Enzyme and Microbial Technology 39 (2006) 74–84 Molecular cloning of a cyclodextrin glucanotransferase gene from alkalophilic Bacillus sp. TS1-1 and characterization of the recombinant enzyme Kamalesh Rahman a , Rosli Md Illias a,∗ , Osman Hassan b , Nik Azmi Nik Mahmood a , Noor Aini Abdul Rashid c a Department of Bioprocess Engineering, Faculty of Chemical and Natural Resources Engineering, Universiti Teknologi Malaysia, 81310 Skudai, Johor, Malaysia b School of Chemical & Food Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43000 Bangi, Selangor, Malaysia c Department of Biology, Faculty of Science, Unversiti Teknologi Malaysia, 81310 Skudai, Johor, Malaysia Received 13 May 2005; received in revised form 15 September 2005; accepted 28 September 2005 Abstract A cyclodextrin glucanotransferase (CGTase) gene from Bacillus sp. TS1-1 was isolated and cloned into Escherichia coli. Starting from TTG codon, there was an open reading frame composed of 2163 bp (721 amino acids). The NH 2 terminal position encoded a 46-amino acid of a signal peptide and followed by the mature enzyme (675 amino acids). The deduced amino acid sequence of the mature CGTase from Bacillus sp. TS1-1 exhibited 98.7% homology with 96% identity to the CGTase sequence from alkalophilic Bacillus sp. 1-1. The recombinant CGTase of Bacillus sp. TS1-1 expressed in E. coli was successfully purified to homogeneity using ammonium sulfate precipitation, followed by -cyclodextrin-bound- epoxy-activated Sepharose 6B affinity chromatography. The purified CGTase enzymes exhibited a single band with molecular weight of 75 kDa on SDS-PAGE. Biochemical characterization of the enzyme shows an optimum temperature of 60 ◦ C and optimum pH of 6.0. The enzyme was stable between pH 7 and 9 and temperature up to 70 ◦ C. The K m and V max values calculated were 0.52 mg/ml and 54.35 mg of -cyclodextrin/ml/min. The yield of the products from soluble starch as the substrate were 86% for -cyclodextrin and 14% for -cyclodextrin after 24 h incubation at 60 ◦ C, without adding any selective agent. The total -CD produced under the conditions mentioned above was 3.65 g/l. © 2005 Published by Elsevier Inc. Keywords: Cyclodextrin glucanotransferase 1. Introduction The cyclodextrin glucanotransferase (CGTase, EC 2.4.1.19) is a member of -amylase family (family 13 of glycosyl hydrolases). Although CGTase is closely related to -amylase, CGTase differs from -amylase in that a-amylase usually cat- alyze hydrolysis reaction using water as acceptor whereby CGTase preferably catalyze transglycosylation reactions in which glucosyl residues are used as acceptor in forming cyclodextrins (CDs) as the main product. CGTase is a multi- functional enzyme [1], besides cyclization it also display inter- molecular transglycosylation (coupling, disproportionation) and hydrolytic activity on starch and CDs. Currently, bacteria are still ∗ Corresponding author. Tel.: +60 7 5535564; fax: +60 7 5581463. E-mail address: [email protected] (R.M. Illias). regarded as an important source of CGTases. Since the discov- ery of Bacillus macerans as the first source that is capable of producing CGTases [2], a wide variety of bacteria have been determined as CGTase producers, namely aerobic mesophilic bacteria, aerobic thermophilic, anaerobic thermophilic and aer- obic alkalophilic bacteria. Various genera of bacteria that are known as CGTase producer includes Bacillus [3], Klebsiella [4], Brevibacterium [5], Thermoanaerobacterium [6] and Micrococ- cus [7]. Most CGTases produce a mixture of -, - and -CD in different ratios, depending on the origin of the CGTase as well as the reaction conditions. CGTase is classified into three different types, -CGTase, -CGTase and -CGTase according to the major CD produced [8]. CD molecules have a unique structure with a hydrophobic cavity and hydrophilic at the outer surface and therefore can form inclusion complexes with a wide variety of hydrophobic guest molecules. Their three-dimensional form and size provide 0141-0229/$ – see front matter © 2005 Published by Elsevier Inc. doi:10.1016/j.enzmictec.2005.09.014

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Enzyme and Microbial Technology 39 (2006) 74–84
Molecular cloning of a cyclodextrin glucanotransferase genefrom alkalophilic Bacillus sp. TS1-1 and characterization
of the recombinant enzyme
Kamalesh Rahman a, Rosli Md Illias a,∗, Osman Hassan b,Nik Azmi Nik Mahmood a, Noor Aini Abdul Rashid c
a Department of Bioprocess Engineering, Faculty of Chemical and Natural Resources Engineering,Universiti Teknologi Malaysia, 81310 Skudai, Johor, Malaysia
b School of Chemical & Food Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43000 Bangi, Selangor, Malaysiac Department of Biology, Faculty of Science, Unversiti Teknologi Malaysia, 81310 Skudai, Johor, Malaysia
Received 13 May 2005; received in revised form 15 September 2005; accepted 28 September 2005
Abstract
cpeTeSbT6©
K
1
ihCaCwcfmh
0d
A cyclodextrin glucanotransferase (CGTase) gene from Bacillus sp. TS1-1 was isolated and cloned into Escherichia coli. Starting from TTGodon, there was an open reading frame composed of 2163 bp (721 amino acids). The NH2 terminal position encoded a 46-amino acid of a signaleptide and followed by the mature enzyme (675 amino acids). The deduced amino acid sequence of the mature CGTase from Bacillus sp. TS1-1xhibited 98.7% homology with 96% identity to the CGTase sequence from alkalophilic Bacillus sp. 1-1. The recombinant CGTase of Bacillus sp.S1-1 expressed in E. coli was successfully purified to homogeneity using ammonium sulfate precipitation, followed by �-cyclodextrin-bound-poxy-activated Sepharose 6B affinity chromatography. The purified CGTase enzymes exhibited a single band with molecular weight of 75 kDa onDS-PAGE. Biochemical characterization of the enzyme shows an optimum temperature of 60 ◦C and optimum pH of 6.0. The enzyme was stableetween pH 7 and 9 and temperature up to 70 ◦C. The Km and Vmax values calculated were 0.52 mg/ml and 54.35 mg of �-cyclodextrin/ml/min.he yield of the products from soluble starch as the substrate were 86% for �-cyclodextrin and 14% for �-cyclodextrin after 24 h incubation at0 ◦C, without adding any selective agent. The total �-CD produced under the conditions mentioned above was 3.65 g/l.
