Myoclonic Epilepsy and Ragged-Red Fibers with Cytochrome Oxldase Deficiency: Neuropathology, Biochemistry, and Molecular Genetics Anne Lombes, MD,"Jerry R. Mendell, MD,? Hirofumi Nakase, MD," Richard J. Barohn, MD,P Eduardo Bonilla, MD," Massimo Zeviani, MD," Allan J. Yates, PhD,t Jennifer Omerta, BS,t Tracy L. Gales, MA,t Keichi Nakahara, MD," Rosario Rizzuto, MD," W. King Engel, MD,$ and Salvatore DiMauro, MD" A 3Gyear-old man with myoclonic epilepsy and ragged-red fibers (MERRF) died after more than 18 years of follow-up study. He was 1 of 3 affected siblings and the offspring of an affected mother, suggesting maternal transmission. At autopsy, there was neuronal loss and gliosis in the dentate nucleus of the cerebellum and in the inferior olivary nucleus. Skeletal muscle showed ragged-red fibers, and paracrystalline inclusions in mitochondria by electron microscopy. Biochemical analysis showed a generalized partial defect of cytochrome c oxidase (COX) in mitochondria isolated from all tissues, including brain, heart, skeletal muscle, kidney, and liver. The Michaelis constant (Km) for cytochrome c was abnormally low, suggesting a defect of the mitochondrially encoded subunit I1 of COX. Immunological studies (enzyme-linked immunosorbent assay, dot-blot, Western blot, and immunohistochemistry) showed that the holoenzyme was decreased but subunit I1 was decreased more than the holocomplex or the nuclearly encoded subunit IV. How- ever, Northern and Southern blots showed that the gene for subunit 11, as well as the genes for subunits I, 111, IV, and VILI, were of normal size and were normally transcribed. A point mutation or a small deletion of mitochondrial DNA, probably affecting the COX-I1 gene, may be responsible for the COX deficiency in this case of MEW. Lombes A, Mendell JR, Nakase H, Barohn RJ, Bonilla E, Zeviani M, Yates AJ, Omerta J, Gales TL, Nakahara K, Rizzuto R, Engel WK, DiMauro S. Myoclonic epilepsy and ragged-red fibers with cytochrome oxidase deficiency: neuropathology, biochemistry, and molecular genetics. Ann Neurol 1989;26:20-33 The term progressive myodonic epilepsy (PM E) charac- terizes a group of secondary generalized epilepsies that have interested clinicians for decades [l). Common features include myoclonic and tonic-clonic seizures associated with progressive neurological dysfunction, especially dementia and ataxia. Investigators have at- tempted to identify specific subgroups of patients with PME on the basis of morphological and biochemical criteria. Recognition of intracytoplasmic inclusions, Lafora bodies, identdied one subgroup 121. Later, biochemical markers distinguished other PME syn- dromes, including neuronal ceroid lipofuscinosis { 31 and sialidosis types I and I1 [4]. More recently, another subgroup of patients has been identified on the basis of mitochondrial abnormalities in skeletal muscle, initially reported by Tsairis and colleagues in 1773 [5]. This variant of PME was later referred to as myoclonic epilepjy with mggedredfibers (MERRF) (61. MERRF is a hereditary mitochondrial encephalo- myopathy apparently transmitted by non-Mendelian maternal inheritance {5- 161. Maternal transmission suggests that the biochemical defect ought to involve 1 of the 13 proteins that are encoded by mitochondrial DNA (mtDNA) and are all components of the mito- chondrial respiratory chain. However, there have been very few biochemical studies of M E W patients: A defect of reducible cytochrome b was reported in 1982 [7] and recently cytochrome c oxidase (COX) defi- ciency was described in 2 unrelated families [ll, 161. The family reported by Tsairis and colleagues C5l was evaluated for more than 18 years and the last af- fected member died recently. We report the clinical From the *H. Houston Merritt Clinical Research Center for Muscu- lar Dystrophy and Related Diseases, Coiumbia University College of Received for publication Sep 5, 1988, and in revised form Dec 7. AcceDted for Dubkation Dec 18. 1988. Physicians-md New NY5 tDeparmenc Of Neurol- Address correspondence to Dr DiMaurq Room 4-420, College of Physicians and Surgeons, 630 West 168th St, New York, NY 10032. ogy, Ohio State University, Columbus, OH, and $University of Southern California School of Medicine, Los Angeles, CA. 20 Copyright 0 1989 by the American Neurological Association

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Myoclonic Epilepsy and Ragged-Red Fibers with Cytochrome Oxldase Deficiency: Neuropathology, Biochemistry, and
Molecular Genetics Anne Lombes, MD,"Jerry R. Mendell, MD,? Hirofumi Nakase, MD," Richard J. Barohn, MD,P
Eduardo Bonilla, MD," Massimo Zeviani, MD," Allan J. Yates, PhD,t Jennifer Omerta, BS,t Tracy L. Gales, MA,t Keichi Nakahara, MD," Rosario Rizzuto, MD," W. King Engel, MD,$
and Salvatore DiMauro, MD"
A 3Gyear-old man with myoclonic epilepsy and ragged-red fibers (MERRF) died after more than 18 years of follow-up study. He was 1 of 3 affected siblings and the offspring of an affected mother, suggesting maternal transmission. At autopsy, there was neuronal loss and gliosis in the dentate nucleus of the cerebellum and in the inferior olivary nucleus. Skeletal muscle showed ragged-red fibers, and paracrystalline inclusions in mitochondria by electron microscopy. Biochemical analysis showed a generalized partial defect of cytochrome c oxidase (COX) in mitochondria isolated from all tissues, including brain, heart, skeletal muscle, kidney, and liver. The Michaelis constant (Km) for cytochrome c was abnormally low, suggesting a defect of the mitochondrially encoded subunit I1 of COX. Immunological studies (enzyme-linked immunosorbent assay, dot-blot, Western blot, and immunohistochemistry) showed that the holoenzyme was decreased but subunit I1 was decreased more than the holocomplex or the nuclearly encoded subunit IV. How- ever, Northern and Southern blots showed that the gene for subunit 11, as well as the genes for subunits I, 111, IV, and VILI, were of normal size and were normally transcribed. A point mutation or a small deletion of mitochondrial DNA, probably affecting the COX-I1 gene, may be responsible for the COX deficiency in this case of M E W .
Lombes A, Mendell JR, Nakase H, Barohn RJ, Bonilla E, Zeviani M, Yates AJ, Omerta J, Gales TL, Nakahara K, Rizzuto R, Engel WK, DiMauro S. Myoclonic epilepsy and ragged-red fibers with cytochrome
oxidase deficiency: neuropathology, biochemistry, and molecular genetics. Ann Neurol 1989;26:20-33
The term progressive myodonic epilepsy (PM E) charac- terizes a group of secondary generalized epilepsies that have interested clinicians for decades [l). Common features include myoclonic and tonic-clonic seizures associated with progressive neurological dysfunction, especially dementia and ataxia. Investigators have at- tempted to identify specific subgroups of patients with PME on the basis of morphological and biochemical criteria. Recognition of intracytoplasmic inclusions, Lafora bodies, identdied one subgroup 121. Later, biochemical markers distinguished other PME syn- dromes, including neuronal ceroid lipofuscinosis { 31 and sialidosis types I and I1 [4]. More recently, another subgroup of patients has been identified on the basis of mitochondrial abnormalities in skeletal muscle, initially reported by Tsairis and colleagues in 1773 [5] . This
variant of PME was later referred to as myoclonic epilepjy with mggedredfibers (MERRF) (61.
