Building 10, Room 9C-101, National Institutes of Health, Bethesda, Maryland 20892-1802, USA. e-mail: StephenM@intra. niddk.nih.gov doi:10.1038/nrc1610 MOLECULAR GENETICS OF MULTIPLE ENDOCRINE NEOPLASIA TYPES 1 AND 2 Stephen J. Marx Abstract | Six multiple endocrine neoplasia (MEN) syndromes have received a level of attention that might seem disproportionate to their low prevalence. The attention has been given because their hormonal excesses cause striking metabolic expressions and because they might clarify pathways disrupted in more common tumours. The recent discovery of the main gene in each MEN syndrome has furthered our understanding of not only hereditary but also sporadic tumours and has fostered new avenues of research. HYPERPARATHYROIDISM The overproduction of parathormone by the parathyroid glands. Usually secondary to an adenoma (an unregulated glandular tumour that produces parathormone in an increased quantity). LOSS OF HETEROZYGOSITY In cells that carry a mutated allele of a tumour-suppressor gene, the gene becomes fully inactivated when the cell loses a large part of the chromosome carrying the wild-type allele. Regions with high frequency of loss of heterozygosity might harbour tumour-suppressor genes. Multiple endocrine neoplasia (MEN) is defined as a disorder with neoplasms in two or more different hormonal tissues. MEN can arise in a range of ways, including the non-hereditary coincidence of two neo- plasms. However, certain MEN tumour patterns recur in seemingly sporadic cases, in several members of a family or in unrelated families — such a repeating pattern is termed a MEN syndrome 1 . Each MEN syn- drome is made up not only of benign hormone-secret- ing tumours but also of some hormonal tumours that can be malignant and/or some tumours that are not even hormone secreting 1 TABLE 1. Therefore, MEN syndromes encompass many aspects of the neoplastic process. Similarly, MEN syndromes can be regarded as a subgroup of the broader category of multiple neo- plasia syndromes. Another subgroup of the multiple neoplasia syndromes is defined by tumours that are multiple and that include only one hormonal tumour type (for example, the hyperparathyroidism–jaw tumour syndrome causes HYPERPARATHYROIDISM; Cowden syndrome causes thyroid cancer; or adenomatous polyposis of the colon causes thyroid cancer 1 ). This review focuses on the molecular genetics of MEN type 1 (MEN1) and MEN type 2 (MEN2). These two syndromes are the most striking in terms of hormonal excesses and the most prevalent of all the MEN syndromes. Furthermore, they are the only MEN syndromes in which a hormonal tumour is the most important cause of morbidity TABLE 1. They are frequently analysed as a pair; unfortunately, they are easily confused because of their similar names 1,2 (FIG. 1). Their most central clinical difference is that the associated cancer can be readily prevented or cured by early treatment in MEN2 but not in MEN1. As a consequence, gene sequencing and genetic counselling have some strikingly different roles in MEN1 versus MEN2. At a mechanistic level, their differences are also remarkable, and these differences are also emphasized in this review. The MEN1 gene and its mutations The multiple endocrine neoplasia I (MEN1) gene is at chromosome 11q13. The gene spans 9.8 kb with 10 exons in the open reading frame. It encodes a 610-amino-acid protein termed menin. MEN1 is a tumour-suppressor gene by several criteria. First, overexpression of menin in tumour cells partially suppresses the tumour phenotype 3,4 . Second, most of the 400 or so unique germline and somatic MEN1 mutations cause menin truncation or menin absence 5,6 . Third, LOSS OF HETEROZYGOSITY at 11q13 is evident in most of the tumours of MEN1 as well as in many sporadic tumours with a MEN1 mutation in the other allele 5,7 . Finally, complete loss of menin has been identified by immunohistology in tumours from patients with MEN1 or from mouse models of MEN1 REFS 8−10. NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 367 REVIEWS

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Building 10, Room 9C-101, National Institutes of Health, Bethesda, Maryland 20892-1802, USA. e-mail: [email protected]:10.1038/nrc1610
MOLECULAR GENETICS OF MULTIPLE ENDOCRINE NEOPLASIA TYPES 1 AND 2Stephen J. Marx
Abstract | Six multiple endocrine neoplasia (MEN) syndromes have received a level of attention that might seem disproportionate to their low prevalence. The attention has been given because their hormonal excesses cause striking metabolic expressions and because they might clarify pathways disrupted in more common tumours. The recent discovery of the main gene in each MEN syndrome has furthered our understanding of not only hereditary but also sporadic tumours and has fostered new avenues of research.
HYPERPARATHYROIDISMThe overproduction of parathormone by the parathyroid glands. Usually secondary to an adenoma (an unregulated glandular tumour that produces parathormone in an increased quantity).
LOSS OF HETEROZYGOSITYIn cells that carry a mutated allele of a tumour-suppressor gene, the gene becomes fully inactivated when the cell loses a large part of the chromosome carrying the wild-type allele. Regions with high frequency of loss of heterozygosity might harbour tumour-suppressor genes.
Multiple endocrine neoplasia (MEN) is defined as a disorder with neoplasms in two or more different hormonal tissues. MEN can arise in a range of ways, including the non-hereditary coincidence of two neo-plasms. However, certain MEN tumour patterns recur in seemingly sporadic cases, in several members of a family or in unrelated families — such a repeating pattern is termed a MEN syndrome1. Each MEN syn-drome is made up not only of benign hormone-secret-ing tumours but also of some hormonal tumours that can be malignant and/or some tumours that are not even hormone secreting1 TABLE 1. Therefore, MEN syndromes encompass many aspects of the neoplastic process. Similarly, MEN syndromes can be regarded as a subgroup of the broader category of multiple neo-plasia syndromes. Another subgroup of the multiple neoplasia syndromes is defined by tumours that are multiple and that include only one hormonal tumour type (for example, the hyperparathyroidism–jaw tumour syndrome causes HYPERPARATHYROIDISM; Cowden syndrome causes thyroid cancer; or adenomatous polyposis of the colon causes thyroid cancer1).
This review focuses on the molecular genetics of MEN type 1 (MEN1) and MEN type 2 (MEN2). These two syndromes are the most striking in terms of hormonal excesses and the most prevalent of all the MEN syndromes. Furthermore, they are the only MEN syndromes in which a hormonal tumour is the
most important cause of morbidity TABLE 1. They are frequently analysed as a pair; unfortunately, they are easily confused because of their similar names1,2 (FIG. 1). Their most central clinical difference is that the associated cancer can be readily prevented or cured by early treatment in MEN2 but not in MEN1. As a consequence, gene sequencing and genetic counselling have some strikingly different roles in MEN1 versus MEN2. At a mechanistic level, their differences are also remarkable, and these differences are also emphasized in this review.
The MEN1 gene and its mutationsThe multiple endocrine neoplasia I (MEN1) gene is at chromosome 11q13. The gene spans 9.8 kb with 10 exons in the open reading frame. It encodes a 610-amino-acid protein termed menin. MEN1 is a tumour-suppressor gene by several criteria. First, overexpression of menin in tumour cells partially suppresses the tumour phenotype3,4. Second, most of the 400 or so unique germline and somatic MEN1 mutations cause menin truncation or menin absence5,6. Third, LOSS OF HETEROZYGOSITY at 11q13 is evident in most of the tumours of MEN1 as well as in many sporadic tumours with a MEN1 mutation in the other allele5,7. Finally, complete loss of menin has been identified by immunohistology in tumours from patients with MEN1 or from mouse models of MEN1 REFS 8−10.
NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 367
R E V I E W S

MEN1 germline mutation causes a pleiomorphic multiple neoplasia syndrome. MEN1 is the gene mutated in the largest range of endocrine tumour types and per-haps in the widest range of all tumour types5,7; however, MEN1 mutation testing in tumours is not used clinically
because it does not have implications for tumour staging. No clear correlation of MEN1 genotype with phenotype has emerged to date.
