© 1995 Oxford University Press Human Molecular Genetics, 1995, Vol. 4, No. 6 1077-1082 Mutations in the third immunoglobulin domain of the fibroblast growth factor receptor-2 gene in Crouzon syndrome M.OIdridge 1 , A.O.M.Wilkie 12 , S.F.SIaney 12 , M.D.Poole 2 , L.J.Pulleyn 3 , P.Rutland 3 , A.D.Hockley 4 , M.J.C.Wake 4 , J.H.Goldin 4 , R.M.Winter 3 , W.Reardon 3 and S.Malcolm 3 * institute of Molecular Medicine, John Radcliffe Hospital, Headington, Oxford OX3 9DU, 2 Oxford Craniofacial Unit, Radcliffe Infirmary, Oxford OX2 6HE, 3 Molecular Genetics Unit and Mothercare Unit of Clinical Genetics and Fetal Medicine, Institute of Child Health, London WC1N 1EH and 4 West Midlands Craniofacial Unit, Queen Elizabeth and Children's Hospitals, Birmingham, UK Received February 24, 1995; Revised and Accepted March 30, 1995 Craniosynostosis, which affects approximately 1 in 2000 children, is the result of the abnormal develop- ment and/or premature fusion of the cranial sutures. Studies of mutations in patients with cranio- synostosis have shown that the family of fibroblast growth factor receptor genes are extremely important in the correct formation of the skull, and digits. Mutations in the third immunoglobulin domain of fibroblast growth factor receptor 2 (FGFR2), in part of the molecule corresponding to a tissue specific isoform (Me), can cause both Crouzon and Pfeiffer syndromes. Two specific mutations in the linking region between the second and third immunoglobulin domains of FGFR2 occur in Apert syndrome. We present here mutations associated with the Crouzon syndrome, also in the third immunoglobulin domain but in an upstream exon. This exon is expressed in both tissue isoforms. Five different mutations were detected in 11 unrelated individuals. A cysteine to phenylalanine change was found in six individuals. This cysteine forms half of the disulphide bridge maintaining the secondary structure of the immuno- globulin domain. The first deletion within an FGFR gene is reported. Together with mutations in exon Illc these account for 25 mutations out of 40 Crouzon patients studied in our combined series (5). INTRODUCTION Fibroblast growth factor receptors are a family of related signalling proteins which span the cell membrane (1,2). They are tyrosine kinases which are activated by fibroblast growth factors binding extracellularly in the presence of proteoglycans and are expressed from embryo gastrulation onwards. The extracellular domain contains three immunoglobulin loops (Fig. 1). In FGFR1, FGFR2 and FGFR3 the third immunoglob- ulin domain consists of two isoforms, Hlb and IIIc, which differ in their carboxyl ends and whose expression is cell-type specific. The two forms are created by alternative splicing in which one of two exons, (also referred to as exon Mb or IIIc in this paper) is spliced to an invariant upstream exon, IIIu (Fig. 1). The family of human FGFR genes contains four members which map to chromosome 8 (FGFR1), 10 (FGFR2), 4 (FGFR3) and 5 (FGFR4). Mutations associated with three members of the FGFR family have been found in human malformation syndromes. Mutation of a single nucleotide in the transmembrane domain of FGFR3 accounts for almost all cases of achondroplasia (3,4,28). Mutations in FGFR2 and FGFR1 have been found in the syndromes Crouzon, Pfeiffer, Apert and Jackson-Weiss (5-9), all of which involve cranio- synostosis as a common feature. Changes in the digits vary from normal development in Crouzon syndrome, through broadening of the thumbs and great toes in Pfeiffer syndrome to severe syndactyly in Apert syndrome. To date mutations causing Crouzon syndrome have been reported only in exon IIIc of FGFR2. In view of the patterns of expression of Iglllb and IIIc (10,11), we had previously predicted, and demonstrated, that mutations in patients with Crouzon syn- drome would occur in exon IIIc (5). In view of the failure to find mutations in 11 of 20 patients in the initial study, we extended the screen to include exon IIIu, which forms the other half of the IgHI loop and is common to both forms. RESULTS Patients All patients showed typical features of Crouzon syndrome, of which the principal features were craniosynostosis, associated proptosis and mid-face hypoplasia. In particular there was no significant broadening of the thumbs or big toes. The diagnosis in all cases was clinical, with occasional radiological confirma- tion of normal limb anatomy. Several patients manifested minor digital abnormalities such as 2/3 syndactyly, commonly found in the normal population and consistent with a Crouzon phenotype (Fig. 2). Two patients have been described pre- viously (C7 in ref. 23, 12529 in ref. 27). Mutations in exon IIIu Thirty-one patients with Crouzon syndrome were analysed for sequence changes in the third immunoglobulin domain of *To whom correspondence should be addressed at University College London on April 23, 2015 http://hmg.oxfordjournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

