CLINICAL STUDY MGMT promoter hypermethylation and its associations with genetic alterations in a series of 350 brain tumors Marta Mellai • Oriana Monzeglio • Angela Piazzi • Valentina Caldera • Laura Annovazzi • Paola Cassoni • Guido Valente • Susanna Cordera • Cristina Mocellini • Davide Schiffer Received: 2 August 2011 / Accepted: 26 December 2011 Ó Springer Science+Business Media, LLC. 2012 Abstract MGMT (O 6 -methylguanine-DNA methyltrans- ferase) promoter hypermethylation is a helpful prognostic marker for chemotherapy of gliomas, although with some controversy for low-grade tumors. The objective of this study was to retrospectively investigate MGMT promoter hypermethylation status for a series of 350 human brain tumors, including 275 gliomas of different malignancy grade, 21 glioblastoma multiforme (GBM) cell lines, and 75 non-glial tumors. The analysis was performed by methylation-specific PCR and capillary electrophoresis. MGMT expression at the protein level was also evaluated by both immunohistochemistry (IHC) and western blotting analysis. Associations of MGMT hypermethylation with IDH1/IDH2 mutations, EGFR amplification, TP53 mutations, and 1p/19q co-deletion, and the prognostic significance of these, were investigated for the gliomas. MGMT promoter hypermethylation was identified in 37.8% of gliomas, but was not present in non-glial tumors, with the exception of one primitive neuroectodermal tumor (PNET). The frequency was similar for all the astrocytic gliomas, with no correlation with histological grade. Significantly higher values were obtained for oligoden- drogliomas. MGMT promoter hypermethylation was sig- nificantly associated with IDH1/IDH2 mutations (P = 0.0207) in grade II–III tumors, whereas it had a borderline association with 1p deletion (P = 0.0538) in oligoden- drogliomas. No other association was found. Significant correlation of MGMT hypermethylation with MGMT protein expression was identified by IHC in GBMs and oligodendrogliomas (P = 0.0001), but not by western blotting. A positive correlation between MGMT protein expression, as detected by either IHC or western blotting, was also observed. The latter was consistent with MGMT promoter hypermethylation status in GBM cell lines. In low-grade gliomas, MGMT hypermethylation, but not MGMT protein expression, was associated with a trend, only, toward better survival, in contrast with GBMs, for which it had favorable prognostic significance. Keywords MGMT promoter hypermethylation Á Genetics Á Immunohistochemistry Á Brain tumors Introduction MGMT (O 6 -methylguanine-DNA methyltransferase) is a DNA repair enzyme involved in the mechanism of resis- tance of human cancers to alkylating agents. MGMT spe- cifically removes mutagenic, carcinogenic, and cytotoxic M. Mellai Á O. Monzeglio Á V. Caldera Á L. Annovazzi Á D. Schiffer (&) Neuro-bio-oncology Center, Policlinico di Monza Foundation, Via Pietro Micca, 29–13100, Vercelli, Italy e-mail: [email protected] A. Piazzi Department of Medical Sciences, University of Piemonte Orientale, Novara, Italy P. Cassoni Department of Biomedical Sciences and Human Oncology, University of Turin, Turin, Italy G. Valente Department of Clinical and Experimental Medicine, University of Piemonte Orientale, Novara, Italy S. Cordera Department of Neurology, Ospedale Regionale, Aosta, Italy C. Mocellini Department of Neurology, Azienda Ospedaliera Santa Croce e Carle, Cuneo, Italy 123 J Neurooncol DOI 10.1007/s11060-011-0787-y

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

CLINICAL STUDY
MGMT promoter hypermethylation and its associationswith genetic alterations in a series of 350 brain tumors
Marta Mellai • Oriana Monzeglio • Angela Piazzi • Valentina Caldera •
Laura Annovazzi • Paola Cassoni • Guido Valente • Susanna Cordera •
Cristina Mocellini • Davide Schiffer
Received: 2 August 2011 / Accepted: 26 December 2011
� Springer Science+Business Media, LLC. 2012
Abstract MGMT (O6-methylguanine-DNA methyltrans-
ferase) promoter hypermethylation is a helpful prognostic
marker for chemotherapy of gliomas, although with some
controversy for low-grade tumors. The objective of this
study was to retrospectively investigate MGMT promoter
hypermethylation status for a series of 350 human brain
tumors, including 275 gliomas of different malignancy
grade, 21 glioblastoma multiforme (GBM) cell lines, and
75 non-glial tumors. The analysis was performed by
methylation-specific PCR and capillary electrophoresis.
MGMT expression at the protein level was also evaluated
by both immunohistochemistry (IHC) and western blotting
analysis. Associations of MGMT hypermethylation with
IDH1/IDH2 mutations, EGFR amplification, TP53
mutations, and 1p/19q co-deletion, and the prognostic
significance of these, were investigated for the gliomas.
MGMT promoter hypermethylation was identified in 37.8%
of gliomas, but was not present in non-glial tumors, with
the exception of one primitive neuroectodermal tumor
(PNET). The frequency was similar for all the astrocytic
gliomas, with no correlation with histological grade.
Significantly higher values were obtained for oligoden-
drogliomas. MGMT promoter hypermethylation was sig-
nificantly associated with IDH1/IDH2 mutations (P =
0.0207) in grade II–III tumors, whereas it had a borderline
association with 1p deletion (P = 0.0538) in oligoden-
drogliomas. No other association was found. Significant
correlation of MGMT hypermethylation with MGMT
protein expression was identified by IHC in GBMs and
oligodendrogliomas (P = 0.0001), but not by western
blotting. A positive correlation between MGMT protein
expression, as detected by either IHC or western blotting,
was also observed. The latter was consistent with MGMT
promoter hypermethylation status in GBM cell lines. In
low-grade gliomas, MGMT hypermethylation, but not
MGMT protein expression, was associated with a trend,
only, toward better survival, in contrast with GBMs, for
which it had favorable prognostic significance.
Keywords MGMT promoter hypermethylation �Genetics � Immunohistochemistry � Brain tumors
Introduction
MGMT (O6-methylguanine-DNA methyltransferase) is a
DNA repair enzyme involved in the mechanism of resis-
tance of human cancers to alkylating agents. MGMT spe-
cifically removes mutagenic, carcinogenic, and cytotoxic
M. Mellai � O. Monzeglio � V. Caldera � L. Annovazzi �D. Schiffer (&)
Neuro-bio-oncology Center, Policlinico di Monza Foundation,
Via Pietro Micca, 29–13100, Vercelli, Italy
e-mail: [email protected]
A. Piazzi
Department of Medical Sciences, University of Piemonte
Orientale, Novara, Italy
P. Cassoni
Department of Biomedical Sciences and Human Oncology,
University of Turin, Turin, Italy
G. Valente
Department of Clinical and Experimental Medicine,
University of Piemonte Orientale, Novara, Italy
S. Cordera
Department of Neurology, Ospedale Regionale, Aosta, Italy
C. Mocellini
Department of Neurology, Azienda Ospedaliera Santa Croce e
Carle, Cuneo, Italy
123
J Neurooncol
DOI 10.1007/s11060-011-0787-y
O6-alkylguanine DNA adducts induced by radiotherapy or
alkylating agents such as temozolomide (TMZ) or nitro-
sourea derivatives. MGMT-mediated resistance to alkyl-
ating drugs correlates with MGMT expression level [1].
During malignant transformation, the MGMT gene may be
epigenetically silenced by hypermethylation of its pro-
moter regions, leading to an increased sensitivity to
alkylating chemotherapy in a variety of tumors [2].
In glioblastoma multiforme (GBM), MGMT promoter
hypermethylation is detected in approximately 32–72% of
cases [3–7] and in 36–50% of gliosarcomas [8]. In long-
term survivors, the values are higher (74–83.3%) [9]. For
GBMs it is an important prognostic and predictive factor in
chemotherapy with TMZ [3, 4, 10].
In low-grade gliomas (LGG), also, hypermethylation of
the MGMT promoter is a frequent event. It occurs in
43–75% of diffuse astrocytomas [6, 11–14]; most tumors
with MGMT hypermethylation contain TP53 mutations,
particularly G:C?A:T transitions [11, 14, 15], and p53
protein accumulation is observed [12]. MGMT hyperme-
thylation and promoter hypermethylation of the p14 gene
are mutually exclusive [14]. In anaplastic astrocytomas, the
frequency is 38–64% [16, 17].
In grade II and III oligodendrogliomas, the frequency of
MGMT promoter hypermethylation is higher (47–92.9 and
70–94.4%, respectively) whereas it is lower in grade II and
III oligoastrocytomas (27–40 and 30–62.5%, respectively)
[13, 18, 19]. In both oligodendrogliomas and oligoastro-
cytomas, it has recently been found to be associated with
1p/19q co-deletion [18–20], although with some exceptions
[11, 13].
Data regarding MGMT hypermethylation status and its
correlation with LGG patients’ prognosis and treatment
response are conflicting. MGMT hypermethylation has
been demonstrated to be associated with longer overall
survival (OS) [21, 22] and progression-free survival (PFS)
[13, 22], with some exceptions [19]. Its significance in
relation to TMZ chemotherapy is still under investigation
in phase III trials. Whereas in grade III astrocytomas
MGMT promoter hypermethylation is associated with
longer PFS in patients treated either by radio–chemother-
apy or radiation alone [23, 24], in grade II gliomas there are
contrasting results. MGMT promoter hypermethylation was
found to be predictive with TMZ treatment [13, 22], but not
for patients with grade II astrocytomas [12] and oligoas-
trocytomas [11] that did not receive alkylating chemo-
therapy. There is evidence of TMZ efficacy at standard
doses [20, 25] with a PFS increase when a protracted daily
TMZ regimen is used [22]; however, this was also
observed in MGMT unmethylated patients [22]. The pro-
longed TMZ regimen could potentially overcome the
MGMT-mediated resistance by progressive depletion of
MGMT activity and by improving sensitivity to TMZ [22].
