Mutual alloying of XAs (X = Ga, In, Al) materials: Tuning the optoelectronic and thermodynamic properties for solar energy applications Bakhtiar Ul Haq a , R. Ahmed a,⇑ , F. El Haj Hassan b , R. Khenata c , Mohd Khalid Kasmin a , Souraya Goumri-Said d,⇑ a Department of Physics, Faculty of Science, Universiti Teknologi Malaysia, UTM Skudai, 81310 Johor, Malaysia b Universite ´ Libanaise, Faculte ´ des sciences (I), Laboratoire de Physique et d’e ´lectronique (LPE), Elhadath, Beirut, Lebanon c Laboratoire de Physique Quantique et de Mode ´lisation Mathe ´matique, Universite ´ de Mascara, Mascara 29000, Algeria d Physical Science Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia Received 18 August 2013; received in revised form 19 November 2013; accepted 22 November 2013 Communicated by: Associate Editor Nicola Romeo Abstract In the present work we did mutual alloying of the versatile XAs (X = Ga, In, Al) materials in order to improve their efficiency and enhance their range of technological applications using state of the art first principles method. We investigate the structural, electronic and thermodynamic properties of Ga 1x Al x As, Ga 1x In x As and In 1x Al x As for x = 0.25, 0.50, and 0.75. Calculations have been per- formed using the density functional theory (DFT) as implemented within the full potential linearized augmented plane wave plus local orbital (FP-LAPW + lo) method. For exchange and correlation energy treatment, we employed the local density approximations (LDA) as proposed by Wang and Perdew and the generalized gradient approximation (GGA) from Perdew et al. proposed. To calculate the accurate band structure, recently modified Becke Johnson (mBJ) potential was suggested as an alternative. Our calculations show a linear fall in the lattice constant in contrast to linear rise in bulk moduli of Ga 1x Al x As and In 1x Al x As with the increase of Al concen- tration. However the change of indium concentration in Ga 1x In x As is displaying a reverse effect. The energy band gap of Ga 1x Al x As and In 1x Al x As was found to be increased, where a crossover from direct to indirect band gap has been observed with the increase of Al concentration. This direct to indirect crossover was found at 93.4% of Al concentration for Ga 1x Al x As and at 84.63% of Al concentration for In 1x Al x As. The effect of the mutual alloying of XAs materials on the thermodynamic properties is comprehensively reported. Ó 2013 Elsevier Ltd. All rights reserved. Keywords: Mutual alloying; Ternary alloys; Critical temperature; FP-LAPW; DFT 1. Introduction III–V semiconductor compounds family especially the common anion XAs (X = Al, Ga, In) have been received considerable attention for their significant role in the semi- conductors industry. These are specially valued for their applications in optoelectronic systems and devices such as lasers, solar cells, and light emitting diodes. Moreover, AlAs and GaAs owing their specific characteristics are used in the formation of epitaxial multilayer structures such as heterojunction bipolar transistors, Bragg reflector super lattices including solid state lasers and high electron mobil- ity transistors (Ferreira et al., 1989). GaAs, as a direct band gap semiconductor material, has shown its promising and 0038-092X/$ - see front matter Ó 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.solener.2013.11.020 ⇑ Corresponding authors. Tel.: +966 562885316 (S. Goumri-Said). E-mail addresses: [email protected], rasofi@hotmail.com (R. Ahmed), [email protected] (S. Goumri-Said). www.elsevier.com/locate/solener Available online at www.sciencedirect.com ScienceDirect Solar Energy 100 (2014) 1–8

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Available online at www.sciencedirect.com
www.elsevier.com/locate/solener
ScienceDirect
Solar Energy 100 (2014) 1–8
Mutual alloying of XAs (X = Ga, In, Al) materials: Tuningthe optoelectronic and thermodynamic properties for solar
energy applications
Bakhtiar Ul Haq a, R. Ahmed a,⇑, F. El Haj Hassan b, R. Khenata c, Mohd Khalid Kasmin a,Souraya Goumri-Said d,⇑
a Department of Physics, Faculty of Science, Universiti Teknologi Malaysia, UTM Skudai, 81310 Johor, Malaysiab Universite Libanaise, Faculte des sciences (I), Laboratoire de Physique et d’electronique (LPE), Elhadath, Beirut, Lebanon
c Laboratoire de Physique Quantique et de Modelisation Mathematique, Universite de Mascara, Mascara 29000, Algeriad Physical Science Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
Received 18 August 2013; received in revised form 19 November 2013; accepted 22 November 2013
Communicated by: Associate Editor Nicola Romeo
Abstract
In the present work we did mutual alloying of the versatile XAs (X = Ga, In, Al) materials in order to improve their efficiency andenhance their range of technological applications using state of the art first principles method. We investigate the structural, electronicand thermodynamic properties of Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs for x = 0.25, 0.50, and 0.75. Calculations have been per-formed using the density functional theory (DFT) as implemented within the full potential linearized augmented plane wave plus localorbital (FP-LAPW + lo) method. For exchange and correlation energy treatment, we employed the local density approximations(LDA) as proposed by Wang and Perdew and the generalized gradient approximation (GGA) from Perdew et al. proposed. To calculatethe accurate band structure, recently modified Becke Johnson (mBJ) potential was suggested as an alternative. Our calculations show alinear fall in the lattice constant in contrast to linear rise in bulk moduli of Ga1�xAlxAs and In1�xAlxAs with the increase of Al concen-tration. However the change of indium concentration in Ga1�xInxAs is displaying a reverse effect. The energy band gap of Ga1�xAlxAsand In1�xAlxAs was found to be increased, where a crossover from direct to indirect band gap has been observed with the increase of Alconcentration. This direct to indirect crossover was found at 93.4% of Al concentration for Ga1�xAlxAs and at 84.63% of Al concentrationfor In1�xAlxAs. The effect of the mutual alloying of XAs materials on the thermodynamic properties is comprehensively reported.� 2013 Elsevier Ltd. All rights reserved.
