Mitochondrial genome data support the basal position of Acoelomorpha and the polyphyly of the Platyhelminthes In ˜ aki Ruiz-Trillo a , Marta Riutort a , H. Matthew Fourcade b , Jaume Bagun ˜a ` a , Jeffrey L. Boore b,c, * a Departament de Gene `tica, Universitat de Barcelona, Av. Diagonal, 645, 08028 Barcelona, Spain b Evolutionary Genomics, DOE Joint Genome Institute and Lawrence Berkeley National Laboratory, Walnut Creek, CA, USA c Department of Integrative Biology, University of California, Berkeley, CA, USA Received 5 November 2003; revised 2 March 2004 Available online 29 July 2004 Abstract We determined 9.7, 5.2, and 6.8 kb, respectively, of the mitochondrial genomes of the acoel Paratomella rubra, the nemertoder- matid Nemertoderma westbladi, and the free-living rhabditophoran platyhelminth Microstomum lineare. The identified gene arrange- ments are unique among metazoans, including each other, sharing no more than one or two single gene boundaries with a few distantly related taxa. Phylogenetic analysis of the amino acid sequences inferred from the sequenced genes confirms that the aco- elomorph flatworms (acoels + nemertodermatids) do not belong to the Platyhelminthes, but are, instead, the most basal extant bi- laterian group. Therefore, the Platyhelminthes, as traditionally constituted, is a polyphyletic phylum. Ó 2004 Elsevier Inc. All rights reserved. Keywords: Platyhelminth; Acoel; Mitochondria; Evolution; Genome; Metazoa 1. Introduction The Acoelomorpha, i.e., Acoela plus Nemertoder- matida, is a group of acoelomate worms that has tradi- tionally been included in the phylum Platyhelminthes. Recent studies of small subunit ribosomal RNA gene (SSU) sequences, however, showed the Platyhelminthes is a polyphyletic assemblage, with the acoelomorphs representing the most basal extant bilaterian clade, and the bulk of the Platyhelminthes (i.e., the Catenulida and the Rhabditophora, which includes all parasitic classes) branching within the Protostomia (Jondelius et al., 2002; Ruiz-Trillo et al., 1999). The basal position of the Acoelomorpha and the consequent polyphyly of the Platyhelminthes have been further tested and corroborated using other mo- lecular characters. First, the Hox clusters of rhabdi- tophoran platyhelminths are now known to have an almost full set (seven or eight) of Hox genes (Bayascas et al., 1998; Orii et al., 1999), some bearing signature peptides indicative of lophotrochozoan and proto- stome affinities (De Rosa et al., 1999). By contrast, re- cent work shows acoels to have a limited set of Hox genes (four or five), which do not bear such signature peptides (Cook et al., 2004). Secondly, these same evo- lutionary relationships have been found in compari- sons of myosin heavy chain type II (myosin II) gene sequences from a large set of metazoans (both in iso- lation and when combined with SSU sequences; Ruiz-Trillo et al., 2002) and in a combined analysis of SSU + LSU gene sequences (Telford et al., 2003). Fi- nally, like all diploblasts examined so far, acoels do not express the heterochronic gene let-7, an essential regulator of developmental timing in all studied bilate- 1055-7903/$ - see front matter Ó 2004 Elsevier Inc. All rights reserved. doi:10.1016/j.ympev.2004.06.002 * Corresponding author. Fax : 1-925-296-5666. E-mail address: [email protected] (J.L. Boore). Molecular Phylogenetics and Evolution 33 (2004) 321–332 MOLECULAR PHYLOGENETICS AND EVOLUTION www.elsevier.com/locate/ympev

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MOLECULAR
Molecular Phylogenetics and Evolution 33 (2004) 321–332
PHYLOGENETICSANDEVOLUTION
www.elsevier.com/locate/ympev
Mitochondrial genome data support the basal positionof Acoelomorpha and the polyphyly of the Platyhelminthes
Inaki Ruiz-Trilloa, Marta Riutorta, H. Matthew Fourcadeb,Jaume Bagunaa, Jeffrey L. Booreb,c,*
a Departament de Genetica, Universitat de Barcelona, Av. Diagonal, 645, 08028 Barcelona, Spainb Evolutionary Genomics, DOE Joint Genome Institute and Lawrence Berkeley National Laboratory, Walnut Creek, CA, USA
c Department of Integrative Biology, University of California, Berkeley, CA, USA
Received 5 November 2003; revised 2 March 2004
Available online 29 July 2004
Abstract
We determined 9.7, 5.2, and 6.8kb, respectively, of the mitochondrial genomes of the acoel Paratomella rubra, the nemertoder-
matid Nemertoderma westbladi, and the free-living rhabditophoran platyhelminthMicrostomum lineare. The identified gene arrange-
ments are unique among metazoans, including each other, sharing no more than one or two single gene boundaries with a few
distantly related taxa. Phylogenetic analysis of the amino acid sequences inferred from the sequenced genes confirms that the aco-
elomorph flatworms (acoels+nemertodermatids) do not belong to the Platyhelminthes, but are, instead, the most basal extant bi-
laterian group. Therefore, the Platyhelminthes, as traditionally constituted, is a polyphyletic phylum.
� 2004 Elsevier Inc. All rights reserved.
Keywords: Platyhelminth; Acoel; Mitochondria; Evolution; Genome; Metazoa
1. Introduction
The Acoelomorpha, i.e., Acoela plus Nemertoder-matida, is a group of acoelomate worms that has tradi-
tionally been included in the phylum Platyhelminthes.
Recent studies of small subunit ribosomal RNA gene
(SSU) sequences, however, showed the Platyhelminthes
is a polyphyletic assemblage, with the acoelomorphs
representing the most basal extant bilaterian clade,
and the bulk of the Platyhelminthes (i.e., the Catenulida
and the Rhabditophora, which includes all parasiticclasses) branching within the Protostomia (Jondelius
et al., 2002; Ruiz-Trillo et al., 1999).
The basal position of the Acoelomorpha and the
consequent polyphyly of the Platyhelminthes have
1055-7903/$ - see front matter � 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.ympev.2004.06.002
* Corresponding author. Fax : 1-925-296-5666.
E-mail address: [email protected] (J.L. Boore).
been further tested and corroborated using other mo-
lecular characters. First, the Hox clusters of rhabdi-
tophoran platyhelminths are now known to have analmost full set (seven or eight) of Hox genes (Bayascas
et al., 1998; Orii et al., 1999), some bearing signature
peptides indicative of lophotrochozoan and proto-
stome affinities (De Rosa et al., 1999). By contrast, re-
cent work shows acoels to have a limited set of Hox
genes (four or five), which do not bear such signature
peptides (Cook et al., 2004). Secondly, these same evo-
lutionary relationships have been found in compari-sons of myosin heavy chain type II (myosin II) gene
sequences from a large set of metazoans (both in iso-
lation and when combined with SSU sequences;
Ruiz-Trillo et al., 2002) and in a combined analysis
of SSU+LSU gene sequences (Telford et al., 2003). Fi-
nally, like all diploblasts examined so far, acoels do
not express the heterochronic gene let-7, an essential
regulator of developmental timing in all studied bilate-

322 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
rians including rhabditophoran Platyhelminthes (Pas-
quinelli et al., 2003).
It remains uncertain whether the Acoelomorpha itself
is a monophyletic group. Molecular data support either
a monophyletic (myosin II; Ruiz-Trillo et al., 2002) or a
paraphyletic (SSU and SSU+LSU; Jondelius et al.,2002; Telford et al., 2003) Acoelomorpha. The mono-
phyly of the Acoelomorpha is supported by some puta-
tive morphological synapomorphies, such as the specific
structure of its basal body–rootlet system of cilia and of
their ciliary tips, and fine structure of frontal organs
(Smith et al., 1986), the lack of a true brain with neuro-
pile (Raikova et al., 1998; Reuter et al., 1998), reduction
of ECM in the body wall (Rieger et al., 1991; Tyler andRieger, 1999) and, potentially, if shown for the nemerto-
dermatids, the duet-spiral type of embryonic cell cleav-
age (Henry et al., 2000).
