-viduals muscle function is close to normal. This observa-tion indicates that a drug that could partially restore the function of dystrophin protein could be of clinical value for the treatment of DMD (reviewed in REFs 33,34).
With 2.4 million base pairs, the dystrophin gene is the largest gene in humans. It comprises 79 exons, 34 of which encode full codons (termed here as in-frame exons). Deletion of an in-frame exon (or exons) does not disrupt the overall reading frame, and allows translation of dystrophin that retains the correct amino acid sequences at the termini of the molecule but is missing its internal portion. Such deletions usually are found in patients with BMD. In the remaining 45 exons the last, first or both terminal codons are split between the two adjacent exons. Most of these exons are out-of-frame and, in contrast to most in-frame exons, their deletion disrupts translation of the dystrophin protein and so causes DMD. The same is true for a deletion that starts at an in-frame exon but ends in an out-of-frame exon33.
It follows, therefore, that SSO-induced skipping of the adjacent out-of-frame exon should restore the reading frame and convert severe DMD to the milder form of the disease, BMD35 (FIG. 3a). This approach would be applicable to about 85% of patients with DMD, as some mutations cannot be corrected by exon skipping.
R E V I E W S
130 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
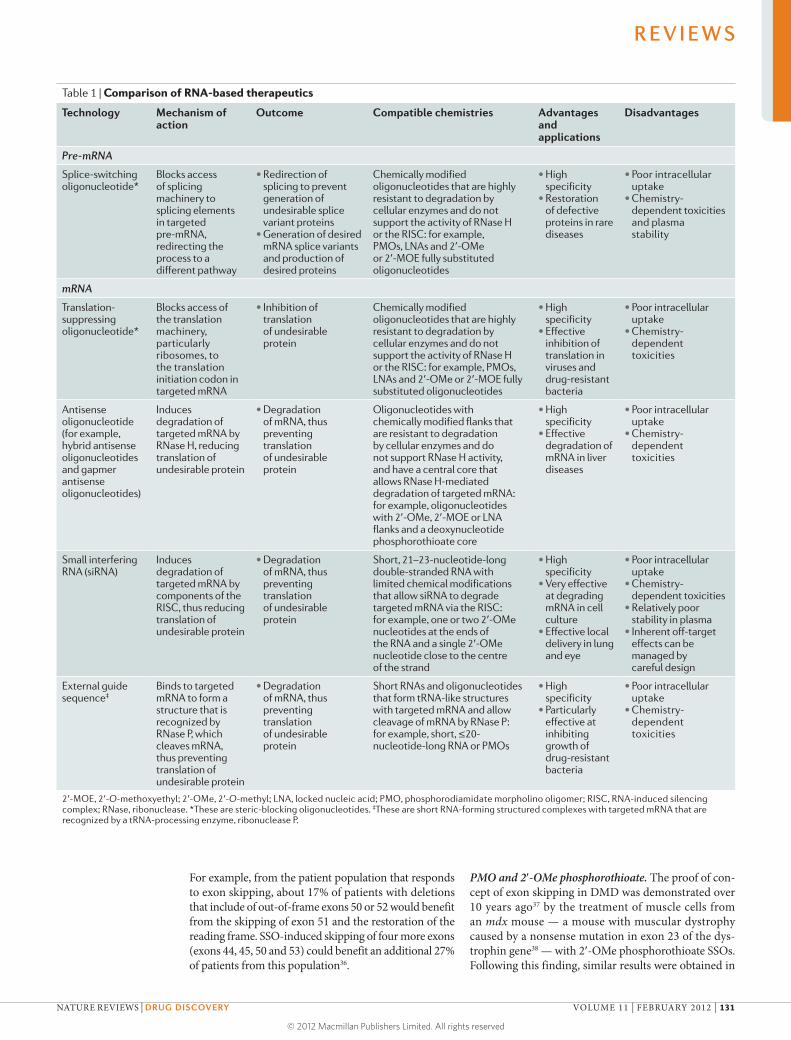
For example, from the patient population that responds to exon skipping, about 17% of patients with deletions that include of out-of-frame exons 50 or 52 would benefit from the skipping of exon 51 and the restoration of the reading frame. SSO-induced skipping of four more exons (exons 44, 45, 50 and 53) could benefit an additional 27% of patients from this population36.
PMO and 2′-OMe phosphorothioate. The proof of con-cept of exon skipping in DMD was demonstrated over 10 years ago37 by the treatment of muscle cells from an mdx mouse — a mouse with muscular dystrophy caused by a nonsense mutation in exon 23 of the dys-trophin gene38 — with 2′-OMe phosphorothioate SSOs. Following this finding, similar results were obtained in
Table 1 | Comparison of RNA-based therapeutics
Technology Mechanism of action
Outcome Compatible chemistries Advantages and applications
Disadvantages
Pre-mRNA
Splice-switching oligonucleotide*
Blocks access of splicing machinery to splicing elements in targeted pre-mRNA, redirecting the process to a different pathway
• Redirection of splicing to prevent generation of undesirable splice variant proteins
• Generation of desired mRNA splice variants and production of desired proteins
Chemically modified oligonucleotides that are highly resistant to degradation by cellular enzymes and do not support the activity of RNase H or the RISC: for example, PMOs, LNAs and 2′-OMe or 2′-MOE fully substituted oligonucleotides
• High specificity
• Restoration of defective proteins in rare diseases
• Poor intracellular uptake
• Chemistry-dependent toxicities and plasma stability
mRNA
Translation-suppressing oligonucleotide*
Blocks access of the translation machinery, particularly ribosomes, to the translation initiation codon in targeted mRNA
• Inhibition of translation of undesirable protein
Chemically modified oligonucleotides that are highly resistant to degradation by cellular enzymes and do not support the activity of RNase H or the RISC: for example, PMOs, LNAs and 2′-OMe or 2′-MOE fully substituted oligonucleotides
• High specificity
• Effective inhibition of translation in viruses and drug-resistant bacteria
• Poor intracellular uptake
• Chemistry-dependent toxicities
Antisense oligonucleotide (for example, hybrid antisense oligonucleotides and gapmer antisense oligonucleotides)
Induces degradation of targeted mRNA by RNase H, reducing translation of undesirable protein
• Degradation of mRNA, thus preventing translation of undesirable protein
Oligonucleotides with chemically modified flanks that are resistant to degradation by cellular enzymes and do not support RNase H activity, and have a central core that allows RNase H-mediated degradation of targeted mRNA: for example, oligonucleotides with 2′-OMe, 2′-MOE or LNA flanks and a deoxynucleotide phosphorothioate core
• High specificity
• Effective degradation of mRNA in liver diseases
• Poor intracellular uptake
• Chemistry-dependent toxicities
Small interfering RNA (siRNA)
Induces degradation of targeted mRNA by components of the RISC, thus reducing translation of undesirable protein
• Degradation of mRNA, thus preventing translation of undesirable protein
Short, 21–23-nucleotide-long double-stranded RNA with limited chemical modifications that allow siRNA to degrade targeted mRNA via the RISC: for example, one or two 2′-OMe nucleotides at the ends of the RNA and a single 2′-OMe nucleotide close to the centre of the strand
• High specificity
• Very effective at degrading mRNA in cell culture
• Effective local delivery in lung and eye
• Poor intracellular uptake
• Chemistry-dependent toxicities
• Relatively poor stability in plasma
• Inherent off-target effects can be managed by careful design
External guide sequence‡
Binds to targeted mRNA to form a structure that is recognized by RNase P, which cleaves mRNA, thus preventing translation of undesirable protein
• Degradation of mRNA, thus preventing translation of undesirable protein
Short RNAs and oligonucleotides that form tRNA-like structures with targeted mRNA and allow cleavage of mRNA by RNase P: for example, short, ≤20-nucleotide-long RNA or PMOs
• High specificity
• Particularly effective at inhibiting growth of drug-resistant bacteria
• Poor intracellular uptake
• Chemistry-dependent toxicities
2′-MOE, 2′-O-methoxyethyl; 2′-OMe, 2′-O-methyl; LNA, locked nucleic acid; PMO, phosphorodiamidate morpholino oligomer; RISC, RNA-induced silencing complex; RNase, ribonuclease. *These are steric-blocking oligonucleotides. ‡These are short RNA-forming structured complexes with targeted mRNA that are recognized by a tRNA-processing enzyme, ribonuclease P.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 131
© 2012 Macmillan Publishers Limited. All rights reserved
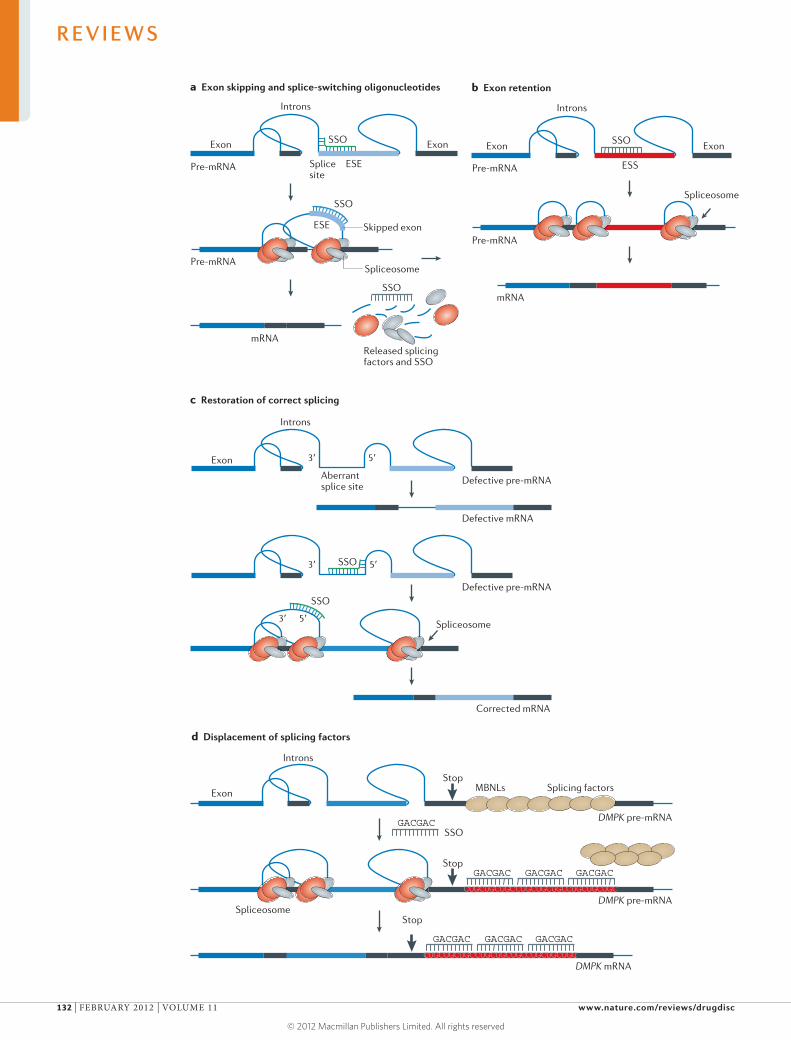
SSO
SSO
SSO
SSO
mRNA
SSO
Released splicingfactors and SSO
SSO
SSO
Pre-mRNA
Pre-mRNA
Exon
Introns
Exon
Introns
Exon
Introns
Exon
a Exon skipping and splice-switching oligonucleotides
c Restoration of correct splicing
b Exon retention
d Displacement of splicing factors
Defective mRNA
Defective pre-mRNA
Pre-mRNA
mRNA
Spliceosome
ESE Skipped exon
Spliceosome
Spliceosome
ESESplice site
Aberrant splice site
3′ 5′
Pre-mRNA
Exon
Introns
Exon
ESS
Nature Reviews | Drug Discovery
Defective pre-mRNA
3′
3′
5′
5′
Corrected mRNA
Stop
DMPK pre-mRNA
Splicing factors
Spliceosome
GACGAC
GACGAC
CUGCUGCUGC
GACGAC
CUGCUGCUGC
GACGAC
CUGCUGCUGC
Stop
DMPK pre-mRNA
MBNLs
Stop
DMPK mRNA
GACGAC
CUGCUGCUGC
GACGAC
CUGCUGCUGC
GACGAC
CUGCUGCUGC
R E V I E W S
132 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

muscle cells taken from patients with DMD and after intramuscular injections of 2′-OMe phosphorothioate SSOs in the mdx mouse model39–41.
2′-OMe phosphorothioate SSOs and PMO SSOs were subsequently tested by systemic administration in mdx mouse and dog models of DMD26,42–44. In mdx mice, PMO SSOs were more effective than 2′-OMe phosphorothioate SSOs at restoring the reading frame of dystrophin mRNA42,43. A single intravenous dose (80 mg per kg) of PMO SSOs induced dystrophin expression in about 25% of muscle fibres in the quadri-ceps, which was the muscle that responded best to treat-ment. Seven weekly injections increased the percentage of muscle fibres that expressed dystrophin to over 80%. A three-dose treatment improved tibialis anterior muscle function to nearly 80% of normal, even though the level of dystrophin in this muscle was only 20% of normal. This is an important result, as it suggests that below-normal dystrophin levels may still be clinically beneficial.
PMO SSOs were also effective in a dog model of DMD44. Importantly, this study demonstrated the effi-cacy of exon skipping in a large animal, showing clear improvements in the mobility of the dog. Although an untreated dog had difficulty finishing a 15-metre run, a dog treated with five weekly injections (at a dose of 120 mg per kg) ran quickly and completed the course without difficulty. This performance was achieved when dystrophin levels in the muscles of the treated animals were — on average — 26% of normal, thus confirming with a more challenging animal model that complete restoration of dystrophin is not essential for clinically
relevant effects. In both the dog and mouse models of DMD, 2′-OMe phosphorothioate SSOs were less effec-tive. For example, a single intravenous injection of the compound (at a dose of ~80 mg per kg) resulted in less than 10% of fibres that were dystrophin-positive in the quadriceps of the mdx mouse. Nevertheless, both 2′-OMe SSOs and PMO SSOs entered into clinical trials as a treatment for DMD. These trials have recently been extensively appraised (reviewed in REF. 45) and so will be discussed here only briefly.
In trials that tested the safety of the drug, which was administered by single, intramuscular injections, 2′-OMe phosphorothioate SSOs (PRO-051)29 and PMO SSOs (AVI-4658)30 were delivered into the tibialis ante-rior and extensor digitorum brevis muscles, respec-tively. Both drugs caused accurate skipping of exon 51: PRO-051 at 0.8 mg, and AVI-4658 at two doses, 0.9 mg and 0.09 mg. Importantly, with both drugs the dys-trophin protein was correctly localized to the muscle fibre membranes, irrespective of the size of the exon deletions in individual patients, which varied from five exons (exons 45–50), to a single exon (exon 50). This indicated that the rescued dystrophin was functional in every patient, as it reconstituted a dystroglycan complex that is normally located in the sarcolemma of healthy muscle. AVI-4658 produced a statistically sig-nificant increase in dystrophin-positive fibres, which was up to 79% of normal and above the background of the control sample from a saline-treated contralateral extensor digitorum brevis muscle. In the PRO-051 trial, up to 97% of muscle fibres were dystrophin-positive. However, this study lacked a negative control, which made quantitative analysis of newly generated dystro-phin questionable.
PRO-051 and AVI-4658, given systemically, were also evaluated in clinical trials31,32. In a Phase I/IIa trial, PRO-051 was initially delivered weekly by subcutane-ous injection for 5 weeks at doses of 0.5–6.0 mg per kg. In a continuation study, all patients were treated 6–15 months later with a weekly dose of 6.0 mg per kg, and results were reported after 12 weeks. In support of the proof of principle for exon-skipping therapy, muscle biopsy samples collected after the last injection showed the presence of dystrophin-positive fibres in most patients. However, because background control pretreat-ment biopsy samples were collected for only a few sub-jects, the validity of the assessment was again debatable. The improvement in the clinical status of the patients was measured by a 6-minute walk test after the addi-tional 12-week treatment. A slight but not statistically significant increase in the distance covered was noted31.
In a Phase II trial, AVI-4658 was systemically deliv-ered by weekly intravenous infusion at doses ranging from 0.5–20.0 mg per kg for 12 weeks; pre- and post-treatment biopsy samples were collected from every patient. Seven patients responded well to treatment; their mean dystrophin fluorescence intensity increased from 8.9% to 16.4% of normal (control). One patient treated with 20 mg per kg of AVI-4658 exhibited 55% of dystrophin-positive fibres over negative control. Overall, there was a significant dose-dependent increase
Figure 3 | Mechanisms of oligonucleotide-induced modulation of gene expression. a | Exon skipping and splice-switching oligonucleotides (SSOs). A chemically modified, RNA-blocking oligonucleotide targeted to a splice site or an exon-internal exonic splicing enhancer (ESE) in pre-mRNA prevents the proper assembly of the spliceosome on the exon and redirects splicing to another pathway, inducing skipping of the targeted exon. Such alternatively spliced mRNA may encode a novel protein with favourable properties, or it may restore translation if exon skipping restores a reading frame, as is the case in Duchenne muscular dystrophy; alternatively, it may change the balance of alternative splice variants. These outcomes cannot be accomplished by antisense oligonucleotides or small interfering RNA, which degrade targeted mRNA. b | Exon retention by SSOs. Some exons are poorly spliced into mRNA because they contain exonic splicing silencer (ESS) elements. An SSO designed to block an ESS interferes with this element’s role in splicing and promotes exon inclusion, as has been demonstrated in the case of spinal muscular atrophy, a genetic disorder. c | Restoration of correct splicing and RNA repair by SSOs. An intron mutation may create and/or activate aberrant splice sites, leading to the inclusion of an intronic fragment into the spliced mRNA, in essence creating a pseudo-exon and interfering with the translational reading frame. An SSO targeted to the aberrant splicing elements restores correct splicing and allows translation of the correct, fully functional protein. d | Displacement of splicing factors from triplet repeats. An extended triplet repeat (CUGCUGCUG) in dystrophia myotonica protein kinase (DMPK) pre-mRNA attracts splicing factors known as muscleblind-like proteins (MBNLs), and titrates them out from the nucleoplasm. As a result, DMPK and several other pre-mRNAs are not properly processed, which prevents the translation of several proteins and causes myotonic dystrophy, a neuromuscular disorder. A modified steric-blocking oligonucleotide displaces MBNL, allowing it to participate in the splicing of appropriate mRNAs and restoring function in the affected muscle.
▶
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 133
© 2012 Macmillan Publishers Limited. All rights reserved

in new dystrophin protein in the muscle biopsy samples. Furthermore, there was a dose-dependent, statistically significant decrease in inflammatory infiltrates in the biopsy material, suggesting that restoration of dystro-phin — even at low levels — ameliorates certain aspects of the disease. Similarly to 2′-OMe phosphorothioate SSOs, PMO SSOs did not result in a statistically signifi-cant increase in distance covered in the 6-minute walk test pre- versus post-treatment32.
The above two clinical studies also highlight the dif-ferences between the PRO-051 and AVI-4658 chemis-tries in terms of tolerability. AVI-4658 was well tolerated and there were no reported drug-related adverse effects at any dose32. However, all patients treated in a continu-ation study with PRO-051 for 12 weeks (at 6 mg per kg) exhibited proteinuria, which is a possible sign of drug-related kidney damage31.
The safety of AVI-4658 was further confirmed in animal studies. At doses of up to 320 mg per kg in monkeys and up to 960 mg per kg in mice, there were no observable toxic effects46. In mdx mice, there was no PMO SSO-induced toxicity after a single injection, even at 3 g per kg of body weight47. A study of the phar-macokinetics and pharmacodynamics of a 2′-OMe phosphorothioate SSO that induced the skipping of exon 23 in mdx mice demonstrated high accumulation
of the compound in kidneys and the liver; however, an analysis of potential toxic effects in these organs was not reported48.
Peptide-conjugated PMOs. PMO SSOs did not gener-ate appreciable levels of dystrophin in the heart of dystrophic dogs44 and, even with high doses, they only generated low levels of dystrophin in the hearts of mdx mice47. Considering that patients with DMD usually die of heart or respiratory failure, this is an important issue. The synthesis of improved, second-generation, peptide-conjugated PMOs (PPMOs) (FIG. 2) was there-fore a significant advancement. In mdx mice, a PPMO (AVI-5225) that included a positively charged, arginine-rich peptide was superior to PMO and other chemistries at generating dystrophin49. When administered at a dose of 12.5 mg per kg, it skipped exon 23 in mdx mice and restored dystrophin protein expression in skeletal mus-cles to levels that were close to normal, which remained unchanged for up to 11 weeks. The PPMO treatment led to a long-term reduction in cardiac hypertrophy50 and also prevented heart failure in mdx mice that were chal-lenged with the β-adrenergic receptor antagonist dobu-tamine; by comparison, 60% of untreated mice died51.
Even more convincing evidence for the high efficacy of PPMO-induced exon skipping was obtained using a double-knockout mouse deficient in both utrophin and dystrophin that displayed very severe muscular dystrophy. As they also lack utrophin, a muscle protein that partially compensates for the lack of dystrophin in mdx mice, these double-knockout mice are largely immobile and have an average lifespan of 8.2 weeks, compared with about 2 years for mdx mice. Treatment with a PPMO targeted to exon 23 and administered at a dose of 25 mg per kg per week for 6 weeks, beginning at 10 days of age, dramatically increased the animals’ mobility, food-seeking behaviour and the turgor of the tail to close to normal values52. Variants of arginine-rich PPMOs that are particularly effective at restoring dystro-phin expression in the heart muscle of mdx mice have been recently reported53.
It should be noted that in a preliminary study in mon-keys, 12 weekly doses of a PPMO targeted to exon 50 of the dystrophin gene resulted in kidney toxicity, which was not observed following a 4-week treatment (see the 25 March 2010 press release on the AVI BioPharma website). As PMO at the core of PPMO is very stable in serum, intracellularly54 and in vivo (R.K. and C. Eckhoff, unpublished observations), it seems possible that less frequent dosing may reduce the accumulation of the compound in the kidneys and alleviate kidney toxicity.
Other indications for SSOsThe therapeutic application of SSOs is most advanced in DMD but, as described in more detail below, these com-pounds are also being studied as potential treatments for two other genetic diseases — SMA and β-thalassaemia — as well as for rheumatoid arthritis, a major inflam-matory condition. Although in each condition a desired outcome is the production of a functional protein, a dif-ferent mechanism of splicing modulation is exploited to
Box 2 | Pre-mRNA splicing as a therapeutic target
The discovery of RNA splicing104,105 was followed by the identification of an intronic mutation in the human β‑globin gene, which corrupted the splicing of β‑globin pre‑mRNA even though the natural splice sites were not mutated and remained potentially functional106,107. This defect prevented the proper translation of the β‑globin protein, causing β‑thalassaemia — a genetic blood disorder. The single point mutation in intron 1 of the β‑globin gene created an additional aberrant 3′ splice site, which redirected the spliceosome away from the natural 3′ splice site. This aberrant splicing pathway, which occurred even though the correct 3′ splice site was still present in the pre‑mRNA, suggested that splice site selection occurs by competition between splice sites for the components of the spliceosome assembling on the pre‑mRNA. If this hypothesis was correct it seemed likely that blocking the aberrant 3′ splice site with an oligonucleotide might redirect the spliceosome back to the natural splice site, thus restoring proper splicing and translation of the β‑globin protein, with obvious potential for clinical application. This ability to repair the RNA and resurrect the missing protein, leading to amelioration of the disease, is the key distinguishing feature of oligonucleotide‑induced modulation of pre‑mRNA splicing. It cannot be achieved by small interfering RNA or classical antisense gapmer oligonucleotides, which lead only to degradation of the targeted RNA25. The discovery of alternative splicing showed that this mechanism of competition between splice sites is not limited to just mutant genes but it also controls the expression of the vast majority of native, properly functioning genes108. Thus, splice‑switching oligonucleotides form a basis for a platform technology with the potential to treat many diseases.
The repair of defective β‑globin pre‑mRNA, which restored the correct splicing of β-globin mRNA, was first demonstrated in splicing extracts from HeLa cells65. Systemically delivered splice‑switching oligonucleotides have been proven to be effective in vivo in animal models of β‑thalassaemia67,68, spinal muscular atrophy61–63 and Duchenne muscular dystrophy26 — three devastating genetic disorders that affect children. More importantly, the application of splice‑switching oligonucleotides as drugs for the treatment of Duchenne muscular dystrophy has already been tested in clinical trials via intramuscular administration29,30, and in recently completed clinical trials via systemic administration31,32.
R E V I E W S
134 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Exonic splicing silencers Sequences present in exons in pre-mRNA that contribute to the modulation of splicing. They can inhibit the inclusion of a given exon into mature mRNA during splicing. Exonic splicing enhancers also exist. Intronic splicing silencers Sequences present in introns in pre-mRNA that contribute to the modulation of splicing. They can inhibit the inclusion of a given intron into mature mRNA during splicing. Intronic splicing enhancers also exist.
achieve this goal. In SMA the SSOs block either exonic splicing silencers or intronic splicing silencers to promote the inclusion of an otherwise skipped exon (FIG. 3b). In β-thalassaemia an aberrant splice site is blocked and splicing is redirected to the correct pathway (FIG. 3c). In rheumatoid arthritis a functional tumour necrosis factor (TNF) receptor is converted by exon skipping into a soluble decoy receptor that inhibits TNF activity. This flexibility in the possible approaches indicates that SSO-induced modulation of splicing has the potential to be clinically beneficial in many diseases.
SMA. SMA is a genetic neuromuscular disease with an incidence of approximately one in 6,000 live births. It is characterized by the loss of lower motor neurons in the spinal cord and results in progressive paralysis and muscle atrophy. There is no treatment for this disorder, which in its severest form leads to early infant death. The disease is caused by mutations in the survival of motor neuron 1 (SMN1) gene, which encodes SMN, a protein that is essential for the preservation of motor neuron integrity55. Humans also carry SMN2, a very closely related variant of the SMN1 gene, but SMN2 produces SMN protein only in low amounts and is insufficient to fully compensate for the loss of SMN1-derived protein. The low activity of the SMN2 gene is caused by a trans-lationally silent mutation that weakens the recognition of exon 7 by the splicing machinery and prevents its inclusion into SMN2 mRNA during splicing56.
A systematic screen of 2′-MOE phosphorothioate SSOs targeted to exon 7 of SMN2 pre-mRNA identified a compound that blocked an exonic splicing silencer located in exon 7, and therefore increased the inclusion of the exon and resulted in the restoration of SMN pro-tein expression in cell culture57 (FIG. 3b). Highly effective 2′-MOE phosphorothioate SSOs targeting intronic splic-ing silencers in the introns flanking exon 7 of SMN2 were also identified58,59. In a mouse model of SMA, 4 weeks of systemic SSO administration increased the inclusion of exon 7 of SMN2 by approximately fivefold in the liver, threefold in the kidney and twofold in the quadriceps of treated mice59. As oligonucleotides do not seem to cross the blood–brain barrier27, it was not surprising that there was no effect in the spinal cord — the key therapeutic target tissue. Nevertheless, the experiments provided an important in vivo proof of principle for the treatment of the disease, and demonstrated that SSOs can be used for both exon skipping and exon retention.
To bypass the blood–brain barrier, 2′-OMe phospho-rothioate SSOs60 or 2′-MOE phosphorothioate SSOs61,62 were directly delivered via intracerebroventricular injec-tions into embryos or newborn pups of mice with SMA. This produced striking results: SSOs were distributed throughout the central nervous system (CNS), including the spinal cord. They also led to improvements in the righting response of the mice and delayed the onset of tail and ear necrosis. In the next step towards clinical trials, intrathecal and intracerebroventricular infusions of 2′-MOE phosphorothioate SSOs delivered the compound to the spinal cord in monkeys. This tissue contained >8 μg per g of the oligonucleotide, which is the level
anticipated to have therapeutic effects based on the results obtained from the mouse models62. If the SSO passes the US Food and Drug Administration (FDA)-mandated safety tests in animals, clinical tolerability and efficacy trials in humans will follow. As there is little evidence that large, charged oligonucleotides can traverse a mature blood–brain barrier, one could assume that the use of SSOs in patients with SMA will require intrathecal delivery. However, an unexpected finding suggests otherwise: a recent study showed that systemic delivery of 2′-MOE phosphorothioate SSOs resulted in a much more striking phenotypic rescue than intracerebroventricular delivery in a mouse model of severe SMA63.
Intracerebroventricular administration of SSOs effi-ciently restored SMN expression in the CNS, including in motor neurons, but only increased mean survival from 10 to 17 days, which was consistent with earlier results in another mouse model62. By contrast, sub-cutaneous SSO administration increased the mean survival to 250 days, with some mice surviving longer than 1 year, even though only limited splicing correc-tion was observed in the CNS. Combined intracerebro-ventricular and systemic administration was slightly better than systemic administration alone. These data suggest that, at least in this mouse model, the defects not only in the motor neurons but also in other tissues contribute to SMA. Low levels of circulating insulin-like growth factor 1 (IGF1) — which is synthesized in the liver — observed in this animal were attributed to reduced mRNA levels of the IGF-binding protein acid labile subunit (IGFALS). Systemic antisense oligonucleo-tide treatment restored IGFALS mRNA and IGF1 pro-tein levels to normal. Given that IGF1 has neurotrophic activity, these results illustrate how pleiotropic peripheral defects in conjugation with SMN defects could contribute to motor neuron disease in SMA.
The extent to which these recent observations in mice with severe SMA are clinically relevant remains to be determined. This in turn will determine whether systemic SSO administration — alone or in combina-tion with intracerebroventricular administration — should be tested in clinical trials. Such an approach will require additional safety and tolerability studies, man-dated by regulatory authorities, in non-human primates before such trials could be conducted. Nevertheless, a treatment for this devastating disease appears to be on the horizon.
β-thalassaemia. β-thalassaemia is caused by mutations in the β-globin gene that decrease the production of β-globin, a subunit of haemoglobin, resulting in insuf-ficient levels of haemoglobin to carry oxygen throughout the body64. This decrease also results in an excess of the α-globin subunit, which contributes to the damage to red blood cells. Blood transfusions are currently the main form of treatment, but in developing countries — where the disease is prevalent — these carry the risk of blood-borne infections. Most common mutations induce aber-rant splicing of β-globin pre-mRNA, which abrogates correct translation of the β-globin protein64. One such mutation in nucleotide 654 of intron 2 (IVS2-654) of
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 135
© 2012 Macmillan Publishers Limited. All rights reserved

