Molecular reorientation of water adsorbed on charged Ag(1 1 1) surfaces Cristi an G. S anchez * Atomistic Simulation Group, School of Mathematics and Physics, Physics Building, QueenÕs University Belfast, University Road, BT7 1NN Belfast, Northern Ireland, UK Received 8 November 2002; accepted for publication 22 January 2003 Abstract In this work we present first principles calculations of water adsorption over charged Ag(1 1 1) surfaces. The ori- entation of the adsorbed water molecule with respect to the surface changes from oxygen pointing away from the surface at negative charges to oxygen pointing towards the surface at positive charges. At zero charge the water molecule is oriented approximately parallel to the surface plane. Complete orientation of the molecule in the direction of the field is achieved for a critical charge density of 15 lC cm 2 for both positive and negative charges. Ó 2003 Elsevier Science B.V. All rights reserved. Keywords: Density functional calculations; Chemisorption; Water; Silver; Low index single crystal surfaces; Solid–liquid interfaces 1. Introduction Double layer modelling plays a fundamental role in theoretical electrochemistry since an accu- rate knowledge of the double layer is needed in any attempt to describe other electrochemical phe- nomena such as charge transfer processes. Many different approaches have been applied to the study of this subject, ranging from first principles mo- lecular dynamics to Monte Carlo simulations. These works have been extensively reviewed re- cently [1–4]. In spite of the effort dedicated there is not a general consensus on a model for the elect- rochemical double layer, and existing models have difficulties to rationalise all of the abundant body of experimental data. This limited success can be ascribed to the inherent complexity of the problem. The interface is a highly anisotropic environment in which large changes of composition occur within a very narrow spatial extent. The abrupt difference in molecular structure between the phases on either side of the interface, that makes them so interest- ing, is extremely challenging from a theoretical point of view. Techniques for the study of both the charged metal surface [5,6] and water from first principles ([7] and references therein) have been made available only recently. No fully ab initio simulation yet exists of the charged interface, only for the neutral case [8,9]. The study of water adsorption over metallic surfaces provides valuable information that may help the understanding of the electrochemical interface and there is abundant experimental in- formation available [10,11]. A number of first * Tel.: +44-28-90273557; fax: +44-28-90241958. E-mail address: [email protected] (C.G. S anchez). 0039-6028/03/$ - see front matter Ó 2003 Elsevier Science B.V. All rights reserved. doi:10.1016/S0039-6028(03)00080-3 Surface Science 527 (2003) 1–11 www.elsevier.com/locate/susc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Molecular reorientation of water adsorbed on chargedAg(1 1 1) surfaces
Cristi�aan G. S�aanchez *
Atomistic Simulation Group, School of Mathematics and Physics, Physics Building, Queen�s University Belfast, University Road,
BT7 1NN Belfast, Northern Ireland, UK
Received 8 November 2002; accepted for publication 22 January 2003
Abstract
In this work we present first principles calculations of water adsorption over charged Ag(1 1 1) surfaces. The ori-
entation of the adsorbed water molecule with respect to the surface changes from oxygen pointing away from the
surface at negative charges to oxygen pointing towards the surface at positive charges. At zero charge the water
molecule is oriented approximately parallel to the surface plane. Complete orientation of the molecule in the direction
of the field is achieved for a critical charge density of 15 lCcm�2 for both positive and negative charges.
� 2003 Elsevier Science B.V. All rights reserved.
Keywords: Density functional calculations; Chemisorption; Water; Silver; Low index single crystal surfaces; Solid–liquid interfaces
1. Introduction
Double layer modelling plays a fundamental
role in theoretical electrochemistry since an accu-
rate knowledge of the double layer is needed in any
attempt to describe other electrochemical phe-
nomena such as charge transfer processes. Manydifferent approaches have been applied to the study
of this subject, ranging from first principles mo-
lecular dynamics to Monte Carlo simulations.
These works have been extensively reviewed re-
cently [1–4]. In spite of the effort dedicated there is
not a general consensus on a model for the elect-
rochemical double layer, and existing models have
difficulties to rationalise all of the abundant body
of experimental data. This limited success can be
ascribed to the inherent complexity of the problem.
The interface is a highly anisotropic environment in
which large changes of composition occur within a
very narrow spatial extent. The abrupt difference in
molecular structure between the phases on either
side of the interface, that makes them so interest-ing, is extremely challenging from a theoretical
point of view. Techniques for the study of both the
charged metal surface [5,6] and water from first
principles ([7] and references therein) have been
made available only recently. No fully ab initio
simulation yet exists of the charged interface, only
for the neutral case [8,9].
The study of water adsorption over metallicsurfaces provides valuable information that may
help the understanding of the electrochemical
interface and there is abundant experimental in-
formation available [10,11]. A number of first
* Tel.: +44-28-90273557; fax: +44-28-90241958.
E-mail address: [email protected] (C.G. S�aanchez).
0039-6028/03/$ - see front matter � 2003 Elsevier Science B.V. All rights reserved.
doi:10.1016/S0039-6028(03)00080-3
Surface Science 527 (2003) 1–11
www.elsevier.com/locate/susc

principles theoretical studies have addressed this
system. Paredes Olivera et al. [14] present some
results on the adsorption energy of water on the
Ag(1 1 1) surface. The aim of this work was the
study of hydronium adsorption. The authors could
not determine the adsorption geometry of the watermolecule due to the small energy difference ob-
tained between the upright and flat configurations.