MERRF is a hereditary mitochondrial encephalo- myopathy apparently transmitted by non-Mendelian maternal inheritance {5- 161. Maternal transmission suggests that the biochemical defect ought to involve 1 of the 13 proteins that are encoded by mitochondrial DNA (mtDNA) and are all components of the mito- chondrial respiratory chain. However, there have been very few biochemical studies of M E W patients: A defect of reducible cytochrome b was reported in 1982 [7] and recently cytochrome c oxidase (COX) defi- ciency was described in 2 unrelated families [ll, 161.
The family reported by Tsairis and colleagues C5l was evaluated for more than 18 years and the last af- fected member died recently. We report the clinical
From the *H. Houston Merritt Clinical Research Center for Muscu- lar Dystrophy and Related Diseases, Coiumbia University College of
Received for publication Sep 5 , 1988, and in revised form Dec 7. AcceDted for Dubkation Dec 18. 1988.
Physicians-md New NY5 tDeparmenc Of Neurol- Address correspondence to Dr DiMaurq Room 4-420, College of Physicians and Surgeons, 630 West 168th St, New York, NY 10032.
ogy, Ohio State University, Columbus, OH, and $University of Southern California School of Medicine, Los Angeles, CA.
20 Copyright 0 1989 by the American Neurological Association

PEDIGREE
1 I
Fig I . Pedigree of family with myoclonic epilepsy with ragged- redfiben. 0 = normal female mbjects, D = normal male stlb- jects, *, H = affected members, 0 a = deceased members, = miscarriages, fl = proband.
t
and neuropathological findings in this patient, as well as the results of biochemical and molecular genetic studies.
Case Reports The family pedigree is shown in Figure 1. The most com- plete data, including biochemistry and neuropathology, were obtained in postmortem studies of Patient IV-7. Findings in other affected family members are summarized in Table 1.
Patient IV-7 The patient (see Fig 1) was normal at birth with normal motor and intellectual milestones. Premature fatigue and oc- casional muscle cramping were noted during childhood. He was seen by a doctor at age 10 for poor concentration, but no diagnosis was established. At age 19 he was re-evaluated because other family members were symptomatic. At that time he had no seizures and the neurological examination was normal. However, a left quadriceps muscle biopsy dem- onstrated many raged-red fibers, and electron microscopy showed excess lipid and abnormal mitochondria with para- crystalline inclusions. Creatine kinase (CK) was elevated (138 IU; normal < 80). Serum lactate and pyruvate were normal. Electroencephalography (EEG) showed disorganized
Patient IV-8 This patient (see Fig 1) was not symptomatic until age 10, when involuntary jerking of head and limbs followed by drop attacks developed. She then showed progressive gait ataxia, weakness, and poor school performance. Within 3 years she showed marked intellectual decline, inability to stand, diffuse muscle weakness and wasting, and bilateral hearing loss. Evaluation at age 14 showed elevated serum lactate and pyruvate and abundant ragged-red fibers in a
Table I . Clinical Featuws of Affected Family Members
background activity and paroxysmal high-voltage discharges in a diffuse distribution associated with spike-and-wave forms. Within 1 year involuntary jerks of limbs and isolated muscle groups developed, as well as difficulty in climbing stairs. Examination showed diffuse, especially proximal, mus- cle weakness, with elevations in serum lactate and pyruvate. The clinical course was characterized by slow deterioration, with progressive gait ataxia, severe limb weakness, and mus- cle atrophy. Myoclonic seizures became more frequent and complex partial seizures developed. In later years, the patient showed intellectual decline and hearing loss. He was con- fined to a wheelchair by age 32.
Examination at age 36 revealed diffuse muscle wasting, more in the legs than in the arms. Myoclonic seizures in- volved limbs and trunk. Ocular movements were intact and no ptosis was present. Mild facial muscle weakness and di- minished gag reflex were seen. Hearing was decreased bilat- erally, and speech was dysarthric. The patient had diffuse weakness, especially in proximal muscles, which was greater in the legs than in the arms. He could not stand without support. Tendon reflexes were normal and plantar responses were flexor. Position and vibration sensation were mildly diminished in the toes.
Laboratory abnormalities were confined to elevated CK, 1,721 IU (normal < 90), and serum lactic acid, 9.3 mmoYL (normal = 0.5-2.2). Magnetic resonance imaging of the brain showed difhse cerebral atrophy. The EEG showed diffuse slowing, consisting of 3- to 5-Hz activity, which was disorganized and associated with bitemporal sharp transients.
Repeated episodes of pneumonia, probably related in part to reduced ventilatory drive, dominated the patient's final year of life. While at home on a portable ventilator, he had cardiopulmonary arrest. Resuscitation by an emergency squad was followed by a brief hospitalization for 36 hours before death at age 36.
Permission for postmortem examination was obtained and the findings are described later.
~ ~ ~ ~
Age at Mitochondrial Generalized Hearing Patienta death (yr) Myopathy Myoclonus Ataxia Seizures Dementia Loss
111-4 47 + + + + b + 0 0 0 IV-7 36 + + + + + + + + + + IV-8 16 + + + + + + + + + + + + + IV-9 12 + + + + + + + + + + + + + "For patients' symbols, see Figure 1. bPhotic induced only.
0 = absent; + = present, mild; + + = present; + + + = present, severe.
Lombes et al: MERRF with Cytochrome Oxidase Deficiency 21

muscle biopsy. She continued to deteriorate, became bedrid- den and unable to swallow, and died at age 16.
Patient IV-9 At 8 years of age, this patient (see Fig 1) developed head tremor and involuntary jerks of trunk and extremities, fol- lowed by gait ataxia, poor coordination, decreasing attention span, and poor school performance. She also had proximal muscle weakness and bilateral hearing loss. At age 10 she had elevated lactate and pyruvate. EEG showed diffuse slow- ing with disorganization. There were frequent paroxysmal bursts of spike-and-wave and polyspikes that were not focal. Muscle biopsies of right and left quadriceps demonstrated ragged-red fibers with mitochondrial abnormalities by elec- tron microscopy. By age 12 the patient was bedridden with generalized weakness and inability to swallow. Death oc- curred after an episode of aspiration pneumonia.
Patient IV-6 This 40-year-old woman (see Fig 1) had no symptoms of progressive myoclonic epilepsy, and she never had seizures. At age 22, EEG and serum lactate, pyruvate, and CK were normal. A muscle biopsy at that time showed a few ragged- red fibers.
