© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 8 855–863 ‘Mitotic drive’ of expanded CTG repeats in myotonic dystrophy type 1 (DM1) Mehrdad Khajavi 1,2 , Ana M. Tari 3 , Nalini B. Patel 4 , Kuniko Tsuji 1,2 , Doris R. Siwak 3 , Marvin L. Meistrich 4 , Nicholas H. A. Terry 4 and Tetsuo Ashizawa 1,2,+ 1 Department of Neurology, Baylor College of Medicine, Houston, TX 77030, USA, 2 Veterans Affairs Medical Center, Houston, TX 77030, USA, 3 Department of Bioimmunotherapy and 4 Department of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA Received 18 December 2000; Revised and Accepted 19 February 2001 In myotonic dystrophy type 1 (DM1), an expanded CTG repeat shows repeat size instability in somatic and germ line tissues with a strong bias toward further expansion. To investigate the mechanism of this expansion bias, 29 DM1 and six normal lympho- blastoid cell lines (LBCLs) were single-cell cloned from blood cells of 18 DM1 patients and six normal subjects. In all 29 cell lines, the expanded CTG repeat alleles gradually shifted toward further expansion by ‘step-wise’ mutations. Of these 29 cell lines, eight yielded a rapidly proliferating mutant with a gain of large repeat size that became the major allele popula- tion, eventually replacing the progenitor allele population. By mixing cell lines with different repeat expansions, we found that cells with larger CTG repeat expansion had a growth advantage over those with smaller expansions in culture. This growth advantage was attributable to increased cell prolifera- tion mediated by Erk1,2 activation, which is negatively regulated by p21 WAF1 . This phenomenon, which we designated ‘mitotic drive’, is a novel mechanism which can explain the expansion bias of DM1 CTG repeat instability at the tissue level, on a basis independent of the DNA-based expansion models. The lifespans of the DM1 LBCLs were significantly shorter than normal cell lines. Thus, we propose a hypothesis that DM1 LBCLs drive themselves to extinction through a process related to increased proliferation. INTRODUCTION Myotonic dystrophy type 1 (DM1, OMIM 160900) is a progressive multisystemic autosomal dominant disorder which shows a phenomenon known as anticipation (1). Anticipation denotes progressively earlier onset of the disease with increasing severity in successive generations. The mutation of DM1 is an unstable CTG repeat expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase (DMPK) gene on chromosome 19q13.3 (2–4). The repeat size of the expanded allele inversely correlates with the age of disease onset and becomes progressively larger in successive genera- tions in DM1 families, providing a molecular basis of anticipa- tion (1,5). Expanded CTG repeat alleles also show a high level of somatic instability, which is evidenced by a smear on Southern and PCR analyses (6,7). Using small pool PCR (SP-PCR), the smear can be resolved into individual alleles with hetero- geneous repeat sizes (8). The somatic instability is also evident as variable repeat sizes among different tissues (9). The expanded CTG repeat in peripheral blood leukocytes (PBLs) is unstable throughout the life of a patient with DM1, with gradual increases in the average repeat size and the repeat size heterogeneity with age (7). Based on these observations, Monckton et al. (8) have proposed a model for the somatic instability of the DM1 CTG repeat. In this model, somatic instability proceeds through a directional pathway which involves ‘step-wise’ gains of a small number of repeat units; thus, as the subject ages, the allele distribution gradually shifts towards expansion with increasing repeat size heterogeneity. Thus, somatic instability of the DM1 CTG repeat involves a dynamic process in which the repeat size increases with age at different rates in various tissues and, therefore, should play important roles in tissue- and age-specific phenotypic varia- bility. To further characterize the somatic instability, we have developed a cell culture system using clonal lymphoblastoid cell lines (LBCLs) prepared from DM1 patients (10). Each clone was derived from a single cell; therefore, any alleles with repeat sizes different from the progenitor allele can be detected as mutant alleles. SP-PCR analyses of these mutant alleles showed two types of mutations in short-term culture: a frequent ‘step-wise’ gain or loss of a small number of repeat units and a ‘gross’ change of repeat size that occurs infre- quently with a bias toward contraction (10). In this study, we demonstrate expansion dynamics of these mutations in long- term cultures, and present evidence for a novel mechanism which can explain the expansion bias observed in DM1 PBLs, based on preferential proliferation of cells with larger CTG repeats. + To whom correspondence should be addressed at: Department of Neurology, SM1801, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA; Tel: +1 713 798 3953; Fax: +1 713 798 3128; Email: [email protected]

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 8 855–863
‘Mitotic drive’ of expanded CTG repeats in myotonicdystrophy type 1 (DM1)Mehrdad Khajavi1,2, Ana M. Tari3, Nalini B. Patel4, Kuniko Tsuji1,2, Doris R. Siwak3,Marvin L. Meistrich4, Nicholas H. A. Terry4 and Tetsuo Ashizawa1,2,+
1Department of Neurology, Baylor College of Medicine, Houston, TX 77030, USA, 2Veterans Affairs Medical Center,Houston, TX 77030, USA, 3Department of Bioimmunotherapy and 4Department of Experimental Radiation Oncology,The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Received 18 December 2000; Revised and Accepted 19 February 2001
In myotonic dystrophy type 1 (DM1), an expandedCTG repeat shows repeat size instability in somaticand germ line tissues with a strong bias towardfurther expansion. To investigate the mechanism ofthis expansion bias, 29 DM1 and six normal lympho-blastoid cell lines (LBCLs) were single-cell clonedfrom blood cells of 18 DM1 patients and six normalsubjects. In all 29 cell lines, the expanded CTG repeatalleles gradually shifted toward further expansion by‘step-wise’ mutations. Of these 29 cell lines, eightyielded a rapidly proliferating mutant with a gain oflarge repeat size that became the major allele popula-tion, eventually replacing the progenitor allelepopulation. By mixing cell lines with different repeatexpansions, we found that cells with larger CTGrepeat expansion had a growth advantage over thosewith smaller expansions in culture. This growthadvantage was attributable to increased cell prolifera-tion mediated by Erk1,2 activation, which is negativelyregulated by p21WAF1. This phenomenon, which wedesignated ‘mitotic drive’, is a novel mechanismwhich can explain the expansion bias of DM1 CTGrepeat instability at the tissue level, on a basisindependent of the DNA-based expansion models.The lifespans of the DM1 LBCLs were significantlyshorter than normal cell lines. Thus, we propose ahypothesis that DM1 LBCLs drive themselves toextinction through a process related to increasedproliferation.
INTRODUCTION
Myotonic dystrophy type 1 (DM1, OMIM 160900) is aprogressive multisystemic autosomal dominant disorder whichshows a phenomenon known as anticipation (1). Anticipationdenotes progressively earlier onset of the disease withincreasing severity in successive generations. The mutation ofDM1 is an unstable CTG repeat expansion in the 3′ untranslated
region of the myotonic dystrophy protein kinase (DMPK) geneon chromosome 19q13.3 (2–4). The repeat size of theexpanded allele inversely correlates with the age of diseaseonset and becomes progressively larger in successive genera-tions in DM1 families, providing a molecular basis of anticipa-tion (1,5). Expanded CTG repeat alleles also show a high level ofsomatic instability, which is evidenced by a smear on Southernand PCR analyses (6,7). Using small pool PCR (SP-PCR), thesmear can be resolved into individual alleles with hetero-geneous repeat sizes (8). The somatic instability is also evidentas variable repeat sizes among different tissues (9).