MEN1 sequencing is usually justified by the potential for information to patient and caregiver, but carries an important limitation. Sequencing of MEN1, unlike that of the gene mutated in MEN2, cannot be the guide for major intervention in most patients. MEN1 sequenc-ing has been done by several laboratories. These detect a germline MEN1 mutation in about 70% of typical MEN1 kindreds5,11. Most of the remaining 30% are false negatives — they have mutations in the MEN1 gene that are not detected by current sequencing methods. The typical familial MEN1 syndrome is probably due to the same gene, as no family with MEN1 has shown disso-ciation from 11q13. An atypical subgroup of patients with sporadic MEN1, manifest by hyperparathyroidism and pituitary tumours, have a much lower yield for detection of MEN1 germline mutation, and likely includes a process (or processes) distinct from MEN1 syndrome12. Two large families with isolated anterior-pituitary tumour have shown linkage to 11q13 REFS 13,14. Neither these large families nor over 50 smaller ones with similar phenotypes have shown any mutation in MEN1; therefore, efforts are directed at identifying other gene(s), particularly one at 11q13 that could be very near to MEN1 REF. 14.
Mouse models of MEN1. Studies in mice have revealed new information about the tissue-specific consequences of Men1 loss of function. Knocking out both Men1 alleles in the mouse germline is lethal in the embryo, causing disorganization in many tissues but not signs of neoplasia9,15. The conditional knockout of both Men1 alleles in the islet β-cells or in the parathyroid
Summary
• Multiple endocrine neoplasia (MEN) type 1 (MEN1) and MEN type 2 (MEN2) are the most striking MEN syndromes in terms of hormonal excesses. They differ in that both are potentially lethal from associated cancer but only MEN2-related cancer can be prevented. Similar names cause confusion between them.
• MEN1 sequencing gives useful information about the MEN1 carrier status. This test is not a clinical requirement for relatives of individuals with MEN1 as it is not a major guide for therapy.
• Men1+/– mice are a good model of MEN1. Features that differ from MEN1 loss in man include a higher penetrance of pheochromocytoma and a stage of polyclonal hyperplasia in pancreatic islet cells as the precursor to insulinoma.
• RET mutations cause three variants of MEN2 with three grades of calcitonin-producing-cell (C-cell) cancer and with a strong correlation of RET genotype to phenotype.
• RET sequencing is recommended in relatives of individuals with MEN2 as a guide for the successful management for C-cell cancer and pheochromocytoma.
• Tumorigenesis after mutation of MEN1 (a tumour suppressor) follows a typical two-hit loss-of-function process.
• Tumorigenesis after mutation of RET (an oncogene) follows a stepwise process; sometimes this involves a second hit in a RET allele. This probably causes further imbalance towards RET gain of function.
• The roles of menin (the protein encoded by MEN1) in tumorigenesis are obscured by its large number of candidate partners and candidate physiologies. A specific anti-MEN1 drug cannot be contemplated until MEN1 function is better understood.
• RET mutations are oncogenic owing to a gain of function in RET’s intrinsic receptor tyrosine kinase activity. Tyrosine-kinase inhibitors are in clinical trials for RET-related neoplasms.
Table 1 | Six multiple endocrine neoplasia syndromes and their main characteristics
Syndrome Benign tumours and/or hormonal excess
Malignant tumours and/or hormonal excess
Non-hormonal neoplasia (benign or malignant)
Multiple endocrine neoplasia type 1
Islet non-secreting, pituitary non-secreting, adrenal cortex, parathyroid hormone*, gastrin*, insulin*, prolactin*, growth hormone*, adrenocorticotropic hormone
Foregut carcinoid*, gastrin‡, glucagon, vasoactive intestinal polypeptide, pancreatic polypeptide
Angiofibroma, collagenoma, lipoma, leiomyoma, meningioma, ependymoma
Multiple endocrine neoplasia type 2 variants
C-cell cancer, adrenal chromaffin, calcitonin*, catecholamine*, parathyroid hormone*
C-cell cancer, adrenal chromaffin, calcitonin‡, catecholamine*
Neuroma (MEN2B)*
Carney complex Cortisol*; thyroxine; growth hormone; prolactin; androgen or oestrogen
Thyroxine Heart myxoma‡, lentigine schwannoma
McCune–Albright syndrome
Androgen or oestrogen*; growth hormone; prolactin; cortisol; thyroxine
– Fibrous dysplasia of bone‡, café-au-lait spots, sarcoma (rare)
Von Hippel–Lindau disease
Catecholamine*, insulin Catecholamine* Renal-cortex cancer‡, haemangioblastoma, retinal angioma
Neurofibromatosis type 1
Duodenal carcinoid, catecholamine
– Neurofibroma‡, café-au-lait spots, Lisch nodules, freckling
*An important clinical feature of the syndrome. ‡Indicates the single most important clinical feature in any multiple endocrine neoplasia (MEN). ACTH, adrenocorticotropic hormone. C-cell, calcitonin-producing cell.
368 | MAY 2005 | VOLUME 5 www.nature.com/reviews/cancer
R E V I E W S

PENETRANCEThe frequency with which individuals who carry a given mutation show the manifestations associated with that mutation. If the penetrance of a disease allele is 100%, then all individuals carrying that allele will express the associated phenotype.
PHEOCHROMOCYTOMAA neuroendocrine tumour that typically arises in the adrenal medulla. These tumours can be benign or malignant. Symptoms often relate to the ability of these tumours to secrete catecholamines.
cells results in the growth of insulinoma or of parathy-roid adenoma10,16,17. However, conditional knockout of Men1 in the hepatocyte has no effect, emphasizing the tissue selectivity of neoplasia from the menin-null status18. Germline heterozygosity for Men1 mutation results in a normal conceptus, and these animals develop endocrine tumours after the age of 9 months10,19. The commonest tumours in these mutant mice are prolac-tinoma, insulinoma and parathyroid adenoma. As these tumours are commonly seen in patients with MEN1, this is an excellent model of MEN1. Three important differences between mouse and human MEN1 have been informative. First, the prevalence of MEN1-associated gastrinoma is lower in the mouse (0−10%) than in man (40%); this is unexplained, other than by species or strain specificity. In this regard, three large human kindreds with MEN1 also show an unusually low (5%) and otherwise unexplained PENETRANCE for gastrinoma20. Second, the Men1-heterozygous mice develop PHEOCHROMOCYTOMA (7%), supporting this as a weakly penetrant MEN1 tumour. The penetrance of pheochromocytoma in human MEN1 is even lower
(below 1%), and it has not been widely recognized as inherent in MEN1 REF. 21. Third, the mice develop giant hyperplasia of the pancreatic islets. This is a precursor stage for the monoclonal insulinoma. The hyperplastic precursor is polyclonal by the criteria of retention of the wild-type Men1 allele and retention of menin by immunohistology10. By contrast, a hyperplastic tumour precursor stage has not been documented in any tissue in patients with MEN1 REF. 22.
THE RET gene and its mutationsThe rearranged during transfection (RET) protoonco-gene is near the centromere of chromosome 10. It spans 60 kb with 21 exons and encodes a protein of approxi-mately 1,100 amino acids. The protein is a transmem-brane receptor tyrosine kinase (RTK), termed RET23–25. Almost all of the germline RET mutations in MEN2 are missense; the rest are small deletions or insertions that also preserve the RET open reading frame. MEN2 is likely to be all from the RET gene, as no MEN2 fam-ily has been identified that excludes the RET locus. The RET germline mutations characterized in MEN2 are concentrated in only a small fraction of the open reading frame (FIG. 2); most are in the cysteine-rich portion of the extracellular domain. Their limitation to missense codons and their highly focused distribu-tion are characteristic of mutations that cause a gain of function, in this case in the RET RTK activity25. This contrasts with the loss-of-function RET mutations that cause many cases of familial HIRSCHSPRUNG DISEASE; such mutations are generally of the stop-codon type and show a much broader distribution across the RET open reading frame5,26. Hirschsprung disease, or megacolon with aganglionosis of the colon, is a phenotype remotely reminiscent of MEN2 only insofar as either can affect the intestinal nerves26,27. Occasional MEN2 families have several affected members that surprisingly also express Hirschsprung disease. Loss of function at the RET pro-moter or its interactors has been suggested to cause the Hirschsprung part of this two-component rarity28.