This effect has also been demonstrated in grade II gliomas
and radiologically verified [26].
MGMT hypermethylation is rarely observed for non-
glial tumors, for example meningiomas, ependymomas,
medulloblastomas, and primitive neuroectodermal tumors
(PNETs) [27–31].
The objective of this study was to investigate MGMT
promoter hypermethylation status and MGMT expression
at the protein level for a series of 311 neuroepithelial
tumors, of which 275 were gliomas and 39 meningiomas.
For gliomas, associations of MGMT hypermethylation with
IDH1/IDH2 mutations, EGFR amplification, TP53 muta-
tions, 1p/19q co-deletion and OS were also studied.
Materials and methods
Patients
Formalin-fixed paraffin-embedded (FFPE) brain tumor
samples were collected from a total of 350 patients
(Table 1) after approval by the relevant Ethics Commit-
tees. Tumors were surgically removed at the Neurosurgery
Table 1 Patients’ demographics
Tumor type WHO
grade
Patients
(n)
Gender
(M/F)
Mean age
(years) and
range
Glial tumors (n = 275)
Pilocytic
astrocytoma
I 20 11/9 44 (19–68)
Diffuse and
gemistocytic
astrocytoma
II 13 5/8 42 (23–68)
Anaplastic
astrocytoma
III 4 3/1 55 (38–75)
Primary GBM IV 161 101/60 61 (23–83)
Secondary GBM IV 2 2/0 46 (42–50)
Gliosarcoma IV 9 6/3 56 (47–73)
Oligoastrocytoma II 3 2/1 38 (28–53)
Anaplastic
oligoastrocytoma
III 1 1/0 58
Oligodendroglioma II 34 20/14 50 (26–79)
Anaplastic
oligodendroglioma
III 28 14/14 54 (31–80)
Non-glial tumors (n = 75)
Meningioma I 30 8/22 63 (23–87)
II 4 2/2 64 (45–73)
III 5 3/2 62 (48–80)
Schwannoma I 15 9/6 58 (25–82)
Ependymoma III 12 8/4 56 (30–84)
Medulloblastoma IV 4 3/1 31 (20–39)
PNET IV 5 3/2 43 (22–59)
J Neurooncol
123

Unit, Department of Neuroscience, University of Turin
(Turin, Italy), Azienda Ospedaliero-Universitaria ‘‘Maggi-
ore della Carita’’ (Novara, Italy), Azienda Ospedaliera
Santa Croce e Carle (Cuneo, Italy), and Clinica Eporediese
(Ivrea, Italy). Histological diagnosis was performed in
accordance with World Health Organization (WHO)
guidelines [32]. All patients underwent either partial or total
resection; their demographics are listed in Table 1. After
informed consent, their tumor and blood samples were pro-
vided for genetic analysis and other research purposes.
One-hundred and four GBMs of the series had already been
investigated for MGMT promoter hypermethylation status [5].
GBMs were regarded as primary (pGBM) or secondary
(sGBM) according to the absence or presence, respectively,
of a previous histologically verified low-grade glioma.
For 21 pGBMs (CV1–21), stabilized cell lines from
primary cultures were also studied.
Glioma patient stratification
Of 172 patients with GBM, 99 received postoperative
standard radiotherapy (RT) (60 Gy total dose in 27–30
fractions by LINAC) and 20 received RT doses of\40 Gy
or died within 1 month after surgery. For 53 patients no
clinical information was available or they were lost to
follow-up. Of the 99 irradiated patients, 55 received stan-
dard TMZ therapy, 75 mg/m2/daily for 6 weeks, followed
by adjuvant TMZ: 200 mg/m2 from day 1 to day 5 every
4 weeks for 6–12 cycles. Treatment and follow-up of
sGBMs were not available.
Of the 3 patients with anaplastic astrocytomas, 1 received
RT and 2 were lost to follow-up. Of the 8 patients with
diffuse astrocytomas and the 7 patients with gemistocytic
astrocytomas, 4 received RT and TMZ, 2 received RT only,
and the others did not receive any treatment. The patients
with pilocytic astrocytomas did not receive any treatment
with the exception of 2 who underwent RT. Of 4 patients
with oligoastrocytomas, 1 received RT only, 2 are still alive
and scheduled for RT, and 1 was lost to follow-up. Of the 34
patients with grade II oligodendrogliomas, 12 received RT
of which 8 also received chemotherapy by PCV, 5 received
PCV only, and 17 did not receive any treatment. Of 28
patients with grade III oligodendrogliomas, 12 received RT
of which 10 also received PCV, 2 did not receive any
treatment, and no information is available for 14.
Molecular genetics
Genomic DNA (gDNA) was extracted from FFPE tumor
samples by use of a standard phenol–chloroform proce-
dure. Before DNA extraction from each sample, only
tumor areas previously identified as proliferating by
hematoxylin and eosin (H&E) staining and microscopic
examination were selected. gDNA from cell lines and
peripheral blood was isolated by use of the QIAmp
DNAMini Kit (Qiagen, Hamburg, Germany) and a salting-
out procedure, respectively.
MGMT promoter hypermethylation status
MGMT promoter hypermethylation status (GenBank
sequence NM_002412) was assessed by methylation-spe-
cific polymerase chain reaction (MS-PCR) followed by
capillary electrophoresis (CE) as reported elsewhere [5].
Sodium bisulfite modification was performed with the
MethylEasyTM
Exceed Rapid DNA Bisulfite Modification
Kit (Human Genetic Signatures, Macquarie Park, Sydney,
Australia) [5]. CpGenomeTM
Universal Methylated DNA
(Chemicon International, Temecula, CA, USA) and normal
lymphocyte DNA were used as methylated and unmethy-
lated controls, respectively. The primer sequences for MS-
PCR and the amplification conditions have already been
reported [2]. After electrophoresis on an ABI� 3130 Genetic
Analyzer (Applied Biosystems), data were collected for
fragment analysis by use of GeneMapper v4.0 software
(Applied Biosystems). A peak height ratio[0.1 was scored
as evidence of the methylated status of the MGMT gene
(mean from two independent experiments) [5].
Because our series contained 2 sGBMs only, for this
analysis 10 supplementary sGBMs were studied, kindly
supplied by Dr Bianca Pollo (Fondazione I.R.C.C.S. Isti-
tuto Neurologico C. Besta, Milan, Italy).
EGFR amplification status
EGFR amplification status was assessed by PCR co-
amplification of both the 110-bp DNA fragment of the
EGFR gene (GenBank sequence NM_005228) and the
85-bp DNA fragment of the INF-c gene (GenBank
sequence NM_000619). INF-c was used as reference
housekeeping gene. The primer sequences and the PCR
conditions have already been reported [33]. After CE, data
were collected by use of GeneMapper v4.0 software for
fragment analysis (Applied Biosystems). The amplification
status of the EGFR gene was determined by measuring the
EGFR/INF-c ratio. A ratio[2.09 was regarded as evidence
of more than two copies of the EGFR gene (mean from two
independent experiments).
IDH1 and IDH2 mutation analysis
Two primer pairs designed on genomic DNA were used to
amplify, by PCR, the IDH1 exon 4 (GenBank sequence
NM_005896), the IDH2 exon 4 (GenBank sequence
NM_002168), and the intron/exon boundaries (including at
least 80 bp of the flanking intronic sequences). The two
J Neurooncol
123

fragments contain, respectively, the arginine residue at IDH1
codon 132 (R132) and the homologous residue at IDH2
codon 172 (R172). The primer sequences are available on
request. PCR conditions have been reported elsewhere [34].
TP53 mutation analysis
Exons 4–8 of the TP53 gene (GenBank sequence
NM_000546) encoding for the highly conserved DNA
binding domain were searched for sequence variations by
direct sequencing. The TP53 gene was amplified from
genomic DNA as six fragments covering the 5 exons and
the intron/exon boundaries (including at least 80 bp of each
flanking intronic sequence). The primer sequences are
available on request. The PCR conditions and thermal
cycling procedure have been described elsewhere [34].
Direct sequencing
All the amplicons for the IDH1, IDH2, and TP53 genes
were analyzed by direct sequencing on an ABI� 3130
Genetic Analyzer by using the BigDye� Terminator v1.1
Cycle Sequencing Kit (Applied Biosystems). Data were
collected by the Sequencing Analysis v.5.3.1 software
(Applied Biosystems). All the identified sequence varia-
tions were confirmed with at least two independent PCR
and sequencing experiments. Mutation nomenclature is in
agreement with http://hgvs.org/mutnomen/recs-prot
(HUGO) recommendations. The reported nucleotide and
amino acid numbering is relative to the transcription start
site (?1) corresponding to the A of the ATG on the cor-
responding GenBank reference sequences.
To establish whether each putative sequence variation
was somatic, i.e. tumor-specific, the corresponding
patient’s constitutional DNA was analyzed when available.
Bioinformatic analysis
Putative functional effects of the identified TP53 missense
mutations were predicted in silico by use of PMUT (http://
mmb.pcb.ub.es/PMut/), PolyPhen (http://genetics.bwh.
harvard.edu/pph/) and SNAP (http://cubic.bioc.columbia.
edu/services/SNAP/) software.
The effect of missense, synonymous, and intronic vari-
ants on splicing was evaluated by use of NNSplice (http://
biologyhelp.awardspace.com/desc7.php?id=14&type=
biotech) and SpliceView (http://bioinfo2.itb.cnr.it/sun/
webgene) software.