Furthermore, although all molecular data clearly
show that the bulk of the Platyhelminthes is associated
with the Lophotrochozoa (Adoutte et al., 2000; Car-
ranza et al., 1997; von Nickisch-Rosenegk et al., 2001;
Ruiz-Trillo et al., 1999, 2002), whether they are derivedor basal lophotrochozoans is still uncertain, since none
of the genes used to infer phylogenetic relationships
has provided resolution at this level. The flatworm body
plan figures prominently in nearly all imagined scenarios
for the evolution of the Metazoa, so solving these issues
addresses questions not only in systematics, but in com-
parative development and in understanding the so-called
Cambrian explosion.Comparing complete mitochondrial genome se-
quences (including the comparison of gene order) can
be very powerful at addressing phylogenetic relation-
ships (Boore and Brown, 1998). Now, over 50 complete
mitochondrial genome sequences from invertebrate an-
imals are available (Boore, 1999; from Evolutionary
Genomics link at http://www.jgi.doe.gov). Nearly all
animal mitochondrial DNAs (mtDNAs) are circularmolecules, about 16kb in size, containing genes for
two ribosomal RNAs, 22 tRNAs, and 13 protein sub-
units. Among triploblast animals, the only gene known
to have been lost is atp8; this has occurred indepen-
dently in several lineages: nematodes (Keddie et al.,
1998; Okimoto et al., 1992), parasitic platyhelminths
(Le et al., 2002; von Nickisch-Rosenegk et al., 2001),
and the bivalve mollusk Mytilus edulis (Hoffmannet al., 1992).
In addition, the study of mitochondrial genomes also
provides additional information useful for phylogenetic
inference, the nucleotide and inferred amino acid se-
quences of genes. The comparison of the concatenated
nucleotide or amino acid sequences of mitochondrial
genes has been used successfully in some studies to solve
deep level relationships (Arnason and Janke, 2002; Bo-ore and Brown, 2000; Boore and Staton, 2002; Inoue
et al., 2003; Miya et al., 2003; Murata et al., 2003). How-
ever, this data set has not been well tested for its power
of resolution at the most basal bilaterian level.
To date, complete or near-complete mtDNA se-
quences are available for 11 species of Platyhelminthes,
all belonging to the parasitic classes (Le et al., 2002; von
Nickisch-Rosenegk et al., 2001). No data are presentlyavailable for any of the free-living Rhabditophora (sev-
en orders), nor for any catenulid or acoelomorph. The
characterization of mitochondrial genomes from free-
living Rhabditophora species and from the basal Acoel-
omorpha is necessary to both elucidate the primitive
traits for the mitochondrial genomes of metazoans, as
well as to assess phylogenetic relationships.
In this study, we describe the partial mitochondrialgenomes from two acoelomorphs, the acoel Paratomella
rubra and the nemertodermatid Nemertoderma westbla-
di, and from a free-living rhabditophoran flatworm,
the macrostomid Microstomum lineare (Order Macro-
stomida), the first representatives of these groups to be
examined. We analyze genomic features in comparison
with mtDNAs of a variety of metazoans and perform
a phylogenetic analysis with their inferred amino acid se-quences. Published mtDNA sequences from cnidarians
(Beagley et al., 1998; Beaton et al., 1998) were used as
outgroups. Our aims are to: (1) further test the phyloge-
netic position of acoels and nemertodermatids being
separated from the rest of the Platyhelminthes, (2) vali-
date the monophyly of the Acoelomorpha, (3) assess the
similarities and differences between free-living and para-
sitic rhabditophoran Platyhelminthes, and (4) test thephylogenetic value of mitochondrial sequences for deep
evolutionary relationships.
2. Materials and methods
2.1. Molecular analysis
Live specimens of P. rubra were obtained from Sitges
(Spain). Specimens of M. lineare and N. westbladi were
kindly provided by Dr. M. Reuter (Abo, Finland) and
Dr. Ulf Jondelius (Uppsala, Sweden). We isolated total
DNA using the Qiagen DNA extraction kit. Initially, we
used universal primers to amplify short fragments of the
genes cox1 (primers LCO1490 and HCO2198; Folmer
et al., 1994), cox3 (primers COIIIF and COIIIB; Booreand Brown, 2000), cob (primers Cytb424 and Cytb876;
Boore and Brown, 2000), and rrnL (primers 16ARL
and 16SBRH; Palumbi, 1996). We used a standard
PCR protocol (50ll, with 1U Dynazyme polymerase
of Finnzimes, 35 cycles of 20s at 94 �C, 45s at 48 �C,and 45s at 72 �C).
The products of those PCRs which amplified bands
of expected size (all in P. rubra, cox1, and rrnL in N.
westbladi, cox1, cob, and rrnL in M. lineare) were puri-
fied using Microcon PCR columns. Purified products

I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332 323
were cycle-sequenced directly from both strands using
BigDye chemistry, precipitated using the DyeEx Spin
Kit (Qiagen) column, and run on ABI Prism 373 or
377 automated sequencers. Contigs were assembled us-
ing SeqEd v1.03. DNA sequences obtained from these
fragments were used to design species-specific oligonu-cleotides that face outwards from the fragments. These
primers were then employed in long-PCR (Barnes,
1994) in all possible combinations with the Advantage
polymerase kit (Clontech). Reaction conditions were
as follows: 35 cycles of 8s at 94 �C and 15min at
70 �C, with 1U Advantage polymerase per 50llreaction.
This generated single fragments of approximately 3.7,6, and 9.7kb (from cox1 to rrnL, rrnL to cob, and cox1
to cob, respectively) in P. rubra; 5.3kb (from cox1 to
rrnL) in N. westbladi; and 3.1 and 3.7kb (from cox1 to
rrnL and from rrnL to cob, respectively) in M. lineare.
Multiple attempts at amplifying the other portions of
these mtDNAs while varying many parameters for the
reactions all failed.
Products were purified and cloned using TOPO XLPCR Cloning kit (Invitrogen). Cloned fragments were
purified by a miniprep extraction (High Pure Plasmid
Isolation Kit, Roche), and sequenced as above, with ad-
ditional primers used to walk through both strands of
each fragment. Sequences were assembled using the Seq-
man II program (DNASTAR).
2.2. Gene annotation
The protein and ribosomal RNA-encoding genes
were identified by comparisons with other pub-
lished sequences using BLAST programs at NCBI
(www.ncbi.nlm.nih.gov/BLAST; Zhang and Madden,
1997). tRNA genes were identified either by using
tRNAscan-SE (version 1.1, www.genetics.wustl.edu/
eddy/tRNAscan-SE; Lowe and Eddy, 1997) or, wheretRNAs were not found using this program, by recogniz-
ing potential secondary structures by eye. The 50 ends of
protein genes were inferred to be at the first legitimate,
in-frame start codon (ATN, GTG, TTG, and GTT),
even if this appeared to overlap by a few nucleotides
with the preceding gene. Protein gene termini were in-
ferred to be at the first in-frame stop codon unless that
codon was located within the sequence of a downstreamgene. Otherwise, a truncated stop codon (T or TA) ad-
jacent to the beginning of the next gene was designated
as the termination codon and was assumed to be com-
pleted by polyadenylation after transcript cleavage (Oj-
ala et al., 1981). The 50 and 30 ends of both rrnL and
rrnS genes were assumed to be adjacent to the ends of
bordering tRNA genes.
The nucleotide sequences reported in this article havebeen deposited in GenBank under Accession Nos.
AY228756, AY228757, and AY228758.
2.3. Alignment
Either the general invertebrate (for P. rubra and N.
westbladi) or the flatworm (M. lineare) mitochondrial
genetic codes were used to infer the amino acid sequence
of the protein-encoding genes. Amino acid and nucleo-tide sequences were aligned first by clustalX as imple-
mented in BioEdit 5.0.6 (http://www.mbio.ncsu.edu/
BioEdit/bioedit.html), and then revised by eye using
the GDE2.0 sequence editor (Smith et al., 1994). The ge-
netic code was tested, by eye, at positions that are clearly
conserved through all protein-encoding genes.
The inferred amino acid sequences of the three taxa
were imported into an aligned matrix that includes 42taxa representing a wide range of published metazoan
mitochondrial genomes (see Table 1 for species and
GenBank accession numbers). Ambiguously aligned po-
sitions and gaps were excluded from the analyses result-
ing in a total of 1383 aligned amino acid positions
(including 1069, 713, and 454 amino acid characters
for P. rubra, M. lineare, and N. westbladi, respectively).
