Copyright © Royal College of pathologists of Australasia. Unauthorized reproduction of this article is prohibited. Melanoma’s connections to the tumour microenvironment JOHANNA M. BRANDNER* AND NIKOLAS K. HAASS{{§ *Department of Dermatology and Venerology, University Hospital Hamurg-Eppendorf, Hamburg, Germany; {The University of Queensland Diamantina Institute, Translational Research Institute, Woolloongabba, Queensland, zThe Centenary Institute, Newtown, and §Discipline of Dermatology, University of Sydney, Camperdown, New South Wales, Australia Summary Melanoma cells interact with and depend on seemingly normal cells in their tumour microenvironment to allow the acquisition of the hallmarks of solid cancer. In general, there are three types of interaction of melanoma cells with their microenvironment. First, there is bilateral communication between melanoma cells and the stroma, which includes fibroblasts, endothelial cells, immune cells, soluble mol- ecules, and the extracellular matrix. Second, while under normal conditions keratinocytes control localisation and proliferative behaviour of melanocytes in the epidermis, once this balance is disturbed and a melanoma has developed, melanoma cells may take over the control of their epidermal tumour microenvironment. Finally, there are subcompart- ments within tumours with different microenvironmental milieu defined by their access to oxygen and nutrients. There- fore, different melanoma cells within a tumour face different microenvironments. Interactions between melanoma cells among each other and with the cell types in their micro- environment happen through endocrine and paracrine communication and/or through direct contact via cell–cell and cell–matrix adhesion, and gap junctional intercellular communication (GJIC). Connexins have been identified as key molecules for direct cell – cell communication and are also thought to be important for the release of signalling molecules from cells to the microenvironment. In this review we provide an update of the alterations in cell–cell communication in melanoma and the tumour microenvironment associated with melanoma development and progression. Abbreviations: Cx, connexin; ECM, extracellular matrix; ETM, epidermal tumour microenvironment; GJ, gap junction; GJIC, gap junctional intercellular communication. Key words: Cell–cell communication, connexins, gap junctions, melanoma, tumour microenvironment. Received 29 April, accepted 29 May 2013 MELANOMA MICROENVIRONMENT Under normal conditions, the state of a cell—quiescence, proliferation, differentiation or cell death—is determined by homeostasis. 1 In human epidermis, this homeostasic balance is maintained in the epidermal melanin unit, a symbiotic relation- ship between a melanocyte and approximately 36 associated keratinocytes. 2,3 Melanocytes are located in the stratum basale of the epidermis, where they keep a life-long stable ratio of 1:5 with basal keratinocytes. 4 This balance is maintained through regulated induction of melanocyte division and is only disturbed during transformation into a naevus or a melanoma. 5 On a molecular level, homeostasis is governed by intercellular communication, which can be endocrine and paracrine via soluble factors (including hormones, growth factors and cyto- kines) and/or by direct contact via cell–cell and cell–matrix adhesion, and gap junctional intercellular communication (GJIC). 6,7 Dysregulation of the homeostasis may cause an imbalance of the epidermal melanin unit and trigger a continuous proliferation of the melanocytes, which may lead to development of melanoma. 