Chromosomal status of the 1p and 19q regions
Multiplex ligation-dependent probe amplification (MLPA)
was used to assess allelic losses on the 1p and 19q
chromosomes, because no constitutive DNA was available
for all the archived tumor samples. Analysis was performed
by use of the SALSA-MLPA Kit P088 (lot number 0608)
(MRC-Holland, The Netherlands) in accordance with the
manufacturer’s instructions. The kit includes a total of 15
probes covering 1p, 8 probes covering 19q, and 15 control
probes located on other chromosomes. Additional probes
were included to verify DNA quantity, quality, and the
denaturation and hybridization steps. MLPA products were
analyzed by CE on an ABI� 3130 Genetic Analyzer
(Applied Biosystems) and data were collected by use of
GeneMapper v4.0 software (Applied Biosystems). In each
run, at least four reference samples were included for
normalization. Data were analyzed by use of Coffalyser
v9.4 software (MRC-Holland).
Chromosomal regions were regarded as deleted if a ratio
\0.75 was observed for two or more consecutive probes on
1p or 19q, whereas a gain of function was defined for ratios
[1.40 (mean from two independent experiments) [35].
Combined loss of 1p/19q was defined as either partial or
complete deletion of both chromosome arms 1p and 19q
[35].
We validated the MLPA Kit P088 for a series of 45
tumor samples by parallel LOH (loss of heterozygosity)
analysis with 7 microsatellite markers for 1p (D1S508,
D1S1612, D1S496, D1S2724, D1S457, D1S534, and
D1S2696 from 1p36.23 to 1p11.1, respectively) and 4 for
19q (D19S908, D19S219, D19S412, and D19S902 from
19q13.3 to 19q13.34, respectively).
MGMT immunohistochemistry (IHC)
Analysis of MGMT protein expression was performed on
5 lm-thick sections by a labeled streptavidin–biotin pro-
cedure after heat-induced epitope retrieval (HIER) as
reported elsewhere [5]. Incubation was with the anti-human
MGMT mouse monoclonal antibody (MAB16200, clone
MT3.1, 1:100; Chemicon International, Temecula, CA,
USA). Diaminobenzidine (DAB; Roche Diagnostics,
Penzberg, Germany) was used for detection. Nuclei were
counterstained with Mayer’s hematoxylin. A negative
control was performed by omission of the primary anti-
body. Nuclear expression in endothelial cells and lym-
phocytes provided positive internal controls for binding of
the primary antibody.
The evaluation was performed by use of a semi-quan-
titative score system considering staining intensity (?, ??,
???), percentage of positive cells (\or[20 and[50%),
and diffuse or focal distribution. Further details of the score
system are given in Table 2. Only nuclear staining was
considered for the evaluation. Infiltrating lymphocytes,
microglial cells, and endothelial cells were not included in
the counts.
J Neurooncol
123

Infiltrating lymphocytes, microglial cells, endothelial
cells, and macrophages were not considered in the counts,
being excluded on the basis of CD68 staining. On parallel
sections, IHC with the anti-human CD68 mouse mono-
clonal antibody (790-2931, clone KP1, prediluted; Ventana
Medical Systems, Tucson, AZ, USA) was performed on a
Ventana Full BenchMark� automatic immunostainer
(Ventana) with the UltraViewTM
Universal DAB Detection
Kit as detection system. HIER was performed in Tris–
EDTA, pH 8 (Ventana).
CD68 immunopositive cells were counted in parallel
sections in areas corresponding to those counted for
MGMT. The number of microglial cells and macrophages
was subtracted from the number of MGMT-positive cells.
Protein extraction and western blotting analysis
Samples drawn from paraffin blocks were deparaffinized
and homogenized in a lysis buffer (50 mM Tris–HCl,
pH 7.4, 150 mM NaCl, 1% v/v Igepal, 2% sodium dodecyl
sulfate (SDS), 0.5% sodium deoxycholate, 10 mM EDTA)
supplemented with a protease inhibitor cocktail (Sigma–
Aldrich, St Louis, MO, USA), 2 mM sodium orthovana-
date, and 10 mM sodium fluoride. Whole protein extracts
were quantified by use of the BCATM
protein assay kit
(Pierce Biotechnology, Rockford, IL, USA) and subjected
to SDS-PAGE (12%). This was followed by immunoblot-
ting analysis as described elsewhere [5] and by probing the
blots with a mouse monoclonal anti-MGMT antibody (#
MS-470-P0, 1:400; NeoMarkers, Fremont, CA, USA).
Immobilon Western Chemiluminescent Substrate (Milli-
pore, Billerica, MA, USA) was used as detection system.
Band intensity was measured and quantified with NIH
Image J software (RSB; NIMH, Bethesda, MD, USA).
Data obtained by densitometric evaluation of MGMT were
expressed relative (arbitrary units) to the signal of a rabbit
polyclonal anti-a-tubulin antibody (# LF-PA0146, 1:5000;
AbFrontier, Seoul, Korea) used for loading and transfer
control.
In-vitro cultures
Tumor surgical tissue was processed as described else-
where [36]. Culture conditions were: Dulbecco’s modified
Eagle’s medium (DMEM)/F-12 with 10 ng/ml bFGF (basic
fibroblast growth factor) and 20 ng/ml EGF (epidermal
growth factor) for neurospheres (NS), and DMEM with
10% fetal bovine serum (FBS) for adherent cells (AC).
Both cultures were maintained in 5% O2/CO2. Human
malignant glioma U87-MG and 010627 cell lines (kindly
supplied by Dr Rossella Galli, DIBIT San Raffaele, Milan,
Italy) were used as reference for both NS and AC.
Statistical methods
Associations between categorical variables were evaluated
by use of 2 9 2 contingency tables and the chi-squared (v2)
or two-tailed Fisher’s exact test, as appropriate.
The correlations between western blotting and immu-
nohistochemistry data were analyzed by use of the two-
sided Pearson’s correlation coefficient test.
OS was defined as the time between diagnosis and death
or last follow-up of the patient. Survival curves were
estimated by use of the Kaplan–Meier method, and the log-
rank test (Mantel–Cox) was performed to compare survival
curves for different groups of individuals. Analysis was
performed by use of SPSS v17.0 software (SPSS, Chicago,
IL, USA).
Results
MGMT methylation status and clinical variables
MS-PCR was performed on 350 brain tumors and MGMT
methylation status was successfully determined in 344
cases (98.3%). MGMT promoter hypermethylation was
detected in 104 of the 275 glial tumors (37.8%). Its fre-
quency in gliomas is reported in Table 3. It was not asso-
ciated with sex, patient age (B50 or [50 years), or tumor
location either for the whole series of gliomas or among
tumor subtypes (P [ 0.05 for all categories).
Among the different types, the frequency of MGMT
hypermethylation was highest for oligodendrogliomas,
with 36 of 62 cases (58.1%) (Table 4). The percentage was
lower for both oligoastrocytic (2 of 4 cases, 50%) and
astrocytic (66 of 207 cases, 31.9%) tumors, with statistical
significance (P = 0.0003) (Table 4).
In oligodendrogliomas, the MGMT gene was hyperme-
thylated in 18 of 34 grade II tumors (52.9%) and in 18 of
28 grade III tumors (64.3%) (Table 3). In astrocytomas, the
frequency of MGMT hypermethylation was as follows:
30% in pilocytic astrocytomas, 38.4% in diffuse and grade
Table 2 Score system for evaluation of MGMT immunostaining
Score Category
Distribution Intensity Percentage
of positive cells
0 No staining - -
1 Heterogeneous ? \20
2 Heterogeneous ??/??? [20
3 Homogeneous ? \20
4 Homogeneous ??/??? [20
5 Homogeneous ??/??? [50
J Neurooncol
123

II gemistocytic astroctyomas, 25% in anaplastic astrocy-
tomas, and 31.5% in pGBMs (Table 3). The frequency of
MGMT hypermethylation in sGBMs was significantly
higher (9 of 12 cases, 75%) (P = 0.0001). In gliosarcomas
the percentage was lower (2 of 9 cases, 22.2%).
In NS, MGMT was hypermethylated in 6 of 9 (66.7%)
cases. Hypermethylated NS originated from hypermethy-
lated GBM primary tumors with the exception of 2 cases.
Four of 6 NS (66.7%) were completely hypermethylated
whereas both hypermethylated and unmethylated MGMT
was observed for 2 cases (CV7 and CV17).
In contrast with NS, in MS-PCR, evidence of a signal
for unmethylated DNA, as a consequence of contamination
of the tumor sample by unmethylated normal and non-
tumor cells, was observed for each hypermethylated and
matched primary tumor. AC were never found to be
hypermethylated.
MGMT promoter hypermethylation was not observed
for non-glial tumors (meningiomas, schwannomas,
ependymomas, medulloblastomas and PNETs), with the
exception of one PNET.
EGFR amplification
EGFR amplification was identified in 57 of 161 GBMs
(35.4%), in one grade III astrocytoma (33.3%), and in one
pilocytic astrocytoma (\1%), but not in grade II astrocy-
tomas (Table 5).
In oligodendrogliomas EGFR amplification was detec-
ted in 2 of 31 (64.6%) grade II and in 8 of 23 (34.8%) grade
III tumors. No gene amplification was identified in
oligoastrocytomas.
IDH1 and IDH2 mutations
Somatic point mutations at IDH1 R132 and IDH2 R172
codons were identified in 53 of 282 gliomas (18.8%). Their
frequency in the different tumor types is depicted in
Table 5. It was higher in oligodendrocytic than in astro-
cytic tumors.
All mutations affected codon R132 with the exception of
one oligodendroglioma with mutation at codon R172.
Details are available elsewhere [34].