The expanded CTG repeat in peripheral blood leukocytes(PBLs) is unstable throughout the life of a patient with DM1,with gradual increases in the average repeat size and the repeatsize heterogeneity with age (7). Based on these observations,Monckton et al. (8) have proposed a model for the somaticinstability of the DM1 CTG repeat. In this model, somaticinstability proceeds through a directional pathway whichinvolves ‘step-wise’ gains of a small number of repeat units;thus, as the subject ages, the allele distribution gradually shiftstowards expansion with increasing repeat size heterogeneity.Thus, somatic instability of the DM1 CTG repeat involves adynamic process in which the repeat size increases with age atdifferent rates in various tissues and, therefore, should playimportant roles in tissue- and age-specific phenotypic varia-bility. To further characterize the somatic instability, we havedeveloped a cell culture system using clonal lymphoblastoidcell lines (LBCLs) prepared from DM1 patients (10). Eachclone was derived from a single cell; therefore, any alleles withrepeat sizes different from the progenitor allele can be detectedas mutant alleles. SP-PCR analyses of these mutant allelesshowed two types of mutations in short-term culture: afrequent ‘step-wise’ gain or loss of a small number of repeatunits and a ‘gross’ change of repeat size that occurs infre-quently with a bias toward contraction (10). In this study, wedemonstrate expansion dynamics of these mutations in long-term cultures, and present evidence for a novel mechanismwhich can explain the expansion bias observed in DM1 PBLs,based on preferential proliferation of cells with larger CTGrepeats.
+To whom correspondence should be addressed at: Department of Neurology, SM1801, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030,USA; Tel: +1 713 798 3953; Fax: +1 713 798 3128; Email: [email protected]

856 Human Molecular Genetics, 2001, Vol. 10, No. 8
RESULTS
Step-wise mutations of expanded CTG repeats towardfurther expansion in DM1 LBCLs
To investigate the instability of CTG repeat size, we estab-lished 22 clonal LBCLs obtained from 18 DM1 patients thatshowed variable sizes of the expanded CTG repeat (Table 1).Additionally, six lines (0313-1, 0313-2, 0313-7, 0313-11,0313-15 and 0313-20) and two lines (1008-1-1 and 1008-1-3)were subcloned from a single cell of parental clonal lines, 0313and 1008-1 (Table 1). Since clonal cells were derived from asingle cell, any alleles that deviate from the progenitor allelemust have resulted from repeat size mutations in culture. Theclones were allowed to grow until the cell number reached 3 × 107,and 1 × 107 cells were passed; this process was repeated as longas the cells stayed viable. Normal alleles of DM1 and normalLBCLs were stable throughout the passages. In contrast,expanded CTG repeat alleles in all 29 DM1 LBCLs showedtwo types of mutations similar to those observed in ourprevious short-term culture experiments (10); frequently-seen‘step-wise’ mutations that result in gains and losses of a smallnumber of repeat units around the progenitor allele, andrelatively rare ‘gross’ mutations that involve large repeat sizechanges (Fig. 1). When we compared two consecutivepassages using the SP-PCR technique (8), changes in themodal size of the expanded alleles by the step-wise mutationswere hardly appreciable, although subtle size heterogeneity ofthese alleles was evident around the progenitor allele. Afterseveral passages, however, we were able to detect a gradualincrease of the modal allele size in all cell lines, indicating an
expansion bias of the step-wise mutation (Fig. 1) similar to thein vivo model based on the data in PBLs of DM1 patients (Fig. 2A)(8); however, repeat size heterogeneity did not dramaticallyincrease over time in most of these LBCLs, giving rise to the‘synchronized’ expansion (Figs 1 and 2B) which was previouslydescribed in cultured dura mater cells and myoblasts derivedfrom DM1 patients (11).
Gross mutations of expanded CTG repeats toward furtherexpansion in DM1 LBCLs
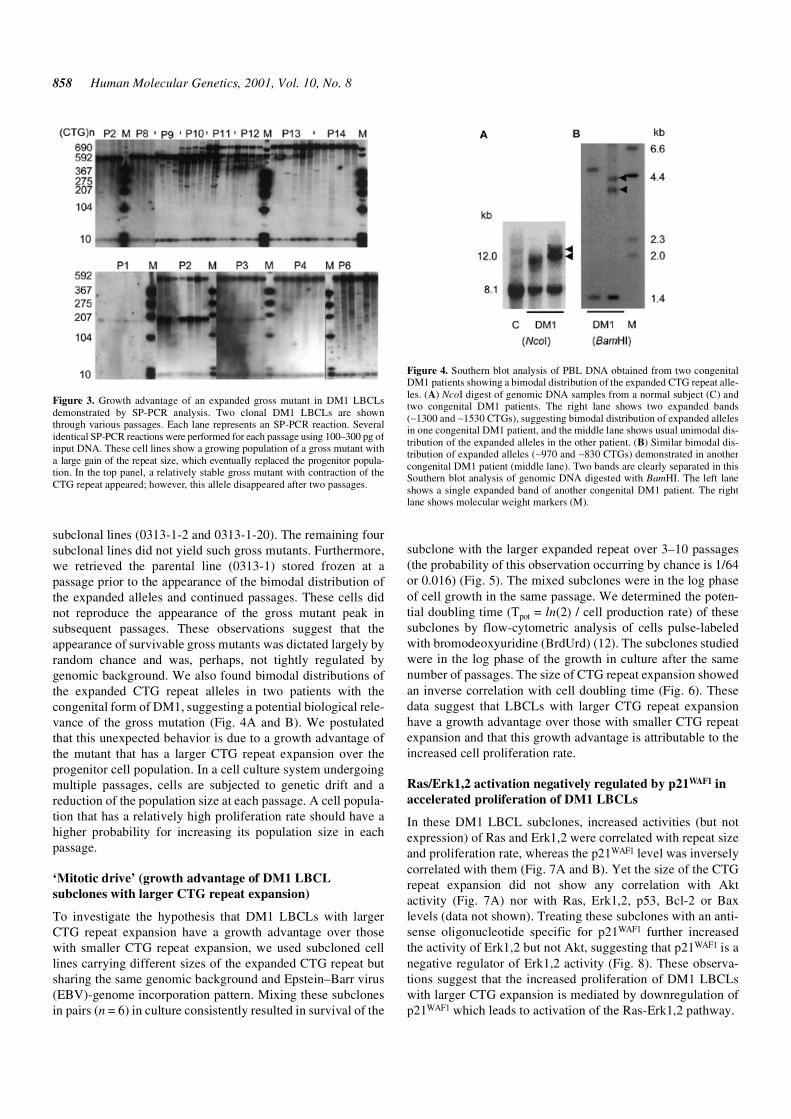
The gross mutants showed unexpected characteristics. Themajority of gross mutants showed contraction of the expandedCTG repeats as we previously reported (10). The grossmutants, which include those with repeat contraction as well asthose relatively rare ones with further expansion, mostlydisappeared in the following passages (Fig. 1). However, arapidly proliferating mutant with a gain of large repeat size (by40–290 repeats) appeared occasionally and became the majorallele population that eventually replaced the progenitor allelepopulation (Fig. 3). This phenomenon was observed in eight ofthe 29 clonal LBCLs (Table 1). Thus, out of eight grossmutants that replaced the progenitor allele population, eightresulted from expansion mutations (the probability of thisobservation occurring by chance is 1/256 or 0.004), whereaswe did not observe contracted gross mutants surviving morethan a few passages. These eight included two of the six
Figure 1. Gradual increases of the expanded CTG repeat size in a DM1 LBCLclone demonstrated by SP-PCR analysis. Each lane represents an SP-PCRreaction. Five identical SP-PCR reactions were performed for each passageusing ∼300 pg of input DNA. The normal allele (N) was stable through pas-sages and the expanded allele (E) showed a slightly increased width of theband. The increased width was dissolved into individual alleles of slightly dif-ferent repeat sizes using ∼6 pg of input DNA (data not shown) due to step-wisemutations as previously demonstrated (10). There were additional grossmutants (arrowheads), mainly with contraction of the expanded repeat. Notethat the main population of expanded alleles gradually shifted toward furtherexpansion through passages 2–8, whereas the gross mutants failed to survivethe passages. The degree of allele size heterogeneity around the modal alleleshowed little change throughout the passages, giving the appearance of ‘syn-chronized’ step-wise expansion.
