F1-Laborpraktikum SS 2013 MOLEKULARE GENETIK DER EUKARYOTEN Institut für Molekulargenetik, gentechnologische Sicherheitsforschung und Beratung Johannes Gutenberg-Universität Mainz Leiter: Prof. Dr. Erwin R. Schmidt Betreuer: Dr. Christiane Kraemer, Dr. Steffen Rapp Dipl. Biol.s, Sarah Brunck, Sabine Fischer, Romina Petersen, Nicholas Bachtadse Tobias Lautwein, Dominik Otto Rudolf Baader, Carola Scholz, Nicole Naumann, Birte Ding, Haris Djozgic, Jens Hammann

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

F1-Laborpraktikum SS 2013
MOLEKULARE GENETIK
DER EUKARYOTEN
Institut für Molekulargenetik, gentechnologische Sicherheitsforschung und Beratung
Johannes Gutenberg-Universität Mainz
Leiter: Prof. Dr. Erwin R. Schmidt
Betreuer: Dr. Christiane Kraemer, Dr. Steffen Rapp
Dipl. Biol.s, Sarah Brunck, Sabine Fischer, Romina Petersen, Nicholas Bachtadse
Tobias Lautwein, Dominik Otto
Rudolf Baader, Carola Scholz, Nicole Naumann, Birte Ding, Haris Djozgic, Jens
Hammann


i
F1-Laborpraktikum SS 2013
Molekulare Genetik der Eukaryoten
Das FI-Praktikum "Molekulare Genetik der Eukaryoten" soll grundlegende molekulargenetische
Arbeitstechniken vermitteln, mit deren Hilfe ein erster Einblick in die Struktur und Funktionsweise der
Genome höherer Eukaryoten möglich wird. Im Vordergrund steht dabei die Analyse sogenannter
repetitiver DNA-Sequenzen. Diese Komponente des Genoms soll im Hinblick auf ihre Organisation, ihre
chromosomale Herkunft und letztlich ihre Primärstruktur (=DNA-Sequenz) untersucht werden.
Versuch 1:
Extraktion und Reinigung von DNA aus tierischen Geweben
a) DNA aus Zellen der Grünen Meerkatze (African Green Monkey)
b) DNA aus Mäusenieren/-leber und/oder Zellkulturzellen
c) DNA aus Chironomus thummi thummi-Larven
d) DNA aus menschlichen Zellen
Versuch 2:
Analyse von Eukaryoten-DNA mit Hilfe von Restriktionsendonukleasen
Bei diesem Versuch werden die isolierten DNAs aus den verschiedenen Quellen mit verschiedenen
Restriktionsenzymen verdaut und die entstandenen Fragmente auf Agarosegelen elektrophoretisch
aufgetrennt. Bei Verwendung von Enzymen, die eine Sequenz in einer tandem-repetitiven DNA-Fraktion
(z.B. Satelliten-DNA) erkennen, erscheint bei einer solchen Analyse ein deutlich sichtbares
Bandenmuster, das eine Bestimmung der Länge der Repetitionseinheit und der relativen Konzentration
der repetitiven DNA-Fraktion zulässt. Die hergestellten Gele werden für die "Southern- Analyse"
(Versuch 3) weiter verwendet.
Versuch 3:
Southern-Analyse repetitiver Satelliten-DNA
Die restringierte und elektrophoretisch aufgetrennte DNA aus Versuch 2 wird durch Transfer auf
Nitrozellulose-Membranen immobilisiert (Southern-Transfer). Die parallel zu Versuch 2 aus dem Gel
isolierte repetitive DNA (oder die isolierte Satelliten-DNA aus Versuch 4) wird radioaktiv markiert. Mit
dieser DNA als Sonde wird anschließend der "Southern-Filter" hybridisiert. Durch anschließende
Autoradiografie wird das Restriktionsmuster der Satelliten-DNA spezifisch und mit hoher
Empfindlichkeit dargestellt.
Versuch 4:
Lokalisation von Genen und tandem-repetitiver DNA durch nichtradioaktive in situ
Hybridisierung
Die aus genomischer DNA isolierten tandem-repetitiven Sequenzen sowie bereitgestellte „single copy“-
Gene werden nicht-radioaktiv markiert und durch in situ Hybridisierung an Speicheldrüsen-
Polytänchromosomen lokalisiert.

ii
wichtige Hinweise:
Während des Praktikums wird u.a. ein gentechnisches Experiment der Sicherheitsstufe S1
durchgeführt. Aus diesem Grund ist es notwendig, dass alle Teilnehmer einen
Kittel mitbringen!!!
Für den Versuch Nr. 4 werden Chromosomenpräparate von Chironomus thummi angefertigt - für
diesen Versuch bitte unbedingt
Präparierbesteck mitbringen!
(insbesondere Präpariernadeln und feine Pinzette)
Viele Substanzen – wie beispielsweise die am ersten Tag isolierte DNA - werden während
des Praktikums immer wieder einmal benötigt. Aus diesem Grund ist eine ausreichende
und dauerhafte Kennzeichnung der verwendeten Reaktionsgefäße sehr wichtig! Hierfür
stehen sowohl „Edding-Stifte“ (bitte nur dicke, schwarze verwenden!) und Klebeetiketten
zur Verfügung. Im Zweifel bitte die Betreuer fragen, wie die Gefäße beschriftet werden
sollen!
Literatur:
Gentechnische Methoden
Eine Sammlung von Arbeitsanleitungen für das molekularbiologische Labor
Schrimpf, Gangolf (Hrsg.)
ISBN: 3-8274-1103-3
3. Aufl. 2002, 512 Seiten, 29,50 Euro
Gentechnologie für Einsteiger
Brown, T. A.
ISBN: 3-8274-2868-8
6. Aufl. 2011, 298 Seiten, 36,95 Euro

iii
Inhaltsverzeichnis
ZEITPLAN ................................................................................................................................... iv
Versuch 1: DNA-Isolation aus tierischen Geweben bzw. Zellkulturen .................................... 1
Versuch 2: Analyse von Eukaryoten-DNA mit Hilfe von Restriktionsendonukleasen .......... 4
Versuch 3: Southern-Analyse repetitiver Satelliten-DNA ....................................................... 13
Versuch 4: Lokalisierung von DNA-Sequenzen durch in situ Hybridisierung an
Polytänchromosomen ............................................................................................... 17
ANHANG 21
1. Puffer und Lösungen: ......................................................................................................... 21
2. Molekulargewichtsstandards für die Gelelektrophorese: ............................................... 24
3. Aufbau Southern Blot ......................................................................................................... 25
4. Restriktionskarte und Multiple Klonierungsschnittstelle von pUC19 und pUC19
........................................................................................... Fehler! Textmarke nicht definiert.
5. Anleitung für den Umgang mit 32
P .................................................................................... 26
6. Einige Tipps und Einheiten ................................................................................................ 30

iv
ZEITPLAN (nicht bindend; Änderungen werden im Kurs bekannt gegeben)
1. Tag Mo. 08.07.2013
DNA-Isolierung & Reinigung (Versuch 1)
Restriktionen (analytisch/präparativ) (Versuch 2)
evtl. präparative Gele und/oder Probegele gießen (Versuch 2)
2. Tag: Di. 09.07.2013
Fällung der restringierten DNA (Versuch 2)
Gelelektrophorese (analytisch und präparativ) (Versuch 2)
Elektroelution von DNA-Fragmenten, Fällung ü.N. (Versuch 2)
Southern Blot aufbauen (Versuch 3)
Probegele vorbereiten (Versuch 2/3)
3. Tag: Mi. 10.07.2013
eluierte DNA-Fragmente lösen und testen (Versuch 2/3)
Sonden-Labelling (radioaktiv) (Versuch 3)
Chromosomenpräparate für FISH (Versuch 4)
evtl. Sondenmarkierung für FISH (Versuch 4)
4. Tag: Do. 11.07.2013
Vorbereitung der Präparate und in situ-Hybridisierung (Versuch 4)
Hybridisierung Southern Blot (Versuch 3)
5. Tag: Fr. 12.07.2013
in situ Hybridisierung – Detektion & Auswertung (Versuch 4)
Hybridisierung abbrechen, Southern Blot waschen & exponieren (Versuch 3)
Autoradiogramme entwickeln (Versuch 3)

Versuch 1. S.1
Versuch 1:
DNA-Isolation aus tierischen Geweben bzw. Zellkulturen
Die DNA-Isolierung erfolgt in folgenden Schritten:
• Gewebeaufschluss
• Kernisolation
• Kernlyse
• Extraktion der Proteine
• Ethanol-Fällung der DNA
• Lösen der DNA
A. Maus-Gewebe bzw. Chironomus-Larven
1) 1-5 g Gewebe/Larven in Potter (auf Eis) überführen
2) pro g Gewebe 2 ml "Homogenisierungspuffer" hinzufügen (eiskalt),
1/10 Vol. 10% Triton-X-100 zugeben
3) unter Eiskühlung homogenisieren
4) optional, bei Chironomus Larven zwingend: Homogenat durch Gaze filtrieren und mit
1/2 Vol. Homogenisierungspuffer Glas und Gaze nachspülen
5) Filtrat in großen Glaszentrifugentuben 15 min, 5.000 Upm, 4°C zentrifugieren
6) Überstand sauber abheben, verwerfen und Sediment in Homogenisierungspuffer resuspendieren,
zentrifugieren (wie 5), diesen Schritt gegebenenfalls ca. 3x wiederholen bis Kern-Pellet „sauber“
erscheint
7) das Kern-Pellet in etwa 1/10 des Ausgangsvolumens eiskaltem Homogenisierungspuffer
resuspendieren (bei Maus-Leber meist ca. 10-15 ml; bei 5 g Chironomus-Larven meist ca. 2 ml;
Betreuer fragen!); die Suspension in 15 ml oder 50 ml Falcon-Röhrchen überführen
8) Proteinase K (eine Spatelspitze) hinzufügen
9) 1/10 Vol. 10% SDS und 1/10 Vol. 10x Dialysepuffer zur Kernlyse hinzufügen – durch Invertieren
vorsichtig mischen (Lösung sollte viskös werden! Warum??)
10) 1-2 Std. (evtl. auch über Nacht) bei 60°C inkubieren (Kernlyse)
11) Inkubationsgemisch auf Raumtemperatur abkühlen (optional)
12) 0,1 Vol. gesättigtes Tris, pH 8,5 hinzufügen (zur pH-Einstellung!)
13) 0,25 Vol. 5 M NaClO4 (Natriumperchlorat) hinzufügen, gut invertieren (es bildet sich ein
weißlicher Niederschlag) 14) gleiches Vol. Phenol/Chloroform/Isoamylalkohol-Gemisch (25:24:1) hinzufügen, mind. 5 min. durch Invertieren gut mischen
15) 10 min., 5000 Upm bei RT zentrifugieren
16) wässrige Phase (immer „oben“!) ohne das Interphase-Material mit abgeschnittener blauer
Pipettenspitze absaugen und mit dieser Phase (enthält die DNA) Schritte 14 + 15 ca. 3 bis 5x
wiederholen, bis die Interphase sauber ist (Betreuer fragen!)
17) zuletzt die DNA-Phase 1x mit gleichem Vol. Chloroform-Isoamylalkohol (24:1) versetzen,
extrahieren (d.h. mind. 5 min invertieren) und zentrifugieren (zur Entfernung sämtlicher
Phenolreste aus der DNA-Phase)
18) nach dem Zentrifugieren obere, wässrige Phase mit einer "weiten" Pipette (abgesägt) ohne
Interphase-Material abheben und in vorgekühltes 50 ml-Falcon überführen - in Eis abkühlen lassen
19) noch einmal 1/10 Vol. 10x Dialysepuffer zugeben und mischen, dann mit 2 Vol. kaltem Ethanol
abs. vorsichtig überschichten

