Perinatal Loss of Nkx2-5 Results in Rapid Conduction and Contraction Defects Laura E. Briggs,* Morihiko Takeda,* Adolfo E. Cuadra,* Hiroko Wakimoto,* Melissa H. Marks, Alexandra J. Walker, Tsugio Seki, Suk P. Oh, Jonathan T. Lu, Colin Sumners, Mohan K. Raizada, Nobuo Horikoshi, Ellen O. Weinberg, Kenji Yasui, Yasuhiro Ikeda, Kenneth R. Chien, Hideko Kasahara Abstract—Homeobox transcription factor Nkx2-5, highly expressed in heart, is a critical factor during early embryonic cardiac development. In this study, using tamoxifen-inducible Nkx2-5 knockout mice, we demonstrate the role of Nkx2-5 in conduction and contraction in neonates within 4 days after perinatal tamoxifen injection. Conduction defect was accompanied by reduction in ventricular expression of the cardiac voltage-gated Na channel pore-forming -subunit (Na v 1.5-), the largest ion channel in the heart responsive for rapid depolarization of the action potential, which leads to increased intracellular Ca 2 for contraction (conduction– contraction coupling). In addition, expression of ryanodine receptor 2, through which Ca 2 is released from sarcoplasmic reticulum, was substantially reduced in Nkx2-5 knockout mice. These results indicate that Nkx2-5 function is critical not only during cardiac development but also in perinatal hearts, by regulating expression of several important gene products involved in conduction and contraction. (Circ Res. 2008;103:580-590.) Key Words: conduction contraction gene targeting transcription C oordinated conduction and contraction is critical for cardiac function. Cardiac conduction is initiated by a spontaneous wave of electricity (action potential) that arises from sinoatrial (SA) nodal cells located in the upper right atrium, followed by sequential spreading of action potential to the atria, atrioventricular (AV) node, His bundles, periph- eral Purkinje fiber, and the ventricles. In the SA and AV nodes, inward Ca 2 currents are primarily responsible for slow depolarization of the action potential, whereas Na currents are primarily responsible for rapid depolarization of the action potential in other cardiomyocytes. 1–5 In both neonatal and adult mouse cardiomyocytes, robust Na currents producing the rapid upstroke action potential are tetrodotoxin-resistant. 6,7 In the mouse heart, tetrodotoxin-re- sistant Na v 1.5 is the primary isoform of the Na channel mRNA throughout development and is most highly expressed in adults; embryonic day (ED)12.5 50% to 100%, neonatal 150% to 200%, and adult 500% to 550%, in comparison to a house keeping gene. 8 Other Na channels, mostly tetrodotoxin-sensitive, are also expressed at lower levels in the heart at the 3 stages. 8 Among them, the highest expression was demonstrated for Na v 2.3 and Na v 1.4 in the adult heart but at 25% to 30% at most compared to the house keeping gene. 8 In adult mouse ventricular cardiomyocytes, 16% of Na channel transcripts are other than Na v 1.5, expressed in the order of Na v 1.4Na v 1.3Na v 1.2Na v 1.1Na v 1.6. 4 After membrane depolarization, L-type Ca 2 channels are activated, followed by Ca 2 release from intracellular Ca 2 stores in sarcoplasmic reticulum (SR) through the cardiac isoform of the ryanodine receptor (RyR2) (Ca 2 -induced Ca 2 release). Sufficient intracellular Ca 2 allows actomyosin in- teractions resulting in cardiac contraction (excitation– con- traction coupling). 1–5 Presumably, ion channels preferentially expressed in heart, such as Na v 1.5 and RyR2, are transcribed in a cardiac-specific manner. Nkx2-5, a homeodomain-containing transcription factor, is identified as a mammalian homolog of Drosophila tinman, 9 critical for cardiac mesoderm formation. Nkx2-5 is highly conserved among species and is among the earliest cardio- genic markers, 10,11 with its expression continuing throughout Original received January 10, 2008; revision received July 29, 2008; accepted July 30, 2008. From the Department of Physiology and Functional Genomics (L.E.B., M.T., A.E.C., M.H.M., A.J.W., T.S., S.P.O., C.S., M.K.R., H.K.), University of Florida College of Medicine, Gainesville; Department of Pediatrics and Developmental Biology (H.W.), Graduate School of Medicine, Tokyo Medical and Dental University, Japan; Cardiology Division (J.T.L.), University of California, San Francisco; Department of Radiation Oncology (N.H.), Washington University School of Medicine, St Louis, Mo; Cardiovascular Research (E.O.W.), Boston University Medical Center, Mass; Department of Bioinformation Analysis (K.Y.), Research Institute of Environmental Medicine, Nagoya University, Aichi, Japan; Department of Molecular Cardiovascular Biology (Y.I.), Yamaguchi University School of Medicine, Ube, Japan; and Cardiovascular Research Center (K.R.C.), Massachusetts General Hospital, Boston. *These authors contributed equally to this work. Correspondence to Hideko Kasahara, MD, PhD, University of Florida College of Medicine, 1600 SW Archer Rd, M-540, Gainesville, FL 32610-0274. E-mail [email protected] © 2008 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.171835 580 Molecular Medicine by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from by guest on May 29, 2018 http://circres.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Perinatal Loss of Nkx2-5 Results in Rapid Conductionand Contraction Defects
Laura E. Briggs,* Morihiko Takeda,* Adolfo E. Cuadra,* Hiroko Wakimoto,* Melissa H. Marks,Alexandra J. Walker, Tsugio Seki, Suk P. Oh, Jonathan T. Lu, Colin Sumners, Mohan K. Raizada,
Nobuo Horikoshi, Ellen O. Weinberg, Kenji Yasui, Yasuhiro Ikeda,Kenneth R. Chien, Hideko Kasahara
Abstract—Homeobox transcription factor Nkx2-5, highly expressed in heart, is a critical factor during early embryoniccardiac development. In this study, using tamoxifen-inducible Nkx2-5 knockout mice, we demonstrate the role ofNkx2-5 in conduction and contraction in neonates within 4 days after perinatal tamoxifen injection. Conduction defectwas accompanied by reduction in ventricular expression of the cardiac voltage-gated Na channel pore-forming-subunit (Nav1.5-), the largest ion channel in the heart responsive for rapid depolarization of the action potential,which leads to increased intracellular Ca2 for contraction (conduction–contraction coupling). In addition, expressionof ryanodine receptor 2, through which Ca2 is released from sarcoplasmic reticulum, was substantially reduced inNkx2-5 knockout mice. These results indicate that Nkx2-5 function is critical not only during cardiac development butalso in perinatal hearts, by regulating expression of several important gene products involved in conduction andcontraction. (Circ Res. 2008;103:580-590.)
Key Words: conduction contraction gene targeting transcription
Coordinated conduction and contraction is critical forcardiac function. Cardiac conduction is initiated by a
spontaneous wave of electricity (action potential) that arisesfrom sinoatrial (SA) nodal cells located in the upper rightatrium, followed by sequential spreading of action potentialto the atria, atrioventricular (AV) node, His bundles, periph-eral Purkinje fiber, and the ventricles. In the SA and AVnodes, inward Ca2 currents are primarily responsible forslow depolarization of the action potential, whereas Na
currents are primarily responsible for rapid depolarization ofthe action potential in other cardiomyocytes.1–5
In both neonatal and adult mouse cardiomyocytes, robustNa currents producing the rapid upstroke action potential aretetrodotoxin-resistant.6,7 In the mouse heart, tetrodotoxin-re-sistant Nav1.5 is the primary isoform of the Na channelmRNA throughout development and is most highly expressedin adults; embryonic day (ED)12.5 50% to 100%, neonatal150% to 200%, and adult 500% to 550%, in comparison to ahouse keeping gene.8 Other Na channels, mostlytetrodotoxin-sensitive, are also expressed at lower levels in
the heart at the 3 stages.8 Among them, the highest expressionwas demonstrated for Nav2.3 and Nav1.4 in the adult heart butat 25% to 30% at most compared to the house keeping gene.8
In adult mouse ventricular cardiomyocytes, 16% of Na
channel transcripts are other than Nav1.5, expressed in theorder of Nav1.4Nav1.3Nav1.2Nav1.1Nav1.6.4
After membrane depolarization, L-type Ca2 channels areactivated, followed by Ca2 release from intracellular Ca2
stores in sarcoplasmic reticulum (SR) through the cardiacisoform of the ryanodine receptor (RyR2) (Ca2-induced Ca2
release). Sufficient intracellular Ca2 allows actomyosin in-teractions resulting in cardiac contraction (excitation–con-traction coupling).1–5 Presumably, ion channels preferentiallyexpressed in heart, such as Nav1.5 and RyR2, are transcribedin a cardiac-specific manner.
Nkx2-5, a homeodomain-containing transcription factor, isidentified as a mammalian homolog of Drosophila tinman,9
critical for cardiac mesoderm formation. Nkx2-5 is highlyconserved among species and is among the earliest cardio-genic markers,10,11 with its expression continuing throughout
Original received January 10, 2008; revision received July 29, 2008; accepted July 30, 2008.From the Department of Physiology and Functional Genomics (L.E.B., M.T., A.E.C., M.H.M., A.J.W., T.S., S.P.O., C.S., M.K.R., H.K.), University
of Florida College of Medicine, Gainesville; Department of Pediatrics and Developmental Biology (H.W.), Graduate School of Medicine, Tokyo Medicaland Dental University, Japan; Cardiology Division (J.T.L.), University of California, San Francisco; Department of Radiation Oncology (N.H.),Washington University School of Medicine, St Louis, Mo; Cardiovascular Research (E.O.W.), Boston University Medical Center, Mass; Department ofBioinformation Analysis (K.Y.), Research Institute of Environmental Medicine, Nagoya University, Aichi, Japan; Department of MolecularCardiovascular Biology (Y.I.), Yamaguchi University School of Medicine, Ube, Japan; and Cardiovascular Research Center (K.R.C.), MassachusettsGeneral Hospital, Boston.
*These authors contributed equally to this work.Correspondence to Hideko Kasahara, MD, PhD, University of Florida College of Medicine, 1600 SW Archer Rd, M-540, Gainesville, FL 32610-0274.
E-mail [email protected]© 2008 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.171835
580
Molecular Medicine
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
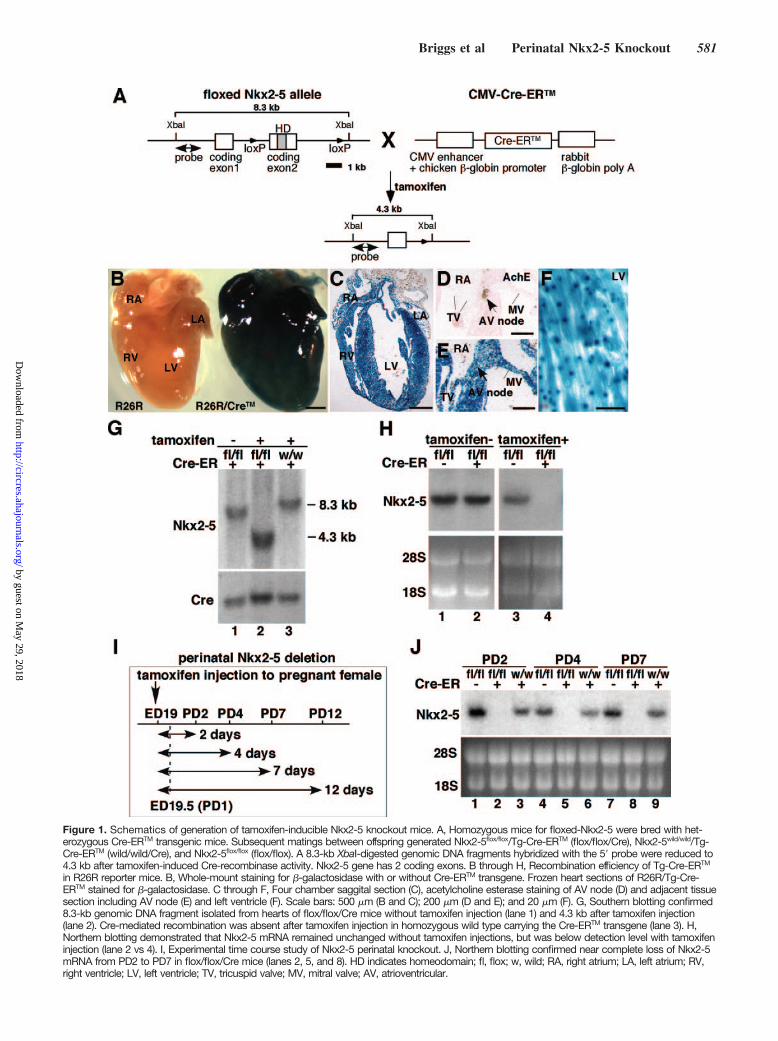
Figure 1. Schematics of generation of tamoxifen-inducible Nkx2-5 knockout mice. A, Homozygous mice for floxed-Nkx2-5 were bred with het-erozygous Cre-ERTM transgenic mice. Subsequent matings between offspring generated Nkx2-5flox/flox/Tg-Cre-ERTM (flox/flox/Cre), Nkx2-5wild/wild/Tg-Cre-ERTM (wild/wild/Cre), and Nkx2-5flox/flox (flox/flox). A 8.3-kb XbaI-digested genomic DNA fragments hybridized with the 5 probe were reduced to4.3 kb after tamoxifen-induced Cre-recombinase activity. Nkx2-5 gene has 2 coding exons. B through H, Recombination efficiency of Tg-Cre-ERTM
in R26R reporter mice. B, Whole-mount staining for -galactosidase with or without Cre-ERTM transgene. Frozen heart sections of R26R/Tg-Cre-ERTM stained for -galactosidase. C through F, Four chamber saggital section (C), acetylcholine esterase staining of AV node (D) and adjacent tissuesection including AV node (E) and left ventricle (F). Scale bars: 500 m (B and C); 200 m (D and E); and 20 m (F). G, Southern blotting confirmed8.3-kb genomic DNA fragment isolated from hearts of flox/flox/Cre mice without tamoxifen injection (lane 1) and 4.3 kb after tamoxifen injection(lane 2). Cre-mediated recombination was absent after tamoxifen injection in homozygous wild type carrying the Cre-ERTM transgene (lane 3). H,Northern blotting demonstrated that Nkx2-5 mRNA remained unchanged without tamoxifen injections, but was below detection level with tamoxifeninjection (lane 2 vs 4). I, Experimental time course study of Nkx2-5 perinatal knockout. J, Northern blotting confirmed near complete loss of Nkx2-5mRNA from PD2 to PD7 in flox/flox/Cre mice (lanes 2, 5, and 8). HD indicates homeodomain; fl, flox; w, wild; RA, right atrium; LA, left atrium; RV,right ventricle; LV, left ventricle; TV, tricuspid valve; MV, mitral valve; AV, atrioventricular.
Briggs et al Perinatal Nkx2-5 Knockout 581
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
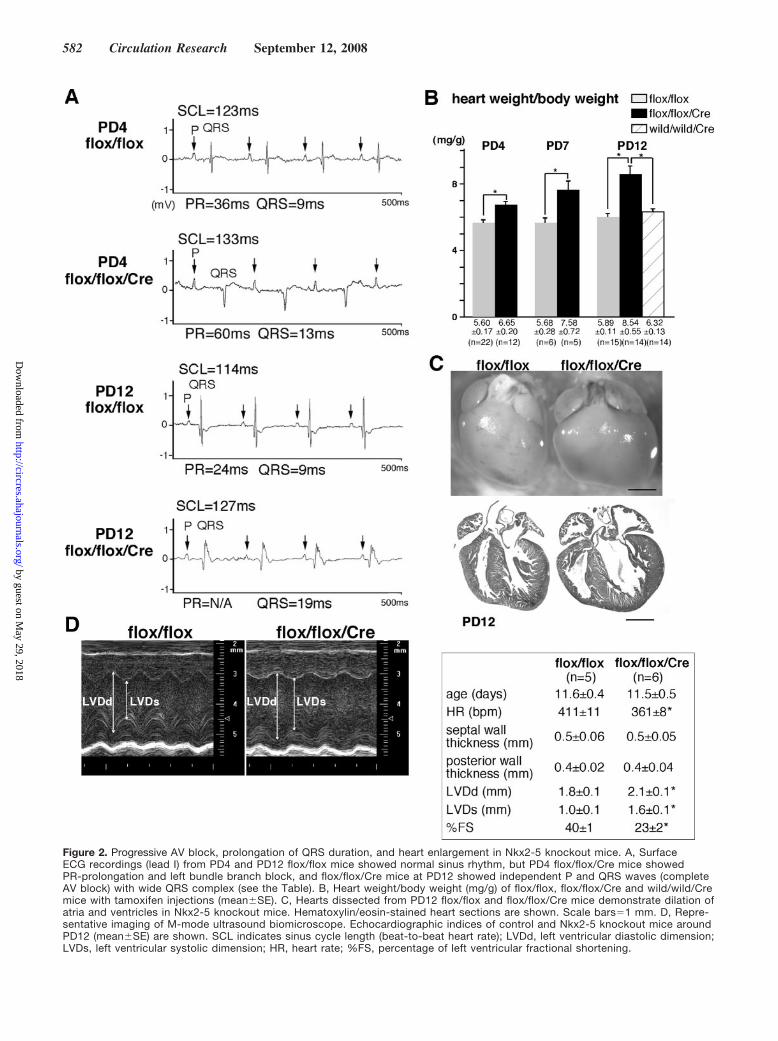
Figure 2. Progressive AV block, prolongation of QRS duration, and heart enlargement in Nkx2-5 knockout mice. A, SurfaceECG recordings (lead I) from PD4 and PD12 flox/flox mice showed normal sinus rhythm, but PD4 flox/flox/Cre mice showedPR-prolongation and left bundle branch block, and flox/flox/Cre mice at PD12 showed independent P and QRS waves (completeAV block) with wide QRS complex (see the Table). B, Heart weight/body weight (mg/g) of flox/flox, flox/flox/Cre and wild/wild/Cremice with tamoxifen injections (meanSE). C, Hearts dissected from PD12 flox/flox and flox/flox/Cre mice demonstrate dilation ofatria and ventricles in Nkx2-5 knockout mice. Hematoxylin/eosin-stained heart sections are shown. Scale bars1 mm. D, Repre-sentative imaging of M-mode ultrasound biomicroscope. Echocardiographic indices of control and Nkx2-5 knockout mice aroundPD12 (meanSE) are shown. SCL indicates sinus cycle length (beat-to-beat heart rate); LVDd, left ventricular diastolic dimension;LVDs, left ventricular systolic dimension; HR, heart rate; %FS, percentage of left ventricular fractional shortening.
582 Circulation Research September 12, 2008
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
adulthood.12–14 Targeted disruption of Nkx2-5 in mice causesembryonic lethality around ED10.5, with retarded cardiacdevelopment.15,16 Ventricular-restricted Nkx2-5 knockoutaround ED8.0 to ED8.5, created by crossing floxed-Nkx2-5mice with myosin light chain 2v-Cre knock-in mice, surviveand demonstrate progressive and advanced conduction de-fects and left ventricular hypertrophy postnatally.17 In hu-mans, mutations in NKX2-5 cause various cardiac anomaliesin an autosomal dominant fashion, as well as progressiveconduction defects and occasional left ventricular dysfunc-tion, which become apparent after birth.18–21
We hypothesize that Nkx2-5 actively regulates a critical setof genes in postnatal cardiomyocytes to maintain propercardiac function. We used tamoxifen-inducible knockoutmice to examine our hypothesis and to elucidate Nkx2-5–dependent regulatory pathways in the perinatal hearts. Wedescribe its effects on cardiomyocyte function, which differfrom recently published results on formation of cardiacconduction systems.22,23
Materials and MethodsAn expanded Materials and Methods section is available in theonline data supplement at http://circres.ahajournals.org and describesthe following: inducible Nkx2-5 knockout mice; Southern, Northern,and Western blotting and immunostaining; surface ECG recordingand ultrasound imaging; histological analyses; gene expressionprofiling; measurement of sodium current and simultaneous record-ing of cardiac contraction and Ca2; real-time RT-PCR; reporterassays; and statistical analyses.
ResultsGeneration of Perinatal Nkx2-5 Knockout MiceTo examine whether Nkx2-5 expression is necessary forperinatal development and maintenance and function of theheart, we generated tamoxifen-inducible Nkx2-5 knockoutmice that carry homozygous floxed-Nkx2-5 alleles17 andheterozygous Cre-ERTM transgene under the control of theCMV enhancer and the chicken -globin promoter (Figure1A).24 Cre-recombinase activities, including tissue prefer-ences, dose-dependent effects of tamoxifen, were extensivelyanalyzed by Hayashi and McMahon using the R26R reportermice.24 Heart was demonstrated as among of the mosteffective organs for Cre-mediated knockout24; however, spa-tial Cre-recombinase activities in the heart have not yet beenanalyzed.
We analyzed cardiac -galactosidase expression in R26Rreporter mice and found it throughout the heart, includingatria, ventricles, valves, and AV node, which expressesacetylcholine esterase in the adjacent tissue section 4 daysafter a single injection of tamoxifen into pregnant mice ongestational day 19 (Figure 1B through 1E and Figure I in theonline data supplement). In ventricles, ubiquitously expressedlacZ staining was detected (Figure 1F), consistent with aprevious report.24
Using this system, Nkx2-5 genes were effectively deletedin the heart, as demonstrated by Southern (Figure 1G), andNorthern (Figure 1H) blotting. To delete floxed-Nkx2-5alleles, both Cre-ERTM transgene and tamoxifen injectionswere required (Figure 1G and 1H). Time course studies andNkx2-5 mRNA expression at postnatal day (PD)2 to PD7 are
shown in Figure 1I and 1J. Of note, this knockout mousemodel is not heart-specific for deletion of Nkx2-5 genes.Although during the fetal period, Nkx2-5 is detected incardiac and extracardiac tissues including, spleen, stomach,liver, tongue, and anterior larynx, after birth, Nkx2-5 expres-sion in extracardiac tissues is markedly downregulated and isessentially limited to cardiomyocytes.12–14 Nkx2-5 expressionappeared to be slightly higher in flox/flox mice compared towild/wild/Cre mice. This could be attributable to the modifi-cation of the Nkx2-5 alleles during insertion of loxP sites inintronic and 3 noncoding sequences, leading to increasedtranscription of Nkx2-5, by modifying transcription factorbinding or alternating microRNA regulation in theseregions.25
Rapid Progressive Conduction Defects and HeartFailure in Perinatal Nkx2-5 Knockout MiceSurface ECG recordings at PD4 demonstrate that Nkx2-5knockout mice exhibited prolonged PR interval and QRSduration (Figure 2A, top traces, and the Table), which wasfurther prolonged and accompanied by occasional 2° to 3°AV blocks at PD12 (Figure 2A, bottom traces, and the Table).Heart weight/body weight was also increased significantly atPD4 and was further increased at PD7 and PD12 (Figure 2B).Enlarged hearts in Nkx2-5 knockout mice at PD12 withdistended ventricular cavities (Figure 2C), accompanied byreduced cardiac contraction using 55 MHz ultrasound imag-ing, are shown (Figure 2D). Survival studies revealed thatapproximately 50% of Nkx2-5 knockout mice died before 3weeks of age (9 of 16 mice). All Nkx2-5 knockout micesurviving at 4 weeks of age demonstrated conduction defectsand enlarged hearts (supplemental Figure II).
These results indicate that perinatal deletion of Nkx2-5results in early conduction and contraction defects withapproximately 50% lethality before 3 weeks of age. Notably,these defects appear sooner and progress more rapidly thanthose seen in mice with embryonic ventricular-restricteddeletion of Nkx2-5, likely attributable to loss of Nkx2-5throughout the heart without compensation during embryoniccardiac development.17
Table. Electrocardiographic data of perinatal knockout ofNkx2-5 (surface ECG)
SCL, ms HR, bpm P, ms PR, ms QRS, ms QTc, ms
PD4
flox/flox(n9)
1069 55146 80 373 102 N/A
flox/flox/Cre(n5)
10513 57576 80 466* 142* N/A
PD12
flox/flox(n6)
11211 53650 80 314 122 504
flox/flox/Cre(n7)
10712 56461 81 365* 174* 509
wild/wild/Cre(n7)
10816 56789 80 282 101 5712
Values are expressed as meanSE *P0.05. SCL, sinus cycle length; HR,heart rate (beat per minute); QTc, rate-corrected QT interval.
Briggs et al Perinatal Nkx2-5 Knockout 583
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
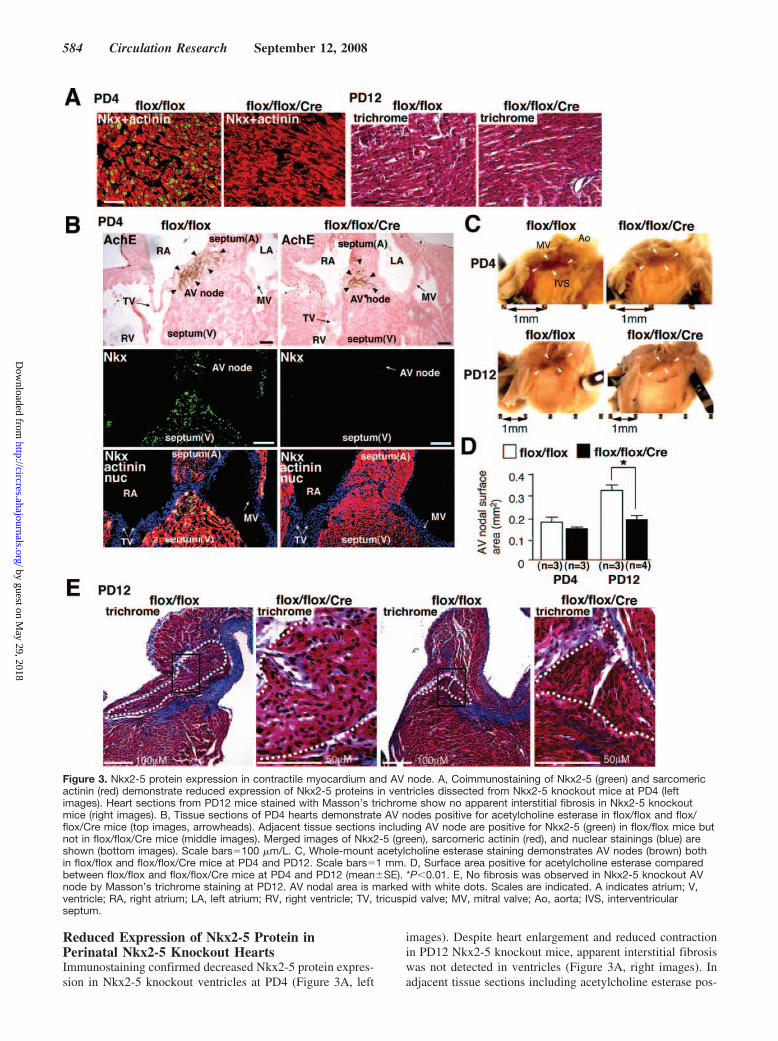
Reduced Expression of Nkx2-5 Protein inPerinatal Nkx2-5 Knockout HeartsImmunostaining confirmed decreased Nkx2-5 protein expres-sion in Nkx2-5 knockout ventricles at PD4 (Figure 3A, left
images). Despite heart enlargement and reduced contractionin PD12 Nkx2-5 knockout mice, apparent interstitial fibrosiswas not detected in ventricles (Figure 3A, right images). Inadjacent tissue sections including acetylcholine esterase pos-
Figure 3. Nkx2-5 protein expression in contractile myocardium and AV node. A, Coimmunostaining of Nkx2-5 (green) and sarcomericactinin (red) demonstrate reduced expression of Nkx2-5 proteins in ventricles dissected from Nkx2-5 knockout mice at PD4 (leftimages). Heart sections from PD12 mice stained with Masson’s trichrome show no apparent interstitial fibrosis in Nkx2-5 knockoutmice (right images). B, Tissue sections of PD4 hearts demonstrate AV nodes positive for acetylcholine esterase in flox/flox and flox/flox/Cre mice (top images, arrowheads). Adjacent tissue sections including AV node are positive for Nkx2-5 (green) in flox/flox mice butnot in flox/flox/Cre mice (middle images). Merged images of Nkx2-5 (green), sarcomeric actinin (red), and nuclear stainings (blue) areshown (bottom images). Scale bars100 m/L. C, Whole-mount acetylcholine esterase staining demonstrates AV nodes (brown) bothin flox/flox and flox/flox/Cre mice at PD4 and PD12. Scale bars1 mm. D, Surface area positive for acetylcholine esterase comparedbetween flox/flox and flox/flox/Cre mice at PD4 and PD12 (meanSE). *P0.01. E, No fibrosis was observed in Nkx2-5 knockout AVnode by Masson’s trichrome staining at PD12. AV nodal area is marked with white dots. Scales are indicated. A indicates atrium; V,ventricle; RA, right atrium; LA, left atrium; RV, right ventricle; TV, tricuspid valve; MV, mitral valve; Ao, aorta; IVS, interventricularseptum.
584 Circulation Research September 12, 2008
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
itive AV nodes, positive Nkx2-5 staining was demonstratedin control but not in Nkx2-5 knockout hearts (Figure 3B).Although formation of the AV node is complete beforebirth,26 whole-mount acetylcholine esterase staining demon-strated that AV nodal surface area size was smaller in Nkx2-5knockout heart at PD12 (Figure 3C and 3D), in agreementwith a previous study using ventricular-specific deletion ofthe Nkx2-5 genes at the embryonic stage.17 In contrast to aprevious study, perinatal Nkx2-5 knockout mice did not showapparent fibrosis in the AV node (Figure 3E).
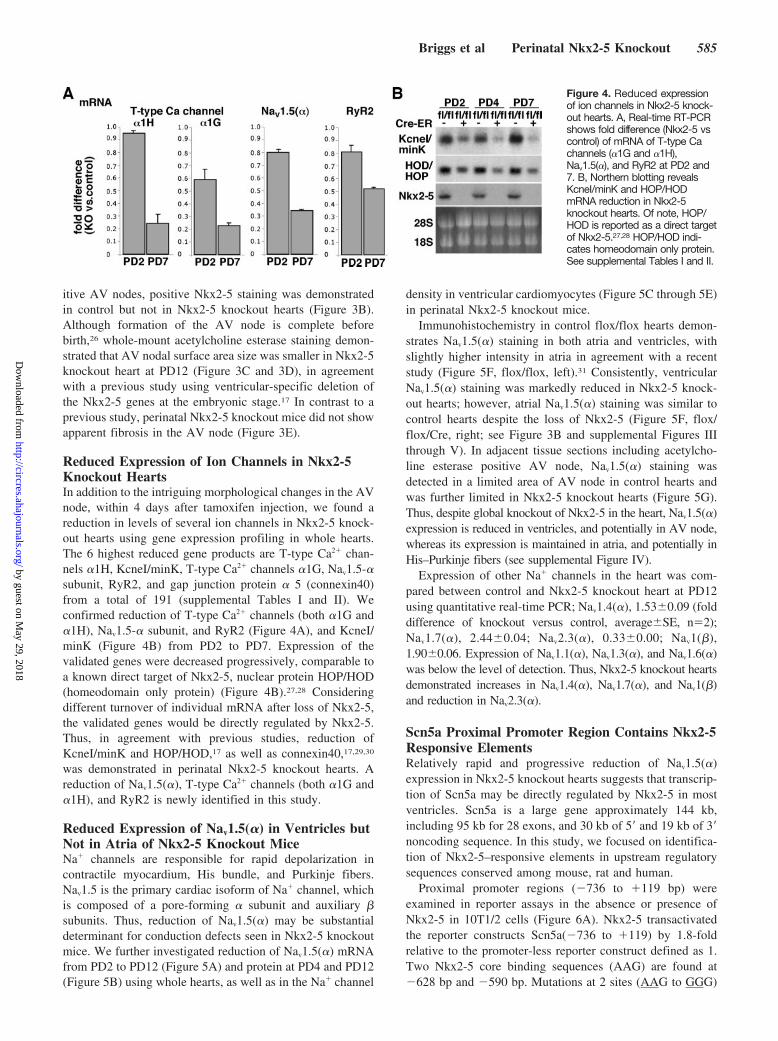
Reduced Expression of Ion Channels in Nkx2-5Knockout HeartsIn addition to the intriguing morphological changes in the AVnode, within 4 days after tamoxifen injection, we found areduction in levels of several ion channels in Nkx2-5 knock-out hearts using gene expression profiling in whole hearts.The 6 highest reduced gene products are T-type Ca2 chan-nels 1H, KcneI/minK, T-type Ca2 channels 1G, Nav1.5-subunit, RyR2, and gap junction protein 5 (connexin40)from a total of 191 (supplemental Tables I and II). Weconfirmed reduction of T-type Ca2 channels (both 1G and1H), Nav1.5- subunit, and RyR2 (Figure 4A), and KcneI/minK (Figure 4B) from PD2 to PD7. Expression of thevalidated genes were decreased progressively, comparable toa known direct target of Nkx2-5, nuclear protein HOP/HOD(homeodomain only protein) (Figure 4B).27,28 Consideringdifferent turnover of individual mRNA after loss of Nkx2-5,the validated genes would be directly regulated by Nkx2-5.Thus, in agreement with previous studies, reduction ofKcneI/minK and HOP/HOD,17 as well as connexin40,17,29,30
was demonstrated in perinatal Nkx2-5 knockout hearts. Areduction of Nav1.5(), T-type Ca2 channels (both 1G and1H), and RyR2 is newly identified in this study.
Reduced Expression of Nav1.5() in Ventricles butNot in Atria of Nkx2-5 Knockout MiceNa channels are responsible for rapid depolarization incontractile myocardium, His bundle, and Purkinje fibers.Nav1.5 is the primary cardiac isoform of Na channel, whichis composed of a pore-forming subunit and auxiliary subunits. Thus, reduction of Nav1.5() may be substantialdeterminant for conduction defects seen in Nkx2-5 knockoutmice. We further investigated reduction of Nav1.5() mRNAfrom PD2 to PD12 (Figure 5A) and protein at PD4 and PD12(Figure 5B) using whole hearts, as well as in the Na channel
density in ventricular cardiomyocytes (Figure 5C through 5E)in perinatal Nkx2-5 knockout mice.
Immunohistochemistry in control flox/flox hearts demon-strates Nav1.5() staining in both atria and ventricles, withslightly higher intensity in atria in agreement with a recentstudy (Figure 5F, flox/flox, left).31 Consistently, ventricularNav1.5() staining was markedly reduced in Nkx2-5 knock-out hearts; however, atrial Nav1.5() staining was similar tocontrol hearts despite the loss of Nkx2-5 (Figure 5F, flox/flox/Cre, right; see Figure 3B and supplemental Figures IIIthrough V). In adjacent tissue sections including acetylcho-line esterase positive AV node, Nav1.5() staining wasdetected in a limited area of AV node in control hearts andwas further limited in Nkx2-5 knockout hearts (Figure 5G).Thus, despite global knockout of Nkx2-5 in the heart, Nav1.5()expression is reduced in ventricles, and potentially in AV node,whereas its expression is maintained in atria, and potentially inHis–Purkinje fibers (see supplemental Figure IV).
Expression of other Na channels in the heart was com-pared between control and Nkx2-5 knockout heart at PD12using quantitative real-time PCR; Nav1.4(), 1.530.09 (folddifference of knockout versus control, averageSE, n2);Nav1.7(), 2.440.04; Nav2.3(), 0.330.00; Nav1(),1.900.06. Expression of Nav1.1(), Nav1.3(), and Nav1.6()was below the level of detection. Thus, Nkx2-5 knockout heartsdemonstrated increases in Nav1.4(), Nav1.7(), and Nav1()and reduction in Nav2.3().
Scn5a Proximal Promoter Region Contains Nkx2-5Responsive ElementsRelatively rapid and progressive reduction of Nav1.5()expression in Nkx2-5 knockout hearts suggests that transcrip-tion of Scn5a may be directly regulated by Nkx2-5 in mostventricles. Scn5a is a large gene approximately 144 kb,including 95 kb for 28 exons, and 30 kb of 5 and 19 kb of 3noncoding sequence. In this study, we focused on identifica-tion of Nkx2-5–responsive elements in upstream regulatorysequences conserved among mouse, rat and human.
Proximal promoter regions (736 to 119 bp) wereexamined in reporter assays in the absence or presence ofNkx2-5 in 10T1/2 cells (Figure 6A). Nkx2-5 transactivatedthe reporter constructs Scn5a(736 to 119) by 1.8-foldrelative to the promoter-less reporter construct defined as 1.Two Nkx2-5 core binding sequences (AAG) are found at628 bp and 590 bp. Mutations at 2 sites (AAG to GGG)
Figure 4. Reduced expressionof ion channels in Nkx2-5 knock-out hearts. A, Real-time RT-PCRshows fold difference (Nkx2-5 vscontrol) of mRNA of T-type Cachannels (1G and 1H),Nav1.5(), and RyR2 at PD2 and7. B, Northern blotting revealsKcneI/minK and HOP/HODmRNA reduction in Nkx2-5knockout hearts. Of note, HOP/HOD is reported as a direct targetof Nkx2-5.27,28 HOP/HOD indi-cates homeodomain only protein.See supplemental Tables I and II.
Briggs et al Perinatal Nkx2-5 Knockout 585
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
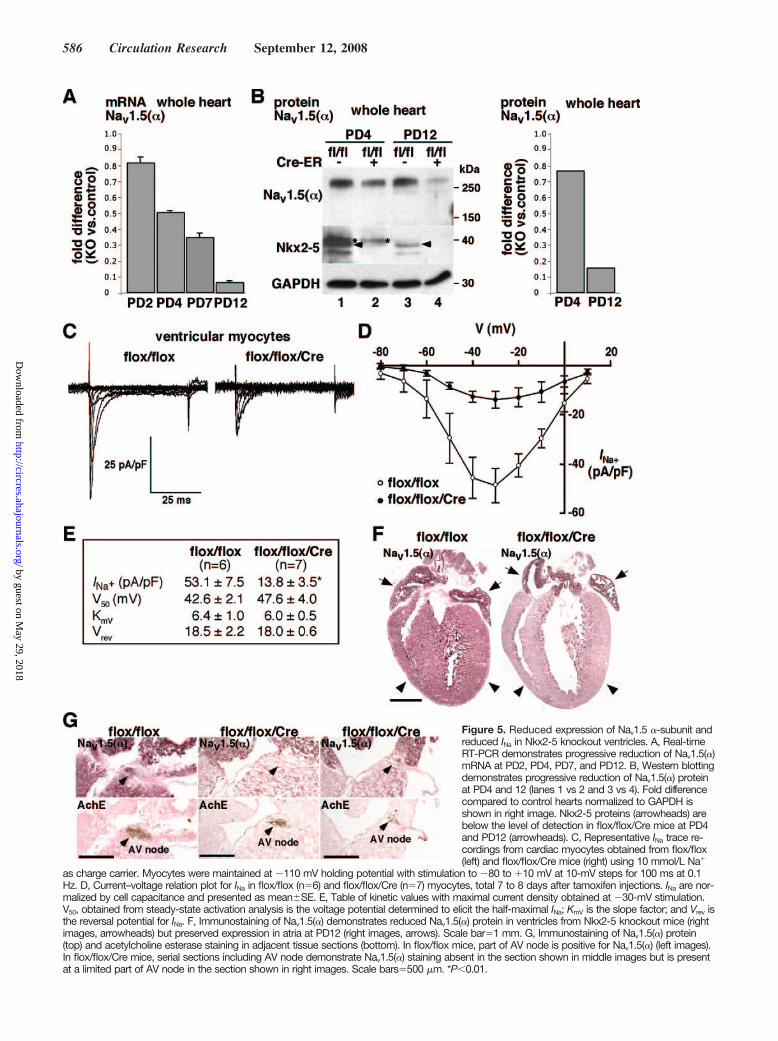
Figure 5. Reduced expression of Nav1.5 -subunit andreduced INa in Nkx2-5 knockout ventricles. A, Real-timeRT-PCR demonstrates progressive reduction of Nav1.5()mRNA at PD2, PD4, PD7, and PD12. B, Western blottingdemonstrates progressive reduction of Nav1.5() proteinat PD4 and 12 (lanes 1 vs 2 and 3 vs 4). Fold differencecompared to control hearts normalized to GAPDH isshown in right image. Nkx2-5 proteins (arrowheads) arebelow the level of detection in flox/flox/Cre mice at PD4and PD12 (arrowheads). C, Representative INa trace re-cordings from cardiac myocytes obtained from flox/flox(left) and flox/flox/Cre mice (right) using 10 mmol/L Na
as charge carrier. Myocytes were maintained at 110 mV holding potential with stimulation to 80 to 10 mV at 10-mV steps for 100 ms at 0.1Hz. D, Current–voltage relation plot for INa in flox/flox (n6) and flox/flox/Cre (n7) myocytes, total 7 to 8 days after tamoxifen injections. INa are nor-malized by cell capacitance and presented as meanSE. E, Table of kinetic values with maximal current density obtained at 30-mV stimulation.V50, obtained from steady-state activation analysis is the voltage potential determined to elicit the half-maximal INa; KmV is the slope factor; and Vrev isthe reversal potential for INa. F, Immunostaining of Nav1.5() demonstrates reduced Nav1.5() protein in ventricles from Nkx2-5 knockout mice (rightimages, arrowheads) but preserved expression in atria at PD12 (right images, arrows). Scale bar1 mm. G, Immunostaining of Nav1.5() protein(top) and acetylcholine esterase staining in adjacent tissue sections (bottom). In flox/flox mice, part of AV node is positive for Nav1.5() (left images).In flox/flox/Cre mice, serial sections including AV node demonstrate Nav1.5() staining absent in the section shown in middle images but is presentat a limited part of AV node in the section shown in right images. Scale bars500 m. *P0.01.
586 Circulation Research September 12, 2008
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from

