CARDIOLOGY/ORIGINAL RESEARCH Misdiagnosis of Long QT Syndrome as Epilepsy at First Presentation Judith M. MacCormick, MBChB Hugh McAlister, FCSANZ Jackie Crawford, NZCS John K. French, PhD Ian Crozier, MD Andrew N. Shelling, PhD Carey-Anne Eddy, MSc (Med) Mark I. Rees, PhD Jonathan R. Skinner, MD From the Green Lane Paediatric and Congenital Cardiac Service, Starship Children’s Hospital, Auckland, New Zealand (MacCormick, Crawford, Skinner); Cardiac Inherited Diseases Group, New Zealand (MacCormick, McAlister, Crawford, French, Crozier, Shelling, Eddy, Rees, Skinner); Department of Cardiology, Waikato Hospital, Hamilton, New Zealand (McAlister); Department of Cardiology, Liverpool Hospital, Sydney, New South Wales, Australia (French); Department of Cardiology, Christchurch Hospital, New Zealand (Crozier); Department of Obstetrics and Gynaecology (Shelling, Eddy) and Department of Molecular Medicine and Pathology, University of Auckland, Auckland, New Zealand (Rees); and the School of Medicine, University of Wales Swansea, Swansea, Wales, United Kingdom (Rees). Study objective: Long QT syndrome has significant mortality, which is reduced with appropriate management. It is known that long QT syndrome masquerades as other conditions, including seizure disorders. We aim to evaluate a series of patients with genetically confirmed long QT syndrome to establish the frequency of delayed recognition. We also examine causes and potential consequences of diagnostic delay. Methods: A consecutive case series of patients with long QT syndrome was identified through the Cardiac Inherited Disease Registry in New Zealand between 2000 and 2005. Detailed retrospective review of 31 cases was undertaken. The primary outcome was the time from first presentation with sudden loss of consciousness to a diagnosis of long QT syndrome. If the diagnosis was not made at the initial presentation, it was considered delayed. For the patients with a delayed diagnosis, the median duration of delay was compared between the subgroup of patients initially misdiagnosed with epilepsy and the others. Results: Genetic mutations in 31 probands were consistent with long QT type 1 in 18 (58%) patients, long QT type 2 in 10 (32%) and long QT type 3 in 3 (10%). Median age at diagnosis was 21 years (1 day to 54 years). Thirteen patients (39%) experienced diagnostic delay after presentation with syncope or seizure: median delay 2.4 years (2 months to 23 years). Electroencephalograms were obtained in 10 patients; 5 were diagnosed with epilepsy. For those labeled epileptic, diagnostic delay was significantly longer than with other misdiagnoses: estimated median difference 9.75 years (95% confidence interval 7.6 to 20.7 years). During the delay period, 4 sudden unexplained deaths reportedly occurred in young relatives. Ten of the 13 had an ECG before diagnosis, with unrecognized pulse rate– corrected QT interval prolongation in 8 cases (range 0.47 to 0.65 seconds). Conclusion: Delayed diagnosis of long QT syndrome is frequent. Symptoms are often attributed to alternative diagnoses, most commonly seizure disorder. Patients labeled as epileptic experience a particularly long diagnostic delay. ECGs were frequently requested, but interpretation errors were common. Given the potentially preventable mortality of long QT syndrome, emergency physicians investigating syncope and seizure should maintain a high index of suspicion. [Ann Emerg Med. 2009;54:26-32.] Provide feedback on this article at the journal’s Web site, www.annemergmed.com. 0196-0644/$-see front matter Copyright © 2008 by the American College of Emergency Physicians. doi:10.1016/j.annemergmed.2009.01.031 INTRODUCTION Background It has been 25 years since the first case report of long QT syndrome being misdiagnosed as epilepsy was published. 1 Several other cases have since appeared in the literature. 2-4 We recently reported the history of a 12-year-old boy diagnosed with and treated for epilepsy who was sub- sequently a victim of sudden death. Although his death was initially ascribed to sudden unexplained death in epilepsy, posthumous genetic screening identified long QT syndrome type 1. 5 Importance The natural history of long QT syndrome suggests mortality greater than 20% in the year after the first syncopal event, with nearly 50% mortality at 5 years. 6 Appropriate intervention can 26 Annals of Emergency Medicine Volume , . : July

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

CARDIOLOGY/ORIGINAL RESEARCH
Misdiagnosis of Long QT Syndrome as Epilepsy at FirstPresentation
Judith M. MacCormick, MBChBHugh McAlister, FCSANZJackie Crawford, NZCSJohn K. French, PhDIan Crozier, MDAndrew N. Shelling, PhDCarey-Anne Eddy, MSc (Med)Mark I. Rees, PhDJonathan R. Skinner, MD
From the Green Lane Paediatric and Congenital Cardiac Service, Starship Children’s Hospital,Auckland, New Zealand (MacCormick, Crawford, Skinner); Cardiac Inherited Diseases Group, NewZealand (MacCormick, McAlister, Crawford, French, Crozier, Shelling, Eddy, Rees, Skinner);Department of Cardiology, Waikato Hospital, Hamilton, New Zealand (McAlister); Department ofCardiology, Liverpool Hospital, Sydney, New South Wales, Australia (French); Department ofCardiology, Christchurch Hospital, New Zealand (Crozier); Department of Obstetrics andGynaecology (Shelling, Eddy) and Department of Molecular Medicine and Pathology, University ofAuckland, Auckland, New Zealand (Rees); and the School of Medicine, University of WalesSwansea, Swansea, Wales, United Kingdom (Rees).
