Publication ofthe International Umon Against Cancer Publication de I Union lnternationale Contre le Cancer L Int. J. Cancer: 60,235-243 (1995) 0 1995 Wiley-Liss, Inc. MODULATION OF CELLULAR CHEMORESISTANCE IN KERATINOCYTES BY ACTIVATION OF DIFFERENT ONCOGENES Ricardo SANCHEZ-PRIETO~, Juan Antonio VARGAS?,Amancio CARNERO~, Elena MARCHETTI’, Jesus ROMERO’, Albert0 DURANTEZ?, Juan Carlos LACAL’ and Santiago RAMON Y CAJAL1,4 Departments of lPathology and ?Internal Medicine. Clinica Puerza de Hierro. Uttcversidad Autdrioma de Madnd; and 31nstitr*to de Investrgacrones BiomPdicas, Madnd, Spurn Response to chemotherapeutic agents in malignant tumors depends on many factors, most of which are as yet unknown. We investigated the correlation between the activation of different oncogenes and protein-kinase-C(PKC) modulation, and the cytotoxicity of some of the most widely used anti-cancer drugs. We transformed the murine keratinocyte cell line PAM 2 12, with different oncogenes (v-H-ras, v-myc and adenovirus €/a) and a mutant p53 suppressor gene (mp53). The cytotoxic effect of cisplatin (CDDP), doxorubicin(D0X) and vincristine (VCR), together with the concomitant action of modulators of PKC, TPA and staurosporine were evaluated by the crystal- violet method, thymidine incorporation and flow cytometry. We report that (a) the oncogene v-H-ras induces resistance to CDDP (> 50%), DOX (> 25%) and VCR (> 20%); (b) the Ela oncogene induces only resistance to VCR (> 40%) and marked sensitivity to CDDP and DOX (c) the mp53 oncogene induces more resistanceto VCR and insignificantresistanceto the other drugs; and (d) activation of PKC by TPA increases the resistance to VCR and DOX in cells transformed by the v-H-ras, while it significantly increases the lethality with CDDP of the Ela- transformed cells. Staurosporine increases the cytoxicity of all the drugs, especially in the E /a-transformed keratinocytes. In the flow-cytometry analysis, the percentage of BUdR incorpora- tion was related to sensitivity to anti-cancer drugs. o 1995 Wiley-Liss, Inc. Chemotherapy is used for the treatment of most malignant tumors with variable clinical response. The marked heteroge- neity in the response of tumors to anti-cancer drugs may be due to several factors, including cell type, degree of differentia- tion, cell-cycle stage, as well as to the different oncogene- induced alterations and other as yet unknown genetic factors. The fact that at least 6 genetic alterations may be involved in tumor development in humans suggests that tumor heterogene- ity may be related to the different oncogenes and suppressor genes activated/mutated (Bishop, 1991; Harnevo and Aguz, 1992). In this regard, we have shown that murine malignant melanomas induced by several oncogenes show different morphological features (Ramon y Cajal et al., 1991). More- over, carcinomas formed by v-H-ras-transformed epithelial cells are inhibited by dermal fibroblasts (DF). and co- transfection with the EZa oncogene can override the inhibitory effect of DF, representing an example of oncogene collabora- tion and tumor formation in vivo (Missero et al., 1991). Correlation between oncogenic activation and resistance to anti-cancer drugs has been reported in NIH-3T3 cells (Niimi et al., 1991; Isonishi et al., 1991; Sklar, 1988) and other cell lines (Sklar and Prochownik, 1991). Drug resistance may be intrinsic or acquired, and one of the most-studied mechanisms of drug resistance is the multidrug-resistance (MDR) gene which encodes a 170-kDa plasma membrane glycoprotein (p- glycoprotein) (Hayes and Wolf, 1990). The promoter of MDR can be modulated by the mutations of the ras and mp53 genes (Chin et al., 1992), and the p-glycoprotein may be activated by protein kinase C (PKC) (Hamada and Tsuruo, 1988). PKC, a ubiquitous kinase family of at least 10 isoenzymes which can be activated by phorbol esters and inhibited by different agents such as staurosporine (Sato et al., 1990), plays an important role in signal transduction mechanisms triggered by the activa- tion of some oncogenes (Lacal et al., 1990). In this paper, we have studied the sensitivity to the most commonly used chemotherapeutic drugs of epithelial cells transformed by different oncogenes, since carcinomas are the most common tumors in human pathology. We transformed a murine keratinocyte cell line PAM 212, with the oncogenes v-H-ras, adenovirus Ela, v-myc and a mutant form of p.53 suppressor gene, and tested their sensitivity to cisplatin (CDDP), doxorubicin (DOX) and vincristine (VCR), in order to determine whether different oncogenic alterations are related to the variable drug sensitivity observed in tumors. To modulate the chemosensitivity of the various transformed keratinocytes, we regulate the PKC activity of the cells with the concomitant addition of TPA and staurosporine, showing that modulation of PKC may increase/decrease the resistance to these drugs in the v-H-ras- and Ela-transformed keratino- cytes. MATERIAL AND METHODS Cells Mouse PAM 212 cells, a spontaneously obtained keratino- cyte cell line, were grown in Dulbecco’s modified essential medium (DMEM) supplemented with 10% FCS (GIBCO BRL, Paisley, UK), and antibiotics, penicillin (200 U/ml) and streptomycin (100 pglml). In vitro transfornation of the cells Transformations were carried out with a retroviral vector MD (Dotto et a/., 1989) containing a cDNA copy of the 13s transcript of the E l a region of adenovirus 5, a MD-p53 carrying a complete cDNA copy of a mutant p.53 gene of murine origin, or VM-myc retrovirus carrying the avian MC29 gag-myc oncogene. A gene for resistance to G418 was inserted in 2 retroviruses, as reported (Dotto et a/., 1989). PAM 212 cells were cultured at 30% confluence and infected with the retroviruses by a 2-hr exposure to undiluted viral stock with the addition of 8 pg/ml polybrene (Aldrich, Milwaukee, WI) as described (Dotto et al., 1989; Missero et al., 1991). The cultures were grown to confluence and then split 1:5 into selective medium containing 800 pg/rnl G418 (GIBCO BRL) to ensure a homogeneously infected population. The infection with Harvey sarcoma virus carrying the viral H-ras oncogene was carried out in similar conditions and the presence of helper virus ensured infection of most, if not all cells. In vitro characterization Characterization of the PAM 212-EZa-transformed and PAM 212 v-H-ras-transformed cells has been described (Mis- sero et al., 1991; Florin-Christensen et al., 1993). Western-blot analysis was performed in the lines generated to verify the expression of Ela and p53 proteins, immunoprecipitation to 4To whom correspondence and reprint requests should be sent, at Departamento de Patologia, Clinica Puerta de Hierro, cisan Martin de Porres 4,28035 Madrid, Spain. Fax: 34-1-3730535. Received: June 14,1994and in revised form October 7,1994.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Publication ofthe International Umon Against Cancer Publication de I Union lnternationale Contre le Cancer L
Int. J. Cancer: 60,235-243 (1995) 0 1995 Wiley-Liss, Inc.
MODULATION OF CELLULAR CHEMORESISTANCE IN KERATINOCYTES BY ACTIVATION OF DIFFERENT ONCOGENES Ricardo SANCHEZ-PRIETO~, Juan Antonio VARGAS?, Amancio CARNERO~, Elena MARCHETTI’, Jesus ROMERO’, Albert0 DURANTEZ?, Juan Carlos LACAL’ and Santiago RAMON Y CAJAL1,4
Departments of lPathology and ?Internal Medicine. Clinica Puerza de Hierro. Uttcversidad Autdrioma de Madnd; and 31nstitr*to de Investrgacrones BiomPdicas, Madnd, Spurn
Response to chemotherapeutic agents in malignant tumors depends on many factors, most of which are as yet unknown. We investigated the correlation between the activation of different oncogenes and protein-kinase-C(PKC) modulation, and the cytotoxicity of some of the most widely used anti-cancer drugs. We transformed the murine keratinocyte cell line PAM 2 12, with different oncogenes (v-H-ras, v-myc and adenovirus € / a ) and a mutant p53 suppressor gene (mp53). The cytotoxic effect of cisplatin (CDDP), doxorubicin(D0X) and vincristine (VCR), together with the concomitant action of modulators of PKC, TPA and staurosporine were evaluated by the crystal- violet method, thymidine incorporation and flow cytometry. We report that (a) the oncogene v-H-ras induces resistance to CDDP (> 50%), DOX (> 25%) and VCR (> 20%); (b) the E l a oncogene induces only resistance to VCR (> 40%) and marked sensitivity to CDDP and DOX (c) the mp53 oncogene induces more resistance to VCR and insignificant resistance to the other drugs; and (d) activation of PKC by TPA increases the resistance to VCR and DOX in cells transformed by the v-H-ras, while it significantly increases the lethality with CDDP of the Ela- transformed cells. Staurosporine increases the cytoxicity of all the drugs, especially in the E /a-transformed keratinocytes. In the flow-cytometry analysis, the percentage of BUdR incorpora- tion was related to sensitivity to anti-cancer drugs. o 1995 Wiley-Liss, Inc.
