MTLA-Veranstaltung Klinikum am Urban, 12.11.2011

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MTLA-Veranstaltung Klinikum am Urban, 12.11.2011

Hämostaseologie
Christoph Sucker
LaboMed Gerinnungszentrum Berlin

Gliederung
• Grundlagen der Blutgerinnung
• venöse Thromboembolie (VTE)
• Gerinnungshemmer (Antithrombotika)
3

Grundlagen der Blutgerinnung
4

• Blutgerinnung – primäre Hämostase
– sekundäre (plasmatische) Hämostase
• antithrombotische Mechanismen – Fibrinolyse
– Protein C-/ Protein S-System
– Antithrombin
Grundlagen der Blutgerinnung
5

Thrombose
Fibrinolyse PC-/PS-System Antithrombin
Verschluss des Gefäßdefektes/Blutstillung
Gefäßdefekt Blutung
Thrombozytenadhäsion Thrombozytenaggregation
primäre Hämostase
Fibrinbildung
(plasmatische) Hämostase
6

Primäre Hämostase
7

Primäre Hämostase
• Definition
– Anlagerung (Adhäsion) und Zusammenlagerung (Aggregation) von Thrombozyten
• benötigt werden
– thrombogene Matrix (Kollagen)
– Thrombozyten
– Von-Willebrand-Faktor (vWF)
8

Funktion der Thrombozyten
• primäre Hämostase Thrombozytenadhäsion, Thrombozytenaggregation
• sekundäre (plasmatische) Hämostase katalytische Oberfläche für die Fibrinbildung (Phospholipide, „Flip-Flop-Mechanismus“)
• Infektabwehr (?)
9

Thrombozytenaktivierung
10

TXR PAR P2Y1 P2Y12 alpha-2
ADP ADP
Epinephrin Thrombin Thromboxan
GP
VI
GP
Ia-IIa
Kollagen
Thrombozytenaktivierung
Formenwechsel
(„Shape-Change“)
Externalisation,
Aktivierung und
Clustering von
Rezeptoren
Präsentation von
Phospholipiden
(„Flip-Flop“)
Freisetzung von
Mediatoren und
Gerinnungsfaktoren
11

Primäre Hämostase
• Definition
– Anlagerung (Adhäsion) und Zusammenlagerung (Aggregation) von Thrombozyten
• benötigt werden
– Kollagenmatrix
– Thrombozyten
– Von-Willebrand-Faktor (vWF)
12

D´ D3 A1 A2 D4 B1-B3 C1 C2
Faktor VIII-Bindung
A3
Thrombozytenadhäsion
GPIb-Bindung Kollagenbindung GPIIb-IIIa-Bindung
Thrombozytenaggregation
Von-Willebrand-Faktor, Monomer 2050 AS
Von-Willebrand-Faktor (vWF)
13

1 Dimer (540 kDa) 1 Monomer
große Multimere (10-20 MDa)
Dimer
4mer
8mer
16mer
32mer
64mer
Von-Willebrand-Faktor (vWF)
14

• Funktionen des von-Willebrand-Faktors:
– primäre Hämostase • Thrombozytenadhäsion
• Thrombozytenaggregation
– sekundäre Hämostase • Faktor VIII-Transport („Carrier-Funktion“)
Von-Willebrand-Faktor (vWF)
15

Primäre Hämostase
16

subendotheliale Matrix (Kollagen)
vWF
GP Ib-V-IX
GP VI GP Ia-IIa
GP IIb-IIIa
Fibrinogen/ vWF
GP IIb-IIIa
Aggregation
Adhäsion
17

Sekundäre (plasmatische) Hämostase
18

Sekundäre (plasmatische) Hämostase
• Definition
– Prozess der Fibrinbildung/ Fibrinstabilisierung
• benötigt werden
– Gerinnungsfaktoren
– Thrombozyten (katalytische Phospholipid-Oberfäche)
19

Sekundäre (plasmatische) Hämostase
• Definition
– Prozess der Fibrinbildung/ Fibrinstabilisierung
• benötigt werden
– Gerinnungsfaktoren
– Thrombozyten (katalytische Phospholipid-Oberfäche)
20

