Mesoscale simulation of spherulite growth during polymer crystallization by use of a cellular automaton D. Raabe * Max-Planck-Institut f€ ur Eisenforschung, Max-Planck-Str. 1, D€ usseldorf 40237, Germany Received 10 June 2003; received in revised form 26 January 2004; accepted 11 February 2004 Available online 16 March 2004 Abstract The paper introduces a 3D cellular automaton model for the spatial and crystallographic prediction of spherulite growth phe- nomena in polymers at the mesoscopic scale. The automaton is discrete in time, real space, and orientation space. The kinetics is formulated according to the Hoffman–Lauritzen secondary surface nucleation and growth theory for spherulite expansion. It is used to calculate the switching probability of each grid point as a function of its previous state and the state of the neighboring grid points. The actual switching decision is made by evaluating the local switching probability using a Monte Carlo step. The growth rule is scaled by the ratio of the local and the maximum interface energies, the local and maximum occurring Gibbs free energy of transformation, the local and maximum occurring temperature, and by the spacing of the grid points. The use of experimental input data provides a real time and space scale. Ó 2004 Acta materialia Inc. Published by Elsevier Ltd. All rights reserved. Keywords: Simulation; Texture; Polymer; Crystallization; Spherulite growth; Microstructure; Transformation; Avrami 1. Motivation for introducing a cellular automaton model for spherulite growth 1.1. Basics of cellular microstructure automata Cellular automata are algorithms that describe the spatial and temporal evolution of complex systems by applying local switching rules to the discrete cells of a regular lattice [1]. Each cell is characterized in terms of state variables which assume one out of a finite set of states (such as crystalline or amorphous), but continu- ous variable states are admissible as well (e.g., crystal orientation) [2]. The opening state of an automaton is defined by mapping the initial distribution of the values of the chosen state variables onto the lattice. The dy- namical evolution of the automaton takes place through the synchronous application of switching rules acting on the state of each cell. These rules determine the state of a lattice point as a function of its previous state and the state of the neighboring sites. The number, arrangement, and range of the neighbor sites used by the switching rule for calculating a state switch determines the range of the interaction and the local shape of the areas which evolve. After each discrete time interval the values of the state variables are updated for all lattice points in syn- chrony mapping the new (or unchanged) values assigned to them through the local rules. Owing to these features, cellular automata provide a discrete method of simu- lating the evolution of complex dynamical systems which contain large numbers of similar components on the basis of their local interactions. The basic rational of cellular automata is to try to describe the evolution of complex systems by simulating them on the basis of the elementary dynamics of the interacting constituents following simple generic rules. In other words, the cel- lular automaton approach pursues the goal to let the global complexity of dynamical systems emerge by the repeated interaction of local rules. The cellular automaton method presented in this paper is a tool for predicting microstructure, kinetics, and texture of crystallizing polymers. It is formulated on Acta Materialia 52 (2004) 2653–2664 www.actamat-journals.com * Tel.: +49-211-679-2278; fax: +49-211-679-2333. E-mail address: [email protected] (D. Raabe). 1359-6454/$30.00 Ó 2004 Acta materialia Inc. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.actamat.2004.02.013

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Acta Materialia 52 (2004) 2653–2664
www.actamat-journals.com
Mesoscale simulation of spherulite growth duringpolymer crystallization by use of a cellular automaton
D. Raabe *
Max-Planck-Institut f€ur Eisenforschung, Max-Planck-Str. 1, D€usseldorf 40237, Germany
Received 10 June 2003; received in revised form 26 January 2004; accepted 11 February 2004
Available online 16 March 2004
Abstract
The paper introduces a 3D cellular automaton model for the spatial and crystallographic prediction of spherulite growth phe-
nomena in polymers at the mesoscopic scale. The automaton is discrete in time, real space, and orientation space. The kinetics is
formulated according to the Hoffman–Lauritzen secondary surface nucleation and growth theory for spherulite expansion. It is used
to calculate the switching probability of each grid point as a function of its previous state and the state of the neighboring grid
points. The actual switching decision is made by evaluating the local switching probability using a Monte Carlo step. The growth
rule is scaled by the ratio of the local and the maximum interface energies, the local and maximum occurring Gibbs free energy of
transformation, the local and maximum occurring temperature, and by the spacing of the grid points. The use of experimental input
data provides a real time and space scale.
� 2004 Acta materialia Inc. Published by Elsevier Ltd. All rights reserved.
Keywords: Simulation; Texture; Polymer; Crystallization; Spherulite growth; Microstructure; Transformation; Avrami
1. Motivation for introducing a cellular automaton model
for spherulite growth
1.1. Basics of cellular microstructure automata
Cellular automata are algorithms that describe the
spatial and temporal evolution of complex systems byapplying local switching rules to the discrete cells of a
regular lattice [1]. Each cell is characterized in terms of
state variables which assume one out of a finite set of
states (such as crystalline or amorphous), but continu-
ous variable states are admissible as well (e.g., crystal
orientation) [2]. The opening state of an automaton is
defined by mapping the initial distribution of the values
of the chosen state variables onto the lattice. The dy-namical evolution of the automaton takes place through
the synchronous application of switching rules acting on
the state of each cell. These rules determine the state of a
lattice point as a function of its previous state and the
* Tel.: +49-211-679-2278; fax: +49-211-679-2333.
E-mail address: [email protected] (D. Raabe).
1359-6454/$30.00 � 2004 Acta materialia Inc. Published by Elsevier Ltd. Al
doi:10.1016/j.actamat.2004.02.013
state of the neighboring sites. The number, arrangement,
and range of the neighbor sites used by the switching
rule for calculating a state switch determines the range
of the interaction and the local shape of the areas which
evolve. After each discrete time interval the values of the
state variables are updated for all lattice points in syn-
chrony mapping the new (or unchanged) values assignedto them through the local rules. Owing to these features,
cellular automata provide a discrete method of simu-
lating the evolution of complex dynamical systems
which contain large numbers of similar components on
the basis of their local interactions. The basic rational of
cellular automata is to try to describe the evolution of
complex systems by simulating them on the basis of the
elementary dynamics of the interacting constituentsfollowing simple generic rules. In other words, the cel-
lular automaton approach pursues the goal to let the
global complexity of dynamical systems emerge by the
repeated interaction of local rules.
The cellular automaton method presented in this
paper is a tool for predicting microstructure, kinetics,
and texture of crystallizing polymers. It is formulated on
l rights reserved.

2654 D. Raabe / Acta Materialia 52 (2004) 2653–2664
the basis of a scaled version of the Hoffman–Lauritzen
rate theory for spherulite growth in partially crystalline
polymers and tracks the growth and impingement of
such spheres at the mesoscopic scale on a real time basis.
The major advantage of a discrete kinetic mesoscalemethod for the simulation of polymer crystallization
is that it considers material heterogeneity in terms of
crystallography, energy, and temperature, as opposed to
classical statistical approaches which are based on the
assumption of material homogeneity [2].
1.2. Cellular automaton models in physical metallurgy
Areas where microstructure-based cellular automa-
ton models have been successfully introduced are pri-
marily in the domain of physical metallurgy. Important
examples are static primary recrystallization and re-
covery [2–9], formation of dendritic grain structures in
solidification processes [10–13], as well as related nu-
cleation and coarsening phenomena [14–16]. Overviews
on cellular automata for materials and metallurgicalapplications are given in [2,17].
