HUMAN MUTATION Mutation in Brief #834 (2005) Online MUTATION IN BRIEF © 2005 WILEY-LISS, INC. Received 26 January 2005; accepted revised manuscript 31 May 2005. Molecular Analysis of the HEXA Gene in Italian Patients With Infantile and Late Onset Tay-Sachs Disease: Detection of Fourteen Novel Alleles Anna Lisa E. Montalvo 1 , Mirella Filocamo 2 , Kristian Vlahoviček 3 , Andrea Dardis 1 , Susanna Lualdi 2 , Fabio Corsolini 2 , Bruno Bembi 1* , and Maria Gabriela Pittis 1 1 Unità di Malattie Metaboliche, I.R.C.C.S. Burlo Garofolo, Trieste, Italy; 2 Laboratorio Diagnosi Pre-Postnatale Malattie Metaboliche, I.R.C.C.S. G Gaslini, Genova, Italy; 3 Protein Structure and Bioinformatics, International Centre for Genetic Engineering and Biotechnology, Trieste, Italy *Correspondence to: Dr. Bruno Bembi, Unità di Malattie Metaboliche I.R.C.C.S. Burlo Garofolo, Via dell’Istria 65/1, (34100) Trieste, Italy; Tel.: +39 040 3785500; Fax: +39 040 3785210; E-mail: [email protected] Communicated by Mark H. Paalman Tay-Sachs disease (TSD) is a recessively inherited disorder caused by the hexosaminidase A deficiency. We report the molecular characterization performed on 31 Italian patients, 22 with the infantile, acute form of TSD and nine patients with the subacute juvenile form, biochemically classified as B1 Variant. Of the 29 different alleles identified, fourteen were due to 15 novel mutations, two being in-cis on a new complex allele. The new alleles caused four frameshifts, three premature stop codons, three amino acid changes, two amino acid deletions and two splicing alterations. As previously reported, the c.533G>A (p.R178H) mutation was present either in homozygosity or as compound heterozygote, in all the patients with the late onset TSD form (B1 Variant); the allele frequency in this group is discussed by comparison with that found in infantile TSD. © 2005 Wiley-Liss, Inc. KEY WORDS: Tay- Sachs; HEXA gene; infantile onset; late onset; B1Variant, mutational analysis INTRODUCTION Human β−hexosaminidases (Hex) A and B (E.C. 3.2.1.52) are dimeric lysosomal glycosidases composed of α/β subunits (Hex A) and β subunits (Hex B). Both isoenzymes remove terminal N-acetylhexosamine residues from G M2 ganglioside that has been bound by the G M2 activator protein; however, in vivo, the G M2 /G M2 activator complex is a substrate only for the Hex A. The active site of the α subunit preferably hydrolyzes substrates with a negative charge, such as β-linked N-acetyl-glucosamine 6-sulfate containing glycosaminoglycans and GM2 ganglioside, although it may also hydrolyze neutral substrates. In contrast, the catalytic site of the ß subunit is only active with neutral substrates and as a result the Hex B isoenzyme is unable to hydrolyze the GM2 ganglioside (Mahuran, 1999; Tutor, 2004). Mutations in either gene encoding its α or β subunits result in a group of recessively inherited disorders, the G M2 Gangliosidosis. In particular, mutations in the HEXA gene (MIM# 606869) encoding the α-subunit of the β-hexosaminidase A lead to Tay-Sachs disease (MIM# 272800), characterized by deficiency of Hex A; mutations in the HEXB gene encoding the β subunit cause Sandhoff disease (MIM# 268800), DOI: 10.1002/humu.9363

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

HUMAN MUTATION Mutation in Brief #834 (2005) Online
MUTATION IN BRIEF
© 2005 WILEY-LISS, INC.
Received 26 January 2005; accepted revised manuscript 31 May 2005.