Gerinnungsfaktoren
• Eiweißmoleküle, die durch ein äußerst komplexes Zusammenspiel letztendlich die Bildung von Fibrin und die Stabilisierung von Fibrin bewirken
• „eigentliche“ Gerinnungsfaktoren
– XIII, XI, X, IX, VII, II, Fibrinogen (I)
• Kofaktoren der Gerinnung
– V, VIII:C
21

subendotheliale Matrix (Kollagen)
F VII
F VIIa
Thrombin
Prothrombin FSAP
Aktivierung
Fibrinogen
XIII
XIIIa
Verstärkung (Amplifikation)
F X
Fibrin
F V F VIII F IX F XI
22

Warum benötigt eine effektive Gerinnung sowohl Thrombozyten als auch Fibrin?
23

Stabile Mauer mit Backsteinen und Zement wie Thrombus aus Thrombozyten und Fibrin!
24

Antithrombotische Mechanismen
25

Antithrombotische Mechanismen
• Auflösung von Gerinnseln
– Fibrinolyse
• Beendigung des Gerinnungsprozesses
– Protein C-/Protein S-System
• Inaktivierung von Thrombin
– Antithrombin (AT)
26

Fibrinolyse
27

Fibrinolyse
• Definition
– physiologische Auflösung von Fibringerinnseln
– benötigt: • Plasminogenaktivatoren
• Plasminogen
28
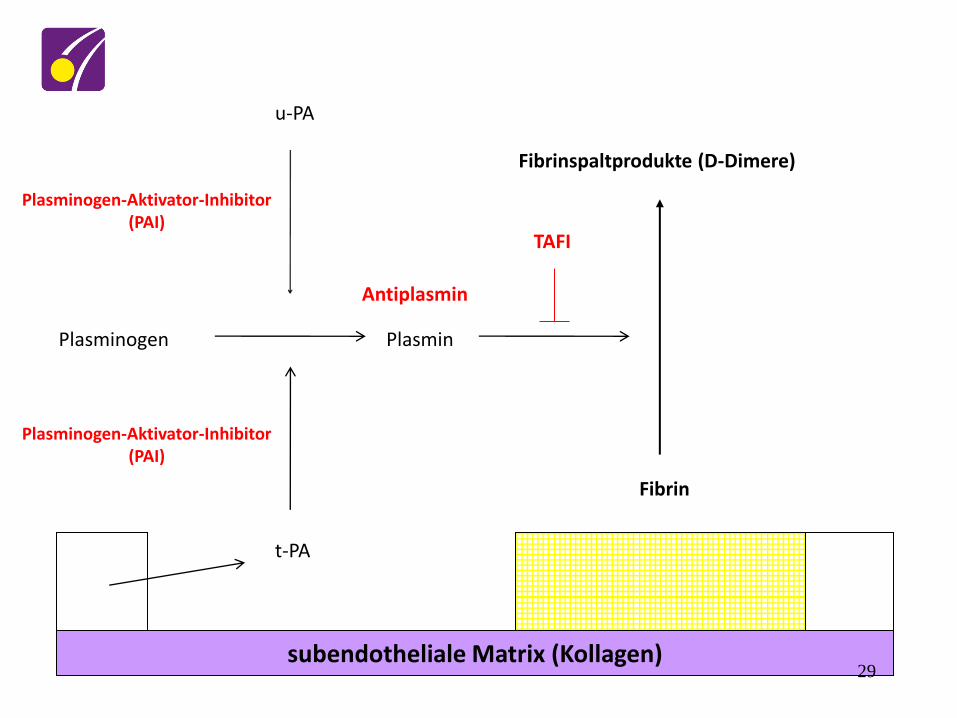
subendotheliale Matrix (Kollagen)
Fibrin
Fibrinspaltprodukte (D-Dimere)
t-PA
Plasminogen Plasmin
u-PA
TAFI
Antiplasmin
Plasminogen-Aktivator-Inhibitor (PAI)
Plasminogen-Aktivator-Inhibitor (PAI)
29

Protein C-/Protein S-System
30

Protein C-/Protein S-System
• das Protein C-/Protein S-System ist der „Ausschalter“ der Gerinnung
• durch Inaktivierung der Faktoren V und VIII:C wird die Amplifikation der Gerinnung unterbrochen und die Thrombinbildung bricht zusammen
31

