Muscle weakness in a mouse model of nemaline myopathy can be reversed with exercise and reveals a novel myofiber repair mechanism Josephine E. Joya 1,{ , Anthony J. Kee 1,{ , Visalini Nair-Shalliker 1 , Majid Ghoddusi 1 , Mai-Anh T. Nguyen 1 , Pradeep Luther 2 and Edna C. Hardeman 1, * 1 Muscle Development Unit, Children’s Medical Research Institute, Westmead, NSW 2145, Australia and 2 Biological Structure and Function Section, Biomedical Sciences Division, Faculty of Medicine, Imperial College, Exhibition Road, London SW7 2AZ, UK Received July 19, 2004; Revised August 25, 2004; Accepted September 4, 2004 Patients with the inherited muscle disease nemaline myopathy experience prolonged muscle weakness following periods of immobility. We have examined endurance exercise as a means of improving recovery following muscle inactivity in our a-tropomyosin slow (Met9Arg)-transgenic mouse model of nemaline myopa- thy. Physical inactivity, mimicked using a hindlimb immobilization protocol, resulted in fiber atrophy and severe muscle weakness. Following immobilization, the nemaline mice (NM) were weaker than WT mice but regained whole-body strength with exercise training. The disuse-induced weakness and the regain of strength with exercise in NM were associated with the respective formation and resolution of nemaline rods, suggesting a role for rods in muscle weakness. Muscles from NM did not show the typical features of muscle repair during chronic stretch-immobilization of the soleus muscle (regeneration occurred with relative lack of centralized nuclei). This indicates that the normal process of regeneration may be altered in nemaline myopathy and may contribute to poor recovery. In conclusion, endurance exercise can alleviate disuse-induced weakness in NM. The altered myofiber repair process in the nemaline mice may be a response to primary myofibrillar damage that occurs in nemaline myopathy and is distinct from the classical repair in muscular dystrophy resulting from plasma membrane defects. INTRODUCTION Nemaline myopathy is a neuromuscular disorder characterized by muscle weakness and the presence of electron dense struc- tures, called nemaline rods, within the muscle fibers and more rarely in the nuclei (1,2). Nemaline rods are thought to be accumulations of sarcomeric proteins, principally a-actinin, formed through an unchecked expansion of the Z-line (3). The clinical presentation of the disease is variable and has been classified into five main sub-types on the basis of the pattern of weakness and age of onset: severe congenital, typical, intermediate congenital, mild form of childhood-onset and adult-onset (2). Mutations in five genes encoding proteins that form or associate with the thin filament of the sarcomere have been identified as causing nemaline myopathy: b-tropomyosin, a-tropomyosin slow (a-Tm slow ), nebulin, a-skeletal actin and troponin T slow (4). A feature of nemaline myopathy that distinguishes it from the muscular dystrophies is the absence of pathological signs of myonecrosis and regeneration (central-nucleated fibers) (5), the principal characteristics of dystrophic muscle. Muscular dystrophies result from mutations in proteins (e.g. dystrophin and sarcoglycans) thought to be involved in maintaining the functional integrity of sarcolemma (6). This leads to increased susceptibility to contraction-induced injury and sarcolemmal damage resulting in necrosis (6). In contrast, membrane damage and muscle degeneration is not a feature of nemaline myopathy (5), presumably because the primary defect is in the sarcomeric thin filament. However, a recent array analysis on muscle from human nemaline patients showed increased Human Molecular Genetics, Vol. 13, No. 21 # Oxford University Press 2004; all rights reserved { The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors. *To whom correspondence should be addressed at: Muscle Development Unit, Children’s Medical Research Institute, Locked Bag 23, Wentworthville NSW 2145, Australia. Tel: þ61 296872800; Fax: þ61 296872120; Email: [email protected] Human Molecular Genetics, 2004, Vol. 13, No. 21 2633–2645 doi:10.1093/hmg/ddh285 Advance Access published on September 14, 2004 by guest on October 8, 2014 http://hmg.oxfordjournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Muscle weakness in a mouse model of nemalinemyopathy can be reversed with exercise andreveals a novel myofiber repair mechanism
Josephine E. Joya1,{, Anthony J. Kee1,{, Visalini Nair-Shalliker1, Majid Ghoddusi1,
Mai-Anh T. Nguyen1, Pradeep Luther2 and Edna C. Hardeman1,*
1Muscle Development Unit, Children’s Medical Research Institute, Westmead, NSW 2145, Australia and2Biological Structure and Function Section, Biomedical Sciences Division, Faculty of Medicine,
Imperial College, Exhibition Road, London SW7 2AZ, UK
Received July 19, 2004; Revised August 25, 2004; Accepted September 4, 2004
Patients with the inherited muscle disease nemaline myopathy experience prolonged muscle weaknessfollowing periods of immobility. We have examined endurance exercise as a means of improving recoveryfollowing muscle inactivity in our a-tropomyosinslow(Met9Arg)-transgenic mouse model of nemaline myopa-thy. Physical inactivity, mimicked using a hindlimb immobilization protocol, resulted in fiber atrophy andsevere muscle weakness. Following immobilization, the nemaline mice (NM) were weaker than WT micebut regained whole-body strength with exercise training. The disuse-induced weakness and the regain ofstrength with exercise in NM were associated with the respective formation and resolution of nemalinerods, suggesting a role for rods in muscle weakness. Muscles from NM did not show the typical featuresof muscle repair during chronic stretch-immobilization of the soleus muscle (regeneration occurred withrelative lack of centralized nuclei). This indicates that the normal process of regeneration may be alteredin nemaline myopathy and may contribute to poor recovery. In conclusion, endurance exercise can alleviatedisuse-induced weakness in NM. The altered myofiber repair process in the nemaline mice may be aresponse to primary myofibrillar damage that occurs in nemaline myopathy and is distinct from the classicalrepair in muscular dystrophy resulting from plasma membrane defects.
INTRODUCTION
Nemaline myopathy is a neuromuscular disorder characterizedby muscle weakness and the presence of electron dense struc-tures, called nemaline rods, within the muscle fibers and morerarely in the nuclei (1,2). Nemaline rods are thought to beaccumulations of sarcomeric proteins, principally a-actinin,formed through an unchecked expansion of the Z-line (3).The clinical presentation of the disease is variable and hasbeen classified into five main sub-types on the basis of thepattern of weakness and age of onset: severe congenital,typical, intermediate congenital, mild form of childhood-onsetand adult-onset (2). Mutations in five genes encoding proteinsthat form or associate with the thin filament of the sarcomerehave been identified as causing nemaline myopathy:
b-tropomyosin, a-tropomyosin slow (a-Tmslow), nebulin,a-skeletal actin and troponin T slow (4).
A feature of nemaline myopathy that distinguishes it fromthe muscular dystrophies is the absence of pathological signsof myonecrosis and regeneration (central-nucleated fibers) (5),the principal characteristics of dystrophic muscle. Musculardystrophies result from mutations in proteins (e.g. dystrophinand sarcoglycans) thought to be involved in maintaining thefunctional integrity of sarcolemma (6). This leads to increasedsusceptibility to contraction-induced injury and sarcolemmaldamage resulting in necrosis (6). In contrast, membranedamage and muscle degeneration is not a feature of nemalinemyopathy (5), presumably because the primary defect is inthe sarcomeric thin filament. However, a recent array analysison muscle from human nemaline patients showed increased
Human Molecular Genetics, Vol. 13, No. 21 # Oxford University Press 2004; all rights reserved
{The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
*To whom correspondence should be addressed at: Muscle Development Unit, Children’s Medical Research Institute, Locked Bag 23, WentworthvilleNSW 2145, Australia. Tel: þ61 296872800; Fax: þ61 296872120; Email: [email protected]
Human Molecular Genetics, 2004, Vol. 13, No. 21 2633–2645doi:10.1093/hmg/ddh285Advance Access published on September 14, 2004
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

expression of genes associated with activated satellite cellsand immature fibers (NCAM1, CDK1 and PAX7 ) (7). Thesignificance of these observations is yet to be established,but it raises the possibility that in nemaline muscle a novelregenerative process may be occurring that does not displaythe traditional features of muscle repair.
We have generated a transgenic mouse model of nemalinemyopathy by expressing the dominant negative a-Tmslow
(Met9Arg) mutant (human TPM3 gene) in skeletal muscle(8). This was the first nemaline-associated mutation to be iden-tified and results in a childhood-onset form of the disease (9).This mouse model has all features of the human diseaseincluding the presence of nemaline rods in skeletal muscleand an increase in slow/oxidative fibers. As has been observedin muscles of a human nemaline patient (10), in the mousemodel the number of rod-containing fibers between musclesvaried significantly. However, the number of rod-containingfibers in individual muscles was consistent within litters ofa-Tmslow(Met9Arg) mice and through generations. Theincrease in number of slow/oxidative fibers was present at1 month of age and was maintained through adulthood,indicating disruption of the early postnatal maturation of thedifferent fiber types.
The hypotonia and muscle weakness in patients with nema-line myopathy often lead to extended periods of immobility(2). Lack of activity exacerbates the problem as muscledisuse in itself leads to myofiber atrophy and loss of musclestrength (11), and the nemaline patients appear to be moresusceptible to the effects of inactivity (12). We have usedour a-Tmslow(Met9Arg) mouse model to examine the follow-ing questions. (i) Does expression of a nemaline myopathymutation exacerbate disuse-induced muscle atrophy andmuscle weakness? (ii) Can endurance exercise improve reco-very of muscle strength following immobilization in nemalinemice? (iii) Does a nemaline mutation impact on the processof recovery following immobilization? We show that thenemaline mice are more sensitive to the effects of chronicinactivity and that endurance exercise was required for reco-very of whole-body strength. Increased nemaline rod densityin the extensor digitorum longus (EDL) muscle correlatedwith muscle weakness raising the possibility that nemalinerods can influence muscle strength. Restoration of myofibrillarstructure (removal of rods) in the EDL with exercise recoveryoccurred without the activation of the normal regenerativeprocess. The process of muscle regeneration followingstretch-induced damage (soleus muscle) was also altered innemaline mice. These two myofiber repair processes (repairwithout regeneration and regeneration with few centralizednuclei) are distinct from sarcolemmal repair in musculardystrophy and repair in normal healthy muscle and may bespecific to diseases of the sarcomeric thin filament.