Study objective: Long QT syndrome has significant mortality, which is reduced with appropriate management. Itis known that long QT syndrome masquerades as other conditions, including seizure disorders. We aim toevaluate a series of patients with genetically confirmed long QT syndrome to establish the frequency of delayedrecognition. We also examine causes and potential consequences of diagnostic delay.
Methods: A consecutive case series of patients with long QT syndrome was identified through the CardiacInherited Disease Registry in New Zealand between 2000 and 2005. Detailed retrospective review of 31 caseswas undertaken. The primary outcome was the time from first presentation with sudden loss of consciousnessto a diagnosis of long QT syndrome. If the diagnosis was not made at the initial presentation, it was considereddelayed. For the patients with a delayed diagnosis, the median duration of delay was compared between thesubgroup of patients initially misdiagnosed with epilepsy and the others.
Results: Genetic mutations in 31 probands were consistent with long QT type 1 in 18 (58%) patients, long QTtype 2 in 10 (32%) and long QT type 3 in 3 (10%). Median age at diagnosis was 21 years (1 day to 54 years).Thirteen patients (39%) experienced diagnostic delay after presentation with syncope or seizure: median delay2.4 years (2 months to 23 years). Electroencephalograms were obtained in 10 patients; 5 were diagnosed withepilepsy. For those labeled epileptic, diagnostic delay was significantly longer than with other misdiagnoses:estimated median difference 9.75 years (95% confidence interval 7.6 to 20.7 years). During the delay period, 4sudden unexplained deaths reportedly occurred in young relatives. Ten of the 13 had an ECG before diagnosis,with unrecognized pulse rate–corrected QT interval prolongation in 8 cases (range 0.47 to 0.65 seconds).
Conclusion: Delayed diagnosis of long QT syndrome is frequent. Symptoms are often attributed to alternativediagnoses, most commonly seizure disorder. Patients labeled as epileptic experience a particularly longdiagnostic delay. ECGs were frequently requested, but interpretation errors were common. Given the potentiallypreventable mortality of long QT syndrome, emergency physicians investigating syncope and seizure shouldmaintain a high index of suspicion. [Ann Emerg Med. 2009;54:26-32.]
Provide feedback on this article at the journal’s Web site, www.annemergmed.com.
0196-0644/$-see front matterCopyright © 2008 by the American College of Emergency Physicians.doi:10.1016/j.annemergmed.2009.01.031
INTRODUCTIONBackground
It has been 25 years since the first case report of long QTsyndrome being misdiagnosed as epilepsy was published.1
Several other cases have since appeared in the literature.2-4
We recently reported the history of a 12-year-old boydiagnosed with and treated for epilepsy who was sub-
sequently a victim of sudden death. Although his death was26 Annals of Emergency Medicine
initially ascribed to sudden unexplained death in epilepsy,posthumous genetic screening identified long QT syndrometype 1.5
ImportanceThe natural history of long QT syndrome suggests mortality
greater than 20% in the year after the first syncopal event, with
nearly 50% mortality at 5 years.6 Appropriate intervention canVolume , . : July

MacCormick et al Misdiagnosis of Long QT Syndrome as Epilepsy
significantly reduce mortality and morbidity, making promptdiagnosis essential.7-10
Goals of This InvestigationConcerned by the ongoing delayed recognition of a
condition with preventable mortality, we aimed to establish thefrequency of this problem in a series of patients with geneticallyconfirmed long QT syndrome. We also explored the factorscontributing to, and the consequences of, diagnostic delay oflong QT syndrome.
MATERIALS AND METHODSBetween 2000 and 2005, 84 families underwent molecular
diagnostic screening through the Cardiac Inherited DiseaseRegistry in New Zealand. A team of physicians interested ininherited causes of arrhythmia and sudden death has formedacross the country, with members in all the major centers. Thesephysicians are involved in the majority of cases of long QTsyndrome nationwide and arrange genetic testing, although theywould not see all case patients. Physicians in the CardiacInherited Disease Group routinely refer their patients to theregistry, but this is at the discretion of the physician and thepatient. A member of each family underwent genetic screeningof 5 long QT syndrome genes: KCNQ1, HERG, SCN5A,KCNE1, and KCNE2. The registry is ethically approved torecord confidential clinical and genetic information for
Editor’s Capsule Summary
What is already known on this topicLong QT syndrome is an uncommon butpotentially lethal condition.
What question this study addressedThis retrospective case series of patients shown tohave long QT syndrome after initially presentingwith loss of consciousness examined how oftendiagnosis was delayed and what diagnoses weremade instead.
What this study adds to our knowledgeThe diagnosis of long QT syndrome may beparticularly delayed when an initial diagnosis ofepilepsy is made. This may increase the mortalityrisk of the patient and their affected familymembers.