Versuch 1. S.2
20) DNA auf sterilisiertem Glasstab aufwickeln (mind. 5 min in dieselbe Richtung drehen)
21) aufgespindelte DNA in 70% Ethanol (5-10 min) stehen lassen (Auswaschen überschüssiger Salze)
22) vor Abwickeln der DNA Glasstab kurz antrocknen, dann DNA in 0,5 – 2 ml HPLC-H2O
"abspulen" und lösen lassen (abhängig von der aufgespulten Menge zunächst in 0,5 bis
1 ml (Chironomuslarven) bzw. 2 ml (Mausleber) abwickeln – Betreuer fragen!)
B. Zellkultur-Zellpellets
(mechanischer Gewebeaufschluss und Kernisolation entfallen)
1) Pellets mit 3 ml Homogenisierungspuffer (eiskalt!!) versehen, vorsichtig mit Pipette
resuspendieren, in 15 ml Falcon-Plastiktube abzentrifugieren (3.500 Upm, 5 min, 4°C).
2) Überstand abheben, zu Pellet ca. 2 ml "Homogenisierungspuffer" hinzufügen (eiskalt)
3) Proteinase K (eine Spatelspitze) hinzufügen
4) 1/10 Vol. 10%SDS und 1/10 Vol. 10x Dialysepuffer zur Zelllyse hinzufügen - durch Invertieren
vorsichtig mischen (Lösung sollte viskös werden! Warum??)
5) 1 - 2 Std. (evtl. auch über Nacht) bei 60°C inkubieren
6) Inkubationsgemisch kurz auf Raumtemperatur abkühlen (optional)
7) 1/10 Vol. gesättigtes Tris, pH 8,5 hinzufügen (zur pH-Einstellung!) 8) 1/4 Vol. 5 M NaClO4 hinzufügen, gut invertieren (es bildet sich ein weißlicher Niederschlag)
9) gleiches Vol. Phenol/Chloroform/Isoamylalkohol- Gemisch (25:24:1) hinzufügen, mind. 5 min.
durch Invertieren gut mischen
10) 10 min., 5000 Upm bei RT zentrifugieren
11) wässrige Phase (immer „oben“!) ohne das Interphase-Material mit abgeschnittener blauer
Pipettenspitze absaugen und mit dieser Phase (enthält die DNA) Schritte 9 + 10 ca. 3 bis 5x
wiederholen, bis die Interphase sauber ist (Betreuer fragen!)
12) zuletzt einmal die DNA-Phase mit gleichem Vol. Chloroform-Isoamylalkohol (24:1) versetzen,
extrahieren (d.h. mind. 5 min invertieren), abzentrifugieren (zur Entfernung sämtlicher Phenolreste
aus der DNA-Phase!)
13) nach dem Zentrifugieren obere, wässrige Phase mit einer "weiten" Pipette (abgesägt) ohne
Interphase-Material abheben und in vorgekühltes 50 ml-Falcon überführen - in Eis abkühlen lassen
14) 1/10 Vol. 10x Dialysepuffer zugeben und vorsichtig invertieren, dann mit 2 Vol. kaltem Ethanol
abs. vorsichtig überschichten
15) DNA auf sterilisierten Glasstab aufwickeln (mind. 5 min in dieselbe Richtung drehen)
16) aufgespindelte DNA kurz in 70% Ethanol stehen lassen (Auswaschen überschüssiger Salze)
17) vor Abwickeln der DNA Glasstab kurz antrocknen, dann DNA in 0,5 – 2 ml HPLC-H2O
"abspulen" und lösen lassen (abhängig von der aufgespulten Menge zunächst in 0,5 bis
1 ml abwickeln)
Sollte sich die DNA nicht aufwickeln lassen (Grund: Molekulargewicht zu klein durch Degradation
während der Aufarbeitung; DNA-Konzentration zu gering), kann die DNA auch durch Mischen von
Ethanol und wässriger Phase gefällt werden. Allerdings sind damit der Reinheitsgrad der DNA sowie das
mittlere Molekulargewicht sehr viel geringer. Es muss dann unter Umständen die RNA enzymatisch
entfernt werden.

Versuch 1. S.3
• Prüfung der Reinheit der DNA:
Das UV-Absorptionsspektrum gibt in gewissem Umfang Auskunft über die Reinheit der DNA. Da
Proteine andere Absorptionsminima/-maxima aufweisen als Nukleinsäuren, lässt sich eine
Verunreinigung der DNA mit Proteinen mit Hilfe des UV-Absorptionsspektrums feststellen. Bei
"sauberer" DNA sollte das Verhältnis
E260nm/280nm = 1,8
sein.
Aufgabe:
Bestimmen Sie photometrisch das E260/280-Absorptionsverhältnis!
• Quantifizierung der DNA mittels Photometrie:
Zur Bestimmung der Konzentration der DNA-Lösung werden je 100 µl einer 1:10- und einer 1:100-
Verdünnung mit Wasser hergestellt und in eine Küvette gegeben. Bestimmt wird der E260-Wert (Wasser
in Referenzküvette).
Zur Ergänzung:
Konz. DNA (µg/ml) = E260 x 47 x Verdünnungsfaktor
Konz. RNA (µg/ml) = E260 x 40 x Verdünnungsfaktor
Konz. Primer (µg/ml) = E260 x 33 x Verdünnungsfaktor
ACHTUNG:
Die genomische DNA wird später für weitere Versuche verwendet: Bitte denken Sie daran, die
gelöste DNA sauber und sorgfältig zu beschriften. Hierzu stehen Ihnen auch Klebeetiketten zur
Verfügung. Beispiel für die Beschriftung:
genom. Maus-DNA (Gr. Ma1) Konz.: 650 µg/ml OD260/OD280: 1,78
12.02.2013

Versuch 2. S.4
Versuch 2:
Analyse von Eukaryoten-DNA
mit Hilfe von Restriktionsendonukleasen
Die aus den verschiedenen Organismen isolierte DNA soll durch Restriktionsanalyse näher charakterisiert
werden. Restriktionsenzyme schneiden die DNA an spezifischen Basensequenzen und zerlegen sie in eine
Vielzahl unterschiedlich langer Fragmente. Diese Restriktionsfragmente werden auf Agarosegelen
elektrophoretisch aufgetrennt und nach Färbung der DNA mit Ethidiumbromid oder SYBR Green
sichtbar gemacht. Enzyme, die eine Sequenz innerhalb einer tandem-repetitiven DNA (z.B. Satelliten-
DNA) erkennen, erzeugen nach Gelelektrophorese ein deutlich sichtbares Bandenmuster, aus dem sich
die Länge der Repetitionseinheit und ihre relative Konzentration innerhalb des Genoms ermitteln lässt.
Aufgabe:
Die Gruppen sollen durch Restriktion mit den Enzymen Cla I (Ch. thummi-DNA; ACHTUNG: Zugabe
von BSA), Sau96I (Maus-DNA), Eco RI (menschliche DNA) und Hind III (Affen-DNA) repetitive DNA-
Sequenzfamilien aus dem jeweiligen Genom herausschneiden, durch Agarosegelelektrophorese
auftrennen und das Molekulargewicht der repetitiven Einheiten bestimmen.
Achtung: Die elektrophoretisch aufgetrennte DNA wird für den Versuch 3 (Southern-Analyse) weiter
verwendet.
Durchführung:
A1. Analytische Restriktionen der isolierten DNA
Jede Gruppe pipettiert folgende Ansätze in beschriftete Eppendorfgefäße:
1 2 3 4 3-5 µg DNA 3-5 µg DNA 3-5 µg DNA 3-5 µg DNA
auf 27µl mit HPLC-H2O
auffüllen auf 26µl mit HPLC-H2O
auffüllen auf 26µl mit HPLC-H2O
auffüllen auf 26µl mit HPLC-H2O
auffüllen + 3 µl 10x Puffer + 3 µl 10x Puffer + 3 µl 10x Puffer + 3 µl 10x Puffer ohne Enzym (unrestringierte
Kontrolle)
+ 1 µl Enzym mix, kurz anzentr.
+ 1 µl Enzym mix, kurz anzentr.
+ 1 µl Enzym mix, kurz anzentr.
bei -20°C aufbewahren
15 min Inkubation bei 37°C
1 h Inkubation bei 37°C
1 h Inkubation bei 37°C
einfrieren
einfrieren + 1 µl Enzym ü.N. bei 37°C
einfrieren
Achtung:
Enzyme stets in Eis stellen, Pipettieren der Enzyme nur mit sauberen, sterilen Eppendorfspitzen;
Betreuer fragen!
Vorbereitung für die Versuche 3 und 5:
• Für Versuch 3 müssen repetitive DNA-Fragmente als „Sonde“ hergestellt werden.
• In Versuch 5 sollen repetitive DNA-Fragmente, die durch Restriktion aus den jeweiligen Genomen
herausgeschnitten worden sind, in Plasmid-Vektoren kloniert werden. Für beide Versuche ist es
erforderlich, eine große Menge genomischer DNA zu restringieren, elektrophoretisch aufzutrennen,
Banden repetitiver Fragmente aus dem Gel auszuschneiden und die DNA aus dem Gelstreifen
wiederzugewinnen.

Versuch 2. S.5
Vor dem Auftragen auf das Gel sollten die analytischen Restriktionsansätze noch gefällt werden!
Restriktionsansatz (30 µl) + 150 µl HPLC-H2O
+ 20 µl 10x Dialysepuffer, gut mischen
+ 400 µl EtOH abs. (kalt), gut mischen
mind. 30 min -20°C
30 min bei 4°C zentrifugieren
Überstand vorsichtig(!) abziehen
+ 400 µl 70 % EtOH (Pellet nicht resuspendieren!)
10 min bei RT zentrifugieren
Überstand vorsichtig abziehen, DNA kurz trocknen lassen (bis kein Alkohol mehr zu riechen ist)
Pellet in 20 µl HPLC-H2O lösen
Durchführung:
A2. Präparative Restriktionsansätze
- ca. 30 µg DNA mit HPLC-H2O auf 135 µl auffüllen
- + 15 µl 10x Puffer
- + 2 µl Enzym
- 2 h 37°C inkubieren
- + 1 µl Enzym
- ü.N. 37°C inkubieren
Vor dem Auftragen auf das Gel muss der präparative Restriktionsansatz noch gefällt werden!
Restriktionsansatz auffüllen auf 180 µl mit HPLC-H2O
+ 20 µl 10x Dialysepuffer, gut mischen
+ 400 µl EtOH abs. (kalt), gut mischen
mind. 30 min -20°C
30 min bei 4°C zentrifugieren
Überstand vorsichtig(!) abziehen
+ 400 µl 70% EtOH (Pellet nicht resuspendieren!)
10 min bei RT zentrifugieren
Überstand vorsichtig abziehen, DNA kurz trocknen lassen (bis kein Alkohol mehr zu riechen ist)
Pellet in 50 µl HPLC- H2O lösen
B. Agarosegelelektrophorese der DNA-Restriktionsfragmente:
Durch Agarosegelelektrophorese werden DNA-Fragmente nach ihrem Molekulargewicht aufgetrennt. Die
DNA kann im Gel durch den interkalierenden Fluoreszenzfarbstoff Ethidiumbromid unter UV-Licht
(312 nm) direkt sichtbar gemacht werden.
In diesem Praktikum wird mit zwei unterschiedlichen Gelkammer-Systemen gearbeitet. Die analytischen
Restriktionen und die präparative Restriktion der Genom-DNA werden auf „großen“ (ca. 13x18 cm)
Agarosegelen aufgetrennt, die von den Praktikanten selbst zusammengebaut werden. Alle folgenden
Gelelektrophoresen (Test- und Probegele, Test der Plasmid-DNA-Restriktionen etc.) werden mit Hilfe
des neuartigen Gelkammersystems der Firma GENterprise durchgeführt.

