This article was downloaded by: [Francisco Lavarda] On: 12 October 2012, At: 07:24 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Molecular Simulation Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/gmos20 Modelling polymers with side chains: MEH-PPV and P3HT A. Batagin-Neto a , E. F. Oliveira a , C. F.O. Graeff a b & F. C. Lavarda a b a UNESP, Univ. Estadual Paulista, POSMAT, Programa de Pós-Graduação em Ciência e Tecnologia de Materiais, Bauru, SP, Brazil b DF-FC, UNESP, Univ. Estadual Paulista, Av. Eng. Luiz Edmundo Carrijo Coube, 14-01, 17033-360, Bauru, SP, Brazil Version of record first published: 12 Oct 2012. To cite this article: A. Batagin-Neto, E. F. Oliveira, C. F.O. Graeff & F. C. Lavarda (): Modelling polymers with side chains: MEH-PPV and P3HT, Molecular Simulation, DOI:10.1080/08927022.2012.724174 To link to this article: http://dx.doi.org/10.1080/08927022.2012.724174 PLEASE SCROLL DOWN FOR ARTICLE Full terms and conditions of use: http://www.tandfonline.com/page/terms-and-conditions This article may be used for research, teaching, and private study purposes. Any substantial or systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form to anyone is expressly forbidden. The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae, and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand, or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article was downloaded by: [Francisco Lavarda]On: 12 October 2012, At: 07:24Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Molecular SimulationPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/gmos20
Modelling polymers with side chains: MEH-PPV andP3HTA. Batagin-Neto a , E. F. Oliveira a , C. F.O. Graeff a b & F. C. Lavarda a ba UNESP, Univ. Estadual Paulista, POSMAT, Programa de Pós-Graduação em Ciência eTecnologia de Materiais, Bauru, SP, Brazilb DF-FC, UNESP, Univ. Estadual Paulista, Av. Eng. Luiz Edmundo Carrijo Coube, 14-01,17033-360, Bauru, SP, Brazil
Version of record first published: 12 Oct 2012.
To cite this article: A. Batagin-Neto, E. F. Oliveira, C. F.O. Graeff & F. C. Lavarda (): Modelling polymers with side chains:MEH-PPV and P3HT, Molecular Simulation, DOI:10.1080/08927022.2012.724174
To link to this article: http://dx.doi.org/10.1080/08927022.2012.724174
PLEASE SCROLL DOWN FOR ARTICLE
Full terms and conditions of use: http://www.tandfonline.com/page/terms-and-conditions
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form toanyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae, and drug doses shouldbe independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims,proceedings, demand, or costs or damages whatsoever or howsoever caused arising directly or indirectly inconnection with or arising out of the use of this material.

Modelling polymers with side chains: MEH-PPV and P3HT
A. Batagin-Netoa, E.F. Oliveiraa, C.F.O. Graeffa,b and F.C. Lavardaa,b*
aUNESP, Univ. Estadual Paulista, POSMAT, Programa de Pos-Graduacao em Ciencia e Tecnologia de Materiais, Bauru, SP, Brazil;bDF-FC, UNESP, Univ. Estadual Paulista, Av. Eng. Luiz Edmundo Carrijo Coube, 14-01, 17033-360 Bauru, SP, Brazil
(Received 17 June 2012; final version received 11 August 2012)
Modelling polymers with side chains is always a challenge once the degrees of freedom are very high. In this study, wepresent a successful methodology to model poly[2-methoxy-5-(20-ethyl-hexyloxy)-p-phenylenevinylene] (MEH-PPV) andpoly[3-hexylthiophene] (P3HT) in solutions, taking into account the influence of side chains on the polymer conformation.Molecular dynamics and semi-empirical quantum mechanical methods were used for structure optimisation and evaluationof optical properties. The methodology allows to describe structural and optical characteristics of the polymers in asatisfactory way, as well as to evaluate some usual simplifications adopted for modelling these systems. Effectiveconjugation lengths of 8-14.6 and 21 monomers were obtained for MEH-PPV and P3HT, respectively, in accordance withexperimental findings. In addition, anti/syn conformations of these polymers could be predicted based on intrinsicinteractions of the lateral branches.
Keywords: MEH-PPV; P3HT; electronic structure calculation; modelling branched polymers
1. Introduction
Polymeric materials have been used in a great variety of
technological applications due to their interesting optical–
electronic and mechanical properties, easy processing
and relatively low cost. In particular, conjugated organic
polymers have attracted great attention in the manufacture
of semiconductor devices such as light-emitting diodes,
solar cells, transistors, optocouplers, photodiodes, triodes,
voltage regulators and lasers [1]. The usefulness of these
materials comes from the fact that small changes in the
constitution of their monomeric units can result in profound
changes in their physicochemical properties, allowing a
wide adjustment range [2].
A significant increase in experimental and theoretical
studies involving not only polymers but also other organic
materials has been recently observed. Molecular model-
ling studies involving computational chemistry and
theoretical physics have enabled significant advances in
understanding the basic properties of these materials, as
well as in the development of more efficient devices/sys-
tems.
Since most of the organic-based devices are structured
in a thin film form, it is interesting that polymeric materials
present good solubility and stability in organic solvents in
order to obtain films with reasonable homogeneity and
good adhesion to substrates [3]. In this sense, polymers with
lateral chains have been employed for the manufacture of
devices.
It is known that polymers’ solubility in organic solvents
depends on several factors mainly associated with the
chemical structure of the polymer, such as molecular
weight, polarity, cross-linking degree, crystallinity and the
presence/absence of side branches [4]. Generally, the
presence of lateral chains renders greater solubility, since it
allows a better interaction between polymer and solvent
molecules. On the other hand, it is also expected that very
long branched chains present high entanglement degree,
hampering the solvation of the polymer and reducing its
solubility. Thus, more soluble materials are supposed to
have relatively short branches.
The presence of these lateral chains, which at the same
time provides higher solubility to the compounds, sets an
additional problem for conformational studies of these
polymers via electronic structure calculations. The high
flexibility of the side chains, combined with possible steric
interactions between branches of adjacent monomeric
units, allows a great variety of distinct conformations
relatively close in energy, resulting in a high number of
structures by considering just one repeating unit.
There is no concern with the conformation search of
these side chains in most of the molecular modelling studies
of polymers reported in the literature. In general, a shorter
lateral chain is chosen to study the conformation of the main
chain [5]. This procedure is based on the fact that the
optical–electronic properties of polymers are directly
associated with the structural characteristics of the main
chain. However, although it is a valid approach, it ignores
situations where there is a significant interaction between
the side chains of adjacent monomeric structures. Such
steric interactions are, in fact, barriers that can influence the
ISSN 0892-7022 print/ISSN 1029-0435 online
q 2012 Taylor & Francis
http://dx.doi.org/10.1080/08927022.2012.724174
http://www.tandfonline.com
*Corresponding author. Email: [email protected]
Molecular Simulation
iFirst article, 2012, 1–13
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2

conformation of the polymer’s main chain and significantly
alter its conjugation length and optical properties.
The work described here presents a methodology for
molecular modelling of polymers by employing semi-
empirical calculations, considering the influence of side
branches in the conformational study of oligomers.
Interactions between distinct polymers chains were not
considered in this model. Although these interactions
presumably influence optical and structural properties of
the system, a secondary hole is expected in solutions
(frequently employed in films manufacturing) where
interchain interactions are not supposed to be relevant. In
addition, interactions between adjacent backbones are
expected to be hindered in polymers with large side chains
[6]. In these systems, stronger interactions are expected
between lateral ramifications of adjacent repeating units
which somewhat shield the main chain.
Two conjugated conducting polymers widely used in
opto-electronic devices were evaluated in order to illustrate
the methodology: poly[2-methoxy-5-(20-ethyl-hexyloxy)-
p-phenylenevinylene] (MEH-PPV) and poly[3-hexylthio-
phene] (P3HT). The method, besides being computation-
ally feasible, has shown to be effective in obtaining
optimised structures and allowing the prediction of some
properties of these polymeric materials in solution.
