Musician’s Dystonia and Comorbid Anxiety: Two Sides of One Coin? Leonie Enders, cand. med., 1 June T. Spector, MD, MPH, 2 Eckart Altenmu ¨ ller, MD, MA, 1 Alexander Schmidt, MD, 3,4 Christine Klein, MD, 3,4 and Hans-Christian Jabusch, MD 5 * 1 Institute of Music Physiology and Musicians’ Medicine, Hannover University of Music, Drama and Media, Hannover, Germany; 2 Occupational & Environmental Medicine Program, University of Washington, Seattle, Washington, USA; 3 Section of Clinical and Molecular Neurogenetics at the Department of Neurology, University of Lu ¨ beck, Lu ¨ beck, Germany; 4 Department of Neurology, University of Lu ¨ beck, Lu ¨ beck, Germany; 5 Institute of Musicians’ Medicine, University of Music Carl Maria von Weber, Dresden, Germany ABSTRACT Background: Psychological abnormalities, including anxiety, have been observed in patients with musician’s dystonia (MD). It is unclear if these conditions develop prior to MD or if they are psychoreactive phenomena. Methods: Psychological conditions were studied in 44 professional musicians with MD, 45 healthy musicians, and 44 healthy nonmusicians using the State-Trait Anxiety Inventory (STAI) and NEO Five-Factor Inventory (NEO-FFI). Results: Musicians with MD had significantly higher STAI state and trait anxiety scores than healthy musicians (P 5 .009 and P 5 .012, respectively) and nonmusicians (P 5 .013 and P 5 .001, respectively) and significantly higher NEO-FFI neuroticism scores than healthy musicians (P 5 .018) and nonmusicians (P 5 .001). Duration of dystonia did not correlate with anxiety or neuroticism scores. Conclusions: Musicians with MD display increased levels of anxiety and neuroticism. The lack of correla- tion between anxiety and the duration of dystonia sug- gests that anxiety may not be a psychoreactive phenomenon and is consistent with the hypothesis that anxiety and MD share a common pathophysiological mechanism. V C 2011 Movement Disorder Society Key Words: zdystonia; musician; psychology; anxiety; State-Trait Anxiety Inventory; NEO-Five-Factors Inventory Focal dystonia (FD)in musicians, also called musi- cian’s dystonia (MD), is a task-specific movement dis- order characterized by painless muscular incoordina- tion of extensively trained movements. 1–4 Involuntary flexion or extension of individual fingers or loss of control of the muscles involved in the embouchure in affected musicians can lead to impaired technical per- formance on the instrument. 1–4 Musicians with FD are therefore often unable to continue their careers as per- forming artists. 5,6 Several published studies have examined psychologi- cal conditions in patients with FD. Increased preva- lence of anxiety, social phobia, and depression has been reported in patients with cervical dystonia, ble- pharospasm, and writer’s cramp. 7–11 Obsessive com- pulsive disorder (OCD) has been reported to be more common in patients with idiopathic focal dystonia and mycoclonus-dystonia than in controls. 12,13 Increased odds of social phobia, agoraphobia, panic disorder, and OCD was found in patients with cervical dystonia and blepharospasm compared with population-based controls. 14 Based on a comparison of self-reported age of onset of psychiatric abnormalities and of FD, the authors concluded that most psychiatric abnormalities were present prior to the onset of FD. 14 Only 2 studies have focused specifically on psycho- logical conditions in musicians with FD, reporting that musicians with FD more commonly exhibited specific phobias, anxiety, and perfectionism than did con- trols. 15,16 However, these studies were limited by their relatively small sample size and lack of use of vali- dated questionnaires to evaluate the mentioned psy- chological characteristics. The present study was designed to investigate psy- chological abnormalities in a larger group of musi- cians with FD compared with healthy musicians and healthy nonmusicians, using validated questionnaires. We hypothesized that a significantly higher degree of anxiety and other psychological conditions are present in musicians with FD. Patients and Methods Patients Patients were recruited from the outpatient clinic of the Hannover Institute of Music Physiology and Musicians’ Medicine. All patients underwent complete neurological evaluations and were diagnosed with MD by at least 1 of the authors (E.A.). Patients with other neurological disorders or secondary dystonias or psy- chiatric diseases were excluded from the study. ------------------------------------------------------------ Leonie Enders and June Spector contributed equally to this article. *Correspondence to: Prof. Dr. Hans-Christian Jabusch, Institute of Musicians’ Medicine, Dresden University of Music Carl Maria von Weber, Wettiner Platz 13, D-01067 Dresden, Germany; [email protected]. Relevant conflicts of interest/financial disclosures: Nothing to report. Full financial disclosures and author roles may be found in the online version of this article. Received: 23 August 2010; Revised: 2 November 2010; Accepted: 29 November 2010 Published online 2 March 2011 in Wiley Online Library (wileyonlinelibrary.com). DOI: 10.1002/mds.23607 BRIEF REPORTS Movement Disorders, Vol. 26, No. 3, 2011 539

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Musician’s Dystonia and ComorbidAnxiety: Two Sides of One Coin?
Leonie Enders, cand. med.,1 June T. Spector, MD, MPH,2
Eckart Altenmuller, MD, MA,1 Alexander Schmidt, MD,3,4
Christine Klein, MD,3,4 and Hans-Christian Jabusch, MD5*
1Institute of Music Physiology and Musicians’ Medicine, HannoverUniversity of Music, Drama and Media, Hannover, Germany;2Occupational & Environmental Medicine Program, University ofWashington, Seattle, Washington, USA; 3Section of Clinical andMolecular Neurogenetics at the Department of Neurology, Universityof Lubeck, Lubeck, Germany; 4Department of Neurology, Universityof Lubeck, Lubeck, Germany; 5Institute of Musicians’ Medicine,University of Music Carl Maria von Weber, Dresden, Germany
ABSTRACTBackground: Psychological abnormalities, includinganxiety, have been observed in patients with musician’sdystonia (MD). It is unclear if these conditions developprior to MD or if they are psychoreactive phenomena.Methods: Psychological conditions were studied in 44professional musicians with MD, 45 healthy musicians,and 44 healthy nonmusicians using the State-Trait AnxietyInventory (STAI) and NEO Five-Factor Inventory (NEO-FFI).Results: Musicians with MD had significantly higher STAIstate and trait anxiety scores than healthy musicians (P 5.009 and P 5 .012, respectively) and nonmusicians (P 5.013 and P 5 .001, respectively) and significantly higherNEO-FFI neuroticism scores than healthy musicians (P 5.018) and nonmusicians (P 5 .001). Duration of dystoniadid not correlate with anxiety or neuroticism scores.Conclusions: Musicians with MD display increasedlevels of anxiety and neuroticism. The lack of correla-tion between anxiety and the duration of dystonia sug-gests that anxiety may not be a psychoreactivephenomenon and is consistent with the hypothesis thatanxiety and MD share a common pathophysiologicalmechanism. VC 2011 Movement Disorder SocietyKey Words: zdystonia; musician; psychology; anxiety;State-Trait Anxiety Inventory; NEO-Five-Factors Inventory
Focal dystonia (FD)in musicians, also called musi-cian’s dystonia (MD), is a task-specific movement dis-order characterized by painless muscular incoordina-tion of extensively trained movements.1–4 Involuntaryflexion or extension of individual fingers or loss ofcontrol of the muscles involved in the embouchure inaffected musicians can lead to impaired technical per-formance on the instrument.1–4 Musicians with FD aretherefore often unable to continue their careers as per-forming artists.5,6
Several published studies have examined psychologi-cal conditions in patients with FD. Increased preva-lence of anxiety, social phobia, and depression hasbeen reported in patients with cervical dystonia, ble-pharospasm, and writer’s cramp.7–11 Obsessive com-pulsive disorder (OCD) has been reported to be morecommon in patients with idiopathic focal dystonia andmycoclonus-dystonia than in controls.12,13 Increasedodds of social phobia, agoraphobia, panic disorder,and OCD was found in patients with cervical dystoniaand blepharospasm compared with population-basedcontrols.14 Based on a comparison of self-reported ageof onset of psychiatric abnormalities and of FD, theauthors concluded that most psychiatric abnormalitieswere present prior to the onset of FD.14
Only 2 studies have focused specifically on psycho-logical conditions in musicians with FD, reporting thatmusicians with FD more commonly exhibited specificphobias, anxiety, and perfectionism than did con-trols.15,16 However, these studies were limited by theirrelatively small sample size and lack of use of vali-dated questionnaires to evaluate the mentioned psy-chological characteristics.The present study was designed to investigate psy-
chological abnormalities in a larger group of musi-cians with FD compared with healthy musicians andhealthy nonmusicians, using validated questionnaires.We hypothesized that a significantly higher degree ofanxiety and other psychological conditions are presentin musicians with FD.
Patients and Methods
Patients
Patients were recruited from the outpatient clinicof the Hannover Institute of Music Physiology andMusicians’ Medicine. All patients underwent completeneurological evaluations and were diagnosed with MDby at least 1 of the authors (E.A.). Patients with otherneurological disorders or secondary dystonias or psy-chiatric diseases were excluded from the study.
------------------------------------------------------------Leonie Enders and June Spector contributed equally to this article.*Correspondence to: Prof. Dr. Hans-Christian Jabusch, Institute ofMusicians’ Medicine, Dresden University of Music Carl Maria von Weber,Wettiner Platz 13, D-01067 Dresden, Germany; [email protected].
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 23 August 2010; Revised: 2 November 2010; Accepted: 29November 2010Published online 2 March 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23607
B R I E F R E P O R T S
Movement Disorders, Vol. 26, No. 3, 2011 539

