Mice Expressing Only the Mutant APOE3Leiden Gene Show Impaired VLDL Secretion Arjen R. Mensenkamp, Bas Teusink, Julius F.W. Baller, Henk Wolters, Rick Havinga, Ko Willems van Dijk, Louis M. Havekes, Folkert Kuipers Abstract—Apolipoprotein E (apoE)-deficient mice develop hepatic steatosis and show impaired very low density lipoprotein (VLDL)-triglyceride (TG) secretion. These effects are normalized on the introduction of the human APOE3 gene. To assess whether this apoE effect is isoform specific, we studied hepatic lipid metabolism in mice expressing either APOE3 or the mutant APOE3Leiden on apoe2/2 or apoe1/2 backgrounds. The transgenes were expressed mainly in periportal hepatocytes, as revealed by in situ hybridization. Mice expressing APOE3Leiden, on the apoe2/2 and apoe1/2 backgrounds, had fatty livers, which were absent in APOE3/apoe2/2 mice. APOE3Leiden/apoe2/2 mice showed a strongly reduced VLDL-TG secretion compared with APOE3/apoe2/2 mice (48614 versus 82610 mmol/kg per hour, respectively). The presence of a single mouse apoe allele increased VLDL-TG secretion in APOE3Leiden/apoe1/2 mice (121643 mmol/kg per hour) compared with APOE3Leiden/apoe2/2 mice. These results show that APOE3Leiden does not prevent development of a fatty liver and does not normalize VLDL-TG secretion in mice with an apoE-deficient background. The presence of a single mouse apoe allele is sufficient to normalize the APOE3Leiden-associated reduction of VLDL-TG secretion but does not prevent steatosis. We conclude that apoE-mediated stimulation of VLDL secretion is isoform specific. (Arterioscler Thromb Vasc Biol. 2001;21:1366- 1372.) Key Words: very low density lipoproteins n apolipoprotein E n lipoprotein assembly n lipoprotein secretion n steatosis A polipoprotein E is a constituent of VLDLs, chylomi- crons, and HDLs and is essential for receptor-mediated uptake of remnant lipoproteins. 1 ApoE deficiency in mice leads to elevated plasma cholesterol levels that are due to the accumulation of remnant lipoproteins, 2–4 and apoE deficiency is associated with the development of atherosclerosis. 3 In addition, these mice develop a fatty liver when fed normal chow 5 and show a decreased VLDL-triglyceride (TG) secre- tion. 4 Recently, we demonstrated that the introduction of human APOE3 in apoE-deficient mice increases VLDL-TG secretion. 6 A role of apoE in control of VLDL-TG secretion in animals and cultured cells has also recently been confirmed by other groups. 7–9 It has recently been shown that humans with the APOE2/E2 genotype have a decreased VLDL secretion compared with secretion in subjects with the APOE3/E3 genotype. 10 In addition, it has been reported that VLDL-apoB secretion is decreased by 30% in humans with the APOE3/E4 genotype compared with those with the APOE3/E3 genotype. 11 In humans, the mutant APOE3Leiden isoform is associated with a dominantly inherited form of familial dysbetalipopro- teinemia. 12–14 The APOE3Leiden gene contains a tandem repeat of codons 120 to 126, yielding a protein of 306 amino acids. 12,13 Transgenic mice expressing APOE3Leiden de- velop hyperlipidemia because of defective binding of E3Leiden-containing remnant lipoproteins to the LDL recep- tor and to the LDL receptor–related protein and are suscep- tible to diet-induced atherosclerosis. 15 We have shown re- cently that mice overexpressing APOE3Leiden in the presence of human APOC-I and the endogenous mouse apoe gene display hepatic lipid accumulation and a mildly de- creased VLDL-TG secretion. 16 Interpretation of these results is hampered by the presence of the APOC-I and apoe genes. To directly establish the effects of APOE3Leiden and the possible compensatory effects of mouse apoe on VLDL-TG secretion and the development of hepatic steatosis, we used mice expressing APOE3Leiden on an apoE-deficient (apoe2/2) or on an apoE-heterozygous (apoe1/2) back- ground. Our data show that expression of only the APOE3Leiden gene is associated with a reduced VLDL-TG secretion and increased hepatic fat content compared with expression of only the human APOE3 gene. In the presence Received February 14, 2001; revision accepted May 23, 2001. From the Groningen University Institute for Drug Exploration (A.R.M., J.F.W.B., H.W., R.H., F.K.), Center for Liver, Digestive, and Metabolic Diseases, Faculty of Medical Sciences and University Hospital Groningen, Groningen, the Netherlands; the Gaubius Laboratory TNO-PG (B.T., L.M.H.), Leiden, the Netherlands; and the Departments of Human Genetics (K.W.v.D.) and General Internal Medicine and Cardiology (L.M.H.), Leiden University, Leiden, the Netherlands. Correspondence to Folkert Kuipers, PhD, Groningen University Institute for Drug Exploration, Center for Liver, Digestive, and Metabolic Diseases, University Hospital Groningen, CMC IV, Room Y2115, Hanzeplein 1, 9713 GZ Groningen, Netherlands. E-mail [email protected] © 2001 American Heart Association, Inc. Arterioscler Thromb Vasc Biol. is available at http://www.atvbaha.org 1366 by guest on November 9, 2015 http://atvb.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Mice Expressing Only the Mutant APOE3Leiden GeneShow Impaired VLDL Secretion
Arjen R. Mensenkamp, Bas Teusink, Julius F.W. Baller, Henk Wolters, Rick Havinga,Ko Willems van Dijk, Louis M. Havekes, Folkert Kuipers
Abstract—Apolipoprotein E (apoE)-deficient mice develop hepatic steatosis and show impaired very low densitylipoprotein (VLDL)-triglyceride (TG) secretion. These effects are normalized on the introduction of the human APOE3gene. To assess whether this apoE effect is isoform specific, we studied hepatic lipid metabolism in mice expressingeither APOE3 or the mutant APOE3Leiden onapoe2/2or apoe1/2backgrounds. The transgenes were expressedmainly in periportal hepatocytes, as revealed by in situ hybridization. Mice expressing APOE3Leiden, on theapoe2/2andapoe1/2backgrounds, had fatty livers, which were absent in APOE3/apoe2/2mice. APOE3Leiden/apoe2/2mice showed a strongly reduced VLDL-TG secretion compared with APOE3/apoe2/2mice (48614 versus 82610mmol/kg per hour, respectively). The presence of a single mouseapoe allele increased VLDL-TG secretion inAPOE3Leiden/apoe1/2mice (121643mmol/kg per hour) compared with APOE3Leiden/apoe2/2mice. These resultsshow that APOE3Leiden does not prevent development of a fatty liver and does not normalize VLDL-TG secretion inmice with an apoE-deficient background. The presence of a single mouseapoeallele is sufficient to normalize theAPOE3Leiden-associated reduction of VLDL-TG secretion but does not prevent steatosis. We conclude thatapoE-mediated stimulation of VLDL secretion is isoform specific.(Arterioscler Thromb Vasc Biol. 2001;21:1366-1372.)