Keywords: Mutual alloying; Ternary alloys; Critical temperature; FP-LAPW; DFT
1. Introduction
III–V semiconductor compounds family especially thecommon anion XAs (X = Al, Ga, In) have been receivedconsiderable attention for their significant role in the semi-
0038-092X/$ - see front matter � 2013 Elsevier Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.solener.2013.11.020
⇑ Corresponding authors. Tel.: +966 562885316 (S. Goumri-Said).E-mail addresses: [email protected], [email protected]
(R. Ahmed), [email protected] (S. Goumri-Said).
conductors industry. These are specially valued for theirapplications in optoelectronic systems and devices such aslasers, solar cells, and light emitting diodes. Moreover,AlAs and GaAs owing their specific characteristics are usedin the formation of epitaxial multilayer structures such asheterojunction bipolar transistors, Bragg reflector superlattices including solid state lasers and high electron mobil-ity transistors (Ferreira et al., 1989). GaAs, as a direct bandgap semiconductor material, has shown its promising and

2 B.U. Haq et al. / Solar Energy 100 (2014) 1–8
efficient role in the photovoltaic system. Overall XAs has awide range of applications including the fabrication ofphotodetectors, photodiodes, infrared spectrum lasers,electro-optic modulators, and frequency-mixing compo-nents (Vurgaftman et al., 2001). However, the rigorousdevelopment in devices and their demanding efficiencieshas enthused researchers to search for appropriate novelbase materials or to efficiently modify the physical proper-ties of existing materials. The advantage of XAs materialsis that the properties of XAs can easily be modified, andnew dimensions can be tailored by their mutual alloyingaccording to the demands of technology. Because of theirnearly same lattice constants and symmetrical cubic struc-tures, XAs compounds may help with their alloying toreach a maximum composition.
To enhance the range of technological applications,mutual alloying of XAs has been the subject of some exper-imental and theoretical studies. Csavinszky and Brownstein(1983, 1984) analytically computed the dielectric function,spatial and site dependent of Ga1�xAlxAs for different con-centrations. Emeny et al. (1991) have investigated thedesorption rate of Indium, using photoluminescence tech-nique, from strained GaInAs alloy system. Zawadzkiet al. (1999) have investigated the cyclotron masses of 2Delectrons in GaAs/GaAlAs hetrostructure. Grenet et al.(1996) treated the surface segregation for GaInAs and AlI-nAs alloys for different concentrations grown via molecularbeam epitaxy. One author of us has investigated the struc-tural, electronic, optical and thermodynamic properties ofGaAlAs alloys using first principles approach (Haj Hassanet al., 2010). Zhang et al. (1999) studied the quaternaryalloys GaAs1�xPx–Al0.3Ga0.7As (001) interface bondswithin DFT in order to determine the band offset. Srivast-ava et al. (2001) used the full potential self consistent linearmuffin tin orbital method within DFT to investigate thestructural and electronic properties of different Ga1�xAlx-
As alloys and they reported a linear variation in the latticeconstant with constituent atoms changes. Merabet et al.(2013) has reported the magnetic behavior of GaAlAswhen doped with Mn element, that can find applicationsin spintronics. Using tight binding and FP-LAPW methodTit et al. (2010) have explored the band gap character ofGaInAs. Recently, Ameri et al. (2012) have studied thestructural and electronic properties of InAlAs withinDFT using LDA and GGA. Though some piecemealreports are found in literature about XAs alloys, a compre-hensive and thorough study of XAs alloys is still elusive.
Being a prospective optoelectronic materials, and toextend their applications over an entire solar spectrum vis-ible to infrared, and to make them viable to the thought ofcheaper and efficient green energy technology, tuning theoptoelectronic and thermodynamic properties by mutualalloying of XAs materials is immeasurably demanded.
In this work, we carried out calculations for structural,electronic and thermodynamic properties of Ga1�xAlxAs,Ga1�xInxAs and In1�xAlxAs (x = 0.25, 0.50 0.75) withinthe framework of DFT (Kohn and Sham, 1965; Hohenberg
and Kohn, 1964) by applying (FP-LAPW + lo) methodSjostedt et al., 2000; Madsen and et al., 2001. For the treat-ment of exchange correlation energy we used Wang andPerdew proposed local density approximations (LDA)Perdew and Wang, 1992 and Perdew and et al. (1992)generalized gradient approximation (GGA). In order toovercome the underestimation on the band gaps from thecommon LDA and GGA approximations, we have usedthe newly suggested and most reliable technique calledmodified Becke–Johnson (mBJ) potential approximation(Tran and Blaha, 2009; Koller et al., 2012), because it iscapable to accurately describe the electronic structure ofsemiconductors and insulators (Anua, 2013).
