MoCalc Interface for Molecular Calculation Version 2.2 - © 2003 - 2006 Windows 95/98/ME/NT/2000/XP User’s Guide Anderson Coser Gaudio Physics Department Federal University of Espírito Santo Brazil

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MoCalc Interface for Molecular Calculation
Version 2.2 - © 2003 - 2006 Windows 95/98/ME/NT/2000/XP
User’s Guide
Anderson Coser Gaudio Physics Department
Federal University of Espírito Santo
Brazil

Copyright notice MoCalc User’s Guide
Copyright notice
Except where otherwise noted, all of the documentation and software included in the MoCalc Setup package is copyrighted by Anderson Coser Gaudio.
Copyright © 2003-2006 Anderson Coser Gaudio. All rights reserved. MoCalc – Interface for Molecular Calculation v. 2.2 is a FREEware (NOT a public domain software) which means that you can use this software for any purpose, including commercial applications, as long as you need, make as many copies as you want and freely distribute it, provided that all the files are included and are unmodified and that no files have been added to the package. Please distribute it by copying the original self-extracting setup file MoCalcSetup.exe. It is FORBIDDEN to ask money or anything else for the distribution. Only the copyright owner may beg for voluntary cash donation that will always be completely independent from the program distribution. Any money sent to the copyright owner will be fully employed in the development of future versions of MoCalc and other free-distributable applications. However, you may NOT attempt to reverse compile, modify, translate or disassemble any parts of the software. NOTE: if you have modified one or more components of MoCalc, you may NOT distribute it as part of our program any longer. If you want to put this component on a freeware CD-ROM or in other compilation, please contact us before doing so, to be sure that you are not including old, incompatible, or incomplete stuff in the compilation. We would also appreciate it if we could get a free copy of the CD-ROM. Also, we need to mention that Gamess, Mopac, Tinker, RasMol, and Babel programs, and the respective user manuals, are NOT our property. Gamess belongs to the Mark Gordon's Quantum Theory Research Group, Ames Laboratory, Iowa State University, which exclusively distributes it. Mopac belongs to the United States of America government. Tinker is property of Jay William Ponder, Biochemistry & Molecular Biophysics, Washington University School of Medicine, USA. Rasmol belongs to Roger Sayle, Glaxo Wellcome Research and Development, Stevenage, Hertfordshire, UK. Babel belongs to Pat Walters and Matt Stahl, Dolata Research Group, Department of Chemistry, University of Arizona, Tucson, AZ 8572. This software is provided "as is". In no event shall the author be liable for any consequential, special, incidental or indirect damages of any kind arising out of the delivery, performance or use of this software. This software has been written with great care but I do not warrant that the software is error free. Dr. Anderson Coser Gaudio Physics Department Federal University of Espírito Santo Campus Universitário "Alaor de Queiroz Araújo" Av. Fernando Ferrari, 514 - Goiabeiras 29075-910 Vitória, ES Brazil [email protected]://www.cce.ufes.br/anderson
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
2

Table of contents MoCalc User’s Guide
Table of contents
Chapter 1. Introduction ....................................................................................................... 5 1.1. Wellcome to MoCalc! ................................................................................................... 5 1.2. MoCalc Team............................................................................................................... 5 1.3. How to get MoCalc....................................................................................................... 6 1.4. Installing MoCalc.......................................................................................................... 7 1.5. How to cite MoCalc .................................................................................................... 10 1.6. MoCalc main form...................................................................................................... 10
1.6.1. Menu bar ........................................................................................................... 11 1.6.2. Tool bar ............................................................................................................. 11 1.6.3. Status bar .......................................................................................................... 12 1.6.4. Pathname bar.................................................................................................... 12 1.6.5. File tree display ................................................................................................. 12 1.6.6. Document space ............................................................................................... 12
Chapter 2. Working with documents................................................................................ 14 2.1. Document types ......................................................................................................... 14 2.2. Creating new documents ........................................................................................... 14 2.3. Opening existing documents...................................................................................... 15 2.4. Opening multiple documents ..................................................................................... 17 2.5. Closing documents .................................................................................................... 18 2.6. Closing all documents................................................................................................ 18 2.7. Closing documents of a given type............................................................................ 18 2.8. Saving documents ..................................................................................................... 18 2.9. Changing the name of documents............................................................................. 19 2.10. Saving all documents................................................................................................. 19 2.11. Converting files .......................................................................................................... 19 2.12. Converting multiple external files ............................................................................... 20 2.13. Importing multiple files ............................................................................................... 22 2.14. Exporting documents to HTML .................................................................................. 23 2.15. Configuring page for printing...................................................................................... 23 2.16. Previewing documents for printing............................................................................. 24 2.17. Printing documents .................................................................................................... 25 2.18. Closing MoCalc .......................................................................................................... 26
Chapter 3. Editing documents .......................................................................................... 27 3.1. Undoing changes in documents................................................................................. 27 3.2. Redoing changes in documents................................................................................. 27 3.3. Cutting pieces of documents...................................................................................... 27 3.4. Copying pieces of documents to the clipboard .......................................................... 28 3.5. Copying the entire content of documents to the clipboard ........................................ 28 3.6. Pasting from the clipboard ......................................................................................... 28 3.7. Clearing the content of documents ............................................................................ 29 3.8. Selecting the entire content of documents................................................................. 29 3.9. Finding strings in documents ..................................................................................... 29 3.10. Replacing strings in documents ................................................................................. 30
Chapter 4. Exhibiting components of MoCalc ................................................................ 32 4.1. Showing and hiding the toolbar.................................................................................. 32 4.2. Showing and hiding the status bar............................................................................. 32 4.3. Showing and hiding the pathname bar ...................................................................... 32 4.4. Showing and hiding the file tree display .................................................................... 32
Chapter 5. Formatting documents ................................................................................... 34 5.1. Formatting the font of a document ............................................................................. 34 5.2. Formatting the background color of a document ....................................................... 34
Chapter 6. Working with Gamess..................................................................................... 36 6.1. Running Gamess input files ....................................................................................... 36 6.2. Editing Gamess keywords ......................................................................................... 36 6.3. Inserting default header in Gamess input documents ............................................... 37 6.4. Setting up the Gamess default input header ............................................................. 38
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
3

Table of contents MoCalc User’s Guide
6.5. Making default this Gamess input header ................................................................. 38 6.6. Importing external structures to Gamess input documents ....................................... 39 6.7. Exporting to Gamess input documents...................................................................... 40 6.8. Showing structures from Gamess documents ........................................................... 41 6.9. Extracting results from Gamess output documents ................................................... 42 6.10. Calculating properties from Gamess output documents............................................ 43 6.11. Comparing properties generated by Gamess............................................................ 44
Chapter 7. Working with Mopac ....................................................................................... 46 7.1. Running Mopac input files.......................................................................................... 46 7.2. Editing Mopac keywords ............................................................................................ 46 7.3. Setting up the Mopac default input header ................................................................ 49 7.4. Inserting default header in Mopac input documents .................................................. 49 7.5. Making default this Mopac input header .................................................................... 50 7.6. Importing external structures to Mopac input documents.......................................... 50 7.7. Showing structures from Mopac documents.............................................................. 51 7.8. Extracting results from Mopac output documents...................................................... 52 7.9. Calculating properties from Mopac output documents .............................................. 53 7.10. Comparing properties generated by Mopac .............................................................. 54
Chapter 8. Working with Tinker ........................................................................................ 57 8.1. Computing properties................................................................................................. 57 8.2. Minimizing the energy ................................................................................................ 58 8.3. Searching the global minimum energy ...................................................................... 60 8.4. Running a vibrational analysis ................................................................................... 61 8.5. Calculating the molecular surface area and volume.................................................. 62 8.6. Importing external structures to Tinker input documents........................................... 64 8.7. Replacing the Tinker input document by the output .................................................. 65 8.8. Showing structures from Tinker documents .............................................................. 66
Chapter 9. Configuring MoCalc ........................................................................................ 67 9.1. Setting up fundamental physical constants ............................................................... 67 9.2. Setting up unit conversion factors.............................................................................. 67 9.3. Setting up warning messages.................................................................................... 68 9.4. Setting up paths ......................................................................................................... 69
Chapter 10. Arranging windows......................................................................................... 71 10.1. Arranging MoCalc document windows ...................................................................... 71
Chapter 11. Getting help ..................................................................................................... 75 11.1. Getting online help for MoCalc................................................................................... 75 11.2. Getting online help for Gamess ................................................................................. 75 11.3. Getting online help for Mopac.................................................................................... 76 11.4. Getting online help for Tinker..................................................................................... 76 11.5. Getting online help for RasMol................................................................................... 77 11.6. Getting online help for Babel...................................................................................... 78 11.7. Visiting the MoCalc web site ...................................................................................... 78
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
4

Chapter 1. Introduction MoCalc User’s Guide
Chapter 1. Introduction
1.1. Wellcome to MoCalc!
MoCalc - Interface for Molecular Calculation is a graphical user interface for the molecular modeling programs Gamess1, Mopac2, Tinker3 Babel4 and Rasmol5. MoCalc helps the user in the preparation of input files, the submission of calculations, the analysis of the results and the visualization of the involved chemical structures. Some properties can also be calculated by MoCalc. The construction of MoCalc system followed the conventional pattern adopted for programs designed for Microsoft Windows environment. The users familiar with this operating system won't have difficulties in most of the tasks MoCalc is capable to execute. The MoCalc development team (Figure 1.1) has made a great effort to create a useful scientific tool. In the coming years, we will try to improve the MoCalc operation capabilities so that it can become more powerful and easier to use. We hope MoCalc can satisfy your expectancy. References: 1. Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.;
Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S.; Windus, T. L.; Dupuis, M.; Montgomery, J. A., Jr.; General Atomic and Molecular Electronic Structure System. Journal of Computational Chemistry, 14, 1347-1363, 1993.
2. Stewart, J. J. P.; MOPAC: A Semiempirical Molecular Orbital Program. Journal of Computer-Aided Molecular Design, 4, 1-105, 1990.
3. TINKER - Software Tools for Molecular Design, 3.7; Jay Ponder Lab, Dept. of Biochemistry & Molecular Biophysics, Washington University School of Medicine: St. Louis, 1999.
4. Walters, P.; Stahl, M.; Babel, Versão V.1.6, 1994. 5. RasMol: Molecular Visualisation Program, Versão 2.7; Glaxo Wellcome Research and
Development, Stevenage, Hertfordshire, 1995.
1.2. MoCalc Team
MoCalc - Interface for Molecular Calculation has been entirely developed in the Physics Department of Federal University of Espírito Santo - UFES. Author: Anderson Coser Gaudio Undergraduate students: Daniela Bertolini Depizzol Marcia Helena Moreira Paiva Thiago Oliveira dos Santos Adress: Physics Department Federal University of Espírito Santo Campus Universitário "Alaor de Queiroz Araújo" Av. Fernando Ferrari, 514 - Goiabeiras Vitória - ES 29075-910 Brazil [email protected] http://www.cce.ufes.br/anderson/
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
5

Chapter 1. Introduction MoCalc User’s Guide
Figure 1.1. MoCalc team: Thiago, Anderson, Daniela and Marcia.
1.3. How to get MoCalc
MoCalc is distributed through a self installing executable file called MoCalcSetup.exe. This file can be downloaded from the MoCalc official web site (see 11.7 - Visiting the MoCalc web site). It runs only in Microsoft Windows 95/98/ME/NT/2000/XP environments.
To get MoCalc:
1. Visit the author’s website: http://www.cce.ufes.br/anderson; 2. Click on the link MoCalc located in the main menu. This will open the MoCalc
Homepage; 3. In the MoCalc Homepage, click on the link Download; 4. Click on the link MoCalcSetup.exe; 5. In the File Download dialog, click on the Save button;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
6
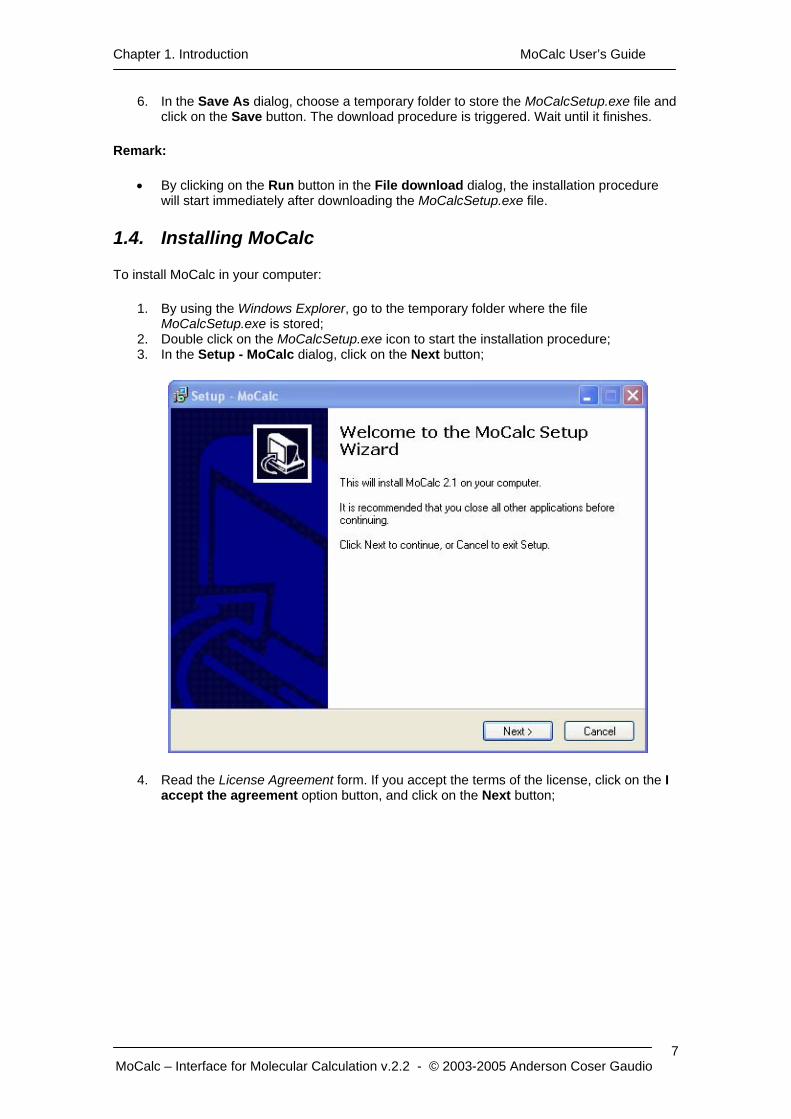
Chapter 1. Introduction MoCalc User’s Guide
6. In the Save As dialog, choose a temporary folder to store the MoCalcSetup.exe file and click on the Save button. The download procedure is triggered. Wait until it finishes.
Remark:
• By clicking on the Run button in the File download dialog, the installation procedure will start immediately after downloading the MoCalcSetup.exe file.
1.4. Installing MoCalc
To install MoCalc in your computer:
1. By using the Windows Explorer, go to the temporary folder where the file MoCalcSetup.exe is stored;
2. Double click on the MoCalcSetup.exe icon to start the installation procedure; 3. In the Setup - MoCalc dialog, click on the Next button;
4. Read the License Agreement form. If you accept the terms of the license, click on the I accept the agreement option button, and click on the Next button;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
7