Key Words: very low density lipoproteinsn apolipoprotein En lipoprotein assemblyn lipoprotein secretionn steatosis
A polipoprotein E is a constituent of VLDLs, chylomi-crons, and HDLs and is essential for receptor-mediated
uptake of remnant lipoproteins.1 ApoE deficiency in miceleads to elevated plasma cholesterol levels that are due to theaccumulation of remnant lipoproteins,2–4and apoE deficiencyis associated with the development of atherosclerosis.3 Inaddition, these mice develop a fatty liver when fed normalchow5 and show a decreased VLDL-triglyceride (TG) secre-tion.4 Recently, we demonstrated that the introduction ofhuman APOE3 in apoE-deficient mice increases VLDL-TGsecretion.6 A role of apoE in control of VLDL-TG secretionin animals and cultured cells has also recently been confirmedby other groups.7–9 It has recently been shown that humanswith the APOE2/E2 genotype have a decreased VLDLsecretion compared with secretion in subjects with theAPOE3/E3 genotype.10 In addition, it has been reported thatVLDL-apoB secretion is decreased by 30% in humans withthe APOE3/E4 genotype compared with those with theAPOE3/E3 genotype.11
In humans, the mutant APOE3Leiden isoform is associatedwith a dominantly inherited form of familial dysbetalipopro-
teinemia.12–14 The APOE3Leiden gene contains a tandemrepeat of codons 120 to 126, yielding a protein of 306 aminoacids.12,13 Transgenic mice expressing APOE3Leiden de-velop hyperlipidemia because of defective binding ofE3Leiden-containing remnant lipoproteins to the LDL recep-tor and to the LDL receptor–related protein and are suscep-tible to diet-induced atherosclerosis.15 We have shown re-cently that mice overexpressing APOE3Leiden in thepresence of human APOC-I and the endogenous mouseapoegene display hepatic lipid accumulation and a mildly de-creased VLDL-TG secretion.16 Interpretation of these resultsis hampered by the presence of the APOC-I andapoegenes.To directly establish the effects of APOE3Leiden and thepossible compensatory effects of mouseapoeon VLDL-TGsecretion and the development of hepatic steatosis, we usedmice expressing APOE3Leiden on an apoE-deficient(apoe2/2) or on an apoE-heterozygous (apoe1/2) back-ground. Our data show that expression of only theAPOE3Leiden gene is associated with a reduced VLDL-TGsecretion and increased hepatic fat content compared withexpression of only the human APOE3 gene. In the presence
Received February 14, 2001; revision accepted May 23, 2001.From the Groningen University Institute for Drug Exploration (A.R.M., J.F.W.B., H.W., R.H., F.K.), Center for Liver, Digestive, and Metabolic
Diseases, Faculty of Medical Sciences and University Hospital Groningen, Groningen, the Netherlands; the Gaubius Laboratory TNO-PG (B.T., L.M.H.),Leiden, the Netherlands; and the Departments of Human Genetics (K.W.v.D.) and General Internal Medicine and Cardiology (L.M.H.), Leiden University,Leiden, the Netherlands.
Correspondence to Folkert Kuipers, PhD, Groningen University Institute for Drug Exploration, Center for Liver, Digestive, and Metabolic Diseases,University Hospital Groningen, CMC IV, Room Y2115, Hanzeplein 1, 9713 GZ Groningen, Netherlands. E-mail [email protected]
© 2001 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol.is available at http://www.atvbaha.org
1366 by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

of a single endogenousapoeallele, VLDL-TG secretion inAPOE3Leiden-expressing mice is normalized, whereas thedominant effect of APOE3Leiden with respect to the devel-opment of steatosis is not overruled.
MethodsAnimalsTransgenic mice expressing human APOE3 were generated accord-ing to Hogan et al.17 Transgenic offspring were identified bypolymerase chain reaction (PCR) analysis and Southern blot analysison genomic tail–derived DNA. One strain, expressing high levels ofhuman APOE3 in the liver, was subsequently bred with C57BL/6Jmice. APOE3Leiden transgenic mice were created as describedpreviously.18,19 The construct used contained the APOE3Leidengene starting from position2650 before exon 1 to 1.4 kb after thepolyA site18 and the so-called hepatic control region.20 The humanAPOE3 or APOE3Leiden gene was introduced into apoE-deficientmice by crossbreeding the respective APOE transgenic mice withapoe2/2mice as described previously.6,19To obtain APOE3Leiden/apoe1/2mice, APOE3Leiden/apoe2/2mice were crossbred withC57BL/6J mice. The resulting offspring were analyzed for thepresence of human APOE by ELISA and the mouseapoegenotypethrough tail-tip DNA analysis, as described earlier.21 APOE expres-sion in the liver was measured by Northern blot as describedpreviously.6 Bands were semiquantified by using the computerprogram Image Master 1D Elite 3.0.
Mice were housed in a light- and temperature-controlled environ-ment. Food and tap water were available ad libitum, and mice werefed a commercial laboratory chow (RMH-B, Hope Farms BV). Theanimals received humane care, and experimental protocols compliedwith local guidelines for the use of experimental animals.