RET-gene sequencing in patients. Oncogenic RET mutations show striking and important correlations with the variant of the MEN2 phenotype (FIGS 2,4). The commonest phenotypic variant is termed MEN2A; this has combinations of CCELL cancer (also termed MEDULLARY THYROID CANCER), pheochromocytoma and hyperparathyroidism. 90% of the RET muta-tions in MEN2A occur in only 6 cysteines in a small 25-amino-acid domain in the extracellular region1. MEN2B is a distinct variant in which C-cell cancer begins at a much earlier age and is more aggres-sive than in MEN2A. Almost all RET mutations in MEN2B are confined to one cytoplasmic amino acid — Met918Thr. A third MEN2 variant is termed FAMILIAL ISOLATED MEDULLARY THYROID CANCER (FMTC); most of its heterozygous RET mutations are in the same amino acids as those in MEN2A. Other mutations seemingly specific to FMTC are in amino acid 533 of the extracellular domain30 and amino acids 791−891 of the cytoplasmic domain1. As a modest-sized kindred
Figure 1 | Endocrine tumours expressed in multiple endocrine neoplasia types 1 and 2. Only the most important tumours are depicted. Similarities and differences of clinical features are tabulated. The difference in the curability of cancers determines the central differences in clinical management. A benign tumour is shown in green. One with high malignant potential is shown in yellow. For multiple endocrine neoplasia type 1 (MEN1), only the commonest tumours are shown.
Pituitary Nerve (MEN2B)
Parathyroid
Bronchial carcinoid
Enteropancreatic
Adrenal chromaffin
Thyroid C-cell
Similarities Differences
High penetrance for hormonal tumourParathyroid adenomas in eachMany good treatments available for most symptomsIncludes life-threatening hormone/metabolicexpression (i.e. gastrin in MEN1 or catecholamines in MEN2) that now is well managed
•••
•
Distinct variants only in MEN2Unique organ grouping for each MEN
••
Metabolic expressions
Similarities Differences
Usually indolentCancer is potentially lethal in 30% of all carriersif not cured
••
Unique organ grouping for each MENGood prevention or cure for an associated cancer by early surgery inMEN2 only
••
Malignant expressions
MEN1 MEN2
NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 369
R E V I E W S
HIRSCHSPRUNG DISEASEHirschsprung disease is characterized by a congenital absence of ganglion cells in the distal colon, resulting in a functional obstruction. Both the myenteric (Auerbach) and submucosal (Meissner) plexus are absent, resulting in reduced bowel peristalsis and function.
CCELLSCalcitonin-producing cells. These form a distinct extrathyroidal gland (the ultimobranchial gland) in all lower vertebrates such as fish and amphibians but are dispersed and entirely within the thyroid gland in mammals. In this latter location, they are also termed parafollicular cells.
MEDULLARY THYROID CANCERMedullary (or C-cell) thyroid cancer. A neoplasm of the calcitonin-producing C-cells in the thyroid gland.
FAMILIAL MEDULLARY THYROID CANCERA variant of multiple endocrine neoplasia type 2 (MEN2) that can also be considered as a very mild extreme of MEN2A.
PENTAGASTRINA synthetic polypeptide that has effects like gastrin. It stimulates the secretion of gastric acid, pepsin and intrinsic factor, and has been used as a diagnostic aid for multiple endocrine neoplasia type 2 by promoting calcitonin secretion.
PERIADRENAL GANGLIONEUROMAA tumour (neuroma) containing ganglion cells that arises from nerves in the periadrenal tissue.
with occult MEN2A could present as FMTC and as several large families with mutations in the cytoplasmic codons typical of FMTC have shown other tumours of MEN2A31, FMTC can also be considered as a disease at the mild end of the MEN2A clinical spectrum. FMTC has also been characterized as a recessive disease in two small kindreds with Val804Met or Ala883Thr32,33. The Val804Met mutation in heterozygotes has also presented as FMTC in other kindreds34,35.
Carrier testing in MEN2 variants has long been a model for management of this familial cancer. It has been successful because of the accurate assessment of the carrier state before tumour development and also because the preneoplastic C-cells all reside in the thyroid, an organ that can be removed and then com-pensated for easily with oral thyroid hormone. Serum calcitonin levels after PENTAGASTRIN challenge had been developed as an index of C-cell hyperfunction and therefore of the MEN2 carrier state, but RET sequenc-ing has almost completely replaced this. In some labo-ratories, over 95% of MEN2 families have been shown to have an identifiable RET germline mutation5,36.
The recommended age by which RET testing and prophylactic thyroidectomy should be completed is highly dependent on MEN2 phenotype and RET genotype2. Although MEN1 and MEN2 have a similar estimated frequency (1 in 30,000)5, there is less use of germline sequencing of MEN1 than of RET. This is mainly because MEN2 presents a stronger clinical justification for gene sequencing; in addition, the more localized mutations (FIG. 2) result in fewer base pairs to be sequenced for RET, and the RET test has a lower false-negative rate (5% versus 30%).
Met918Thr mutations are overrepresented among the somatic RET mutations in sporadic C-cell cancer, and the sporadic C-cell cancers with this mutation have a more aggressive course25. Therefore, RET test-ing might also prove useful for staging of sporadic C-cell neoplasias.
Mouse models of MEN2. There are two major RET iso-forms of 1,072 and 1,114 amino acids that result from alternate splicing distal to glycine 1063. Each isoform has transforming activity, but only the shorter form is essential for embryological development in the mouse37. Two groups expressed a MEN2A RET mutation in the mouse from the calcitonin promoter. In one model, the short RET isoform was mutated and caused multifo-cal C-cell cancer38. In the other, the long isoform was mutated with a comparable result39. When the same mutant RET was expressed on distinct genetic back-grounds, the penetrance for C-cell cancer varied from 0% to 98%; the strain effect might have been specific for C-cell cancer or it might have been specific for the calcitonin promoter39. A more physiologically valid RET promoter is needed for an improved non-human model of MEN2A. Expression in mice of the human mutant allele that is present in MEN2B did not cause any MEN2-like tumour40. When a MEN2B-like murine Ret allele was expressed in mice, the heterozygotes showed C-cell hyperplasia and pheochromocytoma; homozygotes showed only a moderately more severe phenotype that included PERIADRENAL GANGLIONEUROMAS but not C-cell cancer up to the age of 12 months41. So, germline transmission of either RET heterozygous and homozygous mutations is evident in man and mouse. RET (as well as the MET and KIT tyrosine-kinase genes) is one of very few oncogenes that is transmitted through the germline42. This complements the view that most other oncogenes are lethal during embryogenesis.