abolished Nkx2-5 responsiveness similar to baseline lucif-erase reporter activity.
Next, 5 upstream conserved genomic regions were exam-ined for potential enhancer activities (Figure 6B). The10 147-bp site contains the conserved perfect Nkx2-5binding sequence uniquely identified in the entire Scn5agenome using rVista 2.0 program (http://rvista.dcode.org).However, inclusion of a 2-kb fragment (2308 to 119),as well as addition of 4 conserved regions did not enhanceluciferase activities compared to the proximal promoterconstruct (736 to 119). Therefore, Nkx2-5 responsivenesswas demonstrated only in the proximal promoter region inreporter assays.
Reduced Contractility Accompanied byCa2-Handling Defects in Cardiomyocytes IsolatedFrom Nkx2-5 Knockout HeartBecause cardiac conduction and contraction are closely cou-pled, reduction of Nav1.5() alone likely causes contractiondefects as in human patients with SCN5A mutations.32,33 Inaddition, gene expression profiling demonstrated reduction ofRyR2, a cardiac isoform responsible for Ca2 release from SRin Nkx2-5 knockout hearts (supplemental Table I), which wasconfirmed by real-time RT-PCR (Figures 4A and 7A) andWestern blotting (Figure 7B). Expression of other Ca2-handling proteins responsible for restoration of Ca2 into SR,including sarcoendoplasmic reticulum Ca2 ATPase 2a(SERCA2a) and its regulatory protein phospholamban wasmodestly reduced in Nkx2-5 knockout hearts (Figure 7B).
Consistently, Nkx2-5 knockout cardiomyocytes showed re-duced contractile and Ca2-handling parameters (Figures 7Cand 7D). Therefore, altered expression of Ca2-handlingproteins, including RyR2, SERCA2a, and phospholamban, isalso responsible for the depressed contractility of Nkx2-5knockout hearts.
Of note, cardiomyocytes isolated from surviving mice wereexamined at 3 weeks of age for contraction because oftechnical difficulties in isolation when using younger mice.
DiscussionNkx2-5 function is not limited to embryonic heart develop-ment; it is critical for both conduction and contraction in theperinatal heart, within 4 days after tamoxifen injection, asdemonstrated in this study. Initiation of action potential inrapid conduction cardiomyocytes relies on Na inward cur-rents, which were markedly reduced in the ventricles ofNkx2-5 knockout mice, because of reduced expression ofpore-forming subunit Nav1.5(). In addition, Ca2 releasefrom SR was diminished, accompanied by substantial reduc-tion in RyR2 expression. Because excitation and contractionare tightly coupled, reduced handling of Na and Ca2
demonstrated in Nkx2-5 knockout cardiomyocytes may mu-tually lead to rapid conduction and contraction defects.
Mutations in the human SCN5A gene, which encodesNav1.5(), lead to Brugada syndrome and long-QT syndrome,resulting from loss-of-function or gain-of-function mecha-nisms.2,4,5,34 Homozygous knockout of Scn5a resulted inembryonic lethality around ED10.5,35 and heterozygous
Figure 6. Proximal promoter ofScn5a gene contains Nkx2-5responsive elements. A, Sche-matics of 2 Nkx2-5 consensusbinding sites in the Scn5a pro-moter at -628 and –590 bp.Scn5a promoter has CpGislands, but not a TATA box.Scn5a(-736 to 119) (top) andmutated Scn5a(-736 to 119)(bottom), and correspondingrelative luciferase reporteractivities with pcDNA3-Nkx2-5cotransfection normalized to-galactosidase activity andpcDNA empty vector with thevalue in promoter-less lucif-erase reporter defined as 1(means SE). ANOVA demon-strated significant differencebetween 2 groups (F22.1,P0.0003). *P0.0003 byBonferroni’s post hoc test. B,Schematics of conservedgenomic regions amongmouse, rat, and human Scn5agene approximately 27, 17,10, 8, and 2 kb upstreamof the transcriptional start site.Six luciferase reporter con-structs and corresponding rel-ative luciferase reporter activi-ties with pcDNA3-Nkx2-5cotransfection normalized to
-galactosidase activity and pcDNA empty vector with the value in promoter-less luciferase reporter defined as 1 (meansSE). ANOVAdemonstrated no significant difference among groups (F1.08, P0.3966).
Briggs et al Perinatal Nkx2-5 Knockout 587
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from
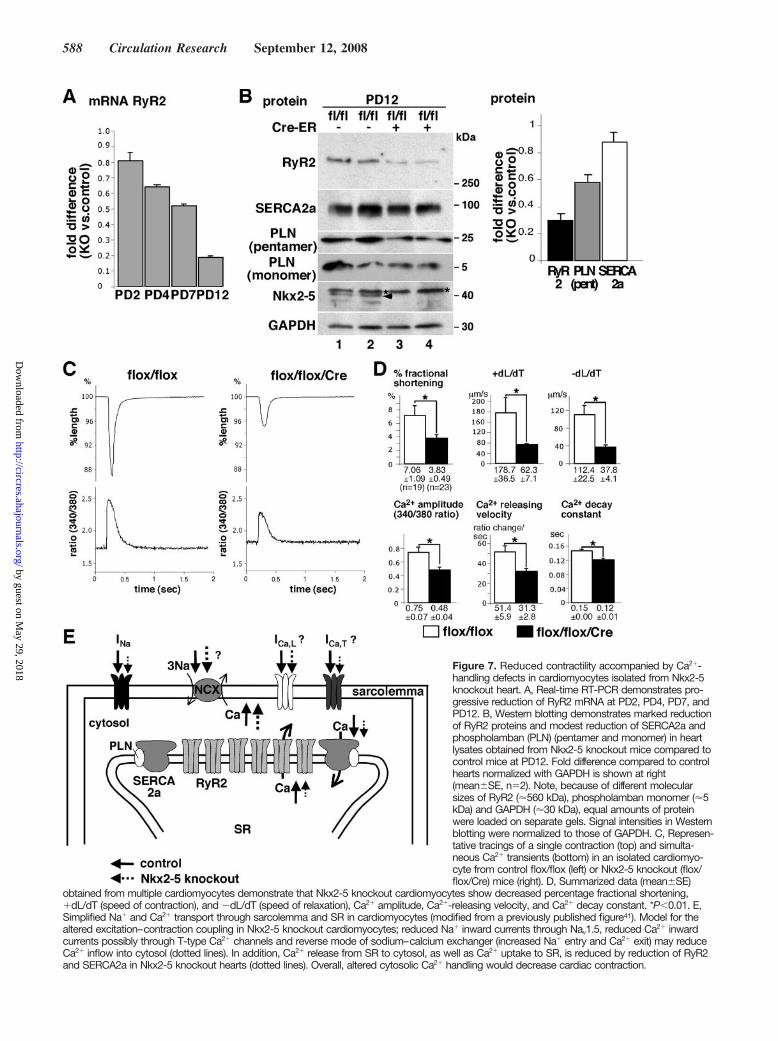
Figure 7. Reduced contractility accompanied by Ca2-handling defects in cardiomyocytes isolated from Nkx2-5knockout heart. A, Real-time RT-PCR demonstrates pro-gressive reduction of RyR2 mRNA at PD2, PD4, PD7, andPD12. B, Western blotting demonstrates marked reductionof RyR2 proteins and modest reduction of SERCA2a andphospholamban (PLN) (pentamer and monomer) in heartlysates obtained from Nkx2-5 knockout mice compared tocontrol mice at PD12. Fold difference compared to controlhearts normalized with GAPDH is shown at right(meanSE, n2). Note, because of different molecularsizes of RyR2 (560 kDa), phospholamban monomer (5kDa) and GAPDH (30 kDa), equal amounts of proteinwere loaded on separate gels. Signal intensities in Westernblotting were normalized to those of GAPDH. C, Represen-tative tracings of a single contraction (top) and simulta-neous Ca2 transients (bottom) in an isolated cardiomyo-cyte from control flox/flox (left) or Nkx2-5 knockout (flox/flox/Cre) mice (right). D, Summarized data (meanSE)
obtained from multiple cardiomyocytes demonstrate that Nkx2-5 knockout cardiomyocytes show decreased percentage fractional shortening,dL/dT (speed of contraction), and dL/dT (speed of relaxation), Ca2 amplitude, Ca2-releasing velocity, and Ca2 decay constant. *P0.01. E,Simplified Na and Ca2 transport through sarcolemma and SR in cardiomyocytes (modified from a previously published figure41). Model for thealtered excitation–contraction coupling in Nkx2-5 knockout cardiomyocytes; reduced Na inward currents through Nav1.5, reduced Ca2 inwardcurrents possibly through T-type Ca2 channels and reverse mode of sodium–calcium exchanger (increased Na entry and Ca2 exit) may reduceCa2 inflow into cytosol (dotted lines). In addition, Ca2 release from SR to cytosol, as well as Ca2 uptake to SR, is reduced by reduction of RyR2and SERCA2a in Nkx2-5 knockout hearts (dotted lines). Overall, altered cytosolic Ca2 handling would decrease cardiac contraction.
588 Circulation Research September 12, 2008
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from