Plasma and Liver Tissue SamplingGroups of mice (n55) were fasted overnight and anesthetized withhalothane. A large blood sample for determination of plasma lipidsand VLDL isolation was collected by cardiac puncture. Subse-quently, the liver was quickly removed, weighed, and frozen inseparate portions for RNA isolation and lipid analysis, respectively.Parts of the liver were stored in paraformaldehyde or slowly frozenin isopentane and used for microscopic examination and in situhybridization.
In Situ HybridizationFrozen liver sections (6mm) were fixed with 8% buffered formalinfor 24 hours at 37°C, followed by washing with PBS treated withdiethyl pyrocarbonate (DEPC)/PBS twice, 2 times with DEPC/H2O,and 1 time with ethanol, and were subsequently dried by air. Sectionswere stored at220°C. Fragments of APOE cDNA were obtainedfrom a pUC-18 vector. The fragments were excised and placed intoa pGEM-3zf(1) vector containing a T7 and Sp6 promoter region. Toproduce digoxigenin-labeled RNA, sense and antisense APOEcDNA was transcribed in vitro with T7 or Sp6 RNA polymerase,respectively, in the presence of digoxigenin-UTP according to themanufacturer’s instructions (Roche Diagnostics). Before hybridiza-tion, sections were treated with 0.25mg/mL proteinase K for 30minutes at 37°C. After overnight hybridization (1 ng probe permicroliter) at 55°C, the sections were treated with RNase (SigmaChemical Co). The hybridized digoxigenin-labeled probes wereimmunodetected with anti-digoxigenin Fab fragments conjugated toalkaline phosphatase, and the bound conjugate was visualized withnitro blue tetrazolium/bromo-4-chloro-3-indolyl phosphate (RocheDiagnostics).
Hepatic Lipid AnalysisLivers were homogenized as described previously.6 Hepatic concen-trations of triglycerides and free and total cholesterol were measuredby using commercial kits (Roche Diagnostics) after lipid extractionaccording to Bligh and Dyer22 and by resolving the lipid in 2%Triton X-100 in water. Phospholipid content of the liver tissue wasdetermined according to Böttcher et al23 after lipid extraction.
Ketone BodiesFive mice per group were fasted for 24 hours. Blood was taken bycardiac puncture, and 250mL of 18% perchloric acid was added to500mL whole blood, mixed thoroughly, and kept on ice for at least10 minutes. The assay was performed as described previously.24
In Vivo VLDL-TG Production RateThe hepatic VLDL-TG production rate in APOE3Leiden/apoe2/2,APOE3Leiden/apoe1/2, and APOE3/apoe2/2mice (n55) wasdetermined as reported previously.4,6 In short, mice were fasted 9hours before the experiment, and 12.5 mg Triton WR-1339 in 100mL PBS was injected via the penile vein. Tail blood samples weretaken under light halothane anesthesia before and at 1, 2, and 3 hoursafter injection of Triton WR-1339 for TG measurements.
VLDL Isolation, Size Determination, and ApoBProduction MeasurementsVLDL/IDL was isolated as previously reported.6,25 ApoB concen-tration was determined by applying VLDL samples to SDS-PAGE inparallel with 4 human LDL-apoB standards.25 Band intensities weredetermined after silver staining by using an image densitometer(Imagemaster, Amersham Pharmacia Biotech) and comparison withstandards, as described previously.6,25 TG and cholesterol contentswere determined as in plasma. Phospholipids were determined byusing a commercial available kit (WAKO Chemicals).
Plasma Lipids and ApoEPlasma TG, free fatty acid, free cholesterol, and total cholesterollevels were determined by using commercially available kits (RocheDiagnostics). ApoE concentrations were determined by ELISA asdescribed previously.21 Lipoproteins were separated by using fastprotein liquid chromatography on a Pharmacia Superose 6B column.
Hepatic RNA IsolationmRNA from '30 mg liver tissue was isolated with the Trizolmethod (GIBCO) followed by the SV Total RNA Isolation System(Promega RNA) to remove potential DNA contamination.
Reverse Transcription–PCRImmediately after RNA isolation, 4.5mg of RNA was used forcDNA synthesis according to manufacturer’s instructions (RocheDiagnostics). PCR results were normalized to 18S RNA levelsdetermined by competitive PCR. PCR was performed in 50mLcDNA preparations by using primers for the genes for ribosomal 18SRNA,26 diacylglycerol acyltransferase (dgat, GenBank accessionNo. AF078752), microsomal triglyceride transfer protein (mttp,GenBank accession No. L47970), and fatty acid synthase (fas,GenBank accession No. X13135).
MiscellaneousProtein concentrations were determined according to the method ofLowry et al27 with BSA used as a standard.
Statistical AnalysisAnalysis of data from the 3 groups was performed by ANOVA,followed by Student-Newman-Keulst test post hoc analysis.
