The Pennsylvania State University The Graduate School College of Medicine MOLECULAR MECHANISMS OF NANOLIPOSOMAL C6-CERAMIDE-INDUCED CELL DEATH IN CHRONIC LYMPHOCYTIC LEUKEMIA A Dissertation in Molecular Medicine by Ushma A. Doshi 2015 Ushma A. Doshi Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2015

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Pennsylvania State University
The Graduate School
College of Medicine
MOLECULAR MECHANISMS OF NANOLIPOSOMAL
C6-CERAMIDE-INDUCED CELL DEATH IN CHRONIC
LYMPHOCYTIC LEUKEMIA
A Dissertation in
Molecular Medicine
by
Ushma A. Doshi
2015 Ushma A. Doshi
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
December 2015

ii
The dissertation of Ushma A. Doshi was reviewed and approved* by the following: Charles Lang Director of the Graduate Program Professor of Cellular and Molecular Physiology Dissertation Co-Adviser Co-Chair of Committee
Mark Kester Professor of Pharmacology, University of Virginia School of Medicine Dissertation Co-Adviser Co-Chair of Committee
Thomas P. Loughran Professor of Medicine, University of Virginia Cancer Center Hong-Gang Wang Lois High Berstler Professor, Pediatrics and Pharmacology Jin-Ming Yang Professor of Pharmacology
David Claxton Special Member Professor of Medicine
Richard R. Young Professor of Supply Chain Management; Business Administration
*Signatures are on file in the Graduate School.

iii
ABSTRACT
Chronic lymphocytic leukemia (CLL) is the most prevalent form of adult
leukemia in Western countries. Despite a high incidence, its pathogenesis is still
poorly understood, hence limiting treatment strategies. Furthermore, since CLL is
predominantly a disease of the elderly, numerous therapeutic strategies are
unsuitable due to limited physical fitness of the patient. Therefore, the CLL
remains incurable for most patients. Further research is needed to develop novel
therapeutic strategies.
Ceramide is a ‘tumor suppressor’ sphingolipid known to regulate
differentiation, senescence and cell cycle arrest. While a large body of work
reveals the mechanism of nanoliposomal ceramide (CNL)-induced cell death in
several types of cancers, the effect in CLL remains unclear. This study
investigates the effect of CNL in CLL and deciphers the key signaling
mechanisms mediating CNL-induced cell death. We have shown that CNL
selectively induces cell death in CLL cells by targeting the Warburg effect
through reducing levels of glyceraldehyde-3-phosphate dehydrogenase
(GAPDH), with no detrimental effects on normal peripheral blood mononuclear
cells. Additionally, CNL treatment results in tumor regression in an in vivo murine
model of CLL. Several reports in the literature have shown that signal transducer
and activator of transcription 3 (STAT3) is constitutively phosphorylated on
serine-727 in CLL and that STAT3 might be a therapeutic target in this disease.
We demonstrate that CNL suppresses STAT3 phosphorylation at both tyrosine-
705 and serine-727 by inhibiting multiple upstream kinases that include Bruton’s
tyrosine kinase, mitogen-activated protein kinase kinase and protein kinase C.

iv
This suppression in STAT3 phosphorylation and the subsequent downregulation
of STAT3 transcriptional activity mediates CNL-induced cell death in CLL. Recent
work in the literature has uncovered that STAT3 phosphorylated at serine-727
associates with mitochondrial components and regulates the respiratory
chain. Overactivation of mitochondrial STAT3 phosphorylated at serine-727
confers viability and stress protection to CLL cells. Our initial results demonstrate
that CNL treatment reduces mitochondrial STAT3 levels, which might also be
critical to the cell death induction.
Taken together, our results suggest that inhibition of glycolytic respiration
and inhibition of STAT3 transcriptional activity are key signaling mechanisms of
CNL-induced cell death in CLL cells. Additionally, we also speculate that
inhibition of STAT3-dependent mitochondrial respiration is also critical for
induction of cell death by CNL treatment. We conclude that CNL could potentially
be an effective therapy for CLL. Overall, this work emphasizes targeting the
sphingolipid pathway and development of sphingolipids-based therapeutics for
cancer.

v
TABLE OF CONTENTS
LIST OF FIGURES ........................................................................................................ vii
ABBREVIATIONS .......................................................................................................... ix
PREFACE ...................................................................................................................... xi
ACKNOWLEDGEMENTS .............................................................................................. xii
CHAPTER 1: Literature Review ................................................................................... 1
Sphingolipids .............................................................................................................. 1
Sphingolipid metabolism .......................................................................................... 1
Functions of Sphingolipids ....................................................................................... 5
Ceramide for cancer therapeutics ............................................................................... 8
Ceramide and induction of cell death ......................................................................11
Chemotherapy-induced ceramide generation by the de novo pathway ...................16
Chemotherapy-induced ceramide generation by sphingomyelinase pathway .........20
Nanotechnology-based drug delivery of ceramide ..................................................23
Chronic Lymphocytic Leukemia (CLL) ........................................................................30
Conclusions ...............................................................................................................34
CHAPTER 2: Nanoliposomal C6-ceramide target the Warburg effect in chronic
lymphocytic leukemia .................................................................................................35
Abstract .....................................................................................................................36
Introduction ................................................................................................................37
Materials and Methods ...............................................................................................40
Results .......................................................................................................................48
Discussion .................................................................................................................63
CHAPTER 3: STAT3 mediates nanoliposomal C6-ceramide-induced cell death in
chronic lymphocytic leukemia ....................................................................................69
Introduction ................................................................................................................69
Materials and Methods ...............................................................................................72
Results .......................................................................................................................78
Discussion ............................................................................................................... 103

vi
CHAPTER 4: Effect of nanoliposomal C6-ceramide on mitochondrial bioenergetics
and mitochondrial STAT3 in chronic lymphocytic leukemia .................................. 109
Introduction .............................................................................................................. 109
Methods and Materials ............................................................................................. 112
Results ..................................................................................................................... 116
Discussion ............................................................................................................... 123
CHAPTER 5 – Conclusions, Future directions and Therapeutic Potential ............ 127
Conclusions ............................................................................................................. 127
Use of nanoliposomal C6-ceramide as a therapy for CLL ..................................... 127
CNL targets the Warburg effect in CLL ................................................................. 129
STAT3 mediates CNL-induced cell death in CLL .................................................. 130
Effect of CNL on mitochondrial STAT3 ................................................................. 131
Effect of CNL on cellular bioenergetics in CLL ...................................................... 132
Future Directions...................................................................................................... 136
Summary and therapeutic potential .......................................................................... 138
References .................................................................................................................. 139
Appendix: Letters of Permission .................................................................................. 153

vii
LIST OF FIGURES
Figure 1-1 Fundamental structures of sphingolipids………...……………………...1
Figure 1-2 Sphingolipid metabolism pathways………………………………………4
Figure 1-3 Compartmentalization of sphingolipid metabolism…………………..…4
Figure 1-4 Strategies to alter the sphingolipid balance in cancer cells to
potentiate cytotoxicity of chemotherapeutic drugs…………………………………10
Figure 1-5 Points of intervention in the sphingolipid pathway……………………29
Figure 2-1 Nanoliposomal C6-ceramide selectively induces cell death in CLL
cells…..………………………………………………………………………………….49
Figure 2-2 Cell death induced by nanoliposomal C6-ceramide occurs through
caspase 3/7-independent necrosis…………………………………………………..52
Figure 2-3 C6-ceramide nanoliposomes target GAPDH in CLL…………………55
Figure 2-4 C6-ceramide targets the glycolytic pathway…………………………..59
Figure 2-5 Pharmacological and molecular confirmation that nanoliposomal C6-
ceramide targets the glycolytic pathway at the level of GAPDH………………….60
Figure 2-6 Nanoliposomal C6-ceramide displays anti-leukemic effect in a CLL
animal model………………………………………………………...…………………62
Figure 3-1A STAT3 is overexpressed in CLL cell lines and patient cells……….79
Figure 3-1B Knockdown of STAT3 induces cell death in CLL cells……………..80
Figure 3-1C STAT3 inhibition reduces viability of CLL cels………………………81
Figure 3-2A CNL suppresses the phosphorylation of STAT3 in CLL cell lines...83
Figure 3-2B CNL suppresses phosphorylation of STAT3 in CLL patient cells…84
Figure 3-2C CNL does affect cellular viability & STAT3 phosphorylation in
HEK293 cells…………………………………………………………………………...85
Figure 3-3ACNL-induced suppression of phosphorylation is specific to STAT3.86
Figure 3-3B Suppression of STAT3 phosphorylation is specifically an effect of
CNL and not other sphingolipids……………………………………………………..87
Figure 3-4A CNL induces necrotic cell death in CLL cell lines……………..……89

viii
Figure 3-4B CNL induces necrotic cell death in CLL patient cells……………….89
Figure 3-4C CNL-induced suppression of p-STAT3 precedes induction of cell
death…………………………………………………………………………………….90
Figure 3-4D CNL-induces early time point suppression of p-STAT3 in CLL
patient cells……………………………………………………………………………..91
Figure 3-5A CNL does not activate phosphatases……………………………......92
Figure 3-5B CNL suppresses the activity of BTK………………………………….94
Figure 3-5C BTK inhibitors suppress phosphorylation of STAT3………………..94
Figure 3-5D CNL suppresses the activity of MEK1/2 kinase…………………….95
Figure 3-5E MEK1/2 inhibitors suppress phosphorylation of STAT3……………96
Figure 3-5F CNL suppresses the activity of PKC………………………………….97
Figure 3-5G PKC inhibitor suppress phosphorylation of STAT3…………………97
Figure 3-6A CNL reduces the levels of STAT3-regulated genes………………..99
Figure 3-6B Reduction of STAT3 phosphorylation precedes reduction of Mcl-1
levels following CNL treatment……………………………………………………….99
Figure 3-6C CNL inhibits expression of luciferase in a STAT3 luciferase reporter
assay…………………………………………………………………………………..100
Figure 3-7 STAT3-C expressing cells are resistant to CNL-induced death…...102
Figure 4-1 CNL treatment results in accumulation of ceramide in the
mitochondria, decreased mitochondrial membrane potential, and generation of
ROS……………………………………………………………………………………118
Figure 4-2 CNL treatment results in cytosolic release of AIF…………………...120
Figure 4-3 CNL treatment dimishes levels of total STAT3 and p-STAT3 in the
mitochondria…………………………………………………………………………..121
Figure 4-4 CNL treatment suppresses the glycolytic flux of JVM-3 cells……...122
Figure 5-1 Molecular mechanisms of CNL-induced cell death in CLL cells…..136

ix
ABBREVIATIONS
AIF Apoptosis inducing factor
ATP Adenosine triphosphate
BCR B-cell receptor
BTK Bruton’s Tyrosine Kinase
CDase Ceramidase
CerS Ceramide synthase
CLL Chronic lymphocytic leukemia
CNL Nanoliposomal C6-ceramide
C6-ceramide D-erythro-hexanoyl-sphingosine
ECAR Extracellular acidification rate
ER Endoplasmic reticulum
ETC Electron transport chain
FB1 Fumonisin B1
GAPDH Glyceraldehyde 3-phosphate dehydrogenase
GCS Glucosylceramide synthase
GlcCer Glucosylceramide
GLUT1 Glucose transporter 1
HIF-1α Hypoxia inducible factor 1 alpha
LGL Large granular lymphocytic leukemia
MEK Mitogen-activated protein kinase kinase
MOMP Mitochondrial outer membrane permeabilization
OA Okadaic acid
OCR Oxygen consumption rate
PKC Protein kinase C

x
PV Pervanadate
ROS Reactive oxygen species
SMase Sphingomyelinase
SMS Sphingomyelin synthase
Sph Sphingosine
SphK Sphingosine kinase
SPT Serine palmitoyl transferase
STAT3 Signal Transducer and Activator of Transcription 3
S1P Sphingosine 1-phosphate
TNF Tumor necrosis factor
TNFR1 Tumor necrosis factor receptor 1
TRAIL TNF-related apoptosis-inducing ligand
TRAILR1 TRAIL receptor 1
TRAILR2 TRAIL receptor 2
UGCG UDP-glucose ceramide glucosyltransferase

xi
PREFACE
I would like to recognize that Chapter 2 of my dissertation titled “C6-ceramide
nanoliposomses target the Warburg effect in chronic lymphocytic leukemia” is
derived from published data (PLoS One, 2013 Dec 19;8(12):e84648) for which I
am co-first author. This project was underway when I joined the Kester lab in
2011. Dr. Lindsay Ryland and I contributed towards the design of the
experiments within this particular chapter. Specifically, I was a major contributor
towards the following figures: Fig 2-1A, Fig. 2-3A, Fig. 2-4A and Fig 2-5. I am in
no way attempting to claim intellectual property over the design of all the
experiments within this particular chapter. Therefore, I would like to acknowledge
Dr. Lindsay Ryland and the other contributing authors for the work presented in
this chapter.
In addition, Dr. Lindsay Ryland performed experiments for Fig. 4-1 in
Chapter 4 of my dissertation titled “Effect of C6-ceramide Nanoliposomes on
mitochondrial bioenergetics and mitochondrial STAT3 in Chronic Lymphocytic
Leukemia”.
The remainder of my dissertation includes part of a published book
chapter for which I am first author (Chapter 1), manuscript for which I am first
author (Chapter 3), and manuscript in preparation for which I am first author
(Chapter 4).

xii
ACKNOWLEDGEMENTS
I would like to thank, first and foremost, my mentor and advisor Dr. Mark
Kester for giving me an opportunity to work in his laboratory and for guiding me
through this memorable journey. I owe my achievements to his incredible
support, guidance and his faith in my work. I would like to express my gratitude
towards him for encouraging me to pursue the MBA degree during the last two
years of my thesis work. Thank you for your patience and flexibility with my
unnatural working hours. I would also like to thank Dr. Thomas Loughran for his
mentorship throughout my graduate years. I would like to specifically thank Dr.
Charles Lang for his immense support and belief in my abilities as a graduate
student. I would not have been a part of Penn State College of Medicine, if it
were not for your conviction in my potential as a graduate student. I would like to
extend special thanks to Dr. HG Wang and Dr. Claxton for always being so
approachable and providing insightful comments on my work. In addition, I would
like to thank Dr. Jin-Ming Yang and Dr. Robert Young for being very supportive
committee members. All my professors at Penn State College of Medicine,
especially Dr. Kent Vrana, Dr. Ralph Keil and Dr. Xin Liu have extended their
support and guidance in my journey. I am indebted to past and present Kester
lab members for making the lab my second home. Special thanks to Dr. Todd
Fox and Dr. Su-Fern Tan for their valuable guidance in my work and their very
special friendship. Special thanks to Dr. Jeremy Haakenson, Dr. Jody Hankins
and Dr. Megan Young for their consistent support. I would like to thank Sriram
Shanmugavelandy for being a superb friend in lab. Thanks to Samuel Linton,

xiii
Tony Brown, Dr. Brian Barth and all other members in the Kester lab for this
spectacular journey. Special thanks to Taryn Dick, members of the Wang lab,
administrative staff of the Graduate Office at College of Medicine and the Penn
State Hershey Security.
I cannot imagine this journey without my friends and family. Vijay Kale,
Rameshwari Kale, Manmeet Raval and Darshan Trivedi – Hershey was indeed
the sweetest place on Earth for me because of your unfailing love and support.
Lastly, and most importantly, I am blessed with the best family. My best friend
and husband, Varun Prabhu, has been my guiding light and the best companion
in the last ten years. Thank you Varun, for your undying motivation, strength and
love. Special thanks to my sister, Kshama Doshi for being the source of my
strength, my best friend and my energy throughout. I heartily thank my parents
and my in laws for their love, constant support, motivation and their
understanding throughout my PhD years. I dedicate this work to my family.
Lastly, I would like to thank God for giving me the strength to dream big and
the grit to achieve them.

1
CHAPTER 1: Literature Review
Sphingolipids
Sphingolipids, a group of bioactive lipids, represent one of the eight major
classes of lipids [1]. This class of lipids are structurally characterized by a
sphingoid base backbone comprising of sphingosine most frequently, and the
presence of an amide-linked fatty acid and/or a headgroup attached to the
hydroxyl on carbon 1 (Fig. 1-1) [1].
Sphingolipid metabolism
Sphingolipid metabolism is a complex, compartmentalized and a highly
inter-connected system comprising of enzymes catalyzing the formation of
different classes of sphilgolipids. Ceramide is considered to be the central hub of
sphingolipid metabolism. The generic ‘ceramide’ is a family of more than 50
distinct molecular species with a base structure consisting of an acyl chain of
variable length attached to the sphingosine backbone [2]. Fig. 1-2 and Fig. 1-3
represent the sphingolipid metabolism pathway and the corresponding cellular
compartments.
Saturated bond forms
dihydroceramides
Figure 1-1 Fundamental structures of sphingolipids: Modified from Merrill et al. [1]. Sphingolipids are defined by having a sphingoid base (shown for sphingosine) that is often derivatized with an amide-linked fatty acid and/or headgroup of the general types shown.

2
Ceramide can be synthesized by an anabolic and a catabolic pathway. De
novo synthesis starts by serine palmitoyl transferase (SPT)-catalyzed
condensation of palmitate and serine to form 3-keto-dihydrosphingosine, which is
subsequently reduced to dihydrosphingosine followed by acylation by ceramide
synthases (CerS). Desaturases catalyze the formation of ceramide from
dihydroceramide. The endoplasmic reticulum (ER) and ER-associated
membranes are the site of de novo synthesis of ceramide [2].
The catabolic pathway of ceramide synthesis involves the conversion of
sphingomyelin to form ceramide by the action of acid sphingomyelinase (SMase)
residing in the outer membrane leaflet or neutral SMase in the inner leaflet of the
bilayer. Sphingomyelin transported to the lysosomes can also be converted to
ceramide by the action of lysosomal SMase. Ceramide can reversibly be
converted to sphingomyelin by the action of sphingomyelin synthase (SMS) in
the Golgi apparatus. The sphingomyelin produced in the Golgi can be
transported to the plasma membrane by vesicular transport where it can be
converted back to ceramide by the action of SMase as described earlier [2].
Ceramide can also be metabolized to glucosylceramide (GlcCer) in the
Golgi apparatus by the action of GlcCer synthase (GCS). GlcCer serves as the
precursor of complex glycosphingolipids in the Golgi. Reversibly, ceramide can
also be synthesized from GlcCer by the action of glucosyl ceramidase localized
in the lysosomal compartment [2]. Ceramide is also metabolized to ceramide-1-
phosphate by the action of ceramide kinase.

3
Another critical metabolism pathway is deacylation of ceramide to
sphingosine (Sph) by the action of ceramidases (CDase). There are five
ceramidases that are products of different genes: neutral CDase, acid CDase
and three forms of alkaline CDase [3]. These enzymes lie at a crucial juncture in
the sphingolipid pathway since, in conjunction with sphingosine kinases (SphK),
they balance the ceramide/sphingosine-1-phosphate (S1P) rheostat in cells.
Lysosomal acid CDase hydrolyses ceramide to form sphingosine (Sph), which is
favorably partitioned into the lysosomes due to its positive charge [2].
Furthermore, conversion of sphingosine to ceramide is mediated by CerS, which
forms a part of the de novo synthesis pathway of ceramide.
Sphingosine kinases 1 and 2 (SphK1 and SphK2) are the two sphingosine
kinase isozymes that catalyze the formation of S1P from sphingosine. It has
been postulated that SphK1 is present just outside the lysosomes ensuring
effective trapping of the Sph within the lysosomes by SphK1-mediated
phosphorylation. The presence of S1P phosphatases in the ER generates Sph
which can move among cellular biomembranes or which is eventually recycled to
form ceramide [2]. S1P lyase, which metabolizes S1P to ethanolamine
phosphate and hexadecenal, is the exit pathway in sphingolipid metabolism.

4
Figure 1-2 Sphingolipid metabolism pathways: Taken from Hannun et al. [2]
Figure 1-3 Compartmentalization of sphingolipid metabolism: Taken from Hannun et al. [2].

5
Functions of Sphingolipids
Sphingolipids function both as structural components of the cell and
mediators of cell signaling. As structural components of cellular biomembranes,
they play a critical role in regulating membrane fluidity and subdomain structure
of the lipid bilayer, especially lipid rafts [4]. Sphingolipids create lateral
differentiation of cellular membranes into a mosaic of structural domains with
unique molecular compositions. Lateral lipid assemblies are formed as a result of
varying miscibility of cell membrane-forming lipids like sphingolipids,
glycerolipids, and sterols. The protein content of such microdomains or rafts
characterize their function and serve as platforms for cellular events like signal
transduction, cell adhesion and protein sorting [5]. Sphingolipids like ceramides
affect the composition and properties of phospholipid bilayers by increasing the
order of the acyl chain in the bilayer, creating phase separation of ceramide-rich
and ceramide-poor domains and facilitating transition from bilayer to non-bilayer
structure [6]. In addition to cellular biomembranes, enzymatically-synthesized
ceramides also alter the properties of lipidic vesicles by inducing destabilization
of lipid bilayers through permeabilization and fusion [6]. Various biological
consequences follow ceramide-induced biophysical changes in cellular
biomembranes. For instance, the change in membrane fluidity after ceramide-
induced raft formation may result in modification of enzymatic activity of
membrane proteins, or may change the protein affinity for the membrane.
Ceramides also bind to specific sites in the target protein and alter enzymatic
activity. These target proteins are both membrane-bound proteins and other

6
cytoplasmic proteins transiently recruited to the bilayer where ceramides are
located [6].
In addition to structural roles, sphingolipids play a critical role in cellular
signaling. Extensive work has been done to elucidate the role of sphingolipids in
modulating cellular signaling. This section will broadly discuss the mechanisms
through which sphingolipids regulate cellular signaling mechanisms. Firstly,
sphingolipids act as ligands to receptors, initiating signaling pathways involved in
cellular processes like growth, adhesion, differentiation and migration. The
ligand-receptor interaction can be triggered in the same cell secreting the
sphingolipids or in neighboring cell types [7]. For instance, S1P is a ligand for a
family of G-protein coupled receptors called S1P receptors that regulate
biological processes such as cell proliferation, angiogenesis, migration, immune
cell trafficking and mitogenesis. Secondly, sphingolipids influence the properties
of receptors via specific lipid-protein interactions and regulating responsiveness
to external stimuli. For instance, it has been shown that the ligand-binding
capacity of human serotonin 1A receptors is impaired under glycosphingolipid-
depleted condition [8]. The authors speculate that this effect is due to a reduction
in the specific interaction of serotonin 1A receptors with membrane
glycosphingolipids [8]. Thirdly, as discussed earlier, sphingolipids alter the
biophysical properties of cellular biomembranes affecting the assembly of
membrane receptors and effector molecules in specific domains called rafts. This
sphingolipid-induced alteration in membrane biophysics regulates cellular
signaling [7]. Lipid rafts create a micro-environment suitable for effective receptor

7
activation by concentrating relevant kinases and protecting proteins from
phosphatases and other molecules that may diminish the signaling processes.
For example, lipid rafts are critical for immunoglobulin E signaling and T-cell
antigen receptor-mediated signaling [9]. Lastly, sphingolipids like ceramides and
S1P can act as direct signaling molecules in processes like proliferation,
differentiation, senescence, cell–cell interaction and transformation [7]. For
instance, intranuclear S1P has been shown to inhibit the enzymatic activities of
histone deacetylases, preventing the removal of acetyl groups from lysine
residues within histone tails. This work has shown the role of intranuclear S1P in
regulating gene expression [10].
Because sphingolipids are bioactive molecules and mediate several
cellular processes like proliferation, differentiation, migration, death and cell-cell
interaction, they have been implicated in the pathogenesis of several human
disorders. There is an abundance of literature reporting the role for sphingolipids
in inflammatory and immune responses, vascular function, neurodegeneration,
insulin signaling and diabetes, microbial pathogenesis and cancer pathogenesis
and therapy [11]. This dissertation is focused on studying the role of short-chain
ceramides as a potential therapy for chronic lymphocytic leukemia (CLL).
Furthermore, this dissertation also focuses on elucidating the key molecular
mechanisms which mediate the effect of short-chain ceramides in CLL cells.

8
Ceramide for cancer therapeutics
Sphingolipids have been implicated in cancer pathogenesis since they
mediate several cellular processes like proliferation, differentiation, migration,
death and cell-cell interaction. Numerous studies in the literature have reported a
dysregulation in sphingolipid metabolism in different types of cancers. The
dysregulated sphingolipid profile in cancer cells contributes to cancer progression
and metastasis, making it an ideal candidate for developing targeted
therapeutics. Moreover, sphingolipids may also serve as vital biomarkers for
cancer progression, as well as guide therapeutic regimens [12].
The role of sphingolipids in cancer pathogenesis and treatment has been
of particular interest to the sphingolipid research community. First reports
describing the involvement of sphingolipids in mediating apoptosis in cancer cells
came in early 1990s. It was reported that synthetic short chain ceramide analogs
like C2-ceramide induced cell death in HL60 leukemic cells and caused
internucleosomal DNA fragmentation [13]. Tumor necrosis factor α (TNFα) and
ionizing radiation induced apoptotic cell death in cancer cells, which was
mediated by sphingomyelin hydrolysis and subsequent ceramide generation [14,
15]. Apoptotic cell death through CD95 crosslinking in U937 cells also utilized the
sphingomyelin pathway and depended on ceramide production [16]. The first
body of work demonstrating the role of sphingolipids in mediating chemotherapy-
induced apoptosis in cancer cells was published in 1995, wherein the authors
showed that daunorubicin, a chemotherapeutic drug, induced apoptosis in P388
and U937 leukemia cells by elevating intracellular ceramide levels. This increase

9
in the intracellular ceramide pool was not due to the action of SMase, but rather
via activation of CerS, which increased de novo ceramide synthesis within cells
[17]. This revelation of a potential role of sphingolipid metabolism in mediating
chemotherapy-induced cytotoxicity generated immense interest in the scientific
community to decipher the connection between chemotherapy and sphingolipid
metabolism. Since then, a large body of research has been conducted to
establish an in-depth understanding of how sphingolipid metabolism mediates,
enhances, or impedes chemotherapy-induced cytotoxicity, with the goal of
identifying critical therapeutic targets and better therapeutic regimens for
management and cure of cancer.
Among the major sphingolipids that play a role in regulating cancer cell
fate, ceramide is termed as a “tumor suppressor lipid” because of its ability to
potentiate signaling cascades that lead to cell death. By contrast, sphingosine-1-
phosphate (S1P) is considered as a pro-survival lipid. Thus, in the context of
sphingolipids, the ceramide-S1P rheostat dictates cancer cell fate. Efforts
directed at altering the sphingolipid balance in cancer cells to induce cell death or
to potentiate cytotoxicity of chemotherapeutic drugs would aim at either elevating
pro-apoptotic sphingolipids, especially ceramide, or down-regulating pro-survival
sphingolipids such as sphingosine-1-phosphate. This can be achieved by: (i)
chemotherapy-induced synthesis of pro-apoptotic ceramides or breakdown of
pro-survival sphingolipids; (ii) disruption of ceramide metabolism to enhance
ceramide accumulation; and (iii) delivery of exogenous ceramides to induce
apoptotic signaling (Fig. 1-4).

10
Figure 1-4 Strategies to alter the sphingolipid balance in cancer cells to potentiate
cytotoxicity of chemotherapeutic drugs. 1) Chemotherapy-induced synthesis of pro-apoptotic
sphingolipids or breakdown of pro-survival sphingolipids; 2) Disruption of metabolism of pro-
apoptotic sphingolipids to enhance accumulation; and 3) Nanoscale-based delivery of exogenous
pro-apoptotic sphingolipids in combination with standard chemotherapeutic drugs to induce
apoptotic signaling.