© 1995 Oxford University Press Human Molecular Genetics, 1995, Vol. 4, No. 6 1077-1082
Mutations in the third immunoglobulin domain of thefibroblast growth factor receptor-2 gene in CrouzonsyndromeM.OIdridge1, A.O.M.Wilkie12, S.F.SIaney12, M.D.Poole2, L.J.Pulleyn3, P.Rutland3, A.D.Hockley4, M.J.C.Wake4, J.H.Goldin4,R.M.Winter3, W.Reardon3and S.Malcolm3*
institute of Molecular Medicine, John Radcliffe Hospital, Headington, Oxford OX3 9DU, 2Oxford Craniofacial Unit, Radcliffe Infirmary,Oxford OX2 6HE, 3Molecular Genetics Unit and Mothercare Unit of Clinical Genetics and Fetal Medicine, Institute of Child Health,London WC1N 1EH and 4West Midlands Craniofacial Unit, Queen Elizabeth and Children's Hospitals, Birmingham, UK
Received February 24, 1995; Revised and Accepted March 30, 1995
Craniosynostosis, which affects approximately 1 in2000 children, is the result of the abnormal develop-ment and/or premature fusion of the cranial sutures.Studies of mutations in patients with cranio-synostosis have shown that the family of fibroblastgrowth factor receptor genes are extremely importantin the correct formation of the skull, and digits.Mutations in the third immunoglobulin domain offibroblast growth factor receptor 2 (FGFR2), in partof the molecule corresponding to a tissue specificisoform (Me), can cause both Crouzon and Pfeiffersyndromes. Two specific mutations in the linkingregion between the second and third immunoglobulindomains of FGFR2 occur in Apert syndrome. Wepresent here mutations associated with the Crouzonsyndrome, also in the third immunoglobulin domainbut in an upstream exon. This exon is expressed inboth tissue isoforms. Five different mutations weredetected in 11 unrelated individuals. A cysteine tophenylalanine change was found in six individuals.This cysteine forms half of the disulphide bridgemaintaining the secondary structure of the immuno-globulin domain. The first deletion within an FGFRgene is reported. Together with mutations in exon Illcthese account for 25 mutations out of 40 Crouzonpatients studied in our combined series (5).
INTRODUCTION
Fibroblast growth factor receptors are a family of relatedsignalling proteins which span the cell membrane (1,2). Theyare tyrosine kinases which are activated by fibroblast growthfactors binding extracellularly in the presence of proteoglycansand are expressed from embryo gastrulation onwards. Theextracellular domain contains three immunoglobulin loops(Fig. 1). In FGFR1, FGFR2 and FGFR3 the third immunoglob-ulin domain consists of two isoforms, Hlb and IIIc, whichdiffer in their carboxyl ends and whose expression is cell-typespecific. The two forms are created by alternative splicing inwhich one of two exons, (also referred to as exon Mb or IIIc
in this paper) is spliced to an invariant upstream exon, IIIu(Fig. 1).
The family of human FGFR genes contains four memberswhich map to chromosome 8 (FGFR1), 10 (FGFR2), 4(FGFR3) and 5 (FGFR4). Mutations associated with threemembers of the FGFR family have been found in humanmalformation syndromes. Mutation of a single nucleotide inthe transmembrane domain of FGFR3 accounts for almost allcases of achondroplasia (3,4,28). Mutations in FGFR2 andFGFR1 have been found in the syndromes Crouzon, Pfeiffer,Apert and Jackson-Weiss (5-9), all of which involve cranio-synostosis as a common feature. Changes in the digits varyfrom normal development in Crouzon syndrome, throughbroadening of the thumbs and great toes in Pfeiffer syndrometo severe syndactyly in Apert syndrome. To date mutationscausing Crouzon syndrome have been reported only in exonIIIc of FGFR2. In view of the patterns of expression ofIglllb and IIIc (10,11), we had previously predicted, anddemonstrated, that mutations in patients with Crouzon syn-drome would occur in exon IIIc (5). In view of the failure tofind mutations in 11 of 20 patients in the initial study, weextended the screen to include exon IIIu, which forms theother half of the IgHI loop and is common to both forms.
RESULTS
PatientsAll patients showed typical features of Crouzon syndrome, ofwhich the principal features were craniosynostosis, associatedproptosis and mid-face hypoplasia. In particular there was nosignificant broadening of the thumbs or big toes. The diagnosisin all cases was clinical, with occasional radiological confirma-tion of normal limb anatomy. Several patients manifestedminor digital abnormalities such as 2/3 syndactyly, commonlyfound in the normal population and consistent with a Crouzonphenotype (Fig. 2). Two patients have been described pre-viously (C7 in ref. 23, 12529 in ref. 27).