ResultsExpression Patterns of APOE3 andAPOE3Leiden TransgenesSteady-state mRNA levels of APOE3Leiden were compara-ble in APOE3Leiden/apoe2/2mice and APOE3Leiden/apoe1/2mice, as shown by Northern blot analysis (Figure1). Expression of APOE3 in APOE3/apoe2/2mice appearedto be somewhat lower than APOE3Leiden expression inAPOE3Leiden mice. In situ hybridization(Figure 2) revealedthat the transgenes were expressed mainly in periportal (zone1) hepatocytes in APOE3Leiden/apoe2/2 (Figure 2A),
Mensenkamp et al Impaired VLDL Secretion in APOE3Leiden Mice 1367
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from
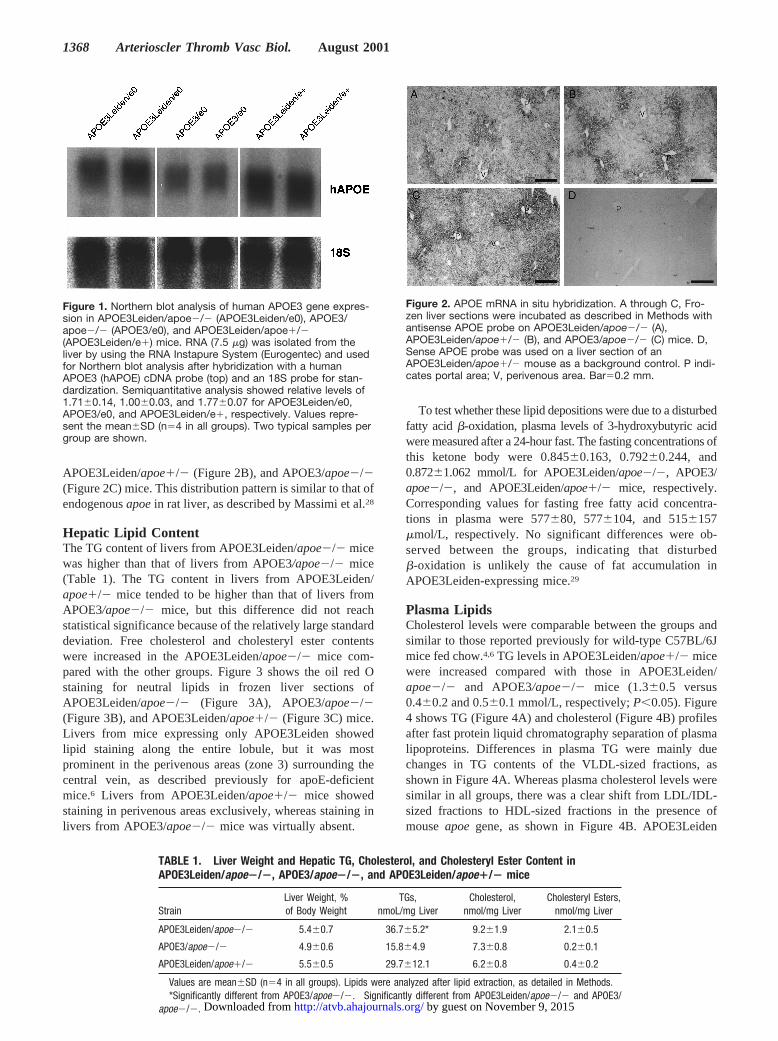
APOE3Leiden/apoe1/2(Figure 2B), and APOE3/apoe2/2(Figure 2C) mice. This distribution pattern is similar to that ofendogenousapoein rat liver, as described by Massimi et al.28
Hepatic Lipid ContentThe TG content of livers from APOE3Leiden/apoe2/2micewas higher than that of livers from APOE3/apoe2/2mice(Table 1). The TG content in livers from APOE3Leiden/apoe1/2mice tended to be higher than that of livers fromAPOE3/apoe2/2mice, but this difference did not reachstatistical significance because of the relatively large standarddeviation. Free cholesterol and cholesteryl ester contentswere increased in the APOE3Leiden/apoe2/2 mice com-pared with the other groups. Figure 3 shows the oil red Ostaining for neutral lipids in frozen liver sections ofAPOE3Leiden/apoe2/2 (Figure 3A), APOE3/apoe2/2(Figure 3B), and APOE3Leiden/apoe1/2(Figure 3C) mice.Livers from mice expressing only APOE3Leiden showedlipid staining along the entire lobule, but it was mostprominent in the perivenous areas (zone 3) surrounding thecentral vein, as described previously for apoE-deficientmice.6 Livers from APOE3Leiden/apoe1/2mice showedstaining in perivenous areas exclusively, whereas staining inlivers from APOE3/apoe2/2mice was virtually absent.
To test whether these lipid depositions were due to a disturbedfatty acidb-oxidation, plasma levels of 3-hydroxybutyric acidwere measured after a 24-hour fast. The fasting concentrations ofthis ketone body were 0.84560.163, 0.79260.244, and0.87261.062 mmol/L for APOE3Leiden/apoe2/2, APOE3/apoe2/2, and APOE3Leiden/apoe1/2 mice, respectively.Corresponding values for fasting free fatty acid concentra-tions in plasma were 577680, 5776104, and 5156157mmol/L, respectively. No significant differences were ob-served between the groups, indicating that disturbedb-oxidation is unlikely the cause of fat accumulation inAPOE3Leiden-expressing mice.29
Plasma LipidsCholesterol levels were comparable between the groups andsimilar to those reported previously for wild-type C57BL/6Jmice fed chow.4,6 TG levels in APOE3Leiden/apoe1/2micewere increased compared with those in APOE3Leiden/apoe2/2 and APOE3/apoe2/2 mice (1.360.5 versus0.460.2 and 0.560.1 mmol/L, respectively;P,0.05). Figure4 shows TG (Figure 4A) and cholesterol (Figure 4B) profilesafter fast protein liquid chromatography separation of plasmalipoproteins. Differences in plasma TG were mainly duechanges in TG contents of the VLDL-sized fractions, asshown in Figure 4A. Whereas plasma cholesterol levels weresimilar in all groups, there was a clear shift from LDL/IDL-sized fractions to HDL-sized fractions in the presence ofmouseapoe gene, as shown in Figure 4B. APOE3Leiden
Figure 2. APOE mRNA in situ hybridization. A through C, Fro-zen liver sections were incubated as described in Methods withantisense APOE probe on APOE3Leiden/apoe2/2 (A),APOE3Leiden/apoe1/2 (B), and APOE3/apoe2/2 (C) mice. D,Sense APOE probe was used on a liver section of anAPOE3Leiden/apoe1/2 mouse as a background control. P indi-cates portal area; V, perivenous area. Bar50.2 mm.
Figure 1. Northern blot analysis of human APOE3 gene expres-sion in APOE3Leiden/apoe2/2 (APOE3Leiden/e0), APOE3/apoe2/2 (APOE3/e0), and APOE3Leiden/apoe1/2(APOE3Leiden/e1) mice. RNA (7.5 mg) was isolated from theliver by using the RNA Instapure System (Eurogentec) and usedfor Northern blot analysis after hybridization with a humanAPOE3 (hAPOE) cDNA probe (top) and an 18S probe for stan-dardization. Semiquantitative analysis showed relative levels of1.7160.14, 1.0060.03, and 1.7760.07 for APOE3Leiden/e0,APOE3/e0, and APOE3Leiden/e1, respectively. Values repre-sent the mean6SD (n54 in all groups). Two typical samples pergroup are shown.