Izvekov and Voth [9] have performed first princi-
ples molecular dynamics of the water/Ag(1 1 1) in-
terface and present some result on the adsorption
of a single molecule. Kua andGoddard [15] address
the problem of methanol oxidation and show re-
sults for water adsorption onPt(1 1 1). Ohwaki et al.[16] have studied water adsorption on Pt(1 1 1) and
Pt(1 0 0). This authors also studied the effect of the
electric field. Two different fields corresponding to
positive and negative charges of about 10 lCcm�2
were used. Their results indicate that for the posi-
tive field the tilting of the water molecule increases
to 41 degrees and for the negative field the water is
normal to the surface with the hydrogen atomspointing to the surface. Jin and Head [17] studied
water adsorption on Al(1 1 1) and Ignaczak and
Gomes [18] on Cu(1 0 0), Ag(1 0 0) and Au(1 0 0)
surfaces. The results presented in this works for
water orientation and adsorption energy are com-
piled in Table 1 together with the model used to
represent the electronic structure of the metal–
water system. Previous work about these and othersurfaces is described by Nazmutdinov and Shapnik
[19].
All these works (with the exception of [9]) have
modelled the metallic surface by means of finite
clusters. The cluster method is a valuable tool for
the study of adsorption phenomena related to the
electrochemical interface [20]. This approach how-
ever, has some limitations that have been ad-dressed by us elsewhere [21] and are discussed by
the authors in some on these works [14,16]. The
most important ones are the oscillatory depen-
dence of adsorption energies on cluster size and
the cluster dipole moment. Although these prob-
lems can be solved in part by carefully preparing
the cluster�s electronic state and geometry, themethod used in the present work which models theinfinite surface with periodic boundary conditions
is more adequate to describe the electronic struc-
ture of the metal.
The presence of water on the electrochemical
interface is often neglected, but it is known to
profoundly affect processes occurring at electrodes
[22]. Since the early flip-flop models [23] the pic-
ture of water molecules close to the electrodechanging their orientation from oxygen-up to oxy-
gen-down in response to the electric field has
dominated our understanding of the electrochem-
ical double layer. These ideas have been confirmed
by experimental data such as X-ray diffraction [24]
and IR spectroscopy [25–27] (see also [10] and
references therein). But, to the best of our knowl-
edge, up to now no first principles results existto support this ideas. In this work we present re-
sults on the relative orientation with respect to the
Table 1
Compilation of first principles results existing in the literature for water adsorption on different metallic surfaces
Surface Method Angle (deg) Eads (eV) Ref.
Ag(1 1 1) Cluster, 28 atoms, MP2 undetermined 0.35 [14]
Ag(1 1 1) Super-cell, PW, BLYP 30 0.53 [9]
Pt(1 1 1) Cluster, 8 atoms, B3LYP 20 0.70 [15]
Pt(1 1 1) Cluster, 7 atoms, B3LYP 16.5 0.56 [16]
Al(1 1 1) Cluster, 10 atoms, ROHF 90 0.56–0.87 [17]
Cu(1 0 0) Cluster, 12 atoms, B3LYP 35 0.31 [18]
Ag(1 0 0) Cluster, 12 atoms, B3LYP 40 0.27 [18]
Au(1 0 0) Cluster, 12 atoms, B3LYP 25 0.30 [18]
Ag(1 1 1) Super-cell, PBE )17 0.2 This work
Positive angles indicate that the oxygen atom is pointing towards the surface and negative ones that the oxygen atom is pointing away
from the surface. The acronyms used in the method column are as follows: MP2 ¼ Moller–Plesset second-order perturbation theory,PW ¼ plane wave basis set, BLYP ¼ density functional theory using BLYP GGA functional, B3LYP ¼ Becke 3 parameter hybrid
Hartree–Fock DFT functional, PBE ¼ DFT using PBE GGA functional.
2 C.G. S�aanchez / Surface Science 527 (2003) 1–11

surface of a water molecule adsorbed on a charged
Ag(1 1 1) surface as a function of charge. The
critical charge necessary to achieve full orientation
of the water molecule in the direction of the field is
obtained. Fully ab initio calculations are compu-
tationally very demanding and hence at present theproblem of a changed electrode in contact with
water containing ions at a non-zero temperature
cannot be calculated exactly. The work described
in this paper represents a first step towards the
fully ab initio description of the charged electro-
chemical interface.
2. The model
The electronic structure is treated quantum me-
chanically using standard state-of-the-art methods
[21,29]. In this section we give the technical details
necessary to reproduce the work. In order to rep-
resent the surface we use a slab geometry and
periodic boundary conditions. The metal is repre-sented by a four layer slab with the lattice param-
eter obtained from the bulk calculation. Water is
adsorbed on both sides of the slab. Although we are
interested in studying an isolated water molecule
adsorbed on the surface our model requires the use
of a periodic system. We require a super-cell of the
lowest possible coverage so as to avoid water–water
interactions. We use a ð2� 2Þ surface unit cell,which is equivalent to a water monolayer with
coverage degree of 1/4. Test calculations made for a
ð3� 3Þ unit cell (coverage degree of 1/9) showed novariation on the adsorption geometries obtained
and hence we do not expect that the presence of
water molecules in neighbouring cells would affect
the results significantly. A calculation of the ð2� 2Þwater monolayer isolated in the vacuum has anenergy per molecule 0.01 eV lower than the isolated
water molecule, and this correction has been taken
into account for the calculation of the adsorption
energy at zero charge. A vacuum space equivalent
to six (1 1 1) layers separates periodic images of the
metal slab on the direction parallel to the surface.
The size of the super-cell in the direction normal to
the surface is 24.3�AA, this gives a separation betweenwater molecules in neighbouring cells in the direc-
tion normal to the surface of 11.6 �AA. The super-cell
used is shown in Fig. 1. The outermost layer of the
substrate was allowed to relax in all optimisations.
To model the electronic structure of the system
we use the generalized gradient approximation
(GGA) to density functional theory (DFT) in the
Perdew–Burke–Ernzerhof (PBE) [28] form. Cal-
culations were carried out using the programSIESTA [30,31] a code designed for DFT calcu-
lations in systems with a large number of atoms.