1.3. Previous models for polymer crystallization
Earlier approaches to the modeling of crystallization
processes in polymers were suggested and discussed in
detail by various groups. For instance Koscher and
Fulchiron [18] studied the influence of shear on poly-propylene crystallization experimentally and in terms of a
kinetic model for crystallization under quiescent condi-
tions. Their approachwas based on a classical topological
Avrami–Johnson–Mehl–Kolmogorov (AJMK) model
for isothermal conditions and on corresponding modifi-
cations such as those of Nakamura et al. [19] and Ozawa
[20] for non-isothermal conditions. The authors found in
part excellent agreement between these predictions andtheir own experimental data. For predicting crystalliza-
tion kinetics under shear conditions the authors used a
AJMK model in conjunction with a formulation for the
number of activated nuclei under the influence of shear.
The latter part of the approach was based on an earlier
study of Eder et al. [21,22] who expressed the number of
activated nuclei as a function of the square of the shear
rate which provides a possibility to predict the thicknessof thread-like precursors. A related approach was pub-
lished byHieber [23] who used theNakamura equation to
establish a direct relation between the Avrami and the
Ozawa crystallization rate constants.
AJMK-based transformation models have been ap-
plied to polymers with great success to cases where their
underlying assumptions of material homogeneity are
reasonably fulfilled [18–23]. It is, however, likely thatfurther progress in understanding and tailoring polymer
microstructures can be made by the introduction of
automaton models which are designed to cope with
more realistic boundary conditions by taking material
heterogeneity into account [24]. Important cases where
the application of cellular automata is conceivable in the
field of polymer crystallization are heterogeneous
spherulite formation and growth; heterogeneous topol-ogies and size distributions of spherulites; topological
and crystallographic effects arising from additives for
heterogeneous nucleation; lateral heterogeneity of the
crystallographic texture; overall crystallization kinetics
under complicated boundary conditions; reduction of
the spherulite size; randomization of the crystallo-
graphic texture; as well as effects arising from impurities
and temperature gradients.
2. Basic design of the cellular automaton model and
treatment of the crystallographic texture
The model for the mesoscale prediction of spherulite
growth in polymers is designed as a 3D cellular au-
tomaton model with a probabilistic switching rule. It isdiscrete in time, real space, and crystallographic orien-
tation space. It is defined on a cubic lattice considering
the three nearest neighbor shells for the calculation of
cell switches. The kinetics is formulated on the basis of
the Hoffman–Lauritzen rate theory for spherulite
growth. It is used to calculate the switching probability
of each grid point as a function of its previous state and
the state of the neighboring grid points. The actual de-cision about a switching event is made by evaluating the
local switching probability using a Monte Carlo step.
The growth rule is scaled by the ratio of the local and
the maximum possible interface properties, the local and
maximum occurring Gibbs free energy of transforma-
tion, the local and maximum occurring temperature,
and by the spacing of the grid points. The use of ex-
perimental input data allows one to make predictions ona real time and space scale. The transformation rule is
scalable to any mesh size and to any spectrum of in-
terface and transformation data. The state update of all
grid points is made in synchrony.
Independent variables of the automaton are time tand space x ¼ ðx1; x2; x3Þ. Space is discretized into an
array of cubic cells. The state of each cell is character-
ized in terms of the dependent field variables. These arethe temperature field, the phase state (amorphous matrix
or partially crystalline spherulite), the transformation
and interface energies, the crystallographic orientation
distribution of the crystalline nuclei f ðhÞ ¼ f ðhÞðu1;/;u2Þ, and the crystallographic orientation distri-
bution f ðgÞ ¼ f ðgÞðu1;/;u2Þ of one spherulite, where hand g are orientation matrices and u1;/;u2 are Euler
angles. The introduction of two variable fields for thetexture, h and g, deserves a more detailed explanation:
When analyzing crystallographic textures of partially
crystalline polymers it is useful to distinguish between

D. Raabe / Acta Materialia 52 (2004) 2653–2664 2655
different orientational scales. These are defined, first, by
the sample coordinate system; second, by the coordinate
system of the crystalline nuclei (where the incipient
crystal orientation is created by the direction of the
alignment of the first lamellae in case of homogeneousnucleation or by epitaxy effects in case of heterogeneous
nucleation); third, by the intrinsic helix angle of the
single lamellae according to the chain conformation;
and fourth, by the complete orientation distribution
function of one partially crystalline spherulite (Figs. 1
and 2). The latter orientation distribution function is
characterized by the orientational degrees of freedom
provided by the lamella helix angle as well as by orien-tational branching creating a complete orientation dis-
Fig. 1. Reference systems for polymer textures (polyethylene as an example
defined by normal, transverse, and longitudinal direction of the macroscopi
defined by the crystallographic orientation of the primary nucleus or by the
cleating agent. Finally, owing to the extensive branching of their internal c
entation but an orientation distribution. Further rotational degrees of fr
single lamella scale due to the molecular conformation of the constituent m
polyethylene).
Fig. 2. Structure and unit cell o
tribution in one spherulite which is described by
f ðgÞ ¼ f ðgÞðu1;/;u2Þ relative to its original nucleus of
orientation h ¼ hðu1;/;u2Þ. The sample coordinate
system is often of orthotropic symmetry as defined by
normal, transverse, and longitudinal direction. The ref-erence system of the spherulite can be defined by the
crystallographic orientation of the primary nucleus or
by the coordinate system inherited by way of epitaxy
from an external nucleus in case of heterogeneous nu-
cleation. The orientation of such a nucleus is described
by the rotation matrix h ¼ hðu1;/;u2Þ as a spin relative
to the sample coordinate system. The orientation
distribution of all nuclei amounts to f ðhÞ ¼ f ðhÞðu1;/;u2Þ. When assuming that all spherulites branch
). The sample coordinate system is often of orthotropic symmetry as
c processing procedure. The reference system of the spherulite can be
coordinate system inherited by way of epitaxy from an external nu-
rystalline lamellae spherulites cannot be characterized by a single ori-
eedom can add to this complicated crystallographic texture at the
onomer and the resulting helix angle of the chains (as in the case of
f crystalline polyethylene.

2656 D. Raabe / Acta Materialia 52 (2004) 2653–2664
in a similar fashion, leading to a single-spherulite tex-
ture, f ðgÞ ¼ f ðgÞðu1;/;u2Þ, the total orientation
distribution of a bulk polymer can then be calculated
according to f ðhÞ � f ðgÞ.The current simulation takes a mesoscopic view at
this multi-scale problem, i.e., it does not provide a ge-
neric simulation of the texture evolution of a single
spherulite but includes only the orientation distribution
f ðhÞ ¼ f ðhÞðu1;/;u2Þ of the texture of the nuclei.
The driving force is the Gibbs free energy Gt per swit-
ched cell associated with the transformation. The starting
data, i.e., the mapping of the amorphous state, the tem-
perature field, and the spatial distribution of the Gibbsfree energy, must be provided by experiment or theory.