Molecular Analysis of the HEXA Gene in Italian Patients With Infantile and Late Onset Tay-Sachs Disease: Detection of Fourteen Novel Alleles Anna Lisa E. Montalvo1, Mirella Filocamo2, Kristian Vlahoviček3, Andrea Dardis1, Susanna Lualdi2, Fabio Corsolini2, Bruno Bembi1*, and Maria Gabriela Pittis1
1Unità di Malattie Metaboliche, I.R.C.C.S. Burlo Garofolo, Trieste, Italy; 2Laboratorio Diagnosi Pre-Postnatale Malattie Metaboliche, I.R.C.C.S. G Gaslini, Genova, Italy; 3Protein Structure and Bioinformatics, International Centre for Genetic Engineering and Biotechnology, Trieste, Italy *Correspondence to: Dr. Bruno Bembi, Unità di Malattie Metaboliche I.R.C.C.S. Burlo Garofolo, Via dell’Istria 65/1, (34100) Trieste, Italy; Tel.: +39 040 3785500; Fax: +39 040 3785210; E-mail: [email protected] Communicated by Mark H. Paalman
Tay-Sachs disease (TSD) is a recessively inherited disorder caused by the hexosaminidase A deficiency. We report the molecular characterization performed on 31 Italian patients, 22 with the infantile, acute form of TSD and nine patients with the subacute juvenile form, biochemically classified as B1 Variant. Of the 29 different alleles identified, fourteen were due to 15 novel mutations, two being in-cis on a new complex allele. The new alleles caused four frameshifts, three premature stop codons, three amino acid changes, two amino acid deletions and two splicing alterations. As previously reported, the c.533G>A (p.R178H) mutation was present either in homozygosity or as compound heterozygote, in all the patients with the late onset TSD form (B1 Variant); the allele frequency in this group is discussed by comparison with that found in infantile TSD. © 2005 Wiley-Liss, Inc. KEY WORDS: Tay- Sachs; HEXA gene; infantile onset; late onset; B1Variant, mutational analysis
INTRODUCTION
Human β−hexosaminidases (Hex) A and B (E.C. 3.2.1.52) are dimeric lysosomal glycosidases composed of α/β subunits (Hex A) and β subunits (Hex B). Both isoenzymes remove terminal N-acetylhexosamine residues from GM2 ganglioside that has been bound by the GM2 activator protein; however, in vivo, the GM2/GM2 activator complex is a substrate only for the Hex A. The active site of the α subunit preferably hydrolyzes substrates with a negative charge, such as β-linked N-acetyl-glucosamine 6-sulfate containing glycosaminoglycans and GM2 ganglioside, although it may also hydrolyze neutral substrates. In contrast, the catalytic site of the ß subunit is only active with neutral substrates and as a result the Hex B isoenzyme is unable to hydrolyze the GM2 ganglioside (Mahuran, 1999; Tutor, 2004). Mutations in either gene encoding its α or β subunits result in a group of recessively inherited disorders, the GM2 Gangliosidosis. In particular, mutations in the HEXA gene (MIM# 606869) encoding the α-subunit of the β-hexosaminidase A lead to Tay-Sachs disease (MIM# 272800), characterized by deficiency of Hex A; mutations in the HEXB gene encoding the β subunit cause Sandhoff disease (MIM# 268800),
DOI: 10.1002/humu.9363

2 Montalvo et al.
characterized by combined deficiency of Hex A and Hex B activities (Mahuran, 1999; Gravel et al., 2001). Still there are mutations in the HEXA gene causing the B1 Variant, associated with the late onset form of Tay-Sachs. This biochemical phenotype is characterized by a Hex A isoenzyme catalytically inactive against the physiological substrate, GM2 ganglioside, but active towards commonly used synthetic substrate (4-methylumbelliferyl β-N-acetylglucosaminide) (Tutor, 2004). Biochemical identification of these patients requires a specific sulfated synthetic substrate, 4-methylumbelliferyl N-acetylglucosamine 6-sulfate (MUGS) (Bayleran, et al., 1984 ).
The clinical picture of TSD is confined to the nervous system and phenotypes range from acute infantile form or classic infantile TSD to subacute, juvenile-adult onset, slowly progressive neurological involvement with little or no effect on the intellect. Classic TSD is characterized by the onset in infancy of developmental retardation, followed by paralysis, dementia and blindness, with death in the second or third year of life. The “cherry-red” spot is a typical funduscopic finding and histopathology verification is provided by the finding of the typically ballooned neurons in the central nervous system, due to the accumulation of GM2 ganglioside, cholesterol and phospholipids (Fernandes Filho & Shapiro, 2004). In contrast, the juvenile TSD appears at a later clinical stage with neurodegenerative symptoms leading to patient deth in the second decade of life (Gravel et al., 2001).
The human HEXA gene (MIM# 606869; GenBank Accession NM_000520.2) is located on chromosome 15q23-q24 and contains 14 exons. More than 100 mutations have been identified to cause TSD disease and its variants, including single base substitutions, small deletions, small duplications/insertions, partial gene deletions, splicing alterations and complex gene rearrangements (http://www.hexdb.mcgill.ca/hexadb; http://www.hgmd.org/; Stenson et al., 2003). Most of these alterations are “private” mutations and have been detected in single or very few families. Others are present in small isolated populations and only a few have been frequently found in diverse populations. In the Ashkenazi Jewish population three distinct HEXA mutations are responsible for 98% of all mutant alleles: the most common four-bases duplication c.1274_1277dupTATC and the splicing mutation c.1421+1G>C (IVS12+1G>C) account for 81% and 15% of alleles, respectively; the late-onset alteration in exon 7 c.805G>A (p.G269S) has been found in approximately 2% of alleles (Kaback et al., 1993). Among the non-Jewish populations the mutation pattern is completely different. Only 30% of the alleles are due to the duplication c.1274_1277dupTATC, none present the IVS12+1G>C and about 5% carry the G269S mutation associated with the adult onset of symptoms (Kaback et al., 1993). By contrast, the abnormal splicing mutation c.1073+1G>A (IVS9+1G>A), absent among the Jewish population, is found in about 15% of the non-Jewish carriers (Akerman et al., 1992). Concerning the HEXA mutations associated with the B1 Variant, the most common is the c.533G>A (p.R178H) at first found predominantly in Portuguese patients (dos Santos et al., 1991; Gravel et al., 2001) and which has been subsequently detected in individuals with different European backgrounds. Finally, about 35% of non-Jewish individuals carry one of the two pseudodeficiency alleles previously described (p.R247W and p.R249W), not associated with neurological disease, since their presence causes the reduction of Hex A activity only towards the artificial substrate but not the natural GM2 Ganglioside (Triggs-Raine et al., 1992; Cao et al., 1993).