11
Ceramide and induction of cell death
Ample evidence in the literature exists that indicate that ceramide is a
tumor-suppressor lipid and halts tumor progression by inducing cell death and by
cell cycle arrest. Both stimulus-induced intracellular ceramide generation and
exogenous cell-permeable short-chain ceramides induce death in cancer cells by
apoptosis, necrosis or autophagy.
Ceramide and apoptosis
Ceramide generation has been linked to both, the extrinsic and the intrinsic
pathway of apoptosis. Ceramide is an important mediator of initiating cell death
by activation of the pro-apoptotic tumor necrosis factor (TNF) receptor
superfamily, including CD95, TNFR1 and the TNF-related apoptosis-inducing
ligand (TRAIL) receptors TRAILR1 and TRAILR2 [18]. Receptor activation leads
to ceramide synthesis at ceramide-enriched membrane platforms by the de novo
pathway or activation of SMases. The ceramide-enriched membrane platforms
act as scaffolds for localization of pro-apoptotic proteins that initiate intracellular
signaling for cell death, some of which possess ceramide-binding domains.
Ceramide-enriched membrane platforms assemble TRAILR2 and CD95, and act
as a scaffold for the formation of the death-inducing signaling complexes [19-21].
Activation of these cell death receptors induce intracellular ceramide generation
by activating specific CerS and SMases [18]. The importance of ceramide
generation in these cell death pathways has been demonstrated by reports that
inhibition of ceramide synthesis diminishes apoptosis. For instance, pretreatment
with dihydroceramide synthase inhibitor fumonisin B1 (FB1) has been shown to

12
diminish CD95-induced apoptosis in leukemia [22]. Similarly, low ceramide levels
have been correlated to resistance to CD-95 induced apoptosis, TNF-induced
and TRAIL-induced cell death in several cancer models [23-25].
Ceramide also mediates intrinsic pathway-driven apoptosis. Ceramide
accumulation affects mitochondrial bioenergetics and induces conformation of
mitochondrial pro-apoptotic proteins to initiate apoptosis. Intensive research on
the effects of ceramide generation in the mitochondria has convincingly
demonstrated that accumulation of ceramide macrodomains in mitochondria
cause formation of ceramide channels that induce mitochondrial outer membrane
permeabilization (MOMP) [26, 27]. Ceramide accumulation either through
exogenously delivered short chain ceramides or endogenous ceramide
generation synergizes with pro-apoptotic proteins like BAX and BID to
permeabilize mitochondrial membrane and the subsequent release of pro-
apoptotic proteins like cytochrome c and activation of the caspase cascade [28-
31]. Ceramide also initiates intrinsic pathway-driven apoptosis by activating
kinases like p38 MAPK, glycogen synthase kinase (GSK) 3β, JUN N-terminal
kinase (JNK), protein kinase C (PKC) δ or inactivating AKT, which eventually
perturb mitochondrial integrity and cause release of pro-apoptotic proteins [18].
Ceramide is linked to several signaling pathways in apoptosis. The role of
ceramide in inactivating the very crucial AKT pathway in cancer cells has been
widely studied. The downstream targets of ceramide to bring about this
inactivation include PP2A, PKCζ and p38 MAPK [18]. Furthermore, ceramide
activates apoptosis signal-regulating kinase 1 (ASK1) which eventually increases

13
p38 and JNK activation [32]. Additionally, ceramide promotes p53 activation in
certain cancer types which causes a reduction in BAX/BCL-2 ratio, eventually
leading to apoptotic cell death [33, 34]. Finally, ceramide has also been shown to
downregulate anti-apoptotic proteins like survivin to induce apoptotic cell death
[35].
Ceramide and necrosis
The role of ceramide in necrotic cell death is not as well characterized as
apoptosis. Treatment with short chain ceramides like C2 and C6-ceramide
induces necrotic cell death in A20 B-, Raji B- and Jurkat T cells. Cell death was
without caspase-3 activation, DNA fragmentation, cell shrinkage, or chromatin
condensation. FasL-dependent delayed elevation of ceramide promoted caspase
8-driven necrotic morphology after treatment. Inhibition of ceramide production
shifted the mechanism of cell death from necrosis to apoptosis [36]. Further
investigation of the necrotic mechanism has revealed that exogenous C6-
ceramide causes necrosis in lymphoid cells by rapid production of reactive
oxygen species (ROS), loss of mitochondrial membrane potential and ATP
depletion [37]. This is supported by data demonstrating that C2-ceramide-
induced oncotic necrosis in mouse epidermal tumor cells is modulated by a
decline in cellular glutathione and an elevation of ROS [38]. Our lab has also
shown that C6-ceramide delivered as a nanoliposomal formulation induces
necrotic cell death in chronic lymphocytic leukemia (CLL) cells by targeting the
Warburg effect through downregulation of glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) enzyme of the glycolysis pathway and ATP depletion,

14
supporting the report that synthetic ceramides induce non-apoptotic and necrotic
cell death in malignant B-lymphocytes [39, 40]. C2-ceramide has also been show
to predominantly induce necrotic cell death in NB16 neuroblastoma cells.
Although combined treatment with TNFα and cycloheximide is mediated by
intracellular ceramide generation, this stimuli induces apoptosis instead of
necrosis. Thus, C2-ceramide does not faithfully mimic the effects of apoptotic
ligands such as TNFα, which are thought to be mediated by an accumulation of
endogenous ceramide. C2-ceramide targets phosphatidylcholine in these cells
and elicits a mixture of cell death mechanisms, including necrosis and apoptosis,
the former being more predominant [41]. Ceramide also induces non-apoptotic,
caspase-independent cell death by inducing ROS generation in A172 human
glioma cells. NF-kappaB is involved in the regulation of ceramide-induced cell
death in human glioma cells [40]. Lastly, it has also been observed that certain
cellular parameters play an important role in determining the mechanism of cell
death after ceramide treatment. For instance, in Hep-G2 cells the mitochondrial
respiratory function determines the mechanism of cell death after treatment with
exogenous short chain ceramides. Herein, C2-ceramide induced necrosis which
was a result of 80% inhibition of the mitochondrial respiratory function leading to
ATP depletion and ROS generation. In contrast, C6-ceramide induced apoptotic
cell death in the same cells since mitochondrial function was not inhibited and
ATP production not diminished [42].

15
Ceramide and autophagy
Despite ceramide being a promoter of autophagy, its role in mediating
autophagic cell death is confounded by the role of autophagy in cancer cells, i.e.
lethal versus survival autophagy. Increased levels of long-chain ceramides
(C14:0 – C20:0 ceramides) and especially dihydroceramides have been
associated with both lethal and survival autophagy in different cancer cell types
[43, 44]. It is believed that the fate of the autolysosome dictates the function of
autophagy as lethal versus survival. Intracellular generation of sphingosine and
S1P in the autolysosomes by the hydrolysis of dihydroceramides and ceramides
promotes pro-survival autophagy. In contrast, accumulation of dihydroceramides
in the autolysosomes can enhance autolysosomal membrane permeability and
cause the release of cathepsins, thereby causing apoptotic cell death [45]. In this
case, ceramides promote lethal autophagy in cancer cells.
Thus, as discussed earlier sphingolipid-based therapeutics aim to alter the
sphingolipid balance to induce cell death by either elevating pro-apoptotic
sphingolipids, especially ceramide, or down-regulating pro-survival sphingolipids
such as S1P. This can be achieved by: (i) chemotherapy-induced synthesis of
pro-apoptotic ceramides or breakdown of pro-survival sphingolipids; (ii) disruption
of ceramide metabolism to enhance ceramide accumulation; and (iii) delivery of
exogenous ceramides to induce apoptotic signaling. The next section discusses
the first strategy as a proof-of-concept to demonstrate that the effectiveness of
current chemotherapies and investigational drugs is partially or completely

16
mediated by endogenous ceramide generation. The discussion will then focus on
the development of exogenous ceramide-based therapeutics for cancer.
Chemotherapy-induced ceramide generation by the de novo pathway
Many chemotherapeutics increase ceramide levels by upregulating the de
novo pathway of ceramide synthesis. This is accomplished by increasing the
activity of serine palmitoyl transferase (SPT), ceramide synthase (CerS), or both.
There are six CerS isoforms (CerS1-6), which are also known as longevity
assurance (LASS) genes, with each isoform corresponding to specific resulting
carbon chain lengths of ceramide [46]. CerS1 produces C18 ceramide, CerS2
C20-C26, CerS3 C22-C26, CerS4 C18 and C20, CerS5 C16, and CerS6 C14
and C16 ceramide [46]. Several inhibitors are routinely used to study the de
novo pathway, including fumonisin B1 (FB1), which inhibits CerS; and L-
cycloserine and myriocin, which inhibit SPT.
The taxanes docetaxel and paclitaxel are two chemotherapeutic agents
that increase ceramide levels by the de novo pathway. Both of these drugs act
by preventing microtubule disassembly, leading to cell cycle arrest [47].
Docetaxel, which is used to treat metastatic ovarian, breast, lung, head and neck,
and prostate cancer, increases CerS1 and CerS2 levels, while decreasing
sphingosine kinase 1 (SK-1) levels in prostate cancer cells [48]. Ceramide is also
a critical regulator of taxane-induced cell death since overexpression of the
ceramide transfer protein, CERT, reduces the sensitivity of cells to taxanes [49].
Similarly, paclitaxel-induced apoptosis in breast cancer cells is dependent on
ceramide produced by the de novo pathway [50].

17
In addition to the taxanes, the anthracyclines doxorubicin and
daunorubicin also affect ceramide metabolism in tumors. In addition to its effects
on DNA, doxorubicin also causes a CerS-dependent increase in ceramide levels
in neuroepithelioma and neuroblastoma cells [51]. However, inhibition of de
novo ceramide generation using FB1 surprisingly has no effect on doxorubicin-
induced apoptosis in these cells [51]. On the other hand, in head and neck
cancer cells, doxorubicin alone or in combination with gemcitabine causes
apoptosis that is dependent on CerS1 and the generation of C18:0 ceramide
[52]. This was confirmed in an animal model of head and neck cancer in which
doxorubicin in combination with gemcitabine decreased C16:0 ceramide in the
tumor, but increased intratumoral CerS1 and C18:0 ceramide [52]. Similar to
doxorubicin, camptothecin also exerts antitumor properties in follicular thyroid
carcinoma through ceramide accumulation via de novo synthesis. Both of these
drugs cause activation of ceramide synthesis without any effects on SMases.
Apoptosis is mediated by ceramide elevation and can be enhanced by the use of
inhibitors of ceramide clearance [53]. Similar to doxorubicin, daunorubicin also
acts by intercalating into DNA [47] and induces apoptosis in leukemia cells by de
novo ceramide generation and ceramide generation by the action of SMase [17].
It is used to treat acute myeloid leukemia (AML) and AIDS-related Kaposi’s
sarcoma (liposomal formulation) [47].
Besides the taxanes and the anthracyclines, vorinostat, a histone
deacetylase inhibitor in combination with sorafenib, a kinase inhibitor also
modulates ceramide metabolism. Vorinostat in combination with sorafenib

18
causes de novo pathway-dependent ROS production and cell death in HCC cells
[54]. This is accompanied by an increase in C16 ceramide, as well as C16, C18,
C22, C24:0, and C24:1 dihydroceramide [54].
Another chemotherapeutic agent that increases ceramide levels is
fludarabine, an inhibitor of DNA synthesis that is used to treat chronic
lymphocytic leukemia (CLL) [47]. Fludarabine causes a de novo pathway-
dependent 2.5- to 3 fold elevation in ceramide levels in CLL cells 6 hours after
treatment. Pretreatment with fumonisin B1 significantly rescues fludarabine-
induced ceramide generation and apoptosis [55]. In addition, CLL cells treated
with non-physiological, short chain C6 ceramide undergo apoptosis and necrosis
[39, 55], suggesting that fludarabine may kill CLL cells via upregulation of
ceramide.
Certain chemotherapeutic drugs generate ceramide by acting on SPT.
Etoposide-induced apoptosis in human leukemia cells is mediated by ceramide
synthesized by the activation of SPT. Ceramide formed by this pathway has
distinct functions in this model system as compared to that formed by the SMase
pathway. Ceramide synthesized by the de novo pathway in this model is not
involved in caspase-induced poly (ADP-ribose) polymerase (PARP) cleavage but
instead perturbs membrane integrity [56].
In addition to current chemotherapies, there are drugs approved by the
United States Food and Drug Administration (FDA) for other uses that are
currently being investigated as potential anti-cancer agents. One of these is the

19
COX-2 inhibitor celecoxib, which is currently used to treat pain, inflammation, and
arthritis, and to prevent polyposis coli [47]. Recent work indicates that celecoxib
decreases cell viability in colorectal carcinoma cells in a CerS6-dependent
manner [57]. This is accompanied by an increase in sphingosine,
dihydrosphingosine, C14:0 ceramide, C16:0 ceramide and C18:0 ceramide, and
a decrease in C24:0 ceramide [57]. Furthermore, C16:0 ceramide is found at
elevated levels in tumors treated with celecoxib in an in vivo model of colorectal
carcinoma [57]. A number of investigational chemotherapeutics also upregulate
ceramide via the de novo pathway. Investigational drugs such as Valspodar,
inostamycin, and spisulosine induce apoptosis in a variety of cancer cell types
via de novo ceramide generation [58-62]. More recently, the endocannabinoid
analog R (+)-methanandamide (RMA) has been shown to increase C16 ceramide
levels in neuroglioma cells via the de novo pathway [63]. RMA, which increases
CerS3 and CerS6 in mantle cell lymphoma (MCL) cells, also increases C16, C24,
and C24:1 ceramide levels via the de novo pathway in MCL cells [64]. This
increase is functionally significant, as RMA-induced cell death in these cells is
also dependent on the de novo pathway [64].
In addition to RMA, another investigational drug that increases ceramide
levels is fenretinide (4-HPR). It is a synthetic retinoid N-(4-hydroxyphenyl) that
induces cell death in various cancers like neuroblastoma [65] and in cell lines
from cervical carcinoma [66] and acute myeloid leukemia (AML) [67]. Studies
have shown that the drug elevates intracellular ceramide levels via de novo
synthesis by increasing the activity of both SPT and CerS [68], and induces p53-

20
and caspase-independent apoptosis in neoplastic cells [69]. 4-HPR also
functions as a dose-dependent inhibitor of ceramide destaurase [70], indicating
that the cytotoxic sphingolipid species may be dihydroceramide or
dihydrosphingosine rather than ceramide [71]. Moreover, 4-HPR-induced
cytotoxicity is synergistic with inhibitors of ceramide metabolism like sphingosine
kinase inhibitors and glucosylceramide synthase inhibitors [72]. Other effects
induced by 4-HPR include generation of ROS and enhanced expression of LC3B
(form II). 4-HPR is currently in clinical trials for ovarian cancer, neuroblastoma,
lymphoma and leukemia [73].
Finally, the investigational AMPK inhibitor Compound C increases CerS5,
C16:0 ceramide, and C18:0 ceramide, and decreases in sphingosine in breast
cancer cells [74]. Clearly, many current and investigational chemotherapeutics
increase ceramide levels by targeting the de novo pathway. This increase in
ceramide, in turn, induces apoptosis in cancer cells.
Chemotherapy-induced ceramide generation by sphingomyelinase pathway
Independent of the de novo pathway, ceramide can also be generated
when SMase hydrolyzes sphingomyelin to produce ceramide. A number of
chemotherapeutics increase ceramide levels in cancer cells by increasing the
activity of SMase. Some drugs, such as daunorubicin, upregulate ceramide by
modulating both the de novo pathway and SMase activity. Besides upregulating
the de novo pathway, it also increases SMase activity in leukemia cells [75]. In
addition, in breast cancer cells, daunorubicin enhances binding of the
transcription factor Sp1 to the nSMase2 gene, leading to increased nSMase2

21
levels and a nSMase-dependent decrease in cell viability [76]. It should be
noted that daunorubicin is currently not approved for the treatment of breast
cancer.
Gemcitabine is another drug that targets both the de novo pathway and
SMase. It increases aSMase activity in pancreatic cancer cells [77]. In addition,
gemcitabine increases aSMase activity in glioma cells, increasing the levels of
C16 and C24 ceramide and an aSMase-dependent decrease in cell survival [78].
One chemotherapeutic that elevates ceramide levels via SMase
independent of the de novo pathway is cytarabine, which increases nSMase
activity and ceramide levels in acute myelogenous leukemia (AML) cells [79].
Cisplatin is another drug that affects SMase activity. Cisplatin, which is a
platinum coordination complex that causes DNA cross-links leading to cell death,
is used to treat testicular, ovarian, bladder, head and neck, cervical, endometrial,
lung, anal, rectal, esophageal, and central nervous system (CNS) cancer, as well
as neoplasms of childhood [47]. It increases ceramide levels and causes
apoptosis in glioma cells in a nSMase-dependent manner, wherein it transiently
increases aSMase activity and causes it to be redistributed to the plasma
membrane [80]. Additionally, a cisplastin-induced increase in SMase activity and
the subsequent accumulation of ceramide levels are essential for cytoskeletal
remodeling following treatment with cisplastin, such as loss of
lamellipodia/filopodia and dephosphorylation and redistribution of the actin-
binding protein ezrin [81].

22
Another chemotherapeutic that is currently used in the clinic and that
affects SMase is rituximab, which is a monoclonal antibody to CD20 that is used
to treat lymphoma and chronic lymphocytic leukemia [47]. This drug increases
aSMase activity in lipid rafts, increasing ceramide levels in B-lymphoma cells,
thereby inhibiting cell growth in these cells [82]. Furthermore, exogenous
treatment with C16 ceramide decreases cell viability in this system [82]. Taken
together, these findings indicate that rituximab may inhibit B-lymphoma cell
proliferation by activating aSMase, thus increasing ceramide levels.
Recently, the investigational drug stichoposide C, a marine triterpine
glycoside, has been shown to cause apoptosis in leukemia and colorectal cancer
cells in an aSMase- and nSMase-dependent manner [83]. It also inhibits tumor
growth in mouse models of leukemia and colorectal cancer [83]. In addition,
stichoposide C-treated tumors contained elevated levels of ceramide [83].
Betuletol 3-methyl ether, a natural phenylbenzo-γ-pyrone, is another
investigational drug that increases ceramide levels in cancer cells [84]. It causes
apoptosis in leukemia cells and increases aSMase activity and ceramide levels
[84]. Finally, the investigational drug withanolide D acts by increasing nSMase
activity, leading to increased ceramide levels and apoptosis in leukemia cells
[85]. It decreases cell viability in leukemia cells but not normal lymphocytes [85].
Furthermore, it induces apoptosis in primary cells from both myeloid and
lymphoid leukemia patients [85]. Withanolide D is a good example of an
investigational therapy that modulates SMase. While several current

23
chemotherapies target SMase, even more such drugs could be added to the
oncology arsenal in the future.
Nanotechnology-based drug delivery of ceramide
Delivering exogenous ceramides to cancer cells is another strategy to
perturb sphingolipid levels for inducing apoptosis. However, this strategy is
greatly limited by the hydrophobicity and insolubility of ceramide molecules,
which in turn restricts delivery in cell culture systems and in vivo. Soluble
ceramide analogs have been developed to circumvent this problem. Short chain
ceramides like C2-, C6- and C8-ceramides are cytotoxic in multiple cancer
models [86]. Chemical modifications of short-chain ceramides improve their
solubility, permeability and pharmacokinetics. Certain chemical modifications that
have been tested in cancer models include uracil-linked ceramides [87],
serinamides [88], serinols [89, 90] and 4,6-diene-ceramides [91]. In addition,
cationic water-soluble pyridinium-ceramides have been developed which
preferentially accumulate in mitochondria and induce cell death by mitochondrial
permeabilization and Bax translocation [92]. These are effective in inducing cell
death in head and neck squamous cell cancer [93] and breast cancer cell lines
[94]. These analogs also synergize with gemcitabine to cause cell cycle arrest in
G0/G1 phase, retard growth and inhibit telomerase activity in human head and
neck squamous cell carcinomas in vitro and in vivo [93, 95]. Other structural
analogs of ceramide that have shown selective cytotoxicity in drug-resistant
human breast cancer cells compared to normal breast epithelial cells include 5R-
OH-3E-C8 ceramide, benzene-C4-ceramide and adamantyl-ceramide [96].

24
Additional novel ceramide analogs including AD2646, AD2672, AD2665, AD2646
and AD2687 have cytotoxic effects on leukemic cells [97].
Ceramide-based therapies face challenges like high insolubility and
difficulties to design formulations. Nanoscale-based formulations have thus been
developed and investigated to deliver these therapies. Nanoemulsions,
nanoliposomes, calcium phosphosilicate nanoparticles and biodegradable linear-
dendritic nanoparticles are used for delivering ceramide-based therapeutics [98].
Ceramide delivered via nanoscale formulations has been shown to induce cell
death selectively in cancer cells while sparing normal cells [98].
Novel oil-in-water nanoemulsions have been evaluated as delivery
vehicles for potential combination therapies in vitro. EGFR-targeted
nanoemulsions containing myrisplatin and C6-ceramide show synergistic in vitro
cytotoxicity in ovarian cancer cells and also possess potential diagnostic
capabilities [99]. Similarly, coadministration of paclitaxel and ceramide in
nanoemulsion formulations induces enhanced cytotoxicity and apoptotic activity
in human glioblastoma cells in vitro [100]. Sustained release of C6-ceramide from
thermoresponsive and biodegradable linear-dendritic nanoparticles induces
apoptosis in breast adenocarcinoma cells in vitro with hyperthermia, thus
presenting a promising formulation to deliver bioactive sphingolipids for treatment
of solid tumors in conjunction with hyperthermia [101]. Biodegradable polymeric
nanoparticles have also been evaluated for modulation of drug resistance in
cancer cells in vitro and in vivo. Paclitaxel and tamoxifen administered in
biodegradable poly (ethylene oxide) - modified poly (epsilon-caprolactone) (PEO-

25
PCL) nanoparticles possess significant antitumor efficacy in ovarian carcinoma in
vitro and in vivo. Tamoxifen in the formulation reverses drug resistance by
inhibiting P-gp and GCS, thus elevating intracellular ceramide levels in cancer
cells [102]. Paclitaxel and C6-ceramide PEO-PCL nanoparticles have also been
used to chemosensitize resistant human ovarian cancer cell lines to induce
apoptotic cell death [103] and suppress growth in xenograft tumor models [104].
Additionally, we have demonstrated the cytotoxic effects of C10-ceramide-loaded
calcium phosphate nanocomposite particles in drug-sensitive and drug-resistant
breast cancer and melanoma cells in vitro [105].
Encapsulation of chemotherapeutic drugs into nanoliposomes has been a
largely successful delivery formulation in cancer models in vitro and in vivo. Our
lab has extensively studied nanoliposomes as a suitable delivery formulation for
ceramides in several cancer models. We have shown that nanoliposomes loaded
with short-chain ceramides suppress tumor growth in models of breast cancer
[106, 107], J774 sarcoma [108], melanoma [109], hepatocellular carcinoma [110],
large granular lymphocytic (LGL) leukemia [35], chronic lymphocytic leukemia
(CLL) [39], natural killer cell leukemia [111] and pancreatic cancer [112].
Mechanistically, the targets of nanoliposomal C6-ceramide include survivin in
LGL leukemia [35], GAPDH in CLL [39] and AKT/PKB and Erk in breast cancer
[107], hepatocellular carcinoma [110], pancreatic cancer [112] and melanoma
models [109]. Our studies and extensive in vivo toxicology studies by National
Cancer Institute sponsored agency, Nanotechnology Characterization Laboratory
(http://ncl.cancer.gov/working_technical_reports.asp) have confirmed that C6-

26
ceramide nanoliposomes have minimum adverse toxicity and elicit apoptosis
selectively in cancer cells [113]. We have established that selectivity of C6-
ceramide nanoliposomes for cancer cells can be attributed to the inhibitory action
of ceramide on the Warburg effect prevalent in cancer cells [39].
Our lab has also evaluated C6-ceramide nanoliposomes as a platform for
combinatorial therapy with other neoplastic agents. We have shown that C6-
ceramide nanoliposomes synergize with encapsulated sorafenib to reduce tumor
development in melanoma and breast cancer cells [109], synergize with
gemcitabine or liposomal PDMP to exhibit antitumor effects on pancreatic tumor
xenografts [112] and synergize with PPMP to induce apoptosis in natural killer
cell leukemia [111]. Recently, in collaboration with the Cabot group we showed
that nanoliposomes loaded with C6-ceramide and tamoxifen served as a
promising regimen for refractory breast cancers like the triple-negative breast
cancer [114]. Tamoxifen amplifies C6-ceramide-induced cytotoxicity in the triple-
negative breast cancer cells by multiple effects like cell cycle arrest, lysosomal
membrane permeability and inhibition of acid ceramidase [114]. C6-ceramide
nanoliposomes also exhibit synergy with the autophagy inhibitor vinblastine to
induce apoptotic cell death in vitro and in vivo in hepatocarcinoma and colorectal
cancer models, potentially mediated by an autophagy mechanism [115]. In
addition to chemotherapeutic drugs, multidrug resistance modulators are also
favorable adjuvants for C6-ceramide nanoliposomes. This strategy is justified by
studies reporting that resistance to C6-ceramide cytotoxicity is a result of
expression of P-gp in some cancer cells [116]. P-gp inhibitors also alter

27
sphingolipid levels in cancer cells. For instance, the multidrug resistance
modulator SDZ PSC 833 elevates intracellular ceramide levels by inducing the
de novo pathway [117]. P-gp antagonists like tamoxifen, verapamil, and
cyclosporine A can also be used in conjunction with cytotoxic drugs like
doxorubicin to decrease GlcCer, subsequently increasing ceramide levels and
sensitizing cells to cytotoxic drugs [59, 118]. In collaboration with the Cabot
group, we have shown that C6-ceramide nanoliposomes-mediated cytotoxicity in
cancer cell lines can be augmented by P-gp antagonists like tamoxifen,
verapamil and VX-710 [119, 120]. C6-ceramide and tamoxifen induced apoptotic
cell death in colorectal cancer cells characterized by PARP cleavage,
mitochondrial membrane permeabilization, caspase-dependent apoptosis and
G1/G2 cell cycle arrest. Moreover, the combinatorial treatment exhibited synergy
and induced upregulation of tumor suppressor p53 [119].
Co-administration of paclitaxel and C6-ceramide exhibit synergy to induce
cytotoxicity in pancreatic cancer cells via transient activation of EGFR and ERK
pathway [121] and ovarian cancer cells [122]. An interesting study revealed that
in the absence of paclitaxel, exogenous C6-ceramide enters the cell through a
predetermined initiation site of mitosis, or diffuses into cells through water
channels and caveolae-mediated endocytosis [122]. However, the combination
induces synergistic cytotoxicity in cancer cells as paclitaxel disrupts cytoskeletal
proteins, thus enabling an even distribution of C6-ceramide in the cytoplasm of
the cells [122]. Other reports have demonstrated that C6-ceramide also
synergizes with histone deacetylase inhibitors like trichostatin A to display

28
anticancer effects in in vivo mice xenograft pancreatic and ovarian cancer
models [123]. The authors have delineated the mechanism of this synergy and
have demonstrated PP1-mediated inactivation of Akt/mTOR and increased α-
tubulin acetylation as events causing cancer cell death. Furthermore, the
combination resulted in a very pronounced elevation in intracellular ceramide
levels and induction of cell death in cancer cells [123]. C6-ceramide also
synergizes in inducing apoptotic cell death in leukemia cells with other neoplastic
agents like the cationic peptide, bovine lactoferricin [124].
In conclusion, Fig. 1-5 summarizes the points of intervention in the
sphingolipid pathway and the drugs being studied for developing sphingolipid-
based cancer therapeutics.

29
Figure 1-5 Points of intervention in the sphingolipid pathway. Enzymes catalyzing ceramide
synthesis or ceramide metabolism can be activated or inhibited respectively to cause ceramide
accumulation and induce death in cancer cells. SPT, serine palmitoyl transferase; CerS,
ceramide synthases; DDase, dihydroceramide desaturase; SMase, sphingomyelinase; SMS,
sphingomyelin synthase; SphK, sphigosine kinase; GCS, glucosyl ceramide synthase; PPMP, 1-
phenyl-2-palmitoylamino-3-morpholino-1-propanol; PDMP, 1-phenyl-2-decanoylamino-3-
morpholino-propanol; 4‑HPR, N‑(4‑hydroxyphenyl) retinamide; DHS, L-threo-dihydrosphingosine;
SKI II, 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole.

30
Chronic Lymphocytic Leukemia (CLL)
CLL is the most prevalent form of adult leukemia in Western countries. As
per 2015 statistics, CLL accounts for approximately 30% of total leukemia cases
diagnosed and it accounts for 20% of deaths from all kinds of leukemia [125].
CLL mainly affects older adults. The average age at the time of diagnosis is
around 71 years. It is rarely seen in people under age 40, and is extremely rare
in children.
CLL is a malignant lymphoproliferative disorder of mature B lymphocytes.
The disease is characterized by an accumulation of mature looking B
lymphocytes in the blood, bone marrow, lymph nodes or other lymphoid tissues.
Leukemic B cells express characteristic surface markers consisting of CD19,
CD20 (weak) and CD23, with co-expression of CD5. Most CLL cells are arrested
in the G0/G1 phase and are highly resistant to apoptosis, eventually leading to
an accumulation of malignant cells [126]. A large body of work has demonstrated
that several factors like microenvironmental stimuli, antigenic drive and
epigenetic and genetic deregulation dictate the pathogenesis of CLL.
CLL is classified into different sub-types that determine the prognosis of
the disease and the treatment strategy. One classification is based on the degree
of somatic hypermutation, i.e. whether the cells express mutated or unmutated
immunoglobulin heavy chain variable region (IGHV) genes. The two groups
follow a different clinical course, with poorer survival in patients exhibiting
unmutated IGHVs. In addition to this determinant, approximately 80% of CLL
cases also show chromosomal aberrations. These genetic aberrations are

31
observed in both IGHV mutated and unmutated CLL, the latter being associated
with higher incidence of high-risk aberrations [125]. Deletion in band 13q14 is the
most common genetic aberration in CLL and has a favorable prognosis. This part
of the chromosome contains mir-15a and mir-16-1 which have been implicated in
CLL pathogenesis [127]. Trisomy 12 is another frequent chromosomal
abnormality in CLL, however, the corresponding molecular implications on
pathogenesis remains unknown. Deletion in band 11q23 is not a very frequent
aberration in early stage disease, but is associated with rapid disease
progression and excessive lymphadenopathy [128, 129]. This deletion usually
corresponds with deletion of the ataxiatelangiectasia-mutated (ATM) gene, which
is an essential component of the cell cycle checkpoint system. Deletion in the
ATM gene is characterized by extreme sensitivity to irradiation, genomic
instability and a predisposition to lymphoid malignancies [125]. Lastly, deletion in
band 17p13, corresponding to p53 deletion and TP53 mutation is associated with
poor prognosis [125].
The microenvironment in the lymphoid organs plays a crucial role in the
pathogenesis of CLL. The microenvironment consists of T-cells, stromal cells and
soluble factors. Soluble factors from the microenvironment provide CLL cells with
a protective environment and provide anti-apoptotic and pro-proliferative stimuli
that are necessary for the progression of the disease. For instance, CLL cells
recruit CD3+ T cells which also express CD40L and CD4. CD40L from these
accessory cells initiate the indispensable B-cell receptor (BCR) signaling in CLL
cells after interaction with CD40 receptor. Such stimuli induce production of anti-

32
apoptotic proteins like survivin, Mcl-1 and Bcl-2 in CLL cells which are crucial for
cell viability [125, 130]. Furthermore, the stromal microenvironment promotes a
metabolic switch in CLL cells from mitochondrial respiration to aerobic glycolysis,
thus conferring growth advantage and conferring chemoresistance to CLL cells
[131]. In conclusion, ample evidence in the literature exists emphasizing the
crucial role of stromal microenvironment in the pathogenesis and
chemoresistance of CLL. A multitude of molecules including integrins, spleen
tyrosine kinase, stromal derived factor-1, Notch, CD44, and thioredoxin have
been identified to be part of the stromal cross talk [125].
The standard treatment regimen for physically fit CLL patients includes a
combination chemo-immunotherapy with fludarabine, cyclophosphamide and
rituximab with an overall response rate of approximately 90% and complete
remission of 72% [132, 133]. A combination of purine analogs, alkylating agents,
monoclonal antibodies (immunotherapy) and BCR pathway and tyrosine kinase
inhibitors is the standard treatment. Novel drugs recently incorporated in the
treatment regimen include ibrutinib, which targets an important component of
BCR signaling, Bruton’s tyrosine kinase (BTK). Another novel orally available
agent is idelalisib, a phosphatidylinositol-3-kinase (PI3K) δ inhibitor. Both of
these drugs have been for relapsed/refractory disease and first-line treatment of
patients with TP53 mutation/deletion [134]. Unfortunately these advances do not
benefit older CLL patients due to their frail health [135]. Overall, CLL is incurable
with the current therapies, with allogeneic stem cell transplantation as the only
potentially curative treatment option in CLL. However, this option is also limited to

33
young and relatively healthy patients. Despite these advances in therapeutics,
eventual drug resistance and relapse ultimately cause CLL to be an incurable
and chronic disease [136]. Further research is needed to develop therapeutic
strategies.
The role of sphingolipids in CLL pathogenesis or treatment has not been
explored yet. Synthetic ceramides have been shown to induce non-apoptotic and
necrotic cell death in malignant B-lymphocytes [137]. Additionally, a few reports
have also demonstrated that sphingolipids mediate cell death in CLL cells. It has
been reported that membrane microdomain sphingolipids are required for anti-
CD20-induced cell death in CLL cells [138]. The authors showed that resistance
to anti-CD20-induced cell death is associated with a defective recruitment of Csk-
binding protein, resulting in a lack of sphingomyelin and ganglioside M1 at the
outer leaflet of the plasma membrane of malignant B cells. Inducing P-
glycoprotein in resistant cells restored sensitivity to anti-CD20 antibody as the
inducer normalized the quantity of sphingomyelin within the membrane [138].
Another recent report has uncovered a novel link between BCR signaling and
sphingolipid metabolism. It has been reported that BCR controls
chemoresistance of primary CLL cells by controlling glucosylation of ceramides
[139]. Specifically, BCR engagement increases levels of anti-apoptotic
glucosylated ceramides via upregulation of UDP-glucose ceramide
glucosyltransferase (UGCG), an enzyme which converts pro-apoptotic ceramide
to anti-apoptotic glucosylceramide. The authors have shown that inhibitors of
BCR signaling sensitize resistant CLL cells towards ABT-737 drug via UGCG

34
inhibition [139]. Another report has demonstrated that sphingolipid metabolism is
a potential novel mechanism of CLL [140]. Taken together, these reports provide
compelling evidence in support of the use of sphingolipid-modulating strategies,
and more specifically, ceramide-based strategies as novel therapeutics in CLL.
Conclusions
The role of nanoliposomal C6-ceramide in inducing cell death in several
types of solid and non-solid cancers is well understood. However, the effect of
nanoliposomal C6-ceramide treatment in CLL remains unclear. This dissertation
primarily focuses on delineating the molecular mechanisms of C6-ceramide-
induced cell death in CLL. Identifying the key signaling pathways inducing cell
death after ceramide treatment is also valuable to uncover additional targets for
potential combination therapies with ceramide nanoliposomes. Encapsulation of
chemotherapeutic drugs into nanoliposomes has been a largely successful
delivery formulation in cancer models in vitro and in vivo. Moreover, the ongoing
success of using ceramide nanoliposomes as a platform for combinatorial
therapy with other neoplastic agents presents a promising future to this endeavor
of developing more effective therapeutics for CLL.