Cryptic splice siteA splice site that is not functional under normal conditions but becomes activated if a mutation modifies its sequence to resemble a functional splice site or if an adjacent normal splice site is inactivated by a mutation.
β-globin pre-mRNA creates a new 5′ splice site and con-comitantly activates a pre-existing 3′ cryptic splice site, leading to the retention of a pseudo-exon in the spliced mRNA (FIG. 3c).
In 1993 it was demonstrated that correct splicing can be restored in vitro by SSOs targeted to β-globin pre-mRNA65, but obtaining results in vivo proved to be diffi-cult to achieve. Several chemistries (for example, 2′-OMe phosphorothioate, PNAs and PMOs) were ineffective in the delivery of the SSO to erythroid progenitor cells in a transgenic mouse model of IVS2-654 β-thalassaemia66. The only effective SSO variant was a PPMO. A 3-week intravenous treatment course with the PPMO caused a sixfold increase in the levels of correctly translated human β-globin mRNA compared with the control. The mRNA was properly translated in circulating red blood cells, and their morphology was improved67.
An elegant follow-up to this work was the treatment of IVS2-654 mice with siRNA, which lowered the pro-duction of α-globin subunits, combined with a vector expressing antisense RNA that modulated the splicing of IVS2-654 β-globin pre-mRNA and increased the levels of β-globin. This combination led to a detectable increase in haemoglobin production in treated IVS2-654 mice68. Clearly, replacing blood transfusions with an oligo nucleotide that restores haemoglobin production would provide a substantial improvement for individuals with β-thalassaemia.
β-thalassaemia is one the most common genetic disorders worldwide, particularly in South East Asia69, and in China IVS2-654 is the most common mutation in the gene encoding β-globin68. However, because of the economic status of the affected countries there is appar-ent little commercial interest in advancing these new treatment options. Given the current high cost of oligo-nucleotides and/or gene therapy, it remains to be seen whether China and other Asian countries will accept treatments that — if developed — would currently be very expensive.
Alternative splicingAlthough the above examples highlight the application of SSOs to the repair of defective mRNA for the pos-sible treatment of genetic disorders, these compounds may also be used to manipulate alternative splicing of genes that are not defective or mutated. As most genes use alternative splicing to express multiple isoforms of mRNA and consequently proteins, this approach could have broad applicability. In early studies, manipula-tion of splicing was tested in vitro in cancer cell lines by switching the splicing pattern of B cell lymphoma X (BCL-X; also known as BCL2L1) pre-mRNA from anti-apoptotic BCL-XL to pro-apoptotic BCL-XS splice variants70,71. This principle was tested in vivo in mouse models of inflammatory disease via oligonucleotide-induced skipping of exon 7 in the pre-mRNA of TNF receptor 2 (TNFR2).
Exon 7 in TNFR2 pre-mRNA encodes a trans-membrane domain in the membrane-bound receptor protein. The removal of this domain has a dual effect: it generates a soluble, secreted form of the receptor
(Δ7TNFR2), which acts as a decoy receptor by binding to TNF and removing it from the bloodstream; and it reduces the level of functional, membrane-bound TNFR2. TNFR2 mediates the activity of TNF, a cytokine that is induced in rheumatoid arthritis, psoriasis and other inflammatory diseases. In mice, injections of exon 7-targeted oligonucleotides with an LNA backbone (FIG. 2) (at a dose of 25 mg per kg) induced high levels of Δ7TNFR2, which persisted in the circulation for 30 days. This treatment prevented TNF-induced acute liver inflammation, and delayed the onset and reduced the severity of collagen-induced arthritis72. LNA SSOs could potentially be injected less frequently than the currently marketed drug etanercept (Enbrel; Amgen/Pfizer), and because patients would express their own Δ7TNFR2 the potential for an immune response would be reduced. Another recent approach involved reduc-ing TNF levels directly via an oligonucleotide that inter-calates into the DNA of the TNF gene and inhibits its expression73.
Non-SSO steric-blocking oligonucleotidesMyotonic dystrophy. Several neuromuscular diseases, including Huntington’s disease, spinocerebellar ataxia and myotonic dystrophy, are caused by the expansion of triplet repeats, most frequently CTG and CAG. In healthy individuals the repeats are 5–35 triplets in length but this number may increase in their descend-ents and cause disease when the expansion reaches 80 to >2,500 repeats. Although Huntington’s disease and ataxias are characterized by defective proteins that cause disease, in myotonic dystrophy the CTG repeat is in the 3′ untranslated region of the gene encoding dystrophia myotonica protein kinase (DMPK), and — in principle — the correct DMPK protein could still be produced. However, the repeat generates a defective, expanded CUG repeat-containing mRNA that is not correctly spliced and accumulates in the cell nucleus in distinct foci, preventing the export of DMPK mRNA to the cytoplasm74. More importantly, the repeat in the accumulated expanded CUG repeat-containing mRNA tightly binds and traps a splicing factor, muscleblind-like protein 1, which is then unavailable for the splicing of pre-mRNA from several other genes, including the muscle-specific chloride channel protein 1 (CLCN1). Lack of this channel results in muscle hyperexcitability, which causes myotonia75.
In a mouse model of myotonia, the injection of a 25-mer PMO-based compound complementary to the CUG repeat displaced bound muscleblind-like protein 1 from the repeat and dispersed RNA foci in the fibres of the treated muscle (FIG. 3d). Moreover, this treatment restored the correct splicing of CLCN1 and other affected genes. Restoration of the correct splicing of CLCN1 mRNA res-cued the production of the chloride channel protein and restored its transmembrane chloride ion conductance. As a result, myotonia of the treated muscle was markedly reduced76. Similar results were obtained by intramuscu-lar injections of 21-mer 2′-OMe phosphorothioate oligo-nucleotides77. Interestingly, oligonucleotide treatment may not only halt the course of the disease but also prevent
R E V I E W S
136 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

its onset. Direct injection of CAG-repeat-targeted LNA oligonucleotides into the muscle of the myotonic mouse suppressed further expansion of the repeat. This suggests that early oligonucleotide treatment may stabilize the repeats at subpathogenic lengths78.
Targeting splicing, as described above, shows that the splicing machinery — in spite of or perhaps because of its complexity and plasticity — provides an amenable and remarkably broad platform for manipulation by oligonucleotides. The achievable effects include repair of splicing defects and restoration of protein func-tion in SMA and β-thalassaemia, or repair of mRNA in which deletions affect protein translation in DMD. These approaches led to the generation of partially functional dystrophin protein in treated animal models and patients with DMD. The possibility that a novel, clinically useful protein can be generated by manipula-tion of alternative splicing is exemplified by Δ7TNFR2, which had anti-inflammatory effects in mice. Finally, blocking the binding of a splicing factor to an extended triplet repeat has pleiotropic effects because it restores the correct splicing and expression of several genes, thus ameliorating myotonic dystrophy, a disorder whose aetiology has only become understood in the past few years.
Antibacterials and antivirals. In addition to inducing the production of functional proteins, RNA-blocking oligonucleotides can be used to downregulate the pro-duction of undesirable proteins via several mechanisms. These include: redirection of splicing to prevent protein production; blocking of protein translation (FIG. 1c); and the generation of an EGS, which guides the tRNA-processing ribozyme RNase P to cleave EGS-targeted mRNA (FIG. 1d). Several reports, highlighted below, have shown the effectiveness of these approaches as antibacte-rial and antiviral treatments.
Oligonucleotides are rarely tested as antibacterials, because the bacterial cell wall presents a formidable obstacle to the entry of the oligonucleotide into the cell. Surprisingly, the administration of very short, 11-mer PMOs to Escherichia coli-infected mice downregu-lated the expression of the acyl carrier protein, which is essential for E. coli lipid biosynthesis79. Further work demonstrated that oligomers with the same sequence that were either conjugated to a cell-penetrating peptide (that is, PPMOs) or contained positively charged sub-units within the backbone (positively charged PMOs; also known as PMOplus) (FIG. 2) were even more effective. All mice treated with either chemistry survived, whereas all control mice died 12 hours post-infection. On a molar basis, PPMO was more effective than ampicillin, a small-molecule antibiotic80,81.
PPMOs were also used to inhibit bacterial growth and the expression of specific genes in cultures of E. coli (Gram-negative) and Bacillus subtilis (Gram-positive) bacteria. A unique feature of this work82 is that PPMOs acted as EGSs, which are short RNAs that are designed to bind to targeted mRNA and form a three-dimensional structure that somewhat resembles tRNA (FIG. 1d). This structure is recognized in bacterial and eukaryotic
cells by a tRNA-processing ribozyme, RNase P, which cleaves the EGS-bound mRNA at the first base pair of the duplex formed by the EGS and the targeted mRNA (reviewed in REF. 83). Surprisingly, when mRNA was hybridized to an EGS with a PMO backbone that was chemically very different from that of RNA (FIG. 2), the complex was still recognized and the mRNA was cleaved by RNase P. Indeed, treatment of chloram-phenicol-resistant E. coli with a mixture of two PPMO–EGSs targeted to mRNA encoding chloram-phenicol-resistance protein reduced bacterial survival by 99.9% in the presence of the antibiotic compared with bacteria treated with chloramphenicol only84. This efficacy suggests that the PPMO–EGS approach may be effective against infections caused by antibiotic-resistant strains of bacteria.
A recent paper summarized the effects of PPMOs conjugated to a different basic peptide84. This peptide, which was derived from a protein from human T cells and had no biological activity on its own, was a remark-able facilitator of the transport of a covalently attached PMO into bacteria. The new conjugate is 10- to 100-fold more effective at killing a variety of bacteria — including E. coli, Staphylococcus aureus and Klebsiella pneumoniae — than the previously tested compounds. The new conjugate is also effective at reducing the survival of drug-resistant bacteria. If the PMO conju-gates that have sequences targeted to eukaryotic mRNAs — and are designed to be recognized as an EGS by eukaryotic RNase P — are delivered to eukaryotic cells, the new agent might also be effective against eukaryotic gene targets such as viral RNAs85.
The effective treatment of monkeys infected with Ebola or Marburg viruses using PMO-based com-pounds provides a striking example of the utility of RNA-blocking morpholino oligomers in inhibiting viral mRNA translation86. Ebola and Marburg viruses are deadly haemorrhagic viruses and in an outbreak they can kill the majority of infected individuals. The only countermeasure is quarantine and prevention of the spread of the virus.
The treatment of Ebola virus-infected (~1000-fold greater than the minimum lethal dose) Rhesus macaque monkeys with the PMOplus drug candidate AVI-6002 resulted in 60% survival at 15 days post-infection, and the treatment of Marburg virus-infected (~1000-fold greater than the minimum lethal dose) Cynomolgus macaque monkeys with the PMOplus drug candidate AVI-6003 resulted in 100% survival at 15 days post-infection. This level of survival has not been observed previously; on average, untreated monkeys survive no more than 10–11 days86 (P. Iversen, personal commu-nication). At 15 days post-infection, both viruses were undetectable in the bloodstream of treated animals. Furthermore, surviving monkeys were resistant to infection when re-challenged with the virus more than 60 days after initial treatment, suggesting that the animals were able to mount an effective immune response once the infection progression was slowed down (P. Iversen, personal communication). The first safety clinical trials (ClinicalTrials.gov identifiers: NCT01353027 and
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 137
© 2012 Macmillan Publishers Limited. All rights reserved

NCT01353040) for AVI-6002 and AVI-6003 have been initiated (see the AVI BioPharma website).
AVI-6002 and AVI-6003 each target two independent genes that are essential for viral survival. This same double-target approach was used against several other viruses, including the clinically important respiratory syncytial virus, herpes simplex virus 1 (REFS 87,88), the picornavirus family89 and influenza viruses90–92. Oligomers were targeted to the AUG sites or the highly conserved terminal sequences of appropriate genes. This double-target design is anticipated to minimize the emergence of resistant mutant viruses86.
The increasing prevalence of bacterial and viral out-breaks as well as drug-resistant strains of these organ-isms indicates that there is a need for the development of novel antibacterial and antiviral drugs. Oligonucleotides may be a rapidly deployable — albeit currently expensive — answer to this problem.
OutlookThe only two approved nucleic acid-based drugs — fomivirsen3, which is an antisense oligonucleotide, and pegaptanib (Macugen; Pfizer/Eyetech Pharmaceuticals), which is an RNA aptamer4 that has not been not dis-cussed in this Review — have reached the market and both are delivered locally by intravitreal injection. It could be argued that oligonucleotide-based drugs cannot be
effective systemically because these large polar mol-ecules do not follow Lipinski’s rules93: empirical rules stating that, among other characteristics, effective drugs invariably have molecular weights below 500 Da and are fairly soluble in both polar and nonpolar solvents. Although oligonucleotides clearly do not meet these criteria, in our opinion — which is supported by the results reviewed above — this field is on the verge of suc-cess. Novel chemistries that enhance the cellular uptake of oligonucleotides are especially promising. Likewise, the potential of RNA-based therapeutics to reach hereto-fore undruggable targets and affect untreatable diseases certainly warrants the efforts to turn these compounds into effective drugs.
A comparison with monoclonal antibodies is instruc-tive. These molecules are as far from Lipinski’s rules as are oligonucleotides yet they have now reached a world-wide market of over $40 billion and are used against seri-ous chronic diseases such as cancer, multiple sclerosis and rheumatoid arthritis. Antigen-specific monoclonal antibodies were first produced in 1975 (REF. 94), and although a single monoclonal antibody was approved by the FDA in 1986 the majority reached the market about 10 years later95. We believe that oligonucleotide- and RNA-based therapeutics have the potential to follow a similar or shorter timeline and reach a similar level of success.
1. Davidson, B. L. & McCray, P. B. Jr. Current prospects for RNA interference-based therapies. Nature Rev. Genet. 12, 329–340 (2011).
2. Goodchild, J. Therapeutic oligonucleotides. Methods Mol. Biol. 764, 1–15 (2011).
3. Crooke, S. T. Vitravene — another piece in the mosaic. Antisense Nucleic Acid Drug Dev. 8, vii–viii (1998).This is a paper on the first — and so far the only — antisense oligonucleotide drug approved for use as a treatment for cytomegalovirus-induced retinitis in patients with advanced AIDS.
4. Moshfeghi, A. A. & Puliafito, C. A. Pegaptanib sodium for the treatment of neovascular age-related macular degeneration. Expert Opin. Investig. Drugs 14, 671–682 (2005).
5. Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
6. de Fougerolles, A., Vornlocher, H. P., Maraganore, J. & Lieberman, J. Interfering with disease: a progress report on siRNA-based therapeutics. Nature Rev. Drug Discov. 6, 443–453 (2007).
7. Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).This was the first demonstration that short double-stranded RNAs, now known as siRNAs can induce mRNA degradation via the RNAi pathway in human cells.
8. Song, E. et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Med. 9, 347–351 (2003).
9. Watts, J. K., Deleavey, G. F. & Damha, M. J. Chemically modified siRNA: tools and applications. Drug Discov. Today 13, 842–855 (2008).
10. DeVincenzo, J. et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res. 77, 225–231 (2008).
11. Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nature Biotech. 28,172–176 (2010).
12. Jackson, A. L. & Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nature Rev. Drug Discov. 9, 57–67 (2010).
13. Bonetta, L. RNA-based therapeutics: ready for delivery? Cell 136, 581–584 (2009).
14. Couzin-Frankel, J. Drug research. Roche exits RNAi field, cuts 4800 jobs. Science 330, 1163 (2010).
15. Krieg, A. M. Is RNAi dead? Mol. Ther. 19, 1001–1002 (2011).
16. Stein, C. A. Does antisense exist? Nature Med. 1, 1119–1121 (1995).
17. Zamecnik, P. C. & Stephenson, M. L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl Acad. Sci. USA 75, 280–284 (1978).
18. Lima, W., Wu, H. & Crooke, S. T. in Antisense Drug Technology: Principles, Strategies, and Applications 2nd edn (ed. Crooke, S. T.) 47–74 (CRC Press, Boca Raton, Florida, 2007).
19. Raal, F. J. et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial. Lancet 375, 998–1006 (2010).
20. Chi, K. N. et al. Randomized Phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 28, 4247–4254 (2010).
21. Chia, S. et al. Phase II trial of OGX-011 in combination with docetaxel in metastatic breast cancer. Clin. Cancer Res. 15, 708–713 (2009).
22. Smith, C. C., Aurelian, L., Reddy, M. P., Miller, P. S. & Ts’o, P. O. Antiviral effect of an oligo(nucleoside methylphosphonate) complementary to the splice junction of herpes simplex virus type 1 immediate early pre-mRNAs 4 and 5. Proc. Natl Acad. Sci. USA 83, 2787–2791 (1986).
23. Stirchak, E. P., Summerton, J. E. & Weller, D. D. Uncharged stereoregular nucleic acid analogs: 2. Morpholino nucleoside oligomers with carbamate internucleoside linkages. Nucleic Acids Res. 17, 6129–6141 (1989).
24. Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nature Rev. Genet. 12, 87–98 (2011).
25. Sazani, P., Graziewicz, M. & Kole, R. in Antisense Drug Technology: Principles, Strategies, and Applications 2nd edn (ed. Crooke, S. T.) 89–114 (CRC Press, Boca Raton, 2007).
26. Lu, Q. L. et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl Acad. Sci. USA 102, 198–203 (2005).
27. Sazani, P. et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nature Biotech. 20, 1228–1233 (2002).
28. Dillman, J. et al. Efficient and persistent splice switching by systemically delivered LNA oligonucleotides in mice. Mol. Ther. 14, 471–475 (2006).
29. van Deutekom, J. C. et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 (2007).
30. Kinali, M. et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928 (2009).
31. Goemans, N. M. et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 364, 1513–1522 (2011).
32. Cirak, S. et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, Phase 2, dose-escalation study. Lancet 378, 595–605 (2011).These two studies showed encouraging results in clinical trials applying two SSO variants, 2′-OMe phosphorothioate (reference 31) and PMO (reference 32), for the systemic treatment of Duchenne muscular dystrophy.
33. Van Ommen, G. J., van Deutekom, J. & Aaartsma-Rus, A. The therapeutic potential of antisense-mediated exon skipping. Curr. Opin. Mol. Ther. 10, 140–149 (2008).
34. Muntoni, F., Torelli, S. & Ferlini, A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2, 731–740 (2003).
R E V I E W S
138 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

35. Matsuo, M. et al. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy kobe. J. Clin. Invest. 87, 2127–2131 (1991).
36. Aartsma-Rus, A. et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 30, 293–299 (2009).
37. Wilton, S. D. et al. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 9, 330–338 (1999).
38. Bulfield, G., Siller, W., Wight, P. & Moore, K. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA 81, 1189–1192 (1984).
39. Lu, Q. L. et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nature Med. 9, 1009–1014 (2003).
40. Takeshima, Y. et al. Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain Dev. 23, 788–790 (2001).
41. van Deutekom, J. C. et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet.10, 1547–1554 (2001).
42. Heemskerk, H. A. et al. In vivo comparison of 2′-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med. 11, 257–266 (2009).
43. Alter, J. et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nature Med. 12, 175–177 (2006).
44. Yokota, T. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 65, 667–676 (2009).
45. Muntoni, F. & Wood, M. J. Targeting RNA to treat neuromuscular disease. Nature Rev. Drug Discov. 10, 621–637 (2011).
46. Sazani, P., Weller, D. L. & Shrewsbury, S. B. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int. J. Toxicol. 29, 143–156 (2010).
47. Wu, B. et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 17, 132–140 (2010).
48. Heemskerk, H. et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 18, 1210–1217 (2010).
49. Jearawiriyapaisarn, N. et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 16, 1624–1629 (2008).
50. Jearawiriyapaisarn, N., Moulton, H. M., Sazani, P., Kole, R. & Willis, M. S. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc. Res. 85, 444–453 (2010).
51. Wu, B. et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl Acad. Sci. USA 105, 14814–14819 (2008).
52. Goyenvalle, A. et al. Prevention of dystrophic pathology in severely affected dystrophin/ utrophin-deficient mice by morpholino-oligomer- mediated exon-skipping. Mol. Ther. 18, 198–205 (2010).
53. Yin, H. et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 19, 1295–1303 (2011).
54. Youngblood, D. S., Hatlevig, S. A., Hassinger, J. N., Iversen, P. L. & Moulton, H. M. Stability of cell-penetrating peptide–morpholino oligomer conjugates in human serum and in cells. Bioconjug. Chem. 18, 50–60 (2007).
55. Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80,155–165 (1995).
56. Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA 96, 6307–6311 (1999).
57. Hua, Y., Vickers, T. A., Baker, B. F., Bennett, C. F. & Krainer, A. R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 5, e73 (2007).
58. Singh, N. K., Singh, N. N., Androphy, E. J. & Singh, R. N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 26, 1333–1346 (2006).
59. Hua, Y., Vickers, T. A., Okunola, H. L., Bennett, C. F. & Krainer, A. R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 82, 834–848 (2008).
60. Williams, J. H. et al. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 29, 7633–7638 (2009).
61. Hua, Y. et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644 (2010).
62. Passini, M. A. et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 3, 72ra18 (2011).
63. Hua, Y. et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126 (2011).This paper reported the unexpected discovery that in a mouse model of spinal muscular atrophy the phenotype can be reversed not only by intrathecal but also by systemic peripheral delivery of an SSO that promotes the correct expression of the SMN2 gene.
64. Gibbons, R., Higgs, D. R., Olivieri, N. F. & Wood, W. G. in The Thalassaemia Syndromes 4th edn Ch. 7 (eds Weatherall, D. J. & Clegg, J. B.) 287–356 (Blackwell Science, Oxford, 2001).
65. Dominski, Z. & Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl Acad. Sci. USA 90, 8673–8677 (1993).This was the first report of oligonucleotide- mediated RNA repair of thalassaemic pre-mRNA by SSOs.
66. Lewis, J. et al. A common human β globin splicing mutation modeled in mice. Blood 91, 2152–2156 (1998).
67. Svasti, S. et al. RNA repair restores hemoglobin expression in IVS2-654 thalassemic mice. Proc. Natl Acad. Sci. USA 106, 1205–1210 (2009).
68. Xie, S. Y. et al. Correction of β654-thalassaemia mice using direct intravenous injection of siRNA and antisense RNA vectors. Int. J. Hematol. 93, 301–310 (2011).
69. Cao, A. & Galanello, R. β-thalassemia. Genet. Med. 12, 61–76 (2010).
70. Taylor, J. K., Zhang, Q. Q., Wyatt, J. R. & Dean, N. M. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nature Biotech. 17, 1097–1100 (1999).
71. Mercatante, D. R., Bortner, C. D., Cidlowski, J. A. & Kole, R. Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. analysis of apoptosis and cell death. J. Biol. Chem. 276, 16411–16417 (2001).
72. Graziewicz, M. A. et al. An endogenous TNF-α antagonist induced by splice-switching oligonucleotides reduces inflammation in hepatitis and arthritis mouse models. Mol. Ther. 16, 1316–1322 (2008).
73. Paquet, J. et al. Alternative for anti-TNF antibodies for arthritis treatment. Mol. Ther. 19,1887–1895 (2011).
74. Miller, J. W. et al. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448 (2000).
75. Lee, J. E. & Cooper, T. A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 37, 1281–1286 (2009).
76. Wheeler, T. M. et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339 (2009).This paper describes the in vivo application of SSO-displacing splicing factors bound to an expanded triplet repeat as a potential treatment for myotonic dystrophy.
77. Mulders, S. A. et al. Triplet-repeat oligonucleotide mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl Acad. Sci. USA 106, 13915–13920 (2009).
78. Nakamori, M., Gourdon, G. & Thornton, C. A. Stabilization of expanded (CTG)•(CAG) repeats by antisense oligonucleotides. Mol. Ther. 19, 2222–2227 (2011).
79. Geller, B. L., Deere, J., Tilley, L. & Iversen, P. L. Antisense phosphorodiamidate morpholino oligomer inhibits viability of Escherichia coli in pure culture and in mouse peritonitis. J. Antimicrob. Chemother. 55, 983–988 (2005).
80. Mellbye, B. L., Puckett, S. E., Tilley, L. D., Iversen, P. L. & Geller, B. L. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob. Agents Chemother. 53, 525–530 (2009).
81. Mellbye, B. L. et al. Cationic phosphorodiamidate morpholino oligomers efficiently prevent growth of Escherichia coli in vitro and in vivo. J. Antimicrob. Chemother. 65, 98–106 (2010).
82. Shen, N. et al. Inactivation of expression of several genes in a variety of bacterial species by EGS technology. Proc. Natl Acad. Sci. USA 106, 8163–8168 (2009).
83. Lundblad, E. W. & Altman, S. Inhibition of gene expression by RNase P. Nature Biotech. 27, 212–221 (2010).
84. Wesolowski, D. et al. Basic peptide–morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc. Natl Acad. Sci. USA 108, 16582–16587 (2011).This paper describes the application of EGS technology using PPMOs as a potential treatment for drug-resistant bacterial infections.
85. Jiang, X. et al. Engineered external guide sequences effectively block viral gene expression and replication in cultured cells. J. Biol. Chem. 286, 322–330 (2011).
86. Warren, T. K. et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nature Med. 16, 991–994 (2010).This study showed that PMO-based translation-suppressing oligomers protect monkeys from lethal haemorrhagic infection by Ebola and Marburg viruses.
87. Lai, S. H. et al. Inhibition of respiratory syncytial virus infections with morpholino oligomers in cell cultures and in mice. Mol. Ther. 16, 1120–1128 (2008).
88. Eide, K. et al. Reduction of herpes simplex virus type-2 replication in cell cultures and in rodent models with peptide-conjugated morpholino oligomers. Antivir. Ther. 15, 1141–1149 (2010).
89. Stone, J. K. et al. A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob. Agents Chemother. 52, 1970–1981 (2008).
90. Gabriel, G., Nordmann, A., Stein, D. A., Iversen, P. L. & Klenk, H. D. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J. Gen. Virol. 89, 939–948 (2008).
91. Lupfer, C. et al. Inhibition of influenza A H3N8 virus infections in mice by morpholino oligomers. Arch. Virol. 153, 929–937 (2008).
92. Paessler, S. et al. Inhibition of alphavirus infection in cell culture and in mice with antisense morpholino oligomers. Virology 376, 357–370 (2008).
93. Lipinski, C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44, 235–249 (2000).This paper describes desirable properties for an effective drug, and leads the reader to the conclusion that oligonucleotides lack such criteria.
94. Köhler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
95. Waldmann, T. A. Immunotherapy: past, present and future. Nature Med. 9, 269–277 (2003).
96. Stephenson, M. L. & Zamecnik, P. C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl Acad. Sci. USA 75, 285–288 (1978).This paper describes the discovery of an oligonucleotide that acts as an antisense compound that is capable of preventing viral replication.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 139
© 2012 Macmillan Publishers Limited. All rights reserved

97. Matsukura, M. et al. Phosphorothioate analogs of oligodeoxynucleotides: inhibitors of replication and cytopathic effects of human immunodeficiency virus. Proc. Natl Acad. Sci. USA 84, 7706–7710 (1987).
98. Agrawal, S., Mayrand, S. H., Zamecnik, P. C. & Pederson, T. Site-specific excision from RNA by RNase H and mixed-phosphate-backbone oligodeoxynucleotides. Proc. Natl Acad. Sci. USA 87,1401–1405 (1990).
99. Geary, R. S., Yu, R. Z. & Levin, A. A. in Antisense Drug Technologies: Principles, Strategies, and Applications 2nd edn (ed. Crooke, S. T.) 183–217 (CRC Press, Boca Raton, Florida, 2007).
100. Krieg, A. M. Therapeutic potential of Toll-like receptor 9 activation. Nature Rev. Drug Discov. 5, 471–484 (2006).
101. Agarwala, S. S. et al. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur. J. Cancer 45, 1807–1814 (2009).
102. Chanan-Khan, A. A. et al. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk. Lymphoma 50, 559–565 (2009).
103. Morrison, T. Genta plunges on failed Phase III survival analysis for Genasense. BioWorld website [online], http://www.bioworld.com/content/genta-plunges-failed-phase-iii-survival-analysis-genasense?c2Vhcm NoX3dvcmQ9YToxOntpOjA7czoxMDoib2JsaW1lcn NlbiI7fQ== (2011).
104. Berget, S. M., Moore, C. & Sharp, P. A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl Acad. Sci. USA 74, 3171–3175 (1977).
105. Chow, L. T., Gelinas, R. E., Broker, T. R. & Roberts, R. J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 (1977).
106. Spritz, R. A. et al. Base substitution in an intervening sequence of a beta+-thalassemic human globin gene. Proc. Natl Acad. Sci. USA 78, 2455–2459 (1981).
107. Busslinger, M., Moschonas, N. & Flavell, R. A. β+ thalassemia: aberrant splicing results from a single point mutation in an intron. Cell 27, 289–298 (1981).
108. Tazi, J., Bakkour, N. & Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 1792, 14–26 (2009).
AcknowledgementsR.K. would like to thank his past and present colleagues at AVI BioPharma for helpful comments on this article. A.R.K. thanks his collaborators at Cold Spring Harbor Laboratory and Isis Pharmaceuticals for helpful discussions.
Competing interests statementThe authors declare competing financial interests: see Web version for details.
FURTHER INFORMATIONAlnylam Pharmaceuticals website — 4 June 2011 press release: http://phx.corporate-ir.net/phoenix.zhtml?c= 148005&p=irol-newsArticle&ID=1570823&highlight=AVI BioPharma website — 25 March 2010 press release: http://investorrelations.avibio.com/phoenix.zhtml?c=64231&p=irol-newsArticle&ID=1406001&highlight=AVI BioPharma website — Hemorrhagic Viruses: http://www.avibio.com/our-programs/infectious-diseases/hemorrhagic-virusesClinicalTrials.gov website: http://www.clinicaltrials.govForbes website — ‘Genta’s Tangled Path’ (29 April 2002): http://www.forbes.com/2002/04/29/0429genta.htmlGenomeWeb website — ‘Pfizer to Shut Down Oligo Therapeutics Unit as Part of Restructuring’ (3 February 2011): http://www.genomeweb.com/rnai/pfizer-shut-down-oligo-therapeutics-unit-part-restructuringOncoGenex website — 30 September 2010 press release: http://ir.oncogenex.com/releasedetail.cfm?releaseid=512563Proactive investors website — ‘Will RNAi Therapeutics Ever Succeed? Roche, Pfizer, Abbott, Merck, Novartis trim commitments’ (15 October 2011): http://www.proactiveinvestors.com/companies/news/19679/will-rnai-therapeutics-ever-succeed-roche-pfizer-abbott-merck-novartis-trim-commitments-19679.html
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
R E V I E W S
140 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Historically, philosophers have subdivided the study of the human mind and behaviour into two broad categories: the cognitive (how we know the world) and the affective (how we feel about it). This division is, how-ever, arbitrary as cognition — a highly complex construct (FIG. 1) — and emotion interact; cognitive status can colour the processing of emotions, and changes in mood affect cognitive function1,2.
It is therefore surprising that changes in emotion are universally recognized as being inherent to psychiatric disorders and their classification, whereas cognitive impairment — which has an equally disabling effect on patients — has been comparatively neglected. Despite this close interrelationship between cognition and mood, the cognitive deficits of psychiatric disorders are not just a secondary consequence of perturbed affect, and their underlying neurobiological substrates differ. Although certain symptoms of psychiatric disorders — such as depression, delusions and anxiety — are allevi-ated by current drugs, cognitive deficits are not usually improved, and may even be worsened3,4. Cognitive dysfunction is, therefore, a poorly controlled and highly
relevant dimension of psychiatric disorders that cuts across traditional diagnostic boundaries, and improved treatment should be a major goal in efforts to enhance quality of life for patients.
Cognitive dysfunction in psychiatric disordersChallenges of defining and characterizing cognitive deficits. Alzheimer’s disease is characterized by poor learning and memory, Parkinson’s disease by motor impairment, depression by melancholy, and schizophre-nia by delusions; however, these and related diagnoses are also accompanied by a range of symptoms involving alterations in mood, motor behaviour, appetite, sleep, diurnal rhythms and, most pertinently, cognitive func-tion. For example, psychosis is common in Alzheimer’s disease, depression can be just as debilitating as motor deficits in Parkinson’s disease, and perturbed cognition is a characteristic of both psychiatric and neurological disorders (TABLE 1).
Defining the precise nature of changes in cognition is challenging. Specificity relative to generalized changes in overall intelligence remains under discussion, in
1Institut de Recherche Servier, 78290 Croissy/Seine, France.Correspondence to M.J.M. e-mail: [email protected]:10.1038/nrd3628
CognitionA suite of interrelated conscious (and unconscious) mental activities, including: pre-attentional sensory gating; attention; learning and memory; problem solving, planning, reasoning and judgment; understanding, knowing and representing; creativity, intuition and insight; ‘spontaneous’ thought; introspection; as well as mental time travel, self-awareness and meta-cognition (thinking and knowledge about cognition).
Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapyMark J. Millan1, Yves Agid2, Martin Brüne3, Edward T. Bullmore4, Cameron S. Carter5, Nicola S. Clayton6, Richard Connor7, Sabrina Davis8, Bill Deakin9, Robert J. DeRubeis10, Bruno Dubois11, Mark A. Geyer12, Guy M. Goodwin13, Philip Gorwood14, Thérèse M. Jay14, Marian Joëls15, Isabelle M. Mansuy16, Andreas Meyer-Lindenberg17, Declan Murphy18, Edmund Rolls19, Bernd Saletu20, Michael Spedding21, John Sweeney22, Miles Whittington23 and Larry J. Young24
Abstract | Studies of psychiatric disorders have traditionally focused on emotional symptoms such as depression, anxiety and hallucinations. However, poorly controlled cognitive deficits are equally prominent and severely compromise quality of life, including social and professional integration. Consequently, intensive efforts are being made to characterize the cellular and cerebral circuits underpinning cognitive function, define the nature and causes of cognitive impairment in psychiatric disorders and identify more effective treatments. Successful development will depend on rigorous validation in animal models as well as in patients, including measures of real-world cognitive functioning. This article critically discusses these issues, highlighting the challenges and opportunities for improving cognition in individuals suffering from psychiatric disorders.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 141
© 2012 Macmillan Publishers Limited. All rights reserved