Mutations in exon IIIuThirty-one patients with Crouzon syndrome were analysed forsequence changes in the third immunoglobulin domain of
*To whom correspondence should be addressed
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

707* Human Molecular Genetics, 1995, Vol. 4, No. 6
IG1 IG2 IG3
or
0.13
EXON 7
Figure 1. Schematic diagram of FGFR2 gene.
FGFR2 using single strand conformation polymorphismanalysis (SSCP).
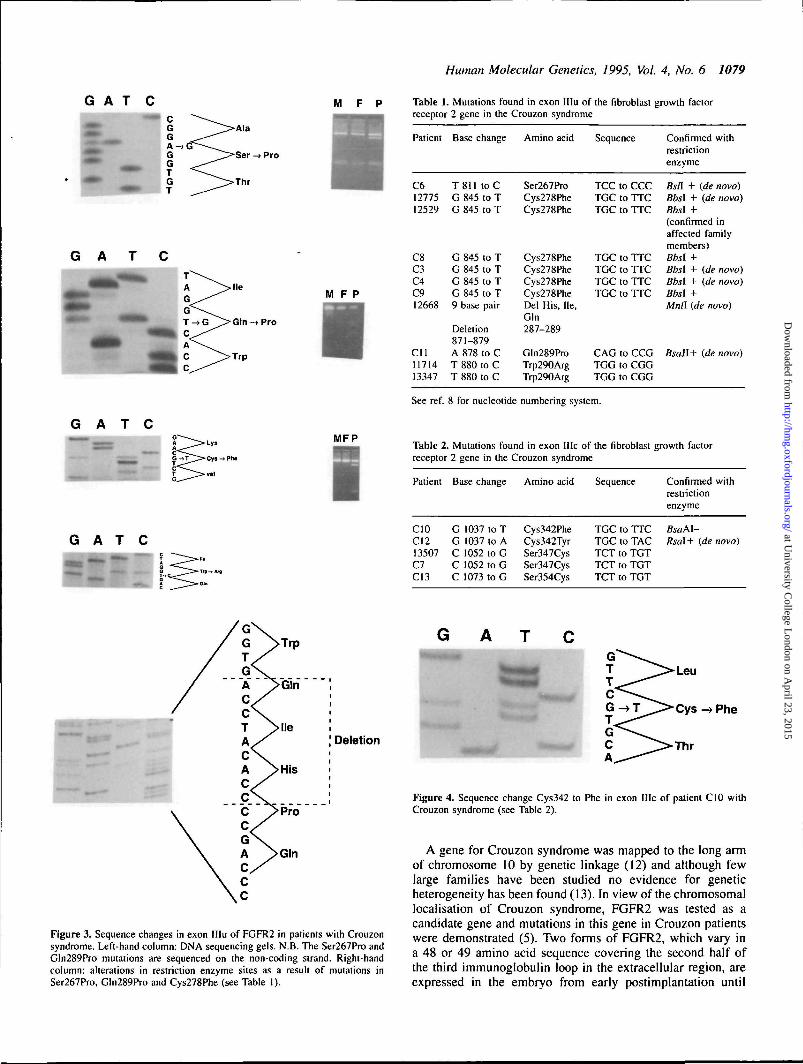
SSCP analysis of exon IIIu revealed a number of differentband shifts. DNA from patients with band shifts was sequencedand five mutations found. Several of the patients appeared tohave an identical band shift. The first three of these patientsto be analysed showed a TGC to TTC mutation, changing acysteine at position 278 to phenylalanine. As this creates aBbsl restriction site, all the remaining samples were tested forcreation of this site. Six patients were found with the mutation(Fig. 3).
A full list of the mutations found is presented in Table 1.The mutations were shown, by restriction enzyme analysis inboth parents and the child, to arise de novo in three of thepatients with a Cys278Phe change, in the patient with aGln289Pro mutation and the Ser267Pro mutation. Themutations include a deletion of nine base pairs resulting in athree amino acid deletion. In this case, which is sporadic, thesmaller fragment which is revealed most clearly after Mnlldigestion, was shown to have arisen de novo. This is the firstdeletion described in the fibroblast growth factor receptorgenes.
One other recurrent mutation is found, Trp290Arg. None ofthe mutations described occur at the sequence CpG.