Patient 111-4 Symptoms of muscle weakness first developed in this patient (see Fig 1) at age 33, when she noticed difficulty in climbing stairs and lifting objects. When she was 38, limb-girdle mus- cular dystrophy was diagnosed. At age 46, CK was 195 (nor- mal < 80), serum lactate and pyruvate were twice normal, and many ragged-red fibers were noted in a muscle biopsy. EEG showed normal background, with epileptiform activity manifesting as bilateral synchronous spike-and-wave dis- charges occurring sporadically. Photic stimulation at 12- and 18-per-second flicker frequency produced myoclonic jerks. N o spontaneous clinical seizures of any type developed. Ap- proximately 3 months after the initial evaluation, a diagnosis of malignant lymphoma was made and the patient died of this malignancy at age 46.
Patient III-3 At age 70, this man (see Fig 1) has no signs of progressive myoclonic epilepsy. An extensive evaluation at age 52 re- vealed normal serum lactate, pyruvate, CK, and EEG. Mus- cle biopsy showed no mitochondrial abnormalities.
Methods Tissues from Patient IV-7, obtained 2 hours after death, were frozen in liquid nitrogen, shipped on solid carbon diox- ide, and stored at -70°C.
Biochemical Studies For crude preparation, samples were homogenized in 9 vol- umes of 0.15 M potassium chloride, 50 mM TRIS hydrochlo- ride (HC1) (pH 7.4) in all-glass, motor-driven homogenizers. Supernatants were obtained after centrifugation at 1,000 gm for 20 minutes. Mitochondria were isolated by the method of Bookelman and associates {17], except for brain mito- chondria, which were isolated by the method of Lai and Clark { 181.
Described spectrophotometric assays were used to mea- sure succinate cytochrome c reductase, nicotinamide adenine dinucleotide (NADH) cytochrome c reductase [19], citrate synthase [20], NADH dehydrogenase [2 13, and succinate dehydrogenase [22}. Cytochrome c oxidase (COX) was also determined spectrophotometrically by measuring the de- crease in absorbance at 550 nm of reduced cytochrome c 1231. Reduced cytochrome c was prepared fresh before each experiment by adding a few grains of sodium hydrosulfite (dithionite) to a 1% cytochrome c solution in 10 m~ potas- sium phosphate buffer (pH 7.0).
Comparative kinetic studies of COX were performed on the same day, with the same fresh reduced cyochrome c preparation, using the spectrophotometric assay. The amount of tissue added to the reaction mixture was adjusted to give approximately the same apparent speed of reaction. Concentration of reduced cytochrome c ranged from 2.4 p , ~ to 57 pM.
Protein was measured by the method of Lowry and as- sociates [24] with bovine serum albumin as standard.
Immunological StudieJ Antibodies against purified bovine heart COX were raised in rabbits as described [25f.
Monoclonal antibodies against subunit IV of COX (COX- IV) were raised in mice as described [26]. These antibodies stained only mitochondria by immunocytochemistry of fro- zen muscle section and recognized only COX-IV in Western blots of mitochondrial preparations.
Polyclonal antibodies against bovine subunit I1 (COX-11) stained only mitochondria in muscle sections and recognized only COX-I1 in Western blots of mitochondrial suspensions.
Control tissues for biochemical assays of the respiratory chain complexes were obtained from autopsies performed 2 to 10 hours after death. Patients presented with various neu- romuscular disorders without defects of the mitochondrial respiratory chain.
Control mitochondria for dot-blot, Western blot, and ki- netic analysis were obtained from the muscle biopsy speci- men of a 15-year-old boy who was eventually deemed free of neuromuscular disease, and from brain and kidney obtained 4 and 6 hours after death from 2 children with encephalopa- thy of unknown origin. The enzyme-linked immunosorbent assay (ELISA) technique was performed as described [25]. The amount of cross-reacting material was roughly evaluated by the antibody dilution necessary to obtain a 50% decrease of maximum optical density.
Dot-blot procedure was performed as described {27), with samples diluted in 50 m~ TRIS HC1, 0.15 M sodium chloride, p H 7.8, containing 0.1% sodium dodecyl sulfate (SDS) using a Bio-Dot microfiltration apparatus (Biorad, Rockville, NY). Decreasing concentrations of mitochondrial suspension and of purified bovine heart COX were blotted onto the membrane (Immobilon, Millipore, Bedford, MA). The intensity of the reaction was measured by densitometry using a Helena Quick-Scan Jr Densitometer equipped with a peak area integrator (Helena Laboratories, Beaumont, TX). For each antibody, we used only points in the linear portion of the optic density curve. The same mitochondrial prepara- tions were used in dot-blots with each first antibody.
For Western blot, we applied Kadenbach and associates’
22 Annals of Neurology Vol 26 No 1 July 1989

Fig 2. Section of dentate nuclew of cerebellum showing neuronal loss and gliosas (A) compared t o ane-matched control subject (B). - - Arrows indicate nerve cells. (Luxol fast blue periodic acid- Schifi x 130 before 4% reduction.)
procedure [28}. Transfer was performed in a Transblot cell (Biorad) as described by Towbin and colleagues [29], using an Immobilon membrane as for the dot-blot procedure. Im- munostaining was performed according to the method of Bresolin and associates 1253. The dilution of the first anti- body was the same as for dot-blot and the intensity of the stain was measured in a Helena Quick-Scan Jr Densitometer.
For immunocytochemical detection of COX holoenzyme, COX-11, and COX-IV, 4-p-thick serial sections from the patient's muscle and from normal muscle were placed on the same coverslip, labeled with the different antibodies, and visualized by immunofluorescence as described [ 30). Ad ja- cent sections were stained for the histochemical demonstra- tion of COX activity. Fluorescence microscopy was carried out with a Zeiss Universal Photomicroscope equipped with epi-illumination.
Molecular Biology Total RNA was prepared from brain by the guanidiuml cesium chloride method [31), and 20-pg aliquots were run on a 1% agarose-formaldehyde gel [32] and transferred to a nylon membrane filter (Magnagraph, MSI, Westborough,
____
Lombes et al: M E W with Cytochrome Oxidase Deficiency 23
MA). Hybridization was performed according to the method of Church and Gilbert [331. The specific activity of the probe was lo8 cpdpg . Mitochondrial DNA was extracted from isolated heart mitochondria according to the procedure of Palva and Palva [341. For Southern analysis, 10 pg mitochon- drial DNA were digested with PvuII, HindIII, and PstI (Boehringer Mannheim, Indianapolis, IN), run on a 0.895 agarose gel, and transferred to nitrocellulose paper [35}. Hy- bridization was performed as described 1361, using as probe human linearized mtDNA radiolabeled according to the method of Feinberg and Vogelstein [37} with a specific activ- ity between 1 and 5 x 10' c p d p g .
Results Autopsy Examination
ination of Patient IV-7 showed only pleural and peri- cardial effusions, ascites, and hemorrhagic areas in both cardiac ventricles and in the septum. Microscopic examination of the heart showed areas of acute trans- mural myocardial infarction (24-40 hours in age) in the anterior and posterior walls of the left ventricle, the septum, and the posterolateral wall of the right ventricle. The lungs showed chronic bronchiolitis and acute bronchopneumonia. Acute hemorrhagic pancre-
NON-NERVOUS SYSTEM TISSUES. Macroscopic exam-

Fig 3. Section of infm.or olivary nucleus showing neuronalloss and gliosis (A) compared to age-matched control subject (B). Ar- rows indicate newe cells. (Luxol fast blue; x 70 befre 4% ye- duction.)
atitis (less than 24 hours) was also present. No abnor- malities were seen in other organs. Ultrastructural ex- amination of myocardium (left atrium, right and left ventricle), kidney, and liver, using methods of fixation and embedding previously described L38 f, failed to reveal abnormal mitochondria.