LearningThe active, experience- and/or training-driven acquisition of information or behaviour. The term ‘conditioning’ is usually used in an experimental context of associative learning. Learning necessitates complementary and distinct processes of encoding and acquisition that can be perturbed and modulated independently.
MemoryPartly separate mechanisms permitting consolidation, retention and retrieval of information from various sensory domains. Short-term memory relates to immediately available information maintained for ~30 seconds. Information retained for longer periods must be consolidated into mechanistically different long-term memory; in principle, this relates to the unlimited (in quantity and in time) capacity to store information.
particular for schizophrenia and autism spectrum dis-orders (ASDs), in which development is abnormal5–8. Furthermore, the precise interrelationship between alterations in cognition and changes in mood, reward, motor performance and effort can be difficult to estab-lish9,10. Finally, apart from treatment, various other factors modify cognitive performance and its measurement in a patient- and disorder-dependent fashion, including: education and age; hormonal status; disease progression; co-morbidity (psychiatric and somatic); whether cog-nitive function is determined in crisis or in remission; motivation; the neuropsychological test used and prac-tice effects; and the means of quantification (self-rating, semi-quantitative scales or informant assessment)6,7,11. Similarities and differences between various disorders are clearly complex — and still being delineated — but several general patterns can be discerned.
Contrasting patterns of cognitive deficits among distinct psychiatric disorders. Cognitive dysfunction does not just signify poor memory — the range of cognitive impairment is broader and more complex (TABLE 1). There are conditions in which a failure to forget or ‘inhibit’ is a characteristic symptom: for example, intrusive thoughts
in obsessive compulsive disorder (OCD)12 and recurrent, unwanted recall (flashbacks) in post-traumatic stress dis-order (PTSD)13,14. The latter state represents a form of ‘hyper-memory’ resulting from defective processes of fear extinction — an active process for suppressing nega-tive emotional memories — rather than just the decay of the mechanisms involved in storage and recall14,15 (FIG. 2). Phobias and social anxiety disorder are likewise typi-fied by blunted fear extinction16,17. Comparatively little cognitive disturbance has been documented for gen-eralized anxiety disorder, despite some subtle changes and a negative cognitive bias to threatening stimuli16,17 (TABLE 1). Cognitive dysfunction in panic disorders is mainly confined to excessive attention and hyperreactivity to threatening — but not emotionally neutral — stimuli. Interestingly, processing speed may actually be acceler-ated in panic disorders16,18. Schizophrenia is charac-terized by a broad pattern of cognitive deficits, from attention and working memory to social cognition and language7,19–22 (BOX 1). Impairments in bipolar disorder, which shares certain genetic risk factors with schizo-phrenia (Supplementary information S1 (figure)), are similar but generally less severe19,23,24 (TABLE 1).
Cognitive impairment is not traditionally associated with depression but it is common, broad-based and often debilitating4,19,25,26. Poor performance in certain tasks reflects reduced reward, low motivation and/or an incapacity for sustained effort — possibly owing to disruption of limbic dopaminergic signalling4,10. This does not, however, provide a satisfactory explanation for overall cognitive impairment. For example, the bias of patients suffering from depression towards affectively negative — and even ambiguous — stimuli (such as facial expressions) involves diminished top-down fronto-cortical cognitive control of emotional processing2,27.
Deficits in attention deficit hyperactivity disorder (ADHD) are not restricted to attention; they affect several other cognitive domains, including an inter-related impairment in working memory and process-ing speed28,29. Among the deficits characterizing OCD, impairment of procedural learning is of particular note12,30. Finally, although disrupted social cognition is a cardi-nal symptom of ASD, several other domains are also affected9,31,32 (TABLE 1).
Changes in specific cognitive domains seen across distinct diagnoses. Determining which cognitive domains are affected in diagnostically discrete disorders is complicated by co-morbidity. Nonetheless, certain cognitive domains can be perturbed in several distinct disorders (TABLE 1).
Most conspicuously, attention is affected in all disor-ders, varying from a cardinal loss of focused attention in ADHD28 to hypervigilance to threatening stimuli in PTSD, panic disorder and even OCD12,13,16–18 (TABLE 1). In ASD, attention to people and their emotions — as well as joint attention with others — is blunted; furthermore, attention towards objects and details is enhanced, while disregarding global aspects (central coherence)8,31.
Perturbed executive function is an additional example of transnosological deficits; however, reflecting their con-trasting integration (FIG. 2), subdimensions are affected
Author addresses2ICM, Pitié-Salpétrière University Hospital, 47 boulevard de l’Hôpital, 75013 Paris, France. 3 Research Department of Cognitive Neuropsychiatry and Psychiatric Preventive Medicine, LWL University Hospital, Ruhr-University Bochum, Alexandrinenstr. 1, 44791 Bochum, Germany.
4 University of Cambridge and GlaxoSmithKline, Cambridge Biomedical Campus, Cambridge CB2 0SZ, UK.
5University of California, Davis, Sacramento, California 95817, USA. 6 Department of Experimental Psychology, University of Cambridge, Cambridge CB2 3EB, UK.
7 Department of Biology, University of Massachusetts Dartmouth, 02747 North Dartmouth, USA.
8 Centre National de la Recherche Scientifique, University of Paris-Sud, 91400 Orsay, France.
9 Neuroscience and Psychiatry Unit, University of Manchester, Manchester M13 9PT, UK. 10University of Pennsylvania, 19104 Philadelphia, USA.11 Institut du Cerveau et de la Moelle Epinière (ICM), Université Pierre et Marie Curie,
Paris 6, UMR-S975 Paris, France. 12University of California San Diego, La Jolla, California 92093-0804, USA.13University of Oxford, Warneford Hospital, Oxford OX3 7JX, UK.14I NSERM; Université Paris, Descartes, Centre de Psychiatrie et Neurosciences U894,
75014 Paris, France.15 Department of Neuroscience and Pharmacology, Rudolf Magnus Institute, University
Medical Center Utrecht, 3584 CG Utrecht, The Netherlands.16Brain Research Institute, University of Zürich and ETHZ, 8057 Zürich, Switzerland.17 Central Institute of Mental Health, Heidelberg University, Medical Faculty Mannheim,
D-68159 Mannheim, Germany.18Institute of Psychiatry, King’s College London, Denmark Hill, London SE5 8AF, UK. 19 Oxford Centre for Computational Neuroscience and Department of Computer
Science, University of Warwick, Coventry CV4 7AL, UK. 20 Department of Psychiatry and Psychotherapy, Medical University of Vienna,
Waehringer Gürtel 18-20, A-1090 Vienna, Austria. 21Les Laboratoires Servier, 50 Rue Carnot, 92284 Suresnes Cedex, France.22 University of Texas Southwestern, Dallas 75235, Texas, USA.23Newcastle University, Newcastle NE2 4HH, UK. 24 Yerkes National Primate Research Center, Emory University, 954 Gatewood Rd,
Atlanta, Georgia 30329, USA.
R E V I E W S
142 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
Universal domains:• Attention, working memory, executive function• Procedural learning and memory• Speed of processing• Fear-extinction learning • Semantic memory
Functional and structural disruption in neurons and/or glia of:• Cellular signalling• Gene transcription and mRNA translation• DNA and/or histone epigenetic codes• Firing rate and patterns (LTP and LTD)• Dendritic spines, synaptic plasticity and neurogenesis• Neuromodulator release
Focal and distributed network perturbation:• Interregional dysconnectivity• Local overconnectivity• Collapse of small-world configurations• Disorganization and desynchronization• Disrupted γ-and θ-oscillations
Higher domains:• Episodic memory• Social cognition• Theory of mind• Verbal learning and memory • Language (use and understanding)
Genetic
Multiple spatial scales: molecules to cerebral circuits Multiple time scales: milliseconds to years
Epigenetic Developmental Environmental
Cognitive impairment
SchizophreniaBipolar disorder
Depression
GAD
Panic disorderPTSD
OCD
ADHD
ASD
ExtinctionThe progressive reduction of a response to a stimulus — for example, owing to discontinuation of reinforcement or loss of association between an unconditioned and conditioned stimulus. Extinction does not just refer to forgetting (a loss or weakening of memory) or ‘un-learning’ (a decay of the processes involved in retention and recall); rather, it refers to a special form of learning that involves active processes of suppression. The extinguished response may reappear following a change of context or exposure to stress.
AttentionThe awareness and attendance to a stimulus or set of stimuli. It depends on the perception, selection and filtering of sensory input and information. Sustained attention (vigilance) is the capacity to maintain attention over an extended period. Selective (focused) attention is the ability to preferentially attend to a subset of stimuli, thus avoiding distraction. Divided attention is the capacity to respond to multiple stimuli simultaneously, and may involve executive shifts in focused attention according to the demands of the situation.
Processing speedThe rapidity with which a cognitive operation is undertaken successfully. Although this is usually related to the speed of information processing, it may also apply to the speed of retrieval. Processing speed affects performance in many tasks and is operationally related to reaction time.
Working memoryPermits the transient ‘online’ evaluation, manipulation and synthesis of newly acquired and/or stored information. Working memory operates in short-term memory but the two terms are not synonymous. Working memory is closely interrelated to, and interacts with, attention and executive function.
differently. Children with ADHD have a poor sense of planning28; autistic individuals are inflexible8,31,32; indi-viduals suffering from depression have problems with decision-making and initiating actions16,26; patients with OCD or bipolar disorder display difficulties with response inhibition12,23; and patients with schizophre-nia have generalized deficits in all these aspects6,7,22. Declarative memory is also affected in psychiatric dis-orders. Of its two basic forms, deficits in semantic memory
are mainly restricted to schizophrenia, whereas impair-ment of episodic memory is common to several disorders as well as schizophrenia9,13,22–24,31 (TABLE 1).
A severe disruption in social cognition, including an impaired theory of mind (BOX 1) and empathy, is proto-typical for ASD8,31,32, and deficient social cognition is also seen in bipolar disorder24, major depression25, ADHD29 and OCD30. In schizophrenia, faulty social cognition is a crucial issue: first, it predicts conversion to full psychosis
Figure 1 | A global view of cognition and its disruption in psychiatric disorders. Psychiatric disorders are associated with complex and disease-specific patterns of cognitive impairment (TABLE 1). Certain domains may be considered to be ‘higher’ in terms of their specialized and sophisticated nature. They are all well represented in humans compared with rodents, and some are prominent both in great apes and — reflecting evolutionary convergence — in higher birds, cetaceans and elephants (Supplementary information S3 (box); Supplementary information S5 (box)). Disruption of cognition is provoked — and countered — by various interacting genetic, epigenetic, developmental and environmental factors. Changes are expressed both at the level of neurons and glia (from altered gene transcription to shifts in neuronal firing) and at the level of neural networks (locally and among interlinked cerebral regions). Dysfunction underlying cognitive impairment is hierarchically and spatially diverse, and enacted over a temporal scale running from milliseconds (for example, cellular firing) to hours (for example, protein synthesis) to years (for example, synaptic architecture). Some susceptibility factors, such as germline and epigenetic factors, can be passed on to offspring. Certain causes of cognitive impairment can be rectified or compensated, but network shifts at the molecular to systems level are not necessarily reversible so prevention and early treatment is crucial. ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder: GAD, generalized anxiety disorder; LTD, long-term depression; LTP, long-term potentiation; OCD, obsessive compulsive disorder; PTSD, post-traumatic stress disorder.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 143
© 2012 Macmillan Publishers Limited. All rights reserved
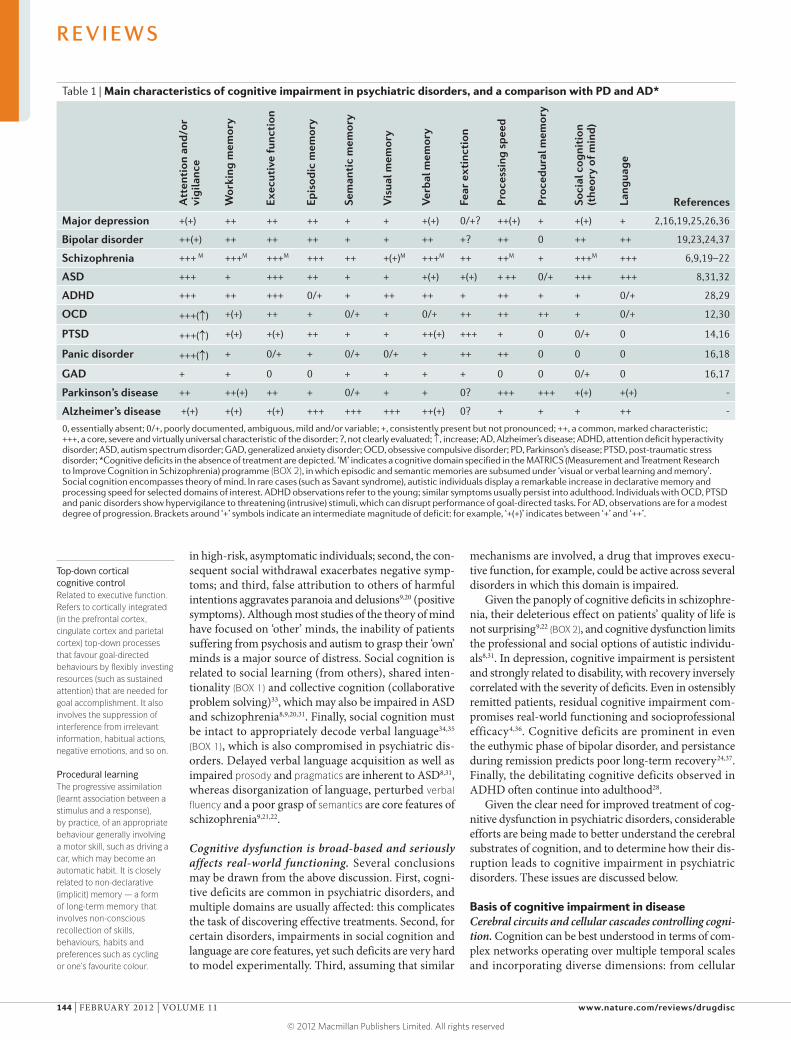
Top-down cortical cognitive controlRelated to executive function. Refers to cortically integrated (in the prefrontal cortex, cingulate cortex and parietal cortex) top-down processes that favour goal-directed behaviours by flexibly investing resources (such as sustained attention) that are needed for goal accomplishment. It also involves the suppression of interference from irrelevant information, habitual actions, negative emotions, and so on.
Procedural learningThe progressive assimilation (learnt association between a stimulus and a response), by practice, of an appropriate behaviour generally involving a motor skill, such as driving a car, which may become an automatic habit. It is closely related to non-declarative (implicit) memory — a form of long-term memory that involves non-conscious recollection of skills, behaviours, habits and preferences such as cycling or one’s favourite colour.
in high-risk, asymptomatic individuals; second, the con-sequent social withdrawal exacerbates negative symp-toms; and third, false attribution to others of harmful intentions aggravates paranoia and delusions9,20 (positive symptoms). Although most studies of the theory of mind have focused on ‘other’ minds, the inability of patients suffering from psychosis and autism to grasp their ‘own’ minds is a major source of distress. Social cognition is related to social learning (from others), shared inten-tionality (BOX 1) and collective cognition (collaborative problem solving)33, which may also be impaired in ASD and schizophrenia8,9,20,31. Finally, social cognition must be intact to appropriately decode verbal language34,35 (BOX 1), which is also compromised in psychiatric dis-orders. Delayed verbal language acquisition as well as impaired prosody and pragmatics are inherent to ASD8,31, whereas disorganization of language, perturbed verbal fluency and a poor grasp of semantics are core features of schizophrenia9,21,22.
Cognitive dysfunction is broad-based and seriously affects real-world functioning. Several conclusions may be drawn from the above discussion. First, cogni-tive deficits are common in psychiatric disorders, and multiple domains are usually affected: this complicates the task of discovering effective treatments. Second, for certain disorders, impairments in social cognition and language are core features, yet such deficits are very hard to model experimentally. Third, assuming that similar
mechanisms are involved, a drug that improves execu-tive function, for example, could be active across several disorders in which this domain is impaired.
Given the panoply of cognitive deficits in schizophre-nia, their deleterious effect on patients’ quality of life is not surprising9,22 (BOX 2), and cognitive dysfunction limits the professional and social options of autistic individu-als8,31. In depression, cognitive impairment is persistent and strongly related to disability, with recovery inversely correlated with the severity of deficits. Even in ostensibly remitted patients, residual cognitive impairment com-promises real-world functioning and socioprofessional efficacy4,36. Cognitive deficits are prominent in even the euthymic phase of bipolar disorder, and persistance during remission predicts poor long-term recovery24,37. Finally, the debilitating cognitive deficits observed in ADHD often continue into adulthood28.
Given the clear need for improved treatment of cog-nitive dysfunction in psychiatric disorders, considerable efforts are being made to better understand the cerebral substrates of cognition, and to determine how their dis-ruption leads to cognitive impairment in psychiatric disorders. These issues are discussed below.
Basis of cognitive impairment in diseaseCerebral circuits and cellular cascades controlling cogni-tion. Cognition can be best understood in terms of com-plex networks operating over multiple temporal scales and incorporating diverse dimensions: from cellular
Table 1 | Main characteristics of cognitive impairment in psychiatric disorders, and a comparison with PD and AD*
Att
enti
on a
nd/o
r vi
gila
nce
Wor
king
mem
ory
Exec
utiv
e fu
ncti
on
Epis
odic
mem
ory
Sem
anti
c m
emor
y
Vis
ual m
emor
y
Ver
bal m
emor
y
Fear
ext
inct
ion
Proc
essi
ng s
peed
Proc
edur
al m
emor
y
Soci
al c
ogni
tion
(t
heor
y of
min
d)
Lang
uage
References
Major depression +(+) ++ ++ ++ + + +(+) 0/+? ++(+) + +(+) + 2,16,19,25,26,36
Bipolar disorder ++(+) ++ ++ ++ + + ++ +? ++ 0 ++ ++ 19,23,24,37
Schizophrenia +++ M +++M +++M +++ ++ +(+)M +++M ++ ++M + +++M +++ 6,9,19–22
ASD +++ + +++ ++ + + +(+) +(+) + ++ 0/+ +++ +++ 8,31,32
ADHD +++ ++ +++ 0/+ + ++ ++ + ++ + + 0/+ 28,29
OCD +++(↑) +(+) ++ + 0/+ + 0/+ ++ ++ ++ + 0/+ 12,30
PTSD +++(↑) +(+) +(+) ++ + + ++(+) +++ + 0 0/+ 0 14,16
Panic disorder +++(↑) + 0/+ + 0/+ 0/+ + ++ ++ 0 0 0 16,18
GAD + + 0 0 + + + + 0 0 0/+ 0 16,17
Parkinson’s disease ++ ++(+) ++ + 0/+ + + 0? +++ +++ +(+) +(+) -
Alzheimer’s disease +(+) +(+) +(+) +++ +++ +++ ++(+) 0? + + + ++ -
0, essentially absent; 0/+, poorly documented, ambiguous, mild and/or variable; +, consistently present but not pronounced; ++, a common, marked characteristic; +++, a core, severe and virtually universal characteristic of the disorder; ?, not clearly evaluated; ↑, increase; AD, Alzheimer’s disease; ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; GAD, generalized anxiety disorder; OCD, obsessive compulsive disorder; PD, Parkinson’s disease; PTSD, post-traumatic stress disorder; *Cognitive deficits in the absence of treatment are depicted. ‘M’ indicates a cognitive domain specified in the MATRICS (Measurement and Treatment Research to Improve Cognition in Schizophrenia) programme (BOX 2), in which episodic and semantic memories are subsumed under ‘visual or verbal learning and memory’. Social cognition encompasses theory of mind. In rare cases (such as Savant syndrome), autistic individuals display a remarkable increase in declarative memory and processing speed for selected domains of interest. ADHD observations refer to the young; similar symptoms usually persist into adulthood. Individuals with OCD, PTSD and panic disorders show hypervigilance to threatening (intrusive) stimuli, which can disrupt performance of goal-directed tasks. For AD, observations are for a modest degree of progression. Brackets around ‘+’ symbols indicate an intermediate magnitude of deficit: for example, ‘+(+)’ indicates between ‘+’ and ‘++’.
R E V I E W S
144 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
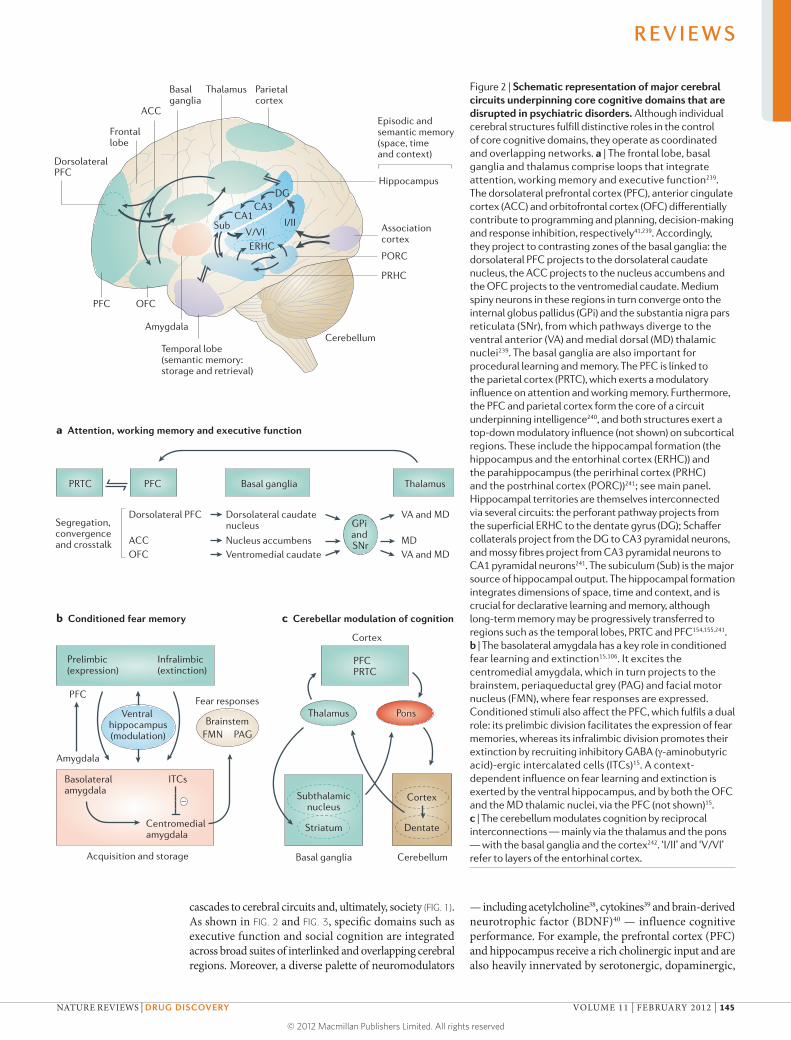
Nature Reviews | Drug Discovery
PRTC PFC Basal ganglia Thalamus
Frontal lobe
PFC OFC
PRHC
PORC
Hippocampus
ACC
DorsolateralPFC
V/VII/II
ERHC
Episodic and semantic memory (space, time and context)
Basal ganglia
Thalamus Parietalcortex
Associationcortex
Amygdala
Temporal lobe(semantic memory:storage and retrieval)
Cerebellum
DGCA3
CA1Sub
Segregation,convergenceand crosstalk
Prelimbic(expression)
Infralimbic(extinction)
Subthalamicnucleus
Striatum
Basal ganglia Cerebellum
Dorsolateral PFC Dorsolateral caudate nucleus
VA and MD
MDVA and MD
ACC Nucleus accumbensOFC Ventromedial caudate
GPi and SNr
a Attention, working memory and executive function
b Conditioned fear memory c Cerebellar modulation of cognition
–
ITCsBasolateral amygdala
Centromedialamygdala
Acquisition and storage
Ventral hippocampus(modulation)
Thalamus PonsBrainstem
Fear responses
FMN PAG
PFC
Amygdala
PFCPRTC
Cortex
Cortex
Dentate
cascades to cerebral circuits and, ultimately, society (FIG. 1). As shown in FIG. 2 and FIG. 3, specific domains such as executive function and social cognition are integrated across broad suites of interlinked and overlapping cerebral regions. Moreover, a diverse palette of neuromodulators
— including acetylcholine38, cytokines39 and brain-derived neurotrophic factor (BDNF)40 — influence cognitive performance. For example, the prefrontal cortex (PFC) and hippocampus receive a rich cholinergic input and are also heavily innervated by serotonergic, dopaminergic,
Figure 2 | Schematic representation of major cerebral circuits underpinning core cognitive domains that are disrupted in psychiatric disorders. Although individual cerebral structures fulfill distinctive roles in the control of core cognitive domains, they operate as coordinated and overlapping networks. a | The frontal lobe, basal ganglia and thalamus comprise loops that integrate attention, working memory and executive function239. The dorsolateral prefrontal cortex (PFC), anterior cingulate cortex (ACC) and orbitofrontal cortex (OFC) differentially contribute to programming and planning, decision-making and response inhibition, respectively41,239. Accordingly, they project to contrasting zones of the basal ganglia: the dorsolateral PFC projects to the dorsolateral caudate nucleus, the ACC projects to the nucleus accumbens and the OFC projects to the ventromedial caudate. Medium spiny neurons in these regions in turn converge onto the internal globus pallidus (GPi) and the substantia nigra pars reticulata (SNr), from which pathways diverge to the ventral anterior (VA) and medial dorsal (MD) thalamic nuclei239. The basal ganglia are also important for procedural learning and memory. The PFC is linked to the parietal cortex (PRTC), which exerts a modulatory influence on attention and working memory. Furthermore, the PFC and parietal cortex form the core of a circuit underpinning intelligence240, and both structures exert a top-down modulatory influence (not shown) on subcortical regions. These include the hippocampal formation (the hippocampus and the entorhinal cortex (ERHC)) and the parahippocampus (the perirhinal cortex (PRHC) and the postrhinal cortex (PORC))241; see main panel. Hippocampal territories are themselves interconnected via several circuits: the perforant pathway projects from the superficial ERHC to the dentate gyrus (DG); Schaffer collaterals project from the DG to CA3 pyramidal neurons, and mossy fibres project from CA3 pyramidal neurons to CA1 pyramidal neurons241. The subiculum (Sub) is the major source of hippocampal output. The hippocampal formation integrates dimensions of space, time and context, and is crucial for declarative learning and memory, although long-term memory may be progressively transferred to regions such as the temporal lobes, PRTC and PFC154,155,241. b | The basolateral amygdala has a key role in conditioned fear learning and extinction15,106. It excites the centromedial amygdala, which in turn projects to the brainstem, periaqueductal grey (PAG) and facial motor nucleus (FMN), where fear responses are expressed. Conditioned stimuli also affect the PFC, which fulfils a dual role: its prelimbic division facilitates the expression of fear memories, whereas its infralimbic division promotes their extinction by recruiting inhibitory GABA (g-aminobutyric acid)-ergic intercalated cells (ITCs)15. A context- dependent influence on fear learning and extinction is exerted by the ventral hippocampus, and by both the OFC and the MD thalamic nuclei, via the PFC (not shown)15. c | The cerebellum modulates cognition by reciprocal interconnections — mainly via the thalamus and the pons — with the basal ganglia and the cortex242. ‘I/II’ and ‘V/VI’ refer to layers of the entorhinal cortex.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 145
© 2012 Macmillan Publishers Limited. All rights reserved

Executive functionA purposeful, goal-directed operation such as planning, decision making, problem solving, reasoning, concept formation, self-monitoring or cognitive flexibility (adaptive alternation between different strategies, responses and behaviours). Executive function reciprocally interacts with attention and working memory. It includes both initiation of appropriate and suppression of inappropriate responses.
Declarative memoryA form of long-term memory that demands conscious learning. It is divided into episodic and semantic memory.
Semantic memoryA form of long-term memory that involves the learning and storing of immutable facts, information, ideas, and so on. In contrast to episodic memory, semantic memory cannot — in principle — be modified by questions and alternative accounts.
noradrenergic and histaminergic neurons. Like the amyg-dala, these key structures contain dense populations of GABA (γ-aminobutyric acid)-ergic interneurons and they communicate with each other — as well as with other territories controlling cognitive function — via glutamatergic projections4,41,42 (FIG. 2).
Pharmacotherapy does not target cerebral circuits per se; rather, it targets G protein-coupled receptors (GPCRs), ion channels, transporters and other pro-teins involved in the actions of neuromodulators. These molecular substrates of cognition43 constitute a vast repertoire of potential drug targets for countering cogni-tive impairment in psychiatric disorders (as discussed below). Mirroring the interlinking of cerebral regions controlling cognition, there is an intricate web of cross-talk among the cellular mediators influencing cognitive processes (Supplementary information S1,S2 (figures)) (FIG. 4), such as the core substrates of neuroplasticity, learning and memory, long-term potentiation (LTP) and long-term depression (LTD)44,45 (BOX 3).
Finally, representing a level of integration that is intermediate between cells and cerebral circuits, neu-rons do not generally act in isolation; rather, they oper-ate as synchronized and rhythmically active assemblies to encode, transmit and modulate information under-pinning cognitive function46,47 (BOX 4).
Disruption of cerebral networks as a cause of cognitive impairment. Networks that modulate cognition display considerable redundancy and pleiotropy at all levels of integration: from intracellular signals, to neurons, to cerebral nuclei4,48,49. The disruption of many elements (known as nodes) can be compensated by others with similar roles; in addition, each element itself has multi-ple functions (Supplementary information S2 (figure)) (FIG. 4). This organization affords considerable resilience to disruption4,48,49. However, the failure of functionally important, highly connected nodes (known as ‘hubs’) has a disruptive effect. For example, a dysfunction in NMDA (N-methyl-d-aspartate) receptors (at the cellu-lar level) and a disruption in frontocortical GABAergic interneurons (at the circuit level) is implicated in the cognitive defects observed in schizophrenia42,50.
Furthermore, multiple ‘hits’ to networks, such as a combination of genetic and developmental or environ-mental factors, are particularly hazardous. For example, when superimposed on a vulnerable genetic background, maternal infection or cannabis use during adolescence increases the risk of schizophrenia and cognitive impair-ment7,11,51,52. Importantly, certain changes in networks (known as phase shifts) may be irreversible, such as the aberrant developmental pruning of neurons in schizo-phrenia7,42,49,53. These network-related concepts can be formally handled by graph theory, which is useful for ana-lysing the perturbation of cognitive circuits in psychiatric disorders4,48,49. For example, information-processing and cognitive performance are enhanced by the small-world features of circuits, which means that key structures are often directly linked to each other, rather than by inter-vening regions. This network attribute is compromised in schizophrenia and ASD4,48,49.
Cognitive deficits observed in schizophrenia have long been ascribed to reduced activation of the dorso-lateral PFC (known as hypofrontality) but many corti-cal and subcortical structures are also affected, with a complex pattern of region-dependent hypo- or hyper-activation9,53–55; increased activity may reflect an attempt to compensate for insufficient performance. Thus, it is arguably more pertinent to consider schizophrenia as a disconnection syndrome55. For example, a disturbance of frontocortical–striatal–thalamic loops (FIG. 2), together with impaired top-down cognitive control from the cortex, contributes to deficits in attention, working memory and executive function54,55. Furthermore, impaired verbal learning and language in schizophrenia can be related to diminished connectivity between the temporal–parietal zone (Wernicke’s area) and frontal lobes (FIG. 3), as well as reduced left hemisphere lateralization of Broca’s area and functionally related regions56.
Altered laterality in language-processing regions is also apparent in ASD57. Altered structure and function of the corpus callosum has been reported in ASD. Although its generality is unclear, a large-scale disconnection among circuits such as frontostriatal, fronto temporal and prefrontal–parietal pathways is a consistent find-ing58,59. Interruption of coupling to the cerebellum has also been reported, together with a disruption of the corticolimbic circuits mediating social and emotional
Box 1 | Social cognition, theory of mind and verbal language
Social cognition refers to processes that are used to acquire and interpret information about others, such as their character, intentions and behaviour. It necessitates: awareness, analysis, choice, sharing and/or avoidance of gaze, recognition of faces, interpretation of facial expressions, as well as scrutiny of head, whole-body and body-part motion34,35,212,213. Social cognition also refers to the understanding (and use) of the rules and concepts governing social interactions by means of gestures, etiquette, touch and proximity (personal space). Social cognition embraces the theory of mind (also known as mental attribution), which is the ability — partly by self-reflection — to infer and internally represent the mental states of others, and hence to attribute and interpret desires, beliefs, intentions and thoughts as determinants and predictors of behaviour20,34,212,214.Cultural context can modify social cognition214, which is indispensable for the full decoding and use of verbal language, especially prosody and pragmatics34,35. Reciprocally, language influences thoughts and feelings related to social cognition215.
Both social cognition and language are disrupted in psychiatric disorders (TABLE 1), and the occurrence of autism spectrum disorder and schizophrenia in humans may be evolutionarily linked to selection for complex social cognition, verbal language, creativity, large brains, an expanded prefrontal cortex and cerebral asymmetry9,20,21,35,216. Sophisticated social cognition is seen in eusocial insects, cetaceans (Supplementary information S3 (box)), some rodents (Supplementary information S4 (box)), great apes, elephants and higher birds102,217–219 (Supplementary information S5 (box)). However, the theory of mind in its fullest expression may be unique to humans, and its unequivocal demonstration in animals is therefore challenging102,214,218. Furthermore, although animals communicate in a sophisticated manner, they lack certain features of human language, such as genuine syntax, full recursion (an infinite palette of meanings generated from a finite set of elements or words) and meta-linguistics (thinking and talking about language)35,103. Hence, it is impossible to fully mimic human language in animals, and to adequately model its disruption in psychiatric disorders. Nonetheless, insights might be gained by studying the communicative role of vocal ultrasonic220, olfactory221 and tactile222 exchanges in rodents and other species, and from both the learning of innate songs and the ‘open-ended’ use of verbal exchanges in birds102,103 (Supplementary information S5 (box)).
R E V I E W S
146 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Episodic memoryThe conscious recollection of experiences linked to times and places in the past — what happened, where and when. It may involve mental time- travel back into a situation (known as autobiographical re-experiencing), mirrored by projection into an imagined future (prospective envisioning). As such, it is related to the theory of mind (‘travel into’ or simulation of other minds). Fully-fledged episodic memory may be a uniquely human trait, but there is evidence for its presence in primates, corvids and even some rodents.
ProsodyThe use (and interpretation) of features such as stress, intonation and rhythm that lend additional meaning and emotion to speech.
PragmaticsThe appropriate social use of spoken language.
Verbal fluencyThe ability to use written and spoken language, to choose the right word at the right time and to make appropriate associations.
processing58,59. Some cortical regions may be more strongly linked, and — at least developmentally — local overconnection (that is, excess neurons and increased dendritic spine density) also exemplifies the brain of autistic individuals59.
Somewhat reminiscent of ASD, poor attention in ADHD is related to a disruption of frontostriatal circuits, and networks interlinking temporal and parietal cor-tices with the cerebellum are also affected60. Although perturbed connectivity of the orbitofrontal cortex and subcortical regions has been consistently related to poor inhibitory control and reduced flexibility in OCD, both increases and decreases in connectivity have been observed depending on the experimental conditions61. Finally, PTSD is triggered by exposure to acute and intense stressors that disrupt PFC–amygdala connectivity, resulting in diminished fear-extinction learning14,15 (FIG. 2). Conversely, the accompanying hypervigilance reflects enhanced coupling of the amygdala to structures modulating attention, such as the anterior cingulate cortex and adrenergic projections62.
Thus, cognitive impairment in psychiatric disorders is characterized by a complex pattern of disconnection and overconnection. An important issue, therefore, is whether the circuits controlling cognition can be recon-stituted once they are disrupted, as certain structural perturbations may be irreversible — as implied by the above-mentioned notion of phase shifts4,63.
Genetic risk factors for cognitive deficits in psychiatric disorders. A full discussion of genetic susceptibility factors is beyond the scope of this article but several
points that are relevant to cognitive dysfunction should be highlighted (Supplementary information S1 (figure)).
First, although psychiatric disorders have a moder-ate to high heritability, genetic risk factors are numerous and only have a small effect; they show low penetrance and epistasis, and they do not necessarily adhere to classical nosological boundaries. For example, schizo-phrenia and bipolar disorder share some susceptibility loci7,11,52,64–66, and the same holds for schizophrenia and ASD (Supplementary information S1 (figure)). Hence, it is difficult to identify genetic risk factors for cognitive dysfunction in psychiatric disorders. Compounding the challenge, for specific psychiatric disorders cognitive impairment is heterogeneous among individuals, with regard to both its causes and characteristics8,9,12,18,23,26,28.
Second, ‘correlated’ does not necessarily imply ‘causal’. If a mutation, deletion or other genetic defect is associated with a psychiatric disorder, this does not necessarily indicate a role in the induction of cognitive impairment. Furthermore, the functional significance of single nucleotide polymorphisms is often uncertain, and some risk loci cover numerous genes7,11,52,64–66.
Third, even if a genetic defect is implicated in the pathological mechanisms that lead to cognitive impair-ment, it is not necessarily an appropriate target for their alleviation, as it may trigger anomalous mechanisms that are no longer under its control. For example, mutations in the gene encoding neuregulin 1 contribute to aber-rant patterns of neuronal migration and synaptogenesis in schizophrenia, but neuregulin 1 has a different func-tional role in the adult brain than in the developing brain, so targeting it is unlikely to reverse such anomalies67.
Fourth, some plasticity-related genes predispose individuals to cognitive deficits under adverse devel-opmental conditions but have the opposite effect in a favourable environment. This complicates analyses of their significance64.
Last, the limited success of even genome-wide studies in finding genes that are major risk factors may also be ascribed to additional layers of epigenetic control that can mask the effects of genetic defects.
Despite these hurdles, with the aid of improved experimental models7,11,68,69 several susceptibility genes for psychiatric disorders have been linked to cell-ular mechanisms that control cognitive processing (Supplementary information S1 (figure)). Furthermore, the future identification of genetic risk factors for cogni-tive deficits will be refined by: pathway analyses based on prior knowledge of protein networks65; multivariate statistics for simultaneous analysis of interacting genes66; and studies of gene associations with heritable, stable and co-segregating cognition-related endophenotypes that are likewise (although less markedly) impaired in healthy relatives11. Examples of such endophenotypes include: verbal learning and memory in bipolar disor-der70; sensorimotor gating and social cognition in schiz-ophrenia71,72; and cerebral circuit disruption in OCD and ASD73,74. Some cognitive endophenotypes may, reflecting similar pathological mechanisms, be common to dis-orders like schizophrenia, bipolar disorders or ASD.
Box 2 | The MATRICS initiative
The recognition that poorly treated cognitive deficits contribute to poor functional outcome in schizophrenia led to the establishment of the ‘MATRICS’ (Measurement and Treatment Research to Improve Cognition in Schizophrenia) initiative, which was sponsored by the National Institute of Mental Health (NIMH) in collaboration with the US Food and Drug Administration, academia and industry. The MATRICS initiative had three aims: first, to build a consensus regarding the nature of cognitive impairment in schizophrenia; second, to improve the evaluation of cognitive deficits; and third, to provide a framework for the formal recognition of treatments that specifically address the cognitive deficits associated with schizophrenia independently of an improvement in psychosis6,69,98,178,179,223.
After identifying the cognitive domains that best characterized schizophrenia (TABLE 1), the MATRICS initiative devised a neuropsychological consensus cognitive battery to support the discovery, clinical assessment and registration of new agents6,178,179. Subsequently, the NIMH funded the selection of potential cognition-enhancing agents and set up a group of academic sites to evaluate their efficacy in proof-of-concept trials. Several compounds tested to date (including a GABA
A
(γ-aminobutyric acid type A) receptor α2 subunit agonist and a dopamine D1 receptor agonist) have not proven to be clearly efficacious (TABLE 2), despite having solid conceptual and preclinical support; this highlights the uncertain predictive utility of cognitive tests in animals98,223. Hence, another programme, titled ‘CNTRICS’ (Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia), was established6,179 to build a consensus on two issues: first, the development of new, more reliable and practical translational paradigms for preclinical and early clinical assessment of drug effects on cognitive processes; and second, the development of imaging biomarkers for parallel use in cognitive trials. Particular efforts are being devoted to a more rigorous evaluation of the impact of therapies on real-world function in patients180,181.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 147
© 2012 Macmillan Publishers Limited. All rights reserved
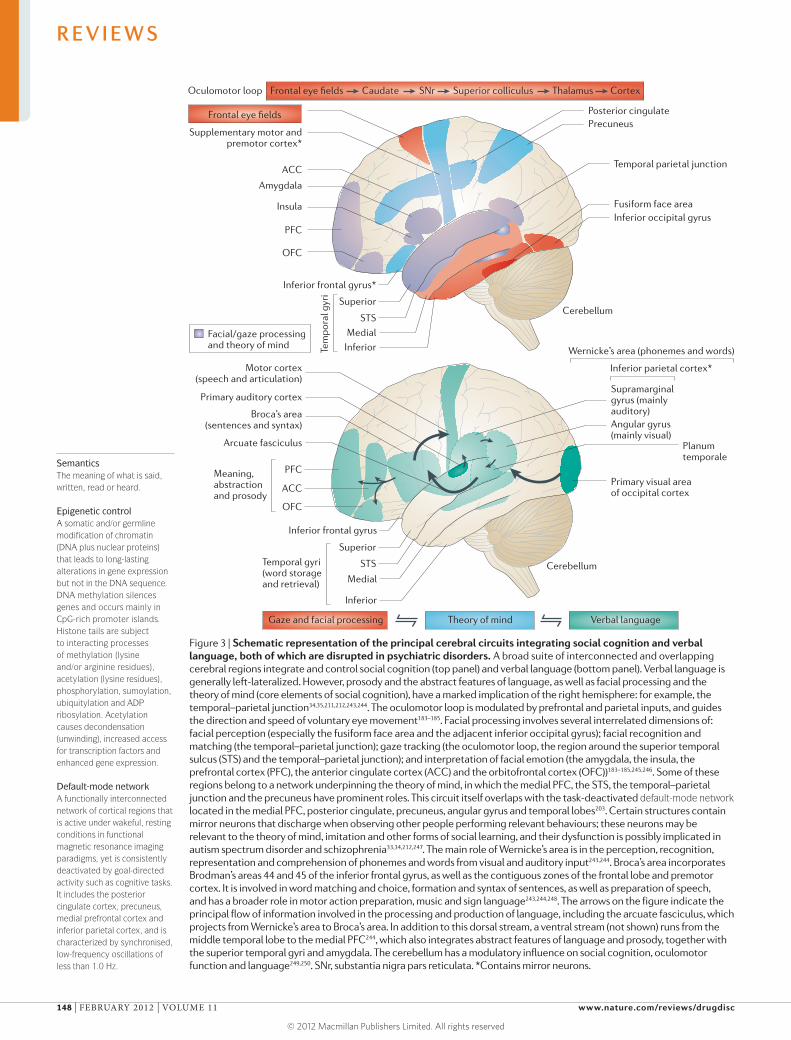
Nature Reviews | Drug Discovery
Amygdala
Cerebellum
Cerebellum
Angular gyrus(mainly visual)
Supramarginalgyrus (mainlyauditory)
Planumtemporale
Primary visual areaof occipital cortexACC
PFC
Broca’s area(sentences and syntax)
Primary auditory cortex
Motor cortex(speech and articulation)
OFC
Inferior frontal gyrus
Inferior parietal cortex*
Meaning, abstractionand prosody
Wernicke’s area (phonemes and words)
ACC
Frontal eye fields
Frontal eye fields Caudate SNr Superior colliculus Thalamus CortexOculomotor loop
PFC
OFC
Medial
Inferior
Temporal gyri(word storageand retrieval)
Medial
Superior
InferiorTem
pora
l gyr
i
Inferior occipital gyrusFusiform face area
Temporal parietal junction
Supplementary motor andpremotor cortex*
Insula
PrecuneusPosterior cingulate
Inferior frontal gyrus*
STS
Superior
STS
Arcuate fasciculus
Gaze and facial processing Theory of mind Verbal language
Facial/gaze processingand theory of mind
SemanticsThe meaning of what is said, written, read or heard.
Epigenetic controlA somatic and/or germline modification of chromatin (DNA plus nuclear proteins) that leads to long-lasting alterations in gene expression but not in the DNA sequence. DNA methylation silences genes and occurs mainly in CpG-rich promoter islands. Histone tails are subject to interacting processes of methylation (lysine and/or arginine residues), acetylation (lysine residues), phosphorylation, sumoylation, ubiquitylation and ADP ribosylation. Acetylation causes decondensation (unwinding), increased access for transcription factors and enhanced gene expression.
Default-mode networkA functionally interconnected network of cortical regions that is active under wakeful, resting conditions in functional magnetic resonance imaging paradigms, yet is consistently deactivated by goal-directed activity such as cognitive tasks. It includes the posterior cingulate cortex, precuneus, medial prefrontal cortex and inferior parietal cortex, and is characterized by synchronised, low-frequency oscillations of less than 1.0 Hz.
Figure 3 | Schematic representation of the principal cerebral circuits integrating social cognition and verbal language, both of which are disrupted in psychiatric disorders. A broad suite of interconnected and overlapping cerebral regions integrate and control social cognition (top panel) and verbal language (bottom panel). Verbal language is generally left-lateralized. However, prosody and the abstract features of language, as well as facial processing and the theory of mind (core elements of social cognition), have a marked implication of the right hemisphere: for example, the temporal–parietal junction34,35,211,212,243,244. The oculomotor loop is modulated by prefrontal and parietal inputs, and guides the direction and speed of voluntary eye movement183–185. Facial processing involves several interrelated dimensions of: facial perception (especially the fusiform face area and the adjacent inferior occipital gyrus); facial recognition and matching (the temporal–parietal junction); gaze tracking (the oculomotor loop, the region around the superior temporal sulcus (STS) and the temporal–parietal junction); and interpretation of facial emotion (the amygdala, the insula, the prefrontal cortex (PFC), the anterior cingulate cortex (ACC) and the orbitofrontal cortex (OFC))183–185,245,246. Some of these regions belong to a network underpinning the theory of mind, in which the medial PFC, the STS, the temporal–parietal junction and the precuneus have prominent roles. This circuit itself overlaps with the task-deactivated default-mode network located in the medial PFC, posterior cingulate, precuneus, angular gyrus and temporal lobes203. Certain structures contain mirror neurons that discharge when observing other people performing relevant behaviours; these neurons may be relevant to the theory of mind, imitation and other forms of social learning, and their dysfunction is possibly implicated in autism spectrum disorder and schizophrenia33,34,212,247. The main role of Wernicke’s area is in the perception, recognition, representation and comprehension of phonemes and words from visual and auditory input243,244. Broca’s area incorporates Brodman’s areas 44 and 45 of the inferior frontal gyrus, as well as the contiguous zones of the frontal lobe and premotor cortex. It is involved in word matching and choice, formation and syntax of sentences, as well as preparation of speech, and has a broader role in motor action preparation, music and sign language243,244,248. The arrows on the figure indicate the principal flow of information involved in the processing and production of language, including the arcuate fasciculus, which projects from Wernicke’s area to Broca’s area. In addition to this dorsal stream, a ventral stream (not shown) runs from the middle temporal lobe to the medial PFC244, which also integrates abstract features of language and prosody, together with the superior temporal gyri and amygdala. The cerebellum has a modulatory influence on social cognition, oculomotor function and language249,250. SNr, substantia nigra pars reticulata. *Contains mirror neurons.
R E V I E W S
148 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