In the following sections, the methodology used for the
polymer modelling is presented, followed by the results of
the study of MEH-PPV and P3HT oligomers employing
the described methodology. It is important to emphasise
that the methodology described here aims to evaluate
medium-sized systems with lateral ramifications; thus, it is
not a general approach to deal with large systems (as
proteins and DNA chains). For this purpose, the reader
may refer to Refs [7–10].
2. Methodology
2.1 Theoretical methods
In this study, we have employed molecular dynamics (MD)
simulations in order to obtain structures with different
conformations of the side chains of the polymers. The
MM þ force field [11] implemented in Hyperchem
computer package was employed.1 Details of MD
simulations are discussed in Section 2.2.
For optimisation of all structures (monomer, dimer,
tetramer and oligomers), we have employed the semi-
empirical parametric methods 3 and 6 (PM3 and PM6)
[12,13] (for MEH-PPV and P3HT, respectively)
implemented in the computational package MOPAC2009
[14,15] in a restricted Hartree–Fock (RHF) approach. The
presence of the solvent was simulated by conductor-like
screening model (COSMO) [16]. Interchain interactions
were not considered.
The choice of a semi-empirical method to structure
optimisation is justified by the size of the considered
systems, since an ab-initio approach would be computa-
tionally expensive, or even unfeasible, given the large
number of conformations evaluated (see Section 2.2). In
addition, it is known that steric interactions, such as van
der Waals interactions (expected to exist between the
lateral chains), are not well described by many ab-initio
and density functional theory approaches [17,18]. PM3
semi-empirical method has been chosen to study MEH-
PPV since barrier torsion studies of poly( p-phenylene-
vinylene) (PPV) employing this method have shown
superior geometries in comparison with similar methods
[19–21]. On the other hand, a relative lack of studies can
be noted on P3HT polymer employing semi-empirical
methods. In this sense, we have opted to use the PM6 semi-
empirical method since it has been already satisfactorily
employed in the study of photovoltaic properties of P3HT
derivatives [22,23].
The theoretical optical absorption spectra of the
structures were all calculated using the ZINDO/S semi-
empirical method in conjunction with configuration
interaction approach for single excitations implemented
in Orca computational package [24,25]. We have
considered the first 10 transitions (roots) for each structure
(considering only singlet state transitions).
MD and geometry optimisations were performed on an
Intel Coree 2 Quad 2.40 GHz computer. Typically, 15 s
were taken for MD simulations of each structure; the
geometry optimisations were performed from 2 s to 42 h
depending on the oligomer size. Optical absorption spectra
calculations were made using Intel Xeon 2.83 GHz
machines, demanding between 24 s and 2 h 30 min for
each structure depending on the oligomer considered.
2.2 Conformational studies approach
Figure 1 shows the repeating units of MEH-PPV and
P3HT polymers.
As can be seen, both structures present side chains
entirely composed of single bonds between carbon atoms.
In fact, high conformation flexibility of these branches can
result in a large number of different conformers with
relatively close energies.
Figure 2 exemplifies possible variations that lateral
chains can perform in these polymers. Since the rotations of
these single bonds require relatively little energy to occur, it
is difficult to obtain unique lower energy structures.
Obtaining geometries that adequately describe the
polymer is extremely important in electronic structure
calculations, since the electronic Hamiltonian depends
parametrically on the position of the atoms of the system
studied. A systematic search of different angles illustrated
in Figure 2 becomes impracticable, since the initial number
of possible conformations would be very high. Consider-
ing, for example, a systematic study with 908 steps, it would
be possible to construct about 65,536 (48) distinct structures
A. Batagin-Neto et al.2
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2

for each monomeric unit of MEH-PPV and 4096 (46)
structures for P3HT.
Given the impossibility of a systematic analysis of the
hyper-surface generated by varying the angles illustrated
in Figure 2, it is an attractive option to scan regions of this
hyper-surface, considering low correlated structures with
different conformations of the side chains.
In order to obtain these initial structures, MD
simulations at high temperatures have been performed.
The main chain of the polymer is maintained (locked) in a
specific configuration (discussed later) while side chains
could change its conformation during MD. The dynamics is
performed supposing the system in contact with a thermal
reservoir at an initial temperature of 1000 K. After 1 ps of
simulation (simulation step of 0.001 ps), the dynamics is
stopped and the resulting structure is stored for further
optimisation. Subsequently, the MD is restarted and
another structure is obtained (after 1 ps of simulation) and
so on. In order to obtain weakly correlated structures, the
reservoir temperature is increased by 500 K for each five
conformations.
The MD simulations were performed only to generate
random structures for further optimisation employing
quantum methods. In this sense, unusual parameters were
employed in the simulations: (i) high temperatures to obtain
weakly correlated conformations and (ii) relatively short
simulation time (just to describe ‘randomic’ bonds’
rotations).
Aiming to ensure that optimisation using quantum
mechanical methods (RHF-PM3/PM6-COSMO) does not
present convergence problems, a pre-optimisation with
molecular mechanics methods was previously performed
for all the initial structures (MMþ with gradient norm#1).
In optimisations via RHF-PM3/PM6-COSMO methods the
gradient norm employed was 0.05, higher than the
commonly used in calculations made in the absence of
solvent [26,27].
The conformational study was divided into topics to
identify relevant aspects of the polymer structure: (i)
monomers, (ii) dimers, (iii) tetramers and (iv) oligomers
(five or more repeating units). In all optimisation
calculations, the same parameters pointed above were
employed.
(1) Monomers study: Given the high conformational
flexibility of polymers’ side chains (Figure 2), a large
variety of conformers is expected. The study of the
monomer was conducted aiming to evaluate possible
lower energy configurations of the isolated branches.
The monomers were obtained by hydrogenating the
ends of the polymer’s repeating unit. Freezing the
main structure on a planar conformation, MD
simulations were performed on the side chains to
obtain distinct structures. Thus, 50 different initial
structures were obtained for further optimisation (for
each polymer). The optimisation was also performed
with the monomer’s main chain locked in a planar
conformation.
(2) Dimers study: A natural step in the search for
equilibrium conformation of a polymer is the study of
dihedral angles formed between adjacent monomers.
In polymers having large lateral branches, this kind of
study is not easy, since a large number of freedom
degrees is expected. With this concern, the study is
generally carried out considering a specific confor-
mation of the side chains. This analysis, however, does
not provide reliable results, since it is restricted to a
specific conformation of the chains. In this approach, it
is not possible to evaluate steric interactions between
them, which probably have a great influence on the
equilibrium conformation of the polymer.
To better evaluate these interactions, we chose to
conduct the study of the angle between adjacent
monomeric structures by sampling, considering the
angles between the adjacent rings of the dimer (see
Figure 3).
Initial structures were obtained by blocking the main
chain of the dimer in four different configurations
(presenting 0, 90, 180 and 2708 for dihedral angles
between adjacent rings) and by employing the MD
approach for obtaining random structures. About 50
different conformations were obtained for each
blocked main chain configuration of MEH-PPV and
Figure 1. Structural formulae of MEH-PPV and P3HT.
Figure 2. Degrees of freedom of side chains’ single bonds forMEH-PPV and P3HT.
Molecular Simulation 3
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2

40 for the P3HT, totaling about 200 structures for
MEH-PPV and 150 for P3HT. The structure optimis-
ation was then carried out (RHF/PM3 or
PM6/COSMO), allowing that all distances, angles
and dihedrals be optimised.
The analysis of the angles was carried out by a specific
program developed in Fortran 90, calculating the angle
observed between the planes defined by adjacent rings
of the optimised dimer (i.e. dimer angle). Figure 3
presents the atoms defining the rings (ring A: atoms 1,
2 and 3; ring B: atoms 4, 5 and 6). The results were
compared with those obtained through similar analysis
using Molden 5.0 [28] and Gabedit 2.3.9 [29]
softwares. The relative angle between the vectors
r21 ¼ r22r1 and r54 ¼ r52r4 was also considered in
order to distinguish between u and 1808 2 u angles.
Finally, to evaluate the influence of the lateral chain
extension in the polymer conformation, we studied the
structures with smaller lateral branches (CH3 group).
In this case, around 100 initial structures were
generated for each polymer using the same approach
described above for obtaining the initial structures and
optimisation.