2005 Published by Elsevier Inc.
eywords: Cyclodextrin glucanotransferase
. Introduction
The cyclodextrin glucanotransferase (CGTase, EC 2.4.1.19)s a member of �-amylase family (family 13 of glycosylydrolases). Although CGTase is closely related to �-amylase,GTase differs from �-amylase in that a-amylase usually cat-lyze hydrolysis reaction using water as acceptor wherebyGTase preferably catalyze transglycosylation reactions inhich glucosyl residues are used as acceptor in forming
yclodextrins (CDs) as the main product. CGTase is a multi-unctional enzyme [1], besides cyclization it also display inter-olecular transglycosylation (coupling, disproportionation) and
ydrolytic activity on starch and CDs. Currently, bacteria are still
∗ Corresponding author. Tel.: +60 7 5535564; fax: +60 7 5581463.E-mail address: [email protected] (R.M. Illias).
regarded as an important source of CGTases. Since the discov-ery of Bacillus macerans as the first source that is capable ofproducing CGTases [2], a wide variety of bacteria have beendetermined as CGTase producers, namely aerobic mesophilicbacteria, aerobic thermophilic, anaerobic thermophilic and aer-obic alkalophilic bacteria. Various genera of bacteria that areknown as CGTase producer includes Bacillus [3], Klebsiella [4],Brevibacterium [5], Thermoanaerobacterium [6] and Micrococ-cus [7]. Most CGTases produce a mixture of �-, �- and �-CDin different ratios, depending on the origin of the CGTase aswell as the reaction conditions. CGTase is classified into threedifferent types, �-CGTase, �-CGTase and �-CGTase accordingto the major CD produced [8].
CD molecules have a unique structure with a hydrophobiccavity and hydrophilic at the outer surface and therefore canform inclusion complexes with a wide variety of hydrophobicguest molecules. Their three-dimensional form and size provide
141-0229/$ – see front matter © 2005 Published by Elsevier Inc.oi:10.1016/j.enzmictec.2005.09.014

K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84 75
an important parameter for complex formation with hydropho-bic compounds. Thus, specific (�-, �- and �-)cyclodextrinsare required for complexation of specific guest molecules. Theformation of inclusion complexes leads to the changes in thechemical and physical properties of the guest molecules. Thesealtered characteristics of encapsulated compounds have led tovarious applications of cyclodextrins in analytical chemistry [9],agriculture [10], biotechnology [11], food, pharmacy and cos-metics [12].
A major disadvantage of cyclodextrin production by CGTaseis that, all known wild type CGTase enzyme produce a mix-ture of �-, �- and �-cyclodextrin and are subjected to inhibitionby these cyclic products. This shows that the availability ofCGTase enzymes capable of producing an increase ratio of oneparticular type of cyclodextrin and with reduced product inhibi-tion is important. This situation has strongly simulated geneticengineering techniques to provide a better CGTase. A CGTaseproducing bacteria; alkalophilic Bacillus sp. TS1-1 has beensuccessfully isolated by our research group. This paper describethe isolation and cloning of the CGTase gene isolated from thebacterium. Characterization of the recombinant enzyme is alsopresented.
2. Materials and methods
2.1. Bacterial strain and plasmids
[rPa
2
BpEwt
2
IsgwplJphsae
2
e
starch in 0.1 M phosphate buffer (pH 6.0) and 0.1 ml enzyme solution was used.The mixture was incubated at 60 ◦C for 10 min in a waterbath. The reactionwas stopped by adding 3.5 ml of 0.03 M NaOH solution. 0.5 ml of 0.02% (w/v)phenolphthalein in 0.005 M Na2CO3 then was added to the reaction mixture.After 15 min, the decrease in colour intensity was measured at 550 nm. Thepercentage of reduction in the original colour intensity was interpreted with astandard curve (% OD reduction versus �-CD in mg produced) for the calculationof CGTase activity. One unit of enzyme activity was defined as the amount ofenzyme that forms l �mol of �-CD from soluble starch in 1 min.
2.5. Nucleotide and protein sequence analysis
The nucleotides and deduced amino acid sequence of Bacillus sp. TS1-1CGTase gene was compared to those available at the GenBank and was alignedby using DNAsis/CLUSTAL X program. The nucleotide sequence reported inthis work has been deposited in the GenBank database under the AccessionNumber AY770576.