Forty-four professional musicians (26 men, 18women; mean age 6 SD, 40.2 6 9.2 years; age range,23–64 years) with MD were enrolled in the study.MD presented as hand dystonias in 40 and as embou-chure dystonia in 4 musicians. At the time of thestudy, the mean duration of the disorder was 9.3 years(SD, 8.1 years; range, 0–37 years).
Controls
Two control groups were enrolled in the study. Thefirst control group consisted of healthy professionalmusicians recruited from German orchestras andmusic schools, and the second group consisted ofhealthy nonmusician university graduates. Potentialcontrols were excluded if they reported a history ofneurological or psychiatric diseases. All groups werematched for age and sex.The healthy musician group consisted of 45 profes-
sional musicians (27 men, 18 women; mean age 6 SD,41.5 6 12.6 years; age range, 20–68 years). This groupwas also matched by instrument family to the MDgroup. The healthy nonmusician group consisted of 44university graduates (27 men, 17 women; mean age 6SD, 41.7 6 16.2 years; age range, 21–76 years).Details of all 3 groups are given in Table 1. Informed
consent was obtained from all subjects before studyparticipation. The study was conducted in accordancewith the principles of the Declaration of Helsinki.
Psychological Assessment
Assessment of personality and anxiety was per-formed using the revised German version17 of theself-administered NEO Five-Factor Inventory (NEO-FFI)18 and the State-Trait-Anxiety Inventory(STAI),19 respectively. Questionnaires were distrib-uted and collected by mail and were accompanied bydetailed written instructions in the German language.All subjects were able to speak, read, and write Ger-man fluently.The NEO-FFI is a multidimensional personality
inventory that consists of 12 items (descriptions ofbehaviors), scored on 5-point Likert scales in eachof 5 personality domains: (1) extraversion, (2)agreeableness, (3) conscientiousness, (4) neuroti-cism, and (5) openness to experience.18 The NEO-FFI has been shown to be reliable, valid, andconsistent.20
The STAI is designed to distinguish chronic, or trait,anxiety (a general propensity to be anxious), fromtemporary, or state anxiety.19 The STAI consists of 20trait and 20 state anxiety statements, scored on4-point Likert scales, that assess how respondents feel‘‘generally’’ and ‘‘right now, at this moment.’’ TheSTAI has been shown to be reliable, valid, and respon-sive to clinical change.21
Statistical Analyses
Comparison of NEO-FFI and STAI scores betweenthe dystonia and control groups was performed usingthe Kruskal–Wallis test and the post hoc Tamhane’sT2 test. The Kruskal–Wallis test was chosen becauseof heterogeneity of variances demonstrated for age andthe NEO-FFI extraversion subscale using the Levenetest. Correlation was determined using Spearman’s r.P < .05 was considered statistically significant.
ResultsAll components of the questionnaires were com-
pleted by all subjects. Musicians with dystonia had sig-nificantly higher NEO-FFI neuroticism subscale scoresthan healthy musicians (P ! .018) or nonmusicians (P! .001); see Figure 1. There was no significant differ-ence in neuroticism scores between healthy musiciansand nonmusicians (P ! .790) or between subjectgroups on any of the other NEO-FFI subscales.Musicians with dystonia had significantly higher
STAI state and trait anxiety subscale scores thanhealthy musicians (P ! .009 and P ! .012, respec-tively) or nonmusicians (P ! .013 and P ! .001,respectively); see Figure 1. There was no significantdifference in state and trait anxiety scores betweenhealthy musicians and nonmusicians (P ! .997 andP ! .614, respectively).There was no significant correlation between the du-
ration of dystonia and NEO-FFI neuroticism subscalescores (Spearman’s r ! 0.005, P ! .976). Negativecorrelation was found between the duration of dysto-nia and the openness subscale (Spearman’s r !"0.268), and this correlation approached statisticalsignificance (P ! .079). There was a negative correla-tion between age and openness in musicians with
Table 1. Patient and control characteristics
Parameter MD HM NM
Number 44 45 44Sex (male/female) 26/18 27/18 27/17Age, y (mean 6 SD) 40.2 6 9.2 41.5 6 12.6 41.7 6 16.2Age, y (min/max) 23/64 20/68 21/76Woodwind instruments (n) 16 15 —String instruments (n) 14 14 —Brass instruments (n) 4 6 —Plucking instruments (n) 5 5 —Keyboard instruments (n) 4 4 —Drums (n) 1 1 —Age at onset of dystonia,y (mean 6 SD)
30.9 6 8.9 — —
Duration of dystonia,y (mean 6 SD)
9.3 6 8.1 — —
MD, musicians with dystonia; HM, healthy musicians; NM, healthynonmusicians; y, years; SD, standard deviation.
E N D E R S E T A L .
540 Movement Disorders, Vol. 26, No. 3, 2011

dystonia (Spearman’s r ! "0.363, P ! .016) but nocorrelation between age and openness in the controlgroups. There was no correlation between duration ofdystonia and state or trait anxiety.
DiscussionIn this study of psychological conditions in MD,
affected musicians showed a higher degree of state andtrait anxiety and neuroticism than did healthy musiciansand nonmusician controls. Our results showingincreased anxiety in musicians with MD are supportedby conclusions of prior publications that reported signifi-cantly greater anxiety in musicians with MD comparedwith healthy musicians.15,16,22 Increased anxiety has alsobeen reported in patients with other dystonias.7,23,24
The cross-sectional nature of this study precludes adefinitive determination of whether or not anxiety orneuroticism was present before the onset of MD.However, trait anxiety and neuroticism scores weresignificantly elevated in MD patients, and there wasno correlation between neuroticism or anxiety scoresand duration of MD. These results are consistent withstudies of nonmusician patients with anxiety and otherforms of FD, which reported that anxiety did not de-velop after the onset of FD, suggesting that anxiety isnot a psychoreactive phenomenon.14
A shared underlying mechanism for the developmentof MD and anxiety may exist, as reported for some
forms of monogenic dystonia such as myoclonus-dys-tonia.25 Transcranial magnetic stimulation and elec-troencephalogram studies have provided evidence ofdecreased cortical inhibition in FD.26–28 Decreasedcortical inhibition has also been observed in subjectswith trait-related anxiety.29 It has been suggested thatreduced cortical inhibition may play a role in thepathophysiology of both FD and anxiety.14,30 Specifi-cally, abnormal neural activity in motor loops linkingthe basal ganglia to the frontal cortex via the thalamusmay additionally influence limbic loops, resulting inboth altered motor and affective processing.14
Strengths of this study include a relatively large sam-ple size and use of reliable and valid questionnairesfor the evaluation of personality18,20 and anxiety.19,21
Limitations include the cross-sectional nature of thisstudy and the resultant difficulty in characterizing tem-poral associations between psychological disordersand FD. The relatively low prevalence of FD in musi-cians1 presents a barrier to the study of FD in musi-cians in a prospective manner.
ConclusionsMusicians with FD showed significantly higher val-
ues of neuroticism, state anxiety, and trait anxietythan did healthy musician and non-musician controls.There was no significant correlation between neuroti-cism or anxiety and duration of focal dystonia, but
FIG. 1. Box plots of NEO-FFI and STAI subscale scores by subject group (N, neuroticism; E, extraversion; O, openness; A, agreeableness; C, con-scientiousness; S, state anxiety; T, trait anxiety; NEO-FFI scores: from 0 5 ‘‘strongly disagree’’ to 4 5 ‘‘strongly agree’’). Overall range of STAIscores: 20–80 points (*P < .05).
A N X I E T Y I N M U S I C I A N S W I T H F O C A L D Y S T O N I A
Movement Disorders, Vol. 26, No. 3, 2011 541

there was a negative correlation between openness toexperience and duration of focal dystonia thatapproached statistical significance. These results raisethe possibility that openness to experience is a psy-choreactive phenomenon as a consequence of MD,whereas other psychological conditions, including neu-roticism and anxiety, do not represent psychoreactivephenomena. The hypothesis of a potential commonpathophysiological mechanism of anxiety and MDshould be further investigated.
References1. Jankovic J, Shale H. Dystonia in musicians. Semin Neurol 1989;9:
131–135.
2. Lederman RJ. Focal dystonia in instrumentalists: clinical features.Med Probl Perform Art 1991:132–136.
3. Altenmuller E. Focal dystonia: advances in brain imaging andunderstanding of fine motor control in musicians. Hand Clin 2003;19:523–538.
4. Conti AM, Pullman S, Frucht SJ. The Hand That Has ForgottenIts Cunning—Lessons from Musicians’ Hand Dystonia. Mov Dis-ord 2008;23:1398–1406
5. Brandfonbrener AG, Robson C. Review of 113 musicians withfocal dystonia seen between 1985 and 2002 at a clinic for perform-ing artists. Adv Neurol 2004;94:255–256.
6. Schuele SU, Lederman RJ. Long-term outcome of focal dystonia ininstrumental musicians. Adv Neurol 2004;94:261–266.
7. Wenzel T, Schnider P, Wimmer A, Steinhoff N, Moraru E, Auff E.Psychiatric comorbidity in patients with spasmodic torticollis.J Psychosom Res 1998;44:687–690.
8. Wenzel T, Schnider P, Griengl H, Birner P, Nepp J, Auff E. Psychi-atric disorders in patients with blepharospasm—a reactive pattern?J Psychosom Res 2000;48:589–591.
9. Windgassen K, Ludolph A. Psychiatric aspects of writer’s cramp.Eur Arch Psychiatry Clin Neurosci 1991;241:170–176.
10. Muller J, Kemmler G, Wissel J, et al. The impact of blepharospasmand cervical dystonia on health-related quality of life and depres-sion. J Neurol 2002;249:842–846.
11. Gundel H, Wolf A, Xidara V, Busch R, Ceballos-Baumann AO.Social phobia in spasmodic torticollis. J Neurol Neurosurg Psychia-try 2001;71:499–504.
12. Saunders-Pullman R, Shriberg J, Heiman G, et al. Myoclonus dys-tonia: Possible association with obsessive-compulsive disorder andalcohol dependence. Neurology 2002;58:242–245.
13. Cavallaro R, Galardi G, Cavallini MC, et al. Obsessive compulsivedisorder among idiopathic focal dystonia patients: an epidemiologi-cal and family study. Biol Psychiatry 2002;52:356–361.
14. Lencer R, Steinlechner S, Stahlberg J, et al. Primary focal dystonia:Evidence for distinct neuropsychiatric and personality profiles.J Neurol Neurosurg Psychiatry 2009;80:1176–1179.
15. Jabusch HC, Muller SV, Altenmuller E. Anxiety in musicians withfocal dystonia and those with chronic pain. Mov Disord 2004;19:1169–1175.
16. Jabusch H, Altenmuller E. Anxiety as an aggravating factor during onsetof focal dystonia in musicians. Med Probl Perform Art 2004;19:75–81.
17. Borkenau P, Ostendorf F. Comparing exploratory and confirma-tory factor analysis: A study on the 5-factor model of personality.Pers Individ Dif 1990;11:515–524.
18. Costa PT, McCrae RR. Revised NEO Personality Inventory (NEO-PI-R) and NEO Five-Factor Inventory (NEO-FFI) Professional Manual.Odessa, FL: Psychological Assessment Resources, Inc.; 1992.
19. Spielberger CD, Gorsuch RL.Manual for the State-Trait AnxietyInventory (Form Y) (‘‘Self-Evaluation Questionnaire’’). Palo Alto,CA: Consulting Psychologists Press; 1983.
20. McCrae RR, John OP. An introduction to the five-factor modeland its applications. J Pers 1992;60:175–215.
21. Quek KF, Low WY, Razack AH, Loh CS, Chua CB. Reliabilityand validity of the Spielberger State-Trait Anxiety Inventory(STAI) among urological patients: a Malaysian study. Med JMalaysia 2004;59:258–267.
22. Altenmuller E, Jabusch HC. Focal hand dystonia in musicians: phe-nomenology, etiology and psychological trigger factors. J HandTher 2009;22:144–155.
23. Scheidt CE, Schuller B, Rayki O, Kommerell G, Deuschl G. Rela-tive absence of psychopathology in benign essential blepharospasmand hemifacial spasm. Neurology 1996;47:43–45.
24. Lauterbach EC, Freeman A, Vogel RL. Differential DSM-III psy-chiatric disorder prevalence profiles in dystonia and Parkinson’sdisease. J Neuropsychiatry Clin Neurosci 2004;16:29–36.
25. Doheny DO, Brin MF, Morrison CE, et al. Phenotypic featuresof myoclonus-dystonia in three kindreds. Neurology 2002;59:1187–1196.
26. Deuschl G, Toro C, Matsumoto J, Hallett M. Movement-relatedcortical potentials in writer’s cramp. Ann Neurol 1995;38:862–868.
27. Ridding MC, Sheean G, Rothwell JC, Inzelberg R, Kujirai T.Changes in the balance between motor cortical excitation and inhi-bition in focal, task specific dystonia. J Neurol Neurosurg Psychia-try 1995;59:493–498.
28. Toro C, Deuschl G, Hallett M. Movement-related electroencephalo-graphic desynchronization in patients with hand cramps: evidence formotor cortical involvement in focal dystonia. Ann Neurol 2000;47:456–461.
29. Wassermann EM, Greenberg BD, Nguyen MB, Murphy DL.Motor cortex excitability correlates with an anxiety-related person-ality trait. Biol Psychiatry 2001;50:377–382.
30. Ron MA. Primary focal dystonia—a disease of brain and mind:Motor and psychiatric manifestations have a common neurobiolog-ical basis. J Neurol Neurosurg Psychiatry 2009;80:1059.
E N D E R S E T A L .
542 Movement Disorders, Vol. 26, No. 3, 2011