35
CHAPTER 2: Nanoliposomal C6-ceramide target the Warburg effect in chronic lymphocytic leukemia
I would like to recognize that Chapter 2 of my dissertation titled “C6-ceramide
nanoliposomses target the Warburg effect in chronic lymphocytic leukemia” is
derived from the following published literature:
Ryland LK*, Doshi UA*, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, Baab KT, Young M, Khan O, Haakenson JK, Jarbadan NR, Liao J, Wang HG, Feith DJ, Loughran TP Jr, Liu X, Kester M. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS One. 2013 Dec 19;8(12):e84648. I am the co-first author for this published work.
This project was underway when I joined the Kester lab in 2011. Dr. Lindsay
Ryland and I contributed towards the design of the experiments within this
particular chapter. Specifically, I was a major contributor towards the following
figures: Fig 2-1A, Fig. 2-3A, Fig. 2-4A and Fig 2-5. I am in no way attempting to
claim intellectual property over the design of all the experiments within this
particular chapter. Therefore, I would like to acknowledge Dr. Lindsay Ryland
and the other contributing authors for the work presented in this chapter.

36
Abstract
Ceramide is a sphingolipid metabolite that induces cancer cell death.
When C6-ceramide is encapsulated in a nanoliposome bilayer formulation, cell
death is selectively induced in tumor models. However, the mechanism
underlying this selectivity is unknown. As most tumors exhibit a preferential
switch to glycolysis, as described in the “Warburg effect”, we hypothesize that
ceramide nanoliposomes selectively target this glycolytic pathway in cancer. We
utilize chronic lymphocytic leukemia (CLL) as a cancer model, which has an
increased dependency on glycolysis. In CLL cells, we demonstrate that C6-
ceramide nanoliposomes, but not control nanoliposomes, induce caspase 3/7-
independent necrotic cell death. Nanoliposomal ceramide inhibits both the RNA
and protein expression of GAPDH, an enzyme in the glycolytic pathway, which is
overexpressed in CLL. To confirm that ceramide targets GAPDH, we
demonstrate that downregulation of GAPDH potentiates the decrease in ATP
after ceramide treatment and exogenous pyruvate treatment as well as GAPDH
overexpression partially rescues ceramide-induced necrosis. Finally, an in vivo
murine model of CLL shows that nanoliposomal C6-ceramide treatment elicits
tumor regression, concomitant with GAPDH downregulation. We conclude that
selective inhibition of the glycolytic pathway in CLL cells with nanoliposomal C6-
ceramide could potentially be an effective therapy for leukemia by targeting the
Warburg effect.

37
Introduction
Sphingolipids are a class of complex cellular lipids that serve both a
structural role in the cellular membrane as well as an intracellular signaling role
within the cell. Several types of sphingolipid metabolites have been shown to
influence the balance between mitogenesis and apoptosis. Of particular interest
is the sphingolipid metabolite, ceramide, which regulates differentiation,
senescence and cell cycle arrest. Induction of cell death by this endogenous
lipid-derived second messenger occurs either via apoptotic, autophagic, or
necrotic cell death pathways [41, 141, 142]. Ceramide inhibits cell proliferation
and induces apoptosis via mechanisms such as dephosphorylation and/or
inactivation of molecules including Akt, phospholipase D, ERK, Bcl-2, survivin,
PKC-α, and pRB [4, 143, 144], as well as activation of JNK kinases [4, 145], or
PKC zeta which, results in suppression of Akt-dependent mitogenesis [146].
Therefore, it is not surprising that dysregulated ceramide metabolism and
signaling has been linked to a variety of human diseases, including cancer.
Based on its ability to selectively block tumor initiation and metastasis, ceramide
has been termed the ‘tumor-suppressor lipid’ [4]. Many cancer chemotherapies
have been shown to generate endogenous ceramide, and when de novo
generation of ceramide is inhibited, the cellular response to cytotoxic
chemotherapeutic agents decreases [4]. In addition the accumulation of
endogenous ceramides or exogenous ceramide treatment is more toxic to tumor
cells than to normal cells [144, 147]. However, the exact mechanism of selectivity
is unknown.

38
One proposed mechanism for how ceramide mediates cell death induction
is through downregulation of nutrient transporter proteins possibly via nutrient
deprivation. [148]. As cancer cells have an increased dependence on glucose,
these nutrient transporters and/or the glycolytic pathway are typically
upregulated. One hallmark of cancer cells is their ability to avidly take up
glucose and convert it to lactate, even in the presence of sufficient oxygen.
Deemed the “Warburg effect,” this altered glycolytic dependency favors less
efficient generation of ATP compared to the oxidative phosphorylation process
which occurs in normal cells [149, 150]. Many human cancers display increased
levels of glycolytic enzymes compared to normal tissue [151]. Consequently, a
variety of chemotherapeutic glycolytic inhibitors or PET modalities are currently
under investigation as potential “Warburg-targeted” therapeutic or diagnostic
imaging tools [152, 153]. Recently, the role of sphingosine kinases in regulating
the Warburg effect in prostate cancer cells has been reported [154]. Treatment of
LNCaP prostate cancer cells with SKi, a non-selective sphingosine kinase
inhibitor, increases intracellular levels of ceramide and sphingosine and indirectly
antagonizes the Warburg effect, resulting in apoptosis of LNCaP cells.
Chronic lymphocytic leukemia (CLL) is the most common B-cell
malignancy in the Western world which presently has no known curative therapy
[155]. Previous studies have demonstrated that treatment with exogenous short-
chain C2-ceramide results in induction of cell death in malignant cells isolated
from CLL patients [137]. Recent advances in nanotechnology have illustrated
the feasibility of generating nanoliposomes that encapsulate hydrophobic

39
compounds, like ceramide, to facilitate treatment of CLL. While it is understood
how nanoliposomal ceramide induces cell death in several types of cancers and
hematological malignancies, the effect of nanoliposomal ceramide treatment in
CLL remains unclear. Currently, several nanoliposomal formulations of anti-
cancer drugs have been approved by the FDA and are the standard of care
[156]. For instance, the efficacy of fludarabine, the cancer chemotherapy
commonly used to treat CLL patients, and which acts via intracellular ceramide
accumulation, is enhanced after being encapsulated in nanoliposomes [157,
158]. Our laboratory has demonstrated that encapsulation of ceramide in a
nanoliposome versus non-liposomal organic formulations increases the cytotoxic
potential with significant less toxicity [159]. Our laboratory has also demonstrated
that the short chain C6-ceramide nanoliposomal formulation displays anti-
proliferative effects in vitro, as well as results in tumor regression in several
animal models of cancer [110, 144, 147, 159].
In this study we sought to elucidate the effect of nanoliposomal C6-
ceramide treatment in CLL. Our data suggest that this treatment is targeting
glucose utilization and results in activation of a caspase-independent, necrotic
cell death mechanism. We have identified glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) as a novel target of ceramide in CLL cells. In the
current study, we conclude that ceramide targets the Warburg effect in cancer
cells and selectively induces necrotic cell death in CLL.

40
Materials and Methods
Reagents
Antibodies specific for caspase 3, poly ADP ribose polymerase (PARP), GAPDH,
-actin and α-tubulin were purchased from Cell Signaling Technology Inc.
(Beverly, MA), glucose transporter 1 (GLUT1) antibody from Abcam (Cambridge,
MA) and lactate degydrogenase (LDH) antibody from Epitomics (Burlingame,
CA). For Western blotting, 12% precasted Nupage electrophoresis gels from
Invitrogen (Carlsbad, CA), and chemiluminescence reagent from Amersham
Biosciences Inc. (Piscataway, NJ) were obtained. Other reagents include zVAD-
fmk and pyruvate from Sigma (St. Louis, MO), dasatinib from Toronto Research
Chemicals Inc. (Ontario, Canada) and 3-bromopyruvate from Enzo Life Sciences
(Farmingdale, NY).
Patient characteristics and preparation of peripheral blood mononuclear
cells
All patients met the clinical criteria of CLL and were not on treatment at the time
of sample acquisition. Peripheral blood specimens from CLL patients were
obtained and informed consents signed for sample collection using a protocol
approved by the Institutional Review Board of Penn State Hershey Cancer
Institute. Buffy coats from normal donors were also obtained from the blood
bank of the Milton S. Hershey Medical Center, Pennsylvania State University,
College of Medicine. Peripheral blood mononuclear cells (PBMCs) were isolated
by Ficoll-Hypaque gradient separation, as described previously [160]. Cell

41
viability was determined by trypan blue exclusion assay with more than 95%
viability in all the samples.
Cell culture
Freshly isolated PBMCs and primary CLL patient cells were cultured using RPMI-
1640 medium supplemented with 10% fetal bovine serum (both from Invitrogen).
JVM3 cells (DSMZ – German Collection of Microorganisms and Cell Cultures,
Braunschweig, Germany), a CLL cell line, were also cultured in this same
medium and cells were grown in 5% CO2 at 37˚C.
Preparation of nanoliposomal ceramide
12% pegylated nanoliposomes (80 ± 15 nm in size) that contain 30 mol%
ceramide were prepared as described previously with lipids 1,2-distearoyl-sn-
glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, N-
hexanoyl-D-erythro-sphingosine (C6-ceramide), 1,2-distearoyl-sn-glycero-3-
phosphoethanolamine-N-[methoxy polyethylene glycol-2000], and N-octanoyl-
sphingosine-1-[succinyl(methoxy polyethylene glycol-750)] (PEG(750)-C8)
combined in chloroform at a molar ratio of 3.75:1.75:3:0.75:0.75 [159]. Combined
lipids were dried under nitrogen gas and resuspended in 0.9% sterile NaCl at
60°C. Following rehydration, resulting solution was sonicated for 5 min followed
by extrusion through a 100-nm polycarbonate membrane using the Avanti Mini
Extruder (Avanti Polar Lipids). Ghost liposomes were prepared in a similar
manner excluding N-hexanoyl-D-erythro-sphingosine (C6). Dihydro-C6-ceramide
liposomes were prepared in a similar manner by replacing N-hexanoyl-D-erythro-

42
sphingosine with N-hexanoyl-D-erythro-sphinganine. Several QA/QC parameters
were evaluated after preparation of nanoliposomes. Nanoliposomes were
formulated within the size range of 85nM – 90nM as measured by dynamic light
scattering. Zeta potentials of the nanoliposomes were measured and these were
between -3 mV to -7 mV, ensuring a neutral charge on the nanoliposomes.
Encapsulation efficiency of the ceramide nanoliposomes was evaluated by
LC/MS/MS.
Cell viability assay
Cell viability was performed using CellTiter 96® Aqueous One Solution assay kit
(Promega) or alamarBlue® assay kit (Invitrogen). Relative viable cell number
was determined by reading the plates at 490 nm or 570 nm wavelength
respectively in Synergy HT Multi-Detection Microplate Reader (Bio-TEK). All
samples were assayed in triplicate and each experiment was repeated at least
three times.
Apoptosis and exclusion assays (Annexin V/7AAD and TUNEL)
Apoptosis was determined in JVM3 cells and in PBMC samples from normal
donors (n=3) by 2-color flow cytometry with annexin-V and 7-amino-actinomycin
D (BD Pharmingen Transduction Laboratories) staining using 5 x 105 cells per
sample. TUNEL assays were also performed using terminal deoxylnucleotidyl
transferase (TdT), biotinylated UTP, and fluorescein isothiocyanate-streptavidin
as described previously [161]. Cell viability was also confirmed by trypan blue
exclusion.

43
Caspase 3/7 assay
For detection of caspase-3 and caspase-7 activation, JVM3 cells were plated in
replicates of six in 96-well plates, and treated with varying doses of ceramide for
24 hours and analyzed using ApoONE Homogeneous Caspase 3/7 Assay
(Promega) according to the manufacturer's instructions.
Western blot analysis
Western blot analyses of caspase 3, PARP, GAPDH, LDH, GLUT1, α-tubulin and
-actin protein expression were performed on whole-cell lysates collected using
RIPA buffer (Sigma). Densitometry analysis was performed using ImageJ
software.
Phase-contrast microscopy
To visualize a morphological necrotic phenotype, JVM3 cells were plated at 1 x
106 cells/ well and treated with 25 µM ghost nanoliposomes for 24 hours or C6-
ceramide nanoliposomes for 2, 6 and 24 hours. Phase contrast microscopy
images were then taken (Olympus CKX41).
GAPDH gene expression: real-time quantitative RT-PCR
Real-time reverse-transcription polymerase chain reaction (RT-PCR) was
performed using primer sets specific for GAPDH and an internal standard, 18S
rRNA, in an ABI PRISM 7900 sequence detector (Applied Biosystems) as
described elsewhere [144]. TRIzoL LS Reagent (Invitrogen) was used for RNA
extraction. Amplification of triplicate cDNA template samples was performed with

44
denaturation for 15 minutes at 95˚C, followed by 45 PCR cycles of denaturation
at 94˚C for 15 seconds, annealing at 55˚C for 30 seconds, and extension at 72˚C
for 30 seconds. A standard curve of cycle thresholds using serial dilutions of
cDNA samples was established and used to calculate the relative abundance of
the target gene. Values were normalized to the relative amount of 18S mRNA.
The relative amount of PCR products generated from each primer set was
determined based on the threshold cycle or threshold cycle value [162]. The
following primers were used for detection: GAPDH sense 5’-
GACCCCTTCATTGACCT CAACTACATG -3’, GAPDH antisense 5’-
GTCCACCACCCTGTTGCTGTAGCC-3’.
Glucose uptake and lactate production
The [3H]-2-DG uptake assay was modified from Kueck et al. [163]. 2-[1,2,-3H
(N)]-Deoxy-D-glucose was purchased from PerkinElmer. Cells were plated in 24-
well plates and 24 hours after experimental treatment were washed with PBS
and resuspended in Krebs-Ringer phosphate buffer + 1% BSA. Glucose uptake
was initiated by adding 1 µCi/mL 3H-2DG and 10 mM unlabeled glucose for 15
minutes. Uptake was terminated by adding ice cold Krebs-Ringer phosphate
buffer + 0.2 mM phloretin. 20 µM cytochalasin B, a potent inhibitor of glucose
transport was used as a control. All experiments were done in triplicate. Lactate
concentrations in culture media were determined after nanoliposomal treatment
using a colorimetric assay kit (Biomedical Research Service Center, SUNY at
Buffalo).

45
Measurement of ATP
Cellular ATP content was measured using a luminescence assay (Cell-Titer Glo
Kit, Promega). JVM3 cells were pretreated in the absence or presence of
pyruvate for 2 hours, and then incubated for 24 hours with nanoliposomal
ceramide. Final luminescence was measured in Synergy HT Multi-Detection
Microplate Reader (Bio-TEK).
shRNA Knockdown of GAPDH
GAPDH shRNA lentiviral clones (Human pLKO.1 vector) were purchased from
Open Biosystems (Huntsville, AL) and used to infect JVM3 cells according to the
manufacturer’s protocol. After selection with puromycin, a pool of infected cells
were expanded and tested for GAPDH expression via Western blotting.
Scrambled shRNA was used as a control in these experiments.
Lentiviral overexpression of GAPDH
Human pLOC vector (Open Biosystems) was used for overexpressing GAPDH in
JVM3 cells. Briefly, viral particles were produced in HEK293-FT cells using
pLOC, VSVG, tat and DR8.2 plasmids and JVM3 cells were transduced thrice
with the viral media according to the manufacturer’s protocol. JVM3 cells were
grown for 72 hours after the last transduction and subsequently harvested for
experiments. GAPDH overexpression was detected 72 hours after last
transduction via Western blotting. pLOC vector containing a red fluorescent
protein (RFP) sequence instead of GAPDH gene was used as a control in these
expreiments.

46
Animal Studies
Animal experimentation was performed according to protocols approved by the
Institutional Animal Care and Use Committee at the Pennsylvania State College
of Medicine and all efforts were made to minimize animal suffering. Female
Balb/c Nu/nu mice of about six weeks of age were obtained from Charles River
Laboratory (Wilmington, MA). Mice were subject to irradiation (600 cGy) one day
prior to inoculation. Ten million JVM3 cells were subcutaneously injected into the
right flank of the mice and treatment began approximately two weeks after
inoculation when tumors reached a volume of 50 to 100 mm3. Based upon prior
studies with multiple animal models [110, 144, 147, 159], leukemic mice were
treated with 40 mg/kg ghost (n=8) or C6-ceramide nanoliposomes (n=8) via IV
injection every other day over a three week period of time. Mice with large or
ulcerated tumors were euthanized.
In a parallel study, leukemic mice treated with an identical dose regimen of ghost
or C6-ceramide nanoliposomes were sacrificed during the course of the study
and tumor tissue was then flash frozen and isolated for protein extraction. Mice
were sacrificed on day 8 of C6-ceramide treatment (n=5), day 14 of C6-ceramide
treatment (n=6) and day 17 of C6-ceramide treatment (n=6). Additionally, mice
treated with the ghost nanoliposomes for 17 days were also sacrificed (n=5) and
protein was extracted from the tumor tissue for further immunoblot analysis.

47
Ethics Statement
A protocol approved by the Institutional Review Board of Penn State Hershey
Cancer Institute (Protocol #29839) was used to collect peripheral blood
specimens from CLL patients. Animal experiments were performed according to
a protocol (Protocol #2009-017) approved by the Institutional Animal Care and
Use Committee at the Pennsylvania State College of Medicine and all efforts
were made to minimize animal pain and discomfort.
Statistical analysis
All data are expressed as mean +/- SEM. All the graphs represent at least three
independent experiments, each replicated in triplicate, unless specified
otherwise. Paired Student t test (2-tail paired) and 2 way analysis of variance test
were used to determine the statistical significance and P value of </= 0.05 was
considered statistically significant.

48
Results
Nanoliposomal C6-ceramide selectively induces cell death in CLL cells
We have previously demonstrated the therapeutic use of C6-ceramide
nanoliposomes in both solid and non-solid tumor models [110, 144, 147, 159]. In
the present study, we investigated the therapeutic efficacy of this nanoliposomal
formulation in CLL. The CLL in vitro JVM3 cell line utilized for these studies was
established by EBV-transformation of human primary B-prolymphocytic leukemic
cells and treatment with phorbol ester, TPA [164]. MTT assay (Fig. 2-1A) and
trypan blue staining (Fig. 2-1B) demonstrated that nanoliposomal C6-ceramide,
but not the ghost nanoliposomes, induced dose-dependent cell death in JVM3
cells. Dihydro-C6-ceramide, a less active analog of C6-ceramide modestly
reduced cell viability at higher doses (Fig. 2-1A); however C6-ceramide was
significantly more toxic to cells in comparison. Concentrations of C6-ceramide
nanoliposomes above 25µM significantly increased percentage of non-viable
cells as shown by annexin V/7AAD staining (Fig. 2-1C) and TUNEL flow
cytometry analysis (Fig. 2-1D). It was further demonstrated that this dose-
dependent cell death induced by nanoliposomal C6-ceramide was also observed
in primary CLL patient cells but not in PBMC isolated from normal donors (Fig. 2-
1E). Flow cytometry analysis confirmed that apoptosis was not induced in these
normal cells (Fig. 2-1F). These results indicate that nanoliposomal C6-ceramide
is preferentially targeting CLL cells and is non-toxic to normal donor cells.

49
Figure 2-1. Nanoliposomal C6-ceramide selectively induces cell death in CLL cells. JVM3
cells were treated with varying doses of ghost or C6-ceramide or dihydro-C6-ceramide
nanoliposomes for 24 hours then A). MTT assay, B). Trypan blue staining was performed.
ANOVA statistical test was used to determine dose dependency between various C6-ceramide
treatment groups. P < .0001. C). JVM3 cells were treated with different doses of ghost or C6-

50
ceramide nanoliposomes for 24 hours, then cells were stained with annexin V and 7AAD for
apoptosis assay. D). Percentage of apoptotic cells was determined via TUNEL analysis after 24
hours. E). PBMC isolated from either CLL patients (n=3) or normal donors (n=3) were treated with
25 µM ghost or C6-ceramide nanoliposome for 2, 16 and 24 hours, then MTT assay was
performed. F). Percentage of apoptotic cells was determined in PBMC from normal donors (n=3)
via annexin V/7AAD staining. Cells were treated with different doses of ghost or C6-ceramide
nanoliposome for 24 hours or treated with 25 µM ghost or C6-ceramide nanoliposome for 2 and
24 hours. * P < 0.05.
Cell death induced by nanoliposomal C6-ceramide is independent of
caspase 3/7
To further characterize the mechanism of cell death being induced in the
JVM3 cell line, we investigated the effect of nanoliposomal C6-ceramide
treatment on caspase cleavage. Caspase 3 or downstream PARP cleavage was
not observed following treatment with the C6-ceramide nanoliposomes (Fig. 2-
2A). Treatment of JVM3 cells with 5 and 10 µM dasatinib, an inducer of apoptosis
in CLL [165], increased cleavage of both caspase 3 and PARP (Fig. 2-2B), thus
confirming that there was no defect in the caspase 3/7 apoptotic pathway in this
cell line. No change in caspase activity was also confirmed by the caspase 3/7
assay (Fig. 2-2C). As controls, 5µM dasatanib was sufficient to stimulate caspase
activity and pretreatment with 15µM zVAD-fmk, a pan-caspase inhibitor, inhibited
this activation. We further confirmed that C6-ceramide-induced cell death was
independent of caspase activation, as pre-treatment with zVAD-fmk did not
rescue cell death (Fig. 2-2D).
We next investigated if this caspase 3/7-independent cell death results in
a necrotic phenotype. To determine the predominant cell death mechanism being

51
induced following nanoliposomal ceramide treatment, JVM3 cells were treated
with 25 µM ghost or C6-ceramide nanoliposomes for varying time points and
visualized using phase contrast microscopy (Fig. 2-2E). Arrows indicate cells
which have increased in cellular volume and resemble a necrotic morphology as
evidenced by early plasma membrane rupture and abundance of cellular debris.
In addition, we confirmed necrotic cell death by flow cytometric analysis and
observed a time-dependent increase in Annexin V positive and 7AAD positive
cell population after treatment with nanoliposomal ceramide for 24 hours (data
not shown). From these results, we conclude that the preferential selectivity of
ceramide treatment appears to target a necrotic cell death mechanism,
consistent with caspase 3/7-independent cell death.