Chapter 1. Introduction MoCalc User’s Guide
5. Select the destination folder where MoCalc will be installed. The default location is C:\Program files\MoCalc. Click on the Next button;
6. Select the start menu folder. The default shortcut for the program in the Windows Start menu is MoCalc. Click on the Next button;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
8

Chapter 1. Introduction MoCalc User’s Guide
7. Click on the Create a desktop icon option box to place a shortcut to MoCalc in the Windows workplace. Click on the Next button;
8. Click on the Next button to install MoCalc in your computer; 9. Click on the Launch MoCalc option box to start MoCalc program after finishing the
installation procedure. Click on the Finish button.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
9
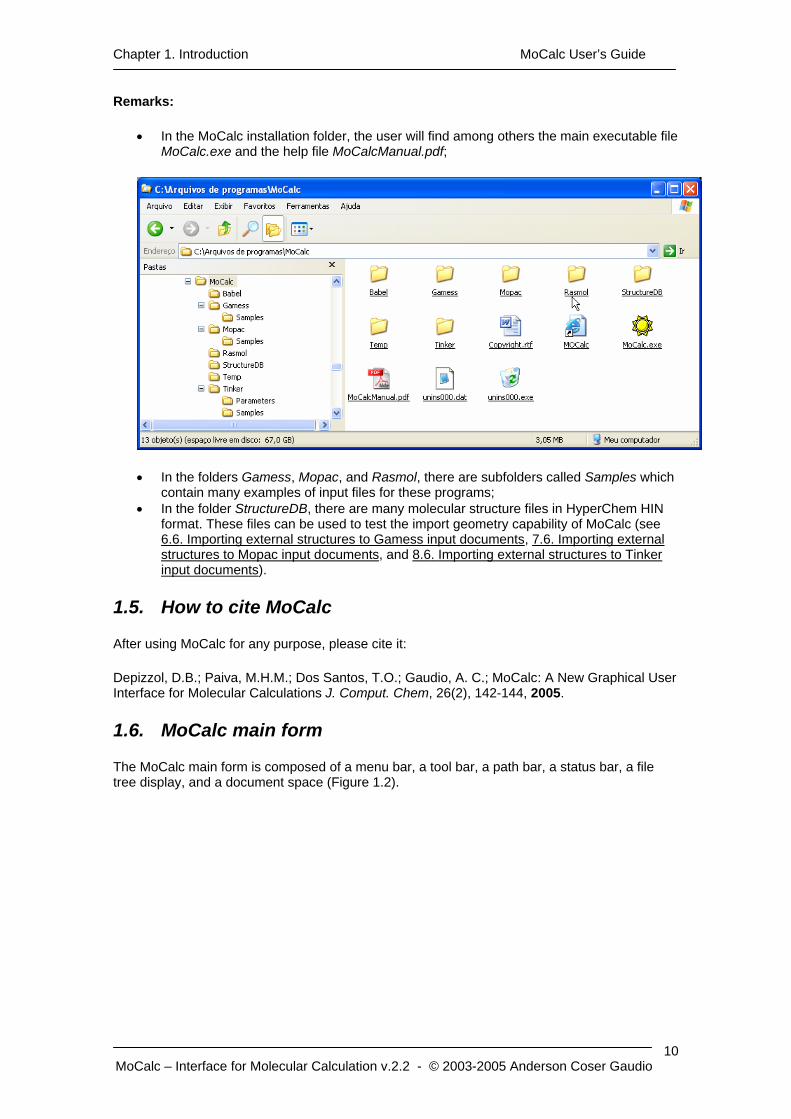
Chapter 1. Introduction MoCalc User’s Guide
Remarks:
• In the MoCalc installation folder, the user will find among others the main executable file MoCalc.exe and the help file MoCalcManual.pdf;
• In the folders Gamess, Mopac, and Rasmol, there are subfolders called Samples which contain many examples of input files for these programs;
• In the folder StructureDB, there are many molecular structure files in HyperChem HIN format. These files can be used to test the import geometry capability of MoCalc (see 6.6. Importing external structures to Gamess input documents, 7.6. Importing external structures to Mopac input documents, and 8.6. Importing external structures to Tinker input documents).
1.5. How to cite MoCalc
After using MoCalc for any purpose, please cite it:
Depizzol, D.B.; Paiva, M.H.M.; Dos Santos, T.O.; Gaudio, A. C.; MoCalc: A New Graphical User Interface for Molecular Calculations J. Comput. Chem, 26(2), 142-144, 2005.
1.6. MoCalc main form
The MoCalc main form is composed of a menu bar, a tool bar, a path bar, a status bar, a file tree display, and a document space (Figure 1.2).
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
10

Chapter 1. Introduction MoCalc User’s Guide
File tree display
Tool bar
Status bar
Path barMenu bar Document space
Figure 1.2. MoCalc main form.
1.6.1. Menu bar
The MoCalc Menu bar contains the menu system. All MoCalc functionality can be accessed by the menu command set.
1.6.2. Tool bar
The MoCalc Tool bar has shortcut buttons to the most common program features, such as create and open documents, print, configure page, etc.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
11
Chapter 1. Introduction MoCalc User’s Guide
1.6.3. Status bar
The MoCalc Status bar, located on the bottom of the MoCalc main window, is used to show useful information to the user.
1.6.4. Pathname bar
The MoCalc Pathname bar displays the full pathname of the active document.
1.6.5. File tree display
The MoCalc File tree display shows the names of the opened files, and can be used to make active one of the opened documents.
1.6.6. Document space
The MoCalc Document space is the place where all the MoCalc documents are opened and can be edited, run, and so on. The form that appears in the foreground is called active
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
12
Chapter 1. Introduction MoCalc User’s Guide
document. Most of the procedures made through the MoCalc menu system affect the active document.
Remark:
• To make active a document, do one of the following: (a) click on its filename in the file tree on the left of the MoCalc main form, (b) click on the document form, (c) press the key combination CTRL+TAB several times until the target document has been brought to front, or (d) in the Window menu, click on the filename in the file list showing the opened documents.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
13

Chapter 2. Working with documents MoCalc User’s Guide
Chapter 2. Working with documents
2.1. Document types
There are nine document types associated to Gamess, Mopac and Tinker programs. They are listed in Table 2.1.
Table 2.1. File types associated to Gamess, Mopac and Tinker programs.
# Program File type Extension Content 1 Gamess Input inp Input data, including initial structure 2 Gamess Output out Calculation results 3 Gamess Punch pun Store $DATA, $HESS, $VEC cards,
among others 4 Mopac Input zmt Input data, including initial structure 5 Mopac Output mno Calculation results 6 Mopac Archive arc Store a summary of the calculation result,
including the last structure 7 Tinker Input xyz Input structure 8 Tinker Geometry geo Output structure 9 Tinker Summary sum Summary of the calculation progress
2.2. Creating new documents
MoCalc lets you to create only three types of documents: Gamess input file (*.inp), Mopac input file (*.zmt), and Tinker input file (*.xyz).
To create a new document:
1. Click on the File/New menu and choose Gamess Input (*.inp), Mopac Input (*.zmt), or Tinker input (*.xyz). A blank document called UntitledGamessInput1.inp, UntitledMopacInput1.zmt, or UntitledTinkerInput1.xyz is created by the MoCalc system.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
14

Chapter 2. Working with documents MoCalc User’s Guide
Alternate method:
1. Click on the button in the toolbar; 2. In the Create new file dialog box, choose the type of the document to be created;
3. Click Ok. A blank document called UntitledGamessInput1.inp, UntitledMopacInput1.zmt, or UntitledTinkerInput1.xyz is created by the MoCalc system.
Remarks:
• On the first time you try to save a new document, MoCalc will show you the Save as dialog box so you can change the temporary filename to a more appropriated one;
• MoCalc do not let the user to create file names containing blank spaces in it; • The preparation of a content of the new document can be much faster through the use
of the Gamess and the Mopac keyword editors (see 6.2. Editing Gamess keywords and 7.2. Editing Mopac keywords) and by importing structure files generated by other programs (see 6.6. Importing external structures to Gamess input documents and 7.6. Importing external structures to Mopac input documents).
2.3. Opening existing documents
To open an existing document:
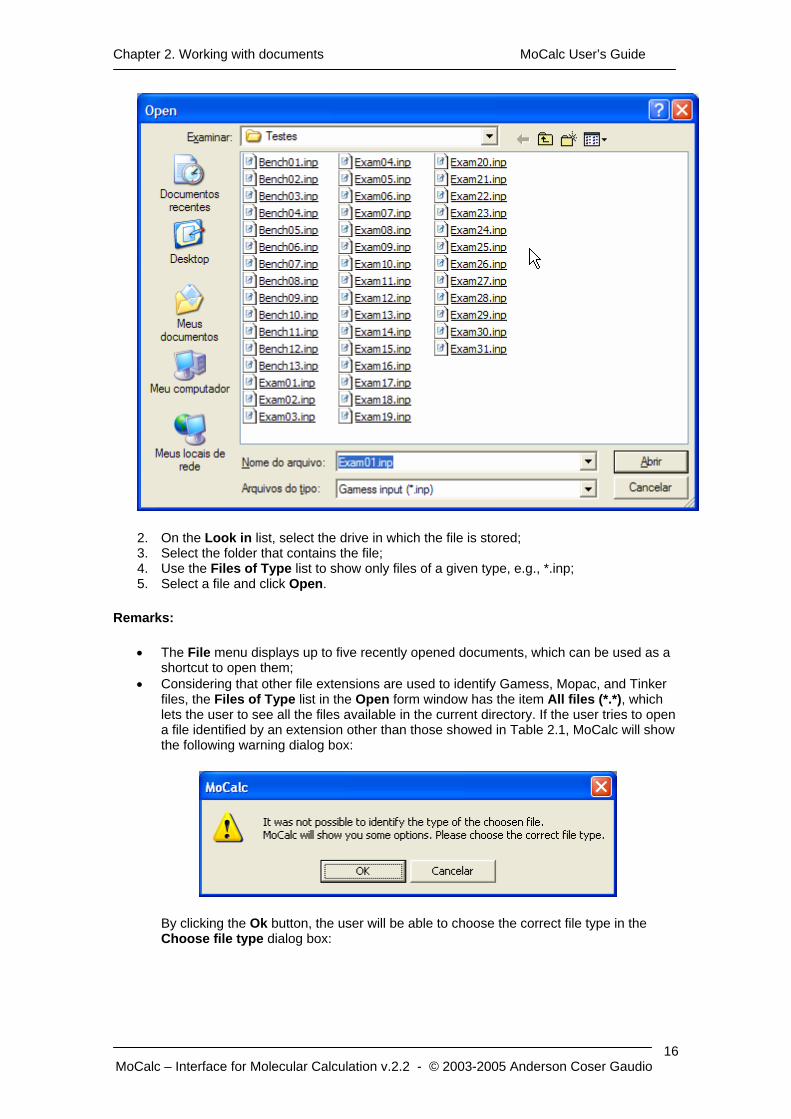
1. Click on the File/Open menu or click on the button in the toolbar. The Open dialog box will be showed;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
15
Chapter 2. Working with documents MoCalc User’s Guide
2. On the Look in list, select the drive in which the file is stored; 3. Select the folder that contains the file; 4. Use the Files of Type list to show only files of a given type, e.g., *.inp; 5. Select a file and click Open.
Remarks:
• The File menu displays up to five recently opened documents, which can be used as a shortcut to open them;
• Considering that other file extensions are used to identify Gamess, Mopac, and Tinker files, the Files of Type list in the Open form window has the item All files (*.*), which lets the user to see all the files available in the current directory. If the user tries to open a file identified by an extension other than those showed in Table 2.1, MoCalc will show the following warning dialog box:
By clicking the Ok button, the user will be able to choose the correct file type in the Choose file type dialog box:
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
16

Chapter 2. Working with documents MoCalc User’s Guide
As MoCalc forces all files to have the standard extensions showed in Table 2.1, by clicking the Ok button, a new warning dialog box is showed;
Click the Ok button to rename the file.
2.4. Opening multiple documents
MoCalc lets the user to open multiple files in just one operation, as long as they have the same file extension.
To open multiple documents at once:
1. Click on the File/Open Multiple Files menu; 2. In the Open multiple files dialog box, select the drive in which the files are stored;
3. Select the folder that contains the files; 4. Choose a proper file filter to display only files of the type of interest;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
17