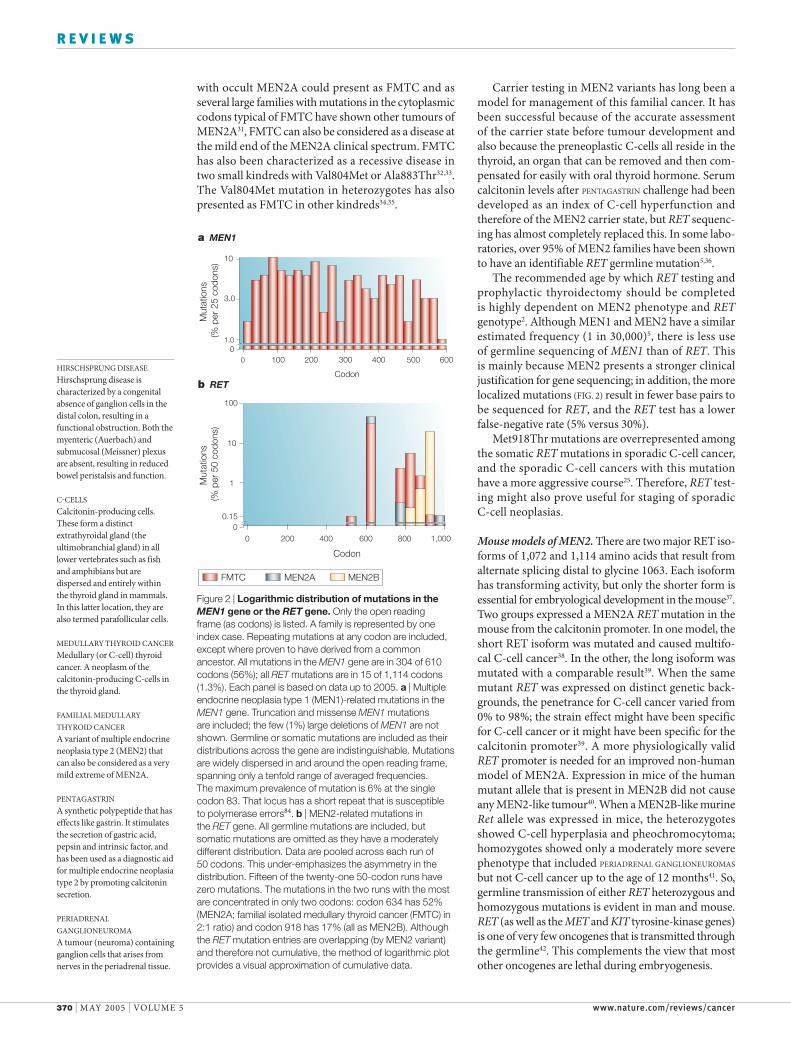
Figure 2 | Logarithmic distribution of mutations in the MEN1 gene or the RET gene. Only the open reading frame (as codons) is listed. A family is represented by one index case. Repeating mutations at any codon are included, except where proven to have derived from a common ancestor. All mutations in the MEN1 gene are in 304 of 610 codons (56%); all RET mutations are in 15 of 1,114 codons (1.3%). Each panel is based on data up to 2005. a | Multiple endocrine neoplasia type 1 (MEN1)-related mutations in the MEN1 gene. Truncation and missense MEN1 mutations are included; the few (1%) large deletions of MEN1 are not shown. Germline or somatic mutations are included as their distributions across the gene are indistinguishable. Mutations are widely dispersed in and around the open reading frame, spanning only a tenfold range of averaged frequencies. The maximum prevalence of mutation is 6% at the single codon 83. That locus has a short repeat that is susceptible to polymerase errors84. b | MEN2-related mutations in the RET gene. All germline mutations are included, but somatic mutations are omitted as they have a moderately different distribution. Data are pooled across each run of 50 codons. This under-emphasizes the asymmetry in the distribution. Fifteen of the twenty-one 50-codon runs have zero mutations. The mutations in the two runs with the most are concentrated in only two codons: codon 634 has 52% (MEN2A; familial isolated medullary thyroid cancer (FMTC) in 2:1 ratio) and codon 918 has 17% (all as MEN2B). Although the RET mutation entries are overlapping (by MEN2 variant) and therefore not cumulative, the method of logarithmic plot provides a visual approximation of cumulative data.
01.0
10
3.0
Mut
atio
ns
(% p
er 2
5 co
dons
)M
utat
ions
(%
per
50
codo
ns)
0 100 200 300 400 500 600
Codon
0
100
10
0 200 400 600 800 1,000
Codon
FMTC MEN2A MEN2B
a MEN1
b RET
1
0.15
370 | MAY 2005 | VOLUME 5 www.nature.com/reviews/cancer
R E V I E W S

SECONDHITIn most medical contexts, this step is a mutation in the remaining normal allele. Gene methylation is an epigenetic step that can have similar consequences.
Hyperplasia of MEN2 tumour precursor cells in chromaffin tissues and C-cell tissues is widely recog-nized for man and mouse43,44. Although tumours are widely assumed to be polyclonal in MEN2, this has not been proven. By contrast, one study suggested that C-cell hyperplasia of human MEN2 was monoclonal and progressed during embryogenesis45.
Multistep tumorigenesisAs for most hereditary cancers, a germline first hit in the MEN1 or RET gene has little or no phenotype for all cells in the conceptus. However, in some tis-sues from mouse models or patients with MEN1 or MEN2 there is evidence for polyclonal hyperplasia as a precursor to monoclonal tumour (see above). It is not clear that this hyperplasia is an obligatory step towards monoclonal expansion. Still, recognition of a polyclonal tumour precursor stage, sometimes termed haploinsufficiency or a dosage effect46, raises the possibility of developing drugs directed at early stages, perhaps even before tumour progression to monoclonal stages that would be more refractory to treatment.
MEN1 is a tumour-suppressor gene. Typically, loss of function of MEN1 occurs as a single-base DNA change within MEN1 for a first hit and a large rear-rangement of chromosome 11 for the SECOND HIT47 (FIG. 3). In contrast to MEN1, which is a tumour suppressor, RET is an oncogene. The first hit at RET is virtually always an in-frame missense mutation at a selected codon. Second-hit mutations at the RET locus have been identified in some MEN2 tumours32,33,48−50 (FIG. 3). It is likely that each of these second hits causes an imbalance in expression of RET alleles with further gain of function of the RET oncogene. It is not clear whether two hits at the RET locus are a requirement for all RET-associated tumours or even what fraction of RET-related tumours have such a process51.
Knudson developed the two-hit model of tumorigenesis to explain the earlier age of onset and the greater number of tumours in hereditary retinoblastoma compared with sporadic cases. This two-hit model can be applied to the early onset age and multiplicity of tumours in MEN1 or in MEN2 REF. 52. That is to say, the model is not restricted to tumour-suppressor genes.
As in most or all cancers, tumorigenesis in MEN1 or MEN2 is a stepwise process. While many tumours show two hits at one locus (in particular, MEN1 or RET), all or most of the tumours also express muta-tional events at loci other than MEN1 or RET. This has been shown by gene sequencing, loss of heterozygosity or comparative genome hybridization1,10,51,53,54.
Menin and RET molecules and pathwaysModelling of tumorigenesis as a multistep process does not assign to the central gene a defect in any molecular pathway or even in any subcellular com-partment. Function has generally been approached, using cell-biology-based methods.
Menin pathways. Molecular partnering studies have identified at least 21 candidate molecules or complexes as possible menin interactors55 TABLE 2. Most, but not all, are nuclear. Documention of two nuclear-localization signals in menin supports the importance of menin’s nuclear localization56. The large number of proteins that bind menin and the number of different cellular pathways that these
Figure 3 | The genetic activation of RET. Stepwise tumorigenesis is sometimes comparable from MEN1, a growth suppressor, or from RET, a growth promoter. a | MEN1 is a tumour-suppressor gene. The first hit at a typical tumour-suppressor gene is commonly in one base. It can cause missense*, splice defect* or premature stop*. Intragenic insertion* or deletion*, and sometimes much larger deletions*, occur. In the precursor cell for somatic tumour, the first hit might be epigenetic (methylation). The second hit is almost always at the same locus (two-hit model) and can be by any of these mechanisms*, but subchromosomal rearrangement* or deletion of an entire chromosome* is selectively more common in the second hit. Starred mechanisms have been identified in MEN1 tumorigenesis. Only one representative mechanism of the first and second hit is pictured. b | RET is an oncogene. The first hit in hereditary and some sporadic RET-related tumours is a single codon missense mutation causing gain of function. Five types of second-hit mutation have been recognized at the RET locus; their prevalence among all RET-related tumours is not known. Data are from C-cell cancer or pheochromocytoma of multiple endocrine neoplasia type 2A (MEN2A), from sporadic C-cell cancer, or from human thyroid tumour cells of C-cell-cancer origin. RET–PTC tumorigenesis is not shown. Germline homozygosity for a missense RET mutation32,33 is technically not a two-hit process, as both mutations are acquired simultaneously as ‘one-hit’ in the germline. The following mechanisms are depicted: somatic duplication of the mutant RET allele in trisomy of chromosome 10 REF. 48; somatic tandem duplication of the mutant RET allele on one copy of chromosome 10 REF. 49; somatic point mutation of the wild-type (WT) RET allele in MEN2A or FMTC50; and somatic loss of the WT RET allele48. Only this latter hit involves loss of a RET allele. Analogous to the four other types of change, it might cause gain of function at the receptor tyrosine kinase through imbalanced expression at the mutant allele.
a MEN1
Loss
b RET
Gain
Gain
Gain
Loss Subchromosome loss
Germlinehomozygote
1st hit 2nd hit
Loss/gain
Trisomy
Tandemamplification
Point mutationon WT allele
Loss ofWT allele
NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 371
R E V I E W S

COMPASSA multisubunit transcriptional complex in yeast with histone-methyltransferase activity. All yeast lack menin, but most of the COMPASS subunits have homologues in mammals. The human proteins include menin, MLL, ASH2L, RBBP5, WDR5, HCF1 and PolII.