2.4. Phylogenetic analyses
A relative rate test on all taxa was performed in
RRTree (Robinson-Rechavi and Huchon, 2000) in
which each taxon was considered to be a separate line-
age. For phylogenetic inference, three different data sets
were analyzed: (1) all taxa; (2) without N. westbladi; and
(3) without the taxa that did not pass the RRT. Thesedata sets were subjected to maximum likelihood (ML)
and neighbor-joining (NJ) analyses. ML analyses were
performed with TREE-PUZZLE 5.0 (Strimmer and
von Haeseler, 1996) using the mtREV24 model (Adachi
and Hasegawa, 1996), the gamma distribution (eight
categories) and 10,000 quartet puzzling (QP) replicates.
NJ analyses were performed in Mega 2.1 (Kumar
et al., 2001) using the gamma model, with the parameteralpha as previously calculated in TREE-PUZZLE and
1000 bootstrap replicates.
3. Results
3.1. Gene content and organization
The 9795nt portion of P. rubra contains a large non-
coding region and 21 genes: nine for proteins (with cox1
and cob being incomplete at the 50 and 30 ends, respec-
tively), 10 for tRNAs, and two for rRNAs. The
5243nt portion of N. westbladi contains 13 genes: four
for proteins (with cox1 being incomplete at the 30 end),
seven for tRNAs, and two for rRNAs (with rrnL being
incomplete at the 50 end). The 6882nt portion ofM. line-
are contains a large non-coding region and 13 genes: five
for proteins (with cob and cox1 being incomplete at the
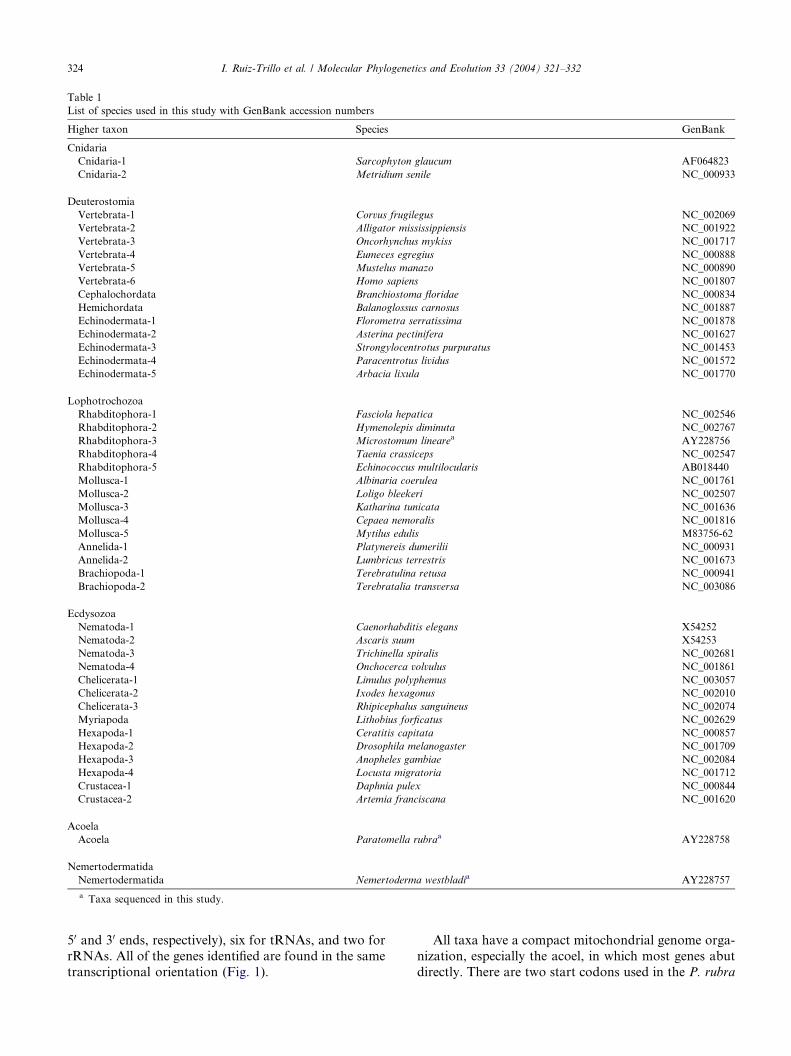
Table 1
List of species used in this study with GenBank accession numbers
Higher taxon Species GenBank
Cnidaria
Cnidaria-1 Sarcophyton glaucum AF064823
Cnidaria-2 Metridium senile NC_000933
Deuterostomia
Vertebrata-1 Corvus frugilegus NC_002069
Vertebrata-2 Alligator mississippiensis NC_001922
Vertebrata-3 Oncorhynchus mykiss NC_001717
Vertebrata-4 Eumeces egregius NC_000888
Vertebrata-5 Mustelus manazo NC_000890
Vertebrata-6 Homo sapiens NC_001807
Cephalochordata Branchiostoma floridae NC_000834
Hemichordata Balanoglossus carnosus NC_001887
Echinodermata-1 Florometra serratissima NC_001878
Echinodermata-2 Asterina pectinifera NC_001627
Echinodermata-3 Strongylocentrotus purpuratus NC_001453
Echinodermata-4 Paracentrotus lividus NC_001572
Echinodermata-5 Arbacia lixula NC_001770
Lophotrochozoa
Rhabditophora-1 Fasciola hepatica NC_002546
Rhabditophora-2 Hymenolepis diminuta NC_002767
Rhabditophora-3 Microstomum linearea AY228756
Rhabditophora-4 Taenia crassiceps NC_002547
Rhabditophora-5 Echinococcus multilocularis AB018440
Mollusca-1 Albinaria coerulea NC_001761
Mollusca-2 Loligo bleekeri NC_002507
Mollusca-3 Katharina tunicata NC_001636
Mollusca-4 Cepaea nemoralis NC_001816
Mollusca-5 Mytilus edulis M83756-62
Annelida-1 Platynereis dumerilii NC_000931
Annelida-2 Lumbricus terrestris NC_001673
Brachiopoda-1 Terebratulina retusa NC_000941
Brachiopoda-2 Terebratalia transversa NC_003086
Ecdysozoa
Nematoda-1 Caenorhabditis elegans X54252
Nematoda-2 Ascaris suum X54253
Nematoda-3 Trichinella spiralis NC_002681
Nematoda-4 Onchocerca volvulus NC_001861
Chelicerata-1 Limulus polyphemus NC_003057
Chelicerata-2 Ixodes hexagonus NC_002010
Chelicerata-3 Rhipicephalus sanguineus NC_002074
Myriapoda Lithobius forficatus NC_002629
Hexapoda-1 Ceratitis capitata NC_000857
Hexapoda-2 Drosophila melanogaster NC_001709
Hexapoda-3 Anopheles gambiae NC_002084
Hexapoda-4 Locusta migratoria NC_001712
Crustacea-1 Daphnia pulex NC_000844
Crustacea-2 Artemia franciscana NC_001620
Acoela
Acoela Paratomella rubraa AY228758
Nemertodermatida
Nemertodermatida Nemertoderma westbladia AY228757
a Taxa sequenced in this study.
324 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
50 and 30 ends, respectively), six for tRNAs, and two for
rRNAs. All of the genes identified are found in the same
transcriptional orientation (Fig. 1).
