Microglia podosomes: Characterization, Ca 2+ regulation and potential role in migration by Tamjeed Ahmed Siddiqui A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Physiology University of Toronto © Copyright by Tamjeed Ahmed Siddiqui 2012

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Microglia podosomes:
Characterization, Ca2+ regulation and potential role in migration
by
Tamjeed Ahmed Siddiqui
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Graduate Department of Physiology
University of Toronto
© Copyright by Tamjeed Ahmed Siddiqui 2012

Microglia podosomes: Characterization, Ca2+ regulation and potential role in
migration
Tamjeed Ahmed Siddiqui
Master of Science
Graduate Department of Physiology University of Toronto
2012
ABSTRACT
Microglia, immune cells of the central nervous system, activate in response to
pathophysiological stimuli. One of their reactive phenotypes is to migrate to site
of injury where they could have either beneficial or detrimental effects. However,
little is known regarding the mechanisms underlying microglial migration and how
they traverse the unique extracellular environment in brain tissue to reach their
destination. Our laboratory first discovered that microglia express structures
called podosomes, which can adhere to as well as degrade extracellular matrix.
In this study, I further characterize microglial podosomes, and show that they
associate with Iba1, Orai1 and calmodulin, proteins not yet observed in
podosomes of other cell types. I also present evidence that podosome formation
depends on Ca2+ and its entry through store-operated Ca2+ channels. The
findings in this thesis contribute to a better understanding of podosome dynamics
and their probable roles in microglia migration.
ii

ACKNOWLEDGEMENTS
I would like to begin by expressing my gratitude towards my supervisor, Dr
Lyanne C Schlichter. Her support, dedication, enthusiasm and advice among
many other attributes made working with her an educational yet fun experience.
Throughout my master’s program, she taught me many valuable lessons that
allowed me to evolve and mature into a better scientist. Dr Schlichter continues
to be a wonderful mentor and I am looking forward to working with her.
I extend thanks to my committee members, Dr Milton Charlton and Dr
Rene Harrison, and examination committee members, especially Dr Christopher
McCulloch, for their time and valued suggestions.
My gratitude also goes to my peers at the lab as well as staff members at
the department who always helped me with any inquiries or issues that I would
face. Specifically, Iska and Star for being patient and kind enough to discuss
topics with me and review my writing. Baosong’s invaluable help with so many
experiments and techniques as well as wonderful “thought experiments” that he
came up with. To Cat, for taking the time to teach me immunocytochemistry
techniques and imaging and Roger being a good person to bounce ideas off of. I
would especially like to thank Xiaoping Zhu. With an infectious smile, she has
been a trusted and reliable colleague, a wonderful friend and someone I can
always count on. I am truly indebted to her for everything she has done and for
teaching me so many techniques.
Special thanks go to my parents for their self-sacrifice to send their
teenage son to Toronto to ensure I received one of the best educations available.
My mother, who provides endless love to the family, taught me that through hard
work anything can be achieved and to never give up. My father, who has always
been patient and understanding, taught me to be honest, tolerant and have an
open mind. They continue to be my role models and are the architects of my
personality. Thanks to both my brothers as well, my partners in crime, for always
being there and sharing the good times with me.
iii

To my one and only Kiruththiga Sures, my companion, my best friend, my
love. Like an angel, she has always been by my side through thick and thin,
always encouraging and motivating me to strive for the best and never settle for
less. She has been so caring, patient and understanding for the countless
number of times that I was late (and after hearing every excuse in the book that I
could imagine). I may never be able to repay her for everything she has done for
my family and especially for me. Thank you.
iv

TABLE OF CONTENTS
Abstract ii
Acknowledgements iii
List of Figures vi List of Tables viii Introduction
Cell migration 1
Invadosomes 4
Introduction to Monocytic Immune Cells 10
Microglia – Immune cells of the CNS 12
Podosomes in microglia 16
Thesis Objectives 20
Materials and Methods
Microglia culture 21
Immunocytochemistry (ICC) 22
Ca2+ and podonuts (podosomes) 24
Image Analysis 26
Results
Previously known podosome components found in microglial podosomes
29
Ca2+ and podosome (podonut) formation 36
Novel expression of several molecules in microglia podosomes 42
Discussion 48
Working Model 59
Future studies 64
References cited 66
v

LIST OF FIGURES
Figure 1. Schematic diagram of migrating cell (Chhabra and Higgs, 2007)
Figure 2. (Top) Electron micrograph showing distinct core and ring structural
arrangement in smooth muscle cell podosome. (Bottom) Schematic diagram
showing localization of common podosome-associated molecules
Figure 3. Schematic diagram illustrating development of immune cells in blood and CNS
Figure 4. Primary rat microglia express podosomes
Figure 5. Functional podosomes expressed in microglia with SK3 localized in podosome
core
Figure 6. Microglia in vitro express podonuts after 20 hr in culture
Figure 7. Heat induced antigen retrieval (HIAR) enhances antibody specificity for
immunogen
Figure 8. Illustration of fluorescence intensity normalization of immunocytochemistry
images
Figure 9. Phosphotyrosine staining show localization to microglial podosomes
Figure 10. Immunostaining of Tks5 show localization to podosome ring in microglia
Figure 11. Phospho-caveolin-1 (pCav1) localizes to podosome ring in microglia
Figure 12. Immunolabeling of Nox1 shows localization to podosome ring in microglia
Figure 13. Ca2+ plays crucial role in podosome (podonut) formation
Figure 14. Importance of Ca2+ for podonut formation
Figure 15. Podosome (podonut) formation required Ca2+ entry through CRAC channels
Figure 16. Blocking CRAC channels results in loss of podosome (podonut) distribution
Figure 17. Immunolabeling of Orai1 show localization to podosome core in microglia
vi

Figure 18. Immunostaining of calmodulin (CaM) mostly shows localization to podosome
ring in microglia
Figure 19. Iba1 staining show localization to core in microglial podosomes
Figure 20. Immunolabeling of TRPM7 and podosome core in microglia
Figure 21. Updated schematic diagram showing podosome-associated molecules in
microglia based on findings in this thesis
vii

LIST OF TABLES
Table 1. Components that constitute podosomes, invadopodia and focal adhesions
Table 2. Summary of podosome components found in microglia and their corresponding
localization
viii

INTRODUCTION
Cell Migration
Cell migration is an important phenomenon that aids in various processes like
development of tissues, immune system function, angiogenesis and wound healing (Alberts
et al., 2008). It can also have a negative influence in pathological processes like tumour
metastasis. Cell migration is the result of a complex interplay between various intracellular
signalling pathways (e.g. Rho family GTPases, Cdc42, protein tyrosine kinases)(Ridley et al.,
2003; Alberts et al., 2008). Most of our understanding comes from studies based on different
cell types such as neutrophils, Dictyostelium, and fibroblasts (see reviews Lauffenburger and
Horwitz, 1996; Weiner, 2002; Le Clainche and Carlier, 2008; Vicente-Manzanares et al.,
2009). It is important to note that although cell migration is a well-studied phenomenon,
detailed mechanisms vary between different cell types and species (Becchetti and Arcangeli,
2010). Migration can be classified into two basic forms: random migration and directed
migration (Ridley et al., 2003; Gilbert, 2006). Random migration (chemokinesis) involves
intrinsic factors being activated that initiate migration without responding to any external
cues, such that cells migrate spontaneously in any direction. Directed migration, however,
adds more complexity to the intricate processes underlying cell migration. Migration
following an external cue, also known as chemotaxis, involves not only regulating the
migration machinery but also responding to the external stimuli via receptor signalling that
continually influences the migration machinery. In either case, cell migration is initiated by
inducing cell polarity and extension of the cell membrane and cytoplasm in the direction of
migration (Ridley et al., 2003). This establishes a leading edge at the cell front and a uropod
at the rear. The leading edge is the site of extensive assembly of cytoskeleton, mainly actin
1

polymerization, protruding lamellipodia and a lamellum (Figure 1). Lamellipodia are sites of
rapid actin polymerization and protrusion that form membrane ruffles that have not yet
adhered to substrate. The lamellum, on the other hand, is located dorsal to lamellipodia, and
provides stability and traction by adhering to the extracellular matrix (ECM). The uropod in
the trailing end of migrating cells is a site of disassembly of cytoskeletal elements like
microtubules, and of retraction and detachment from ECM that allows cells to move forward.
In addition to signalling pathways that regulate cytoskeletal rearrangements,
regulation of intracellular Ca2+ is vital for cell migration (Evans and Falke, 2007). There is
evidence that many migrating cells maintain a descending Ca2+ gradient from rear-to-front of
the cell (Brundage et al., 1991; Laffafian and Hallett, 1995; Xu et al., 2004; Hoffmann et al.,
2009). Removal of extracellular Ca2+ impedes or stops cell migration, illustrating the
importance of local-, global- or spatiotemporally regulated Ca2+ concentrations during
migration (Brundage et al., 1991; Mandeville et al., 1995). Although it is believed that
migrating cells maintain a relatively low Ca2+ concentration at the cell front, Wei and
colleagues showed that directed migration is the result of localized Ca2+ rises in
microdomains (termed Ca2+ flickers) at cell front, which are short-lived and depend on
chemoattractant receptor signalling and membrane stretch (Wei et al., 2009). The authors
suggest that having a low Ca2+ background helps maintain a chemical driving force for Ca2+
entry through Ca2+ permeable channels at the cell front for subsequent initiation of Ca2+
signalling cascades necessary to steer the cells in a particular direction.
2

Figure 1. Schematic diagram of a migrating cell (Chhabra and Higgs, 2007).
Reprinted by permission from Macmillan Publishers Ltd: Nature Cell Biology. Chhabra ES, Higgs HN. The
many faces of actin: matching assembly factors with cellular structures. Copyright 2007
3

As important as regulation of signalling pathways and Ca2+ is to cell migration,
migrating cells also need to remodel the matrix in order to traverse through tissues (Alberts,
2009), which are made up of many cells that are held together by cell-cell interactions (e.g.
tight junctions) as well as cell-ECM contacts (Alberts, 2009). The ECM surrounding cells
serves not only as “glue” that allows cells to adhere for anchorage, but provides additional
support to keep the cells bound together. Characteristics of ECM in different tissues also
differ (Alberts, 2009); the components that make up the overall matrix are variable, leading
to differing rigidity, organization and fluidity. ECM typically consists of fibrillar collagens,
laminin and fibronectin, along with proteoglycans, glycosaminoglycans (GAGs) and
glycoproteins. The brain ECM, however, is very different from peripheral tissues, in that has
very little collagen and other commonly found ECM proteins (Bellail et al., 2004). Instead,
brain ECM primarily consists of glycosaminoglycan hyaluronan, proteoglycans, some of
which are specific for brain ECM (e.g. neurocan), and glycoproteins like tenascin-C. The
highly anionic hyaluronan, which is the major brain ECM component, binds hygroscopic
molecules (e.g., the cations, Na+ and Ca2+) and water (Bellail et al., 2004). The brain matrix
is water-rich and lacks rigid fibrillar collagens, making it softer than the ECM in other tissues
(Bellail et al., 2004). Taken together, this information means that cell migration through the
brain will require that cells degrade a specialized extracellular matrix and navigate through
an environment where cells are densely packed.
Invadosomes
Cell adhesion to the ECM substrate is also important for cell stability, anchorage and
traction during migration. Cell adhesion structures are sites where cells attach to the
4

underlying substratum. Focal complexes and focal adhesions are well-documented and
extensively studied adhesion structures found in many cell types, which under some
circumstances provide traction for cell migration. Integrins at the cell-ECM contact sites are
adhesion molecules that mediate communication with the cell regarding its interaction with
the ECM, and influence many signalling cascades, including cytoskeletal rearrangements
(Caswell et al., 2009). In the 1980s, a new type of adhesion structure was discovered: the
‘podosome’ (David-Pfeuty and Singer, 1980; Wolosewick, 1984; Tarone et al., 1985; Linder
and Aepfelbacher, 2003), which was later proposed to play a role in tissue invasion.
Monocyte-derived cells (e.g. macrophages) (Messier et al., 1993), constitutively active Src-
transformed cell lines (David-Pfeuty and Singer, 1980; Tarone et al., 1985; Abram et al.,
2003), endothelial cells (Osiak et al., 2005), and vascular smooth muscle cells (Burgstaller
and Gimona, 2005) are now known to express podosomes. Invasive cancer cells (e.g. human
breast cancer) (Mandal et al., 2008) possess similar structures, which were named
‘invadopodia’ (the combined term, invadosomes, is often used for invadopodia and
podosomes) (Murphy and Courtneidge, 2011). Podosomes were recently discovered in vivo
in transforming growth factor beta (TGFβ)-stimulated endothelial cells in mice (Rottiers et
al., 2009) and also in 3D cultures of macrophages in vitro (Rottiers et al., 2009; Cougoule et
al., 2010) .
Because many components of invadosomes are also present in other cell-ECM
adhesion structures, it was initially difficult to distinguish invadosomes, and determine
whether they are indeed discrete adhesion structures. It is now clear that they exhibit some
morphological, protein composition, and functional similarities and differences (reviewed in
(Block et al., 2008). Both podosomes and invadopodia are F-actin rich structures that
5

displays a unique ring and core structural arrangement (Figure 2) (Gimona, 2008). The ring
seems to have an adhesive nature and contains most of the integrins and adhesion-associated
molecules (e.g. talin, vinculin, paxillin); whereas, the core is rich in F-actin, and actin-
regulating molecules like Arp2/3 complex, WASP, and cortactin. Besides these cytoskeletal
regulators, other molecules have also been detected in invadosomes (Table 1) (Block et al.,
2008). Src tyrosine kinase signalling, a common signalling pathway involved in many
cellular processes including cell migration, is pivotal for the formation and regulation of
invadosomes (Linder and Aepfelbacher, 2003). Immunostaining for tyrosine phosphorylated
proteins shows localization to both the core and ring, suggesting their presence throughout
the structure. Unlike other adhesion structures, invadosomes can also degrade matrix due to
their association with matrix metalloproteinases (MMPs), and ADAMs (‘a disintegrin and
metalloproteinase’). There are several differences between the two subtypes of invadisomes.
Invadopodia have high degradative capability due to their slow turnover (>1 hr), whereas
podosomes are much more dynamic with turnover rates between 2 and 20 min, and shallower
matrix degradation. Invadopodia are larger structures (~5-8 µm in diameter) compared to
podosomes (~0.5-1 µm). Invadopodia are also sparsely distributed in cells (<10) while
podosomes are more compact and numerous (~20-100) (Murphy and Courtneidge, 2011).
Podosomes are protrusive adhesion structures primarily found at the ventral side of
polarised cells of the monocytic lineage, such as macrophages (Messier et al., 1993),
immature dendritic cells (Binks et al., 1998), and osteoclasts (Zambonin-Zallone et al., 1988;
Calle et al., 2006). Due to their adhesive properties, high turnover rates, and degradative
capacity, podosomes are well designed to aid highly migratory cells like macrophages in
traversing barriers like tissue layers (Carman et al., 2007). Indeed, when podosomes were
6

discovered, the initial opinion was that they functioned in transcellular migration
(Wolosewick, 1984).
7

Figure 2. (Top) Electron micrograph showing the distinct core and ring arrangement in a smooth muscle cell podosome (Gimona, 2008). (Bottom) Schematic diagram showing localization of common podosome-associated molecules (Linder and Aepfelbacher, 2003). Reprinted from The Lancet, Vol. 18, Gimona M, The microfilament system in the formation of invasive adhesions, 23-34, Copyright (2008), with permission from Elsevier Reprinted from The Lancet, Vol. 13, Linder S, Aepfelbacher M, Podosomes: adhesion hot-spots of invasive cells, 376-385, Copyright (2003), with permission from Elsevier
8
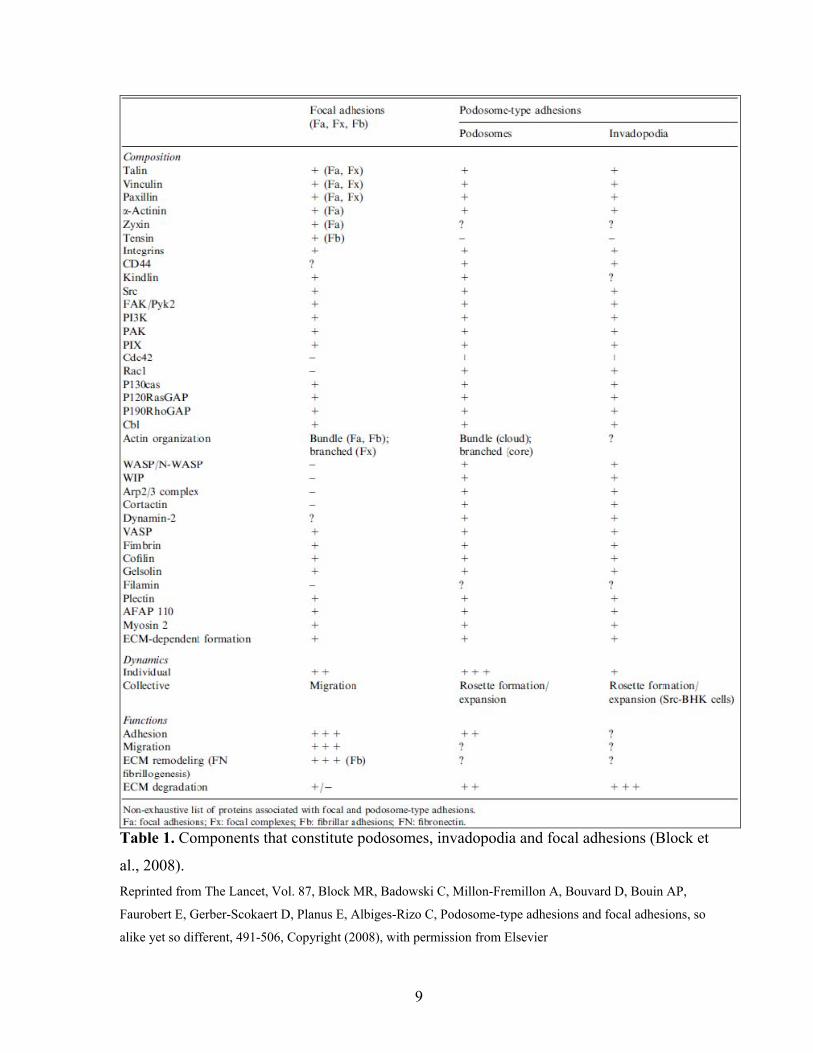
Table 1. Components that constitute podosomes, invadopodia and focal adhesions (Block et
al., 2008). Reprinted from The Lancet, Vol. 87, Block MR, Badowski C, Millon-Fremillon A, Bouvard D, Bouin AP,
Faurobert E, Gerber-Scokaert D, Planus E, Albiges-Rizo C, Podosome-type adhesions and focal adhesions, so
alike yet so different, 491-506, Copyright (2008), with permission from Elsevier
9