knockout of Scn5a, in which Na current density was reducedby approximately 43% (21 pA/pF in Scn5a/ versus 37pA/pF in wild type) with virtually identical channel-gatingcharacteristics, led to prolonged P wave, AV block, andintraventricular conduction defects.35 Na current density inventricular cardiomyocytes was reduced by approximately75% in Nkx2-5 knockout mice compared to wild type (13.8versus 53.1 pA/pF; Figure 5E) at equivalent PD7 to PD8,likely resulting in more profound ventricular conductiondefects than those found in Scn5a/ mice.
Nkx2-5 knockout mice did not demonstrate prolongationof P wave (Table). Consistently, expression of Nav1.5() inatria of Nkx2-5 knockout mice was not reduced despitemarked reduction of Nkx2-5. This indicates that Nkx2-5 iscritical for transcription of Scn5a in the majority of ventric-ular cardiomyocytes but not atrial cardiomyocytes.
Despite a number of studies on functional regulation ofNav1.5,4,5 transcriptional regulation of Scn5a gene is not wellunderstood. It is repressed by the zinc finger protein Snail.5,36
It is unlikely that upregulation of Snail is the mechanism inNkx2-5 knockout hearts, based on gene expression profilingdata (fold difference flox/flox/Cre versus flox/flox is0.980.04 in Snail1; 1.040.03 in Snail2; meanSE; n4).We examined 5 upstream genomic regions of Scn5a andfound that 2 Nkx2-5 binding sites in the proximal promoterseparated by 38 bp are responsive for Nkx2-5–dependenttranscription, whereas 5 potential enhancer elements did notfurther increase transcription in reporter assays.
Cardiomyocytes isolated from Nkx2-5 knockout micedemonstrated substantial Ca2-handling defects, particularlyin Ca2 release, with approximately a 70% reduction of RyR2protein at PD12. Intracellular Ca2 homeostasis is wellbalanced in normal heart but is altered in a number of humanand rodent heart failure models, and it is primarily responsi-ble for the depressed contractility of failing hearts.37–39
Perhaps crosstalk among the Ca2-handling proteins is im-portant, as has been demonstrated in SERCA2a/ mice, inwhich its negative regulator phospholamban is also de-creased, maintaining the constant ratio of SERCA2a andphospholamban.40 Thus, reduction of Ca2-handling proteinsis not specific for Nkx2-5 knockout hearts, except forprofound defects in Ca2 release accompanied by substantialreduction of RyR2 expression. In addition, elevation of theNa inward current through Na channels is usually observedin failing hearts compensatory to increase intracellular Ca2
through the sodium calcium exchanger (Ca2 influx and Na
efflux),41 although this was not likely the case in Nkx2-5knockout hearts.
Shortly after Nkx2-5 knockout, in addition to reduction ofNav1.5() and RyR2 expression, expression of other ionchannels, including T-type Ca2 channels (1G and 1H),was reduced. Because T-type Ca2 channels also play asmall role in the increase of intracellular Ca2 for contrac-tion, and a substantial role in depolarization in slow conduc-tion tissues,1,3,42 reduction in mRNA of T-type Ca2 channelsin Nkx2-5 knockout hearts would also contribute to thephenotype.
We propose a model for the potential mechanisms ofcardiac dysfunction following loss of Nkx2-5 (Figure 7E).
Reduced Na entry into cardiomyocytes may lead to com-pensatory increases in reverse mode of the sodium–calciumexchanger (increased Na entry and Ca2 exit) and a furtherdecrease in intracellular Ca2.33 A reduction of cardiac-specific myosin light chain kinase and phosphorylation ofmyosin light chain 2v was demonstrated in perinatal Nkx2-5knockout mice,43 which likely also contributes to contractiledefects.
Cre-ERTM transgenic mice,24 widely used for Cre-mediatedgene deletion in different tissues, demonstrated near 100%recombination frequency in hearts from R26R-reporter mice;therefore, it is suitable for functional analyses of individualcardiomyocytes. Expression of Nkx2-5 in postnatal stage isnearly limited to cardiomyocytes12–14; thus, global deletion ofNkx2-5 using Cre-ERTM mice does not likely affect cardiacphenotype. An alternative approach would be to use micewith cardiomyocyte-specific expression of tamoxifen-inducible Cre-recombinase through the use of MHC-Cretransgenic mice.44 However, myocyte-specific recombinationfrequency was reported to be approximately 70% to 80%,and, to our knowledge, spatial recombination efficacy in thehighly specialized AV nodal cardiomyocytes has not beenreported in this mouse model.44
In summary, we demonstrate that Nkx2-5 is critical forperinatal conduction and contraction by regulating expressionof several ion channel genes. Our findings may providepotential explanations for progressive conduction and con-traction defects seen after birth in patients with congenitalheart disease associated with NKX2-5 mutations.
AcknowledgmentsWe greatly appreciate C. Ketcham, E. Scott, and E. Chan forvaluable suggestions and technical support.
Sources of FundingThis work was supported by an American Heart AssociationNational Scientific Development Grant and NIH grant HL081577(to H.K.).
DisclosuresNone.
References1. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:
198–205.2. Marban E. Cardiac channelopathies. Nature. 2002;415:213–218.3. Mangoni ME, Couette B, Marger L, Bourinet E, Striessnig J, Nargeot J.
Voltage-dependent calcium channels and cardiac pacemaker activity:from ionic currents to genes. Prog Biophys Mol Biol. 2006;90:38–63.
4. Haufe V, Chamberland C, Dumaine R. The promiscuous nature of thecardiac sodium current. J Mol Cell Cardiol. 2007;42:469–477.
5. Abriel H. Roles and regulation of the cardiac sodium channel Na(v) 1.5:recent insights from experimental studies. Cardiovasc Res. 2007;76:381–389.
6. Benndorf K, Boldt W, Nilius B. Sodium current in single myocardialmouse cells. Pflugers Arch. 1985;404:190–196.
7. Nuss HB, Marban E. Electrophysiological properties of neonatal mousecardiac myocytes in primary culture. J Physiol. 1994;479(pt 2):265–279.
8. Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large-scaleanalysis of ion channel gene expression in the mouse heart duringperinatal development. Physiol Genomics. 2007;28:273–283.
9. Bodmer R. The gene tinman is required for specification of the heart andvisceral muscles in Drosophila. Development. 1993;118:719–729.
10. Harvey RP. NK-2 homeobox genes and heart development. Dev Biol.1996;178:203–216.
Briggs et al Perinatal Nkx2-5 Knockout 589
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from

11. Harvey RP, Rosenthal N, eds. Heart Development. San Diego, Calif:Academic Press; 1999.
12. Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: a novelmurine homeobox gene expressed in early heart progenitor cells and theirmyogenic descendants. Development. 1993;119:419–431.
13. Komuro I, Izumo S. Csx: a murine homeobox-containing gene specif-ically expressed in the developing heart. Proc Natl Acad Sci U S A.1993;90:8145–8149.
14. Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac andextracardiac expression of Csx/Nkx2.5 homeodomain protein. Circ Res.1998;82:936–946.
15. Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP.Myogenic and morphogenetic defects in the heart tubes of murineembryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666.
16. Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiachomeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genesessential for heart development. Development. 1999;126:1269–1280.
17. Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S,Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW,Silberbach M, Shou W, Chien KR. Nkx2-5 pathways and congenital heartdisease; loss of ventricular myocyte lineage specification leads to pro-gressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386.
18. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP,Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused bymutations in the transcription factor NKX2-5. Science. 1998;281:108–111.
19. Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, ZhangY, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, SeidmanCE, Plowden J, Kugler JD. Mutations in the cardiac transcription factorNKX2.5 affect diverse cardiac developmental pathways. J Clin Invest.1999;104:1567–1573.
20. Kasahara H, Benson DW. Biochemical analyses of eight NKX2.5 home-odomain missense mutations causing atrioventricular block and cardiacanomalies. Cardiovasc Res. 2004;64:40–51.
21. Konig K, Will JC, Berger F, Muller D, Benson DW. Familial congenitalheart disease, progressive atrioventricular block and the cardiachomeobox transcription factor gene NKX2.5: identification of a novelmutation. Clin Res Cardiol. 2006;95:499–503.
22. Meysen S, Marger L, Hewett KW, Jarry-Guichard T, Agarkova I,Chauvin JP, Perriard JC, Izumo S, Gourdie RG, Mangoni ME, Nargeot J,Gros D, Miquerol L. Nkx2.5 cell-autonomous gene function is requiredfor the postnatal formation of the peripheral ventricular conductionsystem. Dev Biol. 2007;303:740–753.
23. Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J,Nobrega MA, Yokota Y, Berul C, Izumo S, Seidman JG, Seidman CE. Amolecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiacconduction system development. Cell. 2007;129:1365–1376.
24. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by atamoxifen-inducible form of Cre: a tool for temporally regulated geneactivation/inactivation in the mouse. Dev Biol. 2002;244:305–318.
25. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN.Control of stress-dependent cardiac growth and gene expression by amicroRNA. Science. 2007;316:575–579.
26. Sedmera D, Reckova M, DeAlmeida A, Coppen SR, Kubalak SW,Gourdie RG, Thompson RP. Spatiotemporal pattern of commitment toslowed proliferation in the embryonic mouse heart indicates progressivedifferentiation of the cardiac conduction system. Anat Rec A Discov MolCell Evol Biol. 2003;274:773–777.
27. Chen F, Kook H, Milewski R, Gitler AD, Lu MM, Li J, Nazarian R,Schnepp R, Jen K, Biben C, Runke G, Mackay JP, Novotny J, SchwartzRJ, Harvey RP, Mullins MC, Epstein JA. Hop is an unusual homeoboxgene that modulates cardiac development. Cell. 2002;110:713–723.
28. Shin CH, Liu ZP, Passier R, Zhang CL, Wang DZ, Harris TM, YamagishiH, Richardson JA, Childs G, Olson EN. Modulation of cardiac growthand development by HOP, an unusual homeodomain protein. Cell. 2002;110:725–735.
29. Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T,Huang WY, Manning WJ, Paul D, Lawitts J, Berul CI, Izumo S. Pro-gressive atrioventricular conduction defects and heart failure in miceexpressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001;108:189–201.
30. Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S,Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murinemodel of Holt-Oram syndrome defines roles of the T-box transcriptionfactor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721.
31. Dominguez JN, de la Rosa A, Navarro F, Franco D, Aranega AE. Tissuedistribution and subcellular localization of the cardiac sodium channelduring mouse heart development. Cardiovasc Res. 2008;78:45–52.
32. McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L.SCN5A mutation associated with dilated cardiomyopathy, conductiondisorder, and arrhythmia. Circulation. 2004;110:2163–2167.
33. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ,Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations andsusceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454.
34. Wolf CM, Berul CI. Inherited conduction system abnormalities–onegroup of diseases, many genes. J Cardiovasc Electrophysiol. 2006;17:446–455.
35. Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA,Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI,Colledge WH, Grace AA. Slowed conduction and ventricular tachycardiaafter targeted disruption of the cardiac sodium channel gene Scn5a. ProcNatl Acad Sci U S A. 2002;99:6210–6215.
36. Hesse M, Kondo CS, Clark RB, Su L, Allen FL, Geary-Joo CT, KunnathuS, Severson DL, Nygren A, Giles WR, Cross JC. Dilated cardiomyopathyis associated with reduced expression of the cardiac sodium channelScn5a. Cardiovasc Res. 2007;75:498–509.
37. Houser SR, Piacentino V III, Weisser J. Abnormalities of calcium cyclingin the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607.
38. Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release andcardiac disease. Annu Rev Physiol. 2005;67:69–98.
39. Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2 handling inheart failure. J Clin Invest. 2005;115:556–564.
40. Periasamy M, Huke S. SERCA pump level is a critical determinant ofCa(2)homeostasis and cardiac contractility. J Mol Cell Cardiol. 2001;33:1053–1063.
41. Bers DM, Despa S, Bossuyt J. Regulation of Ca2 and Na in normaland failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177.
42. Yasui K, Niwa N, Takemura H, Opthof T, Muto T, Horiba M, Shimizu A,Lee JK, Honjo H, Kamiya K, Kodama I. Pathophysiological significanceof T-type Ca2 channels: expression of T-type Ca2 channels in fetaland diseased heart. J Pharmacol Sci. 2005;99:205–210.
43. Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N,Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification ofcardiac-specific myosin light chain kinase. Circ Res. 2008;102:571–580.
44. Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, TymitzKM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using atamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25.
590 Circulation Research September 12, 2008
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from

and Hideko KasaharaRaizada, Nobuo Horikoshi, Ellen O. Weinberg, Kenji Yasui, Yasuhiro Ikeda, Kenneth R. Chien
Alexandra J. Walker, Tsugio Seki, Suk P. Oh, Jonathan T. Lu, Colin Sumners, Mohan K. Laura E. Briggs, Morihiko Takeda, Adolfo E. Cuadra, Hiroko Wakimoto, Melissa H. Marks,
Perinatal Loss of Nkx2-5 Results in Rapid Conduction and Contraction Defects
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.108.1718352008;103:580-590; originally published online August 8, 2008;Circ Res.
http://circres.ahajournals.org/content/103/6/580World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2008/09/16/CIRCRESAHA.108.171835.DC1 http://circres.ahajournals.org/content/suppl/2008/09/17/CIRCRESAHA.108.171835.DC2
Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 29, 2018
http://circres.ahajournals.org/D
ownloaded from