Cysteinyl-Glycine Reduction asMarker for Levodopa-InducedOxidative Stress in Parkinson’s
Disease Patients
Thomas Muller, MD1,2* and Siegfried Muhlack, MD2
1Department of Neurology, St. Joseph Hospital Berlin-Weissensee,Berlin, Germany; 2Department of Neurology, St. Josef Hospital, RuhrUniversity Bochum, Bochum, Germany
ABSTRACTOxidative stress is influenced by the thiol homeostasis,which determines the redox milieu. One of its compo-nents is Cysteinyl-glycine (Cys-Gly) generation, as itsmetabolic precursor is the free radicals scavenging gluta-thione. Levodopa is under suspicion to promote oxidative

untreated, 9 were on a chronic drug regimen for treat-ment of PD (L-dopa/DDI [monotherapy, N ! 1] plusvarious dopamine agonists [8]). Exclusion criteriawere prior exposure to neuroleptics or any otherdrugs, which aggravate motor symptoms in PDpatients, clinical signs of dementia, any electrophysio-logical or morphological evidence of additional CNSpathology exceeding PD. Patients fulfilled clinicaldiagnostic UK Brain bank criteria for PD.4
Design
Participants orally received a 200-mg L-dopa/50-mgcarbidopa (CD)-containing tablet at baseline in themorning following an overnight fast and washout oftheir additional PD drug regime, which was initiatedafter the trial again. Blood sampling was performed atbaseline before application of L-dopa/CD applicationand then 60- and 120 min after L-dopa/CDadministration.
Blood Samples
Ten milliliters venous blood specimen were takenfor estimation of L-dopa- and 3-OMD levels from anantecubital vein through an indwelling catheter keptpatent by an infusion of heparin in saline solution(10U/mL). Venous puncture was performed 20 minbefore the baseline investigation, to enable stable con-ditions. Three milliliters of blood were drawn with aseparate syringe and discarded before taking each 10mL specimen. Blood samples were collected in EDTA-test tubes containing 100 lL of 0.5% sodium disulfitesolution. The plasma obtained from rapid centrifuga-tion was immediately frozen at "80#C until analysiswithin 14 days.
MethodsReversed-phase high-performance liquid chromatog-
raphy (HPLC) was used in combination with electro-chemical detection for the measurement of L-dopa and3-OMD concentrations in plasma, which was diluted
with a factor of 1:1.95 before assessment. Free Cys-gly levels were determined by an automated HPLCprocedure with reverse phase separation and fluores-cent detection by NaBH4/mBrB reduction followed bymonobromobimane derivatization.
Statistics
ANCOVAs (covariates: sex, age, body weight, andUPDRS-scores) with repeated measures design wereused for comparisons. The Tukeys HSD-test was usedfor the post hoc analysis. Correlations were done withlinear regression. Its variables were computed differen-ces between moment baseline, 60 and 120 min after L-dopa/CD application of 3-OMD and Cys-Gly levels.Both substrates have a longer half life than L-dopa. 3-OMD accumulates with a certain delay only.
Ethics
All participants gave written informed consent. Thestudy was approved by the local ethical committee ofthe Ruhr University of Bochum.
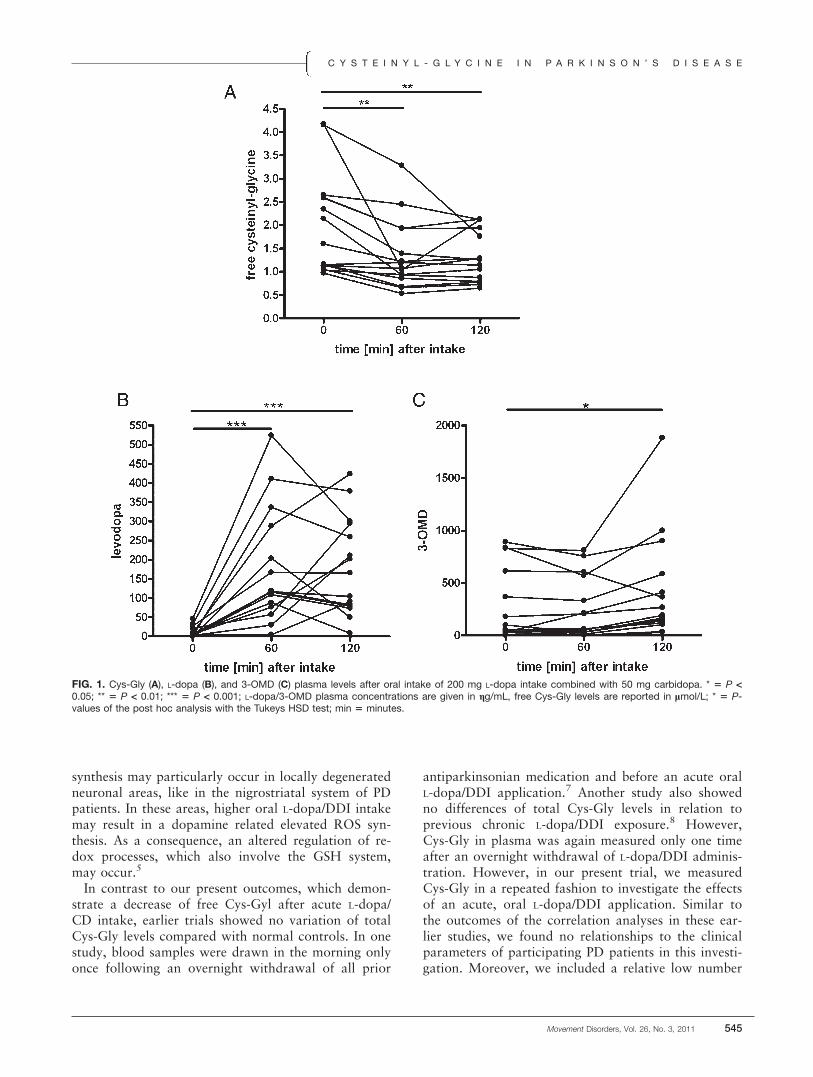
ResultsCys-Gly concentrations decreased following L-dopa/
CD intake (F(df 2, dF 14) ! 9.15, P < 0.0009; Fig. 1A).L-dopa (F(df 2, dF 14) ! 16.88, P < 0.0001; Fig. 1B)and 3-OMD levels (F(df 2, dF 14) ! 4.92, P < 0.015;Fig. 1C) increased. There were inverse relationshipsbetween computed differences of Cys-gly and 3-OMDappearance ([Baseline – 120] R ! "0.52; P ! 0.046;[60 – 120] R ! "0.74; P ! 0.0001). Sex, age, bodyweight, duration of disease, and UPDRS-scores didnot influence our results.
Adverse Events
There were no relevant adverse effects.
DiscussionWe show a decay of free Cys-Gly plasma concentra-
tions following one oral L-dopa/CD tablet intakeunder standardized conditions. As a result, a rise of L-dopa and 3-OMD plasma levels also took place.Hypothetically, these reactions may also occur withinthe brain, as L-dopa is converted to dopamine in tyro-sine hydroxylase-containing dopaminergic neuronsand then is further metabolized in glial cells by MAO-B after leaving the synaptic cleft. This metabolic path-way of dopamine degradation is associated with freeradical generation.3 The observed, inverse correlationsbetween computed differences of the Cys-Gly and 3-OMD levels between various assessments momentssupport this putative link between L-dopa metabolismwith associated excessive dopamine synthesis andrelated oxidative stress.5,6 This increased ROS
Table 1. Clinical characteristics
Age 60.7 6 11.1, 41 – 80
Sex 12 men, 3 womenHoehn and Yahr stage 2.3 6 0.9, I–IIIUPDRS I 2.1 6 1.9, 0 – 6UPDRS II 14.8 6 12.2; 5 – 52UPDRS III 22.3 6 11.6; 7 – 42UPDRS IV 0.8 6 0.8; 0 – 2Duration of disease 5.8 6 5.48; 0 – 13
All data are shown as mean 6 standard deviation, range; UPDRS ! UnifiedParkinson’s disease rating scale; I (mentation, behavior, and mood), II(activities of daily living), III (motor examination), IV (complication oftherapy) ! various parts of the UPDRS; duration of disease, respectivelyinterval since definite clinical diagnosis of PD, is given in months.
M U L L E R E T A L .
544 Movement Disorders, Vol. 26, No. 3, 2011
synthesis may particularly occur in locally degeneratedneuronal areas, like in the nigrostriatal system of PDpatients. In these areas, higher oral L-dopa/DDI intakemay result in a dopamine related elevated ROS syn-thesis. As a consequence, an altered regulation of re-dox processes, which also involve the GSH system,may occur.5
In contrast to our present outcomes, which demon-strate a decrease of free Cys-Gyl after acute L-dopa/CD intake, earlier trials showed no variation of totalCys-Gly levels compared with normal controls. In onestudy, blood samples were drawn in the morning onlyonce following an overnight withdrawal of all prior
antiparkinsonian medication and before an acute oralL-dopa/DDI application.7 Another study also showedno differences of total Cys-Gly levels in relation toprevious chronic L-dopa/DDI exposure.8 However,Cys-Gly in plasma was again measured only one timeafter an overnight withdrawal of L-dopa/DDI adminis-tration. However, in our present trial, we measuredCys-Gly in a repeated fashion to investigate the effectsof an acute, oral L-dopa/DDI application. Similar tothe outcomes of the correlation analyses in these ear-lier studies, we found no relationships to the clinicalparameters of participating PD patients in this investi-gation. Moreover, we included a relative low number
FIG. 1. Cys-Gly (A), L-dopa (B), and 3-OMD (C) plasma levels after oral intake of 200 mg L-dopa intake combined with 50 mg carbidopa. * 5 P <0.05; ** 5 P < 0.01; *** 5 P < 0.001; L-dopa/3-OMD plasma concentrations are given in gg/mL, free Cys-Gly levels are reported in lmol/L; * 5 P-values of the post hoc analysis with the Tukeys HSD test; min 5 minutes.
C Y S T E I N Y L - G L Y C I N E I N P A R K I N S O N ’ S D I S E A S E
Movement Disorders, Vol. 26, No. 3, 2011 545