Versuch 2. S.6
B1. Gelelektrophorese mit „großen“ Agarosegelen:
a) Vorbereitung einer „großen“ Elektrophoresekammer (Demonstration durch Betreuer)
b) Vorbereitung der Restriktionsansätze zum Auftragen auf das Gel:
Jede der insgesamt 4 analytischen Proben einer Gruppe wird mit 5 µl Bromphenolblau-Gemisch
(Ladepuffer) versetzt, gemischt und kurz anzentrifugiert. Der präparative Restriktionsansatz wird mit
10 µl Bromphenolblau-Gemisch (Ladepuffer) versetzt, ebenfalls gemischt und kurz anzentrifugiert.
c) Herstellung eines 1,4% (oder 1,6%) (w/v) -Agarosegels:
- 1,4 g (bzw. 1,6 g) Agarose in 250 ml Erlenmeyer-Kolben einwiegen
- 100 ml 1x TBE zugeben
- in Mikrowellengerät bis zum Kochen erhitzen, ggfs. mit VE-H2O auf 100 ml auffüllen
- mit Pasteurpipette um vorbereitete Vertikalelektrophorese-Kammer herum einen "Sockel" gießen (zur
Abdichtung der Elektrophoresekammer)
- Gellösung auf ca. 60°C abkühlen lassen, dann luftblasenfrei mit Pipette einfüllen
- während der Abkühlungsphase Agaroselösung nach Bedarf nachfüllen!
- nachdem die Agarose erstarrt ist, taschenformenden Kamm vorsichtig aus dem Gel ziehen (Betreuer
fragen!) und Probentaschen ausspülen
- Gelkammer in unteres Pufferreservoir stellen, beide Reservoirs mit 1x TBE füllen
d) Elektrophorese und Ethidiumbromid-Färbung
- Netzgerät an Gelkammer anschließen (Polung!?)
- vor dem Auftragen der Proben kurz prüfen, ob Strom durch das Gel fließt (Netzgerät kurz einschalten)
- Auftragen der Proben auf das Gel (mit Eppendorfpipette); Reihenfolge der Beladung protokollieren!
- Zusätzlich wird eine Spur mit Hind III-restringierter Lambda-DNA und der „100 Bp ladder plus“
(MW-Standard)
- Elektrophorese bei 30 mA (Dauer ca. 2,5 Std.)
- Elektrophorese beenden, wenn Bromphenolblaufront ca. 3 cm vom unteren Gelrand entfernt ist
- Netzgerät abschalten, Kabelverbindungen lösen
- Pufferkammern entleeren, Klammern lösen
- hintere Plexiglasplatte (mit oberer Pufferkammer) abheben und Gel auf vorderer Platte liegend in
Glaswanne überführen
- Ethidiumbromid-Färbelösung (5 µg/ml) zugeben, Gel ca. 5-10 min färben, 2 min wässern
Achtung:
EtBr ist mutagen! Alles, was mit diesem Stoff in Berührung kommt, nur mit Einweghandschuhen
aus Nitril anfassen!
- Gel auf Platte liegend herausnehmen, kurz in VE-H2O wässern (in Plastikschale); DNA-Banden im
UV-Durchlicht (312 nm) fotografieren
Achtung:
Nicht direkt in das UV-Licht hineinschauen und UV-undurchlässige Schutzbrillen tragen oder
hinter Glas arbeiten!
Gele werden für Versuch 3 direkt weiterverwendet!!!
Auswertung:
Bestimmen Sie mit Hilfe des Molekulargewichtsstandards die Größe der genomischen DNA-Banden!
Hierzu wird die Größe der Standardbanden logarithmisch gegen die Wanderungsstrecke (in mm)
aufgetragen (Eichkurve auf halblogarithmischem Papier anfertigen!).
Lässt sich eine Kinetik bezüglich der Enzymwirkung erkennen?
Welches Molekulargewicht haben die repetitiven Einheiten der Eukaryoten-DNA?
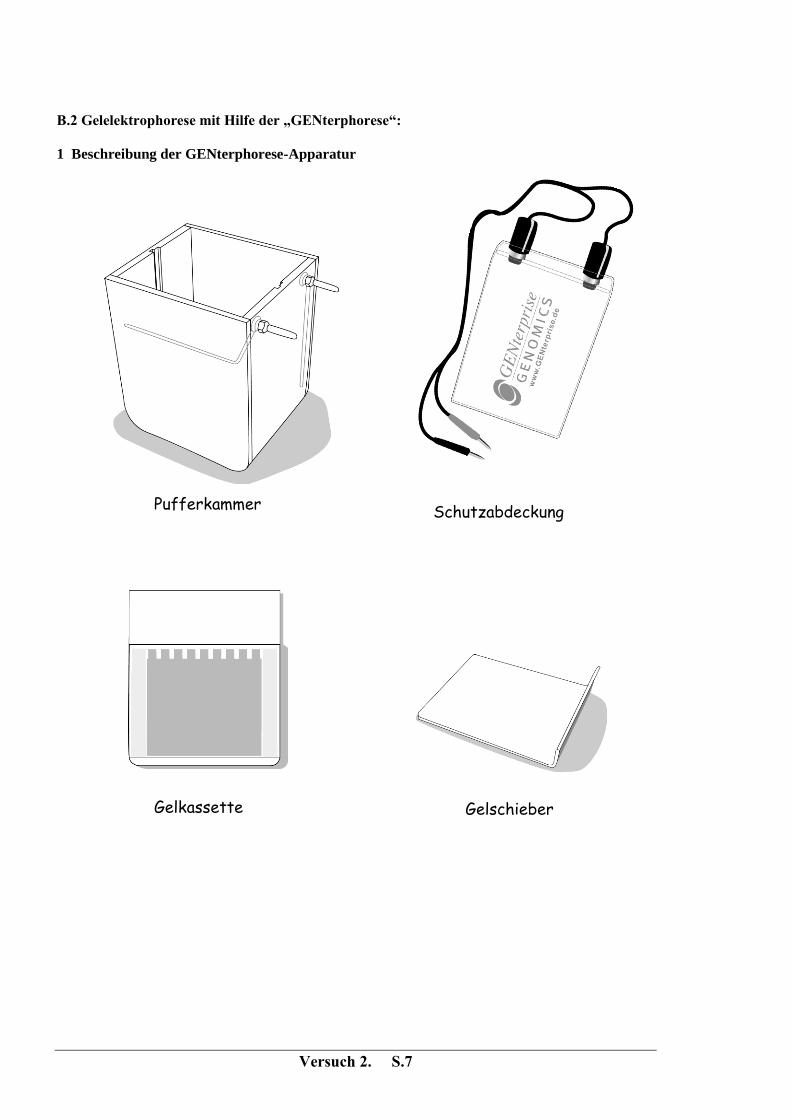
Versuch 2. S.7
B.2 Gelelektrophorese mit Hilfe der „GENterphorese“:
1 Beschreibung der GENterphorese-Apparatur
Gelschieber
Gelkassette
Schutzabdeckung Pufferkammer
Gelkassette

Versuch 2. S.8
2 Vorbereitung der GENterphorese-Apparatur
1. Füllen Sie die Kammer mit 1 x TBE-Laufpuffer bis etwa 3 mm über
die obere Elektrode (Kathode)
2. Setzen Sie das Gel über die Führungsnut in die Kammer ein und
drücken Sie es nach unten (Abb.). Wenn die Anschlüsse der Kammer
nach rechts zeigen, muss sich die kurze Platte der Gelkassette vorne
befinden.
3. Schieben Sie den GENterphorese-Deckel auf die Pufferkammer
4. Verbinden Sie den roten Stecker mit dem roten Anschluss des
Spannungsgerätes und den schwarzen Stecker mit dem schwarzen
Anschluss. Prüfen Sie vor dem Auftragen der Proben, ob Strom
(Einheit: mA) durch das Gel fließt (Netzgerät kurz einschalten;
Betreuer fragen!).
5. Spannungsgerät ausschalten und Deckel entfernen.

Versuch 2. S.9
3 Durchführung der Elektrophorese
1. Die mit Ladepuffer versetzten Proben werden vollständig auf ein 1,4 oder 1,6%iges Agarose-
TBE-Gel aufgetragen (vorzugsweise in der Mitte des Gels). Zusätzlich wird eine Spur mit
Hind III-restringierter Lambda-DNA und einem weiteren Molekulargewichtsstandard für kleine
DNA-Fragmente (z.B. 100 bp-Leiter) beladen.
Achtung!
Auftragsreihenfolge protokollieren!
2. Deckel der GENterphorese-Kammer schließen und Spannungsgerät einschalten.
3. Elektrophorese bei 100 V (Dauer ca. 20 min);
Hinweis: an ein Spannungsgerät können mehrere Gele parallel angeschlossen werden!
4. Während die Elektrophorese läuft, gießen Sie selbst ein Agarosegel (vgl. 4).
5. Elektrophorese durch Ausschalten des Spannungsgerätes beenden, wenn die
Bromphenolblaufront ca. 2-3 cm vom unteren Gelrand entfernt ist. Kabelverbindungen lösen.
6. Gelkassette aus der Pufferkammer ziehen, Puffer in der Pufferkammer belassen (kann mehrfach
verwendet werden).
7. Gel mithilfe des Schiebers aus der Gelkassette herausschieben und dabei
auf eine bereitgestellte Plexiglasplatte gleiten lassen.
8. Gel mit der Plexiglasplatte in eine Schale mit Ethidiumbromid-Färbelösung
(5 µg/ml) legen, Gel ca. 5-10 min färben, 2 min wässern
Achtung:
EtBr ist mutagen ! Alles, was mit diesem Stoff in Berührung kommt, nur
mit Einweghandschuhen aus Nitril anfassen!
9. Gel auf Platte liegend herausnehmen, kurz in VE-H2O wässern
(in Plastikschale);
DNA-Banden im UV-Durchlicht (312 nm) fotografieren
Achtung:
Nicht direkt in das UV-Licht hineinschauen und UV-undurchlässige Schutzbrillen tragen oder
hinter Glas arbeiten!

Versuch 2. S.10
4 Eigene Herstellung von Agarosegelen
1. Herstellen einer 1,4%igen Agarose-Lösung: Für die von uns verwendeten Gelkassetten wird eine
Gelmenge von ca. 20 ml benötigt. Dazu wird 0,28 g Agarose abgewogen und in einen kleinen
Erlenmeyer-Kolben gegeben. Es werden 20 ml 1xTBE-Puffer (NICHT WASSER!!!)
hinzugegeben.
2. Wiegen Sie jetzt ihren Erlenmeyer-Kolben und notieren Sie das Gewicht: _______g.
3. Der Ansatz wird in der Mikrowelle so lange aufgekocht (Achtung, kocht leicht über!), bis in der
Agarose-Lösung keine Partikel oder Schlieren mehr zu sehen sind.
4. Wiegen Sie anschließend Ihren Kolben nochmals und füllen Sie das Gefäß auf das unter 2.
notierte Gewicht mit VE-H2O auf.
5. Gießen der Gellösung: Stellen Sie die Gel-Kassette in die Gelgießvorrichtung und stecken Sie
anschließend den Kamm gerade so tief zwischen die beiden Plexiglasplatten, bis die Taschen des
Kamms komplett ‚verschwunden’ sind.
6. Sobald Sie den Erlenmeyerkolben schmerzlos anfassen können (bei ca. 60°C), beginnen Sie
damit, mit der Pasteurpipette einen ca. 1 cm breiten Sockel zwischen die Plexiglasplatten zu
gießen. Sobald dieser Sockel fest geworden ist (dies ist nach ca. 5 min der Fall) füllen Sie den
Zwischenraum der Gelplatten vollständig mit der noch flüssigen Agaroselösung auf. Sollte die
Agarose im Glas mittlerweile ebenfalls fest geworden sein, kochen Sie diese nochmals kurz in
der Mikrowelle auf.
7. Nach ca. 15 min sollte das Gel ausgehärtet sein (erkennbar an der Trübung des Gels). Die
gefüllte Gel-Kassette lassen Sie dann in der Gießvorrichtung stehen. Bitte lassen Sie den Kamm
im Gel stecken; die Betreuer werden Ihre hergestellten Gele für die weitere Verwendung
archivieren. Die so hergestellten Gele können in Folie eingeschweißt und im Kühlschrank über
mehrere Wochen zur späteren Benutzung gelagert werden. In der Praxis kann daher mit einem
Vorrat an Gelen sehr schnell eine Gelelektrophorese durchgeführt werden, ohne dass jedes Mal
ein Gel neu hergestellt werden muss.
Gelgießständer
Gelkamm
Agarosefront