NeurogenesisThe continuous generation of new neurons from neural precursor cells in humans and other mammals. It is seen mainly in two regions. First, the subventricular zone of the lateral ventricle gives rise to neurons that migrate to become granule neurons and periglomerular neurons mainly in the olfactory bulb. Second, neurogenesis in the subgranular zone of the hippocampal dentate gyrus yields neurons, some of which are integrated into local neural networks once they have matured.
Linking risk genes to network disruption. As empha-sized above, disturbed network synchrony and connec-tivity are implicated in the cognitive deficits observed in psychiatric disorders (BOX 4). From a therapeutic perspective, however, drugs target molecules, so it is crucial to link changes in network operation to events at the cellular and genetic level. Neuroimaging and electrophysiological techniques can help to achieve this goal, and they can be exploited both in humans and in animal models.
One example is the so-called Val158Met polymor-phism (rs4680) in the gene encoding the enzyme cat-echol-O-methyltransferase (COMT), which catabolises dopamine; the Val and Met COMT variants are associ-ated with high and low inactivation of dopamine, respec-tively75,76. In healthy individuals, the Val variant was associated with blunted coupling between hippocampal formation and the PFC during a recognition memory task77. This observation may be related to a role of hippo-campal dopamine D1 receptors in gating hippocampal input to the PFC77. Furthermore, D1 receptor-mediated signalling is modulated by the dopamine- and cyclic AMP-regulated neuronal phosphoprotein (DARPP32; also known as PPP1R1B) (Supplementary information S2 (figure)), and a frequent PPP1R1B haplotype is associated with altered connectivity between the PFC and the stria-tum, as well as cognitive dysfunction and an increased risk of schizophrenia78.
As a second example, a polymorphism (rs1344706)that is associated with the risk of developing schizo-phrenia is located in the gene that encodes zinc finger protein 804A, a transcription factor that affects cogni-tive function72,79. During a working memory procedure, healthy carriers of the polymorphism showed gene dosage-dependent alterations in PFC connectivity across hemispheres, and between the dorsolateral PFC and the hippocampus. Functional anomalies in networks under-pinning theory of mind (FIG. 3) have also been observed72. Interestingly, in patients with schizophrenia this poly-morphism also affects cognition and attention, as well as verbal and/or episodic learning and memory80.
A third example is a rare but penetrant microdele-tion in chromosome 22 (22q11.2) that is associated with learning disabilities, cognitive dysfunction and a 30-fold increased risk of schizophrenia81. Mice with an equiva-lent microdeletion have flawed working memory related to reduced hippocampal–prefrontal synchrony. This in turn reflects a failure of PFC neurons to phase-lock with hippocampal θ-oscillations as a result of aberrant firing of GABAergic interneurons — a deficit seen in psychotic states82.
Stress as a risk factor for cognitive deficits and network disruption. Genetic factors do not fully account for the impaired cognition that is observed in psychiatric disorders. Especially in genetically predisposed individ-uals, exposure to excessive stress is a major risk factor for impaired cognitive function throughout life.
Stress is a familiar but imprecise term for the disrup-tion of homeostasis that occurs following perceived or actual exposure to adverse events, and it harnesses a vast
repertoire of neuromodulators that either promote or counter its effect83,84. An essential feature of pathological stress is hypothalamic–pituitary–adrenal (HPA) axis over-drive: this leads to poorly regulated, sustained and marked increases in levels of corticosterone downstream of the hypophyseal release of corticotropin-releasing hormone. Blockade of forebrain populations of corticotropin- releasing hormone receptor 1 counters the cognitive deficits and dendritic abnormalities elicited by acute stress and early-life adversity85. Nonetheless, most interest has focused on corticosterone. Mirroring the optimal cog-nitive performance seen at moderate levels of arousal, a well-regulated, modest and phasic recruitment of the HPA axis generally favours cognitive performance. However, excessive activation of the HPA axis is detrimental. In other words, there is a bell-shaped curve for the influence of corticosterone on cognition83,84,86,87 (see below).
One explanation for this is that genomic mineralo-corticoid receptors, which are recruited at rest, permit a positive influence over less sensitive glucocorticoid receptors. Conversely, when glucocorticoid receptor stimulation is disproportionate and persistent, cogni-tion is compromised83,88. At least in the hippocampus, this occurs in association with a pronounced release of glutamate and the activation of NMDA receptors mediating LTD45,83,89. However, a diverse pattern of inter actions among corticosterone, mineralocorticoid and glucocorticoid receptors, along with glutamatergic signalling, lead to a complex pattern of influence on cog-nition83,84,86,90,91. Thus, the notion of unitary beneficial and deleterious roles of mineralocorticoid versus gluco-corticoid receptors, respectively, is an oversimplification that complicates their therapeutic exploitation.
Analogous to psychosocial stress in humans, the exposure of adult rodents to adverse events perturbs PFC-derived networks, leading to deficits in LTP, work-ing memory and executive function84,92,93. Chronic stress-induced cognitive deficits are associated with structural remodelling, including dendritic spine retraction and neuronal atrophy in the PFC (BOX 3), reduced LTP and neuro genesis in the hippocampus, and an interference with PFC–hippocampus coupling87,88,90,92,93. Mirroring PTSD in patients14,15, acute stress leads to over-intense encoding of negative emotional memories in PFC–amygdala circuits as well as blunted fear-extinction learning94.
Prenatal and childhood stress triggers long-term changes in adolescents and adults, involving impaired cognitive function and an increased risk of depres-sion and other psychiatric disorders83,84. These delayed effects of stress appear to reflect structural and func-tional changes in corticolimbic circuits. For example, in women suffering from major depression, cognitive impairment was related to a history of early child-hood adversity and reduced hippocampal volume84. Correspondingly, early-life chronic stress in rats is asso-ciated with reductions in hippocampal LTP, dendritic spine complexity, neurogenesis and BDNF expression during adulthood83,95. However, early-life stress is not invariably associated with detrimental consequences. For instance, adult rats that had experienced early-life adversity performed poorly in non-stressful learning
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 149
© 2012 Macmillan Publishers Limited. All rights reserved
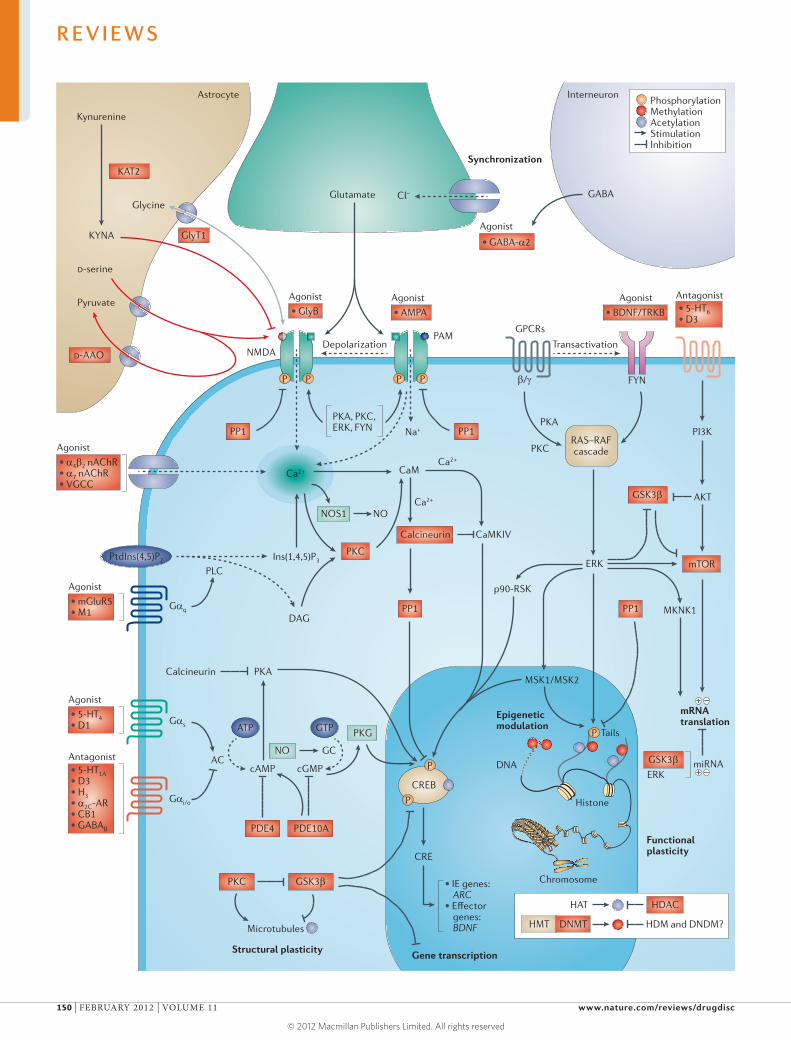
Agonist
• 5-HT4• D1
Agonist
• mGluR5• M1
Agonist
• GlyB
KAT2
GlyT1
D-AAO
Kynurenine
KYNA
Glycine
D-serine
Pyruvate Agonist
PAM
NMDA
• AMPA
Agonist
• GABA-α2
GABA
Agonist
• BDNF/TRKB
• 5-HT1A• D3• H3• α2C-AR• CB1• GABAB
• 5-HT6• D3
Antagonist
GPCRs
Transactivation
FYN
PKA
PKC
β/γ
Antagonist
Gαq
Gαs
Gαi/o
AC
PLC
ATP
cAMP
PKACalcineurin
cGMP
GTP
PtdIns(4,5)P2
CREB
RAS–RAFcascade
CRE
NO
PKG
NOS1 NO
GC
PDE4
Microtubules
Structural plasticity
Functionalplasticity
mRNAtranslation
Epigeneticmodulation
Synchronization
InterneuronAstrocyte
Gene transcription
PDE10A
PKC
GSK3β
GSK3β
PP1
PP1 PP1
PKC
Calcineurin
ERK
GSK3β
mTOR
AKT
PI3K
MKNK1
CaM
Ca2+
Ca2+
ERK
PP1
P
P
P
• IE genes: ARC• Effector genes: BDNF HMT DNMT HDM and DNDM?
HAT HDAC
PhosphorylationMethylationAcetylationStimulationInhibition
Chromosome
miRNA
+ –
+ –DNA
Histone
Tails
MSK1/MSK2
p90-RSK
CaMKIV
Depolarization
Nature Reviews | Drug Discovery
P P P P
Ca2+
Ins(1,4,5)P3
DAG
Na+
PKA, PKC,ERK, FYN
Cl–Glutamate
Agonist
• α4β2 nAChR• α7 nAChR• VGCC
R E V I E W S
150 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

tasks yet performed well under stress, suggesting that the brain had been programmed to operate better under challenging conditions64,96.
Nonetheless, uncontrolled stress and HPA axis over-activity can trigger cognitive dysfunction throughout life84,85,90. The risk of middle-age depression, cogni-tive impairment and metabolic disease followed by dementia is exacerbated by stress, possibly as cortico-sterone and corticotropin-releasing hormone aggravate
glutamatergic neurotoxicity. In elderly patients these hormones worsen the harmful actions of β-amyloid and microtubule-associated protein tau — neurotoxic pro-teins that are implicated in Alzheimer’s disease84,87,90,92,93.
Modelling cognitive deficits. Modelling the genetic, developmental and environmental factors that lead to cognitive impairment in psychiatric disorders is clearly challenging. From the drug discovery perspective, the search for animal models of psychiatric disorders neces-sitates a compromise between fidelity to human pathol-ogy and efficient drug validation8,11,52,68,69. A related key issue is whether cognitive procedures in animal models can efficiently predict the efficacy of drugs in patients (BOX 2). This question is underscored by the concern that numerous pro-cognitive agents and mechanisms have been documented in rodents yet little positive feedback has been acquired in patients.
In fact, if one considers animal models to be for — rather than of — psychiatric disorders, and accepts that they can only reproduce specific aspects (such as individual causes, symptoms, responses, and so on) of a disease (not the psychiatric disorder itself), an array of genetic, developmental and environmental rodent mod-els is available for studying cognitive impairment7,52,68,97. Nonetheless, the familiar adage that ‘the best experimen-tal model is man’ is more applicable to psychiatric dis-orders than to any other field of medicine. Hence, animal models clearly need further refinement, and transgenic strategies only partially mimic human pathology and the attendant cognitive deficits7,52,68. Furthermore, no single procedure is adequate alone, gender and age are insuffi-ciently studied, and inter-individual differences deserve greater attention in view of their prominence in humans and their relevance to personalized medicine6,8,11.
Several other areas also require greater focus, particu-larly where there is a mismatch between the experimental evaluation of drugs and their ultimate use in patients. First, more studies should be undertaken with chronic drug administration to establish the delay to onset of action, long-term efficacy and lack of rebound deterioration in cognition following their discontinuation. Second, the pro-cognitive actions of drugs administered alone in rodents may not be reproduced in patients if they are masked by a deleterious cognitive impact of co-administered agents possessing, for example, antagonist properties at mus-carinic receptors and histamine H1 receptors3,97,98. Thus, mirroring their adjunctive use in humans, the effects of co-administration of pro-cognitive drugs with anti-psychotics and antidepressants should be examined in rodents. Third, many studies examine drug effects on baseline cognition. This is very different to the clinical situation, so a greater focus on drug-induced reversal of cognitive deficits in models of psychiatric disorders is desirable7,11,50,68. Last, the influence of drugs on cognition-related parameters other than behavioural outputs should be studied more intensively, as such mechanisms can be translationally monitored in humans (see below).
Despite these potential advances, many problems will remain. Notably, clinical studies are focusing increasingly on real-world function rather than on
Figure 4 | An overview of molecular substrates targeted by drugs that are designed to enhance cognitive performance in psychiatric disorders. The figure illustrates the complex pattern of crosstalk among the cellular mechanisms influencing cognitive function, of which several (in red boxes and listed in TABLE 2) are potential targets for its improvement in psychiatric disorders. Most mechanisms are depicted for simplicity in a postsynaptic element. Although one specific cell type, such as a prefrontal cortex (PFC)-localized pyramidal projection neuron, might not express all elements, these signalling cascades are widespread. The cell is innervated by a glutamatergic terminal (shown in green) adjacent to an astrocyte (shown in beige) that releases the NMDA (N-methyl-d-aspartate) and glycine B receptor co-agonists d-serine and glycine as well as the antagonist kynurenic acid (KYNA), which is cleaved from kynurenine by kynurenine amino transferase II (KAT2). The GABA (γ-aminobutyric acid)-ergic interneuron synchronizes the activity of glutamatergic neurons and other components of neuronal networks controlling cognition (BOX 4). Notably, there is convergence and divergence in signalling pathways emanating from G protein-coupled receptors (GPCRs), ion channels and tyrosine receptor kinases (TRKs) that are either recruited (agonist properties) or blocked (antagonist properties) by pro-cognitive agents. Drugs may act on downstream intracellular targets: for example, kinases (phosphorylation), the phosphatases protein phosphatase 1 (PP1) and PP2B; also known as calcineurin) (dephosphorylation), and cyclic AMP-specific phosphodiesterase 4D (PDE4D) and PDE10A. They may also act through epigenetic mechanisms of DNA and histone methylation, acetylation and phosphorylation (TABLE 2). Moreover, pharmacotherapy may act upstream via the α2 subunit of GABA
A receptors (GABA
A-α2), or it may control
the availability of glycine (reuptake suppression), d-serine (breakdown inhibition) and kynurenine (synthesis suppression) to NMDA receptors located on pyramidal cells and GABAergic interneurons in the PFC. NMDA receptors mediate rapid changes in cellular excitability, and contribute to long-term potentiation (LTP) and long-term depression (LTD) — core substrates of synaptic plasticity (BOX 3). They are permeable to Ca2+, which affects several mediators controlling cognition, including nitric oxide synthase 1 (NOS1). Changes in cognition are ultimately affected by alterations in: key signals such as extracellular-regulated kinase (ERK) and mammalian target of rapamycin (mTOR); transcription of genes pivotal to cognitive processing, such as cyclic AMP-responsive element binding protein (CREB); epigenetic programming of DNA and histones; microRNA (miRNA)-mediated regulation of mRNA translation; LTP, LTD and dendritic spine plasticity (BOX 3); synaptic architecture; and neurotransmitter release (not shown). CREB recruits CREB-responsive element (CRE) to activate immediate-early (IE) genes such as activity-regulated cytoskeleton-associated protein (ARC) and effector genes like brain-derived neurotrophic factor (BDNF). For a more comprehensive view, see Supplementary information S2 (figure). 5-HT
1A, 5-hydroxytryptamine (serotonin)
receptor 1A; α2C
-AR, α2C
-adrenergic receptor; α4β2 nAChR, α4β2 nicotinic acetylcholine receptor; AC, adenylyl cyclase; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; CaM, calmodulin; CaMKIV, calcium/calmodulin-dependent protein kinase IV; CB1, cannabinoid receptor 1; D1, dopamine D1 receptor; D-AAO, d-amino acid oxidase; DAG, diacylglycerol; DNDM, DNA demethylase; DNMT, DNA methyltransferase; Gα
q, guanine-nucleotide-binding protein Gα
q; GlyB, glycine B;
GSK3β, glycogen synthase kinase 3β; H3, histamine H
3 receptor; HAT, histone
acetyltransferase; HDAC, histone deacetylase; HDM, histone demethylase; HMT, histone methyltransferase; Ins(1,4,5)P
3, inositol-1,4,5-trisphosphate; M1, muscarinic M1 receptor;
mGluR5, metabotropic glutamate receptor 5; MKNK1, MAP kinase interacting serine/threonine kinase 1; MSK1, mitogen- and stress-activated protein kinase 1; NO, nitric oxide; p90-RSK, 90 kDa ribosomal protein S6 kinase; PAM, positive allosteric modulator; PI3K, phosphoinositide 3-kinase; PtdIns(4,5)P
2, phosphatidylinositol-4,5-bisphosphate;
PKA, protein kinase A; PLC, phospholipase C; TRKB, neurotrophic tyrosine kinase receptor type 2; VGCC, voltage-gated calcium channel.
▶
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 151
© 2012 Macmillan Publishers Limited. All rights reserved

neurocognitive test procedures, raising the question of comparability to rodent data (BOX 2). Furthermore, verbal language and human-like social cognition (BOX 1) will presumably remain refractory to study in rodent models.
Non-rodent species may be useful in the search for improved pro-cognitive agents; notable examples include fruitfly models for studying genetics99, Aplysia californica (sea hares)101 for studying synaptic plasticity and Danio rerio (zebrafish)100 for studying developmen-tal processes and behaviour. Moreover, fruitflies and zebrafish are amenable to studies of stress, and to the use of high-throughout protocols99,100. In addition, certain other mammalian species may illuminate the nature and disruption of episodic memory, advanced social cogni-tion and language. These include great apes, elephants, dolphins (Supplementary information S3 (box)), prairie voles (Supplementary information S4 (box)) and higher birds102 (Supplementary information S5 (box)).
Most strikingly, convergent evolution in corvids and parrots has led to alternative neural solutions (including a non-laminar cortex) underpinning genuine episodic memory, sophisticated social cognition and complex vocal communication102. Furthermore, the acquisition of birdsong displays striking parallels to the learning of human language103. Obviously, great apes, elephants and dolphins are unsuitable models for pharmacologi-cal studies, and it remains to be seen whether higher birds will prove to be useful; however, as outlined in
Supplementary information S4 (box), prairie voles are instructive for characterizing the roles of potential drug targets in the control of social cognition.
Strategies to counter cognitive impairmentDirect and indirect modulation of cognitive performance by pharmacotherapy. Increasing awareness of the seri-ousness of cognitive dysfunction in psychiatric disorders, and recent insights into its potential causes, have trig-gered substantial efforts to discover drugs for restor-ing cognitive function104. Studies have focused both on specific domains (such as attention105 and extinction learning106) and on disorders (such as schizophrenia98 and ASD63). The array of concepts under investigation, listed in TABLE 2, is based both on behavioural readouts and on surrogate indexes of cognitive performance, such as cellular signals, LTP and LTD, network synchrony, transmitter release and dendrite spine formation.
As TABLE 2 is limited to targets that directly affect cog-nition, the significance of drug-induced changes in mech-anisms that indirectly modulate cognitive function should be briefly discussed. Agents that enhance sleep quality and architecture, especially slow-wave sleep, should improve hippocampal–cortical mechanisms of consolidation and other components of cognitive processing107. Drugs that normalize disrupted circadian rhythms may favour-ably affect cognitive performance108. Importantly, sleep and diurnal scheduling are often perturbed in psychi-atric disorders4,108. The potential significance of drug-induced changes in appetite and energy balance should also be noted, as glucose is transformed into glutamate and GABA via astrocytes, and diabetes is a risk factor for depression and cognitive impairment90,109. An impact of drugs on immune elements such as cytokines may similarly affect cognitive performance39.
Limited clinical feedback. There has been limited posi-tive clinical feedback so far for many of the putative pro-cognitive drug targets mentioned in TABLE 2. For example, D1 receptor agonists have never been shown to exert pro-cognitive actions in humans98,110, and GABAA recep-tor α2 subunit agonists have yielded mixed findings98,111. Nonetheless, there are some exceptions. Initial clinical studies suggest that α4β2 nicotinic acetylcholine receptor agonists38,112 and 5-hydroxytryptamine (serotonin) recep-tor 6 (5-HT6) antagonists113,114 have positive effects, and substantial data have underscored the role of oxytocin in emotional processing and social cognition115–117. Although its effects may not be entirely unitary, oxytocin consist-ently improves social cognition in volunteers as well as in individuals with ASD or schizophrenia (TABLE 2).
The noradrenaline reuptake inhibitor atomoxetine improves focused attention and executive function in ADHD118. However, noradrenaline reuptake inhibitors have not shown substantial benefits in schizophrenia, and their putative beneficial actions in depression await con-firmation2,4,98. Experimental studies have demonstrated that PFC-localized, pyramidal α2A-adrenergic receptors have a positive influence on working memory. However, the effects of agonists are less robust than those of atomo-xetine in ADHD. Furthermore, a genuine improvement
Box 3 | LTP and LTD: key neuroplastic substrates of cognition
Long-term potentiation (LTP) is the sustained (from hours to months) increase in synaptic strength elicited by a brief period (a few seconds) of patterned, high-frequency (~100 Hz) afferent stimulation. It is a flexible and diverse multiphase mechanism that is involved in many cognitive processes, from declarative learning in the hippocampus to fear-extinction learning in the prefrontal cortex (PFC)15,44. Conversely, long-term depression (LTD) refers to a long-lasting decrease in synaptic response, usually produced by a prolonged sequence (lasting a few minutes) of patterned, low-frequency (~20 Hz) stimulation44,45,134,135. A specific form of LTD (de-potentiation) follows LTP, but LTD does not just serve a homeostatic role as a balancing act for LTP or to improve the signal to noise ratio. Rather, it is also a core mechanism of cognitive plasticity and a legitimate drug target44,45,135. For example, LTD mediated by the NMDA (N-methyl-d-aspartate) receptors and muscarinic M1 receptors in the hippocampus may be implicated in learning45,135. Furthermore, impairment of metabotropic glutamate receptor 5 (mGluR5)-promoted, NMDA-dependent LTP in the PFC and hippocampus may be implicated in the cognitive impairment of schizophrenia50,224–226. Conversely, excessive mGluR5-mediated LTD in the amygdala and other structures contributes to cognitive deficits in fragile X syndrome134,135. The deleterious impact of stress on episodic memory has been related to excessive NMDA receptor-mediated LTD in the hippocampus, possibly as a result of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor endocytosis45. Conversely, stress also impairs cognition by disrupting LTP across a hippocampal–PFC-integrated network88,92. Thus, changes in both LTP and LTD are related to the cognitive deficits observed in psychiatric disorders, and numerous drug targets (such as NMDA receptors, M1 receptors and mGluR5) modulate both of these substrates of neuroplasticity44,45,104,112,135,224,226 (TABLE 2).
Importantly, LTP and LTD are associated with the structural plasticity of dendritic spines — that is, their expansion and formation (LTP), and their contraction and loss (LTD)128 — in several classes of neurons that are important for cognition, including pyramidal neurons in the PFC and medium spinal neurons in the basal ganglia (FIGS 2,4). Spines are regulated in an activity-dependent manner by local protein synthesis and mRNA translation, which is itself subject to modulation by microRNAs168,170. Structural spine plasticity is anomalous in disorders such as schizophrenia and autism spectrum disorder128.
R E V I E W S
152 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