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 1
Materials and Methods:
Inducible Nkx2-5 knockout mice
Floxed-Nkx2-5 homozygous mice (Nkx2-5flox/flox)1 were bred with transgenic mice
carrying the Cre-ERTM gene under CMV promoter, Tg(CMV-Cre-ERTM).2 Subsequent matings
between offsprings generated Nkx2-5flox/flox/Tg(CMV-Cre-ERTM)(flox/flox/Cre), Nkx2-
5wild/wild/Tg(CMV-Cre-ERTM) (wild/wild/Cre) and Nkx2-5flox/flox (flox/flox). For perinatal deletion
of the floxed-Nkx2-5 genes, a single injection of tamoxifen (0.5-1 mg/g body weight, ip) was
administered to pregnant mice on gestation day 19. Male Tg(CMV-Cre-ERTM)2 were also crossed
to females that carried the Rosa reporter allele, R26R3. All animal care protocols fully conformed
to the Association for the Assessment and Accreditation of Laboratory Animal Care, with
approvals from the University of Florida Institutional Animal Care and Use Committees.
Southern, Northern and Western blotting and immunostaining
Southern and Northern blot analyses were performed using the following probes: Nkx2-5
5’ genomic probe, NotI-ClaI fragment of mouse Nkx2-5 genomic DNA; Nkx2-5 coding probe,
PflMI-EcoRI fragment of mouse Nkx2-5 cDNA; Cre-recombinase probe, EcoRI-BamHI
fragment of pCAGGS-nls-Cre plasmid (gift from Dr. H. Xin); RT-PCR products amplified by
the following primer sets: KcneI/minK (476 bp, F’, 5'- CTTGACGCCCAGGATGAGC -3'; R,
5'- GGTGCCCCTACAATAAAGACTATGG -3'); HOD/HOP (703 bp, F’, 5'-
AGCAGACAGGCACCAGCATC -3'; R’- AAGGGAGCACAGGGAAGTGAAC -3'). Western
blot analyses and immunostaining using frozen sections were performed with the following
primary antibodies: anti-Nkx2-5 pAb4, sarcomeric actinin (A7811, SIGMA), Nav1.5(a) (ASC-
005, alomone labs), GAPDH (RDI-TRK5G4-6C5, Research Diagnostic Inc), RyR2 (MA3-916,

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 2
Affinity BioReagents), SERCA2a (sc-8095, Santa Cruz), phospholamban (05-205, Upstate). For
detection of RyR and pentamer of phospholamban, protein samples were heated at 37ºC for 15
min before loading on SDS-PAGE gels.5 Fluorescent microscopic images were obtained using
ZEISS Axiovert200M with or without Apotome.
Surface ECG recording and ultrasound imaging
Recordings of six limb leads surface ECG (ADInstruments, Milford, MA) were
performed without anesthesia as described previously with some modifications.6 ECG recordings
were analyzed using PowerLab software (ADInstruments).6 M-mode ultrasound imaging of the
left ventricle was obtained at the level of the papillary muscle from a parasternal window using
an ultrasound biomicroscope with a single transducer with a frequency 55 MHz (VisualSonics,
Tronto, Canada).
Histological analyses
X-gal staining of whole hearts and frozen sections were performed according to standard
protocol.7 Tissues were fixed in 4% paraformaldehyde/PBS overnight, dehydrated, embedded in
paraffin, sectioned, and stained with hematoxylin/eosin or Masson’s trichrome. Acetylcholine
esterase staining in frozen tissue sections was performed as described previously with some
modifications.8 Whole mount acetylcholine esterase staining was performed with the incisions on
right ventricular and atrial free wall, with addition of 0.1% TritonTM X-100 in all the solutions
except for Cu2+-glycine solution. After fixation with paraformaldehyde, tissue images were
captured by a stereomicroscope (Nikon SMZ800) attached to a CCD camera (QImaging
MicroPublisher3.3, BC, Canada), with scale bars present. The same magnifications and zoom

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 3
were applied for the hearts dissected from flox/flox or flox/flox/Cre mice at the same ages.
Digitalized AV nodal surface size was measured using NIH Image software.
Gene expression profiling
Total RNA pooled from 3-4 hearts of postnatal day 4 (PD4) flox/flox or flox/flox/Cre
mice with tamoxifen administration was analyzed in the CodeLink Bioarrays followed by
CodeLink and GeneSpring software (Genus Biosystems, Northbrook, IL). Four sets of relative
expression values between flox/flox and flox/flox/Cre were averaged and are listed.
Measurement of sodium current and simultaneous recording of cardiac contraction and
Ca2+
After genotyping, neonatal ventricular cardiomyocytes were isolated from flox/flox or
flox/flox/Cre mice side by side at PD3, cultured for 4-5 days before measuring sodium currents
(equivalent to PD7-8). Sodium currents were (INa) obtained by voltage clamp recordings in the
whole cell patch clamp configuration at room temperature (~22°C). Myocytes were bathed in
low sodium buffer containing, in mmol/L: NaCl 10, CsCl 80, CaCl2 2, MgCl2 1, HEPES 10, 4-
aminopyridine 10, tetraethylammonium-Cl 30, dextrose 10, CdCl 0.1; and nifedipine 5 mM with
an osmolarity of 290 mOsm and pH of 7.4 adjusted with CsOH. Recordings were obtained using
Molecular Devices Axon 200A amplifier (Sunnyvale, CA) filtered at 2 kHz and digitized using
Molecular Devices, Digidata 1322A, at 10 kHz. Data were acquired using Molecular Devices
PClamp 9.0 software. Myocytes were impaled with Drummond Wiretrol II capillary pipettes
(Drummond Scientific Company, Broomall, PA) pulled with Sutter instrument P-97 horizontal
pipette puller and polished with Narishige MF-830 microforge with series resistance of 0.5 to 1.5

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 4
mΩ when filled with solution containing, in mmol/L: CsOH 105, glutamic acid 105,
tetraethylammonium-Cl 20, HEPES 10, Cs2-EGTA 5, MgATP 5, NaGTP 0.1 with osmolarity of
285 and pH adjusted to 7.2 with CsOH. A -9.7 mV correction for liquid junction potential,
between the patch pipette and bath solution, was set upon access to the cell cytosol. Cell
capacitance and input series resistance were electronically compensated to approximately 90
percent using online circuitry. Leak currents were subtracted online with P/4 protocol.
Data analysis was performed using Molecular devices Clampfit 9.0, Microsoft Excel, and
GraphPad Prism 4.0 (San Diego, CA). INa current density was derived by dividing current
amplitude from each recording by their respective cell capacitance obtained from amplifier upon
compensation. Experiments that showed inadequate voltage control, such as space clamp error or
inappropriate steep increase in current amplitude in the negative slope of current voltage relation,
were discarded. Steady state activation curves were generated from data used to obtain current
voltage relation plots fitted to a Boltzman equation: I/IMax = 1/[1+exp(V – V1/2 / KV)] where I/IMax
is the current expressed as fraction of maximal current; V1/2 is the voltage to obtain half maximal
current and KmV is the slope factor.
Cardiomyocytes were isolated from 3 weeks old flox/flox or flox/flox/Cre mice. Only
rod-shaped cardiomyocytes with staircase ends, clear cross striations and surface membranes free
from blebs were studied for simultaneous measurements of cell shortening and intracellular free
calcium cardiomyocyte contraction and simultaneous Ca2+ measurement as described
previously.9

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 5
Real-time reverse transcriptase (RT)-PCR
Real-time RT-PCR was performed using the inventoried Taqman Gene Expression
Assays (Applied Biosystems): Scn5a, Mm00451971_m1; RyR2, Mm00465877_m1; T-type Ca
channel a1G, Mm00486549_m1; T-type Ca channel a1H, Mm00445369_m1; Scn1a,
Mm00450580; Scn3a, Mm00658167; Scn4a, Mm00500103; Scn7a, Mm00801952; Scn8a,
Mm00488110; Scn9a, Mm00450762; Scn1b, Mm0041210. Data were normalized with beta-
actin expression (product No, 4352933E). Duplicate experiments were averaged.
Reporter assays
C57BL/6 genomic DNA or BAC clone (Invitrogen, RPC 123.C, Clone ID: 190M7) was
subjected to PCR for amplification of Scn5a genomic DNA fragments using specific primers:
(fragment -736 to +119, F, 5’-TGGCGGTGTGTTTGATTTCAG-3’; R, 5’-
GGGCTCGGTTCGGCGTAG-3’), (fragment -2308 to +119, F, 5’-
GGCTGGAAGTGGTGACATTAGAG-3’; R, 5’-TGGCGGTGTGTTTGATTTCAG-3’),
(fragment -9282 to -8098, F, 5’- TGGCAACCGCAGAACGAC -3’; R, 5’-
CCCTCCTCCCCGCAATCAC -3’), (fragment -10419 to -9917, F, 5’-
TGTGTGGGTGTATGGGTGACC-3’; R, 5’-GGAGGTAGTGCGTCTGTTCCTATC-3’),
(fragment -17405 to -16448, F, 5’-ATGTTCCTGGCATAACCCGAG-3’; R, 5’-
AAACCCTTTCCTCCCCCG-3’), (fragment -27145 to -26589, F, 5’-
CCAAGCCACACTCTCAGGTCAC-3’; R, 5’-GCAAGCACAGGGGGACTGG-3’). PCR
fragments were cloned into pCR-Blunt II-TOPO or pCR2.1 vector (Invitrogen), sequenced, and
inserted into pGL3 basic plasmid (Promega) using appropriate restriction enzyme sites. Nkx2-5
non-binding mutant of -736 to +119 bp fragments were generated by PCR using two primers; F,

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 6
5’-
TGTGGTTCTGGGTGTCCCGGTGTCAGTGTGTCAATGGATGTGTCTCTGGGTCACGTG
GTA-3’; R, 5’-
TACCACGTGACCCAGAGACACATCCATTGACACACTGACACCGGGACACCCAGAAC
CACA-3’.
10T1/2 fibroblast cells cultured in six-well plates were cotransfected with 3 mg of
luciferease reporter constructs, 1 mg of pcDNA3 or pcDNA3-Nkx2-5 expression plasmid and 0.5
mg of Rous sarcoma virus b-galactosidase construct using the calcium phosphate method as
described previously.10
Statistical analyses
Results among groups except for analyses of sodium currents (see above) were compared
using ANOVA and Fisher’s PLSD or Bonferroni’s post-hoc test (StatView version 5.01).