NERVOUS SYSTEM. The brain weighed 1,300 gm. Coronal sections showed softening and grayish dis- coloration of the cerebral cortex. The brainstem, cer- ebellum, spinal cord, and peripheral nerves appeared normal.
Samples of tissue from cerebral cortex, thalamus, basal ganglia, cerebellum, spinal cord, nerve roots, lumbar plexus, sural nerve, and skeletal muscle were removed and prepared for light and electron micros- copy as described i38-j. Skeletal muscle samples from biceps, quadriceps, rectus abdominis, and psoas were also processed for histochemistry C39J
Neuronal loss and gliosis were prominent in the dentate nucleus of the cerebellum (Fig 2 ) and in the inferior olivary nuclei (Fig 3). Similar but milder changes were seen in the dorsal motor nucleus of the vagus and in vestibular nuclei. There was mild pallor of the posterior columns of the spinal cord. Some areas of the brain showed hypoxic changes. These included the frontal, parietal, and occipital cortex; hippocampus; caudate; putamen; and hypothalamus, where scattered eosinophilic neurons with pyknotic nuclei were seen in association with loosening of the neuropil and occa- sional activated microglial cells.
Fresh frozen sections of skeletal muscle from biceps, quadriceps, recms abdominis, and psoas showed ragged-red fibers with the modified Gomori trichrome stain (Fig 4A). These same fibers showed excessive oxidative enzyme reactions with succinic dehydroge- nase and nicotinamide adenine tetrazolium reductase stains (Fig 4B). Ultrastructural examination showed accumulations of abnormal mitochondria with charac- teristic paracrystalline inclusions (Fig 5). No abnor- mal mitochondria were seen in cerebral cortex, thal- amus, basal ganglia, cerebellum, spinal cord, or sural nerve.
24 Annals of Neurology Vol 26 No 1 July 1989

Fig 4. (A) Sectron of muscle showing ragged-redfbers (arrows). (Modified Gomori trichrome stain; X240 before 4% redaction.) (B) Saccinic dehydrogenase itain demonstrates accamulations of oxidative enzyme-positive material (arrows). [ X 240 before 4% reduction.)
Bzocbemistly We assayed the enzyme activities of COX (complex IV), succinate cytochrome c reductase (complexes I1 and III), rotenone-sensitive NADH cytochrome c re- ductase (complexes I and 1111, NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), and citrate synthase (matrix enzyme) in isolated mito- chondria from muscle, heart, brain, liver, and kidney (Fig 6). Cytochrome c oxidase activity was decreased in all tissues whereas other enzymes of the respiratory chain and citrate synthase were essentially normal. Be- cause normal values varied widely for different en- zymes and in different tissues, we expressed our pa- tients enzyme activities as standard deviation from the normal mean, to allow direct comparison of all the data This value is the difference between patient activ- ity and normal mean activity divided by the standard deviation for that particular assay and that particular tissue. Since control brains were obtained relatively
late post mortem (6 hours), COX activity showed large standard deviations (Table 2); this may explain why the patient's COX activity appeared less markedly de- creased in brain than in other tissues. The residual COX activity in isolated mitochondria varied not only in different tissues but also in separate preparations from the same tissue (see Table 2). Different prepara- tions from normal tissues also showed slight variation of COX activity, but this never exceeded 20%.
Kinetic analysis of COX was performed in crude muscle extracts and in mitochondria isolated from muscle and brain, using Lineweaver-Burk plots. In the conditions of the assay, the kinetics were monophasic (Fig 7). In all 3 experiments, we found that the appar- ent Michaelis constant (Km) in M E W tissues was lower than in control tissues. In some cases, variations in apparent Km have been attributed to differences in enzyme concentration: the higher the enzyme concen- tration, the higher the Km [40J However, we ex- cluded this factor by immunotitrating the enzyme con- centration by ELISA in every preparation used for kinetic studies.
Next, we tried to quantify the amount of enzyme
Lombes et al: MERRF with Cytochrome Oxidase Deficiency 25

Muscle 0 Hean A Brain A Liver 4 Kidney
Y f
9
40
n
P ‘4
succinate NADH Cllrate NADH SuccInale
Reduclase Reduclase
4$ COX
cyt. c cyt. c Synlhase Dehydmgonasa Dehydrogenasd
Fig 6. Enzyme activities in isolated mitochondria from mnpack, heart, brain, liver, and kidney. Values expressed as standard deviation from the normal mean (M): patient’s enzyme activity minus mean of normal activitylstandard deviation (see text). (Cyt. c = cytochrome c; NADH = nicotinumid adenine dinu- cleotide; SD = standard deviation; COX = cytochrome c oxi- dase.)
Table 2. Cytochrome c 0xida.re Activity in Patient with Myoclonic Epilepsy and Ragged-Red Fibers
Tissue Patienta Control Subject“
Brain 0.07 t 0.05 0.24 2 0.11 (n = 3)
(n = 6 )
(n = 3 )
(n = 3)
(n = 3 )
(n = 1 5 )
(n = 23)
(n = 11)
(n = 8 )
(n = 7 )
Muscle 0.28 ? 0.24 1.34 5 0.32
Heart 0.12 2 0.04 1.92 2 0.45
Liver 0.08 -t 0.04 0.37 2 0.11
Kidney 0.23 2 0.05 0.43 4 0.09
“Activities are pmol cytochrome c oxidizedmidmg rnitochondrial protein. Values are mean & standard deviation; n = number of determinations (patient) or number of specimens (control subjects).