52
Figure 2-2. Cell death induced by nanoliposomal C6-ceramide occurs through caspase 3/7-
independent necrosis. JVM3 cells were treated with A). different doses of ghost and C6-
ceramide nanoliposome for 24 hours, B) 5 µM and 10 µM dasatinib, as well as DMSO vehicle
control for 24 hours, then Western Blot analysis was performed for caspase 3 and PARP
cleavage. C). Enzymatic activities of caspase 3/7 were measured using caspase 3/7
luminescence kit. The results for cells treated with zVAD-fmk and zVAD-fmk + Dasatinib were
significantly different from untreated cells (NT) and cells treated with Dasatinib. * P < 0.05. D).
JVM3 cells were treated with varying doses of ghost or C6-ceramide nanoliposomes for 24 hours
following a 2 hour pre-treatment or no pre-treatment with zVAD-fmk (15 µM). Cell viability was
assessed via MTT assay. The viability of cells treated with ceramide liposomes and cells treated

53
with ceramide liposomes +zVAD-fmk was significantly different from viability of cells treated with
ghost liposomes * P < 0.05. E). JVM3 cells were treated with 25 µM ghost nanoliposomes for 24
hours or C6-ceramide nanoliposomes for 2, 6 and 24 hours and phase contrast microscopy
images were taken. Arrows indicate morphology of necrotic cell death.
GAPDH as a target for treatment of CLL
There is growing evidence suggesting a link between increased
dependency on glucose metabolism and overexpression of glycolytic enzymes in
cancer cells. Moreover, GAPDH has been shown to be upregulated in many
cancers and is the primary target of some chemotherapeutic drugs [166].
Recently, GAPDH has been implicated in promoting cellular survival,
chemotherapy-resistance and protection from caspase-independent cell death
[167, 168]. We now demonstrate that treatment of JVM3 cells with nanoliposomal
C6-ceramide leads to a significant decrease in GAPDH protein expression (Fig.
2-3A). There was a significant decrease in GAPDH protein following 8 hours
treatment with 25µM nanoliposomal C6-ceramide (Fig. 2-3A(i)).Treatment with
varying concentrations of nanoliposomal C6-ceramide for 24 hours significantly
decreased GAPDH protein expression (Fig. 2-3A(ii)). Based upon these
preliminary studies, we chose to evaluate the physiological and therapeutic
consequences of C6-ceramide-mediated GAPDH reduction at 24 hours as a
function of concentration. It was also apparent that GAPDH protein expression
was decreased following treatment with C6-ceramide nanoliposomes in primary
CLL cells (Fig. 2-3B). In addition, using qRT-PCR analysis it was determined that
mRNA expression of GAPDH in JVM3 cells was decreased after treatment with
nanoliposomal C6-ceramide (Fig. 2-3C). Treatment with nanoliposomal C6-

54
ceramide did not decrease GAPDH protein expression in non-transformed cells
from normal donors (Fig. 2-3D), which again alludes to the preferential specificity
of nanoliposomal C6-ceramide treatment for cancer cells. Reduction in GAPDH
protein preceded induction of cell death. GAPDH reduction was observed as
early as 8 hours after treatment with nanoliposomal C6-ceramide, whereas C6-
ceramide nanoliposomes induced cell death 12 hours after treatment (data not
shown). This indicated that C6-ceramide-induced decrease in GAPDH played a
critical role to induce cell death
A variety of glycolytic genes are ubiquitously overexpressed in human
cancers and are thought to contribute to enhancement of glycolysis [151]. The
differential expression of glycolytic enzymes in CLL was previously unknown, so
we sought to investigate the expression of GAPDH in CLL. The basal protein
level of GAPDH was compared between normal donor PBMC (n=8) and PBMC
isolated from CLL patients (n=14). CLL patients were further stratified according
to their white blood count (WBC) count at the time their plasma was collected.
Patients with WBC counts >50,000 cells/µL were categorized as having high
WBC count (n=5), while patients with counts <50,000 cells/µL were in the low
WBC count group (n=9). It was evident that GAPDH protein expression was
significantly overexpressed in the CLL patient population with high WBC count
(Fig 2-3E). Moreover, treatment with 25µM C6-ceramide nanoliposomes was
significantly more cytotoxic in the subset of CLL patients with the higher WBC
count (Fig 2-3F). Collectively, our data suggest that nanoliposomal C6-ceramide

55
is targeting GAPDH and presents a new mechanism for which ceramide is
selectively targeting cancer cells.
Figure 2-3. C6-ceramide nanoliposomes target GAPDH in CLL. A). JVM3 cells were treated
with (i) 25µM of ghost or C6-ceramide nanoliposomes for varying times, (ii) varying doses of

56
ghost or C6-ceramide nanoliposomes for 24 hours, then Western Blot analysis was performed for
GAPDH. Densitometry analysis from replicate experiments (n=4) was performed via ImageJ
software. In Fig. A(ii) the representative blot demonstrates a decrease in GAPDH at 12.5µM C6-
ceramide, a result not observed in the other two distinct replicate experiments. The protein levels
are also quantified in the graphs. * P < 0.05; ** P <0 .005. B). CLL patient cells (n=3, analyzed
individually) were treated with different doses of ghost or C6-ceramide nanoliposomes for 24
hours, then Western Blot analysis was performed for GAPDH. * P < 0.05. The blot represents the
effect of C6-ceramide nanoliposomes on one patient sample, however, the graph is an average of
the effect on all three patient cells. C). JVM3 cells were treated with different doses of ghost and
C6-ceramide nanoliposomes for 24 hours, then qRT-PCR analysis was performed for expression
of GAPDH mRNA. Expression was normalized to 18S. * P < 0.05. D). PBMC from normal donors
(n=3) were treated with 25 µM ghost or C6-ceramide nanoliposome for 24 hours and then
Western Blot analysis was performed for GAPDH. E). Basal protein expression of GAPDH was
determined via Western Blot analysis on PBMC isolated from normal donors (n=8) or from CLL
patients with either a lower WBC count (n=9) or higher WBC count (n=5). Patients with WBC
counts >50,000 cells/µL were identified as patients with high WBC count. * P < 0.05. F). CLL
patient cells with either a lower WBC count (n=4) or higher WBC count (n=3) were treated with
25µM ghost or C6-ceramide nanoliposomes for 2, 16 and 24 hours then an MTT assay was
performed * P < 0.05.
C6-ceramide targets GAPDH-dependent glycolysis
To further demonstrate that C6-ceramide was targeting the glycolytic
pathway, consistent with our observation of a decrease in GAPDH expression,
we investigated the effects of C6-ceramide upon lactate and ATP production.
Treatment with varying doses of nanoliposomal C6-ceramide for 24 hours
caused a significant decrease in lactate concentration (Fig. 2-4A) and a dose-
dependent decrease in ATP production (Fig. 2-4B) in JVM3 cells. 3-
bromopyruvate, a glycolytic inhibitor was used as a positive control in the ATP
production experiment. To demonstrate that decreased ATP production was
dependent upon ceramide-reduced GAPDH expression we utilized a lentiviral

57
approach to decrease GAPDH in JVM3 cells (Fig. 2-4C inset). We observed a
comparable decrease in ATP production between JVM3 cells treated with
GAPDH shRNA and 25 µM C6-ceramide nanoliposomal treated JVM3 cells. C6-
ceramide nanoliposomal treatment further reduced ATP production in GAPDH-
silenced JVM3 cells (Fig. 2-4C). Taken together, these results suggest that
ceramide decreased ATP production and it may be partially GAPDH-dependent.
Increasing levels of intracellular ceramide correlates with downregulation
of nutrient transporters and leads to cellular starvation [148, 163]. We wanted to
determine if ceramide was also targeting glucose transport in addition to
targeting the GAPDH glycolytic enzyme. Results from a tritiated glucose uptake
assay showed no significant difference between JVM3 cells treated with ghost or
C6-ceramide nanoliposomes (Fig. 2-4D). A significant decrease was observed,
however, in cells treated with cytochalasin B, a potent inhibitor of glucose
transport. Moreover, protein expression of the glucose transporter, GLUT1, was
not altered after treatment with nanoliposomal C6-ceramide (Fig 2-4E). Further
investigation into other glycolytic enzymes, like lactate dehydrogenase (LDH) and
pyruvate kinase M2 (PKM2), revealed no difference between the ghost- and C6-
ceramide nanoliposome treated groups (data not shown).
To illustrate that cell death induced by nanoliposomal C6-ceramide was
dependent on targeting of the glycolytic pathway, we pre-treated JVM3 cells with
pyruvate, the ultimate downstream product of glycolysis and determined its effect
on cell viability and ATP production. Pre-treatment of cells with 10 mM pyruvate
led to a significant rescue of cell viability (Fig. 2-5A) and ATP production (Fig. 2-

58
5B) after C6-ceramide nanoliposome treatment. To further demonstrate that
reduction in GAPDH protein played a critical role in C6-ceramide-induced cell
death, we overexpressed GAPDH in JVM3 cells using a lentiviral transduction
overexpression system (Fig. 2-5C). We showed by Western blotting that
transduction with lentiviral particles containing the GAPDH gene (Lenti-GAPDH)
resulted in overexpression of GAPDH protein (Fig. 2-5C insert). Surprisingly, the
procedure itself (control lentiviral particles containing RFP gene, Lenti-RFP) also
elevated GAPDH protein expression (Fig. 2-5C insert). In both cases,
nanoliposomal C6-ceramide was less effective in inducing cell death when
GAPDH protein was elevated. Taken together, overexpression of GAPDH
significantly rescued cell death in JVM3 cells after treatment with nanoliposomal
C6-ceramide (Fig. 2-5C).
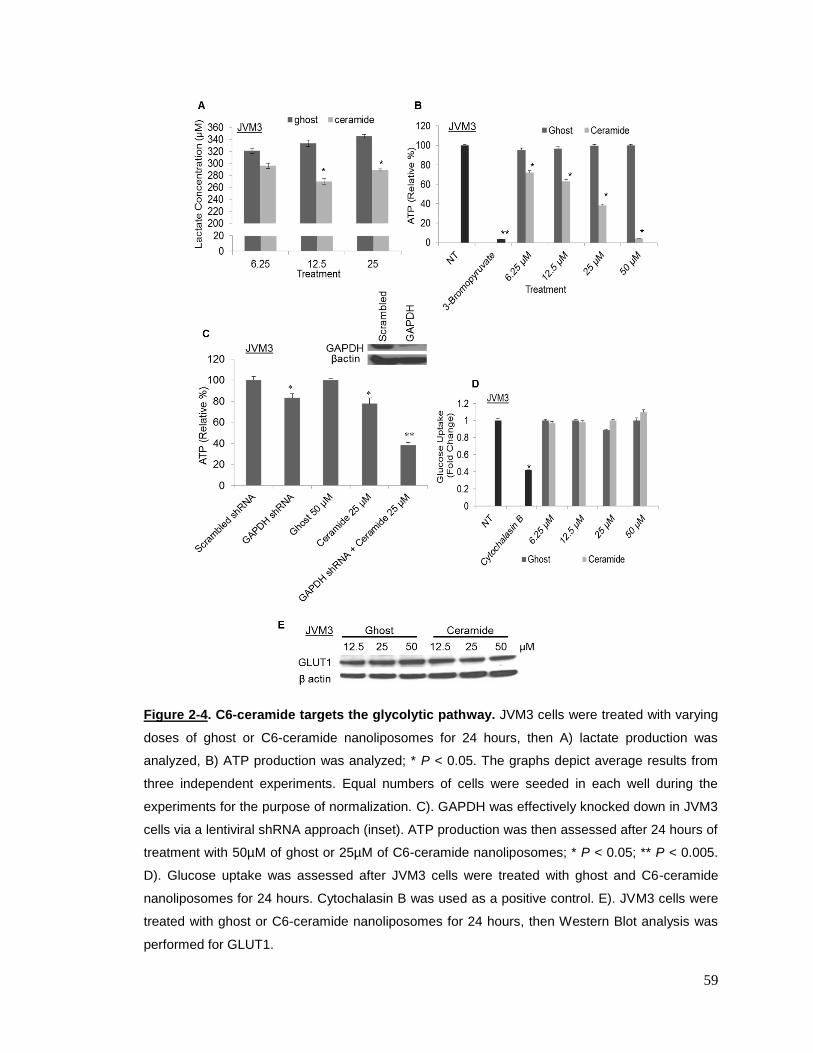
59
Figure 2-4. C6-ceramide targets the glycolytic pathway. JVM3 cells were treated with varying
doses of ghost or C6-ceramide nanoliposomes for 24 hours, then A) lactate production was
analyzed, B) ATP production was analyzed; * P < 0.05. The graphs depict average results from
three independent experiments. Equal numbers of cells were seeded in each well during the
experiments for the purpose of normalization. C). GAPDH was effectively knocked down in JVM3
cells via a lentiviral shRNA approach (inset). ATP production was then assessed after 24 hours of
treatment with 50µM of ghost or 25µM of C6-ceramide nanoliposomes; * P < 0.05; ** P < 0.005.
D). Glucose uptake was assessed after JVM3 cells were treated with ghost and C6-ceramide
nanoliposomes for 24 hours. Cytochalasin B was used as a positive control. E). JVM3 cells were
treated with ghost or C6-ceramide nanoliposomes for 24 hours, then Western Blot analysis was
performed for GLUT1.

60
Figure 2-5. Pharmacological and molecular confirmation that nanoliposomal C6-ceramide
targets the glycolytic pathway at the level of GAPDH. A). JVM3 cells were pre-treated for 2
hours with 10mM pyruvate, then treated with ghost or C6-ceramide nanoliposomes for 24 hours.
MTT cell viability assay was performed. B). ATP production in these cells was also determined;

61
The results are significant between cells treated with ceramide liposomes and cells treated with
ceramide and pyruvate, ** P < 0.005. C). JVM3 cells were transduced with lentiviral particles
overexpressing GAPDH or viral particles expressing RFP (control). Post transduction, Western
blotting analysis was done to determine levels of GAPDH in experimental and control cells
(insert). Cells were treated with ghost or C6-ceramide nanoliposomes for 24 hours. Cell viability
was determined using MTT assay and AlamarBlue assay; * P < 0.005.
Nanoliposomal C6-ceramide displays anti-leukemic effect in CLL animal
model
Given that we have demonstrated selective induction of cell death utilizing
nanoliposomal ceramide in CLL cells in vitro, we next investigated this
therapeutic approach in an in vivo model. Moreover, this model would allow us to
confirm the selective actions of ceramide upon GAPDH-dependent glycolysis in
vivo. Loisel et al. have reported the establishment of a novel human CLL like
xenograft model in nude mice using JVM3 cells [169]. We evaluated the ability of
C6-ceramide nanoliposomes to inhibit tumor growth of leukemic CLL cells in
xenografts in this immunodeficient mouse model. C6-ceramide nanoliposomes
decreased tumor growth beginning on day 13 of treatment (Fig. 2-6A). In
addition, C6-ceramide treated tumors did not progress for the remainder of the
study. Immunoblot analysis showed that C6-ceramide, but not ghost
nanoliposomes, significantly reduced GAPDH protein expression at day 8, 14
and 17 (Fig. 2-6B). ImageJ densitometry analysis for multiple animals is shown,
as well as a representative blot from Day 17 (Fig. 2-6B inset).
Collectively, these results indicate that bioactive ceramide analogues can
be incorporated into pegylated nanoliposomal formulations and elicit potent anti-

62
leukemic efficacy in a mouse model of CLL. In addition, these results
substantiate our previous in vitro data that cell death induction after treatment
with C6-ceramide nanoliposomes involves targeting of GAPDH and the glycolytic
pathway in CLL.
Figure 2-6. Nanoliposomal C6-ceramide displays anti-leukemic effect in a CLL animal
model. A). Two weeks after ten million JVM3 cells were inoculated in the right flank of female
Balb/c Nu/nu mice, animals were treated with 40 mg/kg ghost (n=8) or C6-ceramide (n=8)

63
nanoliposomes via tail vein injection. Treatment regimen was every other day over a three week
period of time. Tumor size was assessed every other day; * P < 0.05; ** P < 0.005. B). Leukemic
mice following an identical dose regimen were sacrificed on day 8 (n=5), day 14 (n=6) and day 17
(n=6) of C6-ceramide treatment and on day 17 for ghost treatment. Immunoblot analysis for
GAPDH protein expression was performed on tumor tissues. A representative blot from day 17 is
shown (Fig. 6B inset); * P < 0.05.
Discussion
The present study identifies dysregulated glucose metabolism as a novel
target in CLL. Furthermore, we found that treatment with C6-ceramide
nanoliposomes led to preferential induction of caspase-independent, necrotic cell
death in CLL cells in vitro. Several investigations support a caspase-
independent, necroptotic cell death mechanism for chemotherapeutics
(tamoxifen, fludarabine) that generate endogenous ceramide [137, 170]. In
addition, short chain ceramides induce necrotic cell death independent of
caspase 3 activation in B cell lymphomas [36]. Several groups, including ours
have studied the metabolism of exogenous short chain ceramides in cancer cells.
Exogenous C6-ceramide is metabolized into natural ceramides through de-
acylation to yield sphingosine followed by subsequent re-acylation with various
fatty acids [171]. Exogenous C6-ceramide is also metabolized to C6-
sphingomyelin, C6-glucosylcreamide and cerebrosides [112, 171, 172]. The
predominant metabolic pathway of exogenous C6-ceramide is specific to the
cancer cell type and concentration of C6-ceramide delivered to the cells.
We observed that treatment of CLL cells with nanoliposomal C6-ceramide
decreased protein and mRNA expression of GAPDH and based upon initial

64
experiments (Fig. 3A) we chose to evaluate the physiological consequences of
ceramide-mediated decrease in GAPDH at 24 hours as a function of various
concentrations of ceramide. GAPDH is known to mediate glycolysis and is
responsible for oxidizing glyceraldehyde-3-phosphate to 1,3-biphosphoglycerate.
Until recently, GAPDH has been considered as a constitutive housekeeping gene
and was used as a control for normalizing changes in expression of protein and
genes. Current studies suggest that GAPDH is upregulated in many cancers and
is the primary target of some chemotherapeutic drugs [166]. The tumor
suppressor TP53 has been shown to increase GAPDH expression in endothelial
cells [173] and significant increases in GAPDH expression were also observed in
breast cancer cells stimulated with growth factors [174]. Interestingly, GAPDH
plays a key role in opposing caspase-independent cell death and promoting
cellular survival [167]. It has been suggested that GAPDH overexpression may
assist cancer cells in evading apoptosis and cell death mechanisms [166]. This
protection from cell death is mediated by an elevation in glycolysis and enhanced
ATP levels [168]. Moreover, it has been demonstrated that overexpression of
GAPDH is sufficient to protect chronic myelogenous leukemia (CML) cells from
imatinib-induced caspase-independent cell death and knockdown of GAPDH
resensitizes cells to imatinib-induced cell death [175]. Therefore, combining a
drug that reduces GAPDH expression (like ceramide) with imatinib could prove to
have therapeutic benefit in overcoming this drug resistance.
In addition to its role in the glycolytic pathway, GAPDH has also been
shown to serve other functions in the cell which remain to be elucidated.

65
Although generally in a cytosolic form, the GAPDH protein can also be found in
the nucleus and nuclear functions of GAPDH include transcriptional regulation,
DNA repair, and maintenance of telomere structure [176]. There is rapid
shortening of telomere length when cells are treated with exogenous, short-chain
ceramides [177]. Ogretmen and colleagues demonstrated that overexpression of
GAPDH results in protection of telomeric DNA in response to exogenous
ceramide treatment [177]. Further analysis showed that ceramide was inhibiting
nuclear localization of GAPDH and showed that a potential mechanism of
ceramide-mediated shortening of telomeres in somatic cells leads to cell
senescence while maintenance of telomeres is associated with immortality of
cancer cells. These data suggest another mechanism by which increased
GAPDH expression contributes to a survival advantage in cancer cells.
Elevation of glucose uptake and glycolysis in cancer cells can depend
heavily on the upregulation of glucose transporters [178]. Others have shown
that nutrient transporter down-regulation is critical in ceramide-induced cell death
[148], so we sought to clarify this hypothesis in CLL. Using a radiolabeled
glucose uptake assay, we demonstrated that the ability of ceramide to induce cell
death in CLL was not due to blocking nutrient transport of glucose into the cell. In
addition, expression of the GLUT1 glucose receptor was not regulated by
ceramide. This lack of effect upon nutrient transport further suggests direct
effects upon glycolytic enzymes overexpressed in cancer.
We identified GAPDH as being overexpressed in a population of CLL
patients. In addition to the Western blot analysis, we also re-analyzed previous

66
microarray data from CLL patients [179] using Gene Expression Omnibus (GEO)
to investigate dysregulation of glycolytic enzymes in CLL. Interestingly, we found
that all of the enzymes in the glycolytic pathway were upregulated in the CLL
patients with a poor prognosis (data not shown). Based on our results and the
independent microarray data, we conclude that glycolysis appears to be
upregulated in a subset of CLL patients with either higher white blood cell counts
and/or a more aggressive clinical course.
Our findings demonstrate that the cytotoxicity of C6-ceramide is due to
targeting of glucose metabolism in CLL. Previous studies reported that inhibitors
of ATP production, like bromopyruvate, effectively induce cell death in cancer
[180]. The abolishment of cellular ATP production proves effective due to the
increased dependence on glycolysis and ATP production in oncogenic cells.
Moreover, treatment with koningic acid, an inhibitor of GAPDH selectively
induces cell death in cancer via ATP deprivation [181]. In a similar fashion, we
also show that nanoliposomal C6-ceramide treatment induces cell death in CLL
through targeting of the glycolytic pathway and results in decreased ATP and
lactate production. Even though our studies indicate that shRNA knock down of
GAPDH reduced ATP production similar to 25 µM C6-ceramide nanoliposomal
treatment, the fact that ceramide augmented this shRNA approach suggests that
ceramide may be targeting the Warburg effect through both decreased GAPDH
as well as an undefined secondary mechanism.
To better understand and confirm if inhibition of the glycolytic pathway was
the means by which ceramide was inducing cell death in CLL, we carried out

67
experiments that involved pre-treatment with a final end product of glycolysis. It
was determined that pre-treatment of CLL cells with pyruvate was sufficient to
rescue ATP depletion and cell death that was otherwise induced after treatment
with the C6-ceramide nanoliposome. In addition, we transiently overexpressed
GAPDH in CLL cells to demonstrate rescue of C6-ceramide-induced cell death in
these cells. Interestingly, we also observed rescue of cell death in control cells
transduced with control viral particles. Yet, protein analysis showed that both, the
experimental and control cells overexpressed GAPDH. This alludes to the fact
that transient overexpression of GAPDH partially protects CLL cells from C6-
ceramide-induced cell death.
Confirming in vitro studies, we provide clear evidence that
nanoliposomal C6-ceramide demonstrated in vivo efficacy via tumor growth
inhibition in a murine xenograft model of CLL. We are aware of a recently
described mouse model for CLL generated by co-injection of primary human CLL
cells and T cells into NOD/ SCID mice, but have chosen to focus our studies on
the mouse xenograft model because we can obtain tissue for ex vivo Western
analysis of GAPDH protein levels [182]. In our in vivo studies we observed that
treatment with nanoliposomal C6-ceramide effectively decreased tumor burden
without systemic side effects. This is consistent with previous studies from our
laboratory and others which show that this C6-ceramide nanoliposome
formulation results in tumor regression in animal models of cancer and
hematological malignancies [144, 147, 159]. In addition, it was expected that this
nanoliposomal formulation would be relatively non-toxic to the animals, as it was

68
previously selected by the Nanotechnology Characterization Laboratory (National
Cancer Institute) for extensive toxicology and stability testing. (Detailed
information on the toxicology studies of the “Ceramide Liposomes” can be found
at http://ncl.cancer.gov/working_technical_reports.asp). In vivo studies also
confirm that ceramide is targeting GAPDH in CLL, as protein isolated from tumor
tissue showed an overall decrease in GAPDH expression following treatment
with C6-ceramide nanoliposomes.
In conclusion, we show C6-ceramide nanoliposomes preferentially inhibit
the altered metabolism of glucose in leukemic cells via downregulation of
GAPDH, resulting in induction of necrotic cell death. We provide the first
evidence that GAPDH is overexpressed in a subset of CLL patients. Our findings
provide a metabolic explanation for the increased sensitivity and selectivity of
cancer cells to ceramide. Taken together, these results suggest that
nanoliposomal C6-ceramide could be an effective novel therapy for patients
whose cancer cells overexpress GAPDH, including those with CLL.

69
CHAPTER 3: STAT3 mediates nanoliposomal C6-ceramide-induced cell death in chronic lymphocytic leukemia
Introduction
Chronic lymphocytic leukemia (CLL) is a slow growing B cell malignancy
and the most prevalent form of adult leukemia in the Western world [125]. It is
characterized by the clonal expansion and accumulation of mature B
lymphocytes in the bone marrow, peripheral blood and often the lymph nodes
[136]. Leukemic B cells express CD5, CD19, CD20 and CD23 on the surface and
are characterized by cell-cycle arrest in the G0/G1 phase and resistance to
programmed cell death [136, 183]. Depending on the degree of somatic
hypermutation and chromosomal abnormalities, the clinical course of CLL ranges
from an indolent and slow progressing disease to rapid disease progression [125,
136]. The standard treatment regimen for CLL includes a combination chemo-
immunotherapy with fludarabine, cyclophosphamide and rituximab with an overall
response rate of approximately 90% and complete remission of 72% [132, 133].
Despite these advances in therapeutics, eventual drug resistance and relapse
ultimately cause CLL to be an incurable and chronic disease with a need for
novel therapies [136].
Sphingolipids, a class of complex cellular lipids, function as structural
components in cellular membranes and as signal transducers regulating cellular
processes like growth, proliferation, response to stress, differentiation,
senescence, autophagy and apoptosis [2, 12]. Ceramide has been termed as a
“tumor-suppressor sphingolipid” because of its ability to potentiate signaling

70
cascades that lead to cell death [184]. Sphingolipid-based cancer therapeutics
are thus gaining increasing attention due to their critical role in cancer initiation,
progression, pathogenesis and metastasis. Sphingolipid-based cancer therapies
aim to perturb the native sphingolipid balance towards anti-proliferative and
apoptotic signaling by elevating levels of intracellular ceramide [18]. Our
laboratory has demonstrated that a nanoliposomal formulation of short chain C6-
ceramide (CNL) is an effective anti-tumorigenic agent in vitro and leads to tumor
regression in vivo in several animal models of cancer [35, 39, 110, 111, 147, 159,
185]. Specifically, we have demonstrated that CNL selectively targets the
Warburg effect in CLL cells by causing downregulation of glyceraldehyde 3-
phosphate dehydrogenase and elicits tumor regression in an in vivo murine
model of CLL [39]. There is also evidence that another short chain ceramide
analog, C2-ceramide, induces non-apoptotic cell death in CLL cells [137].
Interestingly, treatment with fludarabine, a chemotherapeutic agent often used as
first line of treatment for CLL also causes a 2.5 – 3 fold increase in intracellular
ceramide levels 6 hours after treatment and inhibiting accumulation of
intracellular ceramide prevents fludarabine-induced apoptosis [55, 137].
Altogether, these evidence suggest that accumulation of endogenous pro-
apoptotic ceramide species or delivery of pro-apoptotic exogenous ceramide is
induces cell death in CLL cells. Thus, CNL could potentially be an effective anti-
tumorigenic agent for CLL.
Signal transducer and activators of transcription (STAT) are latent
transcription factors that translocate to the nucleus upon activation by receptor or

71
non-receptor tyrosine kinases. The STAT signal transduction pathway plays a
critical role in hematopoietic biology because it mediates the response to a
multitude of cytokines [186]. Among the seven STAT members, STAT3 and
STAT5 have been studied extensively for their role in cancer progression [187].
Specifically in CLL, a report in 1997 demonstrated constitutive phosphorylation of
serine 727 (S727) but not tyrosine 705 (Y705) in STAT1 and STAT3 [188].
However, studies addressing the physiologic significance of this modification
have been conducted only in the last few years. It has been shown STAT3 that is
constitutively phosphorylated at S727 also has the ability to bind DNA and
activate transcription in CLL cells independent of Y705 phosphorylation [189]. In
line with the newly found function of STAT3 phosphorylated at S727 (p-STAT3-
S727) as a regulator of the respiratory chain in mitochondria, it has been
reported that p-STAT3-S727 in CLL cells associates with complex I of the
respiratory chain and imparts viability and stress protection to CLL cells [190,
191]. STAT3 also regulates micro-RNA expression in CLL cells [192].
Interestingly, unphosphorylated STAT3 also promotes CLL cell survival and
proliferation by activating the NF-ΚB pathway [193]. Additionally, ample evidence
in the literature demonstrates the critical role of STAT3 in mediating pro-survival
interactions between CLL cells and the microenvironment [194, 195]. STAT3
inhibitors have shown to sensitize CLL cells to apoptosis and also reverse IL6-
induced resistance to histone deacetylase inhibitors [196, 197]. Taken together, it
is clearly evident that the critical role of STAT3 in CLL pathogenesis makes it a
promising therapeutic target in this disease.

72
In this study, we sought to identify the molecular basis of CNL-induced cell
death in CLL. We demonstrate that CNL specifically suppresses phosphorylation
of STAT3 at S727 and Y705 residues, leading to a reduction in the transcriptional
activity of STAT3 and a subsequent downregulation of critical anti-apoptotic
proteins like Mcl-1 and survivin. Additionally, we have identified the upstream
kinases that are suppressed by CNL, eventually leading to reduction in STAT3
phosphorylation. CNL suppresses the activity of Bruton’s tyrosine kinase (BTK),
mitogen-activated protein kinase kinase (MEK) and protein kinase C (PKC)
resulting in a suppression of STAT3 phosphorylation. These findings collectively
indicate that CNL can potentially be an effective therapy for CLL. Our work
reports two novel targets of CNL. Firstly, these results demonstrate an effect of
CNL on BTK, a critical kinase mediating the B-cell receptor signaling in CLL cells.
Furthermore, the current use of ibrutinib, a BTK inhibitor, in the clinic for CLL
patients imparts clinical relevance to this finding. Secondly, we also demonstrate
an inhibitory effect of CNL on STAT3 phosphorylation.
Materials and Methods
Reagents
Antibodies specific for STAT3, p-STAT3-S727, p-STAT3-Y705, Mcl-1, Ran,
STAT1, p-STAT1-Y701, p-STAT1-S727, STAT2, p-STAT2-Y690, STAT5, Akt-
S473, BTK, p-BTK-Y223, p-ERK, ERK, p-MARCKS, MARCKS, survivin, XIAP,
cyclin D1, p21 and β-actin were purchased from Cell Signaling Technology Inc.
(Beverly, MA). For Western blotting, 2-12% or 12% precasted Nupage
electrophoresis gels were purchased from Invitrogen (Carlsbad, CA) and

73
chemiluminescence reagent was obtained from Thermo Scientific (Waltham,
MA). STAT3 inhibitor - Stattic, MEK inhibitor - U0126 and PKC inhibitor - Bis-I
were purchased from Sigma (St. Louis, MO). BTK inhibitor, ibrutinib was
purchased from MedChem Express (Monmouth Junction, NJ).
Patient characteristics and preparation of peripheral blood mononuclear
cells
All patients met the clinical criteria of CLL and were not on treatment at the time
of sample acquisition. Peripheral blood specimens from CLL patients were
obtained and informed consents signed for sample collection using a protocol
approved by the Institutional Review Board of Penn State Hershey Cancer
Institute. CLL PBMCs were chosen according to the following criteria: CD19+ >
80%, CD20+ > 80%, CD5+ > 90%. These criteria ensured that the PBMCs
isolated from CLL patient blood predominantly consisted of leukemic B-cells.
Buffy coats from normal donors were also obtained from the blood bank of the
Milton S. Hershey Medical Center, Pennsylvania State University, College of
Medicine. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-
Hypaque gradient separation, as described previously [160].
Cell culture
Culture of freshly isolated PBMCs and primary CLL patient cells was carried out
using RPMI-1640 medium supplemented with 10% fetal bovine serum (both from
Invitrogen). JVM-3 cells (DSMZ – German Collection of Microorganisms and Cell
Cultures, Braunschweig, Germany), a CLL cell line with wild type p53, were also

74
cultured in this same medium and cells were grown in 5% CO2 at 37˚C. Mec-2
cells (DSMZ – German Collection of Microorganisms and Cell Cultures,
Braunschweig, Germany), a CLL cell line with mutated p53 were cultured in
Iscove's MDM media supplemented with 10% FBS. HEK-293FT cells (Invitrogen,
Waltham, MA) were cultured in D-MEM media supplemented with 10% FBS and
1X Anti-anti antibiotic (Gibco, Waltham, MA).
Preparation of nanoliposomal ceramide
Preparation of nanoliposomal ceramide has been described in Chapter 2 of the
dissertation.
Preparation of lipid:BSA complexes
Lipids were dried under a stream of nitrogen and then resuspended in DMSO.
The lipid/DMSO solution was then added to a fatty acid-free BSA solution to
achieve a final concentration of 1 mM lipid, 1 mM BSA in 20 mM HEPES with
10% DMSO. Complexes were allowed to form by rocking at room temperature for
30 min, followed by sonication to clarity.
Cell viability assay
A set of experiments were conducted to determine the toxicity of the CNL and
Stattic in JVM-3 cells, Mec-2 cells, CLL patient cells and in normal donor PBMC.
Cell viability was performed using CellTiter 96® Aqueous One Solution assay kit
(Promega) and relative viable cell number was determined by reading the plates
at 490 nm wavelength in Synergy HT Multi-Detection Microplate Reader (Bio-

75
TEK). All samples were assayed in triplicate and each experiment was repeated
at least three times.
Cell death assays (Flow cytometry for AnnexinV/7AAD)
Apoptosis was determined in JVM-3 cells, Mec-2 cells and in CLL patient cells by
2-color flow cytometry with Annexin-V-PE and 7-amino-actinomycin D (BD
Pharmingen Transduction Laboratories) staining using 5 x 105 cells per sample.
Necrosis was quantidied in the Annexin V positive and 7AAD positive quadrant.
Data were collected by flow cytometry.
Western blot analysis
Western blot analysis was performed on whole-cell lysates collected using RIPA
buffer (Sigma). Blots were washed and developed with enhanced
chemiluminescence (Thermo Scientific, Waltham, MA) following the
manufacturer’s instructions. Densitometry analysis was performed using ImageJ
software.
shRNA Knockdown of STAT3
STAT3 shRNA plasmid clones (Human pTRIPZ vector) were purchased from
Open Biosystems (Huntsville, AL) and used to transfect JVM-3 cells.
Nucleofection was performed using the Amaxa Nucleofector I device. JVM-3
cells (3 × 106 ), resuspended in 100μl of Cell line Solution Kit V (Amaxa,
Cologne, Germany) with 6μg of shRNA were transfected with the Amaxa
Nucleofector I device (program X-001), cultured in six-well plates in complete

76
medium for 24, 48, 72 and 96 hours and then examined for STAT3 knockdown
using Western blot analysis. Cells were also analyzed for percent viability using
flow cytometric analysis for AnnexinV and 7AAD at 24, 48, 72 and 96 hours after
nucleofection.
Preparation of pervandate
1mM pervanadate stock was prepared by adding 10µL of 100mM sodium
orthovanate, 0.3% hydrogen peroxide diluted in 20mM HEPES and 940µL of
water. After 5 minutes of incubation, a small amount of catalase was mixed in the
pervanadate stock to remove excess hydrogen peroxide. The pervanadate was
used within 2 hours of preparation.
Luciferase reporter assay
Cignal reporter assay kit from Qiagen (Hilden, Germany) was used for obtaining
plasmids for the luciferase reporter assay. JVM-3 cells (2x106 cells) were
transfected with 4µg of either reporter construct, negative control construct or
positive control construct using the Amaxa Nucleofector I device (X-001
program). Cell were allowed to be in culture for 24 hours post transfection and
then treated for 12 hours with CNL or ghost liposomes. Dual-Glo luciferase assay
system from Promega (Madison, WI) was used to obtain luciferase
luminescence. The assay and quantification was done following the
manufacturer’s instructions.