For the purpose of evaluating the average angle
between the repeating units of the polymers consider-
ing the optimised dihedrals angles (Ai) obtained for
each dimer, we used Equation (1).
Ah i ¼
PiAi exp ð2DEi=kTÞPi exp ð2DEi=kTÞ
; ð1Þ
where kAl represents the average angle between
adjacent units of the polymer, Ai is the dihedral angle
found for ith dimer, exp(2DEi/kT) is the Boltzmann
factor and DEi ¼ Ei 2 E0, whereas Ei represents the
energy of the ith dimer and E0 represents the average
energy obtained by considering all the optimised
dimers. For such calculations, a temperature of 300 K
was adopted.
(3) Tetramers study: The tetramer study was carried out to
obtain a structure in which steric interactions between
the side chains were considered. By blocking the main
chain in a specific configuration (dictated by the dimer
study), a series of MD simulations were performed
with the lateral chains to achieve different structures
(similar to that made for the monomers). Thus, 30
different initial conformations were obtained, which
were optimised in an RHF-PM3/PM6-COSMO
approach. During optimisation, the main chain was
also maintained in the configuration of lower energy
obtained from the dimers study.
(4) Oligomers study: To evaluate whether the approach
allows to reproduce relevant properties of the polymer,
a theoretical optical absorption spectrum study of
different oligomers was carried out. The structures
studied were obtained by joining multiple confor-
mations of the more stable tetramer (following the
pattern established by the dimer study). All structures
were then optimised in an RHF-PM3/PM6-COSMO
approach (allowing the optimisation of all structural
parameters).
In order to evaluate the main peak position of the
absorption spectrum in an infinite chain, the exponential
relationship proposed by Meier et al. [30] was employed:
lðnÞ ¼ l1 2 Dl exp ½2aðn2 1Þ�; ð2Þ
where n represents the number of repeating units of the
polymer; Dl ¼ l1 2 l1 represents the total displacement
of the absorption peak caused by the conjugation extension,
where l1 refers to the optical absorption peak for one
repeating unit (n ¼ 1) and l1 refers to the optical
absorption peak for n ! 1. Dl can also be related to the
overall effect of conjugation [30]. The parameter a
indicates how fast the limit of convergence is achieved.
Using Equation (2) the effective conjugation length
(ECL) of the polymers can also be accessed. Meier et al.
suggest a way to evaluate nef assuming a detection limit of
,1 nm so that:
l1 2 lef # 1nm; ð3Þ
where lef is the value of the optical absorption peak for the
ECL (n ¼ nef).
3. Results and discussion
3.1 Poly[2-methoxy-5-(20-ethyl-hexyloxy)-p-phenylenevinylene]
Figure 4 shows the heat of formation of various monomers
obtained using the methodology outlined above. The
dotted lines subdivide the graph into intervals of kT energy
at room temperature ( , 0.5765 kcal/mol).
A relatively large energy variation can be observed in
the conformers studied (9.7 kcal/mol, ,17 kT at 300 K)
which is exclusively due to the conformations of the side
chains. Figure 5 illustrates the monomers of lower (08, 38
Figure 3. Atoms defining the evaluated ring planes for MEH-PPV and P3HT.
A. Batagin-Neto et al.4
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
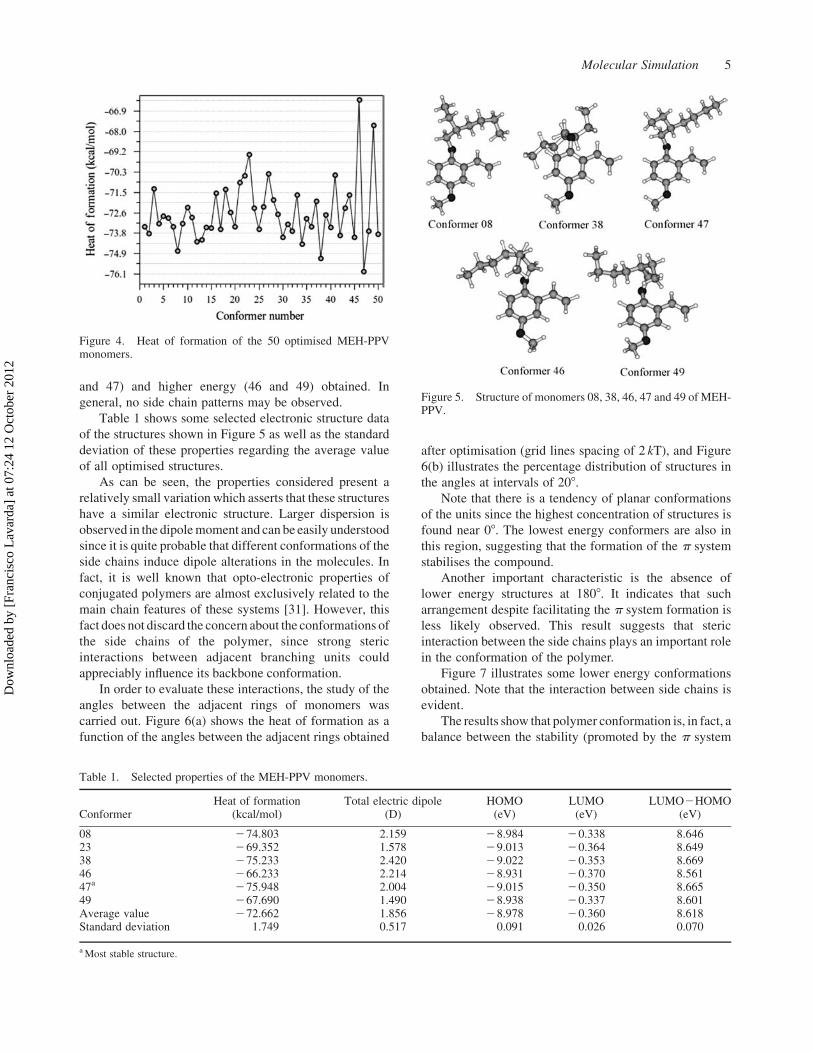
and 47) and higher energy (46 and 49) obtained. In
general, no side chain patterns may be observed.
Table 1 shows some selected electronic structure data
of the structures shown in Figure 5 as well as the standard
deviation of these properties regarding the average value
of all optimised structures.
As can be seen, the properties considered present a
relatively small variation which asserts that these structures
have a similar electronic structure. Larger dispersion is
observed in the dipole moment and can be easily understood
since it is quite probable that different conformations of the
side chains induce dipole alterations in the molecules. In
fact, it is well known that opto-electronic properties of
conjugated polymers are almost exclusively related to the
main chain features of these systems [31]. However, this
fact does not discard the concern about the conformations of
the side chains of the polymer, since strong steric
interactions between adjacent branching units could
appreciably influence its backbone conformation.
In order to evaluate these interactions, the study of the
angles between the adjacent rings of monomers was
carried out. Figure 6(a) shows the heat of formation as a
function of the angles between the adjacent rings obtained
after optimisation (grid lines spacing of 2 kT), and Figure
6(b) illustrates the percentage distribution of structures in
the angles at intervals of 208.
Note that there is a tendency of planar conformations
of the units since the highest concentration of structures is
found near 08. The lowest energy conformers are also in
this region, suggesting that the formation of the p system
stabilises the compound.
Another important characteristic is the absence of
lower energy structures at 1808. It indicates that such
arrangement despite facilitating the p system formation is
less likely observed. This result suggests that steric
interaction between the side chains plays an important role
in the conformation of the polymer.