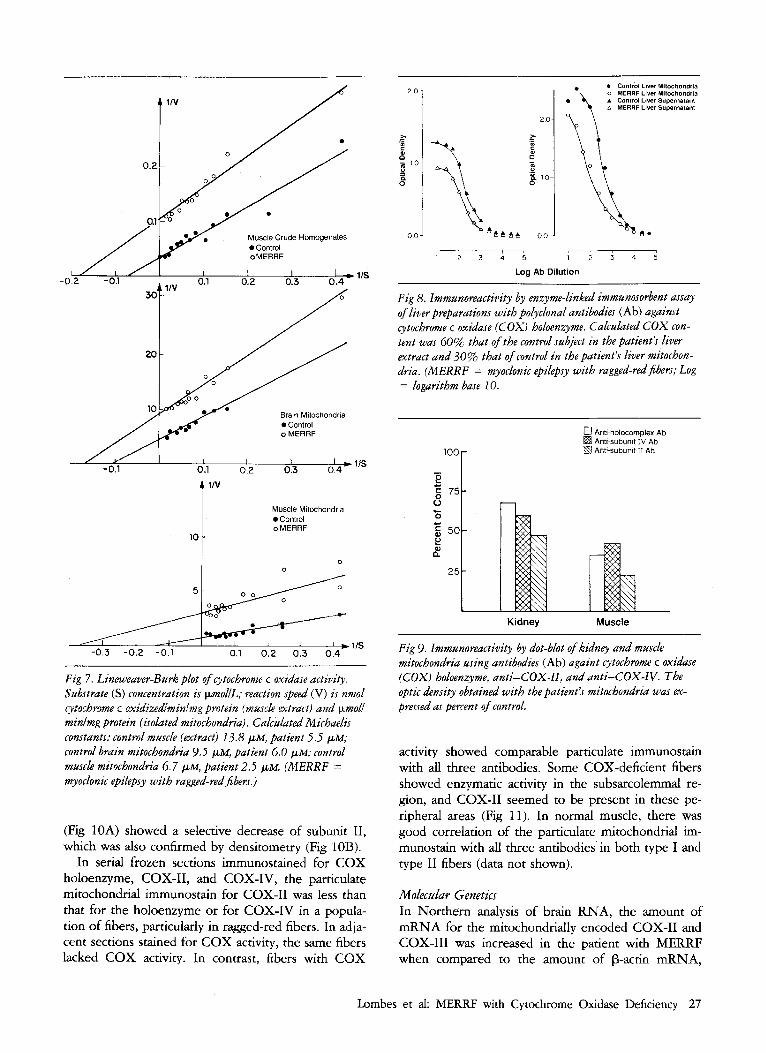
protein in the different tissues and to study the subunit composition of COX. For each tissue, the ELISA tech- nique was used, both with crude extracts and with isolated mitochondria (Fig 8). In muscle ( 5 experi- ments), heart (6 experiments), brain (3 experiments), and liver (9 experiments), a consistent decrease of cross-reacting material was seen using polyclonal anti- bodies against the COX holocomplex, although the decrease varied in different tissues and in different preparations from the same tissue. The results of dot- blot analysis on kidney and muscle mitochondria in- dicated a more marked decrease of CoX-11 than of COX-IV or holoenzyme (Fig 9). Western blots with monoclonal anti-IV and polyclonal anti-I1 antibodies
F i g 5 . Electron micrographs of skeletal mude showing paracrys- talline inclusions in mitochondria at low ( x 45,000 befre 28% reduction) (A) and high { x 94.000) (B) mugnijications. Some postmortem autolytic changes are evident.
26 Annals of Neurology Vol 26 No 1 July 1989
4 1IV
Muscle Crude Homogenates 0 Control oMERRF
I -02 -01 11s
Brain Mitochondria 0 Control o MERRF
Muscle Mitochondria Control I 11" o MERRF
l o t 0 I 0 1 -0.3 -0.2 -0.1 0.1 0.2 0.3 0.4 1 IS
Fig 7. Lineweaver-Burk plot of cytochrome c oxiduse actiuity. Substrate (S) concentvation is pmoliL; reaction speed (V) is nmol qtoihrome c oxidizedfminlmg protein (muscle extract) and p n o l f minimg protein (isolated mitochondriLt). Calcalated M ichaelis constants: control mzrscle (extract) 13.S pM, patient 5.5 pM; control brain mitochondria 9.3 pM, patient 6.0 p ~ ; control muscle mitochondria 6.7 p M , patient 2.5 p ~ . (MERRF = myoclonic epilepsy with ragged-red fibers.)
(Fig 10A) showed a selective decrease of subunit 11, which was also confirmed by densitometry (Fig 10B).
In serial frozen sections immunostained for COX holoenzyme, COX-11, and COX-IV, the particulate mitochondrial immunostain for COX-I1 was less than that for the holoenzyme or for COX-IV in a popula- tion of fibers, particularly in ragged-red fibers. In adja- cent sections stained for COX activity, the same fibers lacked COX activity. In contrast, fibers with COX
2 0
1 2 3 4 5 1 2 3 4 5
Log Ab Dilution
Fig 8. Immunoreactivity by enzyme-linked immunosorbent assay of liver preparations with poiyclonal antibodies (Ab) against cytochrome c oxidase (COX) holoenzyme. Calculated COX con- tent was 60% that of the control subject in the patient's liver extract and 30% that of control in the patient's liver mitochon- dria. (MERRF = myoclonic epilepsy with ragged-red fibers; Log = logarithm base 10.
0 Anti-holocomplex Ab Anti-subunit IV Ah Anti-subunit I1 Ah
Kidney Muscle
Fig 9. Immunoreactivity by dot-blot of kidney and muscle mitochondria using antibodies (Ab) againt cytochrome c oxiduse (COX) holoenzyme, anti-COX-II, and anti-COX-IV. The optic density obtained with the patient's mitochondria was ex- pressed as percent of control.
activity showed comparable particulate immunostain with all three antibodies. Some COX-deficient fibers showed enzymatic activity in the subsarcolemmal re- gion, and COX-I1 seemed to be present in these pe- ripheral areas (Fig 11). In normal muscle, there was good correlation of the particulate mitochondrial im- munostain with all three antibodies in both type I and type I1 fibers (data not shown).
Molecdar Genetics In Northern analysis of brain RNA, the amount of mRNA for the mitochondrially encoded COX-I1 and COX-I11 was increased in the patient with MERRF when compared to the amount of p-actin mRNA,
Lombes et al: M E W with Cytochrome Oxidase Deficiency 27

A
0 Anit-holocornplex An Anti-subunlf IV Ab
R Anti-subunit I I An
P g 75 e
0 - 1
50 e p"
25
Kidney Muscle Brain
B
Fig 10. Western blot using antibodies (Ab) against cytochrome c oxidzse (COXj-II and COX-IV. (A) Lane 1, Purified beef heart COX; 2, patient's kidney mitochondria (65 pg); 3, controlsub- ject's kidnq mitochondria (65 pgj; 4, patient's muscle mitochon- dria (25 ~ g ) ; 5, control's muscle mitochondria (25 kg); 6, pa- tient's brain mitochondria (75 pg); 7, control's brain mitochondria (75 pgj. Marker sizes are shown in kiladultons. (B) Densitometvy of immunoblot. Results expressed as percent of control (see Fig 9).
which was used as internal standard. In contrast, the amount of mRNA for 2 of the nuclearly encoded COX subunits (IV and VIII) was comparable to that of the control subject. No abnormally sized mRNA was seen for any of those subunits (Fig 12).
Southern analysis of cardiac mtDNA excluded any detectable deletion large enough to modify the mtDNA size (Fig 13).
Discussion In 1973, Tsairis and associates { S ] described a family with progressive myoclonic epilepsy associated with ragged-red muscle fibers indicative of a mitochondrial myopathy. Fukuhara and colleagues {b} reported 2 ad- ditional cases and suggested that M E W represented
a separate nosological entity distinguishable from other mitochondrial encephalomyopathies. Additional re- ports supported the notion that M E W represents a rare but identifiable syndrome {7- 161. Features com- mon to all cases are mitochondrial myopathy, myo- clonus, and ataxia. Common but inconsistent features include generalized seizures, hearing loss, and demen- tia, while optic atrophy and neuropathy are less fre- quent.