How this might change clinical practiceEmergency physicians should be cognizant thatpatients with sudden loss of consciousness couldhave a long QT syndrome.
management of and research into inherited cardiac disease.
Volume , . : July
Ethical approval for this registry has been obtained from thenational ethics committee.
We recently described the genetic abnormalities in 40consecutive probands with long QT syndrome (the probandbeing the first member of the family to be diagnosed).11 Thecurrent article is a case series of the 31 New Zealandprobands who had consented for the collection and storageof detailed clinical information when joining the registry.The other 9 subjects either lived outside New Zealand (6cases) or had not provided consent for detailed clinical audit.New Zealand provides every user of the health system with aNational Health Index number, a unique personal identifierused consistently throughout the country. This enabled athorough review of previous hospital records in the NewZealand patients who had consented for detailed clinicalinformation collection.
Our intention was to identify those patients who hadpresented with significant cardiac events before recognition oflong QT syndrome. Therefore we defined a “previouspresentation” as a previous assessment in a hospital emergencydepartment (ED) or specialist outpatient clinic, at which theprimary complaint involved sudden loss of consciousnesswithout obvious precipitating factors. Earlier medicalassessments for other symptoms, such as palpitations, werereviewed but not included as previous presentations because itwas less certain that these events were related to the eventualdiagnosis of long QT syndrome.
Information was abstracted from the clinical records by thefirst author, who was not blinded to the final diagnosis. Datawere collated with a standard abstraction form about history,investigations, diagnosis, and management. ECGs fromprevious presentations were reviewed when available. QTintervals corrected for pulse rate (QTc) were retrospectivelyremeasured and calculated with the Bazett formula12 by the firstauthor and checked independently by the senior author. TheQTc was measured in leads II and V5, which have been shownto correlate best to genotype status in families with long QTsyndrome13 and avoids leads V2 to V4, which often havecomplex repolarization patterns. The QT interval was measuredfrom the onset of the QRS to the intersection of the end of theT wave with baseline or to the end of the U wave if this wasgreater than 50% of the height of the T wave. The Bazettformula is a method of correcting the absolute QT interval forpulse rate and involves dividing the measured absolute QT bythe square root of the preceding RR interval. The QTc intervalwas considered prolonged if one of the measurements wasgreater than or equal to 0.48 and borderline from 0.44 to 0.48.The authors’ ECG findings were subsequently compared withthe interpretation documented by the treating physician in theclinical record at presentation. The majority of ECGs reviewedhad no automated measurement of QTc.
For those patients with a delayed diagnosis of long QTsyndrome, the median duration of delay was compared between
the subgroup of patients initially misdiagnosed with epilepsyAnnals of Emergency Medicine 27
ded.
Misdiagnosis of Long QT Syndrome as Epilepsy MacCormick et al
and the others. Statistical analysis included the Mann-WhitneyU test for the difference between 2 medians.
RESULTSThirty-one gene-positive probands were identified. The
presentation resulting in eventual diagnosis of long QTsyndrome was syncope in 20 (64.5%), resuscitated cardiac deathrequiring defibrillation in 6 (19.5%), incidental ECGabnormality in 3 (10%), palpitations in 1 (3%), and prenatalbradycardia in 1 (3%). The molecular mutations in thesepatients were consistent with LQT1 in 18 (58%), LQT2 in 10(32%), and LQT3 in 3 (10%). Seventy-one percent of probandswere female patients. The median age at diagnosis of long QTsyndrome was 21 years, with a range of 1 day to 54 years.
Eighteen patients (61%) were diagnosed with long QTsyndrome at their first presentation to a hospital or outpatientclinic. Thirteen patients (39%) had been assessed for significantpresentations with syncope or seizure before the diagnosis oflong QT syndrome was made. For these 13 patients, geneticmutations were consistent with LQT1 in 6 and LQT2 in 7.That is, those who were misdiagnosed constituted 33% of theLQT1 probands and 70% of the LQT2 probands. Five of thesepatients had presented on 2 or more occasions with loss ofconsciousness, including seizures, before diagnosis.