TP53 mutations
The TP53 mutation status was investigated in 222 cases of
glioma, and mutations were identified in 57 of these
(25.7%). The incidence of TP53 mutation among tumor
types is reported in Table 5. The range of TP53 mutations
in low and high-grade tumors is illustrated in Fig. 1.
In GBMs, the hotspot codons were Arg158 and Cys176
(exon 5), Tyr220 (exon 6), Cys275, and Pro278 (exon 8); in
low-grade gliomas, the hotspot codons were Ile195 (exon
5), Tyr220 (exon 6), and Arg273 (exon 8). Interestingly, 14
of 222 (6.3%) cases (8 among low-grade gliomas and 6
among GBMs) contained double-nucleotide (doublet) or
multiplet mutations.
Thirty-two of 37 (86.5%) missense mutations are
responsible for either significant or partial loss of p53
protein activity. c.G375A (p.T125T) mutation leads to
aberrant splicing.
Analysis of all point mutations showed that 10 of 46
(21.7%) were G:C?A:T transitions, 8 of which (80%)
affected a CpG site. Of the 10 G:C?A:T transitions, 5 were
identified in methylated tumors and 5 in unmethylated tumors.
Chromosomal status of 1p and 19q regions
This was assessed in 167 astrocytic and 30 oligodendro-
cytic tumors. The frequency of 1p/19q co-deletion in the
different tumor types is reported in Table 5. In
Table 3 Frequency of MGMT promoter hypermethylation according
to WHO grading
Tumor type Patients
(n)
MGMT
hypermethylation
Grade I
Pilocytic astrocytoma 20 6 (30.0%)
Grade II
Diffuse and gemistocytic
astrocytoma
13 5 (38.4%)
Oligodendroglioma 34 18 (52.9%)
Oligoastrocytoma 3 2 (66.7%)
Total 50 25 (49.0%)
Grade III
Anaplastic astrocytoma 4 1 (25%)
Anaplastic oligoastrocytoma 1 0 (0%)
Anaplastic oligodendroglioma 28 18 (64.3%)
Total 33 19 (57.6%)
Grade IV
pGBMs 168 52 (31.0%)
sGBMs 12 9 (75%)
Table 4 Frequency of MGMT promoter hypermethylation among
glioma subtypes
Tumor subtype Patients
(n)
MGMT
hypermethylation
P value
Astrocytic tumors
(Grades I–IV)
207 66 (31.9%) 0.0003
Oligodendrocytic tumors
(Grades II–III)
62 36 (58.1%)
Oligoastrocytic tumors
(Grades II–III)
4 2 (50%) Ns
J Neurooncol
123

oligodendrogliomas, 1p/19q partial or complete co-deletion
was found in 20 of 30 cases (66.7%), and more often in
grade II (80%) than in grade III (53.3%) tumors. Partial
deletion of 1p without 19q loss, no 1p deletion alone, and
no 19q deletion alone were detected in 3, 5, and 2 cases,
respectively. Partial 1p deletions (on 1p36), with or without
19q loss, were detected in 7 of 30 (23.3%) oligodendro-
gliomas, exclusively grade III tumors.
Table 5 Frequency of the genetic alterations investigated in relation to MGMT methylation status
Tumor
type
MGMT
hypermethylation
status
IDH1/IDH2
mutations*
EGFR
amplification
1p
deletion
19q
deletion
1p/19 co-
deletion
TP53
mutations
n n % n % n % n % n % n %
PA Methylated 6 0/6 (0) 1/4 (25) 2/4 (50) 2/4 (50) 2/4 (50) 1/5 (20)
Unmethylated 14 1/14 (7.1) 0/10 (0) 2/11 (18.2) 2/11 (18.2) 2/11 (18.2) 0/14 (0)
DA and GA Methylated 4 3/4 (75) 0/3 (0) 0/1 (0) 1/1 (100) 0/1 (0) 2/2 (100)
Unmethylated 8 0/8 (0) 0/7 (0) 2/4 (50) 1/4 (25) 1/4 (25) 2/4 (50)
AA Methylated 1 1/1 (100) 0/1 (0) 0/1 (0) 0/1 (0) 0/1 (0) –
Unmethylated 3 1/3 (33.3) 1/3 (33.3) 0/1 (0) 0/1 (0) 0/1 (0) 1/3 (33.3)
pGBM Methylated 52 0/52 (0) 22/50 (44) 6/44 (13.6) 2/44 (4.5) 1/44 (2.3) 14/45 (31.1)
Unmethylated 116 1/108 (\1) 35/107(32.7) 10/90 (11.1) 8/90 (8.9) 6/90 (6.7) 19/82 (23.2)
sGBM Methylated 9 7/9 (77.8) 0/2 (0) 1/1 (100) 0/1 (0) 0/1 (0) 1/1 (100)
Unmethylated 3 3/3 (100) – – – – –
OA Methylated 2 2/2 (100) 0/1 (0) 2/2 (100) 1/2 (50) 1/2 (50) 1/2 (50)
Unmethylated 1 1/1 (100) 0/1 (0) 1/1 (100) 1/1 (100) 1/1 (100) 1/1 (100)
AOA Methylated 0 – – – – – –
Unmethylated 1 1/1 (100) 1/1 (100) 1/1 (100) 0/1 (0) 0/1 (0) 1/1 (100)
O Methylated 18 14/18 (77.8) 2/15 (13.3) 7/7 (100) 7/7 (100) 7/7 (100) 3/14 (21.4)
Unmethylated 16 9/16 (56.2) 0/16 (0) 4/7 (57.1) 5/7 (71.4) 4/7 (57.1) 4/14 (28.6)
AO Methylated 18 8/17 (47.1) 5/16 (31.3) 7/9 (77.8) 5/9 (55.6) 4/9 (44.4) 4/16 (25)
Unmethylated 10 2/10 (20) 3/9 (33.3) 4/6 (66.7) 4/6 (66.7) 4/6 (66.7) 1/7 (14.3)
PA pilocytic astrocytoma, DA diffuse astrocytoma, GA gemistocytic astrocytoma, AA anaplastic astrocytoma, GBM glioblastoma multiforme,
O oligodendroglioma, AO anaplastic oligodendroglioma, OA oligoastrocytoma, AOA anaplastic oligoastrocytoma
*Significantly associated with MGMT hypermethylation in grade II–III A (P = 0.0357) and in grade II–III A ? O (P = 0.0207)
Fig. 1 Genomic structure of human TP53 gene. The hatched boxcorresponds to untranslated exon 1. All the identified mutations in low
and high-grade gliomas are reported. Each mutation was verified to be
previously described in gliomas or glioma cell lines in the UMD_TP53
mutation database (R1 release, July 2010, http://p53.free.fr/), in the
Catalogue of Somatic Mutations in Cancer (COSMIC database, R15
release, November 2010, http://www.sanger.ac.uk/genetics/CGP/
cosmic/) and by the International Agency for Research on Cancer
(IARC TP53 database, R15 release, November 2010, http://www.p53.
iarc.fr/). Mutations identified de novo in gliomas in this study are
indicated by asterisks. Mutations responsible for aberrant splicing are
reported in gray. Hotspot codons are in bold
J Neurooncol
123
In the two cases of oligoastrocytomas, one had 1p/19q
co-deletion and the other had 1p deletion only.
MGMT hypermethylation status and molecular markers
After stratification of patients for tumor subtypes and
grades, MGMT promoter hypermethylation was signifi-
cantly associated with IDH1/IDH2 mutations in grade II–
III astrocytomas (P = 0.0357), even more when consid-
ering them together with grade II–III oligodendrocytic
tumors (P = 0.0207), for which the incidence of IDH1/
IDH2 mutation was greater (Table 5).
In grade II–III oligodendrogliomas, no association was
found between MGMT promoter hypermethylation and 1p/
19 co-deletion. However, for 11 of 16 (68.8%) MGMT
hypermethylated tumors 1p/19q co-deletion was observed,
in contrast with 8 of 13 (61.5%) of unmethylated tumors. A
borderline association was identified with 1p deletion only
(P = 0.0538).
In GBMs, neither EGFR amplification nor TP53 muta-
tions was associated with MGMT promoter hypermethyla-
tion (P [ 0.05 for both categories). EGFR amplification was
detected in 22 of 50 (44%) and in 34 of 109 (31.2%) meth-
ylated and unmethylated tumors, respectively. TP53 muta-
tions were identified in 25 of 86 (29.1%) and in 28 of 121
(23.1%) methylated and unmethylated cases, respectively.
MGMT immunohistochemistry
A total of 102 grade I–III gliomas, 172 GBMs, and 96 non-
glial brain tumors were studied. Nuclear staining only was
considered. The evaluation according to the score system is
reported in Table 6. Positive endothelial cells, infiltrating
lymphocytes, microglial cells, and macrophages were
excluded from the counts.