in working memory has yet to be demonstrated and these agonists have a small therapeutic window. Long-term release forms of such agonists may therefore prove to be more useful119,120.
Putative pro-cognitive actions of glycine trans-porter 1 inhibitors in schizophrenia are constrained by motor and autonomic side effects, and results with par-tial agonists at the glycine B co-agonist site on NMDA receptors have been variable98,121. Finally, a vigilance enhancer, modafinil, displayed encouraging effects on cognition (including facial processing and speed of pro-cessing) in patients with schizophrenia, thus supporting studies in volunteers, but the results of more recent, controlled studies have been less compelling98,122,123.
Complex effects on cognition: bell-shaped dose–response curves. Clearly, considerable progress is needed with regard to the clinical profiles of pro-cognitive agents. Their experimental and therapeutic evaluation is com-plicated by the fact that the doses needed to improve cognition depend on several variables, including baseline performance, genotype, test sensitivity and end point. Furthermore, similarly to corticosterone (see above), many agents have ‘inverted U’ dose–response curves in behavioural and mechanistic procedures124–126 (TABLE 2). Biphasic dose–response curves imply a ‘set point’ for optimal performance, such that under- or overactiva-tion of the drug target has a deleterious effect. This is perhaps not surprising, as both deficient and excessive LTP, LTD, ‘plasticity gene’ activity, neurogenesis and dendritic spine generation have a deleterious effect on cognitive processing43–45,64,87,127,128 (BOX 3).
Clinical studies of COMT inhibitors illustrate the significance of this phenonemon75,76. COMT inhibitors enhance extracellular levels of dopamine in the PFC, which improves cognition when basal levels of dopa-mine are low. This has been observed in volunteers and in patients with schizophrenia possessing a Val/Val phe-notype associated with high dopamine metabolism, and also in pathologies in which dopamine levels are reduced in the PFC, such as depression and late-stage Parkinson’s disease. Conversely, cognition deteriorates when basal levels of dopamine are high, as observed in some vol-unteers and in patients with schizophrenia possessing a Met/Met genotype75,76.
Other than off-target, low-potency actions (like muscarinic receptor antagonism) of drugs that perturb cognition, there are several other non-exclusive expla-nations for complex and inverted-U dose–response curves. First, reflecting the homeostatic control of cognition, overactivation of any one mechanism (such as phosphorylation) by a drug may provoke the over-compensatory response of another (such as dephos-phorylation)129 (FIG. 4). Second, a drug may be highly selective for its target but the target itself may exert a complex influence on cognition via spatially distinct receptor populations. For example, activation of post-synaptic α2-adrenergic receptors on PFC pyramidal neurons favours working memory41,119. Conversely, activation of α2-adrenergic receptors that are inhibitory to fronto cortical adrenergic, dopaminergic and cholin-ergic projections is detrimental for working memory and executive function130,131 (TABLE 2). Third, even a single population of sites can mediate a biphasic dose–response curve. Induction of GPCR endocytosis with high concentrations of agonists offers one explanation for this (Supplementary information S2 (figure)), but a more widespread explanation comes from the cou-pling of GPCRs to functionally distinct transduction pathways. For example, 5-HT6 receptors exert a dose-dependent positive and negative effect on cognition via recruitment of cAMP-responsive element binding protein (CREB) and mammalian target of rapamycin (mTOR), respectively114. Accordingly, monotonic, pro-cognitive dose–response relationships could be gen-erated using biased ligands that only recruit specific cellular pathways favouring cognition. A complemen-tary approach would be the exploitation of GPCR-modulatory proteins to direct signalling down specific transduction routes132.
More generally, a prudent approach for enhancing cognition over a broad dose range would be to prior-itize partial rather than full agonists; allosteric modu-lators may also be an option. For intracellular targets such as kinases, partial inhibition is also preferable for safety reasons.
Normalization of pathological processes versus symp-tomatic strategies. There are two complementary ways to restore cognitive performance: first, by countering pathological changes underlying deficits; and second, by recruiting pro-cognitive mechanisms that are inde-pendent of disease aetiology.
Fragile X syndromeA disease that is usually caused by the expansion of a trinucleotide sequence in the 5′-untranslated region of the fragile X mental retardation 1 (FMR1) gene. This leads to FMR1 promoter hyper methylation, transcriptional silencing and loss of the RNA-binding protein FMR1. Abnormal translation of mRNAs, including those regulated by metabotropic glutamate receptor 5, results in excessive long-term depression. Affected individuals have defects in speech, language, attention, working memory and social cognition.
Box 4 | Network synchrony: disruption in psychiatric disorders
Network-coordinated rhythmic activity within and between the regions of the brain controlling cognition is associated with electroencephalographical (EEG) activity that can be quantified non-invasively46,47,88,192,193. Theta (θ; 4–7 Hz) frequencies are related to episodic memory and they are widely used to monitor oscillations driven by hippocampal regions in coordination with the prefrontal cortex (PFC)46,47,88,192,193. Conversely, the activity of GABA (γ-aminobutyric acid)-ergic interneurons in the PFC is reflected in the γ (30–80 Hz) range, which is linked to attention and working memory47,193,195. The synchronization of θ rhythms, γ rhythms and β (12–30 Hz) rhythms across regions of the brain represents a ‘neural code’ that modulates and drives cognitive processes such as top-down cortical control. Accordingly, disruption of EEG-monitored rhythms may reflect cognitive impairment46,47,192,193,227.
Coordinated network activity in the PFC and hippocampus depends on GABAergic interneurons that impose a temporal signature on the firing patterns of neurons controlling cognition42,193,195. Thus, glutamatergic pyramidal cells in the PFC receive trains of fast, inhibitory postsynaptic potentials from parvalbumin-positive GABAergic interneurons that recruit the α2 subunits of GABA
A receptors on their axon hillocks42
(FIG. 4). In schizophrenia, a developmental deficit in excitatory NMDA (N-methyl-d-aspartate) receptors — and possibly AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors — on these GABAergic interneurons leads to a decrease in their activity, anomalous patterns of pyramidal cell firing, perturbed PFC network synchronicity and cognitive impairment42,50,192,228. Frontocortical GABAergic interneurons and pyramidal cells integrate inputs from many modulators controlling cognitive function, including monoamines, acetylcholine and glutamate. Hence, they are a focal point of strategies for enhancing cognition both in schizophrenia and in other psychiatric disorders (FIG. 4). Finally, GABAergic mechanisms in the amygdala are important for fear memory, and a dysfunction of GABAergic intercalated cells (FIG. 2) is implicated in the weakened fear-extinction learning seen in post-traumatic stress disorder15,106 (BOX 1).
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 153
© 2012 Macmillan Publishers Limited. All rights reserved
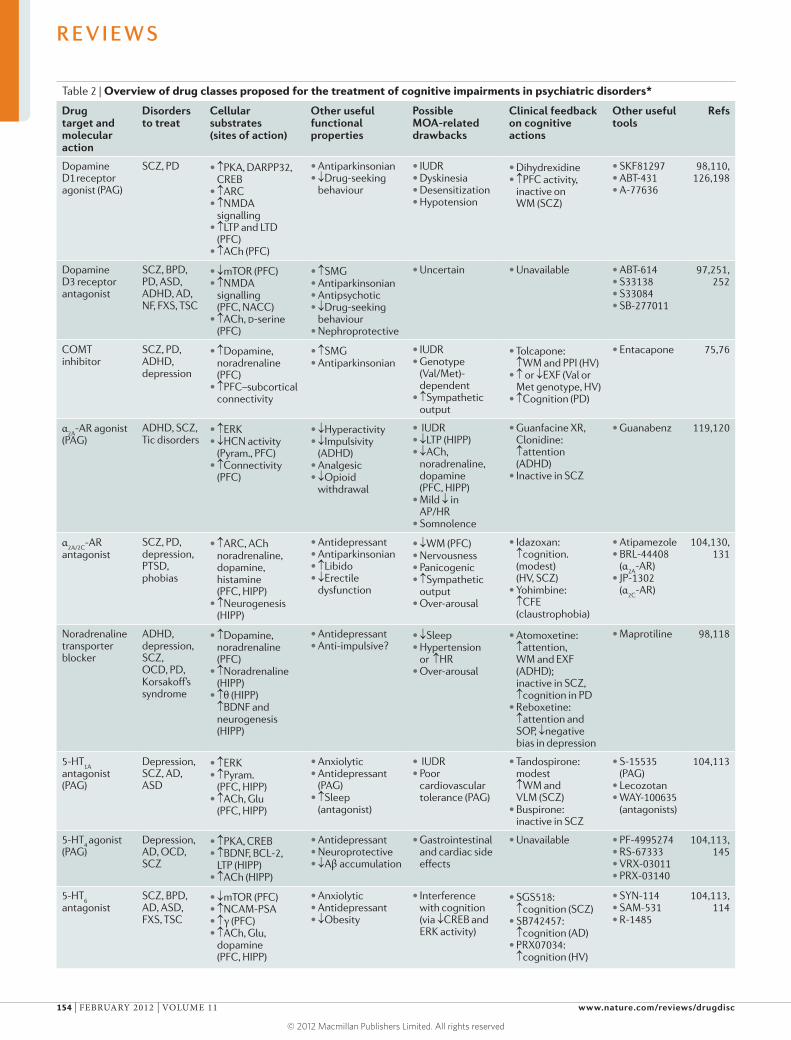
Table 2 | Overview of drug classes proposed for the treatment of cognitive impairments in psychiatric disorders*
Drug target and molecular action
Disorders to treat
Cellular substrates (sites of action)
Other useful functional properties
Possible MOA-related drawbacks
Clinical feedback on cognitive actions
Other useful tools
Refs
Dopamine D1
receptor
agonist (PAG)
SCZ, PD • ↑PKA, DARPP32, CREB
• ↑ARC• ↑NMDA
signalling • ↑LTP and LTD
(PFC) • ↑ACh (PFC)
• Antiparkinsonian• ↓Drug-seeking
behaviour
• IUDR • Dyskinesia • Desensitization • Hypotension
• Dihydrexidine • ↑PFC activity,
inactive on WM (SCZ)
• SKF81297• ABT-431• A-77636
98,110, 126,198
Dopamine D3 receptor antagonist
SCZ, BPD, PD, ASD, ADHD, AD, NF, FXS, TSC
• ↓mTOR (PFC)• ↑NMDA
signalling (PFC, NACC)
• ↑ACh, d-serine (PFC)
• ↑SMG• Antiparkinsonian• Antipsychotic• ↓Drug-seeking
behaviour• Nephroprotective
• Uncertain • Unavailable • ABT-614• S33138• S33084• SB-277011
97,251, 252
COMT inhibitor
SCZ, PD, ADHD, depression
• ↑Dopamine, noradrenaline (PFC)
• ↑PFC–subcortical connectivity
• ↑SMG• Antiparkinsonian
• IUDR• Genotype
(Val/Met)- dependent
• ↑Sympathetic output
• Tolcapone: ↑WM and PPI (HV)
• ↑ or ↓EXF (Val or Met genotype, HV)
• ↑Cognition (PD)
• Entacapone 75,76
α2A
-AR agonist (PAG)
ADHD, SCZ, Tic disorders
• ↑ERK• ↓HCN activity
(Pyram., PFC)• ↑Connectivity
(PFC)
• ↓Hyperactivity• ↓Impulsivity
(ADHD)• Analgesic• ↓Opioid
withdrawal
• IUDR• ↓LTP (HIPP)• ↓ACh,
noradrenaline, dopamine (PFC, HIPP)
• Mild ↓ in AP/HR
• Somnolence
• Guanfacine XR, Clonidine:↑attention (ADHD)
• Inactive in SCZ
• Guanabenz 119,120
α2A/2C
-AR antagonist
SCZ, PD, depression, PTSD, phobias
• ↑ARC, ACh noradrenaline, dopamine, histamine (PFC, HIPP)
• ↑Neurogenesis (HIPP)
• Antidepressant• Antiparkinsonian• ↑Libido• ↓Erectile
dysfunction
• ↓WM (PFC)• Nervousness• Panicogenic• ↑Sympathetic
output• Over-arousal
• Idazoxan: ↑cognition. (modest) (HV, SCZ)
• Yohimbine: ↑CFE (claustrophobia)
• Atipamezole• BRL-44408
(α2A
-AR)• JP-1302
(α2C
-AR)
104,130, 131
Noradrenaline transporter blocker
ADHD, depression, SCZ, OCD, PD, Korsakoff’s syndrome
• ↑Dopamine, noradrenaline (PFC)
• ↑Noradrenaline (HIPP)
• ↑θ (HIPP) ↑BDNF and neurogenesis (HIPP)
• Antidepressant• Anti-impulsive?
• ↓Sleep• Hypertension
or↑HR• Over-arousal
• Atomoxetine:↑attention, WM and EXF (ADHD); inactive in SCZ, ↑cognition in PD
• Reboxetine:↑attention and SOP, ↓negative bias in depression
• Maprotiline 98,118
5-HT1A
antagonist (PAG)
Depression, SCZ, AD, ASD
• ↑ERK • ↑Pyram.
(PFC, HIPP)• ↑ACh, Glu
(PFC, HIPP)
• Anxiolytic• Antidepressant
(PAG)• ↑Sleep
(antagonist)
• IUDR• Poor
cardiovascular tolerance (PAG)
• Tandospirone: modest ↑WM and VLM (SCZ)
• Buspirone: inactive in SCZ
• S-15535 (PAG)
• Lecozotan• WAY-100635
(antagonists)
104,113
5-HT4 agonist
(PAG)Depression, AD, OCD, SCZ
• ↑PKA, CREB • ↑BDNF, BCL-2,
LTP (HIPP)• ↑ACh (HIPP)
• Antidepressant• Neuroprotective• ↓Aβ accumulation
• Gastrointestinal and cardiac side effects
• Unavailable • PF-4995274• RS-67333• VRX-03011• PRX-03140
104,113, 145
5-HT6
antagonistSCZ, BPD, AD, ASD, FXS, TSC
• ↓mTOR (PFC)• ↑NCAM-PSA• ↑γ (PFC) • ↑ACh, Glu,
dopamine (PFC, HIPP)
• Anxiolytic• Antidepressant• ↓Obesity
• Interference with cognition (via ↓CREB and ERK activity)
• SGS518:↑cognition (SCZ)
• SB742457: ↑cognition (AD)
• PRX07034: ↑cognition (HV)
• SYN-114• SAM-531• R-1485
104,113, 114
R E V I E W S
154 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
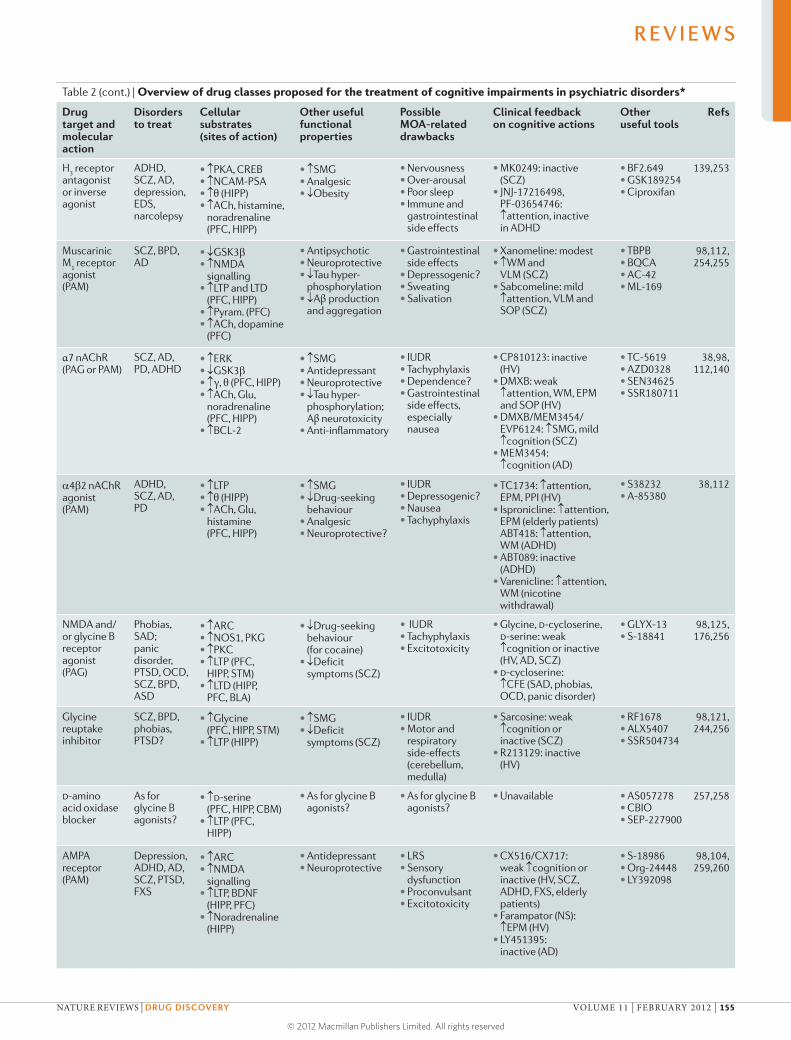
Table 2 (cont.) | Overview of drug classes proposed for the treatment of cognitive impairments in psychiatric disorders*
Drug target and molecular action
Disorders to treat
Cellular substrates (sites of action)
Other useful functional properties
Possible MOA-related drawbacks
Clinical feedback on cognitive actions
Other useful tools
Refs
H3 receptor
antagonist or inverse agonist
ADHD, SCZ, AD, depression, EDS, narcolepsy
• ↑PKA, CREB• ↑NCAM-PSA• ↑θ (HIPP)• ↑ACh, histamine,
noradrenaline (PFC, HIPP)
• ↑SMG• Analgesic• ↓Obesity
• Nervousness• Over-arousal• Poor sleep• Immune and
gastrointestinal side effects
• MK0249: inactive (SCZ)
• JNJ-17216498, PF-03654746:↑attention, inactive in ADHD
• BF2.649• GSK189254• Ciproxifan
139,253
Muscarinic M
1 receptor
agonist (PAM)
SCZ, BPD, AD
• ↓GSK3b• ↑NMDA
signalling • ↑LTP and LTD
(PFC, HIPP) • ↑Pyram. (PFC)• ↑ACh, dopamine
(PFC)
• Antipsychotic• Neuroprotective• ↓Tau hyper-
phosphorylation• ↓Aβ production
and aggregation
• Gastrointestinal side effects
• Depressogenic?• Sweating• Salivation
• Xanomeline: modest• ↑WM and
VLM (SCZ)• Sabcomeline: mild
↑attention, VLM and SOP (SCZ)
• TBPB• BQCA• AC-42• ML-169
98,112, 254,255
α7 nAChR (PAG or PAM)
SCZ, AD, PD, ADHD
• ↑ERK • ↓GSK3b• ↑γ, θ (PFC, HIPP)• ↑ACh, Glu,
noradrenaline (PFC, HIPP)
• ↑BCL-2
• ↑SMG• Antidepressant• Neuroprotective• ↓Tau hyper-
phosphorylation; Aβ neurotoxicity
• Anti-inflammatory
• IUDR• Tachyphylaxis• Dependence?• Gastrointestinal
side effects, especially nausea
• CP810123: inactive (HV)
• DMXB: weak↑attention, WM, EPM and SOP (HV)
• DMXB/MEM3454/EVP6124: ↑SMG, mild ↑cognition (SCZ)
• MEM3454: ↑cognition (AD)
• TC-5619• AZD0328• SEN34625• SSR180711
38,98, 112,140
a4b2 nAChR agonist (PAM)
ADHD, SCZ, AD, PD
• ↑LTP• ↑θ (HIPP) • ↑ACh, Glu,
histamine (PFC, HIPP)
• ↑SMG• ↓Drug-seeking
behaviour• Analgesic• Neuroprotective?
• IUDR• Depressogenic?• Nausea• Tachyphylaxis
• TC1734: ↑attention, EPM, PPI (HV)
• Ispronicline:↑attention, EPM (elderly patients) ABT418: ↑attention, WM (ADHD)
• ABT089: inactive (ADHD)
• Varenicline: ↑attention, WM (nicotine withdrawal)
• S38232• A-85380
38,112
NMDA and/or glycine B receptor agonist (PAG)
Phobias, SAD; panic disorder, PTSD, OCD, SCZ, BPD, ASD
• ↑ARC• ↑NOS1, PKG• ↑PKC• ↑LTP (PFC,
HIPP, STM)• ↑LTD (HIPP,
PFC, BLA)
• ↓Drug-seeking behaviour (for cocaine)
• ↓Deficit symptoms (SCZ)
• IUDR• Tachyphylaxis• Excitotoxicity
• Glycine, d-cycloserine, d-serine: weak ↑cognition or inactive (HV, AD, SCZ)
• d-cycloserine: ↑CFE (SAD, phobias, OCD, panic disorder)
• GLYX-13• S-18841
98,125, 176,256
Glycine reuptake inhibitor
SCZ, BPD, phobias, PTSD?
• ↑Glycine (PFC, HIPP, STM)
• ↑LTP (HIPP)
• ↑SMG• ↓Deficit
symptoms (SCZ)
• IUDR• Motor and
respiratory side-effects (cerebellum, medulla)
• Sarcosine: weak ↑cognition or inactive (SCZ)
• R213129: inactive (HV)
• RF1678 • ALX5407• SSR504734
98,121, 244,256
d-amino acid oxidase blocker
As for glycine B agonists?
• ↑d-serine (PFC, HIPP, CBM)
• ↑LTP (PFC, HIPP)
• As for glycine B agonists?
• As for glycine B agonists?
• Unavailable • AS057278• CBIO• SEP-227900
257,258
AMPA receptor (PAM)
Depression, ADHD, AD, SCZ, PTSD, FXS
• ↑ARC • ↑NMDA
signalling• ↑LTP, BDNF
(HIPP, PFC)• ↑Noradrenaline
(HIPP)
• Antidepressant• Neuroprotective
• LRS• Sensory
dysfunction• Proconvulsant• Excitotoxicity
• CX516/CX717: weak ↑cognition or inactive (HV, SCZ, ADHD, FXS, elderly patients)
• Farampator (NS): ↑EPM (HV)
• LY451395: inactive (AD)
• S-18986• Org-24448• LY392098
98,104, 259,260
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 155
© 2012 Macmillan Publishers Limited. All rights reserved
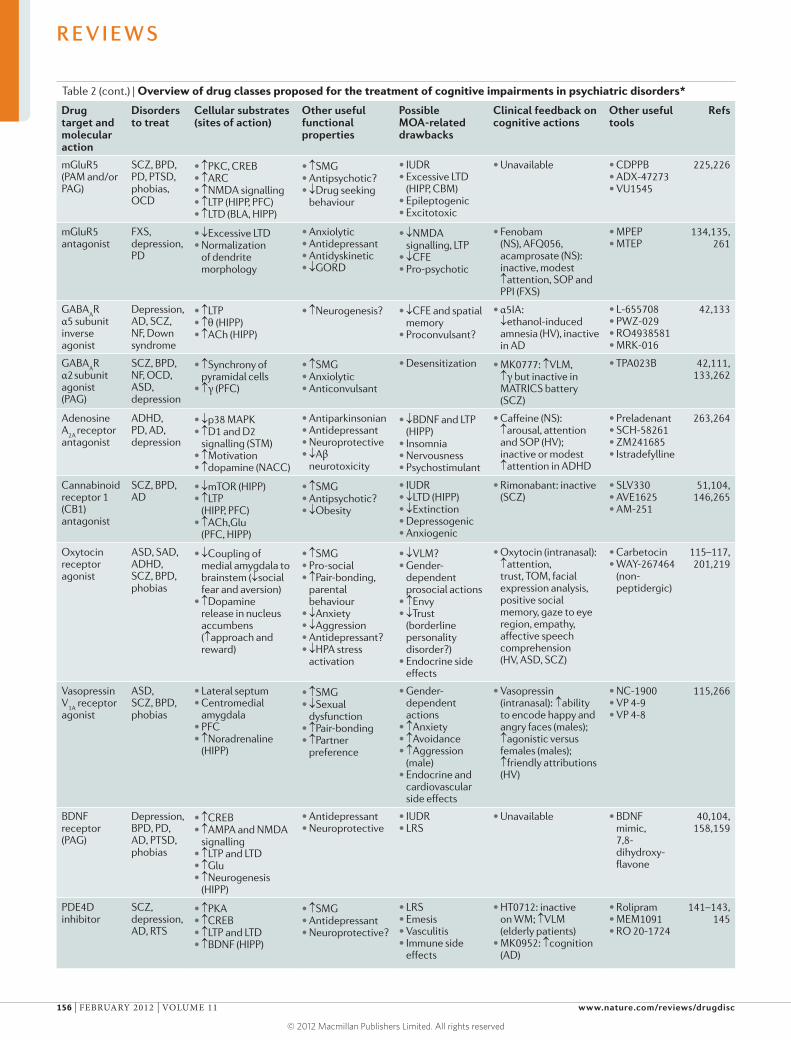
Table 2 (cont.) | Overview of drug classes proposed for the treatment of cognitive impairments in psychiatric disorders*
Drug target and molecular action
Disorders to treat
Cellular substrates (sites of action)
Other useful functional properties
Possible MOA-related drawbacks
Clinical feedback on cognitive actions
Other useful tools
Refs
mGluR5 (PAM and/or PAG)
SCZ, BPD, PD, PTSD, phobias, OCD
• ↑PKC, CREB• ↑ARC• ↑NMDA signalling• ↑LTP (HIPP, PFC)• ↑LTD (BLA, HIPP)
• ↑SMG• Antipsychotic?• ↓Drug seeking
behaviour
• IUDR• Excessive LTD
(HIPP, CBM)• Epileptogenic• Excitotoxic
• Unavailable • CDPPB• ADX-47273• VU1545
225,226
mGluR5
antagonistFXS, depression, PD
• ↓Excessive LTD• Normalization
of dendrite morphology
• Anxiolytic• Antidepressant• Antidyskinetic• ↓GORD
• ↓NMDA signalling, LTP
• ↓CFE• Pro-psychotic
• Fenobam (NS), AFQ056, acamprosate (NS): inactive, modest ↑attention, SOP and PPI (FXS)
• MPEP• MTEP
134,135, 261
GABAAR
α5 subunit inverse agonist
Depression, AD, SCZ, NF, Down syndrome
• ↑LTP• ↑θ (HIPP)• ↑ACh (HIPP)
• ↑Neurogenesis? • ↓CFE and spatial memory
• Proconvulsant?
• α5IA: ↓ethanol-induced amnesia (HV), inactive in AD
• L-655708• PWZ-029• RO4938581• MRK-016
42,133
GABAAR
α2 subunit
agonist (PAG)
SCZ, BPD, NF, OCD, ASD, depression
• ↑Synchrony of pyramidal cells
• ↑γ (PFC)
• ↑SMG• Anxiolytic• Anticonvulsant
• Desensitization • MK0777: ↑VLM, ↑γ but inactive in MATRICS battery (SCZ)
• TPA023B 42,111, 133,262
Adenosine A
2A receptor
antagonist
ADHD, PD, AD, depression
• ↓p38 MAPK• ↑D1 and D2
signalling (STM)• ↑Motivation• ↑dopamine (NACC)
• Antiparkinsonian• Antidepressant• Neuroprotective• ↓Aβ
neurotoxicity
• ↓BDNF and LTP (HIPP)
• Insomnia• Nervousness• Psychostimulant
• Caffeine (NS): ↑arousal, attention and SOP (HV); inactive or modest ↑attention in ADHD
• Preladenant• SCH-58261• ZM241685• Istradefylline
263,264
Cannabinoid receptor 1 (CB1) antagonist
SCZ, BPD, AD
• ↓mTOR (HIPP)• ↑LTP
(HIPP, PFC)• ↑ACh,Glu
(PFC, HIPP)
• ↑SMG• Antipsychotic?• ↓Obesity
• IUDR• ↓LTD (HIPP)• ↓Extinction• Depressogenic• Anxiogenic
• Rimonabant: inactive (SCZ)
• SLV330• AVE1625• AM-251
51,104, 146,265
Oxytocin receptor agonist
ASD, SAD, ADHD, SCZ, BPD, phobias
• ↓Coupling of medial amygdala to brainstem (↓social fear and aversion)
• ↑Dopamine release in nucleus accumbens (↑ approach and reward)
• ↑SMG• Pro-social• ↑Pair-bonding,
parental behaviour
• ↓Anxiety• ↓Aggression• Antidepressant?• ↓HPA stress
activation
• ↓VLM?• Gender-
dependent prosocial actions
• ↑Envy• ↓Trust
(borderline personality disorder?)
• Endocrine side effects
• Oxytocin (intranasal): ↑attention, trust, TOM, facial expression analysis, positive social memory, gaze to eye region, empathy, affective speech comprehension (HV, ASD, SCZ)
• Carbetocin• WAY-267464
(non- peptidergic)
115–117, 201,219
Vasopressin V
1A receptor
agonist
ASD, SCZ, BPD, phobias
• Lateral septum• Centromedial
amygdala• PFC• ↑Noradrenaline
(HIPP)
• ↑SMG• ↓Sexual
dysfunction• ↑Pair-bonding• ↑Partner
preference
• Gender- dependent actions
• ↑Anxiety• ↑Avoidance• ↑Aggression
(male)• Endocrine and
cardiovascular side effects
• Vasopressin (intranasal):↑ability to encode happy and angry faces (males); ↑agonistic versus females (males); ↑friendly attributions (HV)
• NC-1900• VP 4-9• VP 4-8
115,266
BDNF receptor (PAG)
Depression, BPD, PD, AD, PTSD, phobias
• ↑CREB• ↑AMPA and NMDA
signalling• ↑LTP and LTD• ↑Glu• ↑Neurogenesis
(HIPP)
• Antidepressant• Neuroprotective
• IUDR• LRS
• Unavailable • BDNF mimic, 7,8-dihydroxy-flavone
40,104, 158,159
PDE4D inhibitor
SCZ, depression, AD, RTS
• ↑PKA• ↑CREB• ↑LTP and LTD• ↑BDNF (HIPP)
• ↑SMG• Antidepressant• Neuroprotective?
• LRS• Emesis• Vasculitis• Immune side
effects
• HT0712: inactive on WM; ↑VLM (elderly patients)
• MK0952: ↑cognition (AD)
• Rolipram• MEM1091• RO 20-1724
141–143, 145
R E V I E W S
156 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
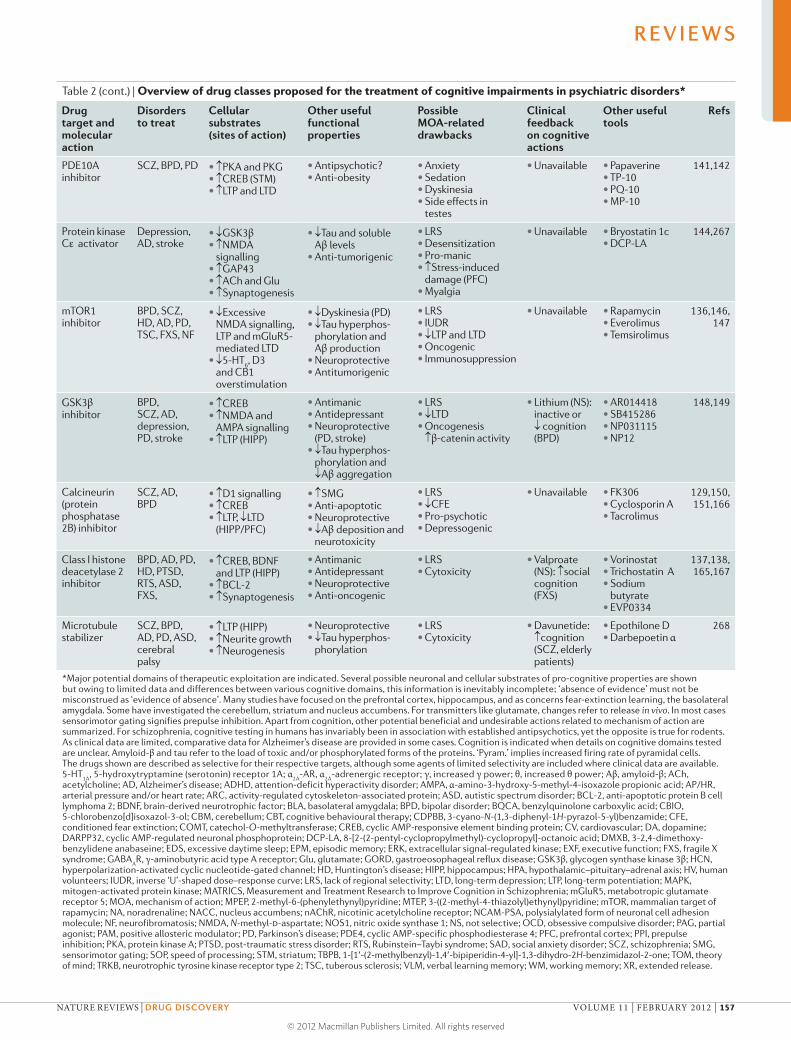
Table 2 (cont.) | Overview of drug classes proposed for the treatment of cognitive impairments in psychiatric disorders*
Drug target and molecular action
Disorders to treat
Cellular substrates (sites of action)
Other useful functional properties
Possible MOA-related drawbacks
Clinical feedback on cognitive actions
Other useful tools
Refs
PDE10A inhibitor
SCZ, BPD, PD • ↑PKA and PKG• ↑CREB (STM)• ↑LTP and LTD
• Antipsychotic?• Anti-obesity
• Anxiety• Sedation• Dyskinesia• Side effects in
testes
• Unavailable • Papaverine• TP-10• PQ-10• MP-10
141,142
Protein kinase Cε activator
Depression, AD, stroke
• ↓GSK3β• ↑NMDA
signalling• ↑GAP43• ↑ACh and Glu• ↑Synaptogenesis
• ↓Tau and soluble Aβ levels
• Anti-tumorigenic
• LRS• Desensitization• Pro-manic• ↑Stress-induced
damage (PFC)• Myalgia
• Unavailable • Bryostatin 1c• DCP-LA
144,267
mTOR1 inhibitor
BPD, SCZ, HD, AD, PD, TSC, FXS, NF
• ↓Excessive NMDA signalling, LTP and mGluR5- mediated LTD
• ↓5-HT6, D3
and CB1 overstimulation
• ↓Dyskinesia (PD)• ↓Tau hyperphos-
phorylation and Aβ production
• Neuroprotective• Antitumorigenic
• LRS• IUDR • ↓LTP andLTD• Oncogenic• Immunosuppression
• Unavailable • Rapamycin• Everolimus• Temsirolimus
136,146, 147
GSK3β inhibitor
BPD, SCZ, AD, depression, PD, stroke
• ↑CREB • ↑NMDA and
AMPA signalling• ↑LTP (HIPP)
• Antimanic• Antidepressant• Neuroprotective
(PD, stroke)• ↓Tau hyperphos-
phorylation and ↓Aβ aggregation
• LRS• ↓LTD• Oncogenesis
↑β-catenin activity
• Lithium (NS): inactive or ↓ cognition (BPD)
• AR014418• SB415286• NP031115• NP12
148,149
Calcineurin (protein phosphatase 2B) inhibitor
SCZ, AD, BPD
• ↑D1 signalling • ↑CREB• ↑LTP, ↓LTD
(HIPP/PFC)
• ↑SMG• Anti-apoptotic• Neuroprotective• ↓Aβ deposition and
neurotoxicity
• LRS• ↓CFE• Pro-psychotic• Depressogenic
• Unavailable • FK306• Cyclosporin A• Tacrolimus
129,150, 151,166
Class I histone deacetylase 2 inhibitor
BPD, AD, PD, HD, PTSD, RTS, ASD, FXS,
• ↑CREB, BDNF and LTP (HIPP)
• ↑BCL-2• ↑Synaptogenesis
• Antimanic• Antidepressant• Neuroprotective• Anti-oncogenic
• LRS• Cytoxicity
• Valproate (NS): ↑social cognition (FXS)
• Vorinostat• Trichostatin A• Sodium
butyrate• EVP0334
137,138, 165,167
Microtubule stabilizer
SCZ, BPD, AD, PD, ASD, cerebral palsy
• ↑LTP (HIPP)• ↑Neurite growth• ↑Neurogenesis
• Neuroprotective• ↓Tau hyperphos-
phorylation
• LRS• Cytoxicity
• Davunetide: ↑cognition (SCZ, elderly patients)
• Epothilone D• Darbepoetin α
268
*Major potential domains of therapeutic exploitation are indicated. Several possible neuronal and cellular substrates of pro-cognitive properties are shown but owing to limited data and differences between various cognitive domains, this information is inevitably incomplete; ‘absence of evidence’ must not be misconstrued as ‘evidence of absence’. Many studies have focused on the prefrontal cortex, hippocampus, and as concerns fear-extinction learning, the basolateral amygdala. Some have investigated the cerebellum, striatum and nucleus accumbens. For transmitters like glutamate, changes refer to release in vivo. In most cases sensorimotor gating signifies prepulse inhibition. Apart from cognition, other potential beneficial and undesirable actions related to mechanism of action are summarized. For schizophrenia, cognitive testing in humans has invariably been in association with established antipsychotics, yet the opposite is true for rodents. As clinical data are limited, comparative data for Alzheimer’s disease are provided in some cases. Cognition is indicated when details on cognitive domains tested are unclear. Amyloid-β and tau refer to the load of toxic and/or phosphorylated forms of the proteins. ‘Pyram.’ implies increased firing rate of pyramidal cells. The drugs shown are described as selective for their respective targets, although some agents of limited selectivity are included where clinical data are available. 5-HT
1A, 5-hydroxytryptamine (serotonin) receptor 1A; α
2A-AR, α
2A-adrenergic receptor; γ, increased γ power; θ, increased θ power; Aβ, amyloid-β; ACh,
acetylcholine; AD, Alzheimer’s disease; ADHD, attention-deficit hyperactivity disorder; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; AP/HR, arterial pressure and/or heart rate; ARC, activity-regulated cytoskeleton-associated protein; ASD, autistic spectrum disorder; BCL-2, anti-apoptotic protein B cell lymphoma 2; BDNF, brain-derived neurotrophic factor; BLA, basolateral amygdala; BPD, bipolar disorder; BQCA, benzylquinolone carboxylic acid; CBIO, 5-chlorobenzo[d]isoxazol-3-ol; CBM, cerebellum; CBT, cognitive behavioural therapy; CDPBB, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; CFE, conditioned fear extinction; COMT, catechol-O-methyltransferase; CREB, cyclic AMP-responsive element binding protein; CV, cardiovascular; DA, dopamine; DARPP32, cyclic AMP-regulated neuronal phosphoprotein; DCP-LA, 8-[2-(2-pentyl-cyclopropylmethyl)-cyclopropyl]-octanoic acid; DMXB, 3-2,4-dimethoxy-benzylidene anabaseine; EDS, excessive daytime sleep; EPM, episodic memory; ERK, extracellular signal-regulated kinase; EXF, executive function; FXS, fragile X syndrome; GABA
AR, γ-aminobutyric acid type A receptor; Glu, glutamate; GORD, gastroeosophageal reflux disease; GSK3β, glycogen synthase kinase 3β; HCN,
hyperpolarization-activated cyclic nucleotide-gated channel; HD, Huntington’s disease; HIPP, hippocampus; HPA, hypothalamic–pituitary–adrenal axis; HV, human volunteers; IUDR, inverse ‘U’-shaped dose–response curve; LRS, lack of regional selectivity; LTD, long-term depression; LTP, long-term potentiation; MAPK, mitogen-activated protein kinase; MATRICS, Measurement and Treatment Research to Improve Cognition in Schizophrenia; mGluR5, metabotropic glutamate receptor 5; MOA, mechanism of action; MPEP, 2-methyl-6-(phenylethynyl)pyridine; MTEP, 3-((2-methyl-4-thiazolyl)ethynyl)pyridine; mTOR, mammalian target of rapamycin; NA, noradrenaline; NACC, nucleus accumbens; nAChR, nicotinic acetylcholine receptor; NCAM-PSA, polysialylated form of neuronal cell adhesion molecule; NF, neurofibromatosis; NMDA, N-methyl-d-aspartate; NOS1, nitric oxide synthase 1; NS, not selective; OCD, obsessive compulsive disorder; PAG, partial agonist; PAM, positive allosteric modulator; PD, Parkinson’s disease; PDE4, cyclic AMP-specific phosphodiesterase 4; PFC, prefrontal cortex; PPI, prepulse inhibition; PKA, protein kinase A; PTSD, post-traumatic stress disorder; RTS, Rubinstein–Taybi syndrome; SAD, social anxiety disorder; SCZ, schizophrenia; SMG, sensorimotor gating; SOP, speed of processing; STM, striatum; TBPB, 1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one; TOM, theory of mind; TRKB, neurotrophic tyrosine kinase receptor type 2; TSC, tuberous sclerosis; VLM, verbal learning memory; WM, working memory; XR, extended release.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 157
© 2012 Macmillan Publishers Limited. All rights reserved