RESULTS
Nemaline mice have greater loss of whole-body strengthfollowing immobilization and require exercise toregain strength
Many patients with nemaline myopathy have prolongedperiods of immobility owing to muscle weakness (2,13).
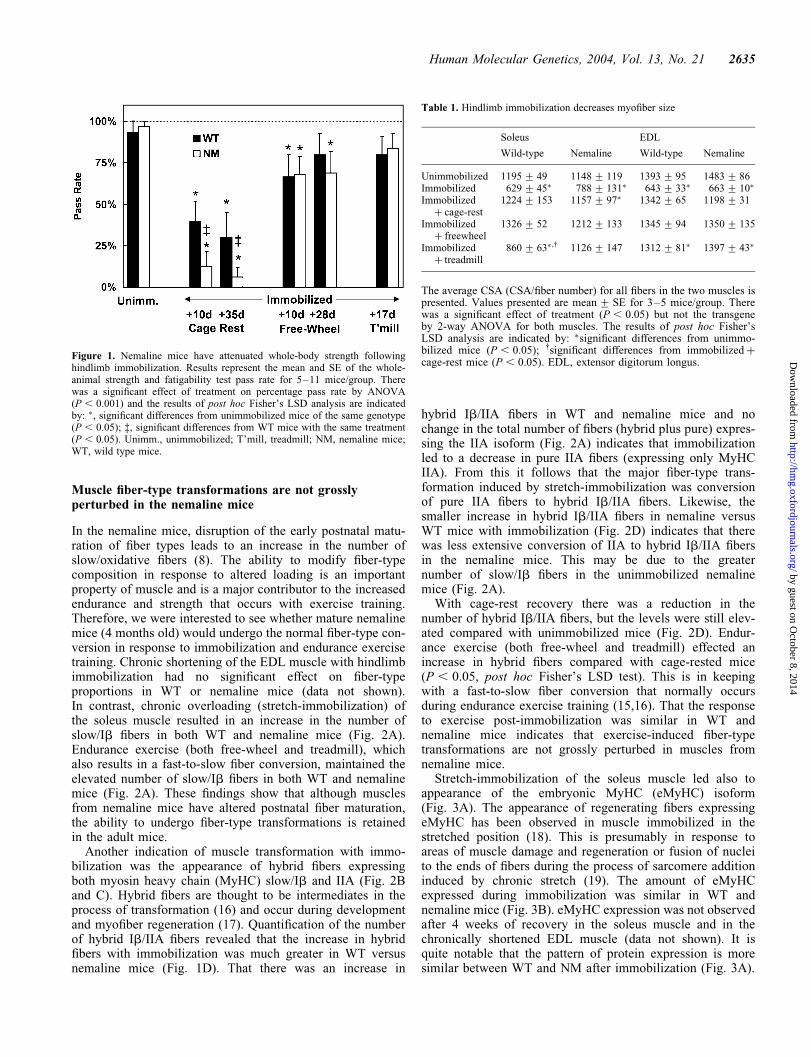
We modeled prolonged inactivity in our transgenic nemalinemouse with a hindlimb immobilization procedure and exam-ined whether exercise could alleviate disuse-induced weaknesswith a standard whole-body strength and fatigability test (seeMaterials and Methods). This procedure tests the overallstrength of the limb and abdominal muscles and mimics clini-cal tests of muscle weakness (14). The results of this test indi-cate that although the nemaline mice were not weak beforeimmobilization, they lost a greater amount of whole-bodystrength following immobilization (Fig. 1). The nemalinemice were weak compared with WT mice after 10 days ofcage-rest recovery and were still severely weak 3–4 weekslater. In contrast, 3–4 weeks of free-wheel or treadmill exer-cise led to almost complete recovery of whole-body strengthin WT and nemaline mice (P , 0.05 versus cage-rest, LSDpost hoc analysis). These results suggest that although thenemaline myopathy mutation (a-TmslowM9R) promotesdisuse-induced muscle weakness it does not alter exercise-induced recovery of muscle strength.
The atrophic effect of immobilization and fiber-sizerecovery was maintained in nemaline mice
As myofiber atrophy (decrease in myofiber size) is a majorcause of muscle weakness, we examined whether the greaterweakness in the nemaline mice after immobilization was dueto myofiber atrophy and the regain of strength with exercisewas the result of fiber-size recovery. Hindlimb immobilizationof WT and nemaline mice lead to a reduction in myofiber sizein both chronically stretched (soleus) and chronically shor-tened (EDL) muscles (Table 1). The decrease in fiber sizewas similar for the two sets of mice. However, the mechanismsfor the decrease in fiber size were different for the two muscles.In the soleus muscle, a decrease in average fiber size was duemainly to the appearance of small regenerating fibers, inducedby chronic muscle extension (see data to follow). While in theshortened EDL a process of true atrophy occurred, there was adecrease in the size of mature fibers without regeneration.This effect may be due to an increase in nemaline rods inresponse to muscle disuse (see data to follow).
Both cage-rest recovery (4 weeks) and free-wheel exer-cise returned fiber size (EDL and soleus muscles) to pre-immobilization levels (Table 1). This occurred to a similarextent for both WT and nemaline mice (Table 1). Fiber sizealso returned to the unimmobilized state with treadmillexercise in the nemaline mice (EDL and soleus muscles),but in the WT soleus fibers retained their immobilized size(Table 1). An atrophic effect of endurance exercise trainingon slow/oxidative fibers has been observed in unimmobilizedmice (15). Thus, it would appear that the atrophic effect ofendurance exercise itself in the present study preventedthe restoration of fiber size in the soleus from WT but notfrom nemaline mice. Overall changes in fiber size (Table 1)did not correlate with muscle weakness (Fig. 1); e.g. micewere weak with 4 weeks cage-rest but fibers were back topre-immobilized size. Thus, fiber size was not primarilyresponsible for the regain of strength with recovery. Moreover,the nemaline myopathy mutation (a-TmslowM9R) had only aminor impact on the trophic response to immobilizationand exercise.
2634 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Muscle fiber-type transformations are not grosslyperturbed in the nemaline mice
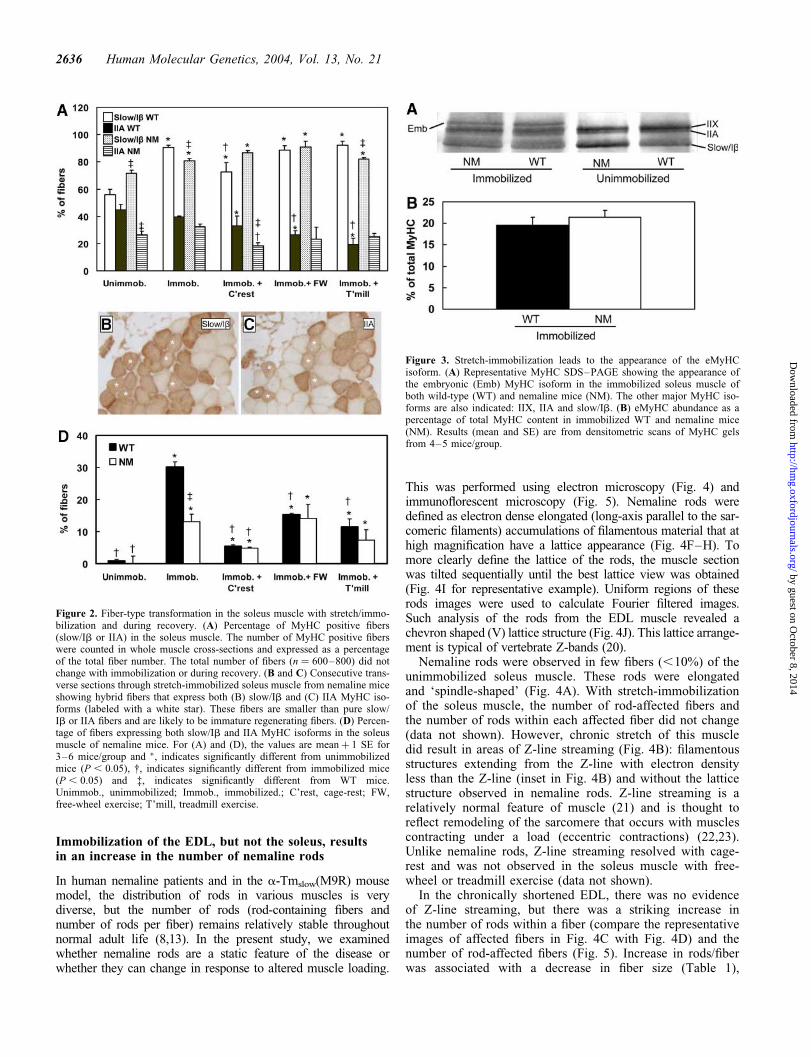
In the nemaline mice, disruption of the early postnatal matu-ration of fiber types leads to an increase in the number ofslow/oxidative fibers (8). The ability to modify fiber-typecomposition in response to altered loading is an importantproperty of muscle and is a major contributor to the increasedendurance and strength that occurs with exercise training.Therefore, we were interested to see whether mature nemalinemice (4 months old) would undergo the normal fiber-type con-version in response to immobilization and endurance exercisetraining. Chronic shortening of the EDL muscle with hindlimbimmobilization had no significant effect on fiber-typeproportions in WT or nemaline mice (data not shown).In contrast, chronic overloading (stretch-immobilization) ofthe soleus muscle resulted in an increase in the number ofslow/Ib fibers in both WT and nemaline mice (Fig. 2A).Endurance exercise (both free-wheel and treadmill), whichalso results in a fast-to-slow fiber conversion, maintained theelevated number of slow/Ib fibers in both WT and nemalinemice (Fig. 2A). These findings show that although musclesfrom nemaline mice have altered postnatal fiber maturation,the ability to undergo fiber-type transformations is retainedin the adult mice.