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 7
References:
1. Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark
B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR.
Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage
specification leads to progressive cardiomyopathy and complete heart block. Cell.
2004;117:373-86.
2. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-
inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the
mouse. Dev Biol. 2002;244:305-18.
3. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet.
1999;21:70-1.
4. Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac
expression of Csx/Nkx2.5 homeodomain protein. Circ Res. 1998;82:936-46.
5. Bhat MB, Hayek SM, Zhao J, Zang W, Takeshima H, Wier WG, Ma J. Expression and
functional characterization of the cardiac muscle ryanodine receptor Ca(2+) release
channel in Chinese hamster ovary cells. Biophys J. 1999;77:808-16.
6. Wakimoto H, Kasahara H, Maguire CT, Izumo S, Berul CI. Developmentally modulated
cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of
DNA nonbinding mutant Nkx2.5. J Cardiovasc Electrophysiol. 2002;13:682-8.
7. Hogan B, Beddingtpm R, Costantini F, Lacy E. Manipulating the mouse embryo. second
ed. New York, NY: Cold Spring Harbor Laboratory Press; 1994.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 8
8. El-Badawi A, Schenk EA. Histochemical methods for separate, consecutive and
simultaneous demonstration of acetylcholinesterase and norepinephrine in cryostat
sections. J Histochem Cytochem. 1967;15:580-8.
9. Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki
H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain
kinase. Circ Res;in press.
10. Kasahara H, Usheva A, Ueyama T, Aoki H, Horikoshi N, Izumo S. Characterization of
homo- and heterodimerization of cardiac Csx/Nkx2.5 homeoprotein. J Biol Chem.
2001;276:4570-80.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 9
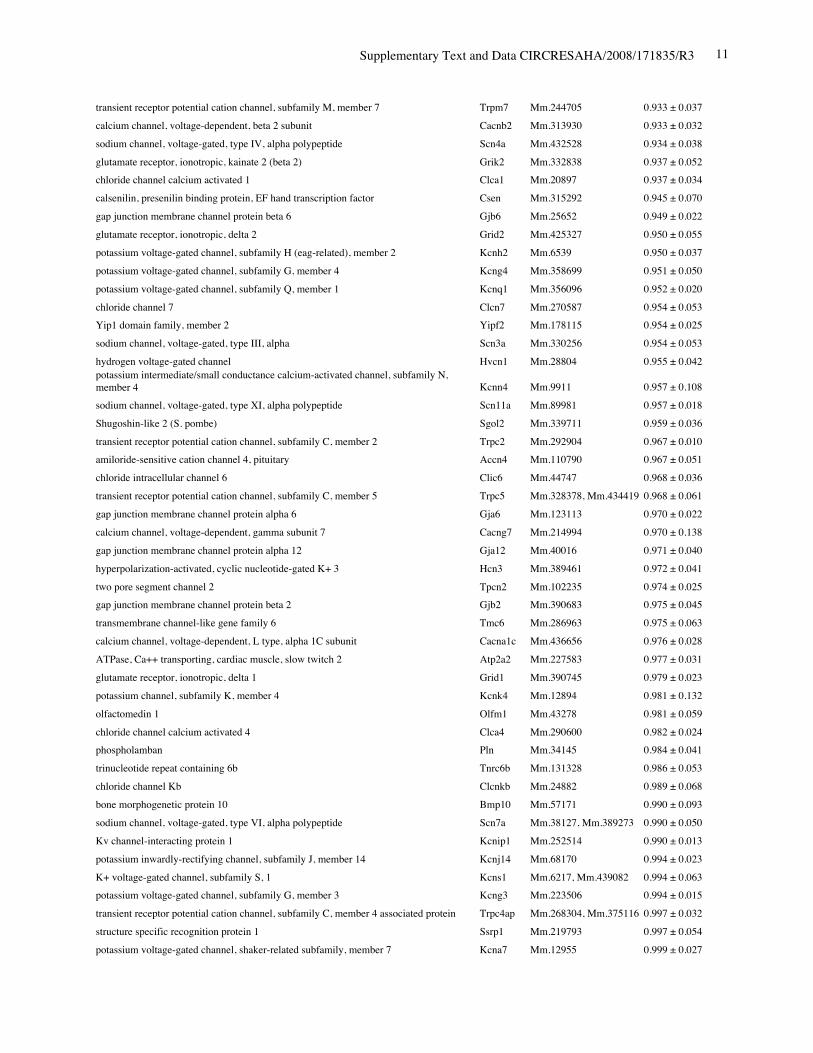
Online Table I. The six most reduced ion channels in Nkx2-5 knockout hearts by gene expressionprofiling.
Protein Gene UniGene No.
fold differenceKO vs. control
(mean ± S.E., n=4)calcium channel, voltage-dependent, T type, alpha 1H subunit Cacna1h Mm.268378 0.321 ± 0.012potassium voltage-gated channel, Isk-related subfamily, member 1 Kcne1 Mm.299425 0.367 ± 0.002calcium channel, voltage-dependent, T type, alpha 1G subunit Cacna1g Mm.29585 0.518 ± 0.018sodium channel, voltage-gated, type V, alpha polypeptide Scn5a Mm.103584 0.745 ± 0.009
gap junction membrane channel protein alpha 5 (connexin 40) Gja5 Mm.281816 0.767 ± 0.029ryanodine receptor 2, cardiac Ryr2 Mm.239871 0.766 ± 0.057
PD4
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 10
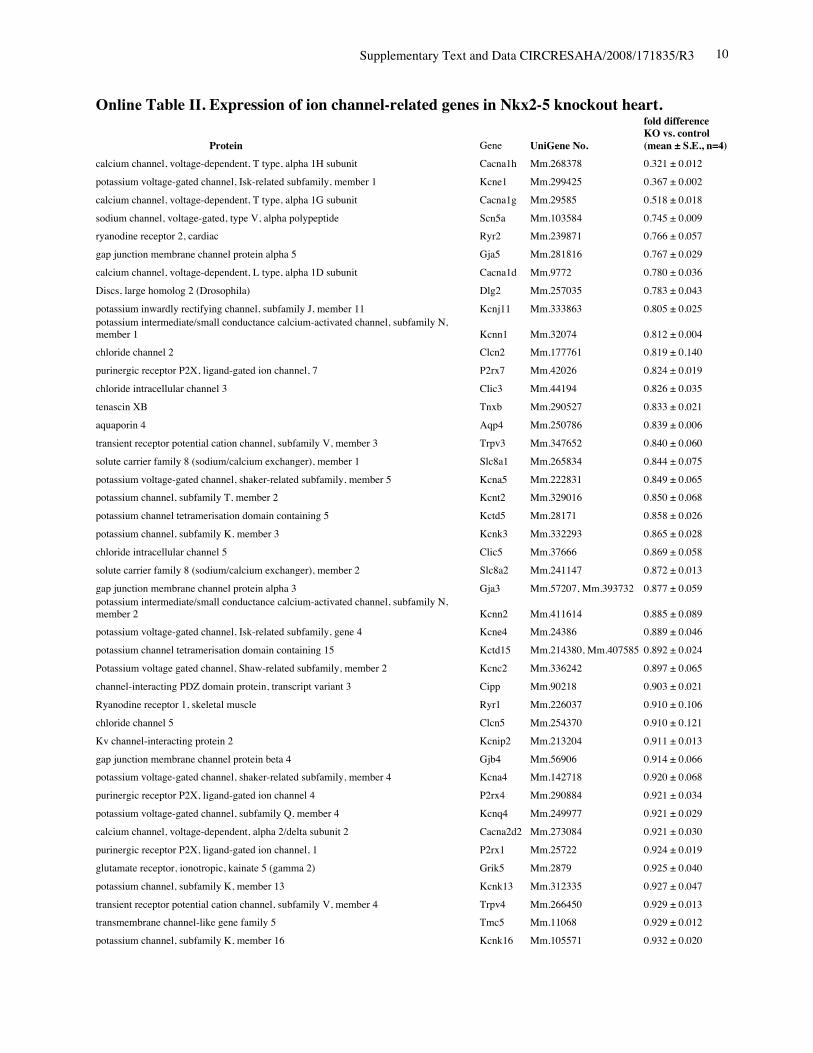
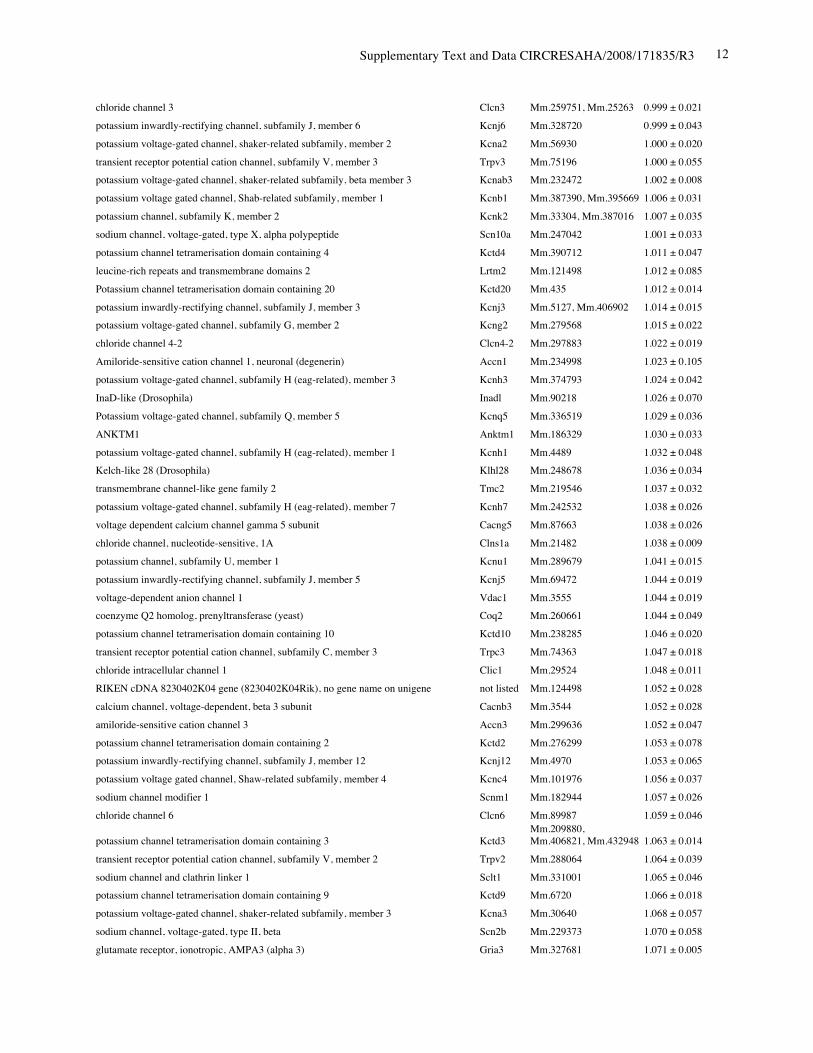
Online Table II. Expression of ion channel-related genes in Nkx2-5 knockout heart.
Protein Gene UniGene No.
fold differenceKO vs. control(mean ± S.E., n=4)
calcium channel, voltage-dependent, T type, alpha 1H subunit Cacna1h Mm.268378 0.321 ± 0.012potassium voltage-gated channel, Isk-related subfamily, member 1 Kcne1 Mm.299425 0.367 ± 0.002calcium channel, voltage-dependent, T type, alpha 1G subunit Cacna1g Mm.29585 0.518 ± 0.018sodium channel, voltage-gated, type V, alpha polypeptide Scn5a Mm.103584 0.745 ± 0.009ryanodine receptor 2, cardiac Ryr2 Mm.239871 0.766 ± 0.057gap junction membrane channel protein alpha 5 Gja5 Mm.281816 0.767 ± 0.029calcium channel, voltage-dependent, L type, alpha 1D subunit Cacna1d Mm.9772 0.780 ± 0.036Discs, large homolog 2 (Drosophila) Dlg2 Mm.257035 0.783 ± 0.043potassium inwardly rectifying channel, subfamily J, member 11 Kcnj11 Mm.333863 0.805 ± 0.025potassium intermediate/small conductance calcium-activated channel, subfamily N,member 1 Kcnn1 Mm.32074 0.812 ± 0.004chloride channel 2 Clcn2 Mm.177761 0.819 ± 0.140purinergic receptor P2X, ligand-gated ion channel, 7 P2rx7 Mm.42026 0.824 ± 0.019chloride intracellular channel 3 Clic3 Mm.44194 0.826 ± 0.035tenascin XB Tnxb Mm.290527 0.833 ± 0.021aquaporin 4 Aqp4 Mm.250786 0.839 ± 0.006transient receptor potential cation channel, subfamily V, member 3 Trpv3 Mm.347652 0.840 ± 0.060solute carrier family 8 (sodium/calcium exchanger), member 1 Slc8a1 Mm.265834 0.844 ± 0.075potassium voltage-gated channel, shaker-related subfamily, member 5 Kcna5 Mm.222831 0.849 ± 0.065potassium channel, subfamily T, member 2 Kcnt2 Mm.329016 0.850 ± 0.068potassium channel tetramerisation domain containing 5 Kctd5 Mm.28171 0.858 ± 0.026potassium channel, subfamily K, member 3 Kcnk3 Mm.332293 0.865 ± 0.028chloride intracellular channel 5 Clic5 Mm.37666 0.869 ± 0.058solute carrier family 8 (sodium/calcium exchanger), member 2 Slc8a2 Mm.241147 0.872 ± 0.013gap junction membrane channel protein alpha 3 Gja3 Mm.57207, Mm.393732 0.877 ± 0.059potassium intermediate/small conductance calcium-activated channel, subfamily N,member 2 Kcnn2 Mm.411614 0.885 ± 0.089potassium voltage-gated channel, Isk-related subfamily, gene 4 Kcne4 Mm.24386 0.889 ± 0.046potassium channel tetramerisation domain containing 15 Kctd15 Mm.214380, Mm.407585 0.892 ± 0.024Potassium voltage gated channel, Shaw-related subfamily, member 2 Kcnc2 Mm.336242 0.897 ± 0.065channel-interacting PDZ domain protein, transcript variant 3 Cipp Mm.90218 0.903 ± 0.021Ryanodine receptor 1, skeletal muscle Ryr1 Mm.226037 0.910 ± 0.106chloride channel 5 Clcn5 Mm.254370 0.910 ± 0.121Kv channel-interacting protein 2 Kcnip2 Mm.213204 0.911 ± 0.013gap junction membrane channel protein beta 4 Gjb4 Mm.56906 0.914 ± 0.066potassium voltage-gated channel, shaker-related subfamily, member 4 Kcna4 Mm.142718 0.920 ± 0.068purinergic receptor P2X, ligand-gated ion channel 4 P2rx4 Mm.290884 0.921 ± 0.034potassium voltage-gated channel, subfamily Q, member 4 Kcnq4 Mm.249977 0.921 ± 0.029calcium channel, voltage-dependent, alpha 2/delta subunit 2 Cacna2d2 Mm.273084 0.921 ± 0.030purinergic receptor P2X, ligand-gated ion channel, 1 P2rx1 Mm.25722 0.924 ± 0.019glutamate receptor, ionotropic, kainate 5 (gamma 2) Grik5 Mm.2879 0.925 ± 0.040potassium channel, subfamily K, member 13 Kcnk13 Mm.312335 0.927 ± 0.047transient receptor potential cation channel, subfamily V, member 4 Trpv4 Mm.266450 0.929 ± 0.013transmembrane channel-like gene family 5 Tmc5 Mm.11068 0.929 ± 0.012potassium channel, subfamily K, member 16 Kcnk16 Mm.105571 0.932 ± 0.020
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 11
transient receptor potential cation channel, subfamily M, member 7 Trpm7 Mm.244705 0.933 ± 0.037calcium channel, voltage-dependent, beta 2 subunit Cacnb2 Mm.313930 0.933 ± 0.032sodium channel, voltage-gated, type IV, alpha polypeptide Scn4a Mm.432528 0.934 ± 0.038glutamate receptor, ionotropic, kainate 2 (beta 2) Grik2 Mm.332838 0.937 ± 0.052chloride channel calcium activated 1 Clca1 Mm.20897 0.937 ± 0.034calsenilin, presenilin binding protein, EF hand transcription factor Csen Mm.315292 0.945 ± 0.070gap junction membrane channel protein beta 6 Gjb6 Mm.25652 0.949 ± 0.022glutamate receptor, ionotropic, delta 2 Grid2 Mm.425327 0.950 ± 0.055potassium voltage-gated channel, subfamily H (eag-related), member 2 Kcnh2 Mm.6539 0.950 ± 0.037potassium voltage-gated channel, subfamily G, member 4 Kcng4 Mm.358699 0.951 ± 0.050potassium voltage-gated channel, subfamily Q, member 1 Kcnq1 Mm.356096 0.952 ± 0.020chloride channel 7 Clcn7 Mm.270587 0.954 ± 0.053Yip1 domain family, member 2 Yipf2 Mm.178115 0.954 ± 0.025sodium channel, voltage-gated, type III, alpha Scn3a Mm.330256 0.954 ± 0.053hydrogen voltage-gated channel Hvcn1 Mm.28804 0.955 ± 0.042potassium intermediate/small conductance calcium-activated channel, subfamily N,member 4 Kcnn4 Mm.9911 0.957 ± 0.108sodium channel, voltage-gated, type XI, alpha polypeptide Scn11a Mm.89981 0.957 ± 0.018Shugoshin-like 2 (S. pombe) Sgol2 Mm.339711 0.959 ± 0.036transient receptor potential cation channel, subfamily C, member 2 Trpc2 Mm.292904 0.967 ± 0.010amiloride-sensitive cation channel 4, pituitary Accn4 Mm.110790 0.967 ± 0.051chloride intracellular channel 6 Clic6 Mm.44747 0.968 ± 0.036transient receptor potential cation channel, subfamily C, member 5 Trpc5 Mm.328378, Mm.434419 0.968 ± 0.061gap junction membrane channel protein alpha 6 Gja6 Mm.123113 0.970 ± 0.022calcium channel, voltage-dependent, gamma subunit 7 Cacng7 Mm.214994 0.970 ± 0.138gap junction membrane channel protein alpha 12 Gja12 Mm.40016 0.971 ± 0.040hyperpolarization-activated, cyclic nucleotide-gated K+ 3 Hcn3 Mm.389461 0.972 ± 0.041two pore segment channel 2 Tpcn2 Mm.102235 0.974 ± 0.025gap junction membrane channel protein beta 2 Gjb2 Mm.390683 0.975 ± 0.045transmembrane channel-like gene family 6 Tmc6 Mm.286963 0.975 ± 0.063calcium channel, voltage-dependent, L type, alpha 1C subunit Cacna1c Mm.436656 0.976 ± 0.028ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 Atp2a2 Mm.227583 0.977 ± 0.031glutamate receptor, ionotropic, delta 1 Grid1 Mm.390745 0.979 ± 0.023potassium channel, subfamily K, member 4 Kcnk4 Mm.12894 0.981 ± 0.132olfactomedin 1 Olfm1 Mm.43278 0.981 ± 0.059chloride channel calcium activated 4 Clca4 Mm.290600 0.982 ± 0.024phospholamban Pln Mm.34145 0.984 ± 0.041trinucleotide repeat containing 6b Tnrc6b Mm.131328 0.986 ± 0.053chloride channel Kb Clcnkb Mm.24882 0.989 ± 0.068bone morphogenetic protein 10 Bmp10 Mm.57171 0.990 ± 0.093sodium channel, voltage-gated, type VI, alpha polypeptide Scn7a Mm.38127, Mm.389273 0.990 ± 0.050Kv channel-interacting protein 1 Kcnip1 Mm.252514 0.990 ± 0.013potassium inwardly-rectifying channel, subfamily J, member 14 Kcnj14 Mm.68170 0.994 ± 0.023K+ voltage-gated channel, subfamily S, 1 Kcns1 Mm.6217, Mm.439082 0.994 ± 0.063potassium voltage-gated channel, subfamily G, member 3 Kcng3 Mm.223506 0.994 ± 0.015transient receptor potential cation channel, subfamily C, member 4 associated protein Trpc4ap Mm.268304, Mm.375116 0.997 ± 0.032structure specific recognition protein 1 Ssrp1 Mm.219793 0.997 ± 0.054potassium voltage-gated channel, shaker-related subfamily, member 7 Kcna7 Mm.12955 0.999 ± 0.027
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 12
chloride channel 3 Clcn3 Mm.259751, Mm.25263 0.999 ± 0.021potassium inwardly-rectifying channel, subfamily J, member 6 Kcnj6 Mm.328720 0.999 ± 0.043potassium voltage-gated channel, shaker-related subfamily, member 2 Kcna2 Mm.56930 1.000 ± 0.020transient receptor potential cation channel, subfamily V, member 3 Trpv3 Mm.75196 1.000 ± 0.055potassium voltage-gated channel, shaker-related subfamily, beta member 3 Kcnab3 Mm.232472 1.002 ± 0.008potassium voltage gated channel, Shab-related subfamily, member 1 Kcnb1 Mm.387390, Mm.395669 1.006 ± 0.031potassium channel, subfamily K, member 2 Kcnk2 Mm.33304, Mm.387016 1.007 ± 0.035sodium channel, voltage-gated, type X, alpha polypeptide Scn10a Mm.247042 1.001 ± 0.033potassium channel tetramerisation domain containing 4 Kctd4 Mm.390712 1.011 ± 0.047leucine-rich repeats and transmembrane domains 2 Lrtm2 Mm.121498 1.012 ± 0.085Potassium channel tetramerisation domain containing 20 Kctd20 Mm.435 1.012 ± 0.014potassium inwardly-rectifying channel, subfamily J, member 3 Kcnj3 Mm.5127, Mm.406902 1.014 ± 0.015potassium voltage-gated channel, subfamily G, member 2 Kcng2 Mm.279568 1.015 ± 0.022chloride channel 4-2 Clcn4-2 Mm.297883 1.022 ± 0.019Amiloride-sensitive cation channel 1, neuronal (degenerin) Accn1 Mm.234998 1.023 ± 0.105potassium voltage-gated channel, subfamily H (eag-related), member 3 Kcnh3 Mm.374793 1.024 ± 0.042InaD-like (Drosophila) Inadl Mm.90218 1.026 ± 0.070Potassium voltage-gated channel, subfamily Q, member 5 Kcnq5 Mm.336519 1.029 ± 0.036ANKTM1 Anktm1 Mm.186329 1.030 ± 0.033potassium voltage-gated channel, subfamily H (eag-related), member 1 Kcnh1 Mm.4489 1.032 ± 0.048Kelch-like 28 (Drosophila) Klhl28 Mm.248678 1.036 ± 0.034transmembrane channel-like gene family 2 Tmc2 Mm.219546 1.037 ± 0.032potassium voltage-gated channel, subfamily H (eag-related), member 7 Kcnh7 Mm.242532 1.038 ± 0.026voltage dependent calcium channel gamma 5 subunit Cacng5 Mm.87663 1.038 ± 0.026chloride channel, nucleotide-sensitive, 1A Clns1a Mm.21482 1.038 ± 0.009potassium channel, subfamily U, member 1 Kcnu1 Mm.289679 1.041 ± 0.015potassium inwardly-rectifying channel, subfamily J, member 5 Kcnj5 Mm.69472 1.044 ± 0.019voltage-dependent anion channel 1 Vdac1 Mm.3555 1.044 ± 0.019coenzyme Q2 homolog, prenyltransferase (yeast) Coq2 Mm.260661 1.044 ± 0.049potassium channel tetramerisation domain containing 10 Kctd10 Mm.238285 1.046 ± 0.020transient receptor potential cation channel, subfamily C, member 3 Trpc3 Mm.74363 1.047 ± 0.018chloride intracellular channel 1 Clic1 Mm.29524 1.048 ± 0.011RIKEN cDNA 8230402K04 gene (8230402K04Rik), no gene name on unigene not listed Mm.124498 1.052 ± 0.028calcium channel, voltage-dependent, beta 3 subunit Cacnb3 Mm.3544 1.052 ± 0.028amiloride-sensitive cation channel 3 Accn3 Mm.299636 1.052 ± 0.047potassium channel tetramerisation domain containing 2 Kctd2 Mm.276299 1.053 ± 0.078potassium inwardly-rectifying channel, subfamily J, member 12 Kcnj12 Mm.4970 1.053 ± 0.065potassium voltage gated channel, Shaw-related subfamily, member 4 Kcnc4 Mm.101976 1.056 ± 0.037sodium channel modifier 1 Scnm1 Mm.182944 1.057 ± 0.026chloride channel 6 Clcn6 Mm.89987 1.059 ± 0.046
potassium channel tetramerisation domain containing 3 Kctd3Mm.209880,Mm.406821, Mm.432948 1.063 ± 0.014
transient receptor potential cation channel, subfamily V, member 2 Trpv2 Mm.288064 1.064 ± 0.039sodium channel and clathrin linker 1 Sclt1 Mm.331001 1.065 ± 0.046potassium channel tetramerisation domain containing 9 Kctd9 Mm.6720 1.066 ± 0.018potassium voltage-gated channel, shaker-related subfamily, member 3 Kcna3 Mm.30640 1.068 ± 0.057sodium channel, voltage-gated, type II, beta Scn2b Mm.229373 1.070 ± 0.058glutamate receptor, ionotropic, AMPA3 (alpha 3) Gria3 Mm.327681 1.071 ± 0.005
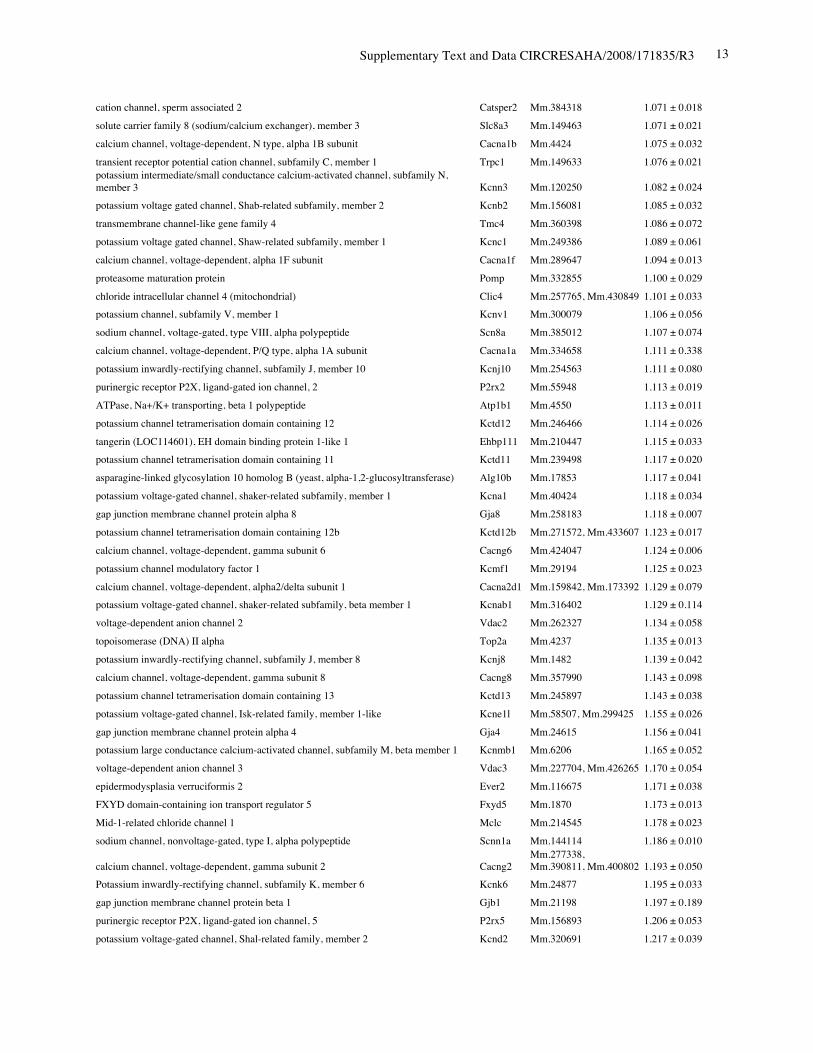
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 13
cation channel, sperm associated 2 Catsper2 Mm.384318 1.071 ± 0.018solute carrier family 8 (sodium/calcium exchanger), member 3 Slc8a3 Mm.149463 1.071 ± 0.021calcium channel, voltage-dependent, N type, alpha 1B subunit Cacna1b Mm.4424 1.075 ± 0.032transient receptor potential cation channel, subfamily C, member 1 Trpc1 Mm.149633 1.076 ± 0.021potassium intermediate/small conductance calcium-activated channel, subfamily N,member 3 Kcnn3 Mm.120250 1.082 ± 0.024potassium voltage gated channel, Shab-related subfamily, member 2 Kcnb2 Mm.156081 1.085 ± 0.032transmembrane channel-like gene family 4 Tmc4 Mm.360398 1.086 ± 0.072potassium voltage gated channel, Shaw-related subfamily, member 1 Kcnc1 Mm.249386 1.089 ± 0.061calcium channel, voltage-dependent, alpha 1F subunit Cacna1f Mm.289647 1.094 ± 0.013proteasome maturation protein Pomp Mm.332855 1.100 ± 0.029chloride intracellular channel 4 (mitochondrial) Clic4 Mm.257765, Mm.430849 1.101 ± 0.033potassium channel, subfamily V, member 1 Kcnv1 Mm.300079 1.106 ± 0.056sodium channel, voltage-gated, type VIII, alpha polypeptide Scn8a Mm.385012 1.107 ± 0.074calcium channel, voltage-dependent, P/Q type, alpha 1A subunit Cacna1a Mm.334658 1.111 ± 0.338potassium inwardly-rectifying channel, subfamily J, member 10 Kcnj10 Mm.254563 1.111 ± 0.080purinergic receptor P2X, ligand-gated ion channel, 2 P2rx2 Mm.55948 1.113 ± 0.019ATPase, Na+/K+ transporting, beta 1 polypeptide Atp1b1 Mm.4550 1.113 ± 0.011potassium channel tetramerisation domain containing 12 Kctd12 Mm.246466 1.114 ± 0.026tangerin (LOC114601), EH domain binding protein 1-like 1 Ehbp111 Mm.210447 1.115 ± 0.033potassium channel tetramerisation domain containing 11 Kctd11 Mm.239498 1.117 ± 0.020asparagine-linked glycosylation 10 homolog B (yeast, alpha-1,2-glucosyltransferase) Alg10b Mm.17853 1.117 ± 0.041potassium voltage-gated channel, shaker-related subfamily, member 1 Kcna1 Mm.40424 1.118 ± 0.034gap junction membrane channel protein alpha 8 Gja8 Mm.258183 1.118 ± 0.007potassium channel tetramerisation domain containing 12b Kctd12b Mm.271572, Mm.433607 1.123 ± 0.017calcium channel, voltage-dependent, gamma subunit 6 Cacng6 Mm.424047 1.124 ± 0.006potassium channel modulatory factor 1 Kcmf1 Mm.29194 1.125 ± 0.023calcium channel, voltage-dependent, alpha2/delta subunit 1 Cacna2d1 Mm.159842, Mm.173392 1.129 ± 0.079potassium voltage-gated channel, shaker-related subfamily, beta member 1 Kcnab1 Mm.316402 1.129 ± 0.114voltage-dependent anion channel 2 Vdac2 Mm.262327 1.134 ± 0.058topoisomerase (DNA) II alpha Top2a Mm.4237 1.135 ± 0.013potassium inwardly-rectifying channel, subfamily J, member 8 Kcnj8 Mm.1482 1.139 ± 0.042calcium channel, voltage-dependent, gamma subunit 8 Cacng8 Mm.357990 1.143 ± 0.098potassium channel tetramerisation domain containing 13 Kctd13 Mm.245897 1.143 ± 0.038potassium voltage-gated channel, Isk-related family, member 1-like Kcne1l Mm.58507, Mm.299425 1.155 ± 0.026gap junction membrane channel protein alpha 4 Gja4 Mm.24615 1.156 ± 0.041potassium large conductance calcium-activated channel, subfamily M, beta member 1 Kcnmb1 Mm.6206 1.165 ± 0.052voltage-dependent anion channel 3 Vdac3 Mm.227704, Mm.426265 1.170 ± 0.054epidermodysplasia verruciformis 2 Ever2 Mm.116675 1.171 ± 0.038FXYD domain-containing ion transport regulator 5 Fxyd5 Mm.1870 1.173 ± 0.013Mid-1-related chloride channel 1 Mclc Mm.214545 1.178 ± 0.023sodium channel, nonvoltage-gated, type I, alpha polypeptide Scnn1a Mm.144114 1.186 ± 0.010
calcium channel, voltage-dependent, gamma subunit 2 Cacng2Mm.277338,Mm.390811, Mm.400802 1.193 ± 0.050
Potassium inwardly-rectifying channel, subfamily K, member 6 Kcnk6 Mm.24877 1.195 ± 0.033gap junction membrane channel protein beta 1 Gjb1 Mm.21198 1.197 ± 0.189purinergic receptor P2X, ligand-gated ion channel, 5 P2rx5 Mm.156893 1.206 ± 0.053potassium voltage-gated channel, Shal-related family, member 2 Kcnd2 Mm.320691 1.217 ± 0.039
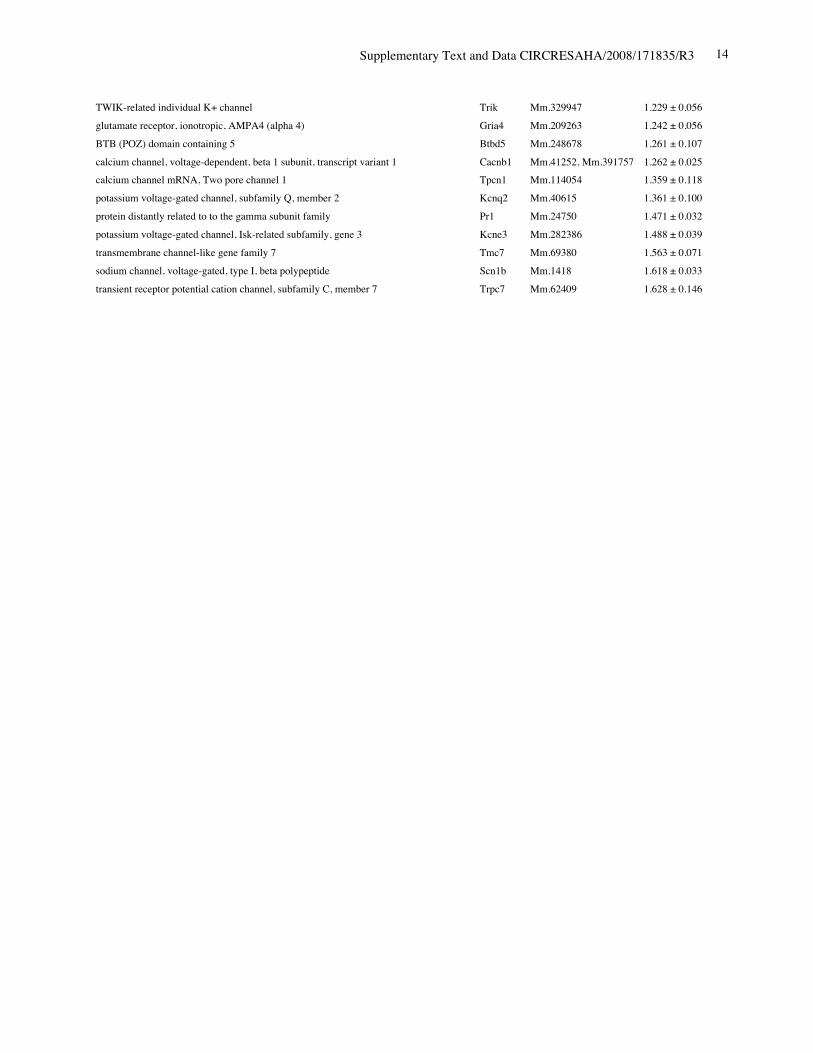
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 14
TWIK-related individual K+ channel Trik Mm.329947 1.229 ± 0.056glutamate receptor, ionotropic, AMPA4 (alpha 4) Gria4 Mm.209263 1.242 ± 0.056BTB (POZ) domain containing 5 Btbd5 Mm.248678 1.261 ± 0.107calcium channel, voltage-dependent, beta 1 subunit, transcript variant 1 Cacnb1 Mm.41252, Mm.391757 1.262 ± 0.025calcium channel mRNA, Two pore channel 1 Tpcn1 Mm.114054 1.359 ± 0.118potassium voltage-gated channel, subfamily Q, member 2 Kcnq2 Mm.40615 1.361 ± 0.100protein distantly related to to the gamma subunit family Pr1 Mm.24750 1.471 ± 0.032potassium voltage-gated channel, Isk-related subfamily, gene 3 Kcne3 Mm.282386 1.488 ± 0.039transmembrane channel-like gene family 7 Tmc7 Mm.69380 1.563 ± 0.071sodium channel, voltage-gated, type I, beta polypeptide Scn1b Mm.1418 1.618 ± 0.033transient receptor potential cation channel, subfamily C, member 7 Trpc7 Mm.62409 1.628 ± 0.146