Identification of further mutations in exon HieExon IIIc (8) was amplified from genomic DNA. Mutationsin five patients, in addition to those previously published, werefound (Table 2; Fig. 4a). All involved the loss or creation ofa cysteine residue. One of them, Cys342Phe, represents anewly reported mutation and three others (one observed twice)have been previously reported. Of these, one also involvesmutation of the critical cysteine (Cys342Tyr) and the othertwo disturb the S-S cross-linking in a different way, bycreating a new cysteine residue (Ser347Cys and Ser354Cys).Cys342Phe destroys a BsaAl site. The Cys342Tyr mutation,
Figure 2. Members of a family with Crouzon syndrome before surgery. Thefamily carry a Cys278Phe mutation in exon IIIu of FGFR2 (27).
which creates an Rsal site, was confirmed by Rsal digestion.In total, of the 40 Crouzon patients analysed, 14 have been
found to have a mutation in this exon (this paper and ref. 5).
DISCUSSION
Crouzon syndrome is an autosomal dominant disorder ofcraniofacial development, characterized clinically by an ab-normal skull shape and prominent eyes. Changes in the handsand/or toes are not observed (20,21). The underlying physicalcause is premature fusion of the cranial sutures or cranio-synostosis. The consequence may be raised pressure on thedeveloping brain requiring surgical intervention. With plannedsurgery a much improved facial appearance is achieved andnormal intellectual development generally follows.
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from
Human Molecular Genetics, 1995, Vol. 4, No. 6 1079
G A T C M F P
G A
G A T C
M F P
Gin -> Pro
G A T C
MFP
Table 1. Mutations found in exon HIu of the fibroblast growth factorreceptor 2 gene in the Crouzon syndrome
Patient
C61277512529
C8C3C4C912668
Cll1171413347
Base change
T811 toCG 845 to TG 845 to T
G 845 to TG 845 to TG 845 to TG 845 to T9 base pair
Deletion871-879A 878 to CT 880 to CT 880 to C
Amino acid
Ser267ProCys278PheCys278Phe
Cys278PheCys278PheCys278PheCys278PheDel His, He,Gin287-289
Gln289ProTrp290ArgTrp290Arg
Sequence
TCC to CCCTGC to TTCTGC to TTC
TGC to TTCTGC to TTCTGC to TTCTGC to TTC
CAG to CCGTGG to CGGTGG to CGG
Confirmed withrestrictionenzyme
fis/I + (de novo)Bbsl + (de novo)Bbsl +(confirmed inaffected familymembers)Bbsl +Bbsl + {de novo)Bbsl + (de novo)Bbsl +Mnll (de novo)
Bsail+ (de novo)
See ref. 8 for nucleotide numbering system.
Table 2. Mutations found in exon Me of the fibroblast growth factorreceptor 2 gene in the Crouzon syndrome
Patient
CIOC1213507C7C13
Base change
G 1037 to TG 1037 to AC 1052 to GC 1052 to GC 1073 to G
Amino acid
Cys342PheCys342TyrSer347CysSer347CysSer354Cys
Sequence
TGC to TTCTGC to TACTCT to TGTTCT to TGTTCT to TGT
Confirmedrestrictionenzyme
BsaM-Rsal+ (de
with
novo)
Deletion
Figure 3. Sequence changes in exon IIIu of FGFR2 in patients with Crouzonsyndrome. Left-hand column: DNA sequencing gels. N.B. The Ser267Pro andGln289Pro mutations are sequenced on the non-coding strand. Right-handcolumn: alterations in restriction enzyme sites as a result of mutations inSer267Pro, Gln289Pro and Cys278Phe (see Table 1).
Figure 4. Sequence change Cys342 to Phe in exon Me of patient C10 withCrouzon syndrome (see Table 2).
A gene for Crouzon syndrome was mapped to the long armof chromosome 10 by genetic linkage (12) and although fewlarge families have been studied no evidence for geneticheterogeneity has been found (13). In view of the chromosomallocalisation of Crouzon syndrome, FGFR2 was tested as acandidate gene and mutations in this gene in Crouzon patientswere demonstrated (5). Two forms of FGFR2, which vary ina 48 or 49 amino acid sequence covering the second half ofthe third immunoglobulin loop in the extracellular region, areexpressed in the embryo from early postimplantation until
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

1080 Human Molecular Genetics, 1995, Vol. 4, No. 6
Ig domain II Illu exon
FGFRlFGFR2FGFR3FGFR4
TCIVENEYGSINHTYQLDWE
» «Y* # • K2r» • • IvQ* • X* • *£*• •
iHRPHiQAGLPAHKTVALGSHVEFMCaCVYSDPQPHIQAJTW.GD. . .vj A. >fll
.Q.AV. . . D . . .H . . . . . .A . . . . .T.AW. . D . . I .L . A. . . . . .