2. Theoretical and computational approaches
In the present first principles study, we have performedour calculations using FP-LAPW + lo method realizedwithin the structure of DFT. In this method of calcula-tions, wave functions and their derivatives are made con-tinuous at the boundary of the spheres, and there is noshape approximations imposed on either the crystallinecharge density or potential. Calculations have been per-formed using WIEN2 k code (Blaha et al., 2001) which isthe practical implementation of FP-LAPW + lo. To treatthe exchange and correlation energy, LDA of Wang andPerdew and GGA of Perdew et al. and, Tran-Blaha’smBJ potential have been used. The model is based on con-sidering the simulated unit cell to be divided into tworegions: Muffin-Tin (MT) and interstitial. In the formerregion, a linear combination of the atomic like wave func-tions times the spherical harmonics are used to expandKohn–Sham wave functions, charge density and potentialwhereas in the later region, plane wave basis set is applied.In our calculations, to expand the wave functions insideMT spheres lmax = 10 and in the interstitial region, energycutoff Kmax = 8.0/RMT (Ryd)1/2 was taken into account.Similarly in MT sphere, charge density and potential areexpanded into lattice harmonics and in the interstitialregion Fourier series is applied. The RMT were chosen forAl and As as 1.94, and for Ga and In as 2.21. The Fourierexpanded charge density was truncated at Gmax = 16 au�1.For well convergence of energy we used 72 k points in thespecial irreducible Brillouin zone (BZ). To obtain betterresults, the total energy was converged to 10�5 Ryd/unitcell in our present self-consistent computations.
3. Discussion of results
To investigate the structural properties of XAs alloys wefirst constructed a supercell by following the special Quasi-Random Scheme approach as suggested by Zunger et al.(1990). In this scheme different structures correspondingto different compositions (0.25, 0.50, and 0.75) have beenarticulated using periodical repeated supercells by takingeight different atomic positions per unit cell. In order tooptimize the structure of different compositions the total

B.U. Haq et al. / Solar Energy 100 (2014) 1–8 3
energy of the corresponding supercell is minimized. Theequilibrium lattice constants, bulk moduli of differentstructures have been determined by fitting the energy ver-sus volume data into the Birch–Murnaghan equation ofstates (Murnaghan, 1944). The computed equilibrium vol-ume and its corresponding total energy of the super cell,the lattice constants, bulk moduli with LDA and GGAare listed in Table 1. From these calculations, we mayobserve that the lattice constant of Ga1�xAlxAs andIn1�xAlxAs decreases with the increase of Al concentra-tion. This is due to the mismatching between the ionic radiiof Ga(1.3 A), In(1.55 A), and Al(1.25 A) atoms. In accor-dance with the well-known relationship (inverse) betweenthe unit cell volume and its bulk modulus, an increasewas found in bulk modulus with decrease in lattice con-stant (Table 1). This behavior may come from the fact thatan increase of Al concentration causes hardness in thesealloys. Similarly, in case of Ga1�xInxAs the lattice constantwas found to be increased with the increase of In concen-tration and the decrease of the bulk modulus as can beenseen from Table 1. The variation of the lattice constantas a function of concentration (x) in host alloys is shownin Fig. 1. Our calculated lattice constants were found tovary almost linearly following the Vegard’s law with a mar-ginal bowing (Vegard, 1921).
For comprehensive understanding of the electronicproperties of Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs,we have computed the electronic band structures and den-sity of states (DOS) of these alloys with LDA, GGA andmBJ. Though pattern of the calculated band structuresand densities of states (DOS) of these materials is similarfor all approximations, to depict the difference in bandgapvalues, we display electronic band structures togetherobtained using LDA and mBJ potential of Ga1�xAlxAs,Ga1�xInxAs and In1�xAlxAs for x = 0.25, 0.50 and 0.75in Fig. 2. Table 2 shows an increase in the band gap valuewith increasing Al concentration in the case of Ga1�xAlxAsand In1�xAlxAs. The physical reason of this increment inthe band gap value is the larger energy gap value of AlAs.The bandgap value of GaInAs was found to be decreasingwith the increase of In concentration because of the small
Table 1The calculated lattice constant (a (A)) and bulk modulus B0 (GPa) ofGa1�xAlxAs, Ga1�xInxAs and In1�xAlxAs for x = 0.25, 0.50, 0.75 withLDA and GGA.
Lattice constant a (A) Bulk modulus B0 (GPa)
LDA GGA LDA GGA
Ga0.75Al0.25As 5.828 5.742 56.040 52.402Ga0.50Al0.50As 5.821 5.737 56.355 54.434Ga0.25Al0.75As 5.809 5.731 58.492 60.808Ga0.75In0.25As 5.740 5.865 72.401 66.923Ga0.50In0.50As 5.848 5.978 57.974 55.435Ga0.25In0.75As 5.951 6.095 55.866 46.516In0.75Al0.25As 5.946 6.094 58.977 74.364In0.50Al0.50As 5.830 5.964 64.631 60.106In0.25Al0.75As 5.732 5.839 70.108 51.645
bandgap of InAs compared to that of GaAs. From theanalysis of band structure, it is found that direct (C–C)band gap value decreases whereas indirect (C–R) bandgap increases with increase of Al concentration. The directto indirect crossover was found at 93.4% of Al concentra-tion for Ga1�xAlxAs and 84.63% of Al concentration forIn1�xAlxAs. Moreover the band gap values calculated withmBJ potential were found to be wide compared to thosecomputed with common GGA and LDA approaches.