Another indication of muscle transformation with immo-bilization was the appearance of hybrid fibers expressingboth myosin heavy chain (MyHC) slow/Ib and IIA (Fig. 2Band C). Hybrid fibers are thought to be intermediates in theprocess of transformation (16) and occur during developmentand myofiber regeneration (17). Quantification of the numberof hybrid Ib/IIA fibers revealed that the increase in hybridfibers with immobilization was much greater in WT versusnemaline mice (Fig. 1D). That there was an increase in
hybrid Ib/IIA fibers in WT and nemaline mice and nochange in the total number of fibers (hybrid plus pure) expres-sing the IIA isoform (Fig. 2A) indicates that immobilizationled to a decrease in pure IIA fibers (expressing only MyHCIIA). From this it follows that the major fiber-type trans-formation induced by stretch-immobilization was conversionof pure IIA fibers to hybrid Ib/IIA fibers. Likewise, thesmaller increase in hybrid Ib/IIA fibers in nemaline versusWT mice with immobilization (Fig. 2D) indicates that therewas less extensive conversion of IIA to hybrid Ib/IIA fibersin the nemaline mice. This may be due to the greaternumber of slow/Ib fibers in the unimmobilized nemalinemice (Fig. 2A).
With cage-rest recovery there was a reduction in thenumber of hybrid Ib/IIA fibers, but the levels were still elev-ated compared with unimmobilized mice (Fig. 2D). Endur-ance exercise (both free-wheel and treadmill) effected anincrease in hybrid fibers compared with cage-rested mice(P , 0.05, post hoc Fisher’s LSD test). This is in keepingwith a fast-to-slow fiber conversion that normally occursduring endurance exercise training (15,16). That the responseto exercise post-immobilization was similar in WT andnemaline mice indicates that exercise-induced fiber-typetransformations are not grossly perturbed in muscles fromnemaline mice.
Stretch-immobilization of the soleus muscle led also toappearance of the embryonic MyHC (eMyHC) isoform(Fig. 3A). The appearance of regenerating fibers expressingeMyHC has been observed in muscle immobilized in thestretched position (18). This is presumably in response toareas of muscle damage and regeneration or fusion of nucleito the ends of fibers during the process of sarcomere additioninduced by chronic stretch (19). The amount of eMyHCexpressed during immobilization was similar in WT andnemaline mice (Fig. 3B). eMyHC expression was not observedafter 4 weeks of recovery in the soleus muscle and in thechronically shortened EDL muscle (data not shown). It isquite notable that the pattern of protein expression is moresimilar between WT and NM after immobilization (Fig. 3A).
Figure 1. Nemaline mice have attenuated whole-body strength followinghindlimb immobilization. Results represent the mean and SE of the whole-animal strength and fatigability test pass rate for 5–11 mice/group. Therewas a significant effect of treatment on percentage pass rate by ANOVA(P , 0.001) and the results of post hoc Fisher’s LSD analysis are indicatedby: �, significant differences from unimmobilized mice of the same genotype(P , 0.05); ‡, significant differences from WT mice with the same treatment(P , 0.05). Unimm., unimmobilized; T’mill, treadmill; NM, nemaline mice;WT, wild type mice.
Table 1. Hindlimb immobilization decreases myofiber size
Soleus EDL
Wild-type Nemaline Wild-type Nemaline
Unimmobilized 1195+ 49 1148+ 119 1393+ 95 1483+ 86Immobilized 629+ 45� 788+ 131� 643+ 33� 663+ 10�
Immobilizedþ cage-rest
1224+ 153 1157+ 97� 1342+ 65 1198+ 31
Immobilizedþ freewheel
1326+ 52 1212+ 133 1345+ 94 1350+ 135
Immobilizedþ treadmill
860+ 63� ,† 1126+ 147 1312+ 81� 1397+ 43�
The average CSA (CSA/fiber number) for all fibers in the two muscles ispresented. Values presented are mean+ SE for 3–5 mice/group. Therewas a significant effect of treatment (P , 0.05) but not the transgeneby 2-way ANOVA for both muscles. The results of post hoc Fisher’sLSD analysis are indicated by: �significant differences from unimmo-bilized mice (P , 0.05); †significant differences from immobilizedþcage-rest mice (P , 0.05). EDL, extensor digitorum longus.
Human Molecular Genetics, 2004, Vol. 13, No. 21 2635
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Immobilization of the EDL, but not the soleus, resultsin an increase in the number of nemaline rods
In human nemaline patients and in the a-Tmslow(M9R) mousemodel, the distribution of rods in various muscles is verydiverse, but the number of rods (rod-containing fibers andnumber of rods per fiber) remains relatively stable throughoutnormal adult life (8,13). In the present study, we examinedwhether nemaline rods are a static feature of the disease orwhether they can change in response to altered muscle loading.
This was performed using electron microscopy (Fig. 4) andimmunoflorescent microscopy (Fig. 5). Nemaline rods weredefined as electron dense elongated (long-axis parallel to the sar-comeric filaments) accumulations of filamentous material that athigh magnification have a lattice appearance (Fig. 4F–H). Tomore clearly define the lattice of the rods, the muscle sectionwas tilted sequentially until the best lattice view was obtained(Fig. 4I for representative example). Uniform regions of theserods images were used to calculate Fourier filtered images.Such analysis of the rods from the EDL muscle revealed achevron shaped (V) lattice structure (Fig. 4J). This lattice arrange-ment is typical of vertebrate Z-bands (20).
Nemaline rods were observed in few fibers (,10%) of theunimmobilized soleus muscle. These rods were elongatedand ‘spindle-shaped’ (Fig. 4A). With stretch-immobilizationof the soleus muscle, the number of rod-affected fibers andthe number of rods within each affected fiber did not change(data not shown). However, chronic stretch of this muscledid result in areas of Z-line streaming (Fig. 4B): filamentousstructures extending from the Z-line with electron densityless than the Z-line (inset in Fig. 4B) and without the latticestructure observed in nemaline rods. Z-line streaming is arelatively normal feature of muscle (21) and is thought toreflect remodeling of the sarcomere that occurs with musclescontracting under a load (eccentric contractions) (22,23).Unlike nemaline rods, Z-line streaming resolved with cage-rest and was not observed in the soleus muscle with free-wheel or treadmill exercise (data not shown).
In the chronically shortened EDL, there was no evidenceof Z-line streaming, but there was a striking increase inthe number of rods within a fiber (compare the representativeimages of affected fibers in Fig. 4C with Fig. 4D) and thenumber of rod-affected fibers (Fig. 5). Increase in rods/fiberwas associated with a decrease in fiber size (Table 1),
Figure 2. Fiber-type transformation in the soleus muscle with stretch/immo-bilization and during recovery. (A) Percentage of MyHC positive fibers(slow/Ib or IIA) in the soleus muscle. The number of MyHC positive fiberswere counted in whole muscle cross-sections and expressed as a percentageof the total fiber number. The total number of fibers (n ¼ 600–800) did notchange with immobilization or during recovery. (B and C) Consecutive trans-verse sections through stretch-immobilized soleus muscle from nemaline miceshowing hybrid fibers that express both (B) slow/Ib and (C) IIA MyHC iso-forms (labeled with a white star). These fibers are smaller than pure slow/Ib or IIA fibers and are likely to be immature regenerating fibers. (D) Percen-tage of fibers expressing both slow/Ib and IIA MyHC isoforms in the soleusmuscle of nemaline mice. For (A) and (D), the values are meanþ 1 SE for3–6 mice/group and �, indicates significantly different from unimmobilizedmice (P , 0.05), †, indicates significantly different from immobilized mice(P , 0.05) and ‡, indicates significantly different from WT mice.Unimmob., unimmobilized; Immob., immobilized.; C’rest, cage-rest; FW,free-wheel exercise; T’mill, treadmill exercise.
Figure 3. Stretch-immobilization leads to the appearance of the eMyHCisoform. (A) Representative MyHC SDS–PAGE showing the appearance ofthe embryonic (Emb) MyHC isoform in the immobilized soleus muscle ofboth wild-type (WT) and nemaline mice (NM). The other major MyHC iso-forms are also indicated: IIX, IIA and slow/Ib. (B) eMyHC abundance as apercentage of total MyHC content in immobilized WT and nemaline mice(NM). Results (mean and SE) are from densitometric scans of MyHC gelsfrom 4–5 mice/group.