In this work we analyzed the HEXA gene in 31 Italian patients including twenty-two with the acute infantile TSD as well as nine presenting the subacute juvenile form, biochemically classified as B1 Variant. We identified fifteen novel mutations (two in-cis on a new complex allele) leading to four frameshifts, three stop codons, three amino-acid changes, two amino-acid deletions and one splicing alteration. The pseudodeficiency alleles p.R247W and p.R249W have not been found in the patients studied. The respective allele frequencies of infantile TSD and B1 Variant are comparatively discussed.
MATERIALS AND METHODS
Patients We studied 31 Italian patients with TSD, 16 females and 15 males. Among this group, 29 patients are unrelated
while subjects 6 and 7 as well as 23 and 24 are consanguineous (Table 1). The diagnosis was suspected on the presence of neurological symptoms and confirmed by the demonstration of reduced Hex A activity in peripheral white blood cells or cell lines (fibroblasts or lymphoblasts). Most of the patients underwent a diagnostic protocol that included: a) laboratory tests (Hb, MCV, g.b., platelets, ferritin, AST, ALT, GGT, alkaline phosphatase, LDH, creatinine, proteinemia, IgA, IgM, IgG), b) electrophysiological studies (EEG, ABR, VEP, EMG) and imaging examinations (encephalic MNR and abdominal ultrasonography). Clinical phenotype and molecular data are summarised in Table 1.

Mutational Analysis in Italian Tay Sachs Patients 3
Table 1. Genotype encountered in Italian TSD patients presenting the acute or subacute clinical phenotype
Genotype * Patient no. (sex)
Age at diagnosis
Clinical phenotype Allele 1 Allele 2
1 (male) 1 yr Acute c.2T>C (p.0) c.1292G>A (p.W431X)
2 (male) 2 yr Acute c.632T>C (p.F211S) c.1495C>T (p.R499C)
3 (male) 4 yr Acute c.380T>G (p.L127R) c.736G>A (p.A246T)
4 (male) 1 yr Acute c.947dupA (p.Y316X) c.947dupA (p.Y316X)
5 (female) 1 yr Acute c.607T>G (p.W203G) c.1061_1063delTCT (p.F354del)
6 (female) ŧ 10 mo Acute c.380T>G (p.L127R) c.380T>G (p.L127R)
7 (female) ŧ 10 mo Acute c.380T>G (p.L127R) c.380T>G (p.L127R)
8 (male) 3 yr Acute c.607T>G (p.W203G) c.1511G>A (p.R504H)
9 (female) 8 mo Acute c.615delG (p.V206X) c.1510C>T (p.R504C)
10 (female) 2 yr Acute c.805+2T>C (r.?) c.805+2T>C (r.?)
11 (male) 1yr Acute c.910_912delTTC (p.F304del) c.1495C>T (p.R499C)
12 (female) 1 yr Acute [c.168C>A; c.170delG] (p.G57AfsX42) c.1123delG (p.E375RfsX6)
13 (male) 1yr Acute c.1121A>G (p.Q374R) c.1121A>G (p.Q374R)
14 (male) 1 yr Acute c.1043_1046delTCAA (p.F348CfsX32) c.1043_1046delTCAA (p.F348CfsX32)
15 (female) 1 yr Acute c.910_912delTTC (p.F304del) c.1510delC (p.R504AfsX4)
16 (female) NA Acute c. 1330+1G>A (r.?) c. 1330+1G>A (r.?)