The method used to include the effects of the
electric field has been described in detail elsewhere
[5,6] in the context of plane wave calculations, and
has been adapted by us to the SIESTA code. The
method relies on the inclusion of a charged plane
at the boundary of the cell parallel to the surface;bearing a charge equal in magnitude but opposite
sign to the charge on the slab. Since overall the
system (metallic slab plus charged plane) is neu-
tral, only dipole–dipole and higher multi-pole
moment interactions can occur between periodic
images as in any slab calculation. The dipole–
dipole interaction is avoided by using a symmetric
slab with no net dipole moment. This is achievedby adsorbing water molecules on both sides of a
symmetric slab placed at the centre of the cell.
Albeit no symmetry restriction is imposed during
the geometry optimisation the symmetry is main-
tained within 0.001 �AA.
Fig. 1. The surface super-cell used in the calculations corre-
sponds to the area enclosed by the black polygon. This is a
ð2� 2Þ surface unit cell with an equivalent water coverage of1/4.
C.G. S�aanchez / Surface Science 527 (2003) 1–11 3

Norm conserving Troullier–Martins [32] pseudo-
potentials were used to represent the atomic cores.
We have used a basis set composed of pseudo-
atomic orbitals for the expansion of the Kohn–
Sham orbitals. This set is constructed using a
generalisation of the method proposed by Sankeyand Niklewski [33], which consists of solving the
problem of the pseudo-atom with the boundary
condition that the electronic valence wave func-
tions vanish beyond a certain cutoff radius. In this
way, the number of non-zero Hamiltonian matrix
elements is dramatically reduced. To give more
flexibility to the basis set, a second group of va-
lence orbitals and a set of polarisation orbitals areadded to the valence set. This corresponds to a
double-f plus polarisation (DZP) basis set in theusual quantum chemistry terminology, with po-
larisation functions for both heavy elements and
hydrogen. The procedure used to construct this set
is described in detail elsewhere [34,35]. The local-
isation radius of the basis set (corresponding to an
energy shift of 0.025 eV [33–35]) was chosen as acompromise between accuracy and computational
efficiency. Further increase of the localisation ra-
dius changed adsorption energies in less than 0.05
eV, the change in forces being smaller than the
tolerance for geometry optimisation (0.005 eV/�AA).In order to check for the accuracy provided by the
basis and pseudo-potential, calculations for bulk
silver and an isolated water molecule were per-formed. For bulk silver we obtained a lattice
constant of 4.21 �AA and a bulk modulus of 0.83Mbar, which can be compared to the experimen-
tal values of 4.09 �AA and 1.04 Mbar respectively.The relatively expanded structure and small bulk
modulus are a result of the application of the
GGA. A plane wave calculation performed with
the Carr–Parrinello moleclar dynamics (CPMD)[36] code with an essentially complete basis set
(using a cutoff of 70 Ry) gives values of 4.20 �AA and0.82 Mbar for the lattice constant and bulk mod-
ulus respectively. The same plane wave calculation
using the local density approximation (LDA) gives
4.05 �AA and 1.29 Mbar, much improving the latticeconstant but not so the bulk modulus, which is off
by a similar magnitude but opposite sign. It is awell know fact that GGA works better for mo-
lecular systems and LDA does a better job for
some condensed matter systems due to a fortuitous
cancellation of errors [37]. However, in order to
describe water properly, which is the main objec-
tive of the present study, a GGA approximation
was required. For the isolated water molecule we
obtain an OH distance of 0.979 �AA and an HOHangle of 103.9 degrees. The experimental values
are 0.957 �AA and 104.5 respectively and the planewave results obtained for a 130 Ry cutoff are 0.969�AA and 104.3 degrees.
3. Results and discussion
The geometry obtained for water adsorbed over
the neutral surface is shown in Fig. 2a and b. No
attempt was made to optimise the structure at
adsorption sites other than top since from our own
calculations [38] and the results published in the
literature [9,11] this is the most stable site. The
geometry reported by Izvekov and Voth [9] was
used as a starting point for the optimisation. In theoptimised structure the oxygen atom is slightly
displaced from the top site and the hydrogen
atoms point slightly towards the surface. The mole-
cular plane forms an angle of 17 degrees with re-
spect to the plane of the substrate. The relaxation
of the metal layer is very small. The silver atom
closest to the water molecule shows an inward
relaxation of about 0.01 �AA. This result is in con-trast with the geometry normally assumed for ex-
perimental evidence [11] for adsorption at UHV,
where the tilt is interpreted to be in the opposite
direction. Similarly, for the electrochemical inter-
face the temperature coefficient of the potential of
zero charge seems to indicate a natural orientation
of water molecules with the oxygen atom pointing
towards the surface [12]. Experimental UHV data,however, is affected by the formation of water
clusters and should be taken with care in order to
predict the structure of water adsorbed at low cov-
erage. With respect to the thermal coefficient of the
potential of zero charge it has been suggested that it
can be explained from the metal electronic contri-
bution [13].
Results obtained from cluster calculations forother surfaces in general agree that the water
molecule is tilted with the hydrogen atoms point-
4 C.G. S�aanchez / Surface Science 527 (2003) 1–11

ing away from the surface [15,16,18] with the ex-
ception of the Al(1 1 1) surface [17] for which theresults indicate orientation normal to the surface.
Previous first principles results for the Ag(1 1 1)
surface are inconclusive with respect to the ad-
sorption geometry. In Ref. [14], as mentioned in
Section 1, the tilting angle could not be obtained
because of the small energy difference between
different orientations, and this was attributed to a
limitation of the cluster model. Most cluster cal-culations avoid full geometry optimisation; nor-
mally the internal geometry of the adsorbate and
the adsorption site are constrained in order to save
computational time and no attempt to relax the
substrate is made. Furthermore, the experimental
lattice constant of the metal is used, which can be
different from the optimum lattice constant for the
model used. These approximations may be im-portant to determine the final water configuration.