The kinetics of the automaton result from changes in
the state of the cells which are hereafter referred to as
cell switches. They occur in accord with a switching rule
which determines the individual switching probability of
each cell as a function of its previous state and the state
of its neighbor cells. The switching rule used in the
simulations discussed below is designed for the simula-tion of static crystallization of a quiescent supercooled
amorphous matrix. It reflects that the state of a non-
crystallized cell belonging to an amorphous region may
change due to the expansion of a crystallizing neighbor
spherulite which grows according to the local tempera-
ture, Gibbs free energy associated with that transfor-
mation, and interface properties. If such an expanding
spherulite sweeps a non-crystallized cell, the energy ofthat cell changes and a new orientation distribution is
assigned to it, namely that of the growing neighbor
spherulite.
3. Basic rate equation for spherulitic growth
In 1957 Keller [25] reported that polyethylene formedchainfolded lamellar crystals from solution. This dis-
covery was followed by the confirmation of the gener-
ality of this morphology, namely, that lamellar crystals
form upon crystallization, both from the solution and
from the melt [26] for a wide variety of polymers.
Most theories of polymer crystallization go back to
the work of Turnbull and Fisher [27] in which the rate of
nucleation is formulated in terms of the product of twoBoltzmann expressions, one quantifying the mobility of
the growth interface in terms of the activation energy for
motion, the other one involving the energy for second-
ary nucleation on existing crystalline lamellae. On the
basis of this pioneering work Hoffman and Lauritzen
developed a more detailed rate equation for spherulite
growth under consideration of secondary surface nu-
cleation [28]. Overviews on spherulite growth kineticswere given by Keller [29], Hoffman et al. [30], Snyder
and Marand [31,32], Hoffman and Miller [33], and Long
et al. [34].
The rate equation by Hoffman and Lauritzen is used
as a basis for the formulation of the switching rule of the
cellular automaton model. It describes growth interface
motion in terms of lateral and forward molecule align-
ment and disalignment processes at a homogeneousplanar portion of an interface segment between a semi-
crystalline spherulite and the amorphous matrix mate-
rial. It is important to note that the Hoffman and
Lauritzen rate theory is a coarse-grained formulation
which homogenizes over two rather independent sets of
very anisotropic mechanisms, namely, secondary nu-
cleation on the lateral surface of the growing lamellae
(creating strong lateral, i.e., out-of-plane volume ex-pansion and new texture components) and lamella
growth (creating essentially two-dimensional, i.e., in-
plane expansion and texture continuation). Although
the two basic ingredients of this rate formulation are
individually of a highly anisotropic character the overall
spherulite equation is typically used in an isotropic
form. The basic rate equation is
_x ¼ _xp exp
�� Q�
RðT � T1Þ
�exp
�� Kg
TDT
�; ð1Þ
where _x is the velocity vector of the interface between
the spherulite and the supercooled amorphous matrix,_xp is the pre-exponential velocity vector, DT the super-
cooling defined by DT ¼ T 0m � T , where T 0
m is the equi-librium melting point (valid for a very large crystal
formed from fully extended chains, no effect of free
surfaces), Q� is the activation energy for viscous mole-
cule flow or, respectively, attachment of the chain to the
crystalline surface, T1 is the temperature below which
all viscous flow stops (glass transition temperature,
temperature at which the viscosity exceeds the value of
1013 Ns/m2), Kg the secondary nucleation exponentand T the absolute temperature of the (computer)
experiment.
According to Hoffman and Lauritzen [28,30,33], the
exponent Kg amounts to
Kg ¼nbrreT 0
m
kBDGf
; ð2Þ
where kB is the Boltzmann constant, n a constant which
equals 4 for growth regimes I and III and 2 for growth
regime II [33,35], b is the thickness of a crystalline lattice
cell in growth direction (see Figs. 1 and 2), r is the free
energy per area for the interface between the lateral
surface and the supercooled melt, re is the free energyper area for the interface between the fold surface where
the molecule chains fold back or emerge from the la-
mella and the supercooled melt, and DGf is the Gibbs
free energy of fusion at the crystallization temperature
which is approximated by DGf � ðDHfDT Þ=Tm [28,
30,33].
The different growth regimes, I (shallow quench
regime), II (deep quench regime), and III (very deep

D. Raabe / Acta Materialia 52 (2004) 2653–2664 2657
quench regime), were discussed by Hoffman and Miller
[33] in the sense that within regime I of spherulite
growth the expansion of the spherulite is dominated by
weak secondary nucleation and preferred forward la-
mellae growth. Nucleation is in this regime dominatedby the formation of few single surface nuclei which are
referred to as stems. It is assumed that when one stem is
nucleated the entire new layer is almost instantaneously
completed relative to the nucleation rate. In regime II
the two rates are comparable. Regime II is the normal
condition for spherulitic growth. It has a weaker DTdependence than regime I. In regime III nucleation of
many nuclei occurs leading to disordered crystal growth.Nucleation is in this regime more important than
growth.
According to the work of Hoffman and Miller [33] the
pre-exponential velocity vector for an interface between
a spherulite and the supercooled amorphous matrix can
be modeled according to
_xp ¼ nN0bJ‘u
kBTbr
�� kBT2brþ abDGf
�; ð3Þ
where N0 is the number of initial stems, soon to be in-
volved in the first stem deposition process, n the unit
normal vector of the respective interface segment, and ‘uthe monomer length. J amounts to
J ¼ 1nz
kBTh
� �; ð4Þ
where 1 is a constant for the molecular friction experi-
enced by the chain as it is reeled onto the growth in-
terface, nz the number of molecular repeat units in the
chain, and h the Planck constant. The fraction kBT=hcan be interpreted as a frequency pre-factor. The pre-
exponential velocity vector can then be written
_xp ¼ nN0b1‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�ð5Þ
yielding a Hoffman–Lauritzen–Miller type version [33]of a velocity-rate vector equation for spherulite growth
under consideration of secondary nucleation
_x ¼ nN0b1‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�
� exp
�� Q�
R T � T1ð Þ
�exp
�� nbrreT 0
m
kBTDTDGf
�: ð6Þ
4. Using rate theory as a basis for the automaton model
4.1. Mapping the rate equation on a cellular automaton
mesh
For dealing with competing state changes affectingthe same lattice cell in a cellular automaton, the statis-
tical rate equation can be rendered into a probabilistic
analogue which allows one to calculate switching
probabilities for the states of the cellular automaton
cells [2]. For this purpose, the rate equation is separated
into a non-Boltzmann part, _x0, which depends com-
paratively weakly on temperature, and a Boltzmannpart, w, which depends exponentially on temperature
_x ¼ _x0w
¼ nN0b1‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�
� exp
�� Q�
R T � T1ð Þ
�exp
�� nbrreT 0
m
kBTDTDGf
�
with _x0 ¼ nN0b1‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�
and w ¼ exp
�� Q�
RðT � T1Þ
�exp
�� nbrreT 0
m
kBTDTDGf
�:
ð7Þ
The Boltzmann factors, w, represent the probability for
cell switches. According to this equation non-vanishing
switching probabilities occur for cells with different
temperatures and/or transformation energies. The au-
tomaton considers the first, second (2D), and third (3D)
neighbor shell for the calculation of the switchingprobability acting on a cell. The local value of the
switching probability may in principal depend on the
crystallographic character of the interfaces entering via
the interface energies in the secondary nucleation term.