2.6. Purification of CGTase
E. coli culture harboring CGTase gene was incubated for 24 h at 37 ◦C,200 rpm in an incubator shaker. The cells were separated from supernatantby centrifugation at 8000 rpm for 10 min at 4 ◦C. Purification steps were car-ried out at 4 ◦C. The recombinant CGTase, was precipitated by the additionof solid ammonium sulfate (NH4)2SO4 to give a 70% saturation. The mixturewere stirred slowly and gently in order to obtain a better dissolution rate ofammonium sulfate and promoting the salting out effect. The mixture was setto stand overnight at 4 ◦C to enhance the precipitation and stabilization of theenzyme. The resulting precipitate was separated from the supernatant by cen-trifugation at 3400 × g for 20 min at 4 ◦C. The precipitate was resuspended in8s1�
m10cebopdlb
2
dts
2
wa(orp
2
t
An alkalophilic bacterium, Bacillus sp. TS1-1 was isolated from the soil13]. Escherichia coli JM109 [endA1, recA1, gyrA96, thi, hsdR17 (rk−, mk+),elA1, supE44, � (lac-proAB), F′ (tra D36, pro AB, lacIqZ � M15)] fromromega was used as the host strain. Plasmid pUC19 from Promega was useds the cloning vector.
.2. Medium and culture conditions
Bacillus sp. TS1-1 was grown overnight at 37 ◦C, 200 rpm in Horikoshiroth [14] which contained 1.0 g/1 of KH2PO4, 0.2 g/1 of MgSO4, 5.0 g/1 ofeptone, 5.0 g/1 of yeast extract and 10.0 g/1 of Na2CO3 (autoclave separately).. coli used as a cloning host was cultured in Luria-Bertani (LB) broth at 37 ◦Chile ampicillin (100 �g/ml) was added to the medium to allow the growth of
he plasmid-carrying strain.
.3. DNA manipulation and cloning procedure
The genomic DNA of Bacillus sp. TS1-1 was prepared according to thesh-Horowitz, method [15]. DNA manipulations were performed according totandard methods as described by Sambrook et al. [16]. Bacillus sp. TS1-1enomic DNA was partially digested with HindIII. The cloning vector, pUC19as also cleaved with HindIII and dephosphorylated with shrimp alkaline phos-hate (SAP). Genomic DNA fragments were then ligated with the dephosphory-ated plasmid pUC19. The ligation products were used to transform into E. coliM 109. The E. coli transformants were plated on LB-ampicillin (100 �g/ml)lates, which contained 1% soluble starch. After growth at 37 ◦C for 24 h, thealo zones that appeared around the colonies after exposure to a KI–I2 indicatorolution suggested the possibility of starch being degraded by the hydrolyticctivity of the �-CGTase, and the diameter of the halos indicated the amount ofnzyme produced.
.4. Assay of enzyme activity
The CGTase activity was measured by the method established by Kanekot al. with modification [17]. The reaction mixture containing 1 ml of 0.04 g
00 ml of 0.01 M acetate buffer, pH 5.5. The mixture was subjected to sub-equent purification procedures. Then the mixture was spun at 8000 rpm for0 min at 4 ◦C to remove any remaining insoluble material before loaded onto-cyclodextrin-bound-epoxy-activated Sepharose 6B affinity column. Twentyillilitres of supernatant containing 0.4 mg/ml of protein was subjected to a
5 mm × 100 mm affinity column, which previously had been equilibrated with.01 M acetate buffer (pH 5.5), at a flow rate of 21 ml/h. The column was suc-essively washed with the same buffer for 4 h. After the unbound protein wasluted, the elution of the desired bound enzyme was carried out with the sameuffer supplemented with 1% �-CD at a flow rate of 19.2 ml/h. Three millilitresf fractions were collected and each one was assayed for CGTase activity androtein content. The fractions that showed CGTase activity were pooled andialyzed overnight against 0.01 M acetate buffer (pH 5.5), in a regenerated cel-ulose dialysis tubing (PIERCE, 10,000 MWCO) at 4 ◦C with three changes ofuffer.
.7. Molecular weight determination
The molecular weight of the purified enzyme was determined by sodiumodecyl-sulphate polyacrylamide gel electrophoresis (SDS-PAGE) accordingo Laemmli [18] on a vertical slab gel using 150 V for 3 h at 25 ◦C. The gel wastained with 1% Coomassie Brilliant Blue R-250.
.8. Effect of pH on purified CGTase enzyme
Optimum pH for the purified enzyme was measured by reacting the enzymeith soluble starch dissolved in different buffers with varying pH. The buffers
re sodium acetate buffer, 0.1 M (pH 4–5), potassium phosphate buffer, 0.1 MpH 6–8) and glycine–NaOH buffer 0.1 M (pH 9–10). The reaction was carriedut using the CGTase assay procedure mentioned before. A pH profile of theelative activity versus pH was drawn by taking the enzyme activity at optimumH as 100%.
.9. Effect of temperature on purified CGTase enzyme
Optimum temperature for the purified CGTase was determined by reactinghe enzyme with soluble starch in 0.1 M phosphate buffer pH 6.0 at different

76 K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84
temperatures, ranging from 40 to 90 ◦C for 10 min. Then, the reaction was doneaccording to the method of CGTase assay described previously. A temperatureprofile of the relative activity versus temperature was drawn by taking the enzymeactivity of optimum temperature as 100%.
2.10. Effect of pH on the stability of purified CGTase
The pH stability of the enzyme was measured by incubating 0.1 ml of pureenzyme with 0.2 ml of 0.1 M sodium acetate buffer (pH 4–5), 0.1 M potassiumphosphate buffer (pH 6–8) and glycine–NaOH buffer (pH 9–10), respectivelyat 60 ◦C, without substrate for 30 min. Then, the residual activity of the enzymewas assayed after reacting 0.1 ml of the enzyme mixture with soluble starch in0.1 M phosphate buffer (pH 6.0) and incubated at 60 ◦C for 10 min. A pH stabilityprofile of the residual activity versus pH was drawn by taking the residual activityof the untreated sample as a control (100% activity).