All taxa have a compact mitochondrial genome orga-
nization, especially the acoel, in which most genes abut
directly. There are two start codons used in the P. rubra
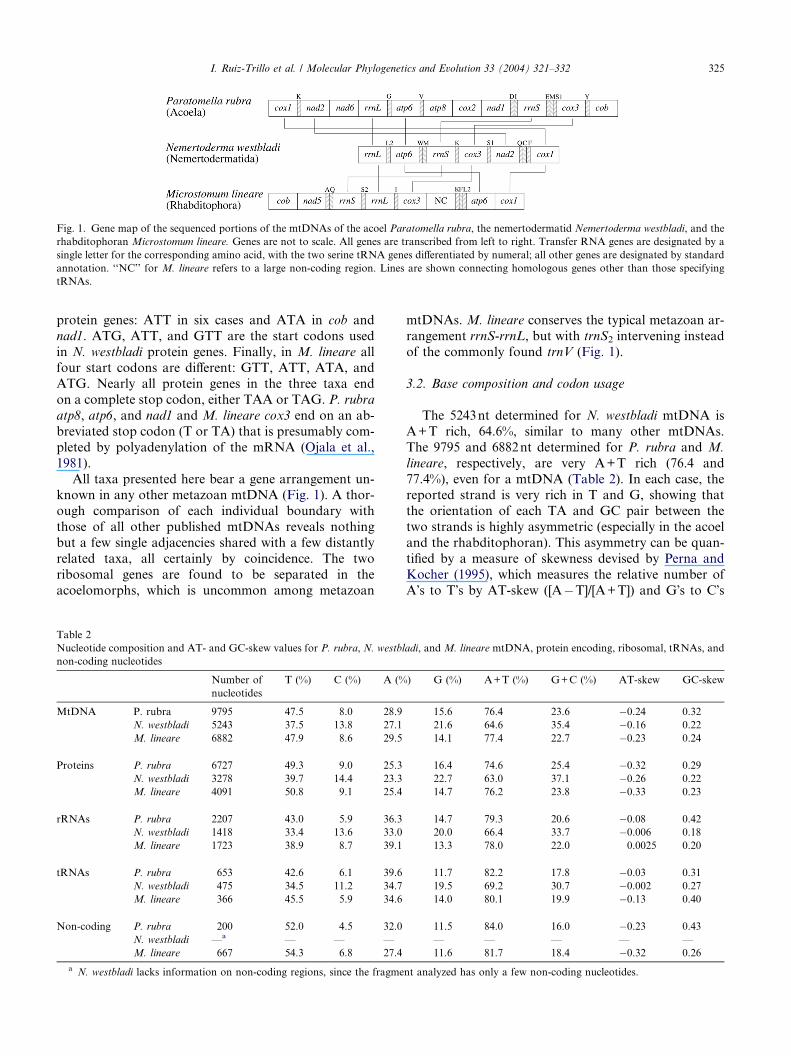
Fig. 1. Gene map of the sequenced portions of the mtDNAs of the acoel Paratomella rubra, the nemertodermatid Nemertoderma westbladi, and the
rhabditophoran Microstomum lineare. Genes are not to scale. All genes are transcribed from left to right. Transfer RNA genes are designated by a
single letter for the corresponding amino acid, with the two serine tRNA genes differentiated by numeral; all other genes are designated by standard
annotation. ‘‘NC’’ for M. lineare refers to a large non-coding region. Lines are shown connecting homologous genes other than those specifying
tRNAs.
I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332 325
protein genes: ATT in six cases and ATA in cob and
nad1. ATG, ATT, and GTT are the start codons used
in N. westbladi protein genes. Finally, in M. lineare allfour start codons are different: GTT, ATT, ATA, and
ATG. Nearly all protein genes in the three taxa end
on a complete stop codon, either TAA or TAG. P. rubra
atp8, atp6, and nad1 and M. lineare cox3 end on an ab-
breviated stop codon (T or TA) that is presumably com-
pleted by polyadenylation of the mRNA (Ojala et al.,
1981).
All taxa presented here bear a gene arrangement un-known in any other metazoan mtDNA (Fig. 1). A thor-
ough comparison of each individual boundary with
those of all other published mtDNAs reveals nothing
but a few single adjacencies shared with a few distantly
related taxa, all certainly by coincidence. The two
ribosomal genes are found to be separated in the
acoelomorphs, which is uncommon among metazoan
Table 2
Nucleotide composition and AT- and GC-skew values for P. rubra, N. westbl
non-coding nucleotides
Number of
nucleotides
T (%) C (%) A (%
MtDNA P. rubra 9795 47.5 8.0 28.9
N. westbladi 5243 37.5 13.8 27.1
M. lineare 6882 47.9 8.6 29.5
Proteins P. rubra 6727 49.3 9.0 25.3
N. westbladi 3278 39.7 14.4 23.3
M. lineare 4091 50.8 9.1 25.4
rRNAs P. rubra 2207 43.0 5.9 36.3
N. westbladi 1418 33.4 13.6 33.0
M. lineare 1723 38.9 8.7 39.1
tRNAs P. rubra 653 42.6 6.1 39.6
N. westbladi 475 34.5 11.2 34.7
M. lineare 366 45.5 5.9 34.6
Non-coding P. rubra 200 52.0 4.5 32.0
N. westbladi —a — — —
M. lineare 667 54.3 6.8 27.4
a N. westbladi lacks information on non-coding regions, since the fragme
mtDNAs. M. lineare conserves the typical metazoan ar-
rangement rrnS-rrnL, but with trnS2 intervening instead
of the commonly found trnV (Fig. 1).
3.2. Base composition and codon usage
The 5243nt determined for N. westbladi mtDNA is
A+T rich, 64.6%, similar to many other mtDNAs.
The 9795 and 6882nt determined for P. rubra and M.
lineare, respectively, are very A+T rich (76.4 and
77.4%), even for a mtDNA (Table 2). In each case, thereported strand is very rich in T and G, showing that
the orientation of each TA and GC pair between the
two strands is highly asymmetric (especially in the acoel
and the rhabditophoran). This asymmetry can be quan-
tified by a measure of skewness devised by Perna and
Kocher (1995), which measures the relative number of
A�s to T�s by AT-skew ([A�T]/[A+T]) and G�s to C�s
adi, and M. lineare mtDNA, protein encoding, ribosomal, tRNAs, and
) G (%) A+T (%) G+C (%) AT-skew GC-skew
15.6 76.4 23.6 �0.24 0.32
21.6 64.6 35.4 �0.16 0.22
14.1 77.4 22.7 �0.23 0.24
16.4 74.6 25.4 �0.32 0.29
22.7 63.0 37.1 �0.26 0.22
14.7 76.2 23.8 �0.33 0.23
14.7 79.3 20.6 �0.08 0.42
20.0 66.4 33.7 �0.006 0.18
13.3 78.0 22.0 0.0025 0.20
11.7 82.2 17.8 �0.03 0.31
19.5 69.2 30.7 �0.002 0.27
14.0 80.1 19.9 �0.13 0.40
11.5 84.0 16.0 �0.23 0.43
— — — — —
11.6 81.7 18.4 �0.32 0.26
nt analyzed has only a few non-coding nucleotides.
326 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
by GC-skew ([G�C]/[G+C]). According to these for-
mulae, skew values can range from �1 to +1, with great-
er compositional asymmetry giving skew values (positive
or negative) closer to 1; a skew value of zero indicates
that the distribution is equal between the strands. AT-
and GC-skew values are, respectively, �0.24 and 0.32for the acoel; �0.16 and 0.22 for the nemertodermatid,
and �0.23 and 0.24 for the rhabditophoran (Table 2).
As expected, GC-skew values are, in general, higher
for tRNAs, ribosomal and non-coding nucleotides and
lower for protein-encoding nucleotides. However AT-
skew values are lower than expected for ribosomal,
Fig. 2. Potential secondary structure of the putative mt tRNAs found for P
tRNAs and non-coding nucleotides (Table 2). A possi-
ble explanation is that among ribosomal and tRNAs
genes a similar number of A�s and T�s are required for
stem structure formation in their products. This may
not have so strong an effect on GC-skew due to the po-
tential formation of GT pairs.