Chapter 2. Working with documents MoCalc User’s Guide
5. To select one or more files, use the [>], [<], [>>], and [<<] buttons to transfer the selected files in the left file list box to the right file list box;
6. Click on the Open button.
Remarks:
• It is possible to select more than one non-contiguous files by pressing the CTRL key while selecting the files in the left list box;
• It is possible to select all files in between two non-contiguous positions by pressing the SHIFT key while selecting the files in the left list box.
2.5. Closing documents
To close a document:
1. Make active the document to be closed; 2. Click on the File/Close menu. If the document has been changed since the last time it
was saved, MoCalc will ask you to save it before closing it.
Remarks:
• To enable this menu option, at least one document of any kind must be opened.
2.6. Closing all documents
To close all opened documents in the MoCalc system:
1. Click on the File/Close All menu. If one or more documents have been changed since the last time they were saved, MoCalc will ask you to save them before closing them.
Remark:
• To enable this menu option, at least one document of any kind must be opened.
2.7. Closing documents of a given type
To close all documents of a given type at once:
1. Make active one of the opened documents of the type to be closed; 2. Click on the File/Close Files Of This Type menu. If the documents have been changed
since the last time they were saved, MoCalc will ask you to save them before closing them.
Remark:
• To enable this menu option, at least one document of any kind must be opened.
2.8. Saving documents
To save a document:
1. Make active the document to be to saved;
2. Click on the File/Save menu or click on the button in the toolbar.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
18

Chapter 2. Working with documents MoCalc User’s Guide
Remarks:
• On the first time the user tries to save a new document, MoCalc will show the Save as dialog box so the user can change the temporary filename by a more appropriated one;
• To enable this menu option, at least one document of any kind must be opened.
2.9. Changing the name of documents
To change the name of a document:
1. Make active the document whose name is to be changed; 2. Click on the File/Save As; 3. In the Save As dialog box, use the Save in list to select the drive letter where to save
the document; 4. Select the folder where the file will be saved; 5. In the Filename box enter the new filename; 6. Click on the Salve button.
Remarks:
• MoCalc do not let the user to create file names containing blank spaces in it; • To enable this menu option, at least one document of any kind must be opened.
2.10. Saving all documents
To save the changes performed in all opened documents:
1. Click on the File/Close All menu.
Remarks:
• If there were new documents opened, i.e., documents created through the File/New menu, MoCalc will show the Save as dialog box so the user can change the temporary filename by a more appropriated one;
• To enable this menu option, at least one document of any kind must be opened.
2.11. Converting files
One of the nice features of MoCalc is the fast conversion between Gamess, Mopac, and Tinker documents. It is possible to convert any Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), and Tinker output (*.geo) documents to Gamess input (*.inp), Mopac input (*.zmt), and Tinker input (*.xyz) documents.
To convert files:
1. Make active a Gamess output (*.out), a Mopac input (*.zmt), a Mopac output (*.mno), a Tinker input (*.xyz), or a Tinker output (*.geo) document;
2. Click on the File/Convert File To menu and choose one of the file types available in the submenus;
3. In the Select files dialog, use the [>], [<], [>>], and [<<] buttons to select the files of the same type to be submitted to the same conversion;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
19

Chapter 2. Working with documents MoCalc User’s Guide
4. Click Ok. The converted files will open as new documents with the same base filename and appropriated file extension.
Remarks:
• If the conversion procedure generates a file whose filename already exist in the same directory, MoCalc will warn the user about the file replacement. However, the warning dialog is shown only if MoCalc is configured to do so. See 9.3. Setting up warning messages for details.
• To enable this menu option, the active document must be a Gamess output (*.out), a Mopac input (*.zmt), a Mopac output (*.mno), a Tinker input (*.xyz), or a Tinker output (*.geo) file.
2.12. Converting multiple external files
MoCalc uses Babel program to convert multiple external files from one format to another, without loading them into MoCalc system. It is possible to convert Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput) files into Gamess input (*.inp), Mopac input (*.zmt), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput) files.
To convert files:
1. Click on the File/Convert Multiple External Files… menu; 2. In the Convert multiple files dialog, use the Convert file type combo box to select the
file type to convert, and the To type combo box to select the file type into which will be converted;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
20

Chapter 2. Working with documents MoCalc User’s Guide
3. In the Convert multiple external files dialog, use the [>], [<], [>>], and [<<] buttons to select the files of the same type to be submitted to the same conversion;
4. Click on the Open button.
Remarks:
• If the procedure is well succeeded, the Conversion completed! message box will be shown and the user will find the converted files in the same folder of the files submitted to conversion;
• In the conversion procedure, the files submitted to conversion are not erased from disk. Instead, they remain untouched while the new files are created. These files have the same base name and an appropriate file extension.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
21
Chapter 2. Working with documents MoCalc User’s Guide
2.13. Importing multiple files
MoCalc uses Babel program to import multiple external files to MoCalc system, converting them from their original format into Gamess input (*.inp), Mopac input (*.zmt), or Tinker input (*.xyz) document. The file formats that can be imported are Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput).
To import files:
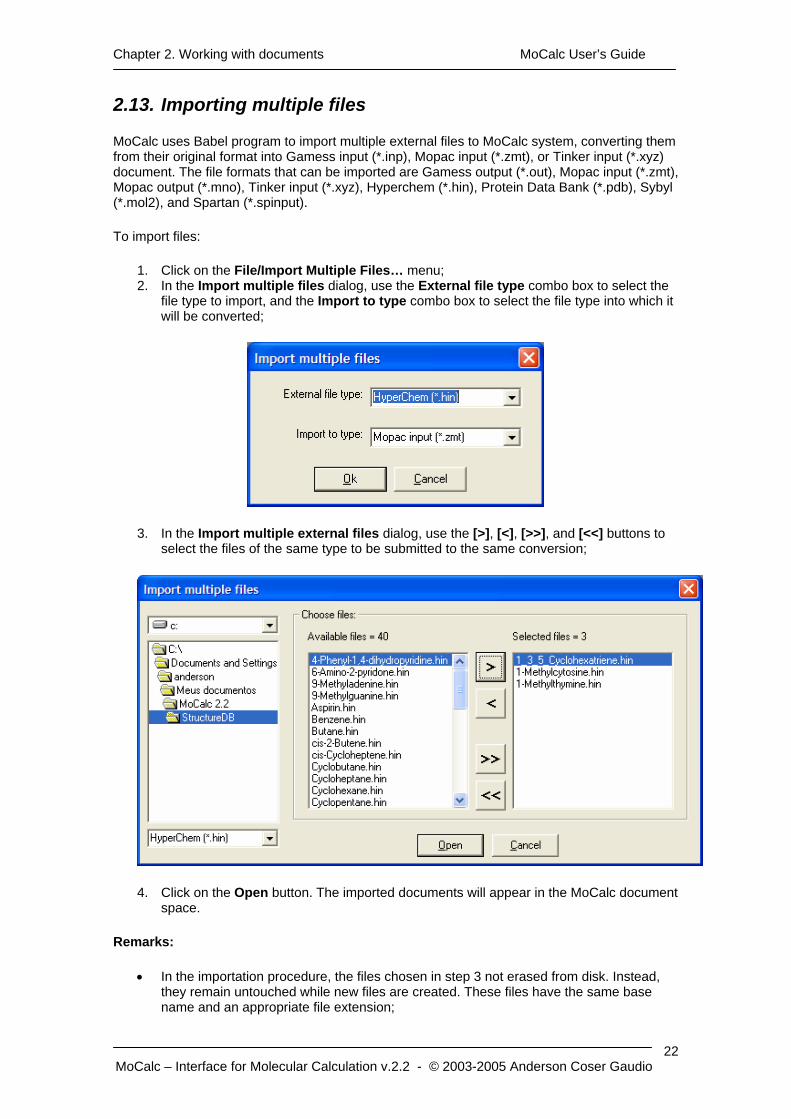
1. Click on the File/Import Multiple Files… menu; 2. In the Import multiple files dialog, use the External file type combo box to select the
file type to import, and the Import to type combo box to select the file type into which it will be converted;
3. In the Import multiple external files dialog, use the [>], [<], [>>], and [<<] buttons to select the files of the same type to be submitted to the same conversion;
4. Click on the Open button. The imported documents will appear in the MoCalc document space.
Remarks:
• In the importation procedure, the files chosen in step 3 not erased from disk. Instead, they remain untouched while new files are created. These files have the same base name and an appropriate file extension;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
22

Chapter 2. Working with documents MoCalc User’s Guide
• MoCalc places the new files in the same folder of the imported files.
2.14. Exporting documents to HTML
MoCalc lets the user to export to HTML input and output Gamess, Mopac, and Tinker text files, that is, Gamess input (*.inp), output (*.out) and punch (*.pun), Mopac input (*.zmt), output (*.mno) and archive (*.arc), and Tinker input (*.xyz), output (*.geo) and summary (*.sum) files.
To export a document to HTML:
1. Make active the document to be exported to HTML; 2. Click on the File/Export To HTML menu; 3. In the Save As dialog box, use the Save in list to select the drive in which the HTML file
will be saved; 4. Select the folder where the file will be saved; 5. In the Filename box enter the name of the HTML file, using the htm suffix; 6. Click on the Save button. A confirmation dialog box will be shown.
Remarks:
• The exported file file.htm can be opened through an Internet browser, such as Microsoft Internet Explorer;
• To enable this menu option, at least one document of any kind must be opened.
2.15. Configuring page for printing
To configure the document page for printing:
1. Click on the File/Page Setup menu or click on the button in the toolbar; 2. In the Page setup dialog box, adjust the parameters that control the margins and the
printing orientation;
3. Click Ok.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
23

Chapter 2. Working with documents MoCalc User’s Guide
Remarks:
• The allowed limits for the top, bottom, left, and right borders are from 0,00 to 5,08 cm, or from 0,00 to 2,00 inches;
• To enable this menu option, at least one document of any kind must be opened.
2.16. Previewing documents for printing
MoCalc lets the user to format the document appearance before printing it.
To preview the printing version of a document:
1. Click on the document you want to preview;
2. Click on the File/Print Preview or click on the button in the toolbar. The document content is transferred to the Print preview form;
3. To change the font type and the font size, select the text and use the font type
and the font size lists in the toolbar to change these properties;
4. To change the font format, select the text and click on the buttons in the toolbar to make the text bold, italic and underlined, respectively;
5. To change the text alignment, click on the buttons to align the paragraph on the left, centered, and on the right, respectively;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
24

Chapter 2. Working with documents MoCalc User’s Guide
6. Click on the button to configure page for printing;
7. Click on the button to print the document.
Remark:
• To enable this menu option, at least one document of any kind must be opened.
2.17. Printing documents
To print a document:
1. Click on the File/Print menu or click on the button in the toolbar; 2. In the Print dialog, select the printer and choose the number print copies;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
25

Chapter 2. Working with documents MoCalc User’s Guide
3. Click Ok.
Remark:
• To enable this menu option, at least one document of any kind must be opened.
2.18. Closing MoCalc
To close MoCalc:
1. Click on the File/Exit menu. If any opened document has been changed since the last time it was saved, MoCalc will ask you to save it before closing it.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
26

Chapter 3. Editing documents MoCalc User’s Guide
Chapter 3. Editing documents
3.1. Undoing changes in documents
The feature Undo lets the user to undo multiple level operations.
To undo alterations in a document:
1. Make active the document in which there are changes to be undone; 2. Click on the Edit/Undo menu or click on the button in the toolbar or enter CTRL+Z
on the keyboard. The last alteration in the document will be undone; 3. By repeating the above action several times, will make previous changes to be undone.
Remarks:
• In the case of many Gamess, Mopac, and Tinker input files opened at the same time, each one with different levels of changes performed, in nothing reduces the ability of MoCalc to undo changes correctly;
• To enable this menu option, a Gamess input file (*.inp), a Mopac input file (*.zmt), or a Tinker (*.xyz) must be the active document and it must be edited.
3.2. Redoing changes in documents
The feature Redo lets the user to redo multiple level operations.
To redo alterations in a document:
1. Make active the document in which there are changes to be redone;
2. Click on the Edit/Redo menu or click on the button in the toolbar or enter CTRL+Y on the keyboard. The last undone change in the document will be redone;
3. By repeating the above action several times, will make previous undone changes to be redone.
Remarks:
• In the case of many Gamess, Mopac, and Tinker input files opened at the same time, each one with different levels of undone changes performed, in nothing reduces the ability of MoCalc to redo changes correctly;
• To enable this menu option, a Gamess input file (*.inp), a Mopac input file (*.zmt), or a Tinker (*.xyz) must be the active document and some change be undone.
3.3. Cutting pieces of documents
The feature Cut erases the selected text of the active document and sends it to the Windows clipboard.
To cut a portion of a text document:
1. Select the text you want to copy;
2. Click on the Edit/Cut menu or click on the toolbar or enter CTRL+X on the keyboard. The selected text is erased from the document and copied to the Windows clipboard, from where it can be pasted elsewhere.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
27