The result that is more comparable to ours in
terms of the model used is that of Ref. [9]. Thegeometry obtained by these authors differs from
ours in that their results show the water molecule
tilted outwards at an angle of 30 degrees with re-
spect to the surface plane. This discrepancy might
be attributed to the lower (less accurate) plane
wave cutoff energy used in Ref. [9] (60 Ry). In our
experience cutoffs of over 90 Ry are needed in
order to ensure proper convergence of the forceswhen norm conserving Troullier–Martins pseudo-
potentials are used for oxygen. We have performed
plane wave calculations using the CPMD code
with a plane wave energy cutoff of 70 Ry and ob-
tained an adsorption geometry very similar to the
one presented here. All of this can be rationalised
from the fact that the potential energy surface for
water adsorption over Ag is a relatively shallowfunction of the tilt angle and molecular position
Fig. 2. Water orientation with respect to the substrate. For the sake of clarity only a ð1� 1Þ surface unit cell of substrate is shown(note that the unit cell used for the calculation is a ð2� 2Þ cell, 4 times larger, as shown in Fig. 1). (a) Zero charge, top view; (b) zerocharge, side view; (c) charge density of 15.7 lCcm�2, top view; (d) charge density of 15.7 lCcm�2, side view; (e) charge density of )15.7lCcm�2, top view; (f) charge density of )15.7 lCcm�2, side view.
C.G. S�aanchez / Surface Science 527 (2003) 1–11 5
and hence it is difficult to obtain a conclusive op-
timum geometry. The high surface diffusion rate
experimentally observed is an evidence of this
characteristic of the potential energy surface. With
respect to the adsorption energy, we can estimate a
value of )0.2 eV, taking into account the basis setsuperposition error by means of the counterpoise
method. The result obtained is comparable to
previous theoretical [9,14,18] and experimental
[10,11] results, the later being somewhat larger
owing to water–water interaction as already men-
tioned.
The most important results in this work concern
the adsorption over charged surfaces. A total of 10different surface charges where studied between
)15.7 and +15.7 lCcm�2 corresponding to an
excess of between 0.0075 and )0.0075 electrons persurface atom. For a Ag(1 1 1) surface in contact
with a 50 mM KClO4 solution the highest negative
and positive charges used correspond to potentials
of approximately )1.5 and )0.5 V vs. SCE re-
spectively [40]. The convergence criterion used forthe optimisation of the charged systems was 0.03
eV/�AA tolerance in the residual forces (for the
neutral surface a tolerance of 0.005 eV/�AA was
used). Once again due to the smooth variation in
energy with orientation the use of the looser con-
vergence criterion gives an uncertainty in the angle
formed with respect to the surface. This uncer-
tainty is less than �1 degree for positive and about�6 degrees for negative charges. For the sake ofconsistency, the results for the neutral surface
shown in Figs. 3, 6 and 7 are those obtained with
the looser criterion.
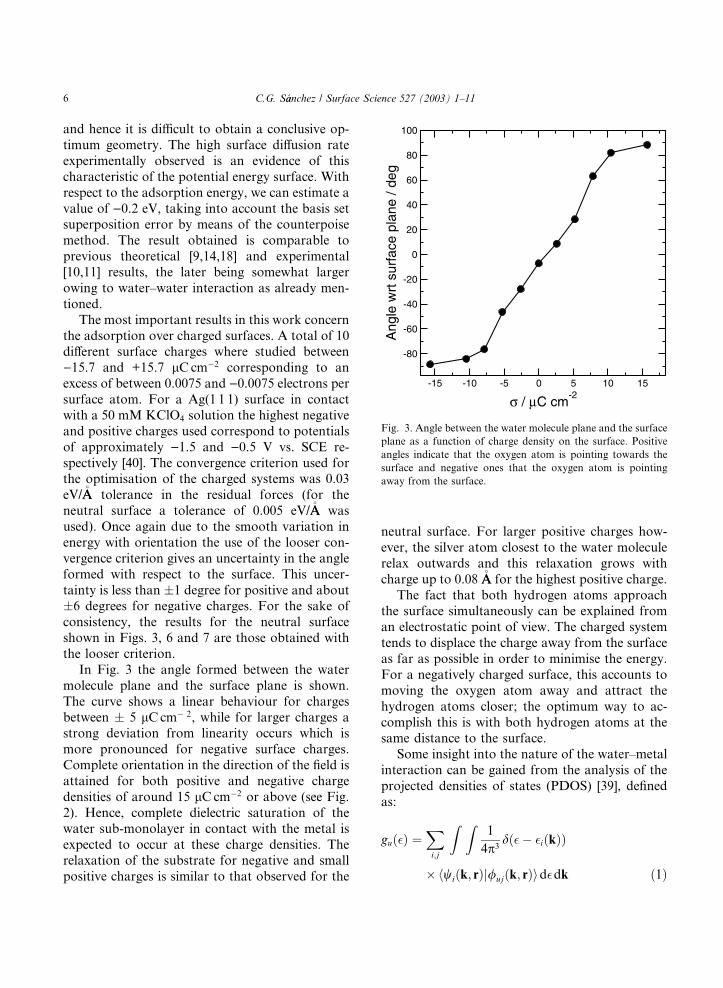
In Fig. 3 the angle formed between the water
molecule plane and the surface plane is shown.
The curve shows a linear behaviour for charges
between � 5 lCcm� 2, while for larger charges astrong deviation from linearity occurs which is
more pronounced for negative surface charges.
Complete orientation in the direction of the field is
attained for both positive and negative charge
densities of around 15 lCcm�2 or above (see Fig.