17 (male) 2 yr Acute c.1360G>A (p.G454S) c.508C>T (p.R170W)
18 (male) 1 yr Acute c.910_912delTTC (p.F304del) c.1470_1474delATCTG (p.S491PfsX5)
19 (female) 18 mo Acute c.1274_1277dupTATC (p.Y427IfsX4) c.756_767del12 (p.V253_E256)
20 (male) 8 mo Acute c.1495C>T (p.R499C) c.1495C>T (p.R499C)
21 (female) 1 yr Acute c.1274_1277dupTATC (p.Y427IfsX4) c.1274_1277dupTATC (p.Y427IfsX4)
22 (female) 9 mo Acute c.1444G>A (p.E482K) c.1444G>A (p.E482K)
23 (male) ŧ 12 yr Subacute c.533G>A (p.R178H) c.533G>A (p.R178H)
24 (male) ŧ 6 yr Subacute c.533G>A (p.R178H) c.533G>A (p.R178H)
25 (female) 5 yr Subacute c.533G>A (p.R178H) c.533G>A (p.R178H)
26 (female) 6 yr Subacute c.533G>A (p.R178H) c.533G>A (p.R178H)
27 (male) 8 yr Subacute c.533G>A (p.R178H) c.533G>A (p.R178H)
28 (female) 3 yr Subacute c.509G>A (p.R170Q) c.533G>A (p.R178H)
29 (female) 3 yr Subacute c.1495C>T (p.R499C) c.533G>A (p.R178H)
30 (female) 3 yr Subacute c.910_912delTTC (p.F304del) c.533G>A (p.R178H)
31 (male) 3 yr Subacute c.1073+1G>A (r.?) c.533G>A (p.R178H)
* New mutations are indicated in bold; RefSeq cDNA NM_000520.2. For cDNA numbering +1 corresponds to the A of the first ATG translation initiation codon. ŧ Consanguinity. NA= not available.
4 Montalvo et al.
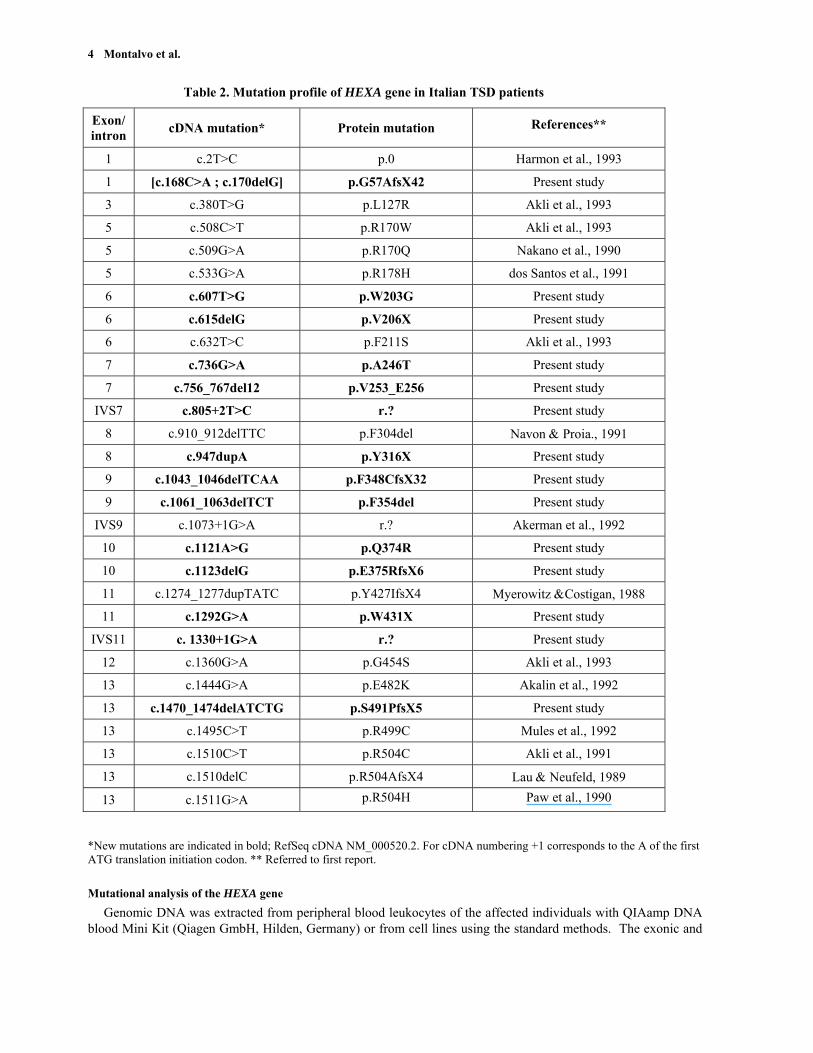
Table 2. Mutation profile of HEXA gene in Italian TSD patients
Exon/ intron cDNA mutation* Protein mutation References**
1 c.2T>C p.0 Harmon et al., 1993
1 [c.168C>A ; c.170delG] p.G57AfsX42 Present study
3 c.380T>G p.L127R Akli et al., 1993
5 c.508C>T p.R170W Akli et al., 1993
5 c.509G>A p.R170Q Nakano et al., 1990
5 c.533G>A p.R178H dos Santos et al., 1991
6 c.607T>G p.W203G Present study
6 c.615delG p.V206X Present study
6 c.632T>C p.F211S Akli et al., 1993
7 c.736G>A p.A246T Present study
7 c.756_767del12 p.V253_E256 Present study
IVS7 c.805+2T>C r.? Present study
8 c.910_912delTTC p.F304del Navon & Proia., 1991
8 c.947dupA p.Y316X Present study
9 c.1043_1046delTCAA p.F348CfsX32 Present study
9 c.1061_1063delTCT p.F354del Present study
IVS9 c.1073+1G>A r.? Akerman et al., 1992
10 c.1121A>G p.Q374R Present study
10 c.1123delG p.E375RfsX6 Present study
11 c.1274_1277dupTATC p.Y427IfsX4 Myerowitz &Costigan, 1988
11 c.1292G>A p.W431X Present study
IVS11 c. 1330+1G>A r.? Present study
12 c.1360G>A p.G454S Akli et al., 1993
13 c.1444G>A p.E482K Akalin et al., 1992
13 c.1470_1474delATCTG p.S491PfsX5 Present study
13 c.1495C>T p.R499C Mules et al., 1992
13 c.1510C>T p.R504C Akli et al., 1991
13 c.1510delC p.R504AfsX4 Lau & Neufeld, 1989
13 c.1511G>A p.R504H Paw et al., 1990
*New mutations are indicated in bold; RefSeq cDNA NM_000520.2. For cDNA numbering +1 corresponds to the A of the first ATG translation initiation codon. ** Referred to first report.
Mutational analysis of the HEXA gene Genomic DNA was extracted from peripheral blood leukocytes of the affected individuals with QIAamp DNA
blood Mini Kit (Qiagen GmbH, Hilden, Germany) or from cell lines using the standard methods. The exonic and

Mutational Analysis in Italian Tay Sachs Patients 5