of participants, who were rather homogenous particu-larly in terms of prior chronic drug treatment. Never-theless, we suggest that this study outcomes allow noconclusions on the effect of continuous L-dopa/DDIapplication on the free Cys-Gly synthesis, as adaptiveoxidative stress reducing processes may occur withrepeat oral L-dopa/DDI application.We assume that the demonstrated free Cys-Gly
reduction is predominantly caused by the redox prop-erty of mitochondrial dopamine synthesis after a singleL-dopa/CD application. Therefore, it will probablyalso occur during repeated L-dopa/DDI applicationwith and without inhibition of the enzyme COMT,which metabolizes L-dopa. But this hypothesis has tobe investigated in future trials. A further shortcomingof the present trial is the fact that our study conclu-sions are based on plasma and not on CSF levels. Thevalue of our trial would also improve with additionalassessment of free radical occurrence.In conclusion, we show that acute L-dopa/CD appli-
cation reduces free Cys-Gly in plasma. This Cys-Glydecline may be linked to prior appearance of oxidativestress with concomitant consumption of antioxidantslike GSH and subsequent conversion of this moleculeto GSSG. Appearance of free radicals in not physio-logical concentrations is known to be involved inirregular harmful cellular metabolism, altered commu-nication between cells and progression of neuronaldegeneration in the brain of PD patients.
Acknowledgment: We thank Tanja Steiner, ChristineStamm, Ulrike Beckmann, Marion Frickmann, andChrista Kraushaar-Szesni for technical assistance. Wethank the participating PD patients.
References1. Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, Lil-
lig CH.Thiol redox control via thioredoxin and glutaredoxin sys-tems. Biochem Soc Trans 2005;33(Pt 6):1375–1377.
2. Asanuma M, Miyazaki I, az-Corrales FJ, Miyoshi K, Ogawa N,Murata M.Preventing effects of a novel anti-parkinsonian agentzonisamide on dopamine quinone formation. Neurosci Res 2008;60:106–113.
3. Riederer P, Gerlach M, Muller T, Reichmann H.Relating mode ofaction to clinical practice: dopaminergic agents in Parkinson’s dis-ease. Parkinsonism Relat Disord 2007;13:466–479.
4. Hughes AJ, Daniel SE, Kilford L, Lees AJ.Accuracy of clinical di-agnosis of idiopathic Parkinson’s disease: a clinico-pathologicalstudy of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184.
5. Chen L, Ding Y, Cagniard B, et al.Unregulated cytosolic dopaminecauses neurodegeneration associated with oxidative stress in mice.J Neurosci 2008;28:425–433.
6. Lee ES, Chen H, King J, Charlton C.The role of 3-O-methyldopain the side effects of L-dopa. Neurochem Res 2008;33:401–411.
7. Kuhn W, Roebroek R, Blom H, et al.Elevated plasma levels of ho-mocysteine in Parkinson’s disease. Eur Neurol 1998;40:225–227.
8. Muller T, Kuhn W.Cysteine elevation in levodopa-treated patientswith Parkinson’s disease. Mov Disord 2009;24:929–932.
Phenotypic Spectrum ofMusician’s Dystonia:
A Task-Specific Disorder?
Alexander Schmidt, MD, MA,1,2,3 Hans-Christian Jabusch, MD,3,4
Eckart Altenmuller, MD, MA,3 Leonie Enders, BSc,3
Rachel Saunders-Pullman, MD, MPH,5,6
Susan B. Bressman, MD,5,6 Alexander Munchau, MD,7
Christine Klein, MD,1,2* and Johann Hagenah, MD1,2
1Section of Clinical and Molecular Neurogenetics; and 2Departmentof Neurology, University of Luebeck, Lubeck, Germany; 3Institute ofMusic Physiology and Musicians’ Medicine, Hanover University of Musicand Drama, Hanover, Germany; 4Institute of Musicians’ Medicine,Dresden University of Music ‘‘Carl Maria von Weber’’, Dresden,Germany; 5Department of Neurology, Albert Einstein College ofMedicine, Bronx, New York, USA; 6Department of Neurology,Beth Israel Medical Center, New York, New York, USA;7Department of Neurology, University Medical CenterHamburg-Eppendorf, Hamburg, Germany
ABSTRACTBackground: Musician’s dystonia (MD) is traditionallyconsidered a sporadic and task-specific movement disor-der. Methods: The phenotypic spectrum of the disorderwas studied in 116 patients suffering from MD includingvideotaping. Results: Based on the movement disordersobserved, we categorized our patients into two differentgroups: (i) 65 patients with isolated MD, that is only pres-ent when playing the instrument and (ii) 51 patients withMD and one or more additional features of primary dys-tonia independent of MD (complex MD). Patients with apositive family history of movement disorders had anincreased risk to develop complex MD [odds ratio 54.80; 95% confidence interval: 1.94–11.92; P 5 0.001].Discussion: In previous studies, we recently identified 22relatives with different types of movement disorders inthe families of 28 MD patients. Taken together, our resultsfurther support a genetic contribution to MD with a broadindividual and familial phenotypic spectrum consisting ofMD, other dystonias and even other, non-dystonic move-ment disorders. VC

Dystonia is characterized by sustained muscle con-tractions causing twisting and repetitive movementsand abnormal postures.1 Musician’s dystonia (MD)has the hallmark features of dystonia and may presentwith painless muscular incoordination when a musi-cian is playing his instrument.2–4 It has been as-sociated with intensive training regimes and thus tradi-tionally been considered a sporadic and focal task-specific form of dystonia (FTSD).2–4 However, wehave recently observed an aggregation of differenttypes of movement disorders in professional musicianssuffering from MD followed at the Hanover Instituteof Music Physiology and Musicians’ Medicine. Thisvideo report aims to illustrate the broad phenotypicspectrum of movement disorder manifestations foundin our patients and challenges the notion of MD as apurely task-specific disorder.
Patients and MethodsAfter obtaining written informed consent, MD
patients were consecutively included in the study.Based on history and clinical features, all were classi-fied as having primary dystonia. They underwent adetailed videotaped neurological examination by amovement disorder specialist (AS, EA). Our patientswere categorized into two different groups on clinicalgrounds: (i) patients with isolated task-specific MDthat is only present when playing the instrument and(ii) patients with MD and one or more additional fea-tures of primary dystonia independent of MD (othertypes of dystonia or tremor in the same or other bodyparts) (complex MD). Statistical multivariate logisticregression analysis adjusting for gender, age of onset, fam-ily history of movement disorders, and type of MD wasperformed to identify significant risk factors for the devel-opment of complex MD using SPSS (SPSS Inc, Chicago).
ResultsA total of 116 MD patients (81 men, 35 women;
age: 43.26 6 12.04 [18–75] years; age of onset: 33.776 10.92 [15–66] years; duration of dystonia: 9.50 68.83 [0–42] years) were included in the study. Ofthese, 94 (81.0%) were affected with upper limbdystonia, mostly hand dystonia and 22 (19.0%) with
embouchure dystonia. A positive family history ofmovement disorders was reported by 38 patients(32.8%). The GAG deletion in the DYT1 gene andmutations in THAP1 (DYT6) were absent in all pro-bands. Isolated MD was present in 65/116 patients(56.0%, video segments 1, 2, 5, and 6), complex MDwith one or more additional FTSD (e.g. writer’s cramp(WC)), other dystonias (e.g. blepharospasm, brachialdystonia; segments 3 and 4) or tremor in 51/116patients (44.0%). In some of the complex cases, addi-tional dystonias were the consequence of spread; con-versely, some patients experienced dystonia outsidethe initially involved segment over the course of thedisease. The observed phenotypic spectrum of MD issummarized in Table 1. Multivariate logistic regres-sion analysis revealed an increased risk to developcomplex MD for patients with a positive family his-tory of movement disorders [odds ratio (OR) ! 4.80;95% confidence interval (CI): 1.94–11.92; P ! 0.001]and patients with upper limb dystonia [OR ! 3.57;95%CI: 1.12–11.39; P ! 0.031].
DiscussionThe traditional concept of MD as a simple task-spe-
cific disorder that is solely ‘‘environmentally’’ acquiredneeds to be reconsidered. About 50% of our patientsshowed additional types of dystonia or tremor asshown in video segments 3 and 4. A recent detailedclinical examination of 101 MD patients revealed a sim-ilar number of patients (54%) suffering from secondarymotor abnormalities in activities other than playing theinstrument.5 Also, in about half of patients presentingwith other forms of focal dystonia (e.g. blepharospasm),dystonia spreads during the first 5 years after disease onset.6
In addition to the described individual aggregationof movement disorders in our patients, we recentlyalso reported a familial clustering.7,8 The families of28 MD index patients were examined using a standar-dized telephone screening interview for dystonia andvideotaped neurological examinations.7,8 Besides the28 index patients, we identified 22 relatives from 21families with one or more different types of dystoniaor even other, non-dystonic movement disorders orabnormalities (MD: n ! 8, other FTSD: n ! 9, other
Table 1. Phenotypic spectrum of musician’s dystonia (MD)
MD phenotype Examples Number/Total Video segment
Isolated MD Only present on the instrument: 65/116hand/arm dystonia 48/65 1, 5, 6embouchure dystonia 17/65 2
Complex MD: MD $ additional dystonia(s) or tremor: 51/116$ other focal task-specific dystonia (FTSD) e.g. writer’s cramp 38/51$ other dystonia e.g. blepharospasm, brachial dystonia 16/51 3, 4$ other movement disorder tremor 4/51Relatives with movement disorders Relatives with FTSD, other dystonia(s) or other movement disorder(s) 38/116 5–8
P H E N O T Y P I C S P E C T R U M O F M U S I C I A N ’ S D Y S T O N I A
Movement Disorders, Vol. 26, No. 3, 2011 547

dystonias: n ! 2, other movement disorders: n ! 3;video segments 5–8).7,8 In light of the observed familialaggregation of movement disorders and the associationof a positive family history with the risk of spread ofsymptoms, a careful evaluation of the family historywith possible examination of relatives may help toidentify musicians at risk to develop (complex) MD.Our results further support a genetic contribution toMD with a broad individual and familial phenotypicspectrum consisting of MD, other dystonias and rarelyeven other, non-dystonic movement disorders.
Legends to the VideoSegment 1. Right-handed 47-year-old pianist with
flexion dystonia of the right third finger and compen-satory intermittent extension of the second and fourthfinger when playing scales (age of onset: 42 years),more pronounced at increased tempo (here and in thefollowing descriptions, the fingers one to five refer tothumb, index, middle, ring, and little finger, respec-tively). Playing scales with the left hand appears to benormal. Octaves and alternating chords played withthe right hand cause hyperextension of the second fin-ger and an abnormal, flexed posture of the third andfourth finger. Dystonic movements and posturesimprove when playing with a special ring on the thirdfinger of the right hand (sensory trick).
Segment 2. Tubist with embouchure dystonia whileplaying the main theme of the prelude of RichardWagner’s Mastersingers (age: 41 years, age of onset:20 years). There is some pulling and also locking ofthe upper lip and some facial grimacing involving theorbicularis oris muscle, and there are also extra move-ments of the chin involving the mentalis muscle.
Segment 3. Right-handed 63-year-old pianist withMD and additional other dystonias (age of onset: 35years). When holding out the arms in front of him,there is right-sided brachial dystonia with a slightlyflexed posture of the fingers four and five. In addition,blepharospasm is present with excessive blinking andoccasional cramping of the orbicularis oculi bilaterally.There is possibly also cervical dystonia with some abnor-mal tilt of the head to the left. While playing scales withthe right hand, he shows flexion dystonia of fingers fourand five and compensatory extension of finger three.
Segment 4. Right-handed 48-year-old pianist withsevere bibrachial dystonia affecting the left arm bothat rest and during different tasks associated with dys-tonic movements and the right hand during writing.At the age of 31 years dystonia first affected the per-formance of the patient, over the course of the diseasedystonic symptoms spread. When playing piano there isa grossly abnormal posture of the left hand with hyper-flexion at the wrist, flexion of the proximal and also
the distal interphalangeal joints of the fingers. He ishardly able to use individual fingers, whereas the righthand is unaffected. When holding out the arms in frontof his face, the left hand immediately adopts a similarhyperflexed dystonic posture, and there are occasionalrapid piano-playing-like dystonic movements of allfingers. There is also some overflow activity involvingthe upper and the lower arm. When spreading hisarms apart, there are again dystonic movements in theleft arm/hand increasing during voluntary movements,e.g., finger-nose testing. Writing with the right hand isabnormal with some ulnar deviation at the wrist andoverflow activity affecting the upper and lower armand also the right shoulder which is elevated andbrought forward. Also, he holds the pen between thesecond and the third finger. Writing and pouringwater into a mug with the left hand are also hamperedby dystonic posturing and movements.
Segment 5. Ambidextrous 43-year-old flutist, withslight flexion dystonia of the left third finger (age ofonset: 35 years). When he is playing very fast, thethird finger adopts a hyperflexed posture in the distalfinger segments leading to subtle rhythmic changesand squalidness.
Segment 6. Monozygotic twin brother of the patientshown in segment 5, a right-handed flutist with thesame type of flexion dystonia of the left third fingerwhen playing scales at increased tempo (age of onset:26 years).
Segment 7. Sister of a guitarist’s cramp patient, 46-years-old, ambidextrous, with right-sided WC and addi-tional task-specific dystonias of the right hand and arm(age of onset: 33 years). There is dystonic posturing ofthe right thumb and index finger and intermittentextension of the wrist that become more severe withincreasing time of writing. Dystonic movements andposturing are also present when typing on the keyboardor when cutting food. Hypertrophy of the wrist exten-sor muscles are observed on the right hand side.
Segment 8. Mother of a MD patient, 69-years-old,right-handed, with unusual dyskinesias and choreicmovements involving facial muscles (age of onset: 40years). When sitting on a chair and during rapid fingermovements of both hands, there are involuntarymouth opening and closing movements and also pout-ing (mirror movements). Voluntary hand movementsare slow, more pronounced on the right side. There isalso slight postural instability and when walkingslightly reduced right-sided armswing.
References1. Albanese A, Lalli S. Is this dystonia? Mov Disord 2009;24:
1725–1731.
S C H M I D T E T A L .
548 Movement Disorders, Vol. 26, No. 3, 2011