Introduction to monocytic immune cells
The immune system is a complex defensive array made up of several cell types with
different phenotypes (Alberts et al., 2008). All immune cells recognise and react to protect
the host from many pathophysiological circumstances, such as cancer, infection, and injury
(Kindt et al., 2007). Blood immune cells develop from a common hematopoietic stem cell,
through a process called hematopoiesis that can give rise to several blood cell types (Figure
3)(Kindt et al., 2007). One type of progenitor immune cell, the granulocyte-monocyte
progenitor cell (Kindt et al., 2007), can give rise to monocytes that can be released into the
blood circulation from the bone marrow. Circulating monocytes are undifferentiated immune
cells that can migrate into various tissues (a process called extravasation) to differentiate into
resident macrophages (as reviewed in Gordon and Taylor, 2005; Kindt et al., 2007). Gordon
and Taylor, in their review, highlight that these tissue macrophages are of heterogeneous
phenotypes, forming subpopulations with unique functions within their corresponding
microenvironments. For example, osteoclasts have the ability to remodel bone tissue,
alveolar macrophages in lungs remove debris and pathogens due to high expression of
pattern recognition receptors and scavenger receptors, and microglia in the adult central
nervous system (CNS) predominantly represent the innate immune system.
In this thesis, I investigated several physiological aspects of rat microglia in vitro.
10

Figure 3. Schematic diagram illustrating development of immune cells in blood and CNS (Ransohoff and Cardona, 2010) Reprinted by permission from Macmillan Publishers Ltd: Nature. Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Copyright 2010
11

Microglia – Immune cells of the CNS Microglia are the resident sentinels of the CNS. Recently it has been shown that
microglia precursor cells of monocyte-lineage develop from mesodermal hematopoietic
cells–originating from the yolk sac– that enter and remain in the developing fetal brain (as
reviewed in Chan et al., 2007). It has been suggested that peri-natally and post-natally,
circulating monocytes in blood can also infiltrate into brain parenchyma (Figure 3)(reviews
Chan et al., 2007; Ransohoff and Cardona, 2010; Kettenmann et al., 2011). These cells then
proliferate, migrate to different regions of the brain and differentiate into microglial cells.
Microglia reside in an immune privileged environment, due to the blood brain barrier (BBB)
that separates the brain anatomically and physiologically, and strictly regulates substances
that cross the barrier (Alberts et al., 2008). Microglia are thought to form 5% to 20% of the
total glial cell population in the CNS (Kaur et al., 2010).
During post-natal development, microglia further differentiate within the brain, and
enter a quiescent state with a ramified morphology (Schlichter et al., 2010; Kettenmann et al.,
2011). In this non-activated state, microglia are non-migratory but have many processes
extending from the soma that are motile, constantly monitoring their environment through
pinocytosis, and interacting with other cells in CNS (Davalos et al., 2005; Nimmerjahn et al.,
2005; Kaur et al., 2010). Microglia respond first to perturbations of CNS homeostasis
(Miyake et al., 1988; Schilling et al., 2003). In response to a pathophysiological event,
microglia undergo a complex transformation process from the non-activated “resting” state to
“activated”. Once activated, microglia can exhibit one or more new phenotypes; e.g.,
increased proliferation, migration, phagocytosis, production of interleukins, cytokines and
chemokines (Hanisch and Kettenmann, 2007; Kaushal et al., 2007; Schlichter et al., 2010;
Kettenmann et al., 2011). The outcome of the microglial response is dependent on its
12

activation state, i.e., microglia react differently depending on the stimuli, factors released by
surrounding neuronal/glial tissue, and the pathophysiological context (Schwartz et al., 2006;
Carson et al., 2007; Colton, 2009; Kettenmann et al., 2011). Microglia are sometimes
referred to as brain macrophages, and can exhibit a wide range of immune functions that are
similar to peripheral macrophages; e.g., phagocytosis, free radical production, secretion of
chemokines and cytokines, and communication with rest of the immune system (Tambuyzer
et al., 2009). However, unlike macrophages, the response of activated microglia can be
immunologically silent, in which a system-wide immune reaction is not activated (Galea et
al., 2007). Along with the versatile nature of microglial activation, there is evidence of
microglial involvement in all neuropathologies (Kreutzberg, 1996; Streit et al., 2005; Block
et al., 2007; Hanisch and Kettenmann, 2007; Davoust et al., 2008; Colton and Wilcock, 2010;
Graeber and Streit, 2010; Kaur et al., 2010).
This thesis will focus on one of the reactive microglial phenotypes, which involves
cells migrating to sites of injury (e.g. to the lesion after intracerebral hemorrhage or ischemic
stroke) (Brockhaus et al., 1996; Zhang et al., 1997; Schlichter et al., 2010). Shortly after
stroke, our lab and other groups have shown that microglia/macrophages respond to the
injury by entering an activated state that includes a change in morphology from ramified to
amoeboid (Brockhaus et al., 1996; Zhang et al., 1997; Wasserman et al., 2008; Moxon-Emre
and Schlichter, 2010). Microglia/macrophages were then observed to progressively migrate
toward the damaged core from surrounding tissue, reaching maximal infiltration by 7 days
after both forms of stroke. Functionally, microglial migration can be either beneficial or
detrimental (Tambuyzer et al., 2009). For instance, beneficial effects include removal of
damaged cells and debris by phagocytosis, and secreting neurotrophic factors that aid in
13

repair processes. It has even been suggested that activated microglia secrete soluble factors
that attract neural precursor cells to the site of injury where they can differentiate to produce
neurons and/or glial cells that aid in the repair process (Aarum et al., 2003). On the other
hand, microglial activation and migration can also be detrimental and cause secondary
damage after the initial insult; e.g., due to production of highly reactive oxygen species
(ROS) and nitric oxide-derived species like peroxynitrite (see review Boje and Arora, 1992;
Kettenmann et al., 2011). Furthermore, production of inflammatory factors, such as
interleukins and tissue necrosis factor (TNF-α), can cause additional neuro-inflammatory
damage that propagates from the site of injury to surrounding healthy tissue (see reviews
Chakraborty et al., 2010; Kettenmann et al., 2011).
Although microglial migration to the site of injury is a well-documented response that
is common to most of its reactive phenotypes, the mechanisms have not been studied
extensively. During development, microglia migrate to different regions of the brain,
(investigated mostly in quails), with migration tangentially at first, and then radially (Cuadros
et al., 1994; Cuadros et al., 1997; Marin-Teva et al., 1998; Rezaie and Male, 1999;
Navascues et al., 2000; Sanchez-Lopez et al., 2004). In their non-activated, quiescent,
ramified state, microglia motility is mainly seen as extension of the many processes arising
from their somata, without overall cell displacement (Davalos et al., 2005; Nimmerjahn et al.,
2005). However, factors like ATP released from necrotic cells in the damaged brain act as
chemo-attractants, activate microglia and alter their morphology to the amoeboid form
(Honda et al., 2001), which is associated with the migratory phenotype (Kettenmann et al.,
2011).
ATP allows Ca2+ entry through receptor signalling via metabotropic P2Y receptors or
14

ionotropic P2X receptors that modulate intracellular pathways, such as PI3K/Akt signalling
(Ohsawa et al., 2007; Lalo et al., 2008). Some other physiological factors that activate and
attract microglia include ectonucleotidase-derived adenosine (Farber et al., 2008), glutamate
(Kim and Ko, 1998; Liu et al., 2009), chemokines (Biber et al., 2001; Liang et al., 2009),
bradykinin (Ifuku et al., 2007), growth factors (Bonifati and Kishore, 2007; Kettenmann et
al., 2011) and beta-amyloid (Stuart et al., 2007). There is evidence that signalling pathways
such as PI3K/Akt, Erk, non-receptor tyrosine kinases (e.g. Pyk2) are involved in microglial
migration (see review Kettenmann et al., 2011). Some ion channels proposed to be involved
in microglial migration are stretch-activated Ca2+ permeable channels, Ca2+-activated
potassium channels, the reversed mode of the Na+/Ca2+ exchanger (NCX), and Cl- channels
(see review Kettenmann et al., 2011). It is interesting that many of the molecules thought to
influence microglial migration are involved in Ca2+ signalling cascades; which suggests a
conserved, vital role for Ca2+ in migration and an important contribution of intracellular Ca2+
regulation (Evans and Falke, 2007), and a complex interplay between ligands, receptors, ion
channels, and the cytoskeleton. Microglia also need to migrate through the unique brain
ECM and tightly packed dense brain tissue without causing damage to normal cells. In this
context, there is very little known about migration mechanisms in microglia.
15

Podosomes in microglia
With Dr. Schlichter, a former graduate student, Catherine Vincent, discovered that
untreated microglia cells grown on glass cover slips show dot-like structures that are
abundant in F-actin, and enriched in the lamella region. Further investigation showed that
these structures possessed a ring pattern of talin staining and F-actin cores, and had
podosome-like dimensions, suggesting that microglia express podosomes in vitro (Figure 4).
Podosomes in macrophages also seem to be localized at the leading edge (Evans et al., 2003).
In addition, microglial podosomes often formed a larger structure that we called a ‘podonut’,
which is made up of many individual podosomes in an open ring formation. Functional
studies showed these structures have the ability to degrade fibronectin, a common non-brain
specific ECM substrate (Figure 5), which is a well-known function of podosomes (Chen et
al., 1985; Seals et al., 2005). Podonuts were observed after microglia were in culture for 20
hours (Figure 6) and formed at the cell-substrate attachment interface. It was also discovered
that SK3, a Ca2+ activated K+ channel, localized to the core of podosomes (Figure 5). In
preliminary studies (Vincent and Schlichter, 2010), blocking SK3 channel activity did not
affect podonut/podosome formation so its function in relation to these structures still requires
further investigation.
Activated microglia that are undergoing migration in the damaged CNS could benefit
from expression of these microscopic structures (podosomes), to provide the means for
localized degradation of brain ECM and to navigate through dense CNS tissue to reach the
site of injury.
16

A B
Figure 4. Primary rat microglia express podosomes. (A) Microglia immunolabeled for the
podosome core marker, Arp2 (red), and the podosome ring marker, talin (green), and the cell
nucleus (DAPI; blue) (B) Microglia immunolabeled for the podosome ring (talin; red), F-
actin in the core (phalloidin; green), and the nucleus (DAPI; blue). Boxes represent areas
chosen for higher magnification and colour separated on the right. Arrows show the
podosome ring and core structural arrangement. Scale bars = 5 µm. (Images from (Vincent
and Schlichter, 2010)
17

Figure 5. Functional podosomes expressed in microglia, with SK3 localized in podosome
core. Microglia stained for SK3 (red) show that it co-localizes with the podosome core
marker, F-actin (phalloidin; blue). Images on the right are the boxed areas, magnified and
colour separated. F-actin dots co-localize with regions of degradation of FITC-labeled
fibronectin (green). Arrowheads show co-localization of SK3, F-actin and fibronectin-
degraded spots. Scale bar = 5 µm. (Images from (Vincent and Schlichter, 2010)
18
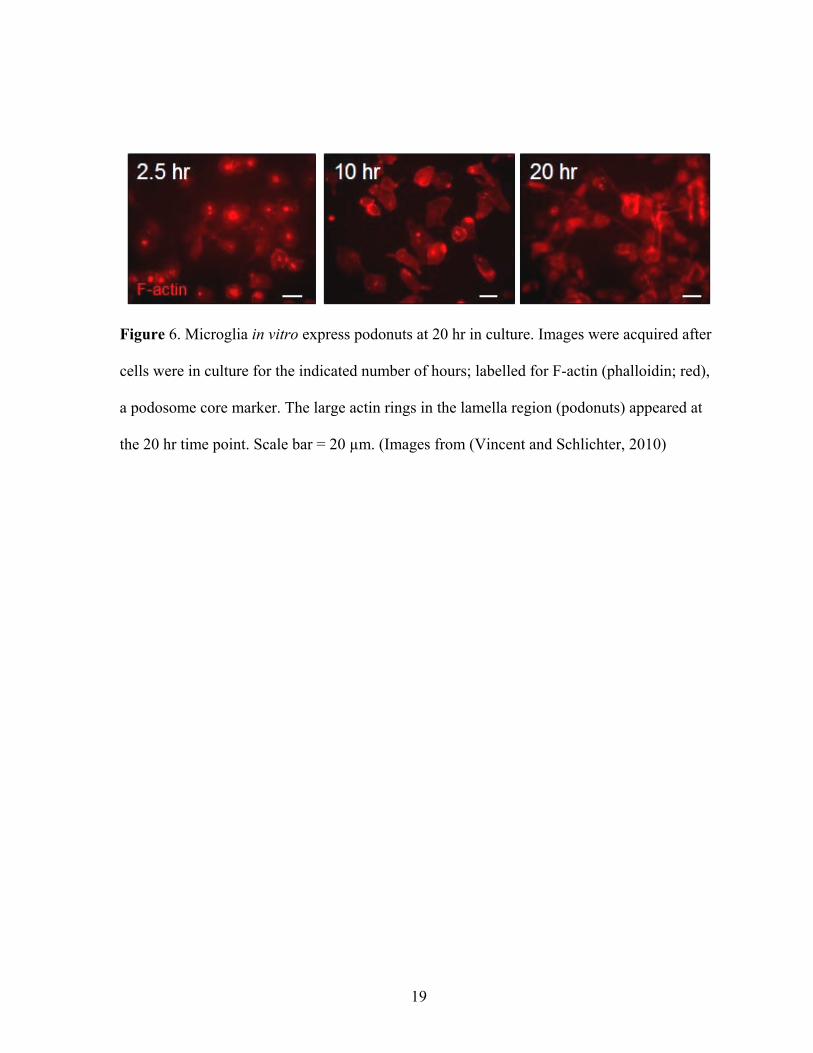
Figure 6. Microglia in vitro express podonuts at 20 hr in culture. Images were acquired after
cells were in culture for the indicated number of hours; labelled for F-actin (phalloidin; red),
a podosome core marker. The large actin rings in the lamella region (podonuts) appeared at
the 20 hr time point. Scale bar = 20 µm. (Images from (Vincent and Schlichter, 2010)
19

Thesis Objectives
Podosome components are fairly conserved among different cell types; however,
some reports have shown apparent differences in their molecular make up. For example, in
smooth muscle cells, SM22α protein was shown to associate with the podosomal core
(Gimona et al., 2003). In dendritic cells, there is evidence that podosome assembly occurs in
a gelsolin-independent manner (Hammarfjord et al., 2011); contrary to the requirement for
gelsolin in osteoclasts (Ma et al., 2008; Crowley et al., 2009; Ma et al., 2010; Van Goethem
et al., 2010). With our lab first discovering podosomes in microglia (Vincent and Schlichter,
2010), and given that podosome components could vary in different cell types, I wanted to
further characterize podosomes in microglia using immunostaining and some functional
studies. In this thesis, I show that although some components are conserved between
podosomes in microglia and other cells; I have identified some molecules not previously
shown to associate with podosomes. Then, because Ca2+ plays a key role in cell migration,
and SK3 —a Ca2+ responsive channel— was found in the podosome core in microglia
(Vincent and Schlichter, 2010), I hypothesized that the regulation of podosomes is dependent
on Ca2+. Overall, the findings in this thesis contribute toward a better understanding of
podosomes, their features in microglia, and their Ca2+-dependence.
20