Chemotherapy is used for the treatment of most malignant tumors with variable clinical response. The marked heteroge- neity in the response of tumors to anti-cancer drugs may be due to several factors, including cell type, degree of differentia- tion, cell-cycle stage, as well as to the different oncogene- induced alterations and other as yet unknown genetic factors. The fact that at least 6 genetic alterations may be involved in tumor development in humans suggests that tumor heterogene- ity may be related to the different oncogenes and suppressor genes activated/mutated (Bishop, 1991; Harnevo and Aguz, 1992). In this regard, we have shown that murine malignant melanomas induced by several oncogenes show different morphological features (Ramon y Cajal et al., 1991). More- over, carcinomas formed by v-H-ras-transformed epithelial cells are inhibited by dermal fibroblasts (DF). and co- transfection with the EZa oncogene can override the inhibitory effect of DF, representing an example of oncogene collabora- tion and tumor formation in vivo (Missero et al., 1991).
Correlation between oncogenic activation and resistance to anti-cancer drugs has been reported in NIH-3T3 cells (Niimi et al., 1991; Isonishi et al., 1991; Sklar, 1988) and other cell lines (Sklar and Prochownik, 1991). Drug resistance may be intrinsic or acquired, and one of the most-studied mechanisms of drug resistance is the multidrug-resistance (MDR) gene which encodes a 170-kDa plasma membrane glycoprotein (p- glycoprotein) (Hayes and Wolf, 1990). The promoter of MDR can be modulated by the mutations of the ras and mp53 genes (Chin et al., 1992), and the p-glycoprotein may be activated by protein kinase C (PKC) (Hamada and Tsuruo, 1988). PKC, a ubiquitous kinase family of a t least 10 isoenzymes which can be activated by phorbol esters and inhibited by different agents such as staurosporine (Sato et al., 1990), plays an important role in signal transduction mechanisms triggered by the activa- tion of some oncogenes (Lacal et al., 1990).
In this paper, we have studied the sensitivity to the most commonly used chemotherapeutic drugs of epithelial cells transformed by different oncogenes, since carcinomas are the most common tumors in human pathology. We transformed a murine keratinocyte cell line PAM 212, with the oncogenes v-H-ras, adenovirus Ela , v-myc and a mutant form of p.53 suppressor gene, and tested their sensitivity to cisplatin (CDDP), doxorubicin (DOX) and vincristine (VCR), in order to determine whether different oncogenic alterations are related to the variable drug sensitivity observed in tumors. To modulate the chemosensitivity of the various transformed keratinocytes, we regulate the PKC activity of the cells with the concomitant addition of TPA and staurosporine, showing that modulation of PKC may increase/decrease the resistance to these drugs in the v-H-ras- and Ela-transformed keratino- cytes.
MATERIAL AND METHODS Cells
Mouse PAM 212 cells, a spontaneously obtained keratino- cyte cell line, were grown in Dulbecco’s modified essential medium (DMEM) supplemented with 10% FCS (GIBCO BRL, Paisley, UK), and antibiotics, penicillin (200 U/ml) and streptomycin (100 pglml).
In vitro transfornation of the cells Transformations were carried out with a retroviral vector
MD (Dotto et a/., 1989) containing a cDNA copy of the 13s transcript of the E l a region of adenovirus 5 , a MD-p53 carrying a complete cDNA copy of a mutant p.53 gene of murine origin, or VM-myc retrovirus carrying the avian MC29 gag-myc oncogene. A gene for resistance to G418 was inserted in 2 retroviruses, as reported (Dotto et a/., 1989).
PAM 212 cells were cultured at 30% confluence and infected with the retroviruses by a 2-hr exposure to undiluted viral stock with the addition of 8 pg/ml polybrene (Aldrich, Milwaukee, WI) as described (Dotto et al., 1989; Missero et al., 1991). The cultures were grown to confluence and then split 1:5 into selective medium containing 800 pg/rnl G418 (GIBCO BRL) to ensure a homogeneously infected population. The infection with Harvey sarcoma virus carrying the viral H-ras oncogene was carried out in similar conditions and the presence of helper virus ensured infection of most, if not all cells.
In vitro characterization Characterization of the PAM 212-EZa-transformed and
PAM 212 v-H-ras-transformed cells has been described (Mis- sero et al., 1991; Florin-Christensen et al., 1993). Western-blot analysis was performed in the lines generated to verify the expression of E l a and p53 proteins, immunoprecipitation to
4To whom correspondence and reprint requests should be sent, at Departamento de Patologia, Clinica Puerta de Hierro, cisan Martin de Porres 4,28035 Madrid, Spain. Fax: 34-1-3730535.
Received: June 14,1994 and in revised form October 7,1994.

236 SANCHEZ ETAL.
check the v-H-ras keratinocytes and Northern blotting with DNA probes for v-rnyc (Dotto et al.. 1989). Briefly, the cells were collected in RIPA buffer (50 mM Tris/HCI. 150 mM NaCI, 0.1% Na-deoxycholate, 1 % triton X-100) with protease inhibitors (10 pg/ml aprotinin and 100 pM PMSF). Protein (30 pg) was dissolved in sample buffer, heated at 90°C for 5 rnin and analyzed by SDS-PAGE in addition to immunoblot- ting with anti-Ela monoclonal antibodies M73 (1:20 dilution) and anti953 PAb 240 (Oncogene Science, Uniondale, NY). The blots were developed using an alkaline-phosphatase- conjugated secondary antibody and revealed by the ECL system (Amersham, Aylesbury, UK).
Immunoprecipitation, using anti-p21(Y 13-259), was carried out with near-confluent dishes of the various cell lines in a methionine-free DMEM supplemented with 2% FCS. Cells were labeled with 150 pCi/ml (35S)-L-methionine (specific activity:1055 Ci/mmol; ICN, Costa Mesa, CA) for 18 hr, cell extracts were collected as described above, analyzed by SDS- PAGE and revealed by film exposure.
Test for sensitivity Doxorubicin (Farmiblastina, Farmitalia Carlo Erba, Milan,
Italy), cisplatin (Neoplatin, Bristol-Myers, Madrid, Spain) and vincristine (DCI) sulfate (Vincrisul, Eli Lilly, Indianapolis, IN) were used at the indicated concentrations.
Cells (4 x 104/well) were plated in 24-multiwell culture plates; subsequently, drugs were added 16 to 24 hr after plating. Drug effect was measured after 72 hr of incubation at 37°C. In time-course experiments, different drug concentra- tions were incubated for 24,48 and 72 hr at 37°C. The cellular resistance was evaluated by the crystal-violet method. Briefly, cells were fixed with 1% glutaraldehyde for 10 min, washed twice in PBS and stained with 1.5 ml of 0.1% crystal-violet solution for 30 min. Wells were rinsed in a beaker with a slow stream of distilled water until the dye was washed off. Wells were left to dry overnight. The absorbance was read at 590 nm by dye uptake in 10% acetic acid. 12-0-tetradecanoyl phorbol- 13-acetate (TPA; Sigma, St. Louis, MO) was dissolved in DMSO, stored at -20°C and used at a final concentration of 30 nM, by dilution with PBS. Staurosporine (Kamiya, Thousand Oaks, CA) was dissolved in dimethylsulfoxide, stored at -20"C, and used at a concentration of 6 nM, by PBS dilution immediately before treatment of cells.
DNA synthesis assay DNA synthesis was measured by [3H]-thymidine (specific
activity: 1 mCi/rnl; Dupont, NEN, Stevenage, UK) incorpora- tion. Cells were incubated overnight in DMEM supplcmented by 0.5% FCS and 1 pCi/ml [3H]-thymidine. Cells were washed twice with PBS, treated with 10% TCA for 2 hr at 4°C and lysed by incubating with 0.5 ml of 0.2 M NaOH for 10 min at room temperature. [3H]-thymidine counts were determined by liquid scintillation counting.