Antithrombin
32

Antithrombin
• Antithrombin bindet überschüssiges Thrombin und hilft damit bei der Lokalisation der Gerinnung auf den Ort mit der höchsten Thrombinkonzentration
• Vorstellung:
– ohne Antithrombin gerinnt bei einer Verletzung das gesamte Blut, wenn genug Thrombin gebildet und mit dem Blutstrom verschleppt wird (Tiermodell)
33

subendotheliale Matrix (Kollagen)
Endothel
Endothel
subendotheliale Matrix (Kollagen)
Endothel
AT
TAT
AT
AT
TAT
AT
Thrombin
Blutfluss
34

Venöse Thromboembolie (VTE)
35

Thrombose
• Bildung eines Gerinnsels (Thrombus) in dem betroffenen Gefäß
• im Allgemeinen bezeichnet der Begriff „Thrombose“ eine Gerinnselbildung in einer Vene
• Thrombophilie bezeichnet im Allgemeinen die venöse Thromboseneigung
36

Häufigkeit von symptomatischen VTE
0
0,05
0,1
0,15
0,2
Glynn et al., 2007
Naess et al., 2007
White et al., 2005
Oger et al., 2000
Silverstein et al., 1998
Nordström et al., 1992
Anderson et al., 1991
(%/Jahr)
N=23,3 Mio. N=366
nach S3-Leitlinie, 2009 37

Risikofaktoren für die Thromboseentstehung
• „Virchow Trias“
– Veränderungen der Gefäßwand
• Arteriosklerose, Entzündung
– veränderte Strömungsgeschwindigkeit des Blutes
• Immobilisation, Gips, etc.
– Veränderungen der Blutbeschaffenheit
• thrombophile Risikofaktoren
Rudolf Virchow (1821-1902)
38

Thromboserisiko
• Disposition – Veranlagung, „intrinsisches Risiko“
• Exposition („Trigger“)
– situationsbedingtes Risiko, „extrinsisches“ Risiko
39

Dispositionelle Risikofaktoren für Thrombosen
• Thrombophilie
• Erkrankungen – Tumorerkrankungen – chronisch-entzündliche Erkrankungen (z.B. CED) – Nierenerkrankungen (nephrotisches Syndrom) – u. v. m.
• Medikamente – orale Kontrazeptiva, Hormon(ersatz)präparate
40

Expositionelle Risikofaktoren für Thrombosen („Trigger“)
• Immobilität – Operationen – Gipsverbände – Bettlägerigkeit
• Entzündungen
• Langstreckenreisen
41

Disposition, Exposition und Thrombose
„kritische Schwelle“ (Thrombose)
Grundrisiko
thrombophile Risikofaktoren
Risikosituation
Disposition
Exposition
Thromboserisiko
42

Disposition, Exposition und Thrombose
Disposition
Exposition
Disposition
Exposition
Disposition
Exposition „kritische Schwelle“ (Thrombose)
43

Thrombophilie
44

Thrombophilie
• genetisch bedingte (hereditäre) Thrombophilie – Faktor V Leiden-Mutation – Prothrombinmutation – Protein C-Mangel – Protein S-Mangel – Antithrombinmangel
– andere (PAI-Mutation, etc.)
• erworbene Thrombophilie
– Antiphospholipidsyndrom (APS)
– chronisch-entzündliche Reaktionen (z.B. CED)
95% der Ursachen einer genetisch-bedingten Thrombophilie
45

Faktor V Leiden-Mutation
• Ursache
– Punktmutation im Faktor V-Gen (G1691A) führt zu einem Aminosäureaustausch (Arg506Glu)
• Konsequenz
– die Inaktivierung von Faktor Va durch aktiviertes Protein C (aPC) wird beeinträchtigt („aPC-Resistenz [APCR])
– es kommt zu einer verlängerten/verstärkten Thrombinwirkung und somit zu einer Thromboseneigung
46

Faktor V Leiden-Mutation
• Epidemiologie
– häufigster genetisch determinierter thrombophiler Risikofaktor
– Normalbevölkerung: • 5-7% heterozygot
• 0,1-0,5% homozygot
• Risikosteigerung für venöse Thrombosen
– 5-7fach (heterozygot)
– 30-50fach (homozygot)
47

Prothrombinmutation
• Ursache
– Mutation in der 3‘-nicht-codierenden Region des Gens für Prothrombin (G20210A) führt zu einer Erhöhung des Plasmaprothrombinspiegels
• Konsequenz
– vermehrte Thrombinbildung führt zu einer Steigerung des Thromboserisikos
48