The current simulation does, however, not account for
this potential source of anisotropy but assumes constant
values for the interface energies of the lateral and of the
fold interface.
4.2. The scaled and normalized switching probability
The cellular automaton is usually applied to starting
data which have a spatial resolution far above the
atomic scale. This means that the automaton grid may
have some mesh size km � b. If a moving boundary
segment sweeps a cell, the spherulite thus grows (orshrinks) by k3m rather than b3. Since the net velocity of
an interface segment (between a partially crystalline
spherulite and the amorphous matrix) must be inde-
pendent of the imposed value of km, an increase of the
jump width must lead to a corresponding decrease of the
grid attack frequency, i.e. to an increase of the charac-
teristic time step, and vice versa. For obtaining a scale-
independent interface velocity, the grid frequency mustbe chosen in a way to ensure that the attempted switch
of a cell of length km occurs with a frequency much
below the atomic attack frequency which attempts to
switch a cell of length b. Mapping the rate equation on
such an imposed grid which is characterized by an ex-
ternal scaling length (lattice parameter of the mesh) kmleads to the equation

2658 D. Raabe / Acta Materialia 52 (2004) 2653–2664
_x ¼ _x0w ¼ nðkmmÞw
with m ¼ N0b1km‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�; ð8Þ
where m is the eigenfrequency of the chosen mesh char-
acterized by the scaling length km.The eigenfrequency given by this equation represents
the attack frequency which is valid for one particular
interface with constant self-reproducing properties
moving in a constant temperature field. In order to in-
troduce the possibility of a whole spectrum of interfaceproperties (due to temperature fields and intrinsic in-
terface energy anisotropy) in one simulation it is nec-
essary to normalize this equation by a general grid
attack frequency m0 which is common to all interfaces in
the system rendering the above equation into
_x ¼ _x0w ¼ nkmm0mm0
� �w ¼ _x0
mm0
� �w ¼ _x0w; ð9Þ
where the normalized switching probability amounts
to
w ¼ mm0
� �exp
�� Q�
RðT � T1Þ
�exp
�� nbrreT 0
m
kBTDTDGf
�
¼ N0b1m0km‘unz
kBTh
� �kBTbr
�� kBT2brþ abDGf
�
� exp
�� Q�
RðT � T1Þ
�exp
�� nbrreT 0
m
kBTDTDGf
�: ð10Þ
The value of the normalization or grid attack frequency
m0 can be identified by using the plausible assumption
that the maximum occurring switching probability
cannot assume a state above one
wmax ¼ N0b1m0km‘unz
kBTmax
h
� �kBTmax
brmin
�� kBTmax
2brmin þ abDGf
�
� exp
�� Q�
R Tmax � T1ð Þ
�
� exp
�� nbrminre;minT 0
m
kBTmaxDTmaxDGf;max
�6
!
1; ð11Þ
where Tmax is the maximum occurring temperature in the
system, DTmax the maximum occurring supercooling,rmin the minimum occurring lateral interface energy (for
instance in cases where a crystallographic orientation
dependence of the interface energy exists), DGf;max the
maximum Gibbs free transformation energy which de-
pends on the local temperature, i.e. DGf ;max �ðDHDTmaxÞ=T 0
m, and re;min the minimum occurring fold
interface energy.
With wmax ¼ 1 in the above equation one obtains thenormalization frequency, mmin
0 , as a function of the upper
bound input data.
mmin0 ¼ N0b1
km‘unz
kBTmax
h
� �kBTmax
brmin
�� kBTmax
2brmin þ abDGf
�
� exp
�� Q�
R Tmax � T1ð Þ
�
� exp
�� nbrminre;minT 0
m
kBTmaxDTmaxDGf ;max
�: ð12Þ
This frequency must only be calculated once per simu-
lation. It normalizes all other switching processes.
Inserting this basic attack frequency of the grid into
Eq. (10) yields an expression for calculating the local
values of the switching probability as a function of
temperature and energy
wlocal¼ TlocalTmax
� � kBTlocalbrlocal
� kBTlocal2brlocalþabDGf ;local
h ikBTmax
brmin� kBTmax
2brminþabDGf ;max
h i�exp
�� Q�
R
� �1
Tlocal�T1ð Þ
��� 1
Tmax�T1ð Þ
���
�exp
�� nbT 0
m
kB
� �rlocalre;local
TlocalDTlocalDGf;local
��
� rminre;min
TmaxDTmaxDGf;max
���: ð13Þ
This expression is the central switching equation of the
algorithm. The cellular automaton generally works in
such a way that an existing (expanding) spherulite in-
fects its neighbor cells at a rate or respectively proba-
bility which is determined by Eq. (13). This means that
wherever a partially crystalline spherulite has the topo-
logical possibility to expand into an amorphous neigh-bor cell the automaton rule uses Eq. (13) to quantify the
probability of that cell switch using the local state var-
iable data which characterize the two neighboring cells.
Eq. (13) reveals that local switching probabilities on
the basis of rate theory can be quantified in terms of
the ratio of the local and the maximum temperature and
the corresponding ratio of the interface properties. The
probability of the fastest occurring interface segment torealize a cell switch is exactly equal to 1. The above
equation also shows that the mesh size of the automaton
does not influence the switching probability but only the
time step elapsing during an attempted switch. The
characteristic time constant of the simulation Dt is
1=mmin0 .
In this context, the local switching probability can
also be regarded as the ratio of the distances that can beswept by the local interface and the interface with
maximum velocity, or as the number of time steps the
local interface needs to wait before crossing the en-
countered neighbor cell [6]. This reformulates the same
underlying problem, namely, that interfaces with dif-
ferent velocities cannot switch the state of the automa-
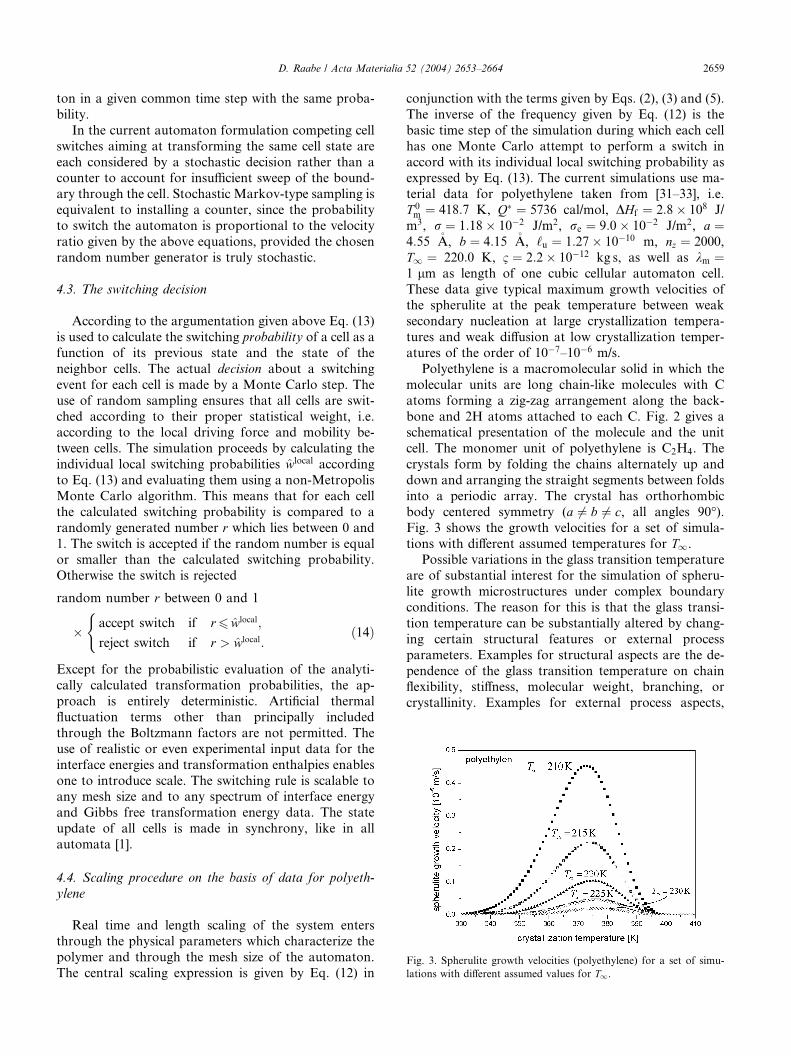
Fig. 3. Spherulite growth velocities (polyethylene) for a set of simu-
lations with different assumed values for T1.