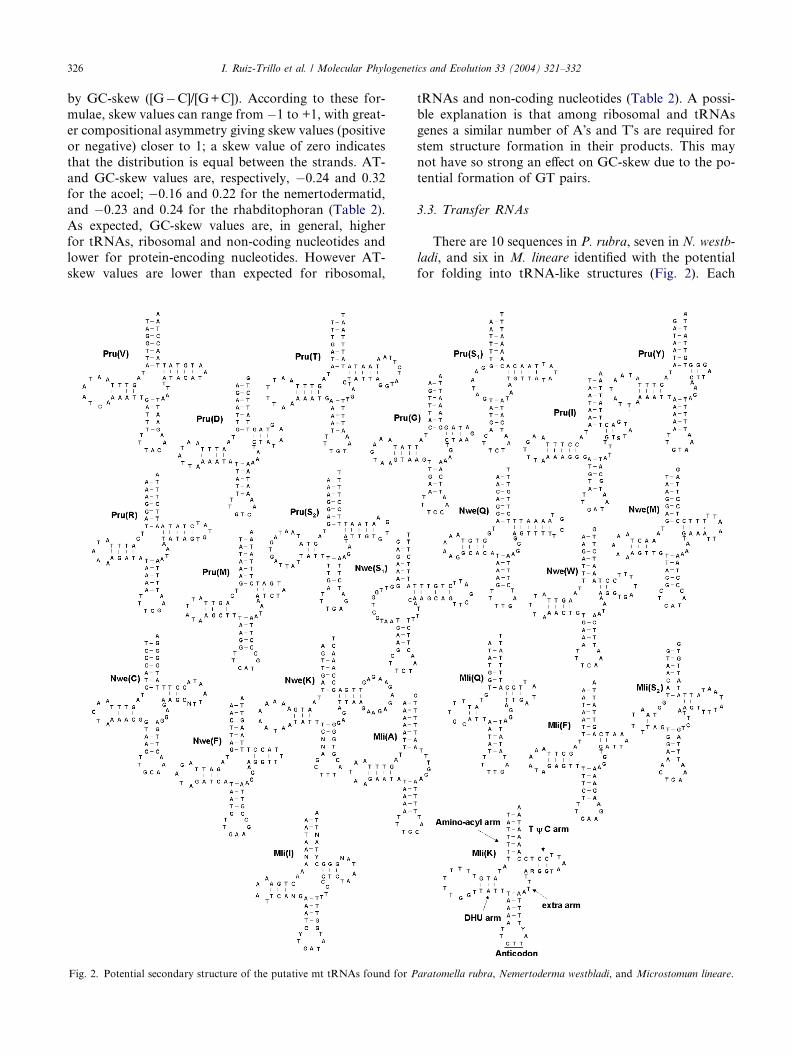
3.3. Transfer RNAs
There are 10 sequences in P. rubra, seven in N. westb-
ladi, and six in M. lineare identified with the potential
for folding into tRNA-like structures (Fig. 2). Each
aratomella rubra, Nemertoderma westbladi, and Microstomum lineare.

I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332 327
has a seven-member amino-acyl acceptor stem and a
five-member anticodon stem, some with one or two mis-
matches. The extra arms have three to five nucleotides.
There are usually three to five nucleotide pairs in both
the DHU and TwC arms. The putative tRNA(K) of
N. westbladi and tRNA(I) of M. lineare have mismatch-es and ambiguously determined nucleotides, and so may
not be real tRNAs. The tRNAs S1 of both acoelomorph
species lack a paired DHU arm. The nemertodermatid
and the macrostomid differ in the anticodon sequence
of tRNA(K), being TTT in N. westbladi (as in most
metazoans) and CTT in M. lineare (as in the flatworm
parasitic species and some others).
3.4. Genetic code
The mitochondrial genetic code for most inverte-
brates appears to deviate from the ‘‘universal’’ genetic
code with regard to the identities of ATA (methionine,
M, instead of isoleucine, I), TGA (tryptophan,W, in-
stead of a stop codon) and AGR codons (serine, S, in-
stead of arginine, R) (Table 3; see Wolstenholme,1992). Diploblasts share only the deviation for the co-
don TGA. Platyhelminthes have been inferred to have
two further differences: AAA encodes asparagine (N)
rather than lysine (K), and ATA encodes I rather than
M (as in the universal code) (Telford et al., 2000).
Our data seem to corroborate previous results (Tel-
ford et al., 2000), in which acoels and nemertodermatids
share the invertebrate mitochondrial genetic code with-out the flatworm variations (Table 3). This has been ob-
served in some amino acid positions throughout the
alignment, such as, for example, at amino acid position
271 (position corresponding to the Homo sequence) of
the Cob gene there is a very conserved I that corresponds
to an ATA codon in the nucleotide sequence from M.
lineare; and at amino acid position 136 of the Cox3 gene
there is a conserved N that corresponds to an AAA co-don in the nucleotide sequence from M. lineare. In the
two positions P. rubra has the codon corresponding to
the invertebrate mitochondrial genetic code for those
amino acids (ATC and AAT respectively; N. westbladi
has AAC for the second site).
However, the use of genetic code changes as phyloge-
netic characters should be used only with great caution,
since: (1) it remains undetermined how common suchchanges are, (2) inference of these deviations is often
Table 3
The amino acid specifications for four codons that commonly vary
TGA
‘‘Universal’’ genetic code Stop
Diploblastica W
Acoelomorpha W
Most invertebrates W
Rhabditophora W
speculative, and (3) there are obvious convergences,
such as those between platyhelminths and echinoderms
for the changes of AAA and ATA.
3.5. Phylogenetic analyses
In order to test whether the clade Acoelomorpha is
valid and whether M. lineare groups with the parasitic
flatworms, we performed initial ML and NJ analyses in-
cluding all of the taxa in Table 1 (see supplementary da-
ta), but the results are poorly supported and clearly
show artifactual associations. P. rubra and N. westbladi
are sister groups, forming the clade Acoelomorpha in
the NJ tree (but with only 60% bootstrap support),but not in the ML tree. The Acoelomorpha appears as
sister group of all the other bilaterians, as found in ear-
lier studies (Ruiz-Trillo et al., 2002). Both NJ and ML
analyses group the rhabditophoran M. lineare with the
parasitic flatworms (53% QP and 89% NJ bootstrap sup-
port). There is an artifactual grouping of nematodes
with platyhelminths and Mytilus within the Lophotro-
chozoa due to the effect of long-branch attraction.The low support for the basal position of Acoelo-
morpha and the difference between NJ and ML analyses
on the Acoelomorpha clade might be due to the fact
that the N. westbladi mitochondrial genome sequence
includes only three complete and one partial protein
coding genes, that is, 454 amino acid positions out of
the total 1383. Thus, we performed an additional anal-
ysis without the nemertodermatid N. westbladi, whichresulted in a similar topology, but with a general in-
crease in branch support (see supplementary data).
Again, nematodes group artifactually with the other
long-branch taxa Platyhelminthes and Mytilus. The
rhabditophoran M. lineare groups with the other platy-
helminths (now with 72% QP and 92% NJ bootstrap
value) and the acoel P. rubra appears as sister group
of all the other bilaterians (now with 83% QP and NJbootstrap support). These analyses corroborate previ-
ous results that acoelomorphs are the most basal extant
bilaterian clade.
We performed a relative rate test for all taxa. At the
1% confidence level, several protostome taxa appear to
have much faster evolutionary rates than the other bila-
terians, and were then eliminated from subsequent phy-
logenetic analysis. Such is the case of the nematodes, theplatyhelminths, the ticks Ixodes and Rhiphicephalus, the
ATA AGR AAA
I R K
I R K
M S K
M S K
I S N

328 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
acoelomorph N. westbladi, and the mollusks Mytilus
and Cepaea. The acoel P. rubra and the lophotrochozo-
ans Albinaria, Loligo, and Terebratalia have only faster
evolutionary rates compared to a few (two to five) short-
er-branched deuterostomes. Thus, they were not consid-
ered fast-clock taxa, and were included in the finalanalysis. A table summarizing this analysis can be found
in the supplementary material.
Fig. 3 shows the results of phylogenetic analysis with
the dataset limited to taxa of more moderate rates of se-
quence change. The ML tree used QP with 10,000 repli-
cates and the NJ tree used 1000 bootstrap replicates for
branch support. The topology corroborates previous
SSU and myosin II analyses (Ruiz-Trillo et al., 1999,2002). Acoela is the most basal extant bilaterian clade
(97 and 96% QP and NJ branch support) and the rest
of the bilaterians, or Eubilateria, are divided into the
three super-clades, Deuterostomia (82% QP and 99%
Fig. 3. Phylogenetic tree based on mitochondrial protein-coding gene
sequences using quartet-puzzling (QP), a form of maximum likelihood
analysis. Numbers above key nodes refers to the percentage obtained
from a QP with 10,000 replicates followed by support from a neighbor-
joining (NJ) analysis with 1000 bootstrap replicates. Asterisks indicate
non-key nodes with more than a 90% for both QP and NJ support.
Taxa with increased evolutionary rates as detected in the RRT have
been excluded. For species names and taxa excluded see Table 1. Ec,
Ecdysozoa; Lo, Lophotrochozoa; De, Deuterostomia; Ac, Acoelo-
morpha; and Di, Diploblastica.