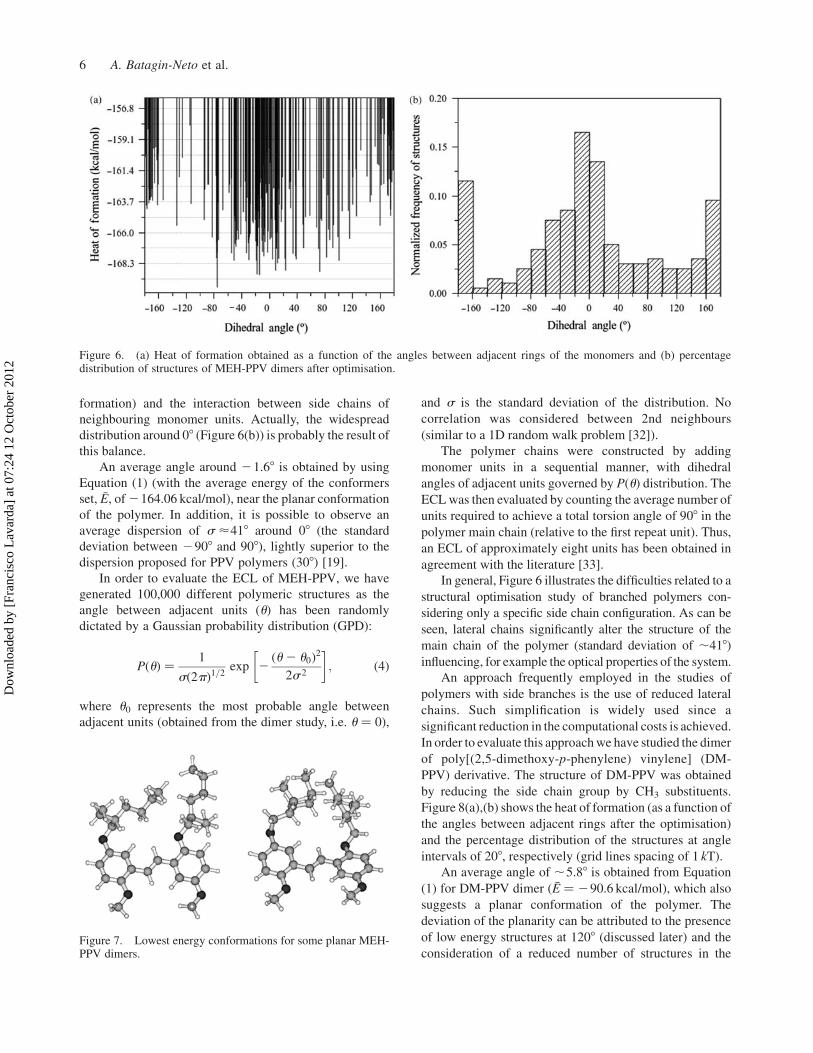
Figure 7 illustrates some lower energy conformations
obtained. Note that the interaction between side chains is
evident.
The results show that polymer conformation is, in fact, a
balance between the stability (promoted by the p system
Table 1. Selected properties of the MEH-PPV monomers.
ConformerHeat of formation
(kcal/mol)Total electric dipole
(D)HOMO
(eV)LUMO
(eV)LUMO2HOMO
(eV)
08 274.803 2.159 28.984 20.338 8.64623 269.352 1.578 29.013 20.364 8.64938 275.233 2.420 29.022 20.353 8.66946 266.233 2.214 28.931 20.370 8.56147a 275.948 2.004 29.015 20.350 8.66549 267.690 1.490 28.938 20.337 8.601Average value 272.662 1.856 28.978 20.360 8.618Standard deviation 1.749 0.517 0.091 0.026 0.070
a Most stable structure.
Figure 4. Heat of formation of the 50 optimised MEH-PPVmonomers.
Figure 5. Structure of monomers 08, 38, 46, 47 and 49 of MEH-PPV.
Molecular Simulation 5
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
formation) and the interaction between side chains of
neighbouring monomer units. Actually, the widespread
distribution around 08 (Figure 6(b)) is probably the result of
this balance.
An average angle around 21.68 is obtained by using
Equation (1) (with the average energy of the conformers
set, E, of 2164.06 kcal/mol), near the planar conformation
of the polymer. In addition, it is possible to observe an
average dispersion of s <418 around 08 (the standard
deviation between 2908 and 908), lightly superior to the
dispersion proposed for PPV polymers (308) [19].
In order to evaluate the ECL of MEH-PPV, we have
generated 100,000 different polymeric structures as the
angle between adjacent units (u) has been randomly
dictated by a Gaussian probability distribution (GPD):
P uð Þ ¼1
s 2pð Þ1=2exp 2
u2 u0ð Þ2
2s2
� �; ð4Þ
where u0 represents the most probable angle between
adjacent units (obtained from the dimer study, i.e. u ¼ 0),
and s is the standard deviation of the distribution. No
correlation was considered between 2nd neighbours
(similar to a 1D random walk problem [32]).
The polymer chains were constructed by adding
monomer units in a sequential manner, with dihedral
angles of adjacent units governed by P(u) distribution. The
ECL was then evaluated by counting the average number of
units required to achieve a total torsion angle of 908 in the
polymer main chain (relative to the first repeat unit). Thus,
an ECL of approximately eight units has been obtained in
agreement with the literature [33].
In general, Figure 6 illustrates the difficulties related to a
structural optimisation study of branched polymers con-
sidering only a specific side chain configuration. As can be
seen, lateral chains significantly alter the structure of the
main chain of the polymer (standard deviation of ,418)
influencing, for example the optical properties of the system.
An approach frequently employed in the studies of
polymers with side branches is the use of reduced lateral
chains. Such simplification is widely used since a
significant reduction in the computational costs is achieved.
In order to evaluate this approach we have studied the dimer
of poly[(2,5-dimethoxy-p-phenylene) vinylene] (DM-
PPV) derivative. The structure of DM-PPV was obtained
by reducing the side chain group by CH3 substituents.
Figure 8(a),(b) shows the heat of formation (as a function of
the angles between adjacent rings after the optimisation)
and the percentage distribution of the structures at angle
intervals of 208, respectively (grid lines spacing of 1 kT).
An average angle of ,5.88 is obtained from Equation
(1) for DM-PPV dimer (E ¼ 290.6 kcal/mol), which also
suggests a planar conformation of the polymer. The
deviation of the planarity can be attributed to the presence
of low energy structures at 1208 (discussed later) and the
consideration of a reduced number of structures in the
Figure 6. (a) Heat of formation obtained as a function of the angles between adjacent rings of the monomers and (b) percentagedistribution of structures of MEH-PPV dimers after optimisation.
Figure 7. Lowest energy conformations for some planar MEH-PPV dimers.
A. Batagin-Neto et al.6
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
calculation (compared to MEH-PPV). In addition, a
smaller dispersion is observed (,16.78) as well as an
absence of structures at 1808, resulting in an ECL of ,36
units (by GPD random walk study), different from that
obtained for larger lateral chains.
The results suggest that the lateral branches’ simplifica-
tion sets a good approximation in the study of the main
chain conformation of PPV derivatives in solution.
However, we must stress the relevance of the optimisation
study by sampling even within this approach. As can be
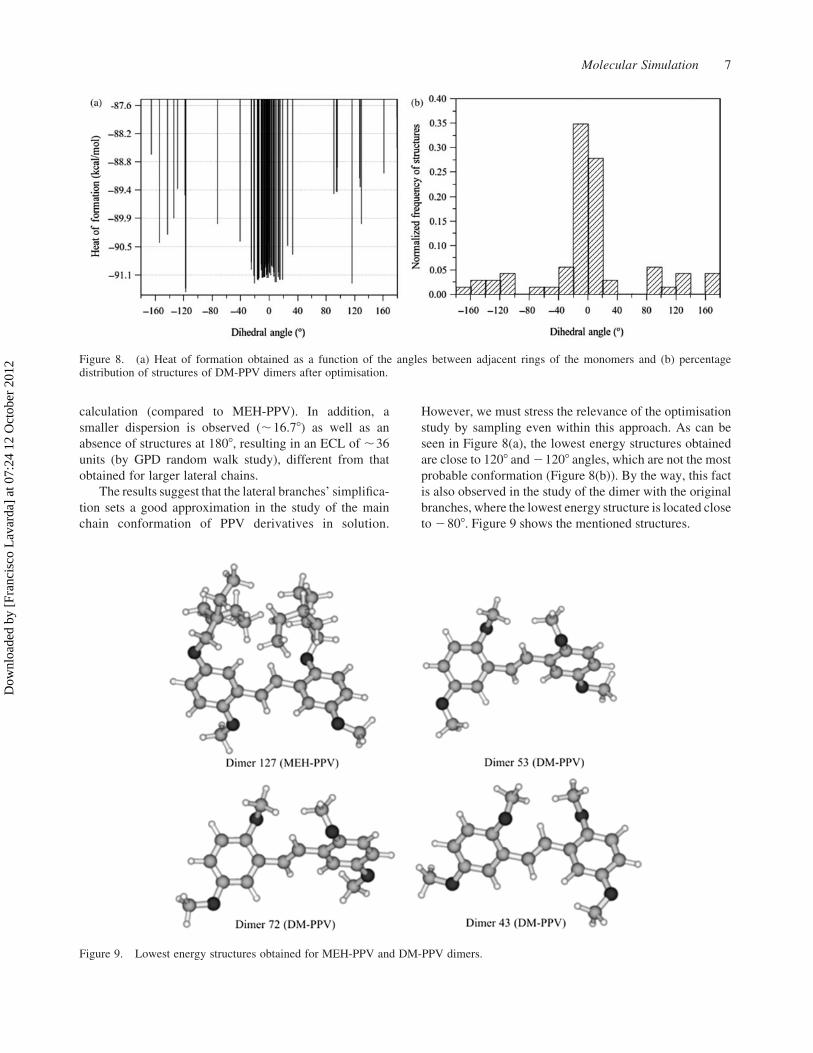
seen in Figure 8(a), the lowest energy structures obtained
are close to 1208 and 21208 angles, which are not the most
probable conformation (Figure 8(b)). By the way, this fact
is also observed in the study of the dimer with the original
branches, where the lowest energy structure is located close
to 2808. Figure 9 shows the mentioned structures.