2). Hence, complete dielectric saturation of the
water sub-monolayer in contact with the metal is
expected to occur at these charge densities. Therelaxation of the substrate for negative and small
positive charges is similar to that observed for the
neutral surface. For larger positive charges how-
ever, the silver atom closest to the water molecule
relax outwards and this relaxation grows with
charge up to 0.08 �AA for the highest positive charge.The fact that both hydrogen atoms approach
the surface simultaneously can be explained from
an electrostatic point of view. The charged system
tends to displace the charge away from the surface
as far as possible in order to minimise the energy.
For a negatively charged surface, this accounts to
moving the oxygen atom away and attract the
hydrogen atoms closer; the optimum way to ac-
complish this is with both hydrogen atoms at thesame distance to the surface.
Some insight into the nature of the water–metal
interaction can be gained from the analysis of the
projected densities of states (PDOS) [39], defined
as:
guð�Þ ¼Xi;j
Z Z1
4p3dð�� �iðkÞÞ
� hwiðk; rÞj/ujðk; rÞid�dk ð1Þ
-15 -10 -5 0 5 10 15
σ / µC cm-2
-80
-60
-40
-20
0
20
40
60
80
100
Ang
le w
rt s
urfa
ce p
lane
/ de
g
Fig. 3. Angle between the water molecule plane and the surface
plane as a function of charge density on the surface. Positive
angles indicate that the oxygen atom is pointing towards the
surface and negative ones that the oxygen atom is pointing
away from the surface.
6 C.G. S�aanchez / Surface Science 527 (2003) 1–11

where /ujðk; rÞ represents the basis set orbital j onatom u, wiðk; rÞ the i Kohn–Sham orbital with ei-genvalue �iðkÞ and the integration on k runs over
the Brillouin zone. In practice, the integration over
the Brillouin zone is replaced by a finite sum, andDirac�s d function is replaced by a Gaussian ofwidth r (0.25 eV was used for the PDOS shownhere):
guð�Þ ¼Xi;j;k
Zwk
1
rffiffiffiffiffiffi2p
p
� exp �� �iðkÞ2r2
� �hwiðk; rÞj/ujðk; rÞid�
ð2Þ
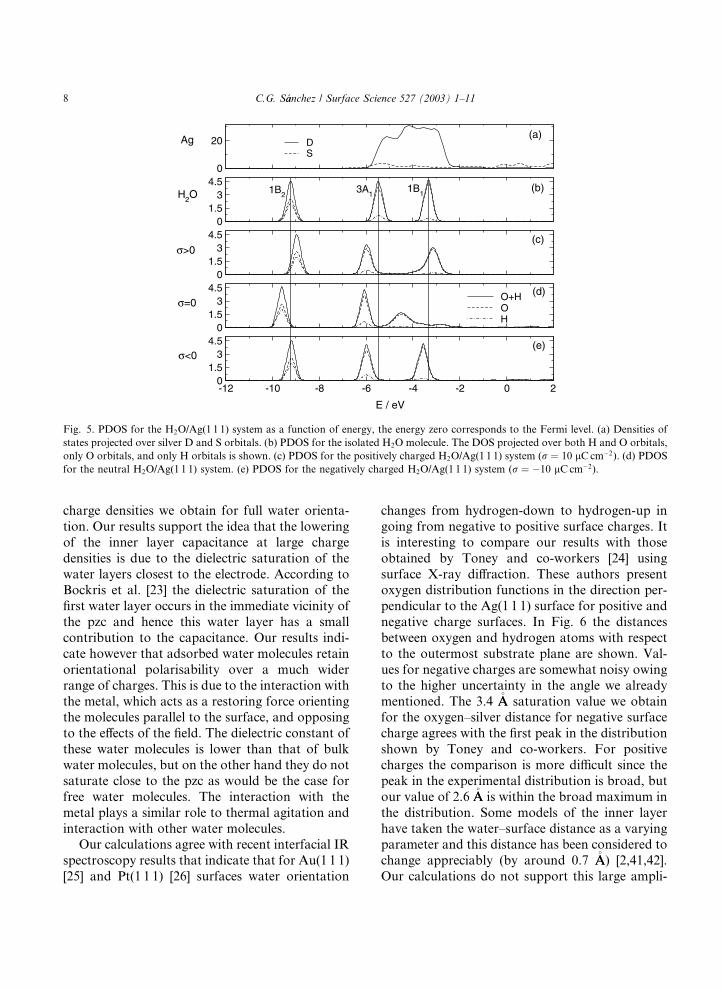
guð�Þ represents the portion of the total density ofstates due to orbitals on atom u. In Fig. 5b thedensity of states of the water molecule is shown,the three highest occupied molecular orbitals can
be seen. Iso-surface plots for these orbitals are
shown in Fig. 4. In Fig. 5b the total DOS is di-
vided into H and O contributions. From this we
can see that orbital 1B2 is formed from almost
equal contributions from oxygen and hydrogen
atomic orbitals. Orbital 3A1 (the r lone pair) has asmall contribution from H orbitals which is evensmaller for 1B1 (the p lone pair). 1B2 is the main rbonding orbital between O and H atoms, orbitals
3A1 and 1B1 are basically oxygen lone pairs. Upon
adsorption on the neutral surface (Fig. 5d) both
1B2 and 3A1 orbitals are stabilised but the most
important changes occur in the 1B1 orbital. This
orbital interacts mainly with the d-band (shown in
Fig. 5a) producing significant stabilisation andwidening. This fact explains that the most stable
orientation of the water molecule over the neutral
surface is almost horizontal. This is the orientation
that allows maximum overlap of the 1B1 orbital
with the surface. Integration of the OþH PDOSfrom )7 eV up to the Fermi level indicates that theoccupation of these orbitals increases, henceforth asmall negative charge (�0.02 electrons) is trans-ferred to the water orbitals. The localisation of this
charge is however, difficult to assess from the
analysis of the PDOS. Upon charging of the sur-
face the interaction with the electric field is even-
tually dominant and a perpendicular orientation is
attained. In this orientations the overlap between
the 1B1 orbital with the surface is smaller and itremains almost unchanged. The interaction is not
equivalent for positive and negative charges (com-
pare 4c and 4e) and some mixing can be appreci-
ated for positive charges when the orbital is closer
to the surface. Other differences exist between
positive and negative charges. While orbital 3A1energy remains almost unchanged the 1B2 orbital
energy is higher with respect to neutral charge andthis effect is more important for positive charges.