TABLE 1. Liver Weight and Hepatic TG, Cholesterol, and Cholesteryl Ester Content inAPOE3Leiden/apoe2/2, APOE3/apoe2/2, and APOE3Leiden/apoe1/2 mice
StrainLiver Weight, %of Body Weight
TGs,nmoL/mg Liver
Cholesterol,nmol/mg Liver
Cholesteryl Esters,nmol/mg Liver
APOE3Leiden/apoe2/2 5.460.7 36.765.2* 9.261.9† 2.160.5†
APOE3/apoe2/2 4.960.6 15.864.9 7.360.8 0.260.1
APOE3Leiden/apoe1/2 5.560.5 29.7612.1 6.260.8 0.460.2
Values are mean6SD (n54 in all groups). Lipids were analyzed after lipid extraction, as detailed in Methods.*Significantly different from APOE3/apoe2/2. †Significantly different from APOE3Leiden/apoe2/2 and APOE3/
apoe2/2.
1368 Arterioscler Thromb Vasc Biol. August 2001
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

concentration in plasma of the APOE3Leiden-expressingmice was higher than that of APOE3 in the APOE3/apoe2/2mice (0.3460.05 versus 0.02060.003 mg/dL, respectively;P,0.05). The presence of a singleapoeallele resulted in a10-fold increase of plasma APOE3Leiden concentration(3.6460.49 mg/dL).
VLDL Production RateThe VLDL-TG production rate in APOE3Leiden/apoe2/2mice, as measured after injection of Triton WR-1339, wasstrongly reduced compared with that in APOE3/apoe2/2mice. The presence of a single mouseapoe allele clearlyincreased the VLDL-TG production in APOE3Leiden-expressing mice compared with that in APOE3-expressingmice (Table 2) and in wild-type (C57BL/6J) mice.6 TheapoB-100 production rate was decreased in APOE3Leiden/
apoe2/2 mice compared with APOE3/apoe2/2 mice,whereas apoB-48 production was not affected. ApoB-100 andapoB-48 production rates in APOE3Leiden/apoe1/2 micewere similar to the rates in APOE3/apoe2/2mice. The poolsize of apoB-100 in APOE3Leiden/apoe1/2 mice wasstrongly elevated compared with both other strains, in accor-dance with elevated TG content of VLDL-sized fractions.
VLDL CompositionVLDL particle composition was determined by measuringlipid composition of VLDL before and 4 hours after TritonWR-1339 injection. Subtraction of these values provided anestimate of the nascent VLDL composition. VLDL compo-sition was similar in all strains (see online Figure I, which canbe accessed at http://www.atvb.ahajournals.org). Therefore,differences in secretion of VLDL-TG are mainly due tochanges in the number of apoB-100–containing lipoproteins.VLDL composition in plasma before Triton WR-1339 injec-tion reflects the average composition of nascent and partiallyhydrolyzed particles. In these particles, the TG content ofmice expressing APOE3Leiden/apoe1/2was 6663% com-pared with 3266% and 3163% for mice expressingAPOE3Leiden/apoe2/2 and APOE3/apoe2/2, respec-tively. This increased TG content is probably due to adecreased lipolysis rate.
mRNA levelsSteady-state mRNA levels of selected enzymes involved inlipogenesis, TG formation, and VLDL assembly in liver wereestimated after an overnight fast by semiquantitative reversetranscription–PCR, including fatty acid synthase (fas), diac-ylglycerol acyltransferase (dgat), and microsomal triglyceride
Figure 3. Oil red O staining for neutral lipids on frozen liver sec-tions from APOE3Leiden/apoe2/2 (A), APOE3/apoe2/2, andAPOE3Leiden/apoe1/2 (C) mice. Bar50.2 mm.
Figure 4. TG (A) and cholesterol (B) profiles after fast proteinliquid chromatography separation of plasma lipoproteins on aSuperose 6B column. Plasma of at least 3 mice per group waspooled, and 0.2 mL was applied to the column and eluted withPBS at a flow rate of 0.5 mL/min. Lipids were measured enzy-matically by using commercially available kits (Roche).
Mensenkamp et al Impaired VLDL Secretion in APOE3Leiden Mice 1369
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

transfer protein (mttp). Expression of all genes was similarbetween the groups when corrected for ribosomal 18S RNA,indicating that differences in VLDL-TG production betweenthe strains are not due to altered expression of these genes(data not shown).