77
Lentiviral transduction for STAT3C overexpression
Human EF.STAT3C.Ubc.GFP vector from Addgene (Cambridge, MA) was used
for expressing STAT3-C in JVM-3 cells [32]. Briefly, viral particles were produced
in HEK293-FT cells using pLOC, VSVG, tat and DR8.2 plasmids and JVM-3
cells were transduced thrice with the viral media according to the manufacturer’s
protocol. JVM-3 cells were grown for 72 hours after the last transduction. The
STAT3-C overexpression vector has an EGFP sequence as a selectable marker
and the transduced cells were sorted for EGFP and grown as a pure population
(JVM3-STAT3C cells). The cells were then harvested for experiments. Human
pLOC overexpression vector (Open Biosystems) containing a RFP sequence
was used as a negative control. Cells were treated with CNL or ghost liposomes.
JVM-3 cells were transduced thrice with media containing the negative control
virus. FACS was not performed for the control JVM3-RFP cells since we
obtained approximately 70-80% transduction efficiency. 72 hours after the last
transduction, JVM3-RFP cells were harvested for experiments. JVM3-STAT3C
cells and JVM3-RFP cells were treated with 20µM and 40µM CNL and ghost
liposomes for 24 hours. 24 hours after treatment, cells death was analyzed using
flow cytometric analysis using AnnexinV-V450 and 7AAD staining.
Statistical analysis
All data are expressed as mean plus or minus SEM. Paired Student t test (2-tail
paired) and 2-way analysis of variance test were used to determine the statistical

78
significance and P value of 0.05 or less was considered statistically significant.
All results are a mean of three independent biological triplicates unless specified.
Results
STAT3 is a potential therapeutic target in CLL
Several reports suggest that STAT3 might play a role in the pathogenesis of
CLL [189, 190, 197]. We compared levels of total STAT3 between normal cells
and CLL cells. STAT3 was highly overexpressed in both, CLL cell lines and
patient cells in comparison to peripheral blood mononuclear cells (PBMC)
obtained from normal blood donors (Fig. 3-1A). Next, we evaluated if inhibiting
STAT3 signaling in CLL cells would induce cell death. Knock down of STAT3 in
JVM3 cells by using an inducible lentiviral STAT3-shRNA significantly increased
the number of Annexin V positive cells 24 hours after doxycycline induction (Fig.
1B). An average of 57% knockdown of STAT3 protein was observed 24 hours
after induction (Fig. 3-1B). Doxycycline was non-toxic to JVM3 cells at doses
used for induction. STAT3 knockdown caused a 108% increase in cell death 24
hours after doxycycline induction as compared to control cells. As the
knockdown weakened at later time points, cell death induction reduced to 42%
48 hours after doxycycline induction, and cell death was further reduced as the
protein levels resumed to normal levels 72 and 96 hours post doxycycline
induction (Fig. 3-1B). Reduction in STAT3 protein levels also corresponded with
a simultaneous reduction in downstream proteins like Mcl-1 that are under the
transcriptional control of STAT3. We also confirmed the role of STAT3 signaling
in CLL cell viability by using a pharmacological inhibitor of STAT3 called Stattic.

79
Stattic is a non-peptidic small molecule selective inhibitor of STAT3 [198]. It
inhibits STAT3 signaling by selectively inhibits activation, dimerization, and
nuclear translocation of STAT3 [198]. We observed that treatment with Stattic for
24 hours caused a dose-dependent reduction in cell viability in two different CLL
cell lines (Fig. 3-1C (i) and (ii)). JVM-3 is a CLL cell line with wild-type p53, while
Mec-2 cells have mutated p53. Furthermore, we tested three different CLL
patient samples and observed a similar reduction in cell viability after treatment
with Stattic, whereas PMBCs from normal blood donors were resistant to
treatment with Stattic (Fig. 3-1C (iii)). Taken together, these results demonstrate
that STAT3 is essential for CLL cell survival. STAT3 signaling inhibited by either
molecular or pharmacological interventions reduced cell viability and induced
death in CLL cells.
Figure 3-1A. STAT3 is overexpressed in CLL cell lines and patient cells. JVM-3 cells, Mec-2
cells, PBMCs from normal blood donors (n=2) and PBMCs from CLL patients (n=4) were lysed
and protein extracted. Western blot analysis was one to determine the level of total STAT3 in the
samples`

80
Figure 3-1B. Knockdown of STAT3 induces cell death in CLL cells. JVM-3 cells
were transfected with several clones of STAT3 shRNA and flow cytometric analysis was
performed to determine the % dead cells 24-96 hours after induction. Western blot
analysis was done at the same time points to determine the knockdown of STAT3 and
the levels of Mcl-1, a protein regulated by the transcriptional activity of STAT3. Cells
nucleofected with TE buffer containing no plasmid were used as a control. 1µg/mL
doxycycline was used to induce the expression of STAT3 shRNA 24 hours after
nucleofection. This concentration was non-toxic to cells. The results are a mean of two
independent triplicates. Students t-test was used for statistical analysis, *** p < 0.0001,
** p < 0.05.

81
Figure 3-1C. STAT3 inhibition reduces viability of CLL cell lines and patient cells.
(i) JVM-3 cells were treated with increasing concentrations of Stattic for 24 hours and
cell viability was determine after 24 hours treatment. Western blotting analysis for p-
STAT3-Y705, p-STAT3-S727 and total STAT3 was performed to confirm the
effectiveness of 24 hours treatment with Stattic. (ii) Mec-2 cells and (iii) PBMCs from

82
normal donors (PBMC n=2) and CLL patients (CLL n=3) were treated with Stattic for 24
hours and cell viability was determined after treatment.
CNL suppresses the phosphorylation of STAT3 at both tyrosine-705 (p-
STAT3-Y705) and serine-727 (p-STAT3-S727) residues in CLL cells
To identify the mechanism of CNL-induced cell death in CLL, we examined
the effect on STAT3 phosphorylation. JVM-3 cells were treated with CNL or
ghost nanoliposomes (negative control) for 24 hours and Western blotting was
performed to detect changes in protein phosphorylation. While the levels of total
STAT3 protein did not change significantly with CNL treatment, phosphorylation
of STAT3 was significantly down regulated. p-STAT3-S727 and p-STAT3-Y705
were suppressed by 45% and 67% respectively after treatment with 40µM CNL
for 24 hours (Fig. 3-2A (i)). Several studies have reported that only S727 residue
of STAT3 is constitutively phosphorylated in CLL [188, 189]. However, we
observed constitutive phosphorylation at both S727 and Y705 sites in both CLL
cell lines and primary cells. This can be attributed to increased activity of
upstream kinases like cAbl that contribute to STAT3 phosphorylation [199]. We
observed a similar trend on STAT3 phosphorylation suppression after CNL
treatment in Mec-2 cells, although after longer treatment with CNL (Fig. 3-2A (ii)).
Similarly, 7 CLL patient cells were treated with 40µM CNL or ghost
nanoliposomes for 24 hours and evaluated for STAT3 phosphorylation.
Consistent with the above results, we observed suppression in STAT3
phosphorylation at both the residues in 6 out of 7 patient samples (Fig. 3-2B).
Although we observed a slight reduction in total STAT3 levels in some patient

83
cells after CNL treatment, the overall change in total STAT3 did not reach
statistical significance in most patient cells. We also tested the effect on CNL
treatment on HEK293 cells. CNL treatment did not affect the viability of HEK293
cells (Fig. 3-2C (i)), nor did it affect the phosphorylation of STAT3 (Fig 3-2C(ii)),
thereby demonstrating that this phenomenon is specific to cancer cells.
Figure 3-2A. CNL suppresses the phosphorylation of STAT3 in CLL cell lines. (i)
JVM3 cells were treated with 20µM and 40µM of ghost nanoliposomes and CNL for 24

84
hours. Western blotting analysis was performed to determine protein levels before and after
treatment. The graphs represent the quantification of Western blotting. Statistical analysis
was performed using Student’s t-test, * p < 0.05, ** p < 0.01(ii) Mec-2 cells were treated
with 40µM nanoliposomes for 48hours and 72 hours and protein levels were determined
using Western blotting analysis.
Figure 3-2B. CNL suppresses phosphorylation of STAT3 in CLL patient cells. CLL
patient cells (n=7) were treated with 40µM of ghost nanoliposomes and CNL for 24 hours.
Western blotting analysis was performed to determine protein levels before and after
treatment. The graphs represent the quantification of Western blotting. Statistical analysis
was performed using Student’s t-test, * p < 0.05.

85
Figure 3-2C. CNL does affect cellular viability & STAT3 phosphorylation in HEK293
cells. (i) Cell viability of HEK293 cells was determined after 24 hour treatment with ghost
nanoliposomes and CNL. (ii) Western blotting analysis was performed to determine levels
of STAT3 phosphorylation in HEK293 cells after 24 hours treatment with ghost
nanoliposomes and CNL.
Suppression of STAT3 phosphorylation is specific to STAT3 and C6-
ceramide
We next evaluated the specificity of CNL-induced suppression of STAT3
phosphorylation. In addition to STAT3, STAT1 is also constitutively
phosphorylated on S727 in CLL patient cells [188]. Although strong evidence
supporting the role of STAT1 in CLL is lacking, some investigations have
reported that fludarabine and JAK kinase inhibitors induce apoptosis in CLL cells
and inhibit STAT1 signaling, thereby suppressing the ability of the cells to
respond to growth signals, interferons and cytokines [200, 201]. STAT1
activation is also critical for differentiation of CLL cells in response to Byrostatin 1
[202]. These reports raise a possibility that STAT1 might play a role in
pathogenesis of CLL by impacting the anti-apoptotic signals in CLL. We

86
observed that treatment with CNL does not significantly affect the
phosphorylation of STAT1 (Fig. 3-3A). The other STATs were either not
constitutively active (STAT2 and STAT5) or were not expressed in JVM-3 cells
(STAT4). We next examined if suppression of STAT3 phosphorylation was
specific to C6-ceramide sphingolipid. We tested three other sphingolipids:
dihydro-C6-ceramide, an inactive analog of C6-ceramide; sphingosine and
sphingosine-1-phosphate. None of the other sphingolipids tested decreased
STAT3 phosphorylation (Fig. 3-3B), thereby demonstrating the specificity of C6-
ceramide. All together, these results prove the specificity of CNL-induced
suppression of STAT3 phosphorylation in CLL.
Figure 3-3A. CNL-induced suppression of phosphorylation is specific to STAT3. JVM-3
cells were treated with 40µM ghost nanoliposomes and CNL for 24 hours and Western blotting
analysis was done. A positive control of STAT2 phosphorylation was also used.

87
Figure 3-3B. Suppression of STAT3 phosphorylation is specifically an effect of CNL and
not other sphingolipids. JVM-3 cells were treated with dihrdro-C6-ceramide nanoliposomes or
BSA:sphingosine complex or BSA:S1P complex for 24 hours. Western blotting analysis was
performed.
CNL induces necrotic cell death in CLL cells
We have previously demonstrated that CNL selectively induces caspase
3/7-independent cell death in CLL cells [39]. Using phase contrast microscopy
we had shown that CLL cells treated with CNL resembled a necrotic morphology
[39]. Here, we confirm these findings using flow cytometric analysis for Annexin V
and a viability dye, 7-AAD. Several reports suggest that necrotic cell death is
quantified in the Annexin V-7AAD double positive quadrant [203, 204]. Necrotic
cell death was induced in both cell lines in a dose-dependent and time-
dependent manner after treatment with CNL (Fig. 3-4A (i) and (ii)). Under the
same conditions, ghost nanoliposomes did not have effect on cell death. Recent
evidence suggests that CLL patients with p53 pathway dysfuntion have poor
prognosis due to reduced response to conventional chemotherapies [133, 205].
We observed that cell death in p53mutated Mec-2 cells was induced after a longer

88
treatment with CNL as compared to p53wild-type JVM-3 cells (Fig. 3-4A). This
preliminary evidence demonstrating the effectiveness of CNL treatment to induce
cell death in Mec-2 cells presents a potential treatment strategy for p53mutated B
malignancies that are in urgent need of better therapeutic approaches. We also
evaluated the effect of CNL on CLL patient cells obtained from 7 different
patients. Treatment with 40µM CNL for 24 hours induced cell death in 5 out of
the 7 patients tested (Fig. 3-4B). Taken together, these results indicate that CNL
increases cell death in both p53mutated and p53wild-type CLL cells. To determine if
suppression of phosphorylation preceded induction of cell death, we examined
phosphorylation levels at early time points after CNL treatment. Significant
necrotic cell death in JVM3 cells was observed 12 hours after treatment with CNL
(Fig 3-4C (i)). However, suppression of STAT3 phosphorylation at both the
residues started about 6 hours after CNL treatment and thus preceded induction
of cell death (Fig. 3-4C (ii)). This suggests that reduction in STAT3
phosphorylation might mediate CNL-induced cell death. Consistent with this, we
also observed suppression of STAT3 phosphorylation in 3 CLL patient cells after
12 hours of treatment with CNL (Fig. 3-4D). Overall, these results indicate that
CNL suppresses STAT3 phosphorylation and this might mediate cell death in
CLL cells.

89
Figure 3-4A. CNL induces necrotic cell death in CLL cell lines (p53 wt and p53 mutated). (i)
JVM-3 cells that have wild type p53 and (ii) Mec-2 cells that have mutated p53 were treated with
20µM and 40 µM ghost nanoliposomes and CNL for indicated time periods. Flow cytometric
analysis for Annexin V and 7AAD stating was performed to determine effect on cell death. Two-
way ANOVA was used to perform statistical analysis * p < 0.01.
Figure 3-4B. CNL induces necrotic cell death in CLL patient cells. CLL patient cells (n=7)
were treated with 40 µM ghost nanoliposomes and CNL for 24 hours. Flow cytometric analysis for
Annexin V and 7AAD stating was performed to determine effect on cell death. Student’s t test
was used to perform statistical analysis ** p < 0.01.

90
Figure 3-4C. CNL-induced suppression of p-STAT3 precedes induction of cell death (i)
JVM-3 cells were treated with ghost nanoliposomes and CNL for indicated time periods and flow
cytometric analysis was performed to determine % cell death. (ii) JVM-3 cells were treated with
40µM ghost nanoliposomes and CNL for indicated time periods and Western blotting was
performed for to determine levels of p-STAT3. Graphical representation of protein levels is also
shown. Statistical analysis was done using Student’s t-test * p < 0.05.

91
Figure 3-4D. CNL-induces early time point suppression of p-STAT3 in CLL patient cells.
CLL patient cells (n=3) were treated for 12 hours with 40µM ghost nanoliposomes or CNL and
Western blotting was done.
CNL suppresses Bruton’s tyrosine kinase (BTK), mitogen-activated protein
kinase kinase (MEK) and protein kinase C (PKC) activity to suppress STAT3
phosphorylation
Suppression in STAT3 phosphorylation can be a result of CNL-induced
activation of downstream phosphatases and/or CNL-induced suppression in
upstream kinases. We studied the effect of CNL on downstream phosphatases
and upstream kinases to determine which enzymes predominantly mediate CNL-
induced suppression in STAT3 phosphorylation. Okadaic acid (OA) was used as
an inhibitor of serine/threonine phosphatases PP1 and PP2A. As shown in Fig.
3-5A (i), pretreatment with OA did not rescue CNL-induced-suppression of p-
STAT3-S727, indicating that suppression of phosphorylation is not a result of
CNL-induced activation of serine/threonine phosphatases. Pretreatment with OA
rescued p-Akt-S473 after CNL treatment, suggesting that the inhibitor was
functional in inhibiting PP2A and PP1. Similarly, we observed that CNL treatment

92
had no effect on the activity of tyrosine phosphatases. Pervanadate (PV) was
used as a functional inhibitor of tyrosine phosphatases. As demonstrated in Fig.
3-5A (ii), pretreatment with PV did not rescue CNL-induced-suppression of p-
STAT3-Y705, indicating that this event is independent of the action of tyrosine
phosphatases. The basal levels of p-STAT3-Y705 increased after pretreatment
with PV confirming that PV was effective in inhibiting tyrosine phosphatases.
Figure 3-5A. CNL does not activate phosphatases. (i) JVM-3 cells were pretreated with 5nM
okadaic acid (OA) for 2 hours, followed by 12 hours of treatment with ghost nanoliposomes and
CNL. (ii) JVM-3 cells were pretreated with 50µM pervanadate (PV) for 2 hours, followed by 24
hours treatment with 40µM ghost nanoliposomes and CNL. Both the inhibitors were non-toxic to
cells at the specific concentration. Western blotting was performed.
After investigating the effect of CNL on downstream phosphatases, we
studied the effect of CNL on upstream kinases. We used two strategies to
determine the effect of CNL on upstream kinases. Firstly, we performed western
blot analysis to study the effect of CNL on the activating phosphorylation of the
kinase or to look at the phosphorylation pattern of the immediate downstream
target of the kinase. These results act as a surrogate for determining the effect

93
on the kinase activity of the enzyme. Additionally, we also treated cells with an
inhibitor of the kinase to determine the direct effect of kinase inhibition on STAT3
phosphorylation. One of our criteria for screening was to obtain inhibition of
kinase activation at early time points after treatment with CNL, which would
thereby indicate that kinase activity suppression preceded suppression in STAT3
phosphorylation. Taken together, these two strategies answer the question of the
effect of CNL on upstream kinases.
We observed that CNL suppresses the activity of BTK, a tyrosine kinase
critical in mediating BCR signaling in CLL cells. As shown in Fig. 3-5B, we
observed a significant reduction in phosphorylation of BTK at Y223 in JVM-3
cells as early as 4-6 hours after treatment with CNL but not ghost liposomes. The
levels of total BTK remained unchanged after treatment. BTK phosphorylation at
Y223 is an activating phosphorylation and is necessary of full activation of BTK
[206, 207]. CNL-induced inhibition of BTK is an exciting observation since BTK is
a promising target in CLL and a BTK inhibitor, ibrutinib, is currently used in the
clinic for CLL therapy [208]. Furthermore, we also observed that treatment with
ibrutinib significantly reduced p-STAT3-Y705, but not p-STAT3-S727 (Fig. 3-5C).
Taken together, these results imply that CNL-induced BTK inhibition mediates
suppression of p-STAT3-Y705.

94
Figure 3-5B. CNL suppresses the activity
of BTK. JVM-3 cells were treated with 40µM
ghost liposomes and CNL for indicated time
periods and Western blotting was performed.
Figure 3-5C. BTK inhibitors suppress phosphorylation of STAT3. JVM-3 cells were treated
with BTK inhibitor, ibrutinib for 6 hours and Western blotting was performed to determine protein
levels. Graphical representation of the Western blot is also shown. Student’s t-test was used to
perform statistical analysis, * p < 0.05.
We also observed that CNL suppresses MEK activity, a serine/threonine
kinase in CLL cells. As shown in Fig. 3-5D, only CNL treatment significantly
suppressed phosphorylation of Erk, a direct downstream target of MEK at early
time points after addition of the liposomes. Furthermore, treatment with U0126, a
MEK inhibitor reduced p-STAT3-S727 and p-STAT3-Y705 in both JVM-3 (Fig. 3-

95
5E (i)) cells and CLL patient cells (Fig. 3-5E (ii)). We also observed that CNL
suppresses PKC activity. CNL treatment, but not ghost liposomes, significantly
suppressed phosphorylation of MARCKS, a direct downstream target of PKC at
early time points, while total MARCKS levels remained unchanged (Fig. 3-5F).
Additionally, treatment with BisI, a PKC inhibitor also suppressed p-STAT3-S727
and p-STAT3-Y705 levels in both JVM-3 cells and CLL patient cells (Fig. 3-5G (i)
and (ii)).It was confounding to obtain inhibition of p-STAT3-Y705 after treatment
with U0126 and BisI, both of which are inhibitors of serine/threonine kinases.
However, some results in the literature have demonstrated that MEK inhibitors
like U0126 and PKC inhibitors like BisI inhibit p-STAT1-Y701 [202]. Since STAT1
and STAT3 pathways share similar upstream signaling, we speculate that MEK
and PKC might also play a role in phosphorylating STAT3-Y705. We conclude
that CNL-induced suppression in phosphorylation of STAT3 is not mediated by
downstream phosphatases, but instead by inhibition of upstream kinases that
include BTK, MEK and PKC.
Figure 3-5D. CNL suppresses the activity of MEK1/2 kinase. JVM-3 and Mec-2 cells were treated with 40µM ghost liposomes and CNL for indicated time periods and Western blotting was performed.

96
Figure 3-5E. MEK1/2 inhibitors suppress phosphorylation of STAT3. (i) JVM-3 cells were
treated for 6 hours and 12 hours with 10µM U0126 and Western blotting was performed to
evaluate protein levels. Blots were probed for p-Erk to confirm that U0126 suppressed MEK
activity. Graphical representation of the blots is also shown. (ii) CLL cells (n=3) were treated for
12 hours with 10µM U0126 and Western blotting was performed to evaluate protein levels.
Graphical representation of the blots is also shown. Student’s t-test was used for statistical
analysis, * p < 0.05.

97
Figure 3-5F. CNL suppresses the
activity of PKC. JVM-3 cells were
treated with 40µM ghost liposomes
and CNL for indicated time periods
and Western blotting was
performed.
Figure 3-5G.
PKC inhibitor
suppress
phosphorylati
on of STAT3
(i) JVM-3 cells
were treated
for 6 hours and
12 hours with
5µM Bis-I and
Western
blotting was
performed to
evaluate
protein levels.
Graphical
representation
of the blots is
also shown.
(ii) CLL cells
(n=3) were
treated for 12
hours with 5µM
Bis-I and Western blotting was performed to evaluate protein levels. Graphical representation of
the blots is also shown. Student’s t-test was used for statistical analysis, * p < 0.05.

98
CNL suppresses the transcriptional activity of STAT3
Having established that CNL suppresses STAT3 phosphorylation, we next
sought to determine if CNL suppressed the transcriptional activity of STAT3.
CNL treatment caused a significant downregulation of STAT3-regulated proteins
like Mcl-1, survivin, XIAP, cyclin D1 and p21 in both JVM-3 and Mec-2 cells (Fig
3-6A). CNL-induced downregulation of critical anti-apoptotic proteins like Mcl-1,
survivin and XIAP was encouraging since ample evidence in the literature has
established that these anti-apoptotic proteins are critical mediators of CLL cell
survival. Mcl-1 levels correlate with poor disease prognosis and chemoresistance
[209, 210]. Additionally, Mcl-1 inhibitors induce apoptosis in CLL cells, thus
making it a relevant therapeutic target [211]. Similarly, survivin levels have been
correlated to poor prognosis in CLL and targeting survivin with a small molecule
inhibitor effectively induces apoptosis in the proliferative subset of CLL [212,
213]. Abnormally high expression of XIAP has also been observed in CLL, thus
conferring resistance from apoptosis in CLL cells [214]. Additionally, XIAP
inhibitors synergize with death receptor ligand tumor necrosis factor–related
apoptosis-inducing ligand (TRAIL) and anti-CD40 therapies such as CD154 gene
therapy to induce apoptosis in CLL [214, 215]. As shown in Fig 3-4C (ii), CNL-
induced suppression of STAT3 phosphorylation started about 6 hours after
treatment. This event preceded reduction in Mcl-1 levels which started
approximately 6-8 hours after CNL treatment (Fig 3-6B). We confirmed these
results using a luciferase reporter assay, wherein we observed a significant
dose-dependent reduction in luciferase units after 12 hours treatment with CNL

99
(Fig. 3-6C). This suggests that CNL suppresses the transcriptional activity of
STAT3.
Figure 3-6A. CNL reduces the
levels of STAT3-regulated
genes. JVM-3 cells and Mec-2
cells were treated with 20µM or
40µM ghost liposomes or CNL
and Western blotting was
performed. JVM3 cells were
treated for 24 hours and Mec-2
cells were treated with 48 hours.
Figure 3-6B. Reduction of STAT3 phosphorylation precedes reduction of Mcl-1 levels
following CNL treatment. JVM-3 cells were treated with 40µM ghost liposomes or CNL for
indicated time periods and Western blotting was performed for determining Mcl-1 levels.

100
Figure 3-6C. CNL inhibits expression of luciferase in a STAT3 luciferase reporter assay.
JVM-3 cells were transfected with different luciferase constructs. 12 hours after transfection, cells
were treated with ghost nanoliposomes or CNL for 12 hours and luciferase assay was performed
according to the manufacturer’s protocol.
Overexpression of STAT3-C rescues CNL-induced cell death in CLL cells
To confirm the role of STAT3 in CNL-induced cell death, we
overexpressed STAT3-C, an oncogenic form of STAT3 that mimics STAT3
dimers and thus acts as a constitutively active STAT3 [216]. Overexpression was
obtained using lentiviral transduction and cells expressing STAT3-C. The STAT3-
C overexpression vector has an EGFP sequence as a selectable marker and the
transduced cells were sorted for EGFP and grown as a pure population (JVM3-
STAT3C cells). STAT3-C expression was confirmed by expression of the FLAG-
tag in JVM3-STAT3C cells (Fig. 3-7A inset). Moreover, as shown in Fig 4-7A
inset, JVM3-STAT3C cells have a higher expression of Mcl-1, thus indicating
higher STAT3 transcriptional activity. An overexpression vector expressing RFP

101
was used as a control for the study (JVM3-RFP cells). Wild type JVM-3 cells
were also used as a negative control (WT-JVM3). As shown in Fig. 3-7A, WT-
JVM3 cells undergo necrotic cell death on treatment with CNL for 24 hours.
Similarly, JVM3-RFP cells undergo cell death after CNL treatment. However,
cells expressing STAT3-C were significantly more resistant to treatment with
CNL as compared to WT-JVM3 and JVM3-RFP cells. Since STAT3-C
overexpressing cells were more resistant to CNL-induced cell death, we
concluded that STAT3 mediates CNL-induced cell death in CLL cells. We also
determined STAT3 phosphorylation levels in JVM-3 xenograft tumors obtained from
a subcutaneous CLL mouse model in Balb/c Nu/nu mice injected with ghost
nanoliposomes (n=1) or CNL (n=2) [39]. Consistent with our in vitro data, we observed a
significant reduction of STAT3 phosphorylation in the xenograft tumors injected with CNL
(Fig. 3-7B). Thus CNL reduces STAT3 phosphorylation in vitro and in vivo.