5 The hallmarks of solid cancer are uncontrolled proliferation, evasion from growth suppres- sors, replicative immortality, escape from immune destruction, tumour-promoting inflammation, invasion and metastasis, induction of angiogenesis, genome instability and mutation, resistance to cell death and deregulation of cellular energetics. Alterations in the interaction between neoplastic cells and their immediate microenvironment play a key role in these processes. 8–10 ‘Tumour microenvironment’ is a broad term, which includes: (1) the tumour stroma composed of fibroblasts, endothelial cells, immune cells, soluble molecules, and the extracellular matrix (ECM); (2) the tissue where the tumour originates from; and (3) different sub-compartments within the tumour itself (Fig. 1). At the molecular level these three tumour microenvironments are characterised by: 1. Signals to and from the stroma via cell – cell and cell – matrix contact and/or via secretion of cytokines and growth factors may lead to a remodelling of the tumour microenvironment and consequently to promotion of melanoma development, growth and metastasis by inducing angiogenesis, invasion and migration. 11,12 2. In addition to the interaction with the tumour stroma, primary melanoma progression as well as cutaneous melanoma metastases impact on the epidermal tumour microenvironment: the multilayered epithelium of the skin. 13 3. Different microenvironmental conditions within the tumour itself are created by differential access to nutrients and oxygen. 14–16 The microenvironment is not only important for the primary tumour, but also for colonisation of a secondary organ. The ‘seed and soil’ hypothesis implies that the metastatic process depends on the tumorigenic capacity of the cells and, again, on their interactions with the microenvironment. 17 Connexins have been identified as key molecules for direct cell–cell communication and are also thought to be important Pathology (August 2013) 45(5), pp. 443–452 REVIEW Print ISSN 0031-3025/Online ISSN 1465-3931 # 2013 Royal College of Pathologists of Australasia DOI: 10.1097/PAT.0b013e328363b3bd

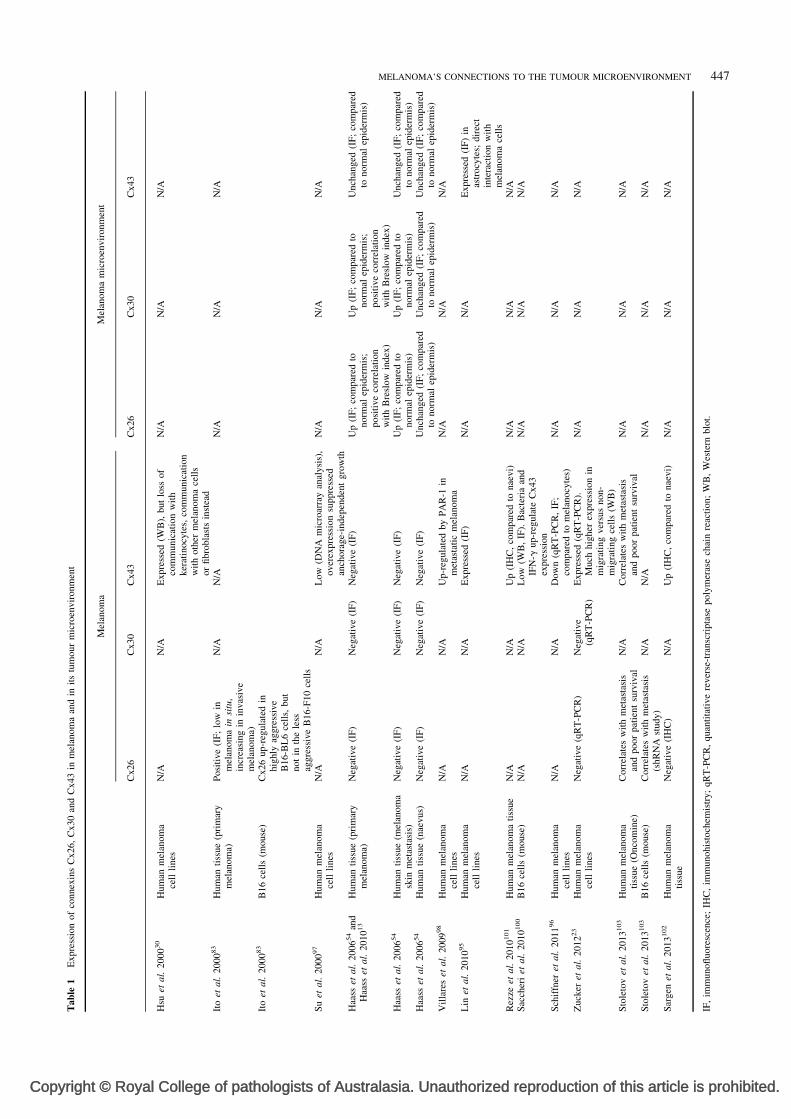
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Copy
Pathology (August 2013) 45(5), pp. 443–452
R E V I E W
right © Roy
Print ISSN 0031
DOI: 10.1097/PA
Melanoma’s connections to the tumour microenvironment
JOHANNA M. BRANDNER* AND NIKOLAS K. HAASS{{§
*Department of Dermatology and Venerology, University Hospital Hamurg-Eppendorf, Hamburg, Germany; {TheUniversity of Queensland Diamantina Institute, Translational Research Institute, Woolloongabba, Queensland,zThe Centenary Institute, Newtown, and §Discipline of Dermatology, University of Sydney, Camperdown, NewSouth Wales, Australia
al College of pathologists of Australasia
-3025/Online ISSN 1465-3931 # 2013 Royal College of Pa
T.0b013e328363b3bd
Summary
Melanoma cells interact with and depend on seeminglynormal cells in their tumour microenvironment to allow theacquisition of the hallmarks of solid cancer. In general, thereare three types of interaction of melanoma cells with theirmicroenvironment. First, there is bilateral communicationbetween melanoma cells and the stroma, which includesfibroblasts, endothelial cells, immune cells, soluble mol-ecules, and the extracellular matrix. Second, while undernormal conditions keratinocytes control localisation andproliferative behaviour of melanocytes in the