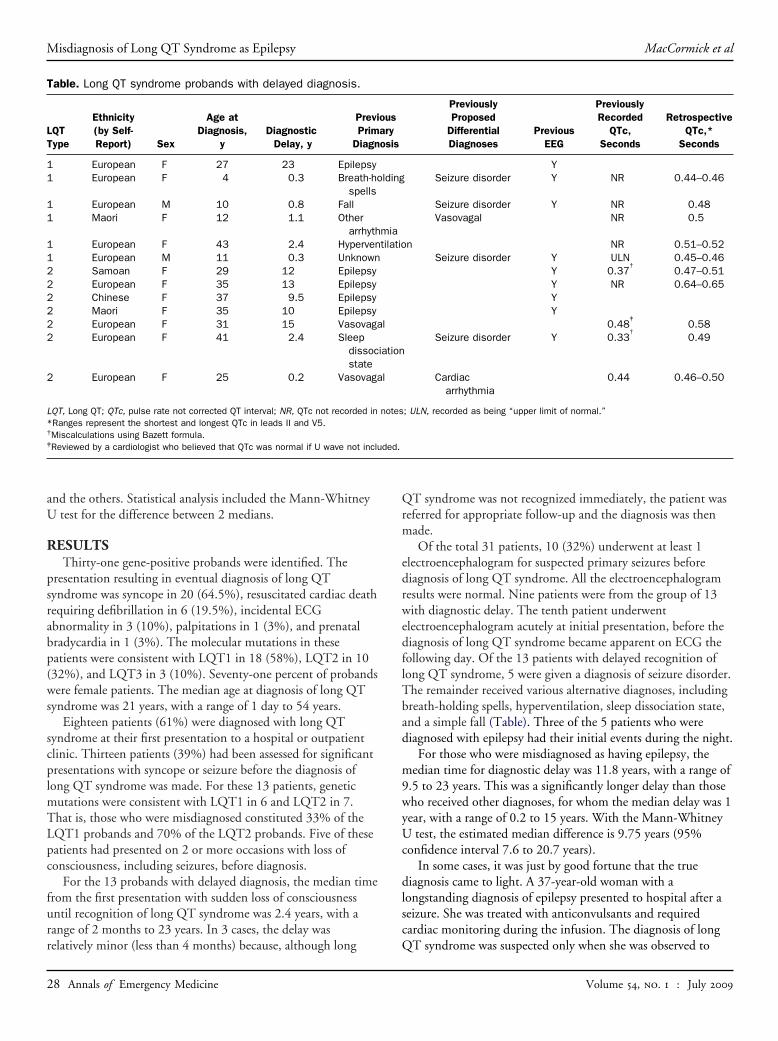
For the 13 probands with delayed diagnosis, the median timefrom the first presentation with sudden loss of consciousnessuntil recognition of long QT syndrome was 2.4 years, with arange of 2 months to 23 years. In 3 cases, the delay was
Table. Long QT syndrome probands with delayed diagnosis.
LQTType
Ethnicity(by Self-Report) Sex
Age atDiagnosis,
yDiagnostic
Delay, y
PreviPrim
Diagn
1 European F 27 23 Epilepsy1 European F 4 0.3 Breath-ho
spells1 European M 10 0.8 Fall1 Maori F 12 1.1 Other
arrhyth1 European F 43 2.4 Hyperven1 European M 11 0.3 Unknown2 Samoan F 29 12 Epilepsy2 European F 35 13 Epilepsy2 Chinese F 37 9.5 Epilepsy2 Maori F 35 10 Epilepsy2 European F 31 15 Vasovaga2 European F 41 2.4 Sleep
dissocstate
2 European F 25 0.2 Vasovaga
LQT, Long QT; QTc, pulse rate not corrected QT interval; NR, QTc not recorded in*Ranges represent the shortest and longest QTc in leads II and V5.†Miscalculations using Bazett formula.‡Reviewed by a cardiologist who believed that QTc was normal if U wave not inclu
relatively minor (less than 4 months) because, although long
28 Annals of Emergency Medicine
QT syndrome was not recognized immediately, the patient wasreferred for appropriate follow-up and the diagnosis was thenmade.
Of the total 31 patients, 10 (32%) underwent at least 1electroencephalogram for suspected primary seizures beforediagnosis of long QT syndrome. All the electroencephalogramresults were normal. Nine patients were from the group of 13with diagnostic delay. The tenth patient underwentelectroencephalogram acutely at initial presentation, before thediagnosis of long QT syndrome became apparent on ECG thefollowing day. Of the 13 patients with delayed recognition oflong QT syndrome, 5 were given a diagnosis of seizure disorder.The remainder received various alternative diagnoses, includingbreath-holding spells, hyperventilation, sleep dissociation state,and a simple fall (Table). Three of the 5 patients who werediagnosed with epilepsy had their initial events during the night.
For those who were misdiagnosed as having epilepsy, themedian time for diagnostic delay was 11.8 years, with a range of9.5 to 23 years. This was a significantly longer delay than thosewho received other diagnoses, for whom the median delay was 1year, with a range of 0.2 to 15 years. With the Mann-WhitneyU test, the estimated median difference is 9.75 years (95%confidence interval 7.6 to 20.7 years).
In some cases, it was just by good fortune that the truediagnosis came to light. A 37-year-old woman with alongstanding diagnosis of epilepsy presented to hospital after aseizure. She was treated with anticonvulsants and requiredcardiac monitoring during the infusion. The diagnosis of long
PreviouslyProposed
DifferentialDiagnoses
PreviousEEG
PreviouslyRecorded
QTc,Seconds
RetrospectiveQTc,*
Seconds
YSeizure disorder Y NR 0.44–0.46
Seizure disorder Y NR 0.48Vasovagal NR 0.5
n NR 0.51–0.52Seizure disorder Y ULN 0.45–0.46
Y 0.37†
0.47–0.51Y NR 0.64–0.65YY
0.48‡
0.58Seizure disorder Y 0.33
†0.49
Cardiacarrhythmia
0.44 0.46–0.50
; ULN, recorded as being “upper limit of normal.”
ousaryosis
lding
miatilatio
l
iation
l
notes
QT syndrome was suspected only when she was observed to
Volume , . : July

MacCormick et al Misdiagnosis of Long QT Syndrome as Epilepsy
have episodes of ventricular tachycardia on the monitor. A 12-lead ECG then confirmed the presence of a prolonged QTinterval.