2. Altenmuller E. Focal dystonia: advances in brain imaging andunderstanding of fine motor control in musicians. Hand Clin 2003;19:523–538,xi.
3. Frucht SJ, Fahn S, Greene PE, et al. The natural history of embou-chure dystonia. Mov Disord 2001;16:899–906.
4. Jankovic J, Ashoori A. Movement disorders in musicians. MovDisord 2008;23:1957–1965.
5. Rosset-Llobet J, Candia V, Fabregas S, Ray W, Pascual-Leone A.Secondary motor disturbances in 101 patients with musician’s dys-tonia. J Neurol Neurosurg Psychiatry 2007;78:949–953.
6. Defazio G, Berardelli A, Hallett M. Do primary adult-onset focaldystonias share aetiological factors?Brain 2007;130 (Part 5):1183–1193.
7. Schmidt A, Jabusch HC, Altenmuller E, et al. Dominantly trans-mitted focal dystonia in families of patients with musician’s cramp.Neurology 2006;67:691–693.
8. Schmidt A, Jabusch HC, Altenmuller E, et al. Etiology of musi-cian’s dystonia: familial or environmental?Neurology 2009;72:1248–1254.
The C.-237_236GA>TT THAP1Sequence Variant Does Not
Increase Risk for Primary Dystonia
Jianfeng Xiao, MD, PhD,1,2 Yu Zhao, MD, PhD,1,2
Robert W. Bastian, MD,3 Joel S. Perlmutter, MD,4,5,6,7,8
Brad A. Racette, MD,4 Samer D. Tabbal, MD,4
Morvarid Karimi, MD,4 Randal C. Paniello, MD,9
Zbigniew K. Wszolek, MD,10 Ryan J. Uitti, MD,10
Jay A. Van Gerpen, MD,10 David K. Simon, MD, PhD,11
Daniel Tarsy, MD,11 Peter Hedera, MD, PhD,12
Daniel D. Truong, MD,13 Karen P. Frei, MD,13
Andrew Blitzer, MD, DDS,14 Monika Rudzinska, MD,15
Ronald F. Pfeiffer, MD,1,2 Carrie Le,1,2 andMark S. LeDoux, MD, PhD,1,2*
1Department of Neurology, University of Tennessee HealthScience Center, Memphis, Tennessee, USA; 2Department ofAnatomy and Neurobiology, University of Tennessee HealthScience Center, Memphis, Tennessee, USA; 3Bastian VoiceInstitute, Downers Grove, Illinois, USA; 4Department of Neurology,Washington University School of Medicine, St. Louis, Missouri,USA; 5Department of Radiology, Washington University Schoolof Medicine, St. Louis, Missouri, USA; 6Department ofNeurobiology, Washington University School of Medicine,St. Louis, Missouri, USA; 7Department of Physical Therapy,Washington University School of Medicine, St. Louis,Missouri, USA; 8Department of Occupational therapy,Washington University School of Medicine, St. Louis,Missouri, USA; 9Department of Otolaryngology-Head andNeck Surgery, Washington University School of Medicine,St. Louis, Missouri, USA; 10Department of Neurology,Mayo Clinic, Jacksonville, Florida, USA; 11Department ofNeurology, Beth Israel Deaconess Medical Center andHarvard Medical School, Boston, Massachusetts, USA;12Department of Neurology, Vanderbilt University, Nashville,Tennessee, USA; 13Parkinson’s & Movement DisorderInstitute, Fountain Valley, California, USA; 14New YorkCenter for Voice and Swallowing Disorders, New York,New York, USA; 15Department of Neurology, JagiellonianUniversity Medical College in Krakow, Krakow, Poland
ABSTRACTBackground: Sequence variants in coding and non-coding regions of THAP1 have been associated withprimary dystonia.Methods: In this study, 1,446 Caucasian subjects withmainly adult-onset primary dystonia and 1,520 controlswere genotyped for a variant located in the 50-untrans-lated region of THAP1 (c.-237_236GA>TT).Results: Minor allele frequencies were 62/2892(2.14%) and 55/3040 (1.81%) in subjects with dystoniaand controls, respectively (P50.202). Subgroup analy-ses by gender and anatomical distribution also failed toattain statistical significance. In addition, there was noeffect of the TT variant on expression levels of THAP1transcript or protein.Discussion: Our findings indicate that the c.-237_236GA>TT THAP1 sequence variant does not increaserisk for adult-onset primary dystonia in Caucasians. VC 2011Movement Disorder Society
Key Words: dystonia; DYT6; high-resolution melting;untranslated region; THAP1
DYT6 dystonia is an autosomal dominant primarydystonia causally associated with sequence variants inTHAP1, which encodes the DNA-binding transcrip-tion factor THAP1.1–3 DYT6 dystonia shows reducedpenetrance and variable expressivity.2–8 In contrast toDYT1 dystonia, DYT6 more commonly remains focalin distribution and often affects the cervical and la-ryngeal musculature.1,4,5 Over 30 sequence variantshave been localized to the coding regions of THAP1and associated with focal, segmental, multifocal, orgeneralized dystonia with age of onset ranging from 2to 62 years.1–8 In addition, several asymptomatic car-riers have been identified in the relatives of probands.The genotypic and phenotypic heterogeneity of DYT6dystonia and variable penetrance of known codingvariants suggest that noncoding variants in THAP1could contribute to the risk of developing adult-onsetprimary dystonia.4,5,7 Given that THAP1 is a
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Mark S. LeDoux, Department of Neurology,University of Tennessee Health Science Center, 855 Monroe Avenue,Memphis 38163, Tennessee; [email protected].
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 6 July 2010; Revised: 24 October 2010; Accepted: 1November 2010Published online 2 March 2011 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23551
T H A P 1 S E Q U E N C E V A R I A N T
Movement Disorders, Vol. 26, No. 3, 2011 549
transcriptional repressor, it is conceivable that non-coding variants which cause minor quantitativechanges in the temporal or spatial patterns of THAP1expression could have broad effects on thetranscriptome.Djarmati et al.4 identified a noncoding sequence vari-
ant (c.-237_236GA>TT) near the transcriptional startsite of THAP1 that might increase the risk of develop-ing primary dystonia. The relative frequency of thispolymorphism in their subjects with dystonia (20/320)relative to controls (7/355) was noteworthy (P !0.0054). Extrapolation of their findings to a broad pop-ulation of late-onset primary dystonia is problematicgiven that the subjects with dystonia were relativelyyoung (mean age of onset ! 38.5 years) and predomi-nantly were Northern German individuals whereas thecontrol group was composed of Caucasian individualsof more diverse European ancestry. Another, relativelysmall case–control study with heterogeneous controland dystonia populations did not find an associationbetween the TT allele and risk for dystonia.7
Herein, we present the results of a large case–controlstudy of c.-237_236GA>TT in primary, mainly adult-onset dystonia. All subjects were Caucasians. Moreover,the effects of the TT variant on overall gene expression(transcript) were interrogated in leukocytes and repli-cated in lymphoblastoid cell lines (transcript and protein)derived from distinct cohorts of cases and controls.Although RNA derived from peripheral blood has beenused to study DYT1 dystonia9 and other movement dis-orders, analysis of gene expression in lymphoblastoidcell lines limits potential exogenous confounds includingthe effects of medications, nutrient intake, and concomi-tant infectious and inflammatory medical conditions.
Patients and Methods
Participants
All human studies were conducted in accordance withthe Declaration of Helsinki with formal approval fromthe institutional review boards at each participatingstudy site. All subjects gave written informed consent.Recruitment of patients with primary dystonia and neu-rologically normal controls is described in the study ofXiao et al.5 Additional Caucasian control samples wereobtained from Sigma-Aldrich (Human Random ControlDNA Panels 1, 3, and 4), Emory Center for Neurodege-nerative Disease Tissue Bank Core, Washington Univer-sity in St. Louis School of Medicine Neuroscience Blue-print Core, and Coriell Institute for Medical Research(Control Panels NDPT020 and NDPT024). All normalcontrols except those from Sigma-Aldrich were exam-ined to exclude dystonia and other neurological disor-ders. Demographics for dystonia and control subjects are
Table
1.Clinicaldiagnose
s,demographics,
genotypes,
andallele
frequenciesin
dystonia
andcontrols
Clinicaldiagnosis
Number(ageofonse
t)*
Family
history**
Minorallele
frequency(TT)
Genotypes
Pva
lue
MF
All
GA/G
AGA/TT
TT/TT
MF
All
Spasmodicdysphonia
464(45.86
16.0,7
–85)
7.8%
4/214(1.9%)
21/714
(2.9%)
25/928
(2.7%)
439/464(94.6%
)25/464
(5.4%)
0/464(0%)
0.501
0.082
0.064
Cervicaldystonia
490(44.56
13.3,4
–76)
9.0%
4/230(1.7%)
12/750
(1.6)
16/980
(1.6%)
474/490(96.7%
)16/490
(3.3%)
0/490(0%)
0.554
0.359
0.420
Blepharospasm
197(58.16
9.5,
20–73)
6.6%
3/122(2.5%)
4/272(1.5%)
7/394(1.8%)
190/197(96.4%
)7/197(3.6%)
0/197(0%)
0.356
0.416
0.581
Hand-fo
rearm
dystonia
52(35.26
15.9,7
–60)
8.3%
0/46
(0%)
2/58
(3.4%)
2/104(1.9%)
50/52(96.2%
)2/52
(3.8%)
0/52
(0%)
0.468
0.317
0.569
Orom
andibulardystonia
17(52.96
11.6,2
0–70)
10.5%
0/8(0%)
0/26
(0%)
0/34
(0%)
17/17(100%)
0/17
(0%)
0/17
(0%)
0.875
0.607
0.539
Otherprimarydystonia
36(42.56
18.3,1
0–74)
13.9%
1/28
(3.6%)
1/44
(2.3%)
2/72
(2.8%)
34/36(94.4%
)2/36
(5.6%)
0/36
(0%)
0.385
0.580
0.382
Segm
entaldystonia
140(47.56
12.2,1
2–74)
13.2%
1/92
(1.1%)
6/188(3.2%)
7/280(2.5%)
134/140(95.7%
)5/140(3.6%)
1/140(0.7%)
0.551
0.179
0.265
Multifocaldystonia
24(32.26
15.8,7
–67)
22.2%
0/16
(0%)
2/32
(6.3%)
2/48
(4.2%)
22/24(91.7%
)2/24
(8.3%)
0/24
(0%)
0.766
0.133
0.222
Generalized
dystonia
26(23.96
19.4,1
–57)
13.3%
1/24
(4.2%)
0/28
(0%)
1/52
(1.9%)
25/26(96.2%
)1/26
(3.8%)
0/26
(0%)
0.342
0.584
0.617
Dystoniatotals
1446
(46.26
13.9,1
–85)
9.1%
14/780
(1.8%)
48/2112(2.3%)
62/2892(2.1%)
1385/1446(95.8%
)60/1446(4.1%)
1/1446
(0.1%)
0.476
0.260
0.202
Neurologicallynorm
alcontrols
1520
(49.36
13.2,2
3–83)
NA22/1320(1.7%)
33/1720(1.9%)
55/3040(1.8%)
1465/1520(96.4%
)55/1520(3.6%)
0/1520
(0%)
M:male;F:female.
*Meanageatstudyenrollm
ent6
standard
errorofthemean(SEM),range(yr).
**First-orse
cond-degreerelativ
ewith
dystonia.
B R I E F R E P O R T S
X I A O E T A L .
550 Movement Disorders, Vol. 26, No. 3, 2011