102
Figure 3-7: A) STAT3-C expressing cells are resistant to CNL-induced cell death. Lentiviral
transduction was performed to express STAT3-C in JVM-3 cells. 72 hours after the last
transduction, FACS was performed to obtain a pure population of cells expressing STAT3-C and
JVM-3
A.
B.JVM-3 xenograft
tumors treated
with:

103
the treatments were done. An overexpression construct expressing RFP was used as a negative
control. 72 hours after the last transduction cycle, cells were treated with ghost liposomes and
CNL for 24 hours. Expression of STAT3-C was confirmed by Western blotting and probing for
Flag-tag. Flow cytometric analysis for Annexin V and 7AAD was performed to quantitate %
necrotic cells. Student’s t-test was used for statistical analysis, * p < 0.05. B) In vivo
confirmation of CNL-induced suppression in STAT3 phosphorylation. JVM-3 xenograft
tumors were obtained from a subcutaneous CLL mouse model in Balb/c Nu/nu mice that were
injected with ghost nanoliposomes (n=1) or CNL (n=2) (from Ryland LK, PLoS One, 8(12)).
Western blotting was performed to determine the levels of STAT3 phosphorylation.
Discussion
In the present study, we have identified STAT3 as a molecular mediator of
CNL-induced cell death in CLL. Using p53wild-type JVM-3 cells, p53mutated Mec-2
cells, cells from CLL patients and JVM-3 xenograft tumors from a murine model
of CLL, this study presents in vitro and in vivo confirmation for CNL-induced
suppression of STAT3 phosphorylation. Reduction in STAT3 phosphorylation is
followed by reduction in levels of critical anti-apoptotic proteins like Mcl-1,
survivin and XIAP, and eventually leading to cell death. The only study in the
literature investigating the effect of ceramide on STAT1 and STAT3
demonstrates that exogenous ceramide or accumulation of endogenous
ceramide in human fibroblasts induces STAT activation through tyrosine
phosphorylation of STAT1 and STAT3 via JAK2, MEK/ERK, JNK and p38
kinases [216]. However, the relation between ceramide and STAT3 in context of
cancer cells has never been studied. A large body of work has delineated the
signaling cascades that are targeted by endogenous or exogenous ceramide to
exert its pro-apoptotic effects in cancer cells. Some of these targets include AKT,

104
ERK, survivin, phospholipase D, p38 MAPK and death receptor to name a few
[18]. This study demonstrates the inhibitory effect of CNL on STAT3 signaling by
suppressing STAT3 phosphorylation. Consistent with some reports in the
literature, we have convincingly demonstrated that STAT3 is a potential
therapeutic target in CLL.
Several studies have reported that STAT3 is constitutively phosphorylated
on S727 and not Y705 in CLL [188, 189]. However, we observed constitutive
phosphorylation of both S727 and Y705 residues of STAT3 in both CLL cell lines
and patient cells. Although the studies mentioned above did not use JVM-3 cells
or Mec-2 cells for their investigations, our observations of dual phosphorylation
may be attributed to increased activity of upstream kinases like cAbl that
contribute to STAT3 phosphorylation at the tyrosine residue as speculated by
Allen et al. [199]. We have demonstrated that CNL suppresses STAT3
phosphorylation at both S727 and Y705 as early as 6 hours to 9 hours after
treatment, thereby preceding induction of cell death. Since this phenomenon
occurs relatively early after treatment with CNL, we believe STAT3 mediates
CNL-induced cell death in CLL. We observed that CNL-induced STAT3
dephosphorylation suppresses protein levels of critical anti-apoptotic proteins like
Mcl-1, survivin and XIAP that are essential for CLL cell proliferation and
resistance to apoptosis, thus confirming that CNL suppresses the transcriptional
activity of STAT3. Results from the STAT3 luciferase reporter assay confirm that
CNL suppresses the transcriptional activity of STAT3 even at early time points,

105
thereby indicating that CNL-induced suppression in anti-apoptotic proteins are
effectors of cell death, rather than just a consequence of cell death.
We have identified that CNL inhibits the activity of BTK, MEK and PKC
that leads to suppression of STAT3 phosphorylation. CNL-induced inhibition of
BTK is an exciting observation since BTK is a promising target in CLL. Multiple
reports have demonstrated that BTK is a critical kinase for CLL development
[217]. BTK inhibitor, ibrutinib, is currently used in the clinic for CLL therapy [208].
We have also demonstrated that CNL inhibits the kinase activity of MEK and
PKC. PKC has also been shown to be important for CLL development and it is
believed that PKC inhibitors may be an attractive option for CLL treatment [218].
We performed rescue experiments to definitely confirm that STAT3
mediates CNL-induced cell death. Since STAT3 is expressed at very high levels
in CLL, it was difficult to overexpress wild type STAT3 in JVM-3 cells. Hence, we
expressed STAT3-C, a constitutively dimerized (activated) form of STAT3 which
is produced via substitution of amino acid residues 661 and 663 near the C
terminus with cysteine. Stat3C monomers form disulfide bonds with each other,
leading to constitutive dimerization [219]. Several publications have
demonstrated that STAT3-C is oncogenic in nature [219, 220]. We observed that
cells expressing STAT3-C were more resistant to CNL-induced cell death as
compared to wild type JVM-3 and JVM-3 cells expressing a control vector.
STAT3-C expression thus rendered partial protection to CNL-induced cell death,
thereby confirming that STAT3 mediates CNL-induced cell death in CLL.

106
We have previously demonstrated that ceramide targets the Warburg
effect in cancer cells [39]. Ceramide causes a decrease in the protein levels of
GAPDH, a glycolytic enzyme resulting in decreased glycolysis. Pretreating CLL
cells with pyruvate, the end product of glycolysis rescued CNL-induced cell death
and CNL-induced ATP depletion. Thus, targeting GAPDH is one mechanism by
which CNL inhibits aerobic glycolysis [39]. In the present study we have
demonstrated that CNL suppresses STAT3 phosphorylation, resulting in
subsequent inhibition of STAT3 transcriptional activity. Our preliminary studies
have demonstrated that GAPDH may be partially regulated by the activity of
STAT3, which establishes a potential link between CNL-induced suppression in
STAT3 phosphorylation and CNL-induced inhibition of Warburg effect. Another
possible link may be established on the basis of evidence provided by Demaria
et al [221]. The authors have shown that STAT3 acts as a master regulator of cell
metabolism by activating HIF-1α-dependent aerobic glycolysis [221]. Activated
STAT3 upregulates HIF-1α expression, which in turn induces the transcription of
different genes involved in glycolysis [221]. In addition to modulating the levels of
HIF-1α, STAT3 can cooperate with HIF-1α by binding to its responsive
promoters, ensuring the formation of a transcriptionally active complex. It is
believed that STAT3 and HIF-1α form a feed forward loop that leads to enhanced
aerobic glycolysis [222]. Thus, we speculate that CNL-induced suppression in
STAT3 phosphorylation might suppress HIF-1α expression and subsequently
suppress HIF-1α-dependent aerobic glycolysis. This is another mechanism by

107
which CNL-induced suppression in STAT3 phosphorylation may be linked to
suppression in the Warburg effect.
The enhanced function of constitutive p-STAT3-Y705 as a transcription
factor contributing to oncogenic processes has been extensively studied in
multiple tumor cells. Recently, an extranuclear pro-oncogenic role of constitutive
p-STAT3-S727 was uncovered, giving rise to the concept of protein moonlighting
in STAT3. It was reported that STAT3 associates with complex I and II of the
electron transport chain and is required for optimal mitochondrial respiration
[191]. Additionally, phosphorylation at the S727 of mitochondrial STAT3 was
identified to be essential for its mitochondrial function and for Ras-dependent
oncogenic transformation [223]. Following this revelation, reports have
documented the pivotal role of mitochondria-associated constitutive p-STAT3-
S727 in pathogenesis of breast cancer, pancreatic cancer, murine
myeloproliferative neoplasms and also CLL [190, 224-226]. Having established
the ability of CNL to suppress p-STAT3-S727 in CLL, it will be interesting to look
at the effect on phosphorylation of the mitochondrial STAT3, effect on electron
transport chain and overall mitochondrial respiration in CLL.
This work is significant because it is the first body of evidence
demonstrating that CNL suppresses STAT3 phosphorylation in cancer and that
STAT3 mediates CNL-induced cell death. As far as our knowledge, we are the
first to report that CNL inhibits BTK activity, thereby suppressing BTK signaling.
This work thus opens up a wide avenue of research directed towards examining
ceramide-STAT3 relation in other cancer models and developing novel

108
combination therapeutics that demonstrate synergism between CNL and STAT3
inhibitors, especially in STAT3-dependent cancers like CLL.

109
CHAPTER 4: Effect of nanoliposomal C6-ceramide on mitochondrial bioenergetics and mitochondrial STAT3 in chronic
lymphocytic leukemia
Introduction
The role of STAT3 as a transcription factor is well established. Upon
ligand binding to the receptor, an intracellular cascade is initiated which begins
by activation of Janus kinase (JAK) residing on the receptor’s cytoplasmic
domain. Activation of JAK is followed by recruitment and subsequent JAK-
mediated phosphorylation of STAT proteins on a specific tyrosine residue.
Phosphorylated STATs form homo or heterodimers, translocate to the nucleus,
bind to specific DNA sequences and regulate transcription of downstream genes.
STATs thus act to transmit transcriptional signals from cell surface to the nucleus
[227]. The tyrosine residue essential for STAT activation is conserved amongst
all STATs. Several reports have also identified the existence of other receptor
and non-receptor tyrosine kinases, predominantly from the Src family, that
phosphorylate the tyrosine residue and activate STATs [228]. In addition to the
conserved tyrosine, all STATs excluding STAT2 also have a conserved and
phosphorylable serine residue at the C-terminal end. Serine kinases upstream of
STATs include MAPK, PKC, NLK and IKKε [229]. The role of serine
phosphorylation as an inhibitor or activator of the transcriptional activity of STATs
is ambiguous and is cell type-dependent and stimulus-dependent [229].
Recent evidence has uncovered a novel cellular localization and a novel
noncanonical function of STAT3. Studies have revealed that STAT3 also

110
localizes in the mitochondria and controls cell respiration and metabolism [191].
Specifically, mitochondrial STAT3 has been shown to promote oxidative
phosphorylation and enhance activity of complex I and complex II of the electron
transport chain (ETC) [191]. Phosphorylation of STAT3 at serine 727 (S727) is
essential for this function and the mitochondrial function of STAT3 is independent
of its function as a transcriptional factor in the nucleus [191]. Specifically in
context of cancer, phosphorylation of STAT3 S727 in the mitochondria is
essential for Ras-expressing cells to generate subcutaneous tumors in nude
mice. The authors observed that in addition to increasing the activity of electron
transport complex II, STAT3 expression also enhanced the activity of ATP
synthase, and unexpectedly, increased lactate dehydrogenase and decreased
mitochondrial membrane potential [223]. Thus mitochondrial STAT3
phosphorylation mediates cellular transformation by enhancing aerobic
glycolysis, ETC activity, ATP abundance and inhibiting overproduction of ROS.
The role of mitochondrial p-STAT3-S727 in cellular transformation has been
confirmed in several malignancies like CLL, prostate cancer, breast cancer and
myeloproliferative neoplasms [222]. One report has reported that the MEK-ERK
pathway phosphorylates mitochondrial STAT3 on S727 and is necessary for
RAS-mediated transformation [230]. Gene associated with retinoid interferon
induced cell mortality 19 (GRIM 19), a complex I subunit imports STAT3 into the
mitochondria and recruits it to complex I of ETC. Furthermore, S727 of STAT3 is
critical for STAT3-GRIM19 interaction [231]. In addition to mitochondrial STAT3,
even nuclear STAT3 has been linked to energy metabolism and cell

111
transformation. Demaria et al. reported that in MEFs expressing the STAT3C
form, nuclear STAT3 triggered Hif-1α-mediated induction of aerobic glycolysis
and Hif-1α-independent down-regulation of mitochondrial activity [221]. STAT3
tyrosine phosphorylation is critical for this metabolic switch since inhibition of
STAT3 tyrosine phosphorylation downregulated glycolysis prior to growth arrest
and cell death [221].
Multiple reports have shown that STAT3 is constitutively phosphorylated
at S727 residue in CLL cells. However, the molecular function of p-STAT3-S727
overactivation and its role in the pathogenesis of the disease was unknown.
Capron C et al. have reported that p-STAT3-S727 localizes to the mitochondria
and associates with complex I of the ETC in CLL B cells but not normal B cells.
The authors also identified that the glutathione-dependent antioxidant pathways
protected CLL cells against high cellular ROS levels and maintained p-STAT3-
S727 overactivation, thereby mediating stromal protection and viability of CLL
cells. Taken together, the authors concluded that overactivation of p-STAT3-
S727 provided stress protection and viability to CLL cells and correlated with CLL
cell resistance to apoptosis [232]. More recently, researchers have focused on
studying the metabolism of CLL cells to understand the effect of
microenvironment on the disease and to identify potential therapeutic targets.
CLL cells display oxidative stress and are characterized by have increased
mitochondrial ROS production and adaptation to intrinsic oxidative stress by
antioxidant pathways [233]. Furthermore, it has been reported that unlike other
malignant cells, CLL cells have increased oxidative phosphorylation but not

112
increased glycolysis and have increased mitochondrial biogenesis that might
contribute to CLL oncogenesis [233]. Thus targeting the respiratory chain and
promoting mitochondrial ROS can be potential targets for therapy. However, the
microenvironment, specifically the stromal cells cause a glycolytic switch in CLL
cells in a Notch-c-myc signaling-dependent manner, thereby making glucose
metabolism a therapeutically exploitable target [131].
In this study, we aim at evaluating the effects of CNL on mitochondrial
structure, function and mitochondrial bioenergetics of CLL cells. Ample evidence
in the literature has reported the effect of ceramides on mitochondrial integrity
and function. We aim at extending these studies in CLL cells and evaluating the
role of CNL-induced suppression in STAT3 phosphorylation in perturbing
mitochondrial function and cellular bioenergetics.
Methods and Materials
Reagents
Antibodies specific for STAT3, p-STAT3-Y705, p-STAT3-S727, cytochrome c,
cyclooxygenase IV, tubulin and β-actin were purchased from Cell Signaling
Technology Inc. (Beverly, MA). For Western blotting, 12% precasted Nupage
electrophoresis gels from Invitrogen (Carlsbad, CA), and chemiluminescence
reagent from Amersham Biosciences Inc. (Piscataway, NJ) were obtained.

113
Patient characteristics and preparation of peripheral blood mononuclear
cells
Patient characteristics and preparation of peripheral blood mononuclear cells
have been described in detail in Chapter 3 of the dissertation.
Cell culture
Cell culture methods have been described in Chapter 3 of the dissertation.
Preparation of nanoliposomal ceramide (CNL)
Preparation of nanoliposomal ceramide has been described in Chapter 2 of the
dissertation.
Cell viability assay
Cell viability assays were performed as described in Chapter 2 of the
dissertation.
Assessment of mitochondrial membrane potential (∆Ψm):DiOC6 Staining
To evaluate ∆Ψm, JVM-3 cells and CLL patient primary cells were treated with
ghost or CNL at a concentration of 25 µM for 24 hours. This dose was selected
based on studies in Chapter 2 of the thesis. Fifteen minutes prior to the end of
incubation, cells were labeled with DiOC6 (40 nM in PBS) at 37°C as described
previously [234]. After washing, cells were analyzed by flow cytometry.

114
Determination of ROS Production
Dihydroethidium (DHE) is a lipophilic cell-permeable dye that can undergo
oxidation to ethidium bromide in the presence of superoxide, and, to a lesser
extent, hydrogen peroxide and hydroxyl radicals. Ethidium then binds irreversibly
to the double-stranded DNA, causing amplification of a red fluorescent signal,
indicating ROS production [235]. DHE fluorescence was analyzed by flow
cytometry (excitation 488 nm and emission 585 nm).
Confocal Studies
To verify accumulation of C6-ceramide in JVM-3 cells, we formulated a
nanoliposomal C6-ceramide vesicle with 10 mol% NBD-C6 ceramide as a marker
for C6. Cells were plated at 2.0 × 104/well in 8-well chamber slides and allowed to
grow overnight. Nanoliposomal-NBD-C6 ceramide was treated at 25μM for a 2-
hour treatment period. Cellular nuclei were counterstained with 4′,6-diamidino-2-
phenylindole, and MitoTracker Deep Red 633 (Molecular Probes) was used as a
marker for mitochondria following the manufacturer's instructions. C6-ceramide
delivery and accumulation was evaluated by confocal microscopy at 63×
magnification (Leica Microsystems).
Extracellular flux assay
Bioenergetics of JVM-3 cells was determined using the XF96e Extracellular Flux
Analyzer (XF Cell Mito Stress Test Kit, Seahorse Bioscience, North Billerica,
MA). Cells were seeded in specialized tissue culture plates at a concentration of
600,000 cells per well in XF media with 25mM glucose onto a Cell-Tak (BD

115
Biosciences)-coated XF96 plate. Oligomycin (1 μM), carbonyl cyanide 4-
(trifluoromethoxy) phenyl hydrazone (2 μM), rotenone (1 μM), and antimycin (1
μM) were added sequentially, and measurements of extracellular flux were
recorded. Glycolytic capacity was calculated from extracellular acidification rate
(ECAR) values (an indicator for lactic acid production or glycolysis) after the
inhibition of ATP synthase by the addition of oligomycin, when cells resort to
meeting their energy demands using their maximum glycolytic capacity. ECAR
data were analyzed using Wave software. All experiments were performed in at
least quadruplets.
Mitochondrial Protein isolation
Mitochondrial protein isolation was performed using the Mitochondria Isolation kit
for cultured cells (Thermo Scientific, Waltham, MA). 20 million cells per sample
were used for isolation of the mitochondrial protein. Cells were added in reagent
A and incubated on ice for 2 minutes. 10µL of reagent B was added and cells
were vortexed. Reagent C was then added to the cells and after a series of
centrifugations, the supernatant contained cytosolic proteins, while the pellet
contained intact mitochondria. To extract mitochondrial proteins, the pellet was
washed in Reagent C and resuspended in 2% CHAPS in TBS. Mitochondrial
protein was obtained after a short centrifugation spin. Protein assay (DC protein
assay, Biorad) was performed to determine the protein concentration of cytosolic
and mitochondrial proteins.

116
Statistical analysis
All data are expressed as mean plus or minus SEM. Paired Student t test (2-tail
paired) and 2 way analysis of variance test were used to determine the statistical
significance and P value of 0.05 or less was considered statistically significant.
Results
CNL increases mitochondrial ceramide, intracellular ROS and decreases
mitochondrial membrane potential
Mitochondrial damage and dysfunction are associated with programmed cell
death and it has been demonstrated that ceramide accumulates and acts on the
mitochondria to initiate antiproliferative effects [159, 236]. Therefore, we
hypothesized that CNL treatment would have a similar effect and also result in
an accumulation of ceramide in the mitochondria of CLL cells. We observed that
25 µM nanoliposomal-NBD-C6-ceramide colocalized with a MitoTracker Deep
Red marker for mitochondria, signifying accumulation of exogenous ceramide in
the mitochondria in CLL (Fig. 4-1 (i)). To determine whether mitochondrial
membrane integrity was damaged by CNL, mitochondrial membrane potential
(∆Ψm) was measured using DiOC6 staining. As shown in Fig. 4-1 (ii), treatment
with 25 µM CNL for 24 hours induced loss of membrane potential in JVM3 cells,
and also in leukemic cells from a patient with CLL. These studies indicate that
CNL localizes within the mitochondria and disrupts the mitochondrial membrane

117
potential in CLL, which could potentially contribute to mitochondrial dysfunction
and resultant cell death.
Ceramide has also been suggested to act on mitochondria and generate
reactive oxygen species (ROS) [237]. ROS can activate apoptosis, but recently
have been reported to also induce autophagy and other cell death processes.
Moreover, ceramide treatment has also been linked necrotic cell death via rapid
formation of ROS. Treatment of A20 B-cell lymphoma cells with exogenous cell
permeable, C6-ceramide, induced necrotic cell death in a dose- and time-
dependent fashion. This was associated with formation of ROS, and was
accompanied by dissipation of mitochondrial membrane potential [36]. In
addition, presence of ROS scavengers, like Tiron, blocked ceramide-induced
necrosis. Ceramide accumulation in these cells was also associated with a
significant depletion of intracellular ATP levels which indicated induction of
necrotic death [37]. Therefore, we asked whether C6-ceramide treatment could
induce ROS production in CLL cells. Indeed, Fig. 4-1 (iii) demonstrated that
treatment of 25 µM CNL for 24 hours led to production of ROS, compared to
treatment with the ghost nanoliposome. In addition, pre-treatment with the
antioxidant and ROS scavenger, N-acetyl-L-cysteine (NAC) was sufficient to
attenuate this increased ROS production associated with ceramide (Fig. 4-1(iii)).
To determine if pre-treatment with NAC was adequate to rescue cell death
induction by CNL, a cell viability assay was performed. It was evident that there
was some rescue of cell death induction when formation of ROS were inhibited

118
(Fig. 4-1 (iv)), suggesting that ROS production was contributing to the cell death
induction process to some degree.
Figure 4-1. CNL treatment results in accumulation of ceramide in the mitochondria,
decreased mitochondrial membrane potential, and generation of ROS A). Confocal
microscopic image of 25 µM (24 hour treatment) nanoliposomal-NBD-C6-ceramide delivery to
JVM3 cells showing that NBD-C6-ceramide (green) colocalized with cellular mitochondria (red);
Areas of co-localization (overlay) were characterized by appearance of the orange color. Cellular
nucleas was stained with Hoechst 33342 (blue). Magnification, 63X. B). DiOC6 was used to
determine changes in mitochondrial membrane potential using fluorscence-activated cell sorter
analysis. After treatment with 25 µM ghost nanolipsomes (black) or CNL (grey) for 24 hours,
JVM3 and CLL patient cells were collected and stained with DiOC6. C6-ceramide induced a shift
to the left in DiOC6 fluorescence, indicative of an impairment in the mitochondrial membrane
potential. C). Dihydroethidium (DHE) was used to determine ROS production. After treatment with
25 µM ghost nanoliposomes (top purple tracing) or CNL (red tracing) nanolipsomes for 24 hours,
JVM3 cells were collected and stained with DHE. CNL induced a shift to the right in ethidium

119
fluorescence, indicative of increased ROS production. Pre-treatment with 1mM of the anti-oxidant
N-acetyl-L-cysteine (NAC) was able to attenuate this increase in ROS production. D). JVM3 cells
pre-treated with 1mM of NAC then treated for 24 hours with various doses of ghost or CNL. MTT
assay was then performed and percentage of non-viable cells was determined.
CNL treatment causes release of apoptosis inducing factor (AIF) from
mitochondria into the cytosol
It is well established that in conjunction with proapoptotic proteins like BAX,
ceramide forms channels in the mitochondrial membrane resulting in
mitochondrial outer membrane permeabilization (MOMP) [18]. MOMP causes
release of apoptogenic factors like cytochrome c and AIF from the mitochondrial
intermembrane space into the cytosol. While we have already demonstrated that
treatment with CNL does not cause release of mitochondrial cytochrome c into
the cytoplasm and does not cause apoptotic cell death in JVM-3 cells [39], we
wanted to evaluate the effect of CNL on AIF release. AIF released into the
cytosol moves to the nucleus and promotes nuclear chromatin condensation and
DNA fragmentation. Several reports in the literature have shown that ceramide
causes release of mitochondrial AIF into cytosol thereby inducing cell death [238-
241]. As shown in Fig. 4-2, we observed release of AIF into the cytosol in JVM-3
cells after treatment with CNL but not ghost nanoliposomes. AIF release has also
been linked to necrosis/necroptsis, which is the predominant form of cell death
observed in CLL cells after CNL treatment [39, 242].

120
Figure 4-2. CNL treatment results in cytosolic release of AIF. JVM-3 cells were treated with
40µM CNL or ghost nanoliposomes for 24 hours. Western blot analysis was performed on
cytosolic and mitochondrial fractions for AIF, cytochrome c (cyto c), tubulin and cyclooxygenase
IV (Cox IV). Cyto c and cox IV are used as mitochondrial markers and tubulin is used as a
cytosolic marker. Absence of cox IV and tubulin in the cytosolic and mitochondrial fractions
respectively indicate purity of the fractions. Results from two independent experiments are shown
here.
CNL diminishes the transport of STAT3 inside the mitochondria and
subsequent STAT3 phosphorylation
We have previously observed that CNL diminishes phosphorylation of
STAT3 in both CLL cell lines and patient cells. Given the critical role of
mitochondrial p-STAT3-S727 in CLL, we next evaluated the effect of CNL on
mitochondrial STAT3. We observed that treatment with CNL but not ghost
liposomes for 14 hours decreased total STAT3 levels in the mitochondrial fraction
(Fig. 4-3). This phenomenon was more pronounced 24 hours after treatment with

121
CNL, as we observed a complete depletion of total STAT3 in the mitochondrial
fraction of cells (Fig. 4-3). There was a reduction in the levels of phosphorylated
forms of STAT3, p-STAT3-S727 and p-STAT3-Y705 in the mitochondrial fraction
of cells after treatment with CNL. The fact that we observed a reduction in total
mitochondrial STAT3 levels at an early time point, i.e. 14 hours after treatment
with CNL indicated that this phenomenon was not just a consequence of
extended treatment times or cell death, but could potentially affect mitochondrial
bioenergetics and could play a role in CNL-induced cell death. These results
were interesting since it indicated that the effect of CNL on cellular bioenergetics
could be a potential mechanism of cell death in CLL cells.
Figure 4-3. CNL treatment dimishes levels of total STAT3 and p-STAT3 in the
mitochondria. JVM-3 cells were treated with 20 µM or 40µM CNL or ghost nanoliposomes for 14
hours and 24 hours. Western blot analysis was performed on cytosolic and mitochondrial
fractions for p-STAT3-Y705, p-STAT3-S727, STAT3,cyclooxygenase IV (Cox IV) and tubulin. Cox
IV is used as a mitochondrial marker and tubulin is used as a cytosolic marker. Absence (or

122
highly reduced levels) of cox IV and tubulin in the cytosolic and mitochondrial fractions
respectively indicate purity of the fractions. Above are representative blots from two independent
experiemnts.
Effect of CNL on cellular bioenergetics in CLL
To study the effect of CNL on cellular bioenergetics, we first performed
experiments to evaluate glycolysis in cells before and after treatment with CNL.
Cellular glycolysis can be measured by quantifying the lactic acid production via
the extracellular proton release (extracellular acidification rate [ECAR]). As
shown in Fig. 4-4, JVM-3 cells a significant reduction in glycolytic capacity after
treatment with CNL. Treatment with 20µM CNL for 6 hours did not significantly
affect ECAR. This corresponds with the fact that significant cell death is not
observed 6 hours after treatment with CNL. However, treatment with 20µM or
40µM CNL for 12 hours or 24 hours significantly reduced the glycolytic capacity
of JVM-3 cells as compared to when treated with ghost liposomes. These results
indicate that CNL treatment suppresses the glycolytic capacity of CLL cells.
These results also serve to validate our conclusion from Chapter 1 of the thesis,
i.e. CNL suppress the Warburg effect in CLL cells.
Figure 4-4. CNL
treatment suppresses
the glycolytic flux of
JVM-3 cells. JVM-3 cells
were treated with 20 µM or
40µM CNL or ghost
nanoliposomes for 6
hours, 14 hours and 24
hours. Mito stress kit was

123
used to measure ECAR in cells treated with ghost liposomes or CNL. Each sample was run in
four replicates. Error bars show mean +/- SEM. ** p ≤ 0.005 (Student’s t-test).
Discussion
Cellular bioenergetics is a crucial system for maintaining cell viability and its
perturbation is an important component of several cell death pathways. In
addition to being the powerhouse of a cell, the mitochondrion is also an effector
organelle of some cell death pathways and a convergence point for other death
mechanisms. The mitochondrion is critical in death because it functions like a
“ticking bomb” once a death stimulus is received by the cell. Firstly, it sequesters
apoptogenic factors in the intermembrane space that can be released under the
influence of certain stimuli. Furthermore, through oxidative phosphorylation, it is
also a dominant source of ROS that can act as a major mediator of cell death.
We investigated the effect of CNL on mitochondrial integrity of CLL cells. As
expected, we observed that CNL treatment resulted in preferential accumulation
of ceramide in the mitochondria. Several reports in the literature have
documented ceramide localization within the mitochondria after treatment with
CNL [189]. Consistent with other reports, we also observed a reduction in
mitochondrial membrane potential, and increased MOMP after treatment with
CNL. This observation is supported by extensive reports that have demonstrated
that ceramide molecules form macrodomains and channels in the mitochondrial
membrane, thereby compromising MOMP and mitochondrial membrane potential
[18]. CNL-induced increased MOMP also released the pro-apoptotic protein, AIF,
which causes DNA fragmentation. Thus, in line with several other reports, we

124
observed that CNL perturbs mitochondrial structure in CLL. Our next step was to
investigate the effect on mitochondrial function and bioenergetics.
Cancer cells are characterized by metabolic reprogramming, i.e.
dependence on aerobic glycolysis or Warburg effect for ATP production rather
than the more efficient oxidative phosphorylation [233]. This glycolytic switch has
emerged as an attractive target in novel chemotherapeutic strategies. The two
papers published in 2009 that revealed the vital role of mitochondrial p-STAT3-
S727 in cellular respiration and in Ras-mediated cellular transformation through
increased oxidative phosphorylation have been seminal in this field of cellular
bioenergetics [191, 223]. Researchers are now focusing their efforts towards
gaining deeper insights into the dynamics between the Warburg effect and
oxidative phosphorylation for ATP synthesis in tumor cells.
Mitochondrial STAT3 phosphorylation mediates cellular transformation by
enhancing aerobic glycolysis, ETC activity, ATP abundance and inhibiting
overproduction of ROS [222]. p-STAT3-S727 is particularly important in CLL
because it is constitutively active in all CLL cells irrespective of the
phosphorylation status of Y705 [188]. Similar to other malignancies, p-STAT3-
S727 localizes to the mitochondria and associates with complex I of the ETC in
CLL cells [232]. Overactivation of p-STAT3-S727 provides stress protection and
viability to CLL cells and correlates with CLL cell resistance to apoptosis,
indicating that its potential function as an enhancer of oxidative phosphorylation
and/or glycolysis might be crucial for CLL cell viability and chemoresistance
[232]. In support of this speculation, recent reports indicate that unlike other

125
malignancies, CLL cells have increased oxidative phosphorylation but not
increased aerobic glycolysis [233]. However, stromal cells have been shown to
cause a glycolytic switch in CLL cells [131].
Since we have previously reported that CNL treatment suppresses levels
of p-STAT3-S727 in CLL cells (Chapter 3), we next investigated the effect of CNL
specifically on mitochondrial STAT3. We observed that CNL suppressed
mitochondrial STAT3 phosphorylation, but more importantly we noticed reduced
levels of total STAT3 in the mitochondria of cells treated with CNL. Several
possibilities can explain this observation. Since STAT3 shuttles between the
mitochondria and cytoplasm, it is likely that dephosphorylation of mitochondrial
STAT3 at S727 enhances its export out of the nucleus, thus causing a reduction
in total STAT3 levels in the mitochondria. Future work in this scenario should
include identifying the mitochondrial kinases or phosphatses regulated by
mitochondrial ceramide to cause this suppression in p-STAT3-S727 and the
eventual export of STAT3 out of the mitochondria. Another possibility is CNL-
induced reduction in the mitochondrial import of STAT3. GRIM-19 is known to
shuttle STAT3 into the mitochondria and phosphorylation of S727 residue of
STAT3 is critical for this interaction [231]. Hence, the effect of CNL on GRIM-19
levels and function is another possibility that should be investigated.
Next, we directly investigated the effects of CNL on cellular bioenergetics,
i.e. aerobic glycolysis and oxidative phosphorylation. Lactic acid release in the
extracellular environment (ECAR) is a measure of glycolysis and glycolytic flux.
We observed a time-dependent reduction in the glycolytic flux of CLL cells after

126
treatment with CNL. This result validates our previously published report that
CNL targets the Warburg effect in CLL cells by reducing levels of GAPDH
(Chapter 2) [39]. It is well accepted that the glycolytic switch is an attractive
target for novel chemotherapeutics. In CLL, targeting the Warburg effect can
prove to be promising, as this strategy will also overcome microenvironment-
mediated CLL cell viability and chemoresistance [131]. We are currently
investigating the effects of CNL on oxidative phosphorylation of CLL cells. We
speculate that CNL treatment will also inhibit oxidative phosphorylation in CLL
cells, since we have consistently observed a reduction in mitochondrial p-STAT3-
S727 on treatment with CNL. Our current and future work includes studying the
effect of CNL on cellular bioenergetics in CLL cells transfected with mitochondria-
targeted STAT3 constructs mutated at S727 and/or Y705 residues. These
experiments will provide a clear picture of the role of mitochondrial p-STAT3-
S727 in CNL-induced alterations in cellular bioenergetics and cell death.
Taken together, this work is crucial for uncovering the exact effect of CNL
on cellular bioenergetics in CLL. This research can identify potential therapeutic
targets in the cellular bioenergetics system that can also be inhibited to improve
the efficiency of CNL as a therapy for CLL.