Another woman in this study had been diagnosed with andtreated for epilepsy for many years before reading an articleabout long QT syndrome in a magazine. The description was soconsistent with her own experience that she was prompted toseek a further medical opinion, at which time the diagnosis oflong QT syndrome was established.
Ten of the 13 patients with delayed diagnosis were foundto have had an ECG recorded at a previous presentation(Table). Retrospective review revealed at least 1 previousECG with a QTc interval that was prolonged in 8 patients(QTc 0.47 to 0.65) and borderline in 2 patients. The ECGsin all 8 patients with prolonged QTc intervals had beeninterpreted as normal by the treating physician. The QTcinterval had not been specifically documented in the originalmedical notes in 4 cases and was incorrectly recorded in 4cases. Two of the latter cases could be identified asmiscalculations with the Bazett formula.
Two further patients were observed to have had previouslyunrecognized QTc prolongation on ECGs requested for otherindications. One of these was a child with Kearns-SayreSyndrome14 (QTc 0.48 to 0.50), and the other was an adultwho had been investigated for palpitations (QTc 0.45 to 0.48).Both of these patients also had ECGs with normal QTcintervals in the past. The total number of patients identifiedwith previously unrecognized QTc interval prolongation was 10of 31 (32%).
We reviewed the recorded family histories of the 13 patientswith diagnostic delay to establish whether there were anysudden unexpected deaths at younger than 40 years. We wereable to identify that in 4 cases, there was a reported history fromthe family of the sudden unexpected and unexplained death of arelative younger than 40 years during the time between theinitial presentation of the proband and eventual recognition oflong QT syndrome. The deceased was a sibling of the probandin 3 cases and a first cousin in the fourth case. In one case, boththe proband and her deceased sibling had congenital deafness,meaning the diagnosis of long QT syndrome in the deceasedwas almost beyond doubt.
LIMITATIONSThis study was subject to all the usual pitfalls of retrospective
review. Data recorded at the time of presentation were notcollected for the purposes of later study. The information wascollected by the first author, who was not blinded to the finaldiagnosis of long QT syndrome in these patients. Patientnumbers are small, not all patients in the registry consented forreview of medical records, and the study was restricted to thosewho had positive genotype testing results. Previouspresentations to a hospital or specialist outpatient clinic werereviewed, and thus we may have missed those who hadpreviously presented with syncope to their family practitioner, a
community emergency clinic or other health professional.Volume , . : July
However, we would propose that on balance these exclusions arelikely to have resulted in an underrepresentation of thefrequency of delayed diagnosis. The period covered by thisreview is long, and there may well have been changes in practicesince the earlier presentations.
DISCUSSIONThe first description of a long QT syndrome in 1957 was of
a recessive form associated with deafness,15 followed in the1960s by reports of a syndrome with autosomal dominantinheritance.16,17 The first study using linkage analysis toestablish an underlying genetic basis was published in 1991.18
To date, we know of 11 genes associated with long QTsyndrome. Within these genes, there are more than 600mutations described thus far, highlighting the significant geneticheterogeneity of the condition.11,19-23
The different genetic subtypes of long QT syndrome resultin individual clinical disorders, with their own characteristicpatterns of presentation, ECG abnormalities, prognosis, andoptimal management.24-27 Current estimates of the prevalenceof long QT syndrome vary from 1 in 2000 to 1 in 7000. Thesemay still be underestimates, given that long QT syndrome isfrequently unrecognized and has variable penetrance.24,28
The propensity for long QT syndrome to imitate otherconditions has been described in the literature. It is wellestablished that cardiac syncope can result in secondary hypoxiaand convulsions, thus mimicking a primary seizure disorder.29
Misdiagnosis of long QT syndrome as epilepsy was described asearly as 1983, and there have been subsequent cases reported inthe literature.1-5 Nevertheless, the diagnosis of long QTsyndrome continues to be missed.
Prompt recognition of long QT syndrome is essentialbecause mortality can be significantly reduced with establishedinterventions. �-Blocker therapy has proven effectiveness inLQT1 and LQT2.6,10 Left-sided cardiac sympatheticdenervation has been shown to be helpful in high-riskpatients,8,9 and overdrive cardiac pacing is useful in somepatients. The introduction of implantable cardioverterdefibrillators for high-risk patients with long QT syndrome hasfurther reduced mortality.7
Our findings show that delayed recognition of long QTsyndrome is still common, with 39% of the patientsexperiencing delay between initial presentation and diagnosis.Although the results and conclusions of this review are based ona New Zealand cohort, they are likely to be applicable to awider population, including the United States. New Zealandhas a high-quality government-funded national health andwelfare service. The health expenditure as a proportion of grossdomestic product is comparable to that of Finland, Spain, andthe United Kingdom.30
The most frequent misdiagnosis in this study was of aprimary seizure disorder. The observation that 31% of thepatients were investigated with electroencephalograms illustratesthe considerable overlap in the clinical presentation of the long
QT syndrome and seizure disorders. Other misdiagnoses notedAnnals of Emergency Medicine 29

Misdiagnosis of Long QT Syndrome as Epilepsy MacCormick et al
in this study included breath-holding spells, hyperventilation,sleep dissociation state, and a simple fall. Misdiagnosis asbreath-holding has also been previously reported in theliterature.31 The authors acknowledge that more than 1condition may exist in an individual; a patient with long QTsyndrome may also have vasovagal syncope or even epilepsy.However, on review of our case series it seems likely that most,if not all, of the previous events in this study were a result oflong QT syndrome.