Figure 2. Schematic models of the DM1 CTG repeat expansion. (A) CTGrepeat instability based on the data obtained in PBLs of DM1 patients in vivo.The size of expanded CTG repeat alleles gradually shifts toward further expan-sion with increasing heterogeneity of the repeat size (8). (B) The step-wisemutations of expanded CTG repeats observed in DM1 LBCLs. The pattern issimilar to (A), but the repeat size heterogeneity does not increase as the peakshifts toward further expansion, giving rise to a ‘synchronized’ pattern ofexpansion. (C) The gross mutation with an expansion bias.
Human Molecular Genetics, 2001, Vol. 10, No. 8 857
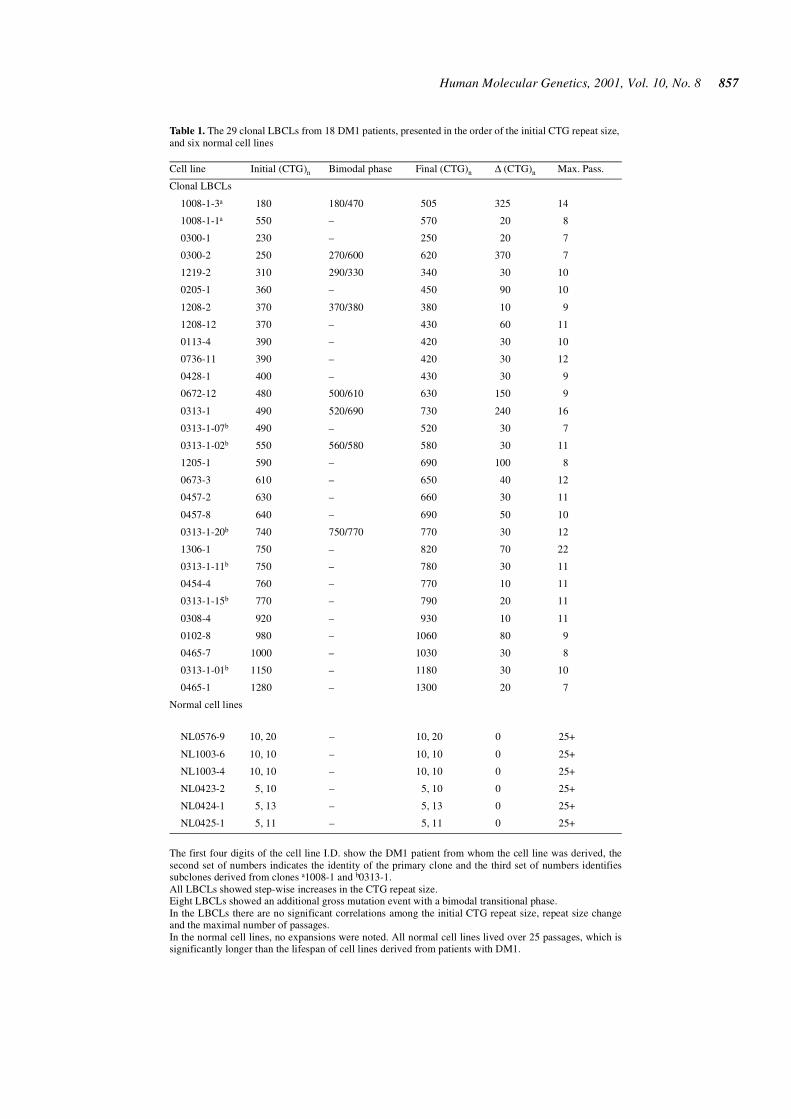
Table 1. The 29 clonal LBCLs from 18 DM1 patients, presented in the order of the initial CTG repeat size,and six normal cell lines
The first four digits of the cell line I.D. show the DM1 patient from whom the cell line was derived, thesecond set of numbers indicates the identity of the primary clone and the third set of numbers identifiessubclones derived from clones a1008-1 and b0313-1.All LBCLs showed step-wise increases in the CTG repeat size.Eight LBCLs showed an additional gross mutation event with a bimodal transitional phase.In the LBCLs there are no significant correlations among the initial CTG repeat size, repeat size changeand the maximal number of passages.In the normal cell lines, no expansions were noted. All normal cell lines lived over 25 passages, which issignificantly longer than the lifespan of cell lines derived from patients with DM1.
Cell line Initial (CTG)n Bimodal phase Final (CTG)n ∆ (CTG)n Max. Pass.