Chapter 3. Editing documents MoCalc User’s Guide
Remarks:
• If some part of the active document is cut by mistake, click on the Edit/Undo menu or enter CTRL+Z to undo the action;
• To enable this menu option, a Gamess input file (*.inp), a Mopac input file (*.zmt), or a Tinker (*.xyz) must be the active document and some text must be selected.
3.4. Copying pieces of documents to the clipboard
The feature Copy transfers the selected text of a document to the Windows clipboard.
To copy text to the clipboard:
1. Select the text you want to copy;
2. Click on the Edit/Copy menu or click on the toolbar or enter CTRL+C on the keyboard. The selected text is copied to the Windows clipboard, from where it can be pasted elsewhere.
Remarks:
• In the case of documents containing graphs, like those generated from the Gamess/Compare Properties menu (see 6.11. Comparing properties generated by Gamess), the action Copy is done without the need of selecting an specific area, even because graphs cannot be selected in the MoCalc environment;
• From the Windows clipboard, the copied part of a document can be pasted in text editors, such as Microsoft Word. In the case of documents containing calculation results from Gamess and Mopac, which are shown in spreadsheets, the cell contents also can be pasted in other spreadsheet programs, such as Microsoft Excel;
• To enable this menu option, some text of the active document must be selected.
3.5. Copying the entire content of documents to the clipboard
The feature Copy all transfers the whole content of a document to the Windows clipboard.
To copy the entire content of a document:
1. Click on the Edit/Copy All menu. All the content of the document will be transferred to the clipboard.
Remarks:
• From the Windows clipboard, the copied part of a document can be pasted in text editors, such as Microsoft Word. In the case of documents containing calculation results from Gamess and Mopac, which are shown in spreadsheets, the cell contents also can be pasted in other spreadsheet programs, such as Microsoft Excel;
• To enable this menu option, at least one document of any kind must be opened.
3.6. Pasting from the clipboard
The feature Paste inserts the content of the Windows clipboard into the document.
To paste the content of the clipboard into a document.
1. Locate the cursor at the place you want to paste;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
28

Chapter 3. Editing documents MoCalc User’s Guide
2. Click on the Edit/Paste menu or click on the toolbar or enter CTRL+V on the keyboard. The content of the Windows clipboard will be inserted into the document.
Remarks:
• If the Paste action is done by mistake, click on the Edit/Undo menu or enter CTRL+Z to undo the action;
• To enable this option, the active document must be a Gamess input file (*.inp), a Mopac input file (*.zmt), or a Tinker input file (*.xyz) and the clipboard must not be empty.
3.7. Clearing the content of documents
The feature Clear erases the whole content of the active document, which must be a Gamess input (*.inp), a Mopac input (*.zmt), or a Tinker input (*.xyz), without transfer it to the Windows clipboard.
To clear a document:
1. Make active a Gamess input (*.inp), a Mopac input (*.zmt), or a Tinker input (*.xyz) document;
2. Click on the Edit/Clear menu.
Remarks:
• If the Clear action is done by mistake, click on the Edit/Undo menu or enter CTRL+Z to undo the action;
• To enable this option, the active document must be a Gamess input file (*.inp), a Mopac input file (*.zmt), or a Tinker input file (*.xyz).
3.8. Selecting the entire content of documents
The feature Select All selects the whole content of a text document.
To select all the document:
1. Click on the Edit/Select All menu or enter CTRL+A on the keyboard.
Remarks:
• To run Copy after Select All is equivalent to Copy All, because in both cases the whole content of the document is transferred to the Windows clipboard;
• To enable this menu option, at least one document of any kind must be opened.
3.9. Finding strings in documents
The feature Find lets the user to find strings inside the active document.
To find a string in a text document:
1. Click on the Edit/Find menu or enter CTRL+F on the keyboard. The Find & Replace dialog is shown;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
29

Chapter 3. Editing documents MoCalc User’s Guide
2. In the combo box Find What, enter the string you want to find; 3. Click on the Find Next button. If the active document had one or more occurrences of
such string, the nearest occurrence will be found and it will appear selected in the document.
Remarks:
• If there was any text selected at the moment the dialog Find & Replace is loaded, it will be transferred to the combo box Find What to speed up the operation;
• Select the check box Match case to make the search case sensitive; • Select the option buttons Up or Down to define the search direction, from the cursor
position, in the document; • Select the option buttons Cursor, Top or Bottom to define the search starting point in
the document. By selecting Cursor, the search starts from the cursor position. By selection Up or Down, the search starts from the beginning or from the end of the document, respectively. In any case, the search direction follows the definition made in the Up or Down option buttons;
• The combo box Find What can store the last ten strings used in previous searches. To use some of these strings in a new search, click on the dropdown button located at the right of the combo and choose one of the available strings;
• See also 3.10. Replacing strings in documents; • To enabled this menu option, the active document must be a text document. MoCalc is
not able to find strings in spreadsheets.
3.10. Replacing strings in documents
The feature Replace lets the user to find strings inside the active document and replace them by other strings.
To find and replace strings in a text document:
1. Click on the Edit/Replace menu or enter CTRL+H on the keyboard. The Find & replace dialog is loaded;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
30

Chapter 3. Editing documents MoCalc User’s Guide
2. In the combo box Find What, enter the string you want to be replaced; 3. In the combo box Replace With, enter the string that will replace the previous one; 4. Click on the Find Next button. If the active document had one or more of such strings,
the nearest string will be found and will appear selected in the document; 5. Click on the Replace button. The selected text will be replaced. If there is more than
one occurrence of the searched text in the document, the nearest one will be select and the system will wait the user’s decision to replace it or not;
6. Click on the Replace All to replace all the occurrences of the searched text in the document.
Remarks:
• If there is any text selected at the moment the dialog Find & Replace is loaded, that text will be transferred to the combo box Find What to speed up the operation;
• Select the check box Match case to make the search case sensitive; • Select the option buttons Up or Down to define the search direction, from the cursor
position, in the document; • Select the option buttons Cursor, Top or Bottom to define the search starting point in
the document. By selecting Cursor, the search starts from the cursor position. By selection Up or Down, the search starts from the beginning or from the end of the document, respectively. In any case, the search direction follows the definition made in the Up or Down option buttons;
• The combo boxes Find What and Replace With can store the last ten strings used in previous searches and replacements. To use some of these strings in a new search, click on the dropdown button located at the right of the combos and choose one of the available strings;
• See also 3.9. Finding strings in documents. • To enable this option, the active document must be a Gamess input file (*.inp), a Mopac
input file (*.zmt), or a Tinker input file (*.xyz).
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
31

Chapter 4. Exhibiting components of MoCalc MoCalc User’s Guide
Chapter 4. Exhibiting components of MoCalc
4.1. Showing and hiding the toolbar
MoCalc has a standard toolbar with shortcut buttons to the most common program features, such as create and open documents, print, configure page, etc. The toolbar can be displayed or hidden, according to the user's preference.
To display or to hide the toolbar:
1. Click on the View/Tool Bar menu. A selection mark will appear next to the menu command when the bar is visible.
4.2. Showing and hiding the status bar
MoCalc has a status bar, located on the bottom of the MoCalc main window, which is used to show system information to the user. The status bar can be displayed or hidden, according to the user's preference.
To display or to hide the status bar:
1. Click on the View/Status Bar menu. A selection mark will appear next to the menu command when the bar is visible.
Remark:
• At the right side of the status bar there are three field boxes used to display the status of the CAPS LOCK, NUM LOCK and INSERT keys.
4.3. Showing and hiding the pathname bar
MoCalc has a pathname bar which displays the pathname of the active document. The pathname bar can be displayed or hidden, according to the user's preference.
To display or to hide the pathname bar:
1. Click on the View/Pathname Bar menu. A selection mark will appear next to the menu command when the bar is visible.
Remark:
• Some documents, such as spreadsheets and graphs, are not stored in disk. In these cases, the bar will show Path: None.
4.4. Showing and hiding the file tree display
MoCalc has a space called File tree display which shows the names of the opened files, and can be used to make active one of the opened documents. The file tree display can be displayed or hidden, according to the user's preference.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
32

Chapter 4. Exhibiting components of MoCalc MoCalc User’s Guide
To display or to hide the tree display:
1. Click on the View/File Tree Display menu. A selection mark will appear next to the menu command when the bar is visible.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
33
Chapter 5. Formatting documents MoCalc User’s Guide
Chapter 5. Formatting documents
5.1. Formatting the font of a document
The user can change the text font of the MoCalc documents. This operation makes easier the identification of the different types of MoCalc documents.
To format the text font of a MoCalc Document:
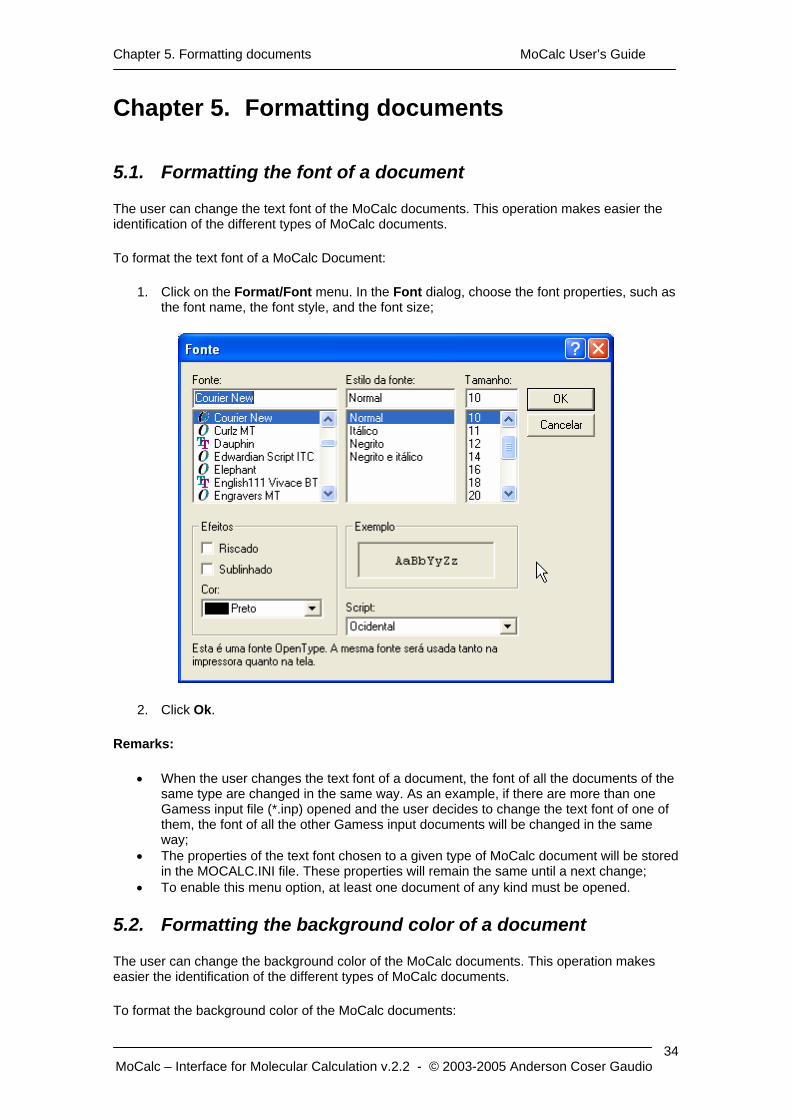
1. Click on the Format/Font menu. In the Font dialog, choose the font properties, such as the font name, the font style, and the font size;
2. Click Ok.
Remarks:
• When the user changes the text font of a document, the font of all the documents of the same type are changed in the same way. As an example, if there are more than one Gamess input file (*.inp) opened and the user decides to change the text font of one of them, the font of all the other Gamess input documents will be changed in the same way;
• The properties of the text font chosen to a given type of MoCalc document will be stored in the MOCALC.INI file. These properties will remain the same until a next change;
• To enable this menu option, at least one document of any kind must be opened.
5.2. Formatting the background color of a document
The user can change the background color of the MoCalc documents. This operation makes easier the identification of the different types of MoCalc documents.
To format the background color of the MoCalc documents:
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
34

Chapter 5. Formatting documents MoCalc User’s Guide
1. Click on the Format/Background menu. In the Color dialog, choose the color you want to apply to the document background;
2. Click Ok.
Remarks:
• When the user changes the background color of a document, the background color of all the documents of the same type are changed in the same way. As an example, if there are more than one Gamess input file (*.inp) opened and the user decides to change the background color of one of them, the background color of all the other Gamess input documents will change in the same way;
• The background color chosen to a given type of MoCalc document will be stored in the MOCALC.INI file. This background color will remain the same until a next change;
• To enable this menu option, at least one document of any kind must be opened.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
35
Chapter 6. Working with Gamess MoCalc User’s Guide
Chapter 6. Working with Gamess
6.1. Running Gamess input files
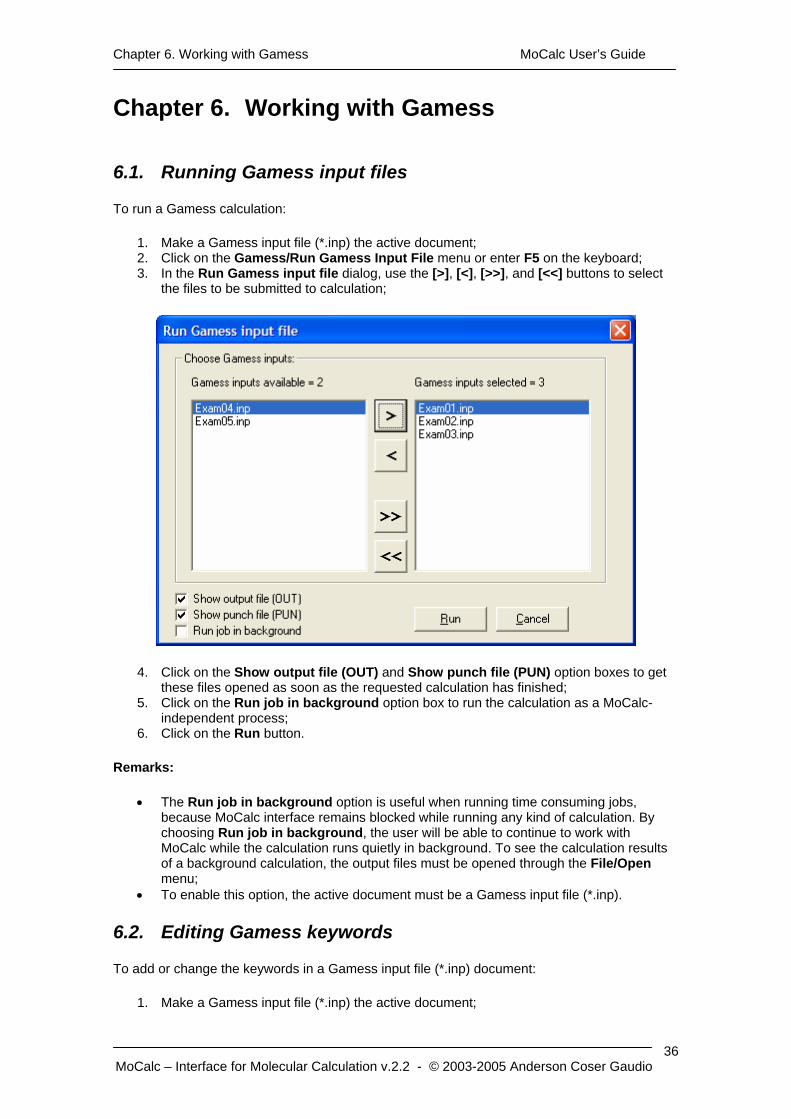
To run a Gamess calculation:
1. Make a Gamess input file (*.inp) the active document; 2. Click on the Gamess/Run Gamess Input File menu or enter F5 on the keyboard; 3. In the Run Gamess input file dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to calculation;
4. Click on the Show output file (OUT) and Show punch file (PUN) option boxes to get these files opened as soon as the requested calculation has finished;
5. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
6. Click on the Run button.
Remarks:
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• To enable this option, the active document must be a Gamess input file (*.inp).
6.2. Editing Gamess keywords
To add or change the keywords in a Gamess input file (*.inp) document:
1. Make a Gamess input file (*.inp) the active document;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
36