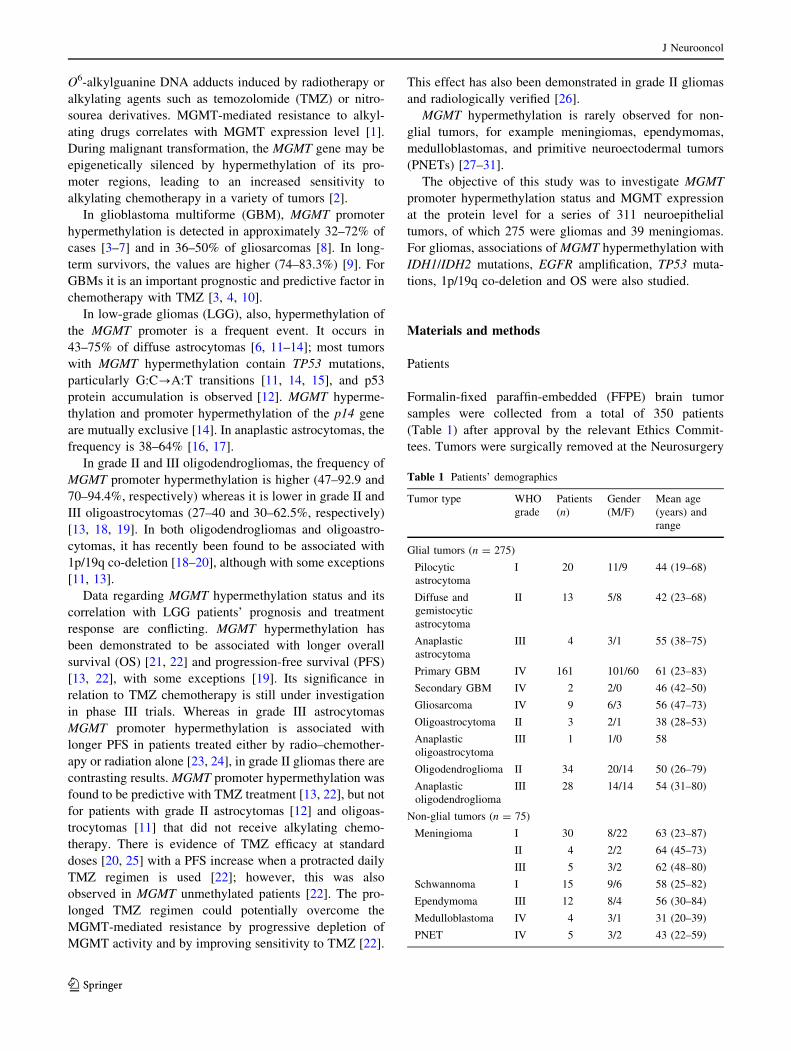
In gliomas, MGMT expression decreases from low to
high-grade tumors without statistical significance. The
percentage of positive tumor cells was highest for pilocytic
astrocytomas (Fig. 2a, b). This percentage was intermedi-
ate and variable for grade II astrocytomas (Fig. 2c, d),
anaplastic astrocytomas, grade II–III oligodendrogliomas
(Fig. 2e, f), and GBMs (Fig. 3a, b). For gemistocytes and
minigemistocytes weak cytoplasmic positivity was
observed. In gliosarcomas, both glial and sarcomatousus
components were positive, with staining intensity slightly
weaker for the latter.
Contamination by macrophages, lymphocytes, and
endothelial cells was regionally heterogeneous (Fig. 3c–e).
After correction for contamination, MGMT protein
expression correlated significantly with MGMT methyla-
tion status in GBMs (P = 0.0001) but not in low-grade
astrocytic tumors (P = 0.2861). It must be emphasized that
most astrocytic tumor samples with MGMT expression had
low MGMT hypermethylation values. In contrast, for oli-
godendrocytic tumors the correlation was statistically sig-
nificant, with 3 of 36 methylated tumors positively stained
and 8 of 23 unmethylated ones without MGMT expression
(P = 0.0001).
Absence of MGMT hypermethylation in meningiomas,
schwannomas, ependymomas, medulloblastomas and
PNETs corresponded to their positive protein expression,
with the exception of one PNET (Fig. 3f).
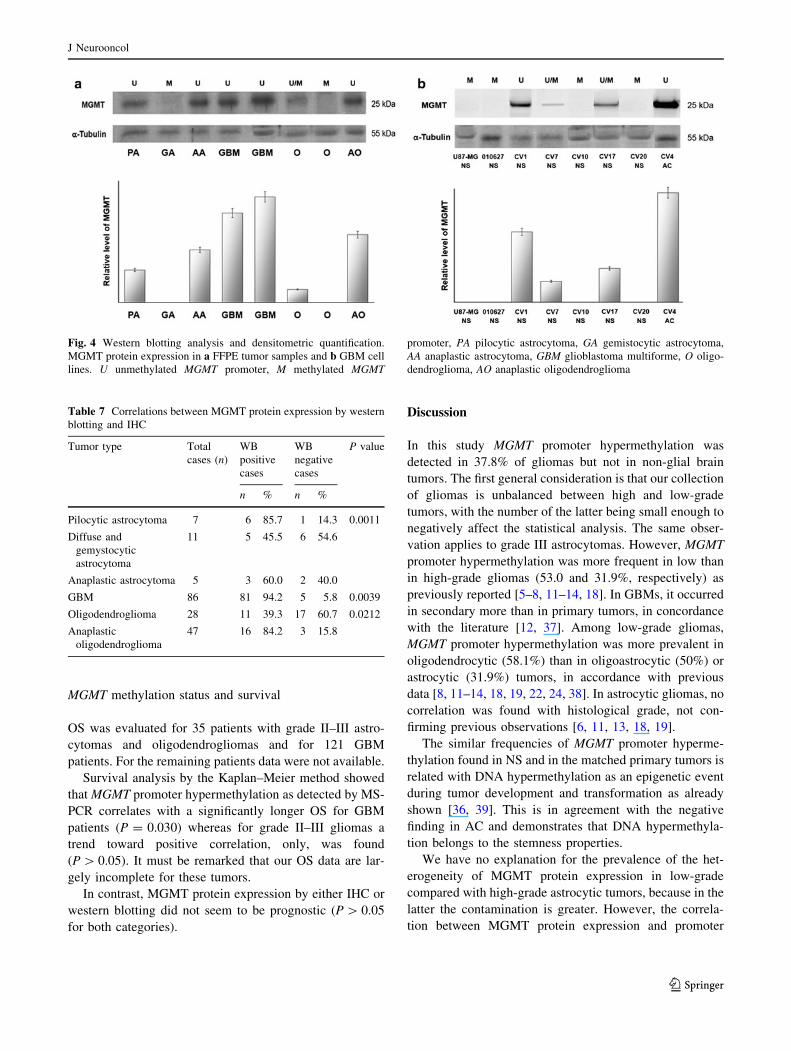
Western blotting
By western blotting analysis tumors were scored as positive
for MGMT protein expression on the basis of a clearly
visible band of molecular size 25 kDa (Fig. 4a). The dis-
tribution is shown in Table 7. A positive linear correlation
with statistical significance was found between MGMT
expression levels detected by IHC (taking into account the
percentage of positive cells, the staining intensity, and type
of distribution) and western blotting analysis, for both the
oligodendrocytic (Pearson’s correlation coefficient
r = 0.328; P = 0.0212) and astrocytic series (Pearson’s
correlation coefficient r = 0.637; P = 0.0011) (Table 7).
Table 6 MGMT immunopositivity in gliomas according to the score system
Score PA
(n = 20)
DA and GA
(n = 13)
AA
(n = 4)
O (n = 34) AO
(n = 27)
OA and AOA
(n = 4)
pGBM
(n = 160)
sGBM
(n = 12)
0 5 (25%) 10 (76.9%) 1 (25%) 19 (55.9%) 16 (59.3%) 2 (50%) 36 (22.6%) 7 (58.5%)
1 2 (10%) 0 (0%) 0 (0%) 5 (14.7%) 4 (14.8%) 1 (25%) 29 (18.1%) 1 (8.3%)
2 3 (15%) 2 (15.4%) 3 (75%) 4 (11.8%) 0 (0%) 1 (25%) 30 (18.8%) 0 (0%)
3 0 (0%) 0 (0%) 0 (0%) 2 (5.9%) 2 (7.4%) 0 15 (9.4%) 2 (16.7%)
4 8 (40%) 0 (0%) 0 (0%) 3 (8.8%) 3 (11.1%) 0 30 (18.8%) 0 (0%)
5 2 (10%) 1 (7.8%) 0 (0%) 1 (2.9%) 2 (7.4%) 0 20 (12.5%) 1 (8.3%)
PA pilocytic astrocytoma, DA diffuse astrocytoma, GA gemistocytic astrocytoma, AA anaplastic astrocytoma, GBM glioblastoma multiforme,
O oligodendroglioma, AO anaplastic oligodendroglioma, OA oligoastrocytoma, AOA anaplastic oligoastrocytoma
J Neurooncol
123
No correlation was found between MGMT hyperme-
thylation status, as detected by MS-PCR, and MGMT
expression by western blotting analysis in primary tumors.
In contrast, in GBM cell lines MGMT protein expression
detected by western blotting was consistent with MGMT
promoter hypermethylation status (Fig. 4b).