Westem blot (immunoblot) analvsis and PDBu-binding analysis of protein kinase C
Cells were grown under normal conditions, washed twice in PBS, lysed by the addition of RIPA buffer, analyzed by SDS-PAGE and by immunoblotting with anti-PKC a and antibody (Antibody M5, Amersham) and revealed by ECL system (Amersham). The ["I-PDBu binding was determined as described by Lacal et al. (1990). The cells were grown in 6-well plates and incubated in 1 ml of DMEM medium without serum plus [3H]-PDBu for 30 rnin (DUPONT NEN; specific activity 20 Ci/mmol) at a final concentration of 1 pC/ml, and unlabeled PDBu was added for a final concentration of 100 nM. For the non-specific binding determination, 3 plates were incubated with the same total radioactivity but with a final concentration of 50 p M unlabeled PDBu, washed 5 times in 1 ml of PBS containing 1 mg of BSA (Sigma), fatty-acid free,
washed twice in PBS without BSA, and dissolved in 500 JLI of 0.5 N NaOH at room temperature for 30 min. Protein content was estimated by the Bradford protein assay (BIO-RAD, Madrid, Spain) and radioactivity was determined by scintilla- tion counting. Specific binding was calculated by subtracting the radioactivity of the samples incubated in 50 p M PDBu from the values obtained in the samples incubated at 100 nM PDBu. The results are represented as the ratio between CPM and mg protein.
Cell-cycle /proliferation assays DNA synthesis was assessed by incorporation of bromode-
oxyuridine (BUdR) and flow-cytometry analysis. The method for preparing BUdR-labelled cells for flow cytometry has been described (Carlton et al., 1991). Briefly, after incubation with 10 JLM BUdR for 20 min, cells were fixed with ethanol 70%, kept at 4°C overnight, and washed 3 times with excess PBS by centrifugation at 480g for 10 rnin. Cell lysis was carried out by incubation with pepsin 0.04%/HCI 0.1 M for 30 min at 37"C, followed by 3 washes with PBS containing 0.5% Tween 20 and 0.5% BSA (PBS-TB). DNA was partially denatured by incuba- tion with 2 M HCI for 10 rnin at 37°C. After neutralizing the samples with 2 volumes of 0.1 M NazB407, the nuclei were washed 3 times with PBS-TB. After incubation with 0.2 ml anti-BUdR-fluorescein (1:20 in PBS-TB for 60 rnin at room temperature; Boehringer Mannheim, Indianapolis, IN) the nuclei were washed twice in PBS-TB. For simultaneous analy- sis of DNA synthesis and cell cycle, nuclei were treated with 45 U/ml RNase (30 min at 3TC, Sigma) and 10 kg/ml propidium iodide (30 rnin at room temperature) in PBS-TB and analyzed by flow cytometry. In order to control non-specific staining, nuclei were prepared as described in the absence of BUdR. Flow-cytometry acquisition was done using a FACScan flow cytometer (Becton Dickinson, San Jose, CA), and the data were analyzed using the Lysis I1 software.
Statistical analysis
Mann-Whitney test for unpaired data. The statistical significance of the data was assessed by the
RESULTS
Mouse PAM 212 keratinocytes are an inmortal cell line spontaneously derived from a primary keratinocyte culture. These cells have lost their normal growth/differentiation response to calcium but have remained sensitive to growth inhibition by TGF-B and other molecules such as corticoste- roids, CAMP and IL-3 (Missero et al., 1991; Florin-Christensen et al., 1993).
Expression of oncogene products The expression of the various oncogene products was
studied by immunoprecipitation and by Western-blot and Northern-blot analysis, as described in Material and Methods (Fig. 1). Only PAM 212 keratinocytes transformed with v-H- ras formed tumors in nude mice after 2 to 3 weeks (data not shown).
Effects of cisplatin on PAM 212 keratinocytes transformed by different oncogenes
Cisplatin is a anti-cancer drug that is very widely used in the treatment of epithelial tumors. CDDP induces DNA-strand breaks and inhibition of DNA synthesis. The highest blood concentration is about 25 x 10 p M for doses of 100 mg/m2.
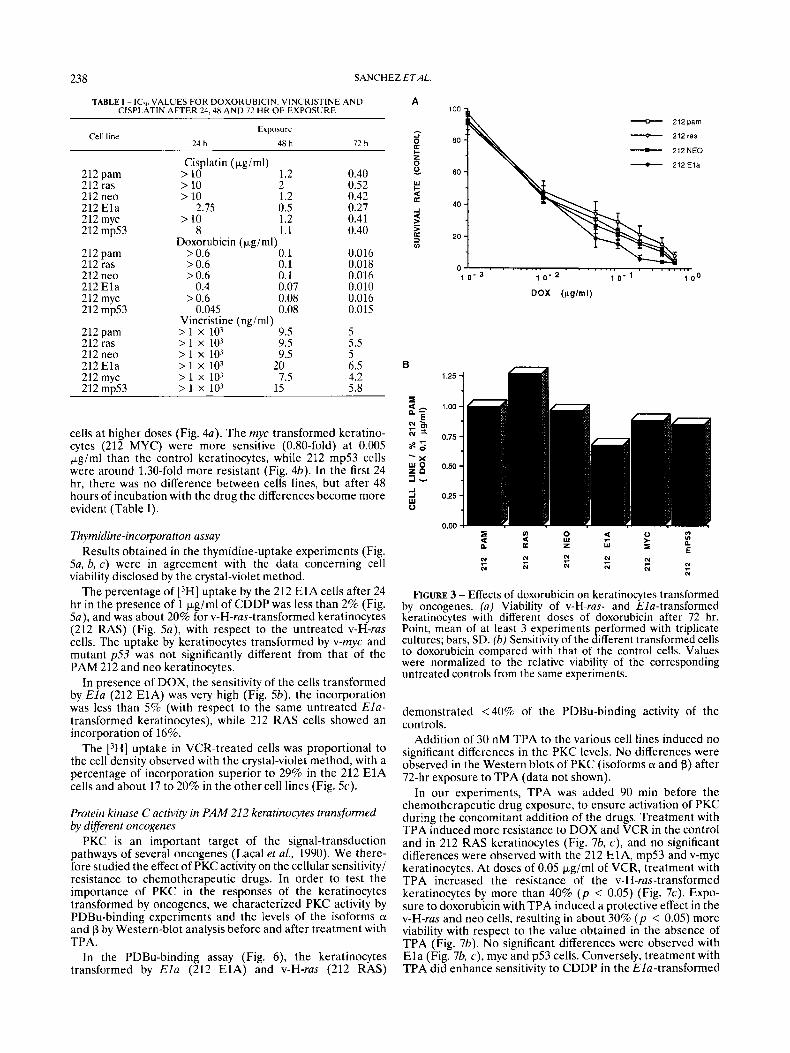
In our experiments, the effects on cell viability of different concentrations of CDDP, ranging from 0.01 pg/ml to 10 pgiml (0.33 to 33.3 pM), were tested in all transformed cell lines. As shown in Figure 2a, the v-H-ras-transformed keratinocytes (212 RAS) proved to be the most resistant cells, while the Ela-PAM 212 cells (212 E1A) were the most sensitive ones in the 72-hr-exposure experiments. v-myc- and mutant-p53-
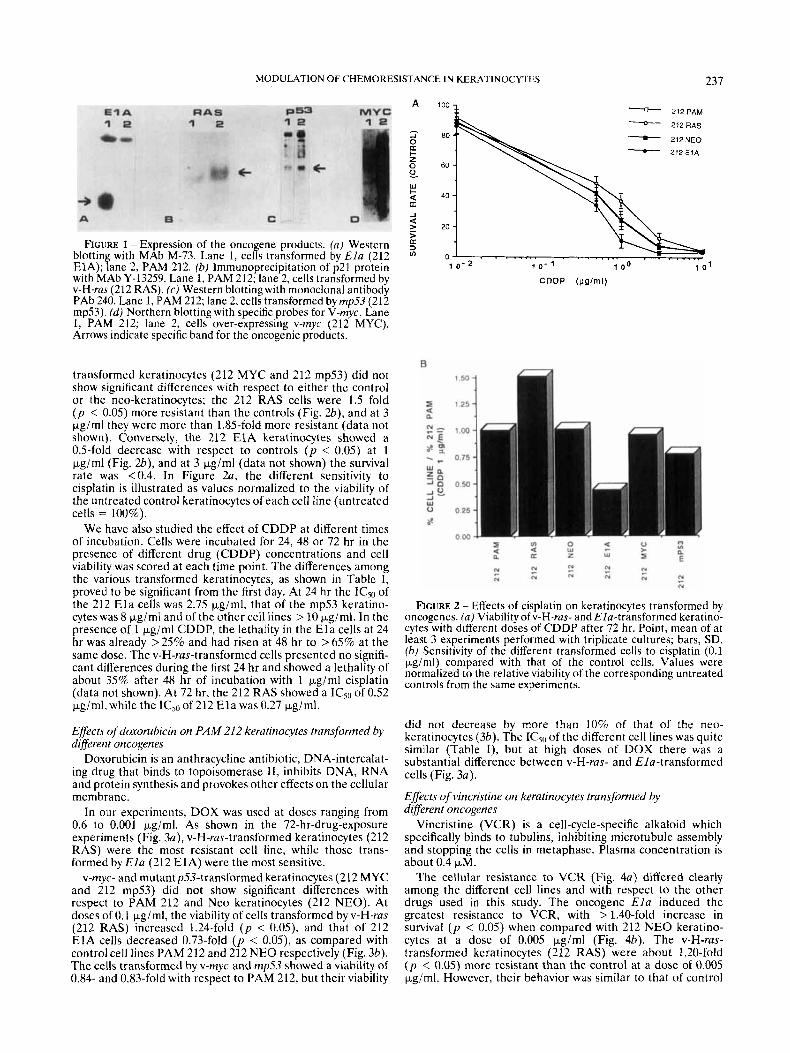
MODULATION OF CHEMORESISTANCE IN KERATINOCYTES 237
FIGURE 1 - Expression of the oncogene products. (a) Western blotting with MAb M-73. Lane 1, cells transformed by E l a (212 ElA); lane 2, PAM 212. (b) Immunoprecipitation of p21 protein with MAb Y-13259. Lane 1, PAM 212; lane 2, cells transformed by v-H-ras (212 RAS). (c) Western blotting with monoclonal antibody PAb 240. Lane 1, PAM 212; lane 2, cells transformed byrnp53 (212 mp53). (d) Northern blotting with specific probes for V-rnyc. Lane I, PAM 212; lane 2, cells over-expressing v-myc (212 MYC). Arrows indicate specific band for the oncogenic products.