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 15
Online Figure I. Frozen sections of R26R/Tg-Cre-ERTM stained for b-galactosidase including aorticvalve (A) and pulmonary valve (B). Bars = 500 um. AoV, aortic valve; PV, pulmonary valve.;LA, left atrium; LV, left ventricle, RV, right ventricle.
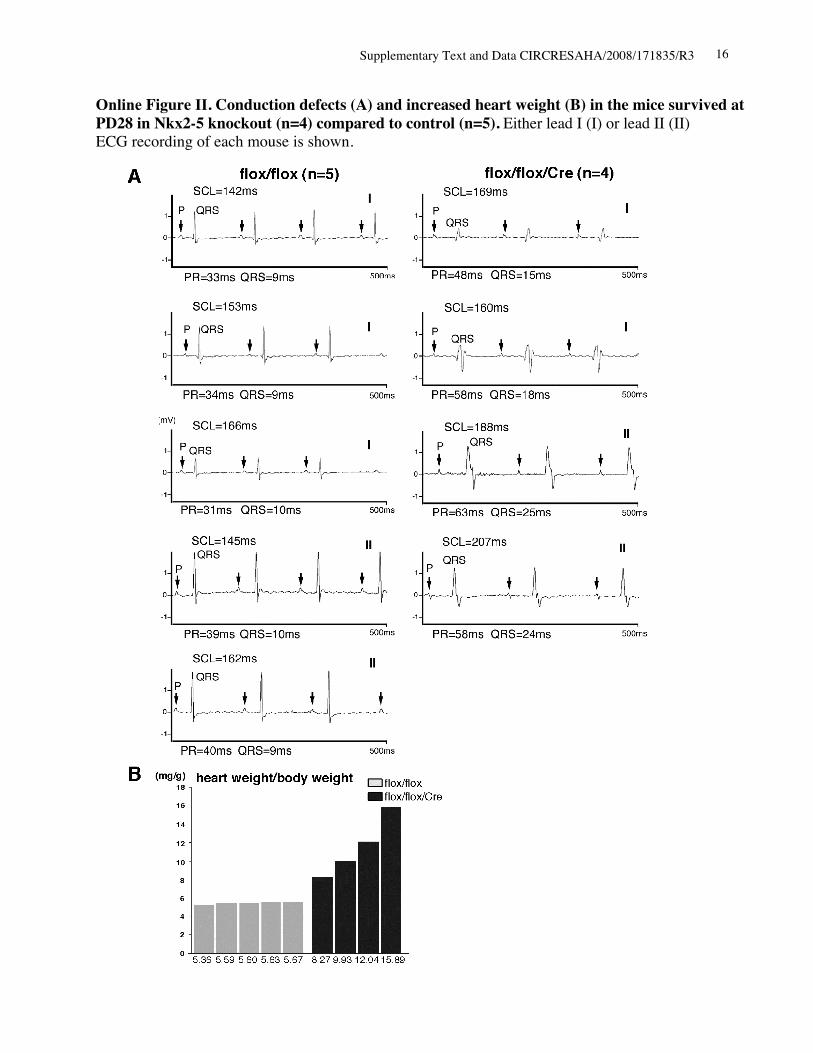
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 16
Online Figure II. Conduction defects (A) and increased heart weight (B) in the mice survived atPD28 in Nkx2-5 knockout (n=4) compared to control (n=5). Either lead I (I) or lead II (II)ECG recording of each mouse is shown.
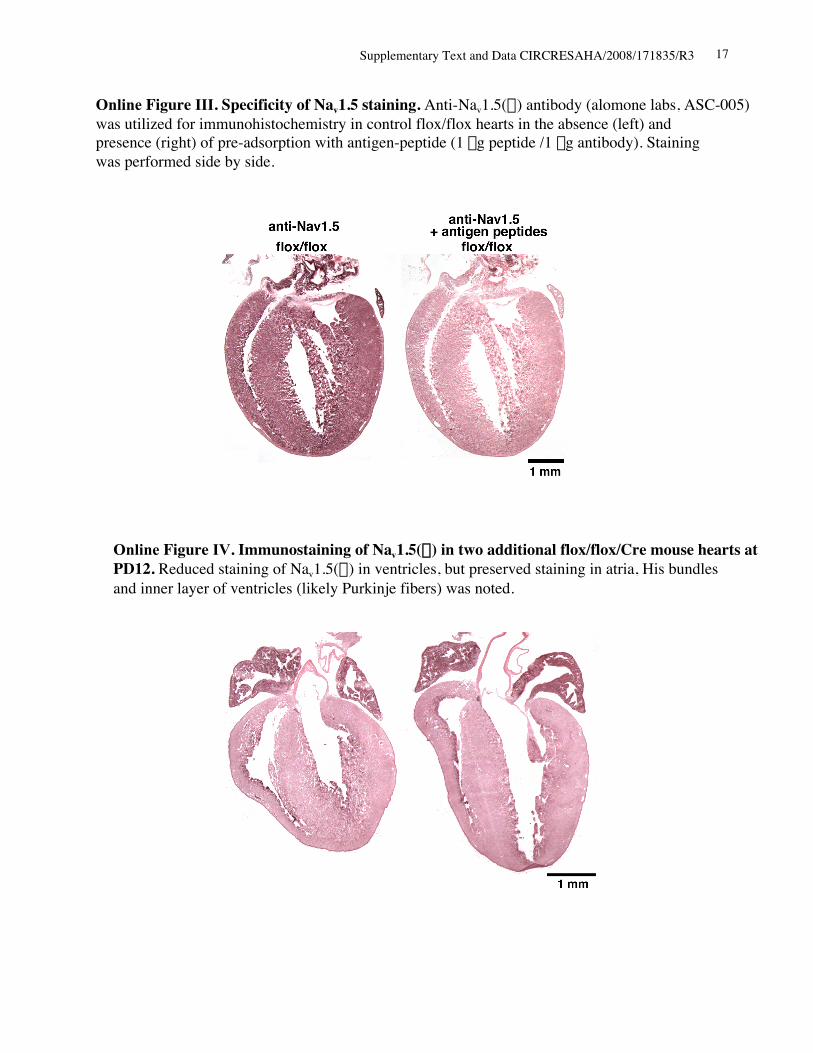
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 17
Online Figure III. Specificity of Nav1.5 staining. Anti-Nav1.5(a) antibody (alomone labs, ASC-005)was utilized for immunohistochemistry in control flox/flox hearts in the absence (left) andpresence (right) of pre-adsorption with antigen-peptide (1 mg peptide /1 mg antibody). Stainingwas performed side by side.
Online Figure IV. Immunostaining of Nav1.5(a) in two additional flox/flox/Cre mouse hearts atPD12. Reduced staining of Nav1.5(a) in ventricles, but preserved staining in atria, His bundlesand inner layer of ventricles (likely Purkinje fibers) was noted.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 18
Nav1.5(a)
Nkx2-5
GAPDH
PD12
1 2
250
40
30
Cre-ER +- fl/flfl/flkDa
proteinNav1.5(a) atria
Online Figure V. Western blotting demonstrates similar levels of Nav1.5(a) protein in atria offlox/flox and flox/flox/Cre mice (lanes 1 vs.2).

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 1
Materials and Methods:
Inducible Nkx2-5 knockout mice
Floxed-Nkx2-5 homozygous mice (Nkx2-5flox/flox)1 were bred with transgenic mice
carrying the Cre-ERTM gene under CMV promoter, Tg(CMV-Cre-ERTM).2 Subsequent matings
between offsprings generated Nkx2-5flox/flox/Tg(CMV-Cre-ERTM)(flox/flox/Cre), Nkx2-
5wild/wild/Tg(CMV-Cre-ERTM) (wild/wild/Cre) and Nkx2-5flox/flox (flox/flox). For perinatal deletion
of the floxed-Nkx2-5 genes, a single injection of tamoxifen (0.5-1 mg/g body weight, ip) was
administered to pregnant mice on gestation day 19. Male Tg(CMV-Cre-ERTM)2 were also crossed
to females that carried the Rosa reporter allele, R26R3. All animal care protocols fully conformed
to the Association for the Assessment and Accreditation of Laboratory Animal Care, with
approvals from the University of Florida Institutional Animal Care and Use Committees.
Southern, Northern and Western blotting and immunostaining
Southern and Northern blot analyses were performed using the following probes: Nkx2-5
5’ genomic probe, NotI-ClaI fragment of mouse Nkx2-5 genomic DNA; Nkx2-5 coding probe,
PflMI-EcoRI fragment of mouse Nkx2-5 cDNA; Cre-recombinase probe, EcoRI-BamHI
fragment of pCAGGS-nls-Cre plasmid (gift from Dr. H. Xin); RT-PCR products amplified by
the following primer sets: KcneI/minK (476 bp, F’, 5'- CTTGACGCCCAGGATGAGC -3'; R,
5'- GGTGCCCCTACAATAAAGACTATGG -3'); HOD/HOP (703 bp, F’, 5'-
AGCAGACAGGCACCAGCATC -3'; R’- AAGGGAGCACAGGGAAGTGAAC -3'). Western
blot analyses and immunostaining using frozen sections were performed with the following
primary antibodies: anti-Nkx2-5 pAb4, sarcomeric actinin (A7811, SIGMA), Nav1.5(a) (ASC-
005, alomone labs), GAPDH (RDI-TRK5G4-6C5, Research Diagnostic Inc), RyR2 (MA3-916,

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 2
Affinity BioReagents), SERCA2a (sc-8095, Santa Cruz), phospholamban (05-205, Upstate). For
detection of RyR and pentamer of phospholamban, protein samples were heated at 37ºC for 15
min before loading on SDS-PAGE gels.5 Fluorescent microscopic images were obtained using
ZEISS Axiovert200M with or without Apotome.
Surface ECG recording and ultrasound imaging
Recordings of six limb leads surface ECG (ADInstruments, Milford, MA) were
performed without anesthesia as described previously with some modifications.6 ECG recordings
were analyzed using PowerLab software (ADInstruments).6 M-mode ultrasound imaging of the
left ventricle was obtained at the level of the papillary muscle from a parasternal window using
an ultrasound biomicroscope with a single transducer with a frequency 55 MHz (VisualSonics,
Tronto, Canada).
Histological analyses
X-gal staining of whole hearts and frozen sections were performed according to standard
protocol.7 Tissues were fixed in 4% paraformaldehyde/PBS overnight, dehydrated, embedded in
paraffin, sectioned, and stained with hematoxylin/eosin or Masson’s trichrome. Acetylcholine
esterase staining in frozen tissue sections was performed as described previously with some
modifications.8 Whole mount acetylcholine esterase staining was performed with the incisions on
right ventricular and atrial free wall, with addition of 0.1% TritonTM X-100 in all the solutions
except for Cu2+-glycine solution. After fixation with paraformaldehyde, tissue images were
captured by a stereomicroscope (Nikon SMZ800) attached to a CCD camera (QImaging
MicroPublisher3.3, BC, Canada), with scale bars present. The same magnifications and zoom

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 3
were applied for the hearts dissected from flox/flox or flox/flox/Cre mice at the same ages.
Digitalized AV nodal surface size was measured using NIH Image software.
Gene expression profiling
Total RNA pooled from 3-4 hearts of postnatal day 4 (PD4) flox/flox or flox/flox/Cre
mice with tamoxifen administration was analyzed in the CodeLink Bioarrays followed by
CodeLink and GeneSpring software (Genus Biosystems, Northbrook, IL). Four sets of relative
expression values between flox/flox and flox/flox/Cre were averaged and are listed.
Measurement of sodium current and simultaneous recording of cardiac contraction and
Ca2+
After genotyping, neonatal ventricular cardiomyocytes were isolated from flox/flox or
flox/flox/Cre mice side by side at PD3, cultured for 4-5 days before measuring sodium currents
(equivalent to PD7-8). Sodium currents were (INa) obtained by voltage clamp recordings in the
whole cell patch clamp configuration at room temperature (~22°C). Myocytes were bathed in
low sodium buffer containing, in mmol/L: NaCl 10, CsCl 80, CaCl2 2, MgCl2 1, HEPES 10, 4-
aminopyridine 10, tetraethylammonium-Cl 30, dextrose 10, CdCl 0.1; and nifedipine 5 mM with
an osmolarity of 290 mOsm and pH of 7.4 adjusted with CsOH. Recordings were obtained using
Molecular Devices Axon 200A amplifier (Sunnyvale, CA) filtered at 2 kHz and digitized using
Molecular Devices, Digidata 1322A, at 10 kHz. Data were acquired using Molecular Devices
PClamp 9.0 software. Myocytes were impaled with Drummond Wiretrol II capillary pipettes
(Drummond Scientific Company, Broomall, PA) pulled with Sutter instrument P-97 horizontal
pipette puller and polished with Narishige MF-830 microforge with series resistance of 0.5 to 1.5

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 4
mΩ when filled with solution containing, in mmol/L: CsOH 105, glutamic acid 105,
tetraethylammonium-Cl 20, HEPES 10, Cs2-EGTA 5, MgATP 5, NaGTP 0.1 with osmolarity of
285 and pH adjusted to 7.2 with CsOH. A -9.7 mV correction for liquid junction potential,
between the patch pipette and bath solution, was set upon access to the cell cytosol. Cell
capacitance and input series resistance were electronically compensated to approximately 90
percent using online circuitry. Leak currents were subtracted online with P/4 protocol.
Data analysis was performed using Molecular devices Clampfit 9.0, Microsoft Excel, and
GraphPad Prism 4.0 (San Diego, CA). INa current density was derived by dividing current
amplitude from each recording by their respective cell capacitance obtained from amplifier upon
compensation. Experiments that showed inadequate voltage control, such as space clamp error or
inappropriate steep increase in current amplitude in the negative slope of current voltage relation,
were discarded. Steady state activation curves were generated from data used to obtain current
voltage relation plots fitted to a Boltzman equation: I/IMax = 1/[1+exp(V – V1/2 / KV)] where I/IMax
is the current expressed as fraction of maximal current; V1/2 is the voltage to obtain half maximal
current and KmV is the slope factor.
Cardiomyocytes were isolated from 3 weeks old flox/flox or flox/flox/Cre mice. Only
rod-shaped cardiomyocytes with staircase ends, clear cross striations and surface membranes free
from blebs were studied for simultaneous measurements of cell shortening and intracellular free
calcium cardiomyocyte contraction and simultaneous Ca2+ measurement as described
previously.9

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 5
Real-time reverse transcriptase (RT)-PCR
Real-time RT-PCR was performed using the inventoried Taqman Gene Expression
Assays (Applied Biosystems): Scn5a, Mm00451971_m1; RyR2, Mm00465877_m1; T-type Ca
channel a1G, Mm00486549_m1; T-type Ca channel a1H, Mm00445369_m1; Scn1a,
Mm00450580; Scn3a, Mm00658167; Scn4a, Mm00500103; Scn7a, Mm00801952; Scn8a,
Mm00488110; Scn9a, Mm00450762; Scn1b, Mm0041210. Data were normalized with beta-
actin expression (product No, 4352933E). Duplicate experiments were averaged.
Reporter assays
C57BL/6 genomic DNA or BAC clone (Invitrogen, RPC 123.C, Clone ID: 190M7) was
subjected to PCR for amplification of Scn5a genomic DNA fragments using specific primers:
(fragment -736 to +119, F, 5’-TGGCGGTGTGTTTGATTTCAG-3’; R, 5’-
GGGCTCGGTTCGGCGTAG-3’), (fragment -2308 to +119, F, 5’-
GGCTGGAAGTGGTGACATTAGAG-3’; R, 5’-TGGCGGTGTGTTTGATTTCAG-3’),
(fragment -9282 to -8098, F, 5’- TGGCAACCGCAGAACGAC -3’; R, 5’-
CCCTCCTCCCCGCAATCAC -3’), (fragment -10419 to -9917, F, 5’-
TGTGTGGGTGTATGGGTGACC-3’; R, 5’-GGAGGTAGTGCGTCTGTTCCTATC-3’),
(fragment -17405 to -16448, F, 5’-ATGTTCCTGGCATAACCCGAG-3’; R, 5’-
AAACCCTTTCCTCCCCCG-3’), (fragment -27145 to -26589, F, 5’-
CCAAGCCACACTCTCAGGTCAC-3’; R, 5’-GCAAGCACAGGGGGACTGG-3’). PCR
fragments were cloned into pCR-Blunt II-TOPO or pCR2.1 vector (Invitrogen), sequenced, and
inserted into pGL3 basic plasmid (Promega) using appropriate restriction enzyme sites. Nkx2-5
non-binding mutant of -736 to +119 bp fragments were generated by PCR using two primers; F,

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 6
5’-
TGTGGTTCTGGGTGTCCCGGTGTCAGTGTGTCAATGGATGTGTCTCTGGGTCACGTG
GTA-3’; R, 5’-
TACCACGTGACCCAGAGACACATCCATTGACACACTGACACCGGGACACCCAGAAC
CACA-3’.
10T1/2 fibroblast cells cultured in six-well plates were cotransfected with 3 mg of
luciferease reporter constructs, 1 mg of pcDNA3 or pcDNA3-Nkx2-5 expression plasmid and 0.5
mg of Rous sarcoma virus b-galactosidase construct using the calcium phosphate method as
described previously.10
Statistical analyses
Results among groups except for analyses of sodium currents (see above) were compared
using ANOVA and Fisher’s PLSD or Bonferroni’s post-hoc test (StatView version 5.01).