As an example of the former strategy, glycine B ago-nists are designed to treat schizophrenia by stimulating hypoactive NMDA receptors localized on GABAergic interneurons in the PFC42,50,98. PFC-integrated cognition can also be restored by agonists acting downstream at the α2 subunit of GABAA receptors42,98,133 (FIG. 4). Another example of this strategy is provided by the mechanism of action of COMT inhibitors, which compensate for low levels of dopamine in depression75,76. The epigenetic developmental disorder fragile X syndrome is character-ized by excessive metabotropic glutamate receptor 5 (mGluR5)-mediated LTD, which leads to cognitive defi-cits that can be countered by mGluR5 antagonists at these sites134,135. Similarly, cognitive deficits associated with the ASD-related disorder tuberous sclerosis are provoked by mutations in the genes encoding tuberous sclerosis pro-tein 1 and tuberous sclerosis protein 2, which interact with mTOR (Supplementary information S2 (figure)); cognitive impairment may therefore be reversed by the mTOR inhibitor rapamycin63,136. More generally, epige-netic reprogramming raises the hope of reversing the cognitive impairment accompanying monogenic ASD and caused by early-life stress (see below)63,137,138.
Although they are conceptually attractive, pathology-driven approaches have limitations. First — as exempli-fied by rare, genetic forms of ASD — they may only be applicable to a subpopulation of patients. Second, molec-ular substrates underlying cognitive deficits are still not generally well understood. Third, it may be impos-sible to retrospectively normalize certain pathological events, such as neonatal insults that trigger alterations in synaptic architecture, and neural circuits that lead to schizophrenia63,67.
Complementary strategies for symptomatic treat-ment do not attempt to normalize a pathological change, such as NMDA receptor hypofunction. Rather, they engage compensatory pro-cognitive mechanisms spared by the disorder in question. For example, there is no evi-dence for 5-HT6 or histamine H3 receptor hyperactiva-tion in schizophrenia, yet antagonists hold promise for correcting a range of cognitive deficits in schizophrenia as well as in other disorders such as depression113,114,139. Pathology-decoupled mechanisms may actually have a broader application than pathology-driven strategies. In addition, some drugs should have beneficial actions not only against cognitive impairment but also against other symptoms (TABLE 2).
Domain-specific and generalized improvements in cognitive performance. A crucial issue is whether drugs will primarily correct one specific cognitive domain or improve several simultaneously. This obviously depends on the drug’s mechanism of action. If a treatment com-pletely reversed the underlying pathology it might — in theory — correct all deficits: one example, as further evoked below, is the blockade of mTOR overactiva-tion in the monogenic disorder tuberous sclerosis63,136. Conversely, for a complex, multifactorial and hetero-geneous disease like schizophrenia, aiming to normal-ize cognitive performance across all domains and in all patients appears to be overambitious.
There may be greater hope of finding a mechanism that could improve one specific cognitive domain in a transnosological manner across numerous diseases. Examples include oxytocin agonists for promoting social cognition115–117, and α7 nicotinic acetylcholine receptor agonists for reinforcing attention and working memory38,112,140 (Supplementary information S4 (box)) (TABLE 2). Clearly, a balance must be sought between the holy grail of ‘one drug, all domains, all disorders’ versus ‘a separate drug for each domain and each disor-der’. Although a pan-cognitive agent that can restore all domains may be unobtainable, multitarget drugs uniting complementary mechanisms of action appear to be the most promising route towards achieving a broad-based improvement in cognition4,104.
Intracellular targets. Intracellular targets are attracting increasing interest as substrates for improving cognitive deficits in psychiatric disorders (FIG. 4) but they are not easy to exploit43,45,57–60,63,212. Such agents do not neces-sarily sidestep the issue of biphasic dose–response rela-tionships, and they raise serious issues of tolerance and regional specificity because of the ubiquity and the mul-tiple roles of most cellular targets. This is illustrated by the contrast between the restricted cerebral localization of 5-HT6 receptors and the broad organismal distribution of phosphodiesterases catalysing cAMP degradation114,141,142 (FIG. 4).
One solution may be to develop drugs that are selec-tive for protein isoforms such as phosphodiesterase 4D or protein kinase Cε.Although achieving selectivity is a for-midable challenge, exploitation of allosteric rather than catalytic sites may help drug design141,143,144. The influence of an inhibitor should be most prominent where and when the activity of its target is aberrantly high. Hence, another approach involves ‘vectoring’ drugs using dual-acting molecules that act on an intracellular protein and an upstream mechanism. For example, coupling inhi-bition of phosphodiesterase 4D to 5-HT4 receptor ago-nism may restrict the facilitation of cAMP-dependent transmission to regions where 5-HT receptors control cognition145. Association of 5-HT6 receptor antagonism with mTOR inhibition could yield similar benefits114,136. However, the design of drugs acting at two or more useful sites is challenging, and it is important to demonstrate an improved therapeutic window of beneficial versus deleterious actions as well as advantages of these drugs compared with selective drugs used in combination.
mTOR is involved in both LTP and LTD; further-more, it favours and — when hyperactive — counters cognitive processes, so it is a particularly interesting intracellular target136. For example, tuberous sclerosis involves loss of the mTOR inhibitory protein partner tuberculosis sclerosis protein, leading to its overstim-ulation (Supplementary information S2 (figure)). Accordingly, the mTOR inhibitor rapamycin attenuated excessive hippocampal LTP and relieved cognitive defi-cits in a mouse model of tuberous sclerosis, and it may also be effective in other forms of ASD136. Rapamycin antagonises cognitive deficits elicited by the stimulation of cannabinoid receptor 1 on inhibitory GABAergic
Tuberous sclerosisAn autosomal dominant disorder, usually caused by sporadic mutations, leading to inactivation of the tumour suppressor genes tuberous sclerosis 1 (TSC1;also known as hamartin) and TSC2 (also known as tuberin), which normally inhibit RHEB (a GTPase that is an activator of mammalian target of rapamycin). Loss of TSC1 or TSC2 leads to disinhibition of cell growth, cortical tubers and giant astrocytomas in the brain. Patients have deficits in attention, executive function and memory, as well as symptoms resembling autism spectrum disorder and attention deficit hyperactivity disorder.
R E V I E W S
158 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

interneurons in the hippocampus146, and by the activa-tion of 5-HT6 and D3 receptors in the PFC14 (TABLE 2), which suggests that mTOR overactivation may be rel-evant in schizophrenia. mTOR is also implicated in hippocampal processes sustaining abnormal fear mem-ory147. Despite these indications that mTOR inhibitors might improve cognitive deficits, their therapeutic use could interfere with physiological LTP, and therefore raises issues of safety and specificity. Such concerns also apply to glycogen synthase kinase-3β inhibitors for pro-moting LTP and long-term memory in depression and bipolar disorder148,149 (TABLE 2).
Protein phosphatase 1 and protein phosphatase 2B (also known as calcineurin) are targets for improving long-term memory as they interfere with the activation of CREB and downstream cognition-related genes such as BDNF129 (FIG. 4). Accordingly, calcineurin inhibitors normalize the biphasic dose–response curves of drugs that enhance CREB activity by phosphorylation, and they promote LTP and learning in the hippocampus129,150. However, inhibition of calcineurin strengthens the for-mation of aversive memories in the amygdala, underscor-ing the multiple effects of phosphatases on CREB activity and cognition129,151 (Supplementary information S2 (fig-ure)). As with kinases, the question of specificity must be addressed before phosphatase inhibitors could be thera-peutically exploited for treating cognitive impairment129.
Neurogenesis. In recent years, considerable attention has been devoted to hippocampal neurogenesis, as its suppression is implicated in the impaired episodic memory associated with depression4,127,152. Adult-born, maturing dentate gyrus cells are especially excitable and plastic, sustain prolonged LTP and are rapidly incorporated into and coordinate neural networks, suggesting that increased neurogenesis favours cogni-tion127. This explains the role of these cells in learning, consolidation and updating of new memories, dif-ferentiating separate memories in the dentate gyrus, transferring hippocampal-dependent memories to extra-hippocampal regions and coupling cognition to external context127,153–155. However, neurogenesis is no exception to the inverted-U dose–response rule, as both the generation and suppression of new neurons is required to optimize cognitive function127. Moreover, although antidepressants consistently enhance neuro-genesis in rodents, they do not generally favour cognitive performance4,156. On balance, therefore, facilitation of neurogenesis is not yet a compelling target for improv-ing cognition in psychiatric disorders.
Further insights may be gained by studying the cell ular signals mediating the influence of neurogen-esis on mood and cognition, such as activity-regulated cytoskeleton-associated protein157 and the upstream driver of neurogenesis, BDNF, which acts via neuro-trophic tyrosine kinase receptor type 2 (NTRK2; also known as TRKB)40,158 (FIG. 4). The decreased expression of BDNF seen in chronic stress may be related to the cognitive deficits associated with depression, as BDNF mediates several forms of hippocampal and PFC plas-ticity, including LTP40,152. Interest in BDNF has been
enhanced by reports showing that TRKB activators block the disruptive influence of stress on hippocam-pus-integrated long-term memory40,158, and that BDNF promotes extinction learning159.
Epigenetics. Stress-induced epigenetic changes in ger-mline cells can be passed on to and alter cognition in offspring, suggesting that environmental risks for cog-nitive deficits might be relevant even before concep-tion63,137,138,160–162. Some changes can be sex-specific. For example, epigenetic imprinting of the maternal allele of genes in the 15q11–13 region is implicated in the aetiology of ASD160.
Of perhaps broader relevance to pharmacotherapy, gene-specific alterations in DNA methylation and his-tone acetylation (FIG. 4) may contribute to long-term impairments in cognition resulting from exposure to stress during early life84,137,138. For example, DNA hypo-methylation (leading to enhanced expression) of the gene encoding corticotropin-releasing hormone may account for HPA axis overdrive in adults who have undergone early-life stress137,138,162. Conversely, perinatal stress leads to hypermethylation-induced silencing of the gene encoding BDNF138. Another example is ASD, in which hypermethylation-induced suppression of the oxytocin receptor gene has been reported161.
Although decreases in DNA methylation are hard to counter pharmacologically, increases could be countered by DNA-N-methyltransferase (DNMT) inhibitors. Apart from their potential utility for correcting epigenetic changes provoked by early-life stress, DNMT inhibitors may also be useful in other disorders: for example, for counteracting the consolidation of fear memories seen in PTSD137,138. Furthermore, DNMT inhibitors may improve cognition in Rett’s syndrome, an X-linked ASD characterized by gene hypermethylation138. In addition, in schizophrenia DNMT inhibitors may normalize cog-nitive deficits resulting from hypermethylation-induced suppression of PFC-localized genes synthesizing GABA and reelin (a developmental glycoprotein that controls synaptic plasticity)163,164.
Reflecting interactions between DNMTs and his-tone deacetylases (HDACs), certain effects of DNMT inhibitors can be mimicked by suppressors of overactive HDACs, such as valproate137,138. Valproate is not selective for HDAC isoforms but selective inhibition of HDAC2 could be especially useful as this isoform negatively reg-ulates hippocampal LTP, dendritic spine density and vis-ual learning, as well as BDNF and CREB gene expression. HDAC2 inhibitors could therefore enhance these pro-cognitive processes and also promote fear-extinction learning, suggesting utility in PTSD137,138,165,166. Turning to ASDs, fragile X syndrome is characterized both by the loss of the mRNA translation regulator fragile X mental retardation 1 protein and by hypoacetylation of several functionally interacting classes of histones134,138. Valproate and a class III HDAC inhibitor increased histone acetylation and reactivated silenced fragile X mental retardation 1 protein in lymphoblastoid cells of patients with fragile X syndrome. Furthermore, val-proate also improved social cognition and attention167.
Rett’s syndromeAn X-linked developmental disorder, mainly seen in females, caused by de novo mutations in the gene encoding methyl CpG binding protein 2 (MECP2). MECP2 normally binds to methylated DNA to transcriptionally repress genes, although some are activated. MECP2 also interacts with histone deacetylases, so its loss leads to gene-dependent histone hypo- and hyperacetylation. Patients with Rett’s syndrome suffer from retardation, loss of verbal learning and speech, and impaired social cognition.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 159
© 2012 Macmillan Publishers Limited. All rights reserved

Rubinstein–Taybi syndrome is an epigenetic ASD-related disorder caused by mutations in CREB-binding proteins that act as histone acetyltransferases (Supplementary information S2 (figure)). Hence, HDAC inhibitors are being studied for the correction of cognitive deficits associated with this syndrome137,138.
Finally, blockade of nuclear protein phosphatase 1 (FIG. 4) should, by promoting histone phosphorylation, favour hippocampal-dependent LTP and memory137,166.
Epigenetic control of gene transcription operates around a set point: DNA hyper- or hypomethylation and histone hypo- or hyperacetylation disrupts cogni-tion. Hence, pharmacotherapy must restore the balanced integration of multiple modes of epigenetic control that are requisite for appropriate cognitive performance. Specificity and safety are also vital issues. A particular concern is the risk of tumorigenic side effects, for example, resulting from hypermethylation-induced inactivation of tumour suppressor genes. Nonetheless, the targeting of methylation, acetylation and other epigenetic markers offers a potentially unique route for the correction of cog-nitive deficits in disorders such as ASD and schizophrenia.
Modulation of miRNA-controlled neural circuits. Further potential opportunities for restoring cognitive function are emerging from studies on brain-enriched microRNAs (miRNAs) that fine-tune cellular networks controlling synaptic plasticity and cognition, both developmen-tally and in adults168. For example, miR-134 inhibits hippocampal LTP and synaptic plasticity in mice by repressing CREB and BDNF synthesis169. The relevance to cognitive deficits in psychiatric disorders is supported by the observation that schizophrenia is associated with the abnormal biogenesis of miRNAs170. More specifi-cally, decreased levels of miR-219 may be due to NMDA receptor hypoactivity171. Furthermore, in fragile X syn-drome altered miRNA processing is implicated in the excessive mGluR5 signalling that contributes to cogni-tive impairment134,135,170.
With regard to the therapeutic exploitation of miR-NAs, one possibility could be to modify their biogen-esis via cellular signals that phosphorylate the proteins involved in their maturation81 (Supplementary informa-tion S2 (figure)) (FIG. 4). Another approach could be to recruit HDACs that suppress the synthesis of miR-134, thereby promoting cognition by preventing the inhibi-tion of CREB and BDNF generation169; furthermore, various classes of modified and stabilized oligonucleo-tides, and of mRNA analogues, have been designed to directly interfere with (or mimic) the activity of specific miRNAs170. Even if miRNA-targeted therapy appears to be a distant prospect at present, miRNA profiling may serve as a biomarker both of cognitive dysfunction and of the actions of pro-cognitive agents170.
Coupling pharmacotherapy with alternative strategies. As mentioned above, highly selective agents acting at a single target may be insufficient for the broad-based correction of cognitive deficits across pathogenetically distinct psychiatric disorders; this has encouraged inter-est in the development of multifunctional agents4. More
generally, it might be questioned whether the admin-istration of pro-cognitive agents (even in combina-tions) will always be adequate. Rather, a combination of pharmacotherapy with alternative strategies (BOX 5) may sometimes be a more effective strategy for palliat-ing cognitive deficits in psychiatric disorders, similarly to the combination of cognitive behavioural therapy and pharmacotherapy for improving the treatment of depression7,172–175.
For example, although they are only modestly active when administered alone, the α2-adrenergic receptor antagonist yohimbine and the NMDA receptor partial agonist d-cycloserine enhance the efficacy of behav-ioural extinction techniques for countering cognitive deficits associated with phobias, panic disorder and OCD106,176. Furthermore, d-cycloserine and oxytocin might be especially effective for treating social cogni-tive deficits in ASD when coupled with behavioural therapies that likewise reinforce social learning176,177 (Supplementary information S4 (box)). It may be instructive to extend such studies to cognitive remedia-tion therapy173 (BOX 5), as the effects of novel agents for relieving cognitive impairments in schizophrenia could best be expressed in synergy with this approach, rather than on top of antipsychotics that even interfere with their actions3,97,98,174.
Clinical development of pro-cognitive agentsAs highlighted above, many concepts are under explora-tion for countering cognitive dysfunction in psychiatric disorders. Their successful development depends on the careful translation of information acquired in animal and cellular models into clinical research6,8,11,68,69,97. Clinical studies of pro-cognitive agents can now use various techniques with experimental counterparts (FIG. 5) for: estimating optimal drug doses for efficacy; tracking cognitive actions in a manner complementary to behavioural rating scales; improving stratification (choice of subpopulations) of patients for drug trials; and exploring cerebral mechanisms of pro-cognitive properties.
Neuropsychological tests for evaluating cognitive func-tion in volunteers and patients. Many procedures are available for characterizing the influence of drugs on cognitive performance, and several test batteries have been proposed7,9,11,68,69,98,178,179 (BOX 2). Although these test batteries are instructive, one drawback is the potential loss of a large domain-specific effect in an overall (aver-age) non-effect. Furthermore, patient performance and test sensitivity can be compromised by poor engage-ment during long, tiring and stressful clinical trials6,9,10. Another issue is that separate cognitive domains may not be genuinely independent, as they interact with and depend on common neuronal substrates5,6,178,179 (FIGS 2,3). Notably, sustained, focused attention — together with high processing speed — is important for success in many procedures. These observations — and a com-mon interrelationship with intelligence — explain why measures of cognitive performance are often corre-lated5,6,97,178,179. It might even be suggested that a single
Rubinstein–Taybi syndromeA rare disorder characterized by autistic features, learning difficulties and poor attention. In approximately 50% of cases, it is caused by de novo mutations or deletions in the genes encoding CREB-binding protein or, rarely, histone acetyltransferase p300. These CREB-binding proteins and transcriptional co-activators are also histone acetylases, so patients display histone hypoacetylation and reduced gene transcription.
MicroRNAs(miRNAs). Small, non-protein- coding sequences (22–24 nucleotides) of RNA, mostly derived from intergenic regions, although some are found in introns. An individual species of miRNA can bind to the 3′-untranslated regions of up to hundreds of different species of mRNA. Translation is usually suppressed but it is sometimes enhanced, and in certain cases mRNA may even be degraded.
R E V I E W S
160 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

Therapeutic approach Influence on emotional symptoms*
Influence on cognitive impairment*
Psychiatric disorders targeted
Currently available pharmacotherapy –/0/++
Schizophrenia, depression, bipolar disorder, anxiety disorders
Deep-brain stimulation or electroconvulsive therapy 0/–+
Major depression
Repetitive transcranial magnetic stimulation 0/+0/+
Mainly depression (autism, schizophrenia)
Cognitive behavioural therapy 0+
Mainly depression (anxiety disorders)
Cognitive remediation therapy +0/+
Mainly schizophrenia (depression)
Exposure therapy for desensitization +0/+
Post-traumatic stress disorder, obsessive compulsive disorder, phobias, social anxiety disorders
Improved drugs (alone and in combination with above strategies)
++Dependent on mechanism of action
*The ‘+’ symbol corresponds to improvement; the ‘–’ symbol corresponds to worsening; and ‘0’ corresponds to no marked change.
dimension, such as working memory, would suffice to predict the overall effect of the drug on cognitive func-tion. However, this is both risky and contentious, so it is best to use multiple procedures to monitor the influence of drugs on baseline cognition and deficits in patients.
The importance of optimizing measurements of cog-nitive performance in volunteers and patients, and of optimizing comparability between preclinical and clini-cal procedures, is exemplified by the establishment of the MATRICS (Measurement and Treatment Research
Savant syndromeA rare syndrome that is closely associated with high- functioning autism spectrum disorder but also found in other developmental disorders and following damage to or disease of the central nervous system. It alludes to ‘islands of genius’ in one or a few cognitive domains such as mathematics despite broader deficits in others, and is usually associated with prodigious memory. Savant-like abilities can partially be reproduced by transcranial magnetic stimulation over the cortex.
Box 5 | Alternative therapeutic strategies to address cognitive impairment
Conventional psychotropic drugs were not specifically designed to target cognitive dysfunction in psychiatric disorders, and they display a range of (usually modest) beneficial, deleterious and neutral actions on cognition (see table). Non-pharmacotherapeutic strategies can be compared in terms of their differential impact on cognitive impairment versus emotional symptoms.
Despite showing efficacy in refractory depression, neither electroconvulsive therapy (which provokes transient retrograde amnesia) nor deep-brain stimulation of the subgenual cortex exerts a long-term, beneficial influence on cognition229,230. Conversely, despite having a less robust influence on depression, repetitive transcranial magnetic stimulation (rTMS) over the left dorsolateral prefrontal cortex (PFC) appears to promote attention, working memory and procedural learning. It probably acts by enhancing GABA (γ-aminobutyric acid)-ergic interneuron activity, γ-oscillations and PFC-driven top-down cognitive control227,230–233. Intriguingly, rTMS of the PFC enhanced γ-activity and cognitive processing in autism spectrum disorder, and elicited Savant syndrome-like cognitive feats in normal individuals233,234. In schizophrenia, enhanced cognition and suppression of auditory hallucinations has been reported230. Beneficial effects are not restricted to the PFC. Application of rTMS to the right parietal cortex favours focused attention, whereas rTMS over Broca’s or Wernickes’s areas improves verbal fluency and language learning; these effects mimic direct transcranial current stimulation — a technique that is used for rehabilitating patients with language and cognitive deficits following brain damage230,235.
Psychotherapy encompasses techniques such as problem solving, behavioural activation and cognitive behavioural therapy (CBT), which encourages patients to shed their negative views on themselves and their life. CBT for depression and anxiety can lead to durable improvements in mood (outlasting treatment), possibly by strengthening PFC-controlled top-down control of emotional processing in dysfunctional limbic structures like the amygdala and the hippocampus172,236. However, CBT has not yet been demonstrated to improve cognitive function. Conversely, cognitive remediation therapy (CRT), which adopts an approach that is broadly similar to cognitive training in elderly patients, can improve cognitive function in schizophrenia173,237 and possibly also in depression238. As for CBT, it is labour-intensive and treatment outcome depends on the therapist, methodology, content and intensity of the programme. Furthermore, CRT cannot be used for all patients and requires a regular commitment despite the low motivation of many patients with schizophrenia. Nonetheless, CRT is instructive as improvement is measured in terms of community functioning, such as salary, time spent at work and normalized social interactions.
Finally, exposure therapy is a cognitive approach for treating post-traumatic stress disorder, phobias, social anxiety disorder and obsessive compulsive disorders. Its application involves repeated contact with an aversive situation or stimulus to encourage desensitization (extinction)106,176,177.
Ideally, a novel pharmacotherapy should target both the emotional and cognitive impairments in psychiatric disorders. Moreover, agents should be developed and evaluated alone, in combination with other pro-cognitive mechanisms as well as in association with alternative therapies as a function of the symptom and patient population concerned. The combination of pharmacotherapy with CBT, CRT or similar approaches may be particularly effective for improving the cognitive impairments associated with psychiatric disorders174,175.
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 161
© 2012 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
EEG of ERPs:• MMN (50–150 milliseconds, pre-attentional)• N170 (170 milliseconds, facial processing)• P300 (300 milliseconds, attentional): amplitude and gating
Electromyography (eye-blink reflex):• Pre-attentional sensorimotor gating• Pre-pulse inhibition
Oculomotor neurophysiology:• Eye movements (saccades and antisaccades)• Smooth pursuit eye movements• Delayed responses
Magnetoencephalography event-related fields:• High spatial and temporal resolution, but mainly sources parallel to skull surface
PET and SPECT imaging:• Cerebral metabolism• Target (for example, GPCR) occupation• Transmitter release
Neuropsychological proceduresand batteries for evaluating drug influence on cognitive function
qEEG: spectral analysis and neuronal synchrony: • γ (30–80 Hz): local, cortical• θ (4–7 Hz): hippocampal and cortico-subcortical
fMRI (BOLD) measures of cerebral activity:• Performance of cognitive tasks• Default mode (resting state: task deactivated)
Magnetic resonance spectroscopy: • Glutamate, ACh and GABA transmission• NAA: energy, neuronal integrity• Fatty acids, neurogenesis
Measures of real-world functioning
to Improve Cognition in Schizophrenia) initiative for schizophrenia6,69,98,178,179 (BOX 2). The insights gained from this initiative could be exploited to set up com-parable programmes dedicated to improving cognition in depression. In an effort to better relate drug actions to everyday living, for both MATRICS and other pro-grammes, co-primary measures of functional capacity are being developed, including patient competence to undertake tasks such as shopping180,181. Improved predic-tion of the social and vocational outcomes of treatment is crucial, as relief of functional disability — not superior performance in test batteries — is the real goal. Notably, however, one tricky question is whether improved real-world functioning can be attributed to the cognitive and/or emotional actions of a specific treatment.
With regard to measures, patient self-assessment is desirable but can be misleading. Furthermore, although informants can reliably assess cognitive abilities of patients, this is not usually practical for drug trials6,180,181.
Finally, the design of long-term studies — for example, from the prodrome to diagnosis in schizophrenia — is far from simple. Shifts from baseline performance rather than changes versus placebo may be preferable, but practice effects following repeated testing must be considered6,182.
All clinical studies in the psychiatric domain com-mence with human volunteers. This population can deliver early feedback on how drugs potentially affect specific cognitive domains, how their effects may best be monitored in subsequent studies, and which under lying cerebral substrates they possibly engage — especially when neurocognitive tests are coupled to electro-encephalography (EEG) and neuroimaging (FIG. 5). Some observations may also be of more direct relevance to patient populations. For example, oxytocin consist-ently enhances social cognition in healthy probands as well as in autistic or schizophrenic individuals115–117. Acute administration of the NMDA receptor antagonist
Figure 5 | Overview of translational models for characterizing and predicting the influence of pharmacological agents on cognitive function in humans. Numerous procedures are available for assessing cognitive domains ranging from attention to social cognition, and several test batteries have been designed, such as the CANTAB (Cambridge Neuropsychological Test Automated Battery) core cognition test and the BACS (Brief Assessment of Cognition in Schizophrenia) test for schizophrenia, as well as the MATRICS (Measurement and Treatment Research to Improve Cognition in Schizophrenia)-derived consensus cognitive battery (BOX 2). Currently, efforts are being directed towards measures that are more closely linked to real-world cognitive functioning. Complementary approaches for exploring the actions of putative pro-cognitive agents in humans, most of which have translational counterparts in rodents, may be broadly classified as follows: quantitative electroencephalographical (qEEG) analysis coupled to spectral analysis of neural circuits, and EEG quantification of auditory event-related potentials (ERPs); magnetoencephalography, which monitors magnetic fields emitted by synchronized neurons with millisecond precision; sensorimotor paradigms, which include measures of eye movement; functional magnetic resonance imaging (fMRI) to estimate neuronal activity in defined brain regions; positron emission tomography (PET) and single-photon emission computed tomography (SPECT) to visualize radioligand binding; and magnetic resonance spectroscopy to quantify the levels of neuromediators. Many informative ERP-based signals can be translationally exploited. Notably, prepulse inhibition refers to the blunting of the startle reflex to an intense auditory stimulus following pre-exposure 100 milliseconds earlier to a subthreshold stimulus. Mismatch negativity (MMN) is a response to an (auditory or visual) oddball stimulus deviating from a regular sequence. Negative deflection (N170) is a 170-millisecond EEG signal that is associated with facial processing, whereas a positive 300-millisecond (P300) signal is associated with attention to a task-relevant, infrequent stimulus. ACh, acetylcholine; BOLD, blood oxygenation level-dependent; GABA, γ-aminobutyric acid; GPCR, G protein-coupled receptor; NAA, N-acetylaspartate.
R E V I E W S
162 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

ketamine to volunteers mimics the NMDA receptor hypoactivity observed in schizophrenia, so this pro cedure is useful for characterizing the influence of putative pro-cognitive antipsychotics on sensorimotor gating (see below) and cognition50,71,98. Furthermore, abnormalities in cognitive function and its underlying neural sub-strates can be probed in healthy individuals displaying variants of susceptibility genes for psychiatric disorders, such as zinc finger protein 804A (see above)72,79,80.
Nonetheless, several important limitations of Phase I studies should be mentioned. The influence of acute drug administration on baseline cognitive function in volunteers — and even its disruption by risk genes or pharmacological agents — may differ from its actions following chronic administration to patients with more complex pathologies. For example, drugs that are designed to normalize the aberrant epigenetic pro-gramming that causes cognitive dysfunction in ASD would be hard to evaluate in volunteers. Furthermore, many drugs developed for schizophrenia are intended for use in combinations, yet studies with other classes of agents cannot be undertaken in Phase I trials. More generally, the influence of drugs on cognitive per-formance in patients will reflect not only their direct impact on cognitive mechanisms but also their ability to relieve emotional symptoms, a facet largely inacces-sible in volunteers. Finally, studies in healthy individuals are inevitably restricted to neurocognitive procedures rather than genuine measures of real-world function. Without neglecting the importance of early-stage clinical studies, these points illustrate the risk of drawing prema-ture conclusions, and highlight that proof of concept can only come from studies in patients.
Oculomotor studies of the control of cognition. Eye-movement studies (FIG. 5) can be exploited to evaluate the influence of drugs on neural processes related to cogni-tion183–186. As few preclinical models of social cognition are recognized, oculomotor-tracking studies of eye gaze in primates as well as in humans — including attention to (and avoidance of) the gaze of others — are of particular interest184. Oculomotor paradigms have also been pro-posed for evaluating the effect of drugs on neural bases involved in sensorimotor gating, attention, working memory, executive function and procedural memory185. For example, oculomotor techniques in primates have been used to explore the influence of PFC populations of D1 receptors on working memory and top-down con-trol of visual processing185,186. Such observations provide a translational platform for clinical studies showing that oculomotor measures are sensitive to both the favourable and unfavourable effects of drugs such as antipsychotics and benzodiazepines on cognition183,185.
Underpinning interest in eye-moment studies in patients, oculomotor deficits have been reported in schiz-ophrenia and several other psychiatric disorders183–185. Eye-movement paradigms can also be coupled to neuro-imaging to characterize the influence of potential pro-cognitive drugs on cerebral circuits187. Although further work is needed to clarify the relationship between eye-movement measures and cognitive mechanisms, they
deserve further characterization as a distinctive trans-lational approach for exploring the actions of putative pro-cognitive drugs, especially with respect to social cognition.
Sensorimotor gating in relation to cognitive perfor-mance. Information processing and sensory gating — pre-attentional and attentional processes that are required for effective cognitive performance — can be monitored by various procedures7,71,98,188,189 (FIG. 5). These include pre-pulse inhibition (FIG. 5), which has marked dopaminergic, serotonergic and glutamatergic components. Accordingly, deficits in prepulse inhibition in patients with schizo-phrenia, and its disruption by psychotomimetic agents in volunteers, can be countered by antipsychotics7,68,71,189. Evaluation of new drugs, such as nicotinic acetylcholine receptor agonists and histamine H3 receptor antagonists, can be guided by their influence on the perturbation of prepulse inhibition in humans and rodents by psychoto-mimetics and (in the latter) by genetic and developmental models of schizophrenia52,71,97,98,139,140,189 (TABLE 2).
An additional pre-attentional response, ‘mismatch negativity’ (BOX 5), has a marked glutamatergic and NMDA receptor component, and deficits have so far been mainly seen in schizophrenia; moreover, impair-ment in high-risk individuals predicts onset of the disease7,71,190. In patients with schizophrenia mismatch negativity was enhanced by N-acetylcysteine, which elevates extracellular levels of glutamate in the PFC191. These observations highlight the interest in sensorimotor gating paradigms for exploring the effects of pro-cognitive drugs on pre-attentional and attentional function in both animals and in humans (FIG. 5).
Quantitative EEG for probing cognitive circuits. By directly probing large-scale electrical activity in the brain with high temporal resolution, quantitative EEG (qEEG) coupled to spectral analysis provides unique insights into the cortical processes underlying cognitive func-tion and affected both in psychiatric disorders and by pharmacotherapy46,47,188 (FIG. 5). EEG monitoring of cortical–subcortical networks in humans and animals is of particular interest because alterations in neural synchronization and connectivity are strongly related to cognitive deficits and their pharmacotherapeutic modu-lation46,47,192,193 (BOX 4). For example, oxytocin can shift cortical resources in volunteers to regions involved in social cognition and emotional processing194. EEG stud-ies are also instructive for characterizing the influence of drugs on arousal, sleep–wake cycles and sleep architec-ture, which can affect memory consolidation and other cognitive processes107.
The related technique of magnetoencephalography picks up magnetic fields generated by intraneuronal cur-rents to provide fine-grained (millisecond) temporal and spatial (superior to EEG) information on coordinated neuronal activity. It can also be used to characterize the relationship of cortical networks to cognition in psychi-atric disorders188. Supporting its use for drug characteri-zation, diazepam modifies θ- and γ-oscillations with a profile that is consistent with its negative influence on
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 163
© 2012 Macmillan Publishers Limited. All rights reserved