In order to understand the physical origin of theobserved composition dependent bowing in GaAlAs,GaInAs and InAlAs alloys, we employ the proceduredeveloped by Bernard and Zunger (1987). In that scheme,the bowing parameter (b) is measured at x = 0.5 usingthe following reaction:
ABðaABÞ þACðaACÞ ! AB0:5C0:5ðaeqÞ ð1Þ
where aAC and aBC are the equilibrium lattice constants ofbinary compounds and aeq is the equilibrium lattice con-stant of the alloy. In Zunger et al. approach, b, has beendefined by three distinct contributions namely volumedeformation, charge transfer and the structural relaxation,where the contribution of compositional effect on the b isconsidered as small. To define these parameters, we decom-pose reaction (1) into three steps:
ABðaABÞ þACðaACÞ!VD
ABðaÞ þACðaÞ ð2Þ
ACðaACÞ þ BCðaBCÞ!CE
AB0:5C0:5ðaÞ ð3Þ
AB0:5C0:5ðaÞ!SR
A0:5B0:5CðaeqÞ ð4Þ
where step (1), (2), and (3) measure the bowing in energygap caused by volume deformation (bVD), charge transfer(bCT) and structural relaxation (bSR) respectively. The termbVD reveals the relative response of the electronic structureof Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs to hydro-static pressure at the two extreme composition i.e. x = 0,1. The term bCT represents the charge transfer effect mayhappen because of the different bonding behavior at latticeconstant “a”. The term bSR indicates the changes whilerelaxing the structure from un-relaxed state. Hence the to-tal bowing may be expressed as, b = bVD + bCT + bSR
where bVD, bCE and bSR are expressed as follow;
bVD ¼ 2½eABðaeqÞ � eABðaÞ þ eACðaeqÞ � eACðaÞ� ð5Þ
bCE ¼ 2½eABðaÞ þ eACðaÞ � 2eABCðaÞ� ð6Þ
bSR ¼ 4½eABCðaÞ � eABCðaeqÞ� ð7Þ
where ‘e’ is the calculated band gap energy. “aeq” repre-sents the equilibrium lattice constant of Ga1�xAlxAs,Ga1�xInxAs and In1�xAlxAs alloys, and “a” is the compo-sition dependent lattice constants calculated from Vegard’srule (Vegard, 1921). Our calculated values of energy gapbowing parameters are listed in Table 3. The values of bcalculated with mBJ potential are comparatively smallerthan those of LDA and GGA. We analyze from our
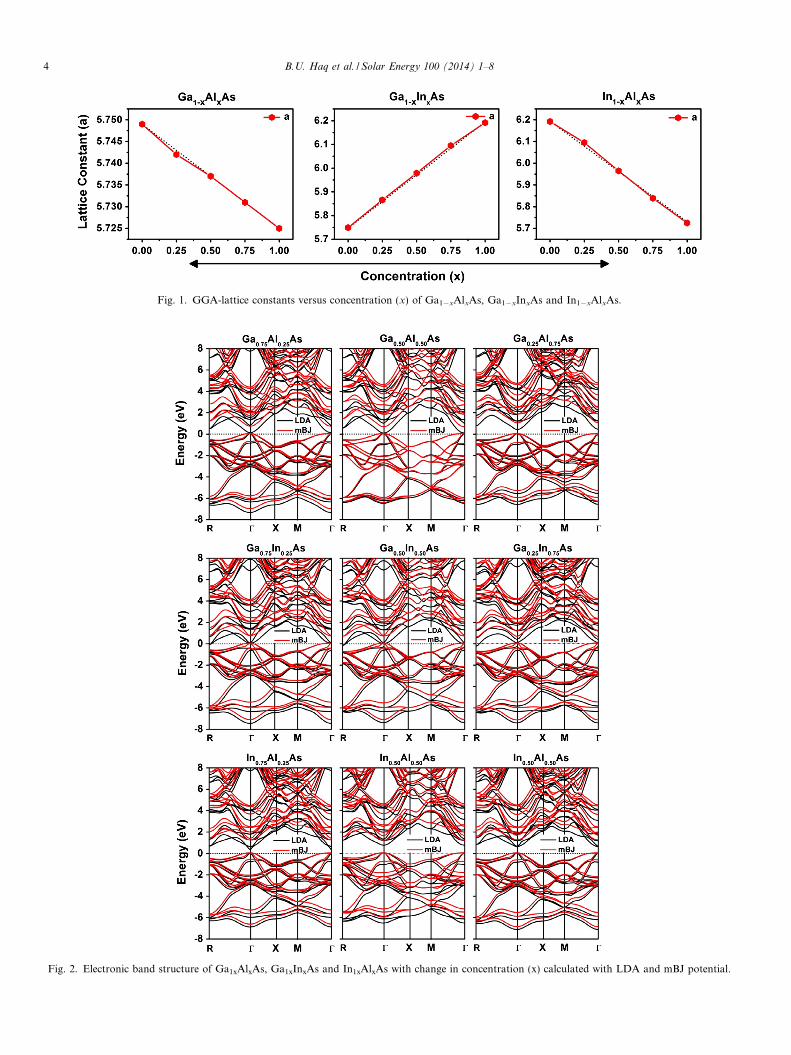
Fig. 1. GGA-lattice constants versus concentration (x) of Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs.
Fig. 2. Electronic band structure of Ga1xAlxAs, Ga1xInxAs and In1xAlxAs with change in concentration (x) calculated with LDA and mBJ potential.