Figure 8. (a) Heat of formation obtained as a function of the angles between adjacent rings of the monomers and (b) percentagedistribution of structures of DM-PPV dimers after optimisation.
Figure 9. Lowest energy structures obtained for MEH-PPV and DM-PPV dimers.
Molecular Simulation 7
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
As can be seen, the structures present a very specific
side chains’ configuration, which promote the stabilisation
of the compound. However, it is reasonable to assume that
these settings are not actually observed at room
temperatures due to the high degree of freedom of these
branches. In this sense, Figure 8(a) illustrates the possible
risk of considering only an initial structure for geometry
optimisation since unrealistic local minima can be
achieved.
Although the results obtained for the study of MEH-
PPV and DM-PPV dimers are reasonable, it is important to
consider that the use of only two monomeric units in the
analysis of dihedral angles overestimates the values
obtained, since the influence of the p system is not
satisfactorily considered in these structures. In this way, the
study of properties of the oligomers is still relevant to
structure evaluation.
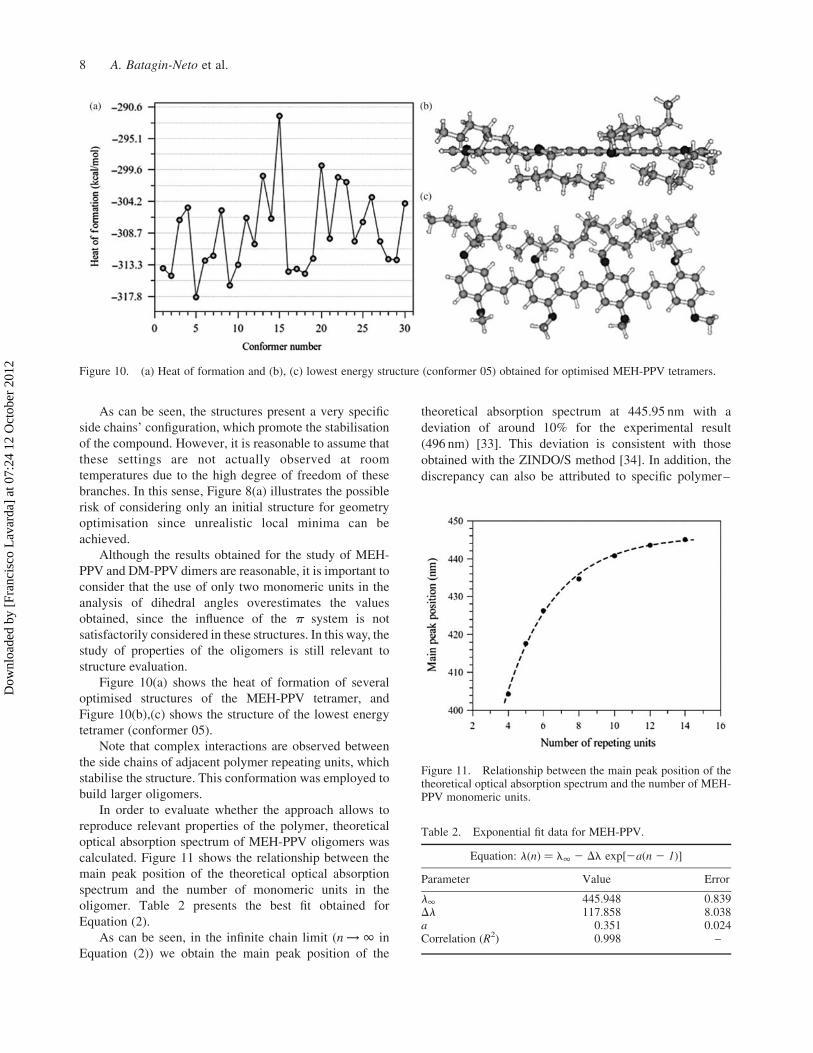
Figure 10(a) shows the heat of formation of several
optimised structures of the MEH-PPV tetramer, and
Figure 10(b),(c) shows the structure of the lowest energy
tetramer (conformer 05).
Note that complex interactions are observed between
the side chains of adjacent polymer repeating units, which
stabilise the structure. This conformation was employed to
build larger oligomers.
In order to evaluate whether the approach allows to
reproduce relevant properties of the polymer, theoretical
optical absorption spectrum of MEH-PPV oligomers was
calculated. Figure 11 shows the relationship between the
main peak position of the theoretical optical absorption
spectrum and the number of monomeric units in the
oligomer. Table 2 presents the best fit obtained for
Equation (2).
As can be seen, in the infinite chain limit (n ! 1 in
Equation (2)) we obtain the main peak position of the
theoretical absorption spectrum at 445.95 nm with a
deviation of around 10% for the experimental result
(496 nm) [33]. This deviation is consistent with those
obtained with the ZINDO/S method [34]. In addition, the
discrepancy can also be attributed to specific polymer–
Table 2. Exponential fit data for MEH-PPV.
Equation: l(n) ¼ l1 2 Dl exp[2a(n 2 1)]
Parameter Value Error
l1 445.948 0.839Dl 117.858 8.038a 0.351 0.024Correlation (R2) 0.998 –
Figure 10. (a) Heat of formation and (b), (c) lowest energy structure (conformer 05) obtained for optimised MEH-PPV tetramers.
Figure 11. Relationship between the main peak position of thetheoretical optical absorption spectrum and the number of MEH-PPV monomeric units.
A. Batagin-Neto et al.8
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
solvent and polymer–polymer steric interactions that have
not been explicitly considered in this study and are
frequently responsible for red shifts in the absorption
spectrum [35].
Finally, to evaluate the ECL of the polymer, we used
Equation (3). Thus, an average value of nef ¼ 14.6 units
can be obtained for MEH-PPV.
Note that the ECL estimation performed employing
both a Gaussian distribution (8 units) and the extrapolation
of the main peak position of the MEH-PPV oligomers (14.6
units) are compatible to other works which suggest the
presence of around 10 planar substructures in the polymer
[33], which reinforces the methodology proposed for
geometry evaluation. Moreover, note that the dimer study
really overestimates the angles between adjacent rings,
resulting in a reduced ECL value (as discussed above).
3.2 Poly[3-hexylthiophene]
In order to better evaluate the proposed methodology, we
also studied the P3HT-conjugated organic polymer using
the same approach described for MEH-PPV.