transformed keratinocytes (212 MYC and 212 mp53) did not show significant differences with respect to either thc control or the neo-keratinocytes; the 212 RAS cells were 1.5 fold ( p < 0.05) more resistant than the controls (Fig. a), and at 3 pg/ml they were more than 1.85-fold more resistant (data not shown). Conversely, the 212 E1A keratinocytes showed a 0.5-fold decrease with respect to controls ( p < 0.05) at 1 pg/ml (Fig. a), and at 3 pg/ml (data not shown) the survival rate was c0.4. In Figure Za, the different sensitivity to cisplatin is illustrated as values normalized to the viability of the untreated control keratinocytes of each cell line (untreated cells = 100%).
We have also studied the effect of CDDP at different times of incubation. Cells were incubated for 24, 48 or 72 hr in the presence of different drug (CDDP) concentrations and cell viability was scored at each time point. The differences among the various transformed keratinocytes, as shown in Table I, proved to be significant from the first day. At 24 hr the IC5" of the 212 E l a cells was 2.75 Kgiml, that of the mp53 keratino- cytes was 8 Fg/ml and of the other cell lines > 10 kg/ml. In the presence of 1 pgiml CDDP, the lethality in the E l a cells at 24 hr was already >25% and had risen at 48 hr to > 65% at the same dose. The v-H-rus-transformed cells presented no signifi- cant differences during the first 24 hr and showed a lethality of about 35% after 48 hr of incubation with 1 &ml cisplatin (data not shown). At 72 hr, the 212 RAS showed a ICso of 0.52 pgiml. while the ICSo of 212 E l a was 0.27 pgiml.
Effects of doxorubicin on PAM 212 keratinocytes transformed by different oncogenes
Doxorubicin is an anthracycline antibiotic, DNA-intercalat- ing drug that binds to topoisomerase 11, inhibits DNA, RNA and protein synthesis and provokes other effects on the cellular membrane.
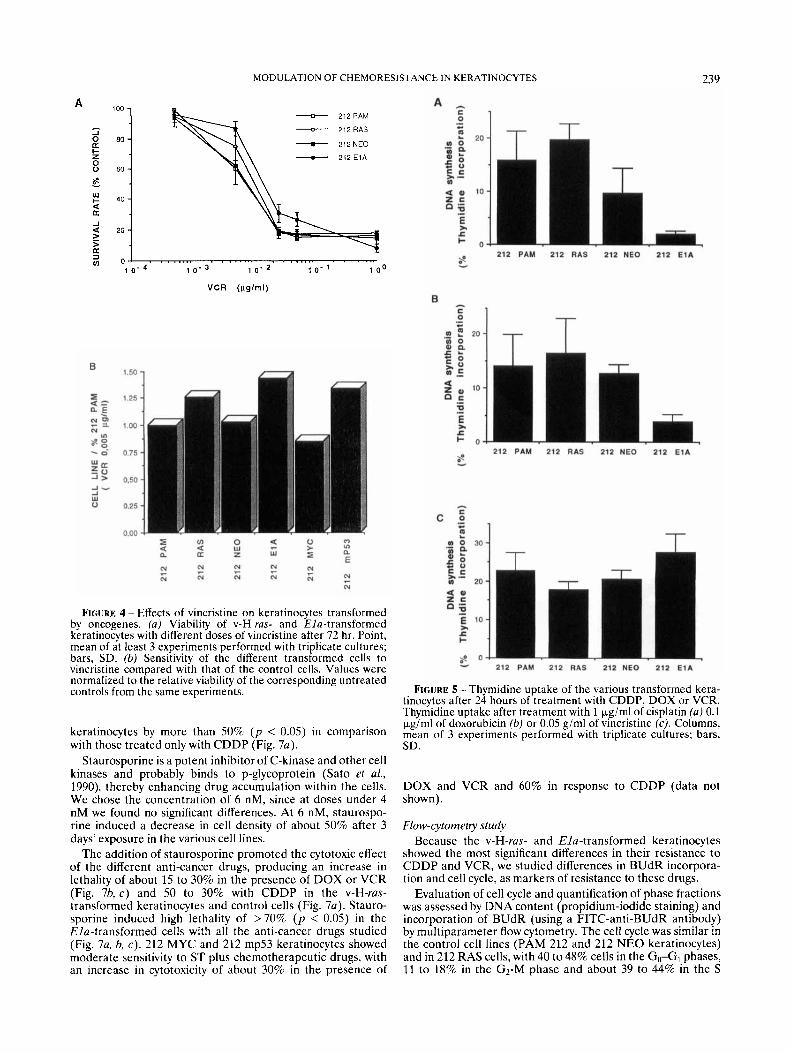
In our experiments, DOX was used at doses ranging from 0.6 to 0.001 pgirnl. As shown in the 72-hr-drug-exposure experiments (Fig. 3a), v-H-rus-transformed keratinocytes (212 RAS) were the most resistant cell line, while those trans- formed by E l a (212 E1A) were the most sensitive.
v-myc- and mutantp53-transformed keratinocytes (212 MYC and 212 mp53) did not show significant differences with respect to PAM 212 and Neo keratinocytes (212 NEO). At doses of 0.1 pg/ml, the viability of cells transformed by v-H-ras (212 RAS) increased 1.24-fold ( p < 0.05), and that of 212 E l A cells decreased 0.73-fold ( p < 0.05), as compared with control cell lines PAM 212 and 212 NEO respectively (Fig. 3b). The cells transformed by v-myc and rnp53 showed a viability of 0.84- and 0.83-fold with respect to PAM 212, but their viability
- 212PAM - 212RAS - 212NEO - 212ElA
40 -
20 -
0 -
1 0 - 2 1 0 - 1 1 0 0 1 0 1
CDDP ($g/ml)
Flcum 2 - EtTects of cisplatin on keratinocytes transformed by oncogenes. (a ) Viability of v-H-ras- and Ela-transformed keratino- cytes with different doses of CDDP after 72 hr. Point, mean of at least 3 experiments performed with triplicate cultures; bars, SD. (b) Sensitivity of the different transformed cells to cisplatin (0.1 Fgiml) compared with that of the control cells. Values were normalized to the relative viability of the corresponding untreated controls from the same experiments.
did not decrease by more than 10% of that of the neo- keratinocytes (36). The IC5,1 of the different cell lines was quite similar (Table I), but at high doses of DOX there was a substantial difference between v-H-ras- and EIu-transformed cells (Fig. 3a).