4 B.U. Haq et al. / Solar Energy 100 (2014) 1–8
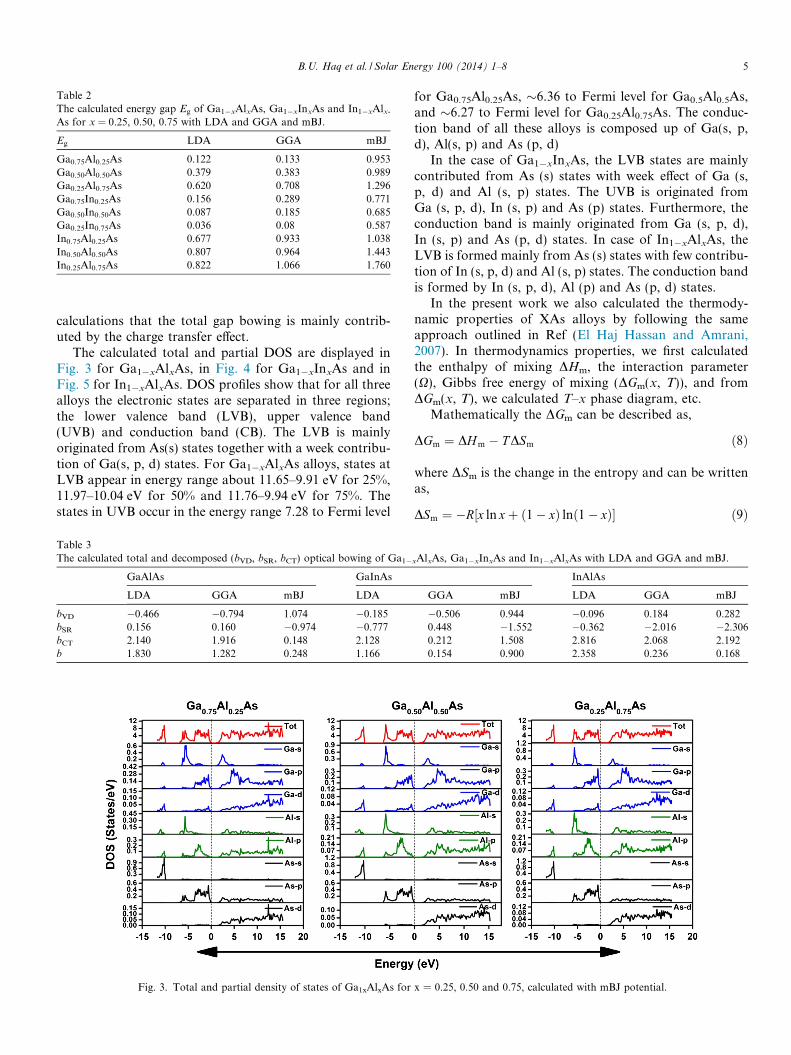
Table 2The calculated energy gap Eg of Ga1�xAlxAs, Ga1�xInxAs and In1�xAlx-
As for x = 0.25, 0.50, 0.75 with LDA and GGA and mBJ.
Eg LDA GGA mBJ
Ga0.75Al0.25As 0.122 0.133 0.953Ga0.50Al0.50As 0.379 0.383 0.989Ga0.25Al0.75As 0.620 0.708 1.296Ga0.75In0.25As 0.156 0.289 0.771Ga0.50In0.50As 0.087 0.185 0.685Ga0.25In0.75As 0.036 0.08 0.587In0.75Al0.25As 0.677 0.933 1.038In0.50Al0.50As 0.807 0.964 1.443In0.25Al0.75As 0.822 1.066 1.760
B.U. Haq et al. / Solar Energy 100 (2014) 1–8 5
calculations that the total gap bowing is mainly contrib-uted by the charge transfer effect.
The calculated total and partial DOS are displayed inFig. 3 for Ga1�xAlxAs, in Fig. 4 for Ga1�xInxAs and inFig. 5 for In1�xAlxAs. DOS profiles show that for all threealloys the electronic states are separated in three regions;the lower valence band (LVB), upper valence band(UVB) and conduction band (CB). The LVB is mainlyoriginated from As(s) states together with a week contribu-tion of Ga(s, p, d) states. For Ga1�xAlxAs alloys, states atLVB appear in energy range about 11.65–9.91 eV for 25%,11.97–10.04 eV for 50% and 11.76–9.94 eV for 75%. Thestates in UVB occur in the energy range 7.28 to Fermi level
Table 3The calculated total and decomposed (bVD, bSR, bCT) optical bowing of Ga1�
GaAlAs GaInAs
LDA GGA mBJ LDA
bVD �0.466 �0.794 1.074 �0.185bSR 0.156 0.160 �0.974 �0.777bCT 2.140 1.916 0.148 2.128b 1.830 1.282 0.248 1.166
Fig. 3. Total and partial density of states of Ga1xAlxAs for
for Ga0.75Al0.25As, �6.36 to Fermi level for Ga0.5Al0.5As,and �6.27 to Fermi level for Ga0.25Al0.75As. The conduc-tion band of all these alloys is composed up of Ga(s, p,d), Al(s, p) and As (p, d)
In the case of Ga1�xInxAs, the LVB states are mainlycontributed from As (s) states with week effect of Ga (s,p, d) and Al (s, p) states. The UVB is originated fromGa (s, p, d), In (s, p) and As (p) states. Furthermore, theconduction band is mainly originated from Ga (s, p, d),In (s, p) and As (p, d) states. In case of In1�xAlxAs, theLVB is formed mainly from As (s) states with few contribu-tion of In (s, p, d) and Al (s, p) states. The conduction bandis formed by In (s, p, d), Al (p) and As (p, d) states.
In the present work we also calculated the thermody-namic properties of XAs alloys by following the sameapproach outlined in Ref (El Haj Hassan and Amrani,2007). In thermodynamics properties, we first calculatedthe enthalpy of mixing DHm, the interaction parameter(X), Gibbs free energy of mixing (DGm(x, T)), and fromDGm(x, T), we calculated T–x phase diagram, etc.
Mathematically the DGm can be described as,
DGm ¼ DH m � TDSm ð8Þ
where DSm is the change in the entropy and can be writtenas,
DSm ¼ �R½x ln xþ ð1� xÞ lnð1� xÞ� ð9Þ
xAlxAs, Ga1�xInxAs and In1�xAlxAs with LDA and GGA and mBJ.