127
CHAPTER 5 – Conclusions, Future directions and Therapeutic Potential
Conclusions
This dissertation has broadly focused on elucidating the molecular
mechanisms of C6-ceramide-induced cell death in chronic lymphocytic leukemia.
C6-ceramide is delivered in a nanoliposomal formulation to overcome the
delivery challenges due to the hydrophobicity of the sphingolipid. It is well
established that CNL is a lucrative delivery formulation for ceramides in several
cancer models and CNL suppresses tumor growth in models of breast cancer,
sarcoma, melanoma, hepatocellular carcinoma and LGL leukemia [18]. This work
aims at testing the use of CNL as a therapy for CLL and also to elucidate the key
molecular signaling pathways mediating CNL-induced cell death in CLL.
Following are the major conclusions presented in this dissertation:
Use of nanoliposomal C6-ceramide as a therapy for CLL
Sphingolipid-based therapeutics is a good strategy for treating CLL. This
conclusion is supported by reports in the literature that demonstrate a
dysregulation of sphingolipid metabolism in CLL. A recent report has uncovered
a novel link between BCR signaling and sphingolipid metabolism. It has been
reported that BCR controls chemoresistance of primary CLL cells by controlling
glucosylation of ceramides via upregulation of UGCG, thereby reducing levels of
proapoptotic ceramide species [139]. Use of BCR signaling inhibitors inhibit
UGCG resulting in an increase intracellular ceramide levels and subsequent

128
chemosensitization of CLL cells [139]. Another report has demonstrated that
sphingolipid metabolism is a potential novel mechanism of CLL [140].
We have demonstrated the effectiveness of CNL in CLL cells both in vitro
and in vivo. We observed that CNL induces caspase-independent, non-
apoptotic, necrotic cell death in CLL cell lines and CLL patient cells. Given the
fact that CNL was also effective in inducing cell death in Mec-2 cells which have
mutated p53, we hypothesize that this therapy, stand-alone or in combination,
would be effective against high risk disease groups like CLL with p53 mutation
cytogenetics.
We obtained promising in vivo evidence that CNL demonstrates in vivo
efficacy via tumor growth inhibition in a murine xenograft model of CLL. We are
aware of a recently described mouse model for CLL generated by co-injection of
primary human CLL cells and T cells into NOD/ SCID mice, but have chosen to
focus our studies on the mouse xenograft model because we can obtain tissue
for ex vivo Western analysis of GAPDH protein levels [182]. In our in vivo studies
we observed that treatment with nanoliposomal C6-ceramide effectively
decreased tumor burden without systemic side effects. Although necrosis is not
the preferred cell death mechanism, as it elicits immune and inflammatory
response that can be detrimental, we did not observe any systemic side-effects
in our animal study. In addition, it was expected that this nanoliposomal
formulation would be relatively non-toxic to the animals, as it was previously
selected by the Nanotechnology Characterization Laboratory (National Cancer
Institute) for extensive toxicology and stability testing. (Detailed information on

129
the toxicology studies of the “Ceramide Liposomes” can be found at
(http://ncl.cancer.gov/working_technical_reports.asp).
CNL targets the Warburg effect in CLL
Cancer cells are characterized by a glycolytic switch, i.e. preference for
aerobic glycolysis or Warburg effect for energy production over the more efficient
oxidative phosphorylation. Several studies have concluded that the Warburg
effect is a therapeutic target for treating cancer and new drugs are being
developed to target different steps of this phenomenon [243]. Specifically in CLL,
a recent paper reported that stromal cells promote a glycolytic switch in CLL cells
and targeting glucose metabolism can be exploited to breach stromal cell-
mediated drug resistance [131]. We have demonstrated that CNL decreases
protein and mRNA expression of GAPDH, an enzyme that oxidizes
glyceraldehyde-3-phosphate to 1,3-biphosphoglycerate. GAPDH is a potent
inhibitor of caspase-independent cell death by enhanced glycolysis and
enhanced autophagy [167]. Several reports have revealed that overexpression of
GAPDH may assist in conferring chemoresistance to cancer cells [166, 175]. We
observed that CNL targets GAPDH-dependent glycolysis. This was further
confirmed by observations like CNL-induced reduction in ATP, lactic acid and
rescue of ATP depletion and cell death by pretreatment with pyruvate which is a
final product of glycolysis. We did not see any changes in glucose uptake or
changes in GLUT1. This indicated that CNL directly targets the glycolytic
pathway. However, the mechanism of CNL-induced GAPDH inhibition is still
unknown. We provide evidence that the glycolytic pathway is an attractive target

130
in CLL that should be explored and our data emphasizes the potential
importance of GAPDH as a target of CNL.
STAT3 mediates CNL-induced cell death in CLL
STAT3 is a well-established therapeutic target in several cancers. Several
reports have demonstrated that STAT3 is constitutively phosphorylated on S727
in CLL cells independent of the phosphorylation status of Y705 residue [188,
189]. p-STAT3-S727 in CLL cells binds to DNA and activates transcription in CLL
cells irrespective of phosphorylation at Y705 [189]. Researchers have long
speculated that STAT3 might be a therapeutic target in CLL and some STAT3
inhibitors have been explored for therapy in CLL [189, 196, 197]. Our data
demonstrates that CNL suppresses STAT3 phosphorylation at both S727 and
Y705 residues. CNL-induced suppression of STAT3 phosphorylation results in
suppressing the transcriptional activity of CLL, subsequently downregulating anti-
apoptotic proteins like Mcl-1, survivin and XIAP. CNL-induced reduction in these
anti-apoptotic proteins is very promising since these proteins are critical for CLL
pathogenesis [209, 213]. We have also identified the upstream kinases that are
modulated by CNL to cause suppression in STAT3 phosphorylation. We have
demonstrated that CNL inhibits the activity of BTK, MEK and PKC kinases to
suppress STAT3 phosphorylation. CNL-induced suppression of MEK/ERK
pathway is consistent with published reports [35]. The role of CNL on PKC
activity is debatable since some reports provide evidence that C6-ceramide
activates PKC in vascular smooth cells, while some demonstrate that C6-
cermaide suppresses tumor metastasis by eliciting PKCζ tumor-suppressive

131
activities [146, 244]. However, our observation that CNL suppresses PKC activity
in CLL supports reports demonstrating that PKC is critical for CLL development
and a potential druggable target [245]. The inhibitory effect of CNL on the kinase
activity of BTK is novel and promising since BTK is a critical kinase for CLL
development. Ibrutinib, a BTK inhibitor, is currently being prescribed for the
treatment of CLL [208]. Our data imply that CNL might be partially inhibiting the
crucial BCR signaling in CLL cells. Taken together, we report novel data that
STAT3 phosphorylation and BTK are targets of CNL.
Effect of CNL on mitochondrial STAT3
The role of ceramide in regulating mitochondrial integrity during the cell
death processes has been extensively studied [27]. Furthermore, with the
discovery of the novel function of STAT3 and specifically p-STAT3-S727 in
regulating mitochondrial oxidative phosphorylation, the next obvious step is study
the effects on mitochondrial function after CNL treatment [191]. We have shown
that CNL treatment reduces mitochondrial membrane potential, induces MOMP
resulting in subsequent AIF release and increases ROS production. Ceramide
has been previously shown to cause such changes in the mitochondria in
cancerous cells [35]. We have studied the effect of CNL treatment on
mitochondrial STAT3 and we observe that treatment with CNL reduces the total
STAT3 levels within the mitochondria which results in lower levels of
mitochondrial phosphorylated STAT3. Since STAT3 shuttles between the
mitochondria and cytoplasm, it is likely that dephosphorylation of mitochondrial
STAT3 at S727 enhances its export out of the nucleus, thus causing a reduction

132
in total STAT3 levels in the mitochondria. Another possibility is CNL-induced
reduction in the mitochondrial import of STAT3, GRIM-19. We have also
examined the effect of CNL on cellular bioenergetics. We have observed a CNL-
induced time-dependent reduction in the glycolytic capacity of CLL cells, which
further validates our conclusion that CNL targets the Warburg effect in CLL.
Effect of CNL on cellular bioenergetics in CLL
In this thesis project, we have discovered three different targets of CNL in
CLL that include the Warburg effect, STAT3 transcriptional activity and STAT3
mitochondrial function. All three components play a significant role in maintaining
cellular bioenergetics. Thus it is intuitive to explore if there exists a link between
these three targets.
Recent work has uncovered a link between glycolytic metabolism and
sphingolipid metabolism. The authors show that glucose availability and
glycolytic metabolism dictate glycosphingolipid levels [246]. Treatment of
leukemic cells that have elevated glycosphingolipids with inhibitors of glycolysis
or the pentose phosphate pathway significantly decreased GlcCer levels and
increased intracellular ceramide levels leading to sensitization to cytotoxic drugs
[246]. However, there is very little evidence suggesting if the same relation exists
in reverse, i.e. if sphingolipid metabolism affects glycolytic metabolism. One
report demonstrated that ceramide starves cells to death by inducing intracellular
nutrient limitation via downregulation of nutrient transporter proteins [148].
However, the authors do not present data about the effect on nutrient transporter
protein downregulation on glycolysis. In this work we have established a novel

133
relation between ceramide and glycolysis. We have demonstrated that ceramide
targets the Warburg effect in cancer cells. Ceramide decreases the protein levels
of GAPDH, a glycolytic enzyme resulting in decreased glycolysis. We have
shown that pretreating CLL cells with pyruvate, the end product of glycolysis
rescues CNL-induced cell death and CNL-induced ATP depletion. Thus,
targeting GAPDH is one mechanism by which CNL inhibits aerobic glycolysis.
We have demonstrated that CNL suppresses STAT3 phosphorylation. It has
been reported that CLL cells are characterized by STAT3 which is constitutively
phosphorylated at S727 [189]. p-STAT3-S727 binds to DNA and exhibits
transcriptional activity in CLL cells independent of Y705 phosphorylation [189]. In
our model, we show that CNL-induced suppression in STAT3 phosphorylation
inhibits STAT3 transcriptional activity. Our initial studies demonstrated that
GAPDH may be partially regulated by the activity of STAT3, which establishes a
link between CNL-induced suppression in STAT3 phosphorylation and CNL-
induced inhibition of Warburg effect. Another possible link may be established on
the basis of evidence provided by Demaria et al. The authors have shown that
STAT3 acts as a master regulator of cell metabolism by activating HIF-1α-
dependent aerobic glycolysis [221]. Activated STAT3 upregulates HIF-1α
expression, which in turn induces the transcription of different genes involved in
glycolysis [221]. In addition to modulating the levels of HIF-1α, STAT3 can
cooperate with HIF-1α by binding to its responsive promoters, ensuring the
formation of a transcriptionally active complex. It is believed that STAT3 and HIF-
1α constitute a feed-forward loop that enhances aerobic glycolysis [222]. Thus,

134
we speculate that CNL-induced suppression in STAT3 phosphorylation might
suppress HIF-1α expression and subsequently suppress HIF-1α-dependent
aerobic glycolysis. This is another mechanism by which CNL-induced
suppression in STAT3 phosphorylation may be linked to suppression in the
Warburg effect.
Ceramide reduces mitochondrial membrane potential, increases MOMP
and subsequently increases ROS production. Furthermore, short-chain
ceramides have also been shown to inhibit the mitochondrial respiratory function,
ETC and oxidative phosphorylation [247]. Although the exact mechanism of
ceramide-induced inhibition of ETC is not clear, it is speculated that ceramide
macrodomains in the mitochondrial membrane may alter the hydrophobicity of
the membrane thus altering protein-lipid interaction. These biophysical alterations
affect the structure of the supercomplexes of the ETC, thus inhibiting function.
Another possibility is that ceramide may modify the activity of ETC by acting as
an allosteric effector by specifically binding to individual ETC complexes [247].
We are currently conducting experiments to provide direct evidence that CNL
reduces mitochondrial respiration but we speculate a CNL-induced reduction in
mitochondrial respiration. Recent reports have confirmed that mitochondrial p-
STAT3-S727 is required for oxidative phosphorylation and for Ras-mediated
cellular transformation. Inhibition or deletion of p-STAT3-S727 suppresses the
activity of complex I, II and/or V of the ETC [191, 223]. Similar to other
malignancies, p-STAT3-S727 localizes to the mitochondria and associates with
complex I of the ETC in CLL cells [232]. Overactivation of p-STAT3-S727

135
provides stress protection and viability to CLL cells and correlates with CLL cell
resistance to apoptosis, indicating that its potential function as an enhancer of
oxidative phosphorylation and/or glycolysis might be crucial for CLL cell viability
and chemoresistance [232]. We have demonstrated that CNL decreases
mitochondrial STAT3 protein and a subsequent decrease in p-STAT3-S727. We
hypothesize that CNL-induced reduction in p-STAT3-S727 is a major player in
CNL-mediated inhibition of mitochondrial respiration.
Taken together, we conclude that CNL treatment has a pronounced effect
on cellular bioenergetics in CLL cells leading to cell death. CNL targets the
Warburg effect and also mitochondrial respiration by targeting GAPDH,
suppressing STAT3 transcriptional activity, suppressing mitochondrial STAT3
function and also perturbing mitochondrial integrity by increased MOMP. This
mechanism of CNL-induced cell death in CLL cells is promising clinically
because recent reports have shown that mitochondrial respiration and stromal-
mediated glycolytic switch are robust therapeutic targets in CLL [131, 233].
Fig. 5-1 represents our proposed mechanisms of CNL-induced cell death
in CLL cells.

136
Figure 5-1. Molecular mechanisms of CNL-induced cell death in CLL cells
Future Directions
Future directions of this research include:
1. Validate the effectiveness of CNL treatment in a leukemic mouse model of
CLL like the recently described mouse model for CLL generated by co-
injection of primary human CLL cells and T cells into NOD/ SCID mice [182].
2. Evaluate the effect of CNL treatment on other components of the glycolytic
metabolism like other glycolytic enzymes and nutrient transporters.
3. Determine other potential mechanisms of CNL-mediated regulation of
GAPDH. Potential mechanisms to be evaluated include regulation at the
transcriptional, translational, post-translational modification level or enzymatic
regulation. CNL-mediated regulation of nuclear GAPDH should also be
assessed.

137
4. Metabolomic studies should be undertaken to obtain a clear picture of CNL-
induced changes in cellular metabolism. This might also shed light on the
effect of CNL treatment on cellular bioenergetics.
5. Determine which of the two phosphorylation sites i.e. S727 or Y705 is
essential for CNL-induced cell death in CLL cells.
6. Elucidate exact mechanism of CNL-induced reduction in mitochondrial
STAT3 levels in CLL cells. Assess the effect of CNL on GRIM-19 and identify
mitochondrial enzymes regulated by CNL that suppress mitochondrial STAT3
phosphorylation.
7. Elucidate the mechanism of CNL-induced suppression in oxidative
phosphorylation in CLL cells and determine if reduction in mitochondrial
p-STAT3-S727 plays a role in reduced oxidative phosphorylation.
8. Assess if CNL affects the autophagic function of cytoplasmic STAT3 and if
this mechanism plays a role in cell death.
9. Investigate if co-treatment of CNL with other inhibitors and/or mimetics of the
sphingolipid pathway enhances this induction of cell death in CLL. Utilization
of a GCS inhibitor (PPMP, PDMP, 4-HPR, etc.), ceramidase inhibitor (NOE,
LCL204, LCL385), or sphingosine kinase inhibitor (etoposide) could
potentially increase endogenous and exogenous ceramide levels and
augment cell death induction in CLL.
10. Investigate if co-treatment of CNL with STAT3 inhibitors enhances the
effectiveness of this therapy.

138
11. Develop targeted delivery of CNL by conjugating the liposome with antibody
against CLL-specific antigens like CD20 and CD74. Rituximab can be used
for targeting CD20 and mitatuzumab can be used for targeting CD74.
Summary and therapeutic potential
The role of CNL as a therapy for tumor regression in several types of solid
and non-solid cancers is extensively studied. However, the effect of CNL
treatment in CLL remains unclear. We have identified key molecular signaling
pathways of CNL-induced cell death in CLL. Identifying the key signaling
pathways inducing cell death following CNL treatment provides insight into the
therapeutic usefulness of this agent and is also valuable to uncover additional
targets for potential combination therapies.
In this dissertation we have provided convincing data demonstrating that
CNL treatment is a multi-targeted treatment strategy. CNL-induced cytotoxicity in
CLL cells is a result of targeting multiple cellular systems including cellular
bioenergetics, mitochondrial integrity and cellular protein expression profile.
Encapsulation of chemotherapeutic drugs into nanoliposomes has been a largely
successful delivery formulation in cancer models in vitro and in vivo. Extensive in
vivo toxicology studies conducted by Nanotechnology Characterization
Laboratory (http://ncl.cancer.gov/working_technical_reports.asp) have confirmed
that CNL have minimum adverse toxicity [113]. Moreover, the ongoing success of
using CNL as a platform for combinatorial therapy with other neoplastic agents
presents a promising and lucrative future to this endeavor of developing more
effective sphingolipid-based therapeutics for CLL.

139
References
1. Merrill, A.H., Jr. and G.M. Carman, Introduction to Thematic Minireview Series: Novel Bioactive Sphingolipids. J Biol Chem, 2015. 290(25): p. 15362-4.
2. Hannun, Y.A. and L.M. Obeid, Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol, 2008. 9(2): p. 139-50.
3. Beckham, T.H., et al., Interdiction of sphingolipid metabolism to improve standard cancer therapies. Adv Cancer Res, 2013. 117: p. 1-36.
4. Ogretmen, B. and Y.A. Hannun, Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer, 2004. 4(8): p. 604-16.
5. Brown, D.A. and E. London, Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem, 2000. 275(23): p. 17221-4.
6. Kolesnick, R.N., F.M. Goni, and A. Alonso, Compartmentalization of ceramide signaling: physical foundations and biological effects. J Cell Physiol, 2000. 184(3): p. 285-300.
7. Holthuis, J.C., et al., The organizing potential of sphingolipids in intracellular membrane transport. Physiol Rev, 2001. 81(4): p. 1689-723.
8. Singh, P., Y.D. Paila, and A. Chattopadhyay, Role of glycosphingolipids in the function of human serotonin(1)A receptors. J Neurochem, 2012. 123(5): p. 716-24.
9. Simons, K. and D. Toomre, Lipid rafts and signal transduction. Nat Rev Mol Cell Biol, 2000. 1(1): p. 31-9.
10. Hait, N.C., et al., Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science, 2009. 325(5945): p. 1254-7.
11. Zeidan, Y.H. and Y.A. Hannun, Translational aspects of sphingolipid metabolism. Trends Mol Med, 2007. 13(8): p. 327-36.
12. Ryland, L.K., et al., Dysregulation of sphingolipid metabolism in cancer. Cancer Biol Ther, 2011. 11(2): p. 138-49.
13. Obeid, L.M., et al., Programmed cell death induced by ceramide. Science, 1993. 259(5102): p. 1769-71.
14. Jarvis, W.D., et al., Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proc Natl Acad Sci U S A, 1994. 91(1): p. 73-7.
15. Haimovitz-Friedman, A., et al., Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med, 1994. 180(2): p. 525-35.
16. Cifone, M.G., et al., Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J Exp Med, 1994. 180(4): p. 1547-52.
17. Bose, R., et al., Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell, 1995. 82(3): p. 405-14.
18. Morad, S.A. and M.C. Cabot, Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer, 2013. 13(1): p. 51-65.
19. Dumitru, C.A. and E. Gulbins, TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene, 2006. 25(41): p. 5612-25.
20. Miyaji, M., et al., Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J Exp Med, 2005. 202(2): p. 249-59.
21. Grassme, H., et al., CD95 signaling via ceramide-rich membrane rafts. J Biol Chem, 2001. 276(23): p. 20589-96.

140
22. Huang, S.T., et al., Phyllanthus urinaria induces the Fas receptor/ligand expression and ceramide-mediated apoptosis in HL-60 cells. Life Sci, 2004. 75(3): p. 339-51.
23. Schaefer, J.T., W. Barthlen, and P. Schweizer, Ceramide induces apoptosis in neuroblastoma cell cultures resistant to CD95 (Fas/APO-1)-mediated apoptosis. J Pediatr Surg, 2000. 35(3): p. 473-9.
24. White-Gilbertson, S., et al., Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene, 2009. 28(8): p. 1132-41.
25. Voelkel-Johnson, C., Y.A. Hannun, and A. El-Zawahry, Resistance to TRAIL is associated with defects in ceramide signaling that can be overcome by exogenous C6-ceramide without requiring down-regulation of cellular FLICE inhibitory protein. Mol Cancer Ther, 2005. 4(9): p. 1320-7.
26. Siskind, L.J. and M. Colombini, The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J Biol Chem, 2000. 275(49): p. 38640-4.
27. Siskind, L.J., Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr, 2005. 37(3): p. 143-53.
28. Ganesan, V., et al., Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis, 2010. 15(5): p. 553-62.
29. von Haefen, C., et al., Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene, 2002. 21(25): p. 4009-19.
30. Darios, F., et al., Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J Neurochem, 2003. 84(4): p. 643-54.
31. Yuan, H., et al., Cytochrome c dissociation and release from mitochondria by truncated Bid and ceramide. Mitochondrion, 2003. 2(4): p. 237-44.
32. Chen, C.L., et al., Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood, 2008. 111(8): p. 4365-74.
33. Kim, S.S., et al., P53 mediates ceramide-induced apoptosis in SKN-SH cells. Oncogene, 2002. 21(13): p. 2020-8.
34. Deng, X., F. Gao, and W.S. May, Protein phosphatase 2A inactivates Bcl2's antiapoptotic function by dephosphorylation and up-regulation of Bcl2-p53 binding. Blood, 2009. 113(2): p. 422-8.
35. Liu, X., et al., Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood, 2010. 116(20): p. 4192-201.
36. Hetz, C.A., et al., Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: onset of necrosis is associated with delayed ceramide increase. J Cell Sci, 2002. 115(Pt 23): p. 4671-83.
37. Villena, J., et al., Ceramide-induced formation of ROS and ATP depletion trigger necrosis in lymphoid cells. Free Radic Biol Med, 2008. 44(6): p. 1146-60.
38. Davis, M.A., et al., Effect of ceramide on intracellular glutathione determines apoptotic or necrotic cell death of JB6 tumor cells. Toxicol Sci, 2000. 53(1): p. 48-55.
39. Ryland, L.K., et al., C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS One, 2013. 8(12): p. e84648.
40. Kim, W.H., et al., Ceramide induces non-apoptotic cell death in human glioma cells. Neurochem Res, 2005. 30(8): p. 969-79.

141
41. Ramos, B., et al., Prevalence of necrosis in C2-ceramide-induced cytotoxicity in NB16 neuroblastoma cells. Mol Pharmacol, 2003. 64(2): p. 502-11.
42. Gentil, B., F. Grimot, and C. Riva, Commitment to apoptosis by ceramides depends on mitochondrial respiratory function, cytochrome c release and caspase-3 activation in Hep-G2 cells. Mol Cell Biochem, 2003. 254(1-2): p. 203-10.
43. Morad, S.A. and M.C. Cabot, Ceramide-orchestrated signalling in cancer cells. Nature reviews. Cancer, 2013. 13(1): p. 51-65.
44. Grosch, S., S. Schiffmann, and G. Geisslinger, Chain length-specific properties of ceramides. Prog Lipid Res, 2012. 51(1): p. 50-62.
45. Zheng, W., et al., Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochimica et biophysica acta, 2006. 1758(12): p. 1864-84.
46. Levy, M. and A.H. Futerman, Mammalian ceramide synthases. IUBMB life, 2010. 62(5): p. 347-56.
47. Brunton, L.L.L., J.S.; Parker, K.L., eds., Goodman and Gilman's The Pharmacological Basis of Therapeutics. Eleventh edition ed. 2006, New York: McGraw-Hill. 2021.
48. Bassoy, E.Y. and Y. Baran, Bioactive sphingolipids in docetaxel-induced apoptosis in human prostate cancer cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2012. 66(2): p. 103-10.
49. Kolesnick, R., D. Altieri, and Z. Fuks, A CERTain role for ceramide in taxane-induced cell death. Cancer Cell, 2007. 11(6): p. 473-5.
50. Charles, A.G., et al., Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer chemotherapy and pharmacology, 2001. 47(5): p. 444-50.
51. Di Bartolomeo, S., et al., Apoptosis induced by doxorubicin in neurotumor cells is divorced from drug effects on ceramide accumulation and may involve cell cycle-dependent caspase activation. Journal of neurochemistry, 2000. 75(2): p. 532-9.
52. Senkal, C.E., et al., Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Molecular cancer therapeutics, 2007. 6(2): p. 712-22.
53. Rath, G., et al., De novo ceramide synthesis is responsible for the anti-tumor properties of camptothecin and doxorubicin in follicular thyroid carcinoma. Int J Biochem Cell Biol, 2009. 41(5): p. 1165-72.
54. Park, M.A., et al., Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer research, 2010. 70(15): p. 6313-24.
55. Biswal, S.S., et al., Changes in ceramide and sphingomyelin following fludarabine treatment of human chronic B-cell leukemia cells. Toxicology, 2000. 154(1-3): p. 45-53.
56. Perry, D.K., et al., Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. The Journal of biological chemistry, 2000. 275(12): p. 9078-84.
57. Schiffmann, S., et al., Activation of ceramide synthase 6 by celecoxib leads to a selective induction of C16:0-ceramide. Biochemical pharmacology, 2010. 80(11): p. 1632-40.
58. Cabot, M.C., et al., SDZ PSC 833, the cyclosporine A analogue and multidrug resistance modulator, activates ceramide synthesis and increases vinblastine

142
sensitivity in drug-sensitive and drug-resistant cancer cells. Cancer research, 1999. 59(4): p. 880-5.
59. Lucci, A., et al., Multidrug resistance modulators and doxorubicin synergize to elevate ceramide levels and elicit apoptosis in drug-resistant cancer cells. Cancer, 1999. 86(2): p. 300-11.
60. Wang, H., A.E. Giuliano, and M.C. Cabot, Enhanced de novo ceramide generation through activation of serine palmitoyltransferase by the P-glycoprotein antagonist SDZ PSC 833 in breast cancer cells. Molecular cancer therapeutics, 2002. 1(9): p. 719-26.
61. Kawatani, M., et al., Involvement of protein kinase C-regulated ceramide generation in inostamycin-induced apoptosis. Experimental cell research, 2000. 259(2): p. 389-97.
62. Sanchez, A.M., et al., Spisulosine (ES-285) induces prostate tumor PC-3 and LNCaP cell death by de novo synthesis of ceramide and PKCzeta activation. European journal of pharmacology, 2008. 584(2-3): p. 237-45.
63. Ramer, R., et al., Ceramide is involved in r(+)-methanandamide-induced cyclooxygenase-2 expression in human neuroglioma cells. Molecular pharmacology, 2003. 64(5): p. 1189-98.
64. Gustafsson, K., et al., Potentiation of cannabinoid-induced cytotoxicity in mantle cell lymphoma through modulation of ceramide metabolism. Molecular cancer research : MCR, 2009. 7(7): p. 1086-98.
65. Ponzoni, M., et al., Differential effects of N-(4-hydroxyphenyl)retinamide and retinoic acid on neuroblastoma cells: apoptosis versus differentiation. Cancer research, 1995. 55(4): p. 853-61.
66. Reynolds, C.P., B.J. Maurer, and R.N. Kolesnick, Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett, 2004. 206(2): p. 169-80.
67. Jiang, L., et al., Preferential involvement of both ROS and ceramide in fenretinide-induced apoptosis of HL60 rather than NB4 and U937 cells. Biochemical and biophysical research communications, 2011. 405(2): p. 314-8.
68. Wang, H., et al., N-(4-hydroxyphenyl)retinamide elevates ceramide in neuroblastoma cell lines by coordinate activation of serine palmitoyltransferase and ceramide synthase. Cancer research, 2001. 61(13): p. 5102-5.
69. Maurer, B.J., et al., Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. Journal of the National Cancer Institute, 2000. 92(23): p. 1897-909.
70. Rahmaniyan, M., et al., Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. The Journal of biological chemistry, 2011. 286(28): p. 24754-64.
71. Kraveka, J.M., et al., Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. The Journal of biological chemistry, 2007. 282(23): p. 16718-28.
72. Apraiz, A., et al., Evaluation of bioactive sphingolipids in 4-HPR-resistant leukemia cells. BMC Cancer, 2011. 11: p. 477.
73. Fabrias, G., et al., Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Prog Lipid Res, 2012. 51(2): p. 82-94.
74. Jin, J., et al., AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. Journal of lipid research, 2009. 50(12): p. 2389-97.
75. Jaffrezou, J.P., et al., Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. The EMBO journal, 1996. 15(10): p. 2417-24.