Versuch 2. S.11
C. Ausschneiden der DNA-Banden auf dem UV-Transilluminator
- Schutzbrille tragen!
- UV-Tisch auf 70% Leistung stellen
- mit einem Skalpell Gelstreifen ausschneiden und diesen mit einer Pinzette in zuvor beschriftete
Eppendorfgefäße geben
D1. Wiedergewinnung von DNA aus Agarose-Gelen durch Elektroelelution
- gewünschte Bande mit Skalpell ausschneiden
- Gel-Stückchen in ca. 5 cm langen Dialyseschlauch einfüllen
- 300 µl TBE-Puffer (1x) hinzufügen, Luftblasen nach oben herausdrücken
- Schläuche luftblasenfrei mit "Spektrum-Clips" verschließen
- in Elektrophoreseschale mit zwei Elektroden so einlegen, dass das Gelstückchen auf der
Kathodenseite (-) liegt (Gel quer zur Stromrichtung)
- 1x TBE-Puffer einfüllen bis Schläuche gerade bedeckt sind
- je nach Molekulargewicht 0,5 -1 Std. bei 100 mA elektrophoretisieren.
- 3 min rückwärts elektrophoretisieren (Pole an der Kammer tauschen)
- Gelstücke entnehmen (Handschuhe tragen!)
- mit Pasteurpipette Flüssigkeit aus dem Schlauch nehmen und in beschriftetes Eppendorfgefäß
überführen
- 200 µl 1x TBE-Puffer in den Dialyseschlauch geben, Dialyseschlauch mit Spektrum-Clip verschließen
und vorsichtig kneten!
- TBE-Puffer mit dem Eluat vereinigen
- dasselbe Volumen (ca. 500 µl) Phenol/Chloroform/Isoamylalkohol (25:24:1) zugeben und mischen
- 5 min. zentrifugieren
- obere Phase abheben und in neues Eppendorfgefäß überführen (Beschriftung nicht vergessen!)
- dasselbe Volumen (ca. 500 µl) Chloroform/Isoamylalkohol (24:1) zugeben und mischen
- 5 min. zentrifugieren
- obere Phase abheben und in neues Eppendorfgefäß überführen (Beschriftung nicht vergessen!)
- Zugabe von 1/10 Vol. 10x Dialyse-Puffer, gut mischen
- + 2 Vol. EtOH abs., gut mischen
- mind. 2 Std. -20°C
- 20 min / 5000 Upm, 4°C
- Pellet in 70% EtOH waschen (3 min zentrifugieren, Pellet vorher nicht resuspendieren)
- am Platz trocknen lassen oder in einer Vakuumzentrifuge (Speedvac) trocknen
- lösen in HPLC-H2O (ca. 20 µl), ca. 1/10 Vol. auf Gel checken
ACHTUNG:
Auch die wiedergewonnenen DNA-Fragmente werden in weiteren Versuchen eingesetzt! Bitte
daher auch diese Eppendorfgefäße sorgfältig beschriften, z.B. folgendermaßen:
Maus (Gr. Ma1) wg. Monomer
21.02.2011

Versuch 2. S.12
D2. Wiedergewinnung von DNA aus Agarosegelen durch Silicamembran-Filtration (wird im Kurs
nicht durchgeführt)
Prinzip :
> Auflösung der Agarose durch chaotrope Ionen
> Bindung der DNA in Anwesenheit von Hochsalz an Silicamembran
> Waschen der Membran zur Entfernung kontaminierender Substanzen
> Elution der DNA durch Niedrigsalz-Lösung (HPLC-H2O)
Durchführung:
- Gelstück ausschneiden und wiegen; das Gewicht sollte 400 mg nicht übersteigen
- zu je 10 mg Gel 30 µl Gelauflösungspuffer (L1; chaotrope Salze!) geben
- mind. 15 min bei 50 °C inkubieren, dabei alle 3 min. mischen bis sich das Gelstück
aufgelöst hat
- Gemisch auf eine Säule pipettieren, die in einem 2 ml-Auffanggefäß steckt
- den Ansatz nun in einer Tischzentrifuge bei 12.000 Upm zentrifugieren
- Flüssigkeit verwerfen, Säule wieder in das Auffanggefäß stecken
- 500 µl Gelauflösungspuffer (L 1) auf das Säulchen geben
- 1 min bei Raumtemperatur inkubieren, dann 1 min bei 12.000 Upm zentrifugieren
- Flüssigkeit verwerfen, Säule wieder in das Auffanggefäß stecken
- 700 µl Waschpuffer (L2) auf die Säule geben
- 5 min bei Raumtemperatur inkubieren
- 1 min in Tischzentrifuge bei 12.000 Upm zentrifugieren.
- Flüssigkeit verwerfen, Säule wieder in das Auffanggefäß stecken und erneut 1 min. leer
zentrifugieren, um Waschpufferreste zu entfernen
- Säulchen in frisches, beschriftetes 1,5 ml Eppendorf-Gefäß stecken
- 50 µl warmes (ca. 70°C) HPLC-H2O. auf die Säule geben
- 1 min bei Raumtemperatur inkubieren
- 2 min. in Tischzentrifuge bei 12000 Upm zentrifugieren
- evtl. 5 µl davon auf Probegel auftragen

Versuch 3. S.13
Versuch 3:
Southern-Analyse repetitiver Satelliten-DNA
Weitgehend homologe, einzelsträngige DNA-Sequenzen können in wässriger Lösung miteinander
hybridisieren. Es lässt sich daher mittels einer solchen Hybridisierungsreaktion feststellen, ob in einem
komplexen Gemisch nicht charakterisierter DNA-Fragmente homologe Sequenzen zu einem bekannten
DNA-Abschnitt vorhanden sind.
Das Prinzip einer Southern-Hybridisierung ist die Kombination von Restriktionsanalyse eines
uncharakterisierten DNA-Gemischs und einer anschließenden Hybridisierung der restringierten DNA mit
einer radioaktiven, bekannten Sonden-DNA. Ein DNA-Gemisch wird mit einem Restriktionsenzym
vollständig verdaut und die entstandenen DNA-Fragmente durch Agarosegelelektrophorese aufgetrennt.
Das entstandene Bandenmuster wird durch Ethidiumbromidfärbung und Betrachtung unter UV-Licht
sichtbar gemacht und durch Fotografieren dokumentiert. Anschließend wird die DNA im Gel durch
Behandlung mit NaOH denaturiert und nach Neutralisierung im einzelsträngigen Zustand auf einen
Nitrozellulosefilter transferiert, wobei das Bandenmuster erhalten bleibt. Es entsteht so ein
maßstabsgetreuer "Abklatsch" des Restriktionsbandenmusters des Gels auf dem Nitrozellulosefilter.
Dieser wird dann unter Hybridisierungsbedingungen mit der bekannten, radioaktiven DNA-Sonde
inkubiert und anschließend autoradiografiert. Diejenigen Banden, die komplementäre DNA-Sequenzen zu
der eingesetzten Hybridisierungssonde enthalten, sind leicht anhand ihrer Radioaktivität zu erkennen.
Durch Vergleich des EtBr-Färbungsergebnisses mit dem Autoradiogramm lässt sich ermitteln, welche der
Banden mit der zur Hybridisierung eingesetzten markierten DNA-Sonde hybridisieren und damit die
gesuchte DNA enthalten.
Aufgabe:
Die restringierte und elektrophoretisch aufgetrennte DNA aus Versuch 2 wird durch Transfer auf
Nitrozellulose- bzw. Nylon-Membranen immobilisiert (Southern-Transfer). Die nach Restriktion
genomischer DNA über Elektrophorese und Gelelution präparativ isolierte repetitive DNA (Versuch 2)
bzw. die durch CsCl-Dichtegradientenzentrifugation isolierte Satelliten-DNA (Versuch 4) wird durch
"Random Primed Oligo-Labelling" radioaktiv oder nicht-radioaktiv markiert. Mit dieser markierten
Sonde wird anschließend der Southern-Filter der restringierten genomischen DNA hybridisiert. Durch
Autoradiografie (bzw. Chemilumineszenz-Nachweis) des hybridisierten Filters wird das
Restriktionsmuster der Satelliten-DNA spezifisch und mit hoher Empfindlichkeit dargestellt.

Versuch 3. S.14
Durchführung der "radioaktiven" Southernhybridisierung: A) Southern-Transfer:
(mit Einweghandschuhen arbeiten!)
- Gele aus Versuch 2 in 0,5 M NaOH - 1,5 M NaCl (Denaturierungslösung) "denaturieren"
- 15 min bei RT stehen lassen, gelegentlich leicht schütteln
- Gele kurz in VE-H2O wässern und in Neutralisierungslösung (3 M NaCl - 0,5 M Tris pH 7,0)
überführen
- mind. 20 min inkubieren, gelegentlich leicht schwenken und Gel untertauchen
- Aufbau des Sandwich-Blots, siehe Anhang; Transfer über Nacht
- Filter nach erfolgtem Transfer kurz in 2 x SSC waschen, auf Filterpapier legen
- auf dem in 2xSSC angefeuchteten Nitrozellulose- bzw. Nylon-Filter können unter UV-Licht die
Probentaschen, starke DNA-Banden und die Gelumrisse erkannt und mit Fettstift markiert werden,
Filter mit Fettstift beschriften (Gruppe, Organismus, Restriktionsenzym, Datum)
- Filter 2 Std. im Vakuumbrutschrank bei 80°C anbacken
- trockene Filter in 3 MM-Papier und Alufolie verpackt im Kühlschrank bis zur
Präinkubation/Hybridisierung aufbewahren
B. Radioaktive Markierung der isolierten repetitiven DNA durch "Random Primed Oligo-
Labelling"
ACHTUNG:
Bitte lesen und beachten Sie unbedingt die Anleitung zum Umgang mit 32
P (Anhang), bevor Sie mit
den radioaktiven Arbeiten beginnen!!!
- DNA (0,1 - 1 µg) in 11 µl HPLC-H2O aufnehmen
- 10 min hitzedenaturieren (94°C)
- sofort auf Eiswasser stellen, kurz anzentrifugieren, danach sofort wieder auf Eis
- + 1 µl dGTP
+ 1 µl dCTP
+ 1 µl dTTP
+ 2 µl React Mix (Puffer + Hexamerprimer) - im Isotopenlabor hinzufügen:
+ 3 µl (30 µCi) α-32
P-dATP (10 µCi/µl)
+ 1 µl Klenow-Enzym
- mischen, evtl. kurz anzentrifugieren
- ca. 1 - 2 h bei RT (evtl.37°C) stehen lassen
- Fällen: + 160 µl HPLC-H2O
+ 20 µl 10x Dialysepuffer, gut mischen
+ 400 µl EtOH abs. (kalt)
mischen, > 15 min. zentrifugieren, mit 70% EtOH waschen
- Einbau mit Geigerzähler messen
- DNA-Pellet in 100 µl HPLC-H2O lösen
- 100 µl (50 µg) heterologe „Carrier“-DNA hinzufügen