Illc exon
FGFRlFGFR2FGFR3FGFR4
WLKHIEVNGSKIGPDNLPYVQILKTAGVNTTDKEMEVLHLRNVSFEDAGEYTCLAGHSIG^ | ~ L • • V • R . • • • • I • • • « 7 • • • J _ U x V • J B j p L » • • • • »HMB a • J L • • * ^ H J - • • • * • • • • • *^^^|^| * B * * ^ H * *
• * • • V • • • • • • V • • • u i » • • T V • • • • • A • • • • • • X J « » * O « U * « X a • • • • • * • • • * » • • » •
* •-* • fiVX • • • S P • A V G F * * m • V # • • » D I • SS—-" # v « • • x • • • » • « • • * « • • « • • « • • » • •
transmembrane
FGFRlFGFR2FGFR3FGFR4
I HHSAI TVIiEALEER-PAVMTSPLYIiEIIIYCTGAFLISCMVGSVIVYKMKSGTKKSDI . F . 1 . . . . . . P . P G R E - K E I T A . . D . . . . A . . . I . V . . . A . . - V T . . L C R . , H T . . . P .F V. . P . E . . L V E . D E A G S V . A J . L S . J V . F . .FH.V.AA.TLCRLR.PP. .GL. .YQ.. .. .. .PEEDPTWT.AAPEAR.TD. .L.AS.SIALAVLLLLAGL.RGQALHGRHP
I
iPfeiffer syndromeApert syndromeCrouzon SyndromeJackson-Weiss syndromeAchondroplasia
| splice site mutation
deletion
Figure 5. Mutations in human fibroblast growth factor receptor genes and malformations.
advanced organogenesis (10). However, the two alternativelyspliced forms showed distinct patterns of spatial expression(11). The form using the Illb exon, which is also calledkeratinocyte growth factor receptor because of its specificbinding to FGF7 or keratinocyte growth factor, is expressedmainly in epithelia and was predicted to have a particular partin the formation of skin and its derivatives, while the formusing exon IIIc is preferentially expressed during osteogenesis(11). Based on this prediction, changes were sought in exonIIIc in patients with Crouzon syndrome. The mutations foundwere believed to be causative because (i) many of theminvolved the cysteine which is essential for the three-dimen-sional structure of the immunoglobulin-like loop; (ii) de novochanges were found in sporadic patients; (iii) the changessegregated with the disorder in familial cases; and (iv) no suchvariants were found in normal individuals (5,6). In the initialstudy (5) mutations were found only in nine of 20 patientsstudied.
Patients with Pfeiffer syndrome have similar craniofacialchanges to those with Crouzon syndrome but additionally havecharacteristic broad thumbs and toes (20-22). A gene forPfeiffer syndrome was mapped to the pericentromeric regionof chromosome 8, in some but not all families, in a linkagestudy (14) and, in view of the findings in Crouzon syndrome,the fibroblast growth factor receptor 1 gene (FGFRl), whichalso maps to that region of chromosome 8, was regarded as acandidate. In contrast to the situation in Crouzon syndrome,there is a common mutation in the Pfeiffer families of a prolineto arginine substitution located in exon IIIu, upstream of thecysteine residue involved in S-S binding, in the region linkingimmunoglobulin-like loops II and III (7). The FGFRl genehas a similar structural organization to the FGFR2 gene withalternative exons for the second half of immunoglobulin-likeloop III (1) and so the Pfeiffer mutation would be expressedin all transcripts from the FGFRl gene locus.
Apert syndrome is a further syndrome involving cranio-
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

Human Molecular Genetics, 1995, Vol. 4, No. 6 1081
synostosis but with severe syndactyly of the hands and feet(20,21). Owing to the severity of the condition, large familiessuitable for linkage studies do not exist, but it was possible toanalyse a number of small families in an exclusion analysisof the loci for the fibroblast growth factor receptor genes 1, 2and 3 (8) which predicted that FGFR2 was again the mostlikely candidate. All 40 cases of Apert syndrome analysedshowed missense mutations in one of two adjacent aminoacids, either serine 252 to tryptophan or proline 253 to arginine(8). This second mutation is exactly equivalent, in positionand sequence, to the Pfeiffer causing mutation on chromosome8 and would, therefore, also be expressed in both isoforms ofthe gene.
In view of these results, exon IIIu of FGFR2, which encodesthe first half of the Iglll domain, was screened, initially bySSCP, for mutations causing Crouzon syndrome. We reporthere five such mutations in 11 unrelated individuals. Theevidence that these changes are disease causing follows thatgiven above for the mutations in exon Me. They are non-conservative and in five cases involve the cysteine involvedin S-S binding, several were observed de novo and no suchchanges have been observed in at least 200 individuals withoutcraniosynostosis.