Prothrombinmutation
• Epidemiologie
– zweithäufigster genetisch determinierter thrombophiler Risikofaktor
– Normalbevölkerung: • 1-3 % heterozygot
• homozygot (?)
• Risikosteigerung für venöse Thrombosen
– 2-4fach (heterozygot)
49

Protein C-Mangel
• Ursache
– Mutationen im Protein C-Gen (2q13-q14), derzeit mehr als 200 bekannte Mutationen
– Verminderung (Typ 1) oder Funktionsstörung (Typ 2) von Protein C
• Konsequenz
– verminderte Inaktivierung von Faktor Va/VIIIa führt zu einer Steigerung des Thromboserisikos
50

Protein C-Mangel
• Epidemiologie
– seltener hereditärer Risikofaktor
– Normalbevölkerung: • 0,2-0,4 %
• Risikosteigerung für venöse Thrombosen
– variabel (je nach Defekt), median ca. 5-7fach (?)
51

Protein S-Mangel
• Ursache
– Mutation im Protein S-Gen (PROS1) auf Chromosom 3 (3q11.2), bislang sind mehr als 100 Mutationen beschrieben
– drei Typen: Typ 1 (Verminderung), Typ 2 (funktioneller Defekt), Typ 3 (funktioneller Defekt des freien Protein S)
• Konsequenz
– verminderte Inaktivierung von Faktor Va/ VIIIa führt zu einer Steigerung des Thromboserisikos
52

Protein S-Mangel
• Epidemiologie
– seltener hereditärer thrombophiler Risikofaktor
– Normalbevölkerung: • 0,03-0,13%
• Risikosteigerung für venöse Thrombosen
– variabel (je nach Defekt), median ca. 2-20fach
• CAVE: labiles Protein, häufige Fehldiagnosen (!)
53

Antithrombinmangel
• Ursache
– Mutationen im Bereich des Antithrombingens auf Chromosom 1 (1q23-25)
• Konsequenz
– Thromboseneigung durch mangelnde Inaktivierung von Thrombin (Thrombinämie)
54

Antithrombinmangel
• Epidemiologie
– schwerer seltener thrombophiler Risikofaktor
– Normalbevölkerung: • 0,02%
• Risikosteigerung für venöse Thrombosen
– variabel (je nach Schweregrad), bis zu 100-200fach
55
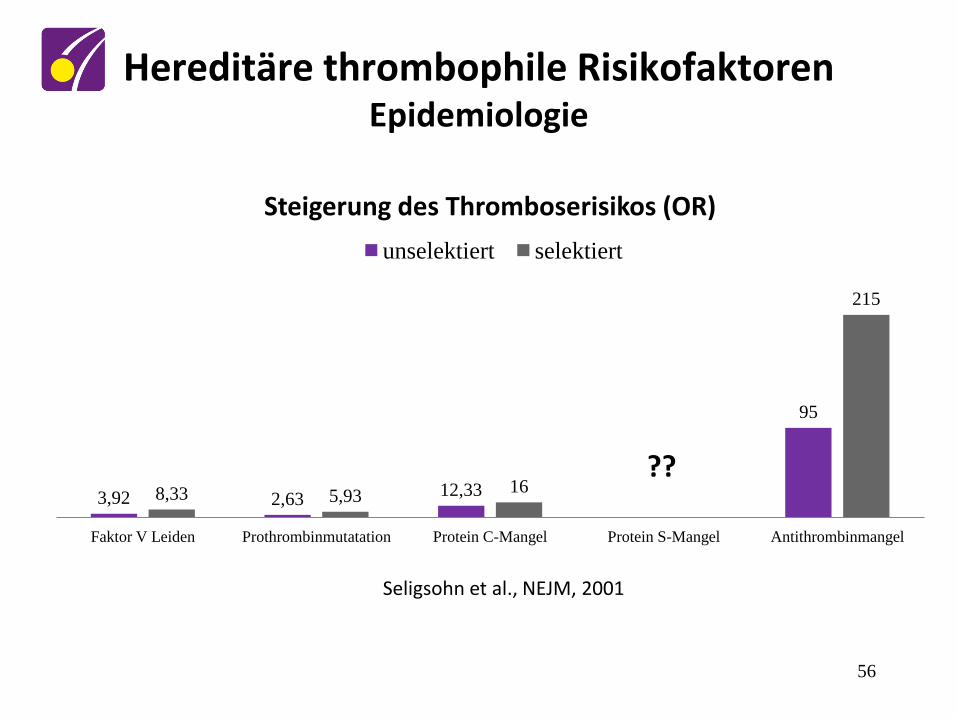
Seligsohn et al., NEJM, 2001
3,92 2,63 12,33
95
8,33 5,93 16
215
Faktor V Leiden Prothrombinmutatation Protein C-Mangel Protein S-Mangel Antithrombinmangel
Steigerung des Thromboserisikos (OR)
unselektiert selektiert
??
Hereditäre thrombophile Risikofaktoren Epidemiologie
56