Clonal LBCLs
1008-1-3a 180 180/470 505 325 14
1008-1-1a 550 – 570 20 8
0300-1 230 – 250 20 7
0300-2 250 270/600 620 370 7
1219-2 310 290/330 340 30 10
0205-1 360 – 450 90 10
1208-2 370 370/380 380 10 9
1208-12 370 – 430 60 11
0113-4 390 – 420 30 10
0736-11 390 – 420 30 12
0428-1 400 – 430 30 9
0672-12 480 500/610 630 150 9
0313-1 490 520/690 730 240 16
0313-1-07b 490 – 520 30 7
0313-1-02b 550 560/580 580 30 11
1205-1 590 – 690 100 8
0673-3 610 – 650 40 12
0457-2 630 – 660 30 11
0457-8 640 – 690 50 10
0313-1-20b 740 750/770 770 30 12
1306-1 750 – 820 70 22
0313-1-11b 750 – 780 30 11
0454-4 760 – 770 10 11
0313-1-15b 770 – 790 20 11
0308-4 920 – 930 10 11
0102-8 980 – 1060 80 9
0465-7 1000 – 1030 30 8
0313-1-01b 1150 – 1180 30 10
0465-1 1280 – 1300 20 7
Normal cell lines
NL0576-9 10, 20 – 10, 20 0 25+
NL1003-6 10, 10 – 10, 10 0 25+
NL1003-4 10, 10 – 10, 10 0 25+
NL0423-2 5, 10 – 5, 10 0 25+
NL0424-1 5, 13 – 5, 13 0 25+
NL0425-1 5, 11 – 5, 11 0 25+
858 Human Molecular Genetics, 2001, Vol. 10, No. 8
subclonal lines (0313-1-2 and 0313-1-20). The remaining foursubclonal lines did not yield such gross mutants. Furthermore,we retrieved the parental line (0313-1) stored frozen at apassage prior to the appearance of the bimodal distribution ofthe expanded alleles and continued passages. These cells didnot reproduce the appearance of the gross mutant peak insubsequent passages. These observations suggest that theappearance of survivable gross mutants was dictated largely byrandom chance and was, perhaps, not tightly regulated bygenomic background. We also found bimodal distributions ofthe expanded CTG repeat alleles in two patients with thecongenital form of DM1, suggesting a potential biological rele-vance of the gross mutation (Fig. 4A and B). We postulatedthat this unexpected behavior is due to a growth advantage ofthe mutant that has a larger CTG repeat expansion over theprogenitor cell population. In a cell culture system undergoingmultiple passages, cells are subjected to genetic drift and areduction of the population size at each passage. A cell popula-tion that has a relatively high proliferation rate should have ahigher probability for increasing its population size in eachpassage.
‘Mitotic drive’ (growth advantage of DM1 LBCLsubclones with larger CTG repeat expansion)
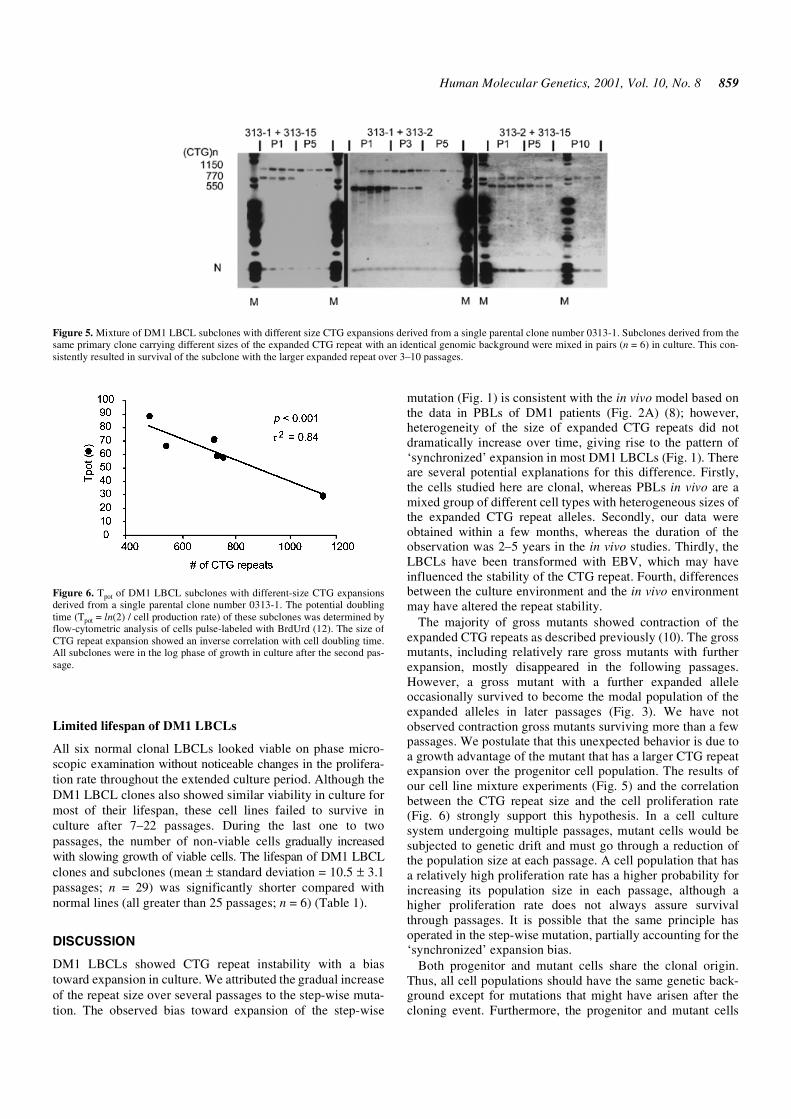
To investigate the hypothesis that DM1 LBCLs with largerCTG repeat expansion have a growth advantage over thosewith smaller CTG repeat expansion, we used subcloned celllines carrying different sizes of the expanded CTG repeat butsharing the same genomic background and Epstein–Barr virus(EBV)-genome incorporation pattern. Mixing these subclonesin pairs (n = 6) in culture consistently resulted in survival of the
subclone with the larger expanded repeat over 3–10 passages(the probability of this observation occurring by chance is 1/64or 0.016) (Fig. 5). The mixed subclones were in the log phaseof cell growth in the same passage. We determined the poten-tial doubling time (Tpot = ln(2) / cell production rate) of thesesubclones by flow-cytometric analysis of cells pulse-labeledwith bromodeoxyuridine (BrdUrd) (12). The subclones studiedwere in the log phase of the growth in culture after the samenumber of passages. The size of CTG repeat expansion showedan inverse correlation with cell doubling time (Fig. 6). Thesedata suggest that LBCLs with larger CTG repeat expansionhave a growth advantage over those with smaller CTG repeatexpansion and that this growth advantage is attributable to theincreased cell proliferation rate.
Ras/Erk1,2 activation negatively regulated by p21WAF1 inaccelerated proliferation of DM1 LBCLs
In these DM1 LBCL subclones, increased activities (but notexpression) of Ras and Erk1,2 were correlated with repeat sizeand proliferation rate, whereas the p21WAF1 level was inverselycorrelated with them (Fig. 7A and B). Yet the size of the CTGrepeat expansion did not show any correlation with Aktactivity (Fig. 7A) nor with Ras, Erk1,2, p53, Bcl-2 or Baxlevels (data not shown). Treating these subclones with an anti-sense oligonucleotide specific for p21WAF1 further increasedthe activity of Erk1,2 but not Akt, suggesting that p21WAF1 is anegative regulator of Erk1,2 activity (Fig. 8). These observa-tions suggest that the increased proliferation of DM1 LBCLswith larger CTG expansion is mediated by downregulation ofp21WAF1 which leads to activation of the Ras-Erk1,2 pathway.