2636 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Figure 4. Immobilization leads to an increase in the number of nemaline rods. Shown are representative electron micrographs from 70 nm longitudinal sectionsof soleus (A and B) and EDL muscle (C–H). (A) Micrograph of unimmobilized soleus showing electron dense elongated spindle-shaped nemaline rods (markedwith white arrow). Note that the nemaline rods often appear as expanded Z-lines and are slightly less electron dense than the Z-lines (marked with black arrow).Scale bar ¼ 2 mm. (B) Micrograph of chronically stretched soleus muscle showing areas of Z-line streaming (arrow head). This was not observed in the shor-tened EDL muscle (not shown). Scale bar ¼ 2 mm. The inset in (B) shows an enlarged image of Z-line streaming. Note that these structures are generally morediffuse and less electron dense than the Z-line (black arrow) and nemaline rods. (C–E) Electron micrographs of EDL muscle showing an increase in the numberof nemaline rods (white arrows) with immobilization (compare rod abundance in C with D) and a decrease with treadmill exercise. A number of adjacent non-affected fibers (white stars in C–E) are also shown in the micrographs. Scale bars ¼ 5 mm. Insets in (C) and (D) are enlarged portions of rod-affected fibers. Notethat, in contrast to the unimmobilized muscle where many Z-lines are visible [arrowheads in main image and inset in (C)], in the immobilized rod-affected fiber(D) there is almost complete absence of Z-line structure [arrowhead in the inset of (D) points to an isolated Z-line]. (F–H) Nemaline rods from EDL muscle athigh magnification. Micrographs from (F) unimmobilized, (G) immobilized and (H) immobilizedþ treadmill exercised nemaline mice are shown. The size,shape and lattice appearance of the rods appeared similar in unimmobilized and immobilized mice (with and without exercise). Black arrowhead in (F)points to a Z-line. The rods in the EDL, in general, were not as elongated as the soleus rods (compare rods in A with rods in F–H); however, the lattice structureof the rods from the two muscles appeared similar (data not shown). Scale bars ¼ 0.5 mm. (I) Representative electron micrograph of a rod from unimmobilizedEDL showing lattice structure. This image was obtained by tilting the muscle section to obtain an optimal view of the lattice. Scale bar ¼ 200 nm. (J) Fourierfiltered image of the boxed region depicted in (J) shows clearly chevron-like (V) links typical of vertebrate Z-bands.
Human Molecular Genetics, 2004, Vol. 13, No. 21 2637
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
indicating that immobilization led to a marked increase inrod density in the EDL. The size and shape of the rodsappeared similar in unimmobilized and immobilized mice(Fig. 4F–H). This suggests that the rods formed with immobil-ization were similar to the constitutive rods that existed in themuscle prior to immobilization. The large accumulations ofrods in the immobilized EDL led to an almost complete break-down of sarcomere structure; Z-line structures were difficult tosee and were very isolated (inset of Fig. 4D). Despite theextensive disruption to the sarcomere, the sarcolemma
appeared to be largely intact in the affected fibers of immobi-lized mice (data not shown).
The nemaline rods formed during immobilization areresolved with endurance exercise without classicalsigns of muscle regeneration
Simple cage-rest had little effect on the number of rods/fiber(data not shown) or the number of rod-affected fibers in theEDL muscle (Fig. 5D). However, exercise (both free-wheel
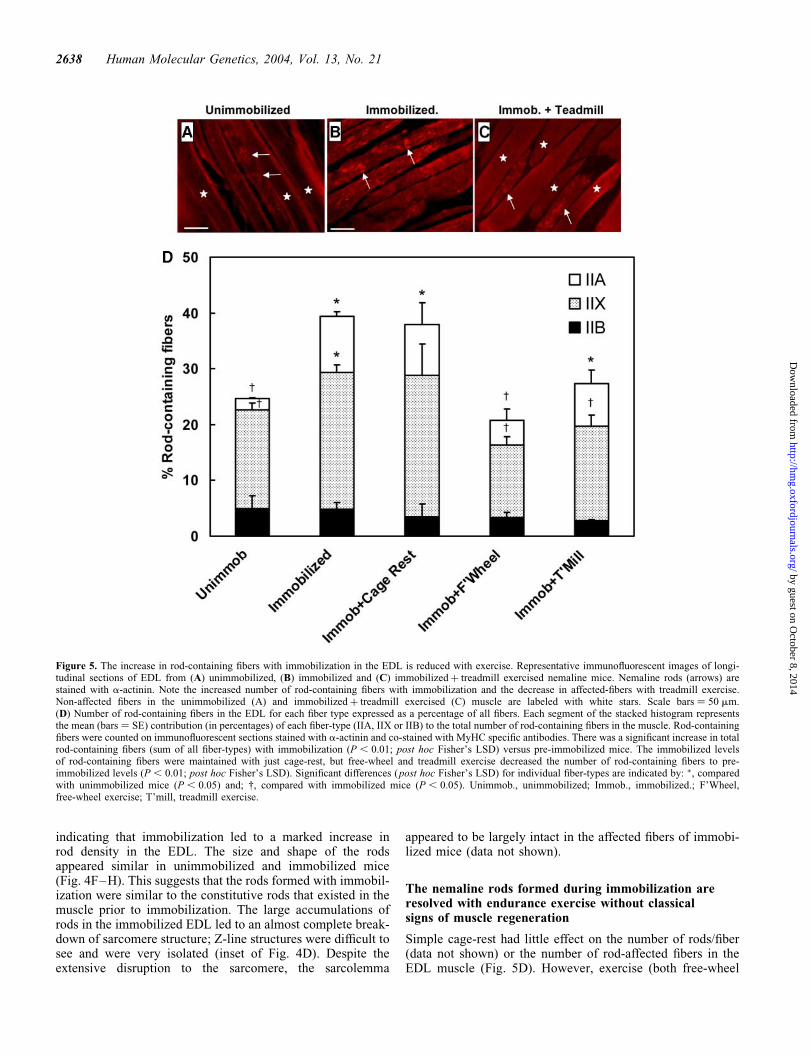
Figure 5. The increase in rod-containing fibers with immobilization in the EDL is reduced with exercise. Representative immunofluorescent images of longi-tudinal sections of EDL from (A) unimmobilized, (B) immobilized and (C) immobilizedþ treadmill exercised nemaline mice. Nemaline rods (arrows) arestained with a-actinin. Note the increased number of rod-containing fibers with immobilization and the decrease in affected-fibers with treadmill exercise.Non-affected fibers in the unimmobilized (A) and immobilizedþ treadmill exercised (C) muscle are labeled with white stars. Scale bars ¼ 50 mm.(D) Number of rod-containing fibers in the EDL for each fiber type expressed as a percentage of all fibers. Each segment of the stacked histogram representsthe mean (bars ¼ SE) contribution (in percentages) of each fiber-type (IIA, IIX or IIB) to the total number of rod-containing fibers in the muscle. Rod-containingfibers were counted on immunofluorescent sections stained with a-actinin and co-stained with MyHC specific antibodies. There was a significant increase in totalrod-containing fibers (sum of all fiber-types) with immobilization (P , 0.01; post hoc Fisher’s LSD) versus pre-immobilized mice. The immobilized levelsof rod-containing fibers were maintained with just cage-rest, but free-wheel and treadmill exercise decreased the number of rod-containing fibers to pre-immobilized levels (P , 0.01; post hoc Fisher’s LSD). Significant differences (post hoc Fisher’s LSD) for individual fiber-types are indicated by: �, comparedwith unimmobilized mice (P , 0.05) and; †, compared with immobilized mice (P , 0.05). Unimmob., unimmobilized; Immob., immobilized.; F’Wheel,free-wheel exercise; T’mill, treadmill exercise.
2638 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

and treadmill) in the post-immobilization period led to adecrease in the number of rods per fiber (Fig. 4E) and thenumber of rod-containing fibers to baseline-unimmobilizedlevels (Fig. 5D). That the increase in rod-affected fibers withimmobilization was similar to the decrease in affected fiberswith exercise (Fig. 5D) is compatible with the possibilitythat the rods formed during immobilization are clearedduring endurance exercise. In addition, the data inFigure 5D display a close, but reciprocal, relationship to theclinical strength measurements in Figure 1.
The increase in the total number of rod-containing fiberswith immobilization and decrease with exercise was due toan increase and decrease, respectively, in the number of rod-containing IIA and IIX fibers (Fig. 5D). This suggests thatthese fibers are more susceptible to rod formation than thefast glycolytic IIB fibers. This is consistent with a previousstudy on these mice where the majority of rods were foundto be in the IIX and IIA fibers in the EDL and the fast fibers(IIA) of the soleus muscle (15). In this earlier study, unimmo-bilized mice underwent the same endurance exercise regimenas the present study, and this had no effect on the total numberof rod-affected fibers in two fast muscles (EDL and extensorcarpi ulnaris). In the present study, exercise decreased thenumber of rod-containing fibers, but not below baselineunimmobilized levels (Fig. 5D). Together the findings ofthese two studies suggest that the constitutive rods presentin the unstressed muscle are more stable thanthe rods formed during immobilization. The decrease in nema-line rod abundance (rods within a fiber and rod-containingfibers) with exercise in the EDL occurred without histologicalevidence of myofiber regeneration, indicating that the repair ofdamaged myofibrils (sarcomere arrays) occurred by processesintrinsic to the muscle fiber.
Muscles of nemaline mice undergo altered myofiberrepair during stretch-immobilization
Muscles under chronic maximal extension immobilizationundergo a cycle of myofiber degeneration and regeneration(24). We were interested to determine whether the processof muscle repair during stretch-immobilization of the soleusmuscle would be altered in the nemaline mice as this mayexplain the poor recovery after immobilization. On hemo-toxylin and eosin (H&E) stained sections, few pathologicalfeatures were observed in muscles from unimmobilizednemaline mice; the myofibers were polygonal in shape withnuclei located at the fiber periphery, and there was littleevidence of myonecrosis or infiltrating mononuclear cells(Fig. 6A and B, respectively). This is consistent with the cat-egorization of this disease as a myopathy without dystrophy.Immobilization of the EDL in the shortened position producedfew histopathological effects; few signs of necrosis, myofiberregeneration or fibrosis and only baseline numbers of central-nucleated fibers (,1% of fibers; data not shown).