MATERIALS AND METHODS
Microglia cell cultures. Giulian and Baker (1986) were the first to develop
isolation and culturing protocols for microglia. The Schlichter lab published it’s first
microglia paper in 1996, and since then has refined the protocol such that after isolation,
neonatal rat microglia remain in a relatively resting state, as judged by very low expression
of many inflammatory mediators (Sivagnanam et al., 2010). Isolation of rat microglia was
done by Dr. Schlichter’s technician, Xiaoping Zhu, following standard protocols as described
previously (Kaushal et al., 2007; Ohana et al., 2009; Sivagnanam et al., 2010). Briefly,
primary microglia cultures were derived from 1 to 2 day old Sprague-Dawley rat pups. After
removing the meninges, the brain was dissected and mashed through a stainless steel sieve in
Minimum Essential Medium (MEM; Invitrogen, Burlington, Canada), and then centrifuged
at 1000g for 10 min. The supernatant was removed and the pellet was re-suspended in MEM
supplemented with 10% fetal bovine serum (FBS; Wisent, St-Bruno, Canada) and 0.05
mg/ml gentamycin (Invitrogen), and plated on flasks to maintain at 37ºC, 5% CO2
atmosphere. After 48 hr, cellular debris, non-adherent cells and supernatant were removed
and fresh medium was added. The mixed cell cultures were then maintained for another 5 to
6 days. Microglial suspensions were then obtained by shaking the mixed cultures on an
orbital shaker for 3-4 hours in 37ºC at 60 to 65 rpm. For my experiments, the suspension
containing microglial cells was then centrifuged for 10 min at 1000g and the cell pellet was
re-suspended in MEM supplemented with 2% FBS and 100 µM gentamycin. Cells were
plated at 60,000 cells per 15 mm diameter glass cover slip, incubated in the 2% FBS
supplemented MEM at 37ºC, 5% CO2, and used within 24 to 48 hr. The Schlichter lab has
repeatedly shown that primary microglia cultures obtained with this protocol are ≥99% pure,
21

based on labelling with several microglia markers: FITC-conjugated tomato lectin, or
antibodies against isolectin B4, ionized Ca2+ binding adaptor-1(Iba1) or CD11b (OX-42)
(Kaushal et al., 2007; Ohana et al., 2009; Sivagnanam et al., 2010).
Chemicals and antibodies. I used the channel blockers: 2-aminoethyl
diphenylborinate (2-APB; Sigma-Aldrich, Oakville, Canada), and spermine
tetrahydrochloride (Calbiochem, San Diego, CA). Several antibodies were used: rabbit
polyclonal anti-phosphotyrosine-14-caveolin 1 (Signalway Antibody; #11090, Pearland, TX),
goat polyclonal anti-Orai1 (Santa Cruz Biotechnology; sc-74778, Santa Cruz, CA), rabbit
polyclonal anti-SK3 (Alomone labs; APC-025, Jerusalem, Israel), rabbit monoclonal anti-
calmodulin (Abcam; ab45689, Cambridge, MA), rabbit polyclonal anti-Arp2 (Santa Cruz
Biotechnology; sc-15389), mouse monoclonal anti-talin 1/2 (Abcam; ab11188), rabbit
polyclonal anti-Tks5 (Santa Cruz Biotechnology; sc-30122), mouse monoclonal anti-
phosphotyrosine (Abcam; ab10321), rabbit polyclonal anti-Iba1 (Wako Chemicals; #019-
19741, Richmond, VA) and goat polyclonal anti-Nox1 (Santa Cruz Biotechnology; sc-5821).
Immunocytochemistry (ICC). Cover slips bearing microglia were fixed in 4%
paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA) in PBS for 10 to 15
min at room temperature, and then permeabilized for 5 min with 0.2% Triton X-100. To
block non-specific staining, cells were incubated in blocking solution (4% donkey serum in
PBS; Jackson Immunoresearch, West Grove, PA) for 1 hr at room temperature. The blocking
solution was replaced and the cells were then incubated overnight at 4ºC with a primary
antibody in blocking solution. After another 1 hr of blocking, a secondary donkey antibody in
22

blocking solution (conjugated to Dylight 488 or Dylight 594; 1:250; Jackson
Immunoresearch) was added to label the corresponding primary antibody, and incubated for
1 hr at room temperature in the dark. F-actin was visualized by incubating cells with Alexa
488-conjugated phalloidin (1:50 in block solution, Invitrogen) for 15 min at room
temperature in the dark. Cell nuclei were stained by incubating with 4',6-diamidino-2-
phenylindole (DAPI; 1:3000 in PBS) for 5 min. Cover slips were mounted on glass slides
with mounting medium (Dako, Glostrup, Denmark) for viewing using epifluorescence
widefield microscopy. Negative controls were prepared for each ICC preparation using the
aforementioned protocol, except that primary antibodies were omitted. Before use, antibodies
in blocking solution were centrifuged at 10,000 rpm for 10 min to remove any aggregates
that might bind non-specifically and introduce artefacts.
For some proteins, heat induced antigen retrieval (HIAR) was required (see example
in Figure 7). That is, when paraformaldehyde fixation immobilizes proteins by chemical
cross-linking, an unwanted side-effect can be that an epitope normally recognized by a
primary antibody becomes masked by the cross-linking (Fox et al., 1985; Mason and
O'Leary, 1991; Dapson, 1993). With HIAR, the chemical masking is essentially removed,
permitting the normal interaction between the antibody and corresponding epitope on the
protein of interest. HIAR was performed before the cell permeabilization step, using a
sodium citrate-based buffer that contained 10 mM trisodium citrate with 0.05% Tween-20
adjusted to pH 6.0 with HCl. The slides were heated in a microwave oven for 4 to 5 min,
with excess citrate buffer to prevent samples from drying, and then allowed to cool to room
temperature before being washed with PBS. Although HIAR is a useful technique to reduce
the undesirable effect of formaldehyde-based fixation, it is challenging, and results can be
23

inconsistent. During my thesis research, I tried several antigen retrieval techniques (e.g.
hydrochloric acid-based, Tris-EDTA-based) and found that citrate-based HIAR yielded the
best results.
Some antibodies did not stain after formaldehyde fixation and/or HIAR. In those
cases, I fixed the microglia with cold methanol, which precipitates proteins by dehydration.
Methanol (HPLC grade) was pre-chilled overnight at -40ºC, and then microglia on cover
slips were incubated for 5 min at -20ºC. Because this fixation technique does not require cell
permeabilization, the immunostaining protocol continued from the first blocking step. Note
that the methanol-fixed cells could not be labelled for F-actin using phalloidin (North, 2006).
Ca2+ and podonuts (podosomes). After microglia were cultured for 24-48 hr,
the tissue culture medium was replaced with either standard bath solution containing (in
mM): 125 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 5 D-glucose and 10 HEPES, adjusted to pH 7.4
(with NaOH) and 285-300 mOsm with sucrose; or Ca2+-free bath solution (prepared by
omitting CaCl2 and adding 1 mM EGTA). Bath solutions were sterilized by filtering through
0.2 µm filters, and all treatments were performed at 37°C. Prior to fixation of treated cells for
immunocytochemistry, cells were washed once with sterile PBS.
24

Figure 7. Heat induced antigen retrieval (HIAR) enhances antibody labelling of the
immunogen. Lower panels: Microglia immunostained for calmodulin (CaM; red), the
microglia marker, tomato lectin (TL, green), and the nucleus (DAPI; blue) show very low
CaM staining and nuclear localization. Upper panels: HIAR-treated microglia show an
improved staining pattern: higher CaM staining intensity, diffuse cytoplasmic localization,
and little nuclear staining. Scale bars = 20 µm (Images from (Vincent and Schlichter, 2010)
25

Image analysis. Microglia were imaged with a Zeiss Axioplan 2 widefield
epifluorescence microscope (Zeiss, Toronto, ON) equipped with a Zeiss Axiocam HR digital
camera. Images were then analyzed and deconvolved using Zeiss Axiovision 4.7 software. I
found that deconvolution was very helpful in analyzing the tiny podosome structures. Images
obtained from widefield microscopes are convoluted due to the thick optical section, in
which the microscope acquires light from the point source (fluorochrome), and from
peripheral ambient light emitting around the fluorochrome. The resulting image is a product
of the true image and the additional noise, which is defined by the point spread function
(PSF); i.e., the additional noise from a source of light above and/or below the plane of focus
(Campisi and Egiazarian, 2007). Deconvolution algorithms reduce the noise and distortions
introduced during image acquisition by using information about the optical system (e.g. type
of objective lens, refractory index of the immersion medium, PSF) (reviewed in Campisi and
Egiazarian, 2007). If the PSF function is unknown, blind deconvolution can be used, in
which the algorithm uses theoretical point-spread functions.
For images to be deconvolved after immunostaining, I acquired high magnification
epifluorescence images through the z-axis at 200 nm increments (z-stacks). The start-to-end
z-stacks corresponded to focal planes that included the entire cell volume. The same
microscope- and acquisition settings were used to obtain images of negative control cover
slips to detect autofluorescence and background signal from non-specific binding of each
fluorophore-conjugated secondary antibody (Figure 8). To remove artefacts introduced
during the staining procedures, this background signal was subtracted when acquiring z-stack
images, which were subsequently deconvolved using a theoretical PSF and the constrained
iterative maximum likelihood deconvolution algorithm in Axiovision 4.7. Automatic z-stack
26

correction was used to compensate for the decay of fluorescence signal due to
photobleaching, because the same sample was imaged along the z-axis numerous times to
acquire the z-stacks. For illustration, an image was extracted from the z-stack in which
podosomes were in focus.
Statistical Analysis. Quantitative data are presented as mean ± standard error of
the mean (SEM). One-way ANOVA statistical test was performed followed by Tukey’s
analysis. Results were considered significant if p<0.01.
27
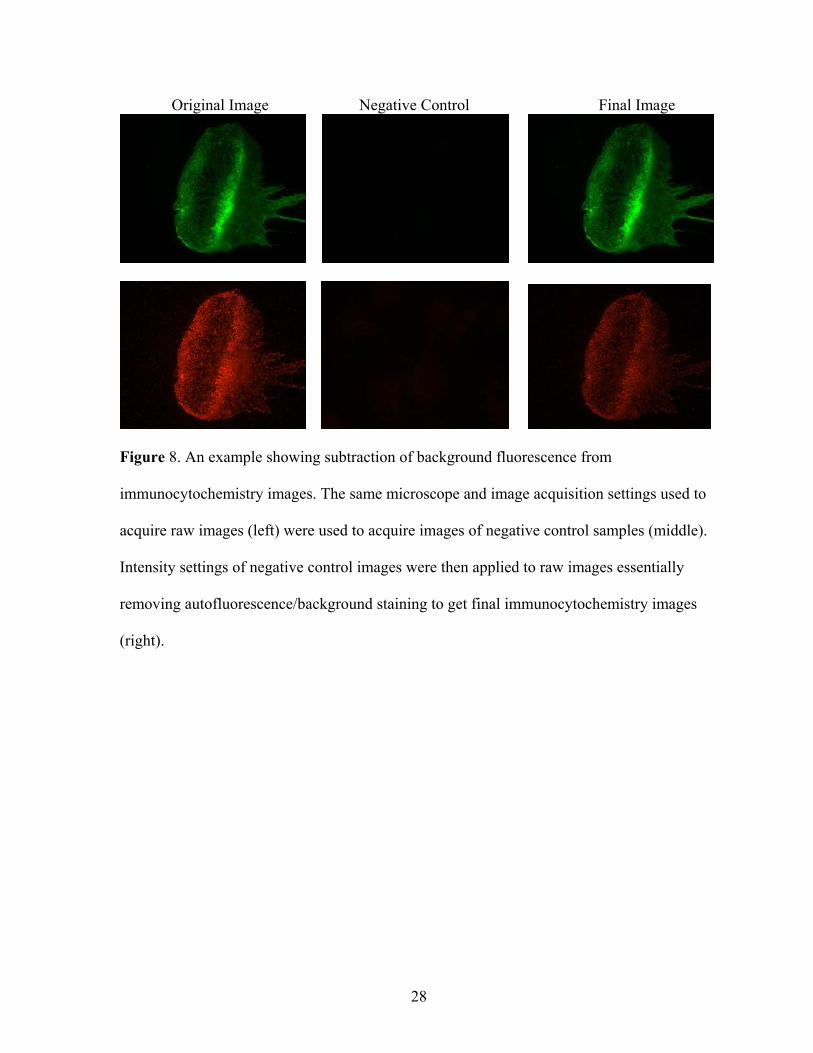
Original Image Negative Control Final Image
Figure 8. An example showing subtraction of background fluorescence from
immunocytochemistry images. The same microscope and image acquisition settings used to
acquire raw images (left) were used to acquire images of negative control samples (middle).
Intensity settings of negative control images were then applied to raw images essentially
removing autofluorescence/background staining to get final immunocytochemistry images
(right).
28

RESULTS
Previously known podosome components found in microglial podosomes
The classical podosome structure consists of a distinct ring and core that differ in
molecular components. In this thesis, talin was used as a marker to identify the podosome
ring, while F-actin or Arp2 were used to identify the core. These markers are commonly used
in the podosome literature (Linder and Aepfelbacher, 2003; Murphy and Courtneidge, 2011).
Src family kinases (SFKs) form a large family of tyrosine kinases involved in
regulating many processes, including angiogenesis, cell proliferation, metastasis, and bone
metabolism (reviewed in Aleshin and Finn, 2010). Src kinase activity is also important for
regulation of podosomes. For instance, expressing constitutively active Src in fibroblasts
causes them to express podosomes (David-Pfeuty and Singer, 1980; Chen et al., 1985;
Tarone et al., 1985; Aleshin and Finn, 2010; Dovas and Cox, 2011); and pharmacological
inhibition of SFKs disrupts podosome formation in macrophages (Linder et al., 2000b;
Cougoule et al., 2005).
As a first test of whether microglial podosomes express similar signalling pathway, I
correlated labelling of some known Src substrates with podosomes. When an anti-
phosphotyrosine antibody was used to label microglia for tyrosine phosphorylated proteins, a
punctate staining pattern was seen at the leading edge of polarized cells. Double labelling
with F-actin and high-resolution imaging showed that phosphotyrosine proteins were
enriched in the podosome core, and in some cases, in the ring-like structure around the F-
actin staining (Figure 9).
29

Figure 9. Phosphotyrosine staining is enriched in microglial podosomes. Colour-separated
images of a representative microglial cell stained for the podosome core marker, F-actin
(phalloidin; green), and phosphotyrosine (pTyr, red), with a merged image shown below. The
boxed area is shown magnified and colour separated on the bottom right. Arrows and
arrowheads show co-localization of F-actin and pTyr. Scale bars = 5 µm n
30

The presence of tyrosine phosphorylated proteins in microglial podosomes suggested
that tyrosine kinase signalling molecules, such as Src, might be involved in podosome
dynamics. To further examine Src substrates in microglial podosomes, cells were
immunolabeled for tyrosine kinase substrate 5 (Tks5): a substrate of Src kinase previously
shown to be enriched in invadopodia and podosomes (Abram et al., 2003; Seals et al., 2005).
It has been suggested that Tks5 acts as an organizer in the initial stages of invadosome
assembly (Seals et al., 2005). In microglia, Tks5 and talin co-localized in the podosome ring
(Figure 10). Caveolin-1 is another tyrosine phosphorylated protein, which can be
phosphorylated at tyrosine 14 residue by the SFKs, Lyn, Hck and c-Src (Li et al., 1996;
Grande-Garcia et al., 2007). Caveolin-1 is a major structural protein specific to specialised
membrane domains, called caveolae (Alberts et al., 2008). In microglia, tyrosine 14
phosphorylated caveolin-1 (pCav1) showed extensive ring-like staining that was similar to
the talin staining pattern (Figure 11).
Together, these findings show that podosomes in microglia express several tyrosine
phosphorylated proteins that are known substrates for Src and Src-family tyrosine kinases.
31

Figure 10. Immunostaining shows localization of Tks5 in the podosome ring in microglia.
Colour-separated images of a representative microglial cell stained for the podosome ring
marker, talin (red) and Tks5 (green); merged image at right. The boxed area is shown
magnified and colour separated at the far right. Arrows and arrowheads show co-localization
of talin with Tks5. Scale bars = 5 µm
32

Figure 11. Phospho-caveolin-1 (pCav1) is enriched in the podosome ring in microglia.
Colour-separated images of a representative microglial cell immunostained for the podosome
ring marker, talin (red) and pCav1 (green); merged image at right. The boxed area is
magnified and colour separated at the top right. Arrows and arrowheads show co-localization
of talin and pCav1. Scale bars = 5 µm
33

There is evidence that Nox1 is localized to invadopodia, and required for their
formation (Seals et al., 2005). The Nox1 enzyme generates reactive oxygen species (ROS),
which have pleiotropic effects, including acting as a second messenger; e.g., inhibiting some
protein phosphatases, activating some protein kinases, and regulating some ion channels
(Bedard and Krause, 2007). Because ROS production by microglia is one important
component of their cytotoxic behaviours, its regulation within podosomes is of interest. In
order to address whether Nox1 is present in microglial podosomes, I found it necessary to use
methanol fixation for immunostaining. The observed co-localization with talin indicates that
Nox1 is present in the podosome ring in microglia (Figure 12).
To summarize this section, I found that podosomes in microglia stained for
phosphotyrosine, Tks5, pCav1 and Nox1. These components have been reported in
invadosomes of at least one other cell type (Tarone et al., 1985; Nakamura et al., 1993; Burns
et al., 2001; Pfaff and Jurdic, 2001; Abram et al., 2003; Colonna and Podesta, 2005; Seals et
al., 2005; Oikawa et al., 2008; Diaz et al., 2009; Gianni et al., 2009; Stylli et al., 2009).
34