DiscussionRecent studies by us4–6 and by others7–9 have demonstratedthat apoE is involved in the regulation of hepatic VLDL-TGsecretion. The present study shows that the mutantAPOE3Leiden isoform, compared with APOE3, does notstimulate VLDL-TG secretion by the apoE-deficient mouseliver. To evaluate the effects of APOE3Leiden on VLDLproduction, we made use of transgenic APOE3Leiden-expressing mice, either without endogenous mouseapoe19
(APOE3Leiden/apoe2/2) or with a single allele of themouseapoe gene (APOE3Leiden/apoe1/2). As a control,we usedapoe2/2mice expressing the human APOE3 gene.We have previously shown that these mice display a ofVLDL-TG secretion rate similar to that observed in wild-typemice.6
In situ hybridization showed that transgene expression inAPOE3Leiden- and APOE3-expressing mice is mainly inperiportal hepatocytes (zone 1). This distribution pattern is inaccordance with a recent report by Massimi et al28 showingthat in rat liver, expression of endogenousapoe is alsoconfined to zone 1. The construct used to create the trans-genic mice contained the hepatic control region18,20 and theupstream region before exon 1, starting at location2650.This region contains a number of potential binding sites fortranscription factors.30–32 It is very likely that a transcriptionfactor(s) regulating hepatic localization of gene expression islocalized in this region. To the best of our knowledge,however, these transcription factors have not yet been iden-tified. In view of the putative role of apoE in VLDL-TGregulation, this observation supports the view that VLDLsecretion is mainly in zone 1,33 and not in zone 3, assuggested by others.34
We have previously shown that apoE-deficient mice4,6 andAPOE3Leiden/APOC-I mice16 develop hepatic steatosis,with fat accumulating most abundantly in perivenously local-ized hepatocytes (zone 3). In the present study, this pattern offat distribution is also observed for mice expressingAPOE3Leiden. This phenomenon is probably partly ex-plained by the impaired VLDL-TG secretion in apoE-deficient and APOE3Leiden-expressing mice, because no
indications for altered hepatic TG synthesis in these micewere found.4 However, mice expressing APOE3Leiden in thepresence ofapoe also develop a fatty liver, whereas theVLDL secretion is similar or even higher than that in miceexpressing APOE3. The observations that synthesis of apoEand probably VLDL secretion are predominantly localized inzone 1 but that TG accumulation is most apparent in zone 3might indicate a dual role for apoE in the development of afatty liver. We speculate that in the presence of functionalapoE, most TGs are removed from the circulation in theperiportal zone of the liver (zone 1), where the LDL receptor–related protein is localized.35 Without functional apoE, uptakein zone 1 is reduced. Consequently, zone 3 hepatocytes facehigh concentrations of lipoproteins and may take them up bymechanisms not yet known. Because VLDL secretion ismainly in zone 1, zone 3 cells might not be able to handlethese excess lipids. In the presence of a single functionalendogenousapoe allele, lipid staining is largely absent inzone 1, probably because VLDL secretion proceeds at anormal rate. Because removal of lipoproteins from the circu-lation is delayed because of the nonfunctional APOE3Leiden(see below), this may lead to accumulation of TGs in zone 3cells.
Fatty liver rapidly develops in situations of defective fattyacid b-oxidation.36 Because apoE localizes to peroxi-somes,6,37 it had to be excluded that the absence of endoge-nous apoE and/or the presence of dysfunctional apoE3Leidenmight result in a decreasedb-oxidation, contributing to thedevelopment of steatosis. All strains showed similarly in-creased concentrations of fatty acids and 3-hydroxybutyricacid after 24 hours of starvation, indicating thatb-oxidation ismost probably not different in the strains used.
In the presence of a single mouseapoeallele, steady-stateplasma concentrations of TGs, APOE, and VLDL-apoB wereincreased in APOE3Leiden transgenic mice compared withAPOE3/apoe2/2mice. This is likely caused by a delayedlipoprotein clearance that is due to the presence ofAPOE3Leiden in high concentrations and a consequentlydecreased hydrolysis of VLDL-TG. Jong et al38 have shownthat high concentrations of apoE inhibit lipolysis, probably bysteric hindrance of lipoprotein binding to heparan sulfateproteoglycans. Reduced lipolysis of TG-rich particles inAPOE3Leiden transgenic mice in the presence of mouseapoe18 is probably due to the lower binding affinity ofAPOE3Leiden to heparan sulfate proteoglycans. In addition,increased levels of APOE3Leiden compared with APOE
TABLE 2. ApoB Pool Sizes and VLDL-ApoB and VLDL-TG PRs
Pool Size, mg/Mouse PR, mg/hVLDL-TG PR,
mmol z kg21 z h21ApoB-100 ApoB-48 ApoB-100 ApoB-48
APOE3Leiden/apoe2/2 561* 2769 1166* 31613 48614*
APOE3/apoe2/2 161 45.1613 3068 4167 81610
APOE3Leiden/apoe1/2 101646† 62616 29617 3368 121642
PR indicates production rate. Values are mean6SD (n54 for each group). VLDL from fasted mice (baseline) andat 4 h after Triton WR-1339 injection was isolated as described in Methods. ApoB concentrations were determinedby applying VLDL samples to SDS-PAGE parallel to 4 concentrations of human LDL-apoB and measuring bandintensities after silver staining. Hepatic VLDL-TG PRs were calculated from the increase in plasma TGs afterintravenous injection of Triton WR-1339.
*Significantly different from APOE3/apoe2/2 and APOE3Leiden/apoe1/2 mice. †Significantly different fromAPOE3/apoe2/2 mice and APOE3Leiden/apoe2/2 mice.
1370 Arterioscler Thromb Vasc Biol. August 2001
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

might also be due to the somewhat higher gene expression ofAPOE3Leiden in these mice. The difference in plasmaAPOE3Leiden levels between both APOE3Leiden-expressing strains is likely due to the increased VLDLsecretion in APOE3Leiden/apoe1/2mice.
VLDL-TG and apoB-100 secretion were markedly im-paired in APOE3Leiden/apoe2/2mice compared withAPOE3/apoe2/2mice, and secretion rates of the formerwere similar to those observed inapoe2/2 mice.4 In thepresence of APOE3Leiden and a single endogenousapoeallele, however, VLDL-TG and apoB-100 secretion wassimilar to that observed in APOE3/apoe2/2mice. Inasmuchas apoB-48 secretion remained constant between the 3 strainsof mice, our data indicate that the additional TGs secreted inthe presence of a functional apoE (mouseapoe or humanAPOE) occurred mainly through a modulation of the produc-tion of apoB-100–containing particles. Inasmuch as VLDLlipid composition was similar between all strains of mice, thisindicates that the decreased VLDL-TG secretion inAPOE3Leiden/apoe2/2is mainly due to a decrease innumber of particles secreted.