Effects of vincristine on keratinocytes transfomed by different oncogenes
Vincristine (VCR) is a cell-cycle-specific alkaloid which specifically binds to tubulins, inhibiting microtubule assembly and stopping the cells in metaphase. Plasma concentration is about 0.4 pM.
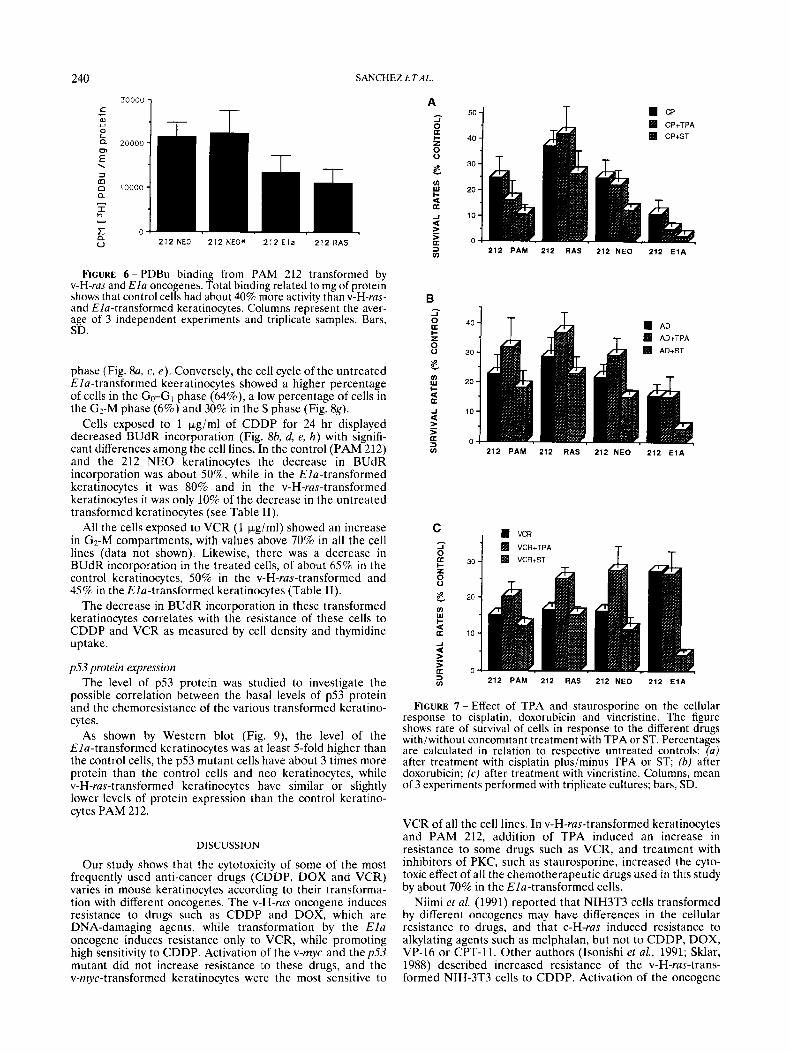
The cellular resistance to VCR (Fig. 4a) differed clearly among the different cell lines and with respect to the other drugs used in this study. The oncogene Ela induced the greatest resistance to VCR, with > 1.40-fold increase in survival ( p < 0.05) when compared with 212 NEO keratino- cytes at a dose of 0.005 pgiml (Fig. 4b). The v-H-ras- transformed keratinocytes (212 RAS) were about 1.20-fold ( p < 0.05) more resistant than the control at a dose of 0.005 pg/ml. However, their behavior was similar to that of control
238 SANCHEZ ETAL.
TABLE I - ICV, VALUES FOR DOXORUBICIN. VINCRISTINE AND CISPLATIN AFTER 24.48 AND 72 HR OF EXPOSURE
A
Exposure Cell line
24 h 48 h 12 h
212 pam 212 ras 212 neo 212 Ela 212 myc 212 mp53
212 pam 212 ras 212 neo 212 Ela 212 myc 212 mp53
212 pam 212 ras 212 neo 212 Ela 212 rnyc 212 mD53
Cisplatin (Fgiml) > 10 1.2 > 10 2 > 10 1.2
2.75 0.5 > 10 1.2
8 1.1
> 0.6 0.1 > 0.6 0.1 > 0.6 0.1
0.4 0.07 > 0.6 0.08
0.045 0.08
Doxorubicin (kg/ml)
Vincristine (ngiml) > I x 103 9.5 > I x 103 9.5 > 1 x 103 9.5 > 1 x 103 20 > 1 x 103 7.5 > 1 x 10-7 15
0.40 0.52 0.42 0.27 0.41 0.40
0.016 0.018 0.016 0.010 0.016 0.015
5 5.5 5 6.5 4.2 5.8
zi a c z 0 0
- 212pam - 212ras - 212NEO
I - 2 1 2 E l a
W
2 a -1
z a 3 VI
O J . . . . . ... 1 0 - 3 1 0 - 2 1 0 - 1 0 0
DOX (pglrnl)
cells at higher doses (Fig. 4a). The myc transformed keratino- cytes (212 MYC) were more sensitive (0.80-fold) at 0.005 kg/ml than the control keratinocytes, while 212 mp53 cells were around 1.30-fold more resistant (Fig. 4b). In the first 24 hr, there was no difference between cells lines, but after 48 hours of incubation with the drug the differences become more evident (Table I).
Thymiditie-incorporation assay Results obtained in the thymidine-uptake experiments (Fig.
Sa, b, c) were in agreement with the data concerning cell viability disclosed by the crystal-violet method.
The percentage of [3H] uptake by the 212 E1A cells after 24 hr in the presence of 1 kg/ml of CDDP was less than 2% (Fig. Sa), and was about 20% for v-H-rus-transformed keratinocytes (212 RAS) (Fig. 5a), with respect to the untreated v-H-ras cells. The uptake by keratinocytes transformed by v-myc and mutant p53 was not significantly different from that of the PAM 212 and neo keratinocytes.
In presence of DOX, the sensitivity of the cells transformed by E l a (212 E1A) was very high (Fig. 5b), the incorporation was less than 5% (with respect to the same untreated Ela- transformed keratinocytes), while 212 RAS cells showed an incorporation of 16%.
The [3H] uptake in VCR-treated cells was proportional to the cell density observed with the crystal-violet method, with a percentage of incorporation superior to 29% in the 212 E1A cells and about 17 to 20% in the other cell lines (Fig. 5c).
Protein kinase C activity in PAM 212 keratinocytes transformed by different oncogenes
PKC is an important target of the signal-transduction pathways of several oncogenes (Lacal et al., 1990). We there- fore studied the effect of PKC activity on the cellular sensitivity/ resistance to chemotherapeutic drugs. In order to test the importance of PKC in the responses of the keratinocytes transformed by oncogenes, we characterized PKC activity by PDBu-binding experiments and the levels of the isoforms ci and p by Western-blot analysis before and after treatment with TPA.
In the PDBu-binding assay (Fig. 6), the keratinocytes transformed by Ela (212 E1A) and v-H-ras (212 RAS)
FIGURE 3 - Effects of doxorubicin on keratinocytes transformed by oncogenes. (a) Viability of v-Hi-ras- and Elu-transformed keratinocytes with different doses of doxorubicin after 72 hr. Point, mean of at least 3 experiments performed with triplicate cultures; bars, SD. (b) Sensitivity of the different transformed cells to doxorubicin compared with that of the control cells. Values were normalized to the relative viability of the corresponding untreated controls from the same experiments.
demonstrated <40% of the PDBu-binding activity of the controls.
Addition of 30 nM TPA to the various cell lines induced no significant differences in the PKC levels. No differences were observed in the Western blots of PKC (isoforms ci and p) after 72-hr exposure to TPA (data not shown).