We observed that an individual might be labeled as epilepticfor many years before the correct diagnosis is made. An initialdiagnosis of seizure disorder was associated with a longer delayin recognition of long QT syndrome than other misdiagnoses.All 5 patients with a diagnosis of epilepsy had a delay of greaterthan 9 years before long QT syndrome was recognized. Thishighlights a need to maintain a high degree of suspicion, even inpatients with a longstanding diagnosis of epilepsy, andparticularly in those with atypical presentations, refractoryseizures, nocturnal seizures, or a normal electroencephalogramresult.
Previous studies have examined cohorts of patients with“epilepsy” and found the diagnosis to be incorrect in 20% to42%.32-34 Many of these patients were found to have“cardiogenic” causes, particularly convulsive vasovagal syncope.The numbers of patients with unrecognized long QT syndromein the epilepsy population may well be small, given the relativelylow prevalence of long QT syndrome. Nevertheless,identification of even a single case of long QT syndromepotentially enables a reduction in mortality and morbidity forboth the individual and the extended family.
Cardiac syncope is typically described as being of suddenonset, without warning symptoms and with rapid return tobaseline level of alertness, as opposed to a postictal state. It iswell recognized that collapse associated with exercise should be acause for concern, prompting careful investigation. Themajority of patients misdiagnosed with epilepsy in this serieshad experienced their events nocturnally. It may be that thisform of presentation is less likely to raise suspicion of cardiacsyncope and thus more prone to misdiagnosis. Patients withLQT1 typically have episodes triggered by exercise orswimming, whereas events during sleep or rest are morecommon in LQT2,24 which might explain the trend toward apreponderance of misdiagnosis in probands with LQT2 overLQT1.
It has been suggested that screening ECGs for long QTsyndrome are indicated in those presenting with their firstafebrile seizure (unless the electroencephalogram result isdiagnostic), those with unexplained syncope, and those withcongenital deafness.35 The current American College ofEmergency Physicians clinical policy for the evaluation andmanagement of adult patients presenting to the ED with new-onset seizures does not recommend an ECG,36 which is incontrast to the English and Scottish guidelines for investigation
of first seizure, both of which recommend ECGs according to30 Annals of Emergency Medicine
the clinical experience of the guideline development group.37,38
The authors of this article would agree that an ECG should beconsidered in all first episodes of seizure and also suggestconsideration in cases in which there is a longstanding diagnosisof epilepsy, resistant to treatment, without a diagnosticelectroencephalogram.
Our case series suggests that even when ECGs areundertaken as part of the assessment for syncope and seizure,interpretation is often suboptimal, which is consistent with arecent study that found that less than 40% of physicians(noncardiologists) and less than 50% of cardiologists were ableto calculate a QTc interval correctly.39 Most current ECGmachines provide an automated QTc interval, but the clinicalexperience of the authors would not recommend relying on this.The QTc should be calculated manually, preferably in leads IIand V5.13 The end of the T wave can be measured at the pointwhere it intersects with the baseline, although a method usingthe intersection of the tangent to the steepest downward slopewith the baseline is also sometimes used. When there is a Uwave present, it is generally recommended that this be includedin the T wave measurement when it is greater than 50% of theheight of the T wave. T wave morphology can provideadditional information when assessing for long QT syndrome,with different repolarization patterns being associated withparticular genotypes.25
The hereditary nature of long QT syndrome means that earlyrecognition of the condition in the proband has implications forthe wider family. Up to approximately 50% of family memberswill be gene carriers and potentially at risk. Clinical and geneticscreening can identify carriers, enabling therapeuticintervention. In our series, 4 family members reportedly diedsuddenly at a young age during the time from initialpresentation of the proband until the diagnosis of long QTsyndrome. Although we cannot confirm that all of these deathsresulted from long QT syndrome, the sudden and unexplainednature of the events at a young age make it highly likely. Wepropose that recognition of long QT syndrome within a family,resulting in appropriate advice and intervention, may lead toreduced mortality.
Population-based studies of young sudden cardiac deathvictims have shown that a third have a negative postmortemresult, with death presumed to be a result of arrhythmia.40,41
Long QT syndrome may account for 20% of such postmortemnegative-result cases.42 Further efforts are needed to identifyindividuals with long QT syndrome. Some researchers haveeven recommended newborn screening with ECGs.43
Additional studies investigating the prevalence of arrhythmicsyncope among those diagnosed with seizure disorders would beuseful.