Figure 3. Growth advantage of an expanded gross mutant in DM1 LBCLsdemonstrated by SP-PCR analysis. Two clonal DM1 LBCLs are shownthrough various passages. Each lane represents an SP-PCR reaction. Severalidentical SP-PCR reactions were performed for each passage using 100–300 pg ofinput DNA. These cell lines show a growing population of a gross mutant witha large gain of the repeat size, which eventually replaced the progenitor popula-tion. In the top panel, a relatively stable gross mutant with contraction of theCTG repeat appeared; however, this allele disappeared after two passages.
Figure 4. Southern blot analysis of PBL DNA obtained from two congenitalDM1 patients showing a bimodal distribution of the expanded CTG repeat alle-les. (A) NcoI digest of genomic DNA samples from a normal subject (C) andtwo congenital DM1 patients. The right lane shows two expanded bands(∼1300 and ∼1530 CTGs), suggesting bimodal distribution of expanded allelesin one congenital DM1 patient, and the middle lane shows usual unimodal dis-tribution of the expanded alleles in the other patient. (B) Similar bimodal dis-tribution of expanded alleles (∼970 and ∼830 CTGs) demonstrated in anothercongenital DM1 patient (middle lane). Two bands are clearly separated in thisSouthern blot analysis of genomic DNA digested with BamHI. The left laneshows a single expanded band of another congenital DM1 patient. The rightlane shows molecular weight markers (M).
Human Molecular Genetics, 2001, Vol. 10, No. 8 859
Limited lifespan of DM1 LBCLs
All six normal clonal LBCLs looked viable on phase micro-scopic examination without noticeable changes in the prolifera-tion rate throughout the extended culture period. Although theDM1 LBCL clones also showed similar viability in culture formost of their lifespan, these cell lines failed to survive inculture after 7–22 passages. During the last one to twopassages, the number of non-viable cells gradually increasedwith slowing growth of viable cells. The lifespan of DM1 LBCLclones and subclones (mean ± standard deviation = 10.5 ± 3.1passages; n = 29) was significantly shorter compared withnormal lines (all greater than 25 passages; n = 6) (Table 1).
DISCUSSION
DM1 LBCLs showed CTG repeat instability with a biastoward expansion in culture. We attributed the gradual increaseof the repeat size over several passages to the step-wise muta-tion. The observed bias toward expansion of the step-wise
mutation (Fig. 1) is consistent with the in vivo model based onthe data in PBLs of DM1 patients (Fig. 2A) (8); however,heterogeneity of the size of expanded CTG repeats did notdramatically increase over time, giving rise to the pattern of‘synchronized’ expansion in most DM1 LBCLs (Fig. 1). Thereare several potential explanations for this difference. Firstly,the cells studied here are clonal, whereas PBLs in vivo are amixed group of different cell types with heterogeneous sizes ofthe expanded CTG repeat alleles. Secondly, our data wereobtained within a few months, whereas the duration of theobservation was 2–5 years in the in vivo studies. Thirdly, theLBCLs have been transformed with EBV, which may haveinfluenced the stability of the CTG repeat. Fourth, differencesbetween the culture environment and the in vivo environmentmay have altered the repeat stability.
The majority of gross mutants showed contraction of theexpanded CTG repeats as described previously (10). The grossmutants, including relatively rare gross mutants with furtherexpansion, mostly disappeared in the following passages.However, a gross mutant with a further expanded alleleoccasionally survived to become the modal population of theexpanded alleles in later passages (Fig. 3). We have notobserved contraction gross mutants surviving more than a fewpassages. We postulate that this unexpected behavior is due toa growth advantage of the mutant that has a larger CTG repeatexpansion over the progenitor cell population. The results ofour cell line mixture experiments (Fig. 5) and the correlationbetween the CTG repeat size and the cell proliferation rate(Fig. 6) strongly support this hypothesis. In a cell culturesystem undergoing multiple passages, mutant cells would besubjected to genetic drift and must go through a reduction ofthe population size at each passage. A cell population that hasa relatively high proliferation rate has a higher probability forincreasing its population size in each passage, although ahigher proliferation rate does not always assure survivalthrough passages. It is possible that the same principle hasoperated in the step-wise mutation, partially accounting for the‘synchronized’ expansion bias.
Both progenitor and mutant cells share the clonal origin.Thus, all cell populations should have the same genetic back-ground except for mutations that might have arisen after thecloning event. Furthermore, the progenitor and mutant cells
Figure 5. Mixture of DM1 LBCL subclones with different size CTG expansions derived from a single parental clone number 0313-1. Subclones derived from thesame primary clone carrying different sizes of the expanded CTG repeat with an identical genomic background were mixed in pairs (n = 6) in culture. This con-sistently resulted in survival of the subclone with the larger expanded repeat over 3–10 passages.
Figure 6. Tpot of DM1 LBCL subclones with different-size CTG expansionsderived from a single parental clone number 0313-1. The potential doublingtime (Tpot = ln(2) / cell production rate) of these subclones was determined byflow-cytometric analysis of cells pulse-labeled with BrdUrd (12). The size ofCTG repeat expansion showed an inverse correlation with cell doubling time.All subclones were in the log phase of growth in culture after the second pas-sage.

860 Human Molecular Genetics, 2001, Vol. 10, No. 8
were grown in the same culture flask; therefore, environmentalfactors were identical. Thus, the putative growth advantageobserved in some gross mutants may be attributable to either oftwo scenarios: (i) a new mutation(s) that arose somewhere inthe genome after cloning, or (ii) the CTG repeat expansionitself. The experimental system we used does not allow us todirectly determine which of these two possibilities is correct.However, if scenario (i) were the case, there would be twopossibilities. One is that such mutations occur preferentially incells with the repeat size larger than that of the progenitor, andthis would suggest that the CTG repeat expansion regulates the
mutability of other genes. The other is that an increased cellcycling by a mutation elsewhere confers an increased chanceof repeat expansion mutations, implying that faster cell replica-tion promotes expansion mutations. The latter explanation iscompatible with models for an expansion bias of the CTGrepeat instability based on unusual structures formed by therepeat involving the Okazaki fragment at the replication fork(13,14).