Versuch 3. S.15
C. Hybridisierung
- Blot-Filter in 2x PM/6x SSC mit 1% SDS (Präinkubationsmedium nach Denhardt, 1966) mindestens
2-3 h bei 60°C inkubieren (Plastikschale)
- Hybridisierungsprobe vorbereiten (Handschuhe, Abschirmung!):
• 10 ml Hybridisierungslösung. (1x PM/3x SSC mit 0,5% SDS) in Eis stellen
• radioaktiv markierte DNA durch Erhitzen denaturieren (10 min 95°C in verschraubtem
Eppendorfständer)
• rasch in Eiswasser abkühlen
• denaturierte Probe mit 10 ml Hybridisierungslösung mischen und in Eiswasser stellen
- Filter aus Präinkubationslösung nehmen und möglichst luftblasenfrei mit Pinzette in Glas-
Hybridisierungsröhre legen
- Hybridisierungslösung einfüllen
- 6-20 Std bei 60°C bzw. 65°C (abhängig vom AT-Gehalt der Probe) inkubieren
- gebrauchte Hybridisierungslösung mit einem Trichter in ein 15 ml-Falcon abgießen (aufheben, kann
2-3x verwendet werden)
- Filter mit 100 ml 2x SSC bei 60°C waschen (mind. 30 min)
- Waschpuffer mit Monitor kontrollieren, evtl. in radioaktiven Abfallbehälter
- diesen Waschvorgang 2-3x wiederholen
- Filter trocknen - auf 3MM-Filterpapier mit Tesastreifen am Rand aufkleben, mit Haushaltsfolie glatt abdecken
- in Expositionskassette Kodak XAR Röntgenfilm auflegen, evtl. Verstärkerfolie (Cronex od. Lanex)
auf den Film legen
- entweder bei RT (ohne Verstärkerfolie) oder bei -80°C (mit Verstärkerfolie) exponieren
- Expositionszeit zwischen 30 min und 2 Wochen, je nach Konzentration homologer Sequenzen,
spezifischer Aktivität der eingesetzten Probe bzw. Menge der eingesetzten Radioaktivität
Aufgabe:
Vergleichen Sie das Muster auf dem Autoradiogramm mit dem Muster des Agarosegels!
Welche Schlussfolgerungen lassen sich aus diesem Experiment ziehen?
Wie lassen sich die Banden erklären?
Bestimmen Sie möglichst exakt die Länge einer monomeren Einheit!

Versuch 3. S.16
• Alternatives Protokoll: nicht radioaktive Southern-Hybridisierung (wird nicht im Kurs
durchgeführt)
Hierbei wird die Sonde nicht-radioaktiv durch Einsatz von Digoxigenin-dUTP markiert (s. Versuch 8,
in situ-Hybridisierung). Die Detektion erfolgt über Anti-Digoxigenin-Antikörper, die wiederum mit dem
Enzym Alkalische Phosphatase gekoppelt sind. Dieses Enzym dephosphoryliert das Chemilumineszenz-
Substrat AMPPD. Die daraus resultierende Lichtemission wird auf einem Röntgenfilm festgehalten.
Achtung: System funktioniert nur gut bei Verwendung von Nylonmembranen (statt Nitrozellulosefilter).
A. Southern-Transfer:
- Denaturierung, Neutralisierung und Aufbau des Southern-Blots wie oben beschrieben
- Nylonfilter nach erfolgtem Transfer kurz in 2 x SSC waschen, lufttrocknen
- 30 min im Vakuumbrutschrank bei 120° anbacken (Nitrozellulose höchstens 80°C)
- trockene Filter in 3 MM-Papier und Alufolie verpackt im Kühlschrank bis zur
Präinkubation/Hybridisierung aufbewahren
B. Hybridisierung:
- Blot-Filter in Prä-Hybridisierungspuffer mindestens 1,5 h bei 60°C präinkubieren
- Hybridisierungsprobe vorbereiten:
• DIG-markierte DNA hitzedenaturieren (10 min 95°C), rasch in Eiswasser abkühlen
• Aliquot der denaturierten Probe mit 15 ml kalter Hybridisierunglösung mischen
- Filter in Hybridisierungsröhre geben
- Hybridisierungslösung einfüllen
- 6 - 20 Std. bei 60° inkubieren - Filter entnehmen, Hybridisierungslösung in Röhrchen laufen lassen, (aufheben, kann mehrfach
verwendet werden)
- Filter mit 200 ml 2x SSC bei 60°C waschen (mind. 2 Std.), Puffer dabei 2x wechseln
- danach Filter kurz (1-5 min) in Waschpuffer waschen (in Plastikschale)
- 30 min oder länger in Puffer 2 inkubieren
- Anti-DIG-AP-Antikörper-Konjugat auf 75 mU/ml (1:10.000) in Puffer 2 frisch verdünnen
- Filter 30 min in 30 ml verdünnter Antikörper-Konjugat-Lösung inkubieren
- 2 x 15 min mit 40-100 ml Wasch-Puffer waschen
- 2-5 min in 20 ml Puffer 3 äquilibrieren
- AMPPD (Substrat)-Vorratslösung (10 mg/ml) 1:100 in Puffer 3 verdünnen
- Membran für 5 min in etwa 10 ml Substrat-Lösung inkubieren (die verdünnte Substratlösung kann bei
-20°C im Dunkeln gelagert und wiederverwendet werden)
- überschüssige Flüssigkeit von der Membran ablaufen lassen, jedoch nicht völlig trocknen lassen
- feuchte Membran zwischen Frischhaltefolie legen
- 5-15 min bei 37°C inkubieren (Brutschrank)
- 15-25 min bei Raumtemperatur exponieren, danach Autoradiogramm entwickeln

Versuch 4. S.17
Versuch 4:
Lokalisierung von DNA-Sequenzen durch
in situ Hybridisierung an Polytänchromosomen
Eine Hybridisierungsreaktion zwischen Nukleinsäuren kann prinzipiell auch in situ durchgeführt werden,
d.h. die DNA/RNA in Chromosomen/Zellkernen kann mit einer DNA/RNA in Lösung unter bestimmten
Bedingungen renaturieren. Mit Hilfe dieser in situ Hybridisierung lassen sich DNA/RNA-Sequenzen in
den Chromosomen lokalisieren. Folgende Voraussetzungen müssen für eine erfolgreiche Lokalisierung
von DNA-Sequenzen erfüllt sein:
• die DNA in den für die Hybridisierung zu verwendenden Chromosomen muss denaturiert
(einzelsträngig) sein.
• die als Hybridisierungsprobe eingesetzte DNA (= die zu lokalisierende DNA) muss entweder
radioaktiv oder anders markiert sein, auch sie muss einzelsträngig sein.
• es muss ein sehr empfindliches Nachweissystem für die markierte DNA verfügbar sein, das einen
mikroskopischen Nachweis erlaubt
• im Übrigen müssen alle Parameter, die auch für die "normale" DNA-Hybridisierung/
-Renaturierung wichtig sind (Cot-Wert, Salzkonzentration, Temperatur, DNA-Konzentration,
Sequenzidentität, pH-Wert u.a.) berücksichtigt werden.
Bei der klassischen Form der in situ Hybridisierung war die zu lokalisierende DNA-Probe radioaktiv,
meistens mit 3H, markiert. Nach der Hybridisierungsreaktion wurde die radioaktive DNA, die
ausschließlich mit den homologen DNA-Abschnitten in den Chromosomen hybridisiert, mit Hilfe eines
sehr dünnen und sehr empfindlichen fotografischen Films durch sog. "Mikroautoradiographie"
nachgewiesen. Obwohl die Methode der Mikroautoradiographie sehr empfindlich ist, hat sie erhebliche
Nachteile:
1. Viele Schritte (Aufbringen des Films auf die Chromosomenpräparate, Entwickeln des Films usw.)
müssen bei dunklem Rotlicht durchgeführt werden.
2. Der Nachweis der markierten DNA ist indirekt: es kommt zu einer Streuung der durch die Strahlung
hervorgerufenen Silberkörner in dem Autoradiographiefilm; der Film selbst kann sich durch die
Manipulationen (Entwicklung, Wässerung, Fixierung, Färbung usw.) verschieben - eine Auswertung
ist dann nicht mehr möglich.
3. Die Expositionszeit des Films kann bis zu mehrere Monate dauern.
Alle diese Nachteile werden durch neuere, nicht-radioaktive Markierungstechniken vermieden. Aus der
inzwischen großen Auswahl an Markierungs- und Nachweistechniken wurden zwei Verfahren
ausgewählt:
1. die direkte Markierung der Hybridisierungssonde mit Biotin-dUTP und der Nachweis der
biotinylierten DNA mit fluoreszenzmarkierten Avidin oder Streptavidin.
2. die direkte Markierung der Sonde unter Einbau von Digoxigenin-dUTP und Nachweis der
digoxigenierten Sonde durch einen fluoreszensmarkierten Anti-DIG-Antikörper.
Beide Nachweise sind sehr empfindlich und können innerhalb eines Tages durchgeführt werden.
Voraussetzung ist die Verfügbarkeit eines Fluoreszenzmikroskops.

Versuch 4. S.18
Durchführung:
A. Präparation von Speicheldrüsenchromosomen aus Larven von Chironomus thummi
1. Kopf und 1. Segment mit 2 Präpariernadeln auf Hohlschliffobjektträger (Objektträger mit Bleistift
beschriften und Platz lassen!) abtrennen. Larvenkörper entfernen.
2. Frisch präparierte Speicheldrüsen auf einem Hohlschliffobjektträger mit Essigsäure (50%) für 1 min
fixieren. Wenn die Drüsen nicht gefärbt werden sollen, sogleich mit einer feinen Pinzette auf einen
planen Objektträger übertragen und wie in Punkt 5 weiterbehandeln (für in situ Hybridisierung dürfen
Chromosomen nicht gefärbt werden). 3. Im Essigsäuretropfen Speicheldrüsenzellen von Sekret befreien
4. Zellen unter einem Deckglas "quetschen", d.h. Deckglas auf den Essigsäuretropfen gleiten lassen,
2 Filterpapierstreifen über den Objektträger legen und mit dem Daumen langsam und gleichmäßig das
Präparat quetschen.
5. Vereisen über Trockeneis: Objektträger mit dem Deckglas nach unten auf Kohlensäureschnee
legen (5 min)
6. Danach Deckglas absprengen (z.B. mit Rasierklingen).
7. Sofort Objektträger mit dem darauf haftendem Objekt in 100% Isopropanol stellen. Wird nicht schnell
genug gearbeitet, verdampft CO2 ehe das Präparat in Alkohol kommt und Objekt trocknet aus!!!
8. Präparate in 100% Isopropanol bei -20°C aufbewahren bis zur Verwendung für in situ Hybridisierung.
B. Vorbereitung der Präparate für die Hybridisierung
- pro Gruppe ca. 10 gute Präparate von Polytänchromosomen (C. thummi) aus Isopropanol die
Alkoholreihe abwärts führen (100% – 70% – 50% – 30% - 0,1x SSC, je ca. 5 min)
- Präparate in 2x SSC überführen
- in 2x SSC 30 min bei 70°C inkubieren (großes Becherglas)
- auf Raumtemperatur abkühlen
- Präparate in 0,1 x SSC überführen
- dann Präparate 60 Sekunden in 0,05 N NaOH eintauchen (Zeit genau einhalten; bei diesem Schritt
wird die DNA in den Chromosomen denaturiert) und anschließend sofort in eiskaltes 0,1x SSC
überführen. Die Qualität der Chromosomen wird dabei stark in Mitleidenschaft gezogen, deshalb nicht
länger als 60 Sek. denaturieren!
- Präparate dann Alkoholreihe aufwärts führen bis zum 100 % Isopropanol (darin aufbewahren)
C1. in situ Hybridisierung unter Verwendung Biotin-markierter Sonden
C1.1. In vitro-Markierung von DNA mit Biotin-dUTP durch "Nick-Translation" (BioNickTM
Labeling System von Invitrogen)
- ca. 1 µg DNA (z.B. isolierte Cla-Elemente) auf 40 µl mit HPLC-H2O auffüllen
- + 5 µl 10x dNTP-Mix
- + 5 µl 10x Enzyme Mix
- gut mischen, kurz anzentrifugieren
- 2,5 Stunde bei RT inkubieren
- 5 µl Stopp-Puffer hinzufügen
- 50 µl 10x SSC hinzufügen
- 1 µl 10%iges SDS hinzufügen
- zum Denaturieren die biotinylierte DNA zusammen mit 4 µl Carrier-DNA (50 µg/ml) 10 min bei 95°C
inkubieren
- schnell in Eiswasser abkühlen
- 8 µl der denaturierten Sonde auf die Chromosomenpräparate aufbringen
- Präparate mit sauberen Deckgläsern (16x16 mm2) eindeckeln; Deckglas mit "Fixogum"-Flüssiggummi
umranden (Abdichtung gegen Austrocknung)
- ca. 30 min bei RT warten bis Fixogum angetrocknet ist
- dann ü.N. bzw. mind. 6 Stunden bei 52°C in einer Plastikschale im Wasserbad inkubieren