However, in the soleus, extensive myofiber degenerationand macrophage infiltration were observed after 10 days ofstretch-immobilization in WT and nemaline mice (Fig. 6Cand D, respectively). The level of necrosis appeared similarin the two mice, and at this stage there were few fibers withcentralized nuclei indicating that active regeneration had not
commenced. After a further 18 days of stretch-immobilization(4 weeks in total), infiltrating macrophages were still evidentin the WT soleus, but myofiber necrosis had essentially disap-peared, replaced by extensive areas of small regeneratingfibers (small central-nucleated fibers) (Fig. 6E). In the nema-line muscle, at the same time point, there were fewer regene-rating fibers; and in contrast to WT muscle, there were veryfew fibers with central nuclei (Fig. 6F). With 4 weeks ofexercise recovery (both free-wheel and treadmill), in theWT soleus muscle, small foci of regenerating fibers (smallcentral-nucleated fibers) were still present and there weremany mature fibers with centralized nuclei (Fig. 6G);whereas in the nemaline mice, central-nucleated maturefibers were virtually absent (Fig. 6H).
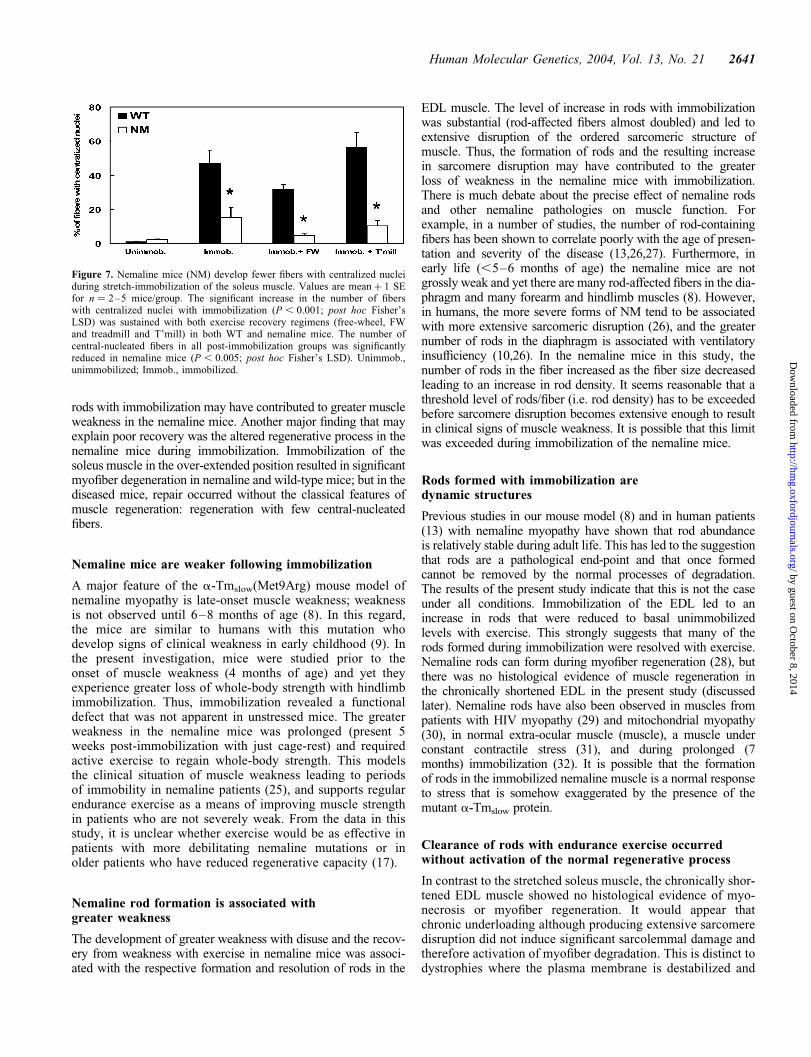
To provide an objective assessment of regeneration in theWT and nemaline mice, the number of fibers with centralizednuclei was counted in whole sections of unstretched andstretched soleus muscles (500–700 fibers/muscle) (Fig. 7).This analysis confirmed the markedly reduced number offibers with centralized nuclei in the soleus muscle of nemalinecompared with WT mice after 4 weeks stretch-immobilized; inWT muscle �50% of the fibers had central nuclei, whereasin the nemaline muscle the number of central-nucleatedfibers was three times less (�15%) (Fig. 7). The number ofcentral-nucleated fibers did not change appreciably witheither freewheel or treadmill exercise, although there was amodest decrease (P ¼ 0.057 and 0.063 for WT and nemalinemice; post hoc Fisher’s LSD) in fibers with central nuclei infreewheel exercise compared with immobilized mice(Fig. 7). In the nemaline mice following both types of exer-cise, the central-nucleated fibers were almost entirely smallregenerating fibers (Fig. 5H) (,1% of mature fibers had cen-tralized nuclei; data not shown), whereas in the WT mice thecentral nuclei were in both immature and mature fibers(Fig. 6G). Together the data suggest that the expression ofthe mutant a-Tmslow protein in muscle alters the normal myo-fiber regenerative process.
DISCUSSION
Muscle weakness is a significant cause of morbidity for patientssuffering from nemaline myopathy and can lead to prolongedperiods of immobility (2,25). Prolonged muscle inactivity maybe particularly detrimental in these patients as an early radio-logical study indicated that nemaline patients experience greatermuscle disuse-atrophy compared with healthy controls (12).The data from our nemaline mouse verifies that a nemalinemutation can indeed increase the sensitivity of muscle toperiods of immobility and that endurance exercise is an effectivemeans of stimulating recovery from such insults. The results ofthe present study also provide important data on potential mech-anisms for poor recovery in nemaline myopathy. Immobilizationof a muscle (EDL) in the shortened position in the nemaline miceled to a large increase in the number of nemaline rods, the defin-ing pathological feature of the disease. Endurance exercise but notcage-rest restored whole-body strength and was associated with areduction in the number of nemaline rods and sarcomere disrup-tion. These observations raise the possibility that nemaline rodabundance can influence muscle strength and the increase in
Human Molecular Genetics, 2004, Vol. 13, No. 21 2639
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
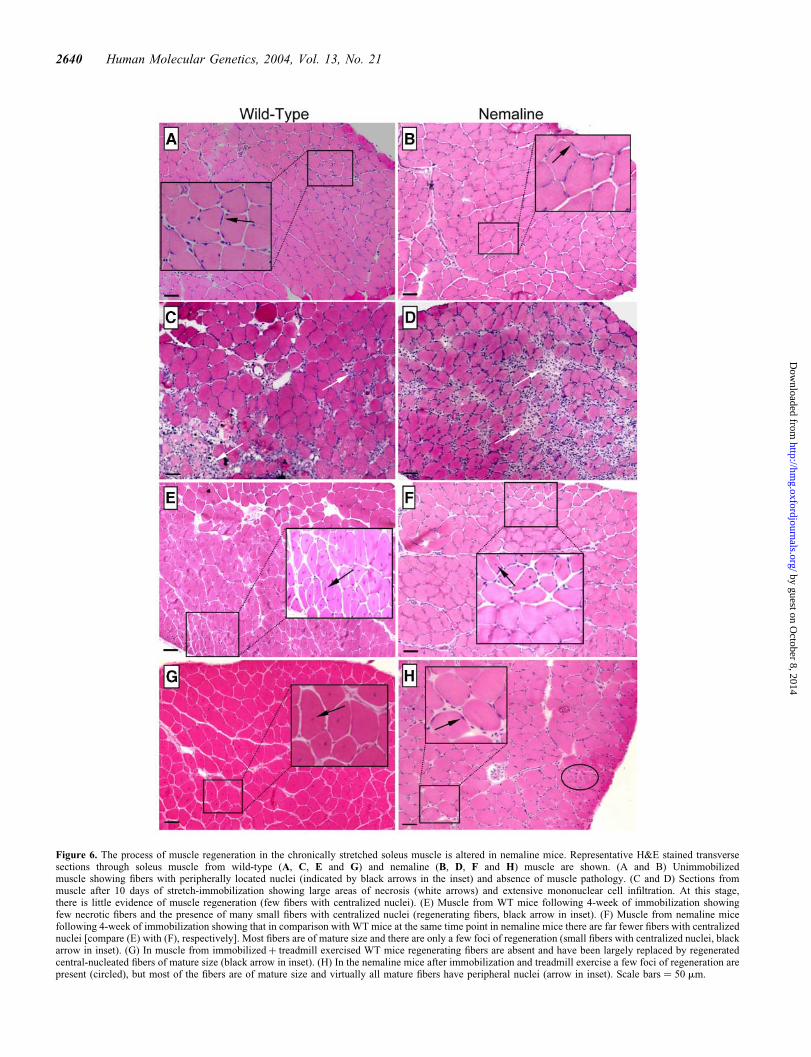
Figure 6. The process of muscle regeneration in the chronically stretched soleus muscle is altered in nemaline mice. Representative H&E stained transversesections through soleus muscle from wild-type (A, C, E and G) and nemaline (B, D, F and H) muscle are shown. (A and B) Unimmobilizedmuscle showing fibers with peripherally located nuclei (indicated by black arrows in the inset) and absence of muscle pathology. (C and D) Sections frommuscle after 10 days of stretch-immobilization showing large areas of necrosis (white arrows) and extensive mononuclear cell infiltration. At this stage,there is little evidence of muscle regeneration (few fibers with centralized nuclei). (E) Muscle from WT mice following 4-week of immobilization showingfew necrotic fibers and the presence of many small fibers with centralized nuclei (regenerating fibers, black arrow in inset). (F) Muscle from nemaline micefollowing 4-week of immobilization showing that in comparison with WT mice at the same time point in nemaline mice there are far fewer fibers with centralizednuclei [compare (E) with (F), respectively]. Most fibers are of mature size and there are only a few foci of regeneration (small fibers with centralized nuclei, blackarrow in inset). (G) In muscle from immobilizedþ treadmill exercised WT mice regenerating fibers are absent and have been largely replaced by regeneratedcentral-nucleated fibers of mature size (black arrow in inset). (H) In the nemaline mice after immobilization and treadmill exercise a few foci of regeneration arepresent (circled), but most of the fibers are of mature size and virtually all mature fibers have peripheral nuclei (arrow in inset). Scale bars ¼ 50 mm.