flanking intronic sequences of the HEXA gene were PCR amplified using specific primers as described. (Triggs-Raine et al., 1991). PCR products were purified with the Genelute PCR DNA Purification Kit (Sigma, St. Louis, MO, USA) and sequenced in the forward and reverse direction. Cycle sequencing was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Warrington, UK) following the manufacturer’s instructions and sequences were analyzed on the ABI PRISM 3700 DNA Analyzer.
Putative mutations were confirmed by sequencing duplicate PCR products as well as by digesting PCR products with the specific restriction endonuclease whose recognition site was consequently altered. If the mutation neither created nor destroyed a restriction site, the amplification was carried out using PCR-mediated site direct mutagenesis that introduced a new cleavage site of restriction (Schwartz et al., 1991). Family studies whenever possible were also carried out.
T
p.Q374R
Figure 1. Positions of HEXA mutations are indicated by space-filled residueE323) are shown in stick representation.
Enzymatic activity assay
Hex A activities were assayed in homogenates of leukocytes and/acetylglucosaminide and 4-methylumbelliferyl β-N-acetylglucosaminconcentration of the samples was determined by the Lowry method.
Table 3. Mutation effects predicted by the stru
Mutation* Description ac.607T>G
(p.W203G) Destabilizes substrate-supporting �-barsite shape
c.736G>A (p.A246T)
Secondary influence on the substrate-bin
c.1121A>G (p.Q374R)
Close proximity of the Q374 and the looE323 could indicate the influence of thisresidue.
* New mutations are named according to RefSeq cDNA NM_000520.2. Forfirst ATG translation initiation codon.
p.W203G
p.A246s in different colors. Active site proton donor (p.
or cell lines using 4-methylumbelliferyl β-N-ide 6 sulfate (MUGS) as substrates. Protein
ctural 3D analysis
nd possible effect rel, possibly distorting the substrate-binding
ding �-barrel
p containing the active-site proton donor mutation on the position of the active-site
cDNA numbering +1 corresponds to the A of the

6 Montalvo et al.
Structural 3D analysis
Theoretical model of the HexA α chain (PDB code 1QBC, residues 109-529; Tews et al., 1996), was used as a basis for the structural analysis. Mutations were analyzed and mapped onto the 3D model using the SwissPDB Viewer v3.7 (http://www.expasy.org/spdbv/; Guex and Peitsch, 1997). Final visualization was prepared using PyMOL (http://www.pymol.org).
Mutation nomenclature All mutations are described according to mutation nomenclature, considering nucleotide +1 the A of the first
ATG translation initiation codon (den Dunnen and Antonarakis, 2000; den Dunnen and Paalman, 2003; http://www.hgvs.org/mutnomen). Nucleotide numbers are derived from cDNA HEXA sequence (RefSeq cDNA NM 000520).
c.1495C>T 9.5%
Infantile TSD
c.533G>A75%
Late Onset TSD
Figure 2. Comparison of the allele frequency between the infantile and late onset TSD. The allelic homogeneity of late onset TSD (B1 Variant) is shown on the right whereas the allelic heterogeneity of infantile TSD is evident on the left . Note that only the two respective most frequent mutations are shown.