Figure 12. Immunolabeling for Nox1 shows localization to the podosome ring in microglia.
Colour-separated images of a representative methanol-fixed microglial cell stained for the
podosome ring marker, talin (red) and Nox1 (green); merged image at right. The boxed area
is shown magnified and colour separated at the top right. Arrows and arrowheads show co-
localization of talin with Nox1. Scale bars = 5 µm.
35

Ca2+ and podosome (podonut) formation
There is little specific information about mechanisms that regulate ECM degradation
and migration of microglia. In several other cell types, migration depends on Ca2+, and is
impeded by removing extracellular Ca2+ (Brundage et al., 1991; Mandeville et al., 1995).
Polarized cells maintain a descending intracellular Ca2+ concentration gradient from the
uropod at the rear to the lamellum at the front (Brundage et al., 1991), and Ca2+
microdomains in lamellipodium has been proposed to aid cell steering during migration (Wei
et al., 2009). Interestingly, our lab discovered that SK3, a Ca2+-calmodulin-gated K+ channel,
localizes to the core of microglial podosomes (Vincent and Schlichter, 2010). Here, I tested
the hypothesis that Ca2+ regulates podosome formation in microglia. Apparently, this aspect
of podosome regulation has not been studied previously in any cell type.
I tested this hypothesis by visualizing the larger podonuts, which are made up of
many individual podosomes (typically >100 (Vincent and Schlichter, 2010). Either
extracellular Ca2+ was removed by replacing the culture medium with a Ca2+-free bath
solution, or intracellular Ca2+ was buffered to an abnormally low level by incubating the cells
with 10 μM BAPTA-AM in standard bath solution. All treatments were carried out for 30
min at 37ºC. Control or untreated cells were maintained in standard bath solution under
identical experimental conditions. An interesting observation when extracellular Ca2+ was
removed or intracellular Ca2+ was buffered, was the loss of the lamellar morphology
displayed by untreated cells (Figure 13).
36
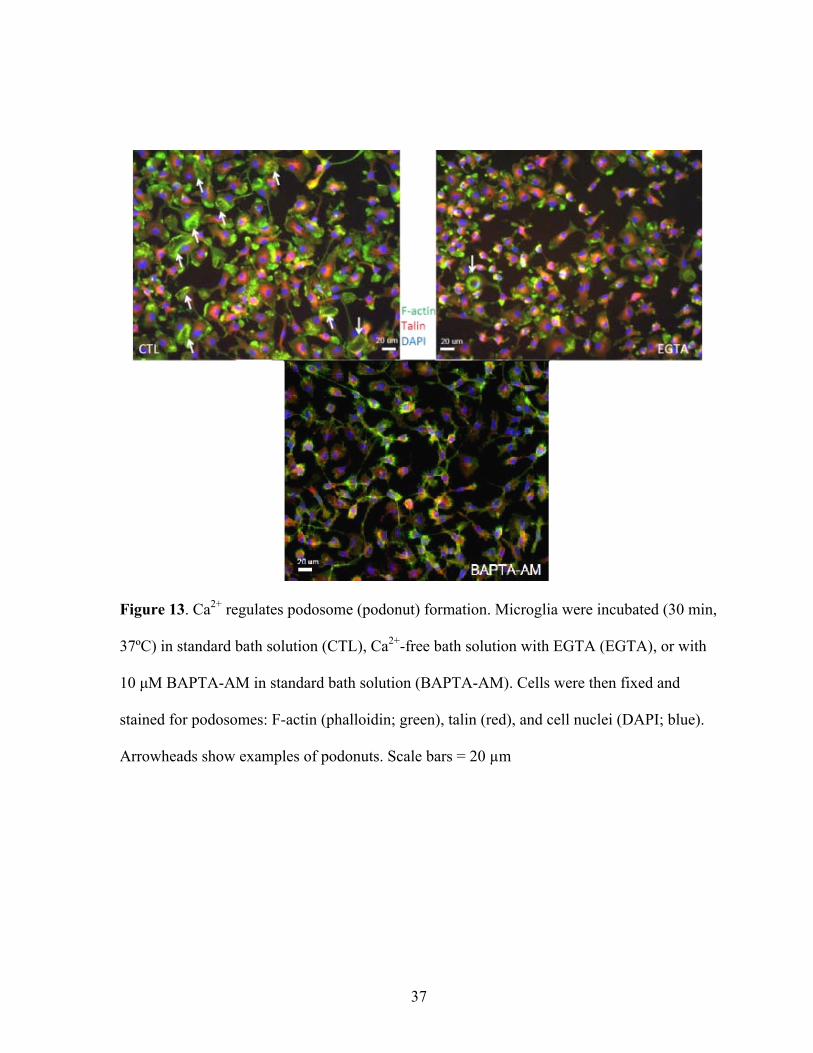
Figure 13. Ca2+ regulates podosome (podonut) formation. Microglia were incubated (30 min,
37ºC) in standard bath solution (CTL), Ca2+-free bath solution with EGTA (EGTA), or with
10 μM BAPTA-AM in standard bath solution (BAPTA-AM). Cells were then fixed and
stained for podosomes: F-actin (phalloidin; green), talin (red), and cell nuclei (DAPI; blue).
Arrowheads show examples of podonuts. Scale bars = 20 µm
37

Figure 14. Ca2+ and Ca2+-permeable ion channels are required for podonut formation. The
percentage of microglial cells expressing podonuts is shown in control microglia (CTL,
untreated), and after treatment with Ca2+-free bath solution (0 Ca2+) or 10 μM BAPTA-AM
as in Figure 13. In addition, two ion-channel blockers were used: 50 μM 2-APB, 100 μM
spermine. Podonuts were counted from three random fields on each immunostained cover
slip across microglia cultures prepared from 4 different animals. Data are shown as mean ±
standard error; and the asterisks indicate statistically significant differences compared to CTL
(untreated); **p<0.01.
38

Removal of external Ca2+ or buffering intracellular Ca2+ dramatically reduced
podonut formation in microglia (Figure 14), implying that their formation and/or persistence
depend on Ca2+. Because both treatments similarly inhibited podonut formation, I
hypothesized that Ca2+ entry, rather than external Ca2+, regulates the podosomes. The
Schlichter lab has shown that rat microglia express two functional Ca2+-permeable ion
channels: the non-selective cation channel, TRPM7, which is blocked by spermine; and
Orai1 (the pore forming subunit of the Calcium Release Activated Calcium [CRAC]
channel), which has a high selectivity for Ca2+ and is blocked by 2-APB (Jiang et al., 2003;
Ohana et al., 2009). Blocking TRPM7 channels with 100 µM or 1 mM spermine did not alter
the prevalence of podonuts (Figure 14, 15B). However, blocking CRAC channels with 50
µM 2-APB not only abolished podonut formation but significantly changed the microglial
morphology (Figure 14, 15A). High magnification images of 2-APB-treated microglia show
a lack of punctate F-actin staining, indicating a loss of podosomes as well as the podonut
superstructure (Figure 16). Control microglia treated with only DMSO (the vehicle for 2-
APB) did not differ from untreated cells. This result indicates that Ca2+ entry via CRAC is
required for expression of podonuts, and hence, podosomes. Interestingly, in time-lapse
images of microglia treated with 50 µM 2-APB (not shown), migration was reduced. These
preliminary data suggest that Ca2+ entry through CRAC channels contributes to microglia
migration.
39

A B
Figure 15. Podonut (podosome) formation requires Ca2+ entry, likely through CRAC
channels. Microglia were treated for 30 min at 37ºC with 2-APB (50 µM, A), a blocker of the
CRAC channel, or the TRPM7 blocker, spermine (100 µM, B) in standard bath solution.
Cells were then fixed and stained for podosomes: F-actin (phalloidin; green), talin (red),
nucleus (DAPI; blue). Arrows show podonuts. Scale bars = 20 µm
40

Figure 16. High-magnification images to show loss of podosomes after 2-APB treatment.
Colour-separated images of microglia cells labelled for podosomes: F-actin (phalloidin;
green) and talin (red). Cells were treated for 30 min at 37ºC with the CRAC channel blocker,
50 µM 2-APB. Scale bars = 5 µm
41

Novel expression of several molecules in microglia podosomes
Although many podosome components are conserved across different cell types, a
unique molecule, SM22α, was identified in podosomes of vascular smooth muscle cells
(Gimona et al., 2003). This suggested that some molecular components of podosomes could
depend on the cell type. Indeed, our lab was the first to identify the SK3 channel as a
component of invadipodia, specifically in microglial podosomes (Vincent and Schlichter,
2010).
Because I found that the Orai1/CRAC channel blocker, 2-APB, reduced number of
podonuts in microglia, I next tested the novel hypothesis that Orai1 is a podosome
component in microglia. After using heat-induced antigen retrieval (HIAR), I observed Orai1
immunolabeling in podonuts, specifically in core of individual podosomes (i.e., co-localized
with Arp2; Figure 17). Orai1 is the pore-forming subunit of the CRAC channel (which
requires the STIM1 protein for functionality). Together, the results in Figures 15–17 suggest
that Ca2+ entry through CRAC channels in podosomes regulates their formation and/or
stability.
The presence of SK3, a Ca2+-activated K+ channel, together with a source of Ca2+ (the
Orai1/CRAC channel) raised the possibility that these two channels interact in podosomes.
However, SK channels require calmodulin (CaM) binding in order to respond to Ca2+;
therefore, I next asked whether CaM is present in microglial podosomes. After performing
HIAR to obtain specific CaM immunolabeling, I found that CaM was enriched in podonuts,
and mainly co-localized with talin in individual podosome rings (Figure 18). Occasionally,
CaM staining was also seen in the centre of talin-stained rings; i.e., in the podosome core.
When using an antibody against ‘ionized calcium binding adaptor molecule 1’ (Iba1)
42

as a specific marker to label microglia, I made the surprising observation that it was enriched
in podonuts (Figure 19). Iba1 is present in individual podosomes, and double labelling for F-
actin (or talin) shows that it co-localizes with F-actin in the core structure. This is intriguing,
because Iba1 is a microglia-specific Ca2+-binding protein that is known to cross-link actin
filaments in the CNS (Imai et al., 1996; Sasaki et al., 2001; Imai and Kohsaka, 2002).
Table 2 summarizes all components found in microglial podosomes in this study, and
their localization to the ring versus core.
43
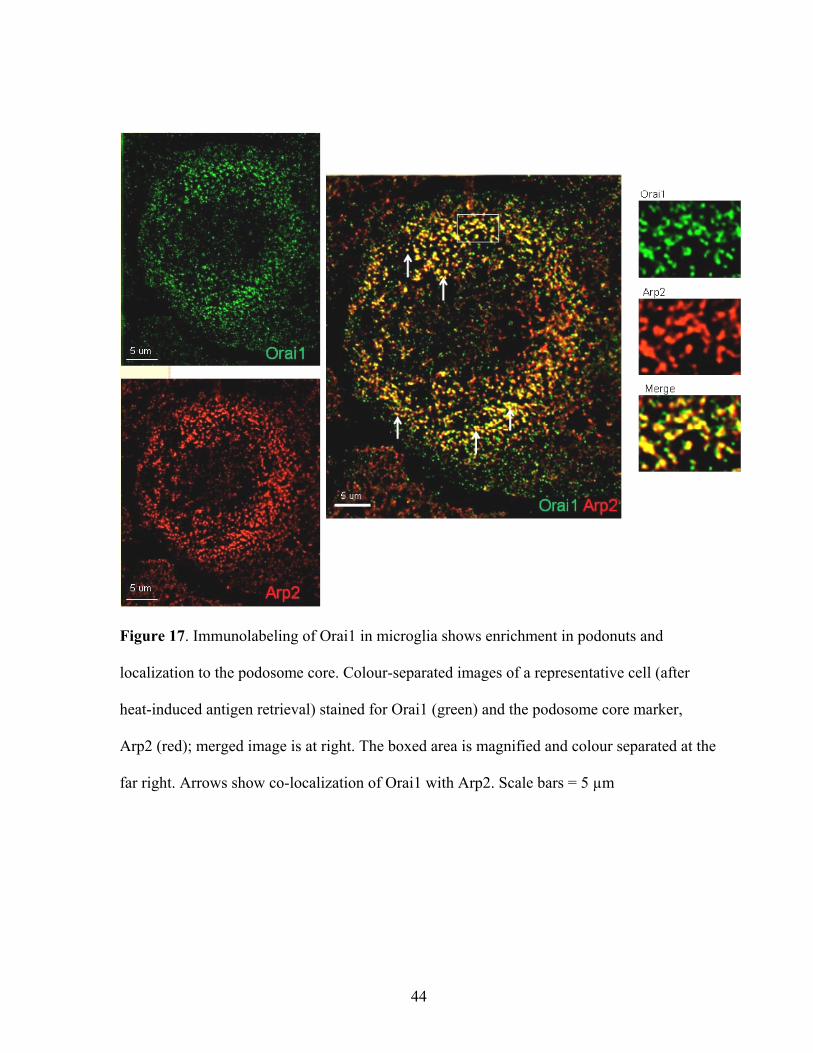
Figure 17. Immunolabeling of Orai1 in microglia shows enrichment in podonuts and
localization to the podosome core. Colour-separated images of a representative cell (after
heat-induced antigen retrieval) stained for Orai1 (green) and the podosome core marker,
Arp2 (red); merged image is at right. The boxed area is magnified and colour separated at the
far right. Arrows show co-localization of Orai1 with Arp2. Scale bars = 5 µm
44

Figure 18. Immunostaining shows calmodulin (CaM) association with microglia podosomes.
Left: After heat-induced antigen retrieval, colour-separated images show CaM (red) and the
podosome core marker, Arp2 (green). The arrows in the merged image (below) show
examples of CaM in podosomes. The boxed area is shown magnified and colour separated at
the right. Right: After HIAR, colour-separated images show staining for CaM (red) and the
podosome ring marker, talin (green). In the merged image (below), arrows show examples of
CaM in individual podosomes. The areas within solid and dotted boxes are shown magnified
and colour separated to the left and right, respectively. CaM was seen mainly in the
podosome ring structure, and less in the core. Scale bars = 5 µm
45
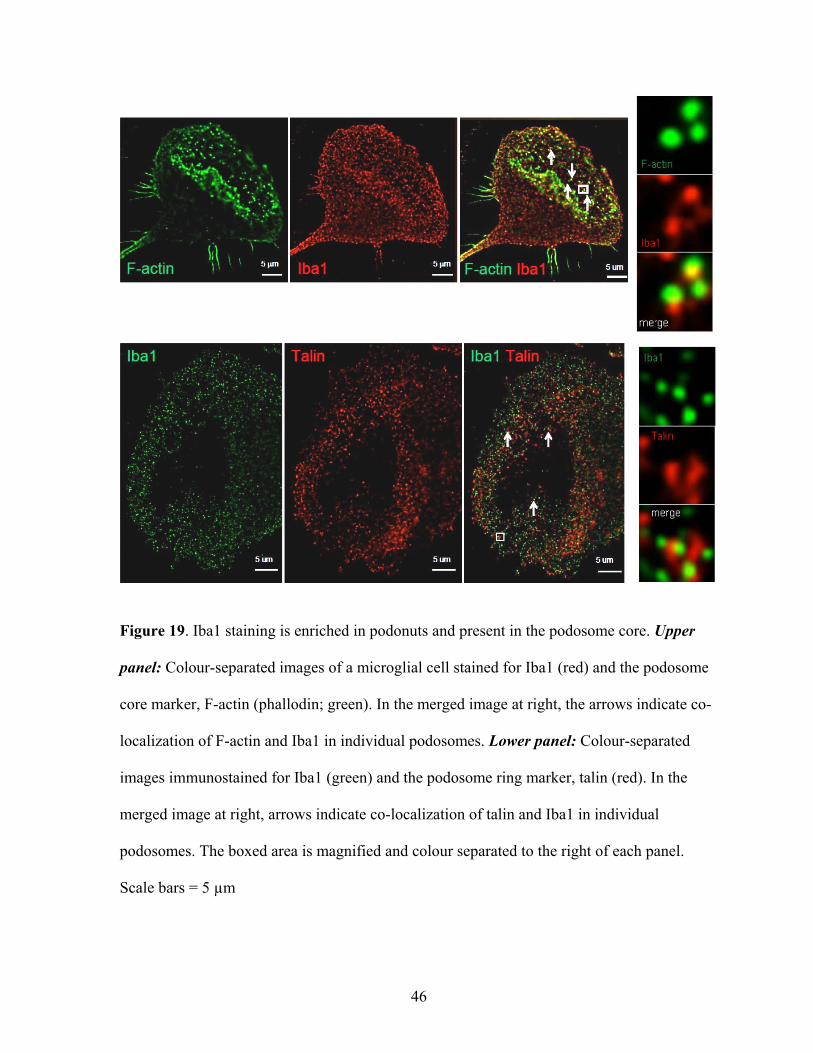
Figure 19. Iba1 staining is enriched in podonuts and present in the podosome core. Upper
panel: Colour-separated images of a microglial cell stained for Iba1 (red) and the podosome
core marker, F-actin (phallodin; green). In the merged image at right, the arrows indicate co-
localization of F-actin and Iba1 in individual podosomes. Lower panel: Colour-separated
images immunostained for Iba1 (green) and the podosome ring marker, talin (red). In the
merged image at right, arrows indicate co-localization of talin and Iba1 in individual
podosomes. The boxed area is magnified and colour separated to the right of each panel.
Scale bars = 5 µm
46
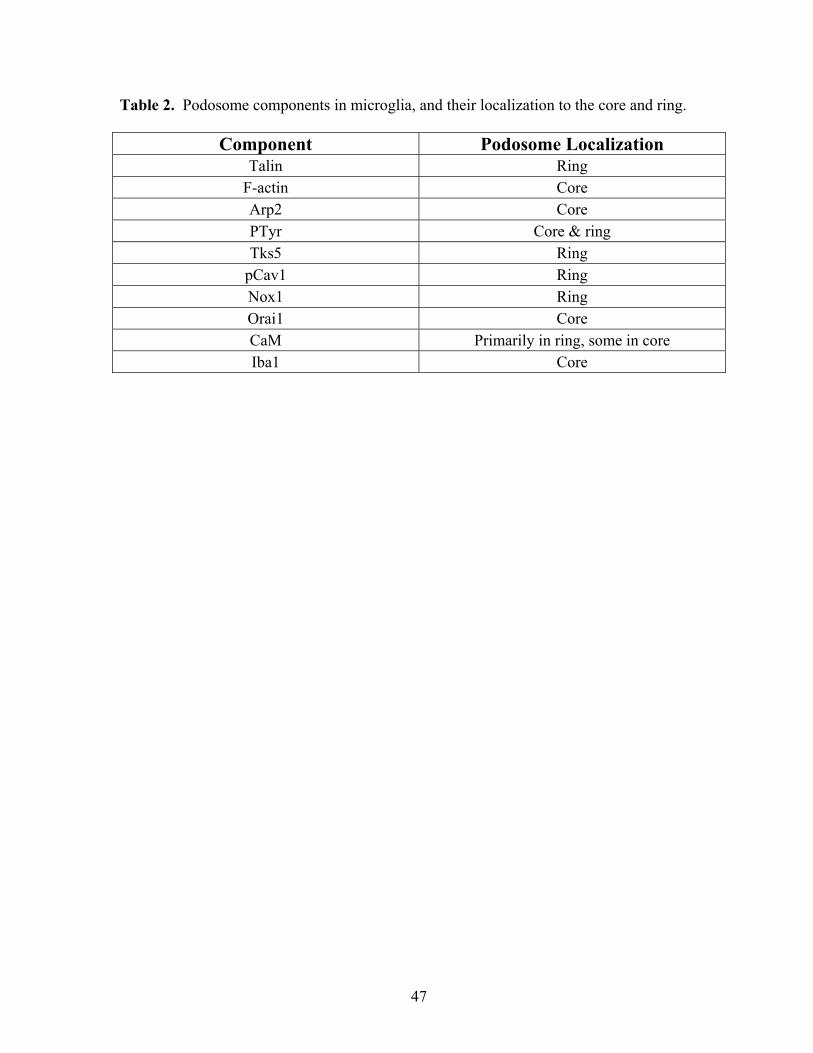
Table 2. Podosome components in microglia, and their localization to the core and ring.i1
Component Podosome Localization Talin Ring
F-actin Core Arp2 Core PTyr Core & ring Tks5 Ring
pCav1 Ring Nox1 Ring Orai1 Core CaM Primarily in ring, some in core Iba1 Core
47