Chapter 6. Working with Gamess MoCalc User’s Guide
2. Click on the Gamess/Edit keywords menu. If the Gamess input had some keyword cards (see the Remarks below), they will be captured and exhibited in the list box on the left of the Gamess keyword editor form;
3. To add new keywords, use the three combo boxes on the editor upper right corner. The first box defines the keyword group, the second exhibits the options of each group, and the third exhibits the parameters available for each option. The user must choose first the group, next the option, and finally the parameter;
4. Click on the Add button to include the keyword chosen on the three combo boxes in the list box on the left.
5. Repeat 3 and 4 until all keywords has been chosen; 6. To remove keywords from the list box, select one of them and click on the Remove
button. To remove all the keywords at once, click on the Clear button; 7. After finish choosing the keyword set, click on the Apply button. The keyword editor will
be closed and the focus will be set back on the Gamess input document. The chosen keywords will be placed on the beginning of the document.
Remarks:
• In the present version of MoCalc, the following keyword groups are available in the keyword editor: $BASIS, $CONTRL, $DRC, $FORCE, $MOROKM, $MP2, $PCM, $SCF, $STATPT, and $SYSTEM;
• To add other keyword groups through the editor, the user can type them on the three combo boxes, one by one and clicking on the Add button;
• When the keyword editor is opened and the existing keywords are imported to the list box, if some keyword is not recognized as being correct, the user will be notified to verify it;
• The MoCalc status bar shows some information about the keyword groups and options when the respective combo box item is selected;
• To enable this menu option, the active document must be a Gamess input file (*.inp).
6.3. Inserting default header in Gamess input documents
To insert the input header:
1. Make a Gamess input file (*.inp) the active document; 2. Place the cursor where you want to insert the input header; 3. Click on the Gamess/Insert Input Header menu.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
37

Chapter 6. Working with Gamess MoCalc User’s Guide
Remarks:
• If nothing happens when the above operation is performed, the MoCalc system may have no default Gamess input header definition;
• To create a new or alter an existing default Gamess input header, see 6.4. Setting up the Gamess default input header;
• The content of the Gamess keyword input header is stored in the MOCALC.INI file; • Depending on the actual content of the default input header stored in the MoCalc
system, it may be necessary to make some manual adjustment to eliminate blank spaces or extra lines added after this operation;
• This menu option is always active.
6.4. Setting up the Gamess default input header
MoCalc lets the user to store a default keyword input header that can be promptly inserted in a Gamess input document.
To create or edit a default keyword input header:
1. Click on the Gamess/Setup Default Input Header menu; 2. In the Setup Gamess input header dialog, insert or change the Gamess keyword
header;
3. Click Ok.
Remarks:
• To insert the keyword input header in a Gamess input document (*.inp) see 6.3. Inserting default header in Gamess input documents. Consult the Gamess manual, through the Help/Gamess Manual menu, to be informed about the correct syntax of the keywords used in the default header;
• The content of the Gamess keyword input header is stored in the MOCALC.INI file; • To enable this menu option, the active document must be a Gamess input (*.inp), an
output (*.out), or a punch (*.pun) document.
6.5. Making default this Gamess input header
To make default a Gamess keyword input header:
1. Make a Gamess input file (*.inp) the active document; 2. By using the mouse or the keyboard (CTRL/SHIFT + ←↑→↓ keys), select the input
header that you want to become the default Gamess input header; 3. Click on the Gamess/Make Default This Input Header menu.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
38

Chapter 6. Working with Gamess MoCalc User’s Guide
Remarks:
• Although no message is shown to the user, the input header is properly stored; • To see and/or edit the default input header, see 6.4. Setting up the Gamess default
input header; • The content of the Gamess keyword input header is stored in the MOCALC.INI file; • To enable this menu option, the active document must be a Gamess input file (*.inp).
6.6. Importing external structures to Gamess input documents
Unfortunately, the present version of MoCalc still does not have a 3D molecular editor. MoCalc uses Babel program to convert structures from Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput) formats to generate an input geometry for Gamess calculation ($DATA group).
To import an external structure to a Gamess input document:
1. Make a Gamess input file (*.inp) the active document; 2. Click on the Gamess/Import External Molecular Geometry menu; 3. In the Import external molecular geometry dialog, choose the type of the file that
contain the external structure;
4. Click on the Insert default input header option box to insert the stored input header (See 6.4. Setting up the Gamess default input header) in the Gamess input document;
5. Click Ok;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
39
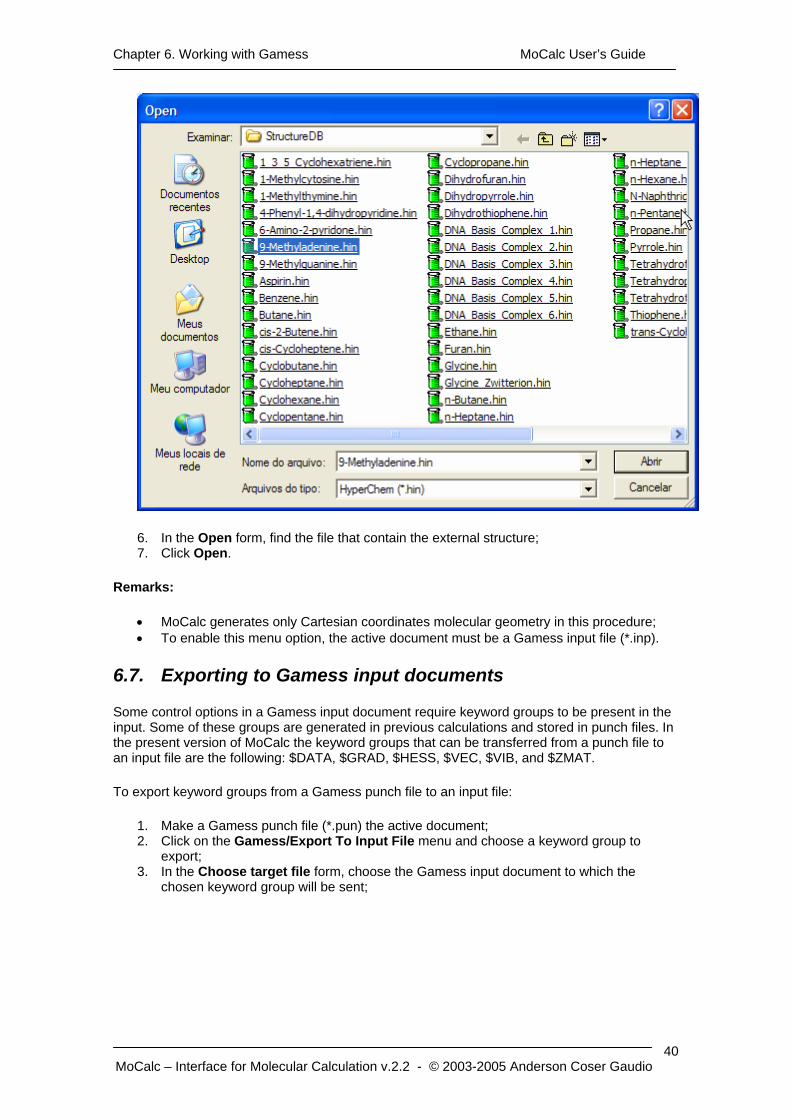
Chapter 6. Working with Gamess MoCalc User’s Guide
6. In the Open form, find the file that contain the external structure; 7. Click Open.
Remarks:
• MoCalc generates only Cartesian coordinates molecular geometry in this procedure; • To enable this menu option, the active document must be a Gamess input file (*.inp).
6.7. Exporting to Gamess input documents
Some control options in a Gamess input document require keyword groups to be present in the input. Some of these groups are generated in previous calculations and stored in punch files. In the present version of MoCalc the keyword groups that can be transferred from a punch file to an input file are the following: $DATA, $GRAD, $HESS, $VEC, $VIB, and $ZMAT.
To export keyword groups from a Gamess punch file to an input file:
1. Make a Gamess punch file (*.pun) the active document; 2. Click on the Gamess/Export To Input File menu and choose a keyword group to
export; 3. In the Choose target file form, choose the Gamess input document to which the
chosen keyword group will be sent;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
40

Chapter 6. Working with Gamess MoCalc User’s Guide
4. Click Ok. The keyword group will be inserted at the end of the chosen file.
Remarks:
• The presence of the keyword groups $GRAD, $HESS, $VEC, $VIB, and $ZMAT in a Gamess input document may requires some changes in existing keyword groups and/or creation of new groups. See the Gamess manual by clicking on the Help/Gamess Manual menu for detailed instructions (See 11.2. Getting online help for Gamess);
• To enable this option, the active document must be a Gamess punch document (*.pun) and one or more Gamess input documents (*.inp) must be opened.
6.8. Showing structures from Gamess documents
MoCalc uses program RasMol as molecular viewer. The only Gamess document that has enough information inside to build a computer molecular model is the Gamess output file (*.out).
To show a molecule from a Gamess document:
1. Make a Gamess output file (*.out) the active document; 2. Click on the Gamess/Show Molecule menu. Program RasMol will start and the last
structure in the Gamess output file will be displayed;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
41

Chapter 6. Working with Gamess MoCalc User’s Guide
3. Use the RasMol resources to change the features concerning the display mode. See the Rasmol manual by clicking on the Help/Rasmol Manual menu for detailed information (See 11.5. Getting online help for RasMol).
Remarks:
• Once program RasMol is initialized from the MoCalc system, it becomes an independent process as any other program running in the Windows environment and, as so, it can be operated and shot down independently from MoCalc;
• To enable this option, the active document must be a Gamess output file (*.out).
6.9. Extracting results from Gamess output documents
In the present version of MoCalc, the results that can be extracted from a Gamess output document are the following: energy, charges, dipole moment, interatomic distances, bond order, valence analysis, eigenvectors, and population analysis. The results that can be extracted from the population analysis are the following: Mulliken population in the molecular orbitals, population in atomic orbitals, Mulliken overlap population, and total Mulliken and Lowdin population. These results are shown in separated grid forms from which they can be copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word.
To extract properties from a Gamess output document:
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
42
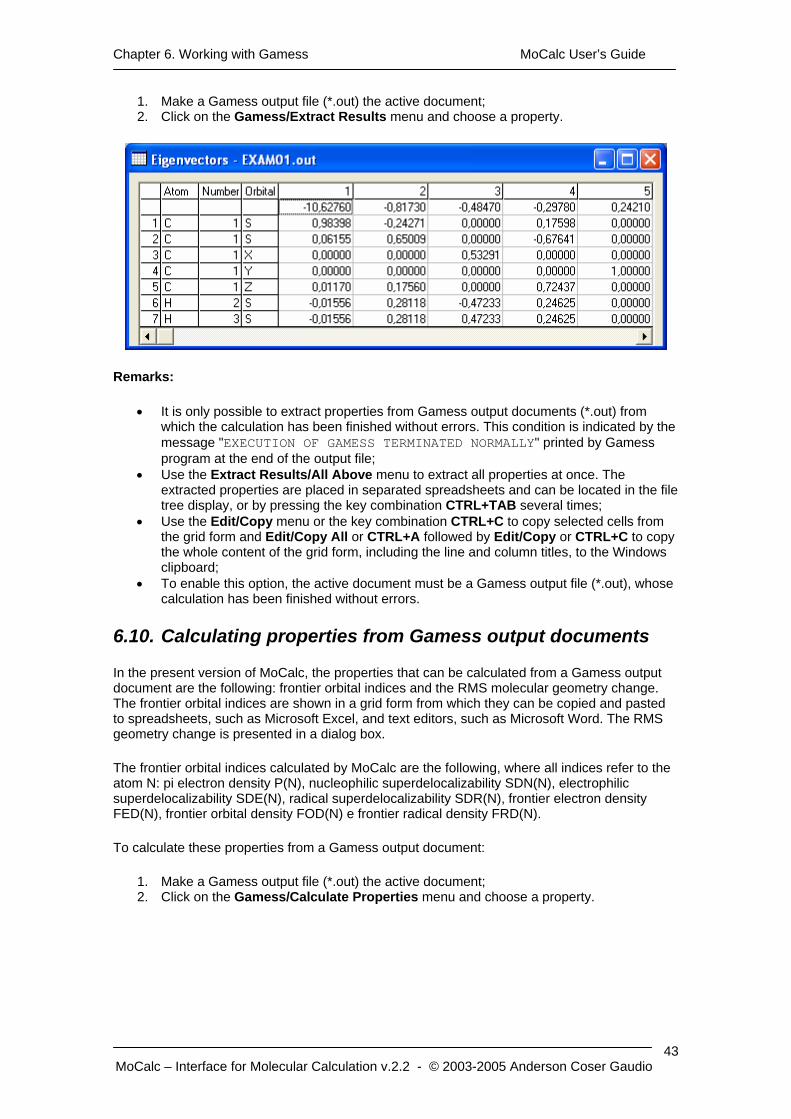
Chapter 6. Working with Gamess MoCalc User’s Guide
1. Make a Gamess output file (*.out) the active document; 2. Click on the Gamess/Extract Results menu and choose a property.
Remarks:
• It is only possible to extract properties from Gamess output documents (*.out) from which the calculation has been finished without errors. This condition is indicated by the message "EXECUTION OF GAMESS TERMINATED NORMALLY" printed by Gamess program at the end of the output file;
• Use the Extract Results/All Above menu to extract all properties at once. The extracted properties are placed in separated spreadsheets and can be located in the file tree display, or by pressing the key combination CTRL+TAB several times;
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the whole content of the grid form, including the line and column titles, to the Windows clipboard;
• To enable this option, the active document must be a Gamess output file (*.out), whose calculation has been finished without errors.
6.10. Calculating properties from Gamess output documents
In the present version of MoCalc, the properties that can be calculated from a Gamess output document are the following: frontier orbital indices and the RMS molecular geometry change. The frontier orbital indices are shown in a grid form from which they can be copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word. The RMS geometry change is presented in a dialog box.
The frontier orbital indices calculated by MoCalc are the following, where all indices refer to the atom N: pi electron density P(N), nucleophilic superdelocalizability SDN(N), electrophilic superdelocalizability SDE(N), radical superdelocalizability SDR(N), frontier electron density FED(N), frontier orbital density FOD(N) e frontier radical density FRD(N).
To calculate these properties from a Gamess output document:
1. Make a Gamess output file (*.out) the active document; 2. Click on the Gamess/Calculate Properties menu and choose a property.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
43