RESULTS AND DISCUSSION
We carried out the complete molecular analysis of the HEXA gene in 31 Italian patients, including twenty-two with the acute infantile TSD as well as nine presenting the subacute juvenile form, biochemically classified as B1 Variant. Overall, we identified 29 different alleles: fourteen are due to 15 novel mutations, two being in-cis on a new complex allele (Table 1). In the presence of novel mutant alleles the supposed disease-causing mutations were confirmed on two independent PCR products and on the parents’ DNA when available. Moreover, the hypothesis of new possible genetic polymorphisms was also excluded ascertaining that none of the 100 control alleles had these alterations.
As shown in Table 2, the mutation profile was characterized by mutations spread over the sequence and consisted of point mutations, small deletions and small duplications causing both missense and nonsense mutations, amino acid deletions, frameshifts and splicing aberrations. It is interesting to note that the new c.1121A>G (p.Q374R) and c.1123delG (p.R375XfsX6) mutations affected exon 10 in which up to date only an inframe 12 bp deletion was reported in the Turkish population (Ozkara & Navon, 1998; Sinici et al., 2004).

Mutational Analysis in Italian Tay Sachs Patients 7
Within the novel mutations, we identified four single base substitutions, c.607T>G, c.736G>A, c.1121A>G and c.1292G>A, that resulted in drastic amino acid changes (p.W203G, p.A246T, p.Q374R) or in a truncated protein (p.W431X). Protein sequence alignment demonstrated that p.W203, p.A246, p.Q374 and p.W431 are highly conserved residues among the human, mouse and rat α-subunits of Hex A. Moreover, these residues are also conserved in both α and β subunits of Hex A, which are 60% identical in their primary structure (Maier et al., 2003). To understand the structural consequences of the novel missense mutations (p.W203G, p.A246T, p.Q374R), we modelled each of the amino acid replacements into the theoretical model of the HexA α subunit (Twes et al., 1996) (Fig. 1). The structural analysis predicts that all three mutations might disturb the substrate-binding site shape; in particular, p.Q374R might disturb the position of the active-site residue p.E323. (Table 3).
The six deletions observed (c.1043_1046delTCAA, c.1123delG, c.1470_1474delATCTG, c.756_767del12, c.1061_1063delTCT, c.615delG) led to three frameshifts (p.F348CfsX32, p.E375RfsX6, p.S491PfsX5), two residue deletions (p.V253_E256, p.F354del) and a premature stop codon (p.V206X), respectively. The duplication of adenine in exon 8 (c.947dupA) also resulted in a stop codon (p.Y316X).
A complex allele was identified in patient 12, characterized by the C>A substitution at nucleotide position 168 and a G deletion at position 170. The presence of both alterations on the same allele causes a shift of the reading frame at codon 57 in exon 1 that introduces a stop codon 32 residues downstream (p. G57AfsX32), leading to an unstable truncated peptide which would be rapidly degraded.
The frameshift mutations were found only in the infantile TSD patients (Table 1). The c.1043_1046delTCAA, found in homozygosity in patient 14, changes the reading frame at codon 348 introducing a premature stop at codon 380 (p.F348CfsX32). The c.1123delG causing an aminoacidic substitution in position 375 and premature protein truncation six residues downstream (p.E375RfsX6), was associated with the complex allele in patient 12. The frameshift in exon 13, starting at codon 491 and ending in a stop at codon 496 (p.S491PfsX5) was found in combination with the frequently reported deletion p.F304del in patient 18.
Finally, two novel mutations affecting mRNA splicing, c.805+2T>C and c. 1330+1G>A, were detected in homozygous state in patient 10 and patient 16, respectively. Several splicing mutations have been reported in both HEXA and HEXB genes. In all cases, mutations involving the first and last two nucleotides of each intron, either in homozygosity or as compound heterozygotes with a second null mutation, resulted in very low mRNA levels or no detectable mRNA. All the splicing mutations found in this work were associated with the acute phenotype of the disease, as it has been previously reported (Mahuran, 1999; Gravel et al., 2001).