MLLEncodes a part of the COMPASS-like complex. It contributes the carboxy-terminal part of an oncogene in a fusion protein, causing leukaemia/lymphoma.
GFRα Glial-cell-derived neurotrophic factor family receptor-α. A glycosylphosphatidyl-inositol-anchored plasma membrane extracellular co-receptor. It binds an extracellular ligand, and it is able to interact directly with RET.
proteins can regulate (see below) could mean that menin is promiscuous. It is more likely though that the important biological partners and affected path-ways will ultimately prove to be one or few. So, herein we focus on JUND and the COMPASS-like complex that appear to be particularly important.
JUND, which belongs to the AP-1 or JUN–FOS family of early-acting transcription factors, was the first menin partner to be identified. Under the same biological environment JUND is unlike other AP-1 family members as it is a growth suppressor, whereas all the other members are growth promot-ers57. Menin binds to the amino-terminus of JUND and can inhibit JUND-activated transcription on an artificial promoter57. There is limited informa-tion about the identities of genes that might be up- or downregulated by this menin−JUND inter-action4. Studies with stable transfected fibroblast lines indicated that menin loss converts JUND from a growth suppressor to a growth promo-tor58,59. Thus, menin interaction can account for JUND’s feature as the only outlier in the AP-1 family, and this interaction also provides a plausible mechanism of how biallelic loss of function at menin can initiate tumours.
Menin co-immunoprecipitates with several mol-ecules such as mixed lineage leukeamia MLL 2 that are homologues of members of a transcriptional complex (COMPASS-like) in yeast60. A similar menin-containing complex was identified with a strategy based on immunoprecipitation of Set1p, a homologue of MLL61. The COMPASS-like complex has several chromatin-related activities, including a histone methyltransferase (HMT) activity for lysine 4 on histone 3. MLL that has lost its HMT activity can be oncogenic when fused to one of several other proteins in cases of human myeloid and lymphoid leukaemia62,63. Menin can recruit members of the COMPASS-like complex to certain promoters. The direct or indirect binding to such a promotor can result in the transcription of HOX genes or the tumour-suppressor genes CDKN2C, which encodes p18 (also known as INK4C), and CDKN1B, which encodes p27 (also known as KIP1). This is one
potential mechanism for menin-mediated inhibition of proliferation8.
Menin has been shown to bind the promoters of several genes8,60,64 TABLE 2. As no specific menin-binding element in the DNA has been identified, menin’s interaction with promotors might be indirect and in complex with other transcription factors.
Downstream actions of menin have been stud-ied less, and very different mechanisms of menin-induced tumorigenesis have been suggested. Menin-null tumours in mice showed increased proliferation without decreased apoptosis9. Menin overexpression in normal fibroblasts led to increased apoptosis65. Several additional studies have indicated that menin stabilizes the genome37,66. Menin knock-out in Drosophila melanogaster resulted in virtually no phenotype, but there was modestly increased sen-sitivity to DNA damage from nitrogen mustard67. By contrast, mouse MEN1 tumours did not show DNA instability68. Therefore, recent data could support a functional role for menin in inhibiting cell pro-liferation, in promoting apoptosis or in increasing genomic stability.
RET pathways. Normal RET is expressed mainly in developing and adult neural ectoderm. The mecha-nisms of RET proximal signalling in tumorigenesis are known in considerable detail, in part from its place in the well-studied RTK family23,24. The RET protein is a subunit in a plasma-membrane signal-ling complex that includes four ligands and four RET co-receptors25,69 (FIG. 4). None of the four lig-ands has been implicated in a MEN2-like state, but mutation of a glial-cell-derived neurotrophic factor family receptor-α4 GFRα4 co-receptor has been sug-gested as a contributor to an MEN2-like state with or without RET mutation70,71. The RET intracellular domain has at least 12 autophosphorylation sites72,73. Phosphotyrosine 1062 in particular is a binding site for many different docking proteins, including SHC, SHCC, IRS1, IRS2, FRS2, DOK1, DOK4, DOK5 and enigma69. Tyrosine 1062 is important for the transforming activity of RET69,73. A MEN2-related mutation that adds a bulky side chain at this site confers resistance to small RTK inhibitors74. MEN2-associated RET mutations cause gain of function at RET tyrosine-kinase signalling in several ways. The extracellular cysteine mutations concentrated in MEN2A cause RET homodimerization, whereas the Met918Thr mutation of MEN2B causes RET auto-phosphorylation. No specific gain-of-function mech-anism has been connected to mutations in FMTC. The various RET–PTC mutations might cause gain of function in RET through the selection of tissue for RET expression, through RET compartmentalization or through RET homodimerization75.
RET tyrosine kinase activates a series of cytoplasmic kinases, including JUN kinase, p38 mitogen activated protein kinase, RAS/extracellular signal-regulated kinase, phophatidylinositol 3-kinase and AKT69. The transmembrane RTKs are all believed to activate a
Table 2 | Protein or DNA partners of menin
Functional category Candidate partner of menin*
Cytoskeleton Non-muscle myosin heavy chain IIA, glial fibrillary acidic protein, vimentin
GTPase activity NM23β
Transcription factor JUND, SIN3A, NF-κB subunits, PEM, SMAD1, SMAD3, SMAD5, RUNX2, COMPASS-like complex
DNA processing RPA2, FANCD2, ASK, double-stranded DNA
Gene promoter TERT, HOXC8, p18 (INK4C), p27 (KIP1)
*Direct binding to menin has been demonstrated for only a subset of these. Detailed references for a similar table are available in REF. 54. ASK, activator of S-phase kinase; COMPASS, complex proteins associated with Set1; FANCD2, Fanconi anaemia complementation group D2; HOXC8, homeobox C8; NF-κB, nuclear factor-κB; RPA2, replication protein A2; RUNX2, Runt-related transcription factor 2; TERT, telomerase reverse transcriptase.
372 | MAY 2005 | VOLUME 5 www.nature.com/reviews/cancer
R E V I E W S
CHROMAFFIN TUMOURA neoplasm composed of chromaffin cells occurring in the medullae of adrenal glands, the organs of Zuckerkandl, or the paraganglia of the thoracolumbar sympathetic chain. Some chromaffin tumours secrete catecholamines.
downstream transcriptional programme, with differ-ing details for each RTK76,77. In the case of RET, the consequence at the level of the whole cell is confus-ing, as there is evidence for increased proliferation, decreased proliferation and increased apoptosis78.
MEN-overlap syndromes in man or mouseCases and small families with various combinations of features from several MENs have been termed MEN-overlap syndromes. With recognition of a broadened tumour spectrum, including sometimes rare tumour types in each MEN syndrome, virtually all such overlaps have recently been reclassified as likely to be an unusual expression of just one MEN syndrome5. For example, MEN1 rarely includes phe-ochromocytoma (see above), or von Hippel−Lindau syndrome rarely includes insulinoma.