Correlation of the phenotype and genotype poses a fascinat-ing problem in the case of FGFRs. In addition to the resultsjust discussed it has been shown that mutations in FGFR2,exon Me, can also cause Pfeiffer syndrome (9), presumablyexplaining at least some and maybe all of the genetic heterogen-eity observed by Robin et al. (14). Surprisingly, two of themutations described, both involving Cys342, are identical tomutations described in the Crouzon syndrome and threeadditional mutations have been found including a splice sitemutation (9,15). A further missense mutation in exon Me,Alanine 344 to Glycine, causes Jackson-Weiss syndrome in asingle family. The findings to date are summarized in Fig. 5.It was impossible to distinguish clinically between the casesof Crouzon syndrome arising from exon Me mutations andthose arising from exon IIIu mutations. Therefore, apparentlythe phenotype arises from disruption of the Me exon form ofFGFR2 with little additional contribution from KGFR (Mb).Conversely, it seems that KGFR mutant molecules may notresult in an abnormal phenotype in the heterozygous state. Italso remains unknown why mutations in a position in themolecule which will affect both forms should have such alimited effect on the phenotype. Fibroblast growth factorreceptors interact with the growth factors via a heparin complex(16,17) and their action in vivo will be modulated by cellularproteoglycans. The Igll-IgM regions of the molecules areinvolved in this interaction (17). It is possible that the clinicaleffects reflect to some extent the exact proteoglycan contentat the site of the sutures or in the digits.
Although, including this work, there are now 14 describedmutations in the third immunoglobulin-like loop which causethe Crouzon syndrome, the distribution is clearly not randomas four different mutations account for 16 cases. None of themutations corresponds to a CpG to TpG change, which hasbeen well documented as having a high frequency and,therefore, the non-random distribution is likely to reflect thephenotypic consequence of the change rather than be anindirect consequence of mutation rate. The recurrent mutationat a specific nucleotide in FGFR3 in achondroplasia occurs
with an estimated frequency of between 1 per 15 000 and 1per 40 000. This represents a remarkably high mutation rate.However, the nucleotide involved is indeed a G to A transition(corresponding to CpG to TpG in the antisense strand).
There is some overall pattern in the range of mutationsfound in Crouzon syndrome patients (5,6, this work). Themajority disrupt the intrachain disulphide bonding typical ofimmunoglobulin-like domains by mutations of either of thetwo cysteines, one from each exon, or by creation of a newcysteine. The disulphide bond stabilizes the whole structureby increasing hydrophobic interactions between amino acidresidues inside the immunoglobulin fold. If the S-S bridgesare removed or reformed a major conformational changeof the secondary structure would be expected. The formalrequirement for cysteine residues for the ligand binding activityof FGFR was shown by site-directed mutagenesis of each ofthe cysteines in FGFR1 (19). In addition the creation of freesulphydryl groups could allow aberrant disulphide bondingwith other mutant FGFR2 molecules. This might lead toconstitutive activation of the mutant homodimer, a mechanismthat has previously been described for erythropoietin, epidermalgrowth factor and RET receptors (24-26).
Two other recurrent mutations, Tyr340His and Trp29OArg,also occur at particularly highly conserved positions of boththe FGFR family and the more extended family of immuno-globulin-like loops (18). This again suggests that they have amajor part in preserving the structure. The difference inphenotypes between the Crouzon and Apert syndromes maybe attributed to mutations which significantly disrupt thesecondary structure and function of the FGFR2 moleculeresulting in the Crouzon syndrome whereas specific mutationssubtly affecting receptor function cause the Apert syndrome.
There remain a number of patients with Crouzon syndrome,all of whom have been fully clinically assessed, in which wehave found no mutation in exon IIIu or exon Me. No SSCPchanges have been found in exon Mb (unpublished results)and there remains the possibility that these cases may becaused by mutations elsewhere in FGFR2, in FGFR1 or evenin one of the ligands for FGFR2, such as FGF1, 2 or 4.
MATERIALS AND METHODS
Exon IIIu analysis
Sequences from exon IIIu were either obtained from cDNA using publishedmethods (8) or from genomic DNA. Primers for PCR amplification of theexon from genomic DNA were designed, using PRIMER program (Version5.0) from the extreme 5' end of the sequence and from the downstream intron.Forward: 5'-GAGCGATCGCCTCACC-3' Reverse: 5'-TGTGGGTACCTT-TAGATTCAGAAAG-3'. The reverse primer was biotinylated at the 5' endand the sequencing carried out using magnetic streptavidin coated beads(Dynal UK). SSCP analysis was carried out as previously described (5,8).