Chapter 6. Working with Gamess MoCalc User’s Guide
Remarks:
• It is only possible to calculate properties from Gamess output documents (*.out) from which the calculation has been finished without errors. This condition is indicated by the message "EXECUTION OF GAMESS TERMINATED NORMALLY" printed by Gamess program at the end of the output file;
• In case of open shell calculation (Unrestricted Hartree-Fock or UHF), in which two sets of eigenvectors, alpha and beta, are produced, MoCalc will show two sets of frontier orbital indices, one for each set of eigenvectors;
• The frontier orbital indices calculation is performed from the last set of eigenvectors printed in the Gamess output file (*.out);
• The RMS geometry change calculation is performed from the first and the last molecular geometries printed in the Gamess output file (*.out);
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the whole content of the grid form, including the line and column titles, to the Windows clipboard;
• To enable this option, the active document must be a Gamess output file (*.out), whose calculation has been finished without errors.
6.11. Comparing properties generated by Gamess
In the present version of MoCalc, the results that can be compared from two or more Gamess output documents are the following: total energy, energy of the highest occupied molecular orbital (HOMO), energy of the lowest unoccupied molecular orbital (LUMO), and dipole moment. The comparison is presented in two ways. A numerical comparison is shown in a grid form from which they can be copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word. A graphical comparison is presented as bar charts, one for each property.
To compare molecular properties from the Gamess results:
1. Make a Gamess output file (*.out) the active document; 2. Click on the Gamess/Compare Molecular Properties menu. A spreadsheet containing
a numerical comparison of the properties and a set of graphs, one for each property, are shown.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
44

Chapter 6. Working with Gamess MoCalc User’s Guide
Remarks:
• When some result to be compared is not found in the Gamess output document (*.out), the consequence is one or more blank cells in the grid form and one or more absent bars in the bar charts;
• Use the file tree display or the key combination CTRL+TAB to navigate between the comparison results;
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the whole content of the grid form, including the line and column titles, to the Windows clipboard;
• Use the Edit/Copy menu or the key combination CTRL+C to copy a bar chart comparison to the Windows clipboard;
• To enable this option, two or more Gamess output documents (*.out), whose calculation has been finished without errors, must be opened, and the active document must be one of these documents.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
45
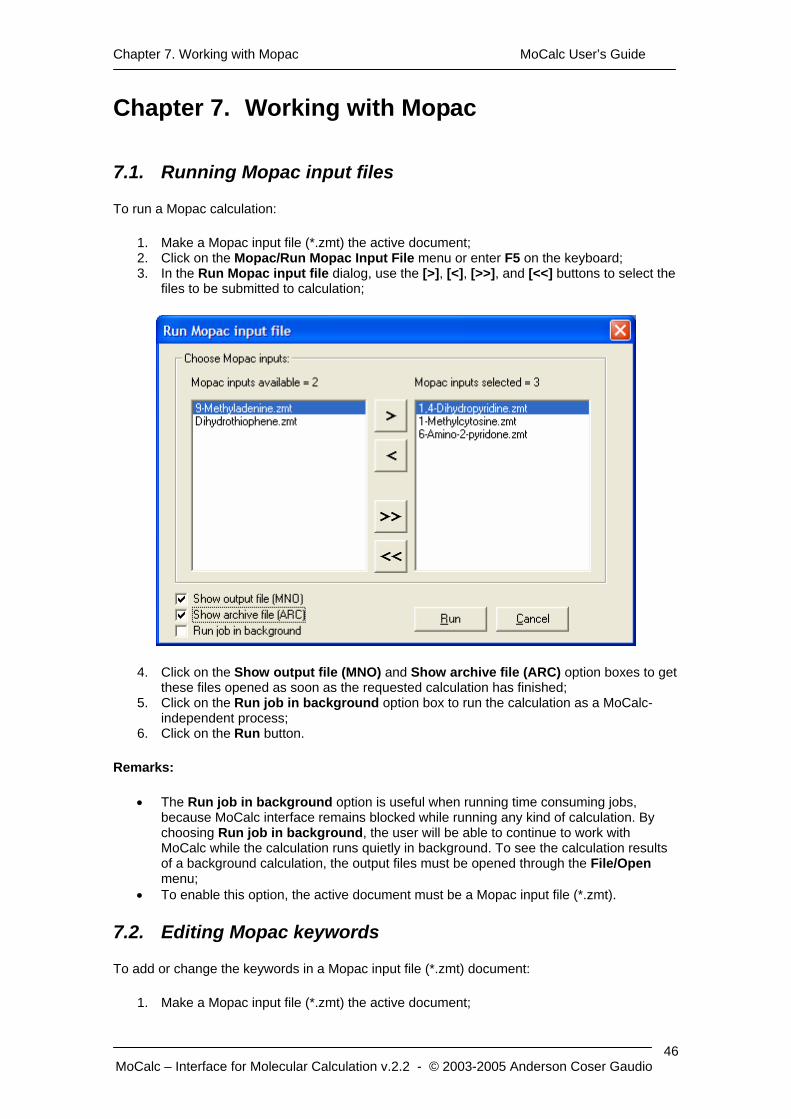
Chapter 7. Working with Mopac MoCalc User’s Guide
Chapter 7. Working with Mopac
7.1. Running Mopac input files
To run a Mopac calculation:
1. Make a Mopac input file (*.zmt) the active document; 2. Click on the Mopac/Run Mopac Input File menu or enter F5 on the keyboard; 3. In the Run Mopac input file dialog, use the [>], [<], [>>], and [<<] buttons to select the
files to be submitted to calculation;
4. Click on the Show output file (MNO) and Show archive file (ARC) option boxes to get these files opened as soon as the requested calculation has finished;
5. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
6. Click on the Run button.
Remarks:
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• To enable this option, the active document must be a Mopac input file (*.zmt).
7.2. Editing Mopac keywords
To add or change the keywords in a Mopac input file (*.zmt) document:
1. Make a Mopac input file (*.zmt) the active document;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
46

Chapter 7. Working with Mopac MoCalc User’s Guide
2. Click on the Mopac/Edit Keywords menu. If the Mopac input document had some set of keywords, they will be captured and exhibited in the list box on the left of the Mopac keyword editor form;
3. To select new keywords, use the combo box on the upper left corner of the keyword editor;
4. Click on the Add button to include the chosen keyword in the list box on the left; 5. Repeat 3 and 4 until the keywords have been chosen; 6. To remove keywords from the list box, select one of them and click on the Remove
button. To remove all the keywords at once, click on the Clear button; 7. After finished choosing the keyword set, click on the Apply button. The keyword editor
will be closed and the focus will be set back to the Mopac input document. The chosen keywords will be placed on the beginning of the document.
Remarks:
• The choice of keywords that require additional parameter, such as CHARGE, is followed by an input form in which the user can enter the parameter value;
• It is not possible to add the key symbols + e &. The Mopac keyword editor recognizes the symbol &, but automatically places back the symbol + in such a way that the number of characters in each line does not exceed 80 characters;
• To add other keyword groups to the editor, the user can type them on the combo box, one by one and clicking on the Add button;
• The first line of a Mopac input document (*.zmt) must contain the keywords that control the calculation procedure. The next two lines are reserved for comments, and the following lines for the molecular geometry.
• When the keyword editor is opened, the existing keywords are imported to the list box on the left of the Mopac keyword editor. If one or more keywords are not recognized as being typed correctly, the user is warned to check it up. Two possible reasons to this problem are syntax error and incorrect number of keyword lines. Consider the following Mopac input document, where there are two misspelled keywords:
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
47

Chapter 7. Working with Mopac MoCalc User’s Guide
By clicking on the Mopac/Edit Keywords menu, MoCalc will show the following dialog box:
By clicking on the OK button, the misspelled keywords appear highlighted in the list box of the Mopac keyword editor:
By clicking on the Correct button, the user will be able to enter the correct all suspicious keywords on the input box showed below:
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
48

Chapter 7. Working with Mopac MoCalc User’s Guide
By pressing the Enter button, the word in the text box will be transferred to the Mopac keyword editor, misspelled or not. To discard the keyword, press the Cancel button:
• To enable this menu option, the active document must be a Mopac input file (*.zmt).
7.3. Setting up the Mopac default input header
MoCalc lets the user to store a default keyword input header that can be promptly inserted in a Mopac input document.
To create or edit a default keyword input header:
1. Click on the Mopac/Setup Default Input Header menu; 2. In the Setup Mopac Input Header, insert or change the Mopac keyword header;
3. Click Ok.
Remarks:
• To insert the keyword input header in a Mopac input document (*.zmt) see 7.4. Inserting default header in Mopac input documents;
• Consult the Mopac manual, through the Help/Mopac Manual menu, to be informed about the correct syntax of the keywords used in the default header (See 11.3. Getting online help for Mopac);
• The content of the Mopac keyword input header is stored in the MOCALC.INI file; • This menu option is always active.
7.4. Inserting default header in Mopac input documents
To insert the input header:
1. Make a Mopac input file (*.zmt) the active document;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
49

Chapter 7. Working with Mopac MoCalc User’s Guide
2. Place the cursor where you want to insert the input header; 3. Click on the Mopac/Insert Input Header menu.
Remarks:
• If nothing happens when the above operation is performed, the MoCalc system may have no default Mopac input header definition;
• To create a new or alter an existing default Mopac input header, see 7.3. Setting up the Mopac default input header;
• The content of the Mopac keyword input header is stored in the MOCALC.INI file; • Depending on the actual content of the default input header stored in the MoCalc
system, it may be necessary to make some manual adjustment to eliminate blank spaces or extra lines added after this operation;
• To enable this menu option, the active document must be a Mopac input file (*.zmt).
7.5. Making default this Mopac input header
To make default a Mopac keyword input header:
1. Make a Mopac input file (*.zmt) the active document; 2. By using the mouse or the keyboard (CTRL/SHIFT + ←↑→↓ keys), select the input
header that you want to become the default Mopac input header; 3. Click on the Mopac/Make Default This Input Header.
Remarks:
• Although no message is shown to the user, the input header is properly stored. • To see and/or edit the default input header, see 7.3. Setting up the Mopac default input
header. • The content of the Mopac keyword input header is stored in the MOCALC.INI file; • To enable this menu option, the active document must be a Mopac input file (*.zmt).
7.6. Importing external structures to Mopac input documents
Unfortunately, the present version of MoCalc still does not have a 3D molecular editor. MoCalc uses Babel program to convert structures from Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput) formats to generate an input geometry for Mopac calculation.
To import an external structure to a Mopac input document:
1. Make a Mopac input file (*.zmt) the active document; 2. Click on the Mopac/Import External Molecular Geometry menu; 3. In the Import external molecular geometry form, choose the type of the file that
contain the external structure and the coordinate style to be inserted in the Mopac input;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
50

Chapter 7. Working with Mopac MoCalc User’s Guide
4. Click on the Insert default input header option box to insert the stored input header (See 7.3. Setting up the Mopac default input header, on page 49) in the Gamess input document;
5. Click Ok; 6. In the Open form, find the file that contain the external structure;
7. Click Open.
Remarks:
• MoCalc generates only internal coordinates molecular geometry in this procedure; • To enable this menu option, the active document must be a Mopac input file (*.zmt).
7.7. Showing structures from Mopac documents
MoCalc uses program RasMol as molecular viewer. The Mopac documents that have enough information inside to build a computer molecular model are the Mopac input (*.zmt) and output (*.mno) documents.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
51