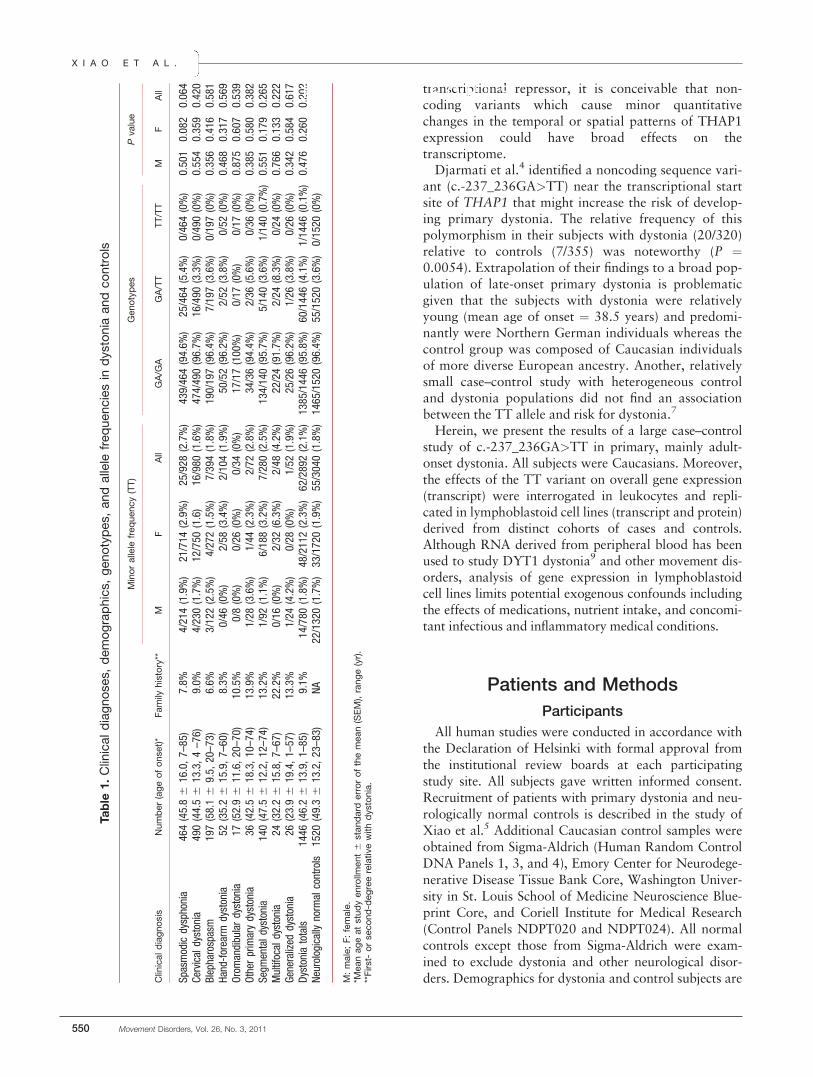
presented in Table 1. Of note, Table 1 does not includefamily members of probands.
High-Resolution MeltingHigh-resolution melting (HRM) analyses were per-
formed with the LightCycler 480 Real-Time PCR sys-tem and High-Resolution Master Mix (Roche India-napolis, IN) in accordance with manufacturer’sinstructions and our laboratory protocol5 using for-ward (acctggcctcagccaatagt) and reverse(ctgcgctcggttggattc) primers designed to amplify the50-untranslated region (UTR) of THAP1. Meltingcurves and difference plots were analyzed by threeinvestigators (J.X., Y.Z., and M.S.L.) blinded to phe-notype. All samples were unambiguously assigned togenotypes by Gene Scanning Software (Roche). Forsamples with shifted melting curves, PCR productswere cleaned using ExoSAP-IT (United States Bio-chemical, Cleveland, OH) and sequenced in the for-ward and reverse directions. To evaluate the sensitivityand specificity of HRM, amplicons from 400 neuro-logically normal controls and 400 subjects with dysto-nia were subjected to Sanger sequencing. Fisher’s exacttest was used to evaluate association of the c.-237_236GA>TT sequence variant with dystonia. TheGenetic Power Calculator, case–control discrete traitsmodule, was used for power analysis.10
THAP1 ExpressionAmbion’s LeukoLOCKTM Total RNA Isolation Sys-
tem and TRI Reagent (Ambion, Inc., Austin, TX,United States) were used to isolate RNA from periph-eral blood leukocytes of dystonia subjects and controls.Leukocyte RNA was used to evaluate the effects of theTT allele on THAP1 expression in dystonia subjectswith the TT allele (n ! 20) and controls without the TTallele (n ! 24). RNA and protein were also extractedfrom lymphoblastoid cells derived from another 5patients with the TT allele and 10 normal controlswithout the TT allele. Sanger sequencing was used toexclude coding, splice-site, 50-UTR, and previouslyreported intronic sequence variants (c.71$9C>A,c.71$126T>C) from the control and dystonia
groups.5,7 Detailed protocols for maintenance of lym-phoblastoid cell lines, quantitative real-time PCR, andquantitative Western blotting are provided in Support-ing Information Methods. Student’s t-tests were used tocompare RNA and protein expression between dystoniaand control samples. G*Power 3 was used for post hocpower analysis.11
ResultsAs depicted in Supporting Information Figure 1,
melting curves robustly discriminated GA/GA homozy-gotes from heterozygotes (GA/TT) and homozygotesof the minor allele (TT/TT). Furthermore, differentplots clustered these genotypes into three discretegroups. Based on follow-up sequencing of samplesexhibiting shifted melting curves and sequencing datafrom 400 neurologically normal controls and 400 sub-jects with dystonia, HRM showed 100% diagnosticspecificity and sensitivity.As detailed in Table 1, only one subject with dysto-
nia was homozygous for the TT allele whereas 60 wereGA/TT heterozygotes. TT allelic frequencies were 62/2892 (2.14%) in subjects with dystonia and 55/3040(1.81%) in controls. A one-tailed Fisher’s exact testfailed to confirm nonrandom associations between theTT allele and dystonia (P ! 0.202). Subgroup analysesfor laryngeal dystonia, cervical dystonia, blepharo-spasm, hand-forearm dystonia, segmental dystonia,multifocal dystonia, and generalized dystonia also failedto attain statistical significance in both male and femalesubjects (P > 0.06, for all). Of note, with a Bonferronicorrection for multiple comparisons, a P value less than0.05/12 (0.0042) would be required to maintain a typeI error rate (a) of 0.05 for the subgroup analyses. Usinga conservative population prevalence for primary dysto-nia of 1/10,00012 and an a of 0.05, our case–controlcohort had over 95% power to detect a twofold rela-tive risk of the TT allele. As seen in Table 2, there wasno effect of the TT allele on THAP1 mRNA expressionlevels in either leukocytes or lymphoblastoid cell lines.Moreover, there was no statistically significant effect ofthe TT allele on expression of THAP1 protein (Sup-porting Information Figure 2). Post hoc power analysis
Table 2. Effect of the c.-237_236GA>TT sequence variant on relative THAP1 expression levels in leukocytes andlymphoblastoid cells
Genotype/phenotype RNA leukocytes RNA lymphoblastoid cell lines Protein lymphoblastoid cell lines
Heterozygous c.-237_236GA >TTPrimary dystonia
1.043 6 0.0361
(n!20)0.973 6 0.065
(n!5)0.811 6 0.146
(n!5)Homozygous GA alleleNeurologically normal controls
1.011 6 0.032(n!24)
1.020 6 0.065(n!10)
1.000 6 0.074(n!10)
P value* 0.508 0.617 0.292
1All values are means 6 SEM.*Difference between dystonia and control samples.
T H A P 1 S E Q U E N C E V A R I A N T
Movement Disorders, Vol. 26, No. 3, 2011 551

with an effect size (d) of 0.20 showed that our compar-ison of leukocyte mRNA expression levels was weaklypowered (1 - b ! 0.10).
DiscussionTHAP1 encodes a 213-residue transcription factor,
which contains a highly conserved DNA sequence-spe-cific zinc-dependent THAP domain (1–81aa), a pro-line-rich region, a nuclear localization signal (146–162aa), and a coiled-coil domain.13,14 Overexpressionof THAP1 in endothelial cells was used by Cayrolet al.15 as an indirect means of identifying THAP1 tar-gets. Downregulated genes were concentrated inclasses related to cell-cycle/cell proliferation and themajority were also regulated by the pRB/E2F pathway.THAP1 knockdown was associated with decreasedexpression of eight pRB/E2F cell-cycle target genes.These data suggest that THAP1 promoter, UTR, andintronic sequence variants associated with alterationsin THAP1 expression may exert deleterious effects onneural function and/or neural development and serveas risk factors for dystonia. However, based on thedata presented herein, the c.-237_236GA>TT THAP1sequence variant has no effect on the risk of develop-ing adult-onset primary dystonia in Caucasians. More-over, the TT allele does not appear to exert importanteffects on either transcription or translation in leuko-cytes and lymphoblastoid cell lines.In contrast to previous investigations of the c.-
237_236GA>TT THAP1 sequence variant, our studywas much larger and focused on late-onset dystonia.4,7
The previously described cohorts from Germany4 andEngland7 contained a higher percentage of subjectswith generalized, segmental, and multifocal dystonia.Although the GA>TT variant did not increase overallrisk in our cohort of Caucasian subjects, we did noteliminate the possibility that this variant could con-tribute to dystonia risk in other populations. Similarly,several of our anatomical subgroups may have beenunderpowered to detect an effect of the TT allele. Inthis regard, the sole individual homozygous for the TTallele manifests segmental craniocervical dystonia16
with blepharospasm and oromandibular dystonia.Finally, our data do not exclude a role for the TT al-lele in epistatic interactions with other genes.Although the results of THAP1 mRNA and protein
expression in leukocytes and lymphoblastoid cell linessuggest that the TT allele does not contribute to dysto-nia risk, our analysis may have been underpowered to
detect a small effect. Moreover, it is possible that theeffect size of the TT allele on THAP1 expression isdevelopmentally regulated in a tissue-specific fashion.Ideally, RNA and protein expression should be exam-ined in several regions of human brain from individu-als with GA/GA and GA/TT genotypes at several timepoints during development. Given the important rolefor THAP1 in primary dystonia, other sequence var-iants in promoter, UTR, and intronic regions ofTHAP1 are also worthy of investigation.
References1. Almasy L, Bressman SB, Raymond D, et al. Idiopathic torsion dys-
tonia linked to chromosome 8 in two Mennonite families. AnnNeurol 1997;42:670–673.
2. Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in theTHAP1 gene are responsible for DYT6 primary torsion dystonia.Nat Genet 2009;41:286–288.
3. Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saun-ders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dysto-nia: a genetic screening study. Lancet Neurol 2009;8:441–446.
4. Djarmati A, Schneider SA, Lohmann K, et al. Mutations inTHAP1 (DYT6) and generalised dystonia with prominent spas-modic dysphonia: a genetic screening study. Lancet Neurol 2009;8:447–452.
5. Xiao J, Zhao Y, Bastian RW, et al. Novel THAP1 sequence var-iants in primary dystonia. Neurology 2010;74:229–238.
6. Paisan-Ruiz C, Ruiz-Martinez J, Ruibal M, et al. Identificationof a novel THAP1 mutation at R29 amino-acid residue insporadic patients with early-onset dystonia. Mov Disord 2009;24:2428–2429.
7. Houlden H, Schneider SA, Paudel R, et al. THAP1 mutations(DYT6) are an additional cause of early-onset dystonia. Neurology2010;74:846–850.
8. Bonetti M, Barzaghi C, Brancati F, et al. Mutation screening of theDYT6/THAP1 gene in Italy. Mov Disord 2009;24:2424–2427.
9. Walter M, Bonin M, Pullman RS, et al. Expression profiling in pe-ripheral blood reveals signature for penetrance in DYT1 dystonia.Neurobiol Dis 2010;38:192–200.
10. Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: designof linkage and association genetic mapping studies of complextraits. Bioinformatics 2003;19:149–150.
11. Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexiblestatistical power analysis program for the social, behavioral, andbiomedical sciences. Behav Res Methods 2007;39:175–191.
12. Defazio G, Abbruzzese G, Livrea P, Berardelli A. Epidemiology ofprimary dystonia. Lancet Neurol 2004;3:673–678.
13. Bessiere D, Lacroix C, Campagne S, et al. Structure–function anal-ysis of the THAP zinc finger of THAP1, a large C2CH DNA-bind-ing module linked to Rb/E2F pathways. J Biol Chem 2008;283:4352–4363.
14. Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Gi-rard JP. The THAP domain of THAP1 is a large C2CH modulewith zinc-dependent sequence-specific DNA-binding activity. ProcNatl Acad Sci USA 2005;102:6907–6912.
15. Cayrol C, Lacroix C, Mathe C, et al. The THAP-zinc finger pro-tein THAP1 regulates endothelial cell proliferation through modu-lation of pRB/E2F cell-cycle target genes. Blood 2007;109:584–594.
16. LeDoux MS. Meige syndrome: what’s in a name. ParkinsonismRelat Disord 2009;15:483–489.
G U I D U B A L D I E T A L .
552 Movement Disorders, Vol. 26, No. 3, 2011