Clinical heterogeneity is common in MERRF fam- ilies, in which some members may have only a few symptoms while others are severely affected, as is exemplified by the cases of Rosing and associates 171. The family we studied also showed remarkable clini- cal heterogeneity. The principal manifestation in the mother (Patient 111-4) was myopathy with insidiously progressive weakness in a limb-girdle distribution. This led to muscle biopsy and to the first description of ragged-red fibers in this disorder {5}. In the daughters (Patients IV-8 and IV-9; see Fig l), the tempo of the disease was more rapid, and the presence of myo- clonus, ataxia, and hearing loss suggested predominant central nervous system (CNS) involvement. Both
28 Annals of Neurology Vol 26 No 1 July 1989

Fig I I . Serial fmzen sections stainedfor cytochrame c oxidase (COX) activity and immunoreactivity to antibodies against COX hobenzyme, COX-IV, andCOX-II. {A) Thesection stained for COX activity contains several COX-deficient fibers (circles), s o w of which show activity confined to the cellperz$h-
late reaction in all fibers, iucluding those with subsarcolemmal collections of mitochondria (arrows). (c) COX-IV immunostain
the spinal cord. In their patients, Berkovic and col- leagues Ell} described foci of necrosis and capillary proliferation in the brainstem similar to the lesions
our case. There was a striking difference between the pathological findings in muscle and brain. While prolif-
ey (arrows). (B) COX holoenzyme immt/nojt& partila- Seen with syndrome; these were not present in
shows mito&oudrial reaction in all fibers. (0) COX-II im- munostain shows diminished reaction in COX-deficient fibers (circles). The reaction is more intense at the peripbey of some of thesejbers (arrows). ( x 220.)
CNS and skeletal muscle were clearly affected in the son, our propositus. Neuropathology has been de- scribed in 4 families with MERRF [lo, ll, 131 and the findings were consistent and similar to those reported here. Neuronal cell loss was confined to the dentate nucleus and inferior olivary complex with an associated Purkinje cell dropout. Findings in other parts of the brain were lunited to pallor of posterior columns of
eration of mitochondria and presence of mitochondrial paracrystalline inclusions were the pathological hall- mark in muscle, no alteration of mitochondria was seen in the brain.
M E W syndrome seems to be transmitted by non- Mendelian maternal inheritance. The family reported by Rosing and associates {7} best exemplifies this pat- tern. Maternal inheritance was also suggested by the pedigree studied by Berkovic and colleagues 1111 and by the present family.
The combination of clinical, neuropathological, and genetic features sets MERRF apart from two other distinctive multisystem mitochondrial disorders with
Lombes et al: MERRF with Cytochrome Oxidase Deficiency 29

Fig 12. Northern blot of brain RNA. Three different mem- branes were used. One (right panel) was hyhdized sequentially with probes for p-actin, cytocbrome c oxidase (COX)-Ill, and COX-11. A second membrane (middle panel) was hybridized sequential& with probesfor P-actin and COX-VlIl. The third membrane (left panel) was hybridized with probes for p-actin and COX-IV. P-Actin was used as an internal standzrdfor the amoant of mRNA loaded on the gel. Relative to p-actin, the amount of RNA in the patient’s brain was comparable or higher than in control brain fop. all COX subunits studied. The size of each band is as foElws: pactin, 1.9 kiloaucleotides; COX-III, 780 nudeotides (nt); COX-11, 680 nt; COX-IV, 700 nt; COX- VIII, 550 nt. (C = control subject; M = patient with myo- clonrc epilepsy with ragged-redfibers.)
ragged-red fibers, the Kearns-Sayre syndrome and mi- tochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) {41]. Although the validity of such distinction has been challenged 1421, accumulating biochemical and molecular genetic data support the concept of distinct causes for the three syndromes. The results of biochemical studies in KSS have been inconsistent 143, 441, but recent evidence indicates that large deletions of mtDNA are fre- quently, perhaps invariably, found in this disorder {45]. Defects of complex I of the respiratory chain have been reported in numerous patients with MELAS 1461, such that complex I deficiency appears to be the major, though not necessarily the only, biochemical cause of this syndrome.
Biochemical studies of M E W patients have been scarce, but the maternal inheritance of this disorder suggests that the biochemical lesion should affect one of the four respiratory chain complexes containing mtDNA-encoded subunits: complex I (NADH- coenzyme Q reductase), complex I11 (reduced coen- zyme Q-cytochrome c reductase), complex 1V (COX), or complex V (mitochondrial ATPase). We found nor- mal activities of complex I and complex 111, but vari- ably decreased activity of cytochrome oxidase in multi- ple tissues from our patient with MERRF. Of the 13 subunits of COX, the 3 largest (I, 11, and 111) are en- coded by mtDNA and synthesized within the mito- chondria 147-501. They subserve the main catalytic functions of the enzyme. Electron transfer from cyto- chrome c to oxygen and proton translocation across the inner mitochondrial membrane are clearly related to subunits I and 11, and binding of cytochrome c to subunit I1 151-531. COX-111 seems to play a role in proton translocation. Two lines of circumstantial evidence suggest that COX deficiency in our patient with MERRF was due to a defect of COX-11, one based on kinetic abnormalities, the other on a selective decrease of COX-I1 relative to the holoenzyme and to COX-IV, a nuclear-encoded subunit.
We found lower than normal Km for cytochrome c in 3 different tissue preparations of our patient. Be- cause we had to use crude tissue extracts or subcellular
30 Annals of Neurology Vol 26 No 1 July 1989

Fig 13. Southern blot of heart mitochondrial DNA (mtDNA). Restriction fragment pattern after digestion with PPU II. Probe was radiolabeled liver human mtDNA. Marker sizes (lambda digested with Hindlll) are shown in kilobases. (C = control subject; M = patient with m.yoclonic epilepsy with ragged-red fibers.)
preparations, our data cannot be directly compared to published Km values for COX, which were obtained with purified enzyme 154-561. However, comparison with many similar preparations from normal tissues suggests that the changes were not artifactual. Further- more, the rate-limiting step in the electron transfer from ferrocytochrome c to oxygen seems to be the dissociation of ferricytochrome c from the enzyme, specifically from subunit I1 of COX [54}. An increased affinity of COX-I1 for cytochrome c could reduce the COX reaction speed and explain the simultaneous de-
crease in maximum velocity and b, a situation rarely encountered in human pathology 1571.
The second line of evidence pointing to an abnor- mality of COX-I1 comes from immunochemical and immunohistochemical data, which indicate a more marked decrease of COX-I1 compared to other sub- units. The selective decrease of COX-I1 was best doc- umented by comparing the immunoreactivity of sub- units I1 and IV in Western blots and dot-blots. Although the selective decrease of COX-11 was appar- ent when compared to the concentration of COX-IV, a nuclear-encoded subunit, a more generalized defect of all 3 mtDNA-encoded COX subunits cannot be excluded by our data because monospecific antibodies against COX-I and COX-111 were not available.
It is unlikely that the decreased amount of COX-I1 could be due to a decreased rate of transcription or to an increased rate of degradation of processed mRNA because Northern analysis showed a normal or in- creased amount of mRNA. A point mutation of the COX-I1 gene could cause altered kinetics and accel- erated degradation of the abnormal protein, somehow resulting in a disproportionate decrease of COX-11. Definition of the molecular defect will require direct sequencing of the mutant gene or of the abnormal protein.