2.11. Effect of temperature on the stability of purified CGTase
The pH stability of the enzyme was measured by incubating 0.1 ml of pureenzyme 0.1 ml of pure CGTase enzyme was diluted with 0.2 ml of 0.1 M phos-phate buffer (pH 6.0) without substrate and incubated at different temperatures,ranging from 40 to 90 ◦C for 30 min. Then, the residual activity of the enzymewas assayed after reacting 0.1 ml of the enzyme mixture with soluble starch in0.1 M phosphate buffer (pH 6.0) and incubated at 60 ◦C for 10 min. A temper-ature stability profile of the residual activity versus temperature was drawn bytaking the residual activity of the untreated sample as a control (100% activity).
2.12. Kinetic parameters of purified CGTase
0vaH
2
pA73ttt
3
3
Tttotrrtn(Ot
initiation codon but the initiation codon appeared to be a TTGcodon near the beginning of a possible signal peptide region.There were two TTG codons (boxed in Fig. 1) near the begin-ning of the possible signal peptide region. Since TTG is knownto function as the initiation codon in E. coli and other prokary-otes [19], either TTG is suspected to function as the translationalinitiation codon. Moreover, a TTG start codon was also observedin CGTase gene from Bacillus sp. A2-5a [20], Bacillus sp. E1[21] and Bacillus ohbensis [22].
However, a typical sequence for the ribosome-binding site(AAGG) was located 5 bp upstream from the second TTG codon,which is between nucleotides −6 and −9 relative to the startcodon based on the Shine-Dalgarno Hypothesis. This revealsthat the second TTG codon is most likely the true initiationcodon. There was a putative Pribnow–Schaller box (TTACGAand ATTAAT) upstream of the mature gene starting codon(Fig. 1). Downstream of the open reading frame, a long invertedrepeated sequences were found, which can form a stable stemand loop structure. It is known to be a �-independent transcrip-tional termination signal. The amino acid sequence from 1 to46 was predicted to be a signal peptide, which is involved insecretion of the protein. The amino acid sequence of this signalpeptide consists several positively charged amino acids followedby a run of hydrophobic amino acid core and a COOH-terminalalanine residue. This is consistent with the characteristic of sig-nal peptides from other Gram-positive bacteria [23,24]. More-omrcwtsB
3
ltChslatlCdtiBrT3fr
The Km and Vmax values for the pure enzyme were determined by incubating.1 ml of purified CGTase (8.3U) in 1 ml 0.1 M phosphate buffer, pH 6.0, atarious concentrations of soluble starch solution, ranging from 0.4 to 6.0 mg/mlt 60 ◦C for 10 min. The values of Km and Vmax were then determined usinganes–Woolf plot.
.13. Analysis of cyclodextrin by HPLC
For all the analyses that involved a high performance liquid chromatogra-hy (HPLC), the conditions were set as followed. The column employed wassahipak amino NH2P-504E from Phenomenex with a length of 300 mm and.8 mm internal diameter. The mobile phase of the system was water/acetonitrile0:70 and the flow rate of the mobile phase was set at 1.0 ml/min. The columnemperature was maintained at 30 ◦C. The eluent from the column was moni-ored by a refractive index detector (Waters 410) and the data was recorded byhe integrated computer system attached to the HPLC (Waters corp.).
. Results and discussion
.1. Molecular cloning of a gene encoding CGTase activity
The partially digested chromosomal DNA of Bacillus sp.S1-1 with HindIII were inserted into HindIII site of pUC19, and
hen transformed into competent cells of E. coli JM109. Twentyhousand colonies of the transformants were screened and onlyne colony showed hydrolytic activity on LB-ampicillin con-aining 1% soluble starch agar plate. The 4.8 kb insert in theecombinant plasmid was sequenced using the universal M13everse sequencing primer. Primer walking was used to sequencehe rest of the insert in both directions. Approximately, 2.4 kbucleotide sequence of the 4.8 kb fragment was determinedFig. 1). The nucleotide sequence analysis revealed a singleRF of 2163 bp encoding 721 amino acid residues. The ini-
iation codon for this unique ORF was not the ATG translational
ver, for a probable cleavage site, the residue in position −1ust be small (either Ala, Ser, Gly, Cys, Thr or Gin) and the
esidue in position −3 must not be aromatic (Phe, His, Tyr, Tip),harged (Asp, Glu, Lys, Arg) or large and polar (Asn, Gin). Itas also suggested that Pro must be absent from positions −3
hrough +1 (Fig. 1). This is the (−3,−l)-rule [25]. The nucleotideequence reported in this work has been deposited in the Gen-ank database under the Accession Number AY770575.