Versuch 4. S.19
Alternativ:
C1.2. In vitro Markierung von DNA mit Biotin-dUTP durch "Random-primed Oligo-Labeling"
- ca. 0,1 µg DNA (z.B. isolierte Cla-Elemente) in 12 µl H2O lösen, in Eppendorf-Gefäß überführen
- DNA 10 min in kochendes Wasser stellen (denaturieren)
- in Eiswasser abkühlen
- je 1 µl dATP, dCTP, dGTP, sowie 2 µl Bio-dUTP hinzufügen
- 2 µl "Hexanukleotid-Mix" hinzufügen
- + 1 µl Klenow-Fragment ("large fragment" der DNA-Polymerase I)
- gut mischen, kurz anzentrifugieren
- 2-4 Stunden oder über Nacht bei Raumtemperatur inkubieren
- dann 30 µl H2O hinzufügen
- 50 µl 10x SSC hinzufügen
- 1 µl 10%iges SDS hinzufügen
- 4 µl Carrier-DNA (50 µg/ml) 10 min bei 95°C inkubieren
- schnell in Eiswasser abkühlen
- 10 µl der denaturierten Sonde auf die Chromosomenpräparate aufbringen
- Präparate mit sauberen Deckgläsern (16x16 mm2) eindeckeln; Deckglas mit "Fixogum"-Flüssiggummi umranden
(Abdichtung gegen Austrocknung)
- ca. 30 min bei RT warten bis Fixogum angetrocknet ist
- dann ü.N. bzw. mind. 6 Stunden bei 52°C in einer Plastikschale im Wasserbad inkubieren
D1. Nachweis-Reaktion (Biotin)
- Präparate nach Hybridisierung in 2x SSC 5 min waschen
- in 1 x PBS für 10 min waschen
- 8 µl FITC-markiertes Avidin (1:75 mit PBS-BSA verdünnt, wobei zu 100 µl verdünnter
Antikörperlösung 4 µl Carrier-DNA gegeben werden) je Chromosomenpräparat aufbringen (Präparate
vorher etwas abtropfen lassen – sie dürfen aber keinesfalls eintrocknen!)
- mit Deckglas eindeckeln
- in feuchter Kammer 2-3 Std. bei 37°C (Wasserbad) inkubieren
- Präparate 10 min in PBS waschen, Deckgläser dabei vorsichtig abschwimmen lassen
- abtropfen (nicht eintrocknen!) lassen
- in "Glycerin/Para-Phenylendiamin"-Mix (10 µl) einbetten (VORSICHT: giftig!), Deckglas auflegen
- mit dem Fluoreszenzmikroskop anschauen (Einweisung durch Lehrperson abwarten), lohnenswerte
Stellen fotografieren (bei einer analogen Kamera muss dazu ein hochempfindlicher Film z.B. Ilford
HP5 oder Kodak Tri-X-Pan verwendet werden – es können aber auch Fotos mit einer Digitalkamera
angefertigt werden!)
C2 in situ Hybridisierung mit Digoxigenin-markierten Sonden
C2.1. In vitro-Markierung von DNA mit Digoxigenin
- DNA in 15 µl HPLC-H2O denaturieren (95°C, 5 min)
- auf Eiswasser stellen
- + 2 µl Hexanucleotid-Mix
- + 2 µl dNTP Mix (mit DIG-dUTP)
- + 1µl Klenow-Enzym
- mix, zentrifugieren, mind. 1 h RT, besser über Nacht inkubieren
- + 30 µl H2O
- + 50 µl 10xSSC
- + 1 µl 10% SDS
- zusammen mit 4 µl Carrier-DNA (50 µg/ml) denaturieren (95°C, 10 min), auf Eiswasser stellen
- Hybridisierung der Präparate wie unter C.1 beschrieben

Versuch 4. S.20
D2. Detektion der digoxigenierten DNA
- Präparate 5 min in 2x SSC, dann 10 min in 1x PBS waschen
- 8 µl Anti-DIG-Antikörper (FITC-gekoppelt, 1:30 verdünnt in PBS/BSA, wobei zu 100 µl verdünnter
Antikörperlösung 4 µl Carrier-DNA gegeben werden) je Chromosomenpräparat aufbringen (Präparate
vorher etwas abtropfen lassen – sie dürfen aber keinesfalls eintrocknen!)
- > 30 min bei 37°C inkubieren
- 10 min in 1x PBS waschen
- mit 10 µl „antifading mix“ einbetten
Aufgabe:
Lokalisierung repetitiver Cla-Elemente an den Polytänchromosomen von Chironomus thummi

Anhang. S.21
ANHANG
1. Puffer und Lösungen
2. Molekulargewichtsstandards für die Gelelektrophorese
3. Aufbau Southern Blot
4. Restriktionskarte und Multiple Klonierungsschnittstelle von pUC19 und pUC19
5. Anleitung für den Umgang mit 32
P
6. EinigeTipps und Einheiten
1. Puffer und Lösungen:
Versuch 1: Puffer zur DNA-Extraktion:
Homogenisierungspuffer:
58 g Saccharose
10 ml 0,1 M Na2 EDTA pH 7,4
2,5 ml 0,1 M Na2 EGTA pH 7,4
50 ml Puffer A (10 x)
auf 500 ml auffüllen mit H2O
Puffer A (10x Stock):
150 ml 2 M KCl
37,5 ml 2 M NaCl
7,5 ml 0,1 M Spermin
25 ml 0,1 M Spermidin
75 ml 1,0 M Tris HCl
auf 500 ml mit H2O auffüllen
pH auf 7,4 (mit HCl) einstellen
Gesättigte Tris-Lösung, pH 8,5:
Zu 50 ml H2O solange Tris unter Rühren zugeben, bis sich ungelöstes Tris absetzt. Dann mit
konz. HCl pH 8,5 einstellen und Lösung filtrieren.
Versuch 2/3: Gelelektrophorese und Southern-Blot-Hybridisierung
TE-Puffer:
10 mM Tris pH 8,0
1 mM EDTA pH8,0
TBE (20x) = Tris-Borat-EDTA (Elektrophorese-Puffer):
108 g Tris
55,6 g Borsäure
4,65 g Na2EDTA
auf 500 ml mit H2O auffüllen

1x TBE:
90 mM Tris
90 mM Borsäure
1,2 mM Na2EDTA
Dialyse-Puffer (10x):
3,00 M NaCl (175,3 g pro Liter)
0,25 M Tris (30,3 g pro Liter)
0,10 M Na2EDTA (37,2 g pro Liter)
"Bromphenolblau-Mix":
4 M Harnstoff
50% Saccharose
0,1 M Na2EDTA
0,1% Bromphenolblau
E-Puffer (10%) =Tris-Phosphat-Elektrophoresepuffer:
0,36 M Tris (86,8 g je 2 Liter)
0,3 M NaH2P04 (95,2 g je 2 Liter)
0,1 M Na2EDTA (7,4 g je 2 Liter)
20x SSC:
3 M NaCl
0,3 M Na-Citrat
6x SSC-2x Prähybridisierungspuffer (Hybridisierungsmedium nach Denhardt)
600 ml 20x SSC
1400 ml H2O
0,8 g Ficoll 400
0,8 g Polyvinylpyrrolidon (PVP)
0,8 g Rinderserumalbumin (BSA)
Denaturierungslösung-Blot:
0,5 M NaOH
1,5 M NaCl
Neutralisierungslösung- Blot:
3,0 M NaCl
0,5 M Tris-HCl pH 7,0
Puffer 1 (nicht-radioaktive Southern-Hybridisierung):
Maleinsäure, 0,1 mol/l
NaCl, 0,15 mol/l
pH 7,5 (mit NaOH einstellen), autoklavieren
Puffer 2 (nicht-radioaktive Southern-Hybridisierung):
Blockierungs-Vorratslösung, verdünnt 1:10 in Puffer 1 (= Endkonzentration von 1% Blocking
Reagenz)
Puffer 3 (nicht-radioaktive Southern-Hybridisierung):
0,1 mol/l Tris-HCl
0,1 mol/l NaCl
50 mmol/l MgCl2
pH 9,5 (20°C), sterilfiltrieren