D. Raabe / Acta Materialia 52 (2004) 2653–2664 2659
ton in a given common time step with the same proba-
bility.
In the current automaton formulation competing cell
switches aiming at transforming the same cell state are
each considered by a stochastic decision rather than acounter to account for insufficient sweep of the bound-
ary through the cell. Stochastic Markov-type sampling is
equivalent to installing a counter, since the probability
to switch the automaton is proportional to the velocity
ratio given by the above equations, provided the chosen
random number generator is truly stochastic.
4.3. The switching decision
According to the argumentation given above Eq. (13)
is used to calculate the switching probability of a cell as a
function of its previous state and the state of the
neighbor cells. The actual decision about a switching
event for each cell is made by a Monte Carlo step. The
use of random sampling ensures that all cells are swit-
ched according to their proper statistical weight, i.e.according to the local driving force and mobility be-
tween cells. The simulation proceeds by calculating the
individual local switching probabilities wlocal according
to Eq. (13) and evaluating them using a non-Metropolis
Monte Carlo algorithm. This means that for each cell
the calculated switching probability is compared to a
randomly generated number r which lies between 0 and
1. The switch is accepted if the random number is equalor smaller than the calculated switching probability.
Otherwise the switch is rejected
random number r between 0 and 1
� accept switch if r6 wlocal;
reject switch if r > wlocal:
(ð14Þ
Except for the probabilistic evaluation of the analyti-
cally calculated transformation probabilities, the ap-
proach is entirely deterministic. Artificial thermal
fluctuation terms other than principally included
through the Boltzmann factors are not permitted. The
use of realistic or even experimental input data for the
interface energies and transformation enthalpies enables
one to introduce scale. The switching rule is scalable toany mesh size and to any spectrum of interface energy
and Gibbs free transformation energy data. The state
update of all cells is made in synchrony, like in all
automata [1].
4.4. Scaling procedure on the basis of data for polyeth-
ylene
Real time and length scaling of the system enters
through the physical parameters which characterize the
polymer and through the mesh size of the automaton.
The central scaling expression is given by Eq. (12) in
conjunction with the terms given by Eqs. (2), (3) and (5).
The inverse of the frequency given by Eq. (12) is the
basic time step of the simulation during which each cell
has one Monte Carlo attempt to perform a switch in
accord with its individual local switching probability asexpressed by Eq. (13). The current simulations use ma-
terial data for polyethylene taken from [31–33], i.e.
T 0m ¼ 418:7 K, Q� ¼ 5736 cal/mol, DHf ¼ 2:8� 108 J/
m3, r ¼ 1:18� 10�2 J/m2, re ¼ 9:0� 10�2 J/m2, a ¼4:55 �A, b ¼ 4:15 �A, ‘u ¼ 1:27� 10�10 m, nz ¼ 2000,
T1 ¼ 220:0 K, 1 ¼ 2:2� 10�12 kg s, as well as km ¼1 lm as length of one cubic cellular automaton cell.
These data give typical maximum growth velocities ofthe spherulite at the peak temperature between weak
secondary nucleation at large crystallization tempera-
tures and weak diffusion at low crystallization temper-
atures of the order of 10�7–10�6 m/s.
Polyethylene is a macromolecular solid in which the
molecular units are long chain-like molecules with C
atoms forming a zig-zag arrangement along the back-
bone and 2H atoms attached to each C. Fig. 2 gives aschematical presentation of the molecule and the unit
cell. The monomer unit of polyethylene is C2H4. The
crystals form by folding the chains alternately up and
down and arranging the straight segments between folds
into a periodic array. The crystal has orthorhombic
body centered symmetry (a 6¼ b 6¼ c, all angles 90�).Fig. 3 shows the growth velocities for a set of simula-
tions with different assumed temperatures for T1.Possible variations in the glass transition temperature
are of substantial interest for the simulation of spheru-
lite growth microstructures under complex boundary
conditions. The reason for this is that the glass transi-
tion temperature can be substantially altered by chang-
ing certain structural features or external process
parameters. Examples for structural aspects are the de-
pendence of the glass transition temperature on chainflexibility, stiffness, molecular weight, branching, or
crystallinity. Examples for external process aspects,

2660 D. Raabe / Acta Materialia 52 (2004) 2653–2664
which in part drastically influence the aforementioned
structural state of the material, are accumulated elastic–
plastic deformation, externally imposed shear rates, or
the hydrostatic pressure.
4.5. Nucleation criteria
Two phenomenological approaches were used in the
simulations to treat nucleation, namely site-saturated
heterogeneous nucleation and constant homogeneous
nucleation. The calculations with site-saturated nucle-
ation condition were based on the assumption of ex-
ternal heterogeneous nucleation sites such as provided att ¼ 0 s by small impurities or nucleating agent particles.
The calculations with constant thermal nucleation were
conducted using the nucleation rate equation of Hoff-
man et al. [28,30].
5. Simulation results and discussion
5.1. Kinetics and spherulite structure for site-saturated
conditions
This section presents some 3D simulation results for
spherulite growth under the assumption of site-saturated
nucleation conditions using a cellular automaton with
10 million lattice cells. These standard conditions are
chosen to study the reliability of the new method withrespect to the kinetic exponents (comparison with the
analytical Avrami–Johnson–Mehl–Kolmogorov solu-
tion), to lattice effects (spatial discreteness), to topology,
and to statistics.