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 7
References:
1. Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark
B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR.
Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage
specification leads to progressive cardiomyopathy and complete heart block. Cell.
2004;117:373-86.
2. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-
inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the
mouse. Dev Biol. 2002;244:305-18.
3. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet.
1999;21:70-1.
4. Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac
expression of Csx/Nkx2.5 homeodomain protein. Circ Res. 1998;82:936-46.
5. Bhat MB, Hayek SM, Zhao J, Zang W, Takeshima H, Wier WG, Ma J. Expression and
functional characterization of the cardiac muscle ryanodine receptor Ca(2+) release
channel in Chinese hamster ovary cells. Biophys J. 1999;77:808-16.
6. Wakimoto H, Kasahara H, Maguire CT, Izumo S, Berul CI. Developmentally modulated
cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of
DNA nonbinding mutant Nkx2.5. J Cardiovasc Electrophysiol. 2002;13:682-8.
7. Hogan B, Beddingtpm R, Costantini F, Lacy E. Manipulating the mouse embryo. second
ed. New York, NY: Cold Spring Harbor Laboratory Press; 1994.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 8
8. El-Badawi A, Schenk EA. Histochemical methods for separate, consecutive and
simultaneous demonstration of acetylcholinesterase and norepinephrine in cryostat
sections. J Histochem Cytochem. 1967;15:580-8.
9. Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki
H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain
kinase. Circ Res;in press.
10. Kasahara H, Usheva A, Ueyama T, Aoki H, Horikoshi N, Izumo S. Characterization of
homo- and heterodimerization of cardiac Csx/Nkx2.5 homeoprotein. J Biol Chem.
2001;276:4570-80.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 9
Online Table I. The six most reduced ion channels in Nkx2-5 knockout hearts by gene expressionprofiling.
Protein Gene UniGene No.
fold differenceKO vs. control
(mean ± S.E., n=4)calcium channel, voltage-dependent, T type, alpha 1H subunit Cacna1h Mm.268378 0.321 ± 0.012potassium voltage-gated channel, Isk-related subfamily, member 1 Kcne1 Mm.299425 0.367 ± 0.002calcium channel, voltage-dependent, T type, alpha 1G subunit Cacna1g Mm.29585 0.518 ± 0.018sodium channel, voltage-gated, type V, alpha polypeptide Scn5a Mm.103584 0.745 ± 0.009
gap junction membrane channel protein alpha 5 (connexin 40) Gja5 Mm.281816 0.767 ± 0.029ryanodine receptor 2, cardiac Ryr2 Mm.239871 0.766 ± 0.057
PD4
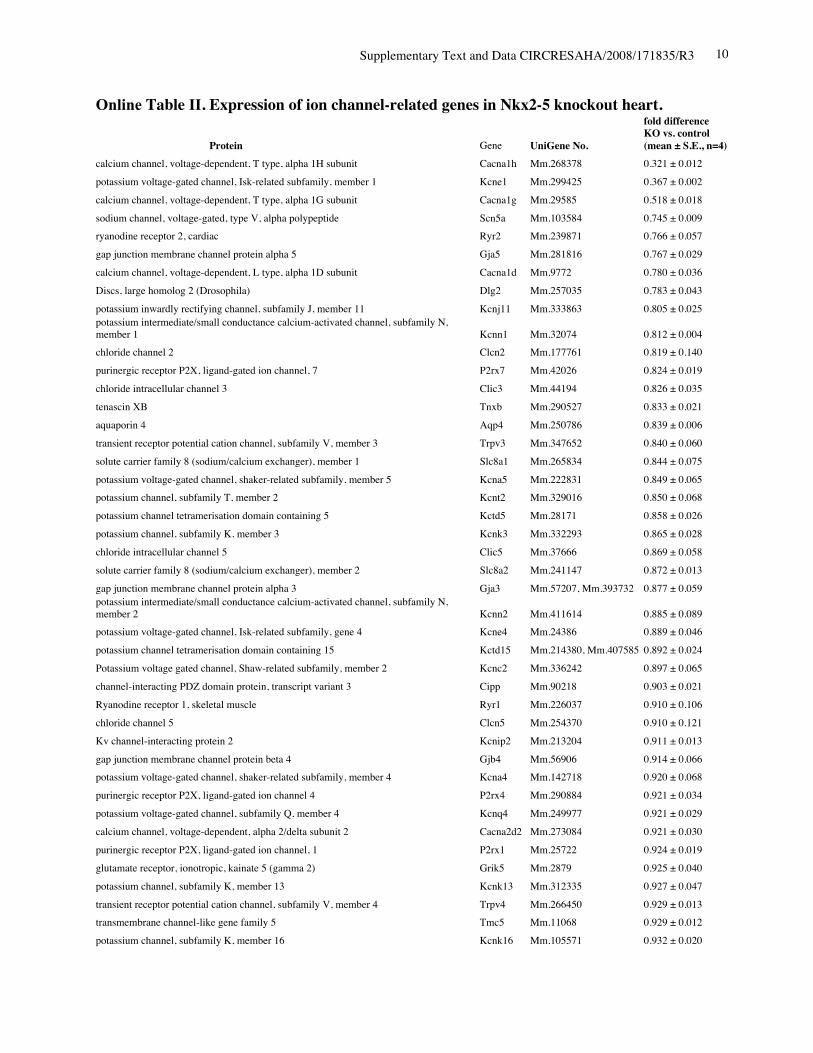
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 10
Online Table II. Expression of ion channel-related genes in Nkx2-5 knockout heart.
Protein Gene UniGene No.
fold differenceKO vs. control(mean ± S.E., n=4)
calcium channel, voltage-dependent, T type, alpha 1H subunit Cacna1h Mm.268378 0.321 ± 0.012potassium voltage-gated channel, Isk-related subfamily, member 1 Kcne1 Mm.299425 0.367 ± 0.002calcium channel, voltage-dependent, T type, alpha 1G subunit Cacna1g Mm.29585 0.518 ± 0.018sodium channel, voltage-gated, type V, alpha polypeptide Scn5a Mm.103584 0.745 ± 0.009ryanodine receptor 2, cardiac Ryr2 Mm.239871 0.766 ± 0.057gap junction membrane channel protein alpha 5 Gja5 Mm.281816 0.767 ± 0.029calcium channel, voltage-dependent, L type, alpha 1D subunit Cacna1d Mm.9772 0.780 ± 0.036Discs, large homolog 2 (Drosophila) Dlg2 Mm.257035 0.783 ± 0.043potassium inwardly rectifying channel, subfamily J, member 11 Kcnj11 Mm.333863 0.805 ± 0.025potassium intermediate/small conductance calcium-activated channel, subfamily N,member 1 Kcnn1 Mm.32074 0.812 ± 0.004chloride channel 2 Clcn2 Mm.177761 0.819 ± 0.140purinergic receptor P2X, ligand-gated ion channel, 7 P2rx7 Mm.42026 0.824 ± 0.019chloride intracellular channel 3 Clic3 Mm.44194 0.826 ± 0.035tenascin XB Tnxb Mm.290527 0.833 ± 0.021aquaporin 4 Aqp4 Mm.250786 0.839 ± 0.006transient receptor potential cation channel, subfamily V, member 3 Trpv3 Mm.347652 0.840 ± 0.060solute carrier family 8 (sodium/calcium exchanger), member 1 Slc8a1 Mm.265834 0.844 ± 0.075potassium voltage-gated channel, shaker-related subfamily, member 5 Kcna5 Mm.222831 0.849 ± 0.065potassium channel, subfamily T, member 2 Kcnt2 Mm.329016 0.850 ± 0.068potassium channel tetramerisation domain containing 5 Kctd5 Mm.28171 0.858 ± 0.026potassium channel, subfamily K, member 3 Kcnk3 Mm.332293 0.865 ± 0.028chloride intracellular channel 5 Clic5 Mm.37666 0.869 ± 0.058solute carrier family 8 (sodium/calcium exchanger), member 2 Slc8a2 Mm.241147 0.872 ± 0.013gap junction membrane channel protein alpha 3 Gja3 Mm.57207, Mm.393732 0.877 ± 0.059potassium intermediate/small conductance calcium-activated channel, subfamily N,member 2 Kcnn2 Mm.411614 0.885 ± 0.089potassium voltage-gated channel, Isk-related subfamily, gene 4 Kcne4 Mm.24386 0.889 ± 0.046potassium channel tetramerisation domain containing 15 Kctd15 Mm.214380, Mm.407585 0.892 ± 0.024Potassium voltage gated channel, Shaw-related subfamily, member 2 Kcnc2 Mm.336242 0.897 ± 0.065channel-interacting PDZ domain protein, transcript variant 3 Cipp Mm.90218 0.903 ± 0.021Ryanodine receptor 1, skeletal muscle Ryr1 Mm.226037 0.910 ± 0.106chloride channel 5 Clcn5 Mm.254370 0.910 ± 0.121Kv channel-interacting protein 2 Kcnip2 Mm.213204 0.911 ± 0.013gap junction membrane channel protein beta 4 Gjb4 Mm.56906 0.914 ± 0.066potassium voltage-gated channel, shaker-related subfamily, member 4 Kcna4 Mm.142718 0.920 ± 0.068purinergic receptor P2X, ligand-gated ion channel 4 P2rx4 Mm.290884 0.921 ± 0.034potassium voltage-gated channel, subfamily Q, member 4 Kcnq4 Mm.249977 0.921 ± 0.029calcium channel, voltage-dependent, alpha 2/delta subunit 2 Cacna2d2 Mm.273084 0.921 ± 0.030purinergic receptor P2X, ligand-gated ion channel, 1 P2rx1 Mm.25722 0.924 ± 0.019glutamate receptor, ionotropic, kainate 5 (gamma 2) Grik5 Mm.2879 0.925 ± 0.040potassium channel, subfamily K, member 13 Kcnk13 Mm.312335 0.927 ± 0.047transient receptor potential cation channel, subfamily V, member 4 Trpv4 Mm.266450 0.929 ± 0.013transmembrane channel-like gene family 5 Tmc5 Mm.11068 0.929 ± 0.012potassium channel, subfamily K, member 16 Kcnk16 Mm.105571 0.932 ± 0.020
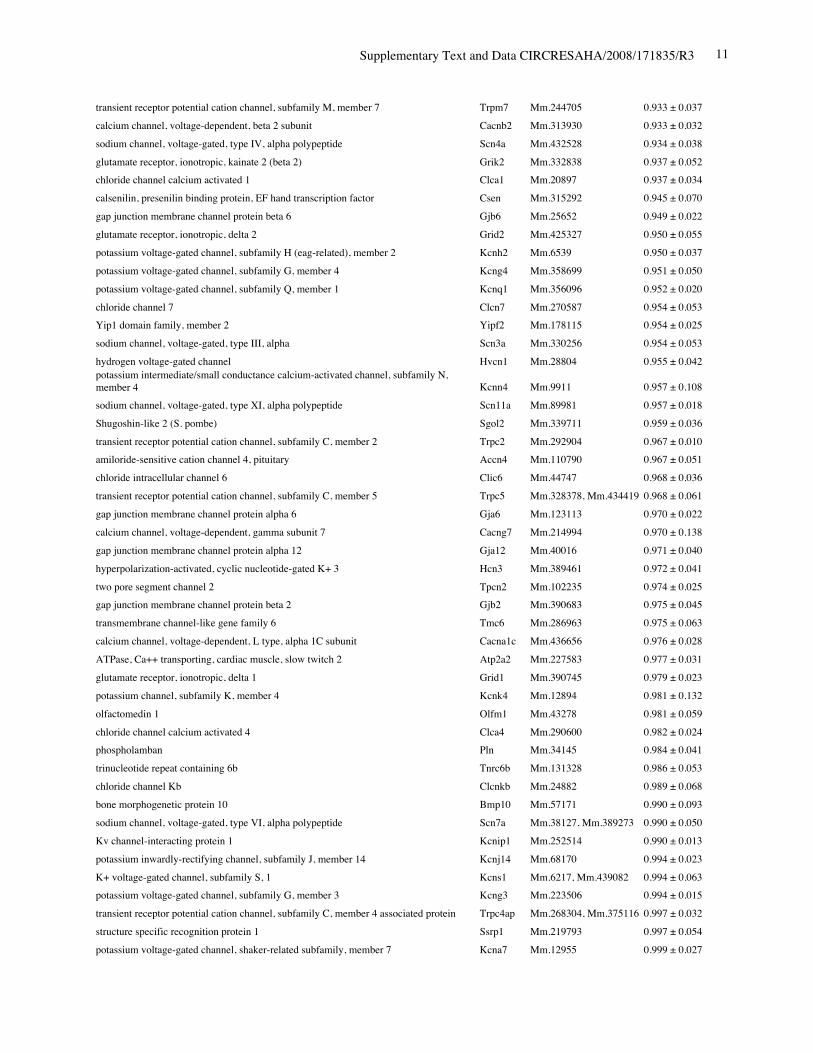
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 11
transient receptor potential cation channel, subfamily M, member 7 Trpm7 Mm.244705 0.933 ± 0.037calcium channel, voltage-dependent, beta 2 subunit Cacnb2 Mm.313930 0.933 ± 0.032sodium channel, voltage-gated, type IV, alpha polypeptide Scn4a Mm.432528 0.934 ± 0.038glutamate receptor, ionotropic, kainate 2 (beta 2) Grik2 Mm.332838 0.937 ± 0.052chloride channel calcium activated 1 Clca1 Mm.20897 0.937 ± 0.034calsenilin, presenilin binding protein, EF hand transcription factor Csen Mm.315292 0.945 ± 0.070gap junction membrane channel protein beta 6 Gjb6 Mm.25652 0.949 ± 0.022glutamate receptor, ionotropic, delta 2 Grid2 Mm.425327 0.950 ± 0.055potassium voltage-gated channel, subfamily H (eag-related), member 2 Kcnh2 Mm.6539 0.950 ± 0.037potassium voltage-gated channel, subfamily G, member 4 Kcng4 Mm.358699 0.951 ± 0.050potassium voltage-gated channel, subfamily Q, member 1 Kcnq1 Mm.356096 0.952 ± 0.020chloride channel 7 Clcn7 Mm.270587 0.954 ± 0.053Yip1 domain family, member 2 Yipf2 Mm.178115 0.954 ± 0.025sodium channel, voltage-gated, type III, alpha Scn3a Mm.330256 0.954 ± 0.053hydrogen voltage-gated channel Hvcn1 Mm.28804 0.955 ± 0.042potassium intermediate/small conductance calcium-activated channel, subfamily N,member 4 Kcnn4 Mm.9911 0.957 ± 0.108sodium channel, voltage-gated, type XI, alpha polypeptide Scn11a Mm.89981 0.957 ± 0.018Shugoshin-like 2 (S. pombe) Sgol2 Mm.339711 0.959 ± 0.036transient receptor potential cation channel, subfamily C, member 2 Trpc2 Mm.292904 0.967 ± 0.010amiloride-sensitive cation channel 4, pituitary Accn4 Mm.110790 0.967 ± 0.051chloride intracellular channel 6 Clic6 Mm.44747 0.968 ± 0.036transient receptor potential cation channel, subfamily C, member 5 Trpc5 Mm.328378, Mm.434419 0.968 ± 0.061gap junction membrane channel protein alpha 6 Gja6 Mm.123113 0.970 ± 0.022calcium channel, voltage-dependent, gamma subunit 7 Cacng7 Mm.214994 0.970 ± 0.138gap junction membrane channel protein alpha 12 Gja12 Mm.40016 0.971 ± 0.040hyperpolarization-activated, cyclic nucleotide-gated K+ 3 Hcn3 Mm.389461 0.972 ± 0.041two pore segment channel 2 Tpcn2 Mm.102235 0.974 ± 0.025gap junction membrane channel protein beta 2 Gjb2 Mm.390683 0.975 ± 0.045transmembrane channel-like gene family 6 Tmc6 Mm.286963 0.975 ± 0.063calcium channel, voltage-dependent, L type, alpha 1C subunit Cacna1c Mm.436656 0.976 ± 0.028ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 Atp2a2 Mm.227583 0.977 ± 0.031glutamate receptor, ionotropic, delta 1 Grid1 Mm.390745 0.979 ± 0.023potassium channel, subfamily K, member 4 Kcnk4 Mm.12894 0.981 ± 0.132olfactomedin 1 Olfm1 Mm.43278 0.981 ± 0.059chloride channel calcium activated 4 Clca4 Mm.290600 0.982 ± 0.024phospholamban Pln Mm.34145 0.984 ± 0.041trinucleotide repeat containing 6b Tnrc6b Mm.131328 0.986 ± 0.053chloride channel Kb Clcnkb Mm.24882 0.989 ± 0.068bone morphogenetic protein 10 Bmp10 Mm.57171 0.990 ± 0.093sodium channel, voltage-gated, type VI, alpha polypeptide Scn7a Mm.38127, Mm.389273 0.990 ± 0.050Kv channel-interacting protein 1 Kcnip1 Mm.252514 0.990 ± 0.013potassium inwardly-rectifying channel, subfamily J, member 14 Kcnj14 Mm.68170 0.994 ± 0.023K+ voltage-gated channel, subfamily S, 1 Kcns1 Mm.6217, Mm.439082 0.994 ± 0.063potassium voltage-gated channel, subfamily G, member 3 Kcng3 Mm.223506 0.994 ± 0.015transient receptor potential cation channel, subfamily C, member 4 associated protein Trpc4ap Mm.268304, Mm.375116 0.997 ± 0.032structure specific recognition protein 1 Ssrp1 Mm.219793 0.997 ± 0.054potassium voltage-gated channel, shaker-related subfamily, member 7 Kcna7 Mm.12955 0.999 ± 0.027
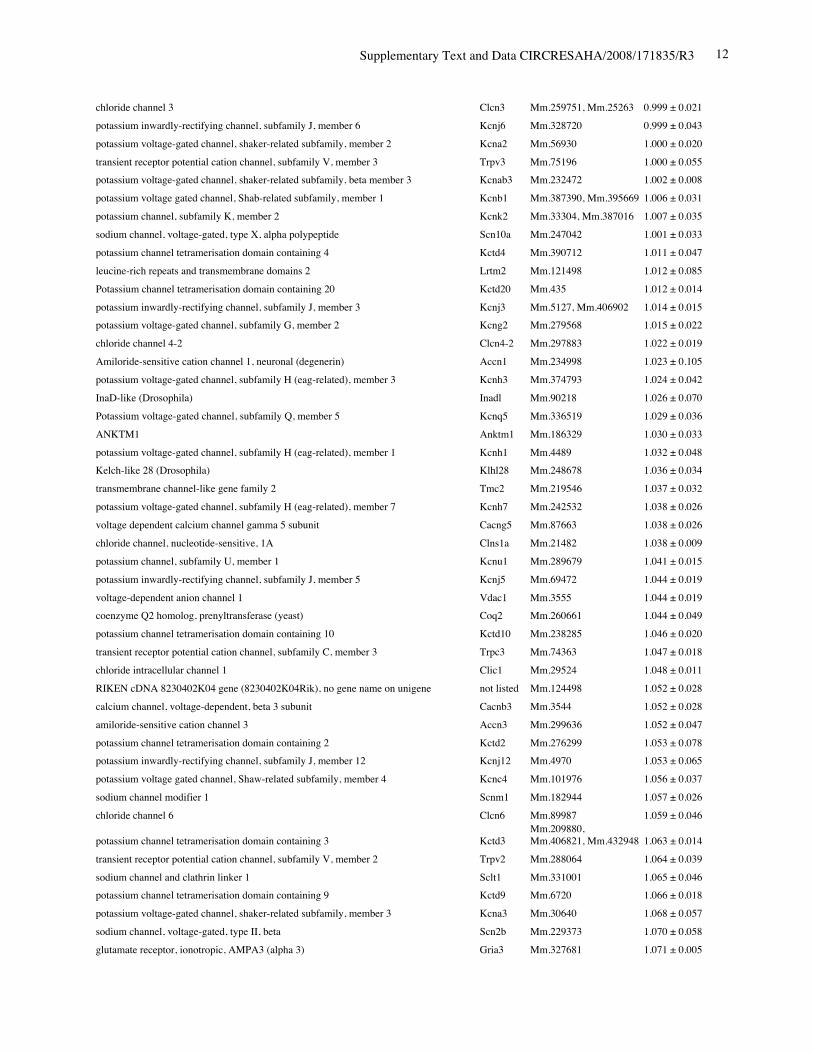
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 12
chloride channel 3 Clcn3 Mm.259751, Mm.25263 0.999 ± 0.021potassium inwardly-rectifying channel, subfamily J, member 6 Kcnj6 Mm.328720 0.999 ± 0.043potassium voltage-gated channel, shaker-related subfamily, member 2 Kcna2 Mm.56930 1.000 ± 0.020transient receptor potential cation channel, subfamily V, member 3 Trpv3 Mm.75196 1.000 ± 0.055potassium voltage-gated channel, shaker-related subfamily, beta member 3 Kcnab3 Mm.232472 1.002 ± 0.008potassium voltage gated channel, Shab-related subfamily, member 1 Kcnb1 Mm.387390, Mm.395669 1.006 ± 0.031potassium channel, subfamily K, member 2 Kcnk2 Mm.33304, Mm.387016 1.007 ± 0.035sodium channel, voltage-gated, type X, alpha polypeptide Scn10a Mm.247042 1.001 ± 0.033potassium channel tetramerisation domain containing 4 Kctd4 Mm.390712 1.011 ± 0.047leucine-rich repeats and transmembrane domains 2 Lrtm2 Mm.121498 1.012 ± 0.085Potassium channel tetramerisation domain containing 20 Kctd20 Mm.435 1.012 ± 0.014potassium inwardly-rectifying channel, subfamily J, member 3 Kcnj3 Mm.5127, Mm.406902 1.014 ± 0.015potassium voltage-gated channel, subfamily G, member 2 Kcng2 Mm.279568 1.015 ± 0.022chloride channel 4-2 Clcn4-2 Mm.297883 1.022 ± 0.019Amiloride-sensitive cation channel 1, neuronal (degenerin) Accn1 Mm.234998 1.023 ± 0.105potassium voltage-gated channel, subfamily H (eag-related), member 3 Kcnh3 Mm.374793 1.024 ± 0.042InaD-like (Drosophila) Inadl Mm.90218 1.026 ± 0.070Potassium voltage-gated channel, subfamily Q, member 5 Kcnq5 Mm.336519 1.029 ± 0.036ANKTM1 Anktm1 Mm.186329 1.030 ± 0.033potassium voltage-gated channel, subfamily H (eag-related), member 1 Kcnh1 Mm.4489 1.032 ± 0.048Kelch-like 28 (Drosophila) Klhl28 Mm.248678 1.036 ± 0.034transmembrane channel-like gene family 2 Tmc2 Mm.219546 1.037 ± 0.032potassium voltage-gated channel, subfamily H (eag-related), member 7 Kcnh7 Mm.242532 1.038 ± 0.026voltage dependent calcium channel gamma 5 subunit Cacng5 Mm.87663 1.038 ± 0.026chloride channel, nucleotide-sensitive, 1A Clns1a Mm.21482 1.038 ± 0.009potassium channel, subfamily U, member 1 Kcnu1 Mm.289679 1.041 ± 0.015potassium inwardly-rectifying channel, subfamily J, member 5 Kcnj5 Mm.69472 1.044 ± 0.019voltage-dependent anion channel 1 Vdac1 Mm.3555 1.044 ± 0.019coenzyme Q2 homolog, prenyltransferase (yeast) Coq2 Mm.260661 1.044 ± 0.049potassium channel tetramerisation domain containing 10 Kctd10 Mm.238285 1.046 ± 0.020transient receptor potential cation channel, subfamily C, member 3 Trpc3 Mm.74363 1.047 ± 0.018chloride intracellular channel 1 Clic1 Mm.29524 1.048 ± 0.011RIKEN cDNA 8230402K04 gene (8230402K04Rik), no gene name on unigene not listed Mm.124498 1.052 ± 0.028calcium channel, voltage-dependent, beta 3 subunit Cacnb3 Mm.3544 1.052 ± 0.028amiloride-sensitive cation channel 3 Accn3 Mm.299636 1.052 ± 0.047potassium channel tetramerisation domain containing 2 Kctd2 Mm.276299 1.053 ± 0.078potassium inwardly-rectifying channel, subfamily J, member 12 Kcnj12 Mm.4970 1.053 ± 0.065potassium voltage gated channel, Shaw-related subfamily, member 4 Kcnc4 Mm.101976 1.056 ± 0.037sodium channel modifier 1 Scnm1 Mm.182944 1.057 ± 0.026chloride channel 6 Clcn6 Mm.89987 1.059 ± 0.046
potassium channel tetramerisation domain containing 3 Kctd3Mm.209880,Mm.406821, Mm.432948 1.063 ± 0.014
transient receptor potential cation channel, subfamily V, member 2 Trpv2 Mm.288064 1.064 ± 0.039sodium channel and clathrin linker 1 Sclt1 Mm.331001 1.065 ± 0.046potassium channel tetramerisation domain containing 9 Kctd9 Mm.6720 1.066 ± 0.018potassium voltage-gated channel, shaker-related subfamily, member 3 Kcna3 Mm.30640 1.068 ± 0.057sodium channel, voltage-gated, type II, beta Scn2b Mm.229373 1.070 ± 0.058glutamate receptor, ionotropic, AMPA3 (alpha 3) Gria3 Mm.327681 1.071 ± 0.005
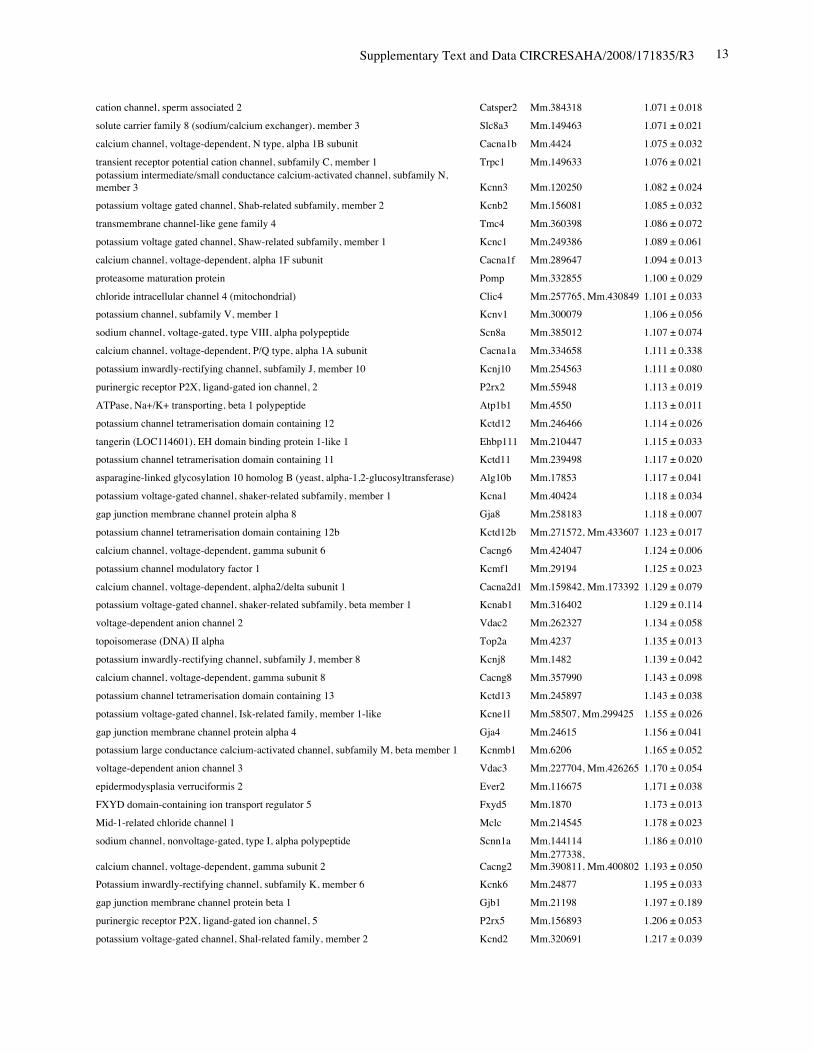
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 13
cation channel, sperm associated 2 Catsper2 Mm.384318 1.071 ± 0.018solute carrier family 8 (sodium/calcium exchanger), member 3 Slc8a3 Mm.149463 1.071 ± 0.021calcium channel, voltage-dependent, N type, alpha 1B subunit Cacna1b Mm.4424 1.075 ± 0.032transient receptor potential cation channel, subfamily C, member 1 Trpc1 Mm.149633 1.076 ± 0.021potassium intermediate/small conductance calcium-activated channel, subfamily N,member 3 Kcnn3 Mm.120250 1.082 ± 0.024potassium voltage gated channel, Shab-related subfamily, member 2 Kcnb2 Mm.156081 1.085 ± 0.032transmembrane channel-like gene family 4 Tmc4 Mm.360398 1.086 ± 0.072potassium voltage gated channel, Shaw-related subfamily, member 1 Kcnc1 Mm.249386 1.089 ± 0.061calcium channel, voltage-dependent, alpha 1F subunit Cacna1f Mm.289647 1.094 ± 0.013proteasome maturation protein Pomp Mm.332855 1.100 ± 0.029chloride intracellular channel 4 (mitochondrial) Clic4 Mm.257765, Mm.430849 1.101 ± 0.033potassium channel, subfamily V, member 1 Kcnv1 Mm.300079 1.106 ± 0.056sodium channel, voltage-gated, type VIII, alpha polypeptide Scn8a Mm.385012 1.107 ± 0.074calcium channel, voltage-dependent, P/Q type, alpha 1A subunit Cacna1a Mm.334658 1.111 ± 0.338potassium inwardly-rectifying channel, subfamily J, member 10 Kcnj10 Mm.254563 1.111 ± 0.080purinergic receptor P2X, ligand-gated ion channel, 2 P2rx2 Mm.55948 1.113 ± 0.019ATPase, Na+/K+ transporting, beta 1 polypeptide Atp1b1 Mm.4550 1.113 ± 0.011potassium channel tetramerisation domain containing 12 Kctd12 Mm.246466 1.114 ± 0.026tangerin (LOC114601), EH domain binding protein 1-like 1 Ehbp111 Mm.210447 1.115 ± 0.033potassium channel tetramerisation domain containing 11 Kctd11 Mm.239498 1.117 ± 0.020asparagine-linked glycosylation 10 homolog B (yeast, alpha-1,2-glucosyltransferase) Alg10b Mm.17853 1.117 ± 0.041potassium voltage-gated channel, shaker-related subfamily, member 1 Kcna1 Mm.40424 1.118 ± 0.034gap junction membrane channel protein alpha 8 Gja8 Mm.258183 1.118 ± 0.007potassium channel tetramerisation domain containing 12b Kctd12b Mm.271572, Mm.433607 1.123 ± 0.017calcium channel, voltage-dependent, gamma subunit 6 Cacng6 Mm.424047 1.124 ± 0.006potassium channel modulatory factor 1 Kcmf1 Mm.29194 1.125 ± 0.023calcium channel, voltage-dependent, alpha2/delta subunit 1 Cacna2d1 Mm.159842, Mm.173392 1.129 ± 0.079potassium voltage-gated channel, shaker-related subfamily, beta member 1 Kcnab1 Mm.316402 1.129 ± 0.114voltage-dependent anion channel 2 Vdac2 Mm.262327 1.134 ± 0.058topoisomerase (DNA) II alpha Top2a Mm.4237 1.135 ± 0.013potassium inwardly-rectifying channel, subfamily J, member 8 Kcnj8 Mm.1482 1.139 ± 0.042calcium channel, voltage-dependent, gamma subunit 8 Cacng8 Mm.357990 1.143 ± 0.098potassium channel tetramerisation domain containing 13 Kctd13 Mm.245897 1.143 ± 0.038potassium voltage-gated channel, Isk-related family, member 1-like Kcne1l Mm.58507, Mm.299425 1.155 ± 0.026gap junction membrane channel protein alpha 4 Gja4 Mm.24615 1.156 ± 0.041potassium large conductance calcium-activated channel, subfamily M, beta member 1 Kcnmb1 Mm.6206 1.165 ± 0.052voltage-dependent anion channel 3 Vdac3 Mm.227704, Mm.426265 1.170 ± 0.054epidermodysplasia verruciformis 2 Ever2 Mm.116675 1.171 ± 0.038FXYD domain-containing ion transport regulator 5 Fxyd5 Mm.1870 1.173 ± 0.013Mid-1-related chloride channel 1 Mclc Mm.214545 1.178 ± 0.023sodium channel, nonvoltage-gated, type I, alpha polypeptide Scnn1a Mm.144114 1.186 ± 0.010
calcium channel, voltage-dependent, gamma subunit 2 Cacng2Mm.277338,Mm.390811, Mm.400802 1.193 ± 0.050
Potassium inwardly-rectifying channel, subfamily K, member 6 Kcnk6 Mm.24877 1.195 ± 0.033gap junction membrane channel protein beta 1 Gjb1 Mm.21198 1.197 ± 0.189purinergic receptor P2X, ligand-gated ion channel, 5 P2rx5 Mm.156893 1.206 ± 0.053potassium voltage-gated channel, Shal-related family, member 2 Kcnd2 Mm.320691 1.217 ± 0.039
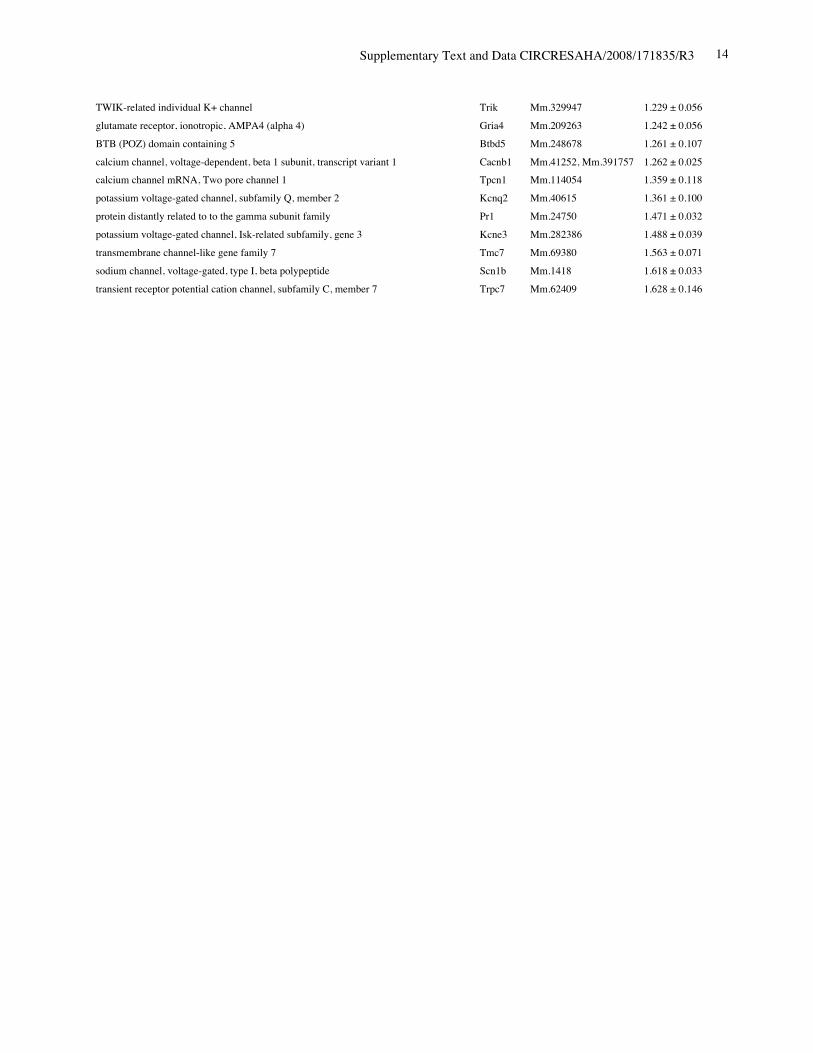
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 14
TWIK-related individual K+ channel Trik Mm.329947 1.229 ± 0.056glutamate receptor, ionotropic, AMPA4 (alpha 4) Gria4 Mm.209263 1.242 ± 0.056BTB (POZ) domain containing 5 Btbd5 Mm.248678 1.261 ± 0.107calcium channel, voltage-dependent, beta 1 subunit, transcript variant 1 Cacnb1 Mm.41252, Mm.391757 1.262 ± 0.025calcium channel mRNA, Two pore channel 1 Tpcn1 Mm.114054 1.359 ± 0.118potassium voltage-gated channel, subfamily Q, member 2 Kcnq2 Mm.40615 1.361 ± 0.100protein distantly related to to the gamma subunit family Pr1 Mm.24750 1.471 ± 0.032potassium voltage-gated channel, Isk-related subfamily, gene 3 Kcne3 Mm.282386 1.488 ± 0.039transmembrane channel-like gene family 7 Tmc7 Mm.69380 1.563 ± 0.071sodium channel, voltage-gated, type I, beta polypeptide Scn1b Mm.1418 1.618 ± 0.033transient receptor potential cation channel, subfamily C, member 7 Trpc7 Mm.62409 1.628 ± 0.146