Other groups have recently shown that overexpression ofAPOE2 or APOE4 by adenovirus-mediated gene transfer inapoE-deficient mice leads to an increased VLDL-TG secre-tion.39,40 These isoforms differ only in a single amino acidfrom APOE3. Because APOE3Leiden is not able to increaseVLDL-TG secretion, our results indicate that incorrect fold-ing of the APOE molecule caused by the introduction of atandem repeat of 7 amino acids is associated with impairedVLDL secretion. This suggests that this site might influencethe lipid-transporting capacity of apoE. Until now, it wasknown that expression ofAPOE3Leiden results in impairedreceptor-mediated uptake19 and impaired hydrolysis ofTG-rich lipoproteins,18 contributing to development ofatherosclerosis in humans14 and mice.41 The present studyshows that APOE3Leiden, in contrast to APOE3, is unableto deter the development hepatic steatosis associated withapoE deficiency16 and to normalize impaired VLDL secre-tion, which has become a characteristic hallmark of thiscondition.4,6
AcknowledgmentThis study was supported by a grant from the Netherlands HeartFoundation (No. 96-011).
References1. Mahley RW. Apolipoprotein E: cholesterol transport protein with
expanding role in cell biology.Science. 1988;240:622–630.2. Quarfordt SH, Oswald B, Landis B, Xu HS, Zhang SH, Maeda N. In vivo
cholesterol kinetics in apolipoprotein E-deficient and control mice.JLipid Res. 1995;36:1227–1235.
3. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hyper-cholesterolemia and arterial lesions in mice lacking apolipoprotein E.Science. 1992;258:468–471.
4. Kuipers F, Lin Y, Havinga R, Bloks V, Verkade HJ, Jong MC, HofkerMH, Moshage H, van Vlijmen BJM, Vonk RJ, et al. Impaired productionof very low density lipid proteins by apolipoprotein E-deficient mousehepatocytes in primary culture.J Clin Invest. 1997;100:2915–2922.
5. Kuipers F, van Ree JM, Hofker MH, Wolters H, in ’t Veld G, Havinga R,Vonk RJ, Princen HMG, Havekes LM. Altered lipid metabolism inapolipoprotein E-deficient mice does not affect cholesterol balance acrossthe liver.Hepatology. 1996;24:241–247.
6. Mensenkamp AR, Jong MC, van Goor H, van Luyn MJA, Bloks V,Havinga R, Voshol PJ, Hofker MH, Willems van Dijk K, Havekes LM,et al. Apolipoprotein E participates in the regulation of very low density
lipoprotein-triglyceride secretion by the liver.J Biol Chem. 1999;274:35711–35718.
7. Huang Y, Ji ZS, Brecht WJ, Rall SC Jr, Taylor JM, Mahley RW.Overexpression of apolipoprotein E3 in transgenic rabbits causescombined hyperlipidemia by stimulating hepatic VLDL production andimpairing VLDL lipolysis. Arterioscler Thromb Vasc Biol. 1999;19:2952–2959.
8. Huang Y, Liu XQ, Rall SC Jr, Taylor JM, von Eckardstein A, AssmannG, Mahley RW. Overexpression and accumulation of apolipoprotein E asa cause of hypertriglyceridemia.J Biol Chem. 1998;273:26388–26393.
9. Maugeais C, Tietge UJF, Tsukamoto K, Glick JM, Rader DJ. Hepaticapolipoprotein E expression promotes very low density lipoprotein-apolipoprotein B production in vivo in mice.J Lipid Res. 2000;41:1673–1679.
10. Demant T, Bedford D, Packard CJ, Shepherd J. Influence of apoli-poprotein E polymorphism on apolipoprotein B-100 metabolism innormolipidemic subjects.J Clin Invest. 1991;88:1490–1501.
11. Welty FK, Lichtenstein AH, Barrett PH, Jenner JL, Dolnikowski GG,Schaefer EJ. Effects of apoE genotype on apoB-48 and apoB-100 kineticswith stable isotopes in humans.Arterioscler Thromb Vasc Biol. 2000;20:1807–1810.
12. van den Maagdenberg AMJM, de Knijff P, Stalenhoef AF, GeversLeuven JA, Havekes LM, Frants RR. Apolipoprotein E3*Leiden alleleresults from a partial gene duplication in exon 4.Biochem Biophys ResCommun. 1989;165:851–857.
13. Wardell MR, Weisgraber KH, Havekes LM, Rall SC Jr. ApolipoproteinE3-Leiden contains a seven-amino acid insertion that is a tandem repeatof residues 121–127.J Biol Chem. 1989;264:339–348.
14. de Knijff P, van den Maagdenberg AMJM, Stalenhoef AF, GeversLeuven JA, Demacker PN, Kuyt LP, Frants RR, Havekes LM. Familialdysbetalipoproteinemia associated with apolipoprotein E*3Leiden in anextended multigeneration pedigree.J Clin Invest. 1991;88:643–655.
15. van Vlijmen BJM, van den Maagdenberg AMJM, Gijbels MJJ, van derBoom H, HogenEsch H, Frants RR, Hofker MH, Havekes LM. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoproteinE3-Leiden transgenic mice.J Clin Invest. 1994;93:1403–1410.
16. Mensenkamp AR, van Luyn MJA, van Goor H, Bloks V, Apostel F,Greeve J, Hofker MH, Jong MC, van Vlijmen BJM, Havekes LM, et al.Hepatic lipid accumulation, altered very low density lipoprotein for-mation and apolipoprotein E deposition in apolipoprotein E3-Leidentransgenic mice.J Hepatol. 2000;33:189–198.
17. Hogan B, Constatini F, Lacey E.Manipulating the Mouse Embryo: ALaboratory Manual. Cold Spring Harbor, New York: Cold Spring HarborLaboratory Press; 1986.
18. Jong MC, Dahlmans VEH, van Gorp PJ, Breuer ML, Mol MJTM, van derZee A, Frants RR, Hofker MH, Havekes LM. Both lipolysis and hepaticuptake of VLDL are impaired in transgenic mice coexpressing humanapolipoprotein E*3Leiden and human apolipoprotein C1.ArteriosclerThromb Vasc Biol. 1996;16:934–940.
19. van Vlijmen BJM, Willems van Dijk K, van ‘t Hof HB, van Gorp PJ, ZeeAvd, van der Boom H, Breuer ML, Hofker MH, Havekes LM. In theabsence of endogenous mouse apolipoprotein E, apolipoprotein E*2(Arg-1583Cys) transgenic mice develop more severe hyperlipoprotein-emia than apolipoprotein E*3-Leiden transgenic mice.J Biol Chem.1996;271:30595–30602.