Anhang. S.23
Blockierungs-Vorratslösung (nicht-radioaktive Southern-Hybridisierung):
Blocking-Reagenz, 10% (w/v) in Puffer 1
autoklavieren
Hybridisierungspuffer:
3x SSC
1x Präinkubationsmedium (nach Denhardt)
Waschpuffer:
Puffer 1 + Tween 20, 0,3% (v/v)
AMPPD (nicht-radioaktive Southern-Hybridisierung):
Vorratslösung: 10 mg/ml; 23,5 mmol/l
die fertige Substratlösung wird frisch 1:100 in Puffer 3 verdünnt
= 0,235 mmol/l
Versuch 4: In situ Hybridisierung
PBS 10x
80 g NaCl
2 g KCl
17,8g Na2HPO4 x 2H2O
2,4g KH2PO4
auf 1000 ml
pH 7,4
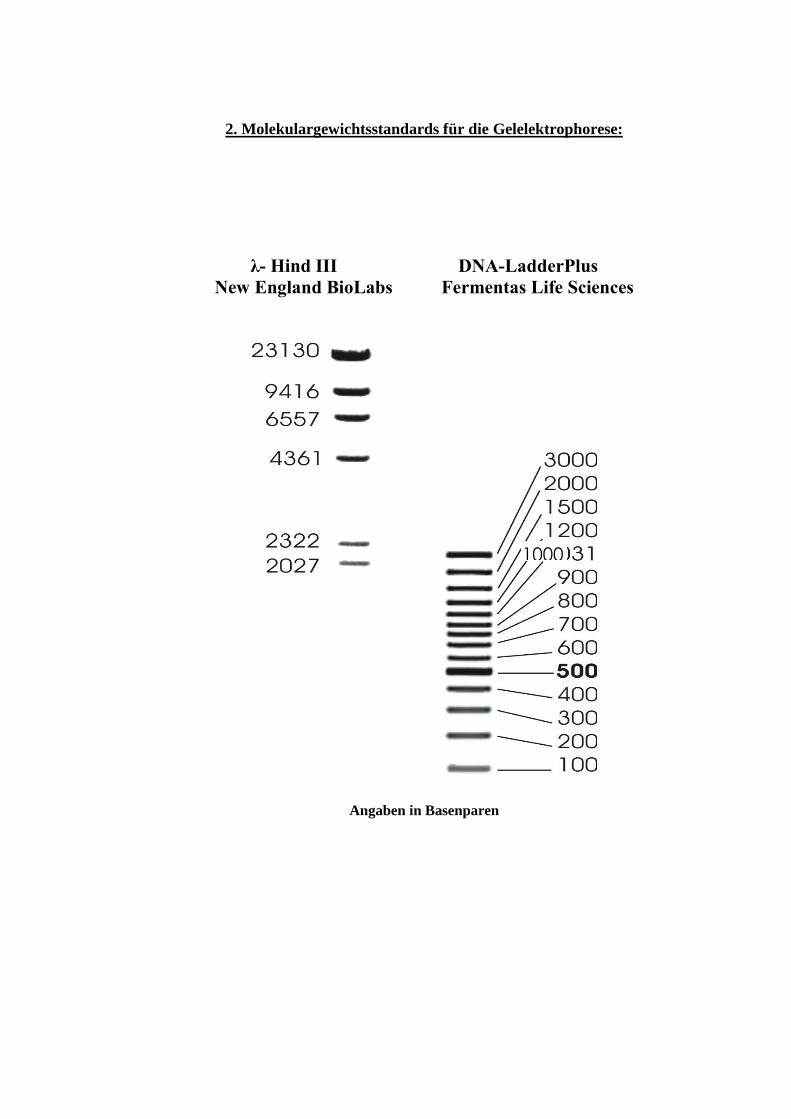
2. Molekulargewichtsstandards für die Gelelektrophorese:
λ- Hind III DNA-LadderPlus
New England BioLabs Fermentas Life Sciences
Angaben in Basenparen
1000
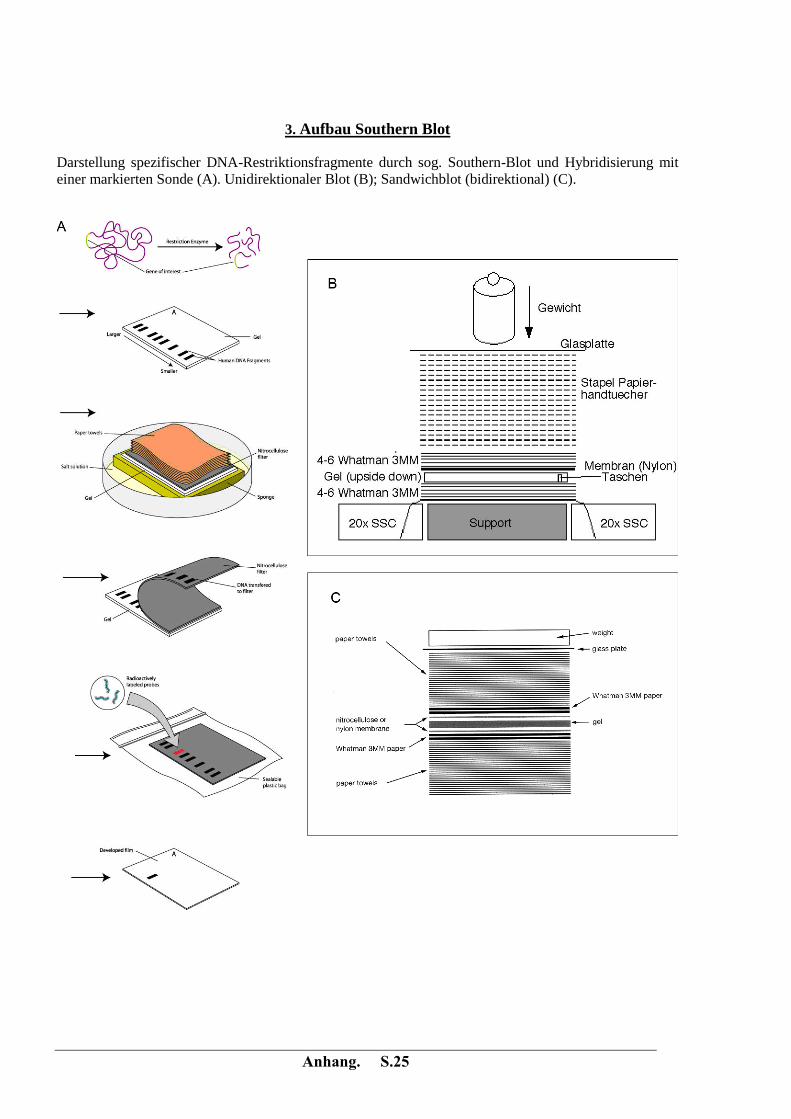
Anhang. S.25
3. Aufbau Southern Blot
Darstellung spezifischer DNA-Restriktionsfragmente durch sog. Southern-Blot und Hybridisierung mit
einer markierten Sonde (A). Unidirektionaler Blot (B); Sandwichblot (bidirektional) (C).

5. Anleitung für den Umgang mit 32
P
Der Einsatz von Phosphor-32 ist heute in der Molekularbiologie weit verbreitet, zum einen, weil 32
P sehr
leicht nachweisbar ist, und zum anderen, da es beim Einbau in Nukleinsäuren sehr hohe spezifische
Aktivitäten ermöglicht. 32
P emittiert jedoch eine hochenergetische ß-Strahlung von 1.71 MeV und muss
deshalb mit besonderer Vorsicht gehandhabt werden. Es ist wichtig, die nachfolgenden Regeln im
Umgang mit 32
P zur eigenen und der Sicherheit anderer strikt einzuhalten.
Achtung: Schwangeren ist das Arbeiten mit Isotopen strikt untersagt!
Schutz vor Strahlung 32
P emittiert ß-Teilchen mit einer maximalen Energie von 1.71 MeV; dies entspricht einer Reichweite in
Luft von ca. 6 Metern. Daher ist es empfehlenswert, immer hinter einer Abschirmung zu arbeiten. Dabei
muß allerdings berücksichtigt werden, dass bei Abschirmung von harter ß-Strahlung immer
"Bremsstrahlung" entsteht, ähnlich der normalen Röntgenstrahlung. Die aus ß-Strahlung entstehende
Bremsstrahlung ist direkt proportional der Energie des ß-Partikels und der Ordnungszahl des getroffenen
Materials. Das abschirmende Material sollte deshalb auf jeden Fall ein Material geringer Dichte sein,
z. B. Plexiglas, Wasser, Plastik usw., um die Erzeugung von Bremsstrahlung so gering wie möglich zu
halten. Soll auch noch die entstehende Bremsstrahlung abgeschirmt werden, kann als "zweite Schicht"
noch ein Material von hoher Dichte hinzugefügt werden (z. B. Blei).
Eine der größten Gefahren bei Verwendung von 32
P ist also die Handhabung ohne Abschirmung. z. B.
beträgt die Dosisrate auf der Oberfläche von 1 ml wässriger Lösung mit 1 mCi 32
P ungefähr 13 Rem/min.
Diese Dosisrate wird auch nur wenig vermindert durch einige Zentimeter Luft. Daraus wird ersichtlich,
dass eine Hand oder ein Gesicht über einer solchen Strahlenquelle eine erhebliche Strahlungsdosis in
einer sehr kurzen Zeit erhält. Eine Übersicht über die Dosisraten von 1 mCi 32
P in den handelsüblichen
Gefäßen bzw. als Kontamination auf nackter Haut gibt die Tabelle 1. Daraus geht hervor, dass unter allen
Umständen ein Kontakt von 32
P mit der Körperoberfläche vermieden werden muss. Auch die
Manipulationen von 32
P in Plastikgefäßen, z. B. Eppendorfzentrifugentuben, müssen auf das absolut
notwendige Maß beschränkt bleiben. Die Eppendorftuben sollen, wenn irgend möglich, in abschirmenden
Plexiglasblöcken gehandhabt werden. Auf der Oberfläche eines Plastikgefäßes (z. B. Eppendorf) mit
1 mCi 32
P in 1 ml wässriger Lösung, ist immerhin noch eine ungefähre Dosisrate von 173.000 mRem pro
Stunde zu erwarten. Die Dosisrate wird drastisch gesenkt, wenn das Lösungsvolumen erhöht wird
(Selbstabsorption); z. B. ist auf der Oberfläche einer Plastikflasche mit 1 mCi 32
P in 50 ml wässriger
Lösung nur noch eine Dosisrate von 3.100 mRem/h, in 100 ml Lösung noch 1950 mREM/h und in 150 ml
Lösung noch 1320 mRem/h messbar.
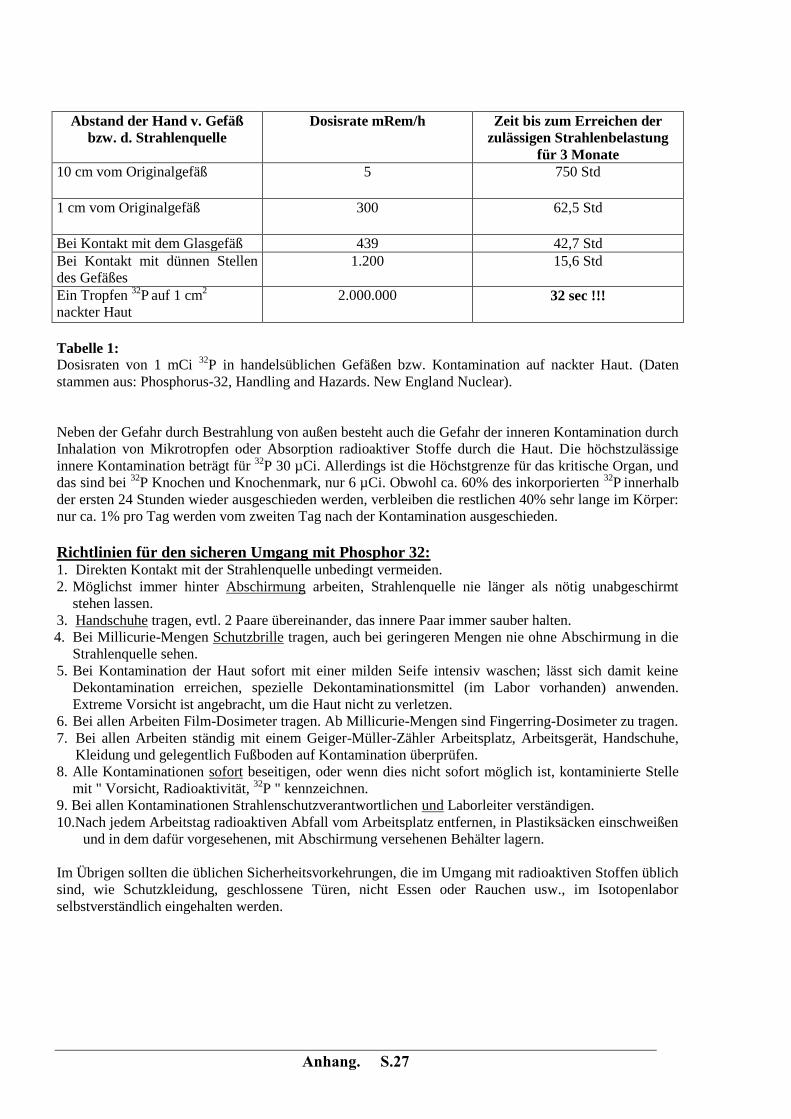
Anhang. S.27
Abstand der Hand v. Gefäß
bzw. d. Strahlenquelle Dosisrate mRem/h
Zeit bis zum Erreichen der
zulässigen Strahlenbelastung
für 3 Monate 10 cm vom Originalgefäß
5 750 Std
1 cm vom Originalgefäß
300 62,5 Std
Bei Kontakt mit dem Glasgefäß 439 42,7 Std Bei Kontakt mit dünnen Stellen
des Gefäßes 1.200 15,6 Std
Ein Tropfen 32
P auf 1 cm2
nackter Haut 2.000.000 32 sec !!!
Tabelle 1:
Dosisraten von 1 mCi 32
P in handelsüblichen Gefäßen bzw. Kontamination auf nackter Haut. (Daten
stammen aus: Phosphorus-32, Handling and Hazards. New England Nuclear).
Neben der Gefahr durch Bestrahlung von außen besteht auch die Gefahr der inneren Kontamination durch
Inhalation von Mikrotropfen oder Absorption radioaktiver Stoffe durch die Haut. Die höchstzulässige
innere Kontamination beträgt für 32
P 30 µCi. Allerdings ist die Höchstgrenze für das kritische Organ, und
das sind bei 32
P Knochen und Knochenmark, nur 6 µCi. Obwohl ca. 60% des inkorporierten 32
P innerhalb
der ersten 24 Stunden wieder ausgeschieden werden, verbleiben die restlichen 40% sehr lange im Körper:
nur ca. 1% pro Tag werden vom zweiten Tag nach der Kontamination ausgeschieden.
Richtlinien für den sicheren Umgang mit Phosphor 32: 1. Direkten Kontakt mit der Strahlenquelle unbedingt vermeiden.
2. Möglichst immer hinter Abschirmung arbeiten, Strahlenquelle nie länger als nötig unabgeschirmt
stehen lassen.
3. Handschuhe tragen, evtl. 2 Paare übereinander, das innere Paar immer sauber halten.
4. Bei Millicurie-Mengen Schutzbrille tragen, auch bei geringeren Mengen nie ohne Abschirmung in die
Strahlenquelle sehen. 5. Bei Kontamination der Haut sofort mit einer milden Seife intensiv waschen; lässt sich damit keine
Dekontamination erreichen, spezielle Dekontaminationsmittel (im Labor vorhanden) anwenden.
Extreme Vorsicht ist angebracht, um die Haut nicht zu verletzen.
6. Bei allen Arbeiten Film-Dosimeter tragen. Ab Millicurie-Mengen sind Fingerring-Dosimeter zu tragen.
7. Bei allen Arbeiten ständig mit einem Geiger-Müller-Zähler Arbeitsplatz, Arbeitsgerät, Handschuhe,
Kleidung und gelegentlich Fußboden auf Kontamination überprüfen.
8. Alle Kontaminationen sofort beseitigen, oder wenn dies nicht sofort möglich ist, kontaminierte Stelle
mit " Vorsicht, Radioaktivität, 32
P " kennzeichnen.
9. Bei allen Kontaminationen Strahlenschutzverantwortlichen und Laborleiter verständigen.
10.Nach jedem Arbeitstag radioaktiven Abfall vom Arbeitsplatz entfernen, in Plastiksäcken einschweißen
und in dem dafür vorgesehenen, mit Abschirmung versehenen Behälter lagern.
Im Übrigen sollten die üblichen Sicherheitsvorkehrungen, die im Umgang mit radioaktiven Stoffen üblich
sind, wie Schutzkleidung, geschlossene Türen, nicht Essen oder Rauchen usw., im Isotopenlabor
selbstverständlich eingehalten werden.
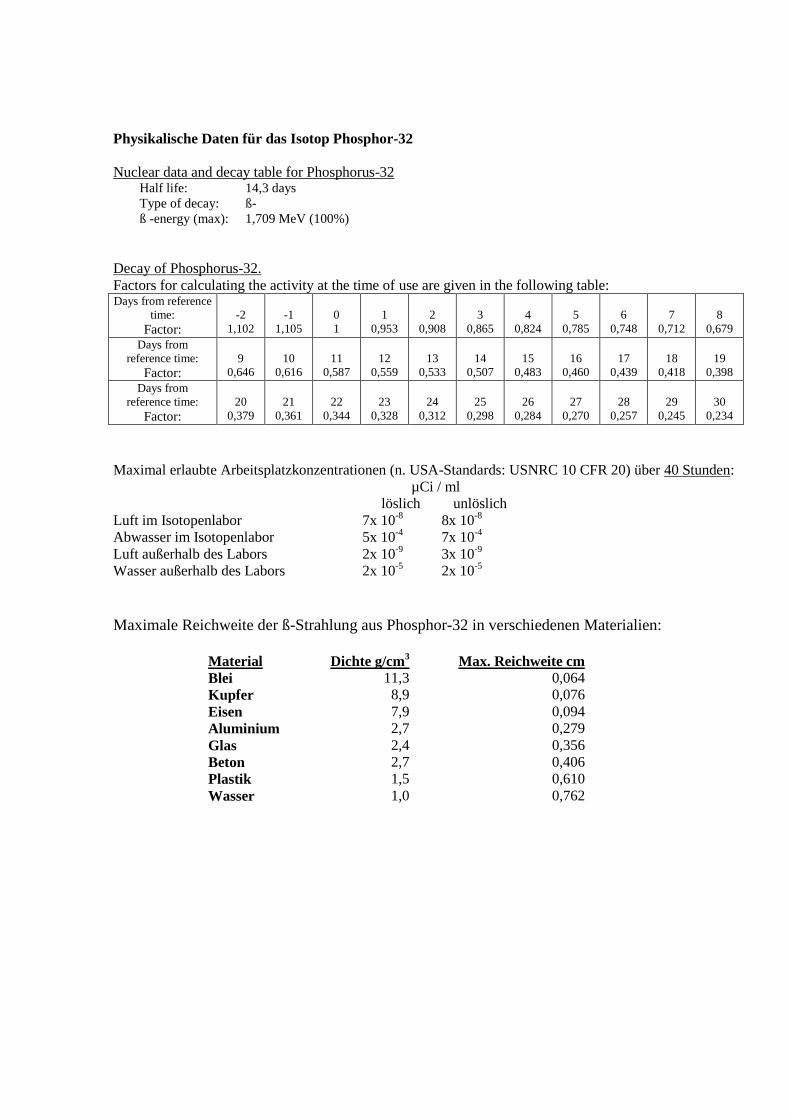
Physikalische Daten für das Isotop Phosphor-32
Nuclear data and decay table for Phosphorus-32 Half life: 14,3 days
Type of decay: ß-
ß -energy (max): 1,709 MeV (100%)
Decay of Phosphorus-32.
Factors for calculating the activity at the time of use are given in the following table: Days from reference
time:
Factor:
-2
1,102
-1
1,105
0
1
1
0,953
2
0,908
3
0,865
4
0,824
5
0,785
6
0,748
7
0,712
8
0,679
Days from
reference time:
Factor:
9
0,646
10
0,616
11
0,587
12
0,559
13
0,533
14
0,507
15
0,483
16
0,460
17
0,439
18
0,418
19
0,398
Days from
reference time:
Factor:
20
0,379
21
0,361
22
0,344
23
0,328
24
0,312
25
0,298
26
0,284
27
0,270
28
0,257
29
0,245
30
0,234
Maximal erlaubte Arbeitsplatzkonzentrationen (n. USA-Standards: USNRC 10 CFR 20) über 40 Stunden:
µCi / ml
löslich unlöslich
Luft im Isotopenlabor 7x 10-8
8x 10-8
Abwasser im Isotopenlabor 5x 10-4
7x 10-4
Luft außerhalb des Labors 2x 10-9
3x 10-9
Wasser außerhalb des Labors 2x 10-5
2x 10-5
Maximale Reichweite der ß-Strahlung aus Phosphor-32 in verschiedenen Materialien:
Material Dichte g/cm
3 Max. Reichweite cm
Blei 11,3 0,064 Kupfer 8,9 0,076 Eisen 7,9 0,094 Aluminium 2,7 0,279 Glas 2,4 0,356 Beton 2,7 0,406 Plastik 1,5 0,610 Wasser 1,0 0,762