DISCUSSION
Podosomes are dynamic microscopic structures that continually undergo assembly,
maturation and disassembly (Murphy and Courtneidge, 2011). Individual podosomes within
a cell might be in different stages of development depending on numerous factors, including
substrate attachment, protein kinase activity, and actin reorganization. Following the
discovery of podosomes in microglia in the Schlichter lab, SK3 was clearly shown to
associate with podosomes (Vincent and Schlichter, 2010). The co-localization of SK3 with
Arp2 and F-actin indicated that it is trafficked to the core region of podosomes. Following
the initial discovery of microglial podosomes, I wanted to determine whether they contain
several components that are conserved in podosomes of other cell types.
In this thesis, I have shown that Ca2+ is involved in podosome regulation in microglia;
and I showed that several known podosome-associated proteins, as well as novel molecules
associate with microglial podosomes. This is a significant discovery because although
migration of activated microglia towards the site of injury in the damaged CNS is a well-
known phenomenon, it is not well understood how microglia perform the difficult task of
migrating through not only distinctive brain ECM but also tightly packed CNS tissue in order
to reach their destination. The expression of podosomes in microglia provides significant
insight into how microglia might move through CNS tissue. Podosomes have several
characteristics that could aid activated migratory microglia: (i) attachment to ECM to provide
anchorage and traction, (ii) fast turnover for quick assembly/disassembly of these structures
for fast migration, and (iii) localized ECM degradation to allow microglia to traverse cell
layers in a controlled manner.
The initial discovery of podosomes in microglia by the Schlichter lab was followed
48

by the discovery that the Ca2+-activated SK3 channel localizes to the podosome core.
Microglial podosomes also collectively form a characteristic superstructure that we have
termed a ‘podonut’; i.e., many podosomes forming a large ring, with a centre that is generally
devoid of podosomes. Osteoclasts form a similar structure, called a sealing zone, in which
podosomes form a belt along the outer edge of the entire cell, resulting in a tight seal (Jurdic
et al., 2006). The sealing zone forms a sealed cavity into which protons and various proteases
are secreted for bone resorption (Sasaki and Hong, 1993; Duong and Rodan, 1998; Mulari et
al., 2003). Podonuts in microglia were found only in the lamella region of polarized cells
(Figure 4; (Vincent and Schlichter, 2010). This places podosomes in the leading edge of
microglia and in an ideal location to help microglia migrate.
Although podosomes may provide microglia with the means to migrate, the
mechanisms that mediate cell migration of microglia is a poorly understood subject. It is a
well-known fact that Ca2+ and its spatiotemporal regulation are important for directed cell
migration (Becker and Showell, 1972; Gallin and Rosenthal, 1974; Wilkinson, 1975; Boucek
and Snyderman, 1976; Estensen et al., 1976; Evans and Falke, 2007). In this study, I tested
the hypothesis that Ca2+ plays a crucial role in regulating podosomes. I provide supporting
evidence that podonut and thus, podosome formation is dependent on Ca2+. The absence of
extracellular Ca2+ or chelation of intracellular Ca2+ with BAPTA significantly reduced the
number of podonuts. Furthermore, microglia in culture tend to show lamellar morphology.
However, removal of Ca2+, intracellular or extracellular, resulted in loss of the lamellae.
Using time-lapse microscopy, our lab showed that microglia undergoing migration all
expressed lamella at their leading edge (Vincent and Schlichter, 2010). Given that lamella
formation is a requirement for microglia migration and Ca2+ chelation leads to loss of
49

lamellae in microglia, this would suggest that microglia migration is dependent on Ca2+.
Further studies, such as live recordings with cell tracking or transwell migration assays need
to be done to confirm this finding.
Because removal of Ca2+ affected podonut stability, I hypothesised that Ca2+ entry
into microglia is required for podonut and podosome formation. Despite attempts by several
laboratories, including ours, only one study has shown any evidence of voltage-gated Ca2+
channels in microglia (Colton et al., 1994). Many laboratories, including ours, have shown
other primary Ca2+ influx pathways, including TRP and CRAC channels (Norenberg et al.,
1997; Hahn et al., 2000; Jiang et al., 2003; Kraft et al., 2004; Fonfria et al., 2006; Kim et al.,
2006; Ohana et al., 2009; see review Kettenmann et al., 2011). While microglia express
transcripts for several TRP channels, TRPM7 expression was shown to be the highest among
them (Ohana et al., 2009), and our lab showed that rat microglia have a large TRPM7 current
(Jiang et al., 2003). Evidence presented in this thesis shows that inhibition of CRAC channels
abolished podonuts and podosomes, while blocking TRPM7 channels had no effect on
podonut numbers. Our present findings suggest that CRAC channel-mediated Ca2+ entry is
needed for formation of podosomes in microglia. Biophysical analysis of the CRAC channel
shows that it is highly selective for Ca2+ (Zweifach and Lewis, 1993). Because Ca2+ is a well-
known second messenger, I speculate that localized Ca2+ entry through Ca2+ permeable
channels in podosomes could modulate downstream effectors that lead to the formation of
podonuts.
In this study of rat microglia, I discovered the novel association of some molecules
with podosomes. Much knowledge regarding podosomes comes from Src-transformed cell
lines and other cell lines (e.g. the monocyte/macrophage RAW cell line). In most of these
50

cases, endogenous factors have been significantly modified (e.g. over-expression of kinases)
that do not reflect the biochemistry of unmodified, untransformed cells. Nonetheless, a key
signalling pathway necessary in formation of podosomes is activation of Src tyrosine kinase
(Linder and Aepfelbacher, 2003). Inhibition of Src kinase activity disrupts podosome
formation in osteoclasts and primary macrophages (Tanaka et al., 1995; Linder et al., 2000a),
while conversely, inhibition of tyrosine phosphatases induces podosome formation in
fibroblasts and monocytes (Marchisio et al., 1988; Cory et al., 2002). In addition, many
substrates of Src were also found to associate with podosomes (e.g. Pyk2 tyrosine kinase,
Tks5, PI3K) indicating further that Src signalling is essential for podosome regulation
(Schaller et al., 1994; Qian et al., 1997; Lock et al., 1998; von Willebrand et al., 1998;
Chellaiah et al., 2001; Abram et al., 2003). Therefore, I tested the hypothesis that similar
signalling molecules are present in microglial podosomes, by staining for substrates of Src
kinases. Immunolabeling for phosphotyrosine showed localization mostly to podosome core,
and to a lesser extent, the podosome ring. This provides the first evidence that tyrosine kinase
signalling might be involved in podosome regulation in primary microglia. Future studies
employing pharmacological modulators of tyrosine kinase signalling can be used to test
whether tyrosine kinases, specifically Src tyrosine kinases, are required for podosome
formation in microglia. I then hypothesized that other substrates of tyrosine kinases might
localize to podosomes. Tks5, a Src kinase substrate, has recently been reported to be an
organizer for podosome formation (Abram et al., 2003; Seals et al., 2005; Burger et al.,
2011). Tks5 consists of 5 SH3 domains that allow it to interact with various proteins,
possibly recruiting them to podosomes. Tks5 also contains a PX domain that can bind to
phosphatidylinositol bisphosphate (PIP2) – a commonly found membrane phospholipid in
51

podosomes that might help Tks5 localize to podosomes (Sechi and Wehland, 2000). In
microglial podosomes, I show that Tks5 localizes to podosomes; specifically to the
podosome ring. Other reports have shown association of Tks5 with podosomes in
transformed cell lines (Abram et al., 2003; Courtneidge et al., 2005), myoblasts (Thompson
et al., 2008), and macrophages (Burger et al., 2011). Tks5 can also associate with other
proteins, including the matrix degrading proteins, ADAMs 12, 15, 19 (Abram et al., 2003)
and MMP9 (Burger et al., 2011), the cytoskeletal regulating proteins, N-WASp, dynamin and
focal adhesion kinase (Courtneidge et al., 2005), and dystroglycan (Thompson et al., 2008).
The structure of Tks5 and its ability to interact with diverse molecules suggests that it
functions as a scaffolding protein that recruits and restricts localization of other proteins to
podosomes, resulting in a signalling nexus for the podosome machinery.
In this thesis, I presented evidence that pCav1 localizes specifically to the podosome
ring component in microglia. Caveolin-1 has been shown to associate with invadopodia in
breast- and melanoma- cancer cell lines (Caldieri et al., 2009; Yamaguchi et al., 2009;
Yamaguchi and Oikawa, 2010). Tyrosine 14 phosphorylated caveolin-1 (pCav1), on the other
hand, localized to podosomes but required ACTH stimulation of Y1 adrenal cells (Colonna
and Podesta, 2005). Caveolin-1 tyrosine phosphorylation can occur in response to growth
factor signalling or oxidative stress, and is thought to be mediated by c-Src (Labrecque et al.,
2004; Parat and Fox, 2004). Subsequent to phosphorylation of caveolin-1, it can interact with
proteins that are involved in cell migration and Src signalling, and with other SFKs, which
suggests that pCav1 serves as an adaptor molecule to recruit other molecules (Labrecque et
al., 2004). In focal adhesions, the phosphorylation state of caveolin-1 determines the turnover
rate of these adhesion structures by regulating signalling cascades downstream of pCav1
52

(Nethe and Hordijk, 2011). In addition, caveolin-1 proteins are major constituents of
specialized structures in the plasma membrane, called caveolae. Caveolae are small flask-
shaped invaginations (50-100 nm) of the membrane that are rich in cholesterol. These
structures have been linked with many physiological functions, such as membrane
trafficking, clathrin-independent endocytosis and cholesterol transport (Fujimoto et al.,
2000). Many cells use caveolae to organize cell-signalling molecules, and even ion channels,
into signalling complexes. Podosomes are hotspots for many signalling molecules and as
such, microglial podosomes might exploit caveolae and its versatile constituent, caveolin-1,
to bring together regulatory molecules that affect podosome formation and expression.
Podosome formation is the result of complex interactions between signalling
pathways (see review Murphy and Courtneidge, 2011), that are themselves regulated by
second messenger molecules. Reactive oxygen species (ROS) can act as second messengers
to modulate signalling pathways. For example, ROS production can inhibit protein tyrosine
phosphatases (PTPs) (Wu et al., 2003; Goldstein et al., 2005; Kwon et al., 2005), activate
kinases (Griendling et al., 2000; Han et al., 2003; Torres and Forman, 2003; Touyz et al.,
2004; Mehdi et al., 2005), and can regulate ion channels involved in K+ permeation and Ca2+
signalling (Zimmerman et al., 2005; Lee et al., 2006; refer review Bedard and Krause, 2007).
Although ROS are also a by-product of the electron transport chain in mitochondria, a family
of NADPH oxidase (Nox) enzymes functions solely to generate ROS (Bedard and Krause,
2007). Nox1, the first identified member of the Nox family, was previously found in
invadopodia, and both the Nox-1 specific blocker, ML171, and the non-specific blocker,
diphenylene iodonium (DPI), disrupted invadopodia formation in human colon cancer cells
(Gianni et al., 2009; Gianni et al., 2010). The authors concluded that localization of
53

functional Nox1 is required for the formation of invadopodia; however, contribution of other
Nox isoforms is possible because the concentration of ML171 used (10 μM) was much
higher than their reported IC50 values: Nox-2 (5 μM), Nox-3 (3 μM) and Nox-4 (5 μM)
(Gianni et al., 2010; Wingler et al., 2011). Gianni and colleagues also stated that Tks
proteins, including Tks5, could serve as organizers of Nox proteins at invadopodia, and they
suggested that inhibiting Nox1-mediated ROS generation increases local tyrosine
phosphatase activity and reduces tyrosine kinase activity. My results show for the first time
that Nox1 localizes to the podosome ring in microglia. Because the adaptor protein, Tks5,
also localised to the microglial podosome ring, it might regulate Nox1 localization. Localized
production of ROS by Nox1 might then facilitate tyrosine kinase signalling in podosomes to
modulate their formation and/or stability. Conversely, ROS generation might be regulated by
podosome components, such as c-Src tyrosine kinase or ion channels, like SK3, for overall
modulation of podosome assembly/disassembly (Khanna et al., 2001; Gianni et al., 2008;
Vincent and Schlichter, 2010).
I have shown that microglial podosome/podonut formation requires Ca2+ entry. I then
hypothesized that podosomes are signalling foci that contain Ca2+ entry channels and other
Ca2+ responders, which might allow formation of Ca2+ nanodomains and local signalling. My
observation of Orai1 in the podosome core supports this hypothesis. The CRAC channel is a
multimeric protein, with Orai1 being the critical pore forming subunit (Smyth et al., 2010).
Our lab showed that store operated Ca2+ entry (SOCE) in rat microglia occurs through the
CRAC channel (Ohana et al., 2009). Its important biophysical properties include inward
rectification, high selectivity for Ca2+, and a very small single-channel conductance (<0.2 pS)
(Zweifach and Lewis, 1993; Ohana et al., 2009). CRAC currents (sometimes referred to as
54