NJ), Lophotrochozoa (90% QP and 81% NJ), and Ec-
dysozoa (100% QP and 100% NJ). The support for a
protostomian clade is very high (91% QP and 98%
NJ). Of course, taxon sampling for many of these
groups remains low, e.g., it is not evaluated in this anal-
ysis whether Ecdysozoa includes the nematodes.
4. Discussion
The results of this study constitute the first attempt to
analyze at the mitochondrial genome level any acoel,
nemertodermatid, or free-living flatworm taxa, the first
molecular evolutionary analysis of their mitochondrialgenome organization, and a test of using mitochondrial
amino acid sequences to address very deep evolutionary
relationships among major animal groups.
4.1. The basal position of the Acoelomorpha and the
polyphyly of the Platyhelminthes
The amino acid sequence comparisons presented here(see Fig. 3 and supplementary data) add to a growing
list of molecular data backing a basal position for the
Acoelomorpha (initially reported using 18S rDNA se-
quences by Ruiz-Trillo et al. (1999) and Jondelius et
al. (2002)) and a derived, lophotrochozoan position,
for the rest of the Platyhelminthes (anticipated in Car-
ranza et al., 1997). These data include SSU sequences,
Hox genes, myosin II and myosin II+SSU sequences,LSU+SSU sequences, and let-7 gene expression (see
Section 1 for a detailed description and references).
How can we reconcile this with the morphological
evidence traditionally used to argue for monophyly of
Platyhelminthes (e.g., Hyman, 1951)? The three mono-
phyletic groups recognized within the Platyhelminthes
as traditionally constructed—Acoelomorpha, Catenul-
ida, and Rhabditophora (Smith et al., 1986)—are eachdefined by well-accepted synapomorphies. However,
lack of robust morphological synapomorphies that
would unite these three major clades led Smith et al.
(1986) to question the monophyly of the Platyhelmin-
thes. Nonetheless, the phylum was generally believed
to be monophyletic because they share a long list of
characters, including having soft bodies, hermaphrodit-
ism, internal fertilization, filiform and biflagellatesperm, lack of acrosomes, and multiciliated epidermal
cells. Other characters, such as spiral cleavage and a
ladder-like nervous system (see Brusca and Brusca,
1990 for general references), have been also considered,
but have been scored for only a limited range of taxa.
However, most of these characters could more
appropriately be argued to be symplesiomorphies or
homoplasies.Considering potential characters that would unite
Acoelomorpha, Catenulida, and Rhabditophora, there

I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332 329
are two others that bear serious consideration: lack of
mitosis in somatic cells (Ehlers, 1985) and presence of
a sack-like gut (which means lack of a proper anus).
The first refers to the inability of most somatic cells to
divide, with new cells being generated from undifferenti-
ated cells which, in Platyhelminthes, are known as neo-blasts (Baguna, 1981; Baguna et al., 1989). However,
this character is imprecisely defined, since all animals
have differentiated cells that lack the ability to divide.
Moreover, it has not been properly scored for many
phyla. As for the second character, it has been suggested
that the sack-like gut of Platyhelminthes (including
Acoelomorpha) has been derived by paedomorphosis
from the one-way through gut of a more complex ances-tor. A parsimonious reinterpretation of this feature on a
tree that places Acoelomorpha as basal to Bilateria is
that it is a symplesiomorphy for the Acoelomorpha
and separately derived for the clade of Catenulida and
Rhabditophora within the Lophotrochozoa.
Alternatively, there are characters that support a
basal position of Acoelomorpha. First, acoels generate
only endomesoderm (Henry et al., 2000), in contrastwith the rest of the Platyhelminthes (and protostomes
in general) that have both ecto- and endomesoderm.
Endomesoderm is considered the ancestral form. Sec-
ond, acoelomorphs have an anterior concentration of
nerve cells without forming a ‘‘true brain’’ with neuro-
pile (Raikova et al., 2000; Reuter et al., 1998; but see
Tyler, 2001 for a contrary view). Moreover, while
other bilaterians, including catenulids and rhabdito-phorans, have longitudinal nerve cords that are dis-
tinctly dorsal or ventral, acoelomorphs have a radial
arrangement, interpreted here as being primitive or
separately derived. Finally, limited and preliminary
study indicates that aceolomorphs have only few (four
to five) Hox genes (Cook et al., 2004), so that the ex-
panded repertoire may be a synapomorphy of the
other bilaterians.This revised phylogeny compels a reinterpretation of
the evolution of some features. First, the lack of proto-
nephridia for Acoelomorpha is regarded in traditional
schemes as having been derived by loss from their
platyhelminth ancestor. However, under the new sce-
nario, this lack of protonephridia may be the retention
of a primitive condition, a state shared with diplo-
blasts. Second, the ‘‘duet-spiral’’ type of embryonic cellcleavage has been usually considered to be derived
from the quartet type spiral cleavage of other Platyhel-
minthes. As discussed by Henry et al. (2000), duet-spir-
al cleavage may have arose alternatively from a form
of radial or biradial cleavage characteristic of the more
primitive programs in the Metazoa, whereas quartet
spiral cleavage would have originated independently
within the Lophotrochozoa. Finally, as mentionedabove, the sack-like gut of the Acoelomorpha may be
a symplesiomorphy shared with the similar state of dip-
loblasts, followed by the appearance of a one-way
through gut in the rest of Bilateria, within which the
Platyhelminthes (without the Acoelomorpha) separate-
ly adopted a similar condition.
Finally, is there enough evidence to support a mono-
phyletic Acoelomorpha? Potential morphological syna-pomorphies of the Acoelomorpha are: (1) a network
formed by interconnecting rootlets of epidermal cilia;
(2) a shaft region in epidermal cilia; (3) similar fine struc-
ture of frontal organs; (4) reduced extracellular matrix
(ECM); and (5) absence of protonephridia (Ax, 1996;
Ehlers, 1985; Smith et al., 1986; Tyler and Rieger,
1999). Another suggested synapomorphy, the spiral-du-
et type of cleavage, has not been unequivocally deter-mined for nemertodermatids, though it seems to be
present (Ulf Jondelius, personal communication). Al-
though the comparison of ribosomal genes results in a
paraphyletic relationship (Jondelius et al., 2002; Telford
et al., 2003) for the Acoelomorpha, phylogenetic analy-
ses of the amino acid sequences of mitochondrial genes
(this work) and myosin II sequences (Ruiz-Trillo et al.,
2002) show a monophyletic Acoelomorpha. However,the fact that acoels and nemertodermatids do not share
any gene boundaries between them and the long branch-
es separating them in the phylogenetic tree (supplemen-
tary data), argue for an extended period since the split of
these lineages or, alternatively, for a very rapidly chang-
ing mode of gene arrangement and nucleotide substitu-
tion. Additional support for an extended period since
acoels and nemertodermatids diverged comes from mor-phology. Acoels and nemertodermatids have significant
differences in the pattern of neurotransmitters (Raikova
et al., 2000), statocyst structure (Ax, 1996) and in the
fact that acoels digestive tract structure is either syncy-
tial or cellular while in nemertodermatids is always epi-
thelial (Rieger et al., 1991; Smith and Tyler, 1985).
Sperm morphology is also different in both lineages:
Nemertodermatids have uniflagellate spermatozoa withthe common 9+2 axonemal pattern (Lundin and Hen-
delberg, 1998), while Acoel spermatozoa bear two flagel-
la with reverse orientation and sometimes a modified
axoneme structure (Raikova et al., 2001). A short com-
mon period followed by a long divergence could explain
the failure of ribosomal genes to recover the Acoelomor-
pha as a monophyletic group. Acoela and Nemertoder-
matida (specially the first) present a high rate ofsubstitutions for these genes hence it makes more prob-
able that any signal that could have accumulated in their
common ancestor would have been erased by the subse-
quent divergence period.
To summarize, our analyses of mitochondrial amino
acid sequences added to morphological characters and a
growing number of molecular evidences argue for a
monophyletic Acoelomorpha as a basal bilateriangroup, occupying a pivotal position between diploblast
and triploblasts. Consequently, under this evolutionary

330 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
scenario, we believe Acoelomorpha merits establishment
as a new phylum.