.2. Amino acid sequence analysis
The deduced amino acid sequence of the CGTase from Bacil-us sp. TS1-1 was compared with other CGTases sequences usinghe BLAST program. The highest similarity was observed withGTase from alkalophilic Bacillus sp. 1-1 (P31746), with 98.8%omology and 97.5% identity. There were a few other CGTasesuch as CGTase from Bacillus sp. KC201 (BAA02380), Bacil-us sp. A2-5a (BAA31539), Brevibacillus CD162 (AAB65420)nd Bacillus ohbensis (P27036) that has a high homology (morehan 70%) with amino acid sequence of CGTase from Bacil-us sp. TS1-1. From the multiple sequence alignment of variousGTase, six highly conserved regions (labeled I–VI) and fiveomains (domains A, B, C, D and E) could be identified inhe sequence of the enzymes (Fig. 2). Whenever referred ton the following text, the residues position are relative to theacillus circulans strain 251 numbering [26], followed by the
espective position in Bacillus sp. TS1-1 CGTase in brackets.hree catalytic residues, Asp-229 (222), Glu-257 (250) and Asp-28 (321) were found at the active site of recombinant CGTaserom Bacillus sp. TS1-1. The catalytic functions of these threeesidues are conserved in all known CGTase. It was suggested

K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84 77
Fig. 1. Nucleotide sequence of the cyclodextrin glucanotransferase (CGTase) gene from Bacillus sp. TS1-1. The flanking regions of the nucleotide sequence ofthe CGTase gene are shown. The signal peptide sequence is shown in italic. The possible promoter region sequences (TTACGA and ATTAAT) and the possibleribosome-binding site, SD, are underlined. Inverted repeats downstream of the stop codon TAA is shown by horizontal arrows below the sequence. A vertical arrowindicates the possible signal peptide cleavage site. This sequence was submitted to the Gen Bank with the Accession Number AY770575.

78 K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84
Fig. 1. (Continued )

K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84 79
Fig. 1. (Continued ).
that these three residues play an important role in the enzymaticreaction catalyzed by CGTase [27]. Glu-257 (250) involved incatalysis of CGTase and is the proton donor in the first step of thecyclization reaction. Asp-229 (222) acts as the general base ornucleophile while Asp-328 (321) is involved in substrate bind-ing.
The specific CGTase residues in Domain A1, which areunique and completely conserved, were also found in CGTasefrom Bacillus sp. TS1-1. These include, Asp27 (23), Asn32 (28),Asn33 (29) and Asp53 (49), which are ligands of a calciumbinding site observed in CGTases but not in �-amylases [28]. InDomain B of Bacillus sp. TS1-1, more unique CGTase residuesare found which is Phel36 (129), Glul53 (146), Glyl65 (158),Tyrl67 (160), Phel75 (168), Hisl77 (170), Glyl80 (173), Ilel90(183), Tyr191 (184) and the stretch 185K(R),N.L.Y.D189. Apartfrom that, several unique residues are found in Domain A2 ofCGTase fromBacillus sp. TS1-1 which is Tyr210 (203), Trp218(211), Ile226 (219) and Trp258 (251). Eleven strictly conservedresidues of the raw-starch binding motif were found in this regionof Domain E in CGTase from Bacillus sp. TS1-1. Four of the11 conserved residues which are Trp616 (604), Lys651 (644),Trp662 (652) and Asn667 (657) are a part of maltose bindingsite one (MB SI) while another three; Thr598 (587), Gly601(590) and Trp636 (625) are a part of MBS2. The remaining fourstrictly conserved residues in CGTase from Bacillus sp. TS1-1;Gly608 (597), Leu613 (602), Gly614 (603) and Pro634 (623)abfp
cyclodextrin product specificities [29]. In position 47, Arg orLys is found to be in CGTases producing mainly �- and/or �-CD while His is found in CGTases with no �-CD produced. Hiscan be found at the same position in CGTase from Bacillus sp.TS1-1 and this implies why Bacillus sp. TS1-1 did not produceany �-CD.
3.3. Enzymatic properties of the purified CGTase
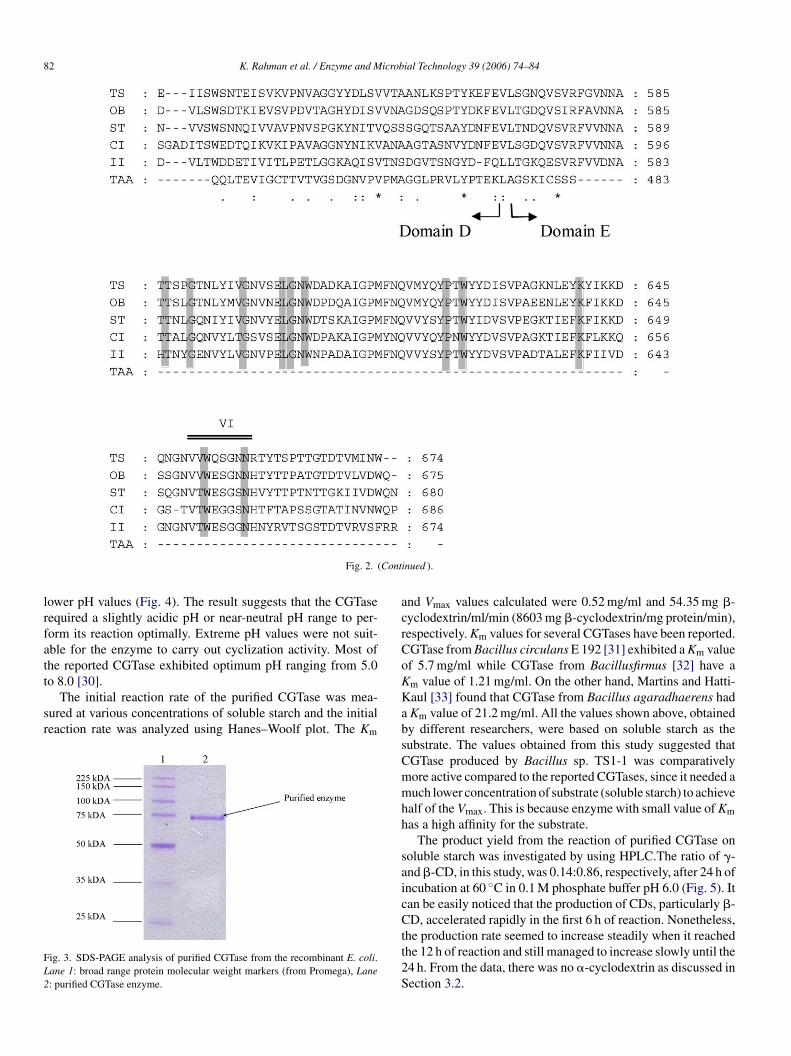
The recombinant CGTase was expressed in E. coli usingpUC19 as a cloning vector. About 17.80 U/ml CGTase activ-ity was observed secreted by the recombinant clone into theculture medium. However this value is lower compared to theparental strain, which exhibited 49.25 U/ml CGTase activity.The recombinant enzyme was successfully purified to homo-geneity in two steps by ammonium sulfate precipitation, fol-lowed by �-cyclodextrin-bound-epoxy-activated Sepharose 6Baffinity chromatography. The purified enzyme showed a singleband of molecular weight of 75 kDa on SDS-PAGE (Fig. 3). Thisis in agreement from deduced molecular weight from aminoacid sequence of CGTase from Bacillus sp. TS1-1, which iscalculated to be 75,139.92 Da. The enzyme eluted from the�-cyclodextrin-bound-epoxy activated Sepharose 6B columnresulted in 280-fold purification with a 5.23% yield.