Figs. 4 and 5 show the kinetics for simulations at 375
and 360 K, respectively. The calculations were con-
ducted with 0.05%, 0.01%, 0.005%, and 0.001% of all
cells as initial nucleation sites (site saturated). The fig-ures show the volume fractions occupied by spherulites
as a function of the isothermal heat treatment time. The
resulting topology of the spherulites is given in Fig. 6. It
Fig. 4. Avrami analysis of the volume fraction occupied by spherulites (fs) as375 K; 107 cells.
is important to note in this context that the depicted
spherulite volume fraction must not be confused with
the crystalline volume fraction, since the spherulites are
two-phase aggregates consisting of heavily branched
crystalline lamellae with amorphous chains betweenthem (Fig. 1). The kinetic anisotropy which is princi-
pally inherent in such structures at the nanoscopic scale
in terms of secondary nucleation events on the lateral
lamellae surfaces and of in-plane lamella growth [36,37]
is homogenized at the mesoscopic scale, where the
growing spherulites typically behave as isotropic
spheres, Eq. (1). In the present study this results in
Avrami-type kinetics with an exponent of 3.0� 0.1%.The small observed deviations of �0.1% from the ideal
kinetic exponent of 3.0 occur at small and at large times
due to the influence of the discreteness of the cubic au-
tomaton lattice at these early and late growth stages,
respectively. The scatter in the data for the spherulite
volumes at large times (particularly above 99% spheru-
lite volume) is even more important than that for short
times (see Figs. 4(b) and 5(b)). This effect is essentiallydue to the fact that the transformation from the amor-
phous to the spherulite state performed by the last cell
switches becomes increasingly discrete owing to the
changing ratio between the final non-transformed vol-
ume and the switched cells. The relevance of the data
predicted for the final stages of the transformation is,
therefore, somewhat overemphazised and should be
treated with some skepticism for instance when analyz-ing kinetical exponents at the end of the transformation.
It should also be mentioned that three simulation
runs were conducted for each set of starting conditions
in order to inspect statistical effects arising from the
Monte Carlo integration scheme, see Section 4.3 and
Eq. (14). Figs. 4 and 5 reveal that the statistical fluctu-
ations arising from this part of the simulation procedure
are very small (note the similarity of three curves on topof each other in both sets of figures).
The simulated spherulite growth kinetics are in good
qualitative accord with experimental data from the
a function of time; site-saturated nucleation conditions; polyethylene;
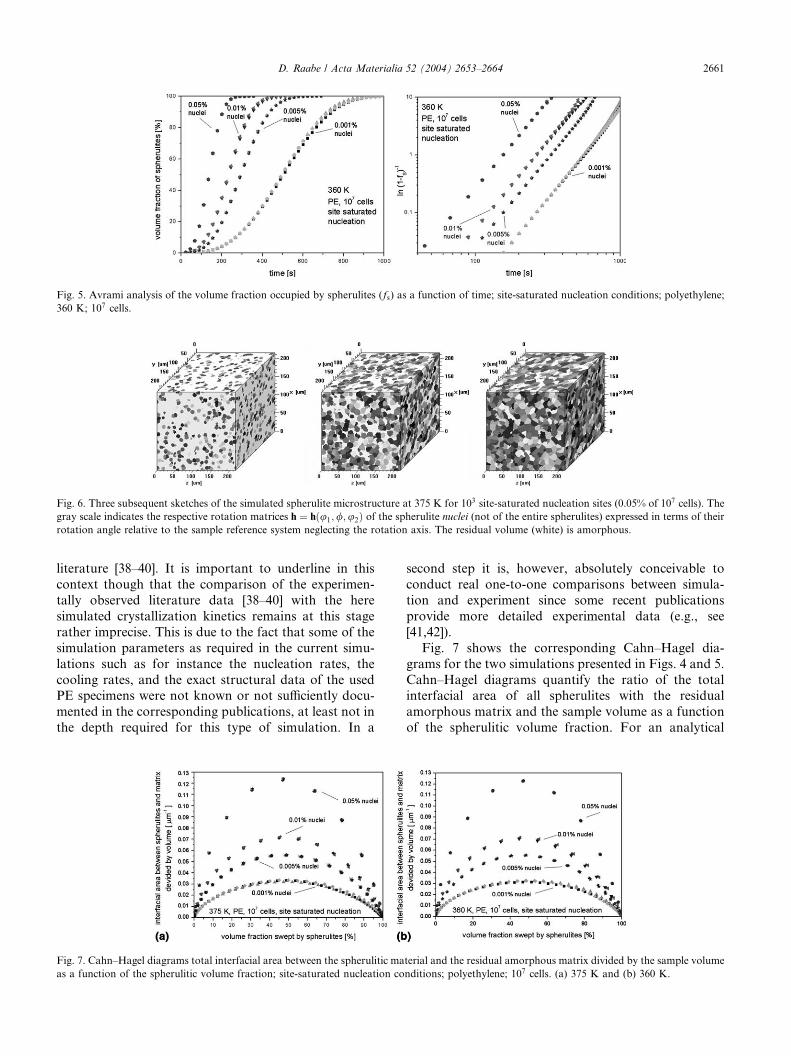
Fig. 5. Avrami analysis of the volume fraction occupied by spherulites (fs) as a function of time; site-saturated nucleation conditions; polyethylene;
360 K; 107 cells.
Fig. 6. Three subsequent sketches of the simulated spherulite microstructure at 375 K for 103 site-saturated nucleation sites (0.05% of 107 cells). The
gray scale indicates the respective rotation matrices h ¼ hðu1;/;u2Þ of the spherulite nuclei (not of the entire spherulites) expressed in terms of their
rotation angle relative to the sample reference system neglecting the rotation axis. The residual volume (white) is amorphous.
D. Raabe / Acta Materialia 52 (2004) 2653–2664 2661
literature [38–40]. It is important to underline in this
context though that the comparison of the experimen-
tally observed literature data [38–40] with the here
simulated crystallization kinetics remains at this stage
rather imprecise. This is due to the fact that some of the
simulation parameters as required in the current simu-
lations such as for instance the nucleation rates, the
cooling rates, and the exact structural data of the usedPE specimens were not known or not sufficiently docu-
mented in the corresponding publications, at least not in
the depth required for this type of simulation. In a
Fig. 7. Cahn–Hagel diagrams total interfacial area between the spherulitic ma
as a function of the spherulitic volume fraction; site-saturated nucleation co
second step it is, however, absolutely conceivable to
conduct real one-to-one comparisons between simula-
tion and experiment since some recent publications
provide more detailed experimental data (e.g., see
[41,42]).