4.2. Rhabditophoran mitochondrial variability
The fact that M. lineare does not share any geneboundaries with any of the parasitic rhabditophoran
flatworms sequenced to date demonstrates that there is
great variability in mitochondrial genome structure
within the Platyhelminthes. This bolsters confidence that
mitochondrial gene arrangements might be a useful tool
for inferring evolutionary relationships within the phy-
lum, many of which remain unresolved (Baguna and
Riutort, 2004; Littlewood and Olson, 2001).
4.3. Phylogenetic value of mitochondrial sequences at
deep evolutionary levels
Our phylogenetic analyses of mitochondrial protein-
coding sequences demonstrate their power for inferring
ancient evolutionary relationships, although they have
difficulty handling problems with long-branch taxa asis the case with all molecular sequence comparisons.
Mitochondrial sequence comparisons support the
monophyly of the three eubilaterian super-clades, Deu-
terostomia, Lophotrochozoa, and Ecdysozoa, at least
so far as the constituent taxa have been sampled. Also,
this analysis well resolves the branching pattern be-
tween the three eubilaterian super-clades, in which
protostomes (ecdysozoans and lophotrochozoans)form a monophyletic group (Fig. 3). Many previous
molecular studies were not able to fully resolve the
branching order among the three super-clades (Ado-
utte et al., 2000; Ruiz-Trillo et al., 1999, 2002; Telford
et al., 2003).
In summary, the high branch support for: (1) the
three bilaterian super-clades, (2) the basal position of ac-
oels, and (3) the branching order of the three eubilateri-an super-clades, indicates mitochondrial sequences may
be indeed a good and promising phylogenetic marker
for deep evolutionary events. This phylogenetic value
of mitochondrial sequences should be carefully tested,
however, with the inclusion of enigmatic taxa, such as
gastrotrichs, rotifers, and chaetognaths.
Acknowledgments
We thank Ulf Jondelius and Maria Reuter for pro-
viding N. westbladi and M. lineare specimens and Clint
Turbeville, Kevin Helfenbein, and Jordi Paps for techni-
cal assistance. We thank the people in the sequencing
unit of the ‘‘Serveis cientıfico-tecnics’’ of the Universitat
de Barcelona. I.R.-T., M.R., and J.B. were supported byCIRIT (Generalitat de Catalunya) Grants 1999SGR-
00026 and 2001SGR-00102 and M.R. and I.R.-T. by
DGICYT (Ministerio de Ciencia y Tecnologıa) Grant
PB97-0937. I.R.-T. was sponsored by a predoctoral
grant from the Universitat de Barcelona. Part of this
work was performed under the auspices of the US De-
partment of Energy, Office of Biological and Environ-
mental Research, in the University of California,Lawrence Berkeley National Laboratory, under Con-
tract No. DE-AC03-76SF00098.
Appendix A. Supplementary material
Supplementary data associated with this article can
be found, in the online version, at doi:10.1016/j.ymp-ev.2004.06.002.
References
Adachi, J., Hasegawa, M., 1996. Model of amino acid substitution in
proteins encoded by mitochondrial DNA. J. Mol. Evol. 42, 459–
468.
Adoutte, A., Balavoine, G., Lartillot, N., Lespinet, O., Prud�homme,
B., de Rosa, R., 2000. The new animal phylogeny: reliability and
implications. Proc. Natl. Acad. Sci. USA 97, 4453–4456.
Arnason, U., Janke, A., 2002. Mitogenomic analyses of eutherian
relationships. Cytogenet. Genome Res. 96, 20–32.
Ax, P., 1996. in: Multicellular Animals. A New Approach to the
Phylogenetic Order in Nature, vol. I. Springer-Verlag, Berlin,
Germany.
Baguna, J., 1981. Planarian neoblasts. Nature 290, 14–15.
Baguna, J., Salo, E., Auladell, C., 1989. Regeneration and pattern
formation in planarians III. Evidence that neoblasts are totipotent
stem cells. Development 107, 77–86.
Baguna, J., Riutort, M., 2004. Molecular phylogeny of the platyhel-
minthes. Can. J. Zool. 82, 168–193.
Barnes, W.M., 1994. PCR amplification of up to 35-kb DNA with high
fidelity and high yield from bacteriophage templates. Proc. Natl.
Acad. Sci. USA 91, 2216–2220.
Bayascas, J.R., Castillo, E., Salo, E., 1998. Platyhelminthes have a
Hox code differentially activated during regeneration, with genes
closely related to those of spiralian protostomes. Dev. Genes Evol.
208, 467–473.
Beagley, C.T., Okimoto, R., Wolstenholme, D.R., 1998. The mito-
chondrial genome of the sea anemone Metridium senile (Cnidaria):
introns, a paucity of tRNA genes, and a near-standard genetic
code. Genetics 148, 1091–1108.
Beaton, M.J., Roger, A.J., Cavalier-Smith, T., 1998. Sequence analysis
of the mitochondrial genome of Sarcophyton glaucum: conserved
gene order among octocorals. J. Mol. Evol. 47, 697–708.
Boore, J.L., Brown, W.M., 1998. Big trees from little genomes:
mitochondrial gene order as a phylogenetic tool. Curr. Opin.
Genet. Dev. 8, 668–674.
Boore, J.L., 1999. Animal mitochondrial genomes. Nucleic Acids Res.
27, 1767–1780.
Boore, J.L., Brown, W.M., 2000. Mitochondrial genomes of Galathe-
olinum, Helobdella, and Platynereis: sequence and gene arrange-
ment comparisons indicate that Pogonophora is not a phylum and
Annelida and Arthropoda are not sister taxa. Mol. Biol. Evol. 17,
87–106.
Boore, J.L., Staton, J.L., 2002. The mitochondrial genome of the
Sipunculid Phascolopsis gouldii supports its association with
Annelida rather than Mollusca. Mol. Biol. Evol. 19, 127–137.

I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332 331
Brusca, R.C., Brusca, G.J., 1990. Invertebrates. Sinauer Associates,
Sunderland.
Carranza, S., Baguna, J., Riutort, M., 1997. Are the Platyhelminthes a
monophyletic primitive group? An assessment using 18S rDNA
sequences. Mol. Biol. Evol. 14, 485–497.
Cook, C.E., Jimenez, E., Akam, M., Salo, E., 2004. The Hox
complement of acoel flatworms, a basal bilaterian clade. Evol.
Dev. 6, 154–163.
De Rosa, R., Grenier, J.K., Andreeva, T., Cook, C.E., Adoutte, A.,
Akam, M., Carroll, S.B., Balavoine, G., 1999. Hox genes in
brachiopods and priapulids and protostome evolution. Nature 399,
772–776.
Ehlers, U., 1985. Phylogenetic relationships within the Platyhelmin-
thes. In: Morris, S.C., George, J.D., Gibson, R., Platt, H.M. (Eds.),
The Origin and Relationships of Lower Invertebrates Groups.
Oxford University Press, Oxford, pp. 143–158.
Folmer, O., Black, M., Hoeh, W., Lutz, R., Vrijenhoek, R., 1994.
DNA primers for amplification of mitochondrial cytochrome c
oxidase subunit I from diverse metazoan invertebrates. Mol. Mar.
Biol. Biotechnol. 3, 294–299.
Henry, J.Q., Martindale, M.Q., Boyer, B.C., 2000. The unique
developmental program of the acoel flatworm, Neochildia fusca.
Dev. Biol. 220, 285–295.
Hyman, L.H., 1951. The Invertebrates. II. Platyhelminthes and
Rhynchocoela. The Acoelomate Bilateria. McGraw-Hill, New
York.
Hoffmann, R.J., Boore, J.L., Brown, W.M., 1992. A novel mitochon-
drial genome organization for the blue mussel, Mytilus edulis.
Genetics 131, 397–412.
Inoue, J.G., Miya, M., Nishida, M., 2003. Basal actinopterygian
relationships: a mitogenomic perspective on the phylogeny of the
ancient fish. Mol. Phylogenet. Evol. 26, 110–120.
Jondelius, U., Ruiz-Trillo, I., Baguna, J., Riutort, M., 2002. The
Nemertodermatida are basal bilaterians and not members of the
Platyhelminthes. Zool. Scr. 31, 201–215.