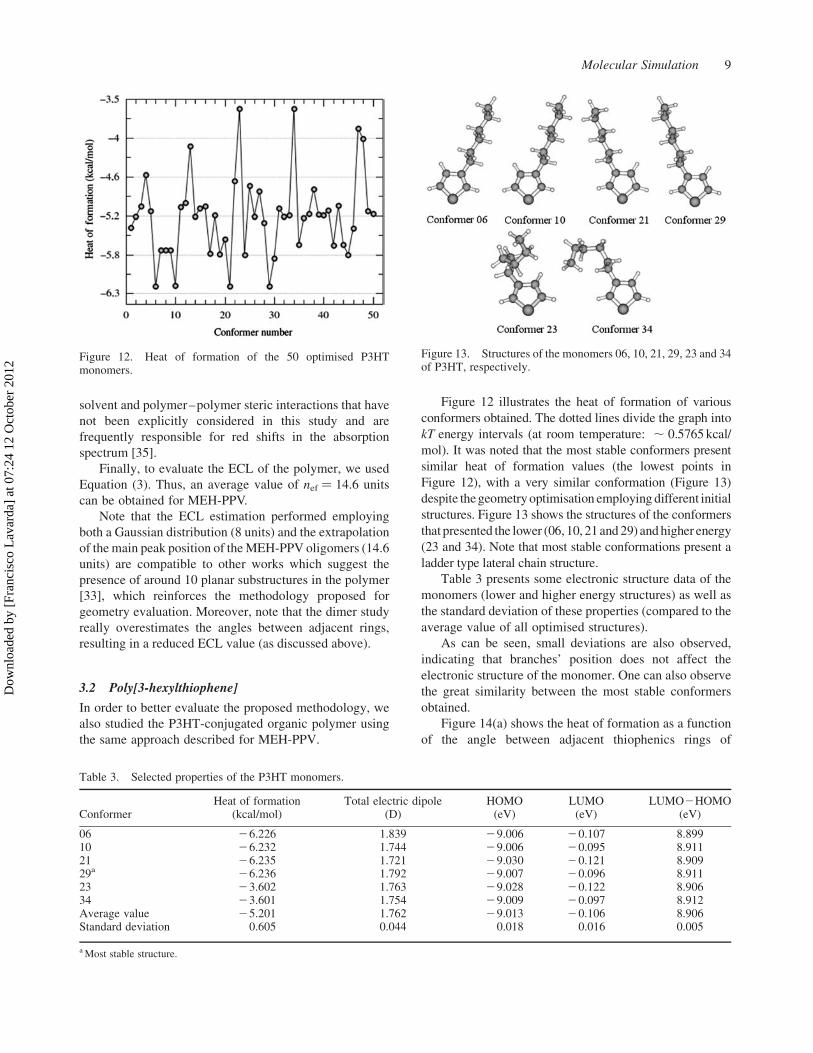
Figure 12 illustrates the heat of formation of various
conformers obtained. The dotted lines divide the graph into
kT energy intervals (at room temperature: , 0.5765 kcal/
mol). It was noted that the most stable conformers present
similar heat of formation values (the lowest points in
Figure 12), with a very similar conformation (Figure 13)
despite the geometry optimisation employing different initial
structures. Figure 13 shows the structures of the conformers
that presented the lower (06, 10, 21 and 29) and higher energy
(23 and 34). Note that most stable conformations present a
ladder type lateral chain structure.
Table 3 presents some electronic structure data of the
monomers (lower and higher energy structures) as well as
the standard deviation of these properties (compared to the
average value of all optimised structures).
As can be seen, small deviations are also observed,
indicating that branches’ position does not affect the
electronic structure of the monomer. One can also observe
the great similarity between the most stable conformers
obtained.
Figure 14(a) shows the heat of formation as a function
of the angle between adjacent thiophenics rings of
Table 3. Selected properties of the P3HT monomers.
ConformerHeat of formation
(kcal/mol)Total electric dipole
(D)HOMO
(eV)LUMO
(eV)LUMO2HOMO
(eV)
06 26.226 1.839 29.006 20.107 8.89910 26.232 1.744 29.006 20.095 8.91121 26.235 1.721 29.030 20.121 8.90929a 26.236 1.792 29.007 20.096 8.91123 23.602 1.763 29.028 20.122 8.90634 23.601 1.754 29.009 20.097 8.912Average value 25.201 1.762 29.013 20.106 8.906Standard deviation 0.605 0.044 0.018 0.016 0.005
a Most stable structure.
Figure 12. Heat of formation of the 50 optimised P3HTmonomers.
Figure 13. Structures of the monomers 06, 10, 21, 29, 23 and 34of P3HT, respectively.
Molecular Simulation 9
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
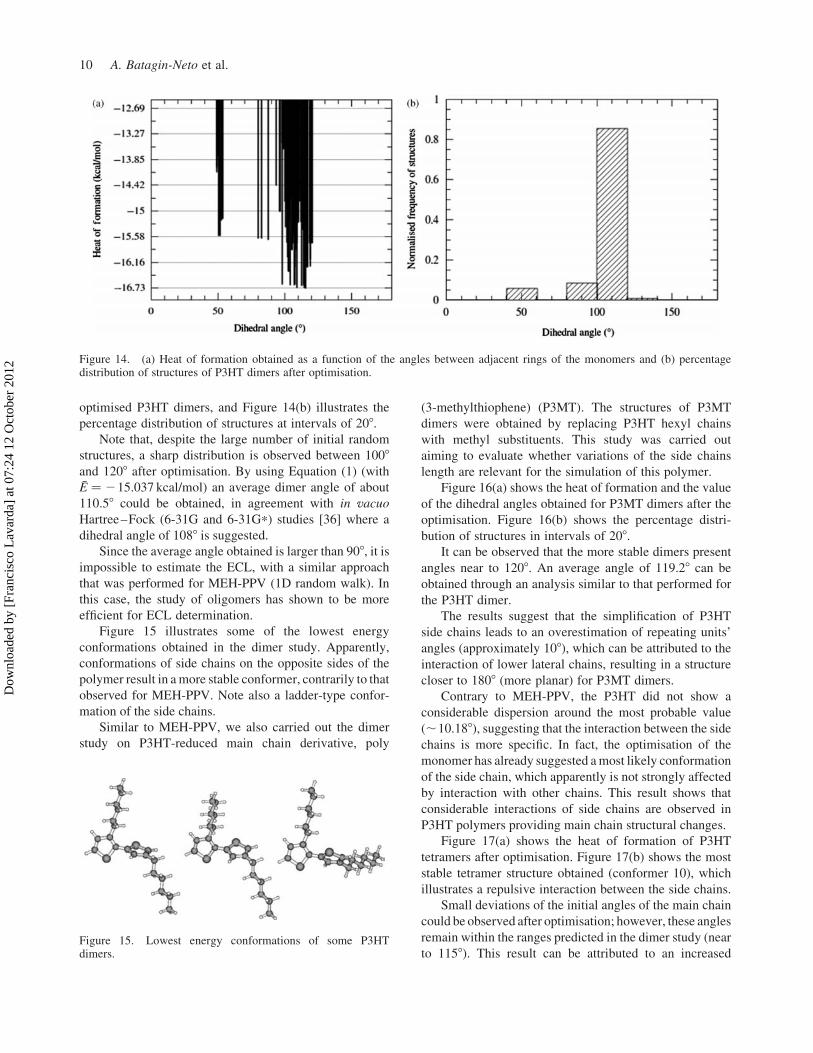
optimised P3HT dimers, and Figure 14(b) illustrates the
percentage distribution of structures at intervals of 208.
Note that, despite the large number of initial random
structures, a sharp distribution is observed between 1008
and 1208 after optimisation. By using Equation (1) (with
E ¼ 215.037 kcal/mol) an average dimer angle of about
110.58 could be obtained, in agreement with in vacuo
Hartree–Fock (6-31G and 6-31G*) studies [36] where a
dihedral angle of 1088 is suggested.
Since the average angle obtained is larger than 908, it is
impossible to estimate the ECL, with a similar approach
that was performed for MEH-PPV (1D random walk). In
this case, the study of oligomers has shown to be more
efficient for ECL determination.
Figure 15 illustrates some of the lowest energy
conformations obtained in the dimer study. Apparently,
conformations of side chains on the opposite sides of the
polymer result in a more stable conformer, contrarily to that
observed for MEH-PPV. Note also a ladder-type confor-
mation of the side chains.