143
76. Ito, H., et al., Transcriptional regulation of neutral sphingomyelinase 2 gene expression of a human breast cancer cell line, MCF-7, induced by the anti-cancer drug, daunorubicin. Biochimica et biophysica acta, 2009. 1789(11-12): p. 681-90.
77. Modrak, D.E., et al., Synergistic interaction between sphingomyelin and gemcitabine potentiates ceramide-mediated apoptosis in pancreatic cancer. Cancer research, 2004. 64(22): p. 8405-10.
78. Dumitru, C.A., et al., Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. Journal of molecular medicine, 2009. 87(11): p. 1123-32.
79. Strum, J.C., et al., 1-beta-D-Arabinofuranosylcytosine stimulates ceramide and diglyceride formation in HL-60 cells. The Journal of biological chemistry, 1994. 269(22): p. 15493-7.
80. Noda, S., et al., Role of ceramide during cisplatin-induced apoptosis in C6 glioma cells. Journal of neuro-oncology, 2001. 52(1): p. 11-21.
81. Zeidan, Y.H., R.W. Jenkins, and Y.A. Hannun, Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J Cell Biol, 2008. 181(2): p. 335-50.
82. Bezombes, C., et al., Rituximab antiproliferative effect in B-lymphoma cells is associated with acid-sphingomyelinase activation in raft microdomains. Blood, 2004. 104(4): p. 1166-73.
83. Yun, S.H., et al., Stichoposide C induces apoptosis through the generation of ceramide in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clinical cancer research : an official journal of the American Association for Cancer Research, 2012. 18(21): p. 5934-48.
84. Rubio, S., et al., Betuletol 3-methyl ether induces G(2)-M phase arrest and activates the sphingomyelin and MAPK pathways in human leukemia cells. Molecular Carcinogenesis, 2010. 49(1): p. 32-43.
85. Mondal, S., et al., Withanolide D induces apoptosis in leukemia by targeting the activation of neutral sphingomyelinase-ceramide cascade mediated by synergistic activation of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. Molecular cancer, 2010. 9: p. 239.
86. Ogretmen, B. and Y.A. Hannun, Biologically active sphingolipids in cancer pathogenesis and treatment. Nature reviews. Cancer, 2004. 4(8): p. 604-16.
87. Macchia, M., et al., Design, synthesis, and characterization of the antitumor activity of novel ceramide analogues. J Med Chem, 2001. 44(23): p. 3994-4000.
88. Liu, J., et al., Novel anti-viability ceramide analogs: design, synthesis, and structure-activity relationship studies of substituted (S)-2-(benzylideneamino)-3-hydroxy-N-tetradecylpropanamides. Bioorg Med Chem, 2010. 18(14): p. 5316-22.
89. Bieberich, E., T. Kawaguchi, and R.K. Yu, N-acylated serinol is a novel ceramide mimic inducing apoptosis in neuroblastoma cells. The Journal of biological chemistry, 2000. 275(1): p. 177-81.
90. Bieberich, E., et al., Selective apoptosis of pluripotent mouse and human stem cells by novel ceramide analogues prevents teratoma formation and enriches for neural precursors in ES cell-derived neural transplants. J Cell Biol, 2004. 167(4): p. 723-34.
91. Struckhoff, A.P., et al., Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J Pharmacol Exp Ther, 2004. 309(2): p. 523-32.
92. Novgorodov, S.A., et al., Positively charged ceramide is a potent inducer of mitochondrial permeabilization. The Journal of biological chemistry, 2005. 280(16): p. 16096-105.

144
93. Senkal, C.E., et al., Potent antitumor activity of a novel cationic pyridinium-ceramide alone or in combination with gemcitabine against human head and neck squamous cell carcinomas in vitro and in vivo. J Pharmacol Exp Ther, 2006. 317(3): p. 1188-99.
94. Hou, Q., et al., Mitochondrially targeted ceramides preferentially promote autophagy, retard cell growth, and induce apoptosis. Journal of lipid research, 2011. 52(2): p. 278-88.
95. Rossi, M.J., et al., Inhibition of growth and telomerase activity by novel cationic ceramide analogs with high solubility in human head and neck squamous cell carcinoma cells. Otolaryngol Head Neck Surg, 2005. 132(1): p. 55-62.
96. Crawford, K.W., et al., Novel ceramide analogues display selective cytotoxicity in drug-resistant breast tumor cell lines compared to normal breast epithelial cells. Cell Mol Biol (Noisy-le-grand), 2003. 49(7): p. 1017-23.
97. Adan-Gokbulut, A., et al., Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr Med Chem, 2013. 20(1): p. 108-22.
98. Barth, B.M., M.C. Cabot, and M. Kester, Ceramide-based therapeutics for the treatment of cancer. Anticancer Agents Med Chem, 2011. 11(9): p. 911-9.
99. Ganta, S., et al., Development of EGFR-Targeted Nanoemulsion for Imaging and Novel Platinum Therapy of Ovarian Cancer. Pharm Res, 2014.
100. Desai, A., T. Vyas, and M. Amiji, Cytotoxicity and apoptosis enhancement in brain tumor cells upon coadministration of paclitaxel and ceramide in nanoemulsion formulations. J Pharm Sci, 2008. 97(7): p. 2745-56.
101. Stover, T.C., et al., Thermoresponsive and biodegradable linear-dendritic nanoparticles for targeted and sustained release of a pro-apoptotic drug. Biomaterials, 2008. 29(3): p. 359-69.
102. Devalapally, H., et al., Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clinical cancer research : an official journal of the American Association for Cancer Research, 2008. 14(10): p. 3193-203.
103. van Vlerken, L.E., et al., Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer research, 2007. 67(10): p. 4843-50.
104. Devalapally, H., et al., Paclitaxel and ceramide co-administration in biodegradable polymeric nanoparticulate delivery system to overcome drug resistance in ovarian cancer. Int J Cancer, 2007. 121(8): p. 1830-8.
105. Kester, M., et al., Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells. Nano Lett, 2008. 8(12): p. 4116-21.
106. Stover, T. and M. Kester, Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther, 2003. 307(2): p. 468-75.
107. Stover, T.C., et al., Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research, 2005. 11(9): p. 3465-74.
108. Shabbits, J.A. and L.D. Mayer, High ceramide content liposomes with in vivo antitumor activity. Anticancer Res, 2003. 23(5A): p. 3663-9.
109. Tran, M.A., et al., Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clinical cancer research : an official journal of the American Association for Cancer Research, 2008. 14(11): p. 3571-81.

145
110. Tagaram, H.R., et al., Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut, 2011. 60(5): p. 695-701.
111. Watters, R.J., et al., Targeting glucosylceramide synthase synergizes with C6-ceramide nanoliposomes to induce apoptosis in natural killer cell leukemia. Leuk Lymphoma, 2013. 54(6): p. 1288-96.
112. Jiang, Y., et al., Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther, 2011. 12(7): p. 574-85.
113. Hankins, J.L., et al., The therapeutic potential of nanoscale sphingolipid technologies. Handbook of experimental pharmacology, 2013(215): p. 197-210.
114. Morad, S.A., et al., Ceramide--antiestrogen nanoliposomal combinations--novel impact of hormonal therapy in hormone-insensitive breast cancer. Molecular cancer therapeutics, 2012. 11(11): p. 2352-61.
115. Adiseshaiah, P.P., et al., Synergistic combination therapy with nanoliposomal C6-ceramide and vinblastine is associated with autophagy dysfunction in hepatocarcinoma and colorectal cancer models. Cancer letters, 2013. 337(2): p. 254-65.
116. Chapman, J.V., V. Gouaze-Andersson, and M.C. Cabot, Expression of P-glycoprotein in HeLa cells confers resistance to ceramide cytotoxicity. Int J Oncol, 2010. 37(6): p. 1591-7.
117. Cabot, M.C., T.Y. Han, and A.E. Giuliano, The multidrug resistance modulator SDZ PSC 833 is a potent activator of cellular ceramide formation. FEBS Lett, 1998. 431(2): p. 185-8.
118. Lavie, Y., et al., Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. The Journal of biological chemistry, 1997. 272(3): p. 1682-7.
119. Morad, S.A., et al., Tamoxifen magnifies therapeutic impact of ceramide in human colorectal cancer cells independent of p53. Biochemical pharmacology, 2013. 85(8): p. 1057-65.
120. Chapman, J.V., et al., P-glycoprotein antagonists confer synergistic sensitivity to short-chain ceramide in human multidrug-resistant cancer cells. Experimental cell research, 2011. 317(12): p. 1736-45.
121. Qiu, L., et al., Paclitaxel and ceramide synergistically induce cell death with transient activation of EGFR and ERK pathway in pancreatic cancer cells. Oncol Rep, 2006. 16(4): p. 907-13.
122. Best, C., et al., Paclitaxel disrupts polarized entry of membrane-permeable C6 ceramide into ovarian cancer cells resulting in synchronous induction of cell death. Oncol Lett, 2013. 5(6): p. 1854-1858.
123. Zhu, Q.Y., et al., C6-ceramide synergistically potentiates the anti-tumor effects of histone deacetylase inhibitors via AKT dephosphorylation and alpha-tubulin hyperacetylation both in vitro and in vivo. Cell Death Dis, 2011. 2: p. e117.
124. Furlong, S.J., J.S. Mader, and D.W. Hoskin, Lactoferricin-induced apoptosis in estrogen-nonresponsive MDA-MB-435 breast cancer cells is enhanced by C6 ceramide or tamoxifen. Oncol Rep, 2006. 15(5): p. 1385-90.
125. Zenz, T., et al., From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer, 2010. 10(1): p. 37-50.
126. Hayden, R.E., et al., Treatment of chronic lymphocytic leukemia requires targeting of the protective lymph node environment with novel therapeutic approaches. Leuk Lymphoma, 2012. 53(4): p. 537-49.

146
127. Hanlon, K., C.E. Rudin, and L.W. Harries, Investigating the targets of MIR-15a and MIR-16-1 in patients with chronic lymphocytic leukemia (CLL). PLoS One, 2009. 4(9): p. e7169.
128. Chavez, J.C., et al., Genomic aberrations deletion 11q and deletion 17p independently predict for worse progression-free and overall survival after allogeneic hematopoietic cell transplantation for chronic lymphocytic leukemia. Leuk Res, 2014. 38(10): p. 1165-72.
129. Dohner, H., et al., Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med, 2000. 343(26): p. 1910-6.
130. Ghia, P., et al., Differential effects on CLL cell survival exerted by different microenvironmental elements. Curr Top Microbiol Immunol, 2005. 294: p. 135-45.
131. Jitschin, R., et al., Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood, 2015. 125(22): p. 3432-6.
132. Tam, C.S., et al., Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood, 2008. 112(4): p. 975-80.
133. Hallek, M., et al., Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet, 2010. 376(9747): p. 1164-74.
134. Smolej, L., Targeted treatment for chronic lymphocytic leukemia: clinical potential of obinutuzumab. Pharmgenomics Pers Med, 2015. 8: p. 1-7.
135. Shanafelt, T., Treatment of older patients with chronic lymphocytic leukemia: key questions and current answers. Hematology Am Soc Hematol Educ Program, 2013. 2013: p. 158-67.
136. Sutton, L.A. and R. Rosenquist, Deciphering the molecular landscape in chronic lymphocytic leukemia: time frame of disease evolution. Haematologica, 2015. 100(1): p. 7-16.
137. Mengubas, K., et al., Ceramide-induced killing of normal and malignant human lymphocytes is by a non-apoptotic mechanism. Oncogene, 1999. 18(15): p. 2499-506.
138. Hammadi, M., et al., Membrane microdomain sphingolipids are required for anti-CD20-induced death of chronic lymphocytic leukemia B cells. Haematologica, 2012. 97(2): p. 288-96.
139. Schwamb, J., et al., B-cell receptor triggers drug sensitivity of primary CLL cells by controlling glucosylation of ceramides. Blood, 2012. 120(19): p. 3978-85.
140. Trojani, A., et al., Gene expression profiling identifies ARSD as a new marker of disease progression and the sphingolipid metabolism as a potential novel metabolism in chronic lymphocytic leukemia. Cancer Biomark, 2011. 11(1): p. 15-28.
141. Kolesnick, R.N. and M. Kronke, Regulation of ceramide production and apoptosis. Annu Rev Physiol, 1998. 60: p. 643-65.
142. Daido, S., et al., Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res, 2004. 64(12): p. 4286-93.
143. Chalfant, C.E., et al., FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem, 2001. 276(48): p. 44848-55.
144. Liu, X., et al., Targeting of survivin by nanoliposomal ceramide induces complete remission in NK-LGL leukemia. Blood, 2010. 116(20): p. 4192-201.

147
145. Clarke, C.J., et al., The extended family of neutral sphingomyelinases. Biochemistry, 2006. 45(38): p. 11247-56.
146. Fox, T.E., et al., Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem, 2007. 282(17): p. 12450-7.
147. Tran, M.A., et al., Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res, 2008. 14(11): p. 3571-81.
148. Guenther, G.G., et al., Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A, 2008. 105(45): p. 17402-7.
149. Vander Heiden, M.G., L.C. Cantley, and C.B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 2009. 324(5930): p. 1029-33.
150. Warburg, O., On the origin of cancer cells. Science, 1956. 123(3191): p. 309-14. 151. Altenberg, B. and K.O. Greulich, Genes of glycolysis are ubiquitously
overexpressed in 24 cancer classes. Genomics, 2004. 84(6): p. 1014-20. 152. Shanmugam, M., S.K. McBrayer, and S.T. Rosen, Targeting the Warburg effect
in hematological malignancies: from PET to therapy. Curr Opin Oncol., 2009. 21(6): p. 531-6.
153. Yeluri, S., et al., Cancer's craving for sugar:an opportunity for clinical exploitation. J Cancer Res Clin Oncol, 2009. 135(7): p. 867-77.
154. Watson, D.G., et al., The roles of sphingosine kinases 1 and 2 in regulating the Warburg effect in prostate cancer cells. Cell Signal, 2013. 25(4): p. 1011-7.
155. Foon, K.A., K.R. Rai, and R.P. Gale, Chronic lymphocytic leukemia: new insights into biology and therapy. Ann Intern Med, 1990. 113(7): p. 525-39.
156. Hofheinz, R.D., et al., Liposomal encapsulated anti-cancer drugs. Anticancer Drugs, 2005. 16(7): p. 691-707.
157. Zhao, X., et al., Liposomal coencapsulated fludarabine and mitoxantrone for lymphoproliferative disorder treatment. J Pharm Sci, 2008. 97(4): p. 1508-18.
158. Biswal, S.S., et al., Changes in ceramide and sphingomyelin following fludarabine treatment of human chronic B-cell leukemia cells. Toxicology, 2000. 154: p. 45-53.
159. Stover, T.C., et al., Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res, 2005. 11(9): p. 3465-74.
160. Yang, J., et al., Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood, 2010. 115(1): p. 51-60.
161. Sato, T., et al., FAP-1: a protein tyrosine phosphatase that associates with Fas. Science, 1995. 268(5209): p. 411-5.
162. Livak, K.J. and T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402-8.
163. Kueck, A., et al., Resveratrol inhibits glucose metabolism in human ovarian cancer cells. Gynecol Oncol, 2007. 107(3): p. 450-7.
164. Melo, J.V., et al., Two new cell lines from B-prolymphocytic leukaemia: characterization by morphology, immunological markers, karyotype and Ig gene rearrangement. Int J Cancer, 1986. 38(4): p. 531-8.

148
165. Veldurthy, A., et al., The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood, 2008. 112(4): p. 1443-52.
166. Rathmell, J.C. and S. Kornbluth, Filling a GAP(DH) in caspase-independent cell death. Cell, 2007. 129(5): p. 861-3.
167. Colell, A., et al., GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell, 2007. 129(5): p. 983-97.
168. Song, S. and T. Finkel, GAPDH and the search for alternative energy. Nat Cell Biol, 2007. 9(8): p. 869-70.
169. Loisel, S., et al., Establishment of a novel human B-CLL-like xenograft model in nude mouse. Leuk Res, 2005. 29(11): p. 1347-52.
170. Scarlatti, F., et al., Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem, 2004. 279(18): p. 18384-91.
171. Ogretmen, B., et al., Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J Biol Chem, 2002. 277(15): p. 12960-9.
172. Chapman, J.V., et al., Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochem Pharmacol, 2010. 80(3): p. 308-15.
173. Bustin, S.A., Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol, 2000. 25(2): p. 169-93.
174. Revillion, F., et al., Glyceraldehyde-3-phosphate dehydrogenase gene expression in human breast cancer. Eur J Cancer, 2000. 36(8): p. 1038-42.
175. Lavallard, V.J., et al., Modulation of caspase-independent cell death leads to resensitization of imatinib mesylate-resistant cells. Cancer Res, 2009. 69(7): p. 3013-20.
176. Sirover, M.A., New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem, 2005. 95(1): p. 45-52.
177. Sundararaj, K.P., et al., Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem, 2004. 279(7): p. 6152-62.
178. Rivenzon-Segal, D., et al., Glucose transporters and transport kinetics in retinoic acid-differentiated T47D human breast cancer cells. Am J Physiol Endocrinol Metab, 2000. 279(3): p. E508-19.
179. Huttmann, A., et al., Gene expression signatures separate B-cell chronic lymphocytic leukaemia prognostic subgroups defined by ZAP-70 and CD38 expression status. Leukemia, 2006. 20(10): p. 1774-82.
180. Geschwind, J.F., et al., Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res, 2002. 62(14): p. 3909-13.
181. Kumagai, S., R. Narasaki, and K. Hasumi, Glucose-dependent active ATP depletion by koningic acid kills high-glycolytic cells. Biochem Biophys Res Commun, 2008. 365(2): p. 362-8.

149
182. Bagnara, D., et al., A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood, 2011. 117(20): p. 5463-72.
183. Caligaris-Cappio, F. and T.J. Hamblin, B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol, 1999. 17(1): p. 399-408.
184. Hankins, J.L., et al., The therapeutic potential of nanoscale sphingolipid technologies. Handb Exp Pharmacol, 2013(215): p. 197-210.
185. Haakenson, J.K., et al., Lysosomal Degradation of CD44 Mediates Ceramide Nanoliposome-induced Anoikis and Diminshed Extravasation in Metastatic Carcinoma Cells. J Biol Chem, 2015.
186. Lin, T.S., S. Mahajan, and D.A. Frank, STAT signaling in the pathogenesis and treatment of leukemias. Oncogene, 2000. 19(21): p. 2496-504.
187. Yu, H., et al., Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer, 2014. 14(11): p. 736-46.
188. Frank, D.A., S. Mahajan, and J. Ritz, B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest, 1997. 100(12): p. 3140-8.
189. Hazan-Halevy, I., et al., STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood, 2010. 115(14): p. 2852-63.
190. Capron, C., et al., Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis, 2014. 5: p. e1451.
191. Wegrzyn, J., et al., Function of mitochondrial Stat3 in cellular respiration. Science, 2009. 323(5915): p. 793-7.
192. Li, P., et al., Signal transducer and activator of transcription-3 induces microRNA-155 expression in chronic lymphocytic leukemia. PLoS One, 2013. 8(6): p. e64678.
193. Liu, Z., et al., STAT-3 activates NF-kappaB in chronic lymphocytic leukemia cells. Mol Cancer Res, 2011. 9(4): p. 507-15.
194. Badoux, X., et al., Cross-talk between chronic lymphocytic leukemia cells and bone marrow endothelial cells: role of signal transducer and activator of transcription 3. Hum Pathol, 2011. 42(12): p. 1989-2000.
195. Giannoni, P., et al., An interaction between hepatocyte growth factor and its receptor (c-MET) prolongs the survival of chronic lymphocytic leukemic cells through STAT3 phosphorylation: a potential role of mesenchymal cells in the disease. Haematologica, 2011. 96(7): p. 1015-23.
196. Lu, K., et al., The STAT3 inhibitor WP1066 reverses the resistance of chronic lymphocytic leukemia cells to histone deacetylase inhibitors induced by interleukin-6. Cancer Lett, 2015. 359(2): p. 250-8.
197. Ishdorj, G., J.B. Johnston, and S.B. Gibson, Inhibition of constitutive activation of STAT3 by curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Mol Cancer Ther, 2010. 9(12): p. 3302-14.
198. Schust, J., et al., Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol, 2006. 13(11): p. 1235-42.
199. Allen, J.C., et al., c-Abl regulates Mcl-1 gene expression in chronic lymphocytic leukemia cells. Blood, 2011. 117(8): p. 2414-22.
200. Martinez-Lostao, L., et al., Role of the STAT1 pathway in apoptosis induced by fludarabine and JAK kinase inhibitors in B-cell chronic lymphocytic leukemia. Leuk Lymphoma, 2005. 46(3): p. 435-42.

150
201. Frank, D.A., S. Mahajan, and J. Ritz, Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med, 1999. 5(4): p. 444-7.
202. Battle, T.E. and D.A. Frank, STAT1 mediates differentiation of chronic lymphocytic leukemia cells in response to Bryostatin 1. Blood, 2003. 102(8): p. 3016-24.
203. Kearney, C.J., et al., Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ, 2015.
204. Miao, B. and A. Degterev, Methods to analyze cellular necroptosis. Methods Mol Biol, 2009. 559: p. 79-93.
205. Gonzalez, D., et al., Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol, 2011. 29(16): p. 2223-9.
206. Varnai, P., K.I. Rother, and T. Balla, Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem, 1999. 274(16): p. 10983-9.
207. Rawlings, D.J., et al., Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science, 1996. 271(5250): p. 822-5.
208. Byrd, J.C., et al., Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med, 2013. 369(1): p. 32-42.
209. Pepper, C., et al., Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood, 2008. 112(9): p. 3807-17.
210. Awan, F.T., et al., Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood, 2009. 113(3): p. 535-7.
211. Hussain, S.R., et al., Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin Cancer Res, 2007. 13(7): p. 2144-50.
212. Grzybowska-Izydorczyk, O., et al., Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur J Cancer, 2010. 46(4): p. 800-10.
213. Purroy, N., et al., Targeting the proliferative and chemoresistant compartment in chronic lymphocytic leukemia by inhibiting survivin protein. Leukemia, 2014. 28(10): p. 1993-2004.
214. Loeder, S., et al., A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res, 2009. 69(23): p. 8977-86.
215. Kater, A.P., et al., Inhibitors of XIAP sensitize CD40-activated chronic lymphocytic leukemia cells to CD95-mediated apoptosis. Blood, 2005. 106(5): p. 1742-8.
216. Maziere, C., M.A. Conte, and J.C. Maziere, Activation of the JAK/STAT pathway by ceramide in cultured human fibroblasts. FEBS Lett, 2001. 507(2): p. 163-8.
217. Woyach, J.A., et al., Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood, 2014. 123(8): p. 1207-13.
218. Kazi, J.U., N.N. Kabir, and L. Ronnstrand, Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med Oncol, 2013. 30(4): p. 757.
219. Chan, K.S., et al., Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene, 2008. 27(8): p. 1087-94.

151
220. Barbieri, I., et al., Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res, 2010. 70(6): p. 2558-67.
221. Demaria, M., et al., A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY), 2010. 2(11): p. 823-42.
222. Poli, V. and A. Camporeale, STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front Oncol, 2015. 5: p. 121.
223. Gough, D.J., et al., Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science, 2009. 324(5935): p. 1713-6.
224. Gough, D.J., et al., STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood, 2014. 124(14): p. 2252-61.
225. Mackenzie, G.G., et al., Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS One, 2013. 8(5): p. e61532.
226. Zhang, Q., et al., Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem, 2013. 288(43): p. 31280-8.
227. Myers, M.G., Jr., Cell biology. Moonlighting in mitochondria. Science, 2009. 323(5915): p. 723-4.
228. Garcia, R., et al., Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene, 2001. 20(20): p. 2499-513.
229. Schindler, C., D.E. Levy, and T. Decker, JAK-STAT signaling: from interferons to cytokines. J Biol Chem, 2007. 282(28): p. 20059-63.
230. Gough, D.J., L. Koetz, and D.E. Levy, The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS One, 2013. 8(11): p. e83395.
231. Tammineni, P., et al., The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem, 2013. 288(7): p. 4723-32.
232. Capron, C., et al., Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis, 2014. 5: p. e1451.
233. Jitschin, R., et al., Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood, 2014. 123(17): p. 2663-72.
234. Zamzami, N., et al., Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med, 1995. 182(2): p. 367-77.
235. Benov, L., L. Sztejnberg, and I. Fridovich, Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med, 1998. 25(7): p. 826-31.
236. Birbes, H., et al., Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv Enzyme Regul, 2002. 42: p. 113-29.
237. Quillet-Mary, A., et al., Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J Biol Chem, 1997. 272(34): p. 21388-95.
238. Scharstuhl, A., et al., Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS One, 2009. 4(8): p. e6688.

152
239. Czubowicz, K. and R. Strosznajder, Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine-1-phosphate. Mol Neurobiol, 2014. 50(1): p. 26-37.
240. Kim, N.H., et al., PKB/Akt inhibits ceramide-induced apoptosis in neuroblastoma cells by blocking apoptosis-inducing factor (AIF) translocation. J Cell Biochem, 2007. 102(5): p. 1160-70.
241. Pardo, J., et al., A role of the mitochondrial apoptosis-inducing factor in granulysin-induced apoptosis. J Immunol, 2001. 167(3): p. 1222-9.
242. Delavallee, L., et al., AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life, 2011. 63(4): p. 221-32.
243. Flaveny, C.A., et al., Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell, 2015. 28(1): p. 42-56.
244. Zhang, P., et al., C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCzeta tumor-suppressive activities and regulating integrin affinity modulation. Sci Rep, 2015. 5: p. 9275.
245. El-Gamal, D., et al., PKC-beta as a therapeutic target in CLL: PKC inhibitor AEB071 demonstrates preclinical activity in CLL. Blood, 2014. 124(9): p. 1481-91.
246. Stathem, M., et al., Glucose availability and glycolytic metabolism dictate glycosphingolipid levels. J Cell Biochem, 2015. 116(1): p. 67-80.
247. Kogot-Levin, A. and A. Saada, Ceramide and the mitochondrial respiratory chain. Biochimie, 2014. 100: p. 88-94.

153
Appendix: Letters of Permission
Chapter 1 and Chapter 2 has material that has been previously published. This
material is used with permission from the respective sources. Copies of the letter
granting permission for use in this dissertation are as follows.

154
Permission letter for Chapter 1
Reprinted (adapted) with permission from the chapter:
Doshi UA, Haakenson JK, Linton SL, Kelly K, Kester M (2015). Chemotherapy
and sphingolipid metabolism. In Bioactive Sphingolipids in Cancer Biology and
Therapy (pp. 401-436), New York, NY: Springer.

155
Permission for Chapter 2
Reprinted (adapted) with permission from:
Ryland LK*, Doshi UA*, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, Baab
KT, Young M, Khan O, Haakenson JK, Jarbadan NR, Liao J, Wang HG, Feith
DJ, Loughran TP Jr, Liu X, Kester M. “C6-ceramide nanoliposomes target the
Warburg effect in chronic lymphocytic leukemia” PLoS One. 2013 Dec 19;
8(12):e84648.
According to the journal policy:
“PLOS applies the Creative Commons Attribution (CC BY) license to works we publish. Using PLOS Content
No permission is required from the authors or the publishers to reuse or
repurpose PLOS content provided the original article is cited. In most cases,
appropriate attribution can be provided by simply citing the original article.”
http://journals.plos.org/plosone/s/content-license

VITA
USHMA A. DOSHI
Education
2015 Ph.D. Molecular Medicine, Pennsylvania State University 2015 MBA Pennsylvania State University 2012 M.S. Biotechnology, University at Buffalo, New York 2008 B.S. Pharmaceutical Sc., Institute of Chemical Technology, Mumbai, India
Publications Doshi UA, Haakenson JK, Linton SL, Kelly K, Kester M (2015). Chemotherapy and
sphingolipid metabolism. In Bioactive Sphingolipids in Cancer Biology and Therapy
(pp. 401-436), New York, NY: Springer.
Ryland LK*, Doshi UA*, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, Baab KT, Young M, Khan O, Haakenson JK, Jarbadan NR, Liao J, Wang HG, Feith DJ, Loughran TP Jr, Liu X, Kester M. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS One. 2013 Dec 19;8(12):e84648. *Co-first authorship
Hankins JL, Doshi UA, Haakenson JK, Young MM, Barth BM, Kester M. The therapeutic potential of nanoscale sphingolipid technologies. Handb Exp Pharmacol. 2013;(215):197-210.
Manuscripts in preparation:
Doshi UA, Fox TE, Loughran TP, Kester M. STAT3 mediates nanoliposomal C6-ceramide-induced cell death in chronic lymphocytic leukemia.
Doshi UA, Fox TE, Loughran TP, Kester M. Nanoliposomal C6-ceramide targets cellular bioenergetics to induce cell death in chronic lymphocytic leukemia.
Awards College of Medicine Class of 1971 Endowed Scholarship, Pennsylvania State
University, 2014
Beta Gamma Sigma Inductee, The Penn State Capital College Chapter of Beta Gamma Sigma scholastic honor society in Business Administration, 2014
Outstanding Graduate Student Award, 7th International Ceramide Conference , 2013
University Graduate Fellowship, Pennsylvania State University, 2010
Related Documents