ICRAC) have been observed in several immune cell types, including microglia (Norenberg et
al., 1997; Ohana et al., 2009), mast cells (Hoth and Penner, 1992), the Jurkat T cell line
(Zweifach and Lewis, 1993), dendritic cells (Hsu et al., 2001); as well as in some non-
immune cells (Fasolato et al., 1993; Delles et al., 1995; Parekh and Penner, 1995;
Somasundaram et al., 1995; Rychkov et al., 2001). ICRAC is generated when intracellular Ca2+
stores in the endoplasmic reticulum (ER) are depleted (Hoth and Penner, 1992; see reviews
Parekh and Putney, 2005; Parekh, 2006; Smyth et al., 2010). Although many different
models have been postulated for the CRAC activation mechanism, the most widely accepted
model involves STIM1-Orai1 interaction after store depletion, as follows. Depletion of ER
Ca2+ is sensed by STIM1, an ER-resident Ca2+ sensor, which then undergoes a
conformational change that allows oligomerization with other STIM1 molecules. This
increases the affinity of STIM1 oligomers for Orai1 in the plasma membrane, and binding of
STIM1 to Orai1 opens the CRAC channel allowing influx of Ca2+. In general, influx through
Ca2+-permeable channels increases the Ca2+ concentration immediately adjacent to the
channel pore; free Ca2+ declines sharply with distance as Ca2+ diffuses away and is buffered
(Allbritton et al., 1992; Baimbridge et al., 1992; Smith et al., 1993; Kasai and Petersen, 1994;
Weber et al., 2010). In future, we want to determine whether Ca2+ microdomains form in
podosomes, and will begin by immunolabeling for STIM1 and podosome markers to see if
STIM1 and Orai1 co-localize in the podosome core.
Calmodulin is another Ca2+-dependent molecule that was present in microglial
podosomes (and throughout the microglia cytoplasm), providing a potential mechanism
whereby Ca2+ entry through CRAC could influence podosome-associated regulatory proteins.
Ca2+-CaM regulates many molecules, including CaM kinases and the gating of SK1–SK4
55

channels (Chin and Means, 2000). In addition, trafficking of SK3 and SK4 to the plasma
membrane depends on CaM-channel interactions, as shown by our lab and others (Khanna et
al., 1999; Joiner et al., 2001; Maylie et al., 2004). Because CaM co-assembles with SK
channels during their biogenesis, it was surprising to find CaM mainly in the microglia
podosome ring and SK3 mainly in the core.
I discovered a third novel Ca2+-binding protein in the microglial podosome core: the
‘ionized calcium-binding adapter-1’ protein (Iba1). Iba1 is a specific marker used to identify
microglia in the CNS. It is involved in binding and cross-linking filamentous actin, and is
regulated by Ca2+ (Sasaki et al., 2001), but has never been identified in podosomes or
invadipodia. Localization of Iba1 to podosomes might facilitate growth of the actin core by
cross-linking filamentous actin and stabilising the core structure. In future, it would be
interesting to determine whether Ca2+-mediated regulation of Iba1 contributes to podosome
expression and microglia migration. In contrast to my results on Iba1, another actin cross-
linking protein, α-actinin, is found in both the podosome ring and core in other cells
(Maruyama and Ebashi, 1965; Mimura and Asano, 1978; Marchisio et al., 1987; Stickel and
Wang, 1987; Imai et al., 1996; Gimona et al., 2003).
Some data that were omitted from this thesis include several molecules that showed
no evidence of localization to podosomes in microglia; i.e., SK4 and TRPM7 channels, and
the matrix metalloproteinases, MMP-9, MMP-12 and MMP-14. Figure 20 is an example
showing punctate TRPM7 staining that was evenly distributed throughout the cytoplasm,
with no podosome-like staining or co-localization with the podosome core marker, F-actin.
This finding is interesting in light of my finding that blocking TRPM7 with spermine did not
affect podonut formation in microglia. The lack of MMP localization to podosomes suggests
56

that the fibronectin degradation by microglial podosomes observed by our lab (Vincent and
Schlichter, 2010) depends on other ECM-degrading proteins. Other proteins with ECM-
degrading capacity (ADAMs, cathepsins) have been reported in invadosomes (Linder, 2007;
Murphy and Courtneidge, 2011), and in future, we plan to assess whether they are present in
microglia podosomes.
In conclusion, this thesis presents evidence that microglial podosomes contain several
components that are common to invadosomes in other cell types: F-actin, talin, Arp2,
tyrosine phosphorylated proteins, Tks5, Tyr-14-phosphorylated caveolin-1 and Nox1. These
findings suggest that signalling pathways that regulate podosomes in other cell types might
also function in microglia. Novel findings in this thesis include the association of the
following molecules with microglial podosomes: SK3, Orai1, CaM, and Iba1. The presence
of a Ca2+ permeable channel (Orai1/CRAC) and several Ca2+ responsive proteins (CaM,
SK3, Iba1) suggests that podosomes might be a hub for local signalling within a Ca2+
microdomain, and is supported by my finding that podonuts/podosomes in microglia depend
on Ca2+ entry through CRAC channels. If so, this would provide migrating cells with
additional regulatory mechanisms for the cell-substrate adhesion and matrix degradation that
is necessary for traversing through tissue. While it will be important to demonstrate that
microglia migration depends on Ca2+, this is very likely the case, and is well established for
many migrating cells.
57

F‐Actin TRPM7 F‐Actin TRPM7
Figure 20. Immunolabeling of TRPM7 channels and F-actin in microglia. Colour-separated
images of a cell stained for the podosome core marker, F-actin (phalloidin; green) and
TRPM7 (red); merged image at right. Scale bars = 5 µm
58

WORKING MODEL
Based on the literature regarding podosomes and the above findings from the
Schlichter lab and in this thesis, I propose a model for the podosome structure, assembly,
disassembly and function (Figure 21). This is by no means complete, but is a culmination of
the literature and our novel findings in microglia.
It is thought that podosome assembly begins when the protruding lamellipodium of a
migrating cell finds, and adheres to a suitable ECM substrate. Upon binding matrix ligands,
integrins undergo a conformational change that leads to changes in cytosolic factors,
including activation of Src kinases, recruitment of phosphoinositide-3-kinase (PI3K), and
interaction with cytoskeletal rearrangement proteins such as talin, vinculin and paxillin (van
der Flier and Sonnenberg, 2001; Tvorogov et al., 2005; Alberts et al., 2008), which modulate
other signalling cascades (Hynes, 2002). This ‘outside-in’ signalling translates integrin
binding to extracellular matrix into effects on cytosolic factors (Hynes, 1992; Alberts et al.,
2008). In addition, integrins mediate ‘inside-out’ signalling, in which the cytoplasmic tail
interacts with proteins (e.g., talin) that can induce a conformational change, and increase
integrin affinity for matrix proteins (Ginsberg et al., 1992; Calderwood et al., 1999; Hynes,
2002; Alberts et al., 2008). Recruitment of adhesion-associated molecules then activates actin
nucleators, such as Arp2/3 complex, WASP and/or N-WASP, to produce filamentous actin
from G-actin monomers in the podosome core (Alberts et al., 2008). Integrin clustering at the
cell-matrix contact site strengthens the adhesion (Alberts et al., 2008). Clustered integrins
might localize to cholesterol-rich domains along with their ligands, such as ECM
components, and bring along adhesion-associated machinery in the cytosol to these domains
(del Pozo et al., 2005). Conversely, integrin clustering at adhesion contact sites might recruit
59

caveolin-1 (Nethe and Hordijk, 2011), a structural component of cholesterol-rich membrane
domains (caveolae) that might serve as the initial organizing center for integrin signalling
(del Pozo et al., 2005; Nethe and Hordijk, 2011).
Once integrins are activated, Src tyrosine kinase activation is thought to promote
phosphorylation and activation of other substrates, including caveolin-1 at Tyr-14 (Lee et al.,
2000; Cao et al., 2002), Tks5 (Lock et al., 1998; Abram et al., 2003), and PLCγ (Kanner et
al., 1993; Clark and Brugge, 1995). Activated PI3K can phosphorylate the membrane lipid,
phosphatidylinositol, to produce PIP2 and PIP3, and PIP2 binds to phosphorylated Tks5,
where it presumably serves as an organizer, bringing other proteins, such as Nox1, to the
maturing podosome (Sechi and Wehland, 2000; Gianni et al., 2010). Nox1 generates ROS,
which can inhibit protein tyrosine phosphatases (PTPs) (Wu et al., 2003; Goldstein et al.,
2005; Kwon et al., 2005), and possibly enhance Src tyrosine kinase signalling in a positive
feedback loop.
Phosphorylation of PLCγ activates it to cleave PIP2, which releases IP3 and DAG
(Nishibe et al., 1990; Tvorogov et al., 2005). IP3, in turn, activates IP3 receptors in the ER
membrane, depletes intracellular Ca2+ stores, and activates Ca2+ influx through CRAC
channels, which might then form a Ca2+ nanodomain around a podosome. Influx of Ca2+
potentially activates other molecules, including the tyrosine kinase, Pyk2 (Lev et al., 1995;
Yu et al., 1996); Iba1, which cross-links actin filaments along with α-actinin (Maruyama and
Ebashi, 1965; Mimura and Asano, 1978; Sasaki et al., 2001); and CaM, which can further
interact with other molecules (Chin and Means, 2000). A relevant Ca2+-CaM interaction
occurs at the SK3 channel, which is opened by Ca2+ binding to CaM and then mediates K+
efflux (Xia et al., 1998). The best known role of SK channels is to hyper-polarize cells,
60

which maintains the large driving force required for Ca2+ influx through CRAC channels
when the ER lumen is depleted of Ca2+ (Lewis and Cahalan, 1995; Fanger et al., 2001;
Ahmmed and Malik, 2005). Once assembly of podosome structure is well underway,
microtubules can traffic molecules, such as matrix-degrading proteins to this structure
(Poincloux et al., 2009; Wiesner et al., 2010).
There is some information regarding disassembly of podosomes. This might begin
with pCav1, if it initially serves to recruit adhesion associated molecules. Tyr-14
phosphorylation of caveolin-1 is required for internalization of cholesterol-enriched
membranes, including caveolae (del Pozo et al., 2005). Along with internalization of
caveolae and podosomes, integrins located in these structures would detach from the matrix,
thereby stopping the integrin-mediated signal required to maintain Src kinase activity. Other
intrinsic factors could also inactivate integrins by reducing their affinity for matrix ligands or
preventing them from binding matrix proteins (Millon-Fremillon et al., 2008; Harburger and
Calderwood, 2009). For example, the intracellular protease, calpain, can break down talin
(Franco et al., 2004), and together with proteolysis of the integrin cytosolic tail (Flevaris et
al., 2007), can interfere with integrin signalling. Without integrin signalling, there would be
dissociation of actin nucleators (e.g. Arp2/3, WASP), actin-association proteins (e.g. α-
actinin), and adhesion-associated proteins (e.g. vinculin, paxillin, talin). PI3K activity in
podosomes would cease, preventing production of PIP2 at the membrane, which would
prevent Tks5 anchoring to the podosome membrane, allowing it to diffuse away. In turn,
Tks5 loss would diminish Nox1-mediated ROS generation in podosomes, and increase PTP
activity. When ER Ca2+ is replenished, the CRAC channel would close, and Ca2+ would be
removed from the nanodomain by buffering (by cytosolic proteins), uptake into organelles, or
61

pumping out due to plasma membrane Ca2+ pumps. The reduction in Ca2+ would close SK3
channels as apoCaM forms. Loss of the Ca2+ signal could influence other molecules; e.g.,
inactivating Pyk2 tyrosine kinase and other Ca2+ regulated molecules, such as Iba1.
Podosome association with the microtubule plus-end associated molecule, KF1C, might also
lead to disassembly of podosomes (Kopp et al., 2006).
62

Figure 21. Updated schematic showing podosome-associated molecules (modified from
(Linder and Aepfelbacher, 2003), with the molecules found in microglia in this thesis shown
in red boxes.
Adapted and reprinted from The Lancet, Vol. 13, Linder S, Aepfelbacher M, Podosomes: adhesion hot-spots of invasive cells, 376-385, Copyright (2003), with permission from Elsevier
63

FUTURE STUDIES
In this thesis, I conducted extensive immunostaining on primary cultures of rat
microglia, and identified both known and novel molecules in podosomes. Knowledge about
podosome components provides us with the foundation to explore some of the signalling
pathway(s) that might be involved in podosome formation and functions in microglia.
Continued characterization of the components of podosomes will help identify potential
pharmacological targets (e.g. SK3, Orai1, CaM, Iba1) for promoting or inhibiting podosome
formation. Selective drugs could then be used with immunocytochemistry and functional
assays to study podosome formation and roles. Both 2-D migration- and 3-D invasion assays
using brain-relevant ECM substrates would shed light on whether active podosomes are
needed for microglial migration or invasion. Because Ca2+ removal disrupted lamellipodia
formation, a structure required for microglia migration (Vincent and Schlichter, 2010), we
expect that microglia invasion through ECM-coated filters in Transwell™ chambers would
be inhibited in the absence of extracellular Ca2+, buffering of intracellular Ca2+, or blocking
specific Ca2+-entry and responding pathways (e.g., Orai1/CRAC and SK3 channels).
Unfortunately, primary microglia are extremely resistant to gene transfection, siRNA-
mediated knockdown or viral-mediated infection (Ohana et al., 2009); therefore, we have to
mainly rely on pharmacological approaches for these proposed studies.
I suggested in this thesis that microglial podosomes represent a nexus for Ca2+- and
ROS signalling pathways. Therefore, I would like to: (i) use Fura2-FF, an analogue of the
fluorescent Ca2+ indicator, Fura-2, to monitor and measure any rapid changes in local Ca2+ in
and around the tiny podosome structures; and (ii) monitor ROS levels with Rhodamine 123
in podonuts, and (iii) determine whether these two signals interact and are interdependent.
64

This would address the possible roles of Ca2+ and ROS as second messengers for podosome
regulation and function.
Our discovery of podosomes in microglia in vitro raises the possibility that migrating
microglia in the brain use these structures to locally degrade ECM and to navigate through
CNS tissue to reach the site of injury. Only a handful of studies have identified podosomes in
vivo, and never in microglia or the CNS. At this point, our in vitro studies have been limited
by using microglia grown in an artificial 2-dimensional environment. Ultimately, to study
podosomes in a 3-dimensional in vivo environment, we would like to use adult rats and our
ischemic and hemorrhagic stroke models, in which massive microglia migration is observed.
This would allow analysis with the naturally occurring complement of ECM molecules, and
factor in changes that occur after acute damage. For instance, Iba1, a microglia/macrophage-
specific marker that has been extensively used in our lab, is strongly up-regulated in
microglia/macrophages in rat brain after stroke. Thus, Iba1 could be used to identify these
cells and simultaneously label podosomes to assess their formation in the CNS in vivo.
In summary, there is evidence that microglia are involved in most neuropathological
processes (Tambuyzer et al., 2009), and microglial migration to the site of injury is a crucial
early response to acute CNS injury. Hence, determining the role of podosomes, and
mechanisms regulating their specific functions, will bring us an important step closer to
developing therapeutic measures that target microglia and brain inflammation.
65

REFERENCES CITED
Aarum J, Sandberg K, Haeberlein SL, Persson MA (2003) Migration and differentiation of
neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A
100:15983-15988.
Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA (2003) The
adaptor protein fish associates with members of the ADAMs family and localizes to
podosomes of Src-transformed cells. J Biol Chem 278:16844-16851.
Ahmmed GU, Malik AB (2005) Functional role of TRPC channels in the regulation of
endothelial permeability. Pflugers Arch 451:131-142.
Alberts B (2009) Essential cell biology, 3rd Edition. New York: Garland Science.
Alberts B, Wilson JH, Hunt T (2008) Molecular biology of the cell, 5th Edition. New York:
Garland Science.
Aleshin A, Finn RS (2010) SRC: a century of science brought to the clinic. Neoplasia
12:599-607.
Allbritton NL, Meyer T, Stryer L (1992) Range of messenger action of calcium ion and
inositol 1,4,5-trisphosphate. Science 258:1812-1815.
Baimbridge KG, Celio MR, Rogers JH (1992) Calcium-binding proteins in the nervous
system. Trends Neurosci 15:303-308.
Becchetti A, Arcangeli A (2010) Integrins and ion channels in cell migration: implications
for neuronal development, wound healing and metastatic spread. Adv Exp Med Biol
674:107-123.
66

Becker EL, Showell HJ (1972) The effect of Ca2+ and Mg2+ on the chemotactic
responsiveness and spontaneous motility of rabbit polymorphonuclear leukocytes. Z
Immunitatsforsch Exp Klin Immunol 143:466-476.
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases:
physiology and pathophysiology. Physiol Rev 87:245-313.
Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG (2004) Microregional extracellular
matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol
36:1046-1069.
Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW (2001) Ischemia-induced neuronal
expression of the microglia attracting chemokine Secondary Lymphoid-tissue
Chemokine (SLC). Glia 34:121-133.
Binks M, Jones GE, Brickell PM, Kinnon C, Katz DR, Thrasher AJ (1998) Intrinsic dendritic
cell abnormalities in Wiskott-Aldrich syndrome. Eur J Immunol 28:3259-3267.
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the
molecular mechanisms. Nat Rev Neurosci 8:57-69.
Block MR, Badowski C, Millon-Fremillon A, Bouvard D, Bouin AP, Faurobert E, Gerber-
Scokaert D, Planus E, Albiges-Rizo C (2008) Podosome-type adhesions and focal
adhesions, so alike yet so different. Eur J Cell Biol 87:491-506.
Boje KM, Arora PK (1992) Microglial-produced nitric oxide and reactive nitrogen oxides
mediate neuronal cell death. Brain Res 587:250-256.
Bonifati DM, Kishore U (2007) Role of complement in neurodegeneration and
neuroinflammation. Mol Immunol 44:999-1010.
67

Boucek MM, Snyderman R (1976) Calcium influx requirement for human neutrophil
chemotaxis: inhibition by lanthanum chloride. Science 193:905-907.
Brockhaus J, Moller T, Kettenmann H (1996) Phagocytozing ameboid microglial cells
studied in a mouse corpus callosum slice preparation. Glia 16:81-90.
Brundage RA, Fogarty KE, Tuft RA, Fay FS (1991) Calcium gradients underlying
polarization and chemotaxis of eosinophils. Science 254:703-706.
Burger KL, Davis AL, Isom S, Mishra N, Seals DF (2011) The podosome marker protein
Tks5 regulates macrophage invasive behavior. Cytoskeleton (Hoboken).
Burgstaller G, Gimona M (2005) Podosome-mediated matrix resorption and cell motility in
vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 288:H3001-3005.
Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE (2001) Configuration of human
dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation.
Blood 98:1142-1149.
Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH (1999) The Talin head
domain binds to integrin beta subunit cytoplasmic tails and regulates integrin
activation. J Biol Chem 274:28071-28074.
Caldieri G, Giacchetti G, Beznoussenko G, Attanasio F, Ayala I, Buccione R (2009)
Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane
cholesterol levels. J Cell Mol Med 13:1728-1740.
Calle Y, Burns S, Thrasher AJ, Jones GE (2006) The leukocyte podosome. Eur J Cell Biol
85:151-157.
Campisi P, Egiazarian K (2007) Blind image deconvolution : theory and applications. Boca
Raton: CRC Press.
68