If the larger expansion of the repeat promotes cell prolifera-tion [scenario (ii)], it may have implications for the disease-causing mechanism in DM1. Delayed differentiation has beendocumented in muscle and brain tissues of congenital DM1patients (15,16) and in myocyte culture models of DM1(17,18). It is tempting to speculate the mechanism of theseobservations in the context of cell proliferation/differentiationcoupling, especially the p21WAF1-cyclin-dependent kinases(Cdks)-retinoblastoma tumor suppressor (Rb) network. Down-regulation of p21WAF1 increases cell proliferation, preventsdifferentiation (19–21) and increases apoptosis (22). Indeed,we demonstrated that the size of CTG repeat expansion showsinverse correlations with cell doubling time and the level ofp21WAF1 and a positive correlation with Erk1,2 activities (Fig. 7A),yet the size of the CTG repeat expansion shows correlationwith neither Akt activity nor Erk1,2, p53, Bcl-2 or Bax levelsin DM1 LBCL subclones. Furthermore, treating the DM1LBCLs with antisense oligonucleotide against p21WAF1 upreg-ulated the activation of Erk1,2 (Fig. 8). These observationsmight suggest that the expansion of the CTG repeat activatesErk1,2 by downregulating the p21WAF1 level, leading toincreased cell proliferation. If mutations elsewhere were acti-vating cell cycling, it would be difficult to explain the correla-tion between the expansion size and specific changes of Erk1,2and p21WAF1, since such mutations are expected to involve vari-able pathways to activate cell cycling. Furthermore, our prelim-
Figure 7. Analysis of Ras and Erk1,2 activity and p21WAF1 expression in DM1 LBCL subclones. (A) Increased activities of Erk1,2 and decreased expression ofp21WAF1 were found in subclones with a larger CTG repeat. Erk1,2 activities were detected by antibodies specific for the dual phosphorylated Erk1,2 (P-Erk1,2)(35). Levels of Akt activity (P-Akt, shown), and expressions of Erk1,2, p53, Bcl-2, Bax and Akt (data not shown) did not significantly differ in these subclones.(B) Activated Ras in cell lysates were co-precipitated with the GST–RBD fusion protein which is specific for activated Ras. The levels of activated Ras were thenanalyzed by western blot (36). Ras activity is increased in DM1 LBCL subclones with larger CTG repeats. Western blot analysis of lysates of the same subclonesdid not show significant differences, suggesting that Ras expression was not altered. NL, normal CTG repeat size; LBCL 1 and LBCL 2, normal control LBCLs;ALL-1, acute lymphocytic leukemia cell line 1.
Figure 8. Treatment of DM1 LBCL subclones with an antisense oligonucleotidespecific for p21WAF1 led to an increase in Erk1,2 activity (P-Erk1,2). Note theantisense oligonucleotide did not alter Akt activity (P-Akt) or the levels ofErk1,2 and Grb2, indicating the specific effect of the antisense oligonucleotideon Erk1,2 activity. A decrease of ∼40% of the p21WAF1 level was observed aftertreatment with the antisense oligonucleotide. These data suggest that p21WAF1
is a negative regulator of Erk1,2 activity in DM1 LBCL subclones.

Human Molecular Genetics, 2001, Vol. 10, No. 8 861
inary study suggested that apoptosis is increased in DM1LBCLs compared with normal LBCLs (data not shown). If thisis confirmed, p21WAF1 upregulation might induce apoptosis by amechanism independent of Bcl-2, Bax and Akt, leading to theshortened lifespan of DM1 LBCLs demonstrated in this study.Further studies are warranted to determine the mechanisminvolved in the correlations between CTG repeat length, cellcycle signal transduction and apoptosis.
An important question is how relevant our observations inLBCLs are to the situations of DM1 patients in vivo. The grossmutations have not been reported in PBLs of DM1 patients.This may be attributable to the artificial environment of ourculture system and the transformation of the cells, which wehave already discussed earlier in this paper. However, largeintergenerational contractions of the expanded CTG repeatallele to the normal range have been reported (23–25) andmutations with large contractions toward the normal rangehave been frequently observed in sperm of DM1 patients (8).Mosaicism consisting of two expanded CTG repeat alleles hasbeen reported in the brain of DM1 patients (26). Furthermore,we have recently encountered two patients with the congenitalform of DM1 who showed a bimodal distribution of theexpanded alleles resembling the bimodal transition phase ofthe gross mutation seen in our LBCLs (Fig. 4A and B). Othershave also found similar cases in transgenic mice (27,28).Although some of these observations could also be explainedby early embryonic repeat size mutations (29) or intratissueheterogeneity of cell populations, we hypothesize that bothfrequent step-wise mutations and rare gross mutations docontribute to the in vivo instability of the expanded CTG repeatin patients with DM1. Further investigations of the bimodalalleles of our patients by examining other tissues and follow-up blood samples would be of interest. Since LBCLs are trans-formed cells derived from an apparently unaffected tissue(i.e. blood) of DM1 patients, we are currently extending ourinvestigations to primary cultures of DM1 muscle cells.
Finally, our data suggest that the association of faster cellproliferation and larger CTG repeat expansion can contributeto the bias of CTG repeat instability toward further expansion;an expansion bias is simply generated by preferential growthof cells with larger alleles over the smaller ones. We havedesignated this phenomenon ‘mitotic drive’ because it resemblesthe meiotic drive (segregation distortion) characterized bypreferential transmissions of a larger CTG repeat allele fromparent to offspring reported in DM1 (30); in either situation,larger CTG repeats are preferentially passed on to the nextgeneration of cells or individuals. Although there have beenconflicting data on meiotic drive in DM1 (30,31), meioticdrive is well documented as a biological phenomenon in otherspecies (32–34). ‘Mitotic drive’ may be unique to the DM1CTG repeat and may not be found with other expandedtrinucleotide repeats. Conventional explanations for the expan-sion bias of the CTG repeat have been based on modelsinvolving unusual DNA structure within the Okazaki fragmentthat consists of a CTG repeat tract (13,14). The ‘mitotic drive’is a mechanism of expansion bias that is independent of thesemodels, although they are not mutually exclusive. In the post-mitotic cells, such as muscle cells, the ‘mitotic drive’ wouldonly be relevant to their stem cell population and their progeniesthat are capable of cell divisions. However, DM1 musclesshow increased proliferation of myogenic stem cells (i.e. satellite
cells) to compensate for muscle loss (16). Thus, ‘mitotic drive’may be an important mechanism of repeat instability in varioustissues of DM1 patients in vivo.
MATERIALS AND METHODS
Patients
Under a consent procedure approved by the local IRB, bloodsamples were drawn from patients who had both the clinicaldiagnosis of DM1 and expanded DM1 CTG repeats, and fromsix normal control subjects (Table 1).
Cloning and passages of LBCLs
PBLs were isolated from blood samples obtained from the 18DM1 patients using the Ficoll–Hypaque gradient and trans-formed into LBCL using EBV. The LBCLs were cultured inRPMI-1640 containing 10% heat-inactivated fetal bovineserum and antimycotics (Gibco BRL) in 5% CO2 at 37°C in a75 cm2 flask (10). Using a hemocytometer, each cell line wassingle-cell cloned by limiting dilutions at the concentration of0.5 cells per well of a 96-well plate. The single cell origin ofeach cell line was assured as previously described (10). Eachclone was transferred to a 25 cm2 flask once the cell numberreached ∼1 × 106 cells. Approximately 300 pg of DNAextracted from each clone was analyzed by PCR to confirm thehomogeneity of the expanded CTG repeat allele. Cell linesshowing two or more major alleles (i.e. multimodal allele sizedistribution) at this stage were excluded from the study. Cloneswere then transferred to 75 cm2 flasks and allowed to growuntil the total cell number reached 3 × 107 cells. Viability ofcells was assessed by tripan blue exclusion. An aliquot washarvested for DNA analyses and another aliquot of 1 × 107
cells was passed on. The remaining cells were frozen in theculture medium containing 10% DMSO. This passage step wasrepeated as long as the cells were viable.