2640 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
rods with immobilization may have contributed to greater muscleweakness in the nemaline mice. Another major finding that mayexplain poor recovery was the altered regenerative process in thenemaline mice during immobilization. Immobilization of thesoleus muscle in the over-extended position resulted in significantmyofiber degeneration in nemaline and wild-type mice; but in thediseased mice, repair occurred without the classical features ofmuscle regeneration: regeneration with few central-nucleatedfibers.
Nemaline mice are weaker following immobilization
A major feature of the a-Tmslow(Met9Arg) mouse model ofnemaline myopathy is late-onset muscle weakness; weaknessis not observed until 6–8 months of age (8). In this regard,the mice are similar to humans with this mutation whodevelop signs of clinical weakness in early childhood (9). Inthe present investigation, mice were studied prior to theonset of muscle weakness (4 months of age) and yet theyexperience greater loss of whole-body strength with hindlimbimmobilization. Thus, immobilization revealed a functionaldefect that was not apparent in unstressed mice. The greaterweakness in the nemaline mice was prolonged (present 5weeks post-immobilization with just cage-rest) and requiredactive exercise to regain whole-body strength. This modelsthe clinical situation of muscle weakness leading to periodsof immobility in nemaline patients (25), and supports regularendurance exercise as a means of improving muscle strengthin patients who are not severely weak. From the data in thisstudy, it is unclear whether exercise would be as effective inpatients with more debilitating nemaline mutations or inolder patients who have reduced regenerative capacity (17).
Nemaline rod formation is associated withgreater weakness
The development of greater weakness with disuse and the recov-ery from weakness with exercise in nemaline mice was associ-ated with the respective formation and resolution of rods in the
EDL muscle. The level of increase in rods with immobilizationwas substantial (rod-affected fibers almost doubled) and led toextensive disruption of the ordered sarcomeric structure ofmuscle. Thus, the formation of rods and the resulting increasein sarcomere disruption may have contributed to the greaterloss of weakness in the nemaline mice with immobilization.There is much debate about the precise effect of nemaline rodsand other nemaline pathologies on muscle function. Forexample, in a number of studies, the number of rod-containingfibers has been shown to correlate poorly with the age of presen-tation and severity of the disease (13,26,27). Furthermore, inearly life (,5–6 months of age) the nemaline mice are notgrossly weak and yet there are many rod-affected fibers in the dia-phragm and many forearm and hindlimb muscles (8). However,in humans, the more severe forms of NM tend to be associatedwith more extensive sarcomeric disruption (26), and the greaternumber of rods in the diaphragm is associated with ventilatoryinsufficiency (10,26). In the nemaline mice in this study, thenumber of rods in the fiber increased as the fiber size decreasedleading to an increase in rod density. It seems reasonable that athreshold level of rods/fiber (i.e. rod density) has to be exceededbefore sarcomere disruption becomes extensive enough to resultin clinical signs of muscle weakness. It is possible that this limitwas exceeded during immobilization of the nemaline mice.
Rods formed with immobilization aredynamic structures
Previous studies in our mouse model (8) and in human patients(13) with nemaline myopathy have shown that rod abundanceis relatively stable during adult life. This has led to the suggestionthat rods are a pathological end-point and that once formedcannot be removed by the normal processes of degradation.The results of the present study indicate that this is not the caseunder all conditions. Immobilization of the EDL led to anincrease in rods that were reduced to basal unimmobilizedlevels with exercise. This strongly suggests that many of therods formed during immobilization were resolved with exercise.Nemaline rods can form during myofiber regeneration (28), butthere was no histological evidence of muscle regeneration inthe chronically shortened EDL in the present study (discussedlater). Nemaline rods have also been observed in muscles frompatients with HIV myopathy (29) and mitochondrial myopathy(30), in normal extra-ocular muscle (muscle), a muscle underconstant contractile stress (31), and during prolonged (7months) immobilization (32). It is possible that the formationof rods in the immobilized nemaline muscle is a normal responseto stress that is somehow exaggerated by the presence of themutant a-Tmslow protein.
Clearance of rods with endurance exercise occurredwithout activation of the normal regenerative process
In contrast to the stretched soleus muscle, the chronically shor-tened EDL muscle showed no histological evidence of myo-necrosis or myofiber regeneration. It would appear thatchronic underloading although producing extensive sarcomeredisruption did not induce significant sarcolemmal damage andtherefore activation of myofiber degradation. This is distinct todystrophies where the plasma membrane is destabilized and
Figure 7. Nemaline mice (NM) develop fewer fibers with centralized nucleiduring stretch-immobilization of the soleus muscle. Values are meanþ 1 SEfor n ¼ 2–5 mice/group. The significant increase in the number of fiberswith centralized nuclei with immobilization (P , 0.001; post hoc Fisher’sLSD) was sustained with both exercise recovery regimens (free-wheel, FWand treadmill and T’mill) in both WT and nemaline mice. The number ofcentral-nucleated fibers in all post-immobilization groups was significantlyreduced in nemaline mice (P , 0.005; post hoc Fisher’s LSD). Unimmob.,unimmobilized; Immob., immobilized.
Human Molecular Genetics, 2004, Vol. 13, No. 21 2641
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

this leads to infiltration of phagocytic cells and myonecrosis(33). The lack of myofiber regeneration in the EDL indicatesthat the decrease in rod abundance and repair of damagedsarcomeres occurred by processes intrinsic to the musclefiber. This repair also appears to be distinct from the processof limited necrosis and autodigestion that occurs after minorfocal injury (28) as necrotic debris was absent in the EDLfrom nemaline mice. Collectively, these observations suggestthat the type of repair that occurs in the nemaline mice isnovel and may be more similar to the normal processes ofremoval of malformed or damaged sarcomeric proteins thatoccur in healthy undamaged muscle.
However, this raises the question of why the rods inducedby immobilization were resolved, while rods in humanpatients and in unstressed nemaline mice remain throughoutlife (8,25). As the number of rod-containing fibers increasedwith immobilization, it would appear that the rods formed inthe fibers that were not affected prior to immobilization.Perhaps there is something different about these fibers or thestructure of the rods formed in these fibers that allowedthem to be cleared with exercise recovery. It is also intriguingthat cage-rest had little effect on rod numbers, whereas bothendurance exercise regimens were able to restore rod numbersto basal levels. Under normal conditions, the turnover ofsarcomeric proteins occurs at a relatively slow rate (half-life,6–9 days depending on the protein) (34). Thus, one couldargue that the breakdown of rods formed during immobili-zation with simple cage-rest may take significantly longerthan the 4-week-period allowed in the present study. In con-trast, exercise leads to an increase in protein turnover andremodeling of the sarcomere (35), and this may have stimu-lated the removal of the rods formed during immobilization.Regardless of the mechanism, this study clearly suggeststhat exercise in adult nemaline mice is effective in resolvingthe sarcomere disruption produced by immobilization.
Muscles from nemaline mice do not exhibit the classicfeatures of muscle repair during immobilization of thesoleus muscle
Central to the normal process of muscle regeneration afterinjury is the activation and proliferation of myogenic satellitecells. Activated satellite cells migrate to the site of injury, fusewith damaged myofibers or to themselves, withdraw from thecell cycle and form new differentiated myotubes with centrallylocated nuclei (17). Normally, as the nascent myofiber maturesit increases in size and the nuclei migrate to the periphery ofthe fiber. However, in rodents, nuclei can remain in thecenter of a regenerated fiber indefinitely and is often used asan index of fibers that have undergone necrosis (28). Theresults of this study indicate that in nemaline myopathy theprocess of regeneration is altered. Regeneration of the soleusmuscle following stretch-induced muscle damage in nemalinemice was associated with far fewer fibers with central nucleicompared with WT mice. This was apparent in the earlyphase of regeneration (at the end of 4 weeks immobilization)and also after 1 month of exercise recovery (both low- andhigh-intensity). Despite the relative lack of centralized nuclei,regeneration did occur in the nemaline muscle; necrotic fiberswere replaced by fibers that appeared morphologically normal.
There are two possible explanations for fewer centralizednuclei in nemaline mice: (i) satellite cells or other myogeniccells fuse to damaged fibers, and nuclei stay at the peripheryor migrate more quickly to the periphery or (ii) myogeniccells are activated but do not fuse as readily with the fibersof nemaline mice. The second mechanism is less likelybecause without fusion of activated satellite cells regenerationwould be attenuated and there is no sign of this in the nemalinemice. With the first mechanism, one could hypothesize that thenemaline myopathy mutation alters the structural or regulatorynetworks responsible for nuclear migration during myofiberregeneration. These networks are still to be described but arelikely to involve similar cytoskeletal systems that mediateorganelle transport in non-muscle cells (microtubules, inter-mediate filaments and actin microfilaments). Whether thealtered regenerative process in nemaline mice impacts on therestoration of myofiber function needs to be established.
Muscle from nemaline patients and the unimmobilizednemaline mouse model do not display the characteristic patho-logical features of muscle regeneration: muscle degeneration,macrophage infiltration and central-nucleated fibers. However,affymetrix array analysis of human nemaline patients (7) andour nemaline mouse (unimmobilized; unpublished data)showed increased expression of genes characteristic of acti-vated satellite cells and immature fibers (e.g. PAX7, MYF6,NCAM1, ANKRD2 ). These observations together with the pre-sented data on the nemaline mice clearly suggest that nemalinemuscle undergoes repair without displaying the traditionalfeatures of regeneration.