Similar to MEH-PPV, we also carried out the dimer
study on P3HT-reduced main chain derivative, poly
(3-methylthiophene) (P3MT). The structures of P3MT
dimers were obtained by replacing P3HT hexyl chains
with methyl substituents. This study was carried out
aiming to evaluate whether variations of the side chains
length are relevant for the simulation of this polymer.
Figure 16(a) shows the heat of formation and the value
of the dihedral angles obtained for P3MT dimers after the
optimisation. Figure 16(b) shows the percentage distri-
bution of structures in intervals of 208.
It can be observed that the more stable dimers present
angles near to 1208. An average angle of 119.28 can be
obtained through an analysis similar to that performed for
the P3HT dimer.
The results suggest that the simplification of P3HT
side chains leads to an overestimation of repeating units’
angles (approximately 108), which can be attributed to the
interaction of lower lateral chains, resulting in a structure
closer to 1808 (more planar) for P3MT dimers.
Contrary to MEH-PPV, the P3HT did not show a
considerable dispersion around the most probable value
(,10.188), suggesting that the interaction between the side
chains is more specific. In fact, the optimisation of the
monomer has already suggested a most likely conformation
of the side chain, which apparently is not strongly affected
by interaction with other chains. This result shows that
considerable interactions of side chains are observed in
P3HT polymers providing main chain structural changes.
Figure 17(a) shows the heat of formation of P3HT
tetramers after optimisation. Figure 17(b) shows the most
stable tetramer structure obtained (conformer 10), which
illustrates a repulsive interaction between the side chains.
Small deviations of the initial angles of the main chain
could be observed after optimisation; however, these angles
remain within the ranges predicted in the dimer study (near
to 1158). This result can be attributed to an increased
Figure 14. (a) Heat of formation obtained as a function of the angles between adjacent rings of the monomers and (b) percentagedistribution of structures of P3HT dimers after optimisation.
Figure 15. Lowest energy conformations of some P3HTdimers.
A. Batagin-Neto et al.10
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
repulsion between the branches due to the additional
neighbouring units. Such interactions, however, are not able
to induce significant alterations on the backbone
conformation.
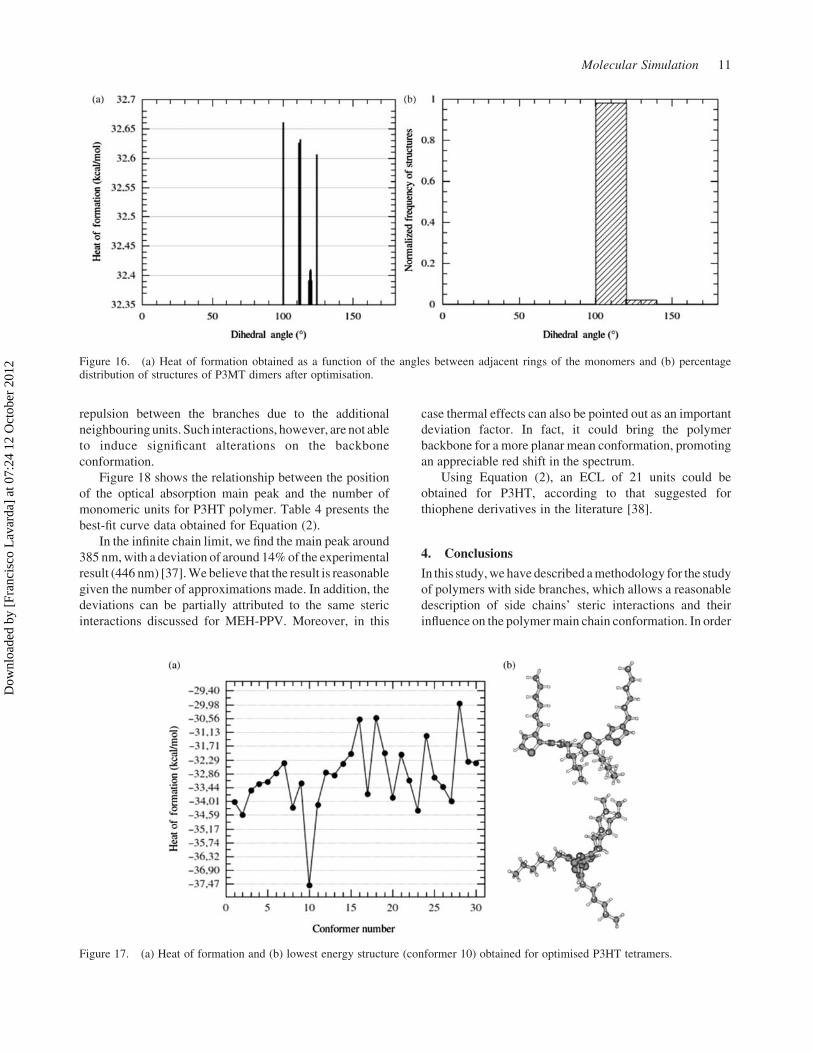
Figure 18 shows the relationship between the position
of the optical absorption main peak and the number of
monomeric units for P3HT polymer. Table 4 presents the
best-fit curve data obtained for Equation (2).
In the infinite chain limit, we find the main peak around
385 nm, with a deviation of around 14% of the experimental
result (446 nm) [37]. We believe that the result is reasonable
given the number of approximations made. In addition, the
deviations can be partially attributed to the same steric
interactions discussed for MEH-PPV. Moreover, in this
case thermal effects can also be pointed out as an important
deviation factor. In fact, it could bring the polymer
backbone for a more planar mean conformation, promoting
an appreciable red shift in the spectrum.
Using Equation (2), an ECL of 21 units could be
obtained for P3HT, according to that suggested for
thiophene derivatives in the literature [38].
4. Conclusions
In this study, we have described a methodology for the study
of polymers with side branches, which allows a reasonable
description of side chains’ steric interactions and their
influence on the polymer main chain conformation. In order
Figure 16. (a) Heat of formation obtained as a function of the angles between adjacent rings of the monomers and (b) percentagedistribution of structures of P3MT dimers after optimisation.
Figure 17. (a) Heat of formation and (b) lowest energy structure (conformer 10) obtained for optimised P3HT tetramers.
Molecular Simulation 11
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
to illustrate it, the structural and optical properties of two
widely studied polymers were evaluated.
The methodology allows to evaluate the properties of
polymers in a satisfactory way as the effective conjugation
length, main peak position of the optical absorption
spectrum as well as inherent structural characteristics.
The dihedral angles’ analysis suggest that the
simplification of the side branches is an interesting approach
for MEH-PPV only if certain precautions are taken during
optimisation. Moreover, significant deviations were
obtained for P3HT, suggesting that such simplification is
inadequate for this polymer.
In summary, the methodology allows to satisfactorily
assess the structural and optical properties of polymers. In
addition, it was possible to evaluate commonly adopted
simplifications of these systems.
Acknowledgements
We would like to thank the Brazilian agencies CAPES (INCTMN),FAPESP (Proc. 2012/03116-7) and CNPq for financial support.This research was also supported by resources supplied by theCenter for Scientific Computing (NCC/GridUNESP) of the SaoPaulo State University (UNESP).
Note
1. Hypercube, HyperChemTM, Hypercube, 2002.
References
[1] S. Gunes, H. Neugebauer, and N.S. Sariciftci, Conjugated polymer-based organic solar cells, Chem. Rev. 107 (2007), pp. 1324–1338.
[2] E. Bundgaard and F.C. Krebs, Low band gap polymers for organicphotovoltaics, Sol. Energy Mater. Sol. Cells. 91 (2007),pp. 954–985.
[3] S. Richter, M. Ploetner, W.-J. Fischer, M. Schneider, P.-T. Nguyen,W. Plieth, N. Kiriy, and H.-J.P. Adler, Development of organic thinfilm transistors based on flexible substrates, Thin Solid Films 477(2005), pp. 140–147.
[4] B.A. Wolf, Solubility of polymers, Pure Appl. Chem. 57 (1985),pp. 323–336.