The clinical presentation of the patients studied at diagnosis was characterized by delayed milestones, hypotonia, seizures, and the presence of cherry-red spots, similarly to what have been reported in other studies (Fernandes Filho & Shapiro, 2004; Nalini & Christopher, 2004). No evident correlation was observed between clinical features and genotype, except for the fact that all the 8 unrelated patients with late onset TSD (B1 Variant) carried the known c.533G>A (p.R178H) mutation, either in homozygosity (four patients) or as compound heterozygotes (4 others in combination with c.509G>A, c.1495C>T, c.910_912delTTC, c.1073+1G>A) (Table 1). The second allele in patients 30 and 31 corresponded to deleterious mutations, c.910_912delTTC and c.1073+1G>A, resulting in a defective subunit assembly and aberrant transcripts, respectively. It is important to note that both patients presented a more severe course of the disease and died at 5 years of age. These findings confirmed the milder nature of the c.533G>A (Gravel et al., 2001), associated with a late onset of the disease. Moreover, as expected, the c.533G>A, which has a widespread geographic and ethnic distribution (dos Santos, et al., 1991; Gravel, et al., 2001), was the most frequent mutation also in our Italian casuistry being responsible for 75% of the patients biochemically classified as B1 Variant. It is interesting to note the impressive contrast between the allelic homogeneity of the B1 Variant group and the allelic heterogeneity of the infantile TSD in which the most frequent mutation, the c.1495C>T, is barely 9.5% of the alleles followed by c.910_912delTTC, c.380T>G and c.1274_1277dupTATC each of them accounting for 7.1%. Still there is a little group of alleles (c.947dupA, c.805+2T>C, c.1444G>A, c.1121A>G, c.1043_1046delTCAA, c. 1330+1G>A) presenting a frequency of 4.1% while the remaining 17 alleles are very rare or “private” with a frequency of 2.4% (Fig. 2). This phenomenon has been observed in other lysosomal storage diseases such as in the Glycogenosis type II or Pompe disease, in which the most frequent mutation named IVS1 –13T>G is restricted to the milder late onset phenotype (Raben et al., 2002).

8 Montalvo et al.
ACKNOWLEDGMENTS
The authors would like to thank Sarah Tripepi for her assistance in the preparation of the manuscript. Samples were obtained from the “Cell Line and DNA Bank from Patients affected by Genetic Disease” collection (http://www.gaslini.org/labdppm.htm) supported by Italian Telethon grants (GTF04002). This work was supported by a grant from Fondazione CRTrieste and the institutional research projects RC56/03 and RF2003/839 “Studio dell’espressione genica e proteomica in malattie rare del metabolismo.”
REFERENCES
Akalin N, Shi HP, Vavougios G, Hechtman P, Lo W, Scriver CR, Mahuran D, Kaplan F.1992. Novel Tay-Sachs disease mutations from China. Hum Mutat 1:40-6.
Akerman BR, Zielenski J, Triggs-Raine BL, Prence EM, Natowicz MR, Lim-Steele JS, Kaback MM, Mules EH, Thomas GH,
Clarke JT, et al.1992. A mutation common in non-Jewish Tay-Sachs disease: frequency and RNA studies. Hum Mutat. 1:303-9.
Akli S, Chelly J, Lacorte JM, Poenaru L, Kahn A.1991. Seven novel Tay-Sachs mutations detected by chemical mismatch
cleavage of PCR-amplified cDNA fragments. Genomics. 11:124-34. Akli S, Boue J, Sandhoff K, Kleijer W, Vamos E, Young E, Gatti R, Di Natale P, Motte J, Vanier MT, et al.1993. Collaborative
study of the molecular epidemiology of Tay-Sachs disease in Europe. Eur J Hum Genet. 1:229-238. Bayleran J, Hechtman P, Saray W. 1984. Synthesis of 4-methylumbelliferyl-beta-D-N-acetylglucosamine-6-sulfate and its use
in classification of GM2 gangliosidosis genotypes. Clin Chim Acta. 143:73-89.Cao Z, Natowicz MR, Kaback MM, Lim-Steele JS, Prence EM, Brown D, Chabot T, Triggs-Raine BL. 1993. A second mutation associated with apparent beta-hexosaminidase A pseudodeficiency: identification and frequency estimation. Am J Hum Genet.53:1198-205.
den Dunnen JT, Antonarakis SE. 2000. Mutation nomenclature extensions and suggestions to describe complex mutations: a
discussion. Hum Mutat 15:7-12. den Dunnen JT, Paalman MH. 2003. Standardizing mutation nomenclature: why bother? Hum Mutat 22:181-182. dos Santos MR, Tanaka A, sá Miranda MC, Ribeiro MG, Maia M, Suzuki K. 1991. GM2-gangliosidosis B1 variant: analysis of
beta-hexosaminidase alpha gene mutations in 11 patients from a defined region in Portugal. Am J Hum Genet. 49:886-90.
Fernandes Filho JA, Shapiro BE. 2004.Tay-Sachs disease.Arch Neurol. 61:1466-8. Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K and Suzuki K. 2001. The GM2 gangliosidoses. In: Scriver CR,
Beaudet AL, Sly WS & Valle D (Editors), The Metabolic and Molecular Bases of Inherited Diseases. McGraw-Hill, New York, 3827-3876.
Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling.
Electrophoresis 18(15):2714-23. Harmon DL, Gardner-Medwin D, Stirling JL. 1993. Two new mutations in a late infantile Tay-Sachs patient are both in exon 1
of the beta-hexosaminidase alpha subunit gene. J Med Genet. 30:123-8. Kaback M, Lim-Steele J, Dabholkar D, Brown D, Levy N, Zeiger K. 1993. Tay-Sachs disease-carrier screening, prenatal
diagnosis, and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA 270:2307-15.

Mutational Analysis in Italian Tay Sachs Patients 9
Lau MM, Neufeld EF. 1989 A frameshift mutation in a patient with Tay-Sachs disease causes premature termination and
defective intracellular transport of the alpha-subunit of beta-hexosaminidase. J Biol Chem 264:21376-80. Mahuran DJ. 1999. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys
Acta.1455:105-138. Mules EH, Hayflick S, Miller CS, Reynolds LW, Thomas GH.1992. Six novel deleterious and three neutral mutations in the gene encoding the alpha-subunit of hexosaminidase A in non-Jewish individuals. Am J Hum Genet 50:834-41.
Maier T, Strater N, Schuette CG, Klingenstein R, Sandhoff K, Saenger W. 2003. The X-ray crystal structure of human beta-
hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 328:669-81. Myerowitz R, Costigan FC. 1988. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for
the alpha-chain of beta-hexosaminidase. J Biol Chem. 263:18587-9. Nakano T, Nanba E, Tanaka A, Ohno K, Suzuki Y, Suzuki K. 1990. A new point mutation within exon 5 of beta-
hexosaminidase alpha gene in a Japanese infant with Tay-Sachs disease. Ann Neurol. 27:465-73. Nalini A, Christopher R. 2004. Cerebral glycolipidoses: clinical characteristics of 41 pediatric patients.J Child Neurol. 19:447-
52. Navon R, Proia RL. 1991. Tay-Sachs disease in Moroccan Jews: deletion of a phenylalanine in the alpha-subunit of beta-
hexosaminidase. Am J Hum Genet 48:412-419Ozkara HA, Navon R. 1998. At least six different mutations in HEXA gene cause Tay-Sachs disease among the Turkish population. Mol Genet Metab. 65:250-3.
Paw BH, Moskowitz SM, Uhrhammer N, Wright N, Kaback MM, Neufeld EF. 1990. Juvenile GM2 gangliosidosis caused by
substitution of histidine for arginine at position 499 or 504 of the alpha-subunit of beta-hexosaminidase. J Biol Chem. 265:9452-7.
Raben N, Plotz P, Byrne BJ.2002.Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med
2:145-66. Schwartz EI, Shevtsov SP, Kuchinski AP, Kovalev Yu P, Plutalov OV, Berlin Yu A. 1991. Approach to identification of a
point mutation in apo B100 gene by means of a PCR-mediated site-directed mutagenesis. Nucleic Acids Res 19:3752. Sinici I, Tropak MB, Mahuran DJ, Ozkara HA. 2004. Assessing the severity of the small inframe deletion mutation in the
alpha-subunit of beta-hexosaminidase A found in the Turkish population by reproducing it in the more stable beta-subunit.J Inherit Metab Dis. 27:747-56.
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. 2003. Human
Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 21(6):577-81. Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. 1996. Bacterial chitobiase structure provides insight into
catalytic mechanism and the basis of Tay-Sachs disease. Nat Struct Biol. 3:638-48. Triggs-Raine BL, Akerman BR, Clarke JT, Gravel RA. 1991. Sequence of DNA flanking the exons of the HEXA gene, and
identification of mutations in Tay-Sachs disease. Am J Hum Genet. 49:1041-54. Triggs-Raine BL, Mules EH, Kaback MM, Lim-Steele JS, Dowling CE, Akerman BR, Natowicz MR, Grebner EE, Navon R,
Welch JP, et al. 1992. A pseudodeficiency allele common in non-Jewish Tay-Sachs carriers: implications for carrier screening. Am J Hum Genet. 51:793-801.
Tutor JC. 2004. Biochemical characterization of the GM2 gangliosidosis B1 variant. Braz J Med Biol Res. 37:777-783.
Related Documents




![Hexa TR Brosur - Masgrup · 2020. 1. 27. · HEXA Serisi Dikey Milli Paslanmaz Çelik Pompalar Genel Bilgiler Performans Aralığı H HEXA Hidrolik Alan - [2900 d/d] (m) HEXA 2 HEXA](https://static.cupdf.com/doc/110x72/60388517a7423666b665e135/hexa-tr-brosur-masgrup-2020-1-27-hexa-serisi-dikey-milli-paslanmaz-elik.jpg)