In summary, we have shown that expression of a nemalinemyopathy mutation exacerbates the effects of chronic inactiv-ity. The data suggests that this may be due to the formation ofnemaline rods and/or altered myofiber repair. In the nemalinemice, endurance exercise alleviated muscle weakness andreduced the number of nemaline rods, supporting its use inpatients with the disease. Two distinct myofiber repair pro-cesses have been identified in the nemaline mice: (i) regene-ration without centralized nuclei and (ii) sarcomere repair(clearance of nemaline rods) without classic signs of regener-ation. These repair processes maybe specific to diseases of thesarcomeric thin filament and are distinct from sarcolemmalrepair in muscular dystrophy and repair in normal healthymuscle.
MATERIALS AND METHODS
This study was approved by the Children’s Medical ResearchInstitute/New Children’s Hospital Animal Care and EthicsCommittee, Westmead.
Transgenic mice
Generation and characterization of the human skeletal actin(HSA)–a-Tmslow(Met9Arg) transgenic mice has beendescribed previously (8). The HSA–a-Tmslow(Met9Arg)construct uses the HSA promoter to drive expression of thea-Tmslow(Met9Arg) mutant cDNA (TPM3 gene) with aSV40 30-UTR sequence specifically in all fibers to varyingdegrees. This approach was taken to achieve a mouse model
2642 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

in which a significant number of fibers would be affected bythe presence of the mutant protein as the majority of mousemuscles have a predominance of fast fibers. Non-transgenicmice arising from the matings of transgenic mice were usedas control WT animals.
Hindlimb immobilization protocol
The hindlimbs of 8-week-old WT and transgenic mice wereimmobilized for 28 days as originally described by Booth(36) with minor modifications. While anaesthetized withketamine/xylazine (100 and 10 mg/kg BW, respectively)both feet were fixed in maximal dorsal flexion using a standardporous adhesive tape. The knees were then bent so that thedistal lower leg were apposed to the proximal upper leg andthe feet were bound in a dorsal-flexed position resultingin the chronic shortening of the tibialis anterior and EDLmuscles and lengthening of the soleus, gastrocnemius andplantaris muscles. The bandages were inspected daily forfraying, soiling or loosening and replaced as required. Follow-ing immobilization, one group of mice were sacrificed(Immobilized group) and the remaining mice were randomlyassigned to one of three recovery regimens: (i) cage-rest(minimal exercise), (ii) free-wheel exercise (low-intensityvoluntary exercise) or (iii) motorized treadmill exercise(high-intensity exercise). Unimmobilized age-matched (12months old) WT and a-Tmslow(Met9Arg) mice were alsostudied.
Free-wheel exercise
Immediately following immobilization, mice were placed inseparate cages containing custom-made (Siwino Pty Ltd,Sydney) exercise free-wheels (30 cm circumference) equippedwith counters to register the number of wheel revolutions.The number of revolutions was recorded every 10 s, 24 h/day,by computer. Following 24 days of free-wheel exercise,strength and fatigability of each mouse were measuredaccording to the test procedures described later.
Endurance exercise training
The exercise-training regimen is similar to that reportedpreviously (15). Following immobilization, the transgenicand WT littermates were allowed to recover from the immo-bilization for 1 week before exercise training commenced.During this period, the mice were housed in a room withreversed light and dark cycle (light off: 7:00; light on:19:00) so that they could be exercised during the period ofmaximum activity (9:00–14:00). The mice were primed toexercise on a motorized treadmill for 1 week (ColumbusExer-4/8, Columbus, OH): a daily 30 min bout of exerciseof increasing intensity (6–15 m min21 on days 1–2 to 15–25 m min21 on day 7) at a 5% incline.
Exercise training was performed over 17 days at a speed of25 m min21. The treadmill incline and length of exercise wasincreased progressively over the training period: 5% and45 min on day 1 to 10% and 90 min on day 17. At the endof the exercise regime, strength and fatigability of each
mouse were measured according to the test proceduresdescribed below.
Strength and fatigability test
Whole-animal strength and fatigability were measured accord-ing to the test procedure of Hubner et al. (14). In brief, this testrequired the mice to pull themselves on top of a suspended rod(3 mm in diameter). The measurement of muscle weaknesswas based on the mean percentage of passes over 15 trialsof the test in a 3 min period. Fatigability was assessed as theaverage pass rate over time for each group of mice.
Muscle collection
Following immobilization and at the end of the recoveryperiods (cage-rest, free-wheel exercise and treadmill exercise)mice were sacrificed by cervical dislocation and the EDL andsoleus muscles were collected. Muscles were coated in tissuefreezing medium (ProSciTech, Brisbane, Australia), frozen inmelting isopentane pre-chilled in liquid nitrogen and stored inliquid nitrogen for immunohistochemical and histopatho-logical analysis.
Fiber morphometry
Quantification of fiber-type and morphometry were performedas described by Nair-Shalliker et al. (15). Fiber-typing wasperformed on transverse sections (8 mm thickness) of musclewith antibodies to specific MyHC isoforms: slow/type I(b)(undiluted BA-F8) (37), type IIB (undiluted BF-F3) (38),type IIA (undiluted SC 71) (38) and IIX (undiluted; giftfrom Dr Joseph Hoh, Sydney) (39). The secondary antibodywas peroxidase-conjugated goat/anti-mouse immunoglobulins(Dako; 1:100 dilution) and detection was carried out usingthe DAB peroxidase substrate kit (Vector Laboratories).Detection of IIX antibody was enhanced using Vectastainw
ABC kit (PK-400) prior to DAB staining. Images from themuscle sections were captured using a digital camera. Fibercross-sectional area (CSA) were obtained from the stained sec-tions using the Image-Pro software (Media Cybernetics) asdescribed previously (15).
Immunohistochemical analysis of nemaline rods
Nemaline rods contain Z-line proteins, principally a-actinin.Antibodies to a-actinin were used to detect rod-containingfibers in longitudinal sections (6 mm thickness) from EDLand soleus muscles. The primary antibody mixture comprisedthe monoclonals that recognize the MyHCs of interest(details of antibodies have been mentioned earlier) and therabbit-polyclonal to a-actinin-2 and -3 (1:200;gift from Alan Beggs, Boston) (40). The sections werewashed thoroughly and incubated in (1:1000 dilution) second-ary antibody (Alexa Fluor 488 goat anti-mouse immunoglobu-lins and Alexa Fluor 594 goat anti-rabbit immunoglobulins;Molecular Probes) for 1 h at RT. The number of rod-contain-ing fibers of specific fiber-types was determined and expressedas a percentage of the total number of fibers (all fiber types)
Human Molecular Genetics, 2004, Vol. 13, No. 21 2643
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

present in the muscle section. At least 300 fibers from eachmuscle were counted.
Electron microscopic analysis of nemaline rods
EDL and soleus muscles were removed from anaesthetizedanimals (100 mg/kg BW ketamine/10 mg/kg BW xylazine)and immediately cut into very thin slices while immersed inmodified Karnovski’s fixative (2.5% glutaraldehyde/4% para-formaldehyde in 1 M cacodylate buffer, pH 7.4). Samples werefurther fixed overnight in the same fixative and post-fixed with2% osmium tetroxide, dehydrated through an ascending seriesof ethanol, and embedded in Spurr’s epoxy resin. Ultrathinsections (70 nm) were cut with a Reichert-Jung ULTRACUTultramicrotome, double contrasted with uranyl acetate andlead citrate, viewed and photographed with a Philips CM120BioTwin transmission electron microscope.
For more detailed analysis of rod ultrastructure sectionswere viewed in a Jeol 1200 electron microscope fitted witha Teitz Fastscan CCD camera. The rod images were captureddirectly onto the 1K � 1K CCD camera. To optimize thelattice view of the nemaline rods, a rotation tilt holder wasused to align the tilt axis along the long axis of a selectedrod and then the section was tilted in sequential steps of 58from 2608 to þ608. The image showing the best latticeview was selected. Uniform regions of rod images wereboxed and in-house software (Imperial College London) wasused to calculate Fourier filtered views.
Histopathology
Transverse sections (8 mm thickness) were cut from themidsection of the muscles. Sections were placed on poly-L-lysine pre-coated glass microscope slides, air-dried, fixedand stained with H&E. The number of fibers with centralizednuclei in the whole muscle section was counted.
Myosin heavy chain SDS–PAGE
MyHC extracts were prepared and SDS–PAGE preformed asdescribed by Nair-Shalliker et al. (15). Briefly the MyHCextract was suspended in Laemmli sample buffer (Biorad),loaded onto an 8% vertical acrylamide gel (3% stacking)(Miniprotean Protean III; Biorad) and electrophoresed at70 V for 40 h at 48C. The gels were stained using a silvernitrate kit (Biorad) and scanned using a Biorad GS800densitometer.
Data and statistical analysis
Results are presented as mean and SEM. The Levene’s testwas performed to assure homogeneity of variance togetherwith the Kolmogorov-Smirnov (Lilliefors) test to check fornormality of the data (41). All data presented satisfied theabove tests and so parametric analysis was employed.
Initially, global differences between groups were deter-mined using two-way ANOVA, the factors being: transgeneand treatment (immobilization, immobilizationþ cage-rest,immobilizationþ free-wheel exercise and immobilizationþtreadmill exercise) (41). If statistically significant differences
were detected by ANOVA, individual comparisons weremade between groups using the post hoc Fisher’s LSD test(a-level ¼ 0.05).
ACKNOWLEDGEMENTS
We gratefully acknowledge Alan Beggs and Joseph Hoh forproviding the a-actinin and MyHC IIX antibodies, respectively.We appreciate work of C. Stephen Robinson in the initial stagesof this project. We would like to thank Peter Gunning, MarkCorbett and Kathy North for their helpful comments aboutaspects of this work. We are indebted to Wendy Thornton andthe other members of the BioServices Unit of CMRI for theirhelp in managing the animals. This investigation was supportedby National Medical Research Council grant no. 185206.