Functional magnetic resonance imaging(fMRI). A technique that exploits the differential paramagnetic properties of oxy- and deoxyhaemoglobin to estimate local cerebral blood oxygenation level-dependent (BOLD) activity. Increased oxygen supply compensates for (and transiently exceeds) energy needs, so the BOLD signal is proportional to neuronal activity. Interpretation of data is challenging as BOLD integrates changes both in neurons and in glia, pre- and postsynaptic changes in excitability, as well as local and upstream effects of drugs. Furthermore, BOLD signals can be affected by energy balance and haemodynamic parameters.
Graph theoryA mathematical approach for modelling complex networks whereby individual elements, like cerebral regions, neurons or cellular proteins, are considered as ‘nodes’ linked by ‘edges’. Brain graphs (derived from neuroimaging data) and cellular graphs (derived from studies of protein networks) reveal non-random topological properties such as modularity (clusters of nodes highly connected to each other) and hubs (nodes with numerous connections). These properties help to optimize network function, including cognitive processing.
cognition195. Low-resolution brain electromagnetic tomography provides three-dimensional information on electrical activity, affording further insights into the actions of pro-cognitive drugs and the allocation of resources to cognitive operations. It revealed a positive influence of modafinil on processing speed, and of psychotropic agents on network oscillations in relation to cognition123,196.
Functional magnetic resonance imaging: cognitive task-related and default modes. An important difference between functional magnetic resonance imaging (fMRI) and qEEG is the high spatial (but lower temporal) resolu-tion of fMRI, emphasizing complementary roles in drug characterization48,49,53,188,197. Although it is challenging to perform in animals (which must be anesthetized)7, fMRI is widely used in humans, and there is increasing interest in pharmacological fMRI to explore the actions of new drugs. It has, for example, been used to probe the influ-ence of antipsychotics and D1 receptor agonists on PFC connectivity in relation to cognition in schizophrenia198,199. Furthermore, α7 nicotinic acetylcholine receptor agonists have been shown to recruit hippocampal GABAergic interneurons200. Recent fMRI studies of the influence of oxytocin on social cognition suggest that it suppresses fear responses in the amygdala while enhancing insular medi-ation of empathy201. Interestingly, fMRI has shown that oxytocin and vasopressin influence cognitive and emo-tional processing via contrasting mechanisms of action, yet in each case involving the amygdala115,202. Despite its limitations7,197, pharmacological fMRI holds considerable promise for the prediction of therapeutic (and undesir-able) effects of pro-cognitive drugs, exploration of mecha-nisms of action and estimation of active doses, particularly when used in parallel with qEEG.
Alhough drug actions can be instructively evaluated while performing cognitive procedures, the human brain has an fMRI-accessible default mode of resting-state oper-ation that is deactivated by goal-directed tasks involving attention and executive function203. This default network includes the medial PFC, posterior cingulate, precuneus, angular gyrus and temporal lobes (FIG. 3), although other related circuits may also be involved203. Default-mode networks mirror introspection, mind-wandering and the theory of mind, as well as social and emotional process-ing, and are related to episodic memory and prospective cognition (Supplementary information S5 (box)). Default-mode network function is disrupted in ASD, schizophre-nia and other psychiatric disorders. In pharmacological fMRI studies, an α7 nicotinic acetylcholine receptor ago-nist enhanced default-mode function in schizophrenia, acting differently to the antipsychotic olanzapine204,205. Furthermore, modafinil enhanced task-related deactiva-tion in volunteers, which was consistent with enhanced processing speed and PFC-mediated cognitive control206.
Proton magnetic resonance spectroscopy. Paralleling neurochemical dialysis studies in rodents, proton mag-netic resonance spectroscopy probes glutamatergic, GABAergic and cholinergic transmission in humans, and also evaluates levels of N-acetylasparate, which is an index of neuronal integrity7,53,207 (FIG. 5). Three examples
illustrate its relevance to cognitive dysfunction in psychi-atric disorders and pro-cognitive drug characterization. First, in patients with schizophrenia, decreased levels of glutamate and N-acetylaspartate were correlated with cognitive deficits207,208. Second, the influence of antipsy-chotics on glutamate levels could be related to their mod-ulation of cognition, such as a modest beneficial effect of clozapine on verbal memory208,209. Third, a distinctive spectral peak of fatty acids that is characteristic of pro-genitor neuronal cells may reflect neurogenetic processes. This offers a parameter for clarifying the relationship of neurogenesis with the influence of drugs on cognition210.
PET and single-photon emission computed tomography. Like fMRI, positron emission tomography (PET) can be used to indirectly evaluate, with high (from seconds to minutes) temporal resolution, the influence of drugs on neuronal activity (glucose and/or oxygen utilization) in the PFC and other structures controlling cognition7,53,211. Alhough challenging to perform in animals7, PET is widely used in clinical trials to study the dose- and time-dependent occupation of regionally defined populations of GPCRs and other targets by drugs in relation to their influence on cognition. Appropriate radioligands are available for many sites controlling cognition, including dopamine D3 receptors, noradrenaline transporters and α4β2 nicotinic acetylcholine receptors211. The related technique of single-photon emission computed tomog-raphy generates three-dimensional images of the brain, although spatial resolution is usually less impressive7,53. It has, for example, been used to evaluate the influence of clozapine on cortico–striatal–thalamic pathways in relation to its influence on cognition209.
Translating translational research. Clearly, there is a pan-oply of techniques for guiding the clinical development of pro-cognitive agents and for optimizing the translational link from animals to the clinic. Time, expense and risk are key factors and it is impossible to perform all con-ceivable studies for any individual drug. Choosing the most appropriate and informative studies should help to reduce attrition by refining the choice of clinical indica-tion, cognitive domain and drug doses, thereby enhanc-ing the success rate of clinical trials. However, despite the sophistication of techniques — including PET, mag-netoencephalography, the insights from fMRI and EEG analyses, and the vital role of neurocognitive batteries — these are all essentially surrogate parameters. As noted above, it is crucial to show that pro-cognitive treatments lead to a functionally relevant enhancement of cognitive performance and an improved quality of life. That is, we must rise to the challenge of ‘translating translational research’ for the more effective development and clini-cal exploitation of improved drugs to treat the cognitive deficits associated with psychiatric disorders.
Concluding commentsAlhough historically there has been an emphasis on the motivational, affective and emotional symptoms of psychiatric disorders, cognitive impairment is just as prominent, persistent and disabling. In recent years we
R E V I E W S
164 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

have witnessed major advances in our understanding of the cellular and neuronal circuits controlling cognition, and of the causes of their perturbation in psychiatric disorders. In this regard, the notion of functional–structural networks and their disruption has emerged to be of particular importance. These insights, along with the improved linking of events integrated at the molecu-lar versus neural level, studies of epigenetic program-ming, identification of novel drug concepts and other advances underpin the hope that it should ultimately be possible to improve the poor cognitive performance in patients with psychiatric disorders. However, rigorous experimental validation of concepts and targets will be required, as well as the imaginative use of translational
procedures to maximize the chances of successful drug development and therapeutic exploitation.
As there is no unitary cause of cognitive impairment, and no single solution for its control, many promising lines of research should be pursued. Furthermore, improved treatment should be articulated around the notions of: uniting complementary mechanisms of pro-cognitive action (for example, with multitarget drugs); combining the benefits of pharmacotherapy with alter-native strategies; and addressing both the emotional and interrelated cognitive deficits associated with psychiatric disorders. Irrespective of the mode of therapy, a focus on genuine improvements in the real-world functioning of patients is essential.
1. Pessoa, L. On the relationship between emotion and cognition. Nature Rev. Neurosci. 9, 148–158 (2008).
2. Harmer, C. J. et al. Effect of acute antidepressant administration on negative affective bias in depressed patients. Am. J. Psychiatry 166, 1178–1184 (2009).
3. Hill, S. K., Bichop, J. R., Palumbo, D. & Sweeney, J. A. Effect of second-generation antipsychotics on cognition: current issues and future challenges. Expert Rev. Neurother. 10, 43–57 (2010).
4. Millan, M. J. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol. Ther. 110, 135–370 (2006).
5. Dickinson, D. & Harvey, P. D. Systemic hypotheses for generalized cognitive deficits in schizophrenia: a new take on an old problem. Schizophr. Bull. 35, 403–414 (2009).
6. Kalkstein, S., Hurford, I. & Gur, R. C. Neurocognition in schizophrenia. Curr. Top. Behav. Neurosci. 4, 373–390 (2010).
7. Millan, M. J. in Animal and Translational Models for CNS Drug Discovery Vol. 1 (eds McArthur, R. A. & Borsini, F.) 1–57 (Academic Press, Burlington, Massachusetts, 2008).
8. Baron-Cohen, S. & Belmonte, M. K. Autism: a window onto the development of the social and the analytic brain. Annu. Rev. Neurosci. 28, 109–126 (2005).
9. Barnett, J. H. et al. Assessing cognitive function in clinical trials of schizophrenia. Neurosci. Biobehav. Rev. 34, 1161–1177 (2010).
10. Hauber, W. & Sommer, S. Prefrontostriatal circuitry regulates effort-related decision making. Cereb. Cortex 19, 2240–2247 (2009).
11. Kas, M. J. H. et al. Advances in multidisciplinary and cross-species approaches to examine the neurobiology of psychiatric disorders. Eur. Neuropsychopharmacol. 21, 532–544 (2011).
12. Burdick, K. E., Robinson, D. G., Malhotra, A. K. & Szeszko, P. R. Neurocognitive profile analysis in obsessive-compulsive disorder. J. Int. Neuropsychol. Soc. 14, 640–645 (2008).
13. McNally, R. J. Cognitive abnormalities in post-traumatic stress disorder. Trends Cogn. Sci. 10, 271–277 (2006).
14. Liberzon, I. & Sripada, C. S. The functional neuroanatomy of PTSD: a critical review. Prog. Brain Res. 167, 151–169 (2008).
15. Quirk, G. J. & Mueller, D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33, 56–72 (2008).
16. Castaneda, A. E., Tuulio-Henriksson, A., Marttunen, M., Suvisaari, J. & Lönnqvist, J. A review on cognitive impairments in depressive and anxiety disorders with a focus on young adults. J. Affect. Disord. 106, 1–27 (2008).
17. Coles, M. E., Turks, C. L. & Heimberg, R. G. Memory bias for threat in generalized anxiety disorder: the potential importance of stimulus relevance. Cogn. Behav. Ther. 36, 65–73 (2007).
18. Gordeev, S. A. Cognitive functions and the state of nonspecific brain systems in panic disorders. Neurosci. Behav. Physiol. 38, 707–714 (2008).
19. Dere, E., Pause, B. M. & Pietrowsky, R. Emotion and episodic memory in neuropsychiatric disorders. Behav. Brain Res. 215, 162–171 (2010).
20. Brüne, M. Theory of mind in schizophrenia: a review of the literature. Schizophr. Bull. 31, 21–42 (2005).
21. Crow, T. J. The big bang theory of the origin of psychosis and the faculty of language. Schizophr. Res. 102, 31–52 (2008).
22. Galderisi, S. et al. Correlates of cognitive impairment in first episode schizophrenia: the EUFEST study. Schizophr. Res. 115, 104–114 (2009).
23. Kurtz, M. M. & Gerraty, R. T. A meta-analytic investigation of neurocognitive deficits in bipolar illness: profile and effects of clinical state. Neuropsychology 23, 551–562 (2009).
24. Wolf, F., Brüne, M. & Assion, H. J. Theory of mind and neurocognitive functioning in patients with bipolar disorder. Bipolar Disord. 12, 657–666 (2010).
25. Zobel, I. et al. Theory of mind deficits in chronically depressed patients. Depress. Anxiety 27, 821–828 (2010).
26. Marazziti, D., Consoli, G., Picchetti, M., Carlini, M. & Faravelli, L. Cognitive impairment in major depression. Eur. J. Pharmacol. 626, 83–86 (2010).
27. Beevers, C. G., Clasen, P., Stice, E. & Schnyer, D. Depression symptoms and cognitive control of emotion cues: a functional magnetic resonance imaging study. Neuroscience 167, 97–103 (2010).
28. Vaidya, C. J. & Stollstorff, M. Cognitive neuroscience of attention deficit hyperactivity disorder: current status and working hypotheses. Dev. Disabil. Res. Rev. 14, 261–267 (2008).
29. Uekermann, J. et al. Social cognition in attention-deficit hyperactivity disorder (ADHD). Neurosci. Biobehav. Rev. 34, 734–743 (2010).
30. Sayin, A., Oral, N., Utku, C. Baysak, E. & Candansayar, S. Theory of mind in obsessive-compulsive disorder: comparison with healthy controls. Eur. Psychiatry 25, 116–122 (2010).
31. Hill, E. L. & Frith, U. Understanding autism: insights from mind and brain. Phil. Trans. R. Soc. Lond. B Biol. Sci. 358, 281–289 (2003).
32. Robinson, S., Goddard, L. Dritschel, B., Wisley, M. & Howlin, P. Executive functions in children with autism spectrum disorders. Brain Cogn. 71, 362–368 (2009).
33. Krause, J., Ruxton, G. D. & Krause, S. Swarm intelligence in animals and humans. Trends Ecol. Evol. 25, 28–34 (2010).
34. Adolphs, R. The social brain: neural basis of social knowledge. Annu. Rev. Psychol. 60, 693–716 (2009).
35. Fitch, W. T., Huber, L. & Bugnyar, T. Social cognition and the evolution of language: constructing cognitive phylogenies. Neuron 65, 795–814 (2010).
36. Gorwood, P., Corruble, E., Falissard, B. & Goodwin, G. M. Toxic effects of depression on brain function: impairment of delayed recall and the cumulative length of depressive disorder in a large sample of depressed outpatients. Am. J. Psychiatry 165, 731–739 (2008).
37. Goodwin, G. M., Martinez-Aran, A., Glahn, D. C. & Vieta, E. Cognitive impairment in bipolar disorder: neurodevelopment of neurodegeneration? An ECNP expert meeting report. Eur. Neuropsychopharmacol. 18, 787–793 (2008).
38. Sarter, M., Parikh, V. & Howe, W. M. nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem. Pharmacol. 10, 658–667 (2009).
39. McAfoose, J. & Baune, B. T. Evidence for a cytokine model of cognitive function. Neurosci. Biobehav. Rev. 33, 355–366 (2009).
40. Cunha, C., Brambilla, R. & Thomas, K. L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 3, 1 (2010).
41. Robbins, T. W. & Arnsten, A. F. T. The neuropsycho-pharmacology of fronto-executive function: monoaminergic modulation. Annu. Rev. Neurosci. 32, 267–287 (2009).
42. Lewis, D. A., Fish, K. N., Arion, D. & Gonzalez-Burgos, G. Perisomatic inhibition and cortical circuit dysfunction in schizophrenia. Curr. Neurobiol. 21, 866–872 (2011).
43. Lee, Y. S. & Silva, A. J. The molecular and cell biology of enhanced cognition. Nature Rev. Neurosci. 10, 126–140 (2009).
44. Neves, G., Cooke, S. F. & Bliss, T. V. P. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nature Rev. Neurosci. 9, 65–75 (2008).
45. Collingridge, G. L., Peineau, S., Howland, J. G. & Wang, Y. T. Long-term depression in the CNS. Nature Rev. Neurosci. 11, 459–473 (2010).
46. Buzsaki, G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron 68, 362–385 (2010).
47. Wang, X. J. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol. Rev. 90, 1195–1268 (2010).
48. Bullmore, E. & Sporns, O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nature Rev. Neurosci. 10, 186–198 (2009).
49. Lynall, M. E. et al. Functional connectivity and brain networks in schizophrenia. J. Neurosci. 30, 9477–9487 (2010).
50. Gilmour, G. et al. NMDA receptors, cognition and schizophrenia — testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology 21 Mar 2011 (doi:10.1016/j.neuropharm.2011.03.015).
51. Heifets, B. D. & Castillo, P. E. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306 (2009).
52. Papaleo, F., Lipska, B. K. & Weinberger, D. R. Mouse models of genetic effects on cognition: relevance to schizophrenia. Neuropharmacology 5 May 2011 (doi:10.1016/j.neuropharm.2011.04.025).
53. McGuire, P., Howes, O. D., Stone, J. & Fusar-Poli, P. Functional neuroimaging in schizophrenia: diagnosis and drug discovery. Trends Pharmacol. Sci. 29, 91–98 (2008).
54. Minzenberg, M. J. et al. Meta-analysis of 41 functional neuroimaging studies of executive function in schizo-phrenia. Arch. Gen. Psychiatry 66, 811–822 (2009).
55. Pettersson-Yeo, W., Allen, P., Benetti, S., McGuire, P. & Mechelli, A. Dysconnectivity in schizophrenia: where are we now? Neurosci. Biobehav. Rev. 35, 1110–1124 (2011).
56. Li, X., Branch, C. A. & DeLisi, L. E. Language pathway abnormalities in schizophrenia: a review of fMRI and other imaging studies. Curr. Opin. Psychiatry 22, 131–139 (2009).
57. Knaus, T. A. et al. Language laterality in autism spectrum disorder and typical controls: a functional, volumetric, and diffusion tensor MRI study. Brain Lang. 112, 113–120 (2010).
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 165
© 2012 Macmillan Publishers Limited. All rights reserved

58. Wass, S. Distortions and disconnections: disrupted brain connectivity in autism. Brain Cogn. 75, 18–28 (2011).
59. Kennedy, D. P. & Courchesne, E. The intrinsic functional organization of the brain is altered in autism. Neuroimage 39, 1877–1885 (2008).
60. Cubillo, A. & Rubia, K. Structural and functional brain imaging in adult attention-deficit/hyperactivity disorder. Expert Rev. Neurother. 10, 603–620 (2010).
61. Harrison, B. J. et al. Altered corticostriatal functional connectivity in obsessive-compulsive disorder. Arch. Gen. Psychiatry 66, 1189–1200 (2009).
62. Van Marle, H. J. F., Hermans, E. J., Qin, S. & Fernandez, G. Enhanced resting-state connectivity of amygdala in the immediate aftermath of acute psychological stress. Neuroimage 53, 348–354 (2010).
63. Ehninger, D., Li, W., Fox, K., Stryker, M. P. & Silva, A. J. Reversing neurodevelopmental disorders in adults. Neuron 60, 950–960 (2008).
64. Belsky, J. et al. Vulnerability genes or plasticity genes? Mol. Psychiatry 14, 746–754 (2009).
65. Penrod, N. M., Cowper-Sallari, R. & Moore, J. H. Systems genetics for drug target discovery. Trends Pharmacol. Sci. 32, 623–630 (2011).
66. Liu, J. et al. Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Hum. Brain Mapp. 30, 241–255 (2009).
67. Jaaro-Peled, H. J. et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495 (2009).
68. Markou, A., Chiamulera, C., Geyer, M. A., Tricklebank, M. & Steckler, T. Removing obstacles in neuroscience drug discovery: the future path for animal models. Neuropsychopharmacology 34, 74–89 (2009).
69. Young, J. W., Powell. S. B., Risbrough, V., Marston, H. M. & Geyer, M. A. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol. Ther. 122, 150–202 (2009).
70. Balanzá-Martínez, V. et al. Neurocognitive endophenotypes (endophenocognotypes) from studies of relatives of bipolar disorder subjects: a systematic review. Neurosci. Biobehav. Rev. 32, 1426–1438 (2008).
71. Turetsky, B. I. et al. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr. Bull. 33, 64–94 (2007).
72. Walter, H. et al., Effects of a genome-wide supported psychosis risk variant on neural activation during a theory-of-mind task. Mol. Psychiatry 16, 462–470 (2011).
73. Mosconi, M. M. et al. Neurobehavioral abnormalities in first-degree relatives of individuals with autism. Arch. Gen. Psychiatry 67, 830–840 (2010).
74. Chamberlain, S. R. & Menzies, L. Endophenotypes of obsessive-compulsive disorder: rationale, evidence and future potential. Expert. Rev. Neurother. 9, 1133–1146 (2009).
75. Apud, J. A. & Weinberger, D. R. Treatment of cognitive deficits associated with schizophrenia: potential role of catechol-O-methyltransferase inhibitors. CNS Drugs 21, 535–557 (2007).
76. Roussos, P., Giakoumaki, S. G. & Bitsios, P. Tolcapone effects on gating, working memory, and mood interact with the synonymous catechol-O-methyltransferase rs4818C/G polymorphism. Biol. Psychiatry 66, 997–1004 (2009).
77. Bertolino, A. et al. Prefrontal–hippocampal coupling during memory processing is modulated by COMT Val158Met genotype. Biol. Psychiatry 60, 1250–1258 (2006).
78. Meyer-Lindenberg, A. et al. Genetic evidence implicating DARPP-32 in human frontostriatal structure, function, and cognition. J. Clin. Invest. 117, 672–682 (2007).
79. Esslinger, C. et al. Cognitive state and connectivity effects of the genome-wide significant psychosis variant in ZNF804A. Neuroimage 54, 2514–2523 (2011).
80. Hashimoto, R. et al. The impact of a genome-wide supported psychosis variant in the ZNF804A gene on memory function in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 5, 153B, 1459–1464 (2010).
81. Karayiorgou, M., Simon, T. J. & Gogos, J. A. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Rev. Neurosci. 11, 402–416 (2010).
82. Sigurdsson, T., Stark, K. L., Karayiorgou, M., Gogos, J. A. & Gordon, J. A. Impaired hippocampal–prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 464, 763–767 (2010).
83. Joëls, M. & Baram, T. Z. The neuro-symphony of stress. Nature Rev. Neurosci. 10, 459–466 (2009).
84. Lupien, S. J., McEwen, B. S., Gunnar, M. R. & Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nature Rev. Neurosci. 10, 434–445 (2009).
85. Wang, X. D. et al. Forebrain CRF1 modulates early-life stress-programmed cognitive deficits. J. Neurosci. 21, 13625–13634 (2011).
86. Schwabe, L., Wolf, O. T. & Oitzl, M. S. Memory formation under stress: quantity and quality. Neurosci. Biobehav. Rev. 34, 584–591 (2010).
87. Howland, J. G. & Wang, Y. T. Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog. Brain Res. 169, 145–158 (2008).
88. Mailliet, F. et al. Protection of stress-induced impairment of hippocampal/prefrontal LTP through blockade of glucocorticoid receptors: implication of MEK signalling. Exp. Neurol. 211, 593–596 (2008).
89. Sandi, C. Glucocorticoids act on glutamatergic pathways to affect memory processes. Trends Neurosci. 34, 165–171 (2011).
90. Sotiropoulos, I. et al. Stress and glucocorticoid footprints in the brain — the path from depression to Alzheimer’s disease. Neurosci. Biobehav. Rev. 32, 1161–1173 (2008).
91. Dorey, R. et al. Membrane mineralocorticoid but not glucocorticoid receptors of the dorsal hippocampus mediate the rapid effects of corticosterone on memory retrieval. Neuropsychopharmacology 36, 2639–2649 (2011).
92. Cerqueira, J. J., Maillet, F., Almeida, O. F., Jay, T. M. & Sousa, N. The prefrontal cortex as a key target of the maladaptive response to stress, J. Neurosci. 27, 2781–2787 (2007).
93. Holmes, A. & Wellman, C. L. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neurosci. Biobehav. Rev. 33, 773–783 (2009).
94. Roozendaal, B., McEwen, B. S. & Chattarji, S. Stress, memory and the amygdala. Nature Rev. Neurosci. 10, 423–433 (2009).
95. Oomen, C. A. et al. Early maternal deprivation affects dentate gyrus structure and emotional learning in adult female rats. Psychopharmacology 214, 249–260 (2011).
96. Champagne, D. L. et al. Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 28, 6037–6045 (2008).
97. Millan, M. J. & Brocco, M. Cognitive impairment in schizophrenia: a review of developmental and genetic models, and pro-cognitive profile of the optimized D3 > D2 antagonist, S33138. Thérapie 63, 187–229 (2008).
98. Barch, D. M. Pharmacological strategies for enhancing cognition in schizophrenia. Curr. Top. Behav. Neurosci. 4, 43–96 (2010).
99. Margulies, C., Tully, T. & Dubnau, J. Deconstructing memory in Drosphila. Curr. Biol. 15, R700–R713 (2005).
100. Hawkins, R. D., Kandel, E. R. & Bailey, C. H. Molecular mechanisms of memory storage in Aplysia. Biol. Bull. 210, 174–191 (2006).
101. Champagne, D. L., Hoefnagels, C. C. M., de Kloet, R. E. & Richardson, M. K. Translating rodent behavioral repertoire to zebrafish (Danio rerio); relevance for stress research. Behav. Brain Res. 214, 332–342 (2010).
102. Emery, N. J. & Clayton, N. S. Comparative social cognition. Annu. Rev. Psychol. 60, 87–113 (2009).
103. Bolhuis, J. J., Okanoya, K. & Scharff, C. Twitter evolution: converging mechanisms in birdsong and human speech. Nature Rev. Neurosci. 11, 747–759 (2010).
104. Wallace, T., Ballard, T. M., Pouzet, B., Riedel, W. J. & Wettstein, J. G. Drug targets for cognitive enhancement in neuropsychiatric disorders. Pharmacol. Biochem. Behav. 99, 130–145 (2011).
105. Levin, E. D., Bushnell, P. J. & Rezvani, A. H. Attention-modulating effects of cognitive enhancers. Pharmacol. Biochem. Behav. 99, 146–154 (2011).
106. Kaplan, G. B. & Moore, K. A. The use of cognitive enhancers in animal models of fear extinction. Pharmacol. Biochem. Behav. 99, 217–228 (2011).
107. Poe, G. R., Walsh, C. M. & Bjorness, T. E. Cognitive neuroscience of sleep. Prog. Brain Res. 185, 1–19 (2010).
108. Kyriacou, C. P. & Hastings, M. H. Circadian clocks: gene, sleep, and cognition. Trends Cogn. Sci. 14, 259–267 (2010).
109. Solas, M. et al. Interactions between age, stress and insulin on cognition: implications for Alzheimer’s disease. Neuropsychopharmacology 35, 1664–1673 (2010).
110. El-Ghundi, M., O’Dowd, B. F. & George, S. R. Insights into the role of dopamine receptor systems in learning and memory. Rev. Neurosci. 18, 37–66 (2007).
111. Buchanan, R. W. et al. A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol. Psychiatry 69, 442–449 (2010).
112. Graef, S., Schönknecht, P., Sabri, O. & Hegerl, U. Cholinergic receptor subtypes and their role in cognition, emotion, and vigilance control: an overview of preclinical and clinical findings. Psychopharmacology, 215, 205–229 (2011).
113. King, M. V., Marsden, C. A. & Fone, K. C. A role for the 5-HT1A, 5-HT4 and 5-HT6 receptors in learning and memory. Trends Pharmacol. Sci. 29, 482–492 (2008).
114. Codony, X., Vela, J. M. & Ramirez, M. J. 5-HT6 receptors and cognition. Curr. Opin. Pharmacol. 11, 94–100 (2011).
115. Goodson, J. L. & Thompson, R. R. Nonapeptide mechanisms of social cognition, behavior and species-specific social systems Curr. Opin. Neurobiol. 20, 784–794 (2010).
116. Insel, T. R. The challenge of translation in social neuroscience: a review of oxytocin, vasopressin, and affiliative behaviour. Neuron 65, 768–779 (2010).
117. Meyer-Lindenberg, A., Domes, G., Kirsch, P. & Heinrichs, M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nature Rev. Neurosci. 12, 524–538 (2011).
118. Garnock-Jones, K. P. & Keating, G. M. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr. Drugs 11, 203–226 (2011).
119. Ramos, B. P. & Arnsten, A. F. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol. Ther. 113, 523–536 (2007).
120. Sallee, F. R. The role of α2-adrenergic agonists in attention-deficit/hyperactivity disorder. Postgrad. Med. 122, 78–87 (2010).
121. Liem-Moolenaar, M. et al. The effects of the glycine reuptake inhibitor R213129 on the central nervous system and on scopolamine-induced impairments in psychomotor and cognitive function in healthy subjects. J. Psychopharmacol. 24, 1671–1679 (2010).
122. Saavedra-Velez, C., Yusim, A., Anbarasan, D. & Lindenmayer, J. P. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J. Clin. Psychiatry 70, 104–112 (2009).
123. Saletu, M. et al. Modafinil improves information processing speed and increases energetic resources for orientation of attention in narcoleptics: double-blind, placebo-controlled ERP studies with low-resolution brain electromagnetic tomography (LORETA). Sleep Med. 10, 850–858 (2009).
124. Uslaner, J. M. et al. Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharmacology 57, 531–538 (2009).
125. Zhang, Z., Gong, N., Wang, W., Xu, L. & Xu, T. L. Bell-shaped d-serine actions on hippocampal long-term depression and spatial memory retrieval. Cereb. Cortex 18, 2391–2401 (2008).
126. Williams, G. V. & Castner, S. A. Under the curve: critical issues for elucidating D1 receptor function in working memory. Neuroscience 139, 263–276 (2006).
127. Deng, W., Aimone, J. B. & Gage, F. H. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nature Rev. Neurosci. 11, 339–350 (2010).
128. Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A. & Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129 (2010).
129. Mansuy, I. M. & Shenolikar, S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 29, 689–696 (2006).
R E V I E W S
166 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

130. Pertovaara, A., Haapalinna, A., Sirviö, J. & Virtanen, R. Pharmacological properties, central nervous system effects, and potential therapeutic applications of atipamezole, a selective α2-adrenoceptor antagonist. CNS Drug Rev. 11, 273–288 (2005).
131. Lapiz, M. D. S. & Morilak, D. A. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience 137, 1039–1049 (2006).
132. Vellano, C. P., Lee, A. E., Dudek, S. M. & Hepler, J. R. RGS14 at the interface of hippocampal signaling and synaptic plasticity. Trends Pharmacol. Sci. 32, 666–674 (2011).
133. Vinkers, C. H. et al. The inhibitory GABA system as a therapeutic target for cognitive symptoms in schizophrenia: investigational agents in the pipeline. Expert Opin. Investig. Drugs 19, 1217–1233 (2010).
134. Dölen, G., Carpenter, R. L., Ocain, T. D. & Bear, M. F. Mechanism-based approaches to treating fragile X. Pharmacol. Ther. 127, 78–93 (2010).
135. Lüscher, C. & Huber, K. M. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459 (2010).
136. Hoeffer, C. A. & Klann, E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 (2010).
137. Franklin, T. & Mansuy, I. M. Epigenetic inheritance in mammals: evidence for the impact of adverse environmental effects. Neurobiol. Dis. 39, 61–65 (2010).
138. Day, J. J. & Sweatt, J. D. Epigenetic treatments for cognitive impairments. Neuropsychopharmacology 18 May 2011 (doi:10.1038/npp.2011.85).
139. Raddatz, R., Tao, M. & Hudkins, R. L. Histamine H3 antagonists for treatment of cognitive deficits in CNS diseases. Curr. Top. Med. Chem. 10, 153–169 (2010).
140. Leiser, S. C., Bowlby, M. R., Comery, T. A. & Dunlop, J. A cog in cognition: how the α7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol. Ther. 122, 302–311 (2009).
141. Reneerkens, O. A. H., Rutten, K., Steinbusch, H. W. M., Blokland, A. & Prickaerts, J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology 202, 419–443 (2009).
142. Schmidt, C. J. Phosphodiesterase inhibitors as potential cognition enhancing agents. Curr. Top. Med. Chem. 10, 222–230 (2010).
143. Burgin, A. B. et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nature Biotech. 28, 63–70 (2010).
144. Sun, M. K. & Alkon, D. L. Pharmacology of protein kinase C activators: cognition-enhancing and antidementic therapeutics. Pharmacol. Ther. 127, 66–77 (2010).
145. Levallet, G., Hotte, M., Boulouard, M. & Dauphin, F. Increased particulate phosphodiesterase 4 in the prefrontal cortex supports 5-HT4 receptor-induced improvement of object recognition memory in the rat. Psychopharmacology 202, 125–139 (2009).
146. Puighemanal, E. et al. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nature Neurosci. 12, 1152–1158 (2009).
147. Gafford, G. M., Parsons, R. G. & Helmstette, F. J. Consolidation and reconsolidation of contextual fear memory requires mammalian target of rapamycin-dependent translation in the dorsal hippocampus. Neuroscience 182, 98–104 (2011).
148. Dewachter, I. et al. GSK3β, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol. Dis. 35, 193–200 (2009).
149. Hooper, C. et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86 (2007).
150. Taglialatela, G., Hogan, D., Zhang, W. R. & Dineley, K. T. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav. Brain Res. 200, 95–99 (2009).
151. Baumgärtel, K. et al. Control of the establishment of aversive memory by calcineurin and Zif268. Nature Neurosci. 11, 572–578 (2009).
152. Pittenger, C. & Duman, R. S. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109 (2008).
153. Lacefield, C. O., Itskov, V., Reardon, T., Hen, R. & Gordon, J. A. Effects of adult-generated granule cells on coordinated network activity in the dentate gyrus. Hippocampus 29 Sep 2010 (doi:10.1002/hipo.20860).
154. Rolls, E. T. A computational theory of episodic memory formation in the hippocampus. Behav. Brain Res. 215, 180–196 (2010).
155. Wixted, J. T. & Squire, L. R. The medial temporal lobe and the attributes of memory. Trends Cogn. Sci. 15, 210–217 (2011).
156. Carolis, N. A. & Eisch, A. J. Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology 58, 884–893 (2011).
157. Bramham, C. R. et al. The Arc of synaptic memory. Exp. Brain Res. 200, 125–140 (2010).
158. Minichiello, L. TrkB signalling pathways in LTP and learning. Nature Rev. Neurosci. 10, 850–860 (2009).
159. Peters, J., Dieppa-Perea, L. M., Melendez, L. M. & Quirk, G. J. Induction of fear extinction with hippocampal-infralimbic BDNF. Science 328, 1288–1290 (2010).
160. Wilkinson, L. S., Davies, W. & Isles, A. R. Genomic imprinting effects on brain development and function. Nature Rev. Neurosci. 8, 832–843 (2007).
161. Gregory, S. G. et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7, 62 (2009).
162. Zhang, T. Y. & Meaney, M. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 61, 439–466 (2010).
163. Guidotti, A. et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology, 60, 1007–1016 (2010).
164. Kundakovic, M., Chen. Y., Guidotti, A. & Grayson, D. R. The reelin of GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol. Pharmacol. 75, 342–354 (2009).
165. Guan, J. S. et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 (2009).
166. Koshibu, K., Gräff, J. & Mansuy, I. M. Nuclear protein phosphate-1: an epigenetic regulator of fear memory and amygdala long-term potentiation. Neuroscience 173, 30–36 (2011).
167. Torrioli, M. et al. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am. J. Med. Genet. A 152A, 1420–1427 (2010).
168. Fischbach, S. J. & Carew, T. J. MiRNAs in memory processing. Neuron 63, 714–716 (2009).
169. Gao, J. et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109 (2010).
170. Hunsberger, J. G., Austin, D. R., Chen, G. & Manji, H. K. MiRNAs in mental health: from biological under-pinnings to potential therapies. Neuromol. Med. 11, 173–182 (2009).
171. Kocerha, J. et al. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc. Natl Acad. Sci. USA 106, 3507–3512 (2009).
172. DeRubeis, R. J., Siegle, G. J. & Hollon, S. D. Cognitive therapy versus medication for depression: treatment outcomes and neural mechanisms. Nature Rev. Neurosci. 9, 788–796 (2008).
173. Medalia, A. & Choi, J. Cognitive remediation in schizophrenia. Neuropsychol. Rev. 19, 353–364 (2009).
174. Swerdlow, N. R. Are we studying and treating schizophrenia correctly? Schizophr. Res. 130, 1–10 (2011).
175. Cuijpers, P., van Staten, A., Hollon, S. D. & Andersson, G. The contribution of active medication to combined treatments of psychotherapy and pharmacotherapy for adult depression: a meta-analysis. Acta Psychiatr. Scand. 19, 1–9 (2009).
176. Myers, K. M., Carlezon, W. A. & Davis, M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology 36, 274–293 (2011).
177. Modi, M. E. & Young, L. J. d-cycloserine facilitates socially reinforced learning in an animal model relevant to autism spectrum disorders. Biol. Psychiatry 70, 298–304 (2011).
178. Keefe, R. S. et al. Characteristics of the MATRICS consensus cognitive battery in a 29-site antipsychotic schizophrenia clinical trial. Schizophr. Res. 125, 161–168 (2010).
179. Carter, C. S. & Barch, D. M. Cognitive neuroscience-based approaches to measuring and improving treatment effects on cognition in schizophrenia: the CNTRICS initiative. Schizophr. Bull. 33, 1131–1137 (2007).
180. Heinrichs, R. W., Ammari, N., Miles, A. A. & McDermid Vaz, S. Cognitive performance and functional competence as predictors of community independence in schizophrenia. Schizophr. Bull. 36, 381–387 (2010).
181. Leifker, F. R., Patterson, T. L., Heaton, R. K. & Harvey, P. D. Validating measures of real-world outcome: the results of the VALERO expert survey and RAND panel. Schizophr. Bull. 37, 334–343 (2009).
182. Green, M. F., Kerns, S. R. & Heaton, R. K. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr. Res. 72, 45–51 (2004).
183. Hill, S. K., Reilly, J. L., Harris, M. S. H., Khine, T. & Sweeney, J. A. Oculomotor and neuropsychological effects of antipsychotic treatment for schizophrenia. Schizophr. Bull. 34, 494–506 (2008).
184. Luna, B., Velanova, K. & Geier, C. F. Development of eye-movement control. Brain Cogn. 68, 293–308 (2008).
185. Reilly, J. L., Lencer, R., Bishop, J., Keedy, S. & Sweeney, J. A. Pharmacological studies of eye movement control. Brain Cogn. 68, 415–435 (2008).
186. Noudoost, B. & Moore, T. Control of visual cortical signals by prefrontal dopamine. Nature 474, 375–379 (2011).
187. Keedy, S. K. et al. An fMRI study of visual attention and sensorimotor function before and after antipsychotic treatment in first episode schizophrenia. Psychiatry Res. 172, 16–23 (2009).
188. Sakkalis, V. Applied strategies towards EEG/MEG biomarker identification in clinical and cognitive research. Biomark. Med. 5, 93–105 (2011).
189. Braff, D. L. & Light, G. A. Preattentional and attentional cognitive deficits as targets for treating schizophrenia. Psychopharmacology 174, 175–185 (2004).
190. Bodatsch, M. et al. Prediction of psychosis by mismatch negativity. Biol. Psychiatry 69, 959–966 (2011).
191. Lavoie, S. et al. Glutathione precursor, N-acetyl-cysteine, improved mismatch negativity in schizophrenia patients. Neuropsychopharmacology 33, 2187–2199 (2008).
192. Fell, J. & Axmacher, N. The role of phase synchronisation in memory processes. Nature Rev. Neurosci. 12, 105–118 (2011).
193. Whittington, M. A., Cunningham, M. O., LeBeau, F. E. N., Racca, C. & Traub, R. D. Multiple origins of the cortical γ rhythm. Dev. Neurobiol. 71, 92–106 (2010).
194. Perry, A. et al. Intranasal oxytocin modulates EEG mu/alpha and beta rhythms during perception of biological motion. Psychoneuroendocrinology 35, 1446–1453 (2010).
195. Hali, S. D., Barnes, G. R., Furlong, P. L., Seri, S. & Hillebrand, A. Neuronal network pharmacodynamics of GABAergic modulation in the human cortex determined using pharmacomagnetoencephalography. Hum. Brain Mapp. 31, 581–594 (2010).
196. Higuchi, Y. et al. Electrophysiological basis for the ability of olanzapine to improve verbal memory and functional outcome in patients with schizophrenia: a LORETA analysis of P300. Schizophr. Res. 101, 320–330 (2008).
197. Murphy, S. E. & Mackay, C. E. Using MRI to measure drug action: caveats and new directions. J. Psychopharmacol. 25, 1168–1174 (2011).
198. Mu, Q. et al. A single 20 mg dose of the full D1 dopamine agonist dihydrexidine (DAR-0100) increases prefrontal perfusion in schizophrenia. Schizophr. Res. 94, 332–341 (2007).
199. Lui, S. et al. Short-term effects of antipsychotic treatment on cerebral function in drug-naive first-episode schizophrenia revealed by “resting state” functional magnetic resonance imaging. Arch. Gen. Psychiatry 67, 783–792 (2010).
200. Tregellas, J. R. et al. Functional magnetic resonance imaging of effects of a nicotinic agonist in schizophrenia. Neuropsychopharmacology 35, 938–942 (2010).
201. Riem, M. M. et al. Oxytocin modulates amygdala, insula, and inferior frontal gyrus responses to infant crying: a randomized controlled trial. Biol. Psychiatry 70, 291–297 (2011).
202. Garner, M., Zurowski, B. & Büchel, C. Different amygdala subregions mediate valence-related and attentional effects of oxytocin in humans. Proc. Natl Acad. Sci. USA 107, 9400–9405 (2010).
R E V I E W S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 11 | FEBRUARY 2012 | 167
© 2012 Macmillan Publishers Limited. All rights reserved