InAlAs
GGA mBJ LDA GGA mBJ
�0.506 0.944 �0.096 0.184 0.2820.448 �1.552 �0.362 �2.016 �2.3060.212 1.508 2.816 2.068 2.1920.154 0.900 2.358 0.236 0.168
x = 0.25, 0.50 and 0.75, calculated with mBJ potential.
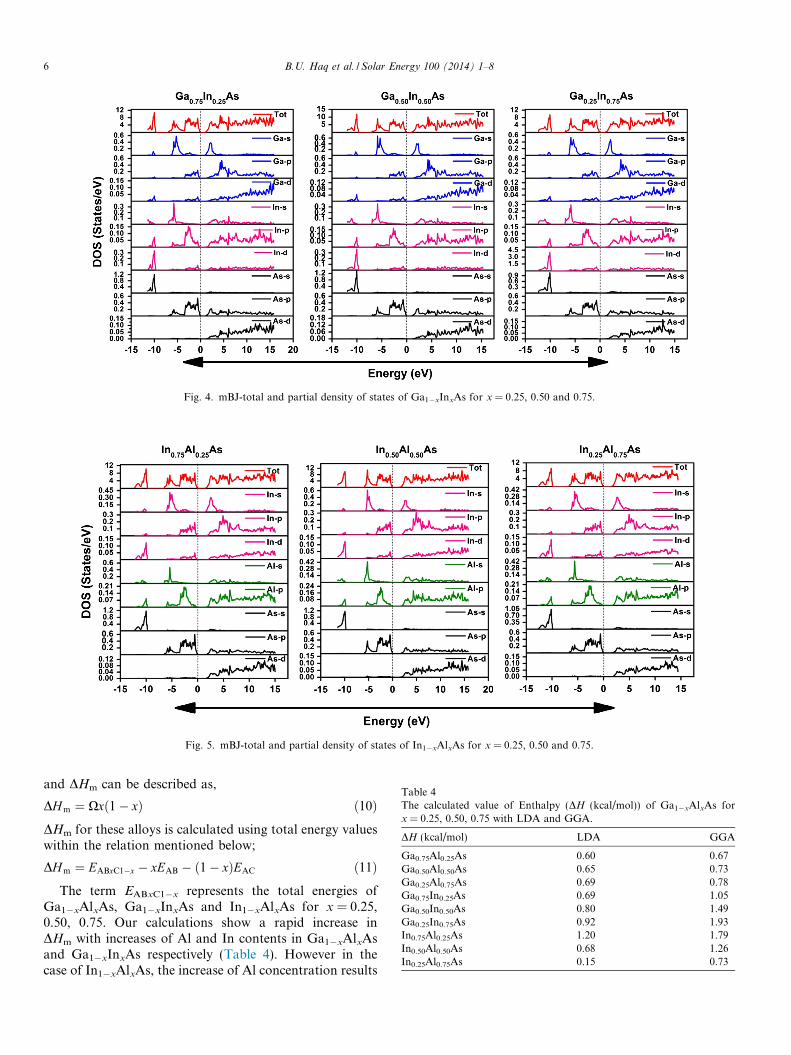
Fig. 4. mBJ-total and partial density of states of Ga1�xInxAs for x = 0.25, 0.50 and 0.75.
Fig. 5. mBJ-total and partial density of states of In1�xAlxAs for x = 0.25, 0.50 and 0.75.
Table 4The calculated value of Enthalpy (DH (kcal/mol)) of Ga1�xAlxAs forx = 0.25, 0.50, 0.75 with LDA and GGA.
DH (kcal/mol) LDA GGA
Ga0.75Al0.25As 0.60 0.67Ga0.50Al0.50As 0.65 0.73Ga0.25Al0.75As 0.69 0.78Ga0.75In0.25As 0.69 1.05Ga0.50In0.50As 0.80 1.49Ga0.25In0.75As 0.92 1.93In0.75Al0.25As 1.20 1.79In0.50Al0.50As 0.68 1.26In0.25Al0.75As 0.15 0.73
6 B.U. Haq et al. / Solar Energy 100 (2014) 1–8
and DHm can be described as,
DH m ¼ Xxð1� xÞ ð10ÞDHm for these alloys is calculated using total energy valueswithin the relation mentioned below;
DH m ¼ EABxC1�x � xEAB � ð1� xÞEAC ð11ÞThe term EABxC1�x represents the total energies of
Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs for x = 0.25,0.50, 0.75. Our calculations show a rapid increase inDHm with increases of Al and In contents in Ga1�xAlxAsand Ga1�xInxAs respectively (Table 4). However in thecase of In1�xAlxAs, the increase of Al concentration results
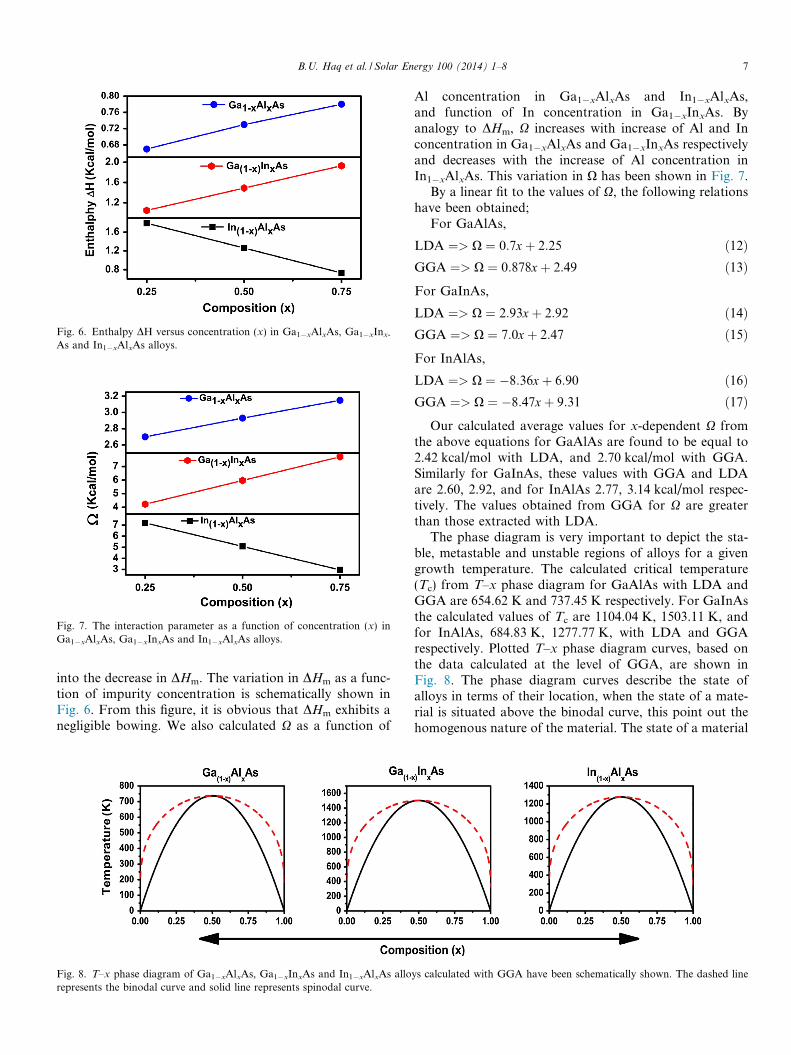
Fig. 6. Enthalpy DH versus concentration (x) in Ga1�xAlxAs, Ga1�xInx-
As and In1�xAlxAs alloys.
Fig. 7. The interaction parameter as a function of concentration (x) inGa1�xAlxAs, Ga1�xInxAs and In1�xAlxAs alloys.
B.U. Haq et al. / Solar Energy 100 (2014) 1–8 7
into the decrease in DHm. The variation in DHm as a func-tion of impurity concentration is schematically shown inFig. 6. From this figure, it is obvious that DHm exhibits anegligible bowing. We also calculated X as a function of
Fig. 8. T–x phase diagram of Ga1�xAlxAs, Ga1�xInxAs and In1�xAlxAs allorepresents the binodal curve and solid line represents spinodal curve.
Al concentration in Ga1�xAlxAs and In1�xAlxAs,and function of In concentration in Ga1�xInxAs. Byanalogy to DHm, X increases with increase of Al and Inconcentration in Ga1�xAlxAs and Ga1�xInxAs respectivelyand decreases with the increase of Al concentration inIn1�xAlxAs. This variation in X has been shown in Fig. 7.
By a linear fit to the values of X, the following relationshave been obtained;
For GaAlAs,
LDA ¼> X ¼ 0:7xþ 2:25 ð12ÞGGA ¼> X ¼ 0:878xþ 2:49 ð13ÞFor GaInAs,
LDA ¼> X ¼ 2:93xþ 2:92 ð14ÞGGA ¼> X ¼ 7:0xþ 2:47 ð15ÞFor InAlAs,
LDA ¼> X ¼ �8:36xþ 6:90 ð16ÞGGA ¼> X ¼ �8:47xþ 9:31 ð17Þ