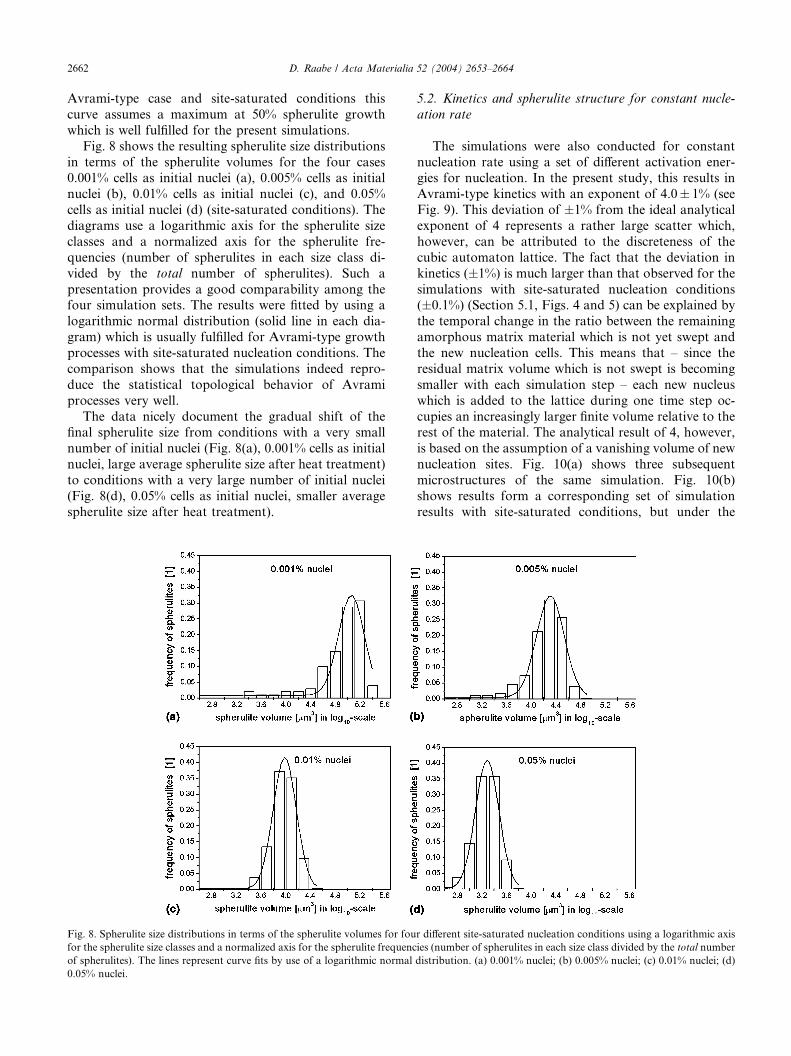
Fig. 7 shows the corresponding Cahn–Hagel dia-
grams for the two simulations presented in Figs. 4 and 5.
Cahn–Hagel diagrams quantify the ratio of the totalinterfacial area of all spherulites with the residual
amorphous matrix and the sample volume as a function
of the spherulitic volume fraction. For an analytical
terial and the residual amorphous matrix divided by the sample volume
nditions; polyethylene; 107 cells. (a) 375 K and (b) 360 K.
2662 D. Raabe / Acta Materialia 52 (2004) 2653–2664
Avrami-type case and site-saturated conditions this
curve assumes a maximum at 50% spherulite growth
which is well fulfilled for the present simulations.
Fig. 8 shows the resulting spherulite size distributions
in terms of the spherulite volumes for the four cases0.001% cells as initial nuclei (a), 0.005% cells as initial
nuclei (b), 0.01% cells as initial nuclei (c), and 0.05%
cells as initial nuclei (d) (site-saturated conditions). The
diagrams use a logarithmic axis for the spherulite size
classes and a normalized axis for the spherulite fre-
quencies (number of spherulites in each size class di-
vided by the total number of spherulites). Such a
presentation provides a good comparability among thefour simulation sets. The results were fitted by using a
logarithmic normal distribution (solid line in each dia-
gram) which is usually fulfilled for Avrami-type growth
processes with site-saturated nucleation conditions. The
comparison shows that the simulations indeed repro-
duce the statistical topological behavior of Avrami
processes very well.
The data nicely document the gradual shift of thefinal spherulite size from conditions with a very small
number of initial nuclei (Fig. 8(a), 0.001% cells as initial
nuclei, large average spherulite size after heat treatment)
to conditions with a very large number of initial nuclei
(Fig. 8(d), 0.05% cells as initial nuclei, smaller average
spherulite size after heat treatment).
Fig. 8. Spherulite size distributions in terms of the spherulite volumes for fou
for the spherulite size classes and a normalized axis for the spherulite frequenc
of spherulites). The lines represent curve fits by use of a logarithmic normal
0.05% nuclei.
5.2. Kinetics and spherulite structure for constant nucle-
ation rate
The simulations were also conducted for constant
nucleation rate using a set of different activation ener-gies for nucleation. In the present study, this results in
Avrami-type kinetics with an exponent of 4.0� 1% (see
Fig. 9). This deviation of �1% from the ideal analytical
exponent of 4 represents a rather large scatter which,
however, can be attributed to the discreteness of the
cubic automaton lattice. The fact that the deviation in
kinetics (�1%) is much larger than that observed for the
simulations with site-saturated nucleation conditions(�0.1%) (Section 5.1, Figs. 4 and 5) can be explained by
the temporal change in the ratio between the remaining
amorphous matrix material which is not yet swept and
the new nucleation cells. This means that – since the
residual matrix volume which is not swept is becoming
smaller with each simulation step – each new nucleus
which is added to the lattice during one time step oc-
cupies an increasingly larger finite volume relative to therest of the material. The analytical result of 4, however,
is based on the assumption of a vanishing volume of new
nucleation sites. Fig. 10(a) shows three subsequent
microstructures of the same simulation. Fig. 10(b)
shows results form a corresponding set of simulation
results with site-saturated conditions, but under the
r different site-saturated nucleation conditions using a logarithmic axis
ies (number of spherulites in each size class divided by the total number
distribution. (a) 0.001% nuclei; (b) 0.005% nuclei; (c) 0.01% nuclei; (d)
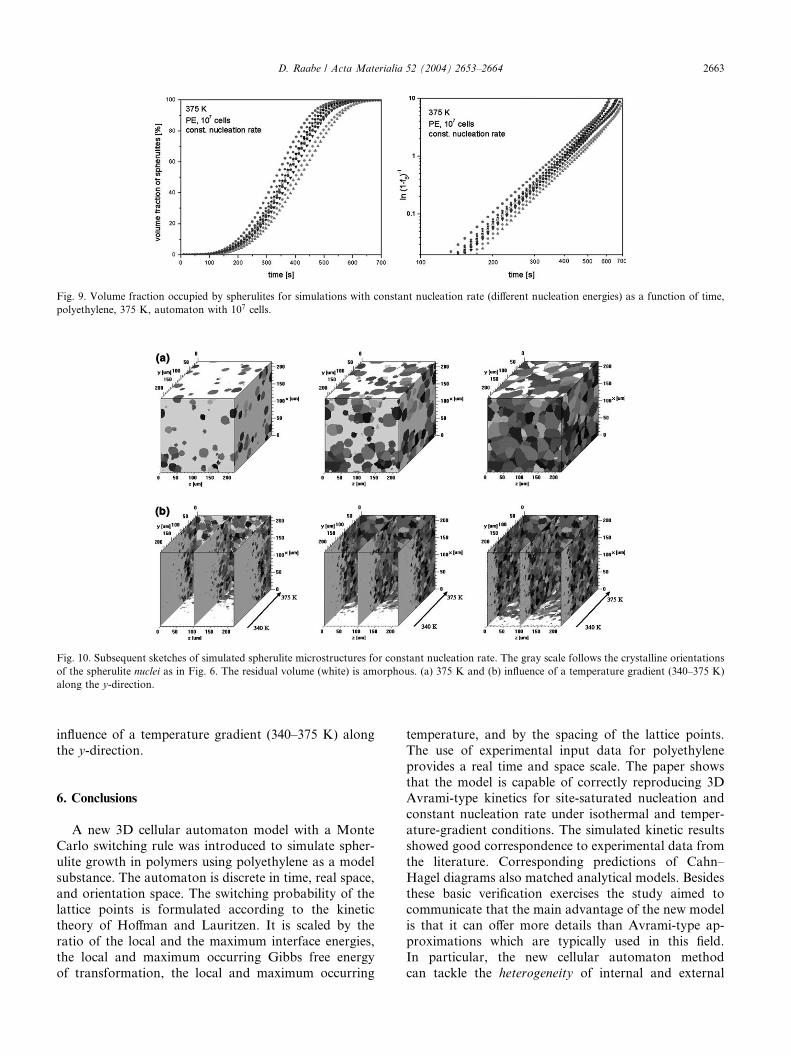
Fig. 9. Volume fraction occupied by spherulites for simulations with constant nucleation rate (different nucleation energies) as a function of time,
polyethylene, 375 K, automaton with 107 cells.