Chapter 7. Working with Mopac MoCalc User’s Guide
To show a molecule from a Mopac document:
1. Make a Mopac input (*.zmt) or output (*.mno) the active document; 2. Click on the Mopac/Show Molecule menu. Program RasMol will start and the last
structure in the Mopac document will be displayed;
3. Use the RasMol resources to change the features concerning the display mode. See the Rasmol manual by clicking on the Help/Rasmol Manual menu for detailed information (See 11.5. Getting online help for RasMol).
Remarks:
• Once program RasMol is initialized from the MoCalc system, it becomes an independent process as any other program running in the Windows environment and, as so, it can be operated and shot down independently from MoCalc;
• To enable this option, the active document must be a Mopac input (*.zmt) or output (*.mno) file.
7.8. Extracting results from Mopac output documents
In the present version of MoCalc, the results that can be extracted from a Mopac output document are the following: heat of formation, charges, dipole moment, interatomic distances, and the eigenvectors. These results are shown in separated grid forms from which they can be
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
52

Chapter 7. Working with Mopac MoCalc User’s Guide
copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word.
To extract properties from a Mopac output document:
1. Make a Mopac output file (*.mno) the active document; 2. Click on the Mopac/Extract Results menu and choose a property.
Remarks:
• It is only possible to extract properties from Mopac output documents (*.mno) from which the calculation has been finished without errors. This condition is indicated by the message "== MOPAC DONE ==" printed by the Mopac program at the end of the output file;
• Use the Extract Results/All Above menu to extract all properties at once. The extracted properties are placed in separated spreadsheets and can be located in the file tree display, or by pressing the key combination CTRL+TAB several times;
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the entire content of the grid form, including the line and column titles, to the Windows clipboard;
• To enable this option, the active document must be a Mopac output file (*.mno), whose calculation has been finished without errors.
7.9. Calculating properties from Mopac output documents
In the present version of MoCalc, the properties that can be calculated from a Mopac output document are the following: frontier orbital indices and the RMS molecular geometry change. The frontier orbital indices are shown in a grid form from which they can be copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word. The RMS geometry change is presented in a dialog box.
The frontier orbital indices calculated by MoCalc are the following, where all indices refer to the atom N: pi electron density P(N), nucleophilic superdelocalizability SDN(N), electrophilic superdelocalizability SDE(N), radical superdelocalizability SDR(N), frontier electron density FED(N), frontier orbital density FOD(N) e frontier radical density FRD(N).
To calculate these properties from a Mopac output document:
1. Make a Mopac output file (*.mno) the active document; 2. Click on the Mopac/Calculate Properties menu and choose a property.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
53

Chapter 7. Working with Mopac MoCalc User’s Guide
Remarks:
• It is only possible to calculate properties from Mopac output documents (*.mno) from which the calculation has been finished without errors. This condition is indicated by the message "== MOPAC DONE ==" printed by the Mopac program at the end of the output file;
• In case of open shell calculation (Unrestricted Hartree-Fock or UHF), in which two sets of eigenvectors, alpha and beta, are produced, MoCalc will show two sets of frontier orbital indices, one for each set of eigenvectors;
• The frontier orbital indices calculation is performed from the last set of eigenvectors printed in the Mopac output file (*.mno);
• The RMS geometry change calculation is performed from the first and the last molecular geometries printed in the Mopac output file (*.mno);
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the entire content of the grid form, including the line and column titles, to the Windows clipboard;
• To enable this option, the active document must be a Mopac output file (*.mno), whose calculation has been finished without errors.
7.10. Comparing properties generated by Mopac
In the present version of MoCalc, the results that can be compared from two or more Mopac output documents are the following: heat of formation, energy of the highest occupied molecular orbital (HOMO), energy of the lowest unoccupied molecular orbital (LUMO), and dipole moment. The comparison is presented in two ways. A numerical comparison is shown in a grid form from which they can be copied and pasted to spreadsheets, such as Microsoft Excel, and text editors, such as Microsoft Word. A graphical comparison is presented as bar charts, one for each property.
To compare molecular properties from the Mopac results:
1. Make a Mopac output file (*.mno) the active document;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
54

Chapter 7. Working with Mopac MoCalc User’s Guide
2. Click on the Mopac/Compare Molecular Properties menu. A spreadsheet containing a numerical comparison of the properties and a set of graphs, one for each property, are shown.
Remarks:
• When some result to be compared is not found in the Mopac output document (*.mno), the consequence is one or more blank cells in the grid form and one or more absent bars in the bar charts;
• Use the file tree display or the key combination CTRL+TAB to navigate between the comparison results;
• Use the Edit/Copy menu or the key combination CTRL+C to copy selected cells from the grid form and Edit/Copy All or CTRL+A followed by Edit/Copy or CTRL+C to copy the whole content of the grid form, including the line and column titles, to the Windows clipboard;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
55

Chapter 7. Working with Mopac MoCalc User’s Guide
• Use the Edit/Copy menu or the key combination CTRL+C to copy a bar chart comparison to the Windows clipboard;
• To enable this option, two or more Mopac output documents (*.mno) , whose calculation has been finished without errors, must be opened, and the active document must be one of these files.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
56

Chapter 8. Working with Tinker MoCalc User’s Guide
Chapter 8. Working with Tinker
8.1. Computing properties
In the present version of MoCalc, the properties that can be calculated from a Tinker input (*.xyz) or output (*.geo) document are the following: total potential energy and its components, energy breakdown over each of the atoms, list of the large individual interactions, details of the large individual interactions, details of all individual interactions, electrostatic, inertial, and virial properties, and force field parameters for interactions.
To compute properties from a Tinker document:
1. Make a Tinker input (*.xyz) or output (*.geo) the active document; 2. Click on the Tinker/Compute Properties menu; 3. In the Tinker properties dialog, select the computations to be run;
4. Click OK; 5. In the Run Tinker calculation dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to computation;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
57

Chapter 8. Working with Tinker MoCalc User’s Guide
6. Click on the Run button. The computation results is stored in a Tinker summary (*.sum) document.
Remark:
• MoCalc uses the Tinker executable file Analyze.exe to compute properties; • The Show output geometry file (GEO) option box is disable because no output
geometry is generated in this procedure; • Click on the Show summary file (SUM) option box to get this file opened as soon as
the requested calculation has finished; • Click on the Run job in background option box to run the calculation as a MoCalc-
independent process. This option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• To enable this option, the active document must be a Tinker input (*.xyz) file.
8.2. Minimizing the energy
The energy of the structure in a Tinker input (*.xyz) document can be minimized in two ways. In the preliminary minimization, final energy gradients from 1.0 to 0.1 can be achieved. This is useful as a first step of a full minimization procedure, where the starting geometry is rough. In the refined minimization, energy gradients from 0.1 to 0.000001 can be achieved. This type o minimization is used as the final step of a full minimization procedure.
To minimize the energy of a Tinker input (*.xyz) document:
1. Make a Tinker input (*.xyz) the active document; 2. Click on the Tinker/Minimize Energy menu; 3. In the Minimize energy dialog, select the type of minimization, the precondition and the
minimization method, and enter RMS gradient criterion. The Ok button becomes enabled only when the gradient value is an acceptable value;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
58

Chapter 8. Working with Tinker MoCalc User’s Guide
4. Click OK; 5. In the Run Tinker calculation dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to computation;
6. Click on the Show output geometry file (GEO) and Show calculation summary (SUM) option boxes to get these files opened as soon as the requested calculation has finished;
7. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
8. Click on the Run button. The computation results are stored in a Tinker summary (*.sum) document.
Remarks:
• MoCalc uses the Tinker executable file Minimize.exe in the preliminary minimization and Newton.exe in the refined minimization;
• By placing the mouse pointer on the RMS gradient criterion label, a popup tip indicates the acceptable interval for the energy gradient value;
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
59

Chapter 8. Working with Tinker MoCalc User’s Guide
choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• Consult the Tinker manual, through the Help/Tinker Manual menu, to get help about the available options for the minimization procedure;
• To enable this option, the active document must be a Tinker input (*.xyz) file.
8.3. Searching the global minimum energy
The global minimum energy search uses the Monte Carlo method to locate the most stable molecular geometry in a Tinker input (*.xyz) document. The success in finding the global minimum energy depends on the search parameters chosen by the user, which in turn depends on the user’s experience.
To search for the global minimum energy geometry of a Tinker input (*.xyz) document:
1. Make a Tinker input (*.xyz) the active document; 2. Click on the Tinker/Global Minimum Energy Search menu; 3. In the Global minimum energy search dialog, enter the number of Monte Carlo steps,
the maximum step (Å) for the atom displacement, the absolute temperature (K), and the RMS gradient criterion. The Ok button becomes enabled only acceptable values are entered for these parameters;
4. Click OK; 5. In the Run Tinker calculation dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to computation;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
60

Chapter 8. Working with Tinker MoCalc User’s Guide
6. Click on the Show output geometry file (GEO) and Show calculation summary (SUM) option boxes to get these files opened as soon as the requested calculation has finished;
7. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
8. Click on the Run button. The final molecular geometry is stored in a Tinker output (*.geo) document and the details of the calculation procedure are stored in a Tinker summary (*.sum) document.
Remarks:
• MoCalc uses the Tinker executable file Monte.exe in the global minimum energy search procedure;
• By placing the mouse pointer on the parameter labels in the Global minimum energy search dialog, popup tips indicate the acceptable interval for the parameter values;
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• Consult the Tinker manual, through the Help/Tinker Manual menu, to get help about the available options for the global minimum energy search procedure;
• To enable this option, the active document must be a Tinker input (*.xyz) file.
8.4. Running a vibrational analysis
A vibrational analysis is performed by computing and diagonalizing the full Hessian matrix, i.e., the second partial derivatives, for the molecular structure in the Tinker input (*.xyz) document.
To perform the vibrational analysis of a Tinker input (*.xyz) document:
1. Make a Tinker input (*.xyz) the active document; 2. Click on the Tinker/Vibrational Analysis menu; 3. In the Run Tinker calculation dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to computation;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
61

Chapter 8. Working with Tinker MoCalc User’s Guide
4. Click on the Show calculation summary (SUM) option boxes to get this file opened as soon as the requested calculation has finished;
5. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
6. Click on the Run button. The calculation result is stored in a Tinker summary (*.sum) document.
Remarks:
• MoCalc uses the Tinker executable file Vibrate.exe in the vibrational analysis procedure;
• The Show output geometry file (GEO) option box is disable because no output structure is generated in this procedure;
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• Consult the Tinker manual, through the Help/Tinker Manual menu, to get help about the details of the vibrational analysis procedure;
• To enable this option, the active document must be a Tinker input (*.xyz) file.
8.5. Calculating the molecular surface area and volume
Calculate the molecular surface area and volume by using a modified version of Connolly’s original analytical description of the molecular surface. The program determines either the van der Waals, accessible or molecular (contact/reentrant) volume and surface area for the molecular structure in the Tinker input (*.xyz) document.
To perform the surface area and volume of a structure in a Tinker input (*.xyz) document:
1. Make a Tinker input (*.xyz) the active document; 2. Click on the Tinker/Surface Area & Volume Calculation menu; 3. In the Surface area & volume calculation dialog, select the type of calculation, enter
the value for the radius of the probe sphere (only for the Accessible area & excluded
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
62

Chapter 8. Working with Tinker MoCalc User’s Guide
volume and Contact-reentrant area & volume calculation), and decide if the hydrogen atoms are to be included in the calculation. The Ok button becomes enabled only if an acceptable value is entered for the probe radius;
4. Click OK; 5. In the Run Tinker calculation dialog, use the [>], [<], [>>], and [<<] buttons to select
the files to be submitted to computation;
6. Click on the Show calculation summary (SUM) option boxes to get this file opened as soon as the requested calculation has finished;
7. Click on the Run job in background option box to run the calculation as a MoCalc-independent process;
8. Click on the Run button. The calculation result is stored in a Tinker summary (*.sum) document.
Remarks:
• MoCalc uses the Tinker executable file Spacefill.exe in the surface area and volume calculation procedure;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
63