Based on rodent models, additional causes for some human cases of overlap remain possible. A syndrome overlapping MEN1 and MEN2 has been described in the rat; it includes C-cell cancer, CHRO
MAFFIN TUMOURS, pituitary tumours and parathyroid hyperplasia. The causative gene is transmitted as a recessive and has not been identified79. Heterozygous
loss of Rb predisposes mice to C-cell thyroid cancer and pituitary tumours of the anterior and middle lobe80. Various combinations of knockouts in the Cdkn2c, Cdkn1a and Cdkn1b genes were engineered in the mouse81. These genes encode cyclin-depend-ent kinase inhibitors; a knockout of any one alone rarely leads to an endocrine tumour. Surprisingly, knockouts of Cdkn2c and Cdkn1b together caused combinations of all the major tumours of MEN1 and MEN2. Thus, MEN1 and MEN2 tumours might develop, in part, through defects in a common cell-cycling pathway, involving RB and/or cyclin-depend-ent-kinase inhibitors.
Tissue selectivity of tumoursTissue selectivity of tumours is at the very essence of the MEN syndromes, as it is for the broader cat-egory of all neoplasias. None of the identified MEN genes directly participates in the processes of hor-mone secretion1. Furthermore, each of the causative genes analysed so far in MEN is quite broadly, if not universally, expressed and thereby does not explain the tissue selectivity of its related syndrome1. RET is expressed in developing neural ectoderm and adult nerve tissue, a pattern that correlates best with its loss-of-function mutations in Hirschsprung disease and in animal models of neural development27. As tis-sue selectivity is mainly the function of promoter ele-ments, this feature in MEN1 and MEN2 will probably be clarified when an appropriate promoter-driven tissue-selective process interacting with MEN1 or RET is identified.
Prospects for specific interventionMost drugs used in MEN1 and MEN2 act on synthe-sis, secretion or action of a hormone; for example, suppressers of catecholamine synthesis, dopamine agonists that target prolactin secretion and proton-pump blockers that inhibit gastrin action. These are effective treatments for the hormonal excess, but, with notable exceptions (dopamine agonists), not for cell growth or progression of the associated malignancies.
The best target for a truly specific intervention in a MEN tumour would seem to be the mutant gene, its mRNA or its protein. Its molecular partners upstream or downstream are possible targets but would be more likely to introduce problems of incomplete action or undesired crossover into other pathways. DNA- or RNA-directed therapies are not evaluated here, as these technologies do not appear sufficiently mature in MEN1 or MEN2 for clinical trials. Targeting of the menin protein has not been attempted because its function and its protein domains that are important in tumorigenesis still need to be identified. Several RTKs have already been successfully targeted with small tyrosine-kinase inhibitors, such as STI571 or imatinib23,82. Related compounds are under explora-tion to inhibit RET83, and trials in humans are under-way in the hope of treating C-cell cancer and several other RET-related neoplasms.
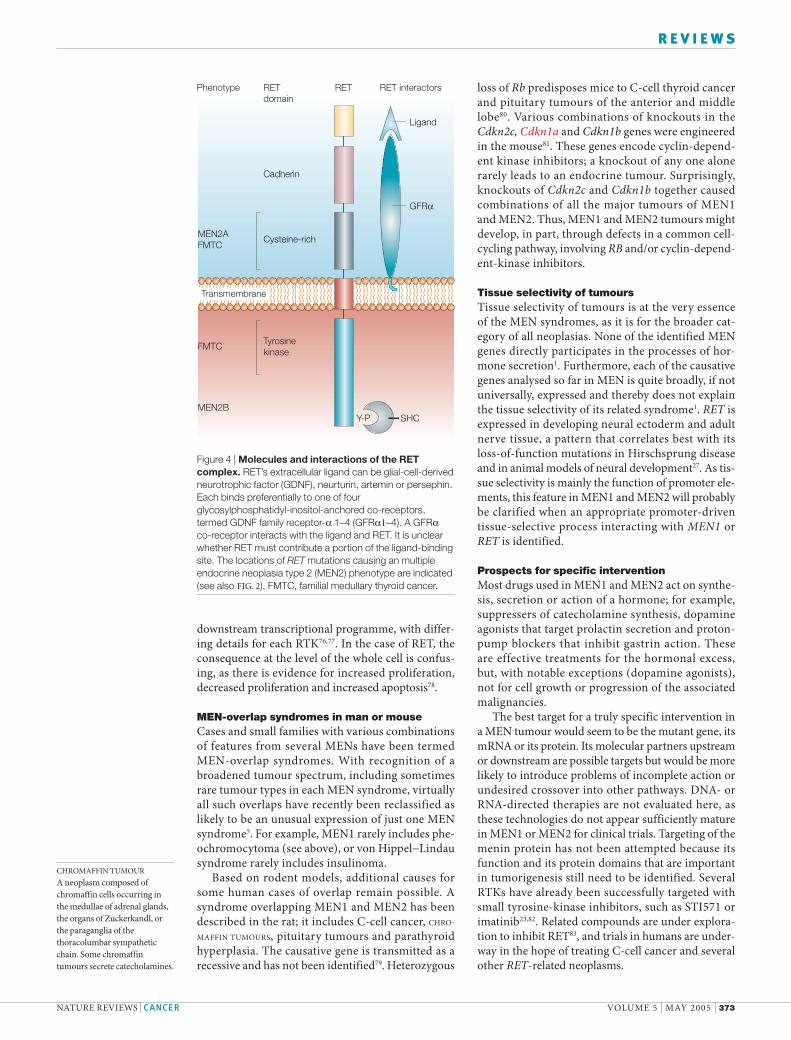
Figure 4 | Molecules and interactions of the RET complex. RET’s extracellular ligand can be glial-cell-derived neurotrophic factor (GDNF), neurturin, artemin or persephin. Each binds preferentially to one of four glycosylphosphatidyl-inositol-anchored co-receptors, termed GDNF family receptor-α 1–4 (GFRα1–4). A GFRα co-receptor interacts with the ligand and RET. It is unclear whether RET must contribute a portion of the ligand-binding site. The locations of RET mutations causing an multiple endocrine neoplasia type 2 (MEN2) phenotype are indicated (see also FIG. 2). FMTC, familial medullary thyroid cancer.
MEN2AFMTC
MEN2B
Cysteine-rich
FMTC Tyrosinekinase
Cadherin
Ligand
GFRα
Y-P SHC
Phenotype RETdomain
RET RET interactors
Transmembrane
NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 373
R E V I E W S

ConclusionDiscovery of the causative genes has changed the management of both MEN1 and MEN2. In particu-lar, RET mutations predict MEN2 phenotype, and identifying these promotes important clinical inter-ventions. Mutation in the MEN1 or RET genes also contributes to many common endocrine tumours. MEN1 is a tumour-suppressor gene, acting through unknown pathways; MEN1 loss of function might
act in tumorigenesis to convert JUND from a growth suppressor to a growth promoter or to regulate gene expression through a multimeric transcription complex. RET is an oncogene by virtue of mutations that induce a gain of function in its RTK activity. In the future a fuller understanding of the function of MEN1 and RET should lead to development of drug thereapies for MEN1- and MEN2-related cancers and for the related sporadic cancers.
1. Marx, S. J. & Simonds W. F. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr. Rev. 4 Jan 2005 (doi:10.1210/er.2003-0037).
2. Brandi, M. L. et al. Guidelines for diagnosis and therapy of multiple endocrine neoplasia type 1 and type 2. J. Clin. Endocrin. Metab. 86, 5658–5671 (2001).Consensus guidelines from an international panel. The guidelines consolidated a large body of information without introducing controversy.
3. Kim, Y. S. et al. Stable overexpession of MEN1 suppresses tumorigenictiy of RAS. Oncogene 18, 5936–5942 (1999).
4. Stalberg, P. et al. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, δ-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J. Clin. Endocrinol. Metab. 89, 2326–2337 (2004).
5. Gagel, R. F. & Marx, S. J. in Williams Textbook of Endocrinology 10th Edn (eds Larsen, P. R., Kronenberg, H., Melmed, S. & Polonsky, K.) 1717–1762 (WB Saunders & Company, Orlando, 2002).
6. Kikuchi, M., Ohkura, N., Yamaguchi, K., Obara, T. & Tsukada, T. Gene dose mapping delineated boundaries of a large germline deletion responsible for multiple endocrine neoplasia type 1. Cancer Lett. 208, 81–88 (2004).