The preventable mortality and hereditary nature of long QTsyndrome make early diagnosis a priority for the individual andthe family. We would suggest that, as well as taking a familyhistory for syncope and young sudden death, an ECG be
considered after all first afebrile seizures, including nocturnalVolume , . : July

MacCormick et al Misdiagnosis of Long QT Syndrome as Epilepsy
seizures. In addition, ECGs should be considered in patientspresenting with seizure who have a longstanding diagnosis ofepilepsy and a negative electroencephalogram result. Accuratecalculation of the QTc interval is essential. The overlap in theclinical presentation of long QT syndrome with other disorders,particularly epilepsy, requires the medical community tomaintain a high index of suspicion.
Supervising editor: Keith A. Marill, MD
Author contributions: All authors participated in the concept,design, analysis, writing, and revision of the article. JS takesresponsibility for the paper as a whole.
Funding and support: By Annals policy, all authors are requiredto disclose any and all commercial, financial, and otherrelationships in any way related to the subject of this article,that might create any potential conflict of interest. See theManuscript Submission Agreement in this issue for examplesof specific conflicts covered by this statement. The CardiacInherited Disease Group is supported by Cure Kids, the GreenLane Research and Education Fund and the Lion Foundation.Dr. MacCormick was supported by a research fellowship grantfrom the Southern Trust.
Publication dates: Received for publication June 22, 2008.Revisions received November 16, 2008, and January 18,2009. Accepted for publication January 26, 2009. Availableonline March 12, 2009.
Reprints not available from the authors.
Address for correspondence: J. R. Skinner, MD, Green LanePaediatric and Congenital Cardiac Services, StarshipChildren’s Hospital, Level 3, Building 32, Private Bag 92 029,Auckland, New Zealand; 64-9-307-4949, fax 64-9-631-0785;E-mail [email protected].
REFERENCES1. Ballardie FW, Murphy RP, Davis J. Epilepsy: a presentation of the
Romano-Ward syndrome. Br Med J (Clin Res Ed). 1983;287:896-897.
2. O’Callaghan CA, Trump D. Prolonged QT syndrome presenting asepilepsy. Lancet. 1993;341:759-760.
3. Herman LL, Stoshak M, Rittenberry TJ. Long QT syndromepresenting as a seizure. Am J Emerg Med. 1992;10:435-438.
4. Moss AJ, Schwartz PJ, Crampton RS, et al. The long QTsyndrome. Prospective longitudinal study of 328 families.Circulation. 1991;84:1136-1144.
5. Skinner J, Chong B, Fawkner M, et al. Use of the newborn screeningcard to define cause of death in a 12-year-old diagnosed withepilepsy. J Paediatr Child Health. 2004;40:651-653.
6. Schwartz PJ. Idiopathic long QT syndrome: progress andquestions. Am Heart J. 1985;109:399-411.
7. Zareba W, Moss AJ, Daubert JP, et al. Implantable cardioverterdefibrillator in high-risk long QT syndrome patients. J CardiovascElectrophysiol. 2003;14:337-341.
8. Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympatheticdenervation in the management of high-risk patients affected bythe long-QT syndrome. Circulation. 2004;109:1826-1833.
9. Schwartz PJ, Locati EH, Moss AJ, et al. Left cardiac sympatheticdenervation in the therapy of congenital long QT syndrome. A
worldwide report. Circulation. 1991;84:503-511.Volume , . : July
10. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitationsof beta-blocker therapy in congenital long-QT syndrome.Circulation. 2000;101:616-623.
11. Chung S-K, MacCormick JM, McCulley CH, et al. Long QT andBrugada syndrome gene mutations in New Zealand. HeartRhythm. 2007;4:1306-1314.
12. Bazett H. An analysis of the time relations of electrocardiograms.Heart. 1920;7:353-367.
13. Monnig G, Eckardt L, Wedekind H, et al. Electrocardiographic riskstratification in families with congenital long QT syndrome. EurHeart J. 2006;27:2074-2080.
14. Skinner JR, Yang T, Purvis D, et al. Coinheritance of long QTsyndrome and Kearns-Sayre syndrome. Heart Rhythm. 2007;4:1568-1572.
15. Jervell A, Lange-Neilson F. Congenital deaf-mutism, functionalheart disease with prolongation of the Q-T interval and suddendeath. Am Heart J. 1957;54:59-68.
16. Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’etapediatrica. II. Accessi sincopali per febrillazione ventricolareparossistica. Clin Pediatr (Bologna). 1963;45:656-683.
17. Ward O. New familial cardiac syndrome in children. J Irish MedAssoc. 1964;54:103-106.
18. Keating M, Atkinson D, Dunn C, et al. Linkage of a cardiacarrhythmia, the long QT syndrome, and the Harvey ras-1 gene.Science. 1991;252:704-706.
19. Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations inlong QT syndrome genes. Circulation. 2000;102:1178-1185.
20. Jongbloed R, Marcelis C, Velter C, et al. DHPLC analysis ofpotassium ion channel genes in congenital long QT syndrome.Hum Mutat. 2002;20:383-391.
21. Liu W, Yang J, Hu D, et al. KCNQ1 and KCNH2 mutationsassociated with long QT syndrome in a Chinese population. HumMutat. 2002;20:475-476.
22. Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiacchannel mutations in 541 consecutive unrelated patients referredfor long QT syndrome genetic testing. Heart Rhythm. 2005;2:507-517.
23. Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in thelong QT syndrome: development and validation of an efficientapproach to genotyping in clinical practice. JAMA. 2005;294:2975-2980.
24. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotypecorrelation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
25. Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST-T-wavepatterns and repolarization parameters in congenital long-QTsyndrome: ECG findings identify genotypes. Circulation. 2000;102:2849-2855.
26. Napolitano C, Bloise R, Priori SG. Gene-specific therapy for inheritedarrhythmogenic diseases. Pharmacol Ther. 2006;110:1-13.
27. Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype onthe clinical course of the long-QT syndrome. International Long-QTSyndrome Registry Research Group. N Engl J Med. 1998;339:960-965.
28. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the Long-QT syndrome: clinical impact. Circulation. 1999;99:529-533.
29. Schott GD, McLeod AA, Jewitt DE. Cardiac arrhythmias thatmasquerade as epilepsy. Br Med J. 1977;1:1454-1457.
30. New Zealand health and disability sector overview. Ministry ofHealth. Available at: http://www.moh.govt.nz. AccessedSeptember 4, 2007.
31. Franklin WH, Hickey RW. Long-QT syndrome. N Engl J Med. 1995;
333:355.Annals of Emergency Medicine 31

Misdiagnosis of Long QT Syndrome as Epilepsy MacCormick et al
32. Zaidi A, Clough P, Cooper P, et al. Misdiagnosis of epilepsy:many seizure-like attacks have a cardiovascular cause. J Am CollCardiol. 2000;36:181-184.
33. McDade G, Brown SW. Non-epileptic seizures: management andpredictive factors of outcome. Seizure. 1992;1:7-10.
34. Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsyand the management of refractory epilepsy in a specialist clinic.QJM. 1999;92:15-23.
35. Davis AM, Wilkinson JL. The long QT syndrome and seizures inchildhood. J Paediatr Child Health. 1998;34:410-411.
36. American College of Emergency Physicians. Clinical policy: criticalissues in the evaluation and management of adult patientspresenting to the emergency department with seizures. AnnEmerg Med. 2004;43:605-625.
37. The diagnosis and management of the epilepsies in adults andchildren in primary and secondary care. NHS National Institute forClinical Excellence. Available at: http://www.nice.org.uk/nicemedia/
pdf/CG020fullguideline.pdf. Accessed September 20, 2008.32 Annals of Emergency Medicine
38. Diagnosis and management of epilepsies in children and youngpeople. Scottish Intercollegiate Guidelines Network. Available at:http://www.sign.ac.uk/pdf/sign81.pdf. Accessed September 20,2008.
39. Viskin S, Rosovski U, Sands AJ, et al. Inaccurateelectrocardiographic interpretation of long QT: the majority ofphysicians cannot recognize a long QT when they see one. HeartRhythm. 2005;2:569-574.
40. Puranik R, Chow CK, Duflou JA, et al. Sudden death in the young.Heart Rhythm. 2005;2:1277-1282.
41. Doolan A, Langlois N, Semsarian C. Causes of sudden cardiacdeath in young Australians. Med J Aust. 2004;180:110-112.
42. Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetictesting for sudden unexplained death in the young. J Am CollCardiol. 2007;49:240-246.
43. Quaglini S, Rognoni C, Spazzolini C, et al. Cost-effectiveness ofneonatal ECG screening for the long QT syndrome. Eur Heart J.
2006;27:1824-1832.Residents’ Perspective: Call for Submissions
The Residents’ Perspective section of Annals of Emergency Medicine has been a fixture in the journal since 1993 andprovides a peer-reviewed venue for the unique perspective of the resident physician. We publish brief articles authored orco-authored by residents, including data-based reviews of important topics that have not been well covered elsewhere,informative instructional pieces of particular interest to residents, and occasionally, well-referenced position papers. Wealso welcome small-scale original research articles, especially those that address educational innovations and arepresented in the context of a broader discussion of the current literature. We do not publish individual opinion pieces.
We are particularly, though not exclusively, interested in pieces co-authored by residents and expert faculty in the field. Ifyou have a topic you would like to cover and are not able to find a co-author, please contact us, as we may be able tosuggest one.
To develop a manuscript for the Residents’ Perspective section, please complete a brief literature review on your chosentopic to ensure that it has not recently been covered elsewhere and then submit a 300-word structured abstract. Theabstract should include the following information: the proposed title and authors (not included in the 300 words); a briefbackground of the topic, including its significance to emergency medicine practice; an outline of the proposed structure ofthe article; and any pertinent references. If you are interested in submitting a well-referenced original manuscript that isalready completed, please contact us by e-mail.
All submitted abstracts should be received by July 14th, 2009. Once abstracts have been approved, final manuscriptsshould be received within 2 months of approval date.
Submit abstracts by email to Aaron Brown, MD and Suzanne Lippert, MD, MS, Resident Fellows, at [email protected].
Volume , . : July
Related Documents