SP-PCR analysis
The genomic DNA was extracted using Wizard Genomic DNAPurification Kit (Promega) at each passage. In typical experi-ments, after amplifiable genome equivalence (a.g.e.) of inputDNA was determined by SP-PCR using 3–6 pg of DNA, 20–200a.g.e. (∼60–600 pg) of DNA was analyzed by SP-PCR usingprimers flanking the DM1 CTG repeat, DMA and DMBR,under PCR conditions previously described (8). The SP-PCRproducts were subjected to 1.8% agarose gel electrophoresis.After Southern blotting to a nylon membrane (Hybond N;Gibco BRL), the products were hybridized with a 32P-γ-ATPendlabeled (CAG)10 oligonucleotide probe in 5× SSPE with0.5% SDS at 42°C overnight.
Southern blot analysis
Southern blot analysis was performed as described elsewhere(1,2).
Mixture experiment
Subclones derived from the same primary clone carryingdifferent sizes of the expanded CTG repeat with an identicalgenomic background were mixed in pairs (n = 6) in culture. An

862 Human Molecular Genetics, 2001, Vol. 10, No. 8
aliquot of 5 × 106 of each subclone was mixed and passageswere conducted as described earlier.
Flow cytometry
To determine the potential doubling time (Tpot = ln(2) / cellproduction rate), lymphoblastoid cells were pulse labeled by1 µMBrdUrd for 20 min followed by a chase period of 6 h. Cellswere fixed in a final concentration of 2 × 106 cells per 2 ml ofsolution in 65% ethanol in phosphate-buffered saline at 4°Covernight, and then stained with anti-BrdUrd monoclonal anti-body in PBS with 0.5% Tween-20 at 1:100 dilution andincubated for 60 min at room temperature in the dark. UsingGoat antimouse-fluorescein isothiocyanate (FITC) as a secondantibody, the flow cytometry was performed to measure bivariatedistributions of BrdUrd content versus DNA content. Thelength of S phase (Ts) and Tpot of the cell population was calcu-lated by a method previously described (12).
Western blot and Ras/Raf-GST co-precipitation analyses
Protein lysates (35 µg) were loaded onto 12% polyacrylamidegel followed by transfer to a nylon membrane. Levels ofErk1,2, p53, Bcl-2, Bax and Akt were analyzed using anti-bodies against the respective proteins and Erk1,2 and Aktactivities were detected by phospho-specific antibodies (35).Phospho-Erk1,2 antibodies (Thr202/Tyr204) and phospho-Aktantibodies (Ser473) were purchased from New EnglandBiolabs. Activated Ras was co-precipitated from 50 µg ofprotein lysates with Raf-GST fusion protein and analyzed bywestern blot as described by Warne et al. (36).
Antisense oligonucleotide experiments
p21WAF1 antisense oligonucleotides were made of nuclease-resistant P-ethoxy oligos (Oligos Etc). The sequence of thep21WAF1 antisense is 5′-GCCGCATGGGTTCTGACG-3′.Oligos were incorporated into liposomes as described by Tari(37). Cells were incubated with liposomal p21WAF1 antisenseoligos for 3 days.
ACKNOWLEDGEMENTS
We are grateful to Dr J. Downward (Imperial CancerResearch Fund) for providing us with Escherichia coliexpressing GST-RBD. This study was supported by VeteransAffair Merit Review (T.A.), the Hunter Research Fund (T.A.),the Elsa Pardee Foundation grant (A.M.T.), NIH/NCI PO1grant CA-06294 (N.H.A.T.), NIH/NCI CA78973-02 (M.L.M.)and a donation from John and Marge Vasku (T.A.). K.T.’sfellowship was supported by Osaka Medical Research Founda-tion for Incurable Diseases.
REFERENCES
1. Ashizawa, T., Dubel, J.R., Dunne, P.W., Dunne, C.J., Fu, Y.H., Pizzuti,A., Caskey, C.T., Boerwinkle, E., Perryman, M.B., Epstein, H.F. andHejtmancik, J.F. (1992) Anticipation in myotonic dystrophy. II. Complexrelationships between clinical findings and structure of the GCT repeat.Neurology, 42, 1877–1883.
2. Fu, Y.H., Pizzuti, A., Fenwick, R.G., Jr, King, J., Rajnarayan, S., Dunne,P.W., Dubel, J., Nasser, G.A., Ashizawa, T., de Jong, P. et al. (1992) Anunstable triplet repeat in a gene related to myotonic muscular dystrophy.Science, 255, 1256–1258.
3. Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C.,Jansen, G., Neville, C., Narang, M., Barcelo, J., O’Hoy, K. et al. (1992)Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untrans-lated region of the gene. Science, 255, 1253–1255.
4. Brook, J.D., McCurrach, M.E., Harley, H.G., Buckler, A.J., Church, D.,Aburatani, H., Hunter, K., Stanton, V.P., Thirion, J.P., Hudson, T. et al.(1992) Molecular basis of myotonic dystrophy: expansion of a trinucle-otide (CTG) repeat at the 3′ end of a transcript encoding a protein kinasefamily member. Cell, 68, 799–808.
5. Lavedan, C., Hofmann-Radvanyi, H., Shelbourne, P., Rabes, J.P., Duros,C., Savoy, D., Dehaupas, I., Luce, S., Johnson, K. and Junien, C. (1993)Myotonic dystrophy: size- and sex-dependent dynamics of CTG meioticinstability and somatic mosaicism. Am. J. Hum. Genet., 52, 875–883.
6. Buxton, J., Shelbourne, P., Davies, J., Jones, C., Van Tongeren, T.,Aslanidis, C., de Jong, P, Jansen, G., Anvret, M., Riley, B. et al. (1992)Detection of an unstable fragment of DNA specific to individuals withmyotonic dystrophy. Nature, 355, 547–548.
7. Wong, L.J.C., Ashizawa, T., Monckton, D.G., Caskey, C.T. and Richards,C.S. (1995) Somatic heterogeneity of the CTG repeat in myotonic dystro-phy is age and size dependent. Am. J. Hum. Genet., 56, 114–122.
8. Monckton, D.G., Wong, L.J.C., Ashizawa, T. and Caskey, C.T. (1995)Somatic mosaicism, germline expansions, germline reversions and inter-generational reductions in myotonic dystrophy males: small pool PCRanalyses. Hum. Mol. Genet., 4, 1–8.
9. Ashizawa, T., Dubel, J.R. and Harati, Y. (1993) Somatic instability ofCTG repeat in myotonic dystrophy. Neurology, 43, 2674–2678.
10. Ashizawa, T., Monckton, D.G., Vaishnav, S., Patel, B.J., Voskova, A. andCaskey, C.T. (1996) Instability of the expanded (CTG)n repeats in themyotonin protein kinase gene in cultured lymphoblastoid cell lines frompatients with myotonic dystrophy. Genomics, 36, 47–53.