Fig. 2 MGMT immunohistochemistry in low-grade gliomas. a Pilo-
cytic astrocytoma (unmethylated MGMT promoter), [75% positive
nuclei. b Pilocytic astrocytoma (slightly methylated MGMT pro-
moter), 20% positive nuclei. c Diffuse astrocytoma (unmethylated
MGMT promoter), [75% positive nuclei. d Diffuse astrocytoma
(highly methylated MGMT promoter), non-positive nuclei. e Anaplas-
tic oligodendroglioma (unmethylated MGMT promoter), [75%
positive nuclei, and f Anaplastic oligodendroglioma (highly methyl-
ated MGMT promoter), non-positive nuclei. All DAB. Scale bar50 lm
J Neurooncol
123
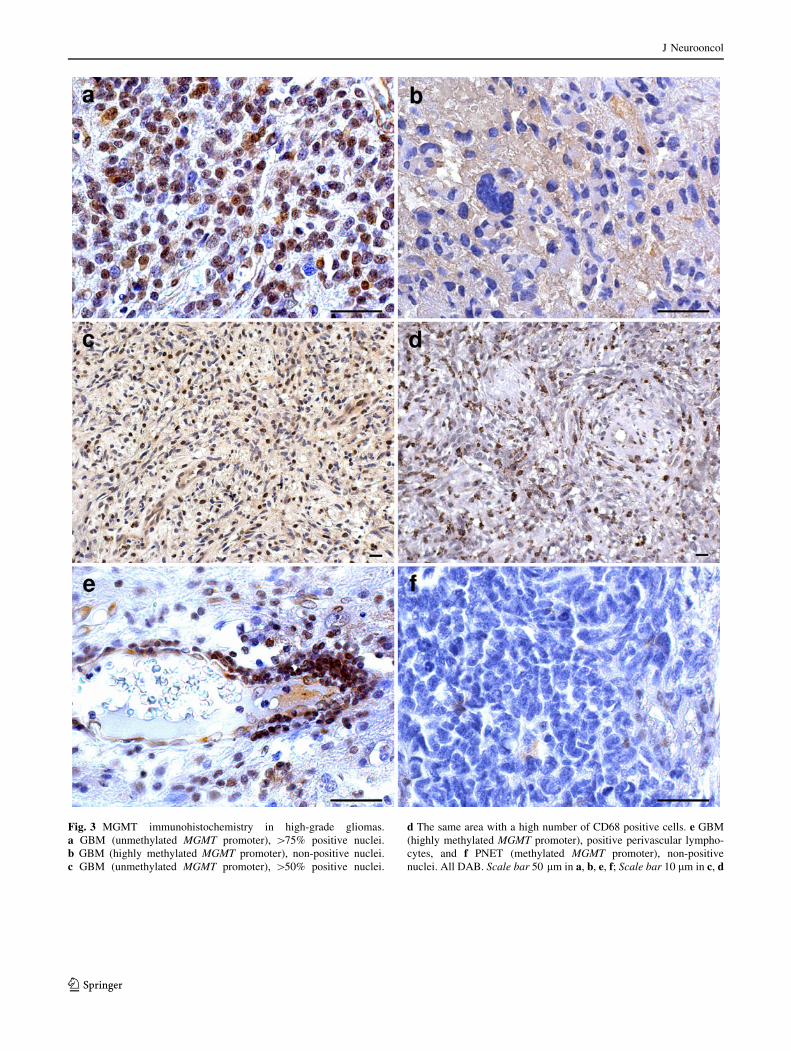
Fig. 3 MGMT immunohistochemistry in high-grade gliomas.
a GBM (unmethylated MGMT promoter), [75% positive nuclei.
b GBM (highly methylated MGMT promoter), non-positive nuclei.
c GBM (unmethylated MGMT promoter), [50% positive nuclei.
d The same area with a high number of CD68 positive cells. e GBM
(highly methylated MGMT promoter), positive perivascular lympho-
cytes, and f PNET (methylated MGMT promoter), non-positive
nuclei. All DAB. Scale bar 50 lm in a, b, e, f; Scale bar 10 lm in c, d
J Neurooncol
123
MGMT methylation status and survival
OS was evaluated for 35 patients with grade II–III astro-
cytomas and oligodendrogliomas and for 121 GBM
patients. For the remaining patients data were not available.
Survival analysis by the Kaplan–Meier method showed
that MGMT promoter hypermethylation as detected by MS-
PCR correlates with a significantly longer OS for GBM
patients (P = 0.030) whereas for grade II–III gliomas a
trend toward positive correlation, only, was found
(P [ 0.05). It must be remarked that our OS data are lar-
gely incomplete for these tumors.
In contrast, MGMT protein expression by either IHC or
western blotting did not seem to be prognostic (P [ 0.05
for both categories).
Discussion
In this study MGMT promoter hypermethylation was
detected in 37.8% of gliomas but not in non-glial brain
tumors. The first general consideration is that our collection
of gliomas is unbalanced between high and low-grade
tumors, with the number of the latter being small enough to
negatively affect the statistical analysis. The same obser-
vation applies to grade III astrocytomas. However, MGMT
promoter hypermethylation was more frequent in low than
in high-grade gliomas (53.0 and 31.9%, respectively) as
previously reported [5–8, 11–14, 18]. In GBMs, it occurred
in secondary more than in primary tumors, in concordance
with the literature [12, 37]. Among low-grade gliomas,
MGMT promoter hypermethylation was more prevalent in
oligodendrocytic (58.1%) than in oligoastrocytic (50%) or
astrocytic (31.9%) tumors, in accordance with previous
data [8, 11–14, 18, 19, 22, 24, 38]. In astrocytic gliomas, no
correlation was found with histological grade, not con-
firming previous observations [6, 11, 13, 18, 19].
The similar frequencies of MGMT promoter hyperme-
thylation found in NS and in the matched primary tumors is
related with DNA hypermethylation as an epigenetic event
during tumor development and transformation as already
shown [36, 39]. This is in agreement with the negative
finding in AC and demonstrates that DNA hypermethyla-
tion belongs to the stemness properties.
We have no explanation for the prevalence of the het-
erogeneity of MGMT protein expression in low-grade
compared with high-grade astrocytic tumors, because in the
latter the contamination is greater. However, the correla-
tion between MGMT protein expression and promoter
Fig. 4 Western blotting analysis and densitometric quantification.
MGMT protein expression in a FFPE tumor samples and b GBM cell
lines. U unmethylated MGMT promoter, M methylated MGMT
promoter, PA pilocytic astrocytoma, GA gemistocytic astrocytoma,
AA anaplastic astrocytoma, GBM glioblastoma multiforme, O oligo-
dendroglioma, AO anaplastic oligodendroglioma
Table 7 Correlations between MGMT protein expression by western
blotting and IHC
Tumor type Total
cases (n)
WB
positive
cases
WB
negative
cases
P value
n % n %
Pilocytic astrocytoma 7 6 85.7 1 14.3 0.0011
Diffuse and
gemystocytic
astrocytoma
11 5 45.5 6 54.6
Anaplastic astrocytoma 5 3 60.0 2 40.0
GBM 86 81 94.2 5 5.8 0.0039
Oligodendroglioma 28 11 39.3 17 60.7 0.0212
Anaplastic
oligodendroglioma
47 16 84.2 3 15.8
J Neurooncol
123

hypermethylation is poorer in the former than in the latter
[40].
No MGMT promoter hypermethylation was identified in
non-glial brain tumors (meningiomas, schwannomas,
ependymomas, medulloblastomas, and PNETs) by MS-
PCR, with the exception of one PNET, in agreement with
previous observations [27–31]. The unmethylated status of
MGMT promoter was confirmed by both IHC and western
blotting analysis.
Overall, there is variability in assessment of MGMT
hypermethylation status among the different series in the
literature. This might be because of the different sensitiv-
ities of the methods used to assess MGMT status. The
different methods currently used can generate inter-labo-
ratory inconsistencies and, consequently, different selec-
tions of patients for treatment [7, 41]. In our and others’
experience, MS-PCR is a reproducible and accurate semi-
quantitative method [5, 42], particularly when followed by
capillary electrophoretic analysis [5], with sensitivity of
0.1% methylated tumor cells in a heterogeneous cell pop-
ulation [43]. As a reference test in clinical practice it cor-
relates with OS [2, 4, 5, 44], but this does not exclude
alternative methods from being reliable in patient selection.
By IHC, there was intratumoral heterogeneity in MGMT
protein expression in all FFPE tumor samples, both
because of the wide heterogeneity of gliomas and the
clonal origin of MGMT hypermethylation. However, hy-
permethylation was found in a previous study analyzing
multiple samples from the same tumor by stereotactic
procedures, intratumoral homogeneity of MGMT [45].
Contamination by non-neoplastic cells (microglial cells,
macrophages, endothelial cells, and infiltrating lympho-
cytes) expressing MGMT is involved [5, 46]. Contamina-
tion is important in phenotypically homogeneous and
heterogeneous tumors. IHC can also be invalidated by the
up-regulation of protein expression by either radio or
chemotherapy or steroid treatment [4, 47]. Its altering
effect on the number of positive cells can be nullified by
identifying contaminating cells with CD68. This obviously
applies to IHC, but not to western blotting.
On the whole, our data are in favour of a correlation
between assessment of MGMT hypermethylation status by
MS-PCR and the MGMT protein expression as detected by
IHC, but not in low-grade astrocytomas. These results are
partially inconsistent with previous observations [40, 44,
46, 48, 49]. However, the lack of a significant association
between the immunoistochemical MGMT protein expres-
sion and patient outcome advises against the use of anti-
MGMT immunohistochemistry as a clinical biomarker for
routine diagnostic purposes [40]. A recent systematic
review and meta-analysis demonstrates that evaluation of
MGMT protein expression by IHC alone fails to reflect
promoter hypermethylation status and to predict patient
survival or glioma chemosensitivity in a way that is
interchangeable with MS-PCR [50]. The two methods
select different groups of patients [51].
A significant correlation was observed between IHC and
western blotting analysis, both in astrocytic and oligo-
dendrocytic tumors [5]. MGMT hypermethylation detected
by MS-PCR does not correlate with western blotting for
primary tumors, but it does for GBM cell lines [36]. On the
whole, most observations favor the hypothesis that MGMT
protein expression, whether by IHC or western blotting, has
poor prognostic significance, as already described [52].
Our MGMT promoter hypermethylation data confirm the
statistically significant association with IDH1/IDH2 muta-
tions for grade II–III astrocytic and oligodendrocytic
tumors [38, 53] but not for GBMs [6, 54].
For oligodendrogliomas and oligoastrocytomas, MGMT
promoter hypermethylation has previously been associated
with 1p/19q co-deletion [18–20, 53], with some exceptions
[6, 11, 13]. In our study, because of the small number of
cases, we did not make a distinction between partial or
complete 1p deletions, even though this might be important
for prognosis. Complete and partial 1p deletions associated
with 19q deletions may [55, 56] or may not [35] have
prognostic significance. Because partial 1p deletions may
have an unfavorable effect, and not only in oligodendro-
gliomas [55], it should be mandatory to distinguish partial
from complete deletions. This can be achieved only by
multiple loci techniques, for example CGH or MLPA [57].