20. Dang Q, Walker D, Taylor S. Structure of the hepatic control region of thehuman apolipoprotein E/C-I gene locus.J Biol Chem. 1995;270:22577–22585.
21. van Ree JM, van den Broek WJAA, Dahlmans VEH, Groot PHE,Vidgeon-Hart M, Frants RR, Wierenga B, Havekes LM, Hofker MH.Diet-induced hypercholesterolemia and atherosclerosis in heterozygousapolipoprotein E-deficient mice.Atherosclerosis. 1994;111:25–37.
22. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purifi-cation.Can J Biochem Physiol. 1959;37:911–917.
23. Böttcher CFJ, van Gent CM, Pries C. A rapid and sensitive sub-micro-phosphorus determination.Anal Chim Acta. 1961;24:203–204.
24. Williamson DH, Mellanby J.D-(-)-3-Hydroxybutyrat. In: Bergmeyer HU,ed. Methoden der enzymatischen analyse. Weinheim: Verlag Chemie;1970:1772–1779.
25. Li X, Catalina F, Grundy SM, Patel S. Method to measure apolipoproteinB-48 and B-100 secretion rates in an individual mouse: evidence for avery rapid turnover of VLDL and preferential removal of B-48-relative toB-100-containing lipoproteins.J Lipid Res. 1996;37:210–220.
26. Bloks V, Plösch T, van Goor H, Roelofsen H, Baller J, Havinga R,Verkade HJ, van Tol A, Jansen PLM, Kuipers F. Hyperlipidemia andatherosclerosis associated with liver disease in ferrochelatase deficientmice.J Lipid Res. 2001;42:41–50.
Mensenkamp et al Impaired VLDL Secretion in APOE3Leiden Mice 1371
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

27. Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurementwith Folin reagent.J Biol Chem. 1951;193:265–275.
28. Massimi M, Lear SR, Williams DL, Jones AL, Erickson SK. Differentialexpression of apolipoprotein E messenger RNA within the rat lobuledetermined byin situ hybridization.Hepatology. 1999;29:1549–1555.
29. Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisomeproliferator-activated receptor alpha (PPARalpha) in the cellular fastingresponse: the PPARalpha-null mouse as a model of fatty acid oxidationdisorders.Proc Natl Acad Sci U S A. 1999;96:7473–7478.
30. Salero E, Perez-Sen R, Aruga J, Gimenez C, Zafra F. Transcriptionfactors Zic1 and Zic2 bind and transactivate the apolipoprotein E genepromoter.J Biol Chem.2001;276:1881–1888.
31. Berg DT, Calnek DS, Grinnell BW. The human apolipoprotein E gene isnegatively regulated in human liver HepG2 cells by the transcriptionfactor BEF-1.J Biol Chem. 1995;270:15447–15450.
32. Chang DJ, Paik YK, Leren TP, Walker DW, Howlett GJ, Taylor JM.Characterization of a human apolipoprotein E gene enhancer element andits associated protein factors.J Biol Chem. 1990;265:9496–9504.
33. Franke H, Potratz I, Dargel R. Zonal differences in lipoprotein formationin the thioacetamide-induced micronodular-cirrhotic rat liver.Exp ToxicolPathol. 1994;46:503–511.
34. Guzmán M, Castro J. Zonation of fatty acid metabolism in rat liver.Biochem J. 1989;264:107–113.
35. Voorschuur AH, Kuiper J, Neelissen JA, Boers W, Van Berkel TJ.Different zonal distribution of the asialoglycoprotein receptor, the alpha
2-macroglobulin receptor/low-density-lipoprotein receptor-relatedprotein and the lipoprotein-remnant receptor of rat liver parenchymalcells.Biochem J. 1994;303:809–816.
36. Pessayre D, Mansouri A, Haouzi D, Fromenty B. Hepatotoxicity due tomitochondrial dysfunction.Cell Biol Toxicol. 1999;15:367–373.
37. Hamilton RL, Wong JS, Guo LSS, Krisans S, Havel RJ. ApolipoproteinE localization in rat hepatocytes by immunogold labeling of cryothinsections.J Lipid Res. 1990;31:1589–1603.
38. Jong MC, Dahlmans VEH, Hofker MH, Havekes LM. Nascent very-low-density lipoprotein triacylglycerol hydrolysis by lipoprotein lipase isinhibited by apolipoprotein E in a dose-dependent manner.Biochem J.1997;328:745–750.
39. Tsukamoto K, Maugeais C, Glick JM, Rader DJ. Markedly increasedsecretion of VLDL triglycerides induced by gene transfer of apoli-poprotein E isoforms in apoE-deficient mice.J Lipid Res. 2000;41:253–259.
40. de Beer F, Willems van Dijk K, Jong MC, van Vark LC, van Der ZA,Hofker MH, Fallaux FJ, Hoeben RC, Smelt AH, Havekes LM. Apoli-poprotein E2 (Lys1463Gln) causes hypertriglyceridemia due to an apo-lipoprotein E variant-specific inhibition of lipolysis of very low densitylipoproteins-triglycerides.Arterioscler Thromb Vasc Biol. 2000;20:1800–1806.
41. van den Maagdenberg AMJM, Hofker MH, Krimpenfort PJA, de BruijnIH, van Vlijmen BJM, van der Boom H, Havekes LM, Frants RR.Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibithyperlipoproteinemia.J Biol Chem. 1993;268:10540–10545.
1372 Arterioscler Thromb Vasc Biol. August 2001
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from

Willems van Dijk, Louis M. Havekes and Folkert KuipersArjen R. Mensenkamp, Bas Teusink, Julius F.W. Baller, Henk Wolters, Rick Havinga, Ko
Mice Expressing Only the Mutant APOE3Leiden Gene Show Impaired VLDL Secretion
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2001 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/hq0801.0938642001;21:1366-1372Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/21/8/1366World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2001/08/10/21.8.1366.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on November 9, 2015http://atvb.ahajournals.org/Downloaded from
Related Documents