Cao H, Courchesne WE, Mastick CC (2002) A phosphotyrosine-dependent protein
interaction screen reveals a role for phosphorylation of caveolin-1 on tyrosine 14:
recruitment of C-terminal Src kinase. J Biol Chem 277:8771-8774.
Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak
AM, Springer TA (2007) Transcellular diapedesis is initiated by invasive podosomes.
Immunity 26:784-797.
Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM (2007) A rose
by any other name? The potential consequences of microglial heterogeneity during
CNS health and disease. Neurotherapeutics 4:571-579.
Caswell PT, Vadrevu S, Norman JC (2009) Integrins: masters and slaves of endocytic
transport. Nat Rev Mol Cell Biol 10:843-853.
Chakraborty S, Kaushik DK, Gupta M, Basu A (2010) Inflammasome signaling at the heart
of central nervous system pathology. J Neurosci Res 88:1615-1631.
Chan WY, Kohsaka S, Rezaie P (2007) The origin and cell lineage of microglia: new
concepts. Brain Res Rev 53:344-354.
Chellaiah MA, Biswas RS, Yuen D, Alvarez UM, Hruska KA (2001) Phosphatidylinositol
3,4,5-trisphosphate directs association of Src homology 2-containing signaling
proteins with gelsolin. J Biol Chem 276:47434-47444.
Chen WT, Chen JM, Parsons SJ, Parsons JT (1985) Local degradation of fibronectin at sites
of expression of the transforming gene product pp60src. Nature 316:156-158.
Chhabra ES, Higgs HN (2007) The many faces of actin: matching assembly factors with
cellular structures. Nat Cell Biol 9:1110-1121.
69

Chin D, Means AR (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol
10:322-328.
Clark EA, Brugge JS (1995) Integrins and signal transduction pathways: the road taken.
Science 268:233-239.
Colonna C, Podesta EJ (2005) ACTH-induced caveolin-1 tyrosine phosphorylation is related
to podosome assembly in Y1 adrenal cells. Exp Cell Res 304:432-442.
Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in
the brain. J Neuroimmune Pharmacol 4:399-418.
Colton CA, Wilcock DM (2010) Assessing activation states in microglia. CNS Neurol Disord
Drug Targets 9:174-191.
Colton CA, Jia M, Li MX, Gilbert DL (1994) K+ modulation of microglial superoxide
production: involvement of voltage-gated Ca2+ channels. Am J Physiol 266:C1650-
1655.
Cory GO, Garg R, Cramer R, Ridley AJ (2002) Phosphorylation of tyrosine 291 enhances the
ability of WASp to stimulate actin polymerization and filopodium formation.
Wiskott-Aldrich Syndrome protein. J Biol Chem 277:45115-45121.
Cougoule C, Carreno S, Castandet J, Labrousse A, Astarie-Dequeker C, Poincloux R, Le
Cabec V, Maridonneau-Parini I (2005) Activation of the lysosome-associated p61Hck
isoform triggers the biogenesis of podosomes. Traffic 6:682-694.
Cougoule C, Le Cabec V, Poincloux R, Al Saati T, Mege JL, Tabouret G, Lowell CA,
Laviolette-Malirat N, Maridonneau-Parini I (2010) Three-dimensional migration of
macrophages requires Hck for podosome organization and extracellular matrix
proteolysis. Blood 115:1444-1452.
70

Courtneidge SA, Azucena EF, Pass I, Seals DF, Tesfay L (2005) The SRC substrate Tks5,
podosomes (invadopodia), and cancer cell invasion. Cold Spring Harb Symp Quant
Biol 70:167-171.
Crowley JL, Smith TC, Fang Z, Takizawa N, Luna EJ (2009) Supervillin reorganizes the
actin cytoskeleton and increases invadopodial efficiency. Mol Biol Cell 20:948-962.
Cuadros MA, Moujahid A, Quesada A, Navascues J (1994) Development of microglia in the
quail optic tectum. J Comp Neurol 348:207-224.
Cuadros MA, Rodriguez-Ruiz J, Calvente R, Almendros A, Marin-Teva JL, Navascues J
(1997) Microglia development in the quail cerebellum. J Comp Neurol 389:390-401.
Dapson RW (1993) Fixation for the 1990's: a review of needs and accomplishments. Biotech
Histochem 68:75-82.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan
WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat
Neurosci 8:752-758.
David-Pfeuty T, Singer SJ (1980) Altered distributions of the cytoskeletal proteins vinculin
and alpha-actinin in cultured fibroblasts transformed by Rous sarcoma virus. Proc
Natl Acad Sci U S A 77:6687-6691.
Davoust N, Vuaillat C, Androdias G, Nataf S (2008) From bone marrow to microglia:
barriers and avenues. Trends Immunol 29:227-234.
del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-Garcia A, Anderson
RG, Schwartz MA (2005) Phospho-caveolin-1 mediates integrin-regulated membrane
domain internalization. Nat Cell Biol 7:901-908.
71

Delles C, Haller T, Dietl P (1995) A highly calcium-selective cation current activated by
intracellular calcium release in MDCK cells. J Physiol 486 ( Pt 3):557-569.
Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA (2009) Tks5-
dependent, nox-mediated generation of reactive oxygen species is necessary for
invadopodia formation. Sci Signal 2:ra53.
Dovas A, Cox D (2011) Signaling networks regulating leukocyte podosome dynamics and
function. Cell Signal 23:1225-1234.
Duong LT, Rodan GA (1998) Integrin-mediated signaling in the regulation of osteoclast
adhesion and activation. Front Biosci 3:d757-768.
Estensen RD, Reusch ME, Epstein ML, Hill HR (1976) Role of Ca2+ and Mg2+ in some
human neutrophil functions as indicated by ionophore A23187. Infect Immun 13:146-
151.
Evans JG, Correia I, Krasavina O, Watson N, Matsudaira P (2003) Macrophage podosomes
assemble at the leading lamella by growth and fragmentation. J Cell Biol 161:697-
705.
Evans JH, Falke JJ (2007) Ca2+ influx is an essential component of the positive-feedback
loop that maintains leading-edge structure and activity in macrophages. Proc Natl
Acad Sci U S A 104:16176-16181.
Fanger CM, Rauer H, Neben AL, Miller MJ, Rauer H, Wulff H, Rosa JC, Ganellin CR,
Chandy KG, Cahalan MD (2001) Calcium-activated potassium channels sustain
calcium signaling in T lymphocytes. Selective blockers and manipulated channel
expression levels. J Biol Chem 276:12249-12256.
72

Farber K, Markworth S, Pannasch U, Nolte C, Prinz V, Kronenberg G, Gertz K, Endres M,
Bechmann I, Enjyoji K, Robson SC, Kettenmann H (2008) The ectonucleotidase
cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia 56:331-
341.
Fasolato C, Hoth M, Penner R (1993) A GTP-dependent step in the activation mechanism of
capacitative calcium influx. J Biol Chem 268:20737-20740.
Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X (2007) A molecular switch that
controls cell spreading and retraction. J Cell Biol 179:553-565.
Fonfria E, Mattei C, Hill K, Brown JT, Randall A, Benham CD, Skaper SD, Campbell CA,
Crook B, Murdock PR, Wilson JM, Maurio FP, Owen DE, Tilling PL, McNulty S
(2006) TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated,
and functionally expressed in C13 microglia. J Recept Signal Transduct Res 26:179-
198.
Fox CH, Johnson FB, Whiting J, Roller PP (1985) Formaldehyde fixation. J Histochem
Cytochem 33:845-853.
Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A (2004)
Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol
6:977-983.
Fujimoto T, Kogo H, Nomura R, Une T (2000) Isoforms of caveolin-1 and caveolar
structure. J Cell Sci 113 Pt 19:3509-3517.
Galea I, Bechmann I, Perry VH (2007) What is immune privilege (not)? Trends Immunol
28:12-18.
73

Gallin JI, Rosenthal AS (1974) The regulatory role of divalent cations in human granulocyte
chemotaxis. Evidence for an association between calcium exchanges and microtubule
assembly. J Cell Biol 62:594-609.
Gianni D, Bohl B, Courtneidge SA, Bokoch GM (2008) The involvement of the tyrosine
kinase c-Src in the regulation of reactive oxygen species generation mediated by
NADPH oxidase-1. Mol Biol Cell 19:2984-2994.
Gianni D, Diaz B, Taulet N, Fowler B, Courtneidge SA, Bokoch GM (2009) Novel
p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity.
Sci Signal 2:ra54.
Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown
SJ, Bokoch GM, Rosen H (2010) A novel and specific NADPH oxidase-1 (Nox1)
small-molecule inhibitor blocks the formation of functional invadopodia in human
colon cancer cells. ACS Chem Biol 5:981-993.
Gilbert SF (2006) Developmental biology, 8th Edition. Sunderland, Mass.: Sinauer
Associates, Inc. Publishers.
Gimona M (2008) The microfilament system in the formation of invasive adhesions. Semin
Cancer Biol 18:23-34.
Gimona M, Kaverina I, Resch GP, Vignal E, Burgstaller G (2003) Calponin repeats regulate
actin filament stability and formation of podosomes in smooth muscle cells. Mol Biol
Cell 14:2482-2491.
Ginsberg MH, Du X, Plow EF (1992) Inside-out integrin signalling. Curr Opin Cell Biol
4:766-771.
74

Goldstein BJ, Mahadev K, Wu X (2005) Redox paradox: insulin action is facilitated by
insulin-stimulated reactive oxygen species with multiple potential signaling targets.
Diabetes 54:311-321.
Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Immunol
5:953-964.
Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol 119:89-
105.
Grande-Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman-Storer CM, Valdivielso
JM, del Pozo MA (2007) Caveolin-1 regulates cell polarization and directional
migration through Src kinase and Rho GTPases. J Cell Biol 177:683-694.
Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M (2000) Modulation of protein kinase
activity and gene expression by reactive oxygen species and their role in vascular
physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20:2175-2183.
Hahn J, Jung W, Kim N, Uhm DY, Chung S (2000) Characterization and regulation of rat
microglial Ca(2+) release-activated Ca(2+) (CRAC) channel by protein kinases. Glia
31:118-124.
Hammarfjord O, Falet H, Gurniak C, Hartwig JH, Wallin RP (2011) Gelsolin-independent
podosome formation in dendritic cells. PLoS One 6:e21615.
Han MJ, Kim BY, Yoon SO, Chung AS (2003) Cell proliferation induced by reactive oxygen
species is mediated via mitogen-activated protein kinase in Chinese hamster lung
fibroblast (V79) cells. Mol Cells 15:94-101.
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the
normal and pathologic brain. Nat Neurosci 10:1387-1394.
75

Harburger DS, Calderwood DA (2009) Integrin signalling at a glance. J Cell Sci 122:159-
163.
Hoffmann EK, Lambert IH, Pedersen SF (2009) Physiology of cell volume regulation in
vertebrates. Physiol Rev 89:193-277.
Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S (2001)
Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-
coupled P2Y receptors. J Neurosci 21:1975-1982.
Hoth M, Penner R (1992) Depletion of intracellular calcium stores activates a calcium
current in mast cells. Nature 355:353-356.
Hsu S, O'Connell PJ, Klyachko VA, Badminton MN, Thomson AW, Jackson MB, Clapham
DE, Ahern GP (2001) Fundamental Ca2+ signaling mechanisms in mouse dendritic
cells: CRAC is the major Ca2+ entry pathway. J Immunol 166:6126-6133.
Hynes RO (1992) Integrins: versatility, modulation, and signaling in cell adhesion. Cell
69:11-25.
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673-687.
Ifuku M, Farber K, Okuno Y, Yamakawa Y, Miyamoto T, Nolte C, Merrino VF, Kita S,
Iwamoto T, Komuro I, Wang B, Cheung G, Ishikawa E, Ooboshi H, Bader M, Wada
K, Kettenmann H, Noda M (2007) Bradykinin-induced microglial migration mediated
by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the
Na+/Ca2+ exchanger. J Neurosci 27:13065-13073.
Imai Y, Kohsaka S (2002) Intracellular signaling in M-CSF-induced microglia activation:
role of Iba1. Glia 40:164-174.
76

Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S (1996) A novel gene iba1 in the major
histocompatibility complex class III region encoding an EF hand protein expressed in
a monocytic lineage. Biochem Biophys Res Commun 224:855-862.
Jiang X, Newell EW, Schlichter LC (2003) Regulation of a TRPM7-like current in rat brain
microglia. J Biol Chem 278:42867-42876.
Joiner WJ, Khanna R, Schlichter LC, Kaczmarek LK (2001) Calmodulin regulates assembly
and trafficking of SK4/IK1 Ca2+-activated K+ channels. J Biol Chem 276:37980-
37985.
Jurdic P, Saltel F, Chabadel A, Destaing O (2006) Podosome and sealing zone: specificity of
the osteoclast model. Eur J Cell Biol 85:195-202.
Kanner SB, Grosmaire LS, Ledbetter JA, Damle NK (1993) Beta 2-integrin LFA-1 signaling
through phospholipase C-gamma 1 activation. Proc Natl Acad Sci U S A 90:7099-
7103.
Kasai H, Petersen OH (1994) Spatial dynamics of second messengers: IP3 and cAMP as
long-range and associative messengers. Trends Neurosci 17:95-101.
Kaur G, Han SJ, Yang I, Crane C (2010) Microglia and central nervous system immunity.
Neurosurg Clin N Am 21:43-51.
Kaushal V, Koeberle PD, Wang Y, Schlichter LC (2007) The Ca2+-activated K+ channel
KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent
neurodegeneration. J Neurosci 27:234-244.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia.
Physiol Rev 91:461-553.
77

Khanna R, Roy L, Zhu X, Schlichter LC (2001) K+ channels and the microglial respiratory
burst. Am J Physiol Cell Physiol 280:C796-806.
Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC (1999) hSK4/hIK1, a
calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and
volume regulation. J Biol Chem 274:14838-14849.
Kim SR, Kim SU, Oh U, Jin BK (2006) Transient receptor potential vanilloid subtype 1
mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial
damage and cytochrome c release. J Immunol 177:4322-4329.
Kim WK, Ko KH (1998) Potentiation of N-methyl-D-aspartate-mediated neurotoxicity by
immunostimulated murine microglia. J Neurosci Res 54:17-26.
Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007) Kuby immunology, 6th Edition. New
York: W.H. Freeman.
Kopp P, Lammers R, Aepfelbacher M, Woehlke G, Rudel T, Machuy N, Steffen W, Linder S
(2006) The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in
macrophages. Mol Biol Cell 17:2811-2823.
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G,
Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated
calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286:C129-
137.
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends
Neurosci 19:312-318.
78

Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS (2005) Receptor-stimulated
oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. Embo J
24:2331-2341.
Labrecque L, Nyalendo C, Langlois S, Durocher Y, Roghi C, Murphy G, Gingras D,
Beliveau R (2004) Src-mediated tyrosine phosphorylation of caveolin-1 induces its
association with membrane type 1 matrix metalloproteinase. J Biol Chem 279:52132-
52140.
Laffafian I, Hallett MB (1995) Does cytosolic free Ca2+ signal neutrophil chemotaxis in
response to formylated chemotactic peptide? J Cell Sci 108 ( Pt 10):3199-3205.
Lalo U, Pankratov Y, Wichert SP, Rossner MJ, North RA, Kirchhoff F, Verkhratsky A
(2008) P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical
astrocytes. J Neurosci 28:5473-5480.
Lauffenburger DA, Horwitz AF (1996) Cell migration: a physically integrated molecular
process. Cell 84:359-369.
Le Clainche C, Carlier MF (2008) Regulation of actin assembly associated with protrusion
and adhesion in cell migration. Physiol Rev 88:489-513.
Lee H, Volonte D, Galbiati F, Iyengar P, Lublin DM, Bregman DB, Wilson MT, Campos-
Gonzalez R, Bouzahzah B, Pestell RG, Scherer PE, Lisanti MP (2000) Constitutive
and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site
(Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol
Endocrinol 14:1750-1775.
Lee YM, Kim BJ, Chun YS, So I, Choi H, Kim MS, Park JW (2006) NOX4 as an oxygen
sensor to regulate TASK-1 activity. Cell Signal 18:499-507.
79

Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B,
Schlessinger J (1995) Protein tyrosine kinase PYK2 involved in Ca(2+)-induced
regulation of ion channel and MAP kinase functions. Nature 376:737-745.
Lewis RS, Cahalan MD (1995) Potassium and calcium channels in lymphocytes. Annu Rev
Immunol 13:623-653.
Li S, Seitz R, Lisanti MP (1996) Phosphorylation of caveolin by src tyrosine kinases. The
alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem
271:3863-3868.
Liang KJ, Lee JE, Wang YD, Ma W, Fontainhas AM, Fariss RN, Wong WT (2009)
Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Invest
Ophthalmol Vis Sci 50:4444-4451.
Linder S (2007) The matrix corroded: podosomes and invadopodia in extracellular matrix
degradation. Trends Cell Biol 17:107-117.
Linder S, Aepfelbacher M (2003) Podosomes: adhesion hot-spots of invasive cells. Trends
Cell Biol 13:376-385.
Linder S, Hufner K, Wintergerst U, Aepfelbacher M (2000a) Microtubule-dependent
formation of podosomal adhesion structures in primary human macrophages. J Cell
Sci 113 Pt 23:4165-4176.
Linder S, Higgs H, Hufner K, Schwarz K, Pannicke U, Aepfelbacher M (2000b) The
polarization defect of Wiskott-Aldrich syndrome macrophages is linked to
dislocalization of the Arp2/3 complex. J Immunol 165:221-225.
Liu GJ, Nagarajah R, Banati RB, Bennett MR (2009) Glutamate induces directed chemotaxis
of microglia. Eur J Neurosci 29:1108-1118.
80

Lock P, Abram CL, Gibson T, Courtneidge SA (1998) A new method for isolating tyrosine
kinase substrates used to identify fish, an SH3 and PX domain-containing protein, and
Src substrate. Embo J 17:4346-4357.
Ma T, Samanna V, Chellaiah MA (2008) Dramatic inhibition of osteoclast sealing ring
formation and bone resorption in vitro by a WASP-peptide containing pTyr294 amino
acid. J Mol Signal 3:4.
Ma T, Sadashivaiah K, Madayiputhiya N, Chellaiah MA (2010) Regulation of sealing ring
formation by L-plastin and cortactin in osteoclasts. J Biol Chem 285:29911-29924.
Mandal S, Johnson KR, Wheelock MJ (2008) TGF-beta induces formation of F-actin cores
and matrix degradation in human breast cancer cells via distinct signaling pathways.
Exp Cell Res 314:3478-3493.
Mandeville JT, Ghosh RN, Maxfield FR (1995) Intracellular calcium levels correlate with
speed and persistent forward motion in migrating neutrophils. Biophys J 68:1207-
1217.
Marchisio PC, Cirillo D, Teti A, Zambonin-Zallone A, Tarone G (1987) Rous sarcoma virus-
transformed fibroblasts and cells of monocytic origin display a peculiar dot-like
organization of cytoskeletal proteins involved in microfilament-membrane
interactions. Exp Cell Res 169:202-214.
Marchisio PC, D'Urso N, Comoglio PM, Giancotti FG, Tarone G (1988) Vanadate-treated
baby hamster kidney fibroblasts show cytoskeleton and adhesion patterns similar to
their Rous sarcoma virus-transformed counterparts. J Cell Biochem 37:151-159.
81

Marin-Teva JL, Almendros A, Calvente R, Cuadros MA, Navascues J (1998) Tangential
migration of ameboid microglia in the developing quail retina: mechanism of
migration and migratory behavior. Glia 22:31-52.
Maruyama K, Ebashi S (1965) Alpha-actinin, a new structural protein from striated muscle.
II. Action on actin. J Biochem 58:13-19.
Mason JT, O'Leary TJ (1991) Effects of formaldehyde fixation on protein secondary
structure: a calorimetric and infrared spectroscopic investigation. J Histochem
Cytochem 39:225-229.
Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP (2004) Small conductance Ca2+-
activated K+ channels and calmodulin. J Physiol 554:255-261.
Mehdi MZ, Pandey NR, Pandey SK, Srivastava AK (2005) H2O2-induced phosphorylation
of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src.
Antioxid Redox Signal 7:1014-1020.
Messier JM, Shaw LM, Chafel M, Matsudaira P, Mercurio AM (1993) Fimbrin localized to
an insoluble cytoskeletal fraction is constitutively phosphorylated on its headpiece
domain in adherent macrophages. Cell Motil Cytoskeleton 25:223-233.
Millon-Fremillon A, Bouvard D, Grichine A, Manet-Dupe S, Block MR, Albiges-Rizo C
(2008) Cell adaptive response to extracellular matrix density is controlled by ICAP-1-
dependent beta1-integrin affinity. J Cell Biol 180:427-441.
Mimura N, Asano A (1978) Actin-related gelation of Ehrlich tumour cell extracts is
reversibly inhibited by low concentrations of Ca2+. Nature 272:273-276.
82

Miyake T, Hattori T, Fukuda M, Kitamura T, Fujita S (1988) Quantitative studies on
proliferative changes of reactive astrocytes in mouse cerebral cortex. Brain Res
451:133-138.
Moxon-Emre I, Schlichter LC (2010) Evolution of inflammation and white matter injury in a
model of transient focal ischemia. J Neuropathol Exp Neurol 69:1-15.
Mulari MT, Zhao H, Lakkakorpi PT, Vaananen HK (2003) Osteoclast ruffled border has
distinct subdomains for secretion and degraded matrix uptake. Traffic 4:113-125.
Murphy DA, Courtneidge SA (2011) The 'ins' and 'outs' of podosomes and invadopodia:
characteristics, formation and function. Nat Rev Mol Cell Biol 12:413-426.
Nakamura N, Tanaka J, Sobue K (1993) Rous sarcoma virus-transformed cells develop
peculiar adhesive structures along the cell periphery. J Cell Sci 106 ( Pt 4):1057-
1069.
Navascues J, Calvente R, Marin-Teva JL, Cuadros MA (2000) Entry, dispersion and
differentiation of microglia in the developing central nervous system. An Acad Bras
Cienc 72:91-102.
Nethe M, Hordijk PL (2011) A model for phospho-caveolin-1-driven turnover of focal
adhesions. Cell Adh Migr 5:59-64.
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic
surveillants of brain parenchyma in vivo. Science 308:1314-1318.
Nishibe S, Wahl MI, Hernandez-Sotomayor SM, Tonks NK, Rhee SG, Carpenter G (1990)
Increase of the catalytic activity of phospholipase C-gamma 1 by tyrosine
phosphorylation. Science 250:1253-1256.
83

Norenberg W, Cordes A, Blohbaum G, Frohlich R, Illes P (1997) Coexistence of purino- and
pyrimidinoceptors on activated rat microglial cells. Br J Pharmacol 121:1087-1098.
North AJ (2006) Seeing is believing? A beginners' guide to practical pitfalls in image
acquisition. J Cell Biol 172:9-18.
Ohana L, Newell EW, Stanley EF, Schlichter LC (2009) The Ca2+ release-activated Ca2+
current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels
(Austin) 3:129-139.
Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S (2007) Involvement of
P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia 55:604-616.
Oikawa T, Itoh T, Takenawa T (2008) Sequential signals toward podosome formation in
NIH-src cells. J Cell Biol 182:157-169.
Osiak AE, Zenner G, Linder S (2005) Subconfluent endothelial cells form podosomes
downstream of cytokine and RhoGTPase signaling. Exp Cell Res 307:342-353.
Parat MO, Fox PL (2004) Oxidative stress, caveolae and caveolin-1. Subcell Biochem
37:425-441.
Parekh AB (2006) On the activation mechanism of store-operated calcium channels. Pflugers
Arch 453:303-311.
Parekh AB, Penner R (1995) Depletion-activated calcium current is inhibited by protein
kinase in RBL-2H3 cells. Proc Natl Acad Sci U S A 92:7907-7911.
Parekh AB, Putney JW, Jr. (2005) Store-operated calcium channels. Physiol Rev 85:757-810.
Pfaff M, Jurdic P (2001) Podosomes in osteoclast-like cells: structural analysis and
cooperative roles of paxillin, proline-rich tyrosine kinase 2 (Pyk2) and integrin
alphaVbeta3. J Cell Sci 114:2775-2786.
84

Poincloux R, Lizarraga F, Chavrier P (2009) Matrix invasion by tumour cells: a focus on
MT1-MMP trafficking to invadopodia. J Cell Sci 122:3015-3024.
Qian D, Lev S, van Oers NS, Dikic I, Schlessinger J, Weiss A (1997) Tyrosine
phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J Exp
Med 185:1253-1259.
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system
parenchyma. Nature 468:253-262.
Rezaie P, Male D (1999) Colonisation of the developing human brain and spinal cord by
microglia: a review. Microsc Res Tech 45:359-382.
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT,
Horwitz AR (2003) Cell migration: integrating signals from front to back. Science
302:1704-1709.
Rottiers P, Saltel F, Daubon T, Chaigne-Delalande B, Tridon V, Billottet C, Reuzeau E,
Genot E (2009) TGFbeta-induced endothelial podosomes mediate basement
membrane collagen degradation in arterial vessels. J Cell Sci 122:4311-4318.
Rychkov G, Brereton HM, Harland ML, Barritt GJ (2001) Plasma membrane Ca2+ release-
activated Ca2+ channels with a high selectivity for Ca2+ identified by patch-clamp
recording in rat liver cells. Hepatology 33:938-947.
Sanchez-Lopez A, Cuadros MA, Calvente R, Tassi M, Marin-Teva JL, Navascues J (2004)
Radial migration of developing microglial cells in quail retina: a confocal microscopy
study. Glia 46:261-273.
Sasaki T, Hong MH (1993) Localization of endothelin-1 in the osteoclast. J Electron Microsc
(Tokyo) 42:193-196.
85

Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S, Imai Y (2001) Iba1 is an actin-cross-linking
protein in macrophages/microglia. Biochem Biophys Res Commun 286:292-297.
Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT (1994)
Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-
dependent binding of pp60src. Mol Cell Biol 14:1680-1688.
Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R (2003)
Microglial activation precedes and predominates over macrophage infiltration in
transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone
marrow chimeric mice. Exp Neurol 183:25-33.
Schlichter LC, Kaushal V, Moxon-Emre I, Sivagnanam V, Vincent C (2010) The Ca2+
activated SK3 channel is expressed in microglia in the rat striatum and contributes to
microglia-mediated neurotoxicity in vitro. J Neuroinflammation 7:4.
Schwartz M, Butovsky O, Bruck W, Hanisch UK (2006) Microglial phenotype: is the
commitment reversible? Trends Neurosci 29:68-74.
Seals DF, Azucena EF, Jr., Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge
SA (2005) The adaptor protein Tks5/Fish is required for podosome formation and
function, and for the protease-driven invasion of cancer cells. Cancer Cell 7:155-165.
Sechi AS, Wehland J (2000) The actin cytoskeleton and plasma membrane connection:
PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J
Cell Sci 113 Pt 21:3685-3695.
Sivagnanam V, Zhu X, Schlichter LC (2010) Dominance of E. coli phagocytosis over LPS in
the inflammatory response of microglia. J Neuroimmunol 227:111-119.
86

Smith SJ, Buchanan J, Osses LR, Charlton MP, Augustine GJ (1993) The spatial distribution
of calcium signals in squid presynaptic terminals. J Physiol 472:573-593.
Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW (2010) Activation and
regulation of store-operated calcium entry. J Cell Mol Med 14:2337-2349.
Somasundaram B, Norman JC, Mahaut-Smith MP (1995) Primaquine, an inhibitor of
vesicular transport, blocks the calcium-release-activated current in rat
megakaryocytes. Biochem J 309 ( Pt 3):725-729.
Stickel SK, Wang YL (1987) Alpha-actinin-containing aggregates in transformed cells are
highly dynamic structures. J Cell Biol 104:1521-1526.
Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL (2005) Role of microglia in the
central nervous system's immune response. Neurol Res 27:685-691.
Stuart LM, Bell SA, Stewart CR, Silver JM, Richard J, Goss JL, Tseng AA, Zhang A, El
Khoury JB, Moore KJ (2007) CD36 signals to the actin cytoskeleton and regulates
microglial migration via a p130Cas complex. J Biol Chem 282:27392-27401.
Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P (2009) Nck
adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J
Cell Sci 122:2727-2740.
Tambuyzer BR, Ponsaerts P, Nouwen EJ (2009) Microglia: gatekeepers of central nervous
system immunology. J Leukoc Biol 85:352-370.
Tanaka S, Takahashi N, Udagawa N, Murakami H, Nakamura I, Kurokawa T, Suda T (1995)
Possible involvement of focal adhesion kinase, p125FAK, in osteoclastic bone
resorption. J Cell Biochem 58:424-435.
87

Tarone G, Cirillo D, Giancotti FG, Comoglio PM, Marchisio PC (1985) Rous sarcoma virus-
transformed fibroblasts adhere primarily at discrete protrusions of the ventral
membrane called podosomes. Exp Cell Res 159:141-157.
Thompson O, Kleino I, Crimaldi L, Gimona M, Saksela K, Winder SJ (2008) Dystroglycan,
Tks5 and Src mediated assembly of podosomes in myoblasts. PLoS One 3:e3638.
Torres M, Forman HJ (2003) Redox signaling and the MAP kinase pathways. Biofactors
17:287-296.
Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL (2004) Angiotensin II and endothelin-1
regulate MAP kinases through different redox-dependent mechanisms in human
vascular smooth muscle cells. J Hypertens 22:1141-1149.
Tvorogov D, Wang XJ, Zent R, Carpenter G (2005) Integrin-dependent PLC-gamma1
phosphorylation mediates fibronectin-dependent adhesion. J Cell Sci 118:601-610.
van der Flier A, Sonnenberg A (2001) Function and interactions of integrins. Cell Tissue Res
305:285-298.
Van Goethem E, Guiet R, Balor S, Charriere GM, Poincloux R, Labrousse A, Maridonneau-
Parini I, Le Cabec V (2010) Macrophage podosomes go 3D. Eur J Cell Biol 90:224-
236.
Vicente-Manzanares M, Choi CK, Horwitz AR (2009) Integrins in cell migration--the actin
connection. J Cell Sci 122:199-206.
Vincent C, Schlichter LC (2010) Expression of podosomes and small-conductance Ca2+-
activated K+ channels in cultures microglia. In: Department of Physiology, p 122.
Toronto, Canada: University of Toronto.
88

von Willebrand M, Williams S, Saxena M, Gilman J, Tailor P, Jascur T, Amarante-Mendes
GP, Green DR, Mustelin T (1998) Modification of phosphatidylinositol 3-kinase SH2
domain binding properties by Abl- or Lck-mediated tyrosine phosphorylation at Tyr-
688. J Biol Chem 273:3994-4000.
Wasserman JK, Yang H, Schlichter LC (2008) Glial responses, neuron death and lesion
resolution after intracerebral hemorrhage in young vs. aged rats. Eur J Neurosci
28:1316-1328.
Weber AM, Wong FK, Tufford AR, Schlichter LC, Matveev V, Stanley EF (2010) N-type
Ca2+ channels carry the largest current: implications for nanodomains and transmitter
release. Nat Neurosci 13:1348-1350.
Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H (2009) Calcium flickers steer cell
migration. Nature 457:901-905.
Weiner OD (2002) Regulation of cell polarity during eukaryotic chemotaxis: the chemotactic
compass. Curr Opin Cell Biol 14:196-202.
Wiesner C, Faix J, Himmel M, Bentzien F, Linder S (2010) KIF5B and KIF3A/KIF3B
kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix
degradation in primary macrophages. Blood 116:1559-1569.
Wilkinson PC (1975) Leucocyte locomotion and chemotaxis. The influence of divalent
cations and cation ionophores. Exp Cell Res 93:420-426.
Wingler K, Hermans JJ, Schiffers P, Moens A, Paul M, Schmidt HH (2011) NOX1, 2, 4, 5:
counting out oxidative stress. Br J Pharmacol 164:866-883.
Wolosewick JJ (1984) Distribution of actin in migrating leukocytes in vivo. Cell Tissue Res
236:517-525.
89

Wu X, Hardy VE, Joseph JI, Jabbour S, Mahadev K, Zhu L, Goldstein BJ (2003) Protein-
tyrosine phosphatase activity in human adipocytes is strongly correlated with insulin-
stimulated glucose uptake and is a target of insulin-induced oxidative inhibition.
Metabolism 52:705-712.
Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B,
Bond CT, Lutsenko S, Maylie J, Adelman JP (1998) Mechanism of calcium gating in
small-conductance calcium-activated potassium channels. Nature 395:503-507.
Xu HT, Yuan XB, Guan CB, Duan S, Wu CP, Feng L (2004) Calcium signaling in
chemorepellant Slit2-dependent regulation of neuronal migration. Proc Natl Acad Sci
U S A 101:4296-4301.
Yamaguchi H, Oikawa T (2010) Membrane lipids in invadopodia and podosomes: key
structures for cancer invasion and metastasis. Oncotarget 1:320-328.
Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K (2009) Lipid rafts
and caveolin-1 are required for invadopodia formation and extracellular matrix
degradation by human breast cancer cells. Cancer Res 69:8594-8602.
Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ,
Graves LM, Earp HS (1996) Activation of a novel calcium-dependent protein-
tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated
protein kinase activation. J Biol Chem 271:29993-29998.
Zambonin-Zallone A, Teti A, Carano A, Marchisio PC (1988) The distribution of podosomes
in osteoclasts cultured on bone laminae: effect of retinol. J Bone Miner Res 3:517-
523.
90

Zhang Z, Chopp M, Powers C (1997) Temporal profile of microglial response following
transient (2 h) middle cerebral artery occlusion. Brain Res 744:189-198.
Zimmerman MC, Sharma RV, Davisson RL (2005) Superoxide mediates angiotensin II-
induced influx of extracellular calcium in neural cells. Hypertension 45:717-723.
Zweifach A, Lewis RS (1993) Mitogen-regulated Ca2+ current of T lymphocytes is activated
by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A 90:6295-6299.
91
Related Documents