Novel Mutations in SPG11 CauseHereditary Spastic ParaplegiaAssociated with Early-Onset
Levodopa-ResponsiveParkinsonism
Arianna Guidubaldi, MD,1 Carla Piano, MD,1
Filippo M. Santorelli, MD,2 Gabriella Silvestri, MD, PhD,1,3
Martina Petracca, MD,1 Alessandra Tessa, PhD,2 andAnna Rita Bentivoglio, MD, PhD1*
1Department of Neurology, Universita Cattolica del Sacro Cuore,Rome, Italy; 2Division of Molecular Medicine & NeurodegenerativeDisorders, IRCCS Foundation Stella Maris, Pisa, Italy; 3Don CarloGnocchi Foundation, ONLUS, Milano, Italy
ABSTRACTBackground: Autosomal recessive hereditary spasticparaplegia with thin corpus callosum is a neurodege-nerative disorder characterized by spastic paraparesis,cognitive impairment, and peripheral neuropathy. Theneuroradiologic hallmarks are thin corpus callosum andperiventricular white matter changes. Mutations in theSPG11 gene have been identified to be a major causeof autosomal recessive hereditary spastic paraplegiawith thin corpus callosum and recently also proven tobe responsible for juvenile parkinsonism associatedwith spastic paraplegia.Methods: We describe one Italian autosomal recessivehereditary spastic paraplegia with thin corpus callosumpatient who unusually presented at onset, 16 years,with parkinsonism-like features, responsive to dopami-nergic therapy. Then the clinical picture evolved andbecame more complex. A brain magnetic resonanceimaging scan showed thin corpus callosum and hyper-intense T2-weighted lesions in periventricular regions,and the 123I-ioflupane single-photon emission coupledtomography was abnormal.Results: Genetic analysis detected two novel muta-tions, a c.3664insT variant in compound heterozygositywith a c.6331insG mutation, in SPG11.Discussion: This case confirms the high genetic andclinical heterogeneity associated with SPG11 muta-tions. It also offers further evidence that parkinsonismmay initiate autosomal recessive hereditary spastic par-aplegia with thin corpus callosum and that parkinso-nian symptoms can have variable dopaminergicresponse in these patients.VC 2011 Movement Disorder SocietyKey Words: parkinsonism; levodopa; spastic para-plegia; SPG11; thin corpus callosum
Hereditary spastic paraplegias (HSPs) are heteroge-neous neurodegenerative disorders characterized by awide clinical and genetic heterogeneity.1,2 Both pureand complicated forms are recognized, depending onwhether the spastic paraplegia occurs in isolation or iscombined with other neurological or extraneurologicalfeatures.3
Among the rare recessive HSP forms, autosomal re-cessive (AR)-HSP with thin corpus callosum (TCC)(MIM 604360) presents with early-onset involvementof corticospinal tract and muscle stiffness followed bya slow development of progressive paraparesis, cogni-tive deterioration, and predominantly axonal motor orsensorimotor peripheral neuropathy.4 Thinning of theanterior corpus callosum (TCC) and white matterabnormalities are the most useful neuroradiologicalcriteria to propose a diagnosis of AR-HSP-TCC.5 Sofar, mutations in SPG11 on chromosome 15q(MIM610844) represent the most common cause ofAR-HSP, with or even without TCC,6 with a higherthan expected frequency in the Mediterranean area.7,8
In AR-HSP-TCC, learning disabilities or stiff legs orboth represent the commonest symptoms at onset, butatypical presentations,9 also including juvenile parkin-sonism,10 have been recently described.Here we present the clinical features and the molecular
genetic data of a patient affected by SPG11 who initiallydisplayed a juvenile levodopa-responsive parkinsonismin association with HSP.
Patients and Methods
Case
The patient, a 32-year-old Italian woman, offspringof a second-degree consanguineous marriage, wasborn by spontaneous delivery and had a normal, earlypsychomotor development. Her parents report poorscholastic performances even since primary school.
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
*Correspondence to: Dr. Anna Rita Bentivoglio, Istituto di Neurologia,Universita Cattolica del Sacro Cuore Largo Agostino Gemelli, 8 - 00168Roma, Italia; [email protected].
Relevant conflicts of interest/financial disclosures: Nothing to report.Full financial disclosures and author roles may be found in the onlineversion of this article.This study was funded in part by research grants from the Italian Ministryof Health, Fondazione Telethon (GGP10121A) and an E-RARE grant fromthe European Union (EUROSPA network) to FMS.
Received: 23 June 2010; Revised: 27 October 2010; Accepted: 1November 2010Published online in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.23552
S P A N D P A R K I N S O N I S M I N S P G 1 1 M U T A T I O N S
Movement Disorders, Vol. 26, No. 3, 2011 553
At age 14, the patient was referred to a neurologistbecause of abnormal gait and progressive rigidity in herlower limbs. Later on, she presented dysarthria, bilat-eral resting tremor, and mild bradykinesia in her upperlimbs. The disease rapidly progressed, so that 2 yearsfrom onset she could no longer walk without support.At age 16, she started a treatment with levodopa
(titrated up to 100 mg t.i.d.) with significant improve-ment of parkinsonism. A year later, the patient devel-oped the wearing off phenomenon and ‘‘peak-dose’’dyskinesias, featuring facial, particularly lingual, trun-cal and limbs choreo-dystonic movements.At age 23, when the patient was referred to our cen-
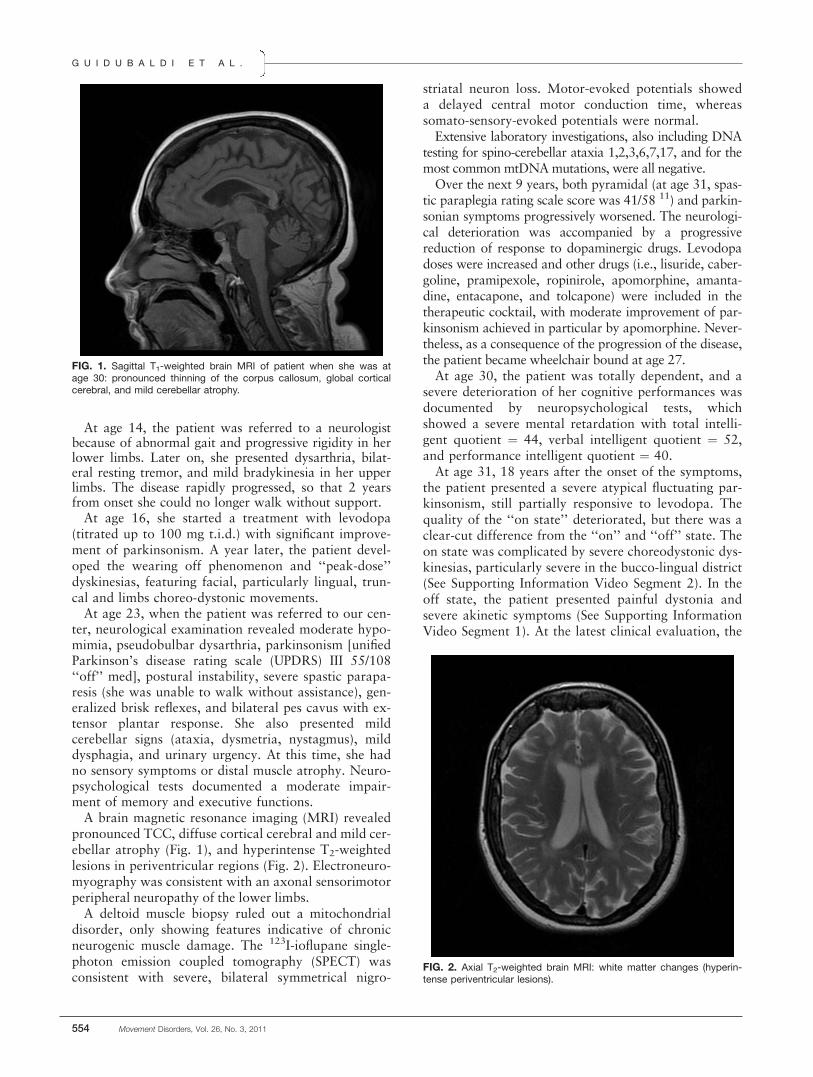
ter, neurological examination revealed moderate hypo-mimia, pseudobulbar dysarthria, parkinsonism [unifiedParkinson’s disease rating scale (UPDRS) III 55/108‘‘off’’ med], postural instability, severe spastic parapa-resis (she was unable to walk without assistance), gen-eralized brisk reflexes, and bilateral pes cavus with ex-tensor plantar response. She also presented mildcerebellar signs (ataxia, dysmetria, nystagmus), milddysphagia, and urinary urgency. At this time, she hadno sensory symptoms or distal muscle atrophy. Neuro-psychological tests documented a moderate impair-ment of memory and executive functions.A brain magnetic resonance imaging (MRI) revealed
pronounced TCC, diffuse cortical cerebral and mild cer-ebellar atrophy (Fig. 1), and hyperintense T2-weightedlesions in periventricular regions (Fig. 2). Electroneuro-myography was consistent with an axonal sensorimotorperipheral neuropathy of the lower limbs.A deltoid muscle biopsy ruled out a mitochondrial
disorder, only showing features indicative of chronicneurogenic muscle damage. The 123I-ioflupane single-photon emission coupled tomography (SPECT) wasconsistent with severe, bilateral symmetrical nigro-
striatal neuron loss. Motor-evoked potentials showeda delayed central motor conduction time, whereassomato-sensory-evoked potentials were normal.Extensive laboratory investigations, also including DNA
testing for spino-cerebellar ataxia 1,2,3,6,7,17, and for themost common mtDNAmutations, were all negative.Over the next 9 years, both pyramidal (at age 31, spas-
tic paraplegia rating scale score was 41/58 11) and parkin-sonian symptoms progressively worsened. The neurologi-cal deterioration was accompanied by a progressivereduction of response to dopaminergic drugs. Levodopadoses were increased and other drugs (i.e., lisuride, caber-goline, pramipexole, ropinirole, apomorphine, amanta-dine, entacapone, and tolcapone) were included in thetherapeutic cocktail, with moderate improvement of par-kinsonism achieved in particular by apomorphine. Never-theless, as a consequence of the progression of the disease,the patient became wheelchair bound at age 27.At age 30, the patient was totally dependent, and a
severe deterioration of her cognitive performances wasdocumented by neuropsychological tests, whichshowed a severe mental retardation with total intelli-gent quotient ! 44, verbal intelligent quotient ! 52,and performance intelligent quotient ! 40.At age 31, 18 years after the onset of the symptoms,
the patient presented a severe atypical fluctuating par-kinsonism, still partially responsive to levodopa. Thequality of the ‘‘on state’’ deteriorated, but there was aclear-cut difference from the ‘‘on’’ and ‘‘off’’ state. Theon state was complicated by severe choreodystonic dys-kinesias, particularly severe in the bucco-lingual district(See Supporting Information Video Segment 2). In theoff state, the patient presented painful dystonia andsevere akinetic symptoms (See Supporting InformationVideo Segment 1). At the latest clinical evaluation, the
FIG. 1. Sagittal T1-weighted brain MRI of patient when she was atage 30: pronounced thinning of the corpus callosum, global corticalcerebral, and mild cerebellar atrophy.
FIG. 2. Axial T2-weighted brain MRI: white matter changes (hyperin-tense periventricular lesions).
G U I D U B A L D I E T A L .
554 Movement Disorders, Vol. 26, No. 3, 2011