Restriction enzyme digestion
PCR products from exon IIIu were digested with Bbs\ (New England Biolabs),Bsfl. (New England Biolabs), Bsall (New England Biolabs) or Mnh (NewEngland Biolabs) according to the manufacturer's instructions and the resultingproduct separated on a 2% agarose or 4.5% Metaphor (Flowgen) gel. PCRproducts from exon IIIc were digested with BsaAl (New England Biolabs)and separated on a 2% agarose gel.
Exon IIIc analysis
SSCP and DNA sequencing was carried out as previously described (5).
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

1082 Human Molecular Genetics, 1995, Vol. 4, No. 6
ACKNOWLEDGEMENTS
This work was supported by the Medical Research Council (UP, WR, RMW,SM) and the Wellcome Trust (MO, AOMW, SFS). We are grateful to AdrianSugar for referring two of the patients and John Heath for discussions. Figure2 is reproduced by kind permission of British Journal of Hospital Medicine(ref. 27).
REFERENCES1. Johnson, D.E., Williams, L.T.(1993) Structural and functional diversity
in the FGF receptor multigene family. Adv. Cane. Res., 60: 1-41.2. Mason, I.J. (1994) The ins and outs of fibroblast growth factors. Cell,
78: 547-552.3. Shiang, R., Thompson, L.M., Zhu, Y.Z., Church, D.M., Fielder, T.J.,
Bocian, M., Winokur, S.T., Wasmuth, J.J. (1994) Mutations in thetransmembrane domain of FGFR3 cause the most common form ofgenetic dwarfism, achondroplasia. Cell, 78: 335-342.
4. Rousseau, F., Bonaventure, J., Legeai-Mallet, L., Pelet, A., Rozet, J-M.,Maroteaux, P., LeMerre, M., Munnich, A. (1994) Mutation in the geneencoding fibroblast growth factor receptor-3 in achondroplasia. Nature,371: 252-254.
5. Reardon, W., Winter, R.M., Rutland, P., Pulleyn, L.J., Jones, B., Malcolm,S. (1994) Mutations in the fibroblast growth factor receptor-2 gene causeCrouzon syndrome. Nature Genet., 8: 98-103.
6. Jabs, E.W., Li, X., Scott, A.F., Meyers, G., Chen, W., Eccles, M., Mao,J., Charnas, L.R., Jackson, C.E., Jaye, M. (1994) Jackson-Weiss andCrouzon syndromes are allelic with mutations in fibroblast growth factorreceptor 2. Nature Genet., 8: 275-279.
7. Muenke, M., Schell, U., Hehr, A., Robin, N.H., Losken, H.W., Schinzel,A., Pulleyn, L.J., Rutland, P., Reardon, W., Malcolm, S., Winter, R.M.(1994) A common mutation in the fibroblast growth factor receptor 1gene in Pfeiffer syndrome. Nature Genet., 8: 269-273.
8. Wilkie, A.O.M., Slaney, S.F., Oldridge, M., Poole, M.D., Ashworth, G.,Hockley, A.D., Hayward, R.D., David, D.J., Pulleyn, L.J., Rutland, P.,Malcolm, S., Winter, R.M., Reardon, W. (1995) Apert syndrome resultsfrom localised mutations of FGFR2 and is allelic with Crouzon syndrome.Nature Genet., 9: 165-172.
9. Rutland, P., Pulleyn, L.J., Reardon., W., Baraitser, M., Hayward, R.,Jones, B., Malcolm, S., Winter, R.M., Oldridge, M., Slaney, S.F., Poole,M.D., Wilkie, A.O.M. (1995) Identical mutations in the FGFR2 genecause both Pfeiffer and Crouzon syndrome. Nature Genet., 9: 173-176.
10. Orr-Urtreger, A., Givol, D., Yayon, A., Yarden, Y, Lonai, P. (1991)Developmental expression of two murine fibroblast growth factorreceptors, fig and bek. Development, 113: 1419-1434.
11. Orr-Urtreger, A., Bedford, M.T., Burakova, T, Arman, E., Zimmer, Y,Yayon, A., Givol, D., Lonai, P. (1993) Developmental localization of thesplicing alternatives of fibroblast growth factor receptor 2 (FGFR2). Dev.Bioi, 158: 475^186.
12. Preston, R.A., Post, J.C., Keats, B.J.B., Aston, C.E., Ferrell, R.E., Priest,J., Nouri, N., Losken, H.W., Morris, C.A., Hum, MR., Mulvihill, J.J.,Ehrlich, G.D. (1994) A gene for Crouzon craniofacial dysostosis mapsto the long arm of chromosome 10. Nature Genet., 7: 149-153.