Our calculated average values for x-dependent X fromthe above equations for GaAlAs are found to be equal to2.42 kcal/mol with LDA, and 2.70 kcal/mol with GGA.Similarly for GaInAs, these values with GGA and LDAare 2.60, 2.92, and for InAlAs 2.77, 3.14 kcal/mol respec-tively. The values obtained from GGA for X are greaterthan those extracted with LDA.
The phase diagram is very important to depict the sta-ble, metastable and unstable regions of alloys for a givengrowth temperature. The calculated critical temperature(Tc) from T–x phase diagram for GaAlAs with LDA andGGA are 654.62 K and 737.45 K respectively. For GaInAsthe calculated values of Tc are 1104.04 K, 1503.11 K, andfor InAlAs, 684.83 K, 1277.77 K, with LDA and GGArespectively. Plotted T–x phase diagram curves, based onthe data calculated at the level of GGA, are shown inFig. 8. The phase diagram curves describe the state ofalloys in terms of their location, when the state of a mate-rial is situated above the binodal curve, this point out thehomogenous nature of the material. The state of a material
ys calculated with GGA have been schematically shown. The dashed line

8 B.U. Haq et al. / Solar Energy 100 (2014) 1–8
below the binodal curve shows that this material is unstablein homogenous states. The material is considered as meta-stable between the spinodal and binodal curves. Our resultsshow that XAs alloys exhibit a broad miscibility gap sur-rounded by binodal line, and are stable at hightemperature.
In summary, the effect of mutual alloying XAs on thephysical properties has been investigated in the presentwork. We find a linear decrease in lattice constant andincrease in bulk moduli with increase of Al concentrationin Ga1�xAlxAs and In1�xAlxAs. On the other hand, theincrease of In concentration in Ga1�xInxAs has induced adecrease in lattice constant. The increase of Al concentra-tion in Ga1�xAlxAs and In1�xAlxAs also lead to anincrease of the energy gap values. Moreover it leads alsoto a crossover from direct to indirect band gap. The bandgap bowing was found to be caused mainly by chargetransfer effect with a comparatively smaller contributionof the volume deformation and the structural relaxation.Enthalpy and interaction parameter were found to increasefor Ga1�xAlxAs and Ga1�xInxAs with increase in concen-tration of Al and In respectively. Whereas they decreasefor In1�xAlxAs with increase of Al contents. The calculatedphase diagrams reveal that all these alloys exhibit a signif-icant phase miscibility gap. The calculated high criticaltemperature indicates that these alloys are stable at hightemperature.