[5] B. Milian Medina, A. Van Vooren, P. Brocorens, J. Gierschner,M. Shkunov, M. Heeney, I. McCulloch, R. Lazzaroni, and J. Cornil,Electronic structure and charge-transport properties of polythio-phene chains containing thienothiophene units: A joint experimentaland theoretical study, Chem. Mater. 19 (2007), pp. 4949–4956.
[6] L.-O. Palsson, C. Wang, D.L. Russell, A.P. Monkman, M.R. Bryce,G. Rumbles, and I.D.W. Samuel, Photophysics of a fluorene co-polymer in solution and films, Chem. Phys. 279 (2002),pp. 229–237.
[7] S.A. Adcock and J.A. McCammon, Molecular dynamics: Survey ofmethods for simulating the activity of proteins, Chem. Rev. 106(2006), pp. 1589–1615.
[8] A. Perez, F.J. Luque, and M. Orozco, Frontiers in moleculardynamics simulations of DNA, Acc. Chem. Res. 45 (2012),pp. 196–205.
[9] S. Shenoy, W. Bates, H. Frisch, and G. Wnek, Role of chainentanglements on fiber formation during electrospinning of polymersolutions: Good solvent, non-specific polymer–polymer interactionlimit, Polymer 46 (2005), pp. 3372–3384.
[10] G.M. Foo and R.B. Pandey, Conformation of interacting polymerchains: Effects of temperature, bias, polymer concentration, andporosity, Phys. Rev. E. 55 (1997), pp. 4433–4441.
[11] A. Hocquet and M. Langgard, An evaluation of the MMþ forcefield, J. Mol. Model. 4 (1998), pp. 94–112.
[12] J.J.P. Stewart, Optimization of parameters for semiempiricalmethods I. Method, J. Comput. Chem. 10 (1989), pp. 209–220.
[13] J.J.P. Stewart, Optimization of parameters for semiempiricalmethods V: Modification of NDDO approximations and applicationto 70 elements, J. Mol. Model. 13 (2007), pp. 1173–1213.
[14] J.J.P. Stewart, MOPAC Molecular orbital package, StewartComputational Chemistry, 2009; Available at http://www.openmopac.net/MOPAC2009.html (2009).
[15] J.J.P. Stewart, MOPAC: A semiempirical molecular orbitalprogram, J. Comput.-Aided Mol. Des. 4 (1990), pp. 1–103.
[16] A. Schafer, A. Klamt, D. Sattel, J.C.W. Lohrenz, and F. Eckert,COSMO implementation in TURBOMOLE: Extension of an efficientquantum chemical code towards liquid systems, Phys. Chem. Chem.Phys. 2 (2000), pp. 2187–2193.
[17] N. Kurita and H. Sekino, Ab initio and DFT studies for accuratedescription of van der Waals interaction between He atoms, Chem.Phys. Lett. 348 (2001), pp. 139–146.
[18] R. Peverati and K.K. Baldridge, Implementation and performance ofDFT-D with respect to basis set and functional for study ofdispersion interactions in nanoscale aromatic hydrocarbons,J. Chem. Theory Comput. 4 (2008), pp. 2030–2048.
[19] D.S. Galvao, Z.G. Soos, S. Ramasesha, and S. Etemad, A parametricmethod 3 (PM3) study of trans-stilbene, J. Chem. Phys. 98 (1993),pp. 3016–3021.
[20] R. Vendrame, V.R. Coluci, and D.S. Galvao, Comparativeparametric method 5 (PM5) study of trans-stilbene, J. Mol. Struct.686 (2004), pp. 103–108.
[21] A. Camilo, R.P.B. dos Santos, V.R. Coluci, and D.S. Galvao,Comparative parametric method 6 (PM6) and Recife model 1(RM1) study of trans-stilbene, Mol. Simul. 38 (2011), pp. 1–7.
[22] R.C. Hiorns, E. Cloutet, E. Ibarboure, L. Vignau, N. Lemaitre,S. Guillerez, C. Absalon, and H. Cramail, Main-chain fullerenepolymers for photovoltaic devices, Macromolecules 42 (2009),pp. 3549–3558.
[23] R.C. Hiorns, P. Iratcabal, D. Begue, A. Khoukh, R. De Bettignies, J.Leroy, M. Firon, C. Sentein, H. Martinez, H. Preud’homme, and
Table 4. Exponential fit data for P3HT.
Equation: l(n) ¼ l12Dl exp[2a(n 2 1)]
Parameter Value Error
l1 385.335 0.408Dl 68.464 1.629a 0.211 0.008Correlation (R 2) 0.998 –
Figure 18. Relationship between the main peak position of thetheoretical optical absorption spectrum and the number of P3HTmonomeric units.
A. Batagin-Neto et al.12
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2

C. Dagron-Lartigau, Alternatively linking fullerene and conjugatedpolymers, J. Polym. Sci. Part A: Polym. Chem. 47 (2009),pp. 2304–2317.
[24] F. Neese, ORCA: An ab initio, DFT and semiempirical SCF-MO,Available at http://www.thch.uni-bonn.de/tc/orca (2010).
[25] F. Neese, The ORCA program system, Wiley Interdiscip. Rev.Comput. Mol. Sci. 2 (2012), pp. 73–78.
[26] J. Tomasi, B. Mennucci, and R. Cammi, Quantum mechanicalcontinuum solvation models, Chem. Rev. 105 (2005),pp. 2999–3093.
[27] M.A. Watson, D. Rappoport, E.M.Y. Lee, R. Olivares-Amaya, andA. Aspuru-Guzik, Electronic structure calculations in arbitraryelectrostatic environments, J. Chem. Phys. 136 (2012), pp. 024101-1–024101-14.
[28] G. Schaftenaar and J.H. Noordik, Molden: A pre- and post-processing program for molecular and electronic structures,J. Comput.-Aided Mol. Des. 14 (2000), pp. 123–134.
[29] A.-R. Allouche, Gabedit-A graphical user interface for compu-tational chemistry softwares, J. Comput. Chem. 32 (2011),pp. 174–182.
[30] H. Meier, U. Stalmach, and H. Kolshorn, Effective conjugationlength and UV/vis spectra of oligomers, Acta Polym. 48 (1997),pp. 379–384.
[31] J. Gierschner, J. Cornil, and H.-J. Egelhaaf, Optical bandgaps of p-conjugated organic materials at the polymer limit: Experiment andtheory, Adv. Mater. 19 (2007), pp. 173–191.
[32] F. Reif, Introduction to statistical methods, in Fundamentals of
Statistical and Thermal Physics, 2nd ed., Boston, McGraw-Hill,
1965, pp. 1–37.
[33] B.J. Schwartz, Conjugated polymers as molecular materials: How
chain conformation and film morphology influence energy transfer
and interchain interactions, Annu. Rev. Phys. Chem. 54 (2003),
pp. 141–172.
[34] L.E. Friedrich and J.E. Eilers, Progress toward calculation of the
hues of azomethine dyes, J. Imaging Sci. Technol. 38 (1994),
pp. 24–27.
[35] A.K. Baveja and V.K. Gupta, Spectrophotometric determination of
vanadium in complex materials using N-m-TMBHA and Thiocya-
nate, Int. J. Environ. Anal. Chem. 17 (1984), pp. 299–306.
[36] M.A. de Oliveira, W.B. Almeida, and H.F. dos Santos, Structure and
electronic properties of alkylthiophenes coupled by Head-to-Tail
and Head-to-Head regioselectivity, J. Braz. Chem. Soc. 15 (2004),
pp. 832–838.
[37] L. Huo, Y. Zhou, and Y. Li, Alkylthio-substituted polythiophene:
Absorption and photovoltaic properties, Macromol. Rapid Com-
mun. 30 (2009), pp. 925–931.
[38] H. Nakanishi, N. Sumi, Y. Aso, and T. Otsubo, Synthesis and
properties of the longest Oligothiophenes: The Icosamer and
Heptacosamer, J. Org. Chem. 63 (1998), pp. 8632–8633.
Molecular Simulation 13
Dow
nloa
ded
by [
Fran
cisc
o L
avar
da]
at 0
7:24
12
Oct
ober
201
2
Related Documents