In our experiments, TPA was added 90 min before the chemotherapeutic drug exposure, to ensure activation of PKC during the concomitant addition of the drugs. Treatment with TPA induced more resistance to DOX and VCR in the control and in 212 RAS keratinocytes (Fig. 76, c), and no significant differences were observed with the 212 E l A , mp53 and v-myc keratinocytes. At doses of 0.05 kg/ml of VCR, treatment with TPA increased the resistance of the v-H-ras-transformed kcratinocytes by more than 40% ( p < 0.05) (Fig. 7c). Expo- sure to doxorubicin with TPA induced a protective effect in the v-H-ras and neo cells, resulting in about 30% ( p < 0.05) more viability with respect to the value obtained in the absence of TPA (Fig. 76). No significant differences were observed with E l a (Fig. 7b, c), myc and p53 cells. Conversely, treatment with TPA did enhance sensitivity to CDDP in the Ela-transformed
MODULATION OF CHEMORESISTANCE IN KERATINOCYTES 239
A
3 0 E l- z 0 0
w l-
U a
100 -
80 -
60 -
40 -
20 -
c - 212PAM - 2 i 2 R A S - 2iPNEO - 2 i 2 E i A
FIGURE 4 - Effects of vincristine on keratinocytes transformed by oncogenes. (a) Viability of v-H-ras- and Ha-transformed keratinocytes with different doses of vincristine after 72 hr. Point, mean of at least 3 experiments performed with triplicate cultures; bars, SD. (b) Sensitivity of the different transformed cells to vincristine compared with that of the control cells. Values were normalized to the relative viability of the corresponding untreated controls from the same experiments.
keratinocytes by more than 50% ( p < 0.05) in comparison with those treated only with CDDP (Fig. 7a).
Staurosporine is a potent inhibitor of C-kinase and other cell kinases and probably binds to p-glycoprotein (Sato et al., 1990), thereby enhancing drug accumulation within the cells. We chose the concentration of 6 nM, since at doses under 4 nM we found no significant differences. At 6 nM, staurospo- rine induced a decrease in cell density of about 50% after 3 days' exposure in the various cell lines.
The addition of staurosporine promoted the cytotoxic effect of the different anti-cancer drugs, producing an increase in lethality of about 15 to 30% in the presence of DOX or VCR (Fig. 76, c) and 50 to 30% with CDDP in the v-H-ras- transformed keratinocytes and control cells (Fig. 7a). Stauro- sporine induced high lethality of >70% ( p < 0.05) in the Ela-transformed cells with all the anti-cancer drugs studied (Fig. 7a, b, c). 212 MYC and 212 mp53 keratinocytes showed moderate sensitivity to ST plus chemotherapeutic drugs, with an increase in cytotoxicity of about 30% in the presence of
FIGURE 5 - Thymidine uptake of the various transformed kera- tinocytes after 24 hours of treatment with CDDP, DOX or VCR. Thymidine uptake after treatment with 1 p,g/ml of cisplatin (a) 0.1 pg/ml of doxorubicin (b) or 0.05 g/ml of vincristine (c). Columns, mean of 3 experiments performed with triplicate cultures: bars, SD.
DOX and VCR and 60% in response to CDDP (data not shown).
Flow-cytometiy study Because the v-H-ras- and Ela-transformed keratinocytes
showed the most significant differences in their resistance to CDDP and VCR, we studied differences in BUdR incorpora- tion and cell cycle, as markers of resistance to these drugs.
Evaluation of cell cycle and quantification of phase fractions was assessed by DNA content (propidium-iodide staining) and incorporation of BUdR (using a FITC-anti-BUdR antibody) by multiparameter flow cytometry. The cell cycle was similar in the control cell lines (PAM 212 and 212 NEO keratinocytes) and in 212 RAS cells, with 40 to 48% cells in the G&, phases, 11 to 18% in the G2-M phase and about 39 to 44% in the S
240 SANCHEZ ETAL.
FIGURE 6-PDBu binding from PAM 212 transformed by v-H-ras and EIa onco enes Total binding related to mg of protein shows that control celfs hadabout 40% more activity than v-H-rus- and Elu-transformed keratinocytes. Columns represent the aver- age of 3 independent experiments and triplicate samples. Bars, SD.
phase (Fig. 8a, c, e). Conversely, the cell cycle of the untreated Elu-transformed keeratinocytes showed a higher percentage of cells in the Go-GI phase (64%), a low percentage of cells in the G2-M phase (6%) and 30% in the S phase (Fig. 8g).
Cells exposed to 1 kg/ml of CDDP for 24 hr displayed decreased BUdR incorporation (Fig. 8b, d, e, h) with signifi- cant differences among the cell lines. In the control (PAM 212) and the 212 NEO keratinocytes the decrease in BUdR incorporation was about 50%. while in the Ela-transformed keratinocytes it was 80% and in the v-H-ras-transformed keratinocytes it was only 10% of the decrease in the untreated transformed keratinocytes (see Table 11).
All the cells exposed to VCR (1 kglml) showed an increase in G2-M compartments, with values above 70% in all the cell lines (data not shown). Likewise, there was a decrease in BUdR incorporation in the treated cells, of about 65% in the control keratinocytes, 50% in the v-H-rus-transformed and 45% in the Ela-transformed keratinocytes (Table 11).
The decrease in BUdR incorporation in these transformed keratinocytes correlates with the resistance of these cells to CDDP and VCR as measured by cell density and thymidine uptake.
p53 protein expression The level of p53 protein was studied to investigate the
possible correlation between the basal levels of p53 protein and the chemoresistance of the various transformed keratino- cytes.
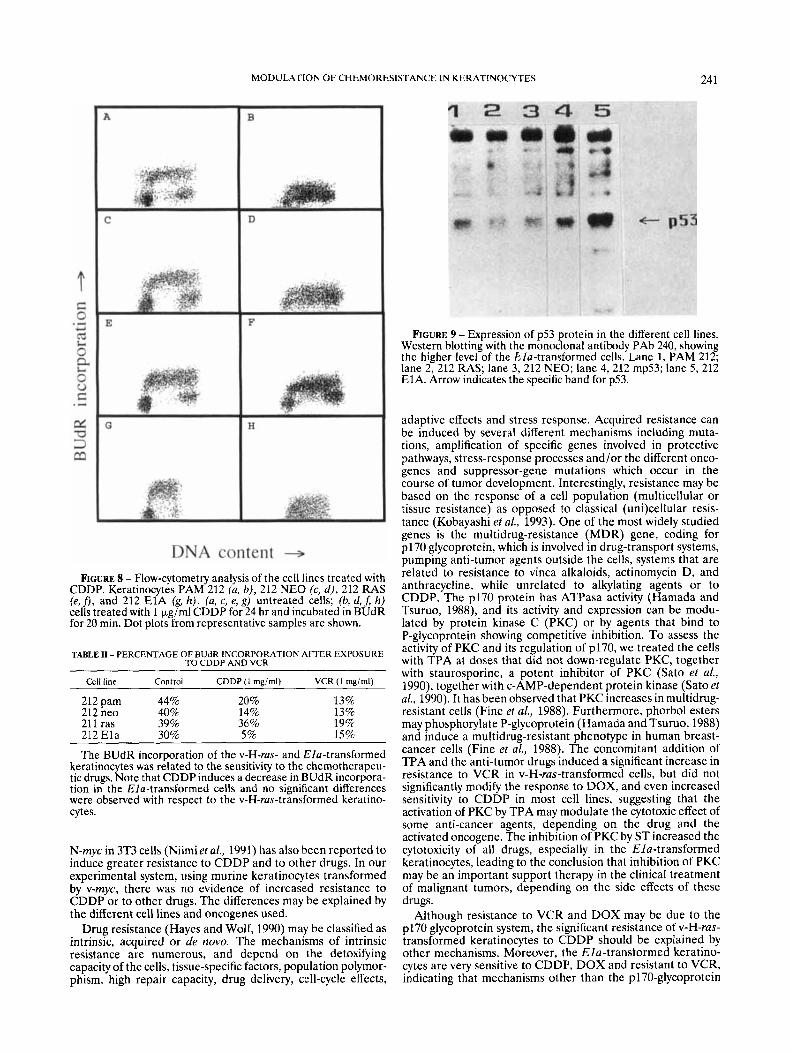
As shown by Western blot (Fig. 9), the level of the Elu-transformed keratinocytes was at least 5-fold higher than the control cells, the p53 mutant cells have about 3 times more protein than the control cells and neo keratinocytes, while v-H-rus-transformed keratinocytes have similar or slightly lower levels of protein expression than the control keratino- cytes PAM 21 2.
DISCUSSION
Our study shows that the cytotoxicity of some of the most frequently used anti-cancer drugs (CDDP, DOX and VCR) varies in mouse keratinocytes according to their transforma- tion with different oncogenes. The v-H-rus oncogene induces resistance to drugs such as CDDP and DOX, which are DNA-damaging agents, while transformation by the E l u oncogene induces resistance only to VCR, while promoting high sensitivity to CDDP. Activation of the v-myc and thep53 mutant did not increase resistance to these drugs, and the v-rnyc-transformed keratinocytes were the most sensitive to
FIGURE 7 - Effect of TPA and staurosporine on the cellular response to cisplatin, doxorubicin and vincristine. The figure shows rate of survival of cells in response to the different drugs withiwithout concomitant treatment with TPA or ST. Percentages are calculated in relation to respective untreated controls: (a) after treatment with cisplatin plusiminus TPA or ST; (b) after doxorubicin; ( c ) after treatment with vincristine. Columns, mean of 3 experiments performed with triplicate cultures; bars, SD.