Irrespective of the exact nature of the molecular defect, any biochemical error caused by a mutation of mtDNA should be generalized but variably expressed in different tissues, depending on the relative propor- tion of normal and mutant mtDNA in each tissue. In our patient, the defect of COX activity and COX im- munoreactivity was generalized, partial, and variably expressed in different tissues. Immunohistochemistry showed the presence of two populations of mitochon- dria in individual muscle fibers, a population with nor- mal COX activity and COX-I1 immunoreactivity and a population that showed decreased enzyme activity and decreased COX-I1 immunostain. This dual population of mitochondria can explain the variability of COX activity and COX immunoreactivity that was observed in different tissues. The existence in muscle of two mitochondrial populations has been shown by Holt and colleagues [58} and Zeviani and associates 1451 in patients with mitochondrial myopathies and Kearns- Sayre syndrome associated with mtDNA deletions. Although our MERRF patient had no apparent dele- tion of mtDNA, he also showed two populations of mitochondria based on COX histochemistry and im- munocytochemistry .
Addendum In a recent paper Wallace and co-workers 1591 re- ported a combined defect of complex I and complex IV in muscle biopsies from 2 members of the pedigree described by Rosing and associates 171. A dual popula-
Lombes et al: MERRF with Cytochromc Oxidase Deficiency 31

tion of mtDNAs (heteroplasmy) was suggested by variation in the mitochondrial energetic capacity be- tween family members. N o deletion of mtDNA was detected by Southern blot analysis.
Supported by Center grants from NINCDS (NS 11766) and the Muscular Dystrophy Association, and by a generous donation from kbero and Graziella Danes1 Dr Lombes was supported by postdoc- toral fellowships from the Fondation pour la Recherche Medicale and from the Mirustkre Franps des Relations Extkrieures, and from the Muscular Dystrophy Associanon. Dr Nakase was supported by the Uehara Scienufic Foundation and Dr Zeviam by the As- sociazione per la Promozione delle hcerche Neurologche (ARIN) Drs &zzuto and Nakahara are fellows of the Muscular Dystrophy Association
Punfied beef heart COX was a generous gift of Dr Roberto Bisson, University of Padova, Italy. Polyclonal antibodies agamst bovine subunit I1 (COX-11) were kindly provided by Dr Roderick A. Capaldi, Utuversity of Oregon, Eugene Mitochondrial cDNA probes were kindly provided by Dr Giuseppe Attardi, California Institute of Technology, Pasadena, and the p-actin probe was a gift from Drs Peter Gunning and Lawrence Kedes, Stanford University, Stanford, California
We thank Dr Eric A. Schon for his comments and Ms Mary Tor- torelis for typing the manuscript
References 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
32
Berkovic SF, Andermann F, Carpenter S, et al. Progressive myoclonus epilepsies: specific causes of diagnosis. N Engl J Med 1986;3 15296-305 Lafora G R Uber das borkommen amyloider korperchen imm innern der ganglienzellen; zugleich ein beitrag zum studium der amyloiden substanz in nervensystem. Virchows Arch (Path01 Anat) 1911;205:295-303 Zeman W, Donahue S, Kyken P, et al. The neuronal ceroid- lipofuscinoses (Batten-Voget syndrome). In: Vinken PJ, Bruyn GW, eds. Hbdbook of clinical neurology, Vol 10. Amsterdam: North Holland, 1970;588-679 Lowden JA, OBrien JS. Sialidosis: a review of human neuraminidee deficiency. Am J Hum Genet 1979;31:1-18 Tsairis P, Engel WK, Kark P. Familial myoclonic epilepsy syn- drome associated with skeletal muscle mitochondrial' abnor- malities. Neurology 1973;23:408 Fukuhara N, Tokguchi S, Shirakawa K, et al. Myoclonus epilepsy associated with ragged-red fibers (mitochondrial abnor- malities): disease entity or syndrome? J Neurol Sci 1980;47:
Rosing HS, Hophns LC, Wallace DC, et al. Maternally inher- ited mitochondrial myopathy and myoclonic epilepsy. Ann Neurol 1981;17:228-237 Fitzsimons R, ClifronlBligh P, Wolfenden W. Mitochondrial myopathy and lactic acidemia with myoclonic epilepsy, ataxia and hypothalamic infertility: a variant of Ramsay-Hunt syn- drome? J Neurol Neurosurg Psychiatry 1981;44:79-82 Morgan-Hughes J, Hayes D, Clark J, et al. Mitochondrial en- cephalomyopathies. Biochemical studies in' two cases revealing defects in the respiratory chain. Brain 1982;105:553-582 Sasaki H, Kuzuhara S, Kanazawa I, et al. Myoclonus, cerebellar disorder, neuropathy, mitochondrial myopathy and ACTH deficiency. Neurology 1983;33:1288-1293
117-133
Annals of Neurology Vol 26 No 1 July 1989
11. Berkovic SF, Carpenter S, Karpati G, et al. Cytochrome c oxi- dase deficiency: a remarkable spectrum of clinical and neuro- pathologic findings in a single family. Neurology 1987;37: 223 (abstract)
12. May DL, White HH. Familial myoclonus, cerebellar ataxia and deafness. Specific genetically determined disease. Arch Neurol
13. Nakano T, Sakai H, Naoji A, et al. An autopsy case of de- generative type myoclonus epilepsy associated with Friedreich's ataxia and mitochondrial myopathy. Brain and Nerve 1982;
14. Feit H, Kirkpatrick J, Vanwoert MH, Pandian G. Myoclonus, ataxia and hypoventilation: response to L5 hydrotryptophan. Neurology 1983;33:105-112
15. Rims JE, Schochet SS, Fakadej AV, et al. Mitochondrial en- cephalomyopathy with decreased succinate cytochrome c reduc- tase activity. Neurology 1984;34:48-53
16. Mendell JR, Barohn RJ, Yates AJ, et al. Autopsy case of myo- clonic epilepsy and ragged-red fibers with cytochrome c oxidase deficiency: evidence for a maternally inherited biochemical de- fect. Ann Neurol 1987;22:28 (abstract)
17. Bookelman H, Trijbels JMF, Sengers RCA, Janssen AJM. Mea- surement of cytochromes in human skeletal muscle mitochon- dria isolated from fresh and frozen stored muscle specimens. Biochem Med 1978;19:366-373
18. Lai JCK, Clark JB. Preparation of synaptic and non-synaptic mitochondria from mammalian brain. Methods Enzymol 1979;
19. Sottocasa GL, Kuylenstierna B, Ernster L, Begstrand A. An electron transport system associated with the outer membrane of the mitochondria. J Cell Biol 1967;32:415-438
1968;19:331-338
34:321-332
55!51-53
20. Srere PA. Citrate synthase. Methods Enzymol 1369;13:3-11 21. King TE, Howard RL Preparation and properties of soluble