Keddie, E.M., Higazi, T., Unnasch, T.R., 1998. The mitochondrial
genome of Onchocerca volvulus: sequence, structure and phyloge-
netic analysis. Mol. Biochem. Parasitol. 95, 111–127.
Kumar, S., Tamura, K., Jakobsen, I.B., Nei, M., 2001. MEGA2:
molecular evolutionary genetics analysis software. Bioinformatics
17, 1244–1245.
Le, T.H., Blair, D., McManus, D.P., 2002. Mitochondrial genomes of
parasitic flatworms. Trends Parasitol. 18, 206–213.
Littlewood, D.T.J., Olson, P.D., 2001. Small subunit rDNA and the
Platyhelminthes: signal, noise and compromise. In: Littlewood,
D.T.J., Bray, R.D. (Eds.), The Interrelationships of the Platyhel-
minthes. Taylor and Francis, London, pp. 252–278.
Lowe, T.M., Eddy, S.R., 1997. tRNAscan-SE: a program for
improved detection of transfer RNA genes in genomic sequence.
Nucleic Acids Res. 25 (5), 955–964.
Lundin, K., Hendelberg, J., 1998. Is the sperm type of the Nemerto-
dermatida close to that of the ancestral Platyhelminthes?. Hydro-
biologia 383, 197–205.
Miya, M., Takeshima, H., Endo, H., Ishiguro, N.B., Inoue, J.G.,
Mukai, T., Satoh, T.P., Yamaguchi, M., Kawaguchi, A., Mabuchi,
K., Nishida, M., 2003. Major patterns of higher teleostean
phylogenies: a new perspective based on 100 complete mitochon-
drial DNA sequences. Mol. Phylogenet. Evol. 26, 121–138.
Murata, Y., Nikaido, M., Sasaki, T., Cao, Y., Fukumoto, Y., Okada,
N., 2003. Afrotherian phylogeny as inferred from complete
mitochondrial genomes. Mol. Phylogenet. Evol. 28, 253–260.
von Nickisch-Rosenegk, M., Brown, W.M., Boore, J.L., 2001.
Complete sequence of the mitochondrial genome of the tapeworm
Hymenolepis diminuta: gene arrangements indicate that platyhel-
minthes are eutrochozoans. Mol. Biol. Evol. 18, 721–730.
Ojala, D., Montoya, J., Attardi, G., 1981. tRNA punctuation model of
RNA processing in human mitochondria. Nature 290, 470–474.
Okimoto, R., MacFarlane, J.L., Clary, D.O., Wolstenholme, D.R.,
1992. The mitochondrial genomes of two nematodes, Caenorhabd-
itis elegans and Ascaris suum. Genetics 130, 471–498.
Orii, H., Kato, K., Umesono, Y., Sakurai, T., Agata, K., Watanabe,
K., 1999. The planarian HOM/HOX homeobox genes (Plox)
expressed along the anteroposterior axis. Dev. Biol. 210, 456–468.
Palumbi, S.R., 1996. Nucleic acids II: the polymerase chain reaction.
In: Hillis, D.M., Moritz, C., Mable, B.K. (Eds.), Molecular
Systematics. Sinauer, Sunderland, MA, pp. 205–247.
Pasquinelli, A.E., McCoy, A., Jimenez, E., Salo, E., Ruvkun, G.,
Martindale, M.Q., Baguna, J., 2003. Expression of the 22 nucle-
otide let-7 heterochronic RNA throughout the Metazoa: a role in
life history evolution?. Evol. Dev. 5, 372–378.
Perna, N.T., Kocher, T.D., 1995. Patterns of nucleotide composition
at fourfold degenerate sites of animal mitochondrial genomes. J.
Mol. Evol. 41, 353–358.
Raikova, O.I., Reuter, M., Kotikova, E.A., Gustafsson, M.K.S., 1998.
A commisural brain! The pattern of 5-HT immunoreactivity in
Acoela (Platyhelminthes). Zoomorphology 118, 69–77.
Raikova, O.I., Reuter, M., Jondelius, U., Gustafsson, M.K.S., 2000.
The brain of the Nemertodermatida (Platyhelminthes) as revealed
by anti-5HT and anti-FMRMamide immunostainings. Tissue Cell
32, 358–365.
Raikova, O.I., Reuter, M., Justine, J.L., 2001. Contributions to the
phylogeny and systematics of the Acoelomorpha. In: Littlewood,
D.T.J., Bray, R.A. (Eds.), Interrelationships of the Platyhelmin-
thes. Taylor and Francis, London, pp. 13–23.
Reuter, M., Raikova, O.I., Gustafsson, M.K., 1998. An endocrine
brain? The pattern of FMRF-amide immunoreactivity in Acoela
(Plathelminthes). Tissue Cell 30, 57–63.
Rieger, R.M., Tyler, S., Smith III, J.P.S., Rieger, G.E., 1991.
Platyhelminthes: Turbellaria. In: Harrison, F.W., Bogitsh, B.J.
(Eds.), Microscopic Anatomy of Invertebrates. vol. 3: Platyhel-
minthes and Nemertinea. Wiley-Liss, New York, pp. 7–140.
Robinson-Rechavi, M., Huchon, D., 2000. RRTree: relative-rate tests
between groups of sequences on a phylogenetic tree. Bioinformatics
16, 296–297.
Ruiz-Trillo, I., Riutort, M., Littlewood, D.T.J., Herniou, E.A.,
Baguna, J., 1999. Acoel flatworms: earliest extant bilaterian
metazoans, not members of the Platyhelminthes. Science 283,
1919–1923.
Ruiz-Trillo, I., Paps, J., Loukota, M., Ribera, C., Jondelius, U.,
Baguna, J., Riutort, M., 2002. A phylogenetic analysis of myosin
heavy chain type II sequences corroborates that Acoela and
Nemertodermatida are basal bilaterians. Proc. Natl. Acad. Sci.
USA 99, 11246–11251.
Smith, J.P.S., Tyler, S., 1985. The acoel turbellarians: kingpins of
metazoan evolution or a specialized offshoot. In: Conway Morris,
C., George, J.D., Gibson, R., Platt, H.M. (Eds.), The Origins and
Relationships of Lower Invertebrates. Oxford University Press,
Oxford, pp. 123–142.
Smith, J.P.S., Tyler, S., Rieger, R.R., 1986. Is the Turbellaria
polyphyletic?. Hydrobiologia 132, 13–21.
Smith, S.W., Overbeek, R., Woese, C.R., Gilbert, W., Gillevet, P.M.,
1994. The genetic data environment: an expandable GUI for
multiple sequence-analysis. Comp. Appl. Biosci. 10, 671–675.
Strimmer, K., von Haeseler, A., 1996. Quartet puzzling: a quartet
maximum likelihood method for reconstructing tree topologies.
Mol. Biol. Evol. 13, 964–969.
Telford, M.J., Herniou, E.A., Russell, R.B., Littlewood, D.T.J., 2000.
Changes in mitochondrial gene tic code as phylogenetic characters:
two examples from the flatworms. Proc. Natl. Acad. Sci. USA 97,
11359–11364.
Telford, M.J., Lockyer, A.E., Cartwright-Finch, C., Littlewood,
D.T.J., 2003. Combined large and small subunit ribosomal RNA
phylogenies support a basal position of the acoelomorph flat-
worms. Proc. R. Soc. (Lond.) B 270, 1077–1083.

332 I. Ruiz-Trillo et al. / Molecular Phylogenetics and Evolution 33 (2004) 321–332
Tyler, S., 2001. The early worm: origins and relationships of the lower
flatworms. In: Littlewood, D.T.J., Bray, R. (Eds.), Interrelation-
ships of the Platyhelminthes. Taylor & Francis, London, pp. 3–12.
Tyler, S., Rieger, R.M., 1999. Functional morphology of musculature
in the acoelomate worm, Convoluta pulchra (Platyhelminthes).
Zoomorphology 119, 127–141.
Wolstenholme, D.R., 1992. Genetic novelties in mitochondrial ge-
nomes of multicellular animals. Curr. Opin. Genet. Dev. 2, 918–
925.
Zhang, J., Madden, T.L., 1997. Power BLAST: a new network BLAST
application for interactive or automated sequence analysis and
annotation. Genome Res. 7, 649–656.
Related Documents