Fig. 10. Subsequent sketches of simulated spherulite microstructures for constant nucleation rate. The gray scale follows the crystalline orientations
of the spherulite nuclei as in Fig. 6. The residual volume (white) is amorphous. (a) 375 K and (b) influence of a temperature gradient (340–375 K)
along the y-direction.
D. Raabe / Acta Materialia 52 (2004) 2653–2664 2663
influence of a temperature gradient (340–375 K) along
the y-direction.
6. Conclusions
A new 3D cellular automaton model with a Monte
Carlo switching rule was introduced to simulate spher-
ulite growth in polymers using polyethylene as a model
substance. The automaton is discrete in time, real space,
and orientation space. The switching probability of thelattice points is formulated according to the kinetic
theory of Hoffman and Lauritzen. It is scaled by the
ratio of the local and the maximum interface energies,
the local and maximum occurring Gibbs free energy
of transformation, the local and maximum occurring
temperature, and by the spacing of the lattice points.
The use of experimental input data for polyethylene
provides a real time and space scale. The paper showsthat the model is capable of correctly reproducing 3D
Avrami-type kinetics for site-saturated nucleation and
constant nucleation rate under isothermal and temper-
ature-gradient conditions. The simulated kinetic results
showed good correspondence to experimental data from
the literature. Corresponding predictions of Cahn–
Hagel diagrams also matched analytical models. Besides
these basic verification exercises the study aimed tocommunicate that the main advantage of the new model
is that it can offer more details than Avrami-type ap-
proximations which are typically used in this field.
In particular, the new cellular automaton method
can tackle the heterogeneity of internal and external

2664 D. Raabe / Acta Materialia 52 (2004) 2653–2664
boundary and starting conditions which is not possible
for Avrami-models because they are statistical in nature.
The new automaton approach can for instance predict
intricate spherulite topologies, kinetic details, and crys-
tallographic textures in homogeneous or heterogeneousfields (the paper gives an example of spherulite growth
in a temperature gradient field). Furthermore, it can be
coupled to forming and processing models making use
of local rather than only global boundary conditions.
References
[1] von Neumann J. The general and logical theory of automata.
1963. In: Aspray W, Burks A, editors. Papers of John von
Neumann on Computing and Computer Theory. The Charles
Babbage Institute Reprint Series for the History of Computing,
vol. 12. MIT Press; 1987.
[2] Raabe D. Ann Rev Mater Res 2002;32:53.
[3] Hesselbarth HW, G€obel IR. Acta Metall 1991;39:2135.
[4] Pezzee CE, Dunand DC. Acta Metall 1994;42:1509.
[5] Davies CHJ. Scri Metall Mater 1995;33:1139.
[6] Marx V, Reher F, Gottstein G. Acta Mater 1998;47:1219.
[7] Raabe D. Philos Mag A 1999;79:2339.
[8] Raabe D, Becker R. Model Simul Mater Sci Eng 2000;8:445.
[9] Janssens KGF. Model Simul Mater Sci Eng 2003;11:157.
[10] Cortie MB. Metall Trans B 1993;24:1045.
[11] Brown SGR, Williams T, Spittle JA. Acta Metall 1994;42:2893.
[12] Gandin CA, Rappaz M. Acta Metall 1997;45:2187.
[13] Gandin CA, Desbiolles JL, Thevoz PA. Metall Mater Trans A
1999;30:3153.
[14] Spittle JA, Brown SGR. Acta Metall 1994;42:1811.
[15] Geiger J, Roosz A, Barkoczy P. Acta Mater 2001;49:623.
[16] Young MJ, Davies CHJ. Scri Mater 1999;41:697.
[17] Raabe D. Computational materials science. Weinheim: Wiley-
VCH; 1998.
[18] Koscher E, Fulchiron R. Polymer 2002;43:6931.
[19] Nakamura K, Watanabe K, Katayama K, Amano T. J Appl
Polym Sci 1972;16:1077.
[20] Ozawa T. Polymer 1971;12:150.
[21] Eder G, Janeschitz-Kriegl H. In: Cahn RW, Haasen P, Kramer
EJ, editors. Processing of polymers. Meijer HEH, editor. Mater
Sci Technol, vol. 18. VCH; 1997. p. 269 [chapter 5].
[22] Eder G, Janeschitz-Kriegl H, Krobath G. Progr Colloid Polym Sci
1989;80:1.
[23] Hieber CA. Polymer 1995;36:1455.
[24] Michaeli W, Hoffmann S, Cramer A, Dilthey U, Brandenburg A,
Kretschmer M. Adv Eng Mater 2003;5:133.
[25] Keller A. Philos Mag 1957;2:1171.
[26] Toda A, Keller A. Colloid Polym Sci 1993;271:328.
[27] Turnbull D, Fisher JC. J Chem Phys 1949;17:71.
[28] Lauritzen JI, Hoffman JD. J Res Natl Bur Stand 1960;64:73.
[29] Keller A. Rep Progr Phys 1968;31:623.
[30] Hoffman JD, Davis GT, Lauritzen JI. In: Hanney NB, editor.
Treatise of solid state chemistry. New York: Plenum Press; 1976.
p. 497 [chapter 7].
[31] Snyder CR, Marand H, Mansfield ML. Macromolecules
1996;29:7508.
[32] Snyder CR, Marand H. Macromolecules 1997;30:2759.
[33] Hoffman JD, Miller RL. Polymer 1997;38:3151.
[34] Long Y, Shanks RA, Stachurki ZH. Progr Polym Sci 1995;20:
651.
[35] Point JJ, Janimak JJ. J Cryst Growth 1993;131:501.
[36] Hobbs JK, Humphris ADL, Miles MJ. Macromolecules
2001;34:5508.
[37] Hobbs JK, McMaster TJ, Miles MJ, Barham PJ. Polymer
2001;39:2437.
[38] Cooper M, Manley RSJ. Macromolecules 1975;8:219.
[39] Leung WM, Manley RSJ, Panaras AR. Macromolecules
1985;18:760.
[40] Ergoz E, Fatou JG, Mandelkern L. Macromolecules 1972;5:
147.
[41] Supaphol P, Spruiell JE. J Appl Polym Sci 2002;86:1009.
[42] Zhu X, Li Y, Yan D, Fang Y. Polymer 2001;42:9217.
Related Documents