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 15
Online Figure I. Frozen sections of R26R/Tg-Cre-ERTM stained for b-galactosidase including aorticvalve (A) and pulmonary valve (B). Bars = 500 um. AoV, aortic valve; PV, pulmonary valve.;LA, left atrium; LV, left ventricle, RV, right ventricle.
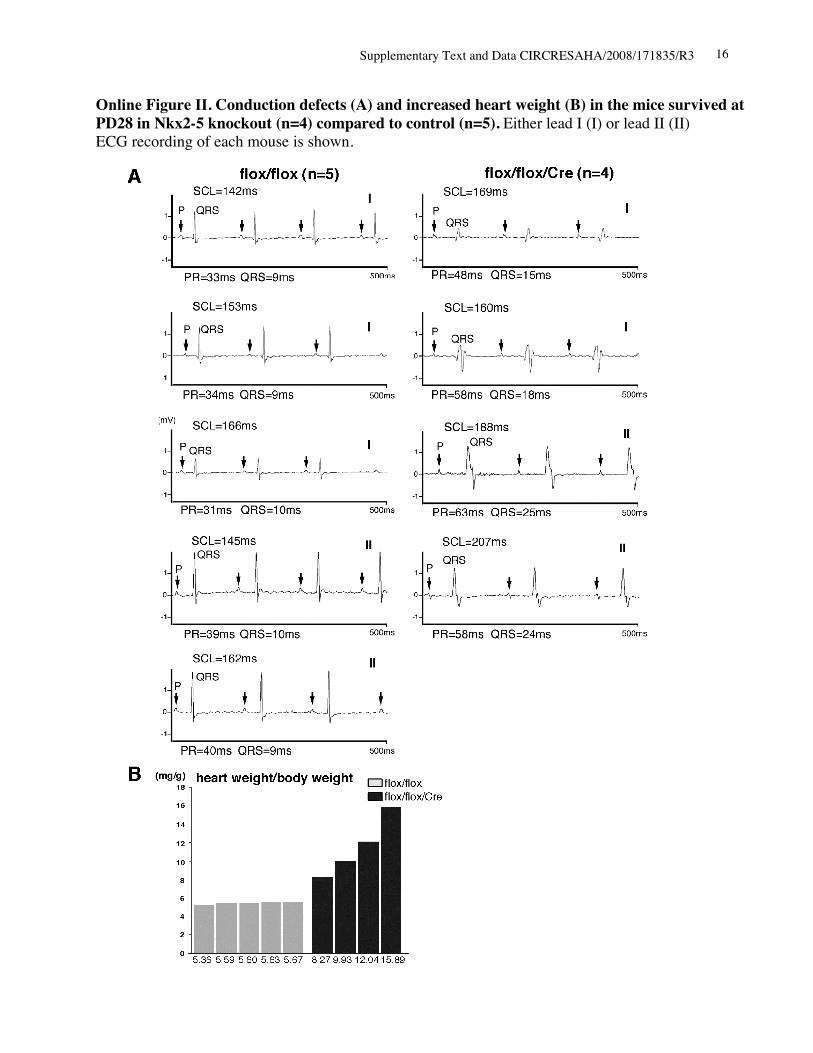
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 16
Online Figure II. Conduction defects (A) and increased heart weight (B) in the mice survived atPD28 in Nkx2-5 knockout (n=4) compared to control (n=5). Either lead I (I) or lead II (II)ECG recording of each mouse is shown.
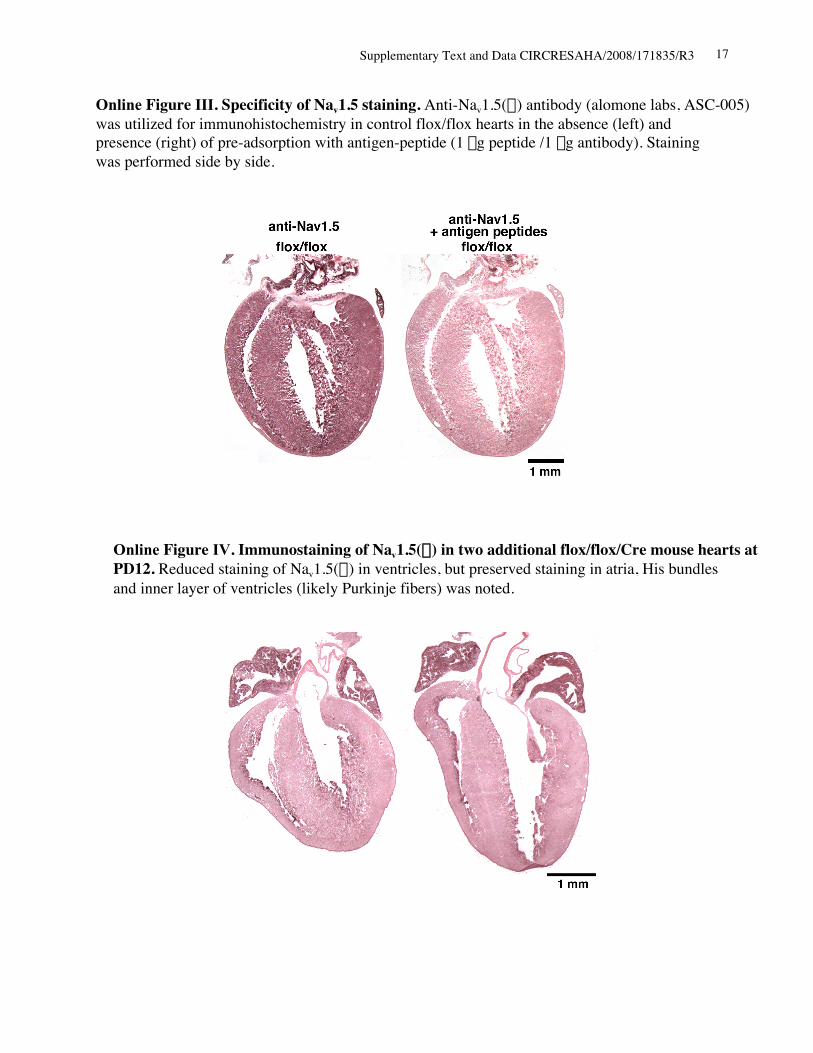
Supplementary Text and Data CIRCRESAHA/2008/171835/R3 17
Online Figure III. Specificity of Nav1.5 staining. Anti-Nav1.5(a) antibody (alomone labs, ASC-005)was utilized for immunohistochemistry in control flox/flox hearts in the absence (left) andpresence (right) of pre-adsorption with antigen-peptide (1 mg peptide /1 mg antibody). Stainingwas performed side by side.
Online Figure IV. Immunostaining of Nav1.5(a) in two additional flox/flox/Cre mouse hearts atPD12. Reduced staining of Nav1.5(a) in ventricles, but preserved staining in atria, His bundlesand inner layer of ventricles (likely Purkinje fibers) was noted.

Supplementary Text and Data CIRCRESAHA/2008/171835/R3 18
Nav1.5(a)
Nkx2-5
GAPDH
PD12
1 2
250
40
30
Cre-ER +- fl/flfl/flkDa
proteinNav1.5(a) atria
Online Figure V. Western blotting demonstrates similar levels of Nav1.5(a) protein in atria offlox/flox and flox/flox/Cre mice (lanes 1 vs.2).
Related Documents