13. Li, X., Lewanda, A.F., Eluma, F, Jerald, H., Choi, H., Alozie, I., Proukakis,C , Talbot, C.C., Kolk, C.V., Bird, L.M., Jones, M.C., Cunningham, M.,Clarren, S.K., Pyeritz, R.E., Weissenbach, J., Jackson, C.E., Jabs, E.W.(1994) Two craniosynostotic syndrome loci, Crouzon and Jackson-Weiss,map to chromosome 10q23-q26. Genomics, 22: 418-424.
14. Robin, N.H., Feldman, G.J., Mitchell, H.F., Lorenz, P., Wilroy, R.S.,Zackal, E.H., Allanson, J.E., Reich, E.W, Pfeiffer, R.A., Clarke, L.A.,Warman, M.L., Mulliken, J.B., Brueton, L.A., Winter, R.M., Price, R.A.,Gasser, D.L., Muenke, M. (1994) Linkage of Pfeiffer syndrome tochromosome 8 centromere and evidence for genetic heterogeneity. Hum.Mol. Genet., 3: 2153-2158.
15. Lajeunie, E., Ma, H.W., Bonaventure, J., Munnich, A., Le Merrer, M.,Renier, D. (1995) FGFR2 mutations in Pfeiffer syndrome. Nature Genet.,9: 108.
16. Spivak-Kroizman, T., Lemmon, M.A., Dikic, I., Ladbury, J.E., Pinchael,D., Huang, J., Jaye, M., Crumley, G., Schlessinger, J., Lax, I. (1994)Heparin-induced oligomerization of FGF molecules is responsible forFGF receptor dimerization, activation and cell proliferation. Cell, 79:1015-1024.
17. Kan, M., Wang, F., Xu, J., Crabb, J.W., Hou, J., McKeehan, W.L. (1993)
An essential heparin-binding domain in the fibroblast growth factorreceptor kinase. Science, 259: 1918-1921.
18. Williams, A.F., Barclay, A.N. (1988) The immunoglobulin superfamily—domains for cell surface recognition. Annu. Rev. Immunol., 6: 381—405.
19. Hou, J., Kan, M., Wang, F, Xu, J., Nakahara, M., McBride, G., McKeehan,K., McKeehan, W.L. (1992) Substitution of putative half-cystine residuesin heparin-binding fibroblasl growth factor receptors. J. Biol. Chem., 267:17804-17808.
20. Winter, R.M., Baraitser, M. (1994) The London Dysmorphology Database.Oxford University Press, Oxford.
21. Cohen, M.M. Jr. (1986) Craniosynostosis: Diagnosis, Evaluation andManagement. Raven Press, New York.
22. Pfeiffer, R.A. (1964) Dominant erbliche Akrocephalosyndaktylie. Z.Kinderheilk., 90: 301-320.
23. Sugar, A.W., Walker, D.M., Bounds, G.A. (1990) Surgical ciliated(postoperative maxillary) cysts following mid-face osteotomies. Br. J.Oral Maxillofacial Surg., 28: 264-267.
24. Watowich, S.S., Yoshimura, A., Longmore, CD., Hilton, D.J., Yoshimura,Y, Lodish, H.F. (1994) Homodimerization and constitutive activation ofthe erythropoietin receptor. Proc. Natl Acad. Sci. USA, 89: 214-244.
25. Sorokin, A., Lemmon, M.A., Ullrich, A., Schlessinger, J. (1994)Stabilisation of an active dimeric form of the epidermal growth factor byintroduction of an inter-receptor disulfide bond. /. Biol. Chem., 269:9752-9759.
26. Santoro, M., Carlomagno, F, Romano, A., Bottaro, D.P., Dothan, N.A.,Grieco, M., Frisco, A., Vecchio, G., Matoskova, B., Kraus, M.H., DiFiore, P.P. (1995) Activation of RET as a dominant transforming geneby germline mutations of MEN2A and MEN2B. Science, 267: 381-383.
27. Goldin, H., Hockley, A., Wake, M., Beasley, J. (1986) Craniofacialsurgery. Br. J. Hosp. Med., 41: 368-373.
28. Superti-Furga, A., Eich,G., Bucher.H.U., Wisser.J., Giedion.A.,Gitzelmann.R., Steinmann.B. (1995) A glycine 375 to cysteine substitutionin the transmembrane domain of the fibroblast growth factor receptor 3in a new born with achondroplasia. Eur. J. Paediatr., 154: 215-219.
at University C
ollege London on A
pril 23, 2015http://hm
g.oxfordjournals.org/D
ownloaded from
Related Documents