203. Broyd, S. J. et al. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci. Biobehav. Rev. 33, 279–296 (2009).
204. Sambataro, P. et al. Treatment with olanzapine is associated with modulation of the default mode network in patients with schizophrenia. Neuropsychopharmacology 35, 904–912 (2010).
205. Tregellas, J. R. et al. Effects of an α7-nicotinic agonist on default network activity in schizophrenia. Biol. Psychiatry 69, 7–11 (2011).
206. Minzenberg, M. J., Yoon, J. H. & Carter, C. S. Modafinil modulation of the default mode network. Psychopharmacology 215, 23–31 (2011).
207. Szulc, A. et al. Proton magnetic resonance spectroscopy study of brain metabolite changes after antipsychotic treatment. Pharmacopsychiatry 44, 148–157 (2011).
208. Bustillo, J. R. Glutamate as a marker of cognitive function in schizophrenia: a proton spectroscopic imaging study at 4 Tesla. Biol. Psychiatry 69, 19–27 (2011).
209. Ertugrul, A. et al. The effect of clozapine on regional cerebral blood flow and brain metabolite ratios in schizophrenia: relationship with treatment response. Psychiatry Res. 174, 121–129 (2009).
210. Manganas, L. N. et al. Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science 318, 980–985 (2007).
211. Vyas, N. S., Patel, N. H., Nijran, K. S., Al-Nahhas, A. & Puri, B. K. The use of PET imaging in studying cognition, genetics and pharmacotherapeutic interventions in schizophrenia. Expert Rev. Neurother. 11, 37–51 (2011).
212. Van Overwalle, F. & Baetens, K. Understanding others’ actions and goals by mirror and mentalizing systems: a meta-analysis. Neuroimage 48, 564–584 (2009).
213. Young, L. J. & Wang, Z. The neurobiology of pair bonding. Nature Neurosci. 7, 1048–1054 (2004).
214. Hermann, E., Call, J., Hernandez-Lloreda, M. V., Hare, B. & Tomasello, M. Humans have evolved specialized skills to social cognition: the cultural intelligence hypothesis. Science 317, 1360–1366 (2007).
215. Carruthers, P. The cognitive functions of language. Behav. Brain Sci. 25, 657–674 (2002).
216. Nelson, B. & Rawlings, D. Relating schizotypy and personality to the phenomenology of creativity. Schizophr. Bull. 36, 388–399 (2010).
217. Hart, B. L., Hart, L. A. & Pinter-Wollman, N. Large brains and cognition: where do elephants fit in? Neurosci. Biobehav. Rev. 32, 86–98 (2008).
218. Premack, D. Human and animal cognition: continuity and discontinuity. Proc. Natl Acad. Sci. USA 104, 13861–13867 (2007).
219. McGraw, L. A. & Young, L. J. The prairie vole: an emerging model organism for understanding the social brain. Trends Neurosci. 33, 106–109 (2009).
220. Scattoni, M. L., Crawley, J. & Ricceri, L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 33, 508–515 (2009).
221. Arakawa, H., Blanchard, D. C., Arakawa, K., Dunlap, C. & Blanchard, R. J. Scent marking behavior as an odorant communication in mice. Neurosci. Biobehav. Rev. 32, 1236–1248 (2008).
222. Dunbar, R. I. The social role of touch in humans and primates: behavioural function and neurobiological mechanisms. Neurosci. Biobehav. Rev. 34, 260–268 (2010).
223. Buchanan, R. W. et al. The FDA-NIMH-MATRICS guidelines for clinical trial design of cognitive-enhancing drugs: what do we know 5 years later? Schizophr. Bull. 37, 1209–1217 (2010).
224. Manahan-Vaughan, D., Widlfôrster, V. & Thomsen, C. Rescue of hippocampal LTP and learning deficits in a rat model of psychosis by inhibition of glycine transporter-1 (GlyT1). Eur. J. Neurosci. 28, 1342–1350 (2008).
225. Stefani, M. R. & Moghaddam, B. Activation of type 5 metabotropic glutamate receptors attenuates deficits in cognitive flexibility induced by NMDA receptor blockade. Eur. J. Pharmacol. 639, 26–32 (2010).
226. Ayala, J. E. et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071 (2009).
227. Zanto, F. P., Rubens, M. T., Thangavel, A. & Gazzaley, A. Causal role of the prefrontal cortex in top-down modulation of visual processing and working memory Nature Neurosci. 14, 656–662 (2011).
228. Belforte, J. E. et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nature Neurosci. 13, 76–83 (2010).
229. Moreines, J. L., McClintock, S. M. & Holtzheimer, P. E. Neuropsychologic effects of neuromodulation technique for treatment-resistant depression: a review. Brain Stimul. 4, 17–27 (2010).
230. Matheson, S. L., Green, M. J., Loo, C. & Carr, V. J. Quality assessment and comparison of evidence for electroconvulsive therapy and repetitive transcranial magnetic stimulation for schizophrenia: a systematic meta-review. Schizophr. Res. 118, 201–210 (2010).
231. Vanderhasselt, M. A., De Raedt, R., Baeken, C., Leyman, L. & D’Haanen, H. A single session of rTMS over the left dorsolateral prefrontal cortex influences attentional control in depressed patients. World J. Biol. Psychiatry 10, 34–42 (2009).
232. Barr, M. S. et al. Potentiation of γ oscillatory activity through repetitive transcranial magnetic stimulation of the dorsolateral prefrontal cortex. Neuropsychopharmacology 34, 2359–2367 (2009).
233. Sokhadze, E. M. et al. Effects of low frequency repetitive transcranial magnetic stimulation (rTMS) on γ frequency oscillations and event-related potentials during processing of illusory figures in autism. J. Autism Dev. Disord. 39, 619–634 (2009).
234. Treffart, D. A. The savant syndrome: an extraordinary condition. A synopsis: past, present, future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 1351–1358 (2009).
235. Cattaneo, Z., Pisoni, A. & Papagno, C. Transcranial direct current stimulation over Broca’s region improves phonemic and semantic fluency in healthy individuals. Neuroscience 183, 64–70 (2011).
236. De Carvalho, M. R., Rozenthal, M. & Nardi, A. E. The fear circuitry in panic disorder and its modulation by cognitive-behaviour therapy interventions. World J. Biol. Psychiatry 11, 188–198 (2009).
237. Wykes, T., Huddy, V., Cellard, C., McGurk, S. R. & Czobor, P. A meta-analysis of cognitive remediation for schizophrenia: methodology and effect sizes. Am. J. Psychiatry 168, 472–485 (2011).
238. Naismith, S. L. Enhancing memory in late-life depression: the effects of a combined psychoeducation and cognitive training program. Am. J. Psychiatry 19, 240–248 (2011).
239. Alexander, G. E., DeLong, M. R. & Strick, P. L. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 9, 357–381 (1986).
240. Jung, R. E. & Haier, R. J. The parieto-frontal integration theory (P-FIT) of intelligence: converging neuroimaging evidence. Behav. Brain Sci. 30, 135–187 (2007).
241. Van Strien, N. M., Cappaert, N. L. M. & Witter, M. P. The anatomy of memory: an interactive overview of the parahippocampal–hippocampal network. Nature Rev. Neurosci. 10, 272–282 (2009).
242. Strick, P. L., Dum, R. P. & Fiez, J. A. Cerebellum and non-motor function. Annu. Rev. Neurosci. 32, 413–434 (2009).
243. Price, C. J. The anatomy of language: a review of 100 fMRI studies published in 2009. Ann. NY Acad. Sci. 1191, 62–88 (2010).
244. Saur, D. et al. Ventral and dorsal pathways for language. Proc. Natl Acad. Sci. USA 105, 18035–18040 (2008).
245. Fusar-Poli, P. et al. Functional atlas of emotional faces processing: a voxel-based meta-analysis of 105 functional magnetic resonance imaging studies. J. Psychiatry Neurosci. 34, 418–432 (2009).
246. Ishai, A., Schmidt, C. F. & Boesiger, P. Face perception is mediated by a distributed cortical network. Brain Res. Bull. 67, 87–93 (2005).
247. Cattaneo, L. & Rizzolatti, G. The mirror neuron system. Arch. Neurol. 66, 557–560 (2009).
248. Fadiga, L., Craighero, L. & D’Ausilio, A. Broca’s area in language, action, and music. Ann. NY Acad. Sci. 1169, 448–458 (2009).
249. Doron, K. W., Funk, C. M. & Glickstein, M. Fronto-cerebellar circuits and eye movement control: a diffusion imaging tractography study on human cortico-pontine projections. Brain Res. 1307, 63–71 (2010).
250. Beaton, A. & Mariën P. Language, cognition and the cerebellum: grappling with an enigma. Cortex 46, 811–820 (2010).
251. Watson, D. J. G., Marsden, M. A., Millan, M. J. & Fone, K. F. C. Blockade of dopamine D3 but not D2 receptors reverses the novel object discrimination impairment produced by post-weaning social isolation: implications for schizophrenia and its treatment. Int. J. Neuropsychopharmacol. 18 Mar 2011 (doi:10.1017/S1461145711000435).
252. Loiseau F. & Millan M. J. Blockade of dopamine D3 receptors in frontal cortex, but not in sub-cortical structures, enhances social recognition in rats: similar actions of D1 receptor agonists, but not of D2 antagonists. Eur. Neuropsychopharmacol. 19, 23–33 (2009).
253. Brioni, J. D., Esbenshade, T. A., Garrison, T. R., Bitner, S. R. & Cowart, M. D. Discovery of histamine H3 antagonists for the treatment of cognitive disorders and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 336, 38–46 (2011).
254. Sellin, A. K., Shad, M. & Tamminga, C. Muscarinic agonists for the treatment of cognition in schizophrenia. CNS Spectr. 13, 985–996 (2008).
255. McArthur, R. A., Gray, J. & Schreiber, R. Cognitive effects of muscarinic M1 functional agonists in non-human primates and clinical trials. Curr. Opin. Invest. Drugs 11, 740–760 (2011).
256. Shimazaki, T., Kaku, A. & Chaki, S. d-serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology 20, 263–270 (2010).
257. Smith, S. M., Uslaner, J. M. & Hutson, P. H. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med. Chem. J. 4, 3–9 (2010).
258. Labrie, V. et al. Genetic inactivation of d-amino acid oxidase enhances extinction and reversal learning in mice. Learn. Mem. 16, 28–37 (2009).
259. Roberts, B. M. et al. Prevention of ketamine-induced working memory impairments by AMPA potentiators in a nonhuman primate model of cognitive dysfunction. Behav. Brain Res. 212, 41–48 (2010).
260. O’Neill, M. J. & Dix, S. AMPA receptor potentiatiors as cognitive enhancers. IDrugs 10, 185–192 (2007).
261. Simonyi, A., Schachtman, T. R. & Christoffersen, G. R. J. Metabotropic glutamate receptor subtype 5 antagonism in learning and memory. Eur. J. Pharmacol. 639, 17–25 (2010).
262. Castner, S. A. et al. Reversal of ketamine-induced working memory impairments by the GABAAα2/3 agonist TPA023. Biol. Psychiatry 15, 998–1001 (2010).
263. Takahashi, R. N., Pamplona, F. A. & Prediger, R. D. Adeno sine receptor antagonists for cognitive dysfunction: a review of animal studies. Front. Biosci. 13, 2614–2632 (2008).
264. Wei, C. J. et al. Selective inactivation of adenosine A2A receptors in striatal neurons enhances working memory and reversal learning. Learn. Mem. 21, 459–474 (2011).
265. de Bruin, N. M. W. et al. SVL330, a cannabinoid CB1 receptor antagonist, ameliorates deficits in the T-maze, object recognition and social recognition tasks in rodents. Neurobiol. Learn. Mem. 217, 408–415 (2010).
266. Egashira, N., Mishima, K., Iwasaki, K., Oishi, R. & Fujiwara, M. New topics in vasopressin receptors and approach to novel drugs: role of the vasopressin receptor in psychological and cognitive functions. J. Pharmacol. Sci. 109, 44–49 (2009).
267. Hongpaisan, J., Sun, M. K. & Alkon, D. L. PKCε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 12, 630–643 (2011).
268. Gozes, I. Microtubules, schizophrenia and cognitive behaviour: preclinical development of davunetide (NAP) as a peptide-drug candidate. Peptides 32, 428–431 (2010).
AcknowledgementsM. Soubeyran is thanked for excellent secretarial assistance, S.-M. Rivet for the excellent graphics, and A. Gobert and A. Dekeyne are likewise thanked for their logistical help. We would like to thank three anonymous reviewers for their insightful comments that helped to improve the manuscript. This paper emerged from a Congress that took place in France, 2009, organized by ‘Advances in Neuroscience for Medical Innovation’ and supported by an educational grant from Institut de Recherche Servier.
Competing interests statementThe authors declare competing financial interests: see Web version for details.
SUPPLEMENTARY INFORMATIONSee online article: S1 (figure) | S2 (figure) | S3 (box) | S4 (box) | S5 (box)
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
R E V I E W S
168 | FEBRUARY 2012 | VOLUME 11 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved

L I N K TO O R I G I N A L A RT I C L E
As summarized in a recent article (Antibacterial R&D incentives. Nature Rev. Drug Discov. 10, 727–728 (2011))1, numerous ‘push’ and ‘pull’ economic incentives have been proposed to rekindle the antibiotic pipeline2–6. These have been the subject of discussions in both the United States and Europe1,4.
Push incentives act early in research and development (R&D) and are generally of smaller monetary amounts than pull incentives, which do not become operative until after a drug is approved for use in humans. Until recently, the relative potency of push and pull incentives at promoting new R&D in the antibiotic area had been uncertain. However, an analysis by Sharma and Towse6 found that a key pull incentive — extended market exclusivity — had minimal impact on improving the net present value (NPV) of antibiotics. By contrast, push incentives may be of greater value. Because pull incentives should result in larger amounts of revenue than push incentives, the assertion that pull incentives are less economically valuable than push incentives may be counterintuitive. However, as we show below, push incentives may be 95% smaller than pull incentives and still yield similar value.
To explain this paradox, we developed a model to determine the present value (PV) of push and pull incentives. In the base case model, the extended exclusivity incentive began after patent expiration, which included the 20 years of patent term and 5 years of patent term restoration (a total of 25 years) afforded to drugs in the United States under the Hatch–Waxman Act (Supplementary information S1 (figure); panel a). As is standard, the model incorporated time discounting in calculating the NPV7,8. Because a discount rate of 10–11% has become standard in pharmaceutical economic models as a way to adjust for risk5,9–11, a discount rate of 10.5% was used in the current model, with sensitivity analyses run at 7% and 20%12,13. Thus, in the base case estimate, future revenue from the extended exclusivity incentive was discounted by 10.5% compounded annually for 25 years before the incentive began, which was consistent with a
timeline including: preclinical development (4 years); clinical development (8 years) plus time for filing and approval of a new drug application (NDA) (1 year); and sales protected for a period of 12 years after initial approval by the initial patent on the compound (7 years remaining at the time of NDA filing) plus 5 years of patent term restoration5,10,12 (Supplementary information S1 (figure); panel a). Discounting continued during the period of extended exclusivity.
To model the impact of discounting on pull incentives, the base case estimate of annual antibiotic sales during the period of extended exclusivity was US$400 million5,10. Time discounting was applied to the difference between the total sales with extended exclusivity and the sales without extended exclusivity. To calculate this difference, a base case estimate of a 15% annual decline in sales following the expiration of exclusivity was modelled5. Total sales resulting from the extended exclusivity incentive were calculated by summing up the difference between the sales during the extended exclusivity incentive period ($400 million per year in the base case model) and sales starting at $400 million per year but declining at 15% per year without the incentive.
To model the impact of discounting on push incentives, a separate base case scenario was developed. The model did not distinguish among the different types of push incentives; it could equally apply to grants, contracts, R&D tax credits, matching funds, other forms of public–private partnerships (PPPs) or any other push incentive of the same dollar amounts and time period modelled.
In the base case pull incentive model, the total revenue generated during the 5 years of extended exclusivity was $739 million (TABLE 1). However, this revenue did not become available until 26–30 years after the discovery of the drug (Supplementary information S1 (figure); panel b). Because of time discounting, the PV of these future total revenues was only $43.1 million (TABLE 1).
In sensitivity analyses, changing the discount rate to 7%, which was reflective of a value published 20 years ago14, still resulted
in an 86% erosion in the PV of the incentive (TABLE 1). Increasing the discount rate to 20%, which was more consistent with the higher R&D risk at small companies12–14, resulted in a remarkable 99.4% erosion in the PV of the incentive (TABLE 1). However, acquisition of a drug from another company after some period of development had already occurred shortened the period of discounting. For example, drug acquisition after completion of preclinical development (filing an investigational new drug (IND) application), Phase I trials or Phase II trials led to discount periods of 21, 19 or 17 years, respectively, before the exclusivity extension began (Supplementary information S1 (figure); panel b), resulting in 91%, 89% or 87% PV erosions, respectively (TABLE 1).
Doubling the length of the extended exclusivity period from 5 to 10 years (Supplementary information S1 (figure); panel b) more than doubled the PV of the incentive to $96 million (TABLE 1) because the annual 15% decline in sales after patent expiration is cumulative. Thus, the difference in sales with versus without extended exclusivity increases by 15% annually for each year of the exclusivity incentive (that is, it is 15% greater in year 30 than in year 29, and so on). For the same reason, combining a 10year extended exclusivity incentive with a shortened duration of discounting due to acquisition of the drug after completion of preclinical development (filing an IND application), Phase I trials or Phase II trials resulted in synergistically higher absolute PVs (of $143 million, $175 million or $214 million, respectively).
Changing the rate of declining sales after expiration of exclusivity had a minimal impact on the PV (TABLE 1). By contrast, the overall value of the extended exclusivity incentive was affected by changing the peak antibiotic sales at the time the extended exclusivity began (TABLE 1). Finally, initiating extended exclusivity after the expiration of the 5year Hatch–Waxman data exclusivity period (which begins once the drug is approved), rather than after patent expiration resulted in the extended exclusivity expiring 2 years before patent expiration (Supplementary information S1 (figure); panel c). The net effect was no period of extended exclusivity, and hence no additional revenue, resulting in a PV of $0. R&D had to be prolonged beyond 15 years for the extended exclusivity to generate any value if it began after the expiration of data exclusivity.
In the base case push incentive model, the full amount of the push incentive was applied to year 1, which is consistent with an incentive that is intended to encourage a new antibiotic
The critical impact of time discounting on economic incentives to overcome the antibiotic market failureBrad Spellberg, Priya Sharma and John H. Rex
CORRESPONDENCE
NATURE REVIEWS | DRUG DISCOVERY www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
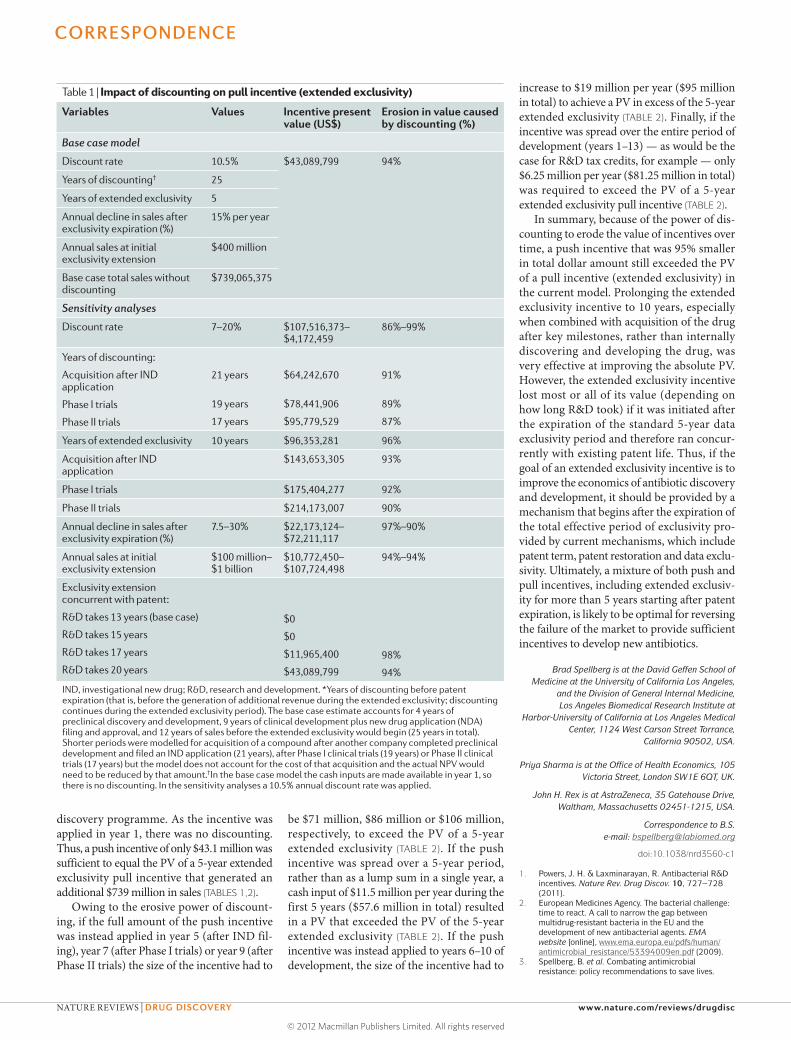
discovery programme. As the incentive was applied in year 1, there was no discounting. Thus, a push incentive of only $43.1 million was sufficient to equal the PV of a 5year extended exclusivity pull incentive that generated an additional $739 million in sales (TABLES 1,2).
Owing to the erosive power of discounting, if the full amount of the push incentive was instead applied in year 5 (after IND filing), year 7 (after Phase I trials) or year 9 (after Phase II trials) the size of the incentive had to
be $71 million, $86 million or $106 million, respectively, to exceed the PV of a 5year extended exclusivity (TABLE 2). If the push incentive was spread over a 5year period, rather than as a lump sum in a single year, a cash input of $11.5 million per year during the first 5 years ($57.6 million in total) resulted in a PV that exceeded the PV of the 5year extended exclusivity (TABLE 2). If the push incentive was instead applied to years 6–10 of development, the size of the incentive had to
increase to $19 million per year ($95 million in total) to achieve a PV in excess of the 5year extended exclusivity (TABLE 2). Finally, if the incentive was spread over the entire period of development (years 1–13) — as would be the case for R&D tax credits, for example — only $6.25 million per year ($81.25 million in total) was required to exceed the PV of a 5year extended exclusivity pull incentive (TABLE 2).
In summary, because of the power of discounting to erode the value of incentives over time, a push incentive that was 95% smaller in total dollar amount still exceeded the PV of a pull incentive (extended exclusivity) in the current model. Prolonging the extended exclusivity incentive to 10 years, especially when combined with acquisition of the drug after key milestones, rather than internally discovering and developing the drug, was very effective at improving the absolute PV. However, the extended exclusivity incentive lost most or all of its value (depending on how long R&D took) if it was initiated after the expiration of the standard 5year data exclusivity period and therefore ran concurrently with existing patent life. Thus, if the goal of an extended exclusivity incentive is to improve the economics of antibiotic discovery and development, it should be provided by a mechanism that begins after the expiration of the total effective period of exclusivity provided by current mechanisms, which include patent term, patent restoration and data exclusivity. Ultimately, a mixture of both push and pull incentives, including extended exclusivity for more than 5 years starting after patent expiration, is likely to be optimal for reversing the failure of the market to provide sufficient incentives to develop new antibiotics.
Brad Spellberg is at the David Geffen School of Medicine at the University of California Los Angeles,
and the Division of General Internal Medicine, Los Angeles Biomedical Research Institute at
Harbor-University of California at Los Angeles Medical Center, 1124 West Carson Street Torrance,
California 90502, USA.
Priya Sharma is at the Office of Health Economics, 105 Victoria Street, London SW1E 6QT, UK.
John H. Rex is at AstraZeneca, 35 Gatehouse Drive, Waltham, Massachusetts 02451-1215, USA.
Correspondence to B.S. e-mail: [email protected]
doi:10.1038/nrd3560-c1
1. Powers, J. H. & Laxminarayan, R. Antibacterial R&D incentives. Nature Rev. Drug Discov. 10, 727–728 (2011).
2. European Medicines Agency. The bacterial challenge: time to react. A call to narrow the gap between multidrug-resistant bacteria in the EU and the development of new antibacterial agents. EMA website [online], www.ema.europa.eu/pdfs/human/antimicrobial_resistance/53394009en.pdf (2009).
3. Spellberg, B. et al. Combating antimicrobial resistance: policy recommendations to save lives.
Table 1 | Impact of discounting on pull incentive (extended exclusivity)
Variables Values Incentive present value (US$)
Erosion in value caused by discounting (%)
Base case model
Discount rate 10.5% $43,089,799 94%
Years of discounting† 25
Years of extended exclusivity 5
Annual decline in sales after exclusivity expiration (%)
15% per year
Annual sales at initial exclusivity extension
$400 million
Base case total sales without discounting
$739,065,375
Sensitivity analyses
Discount rate 7–20% $107,516,373–$4,172,459
86%–99%
Years of discounting:
Acquisition after IND application
Phase I trials
Phase II trials
21 years
19 years
17 years
$64,242,670
$78,441,906
$95,779,529
91%
89%
87%
Years of extended exclusivity 10 years $96,353,281 96%
Acquisition after IND application
$143,653,305 93%
Phase I trials $175,404,277 92%
Phase II trials $214,173,007 90%
Annual decline in sales after exclusivity expiration (%)
7.5–30% $22,173,124–$72,211,117
97%–90%
Annual sales at initial exclusivity extension
$100 million– $1 billion
$10,772,450–$107,724,498
94%–94%
Exclusivity extension concurrent with patent:
R&D takes 13 years (base case)
R&D takes 15 years
R&D takes 17 years
R&D takes 20 years
$0
$0
$11,965,400
$43,089,799
98%
94%
IND, investigational new drug; R&D, research and development. *Years of discounting before patent expiration (that is, before the generation of additional revenue during the extended exclusivity; discounting continues during the extended exclusivity period). The base case estimate accounts for 4 years of preclinical discovery and development, 9 years of clinical development plus new drug application (NDA) filing and approval, and 12 years of sales before the extended exclusivity would begin (25 years in total). Shorter periods were modelled for acquisition of a compound after another company completed preclinical development and filed an IND application (21 years), after Phase I clinical trials (19 years) or Phase II clinical trials (17 years) but the model does not account for the cost of that acquisition and the actual NPV would need to be reduced by that amount.†In the base case model the cash inputs are made available in year 1, so there is no discounting. In the sensitivity analyses a 10.5% annual discount rate was applied.
CORRESPONDENCE
NATURE REVIEWS | DRUG DISCOVERY www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
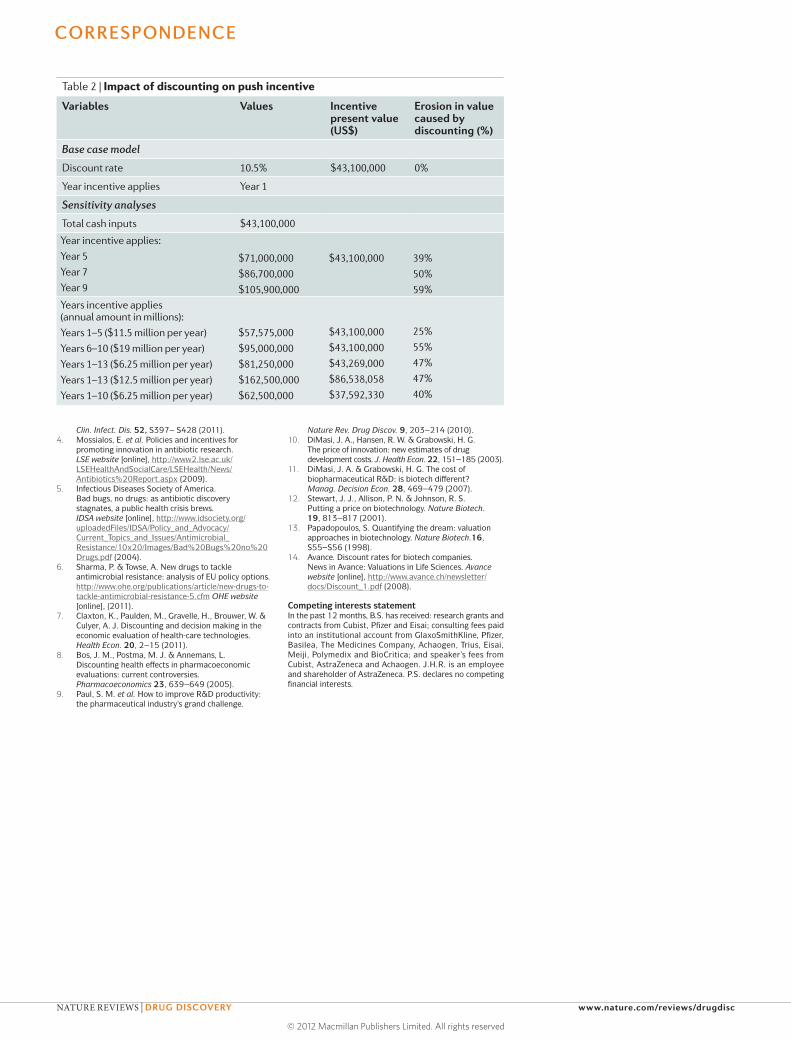
Clin. Infect. Dis. 52, S397– S428 (2011).4. Mossialos, E. et al. Policies and incentives for
promoting innovation in antibiotic research. LSE website [online], http://www2.lse.ac.uk/LSEHealthAndSocialCare/LSEHealth/News/Antibiotics%20Report.aspx (2009).
5. Infectious Diseases Society of America. Bad bugs, no drugs: as antibiotic discovery stagnates, a public health crisis brews. IDSA website [online], http://www.idsociety.org/uploadedFiles/IDSA/Policy_and_Advocacy/ Current_Topics_and_Issues/Antimicrobial_Resistance/10x20/Images/Bad%20Bugs%20no%20Drugs.pdf (2004).
6. Sharma, P. & Towse, A. New drugs to tackle antimicrobial resistance: analysis of EU policy options. http://www.ohe.org/publications/article/new-drugs-to-tackle-antimicrobial-resistance-5.cfm OHE website [online], (2011).
7. Claxton, K., Paulden, M., Gravelle, H., Brouwer, W. & Culyer, A. J. Discounting and decision making in the economic evaluation of health-care technologies. Health Econ. 20, 2–15 (2011).
8. Bos, J. M., Postma, M. J. & Annemans, L. Discounting health effects in pharmacoeconomic evaluations: current controversies. Pharmacoeconomics 23, 639–649 (2005).
9. Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge.
Nature Rev. Drug Discov. 9, 203–214 (2010).10. DiMasi, J. A., Hansen, R. W. & Grabowski, H. G.
The price of innovation: new estimates of drug development costs. J. Health Econ. 22, 151–185 (2003).
11. DiMasi, J. A. & Grabowski, H. G. The cost of biopharmaceutical R&D: is biotech different? Manag. Decision Econ. 28, 469–479 (2007).
12. Stewart, J. J., Allison, P. N. & Johnson, R. S. Putting a price on biotechnology. Nature Biotech. 19, 813–817 (2001).
13. Papadopoulos, S. Quantifying the dream: valuation approaches in biotechnology. Nature Biotech.16, S55–S56 (1998).
14. Avance. Discount rates for biotech companies. News in Avance: Valuations in Life Sciences. Avance website [online], http://www.avance.ch/newsletter/docs/Discount_1.pdf (2008).
Competing interests statementIn the past 12 months, B.S. has received: research grants and contracts from Cubist, Pfizer and Eisai; consulting fees paid into an institutional account from GlaxoSmithKline, Pfizer, Basilea, The Medicines Company, Achaogen, Trius, Eisai, Meiji, Polymedix and BioCritica; and speaker’s fees from Cubist, AstraZeneca and Achaogen. J.H.R. is an employee and shareholder of AstraZeneca. P.S. declares no competing financial interests.
Table 2 | Impact of discounting on push incentive
Variables Values Incentive present value (US$)
Erosion in value caused by discounting (%)
Base case model
Discount rate 10.5% $43,100,000 0%
Year incentive applies Year 1
Sensitivity analyses
Total cash inputs $43,100,000
Year incentive applies:Year 5Year 7Year 9
$71,000,000$86,700,000$105,900,000
$43,100,000 39%50%59%
Years incentive applies (annual amount in millions):Years 1–5 ($11.5 million per year)Years 6–10 ($19 million per year)Years 1–13 ($6.25 million per year)Years 1–13 ($12.5 million per year)Years 1–10 ($6.25 million per year)
$57,575,000$95,000,000$81,250,000$162,500,000$62,500,000
$43,100,000$43,100,000$43,269,000$86,538,058$37,592,330
25%55%47%47%40%
CORRESPONDENCE
NATURE REVIEWS | DRUG DISCOVERY www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
Related Documents