In our experience, MLPA is confirmed by LOH analysis
with 7 microsatellites on 1p and it can resolve the problem
of allelic imbalance (loss vs. gain). In anaplastic oligo-
dendrogliomas with 1p/19q co-deletion, unfavorable
prognostic significance may be associated with a polisomy
of 1q and 19p detected by FISH [58].
In our series, partial and complete 1p/19q co-deletions
did not correlate significantly with MGMT promoter hy-
permethylation; however, 68.8% of 1p/19q co-deleted
tumors with MGMT promoter hypermethylation may
indicate a trend, with all the limitations discussed above.
The association between MGMT promoter hyperme-
thylation and TP53 mutations is controversial. A positive
association was found for diffuse astrocytomas [11, 14,
15], with greater occurrence of G:C?A:T transitions in
MGMT methylated tumors [15], and with p53 protein
accumulation [12]. The p53 immunopositivity of methyl-
ated GBMs is of little significance because GBMs with
negative MGMT immunohistochemical expression had a
significantly higher number of TP53 mutations [59]. In
contrast, no association was found by others with either
TP53 mutations in low and high-grade astrocytic tumors [6,
13, 17] or p53 immunopositivity [53]. In our series, we did
not find any correlation with TP53 mutation status and
EGFR amplification, but there are exceptions.
J Neurooncol
123

Together with IDH1/IDH2 mutations, MGMT promoter
hypermethylation is a frequent and early epigenetic event
during gliomagenesis of both astrocytic and oligodendro-
cytic tumors [17, 60], preceding the differentiation of
precursors. Its prevalence among diffuse astrocytomas and
grade II–III oligodendrogliomas and oligoastrocytomas
suggests that these tumors may originate from a common
glial precursor cell population of NCSCs (neural cancer
stem cells) through two different IDH-dependent or inde-
pendent pathways [61].
Promoter-associated CpG island hypermethylation in
general has widely been reported for human GBMs and
other glioma subtypes [62]. Our observations of MGMT
promoter hypermethylation are in agreement with the
suggestion that a glioma-CpG island methylator phenotype
(G-CIMP) does exist [63]. This phenotype would prevail
among low-grade gliomas and have distinct copy number
alterations that are closely associated with IDH1/IDH2
somatic mutations and improved patient survival [63]. As a
matter of fact, a significant association between IDH1/
IDH2 mutations and MGMT promoter hypermethylation
was found in this series of low-grade gliomas [34], and this
has recently been regarded as useful in the molecular
subclassification of gliomas [64]. Together with 1p/19q co-
deletion and the newly identified G-CIMP phenotype,
MGMT promoter hypermethylation should belong to the
transcriptionally defined proneuronal glioma subclass
which characterizes low rather than high-grade gliomas,
the latter belonging to the mesenchymal glioma subclass
[65]. Therefore, this epigenetic alteration may be used as a
stratification marker to identify subgroups of glioma
patients with a better survival according to their methyla-
tion profile [66].
MGMT hypermethylation as detected by MS-PCR con-
fers a survival benefit on GBM patients [3–5, 10, 24, 38],
as confirmed by us, whereas its prognostic and predictive
significance for low-grade gliomas is still debated. On this
matter, our data are still insufficient for definite ascertain-
ment; however, our preliminary results suggests MGMT
hypermethylation is not prognostic for these tumors.
Conclusions
MGMT hypermethylation has prognostic significance for
high-grade but not low-grade gliomas. The reliability of
IHC for evaluation of MGMT protein expression and
therefore, indirectly, of MGMT hypermethylation status, is
lower than that of MS-PCR, mainly because of contami-
nation. Contamination also affects western blotting
analysis.
MGMT promoter hypermethylation is significantly
associated with IDH1/IDH2 mutations in grade II–III gli-
omas. It has a borderline association with 1p deletion for
oligodendrogliomas. No correlation is found with either
TP53 mutations or EGFR amplification.
Acknowledgments This work was supported by a Grant from
Compagnia di San Paolo, Turin. We are greatly indebted to Dr Bianca
Pollo (Fondazione I.R.C.C.S. Istituto Neurologico C. Besta, Milan,
Italy) for providing sGBMs.
References
1. Kaina B, Christmann M, Naumann S et al (2007) MGMT: key
node in the battle against genotoxicity, carcinogenicity and
apoptosis induced by alkylating agents. DNA Repair (Amst)
6:1079–1099
2. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG
(1999) Inactivation of the DNA repair gene O6-methylguanine-
DNA methyltransferase by promoter hypermethylation is a
common event in primary human neoplasia. Cancer Res
59:793–797
3. Hegi ME, Diserens AC, Godard S et al (2004) Clinical trial
substantiates the predictive value of O6-methylguanine-DNA
methyltransferase promoter methylation in glioblastoma patients
treated with temozolomide. Clin Cancer Res 10:1871–1874
4. Hegi ME, Diserens AC, Gorlia T et al (2005) MGMT gene
silencing and benefit from temozolomide in glioblastoma. N Engl
J Med 352:997–1003
5. Mellai M, Caldera V, Annovazzi L et al (2009) MGMT promoter
hypermethylation in a series of 104 glioblastomas. Cancer
Genomics Proteomics 6:219–227
6. Jha P, Suri V, Jain A et al (2010) O6-methylguanine DNA
methyltransferase gene promoter methylation status in gliomas
and its correlation with other molecular alterations: first Indian
report with review of challenges for use in customized treatment.
Neurosurgery 67:1681–1691
7. Suri V, Jha P, Sharma MC, Sarkar C (2011) O6-methylguanine
DNA methyltransferase gene promoter methylation in high-grade
gliomas: a review of current status. Neurol India 59:229–235
8. Kang SH, Park KJ, Kim CY et al (2011) O6-methylguanine DNA
methyltransferase status determined by promoter methylation and
immunohistochemistry in gliosarcoma and their clinical impli-
cations. J Neurooncol 101:477–486
9. Das P, Puri T, Jha P et al (2011) A clinicopathological and
molecular analysis of glioblastoma multiforme with long-term
survival. J Clin Neurosci 18:66–70
10. Olson RA, Brastianos PK, Palma DA (2011) Prognostic and
predictive value of epigenetic silencing of MGMT in patients
with high grade gliomas: a systematic review and meta-analysis.
J Neurooncol 105:325–335
11. Watanabe T, Nakamura M, Kros JM et al (2002) Phenotype
versus genotype correlation in oligodendrogliomas and low-grade
diffuse astrocytomas. Acta Neuropathol 103:267–275
12. Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T,
Fukushima T (2003) Promoter hypermethylation of the DNA
repair gene O6-methylguanine-DNA methyltransferase is an
independent predictor of shortened progression-free survival in
patients with low-grade diffuse astrocytomas. Brain Pathol
13:176–184
13. Everhard S, Kaloshi G, Criniere E et al (2006) MGMT methyl-
ation: a marker of response to temozolomide in low-grade glio-
mas. Ann Neurol 60:740–743
14. Watanabe T, Katayama Y, Yoshino A et al (2007) Aberrant hy-
permethylation of p14ARF and O6-methylguanine-DNA meth-
yltransferase genes in astrocytoma progression. Brain Pathol
17:5–10
J Neurooncol
123

15. Bello MJ, Alonso ME, Aminoso C et al (2004) Hypermethylation
of the DNA repair gene MGMT: association with TP53 G:C to
A:T transitions in a series of 469 nervous system tumors. Mutat
Res 554:23–32
16. Watanabe T, Katayama Y, Komine C et al (2005) O6-methyl-
guanine-DNA methyltransferase methylation and TP53 mutation
in malignant astrocytomas and their relationships with clinical
course. Int J Cancer 113:581–587
17. Groenendijk FH, Taal W, Dubbink HJ et al (2011) MGMT pro-
moter hypermethylation is a frequent, early, and consistent event
in astrocytoma progression, and not correlated with TP53 muta-
tion. J Neurooncol 101:405–417
18. Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G
(2005) Frequent promoter hypermethylation and low expression
of the MGMT gene in oligodendroglial tumors. Int J Cancer
113:379–385
19. Brandes AA, Tosoni A, Cavallo G et al (2006) Correlations
between O6-methylguanine DNA methyltransferase promoter
methylation status, 1p and 19q deletions, and response to tem-
ozolomide in anaplastic and recurrent oligodendroglioma: a
prospective GICNO study. J Clin Oncol 24:4746–4753
20. Levin N, Lavon I, Zelikovitsh B et al (2006) Progressive low-
grade oligodendrogliomas: response to temozolomide and
correlation between genetic profile and O6-methylguanine
DNA methyltransferase protein expression. Cancer 106:1759–
1765
21. Tosoni A, Franceschi E, Ermani M et al (2008) Temozolomide
three weeks on and one week off as first line therapy for patients
with recurrent or progressive low grade gliomas. J Neurooncol
89:179–185
22. Kesari S, Schiff D, Drappatz J et al (2009) Phase II study of
protracted daily temozolomide for low-grade gliomas in adults.