VCR of all the cell lines. In v-H-ras-transformed keratinocytes and PAM 212, addition of TPA induced an increase in resistance to some drugs such as VCR, and treatment with inhibitors of PKC, such as staurosporine, increased the cyto- toxic effect of all the chemotherapeutic drugs used in this study by about 70% in the Ela-transformed cells.
Niimi et al. (1991) reported that NIH3T3 cells transformed by different oncogenes may have differences in the cellular resistance to drugs, and that c-H-rus induced resistance to alkylating agents such as melphalan, but not to CDDP, DOX, VP-16 or CPT-11. Other authors (Isonishi et al., 1991; Sklar, 1988) described increased resistance of the v-H-rus-trans- formed NIH-3T3 cells to CDDP. Activation of the oncogene
MODULATION OF CHEMORESISTANCE IN KERATINOCYTES 241
DNA content --+
RGURE 8 - Flow-cytometry analysis of the cell lines treated with CDDP. Keratinocytes PAM 212 (a, b), 212 NEO (c, d) , 212 RAS (e, f), and 212 E1A (g, h) . {a, c, e, g) untreated cells; (b, d, $ h) cells treated with 1 kg/ml CDDP for 24 hr and incubated in BUdR for 20 min. Dot plots from representative samples are shown.
TABLE I1 - PERCENTAGE OF BUdR INCORPORATION AFTER EXPOSURE TO CDnP A N D VCR
Cell line Control CDDP (I mgiml) VCR (1 mgiml)
212 pam 44% 20% 13% 212 neo 40 % 14% 13% 211 ras 39% 36% 19% 212 Ela 30% 5% 15%
The BUdR incorporation of the v-H-ras- and Ela-transformed keratinocytes was related to the sensitivity to the chemotherapeu- tic drugs. Note that CDDP induces a decrease in BUdR incorpora- tion in the Ela-transformed cells and no significant differences were observed with respect to the v-H-ras-transformed keratino- cytes.
N-myc in 3T3 cells (Niimi et af., 199 1) has also been reported to induce greater resistance to CDDP and to other drugs. In our experimental system, using murine keratinocytes transformed by v-myc, there was no evidence of increased resistance to CDDP or to other drugs. The differences may be explained by the different cell lines and oncogenes used.
Drug resistance (Hayes and Wolf, 1990) may be classified as intrinsic, acquired or de novo. The mechanisms of intrinsic resistance are numerous, and depend on the detoxifying capacity of the cells, tissue-specific factors, population polymor- phism, high repair capacity, drug delivery, cell-cycle effects,
FIGURE 9 - Expression of p53 protein in the different cell lines. Western blotting with the monoclonal antibody PAb 240, showing the higher level of the Ela-transformed cells. Lane 1, PAM 212; lane 2,212 RAS; lane 3,212 NEO; lane 4,212 mp53; lane 5,212 E1A. Arrow indicates the specific band for p53.
adaptive effects and stress response. Acquired resistance can be induced by several different mechanisms including muta- tions, amplification of specific genes involved in protective pathways, stress-response processes and/or the different onco- genes and suppressor-gene mutations which occur in the course of tumor development. Interestingly, resistance may be based on the response of a cell population (multicellular or tissue resistance) as opposed to classical (uni)cellular resis- tance (Kobayashi et al., 1993). One of the most widely studied genes is the multidrug-resistance (MDR) gene, coding for p170 glycoprotein, which is involved in drug-transport systems, pumping anti-tumor agents outside the cells, systems that are related to resistance to vinca alkaloids, actinomycin D, and anthracycline, while unrelated to alkylating agents or to CDDP. The p170 protein has ATPasa activity (Hamada and Tsuruo, 1988), and its activity and expression can be modu- lated by protein kinase C (PKC) or by agents that bind to P-glycoprotein showing competitive inhibition. To assess the activity of PKC and its regulation of p170, we treated the cells with TPA at doses that did not down-regulate PKC, together with staurosporine, a potent inhibitor of PKC (Sato et al., 1990), together with c-AMP-dependent protein kinase (Sato et af. , 1990). It has been observed that PKC increases in multidrug- resistant cells (Fine et al., 1988). Furthermore, phorbol esters may phosphorylate P-glycoprotein (Hamada and Tsuruo, 1988) and induce a multidrug-resistant phenotype in human breast- cancer cells (Fine et al., 1988). The concomitant addition of TPA and the anti-tumor drugs induced a significant increase in resistance to VCR in v-H-ras-transformed cells, but did not significantly modify the response to DOX, and even increased sensitivity to CDDP in most cell lines, suggesting that the activation of PKC by TPA may modulate the cytotoxic effect of some anti-cancer agents, depending on the drug and the activated oncogene. The inhibition of PKC by S T increased the cytotoxicity of all drugs, especially in the Ela-transformed keratinocytes, leading to the conclusion that inhibition of PKC may be an important support therapy in the clinical treatment of malignant tumors, depending on the side effects of these drugs.
Although resistance to VCR and DOX may be due to the p170 glycoprotein system, the significant resistance of v-H-ras- transformed keratinocytes to CDDP should be explained by other mechanisms. Moreover, the Ela-transformed keratino- cytes are very sensitive to CDDP, DOX and resistant to VCR, indicating that mechanisms other than the pl70-glycoprotein

242 SANCHEZ ETAL
system, eg., changes in drug targets such as the tubulins (Stein and Ziff, 1984), DNA-repair systems or other factors as yet unknown, are involved in that resistance. Likewise, these differences among oncogenes imply that the general mecha- nisms of resistance, such as MDR, cannot always explain the variations in drug resistance among malignant cells. Secondary genetic alterations promoted by transformation with onco- genes can modify the biochemical targets of the anti-cancer drugs, thereby inducing cellular resistance.
The greater resistance to CDDP of the v-H-ras-transformed cells could be explained by the GSH-transferase system (Tsu- chida and Sato, 1992), though neither the activation of the GSH-transferase system nor the increased detoxifying cell enzyme activity have been probed in the v-H-ras-transformed cells (Niimi et a/., 1991; Isonishi et al., 1991). Increases in oxidized GSH have been described in src- and ras-transformed NIH3T3 cells, but not after transformation of NIH3T3 cells by sis, erbB, dbl and raf (Vincenzimi C t al., 1993). The fact that v-H-ras-transformed cells are also more resistant to the effects of radiation (Sklar, 1988) suggests “activated” DNA-repair systems in the v-H-ras-transformed cells.
Flow-cytometry studies of cells treated with CDDP and other drugs have been described by Demarq et al. (1992). CDDP exerts its action at the S and GrM phases, with an accumulation of cells at the SIG? transition of the cell cycle and decrease in BUdR incorporation (Demarcq et al., 1992). Correlation of BUdR incorporation, cell density, evaluated by the crystal-violet method, and thymidine uptake suggest that flow-cytometry studies may be useful to study the sensitivity of CDDP and other anti-cancer drugs.
The resistance to anti-cancer agents has been related top%’, a suppressor gene involved in regulation of the checkpoint following certain types of DNA damage (Kuerbitz et al., 1992; Lowe er al., 1993). Moreover, accumulation of p53 after treatment with anti-cancer drugs has been described in some cell lines, also induction of apoptosis (Lowe et al., 1993; Clarke et a/., 1993), suggesting that involvement of p53 in the apop- totic response may be a mechanism whereby tumor cells can acquire cross-resistance to anti-cancer agents (Lowe et a/., 1993). In our experimental system, the cells transformed by Ela have high levels of p53 (Braitwaite et a/.. 1990), much higher than those of the v-H-ras-transformed keratinocytes PAM 212 and the other cell lines. Our results with EZa-
transformed keratinocytes would be in keeping with the relation between p53-dependent apoptosis and resistance to anti-cancer drugs, though no significant differences could be observed in the basal levels of the v-H-ras-transformed and the control keratinocytes to justify the different resistance to anti-cancer drugs. In addition, although VCR increases the levels of p53 in the Ela-transformed keratinocytes (data not shown), the fact that these cells are resistant to VCR suggests that other mechanisms collaborate with p53, triggering the cells to “commit suicide”. Protein-kinase-C activators, such as phorbol-12-myristate-13-acetate (PMA), and analogs to endog- enous diacylglycerols, such as dioctanoylglycerol (DiC8), have been associated with apoptosis in lymphocytes after the concomitant treatment of the cells with calcium ionophores, independently of p53 (Clarke et a/., 1993). The fact that we obtained high cytotoxicity in the Ela-transformed keratino- cytes treated with TPA only after addition of CDDP, and not after addition of VCR and DOX, suggests the existence of p53-dependent and of independent apoptosis pathways.