The same trend is observed for the 1B1 orbital.
This changes in orbital energies may be a result of
the combination of both orientation change and
charge effects. From the integration of the PDOS
in Fig. 5a change in occupation of around 0.1
electrons can be observed for both 3A1 and 1B1orbitals. This occupation increase is positive fornegative charge and the opposite holds for positive
surface charge.
In Fig. 7 of Ref. [40] Valette shows a plot of the
inner layer capacity of the water/Ag(1 1 1) interface
as a function of charge density. The inner layer
capacitance has a maximum at the potential of
zero charge (pzc) and levels off at about the same
Fig. 4. The three highest occupied molecular orbitals of H2O. The energy order is 1B1 > 3A1 > 1B2.
C.G. S�aanchez / Surface Science 527 (2003) 1–11 7
charge densities we obtain for full water orienta-
tion. Our results support the idea that the lowering
of the inner layer capacitance at large charge
densities is due to the dielectric saturation of the
water layers closest to the electrode. According toBockris et al. [23] the dielectric saturation of the
first water layer occurs in the immediate vicinity of
the pzc and hence this water layer has a small
contribution to the capacitance. Our results indi-
cate however that adsorbed water molecules retain
orientational polarisability over a much wider
range of charges. This is due to the interaction with
the metal, which acts as a restoring force orientingthe molecules parallel to the surface, and opposing
to the effects of the field. The dielectric constant of
these water molecules is lower than that of bulk
water molecules, but on the other hand they do not
saturate close to the pzc as would be the case for
free water molecules. The interaction with the
metal plays a similar role to thermal agitation and
interaction with other water molecules.Our calculations agree with recent interfacial IR
spectroscopy results that indicate that for Au(1 1 1)
[25] and Pt(1 1 1) [26] surfaces water orientation
changes from hydrogen-down to hydrogen-up in
going from negative to positive surface charges. It
is interesting to compare our results with those
obtained by Toney and co-workers [24] using
surface X-ray diffraction. These authors presentoxygen distribution functions in the direction per-
pendicular to the Ag(1 1 1) surface for positive and
negative charge surfaces. In Fig. 6 the distances
between oxygen and hydrogen atoms with respect
to the outermost substrate plane are shown. Val-
ues for negative charges are somewhat noisy owing
to the higher uncertainty in the angle we already
mentioned. The 3.4 �AA saturation value we obtainfor the oxygen–silver distance for negative surface
charge agrees with the first peak in the distribution
shown by Toney and co-workers. For positive
charges the comparison is more difficult since the
peak in the experimental distribution is broad, but
our value of 2.6 �AA is within the broad maximum inthe distribution. Some models of the inner layer
have taken the water–surface distance as a varyingparameter and this distance has been considered to
change appreciably (by around 0.7 �AA) [2,41,42].Our calculations do not support this large ampli-
0
20 DS
01.5
34.5
01.5
34.5
01.5
34.5
O+HOH
-12 -10 -8 -6 -4 -2 0 2
E / eV
01.5
34.5
Ag
H2O
σ>0
σ=0
σ<0
1B23A1
1B1
(a)
(b)
(c)
(d)
(e)
Fig. 5. PDOS for the H2O/Ag(1 1 1) system as a function of energy, the energy zero corresponds to the Fermi level. (a) Densities of
states projected over silver D and S orbitals. (b) PDOS for the isolated H2O molecule. The DOS projected over both H and O orbitals,
only O orbitals, and only H orbitals is shown. (c) PDOS for the positively charged H2O/Ag(1 1 1) system (r ¼ 10 lCcm�2). (d) PDOS
for the neutral H2O/Ag(1 1 1) system. (e) PDOS for the negatively charged H2O/Ag(1 1 1) system (r ¼ �10 lCcm�2).
8 C.G. S�aanchez / Surface Science 527 (2003) 1–11
tude, but the tendency obtained is the same. Even
if the O–Ag distance experiences variations of the
order of 0.8 �AA, the centre of charge of the watermolecule remains quite still, varying about 0.2 �AAon going from negative to positive charges. The
molecule is closer to the surface for positive
charges.