REFERENCES
1. Ilkovski, B., Nowak, K.J., Domazetovska, A., Maxwell, A.L., Clement, S.,Davies, K.E., Laing, N.G., North, K.N. and Cooper, S. (2004)Evidence for a dominant-negative effect in ACTA1 nemaline myopathycaused by abnormal folding, aggregation and altered polymerization ofmutant actin isoforms. Hum. Mol. Genet., 13, 1727–1743.
2. North, K.N., Laing, N.G. and Wallgren-Pettersson, C. (1997) Nemalinemyopathy: current concepts. The ENMC International Consortium andNemaline Myopathy. J. Med. Genet., 34, 705–713.
3. Morris, E.P., Nneji, G. and Squire, J.M. (1990) The three-dimensionalstructure of the nemaline rod Z-band. J. Cell Biol., 111, 2961–2978.
4. Goebel, H.H. and Laing, N.G. (2002) In Karpati, G. (ed.), Structuraland Molecular Basis of Skeletal Muscle Diseases. ISN Neuropath,Basel, pp. 74–77.
5. Fardeau, M. and Tome, F.M.S. (1994) In Engel, A.G. andFranzini-Armstrong C. (eds), Myology. Basic and Clinical.McGraw Hill, Inc., New York, pp. 1487–1532.
6. Dalkilic, I. and Kunkel, L.M. (2003) Muscular dystrophies: genes topathogenesis. Curr. Opin. Genet. Dev., 13, 231–238.
7. Sanoudou, D., Haslett, J.N., Kho, A.T., Guo, S., Gazda, H.T.,Greenberg, S.A., Lidov, H.G., Kohane, I.S., Kunkel, L.M. and Beggs,A.H. (2003) Expression profiling reveals altered satellite cell numbersand glycolytic enzyme transcription in nemaline myopathy muscle.Proc. Natl Acad. Sci. USA, 100, 4666–4671.
8. Corbett, M.A., Robinson, C.S., Dunglison, G.F., Yang, N.,Joya, J.E., Stewart, A.W., Schnell, C., Gunning, P.W., North, K.N.and Hardeman, E.C. (2001) A mutation in alpha-tropomyosin(slow)affects muscle strength, maturation and hypertrophy in a mousemodel for nemaline myopathy. Hum. Mol. Genet., 10, 317–328.
9. Laing, N.G., Wilton, S.D., Akkari, P.A., Dorosz, S., Boundy, K.,Kneebone, C., Blumbergs, P., White, S., Watkins, H., Love, D.R. andHaan, E. (1995) A mutation in the alpha tropomyosin gene TPM3associated with autosomal dominant nemaline myopathy. Nat. Genet., 9,75–79.
10. Shafiq, S.A., Dubowitz, V., Peterson, H.C. and Milhorat, A.T. (1967)Nemaline myopathy: report of a fatal case, with histochemical andelectron microscopic studies. Brain, 90, 817–828.
11. Baldwin, K.M. and Haddad, F. (2002) Skeletal muscle plasticity:cellular and molecular responses to altered physical activity paradigms.Am. J. Phys. Med. Rehabil., 81, S40–S51.
12. Wallgren-Pettersson, C., Kivisaari, L., Jaaskelainen, J.,Lamminen, A. and Holmberg, C. (1990) Ultrasonography, CT, andMRI of muscles in congenital nemaline myopathy.Pediatr. Neurol., 6, 20–28.
13. Wallgren-Pettersson, C., Rapola, J. and Donner, M. (1988) Pathologyof congenital nemaline myopathy. A follow-up study.J. Neurol. Sci., 83, 243–257.
14. Hubner, C., Lehr, H.A., Bodlaj, R., Finckh, B., Oexle, K., Marklund, S.L.,Freudenberg, K., Kontush, A., Speer, A., Terwolbeck, K. et al. (1996)Wheat kernel ingestion protects from progression of muscle weaknessin mdx mice, an animal model of Duchenne muscular dystrophy.Pediatr. Res., 40, 444–449.
2644 Human Molecular Genetics, 2004, Vol. 13, No. 21
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

15. Nair-Shalliker, V., Kee, A.J., Joya, J.E., Lucas, C.A., Hoh, J.F. andHardeman, E.C. (2004) Myofiber adaptational response to exercise in amouse model of nemaline myopathy. Muscle Nerve, 30, 470–480.
16. Pette, D. (1998) Training effects on the contractile apparatus.Acta Physiol. Scand., 162, 367–376.
17. Charge, S.B.P. and Rudnicki, M.A. (2004) Cellular and molecularregulation of muscle regeneration. Physiol. Rev., 84, 209–238.
18. Loughna, P.T., Izumo, S., Goldspink, G. and Nadal-Ginard, B. (1990)Disuse and passive stretch cause rapid alterations in expression ofdevelopmental and adult contractile protein genes in skeletal muscle.Development, 109, 217–223.
19. Dix, D.J. and Eisenberg, B.R. (1990) Myosin mRNA accumulation andmyofibrillogenesis at the myotendinous junction of stretched musclefibers. J. Cell Biol., 111, 1885–1894.
20. Luther, P.K., Barry, J.S., and Squire, J.M. (2002) The three-dimensionalstructure of a vertebrate wide (slow muscle) Z-band: lessons on Z-bandassembly. J. Mol. Biol., 315, 9–20.
21. Meltzer, H.Y., Kuncl, R.W. and Yang, V. (1976) Incidence of Z bandstreaming and myofibrillar disruptions in skeletal muscle from healthyyoung people. Neurology, 26, 853–857.
22. Yu, J.G., Furst, D.O. and Thornell, L.E. (2003) The mode ofmyofibril remodelling in human skeletal muscle affected byDOMS induced by eccentric contractions. Histochem.Cell Biol., 119, 383–393.
23. Yu, J.G., Carlsson, L. and Thornell, L.E. (2004) Evidence for myofibrilremodeling as opposed to myofibril damage in human muscles withDOMS: an ultrastructural and immunoelectron microscopic study.Histochem. Cell Biol., 121, 219–227.
24. Sjostrom, M., Wahlby, L. and Fugl-Meyer, A. (1979) Achilles tendoninjury. 3. Structure of rabbit soleus muscles after immobilization atdifferent positions. Acta Chir. Scand., 145, 509–521.
25. Wallgren-Pettersson, C. (1989) Congenital nemaline myopathy. Aclinical follow-up of twelve patients. J. Neurol. Sci., 89, 1–14.
26. Ryan, M.M., Ilkovski, B., Strickland, C.D., Schnell, C., Sanoudou, D.,Midgett, C., Houston, R., Muirhead, D., Dennett, X., Shield, L.K. et al.(2003) Clinical course correlates poorly with muscle pathology innemaline myopathy. Neurology, 60, 665–673.
27. Shimomura, C. and Nonaka, I. (1989) Nemaline myopathy:comparative muscle histochemistry in the severe neonatal, moderatecongenital, and adult-onset forms. Pediatr. Neurol., 5, 25–31.
28. Engel, A.G. and Banker, B.Q. (1994) In Engel, A.G. andFranzini-Armstrong C. (eds), Myology. Basic and Clinical.McGraw Hill, Inc., New York, pp. 889–1017.
29. Feinberg, D.M., Spiro, A.J. and Weidenheim, K.M. (1998)Distinct light microscopic changes in human immunodeficiencyvirus-associated nemaline myopathy. Neurology, 50, 529–531.
30. Fukunaga, H., Osame, M. and Igata, A. (1980) A case of nemalinemyopathy with ophthalmoplegia and mitochondrial abnormalities.J. Neurol. Sci., 46, 169–177.
31. Martinez, A.J., Hay, S., and McNeer, K.W. (1976) Extraocular muscles:light microscopy and ultrastructural features. Acta Neuropathol. (Berl.),34, 237–253.
32. Baranska, B. (1999) Formation of the nemaline structures in soleusmuscle of rats subjected to long-lasting immobilization.Folia Morphol (Warsz.), 58, 207–214.
33. Spence, H.J., Chen, Y.J. and Winder, S.J. (2002) Muscular dystrophies,the cytoskeleton and cell adhesion. Bioessays, 24, 542–552.
34. Zak, R., Martin, A.F., Prior, G. and Rabinowitz, M. (1977) Comparisonof turnover of several myofibrillar proteins and critical evaluation ofdouble isotope method. J. Biol. Chem., 252, 3430–3435.
35. Rennie, M.J. and Tipton, K.D. (2000) Protein and amino acidmetabolism during and after exercise and the effects of nutrition.Annu. Rev. Nutr., 20, 457–483.
36. Booth, F.W. (1977) Time course of muscular atrophy duringimmobilization of hindlimbs in rats. J. Appl. Physiol., 43, 656–661.
37. Borrione, A.C., Zanellato, A.M., Saggin, L., Mazzoli, M., Azzarello,G. and Sartore, S. (1988) Neonatal myosin heavy chains are notexpressed in Ni-induced rat rhabdomyosarcoma. Differentiation,38, 49–59.
38. Schiaffino, S., Gorza, L., Sartore, S., Saggin, L., Ausoni, S.,Vianello, M., Gundersen, K. and Lomo, T. (1989) Three myosinheavy chain isoforms in type 2 skeletal muscle fibres. J. MuscleRes. Cell Motil., 10, 197–205.
39. Lucas, C.A., Kang, L.H. and Hoh, J.F. (2000) Monospecific antibodiesagainst the three mammalian fast limb myosin heavy chains.Biochem. Biophys. Res. Commun., 272, 303–308.
40. North, K.N. and Beggs, A.H. (1996) Deficiency of a skeletal muscleisoform of alpha-actinin (alpha-actinin-3) in merosin-positivecongenital muscular dystrophy. Neuromuscul. Disord., 6, 229–235.
41. SPSS Inc. (1997) SPSS Advanced Statistics Manual. SPSS Inc., Chicago.
Human Molecular Genetics, 2004, Vol. 13, No. 21 2645
by guest on October 8, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Related Documents