7. Heppner, C. et al. Somatic mutation of the MEN1 gene in parathyroid tumors. Nature Gen. 16, 375–378 (1997).
8. Milne, T. A. et al. Menin and MLL cooperativelyy regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl Acad. Sci. USA 102, 749–754 (2005).
9. Crabtree, J. S. et al. A mouse model of MEN1 develops multiple endocrine tumors. Proc. Natl Acad. Sci. USA 98, 1118–1123 (2001).
10. Crabtree, J. S. et al. Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 23, 6075–6085 (2003).This paper documents the successful development of immunohistochemistry for menin and showed that islet hyperplasia is a MEN1 haploinsufficiency state.
11. Klein, R. D., Salih, S., Bessoni, J. & Bale, A. E. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet. Med. 7, 131–138 (2005).
12. Hai, N., Aoki, N., Shimatsu, A., Mori, T. & Kosugi, S. Clinical features of multiple endocrine neoplasia type 1 (MEN1) phenocopy without germline MEN1 gene mutations: analysis of 20 Japanese sporadic cases with MEN1. Clin. Endocrinol. 52, 509–518 (2000).
13. Gadelha, M. R., Kineman, R. D. & Frohman, L. A. Familial somatotropinomas: clinical and genetic aspects. Endocrinologist 9, 277–285 (1999).
14. Beckers, A. Familial isolated pituitary adenomas. J. Int. Med. 255, A698 (2004)
15. Bertolino, P. et al. Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech. Dev. 120, 549–560 (2003).
16. Libutti, S. K. et al. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 63, 8022–8028 (2003).
17. Biondi, C. A. et al. Conditional inactivation of the Men1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Mol. Cell. Biol. 24, 3125−3131 (2004).
18. Scacheri, P. C. et al. Homozygous loss of menin is well tolerated in liver, a tissue not affected in MEN1. Mam. Genome 15, 872–877 (2004).
19. Bertolino, P., Tong, W. M., Galendo, D., Wang, Z. Q. & Zhang, C. X. Heterozygous mutant Men1 mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 17, 1880–1892 (2003).
20. Hao, W. et al. MEN1 variant with frequent prolactinoma and rare gastrinoma. J. Clin. Endocrinol. Metab. 89, 3776–3784 (2004).
21. Cote, G. J et al. The spectrum of mutations in MEN1 variant syndromes. Program Abstr. Endocr. Soc. Annu. Meet. 106–107 (1998).
22. LeBodic, M. F. et al. Immunohistochemical study of 100 pancreatic tumors in 28 patients with multiple endocrine neoplasia, type I. Am. J. Surg. Pathol. 20, 1378–1384 (1996).
23. Gschwind, A., Fischer, O. M. & Ullrich, A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature Rev. Cancer 4, 361–370 (2004).
24. Blume-Jensen, P. & Hunter, T. Oncogenic kinase signalling. Nature 411, 355–365 (2001).
25. Santoro, M., Melillo, R. M., Carlomagno, F., Vecchio, G. & Fusco, A. RET: normal and abnormal functions. Endocrinology 145, 5448–5451 (2004).A concise review of RET gene pathophysiology.
26. McCallion, A. S. et al. Genomic variation in multigenic traits: Hirschsprung disease. Cold Spring Harb. Symp. Quant. Biol. 68, 373–381 (2003).
27. Manie, S., Santoro, M., Fusco, A. & Billaud, M. The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet. 17, 420–429 (2001).
28. Garcia-Barcelo, M. et al. TTF-1 and RET promoter SNPs: regulation of RET transcription in Hirschsprung’s disease. Hum. Mol. Genet. 14, 191–204 (2005).
29. Nikiforova, M. N. et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 290, 138−141 (2000).
30. Alvares da Silva, A. M. et al. A novel germ-line point mutation in RET exon 8 (Gly553Cys) in a large kindred with familial medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 88, 5438–5443 (2003).
31. Nilsson, O. et al. Adrenal and extra-adrenal pheochromocytomas in a family with germline V804L mutation. J. Am. Med. Assoc. 281, 1587–1588 (1999).
32. Lecube, A. et al. Recessive V804M RET mutation and familial medullary thyroid carcinoma: report of a large family with expression of the disease only in the homozygous gene carriers. Surgery 131, 509−514 (2002).
33. Elisei, R. et al. Identification of a novel point mutation in the RET gene (Ala883Thr), which is associated with medullary throid carcinoma phenotype only in homozygous condition. J. Clin. Endocrinol. Metab. 89, 5823–5827 (2004).
34. Jackson, C. E. et al. Variable expressivity of familial medullary thyroid carcinoma (FMTC) due to a RET V804M (GTG→ATG) mutation. Surgery 128, 93–98 (2000).
35. Lombardo, F. et al. Familial medullary thyroid carcinoma: clinical variability and low aggressiveness associated with RET mutation at codon 804. J. Clin. Endocrinol. Metab. 87, 1674–1680 (2002).
36. Machens, A. et al. European Multiple Endocrine Neoplasia (EUROMEN) Study Group Early malignant progression of hereditary medullary thyroid cancer. N. Engl. J. Med. 349, 1517–1525 (2003).
37. Sakurai, A. et al. Premature centromere division in patients with multiple endocrine neoplasia type 1. Cancer Genet. Cytogenet. 109, 138–140 (1999).
38. Michels, F. M. et al. Development of medulary thyroid carcinoma in transgenic mice expressing the RET protooncogene altered by a multiple endocrine neoplasia type 2A mutation. Proc. Natl Acad. Sci. USA 94, 3330–3335 (1997).
39. Cranston, A. N. & Ponder, B. J. Modulation of medullary thyroid carcinoma penetrance suggests the presence of modifier genes in a RET transgenic mouse model. Cancer Res. 63, 4777–4780 (2003).
40. Skinner, M. A. et al. A human yeast artificial chromosome containing the multiple endocrine
neoplasia type 2B Ret mutation does not induce medullary thyroid carcinoma but does support the growth of kidneys and partially rescues enteric nervous system development in ret-deficient mice. Am. J. Pathol. 166, 265–274 (2005).
41. Smith-Hicks, C. L., Sizer, K. C., Powers, J. F., Tischler, A. S. & Costantini, F. C-cell hyperplasia, pheochromocytoma, and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. EMBO J. 19, 612–622 (2000).
42. Futreal, P. A. et al. A census of human cancer genes. Nature Rev. Cancer 4, 177–183 (2004)
43. Koch, C. A., Pacak, K. & Chrousos, G. P. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J. Clin. Endocrinol. Metab. 87, 5367–5384 (2002).
44. Cranston, A. Howard, L. & Howard, V. Quantitative phenotyping as an efficient means to estimate C-cell number in a knock-in model of MEN2B. Transgenic Res. 13, 339–348 (2004).
45. Diaz-Cano, S. J., de Miguel, M., Blanes, A., Tashjian, R. & Wolfe, H. Germline RET 634 mutation positive MEN2A-related C-cell hyperplasias have genetic features consistent with intraepithelial neoplasia. J. Clin. Endocrinol. Metab. 86, 3948–3957 (2001).
46. Santarosa, M. & Ashworth, A. Haploinsufficiency for tumor suppressor genes: when you don’t need to go all the way. Biochem. Biophys. Acta 1654, 105–122 (2004).
47. Pannett, A. A. & Thakker, R. V. Somatic mutations in MEN type 1 tumors, consistent with the Knudson ‘two-hit’ hypothesis. J. Clin. Endocrin. Metab. 86, 4371–4374 (2001).
48. Huang, S. C. et al. Duplication of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine neoplasia type 2-associated pheochromocytomas. Cancer Res. 60, 6223–6226 (2000).
49. Huang, S. C. et al. Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 88, 459–463 (2003).
50. Lombardo, F. et al. Familial medullary thyroid carcinoma: clinical variability and low aggressiveness associated with RET mutation at codon 804. J. Clin. Endocrinol. Metab. 87, 1674–1680 (2002).