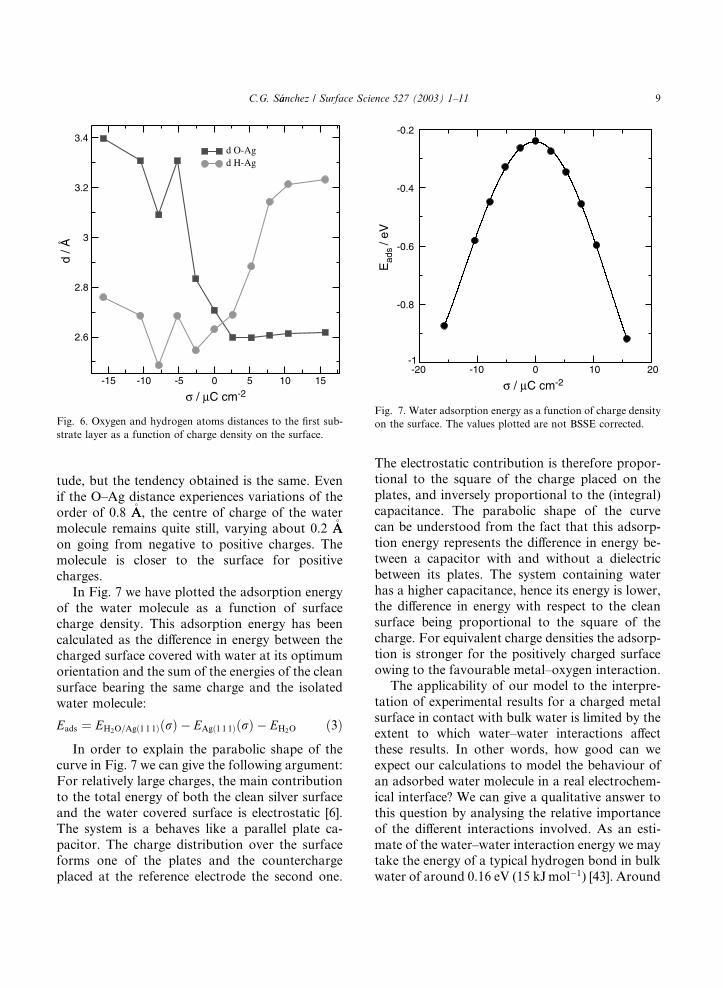
In Fig. 7 we have plotted the adsorption energy
of the water molecule as a function of surface
charge density. This adsorption energy has been
calculated as the difference in energy between thecharged surface covered with water at its optimum
orientation and the sum of the energies of the clean
surface bearing the same charge and the isolated
water molecule:
Eads ¼ EH2O=Agð1 1 1ÞðrÞ � EAgð1 1 1ÞðrÞ � EH2O ð3Þ
In order to explain the parabolic shape of thecurve in Fig. 7 we can give the following argument:
For relatively large charges, the main contribution
to the total energy of both the clean silver surface
and the water covered surface is electrostatic [6].
The system is a behaves like a parallel plate ca-
pacitor. The charge distribution over the surface
forms one of the plates and the countercharge
placed at the reference electrode the second one.
The electrostatic contribution is therefore propor-tional to the square of the charge placed on the
plates, and inversely proportional to the (integral)
capacitance. The parabolic shape of the curve
can be understood from the fact that this adsorp-
tion energy represents the difference in energy be-
tween a capacitor with and without a dielectric
between its plates. The system containing water
has a higher capacitance, hence its energy is lower,the difference in energy with respect to the clean
surface being proportional to the square of the
charge. For equivalent charge densities the adsorp-
tion is stronger for the positively charged surface
owing to the favourable metal–oxygen interaction.
The applicability of our model to the interpre-
tation of experimental results for a charged metal
surface in contact with bulk water is limited by theextent to which water–water interactions affect
these results. In other words, how good can we
expect our calculations to model the behaviour of
an adsorbed water molecule in a real electrochem-
ical interface? We can give a qualitative answer to
this question by analysing the relative importance
of the different interactions involved. As an esti-
mate of the water–water interaction energy we maytake the energy of a typical hydrogen bond in bulk
water of around 0.16 eV (15 kJmol�1) [43]. Around
-15 -10 -5 0 5 10 15
σ / µC cm-2
2.6
2.8
3
3.2
3.4
d / Å
d O-Agd H-Ag
Fig. 6. Oxygen and hydrogen atoms distances to the first sub-
strate layer as a function of charge density on the surface.
-20 -10 0 10 20
σ / µC cm-2
-1
-0.8
-0.6
-0.4
-0.2
Ead
s / e
V
Fig. 7. Water adsorption energy as a function of charge density
on the surface. The values plotted are not BSSE corrected.
C.G. S�aanchez / Surface Science 527 (2003) 1–11 9

the point of zero charge, the water–metal interac-
tion energy is comparable to the water–water in-
teraction and hence we expect the interaction with
other adsorbed molecules, and molecules from the
solution side, to be important in determining the
structure. At higher charges however, the interac-tion with the metal and the electric field is much
stronger than the interaction with other water
molecules. Therefore we argue that our results
better represent the behaviour of water at large
surface charge densities. The problem with bulk
water is that it cannot be modelled by a single
distribution of water molecules, but a statistical
sampling is necessary. First principles moleculardynamics or Monte Carlo simulations of the
charged metal–water interface would provide a
definitive answer about the extent to which water–
water and water–metal interactions and thermal
agitation determine the structure of the inner layer.
Work in this direction is underway.
4. Conclusions
We have studied the adsorption of a water
molecule on charged Ag(1 1 1) surfaces. The ori-
entation relative to the surface for different surface
charge densities was obtained. The metal–water
bond is due to the interaction of the 1B1 oxygen
lone pair with the surface, which is optimal whenthe water molecule is parallel to the surface. The
results agree with experimental findings that water
orientation in the electrochemical interface chan-
ges from oxygen-up to oxygen-down on going
from negative to positive charges. The critical
surface charge needed for full orientation with the
field has been found to be around 15 lCcm�2. This
supports the idea that the lowering and levellingof the inner layer capacitance at this charge den-
sities is due to dielectric saturation of the water
layer closest to the electrode. These results pro-
vide new and important information about the
role of the water–metal interaction in determining
the properties of the inner layer. Results concern-
ing the internal geometry of the water molecule,
vibrational frequencies and the nature of the me-tal–water bond will be published in detail else-
where.
Acknowledgements
The author is grateful to Ruth M. Lynden-Bell,
Alexander Y. Lozovoi and Jorge J. Kohanoff for
fruitful discussions and to the authors of the SI-ESTA program who kindly provided their code.
This work was supported by EPSRC through
grant GR/M03931.
References
[1] R. Parsons, Chem. Rev. 90 (1990) 813.
[2] W. Schmickler, Chem. Rev. 96 (1996) 3177.