Erworbene Thrombophilie
• entzündliche Reaktionen
– eine Entzündung führt zur Aktivierung der Gerinnung
– Entzündungen führen somit zu einem erhöhten Risiko für Thrombosen
• Antiphospholipidsyndrom
– erworbene Gerinnungsstörung • Neigung zu venösen (70%) und arteriellen Thrombosen (70%)
• Abortneigung, Schwangerschaftskomplikationen
57

Antiphospholipidsyndrom
• klinische Kriterien
– venöse Thrombosen
– arterielle Thrombosen
– Aborte, Schwangerschaftskomplikationen
• Laborkriterien
– Nachweis von „gerinnungsaktiven“ und „nicht-gerinnungsaktiven“ Antiphospholipidantikörpern
• Lupusantikoagulanzien
• Cardiolipin- und ß2-Glykoprotein I-Antikörper
58

Klinische Relevanz thrombophiler Risikofaktoren
• entscheidend für die Bewertung thrombophiler Risikofaktoren ist nicht das relative Risiko, sondern das absolute jährliche Risiko (z.B. %/Jahr)
• die klinische Relevanz nachgewiesener thrombophiler Risikofaktoren kann nur unter Berücksichtigung von klinischen Angaben bestimmt werden
59

Bewertung der Relevanz thrombophiler Risikofaktoren
• Eigenanamnese
– Thromboseneigung ?
• Familienanamnese
– Thromboseneigung ?
• Begleiterkrankungen
– thrombogene Wirkung ?
• Lebensalter
60

Lebensalter und Thromboserisiko
Lebensalter
Thromboserisiko
altersabhängiges Thromboserisiko
„kritische Schwelle“
zusätzliches expositionelles Risiko
zusätzliches expositionelles Risiko
0,01%/Jahr 0,1%/Jahr 1%/Jahr
61

Gerinnungshemmer (Antithrombotika)
62

Klassifikation von Antithrombotika
63

Antithrombotika
Thrombozytenfunktionshemmer Antikoagulanzien
Hemmung der Thrombozyten
Hemmung der Fibrinbildung
64

Thrombozytenfunktionshemmer
65

thrombogene Matrix
Arachidonsäure Thromboxan A2 Thromboxan A2
TXA2R P2Y1
PAR1
Thrombin
ADP
GP Ib-V-IX
GP IIb/IIIa
GP IIb/IIIa
Ticlopidin Clopidogrel Prasugrel Ticagrelor Cangrelor
(E5555)
Abciximab Eptifibatid Tirofiban
(andere Fibane)
(Terutroban)
(Tokaracetin)
Aspirin (Nitroaspirin)
66

Verfügbare Thrombozytenfunktionshemmer
• Thrombozytenfunktionshemmer
– Acetylsalicylsäure (Aspirin)
– Thienopyridine (Ticlopidin, Clopidogrel, Prasugrel)
– Cyclopentyltriazolopyrimidine (Ticagrelor)
• spezifische Thrombozytenaggregationshemmer
– Abciximab
– Eptifibatid
– Tirofiban
67

Acetylsalicylsäure (Aspirin)
• Wirkungsmechanismus
– Hemmung der thrombozytären Thromboxan-Synthese durch Inhibition der Cyclooxygenase 1 (COX-1)
• irreversible Blockade des katalytischen Zentrums von COX-1 durch Acetylierung eines Serinrestes (Serin 350)
68