NADH dehydrogenase from cardiac muscle. Methods Enzymol
22. King TE. Preparation of succinate dehydrogenase and recon- stitution of succinate oxidase. Methods Enzymol 1967;10:322- 331
23. Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 1967;10:245-250
24. Lowry OH, Rosenbrough NJ, Farr AL, Randali RJ. Protein measurement with the folin phenol reagent. J Biol Chem
25. Bresolin N, Zeviani M, Bonilla E, et al. Fatal infantile cyto- chrome c oxidase deficiency: decrease of immunologically de- tectable enzyme in muscle. Neurology 1985;35:802-812
26. Nakagawa M, Miranda AF. A monoclonal antibody against cytochrome c oxidase distinguishes cardiac and skeletal muscle mitochondria. Exp Cell Res 1987;168:44-52
27. Hawkes R, Niday E, Gordon J. A dot-immunobinding assay for monoclonal and other antibodies. Anal Biochem 1982;119:
28. Kadenbach B, Jarauch J, Hartmann R, Merle P. Separation of mammalian cytochrome c oxidase into 13 polypeptides by a sodium dodecylsulfate-gel electrophoretic procedure. Anal Biochem 1983;129:5 17-52 1
29. Towbin H, Stachelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: pro- cedure and some applications. Proc Natl A c d Sci USA 1979;76:43 50-43 54
30. Bonilla E, Miranda AF, Prelle A, et al. Immunocycochemical study of nebulin in Duchenne muscular dystrophy. Neurology
3 1. Ulrich A, Shine J, Chirgwin J, et al. Rat insulin genes: construe-
1967;10:275-294
1951;193:265-275
142-147
1988;38: 1600-1603 -
tion of plasmids containing the coding sequences. Science 1977;196 1313-1319
32. Lehrach H, Diamond D, Wozney JM, Boedtker H. RNA

molecular weight determinations by gel electrophoresis under denaturing conditions: a critical reexamination. Biochemistry 1977;16:4743-475 1
33. Church G, Gilbert W. Genomic sequenciqq. Proc Natl Acad Sci USA 1984;81:1991-1995
34. Palva TK, Palva ET. Rapid isolation of animal mitochondrial DNA by alkaline extraction. FEBS Lett 1984;192:267-270
35. Southern E. Detection of specific sequences among DNA frag- ments separated by gel electrophoresis. J Mol Biol 1975;98:
36. Benton WD, Davis RW. Screening Agt recombinant clones by hybridization of single plaques in situ. Science 1977;196:180- 183
37. Feinberg AP, Vogelstein B. A technique for radiolabelling DNA restriction endonuclwe fragments to high specific activ- ity. Anal Biochem 1984;137:266-267
38. Mendell JR, Sahenk Z, Whitaker JN, et al. Polyneuropdthy and IgM monoclorial gammopathy: studies on the pathogenetic role of anti-myelin-associated glycoprotein antibody. Ann Neurol
39. Warmola JR, Mendell Jk Open-biopsy EMG: direct correla- tion of a pattern of excessively recruited, pathologically s m d motor unit potentials with histologic evidence of neuropathy. Arch Neurol 1979;36:406-409
40. Ferguson-Miller S, Brautigan DI, Margoliash E. Correlation of the kinetics of electron transfer activity of ,v;irious eukaryotic cytochromes c with binding to mitochondrial cytochrome c oxi- dase. J Biol Chem 1976;251:1104-1115
41. Pavlakis SG, Phillips PC; DiMauro S, er al. Mitochondrial my- oparhy, encephalopathy, lactic acidosis, and stroke-like epi- sodes: a distinctive clinical syndrome. Ann Neurol 1984;16: 481-488
42. Petty RKH, Harding AE, Morgan-Hughes JA. The clinical fea- tures of mitochondrial myopathy. Brain 1986;103:915-938
43. Muller-Hocker J, Johannes A, Drosto M. Fatal mitochondrial cadiomyopathy in Kearns-Sayre syndrome with deficiency in cytochrome c oxidase in cardiac and skeletal muscle. Virchows Arch (Cell Pathol) 1986;52:353-367
44. Bresolin N, Moggio M, Bet L, et al. Progressive cytochrome c oxidase deficiency in a case of Kearns-Sayre syndrome: mor- phological, immunological, and biochemical studies in muscle biopsies and autopsy tissues. Ann Neurol 1987;21:564-572
45. Zeviani M, Moraes CT, DiMauro S , et al. Deletions of mite chondrial DNA in Kearns-Sayrc syndrome. Neurology 1988;
503-517
1985;17:243-254
38~1339-1346
46. Koga W, Nonaka I, Kobayashi M, et al. Muscle pathology in complex I (NADH-CoQ reductase) deficiency. Ann Neurol
47. Tzagoloff A. Cytochrome oxidase: model of a membrane en- zyme. In: Tzagoloff A, ed. Mitochondria. New York: Plenum Press, 1982:lll-130
48. Azzi A, Cytochrome c oxidase. Towards a clarification of its structure, interactions and mechanism. Biochim Biophys Acta
49. Capaldi RA, Malatesta F, Darley-Usmar VM. Structure of cyttr chrome c oxidase. Biothim Biophys Acta 1983;726: 135-148
50. Kadenbach B, Kuhn-Nentwig L, Buge U. Evolution of a regula- tory enzyme: cytochrome t oxidase (complex 1V). In Lee CP, ed. Current topics in bioenergetics, Vol 15. New York: Aca- demic, 1987:114-162
51. Bisson R, Azzi A, Gutiveniger H, et al. Interaction of cyto- chrome c with cytochrome c oxidase. J Biol Chem 1978;253:
52. Millett F, Darley-Usmar V, Capaldi RA. Cytochrome c is cross- linked to subunit I1 of cytochrbme c oxidase by a water-soluble carbodiimide. Biochemistry 1982 ;2 1 : 3 85 7- 3 862
53. Millett F, de Jong C, Paulson L, Capaldi RA. Identification of specific carboxylate groups on cytochrome c oxidase that are involved in binding cytochrome c. Biochemistry 1983;22:546- 552
54. Gibson QH, Greenwood C, Wharton IX , Palmer G. The reac- tion of cytochrome oxidase with cytochrome c. J Biol Chem
55. Wilms J, Veerman ECI, Konig BW, et al. Ipnic strength effects on cytochrome aaj kinetics. Biochim Biophys Acta 1981; 635: 13-23
56. Hasinoff BB, Davey JP. The kinetics of the aerobic oxidation of ferrocytochrome c by cytochrome c oxidase in solvents of in- creased viscosity are pixtially diffusion controlled. Biochim Biophys Acta 1987;192:1-9
57. Bresolin N, Miranda A, Chang HW, et al. Phosphoglycerate kinase deficiency myopathy: biochemical and immunological studies of the murant enzyme. Muscle Nerve 1984;7:542-551
58. Holt IJ, Harding AE, Morgan-Hughes JA.,Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988;33 1:717-7 19
59. Wallace DC, Zheng X, Lon MT, et al. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological and biochemical characterization of a mitochondrial DNA disease. Cell 1988;55:60 1-610
1988;24:749-756
1980;594:231-252
1874-1 880
1965 ;240:888-894
Lombes et al: M E W with Cytochrorne Oxidase Deficiency 33
Related Documents