51. Marsh, D. J. et al. Genome-wide copy number imbalances identified in familial and sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 88, 1866–1872 (2003).
52. Knudson, A. G. Hereditary cancer: two hits revisited. J. Cancer Res. Clin. Oncol. 122, 134–140 (1996).
53. Koch, C. A. et al. Somatic VHL gene deletion and point mutation in MEN2A-associated pheochromocytoma. Oncogene 21, 479–482 (2002).
54. Farnebo F. et al. Alternative genetic pathways in parathyroid tumorigenesis. J. Clin. Endocrinol. Metab. 84, 3775–3780 (1999).
55. Agarwal, S. K. et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm. Metab. Res. (in the press).
56. Guru, S. C. et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl Acad. Sci. USA 95, 1630–1634 (1998).
57. Agarwal, S. K. et al. Menin interacts with the AP1 transcription factor JunD and represses JunD activated transcription. Cell 96, 143–152 (1999).
58. Knapp, J. L. et al. Identification and characterization of junD missense mutants that lack menin binding. Oncogene 19, 4706–4712 (2000).
59. Agarwal, S. K. et al. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc. Natl Acad. Sci. USA 100, 10770–10775 (2003).Deprivation of menin converts JUND from a growth suppressor to a growth promoter, highlighting a possible pathway for tumorigenesis by menin.
374 | MAY 2005 | VOLUME 5 www.nature.com/reviews/cancer
R E V I E W S

60. Hughes, C. M. et al. Menin associates with a Trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol. Cell 13, 587–597 (2004).Menin might act by binding to a multi-unit complex, homologous to the yeast COMPASS complex.
61. Yokoyama, A. et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 24, 5639–5649 (2004).
62. Daser, A. & Rabbitts, T. H. Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev. 18, 965–974 (2004).
63. Hess, J. L. Mechanisms of transformation by MLL. Crit. Rev. Eukaryot. Gene Expr. 14, 235–254 (2004).
64. Lin, S. Y. & Elledge, S. J. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 113, 881–889 (2003).
65. Hua, X. X. et al. Menin induces apoptosis in murine embryonic fibroblasts. J. Biol. Chem. 279, 10685–10691 (2004).
66. Sukhodolets, K. E. et al. The 32-kDa subunit of replication protein protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol. Cell. Biol. 223, 493–509 (2003).
67. Busygina, V. et al. Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum. Mol. Genet. 13, 2399–2408 (2004).
68. Scacheri, P. C. et al. Pancreatic insulinomas in multiple endocrine neoplasia, type 1 knockout mice can develop in the absence of chromosome instability or microsatellite instability. Cancer Res. 64, 7039–7044 (2004).
69. Ichihara, M., Murakumo, Y. & Takahashi, M. RET and neuroendocrine tumors. Cancer Lett. 204, 197–211 (2004).
70. Borrego, S. et al. Evaluation of germline sequence variants of GFRA1, GFRA2, and GFRA3 genes in a cohort of Spanish patients with sporadic medullary thyroid cancer. Thyroid 12, 1017–1022 (2002).
71. Vanhorne, J. B. et al. A model for GFRα4 function and a potential modifying role in multiple endocrine neoplasia 2. Oncogene 24, 1091–1097 (2005).
72. Schlessinger, J. & Lemmon, M. A. SH2 and PTB domains in tyrosine kinase signalling. Sci. STKE 191, RE12 (2003).
73. Kawamoto, Y. et al. Identification of RET autophosphorylation sites by mass spectrometry. J. Biol. Chem. 279, 14213–14224 (2004).
74. Carlomagno, F. et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23, 6056–6063 (2004).One of a series of articles suggesting that tyrosine-kinase inhibition should be pursued as a drug target against RET-related neoplasia.
75. Santoro, M. et al. Molecular mechanisms of RET activation in human cancer. Ann. NY Acad. Sci. 963, 116–121 (2002).
76. Myers, S. M. & Mulligan, L. M. The RET receptor is linked to stress response pathways. Cancer Res. 64, 4453–4463 (2004).
77. Kung, C. et al. Chemical genomic profiling to identify intracellular targets of multiplex kinase inhibitor. Proc. Natl Acad. Sci. USA 102, 3587–3592 (2005).
78. Castellone, M. D. et al. Ras-mediated apoptosis of PC CL3 rat thyroid cells induced by RET/PTC oncogenes. Oncogene 22, 246–255 (2003).
79. Fritz, A. et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 62, 3048–3051 (2002).
80. Zhou, Z. et al. Suppression of melanotroph carcinogenesis leads to accelerated progression of anterior lobe tumors and medullary thyroid carcinomas in Rb+/– mice. Cancer Res. 65, 1–10 (2005).
81. Franklin, D. S. et al. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol. Cell. Biol. 20, 6147–6158 (2000).Double knockout of Cdkn2c and Cdkn1b in mice gives a phenotype with extensive overlap between MEN1 and MEN2. The downstream mechanism for each of these syndromes might pass through this pathway.
82. Noble, M. E. M., Endicott, J. A. & Johnson, L. N. Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805 (2004).
83. Carlomagno, F. et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 62, 7284–7290 (2002).
84. Agarwal, S. K. et al. Analysis of recurrent germline mutations in the MEN1 gene encountered in apparently unrelated families. Hum. Mutat. 12, 75–82 (1998).
AcknowledgementsI thank many colleagues for collaborations and discussions. In partic-ular, these include the participants in the National Institutes of Health (NIH) Campus Collaborative Group on MEN1 and the participants in the NIH Interinstitute Endocrine Training Program. I also thank S. Agarwal and L. Mulligan for analysing data to develop figure 2.
Competing interests statementThe author declares no competing financial interests.
Online links
DATABASESThe following terms in this article are linked online to:Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneCdkn1a | CDKN2C | JUND | MEN1 | RETOMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIMHirschsprung disease | RET–PTCs | MEN type 1 | MEN type 2 | MEN2B
FURTHER INFORMATIONAn academic type review of MEN2 from GeneTests: http://www.genetests.org/servlet/access?db=geneclinics&site=gt&id=8888891&key=8bwk-–DS9brd9&gry=&fcn=y&fw=BCo7&filename=/profiles/men2/index.htmlNIDDK page on MEN1: http://www.niddk.nih.gov/health/endo/pubs/men1/men1.htmThe Association for Multiple Endocrine Neoplasia Disorders: http://www.mensociety.com/Access to this interactive links box is free online.
NATURE REVIEWS | CANCER VOLUME 5 | MAY 2005 | 375
R E V I E W S

BiographyStephen Marx received a B.A. from Yale College, an M.D. from Johns Hopkins Medical School, and house-staff training at Massachussets General Hospital. He has worked at National Institutes of Health (NIH) for over 30 years in the field of parathyroid and other endo-crine hyperfunctions. He characterized familial hypocalciuric hypercalcaemia and its homozygous severe variant and helped show CASR (calcium-sensing receptor) mutation in those two syndromes. He co-directed the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) component in the cloning of the HRPT2 (hyperparathyroidism 2) gene, cause of the hyperparathyroidism-jaw tumour syndrome. He co-directs the NIH team that cloned the mul-tiple endocrine neoplasia I (MEN1) gene and that studied the MEN1 mutations in kindreds and in tumours. They more recently studied mouse models of multiple endocrine neoplasia type 1 and actions of the menin protein, encoded by MEN1. His group identified several menin partners and extensively studied JUND as a menin partner that could contribute to MEN1-induced tumorigenesis. He is in NIDDK/NIH as Chief of the Metabolic Diseases Branch and Chief of its Genetics and Endocrinology Section.
LINKSLINKS
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
Cdkn1ahttp://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=12575
CDKN2Chttp://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=1031
JUND http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=3727
MEN1http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=4221
REThttp://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=5979
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
Hirschsprung diseasehttp://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=142623
RET-PTCshttp://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=188550
MEN type 1http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=131100
MEN type 2http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=171400
MEN2Bhttp://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=162300
O N L I N E O N LY
Related Documents