COX-1
Arachidonsäure
Thromboxan Thromboxan
TXA2R
Plättchenaktivierung
ASS
69

Acetylsalicylsäure (Aspirin)
• Indikationen – Prophylaxe der arteriellen Thrombose
• koronare Herzkrankheit
• periphere arterielle Verschlusskrankheit
• Verschlusskrankheit der extrakraniellen Hirngefäße
– (Augenvenenthrombose ?)
– Andere
• Dosierung – 100 mg/d
70

Thienopyridine und Cyclopentyltriazolopyrimidine
71

Thienopyridine
• 1. Generation: – Ticlopidin (Tiklyd®, Desitic®)
• 2. Generation: – Clopidogrel (Plavix®, Iscover®, diverse Generika
[Besilate])
• 3. Generation: – Prasugrel (Efient®)
72

• 1. Generation: – Ticagrelor (Brilique®)
Cyclopentyltriazolopyrimidine
73

Thienopyridine und Cyclopentyltriazolopyrimidine
Wirkungsmechanismus
74

ADP-Rezeptor (P2Y12)
AC
cAMP
PKA
VASP VASP-P
Plättchen aktiviert Plättchen ruhend
ADP Thienopyridine (inaktiv)
Aktivierung
Cytochrome (Leber)
Cyclopentyltriazolopyrimidine (aktiv)
75

Antikoagulanzien
76

thrombogene Matrix
Faktor VII
Faktor VIIa Thrombin
Fibrin
Amplifikation
Faktor Xa
XI IX VIII:C
Faktor Va Antihrombin
Heparine Heparinoide
Hirudin Desirudin
Dabigatran Bivalirudin Argatroban
Fondaparinux Idraparinux Rivaroxaban (Apibaxan)
(Otamixaban) (Betrixaban) (LY517717)
(YM150) (DI-176b) (TAK442)
(DX-9065a)
(NAPc2) TFPI (Tifacogin)
rFVIIai
dTM (ART-123) Drotrecogin (aPC)
dTM (ART-123) Drotrecogin (aPC)
Faktor IX-Aptamere (TTP-889)
Kumarine Phenprocoumon Acenocoumarol
Warfarin (Coumadin)
77

Verfügbare Antikoagulanzien
• Kumarine (Vitamin K-Antagonisten) – Phenprocoumon, Acenocoumarol, Warfarin
(Coumadin)
• Heparine – niedermolekulare Heparin, unfraktionierte Heparine
• Heparinoide – Danaparoid
• Thrombininhibitoren – Lepirudin, Desirudin, Dabigatran, Argatroban,
Bivalirudin
• Xa-Inhibitoren – Fondaparinux, Rivaroxaban, Apixaban
78

Unterschiede der Antikoagulanzien (I)
• Wirkungsmechanismus – direkt/indirekt – Xa-Inhibitoren, Thrombininhibitoren, u. a.
• Applikationsform – subkutan (s. c.), intravenös (i. v.), peroral
• Elimination – renal, hepatisch
• Plazentagängigkeit, Übergang in die Muttermilch
79

Unterschiede der Antikoagulanzien (II)
• Nebenwirkungsspektrum
• Arzneimittelindikationen
• spezielle Indikationen – Therapie bei heparininduzierter Thrombozytopenie
(HIT) – Therapie in Schwangerschaft und Stillzeit
• Plazentagängigkeit, Übergang in die Muttermilch
• Zulassungsstatus
• Kosten
80

Das ideale Antikoagulans (I)
• schneller Wirkungseintritt, schnelles Abklingen
• große therapeutische Breite
• keine Kumulation bei Leber- und Niereninsuffizienz
• lineare Dosis-Wirkungs-Beziehung
• Monitoring möglich, aber nicht erforderlich
81

Das ideale Antikoagulans (II)
• oral verfügbar (gute Bioverfügbarkeit)
• keine Nebenwirkungen (Blutungsneigung, Allergie, HIT, Osteoporose, etc.)
• keine Interaktionen
• Antidot verfügbar
• kostengünstig
82

Das gibt es nicht !
83

Korrespondenzadresse
Priv.-Doz. Dr. med. habil. Christoph Sucker LaboMed Gerinnungszentrum Berlin
Tauentzienstrasse 7b/c 10789 Berlin
Telefon: 030-2128088-0
E-Mail: [email protected]
84
Related Documents