UPDRS motor score improved from 77 to 69 followingtreatment with 250 mg levodopa.
Molecular Genetic AnalysesGenomic DNA from peripheral blood samples was
extracted using a common salting out procedure. The40 coding exons of SPG11 and at least 50 base pairsof flanking intronic sequence were PCR-amplified andsubjected to d-high-performance liquid chromatogra-phy analysis using described oligonucleotide primerpairs.12 Amplicons showing abnormal elution profileswere agarose gel purified and directly sequenced usinga BigDye 3.1 chemistry protocol on a 3130XL GeneticAnalyzer (Applied Biosystems, Foster City, CA; http://www.appliedbiosystems.com).
ResultsDirect sequencing of SPG11 in blood DNA from the
proband detected a c.3664insT (p.K1222IfsX13) in com-pound heterozigosity with a c.6331insG (p.E2111GfsX36)mutation. Segregation of the new mutations in peripheralblood DNA from the healthy parents was performed bydirect sequencing. The two novel mutations were notfound in 200 Italian control chromosomes.
DiscussionWe report a sporadic Italian case with early parkin-
sonism and AR-HSP-TCC associated with two novelmutations in SPG11. SPG11 encodes spatacsin, a pro-tein with still unknown specific biological function.Spatacsin is expressed ubiquitously, including the ba-sal ganglia, and it appears to be essential to the func-tion and the survival of many neurons.13
Patients with SPG11 most commonly present clini-cal involvement of the pyramidal tract in associationwith axonal polyneuropathy, mental retardation, ormoderate cognitive impairment. Some patients also de-velop pseudobulbar dysarthria, cerebellar ataxia, uri-nary incontinence, or cataract. However, unusual phe-notypes are increasingly been recognized, includingpure spastic paraplegia14 and juvenile, slowly progres-sive motor neuron disorder.15
The patient described in this article presented acomplex neurological phenotype dominated by parkin-sonism at its onset, with dopaminergic denervationdocumented by 123I-ioflupane SPECT, which is quiteunusual for SPG11-related phenotypes.10,14,16–18
An extensive diagnostic panel had ruled out other eti-ologies, such as hereditary SpinoCerebellar Ataxias(SCAs), mitochondrial encephalomyopathies, and dis-orders of copper metabolism.We also considered neurodegeneration with brain iron
accumulation type 1, due to mutations in PANK2 gene,that can present with parkinsonism, pyramidal signs, andcognitive impairement.19 Mutations in other genes, char-
acterized by AR inheritance, ATP13A2 (PARK9, Kufor-Rakeb syndrome), PLA2G6 (PARK 14), and FBXO7(PARK 15) also produce clinical similar phenotype withrapidly progressive parkinsonism, with transient responseto levodopa, pyramidal signs, and cognitive decline.20–23
Most of these conditions present radiological signs of neu-rodegeneration with progressive cortical atrophy.As in our patient, brain MRI showed the presence
of TCC and periventricular white matter changes, typ-ical, albeit not exclusive, features of AR-HSP linked toSPG11 mutations, we oriented the molecular study onSPG11 gene, and indeed two novel heterozygousmutations have been found.This is the second report of a molecularly-proven
SPG11-linked AR-HSP-TCC associated with atypicaljuvenile parkinsonism and abnormal 123I-ioflupaneSPECT. Our patient, who carried different mutationsfrom the cases initially reported by Anheim et al.,10
showed some distinctive features compared with thesepatients. Indeed, unlike them, she still conserves a lev-odopa response after 18 years since the onset of thedisease; moreover our patient presents symmetricalnigro-striatal neuron loss and showed good toleranceand positive response to levodopa and other dopami-nerigic drugs. However, she soon developed the‘‘wearing off ’’ phenomenon and dyskinesias, thatcomplicated therapy management.Taken together, these findings suggest that different
mutations in SPG11 may affect dopaminergic neuronsin a distinct way. In conclusion, our report confirmsthat SPG11 mutations may cause early-onset parkin-sonism. Other two descriptions14,17 closely resembleour patient: in one of them, a clinical response to levo-dopa was reported, in the other one complicated bypeak-dose dyskinesias.14 However, SPG11 gene testingwas not performed in these patients.Therefore we suggest that (1) mutations in SPG11
should be considered in patients with atypical juve-nile-onset parkinsonism accompanied by limited orgood response to levodopa and (2) in similar casestreatment with dopaminergic drugs should beattempted as parkinsonian symptoms and so the qual-ity of life can be improved.
Legends to the VideoSegment 1. (1) Off-state evaluation. A severe par-
kinsonism is evident: marked dysarthria, hypomimia,bradykinesia with very slow finger tapping, plastic ri-gidity in the upper limbs, upper limb resting tremor,more severe on the right side. (2) Patient is unable towalk and to stand without assistance for severe spasticparaparesis.Segment 2. On-state evaluation. After a standard
dose of levodopa/carbidopa (250/25 mg), the neuro-logical examination reveals a mild improvement ofparkinsonian symptoms with ‘‘peak-dose’’ lingual dys-kinesias.
S P A N D P A R K I N S O N I S M I N S P G 1 1 M U T A T I O N S
Movement Disorders, Vol. 26, No. 3, 2011 555

Acknowledgments: We thank Mrs. Katherine McKullen for revising the English text of he manuscript.
References1. Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11,
encoding spatacsin, are a major cause of spastic paraplegia withthin corpus callosum. Nat Genet 2007;39:366–372.
2. Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spasticparaplegia: clinical features and pathogenic mechanisms. LancetNeurol 2008;7:1127–1138.
3. Harding AE. Classification of the hereditary ataxias and paraple-gias. Lancet 1983;1:1151–1155.
4. Winner B, Uyanik G, Gross C, et al. Clinical progression and geneticanalysis in hereditary spastic paraplegia with thin corpus callosum inspastic gait gene 11 (SPG11). Arch Neurol 2004;61:117–121.
5. Iwabuchi K, Kubota Y, Hanihara T, Nagatomo H. Three patientsof complicated form of autosomal recessive hereditery spastic para-plegia associated with hypoplasia of the corpus callosum. No toshinkei! Brain and Nerve 1994;46:941–947.
6. Crimella C, Arnoldi A, Crippa F, et al. Point mutations and a largeintragenic deletion in SPG11 in complicated spastic paraplegiawithout thin corpus callosum. J Med Genet 2009;46:345–351.
7. Casali C, Valente EM, Bertini E, et al. Clinical and genetic studiesin hereditary spastic paraplegia with thin corpus callosum. Neurol-ogy 2004;62:262–268.
8. Stevanin G, Montagna G, Azzedine H, et al. Spastic paraplegiawith thin corpus callosum: description of 20 new families, refine-ment of SPG 11 locus, candidate gene analysis and evidence ofgenetic heterogeneity. Neurogenetics 2006;7:149–156.
9. Paisanr-Ruiz C, Nath P, Wood NW, Singleton A, Houlden H.Clinical heterogeneity and genotype–phenotype correlation in he-reditary spastic paraplegia because of Spatacsin mutations(SPG11). Eur J Neurol 2008;15:1065–1070.
10. Anheim M, Lagier-Tourenne C, Stevanin G, et al. SPG11 spasticparaplegia. A new cause of juvenile parkinsonism. J Neurol 2009;256:104–108.
11. Schule R, Holland-Letz T, Klimpe S. The Spastic Paraplegia RatingScale (SPRS): a reliable and valid measure of disease severity. Neu-rology 2006;67:430–434.
12. Denora PS, Schlesinger D, Santorelli FM, et al. Sceening ofARHSP-TCC patients expands the spectrum of SPG11 mutationsand includes a large scale gene deletion. Hum Mutat 2009;30:E500–E519.
13. Del Bo R, Fonzo A, Ghezzi S, et al. SPG11: a consistent clinicalphenotype in a family with homozygous spatacsin truncating muta-tion. Neurogenetics 2007;8:301–305.
14. Micheli F, Cersosimo MG, Zuniga Ramırez C. Hereditary spasticparaplegia associated with dopa-responsive parkinsonism. MovDisord 2006;21:716–717.
15. Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutationscause autosomal recessive juvanile amyotrophic lateral sclerosis.Brain 2010;133:591–598.
16. Iwabuchi K, Yagishita S, Amano N, et al. An autopsy case of com-plicated form of spastic paraplegia with amyotrophy, mental defi-ciency, sensory impairement, and parkinsonism. No to Shinkei1990;1:1075–1083.
17. Kang SY, Lee MH, Lee SK, Sohn YH. Levodopa-responsive par-kinsonism in hereditary spastic paraplegia with thin corpus cal-losum. Parkinsonism Relat Disord 2004;10:425–427.
18. Hehr U, Bauer P, Winkler J, et al. Longterm course and mutationalspectrum of spatacsin-linked spastic paraplegia. Ann Neurol 2007;62:656–665.
19. Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neu-rodegeneration with brain iron accumulation (Hallervorden-Spatzsyndrome) and pantothenate kinase-associated neurodegeneration.Mov Disord 2004;19:36–42.
20. Williams DR, Hadeed A, al-Din AS, et al. Kufor Rakeb disease:autosomal recessive, levodopa-responsive parkinsonism with py-ramidal degeneration, supranuclear gaze palsy, and dementia. MovDisord 2005;20:1264–1271.
21. Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization ofPLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol2009;65:19–23.
22. Di Fonzo A, Dekker MC, Montagna P, et al. FBXO7 mutationscause autosomal recessive, early-onset parkinsonian-pyramidal syn-drome. Neurology 2008;72:240–245.
23. Paisan-Ruiz C, Guevara R, Federoff M, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2,PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 2010;25:1791–1800.
G U I D U B A L D I E T A L .
556 Movement Disorders, Vol. 26, No. 3, 2011
Related Documents