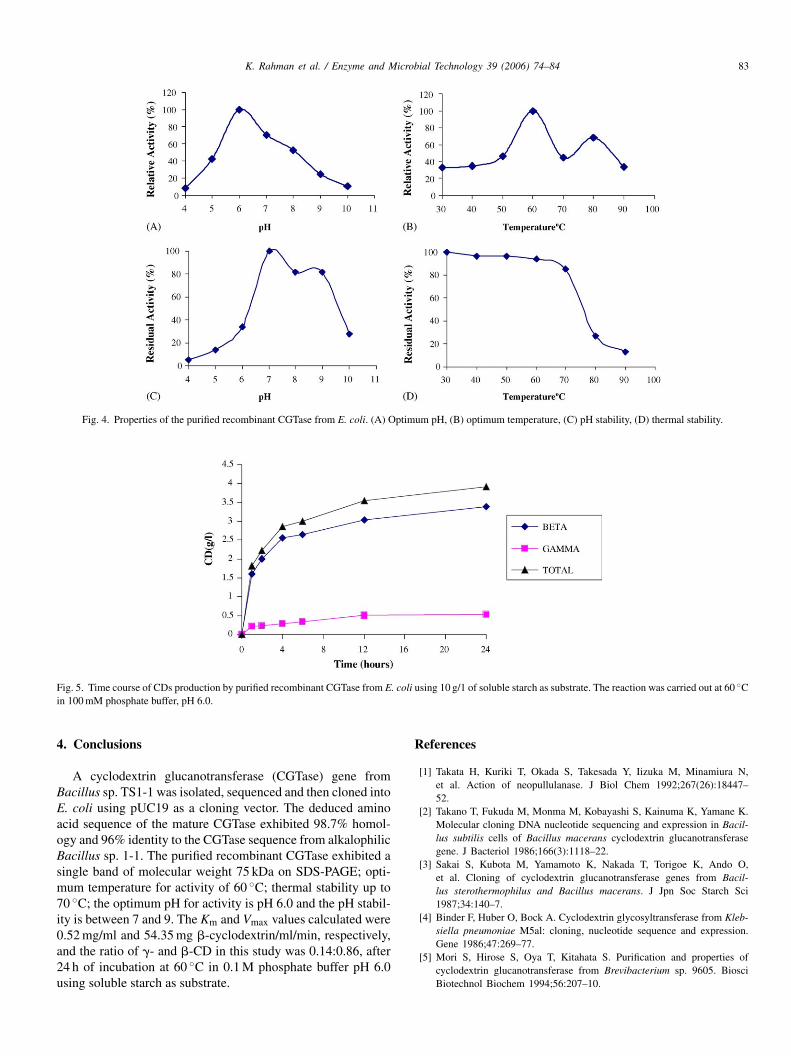
The enzyme displayed optimum activity at 60 ◦C and werestable up to 70 ◦C, retaining 90% of the original activity afterhCep
re probably required for structural support of the raw starch-inding domain. Sequence comparisons of different CGTasesrom different sources also suggest that amino acid residue atosition 47 (Bacillus circulans 251 CGTase numbering) affects
eat treatment at 60 ◦C for 30 min. The effect of pH on theGTase activity and stability were also determined. The enzymexhibited an optimum activity at pH 6.0 and was stable fromH 7.0 to 9.0 with a gradual loss of activity at higher and

80 K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84
Fig. 2. Comparison of the deduced amino acid sequence of Bacillus sp. TS1-1 with other typical �-, �-, �-CGTases and �-amylase. The six highly conserved regionsin different CGTases are overlined. The important amino acids are shaded. The numbering starts after the respective signal sequence, with identity (*), stronglysimilar (:) and weakly similar (·). TS: CGTase from Bacillus sp. TS1-1 (�/�-CGTase); OB: CGTase from Bacillus ohbensis (�/�-CGTase); ST: CGTase from Bacillusstearothermophilus (�/�-CGTase); CI: CGTase from Bacillus circulans strain 251(�-CGTase); II: CGTase from Bacillus clarkii 7364 (�-CGTase); TAA: �-amylasefrom Taka-amylase A.