Clin Cancer Res 15:330–337
23. van den Bent MJ, Dubbink HJ, Sanson M et al (2009) MGMT
promoter methylation is prognostic but not predictive for out-
come to adjuvant PCV chemotherapy in anaplastic oligoden-
droglial tumors: a report from EORTC Brain Tumor Group Study
26951. J Clin Oncol 27:5881–5886
24. Wick W, Hartmann C, Engel C et al (2009) NOA-04 randomized
phase III trial of sequential radiochemotherapy of anaplastic
glioma with procarbazine, lomustine, and vincristine or tem-
ozolomide. J Clin Oncol 27:5874–5880
25. Kaloshi G, Benouaich-Amiel A, Diakite F et al (2007) Tem-
ozolomide for low-grade gliomas: predictive impact of 1p/19q
loss on response and outcome. Neurology 68:1831–1836
26. Ochsenbein AF, Schubert AD, Vassella E, Mariani L (2011)
Quantitative analysis of O6-methylguanine DNA methyltrans-
ferase (MGMT) promoter methylation in patients with low-grade
gliomas. J Neurooncol 103:343–351
27. de Robles P, McIntyre J, Kalra S et al (2008) Methylation status
of MGMT gene promoter in meningiomas. Cancer Genet Cyto-
genet 187:25–27
28. Brokinkel B, Fischer BR, Peetz-Dienhart S et al (2010) MGMT
promoter methylation status in anaplastic meningiomas. J Neu-
rooncol 100:489–490
29. Buccoliero AM, Castiglione F, Rossi Degl’Innocenti D et al
(2008) O6-Methylguanine-DNA-methyltransferase in recurring
anaplastic ependymomas: PCR and immunohistochemistry.
J Chemother 20:263–268
30. Faoro D, von Bueren AO, Shalaby T et al (2011) Expression of
O(6)-methylguanine-DNA methyltransferase in childhood
medulloblastoma. J Neurooncol 103:59–69
31. Oh J, Bilbao JM, Tsao MN et al (2009) Recurrent PNET with
MGMT methylation responds to temozolomide. Can J Neurol Sci
36:654–657
32. Louis DN, Ohgaki H, Wiestler OD et al (2007) WHO classifi-
cation of tumors of the central nervous systems, 4th edn. Inter-
national Agency for Research on Cancer (IARC), Lyon
33. Waha A, Rollbrocker B, Wiestler OD, von Deimling A (1996) A
polymerase chain reaction-based assay for the rapid detection of
gene amplification in human tumors. Diagn Mol Pathol
5:147–150
34. Mellai M, Piazzi A, Caldera V et al (2011) IDH1 and IDH2
mutations, immunohistochemistry and associations in a series of
brain tumors. J Neurooncol 105:345–357
35. Weller M, Berger H, Hartmann C et al (2007) Combined 1p/19q
loss in oligodendroglial tumors: predictive or prognostic bio-
marker? Clin Cancer Res 13:6933–6937
36. Caldera V, Mellai M, Annovazzi L, Piazzi A, Cassoni P, Schiffer D
(2011) Antigenic and genotypic similarity between primary glio-
blastomas and their derived neurospheres. J Oncol 2011:314962
37. Ohgaki H, Kleihues P (2007) Genetic pathways to primary and
secondary glioblastoma. Am J Pathol 170:1445–1453
38. van den Bent MJ, Dubbink HJ, Marie Y et al (2010) IDH1 and
IDH2 mutations are prognostic but not predictive for outcome in
anaplastic oligodendroglial tumors: a report of the European
Organization for Research and Treatment of Cancer Brain Tumor
Group. Clin Cancer Res 16:1597–1604
39. Sciuscio D, Diserens AC, van Dommelen K et al (2011) Extent
and patterns of MGMT promoter methylation in glioblastoma-
and respective glioblastoma-derived spheres. Clin Cancer Res
17:255–266
40. Preusser M, Janzer RC, Felsberg J et al (2008) Anti-O6-methyl-
guanine-methyltransferase (MGMT) immunohistochemistry in
glioblastoma multiforme: observer variability and lack of asso-
ciation with patient survival impede its use as clinical biomarker.
Brain Pathol 18:520–532
41. Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G
(2010) Molecular diagnostics of gliomas: state of the art. Acta
Neuropathol 120:567–584
42. Preusser M, Elezi L, Hainfellner JA (2008) Reliability and
reproducibility of PCR-based testing of O6-methylguanine-DNA
methyltransferase gene (MGMT) promoter methylation status in
formalin-fixed and paraffin-embedded neurosurgical biopsy
specimens. Clin Neuropathol 27:388–390
43. Esteller M (2002) CpG island hypermethylation and tumor sup-
pressor genes: a booming present, a brighter future. Oncogene
21:5427–5440
44. Karayan-Tapon L, Quillien V, Guilhot J et al (2010) Prognostic
value of O6-methylguanine-DNA methyltransferase status in
glioblastoma patients, assessed by five different methods. J Neu-
rooncol 97:311–322
45. Grasbon-Frodl EM, Kreth FW, Ruiter M (2007) Intratumoral
homogeneity of MGMT promoter hypermethylation as demon-
strated in serial stereotactic specimens from anaplastic astrocy-
tomas and glioblastomas. Int J Cancer 121:2458–2464
46. Brell M, Tortosa A, Verger E et al (2005) Prognostic significance
of O6-methylguanine-DNA methyltransferase determined by
promoter hypermethylation and immunohistochemical expression
in anaplastic gliomas. Clin Cancer Res 11:5167–5174
47. Stupp R, Dietrich PY, Ostermann Kraljevic S et al (2002)
Promising survival for patients with newly diagnosed glioblas-
toma multiforme treated with concomitant radiation plus tem-
ozolomide followed by adjuvant temozolomide. J Clin Oncol
20:1375–1382
48. Idbaih A, Omuro A, Ducray F, Hoang-Xuan K (2007) Molecular
genetic markers as predictors of response to chemotherapy in
gliomas. Curr Opin Oncol 19:606–611
49. Jeuken JW, Cornelissen SJ, Vriezen M et al (2007) MS-MLPA:
an attractive alternative laboratory assay for robust, reliable, and
J Neurooncol
123

semiquantitative detection of MGMT promoter hypermethylation
in gliomas. Lab Invest 87:1055–1065
50. Brell M, Ibanez J, Tortosa A (2011) O6-Methylguanine-DNA
methyltransferase protein expression by immunohistochemistry
in brain and non-brain systemic tumours: systematic review and
meta-analysis of correlation with methylation-specific polymer-
ase chain reaction. BMC Cancer 11:35
51. Everhard S, Tost J, El Abdalaoui H et al (2009) Identification of
regions correlating MGMT promoter methylation and gene
expression in glioblastomas. Neuro Oncol 11:348–356
52. Capper D, Mittelbronn M, Meyermann R, Schittenhelm J (2008)
Pitfalls in the assessment of MGMT expression and in its cor-
relation with survival in diffuse astrocytomas: proposal of a
feasible immunohistochemical approach. Acta Neuropathol
115:249–259
53. Sanson M, Marie Y, Paris S et al (2009) Isocitrate dehydrogenase
1 codon 132 mutation is an important prognostic biomarker in
gliomas. J Clin Oncol 27:4150–4154
54. Weller M, Felsberg J, Hartmann C et al (2009) Molecular pre-
dictors of progression-free and overall survival in patients with
newly diagnosed glioblastoma: a prospective translational study
of the German Glioma Network. J Clin Oncol 27:5743–5750
55. Idbaih A, Marie Y, Pierron G et al (2005) Two types of chro-
mosome 1p losses with opposite significance in gliomas. Ann
Neurol 58:483–487
56. Vogazianou AP, Chan R, Backlund LM et al (2010) Distinct
patterns of 1p and 19q alterations identify subtypes of human
gliomas that have different prognoses. Neuro Oncol 12:664–678
57. Idbaih A, Kouwenhoven M, Jeuken J et al (2008) Chromosome
1p loss evaluation in anaplastic oligodendrogliomas. Neuropa-
thology 28:440–443
58. Snuderl M, Eichler AF, Ligon KL et al (2009) Polysomy for
chromosomes 1 and 19 predicts earlier recurrence in anaplastic
oligodendrogliomas with concurrent 1p/19q loss. Clin Cancer Res
15:6430–6437
59. Lotfi M, Afsharnezhad S, Raziee HR et al (2011) Immunohisto-
chemical assessment of MGMT expression and p53 mutation in
glioblastoma multiforme. Tumori 97:104–108
60. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1
mutations are early events in the development of astrocytomas
and oligodendrogliomas. Am J Pathol 174:1149–1153
61. Yan H, Bigner DD, Velculescu V, Parsons DW (2009) Mutant
metabolic enzymes are at the origin of gliomas. Cancer Res
69:9157–9159
62. Martinez R, Martin-Subero JI, Rohde V et al (2009) A micro-
array-based DNA methylation study of glioblastoma multiforme.
Epigenetics 4:255–264
63. Noushmehr H, Weisenberger DJ, Diefes K et al (2010) Identifi-
cation of a CpG island methylator phenotype that defines a dis-
tinct subgroup of glioma. Cancer Cell 17:510–522
64. Huse JT, Phillips HS, Brennan CW (2011) Molecular subclassi-
fication of diffuse gliomas: seeing order in the chaos. Glia
59:1190–1199
65. Phillips HS, Kharbanda S, Chen R et al (2006) Molecular sub-
classes of high-grade glioma predict prognosis, delineate a pat-
tern of disease progression, and resemble stages in neurogenesis.
Cancer Cell 9:157–173
66. Laffaire J, Everhard S, Idbaih A et al (2011) Methylation pro-
filing identifies 2 groups of gliomas according to their tumori-
genesis. Neuro Oncol 13:84–98
J Neurooncol
123
Related Documents




![Promoter hypermethylation profiling of distant breast ... · phenotype of distant breast cancer metastases [14–16]. Extensive knowledge of the hypermethylation status of tumor suppressor](https://static.cupdf.com/doc/110x72/5d21f00788c993722e8c67ea/promoter-hypermethylation-profiling-of-distant-breast-phenotype-of-distant.jpg)