In summary, in murine PAM 212 keratinocytes, there are striking differences in the resistance/sensitivity to cytotoxic drugs, depending on the oncogene activation. Transformation by v-H-ras induces significant resistance to CDDP, VCR and DOX, while the Ela oncogene and the p53 mutant induce resistance only to VCR, while markedly enhancing sensitivity to CDDP and DOX. Inhibition of PKC may increase the sensitivity of transformed cells, depending on oncogenic activa- tion. The analysis of oncogene and suppressor genes in human tumors may constitute the “cornerstone” of oncological treat- ment in future attempts to apply more appropriate chemo- therapy. While no specific inhibitors for malignant transforma- tion or oncogene pathways are in clinical use, anti-cancer drugs used in malignant tumors could be correlated with the different oncogenes in each tumor and in each patient.
ACKNOWLEDGEMENTS
We thank Dr. G.P. Dotto for his expert advice and for providing the cells, Ms. C. Lorences, Ms. G. Peraile and Ms. M.A. Ramos for their expert technical assistance. E.M. is a doctoral fellow of the University of Florence. This work was supported by grants from the “Fondo Espafiol de Investiga- ciones Sanitarias”.
REFERENCES
BISHOP, J.M.. Molecular themes in oncogenesis. Cell, 64, 235-248 (1991).
PIGOIIT. D. and JENKINS, J., Transactivation of the p53 oncogene by E l a gene products. Hrologv, 177,595-605 (1990). CARLTON, J.B.. TERRY. N.A. and WHITE, R.A.. Measuring potential doubling times of murine tumors using flow cytometry. Cytometty, 12,
CHIN, K.V.. UEDA, K., PASTAN. I . and GOTTESMAN, M.M., Modulation of activity of the promoter of the human mdrl gene by ras and pS3. Science. 255, 459-462 (1992). CLARKE. A.R.. PURDIE. C.A., HARRISON. D.J., MORRIS, R.G., BIRD, C.C.. HOOPER, M.L. and WYLLIE, T., Thymocyte apoptosis induced by pS3-dependent and independent pathways. Nafirre (Lond.), 362,849- 8.52 (1993). DEMARCO, C.. BASTIAN, G. and REMVIKOS, Y. . BrdUrdiDNA flow- cytometry analysis demonstrates cis-diamminedichloroplatinum(I1)- induced multiple cell-cycle modifications on human lung-carcinoma cells. Cvrometty. 13,416-422 (1992). DOTTO, G.P., MOELLMAN, G., GHOSH, S. , EDWARDS, M. and HALABAN, R., Transformation of murine melanocytes by basic fibroblast-growth- factor cDNA and oncogenes and selective suppression of the trans- formed phenotype in a reconstituted cutaneous environment. J. Cell Biol., 109,3115-3128 (1989).
BRAITWAITt, A,, NELSON, c . , SKULIMOWSKI. A, , MCGOVERN, J.,
645-650 (1991).
FINE, R.L., PATEL, J. and CHABNER. B.A., Phorbol esters induce multidrug resistance in human breast-cancer cells. Proc. nat. Acad. Sci. (Wash.), 85,582-586 (1988). FLORIN-CHRISTENSEN, M., MISSERO, C. , FLORIN, J., TRANQUE, P.. RAMON Y CAJAL. S. and DOTTO, G.P.. Counteracting effects of E l a transformation on CAMP growth inhibition. Exp. Cell Res., 207, 57-61 (1993). HAMADA, H. and TSURUO, T., Characterization of the ATpase activity of the M,-170,000 to -180,000 membrane glycoprotein (P-glycoprotein) associated with multidrug resistance in K562iADM cells. Cancer Rex. 48,4926-4932 (1988). HARNEVO. L.E. and AGUR, Z., Drug resistance as a dynamic process in a model for multistep gene amplification under various levels of selection stringency. Cancer Chemother. Pharmucol., 30, 469476 (1992). HAYES, J.D. and WOLF, R., Molecular mechanisms of drug resistance. Biochem. J.. 272,281-295 (1990).
BASU, A,, LAZO, J.S., EASTMAN, A. and HOWELL, S.B., Expression of the c -Hams oncogene in mouse NIH 3T3 cells induces resistance to cisplatin. Cancer Res., 51, S903-5909 (1991). KORAYASHI, H., MAN, S., GRAHAM. C.H., KAPITAIN, S.J., TEICHER, B.A. and KERBEL, R.S., Acquired multicellular-mediated resistance to
ISONISHI, S., HOM, D.K., THIEBAUT, F.B., MA”, S.C., ANDREWS, P.A..

MODULATION OF CHEMORESISTANCE IN KERATINOCYTES 243
alkylating agents in cancer. Proc. nat. Acad. Sci. (Wash.), 90,3294-3298 (1993). KUERBITZ, S.J., PLUMKETT, B.S., WALSH, W.V. and KASTAN. M.B., Wild-typep53 is a cell-cycle-checkpoint determinant following irradia- tion. Proc. nat. Acad. Sci. (Wash.), 89,7491-7495 (1992). LACAL. J.C., CUADRADO, A,, JONES, J.E., TROTTA, R., BURSTEIN, D.E., THOMSON, T. and PELLICER, A,, Regulation of protein kinase-C activity in neuronal differentiation induced by the N-ras oncogene in PC-12 cells. Mol. cell. B i d , 10,2983-2990 (1990). LOWE, S.W., RULEY. H.E., JACKS. T. and HOUSMAN, D.E., $3- de endent apoptosis modulates the cytotoxicity of anti-cancer drugs. Ceg, 74,957-967 (1993). MISSERO, C., RAMON Y CAJAL. S. and DOTTO. G.P., Escape from transforming-growth-factor-B control and oncogene cooperation in skin-tumor development. Proc. nat. Acad. Sci. (Wash.), 88, 1295-1300 (1991). NIIMI, S., NAKAGAWA, K., YOKOTA. J., TSUNOKAWA, Y. , NISHIO, K., TERASHIMA, Y., SHIBUYA, M., TERADA, M. and SAIJO, N., Resistance to anti-cancer drugs in NIH3T3 cells transfected with c-myc and/or c-H-ras genes. Brit. J. Cancer, 63,237-241 (1991). RAMON Y CAJAL, S., SUSTER, S., HALABAN, R., FILVAROFF, E. and DOTTO, G.P., Induction of different morphologic features of malignant
melanoma and pigmented lesions after transformation of murine melanocytes with bFGF-cDNA and H-ras, myc, neu and E la onco- genes. Amer. J. Pathol., 138,349-358 (1991). SATO, W., YUSA, K., NAITO, M. and TSURUO. T., Staurosporine, a potent inhibitor of C-kinase, enhances drug accumulation in multidrug- resistant cells. Biochem. Biophys. Res. Comm., 173, 1252-1256 (1990). SKLAR, M.D., Increased resistance to cis-diamminedichloro platinum (11) in NIH 3T3 cells transformed by ras oncogenes. Cancer Res., 48, 793-797 (1988). SKLAR, M.D., Ras oncogenes increase the intrinsic resistance NIH 3T3 cells to ionizing radiation. Science. 239,645-647 (1988). SKLAR, M.D. and PROCHOWNIK, E., Modulation of cis-platinum resis- tance in Friend erythroleukemia cells by c-myc. Cancer Res., 51, 21 18-2123 (1991). STEIN, R. and ZIFF, E.B.. E la and tubulin. Mol. cell. Bid. , 4,2792-2801 (1984). TSUCHIDA, S. and SATO, K., Glutathione transferases and cancer. Crit. Rev. Biochem. mol. Biol., 27,337-384 (1992). VINCENZIMI, M.T., MARRACINI, P.. IANTOMASI. T., FAVILLI. F., PACINI, S. and RUGGIERO, M.. Altered metabolism of gluthatione in cells transformed by oncogenes which cause resistance to ionizing radia- tion. FEBS Lett., 320,219-223 (1993).
Related Documents