K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84 81
Fig. 2. (Continued )
82 K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84
Fig. 2. (Continued ).
lower pH values (Fig. 4). The result suggests that the CGTaserequired a slightly acidic pH or near-neutral pH range to per-form its reaction optimally. Extreme pH values were not suit-able for the enzyme to carry out cyclization activity. Most ofthe reported CGTase exhibited optimum pH ranging from 5.0to 8.0 [30].
The initial reaction rate of the purified CGTase was mea-sured at various concentrations of soluble starch and the initialreaction rate was analyzed using Hanes–Woolf plot. The Km
Fig. 3. SDS-PAGE analysis of purified CGTase from the recombinant E. coli.Lane 1: broad range protein molecular weight markers (from Promega), Lane2: purified CGTase enzyme.
and Vmax values calculated were 0.52 mg/ml and 54.35 mg �-cyclodextrin/ml/min (8603 mg �-cyclodextrin/mg protein/min),respectively. Km values for several CGTases have been reported.CGTase from Bacillus circulans E 192 [31] exhibited a Km valueof 5.7 mg/ml while CGTase from Bacillusfirmus [32] have aKm value of 1.21 mg/ml. On the other hand, Martins and Hatti-Kaul [33] found that CGTase from Bacillus agaradhaerens hada Km value of 21.2 mg/ml. All the values shown above, obtainedby different researchers, were based on soluble starch as thesubstrate. The values obtained from this study suggested thatCGTase produced by Bacillus sp. TS1-1 was comparativelymore active compared to the reported CGTases, since it needed amuch lower concentration of substrate (soluble starch) to achievehalf of the Vmax. This is because enzyme with small value of Kmhas a high affinity for the substrate.
The product yield from the reaction of purified CGTase onsoluble starch was investigated by using HPLC.The ratio of �-and �-CD, in this study, was 0.14:0.86, respectively, after 24 h ofincubation at 60 ◦C in 0.1 M phosphate buffer pH 6.0 (Fig. 5). Itcan be easily noticed that the production of CDs, particularly �-CD, accelerated rapidly in the first 6 h of reaction. Nonetheless,the production rate seemed to increase steadily when it reachedthe 12 h of reaction and still managed to increase slowly until the24 h. From the data, there was no �-cyclodextrin as discussed inSection 3.2.
K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84 83
Fig. 4. Properties of the purified recombinant CGTase from E. coli. (A) Optimum pH, (B) optimum temperature, (C) pH stability, (D) thermal stability.
Fig. 5. Time course of CDs production by purified recombinant CGTase from E. coli using 10 g/1 of soluble starch as substrate. The reaction was carried out at 60 ◦Cin 100 mM phosphate buffer, pH 6.0.
4. Conclusions
A cyclodextrin glucanotransferase (CGTase) gene fromBacillus sp. TS1-1 was isolated, sequenced and then cloned intoE. coli using pUC19 as a cloning vector. The deduced aminoacid sequence of the mature CGTase exhibited 98.7% homol-ogy and 96% identity to the CGTase sequence from alkalophilicBacillus sp. 1-1. The purified recombinant CGTase exhibited asingle band of molecular weight 75 kDa on SDS-PAGE; opti-mum temperature for activity of 60 ◦C; thermal stability up to70 ◦C; the optimum pH for activity is pH 6.0 and the pH stabil-ity is between 7 and 9. The Km and Vmax values calculated were0.52 mg/ml and 54.35 mg �-cyclodextrin/ml/min, respectively,and the ratio of �- and �-CD in this study was 0.14:0.86, after24 h of incubation at 60 ◦C in 0.1 M phosphate buffer pH 6.0using soluble starch as substrate.
References
[1] Takata H, Kuriki T, Okada S, Takesada Y, Iizuka M, Minamiura N,et al. Action of neopullulanase. J Biol Chem 1992;267(26):18447–52.
[2] Takano T, Fukuda M, Monma M, Kobayashi S, Kainuma K, Yamane K.Molecular cloning DNA nucleotide sequencing and expression in Bacil-lus subtilis cells of Bacillus macerans cyclodextrin glucanotransferasegene. J Bacteriol 1986;166(3):1118–22.
[3] Sakai S, Kubota M, Yamamoto K, Nakada T, Torigoe K, Ando O,et al. Cloning of cyclodextrin glucanotransferase genes from Bacil-lus sterothermophilus and Bacillus macerans. J Jpn Soc Starch Sci1987;34:140–7.
[4] Binder F, Huber O, Bock A. Cyclodextrin glycosyltransferase from Kleb-siella pneumoniae M5al: cloning, nucleotide sequence and expression.Gene 1986;47:269–77.
[5] Mori S, Hirose S, Oya T, Kitahata S. Purification and properties ofcyclodextrin glucanotransferase from Brevibacterium sp. 9605. BiosciBiotechnol Biochem 1994;56:207–10.

84 K. Rahman et al. / Enzyme and Microbial Technology 39 (2006) 74–84
[6] Wind RD, Liebl W, Buitelaar RM, Penninga D, Spreinat A, DijkhuizenL, et al. Cyclodextrin formation by the thermostable oc-Amylase ofThermoanaerobacterium thermosulfurigenes EMI and reclassification ofthe enzyme as a cyclodextrin glycosyltransferase. Appl Environ Micro-biol 1995;61(4):1257–65.
[7] Yagi Y, Kouno K, Inui TA. A process producing cyclodextrin. USAPatent No. 4,317,881; 1987.
[8] Tonkova A. Bacterial cyclodextrin glucanotransferase. Enzyme MicrobTechnol 1998;22:678–86.