[3] W. Schmickler, Ann. Rep. Prog. Chem. Sect. C 95 (1999)
117.
[4] R. Guidelli, W. Schmickler, Electrochim. Acta 45 (1999)
2317.
[5] A.Y. Lozovoi, A. Alavi, J. Kohanoff, R.M. Lynden-Bell, J.
Chem. Phys. 115 (2001) 1661.
[6] A.Y. Lozovoi, A. Alavi, submitted for publication.
[7] P. Vassilev, C. Hartnig, M.T.M. Koper, F. Frechard, A.R.
van Santen, J. Chem. Phys. 115 (2001) 9815.
[8] S. Izvekov, A. Mazzolo, K. VanOpdorp, G. Voth, J. Chem.
Phys. 114 (2001) 3248.
[9] S. Izvekov, G.A. Voth, J. Chem. Phys. 115 (2001) 7196.
[10] M.A. Henderson, Surf. Sci. Rep. 46 (2002) 1.
[11] P.A. Thiel, T.E. Madey, Surf. Sci. Rep. 7 (1987) 211.
[12] A. Hamelin, L. Doubova, L. Stoicoviciu, S. Trasatti, J.
Electroanal. Chem. 244 (1988) 133.
[13] R. Guidelli, G. Aloisi, E. Leiva, W. Schmickler, J. Phys.
Chem. 92 (1988) 6671.
[14] P. Paredes Olivera, A. Ferral, E.M. Patrito, J. Phys. Chem.
B 105 (2001) 7227.
[15] J. Kua, W.A. Goddard III, J. Am. Chem. Soc. 121 (1999)
10928.
[16] T. Ohwaki, K. Kamegai, K. Yamashita, Bull. Chem. Soc.
Jpn. 74 (2001) 1021.
[17] S. Jin, J.D. Head, Surf. Sci. 318 (1994) 204.
[18] A. Ignaczak, J.A.N.F. Gomes, J. Electroanal. Chem. 420
(1997) 209.
[19] R.R. Nazmutdinov, M.S. Shapnik, Electrochim. Acta 41
(1996) 2253.
[20] C. S�aanchez, E.P.M. Leiva, in: W. Vielstich, A. Lamm, H.
Gasteiger (Eds.), Handbook of Fuel Cells: Fundamentals,
Technology and Applications, vol. 2, John Wiley & Sons,
Chichester, 2003 (Chapter 12).
[21] C. S�aanchez, E.P.M. Leiva, in: W. Vielstich, A. Lamm, H.
Gasteiger (Eds.), Handbook of Fuel Cells: Fundamentals,
Technology and Applications, vol. 2, John Wiley & Sons,
Chichester, 2003 (Chapter 3).
[22] C. S�aanchez, E.P.M. Leiva, in: W. Vielstich, A. Lamm, H.
Gasteiger (Eds.), Handbook of Fuel Cells: Fundamentals,
Technology and Applications, vol. 2, John Wiley & Sons,
Chichester, 2003 (Chapter 2).
10 C.G. S�aanchez / Surface Science 527 (2003) 1–11

[23] J.O�M. Bockris, A.K.N. Reddy, M. Gaboa-Aldeco, Mod-ern electrochemistry, Fundamentals of Electrodics, vol.
2A, second ed., Kluwer Academic, New York, 2000.
[24] J.G. Gordon, O.R. Melroy, M.F. Toney, Electrochim.
Acta 40 (1995) 3.
[25] K. Ataka, T. Yotsuyanagi, M. Osawa, J. Phys. Chem. 100
(1996) 10664.
[26] T. Iwasita, X. Xia, J. Electroanal. Chem. 411 (1996) 95.
[27] T. Iwasita, X.H. Xia, H.-D. Liess, W. Vielstich, J. Phys.
Chem. B 101 (1997) 7542.
[28] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77
(1996) 3865.
[29] R. Parr, W. Yang, Density Functional Theory of Atoms
and Molecules, Oxford University Press, 1989.
[30] P. Ordej�oon, E. Artacho, R. Cachau, J. Gale, A. Garc�ııa, J.
Junquera, J. Kohanoff, M. Machado, D. S�aanchez-Portal,
J.M. Soler, R. Weht, Mat. Res. Soc. Symp. Proc. 677
(2001) AA9.6.1.
[31] http://www.uam.es/departamentos/ciencias/fismateriac/
siesta/.
[32] N. Troullier, J.L. Martins, Phys. Rev. B 43 (1991) 1993.
[33] O.F. Sankey, D.J. Niklewski, Phys. Rev. B 40 (1998)
3979.
[34] D. S�aanchez-Portal, P. Ordej�oon, E. Artacho, J.M. Soler, Int.
J. Quant. Chem. 65 (1997) 453.
[35] E. Artacho, D. S�aanchez-Portal, P. Ordej�oon, A. Garc�ııa,
J.M. Soler, Phys. Status Solidi 215 (1999) 809.
[36] CPMD, version 3.3, written by J. Hutter, A. Alavi, T.
Deutsch, and the group of M. Parrinello at the MPI in
Stutgart and IBM Research Laboratory in Z€uurich.
[37] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 80
(1998) 891.
[38] C.G. S�aanchez, J.J. Kohanoff, unpublished results, 1999.
[39] R. Hoffmann, Rev. Mod. Phys. 60 (1988) 601.
[40] G. Valette, J. Electroanal. Chem. 269 (1989) 191.
[41] S. Amokrane, J.P. Badiali, Electrochim. Acta 34 (1989)
39.
[42] S. Amokrane, J.P. Badiali, J. Electroanal. Chem. 150
(1989) 315.
[43] A. Rahman, F.H. Stilinger, J. Chem. Phys. 60 (1974) 154.
C.G. S�aanchez / Surface Science 527 (2003) 1–11 11
Related Documents