Acknowledgement
Authors would like to thank the MOHE of Malaysiaand UTM for financial support of this research throughGrant Nos. R.J130000.7726.4D034, Q.J130000.2526.02H89 and Q.J130000.2526.04H14. Author (R. Khenata)acknowledges financial support by the Deanship of Scien-tific Research at King Saud University through researchgroup project RPG-VPP-088.
References
Ameri, M. et al., 2012. Ab Initio Calculations Study of Structural andElectronic Properties of Ternary Alloy AlxIn1�xAs 3, 674–683.
Anua, N.N., Ahmed, R., Shaari, A., Saeed, M.A., Ul Haq, B., Goumri-Said, S., 2013. Non-local exchange correlation functionals impact onthe structural, electronic and optical properties of III–V arsenides.Semicond. Sci. Technol. 28 (10), 105015.
Bernard, J.E., Zunger, A., 1987. Electronic structure of ZnS, ZnSe, ZnTe,and their pseudobinary alloys. Phys. Rev. B 36 (6), 3199.
Blaha, P., et al., 2001. WIEN2k. An augmented plane wave+ local orbitalsprogram for calculating crystal properties.
Csavinszky, P., Brownstein, K., 1983. Concentration-dependent dielectricresponse of Ga1�xAlx As. Phys. Rev. B 28 (4), 2226.
Csavinszky, P., Brownstein, K., 1984. Site-dependent and composition-dependent spatial dielectric functions for Ga1�xAlxAs. J. Phys. Chem.Solids 45 (5), 567–570.
Emeny, M. et al., 1991. A photoluminescence study of indium desorptionfrom strained Ga1�xInxAs/GaAs. J. Cryst. Growth 111 (1), 413–418.
El Haj Hassan, F. et al., 2010. First-principles study of the ternarysemiconductor alloys (Ga, Al)(As, Sb). J. Alloys Compd. 499 (1), 80–89.
El Haj Hassan, F., Amrani, B., 2007. Structural, electronic and thermo-dynamic properties of magnesium chalcogenide ternary alloys. J.Phys.: Condens. Matter 19 (38), 386234.
Ferreira, L., Wei, S.H., Zunger, A., 1989. First-principles calculation ofalloy phase diagrams: the renormalized-interaction approach. Phys.Rev. B 40 (5), 3197.
Grenet, G. et al., 1996. In situ XPS investigation of indium surfacesegregation for Ga1�xInxAs and Al1�xInx As alloys grown by MBE onInP (001). Surf. Sci. 352, 734–739.
Hohenberg, P., Kohn, W., 1964. Inhomogeneous electron gas. Phys. Rev.136 (3B), B864.
Kohn, W., Sham, L.J., 1965. Self-consistent equations including exchangeand correlation effects, APS.
Koller, D., Tran, F., Blaha, P., 2012. Improving the modified Becke-Johnson exchange potential. Phys. Rev. B 85 (15), 155109.
Madsen, G.K. et al., 2001. Efficient linearization of the augmented plane-wave method. Phys. Rev. B 64 (19), 195134.
Merabet, B. et al., 2013. Electronic and optical properties of (AlxGa1�x1�y
Mny As single crystal: a new candidate for integrated optical isolatorsand spintronics. J. Mater. Sci. 48 (2), 758–764.
Murnaghan, F., 1944. The compressibility of media under extremepressures. Proc. Nat. Acad. Sci. U.S.A. 30 (9), 244.
Perdew, J.P. et al., 1992. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchangeand correlation. Phys. Rev. B 46 (11), 6671.
Perdew, J.P., Wang, Y., 1992. Accurate and simple analytic representationof the electron-gas correlation energy. Phys. Rev. B 45 (23), 13244.
Sjostedt, E., Nordstrom, L., Singh, D., 2000. An alternative way oflinearizing the augmented plane-wave method. Solid State Commun.114 (1), 15–20.
Srivastava, R. et al., 2001. A first-principles study of structural andelectronic properties of Ga1�xAlxAs alloys. Solid State Commun. 118(9), 479–484.
Tit, N., Amrane, N., Reshak, A., 2010. Bandgap characters in GaAs-based ternary alloys. Cryst. Res. Technol. 45 (1), 59–69.
Tran, F., Blaha, P., 2009. Accurate band gaps of semiconductors andinsulators with a semilocal exchange-correlation potential. Phys. Rev.Lett. 102 (22), 226401.
Vegard, L., 1921. The constitution of mixed crystals and the spaceoccupied by atoms. Z. Phys. 5 (17), 17–26.
Vurgaftman, I., Meyer, J., Ram-Mohan, L., 2001. Band parameters forIII–V compound semiconductors and their alloys. J. Appl. Phys. 89(11), 5815–5875.
Zawadzki, W. et al., 1999. Cyclotron emission study of electron masses inGaAs–GaAlAs heterostructures. Semicond. Sci. Technol. 9 (3), 320.
Zhang, X. et al., 1999. First-principles calculations of band offsets. J.Phys.: Condens. Matter 10 (3), 577.
Zunger, A. et al., 1990. Special quasirandom structures. Phys. Rev. Lett.65 (3), 353–356.
Related Documents