Anhang. S.29
Strahlenschutzbelehrung gemäß § 39 StrlSchV
Strahlenschutzanweisung bei Arbeiten mit offenen radioaktiven Stoffen
1. Schutz vor Strahlenbelastung von außen
Zum Schutz vor zu hohen Dosen durch äußere Bestrahlung müssen folgende vorsorgliche Maßnahmen
getroffen werden:
a) Benutzung der in den Labors vorhandenen Abschirmungen bei Arbeiten mit 32
P
b) Lagerung von 32
P in strahlensicheren Blei- oder Kunststoffbehältern
c) Einschränkung der Arbeitszeit im Strahlenfeld auf das unbedingt notwendige Maß
d) Besonders vorsichtige Arbeitsweise
e) Tragen von Schutzhandschuhen und Labormänteln
2. Schutz vor Strahlenbelastung von innen
Die nachfolgend angeführten Schutzmaßnahmen dienen dazu, die Aufnahme radioaktiver Stoffe in den
Körper zu verhindern. Inkorporationen können durch Ingestion, Inhalation und durch Eindringen von
Radionukliden in Wunden erfolgen.
Daher sind folgende Gebote beim Arbeiten mit offenen radioaktiven Stoffen unbedingt zu beachten:
a) Arbeiten unter Abzügen
b) Essen, Trinken und Rauchen unterlassen
c) Laborgegenstände (Pipetten usw.) nicht an den Mund führen
d) Nicht mit offenen, ungeschützten Wunden arbeiten
e) Nach Entnahme der radioaktiven Substanz den Lagerbehälter sofort wieder dicht verschließen
f) Hautoberfläche (Hände) regelmäßig auf Kontamination überprüfen; als Grenzwerte im
Kontrollbereich gelten:
• α-Strahlen 1x10-5
µCi/cm2
• β-Strahlen 1x10-4
µCi/cm2
3. Schutz der Umgebung vor Strahlenbelastung
Zum Schutz der Umgebung muss eine unkontrollierte Ableitung oder Ausbreitung der Radioaktivität
unbedingt vermieden werden. Die nicht vermeidbare Ableitung durch Luft muss so gering wie möglich
sein.
Folgende Vorsorgepflichten sind zu beachten:
a) Isotopenlabortüren und Fenster müssen stets geschlossen sein
b) Radioaktive Abfälle müssen in geeigneten Behältern gesammelt und zur Beseitigung einer behördlich
zugelassenen Einrichtung zugeführt werden
c) Radioaktiv verunreinigte Kleidungsstücke nicht in öffentliche Waschanstalten geben oder mit sonst
anfallender Wäsche waschen
Grenzwert der Organdosis für beruflich strahlenexponierte Personen:
Körperbereich
1. Augenlinse 150 Millisievert
2. Haut, Hände, Unterarme, Füße, Knöchel 500 Millisievert
3. Keimdrüsen, Gebärmutter und Knochenmark 50 Millisievert
4. Knochenoberfläche, Schilddrüse 300 Millisievert
5. Andere Organe wie z.B. Dickdarm, Lunge, Magen,
Blase, Brust, Leber
150 Millisievert

6. Einige Tipps und Einheiten
1 A260 Einheit doppelsträngiger DNA = 50 µg/ml
E260 für 1 mg/ml doppelsträngiger DNA = 20,3
E260 für 1 mM (in Nukleotiden) doppelsträngiger DNA = 6,7
1 A260 doppelsträngiger DNA = 0,15 mM
1 A260 Einheit einzelsträngiger DNA = 33 µg/ml
E260 für 1 mg/ml einzelsträngiger DNA = 25,5
E260 für 1 mM (in Nukleotiden) einzelsträngiger DNA = 8,5
1 A260 einzelsträngiger DNA = 0,12 mM
1 A260 Einheit einzelsträngiger RNA = 40 µg/ml
1 µg pBR322 = 0,36 pmol
1 pmol pBR322 = 2,8 µg
1 pmol des linearisierten pBR322 = 1,4 µg
1 nmol einer 1000 Basenpaar langen doppelsträngigen DNA = 0,66 µg
1 kb DNA = 6,5x 105 Dalton doppelsträngiger DNA (NaCl)
1 kb DNA = 3,3x 105 Dalton einzelsträngiger DNA (NaCl)
1 kb RNA = 3,4x 105 Dalton einzelsträngiger RNA (NaCl)
Durchschnittliches Molekulargewicht einer deoxynukleotid Base = 324,5 Dalton
Durchschnittliches Molekulargewicht eines deoxynukleotid Basepaares = 649 Dalton
1 µg/ml DNA = 3,08 µM Phosphat
1 µg/ml einer 1kb DNA = 3,08 nM 5' Enden
Mol der Enden (5' oder 3') zirkulärer DNA
= Mol der DNA x Anzahl der Schnitte x 2 (Enden pro Schnitt)
Mol der Enden (5' od 3') linearer DNA
= Mol der DNA x 2 (Enden pro Schnitt) + 2 (Enden der linearen DNA)
10.000 Dalton Protein = 0,27 kb DNA
50.000 Dalton Protein = 1,35 kb DNA
100.000 Dalton Protein = 2,7 kb DNA
Becquerel (Bq) = 1 Disintegration/sec
1 Curie (Ci) = 3,7 x 1010
Bq = 2,2 x 1012
Disintegration/min (dpm)
1 mCi = 3,7 x 107 Bq = 2,2 x 10
9 dpm
1 µCi = 3,7 x 104 Bq = 2,2 x 10
6 dpm
Related Documents