11. Wöhrle, D., Kennerknecht, I., Wolf, M., Enders, H., Schwemmle, S. andSteinbach, P. (1995) Heterogeneity of DM kinase repeat expansion in dif-ferent fetal tissues and further during cell proliferation in vitro: evidencefor a causal involvement of methyl-directed DNA mismatch repair in tri-plet repeat stability. Hum. Mol. Genet., 4, 1147–1153.
12. Terry, N.H., White, R.A., Meistrich, M.L. and Calkins, D.P. (1991) Eval-uation of flow cytometric methods for determining population potentialdoubling times using cultured cells. Cytometry, 12, 234–241.
13. Richards, R.I. and Sutherland, G.R. (1994) Simple repeat DNA is not rep-licated simply. Nature Genet., 6, 114–116.
14. Freudenreich, C.H., Kantrow, S.M. and Zakian, V.A. (1998) Expansionand length-dependent fragility of CTG repeats in yeast. Science, 279, 853–856.
15. Harper, P.S. (1989) Myotonic Dystrophy, 2nd edn. Saunders, London.16. Iannaccone, S.T., Bove, K.E., Vogler, C., Azzarelli, B. and Muller, J.
(1986) Muscle maturation delay in infantile myotonic dystrophy. Arch.Pathol. Lab. Med., 110, 405–411.
17. Sabourin, L.A., Tamai, K., Narang, M.A. and Korneluk, R.G. (1997)Overexpression of 3′-untranslated region of the myotonic dystrophykinase cDNA inhibits myoblast differentiation in vitro. J. Biol. Chem.,272, 29626–29635.
18. Amack, J.D., Paguio, A.P. and Mahadevan, M.S. (1999) Cis and transeffects of the myotonic dystrophy (DM) mutation in a cell culture model.Hum. Mol. Genet., 8, 1975–1984.
19. Halevy, O., Novitch, B.G., Spicer, D.B., Skapek, S.X., Rhee, J., Hannon,G.J., Beach, D. and Lassar, A.B. (1995) Correlation of terminal cell cyclearrest of skeletal muscle with induction of p21 by MyoD. Science, 267,1018–1021.
20. Skapek, S.X., Rhee, J., Spicer, D.B. and Lassar, A.B. (1995) Inhibition ofmyogenic differentiation in proliferating myoblasts by cyclin D1-depend-ent kinase. Science, 267, 1022–1024.
21. Parker, S.B., Eichele, G., Zhang, P., Rawls, A., Sands, A.T., Bradley, A.,Olson, E.N., Harper, J.W. and Elledge, S.J. (1995) p53-independentexpression of p21Cip1 in muscle and other terminally differentiating cells.Science, 267, 1024–1027.
22. Wang, J. and Walsh, K. (1996) Resistance to apoptosis conferred by Cdkinhibitors during myocyte differentiation. Science, 273, 359–361.
23. Brunner, H.G., Jansen, G., Nillesen, W., Nelen, M.R., de Die, C.E.,Howeler, C.J., van Oost, B.A., Wieringa, B., Ropers, H.H. and Smeets,H.J. (1993) Brief report: reverse mutation in myotonic dystrophy. N. Engl.J. Med., 328, 476–480.
24. O’Hoy, K.L., Tsilfidis, C., Mahadevan, M.S., Neville, C.E., Barcelo, J.,Hunter, A.G. and Korneluk, R.G. (1993) Reduction in size of the

Human Molecular Genetics, 2001, Vol. 10, No. 8 863
myotonic dystrophy trinucleotide repeat mutation during transmission.Science, 259, 809–812.
25. Shelbourne, P., Winqvist, R., Kunert, E., Davies, J., Leisti, J., Thiele, H.,Bachmann, H., Buxton, J., Williamson, B. and Johnson, K. (1992) Unsta-ble DNA may be responsible for the incomplete penetrance of the myot-onic dystrophy phenotype. Hum. Mol. Genet., 1, 467–473.
26. Jansen, G., Willems, P., Coerwinkel, M., Nillesen, W., Smeets, H., Vits,L., Howeler, C., Brunner, H. and Wieringa, B. (1994) Gonosomal mosai-cism in myotonic dystrophy patients: involvement of mitotic events in(CTG)n repeat variation and selection against extreme expansion in sperm.Am. J. Hum. Genet., 54, 575–585.
27. Fortune, M.T., Vassilopoulos, C., Coolbaugh, M.I., Siciliano, M.J. andMonckton, D.G. (2000) Dramatic, expansion-biased, age-dependent, tis-sue-specific somatic mosaicism in a transgenic mouse model of tripletrepeat instability. Hum. Mol. Genet., 9, 439–445.
28. Seznec, H., Lia-Baldini, A.S., Duros, C., Fouquet, C., Lacroix, C.,Hofmann-Radvanyi, H., Junien, C. and Gourdon, G. (2000) Transgenicmice carrying large human genomic sequences with expanded CTG repeatmimic closely the DM CTG repeat intergenerational and somatic instabil-ity. Hum. Mol. Genet., 9, 1185–1194.
29. Gibbs, M., Collick, A., Kelly, R.G. and Jeffreys, A.J. (1993) A tetranucleotiderepeat mouse minisatellite displaying substantial somatic instabilityduring early preimplantation development. Genomics, 17, 121–128.
30. Carey, N., Johnson, K., Nokelainen, P., Peltonen, L., Savontaus, M.L.,Juvonen, V., Anvret, M., Grandell, U., Chotai, K., Robertson, E. et al.(1994) Meiotic drive at the myotonic dystrophy locus? Nature Genet., 6,117–118.
31. Chakraborty, R., Stivers, D.N., Deka, R., Yu, L.M., Shriver, M.D. andFerrell, R.E. (1996) Segregation distortion of the CTG repeats at the myo-tonic dystrophy locus. Am. J. Hum. Genet. 59, 109–118.
32. Merrill, C., Bayraktaroglu, L., Kusano, A. and Ganetzky, B. (1999) Trun-cated RanGAP encoded by the Segregation Distorter locus of Drosophila.Science, 283, 1742–1745.
33. Schimenti, J. (2000) Segregation distortion of mouse t haplotypes themolecular basis emerges. Trends Genet. 16, 240–243.
34. Dawe, R.K. and Cande, W.Z. (1996) Induction of centromeric activity inmaize by suppressor of meiotic drive 1. Proc. Natl Acad. Sci. USA, 93,8512–8517.
35. Tari, A.M., Hung, M.C., Li, K. and Lopez-Berestein, G. (1999) Growthinhibition of breast cancer cells by Grb2 downregulation is correlated withinactivation of mitogen-activated protein kinase in EGFR, but not inErbB2, cells. Oncogene, 18, 1325–1332.
36. Warne, P.H., Viciana, P.R. and Downward, J. (1993) Direct interaction ofRas and the amino-terminal region of Raf-1 in vitro. Nature, 364, 352–355.
37. Tari, A.M. (2000) Preparation and application of liposome-incorporatedoligodeoxynucleotides. Methods Enzymol., 313, 372–388.

864 Human Molecular Genetics, 2001, Vol. 10, No. 8
Related Documents