Chapter 8. Working with Tinker MoCalc User’s Guide
• The Show output geometry file (GEO) option box is disable because no output structure is generated in this procedure;
• The Run job in background option is useful when running time consuming jobs, because MoCalc interface remains blocked while running any kind of calculation. By choosing Run job in background, the user will be able to continue to work with MoCalc while the calculation runs quietly in background. To see the calculation results of a background calculation, the output files must be opened through the File/Open menu;
• Consult the Tinker manual, through the Help/Tinker Manual menu, to get help about the details of the surface area and volume calculation procedure;
• To enable this option, the active document must be a Tinker input (*.xyz) file.
8.6. Importing external structures to Tinker input documents
Unfortunately, the present version of MoCalc still does not have a 3D molecular editor. MoCalc uses Babel program to convert structures from Gamess output (*.out), Mopac input (*.zmt), Mopac output (*.mno), Tinker input (*.xyz), Hyperchem (*.hin), Protein Data Bank (*.pdb), Sybyl (*.mol2), and Spartan (*.spinput) formats to generate an input geometry for Tinker calculation.
To import an external structure to a Tinker input document:
1. Make a Tinker input file (*.xyz) the active document; 2. Click on the Tinker/Import External Molecular Geometry menu; 3. In the Import external molecular geometry form, choose the type of the file that
contain the external structure;
4. Click Ok; 5. In the Open form, find the file that contain the external structure;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
64
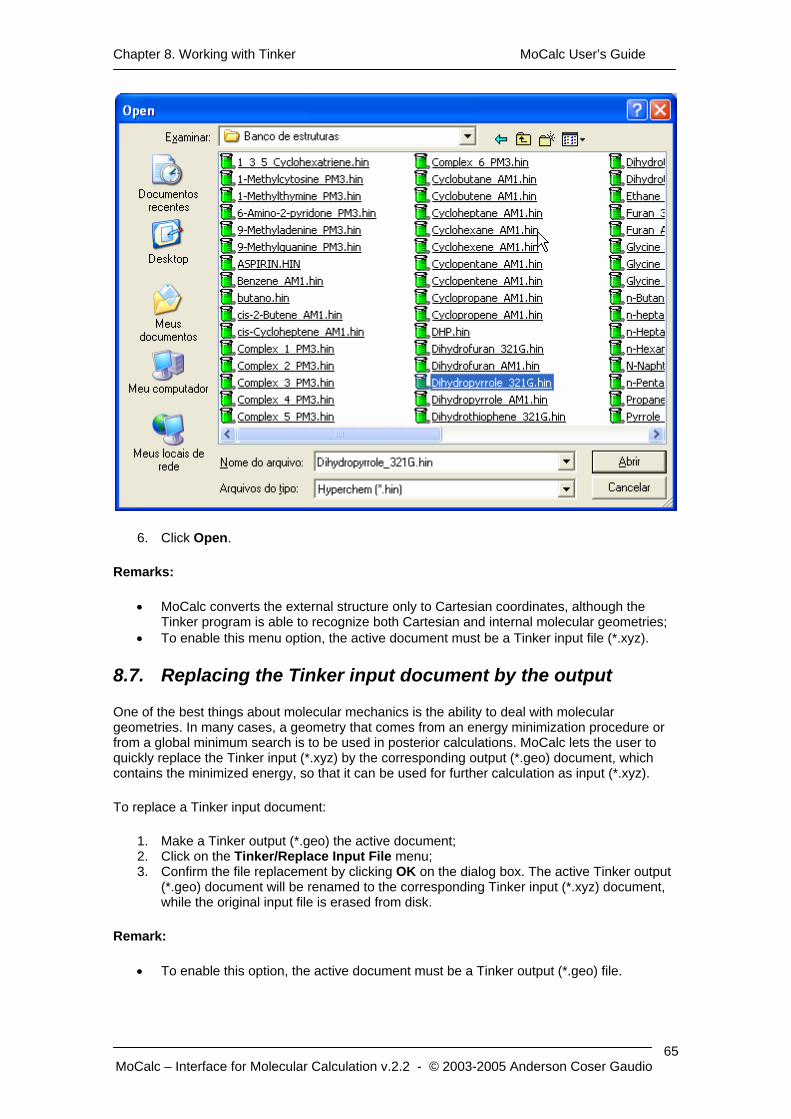
Chapter 8. Working with Tinker MoCalc User’s Guide
6. Click Open.
Remarks:
• MoCalc converts the external structure only to Cartesian coordinates, although the Tinker program is able to recognize both Cartesian and internal molecular geometries;
• To enable this menu option, the active document must be a Tinker input file (*.xyz).
8.7. Replacing the Tinker input document by the output
One of the best things about molecular mechanics is the ability to deal with molecular geometries. In many cases, a geometry that comes from an energy minimization procedure or from a global minimum search is to be used in posterior calculations. MoCalc lets the user to quickly replace the Tinker input (*.xyz) by the corresponding output (*.geo) document, which contains the minimized energy, so that it can be used for further calculation as input (*.xyz).
To replace a Tinker input document:
1. Make a Tinker output (*.geo) the active document; 2. Click on the Tinker/Replace Input File menu; 3. Confirm the file replacement by clicking OK on the dialog box. The active Tinker output
(*.geo) document will be renamed to the corresponding Tinker input (*.xyz) document, while the original input file is erased from disk.
Remark:
• To enable this option, the active document must be a Tinker output (*.geo) file.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
65
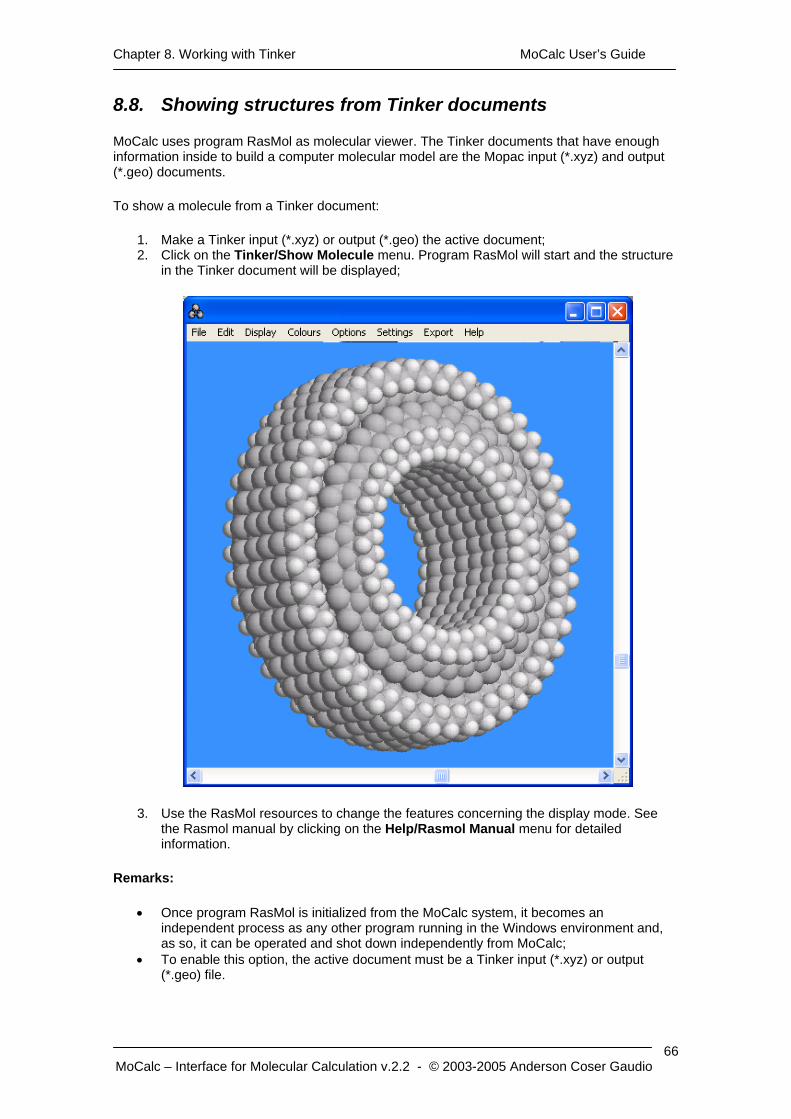
Chapter 8. Working with Tinker MoCalc User’s Guide
8.8. Showing structures from Tinker documents
MoCalc uses program RasMol as molecular viewer. The Tinker documents that have enough information inside to build a computer molecular model are the Mopac input (*.xyz) and output (*.geo) documents.
To show a molecule from a Tinker document:
1. Make a Tinker input (*.xyz) or output (*.geo) the active document; 2. Click on the Tinker/Show Molecule menu. Program RasMol will start and the structure
in the Tinker document will be displayed;
3. Use the RasMol resources to change the features concerning the display mode. See the Rasmol manual by clicking on the Help/Rasmol Manual menu for detailed information.
Remarks:
• Once program RasMol is initialized from the MoCalc system, it becomes an independent process as any other program running in the Windows environment and, as so, it can be operated and shot down independently from MoCalc;
• To enable this option, the active document must be a Tinker input (*.xyz) or output (*.geo) file.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
66

Chapter 9. Configuring MoCalc MoCalc User’s Guide
Chapter 9. Configuring MoCalc
9.1. Setting up fundamental physical constants
MoCalc lets the user to update the values of the fundamental physical constants, eventually used internally. This feature can be useful when new constant measurements, performed with more precision, are published in the literature.
To change the value of the physical constants used by MoCalc:
1. Click on the Tools/Setup Constants menu; 2. In the Setup constants dialog, change one or more physical constant values shown in
the text boxes;
3. Click Ok;
Remarks:
• To recovery the original values of all physical constants, stored in the source code of MoCalc, click on the Reset button;
• The original values of the physical constants were extracted from the homepage of National Institute of Standards and Technology (NIST), Gaithersburg, MD, USA, (http://physics.nist.gov/cuu/), on September 4th, 2003;
• The changes made by the user in the physical constants are stored in the MOCALC.INI file.
9.2. Setting up unit conversion factors
MoCalc lets the user to update the values of the unit conversion factors, eventually used internally. This feature can be useful when updated conversion factors are published in the literature.
To change the unit conversion factors:
1. Click on the Tools/Setup Unit Conversion Factors; 2. In the Setup unit conversion factors dialog, change one or more conversion factors
shown in the text boxes;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
67

Chapter 9. Configuring MoCalc MoCalc User’s Guide
3. Click Ok;
Remarks:
• To recovery the original values of all conversion factors, stored in the source code of MoCalc, click on the Reset button;
• The original values of the unit conversion factors extracted from the homepage of National Institute of Standards and Technology (NIST), Gaithersburg, MD, USA, (http://physics.nist.gov/cuu/), on September 4th, 2003;
• The changes made by the user in the convertion factors are stored in the MOCALC.INI file.
9.3. Setting up warning messages
MoCalc lets the user to decide if some warning messages should be displayed. This feature can be useful to speed up the normal use of MoCalc, especially for experienced users.
To setup some of the MoCalc warning messages:
1. Click on the Tools/Setup Warning Messages menu; 2. In the Setup warning messages dialog, click on the Show or Don't show option
boxes to display the corresponding warning message or not;
3. Click Ok;
Remarks:
• The Replace an existing file warning message occurs when the user clicks on the File/Convert File To menu and the file that will be created in this operation already
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
68

Chapter 9. Configuring MoCalc MoCalc User’s Guide
exists in the same directory. If the user chooses Don't show, the existing file will be automatically replaced by the new one without warning;
• The Save document before displaying molecule warning message occurs when the user clicks on the Display molecule submenu of the Gamess or Mopac menus and the active file has been changed and not saved. If the user chooses Don't show, the active document will be automatically saved;
• The Save document before converting files warning message occurs when the user clicks on the File/Convert File To menu and one or more files that will be converted have been changed and not saved. If the user chooses Don't show, these document will be automatically saved;
• The Close DOS window after running a calculation warning message occurs after running calculations with MoCalc. In some versions of the Windows operating system the DOS window in which the calculation actually takes place does not automatically close after the calculation had finished.
9.4. Setting up paths
MoCalc lets the user to change the path of many files that are used by the MoCalc system. The usefulness of this feature can be exemplified by the case in which the user has two versions of the Gamess executable files, each of which in different paths. In order to use one or the other to perform a given calculation it is only necessary to adjust the path of the Gamess.exe file.
To setup paths:
1. Click on the Tools/Setup Paths; 2. In the Setup paths dialog, select the file whose path is to be changed and click on the
Change button;
3. In the Setup directory dialog box, choose the new path for that file, and click Ok.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
69

Chapter 9. Configuring MoCalc MoCalc User’s Guide
4. Click Ok;
Remarks:
• To recovery the original paths of all files, stored in the source code of MoCalc, click on the Reset button;
• The changes made by the user in the paths are stored in the MOCALC.INI file.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
70
Chapter 10. Arranging windows MoCalc User’s Guide
Chapter 10. Arranging windows
10.1. Arranging MoCalc document windows
To arrange the document windows in the MoCalc system:
1. Click on the Window/Cascade menu to arrange the opened documents in cascade; 2. Click on the Window/Tile Horizontal menu to arrange the opened documents as a grid
in which the number of columns is the smallest possible; 3. Click on the Window/Tile Vertical menu to arrange the opened documents as a grid in
which the number of lines is the smallest possible; 4. Click on the Window/Arrange Icons menu to arrange the icons of the minimized
documents;
Cascade arrangement
Horizontal arrangement
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
71

Chapter 10. Arranging windows MoCalc User’s Guide
Vertical arrangement
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
72

Chapter 10. Arranging windows MoCalc User’s Guide
Icons arrangement
Remarks:
• The menu options Cascade, Tile Horizontal, and Tile Vertical work only when the document windows are not maximized;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
73

Chapter 10. Arranging windows MoCalc User’s Guide
• The menu option Arrange Icons works only when at least one of the opened document window is minimized and the other are not maximized.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
74
Chapter 11. Getting help MoCalc User’s Guide
Chapter 11. Getting help
11.1. Getting online help for MoCalc
MoCalc offers to the user online help files in PDF format.
To get online help for MoCalc:
1. Clique on the Help/Content menu to open the MoCalc online manual;
Remark:
• In order to open the online MoCalc manual, it is necessary to have the Adobe Acrobat Reader® installed in your computer.
11.2. Getting online help for Gamess
To get online help for Gamess:
1. Click on the Help/Gamess Manual menu to open the Gamess online manual;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
75
Chapter 11. Getting help MoCalc User’s Guide
11.3. Getting online help for Mopac
To get online help for Mopac:
1. Click on the Help/Mopac Manual menu to open Mopac online manual;
11.4. Getting online help for Tinker
To get online help for Tinker:
1. Click on the Help/Tinker Manual menu to open Tinker online manual;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
76

Chapter 11. Getting help MoCalc User’s Guide
11.5. Getting online help for RasMol
To get online help for RasMol:
1. Click on the Help/RasMol Help menu to open the RasMol online manual;
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
77

Chapter 11. Getting help MoCalc User’s Guide
11.6. Getting online help for Babel
To get online help for Babel:
1. Click on the Help/Babel Help menu to open the Babel online manual;
11.7. Visiting the MoCalc web site
The MoCalc homepage has all the information about the program, including download, updates, manuals, FAQs, etc. The address is http://www.cce.ufes.br/anderson. The homepage can be easier accessed through the MoCalc Help menu.
To access the MoCalc homepage:
1. Click on the Help/Visit MoCalc Web Site menu. The default Internet browser will point to the author's homepage, whose address is http://www.cce.ufes.br/anderson, where the user will find a proper link to MoCalc.
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
78
Chapter 11. Getting help MoCalc User’s Guide
MoCalc – Interface for Molecular Calculation v.2.2 - © 2003-2005 Anderson Coser Gaudio
79
Related Documents

![Windows 版GAMESS インストールマニュアル...Windows版GAMESSインストールマニュアル 2020/10/28 [64bit Windows の場合] [32bit Windows の場合] [64bit Windows](https://static.cupdf.com/doc/110x72/607a0facf2b70c37922575be/windows-cgamess-ffffffff-windowscgamessffffffff.jpg)