[9] Luong JH, Brown RS, Male KB, Cattaneo MV, Zhao S. Enzyme reactionin the presence of cyclodextrins: biosensors and enzyme assays. TrendsBiotechnol 1995;13:457–63.
[10] Saenger W. Cyclodextrin inclusion compounds in research and industry.Angew Chem 1980;19:344–62.
[11] Szejtli J. Medicinal application of cyclodextrin. Med Care Res Rev1994;14:353–86.
[12] Allegre M, Deratani A. Cyclodextrin uses: from concept to industrialreality. Agro-Food Ind 1994;(Jan/Feb):9–17.
[13] Khairizal M. Cyclodextrin glucanotransferase from alkalophilic Bacillussp. TS1 bacterium. Master thesis, Universiti Teknologi Malaysia; 2000.
[14] Horikoshi K, Nakamura N. Purification and properties of neutral-cyclodextrin glucanotransferase of an alkalophilic Bacillus sp. Agric BiolChem 1976;40:1053–62.
[15] Ish-Horowitz D, Burke JF. Rapid and efficient cosmid cloning. NucleicAcids Res 1981;9(13):2989–998.
[16] Sambrook J, Fritsch EF, Maniatis T. Molecular cloning a laboratorymanual. 2nd ed. USA: Cold Spring Harbor Laboratory Press; 1989.
[17] Kaneko T, Kato T, Nakamura N, Horikoshi K. Spectrophotometricdetermination of cyclization activity of �-cyclodextrin-forming cyclo-maltodextrin glucanotransferase. J Jpn Soc Starch Sci 1987;34(1):45–8.
[18] Laemmli UK. Cleavage of structural proteins during the assembly of the
[
[
[
[22] Sin K, Nakamura A, Kobayashi K, Masaki H, Uozumi T. Cloningsequencing of a cyclodextrin glucanotransferase gene from Bacillusohbensis and its expression in Escherichia coli. Appl Microbiol Biotech-nol 1991;35:600–5.
[23] Kaneko T, Hamamoto T, Horikoshi K. Molecular cloning nucleotidesequencing of the cyclomaltodextrin glucanotransferase gene from thealkalophilic Bacillus sp. strain no. 38-2. J Gen Microbiol 1988;134:97–105.
[24] Kimura K, Takano T, Yamane K. Molecular cloning of the P-cyclodextrin synthestase gene from an alkalophilic Bacillus and itsexpression in Escherichia coli and Bacillus subtilis. Appl MicrobiolBiotechnol 1987;26:149–53.
[25] von Heijne GA. New method for predicting signal sequence cleavagesites. Nucleic Acids Res 1986;14(11):4683–90.
[26] van der Veen BA, Uitdehaag JCM, Dijkstra BW, Dijkhuizen L. Engineer-ing of cyclodextrin glycosyltransferase reaction and product specificity.Biochem Biophys Acta 2000;1543:336–60.
[27] Nakamura A, Haga K, Ogawa S, Kuwano K, Kimura K, Yamane K.Functional relationship between cyclodextrin glucanotransferase from analkalophilic Bacillus and �-amylases: site-directed mutagenesis of theconserved two Asp and one Glu residues. Federation Eur Biochem Soc1992;296(1):37–40.
[28] Lawson CL, van Montfort R, Strpkoptov B, Rozeboom HJ, Kalk KH, deVries G, et al. Nucleotide sequence and X-ray structure of cyclodextringlycosyltransferase from Bacillus circulans strain 251 in a maltose-dependent crystal form. J Mol Biol 1994;236:590–600.
[29] van der Veen B. Engineering reaction and product specificity ofcyclodextrin glycosyltransferase from Bacillus circulans strain 251. Uni-versity of Groningen. PhD thesis; 2000.
[30] Sohn CB, Kim SA, Park YA, Kim MH, Moon SK, Jang SA,et al. Purification and characterization of cyclodextrin glycosyltrans-
[
[
[
head of bacteriophage T4. Nature 1970;227:680–5.19] Schmidt AK, Cottaz S, Driguez H, Schulz GE. Structure of cyclodextrin
glucosyltransferase complexed with a derivative of its main product �-cyclodextrin. J Biochem 1998;37:5909–15.
20] Ohdan K, Kuriki T, Takata H, Okada S. Cloning of the cyclodex-trin glucanotransferase gene from alkalophilic Bacillus sp. A2-5a andanalysis of the raw starch-binding domain. Appl Microbiol Biotechnol2000;53:430–4.
21] Yong J, Choi JN, Park SS, Park CS, Park KH, Choi YD. Secretion ofheterologous cyclodextrin glycosyltransferase of Bacillus sp. E1 fromEscherichia coli. Biotechnol Lett 1996;18:1223–8.
ferase from Bacillus firmus. J-Korean Soc Food Sci Nutr 1997;26(2):351–7.
31] Bovetto LJ, Backer DP, Villette JR, Sicard PJ, Bouquelet SJL. Cycloma-ltodextrin glucanotransferase from Bacillus circulans E 192. BiotechnolAppl Biochem 1992;5:48–58.
32] Gawande BN, Goel A, Patkar AY, Nene SN. Purification and propertiesof a novel raw starch degrading cyclomaltodextrin glucanotransferasefrom Bacillus firmus. Appl Microbiol Biotechnol 1999;51:504–9.
33] Martins RF, Hatti-Kaul R. A new cyclodextrin glycosyltransferase froman alkalophilic Bacillus agaradhaerens isolate: purification and charac-terisation. Enzyme Microb Technol 2002;30:116–24.
Related Documents