epidermis, oncethis balance is disturbed and a melanoma has developed,melanoma cells may take over the control of their epidermaltumour microenvironment. Finally, there are subcompart-ments within tumours with different microenvironmentalmilieu defined by their access to oxygen and nutrients. There-fore, different melanoma cells within a tumour face differentmicroenvironments. Interactions between melanoma cellsamong each other and with the cell types in their micro-environment happen through endocrine and paracrinecommunication and/or through direct contact via cell–celland cell–matrix adhesion, and gap junctional intercellularcommunication (GJIC). Connexins have been identified askey molecules for direct cell–cell communication and are alsothought to be important for the release of signalling moleculesfrom cells to the microenvironment. In this review we providean update of the alterations in cell–cell communication inmelanoma and the tumour microenvironment associated withmelanoma development and progression.
Abbreviations: Cx, connexin; ECM, extracellular matrix; ETM, epidermal
tumour microenvironment; GJ, gap junction; GJIC, gap junctional intercellular
communication.
Key words: Cell–cell communication, connexins, gap junctions, melanoma,
tumour microenvironment.
Received 29 April, accepted 29 May 2013
MELANOMA MICROENVIRONMENT
Under normal conditions, the state of a cell—quiescence,proliferation, differentiation or cell death—is determined byhomeostasis.1 In human epidermis, this homeostasic balance ismaintained in the epidermal melanin unit, a symbiotic relation-ship between a melanocyte and approximately 36 associatedkeratinocytes.2,3 Melanocytes are located in the stratum basaleof the epidermis, where they keep a life-long stable ratio of 1:5with basal keratinocytes.4 This balance is maintained through
regulated induction of melanocyte division and is onlydisturbed during transformation into a naevus or a melanoma.5
On a molecular level, homeostasis is governed by intercellularcommunication, which can be endocrine and paracrine viasoluble factors (including hormones, growth factors and cyto-kines) and/or by direct contact via cell–cell and cell–matrixadhesion, and gap junctional intercellular communication(GJIC).6,7 Dysregulation of the homeostasis may cause animbalance of the epidermal melanin unit and trigger acontinuous proliferation of the melanocytes, which may leadto development of melanoma.5 The hallmarks of solid cancerare uncontrolled proliferation, evasion from growth suppres-sors, replicative immortality, escape from immune destruction,tumour-promoting inflammation, invasion and metastasis,induction of angiogenesis, genome instability and mutation,resistance to cell death and deregulation of cellular energetics.Alterations in the interaction between neoplastic cells andtheir immediate microenvironment play a key role in theseprocesses.8–10 ‘Tumour microenvironment’ is a broad term,which includes: (1) the tumour stroma composed of fibroblasts,endothelial cells, immune cells, soluble molecules, and theextracellular matrix (ECM); (2) the tissue where the tumouroriginates from; and (3) different sub-compartments within thetumour itself (Fig. 1). At the molecular level these three tumourmicroenvironments are characterised by:
1. S
. Un
thol
ignals to and from the stroma via cell–cell and cell–matrix contact and/or via secretion of cytokines andgrowth factors may lead to a remodelling of thetumour microenvironment and consequently to promotionof melanoma development, growth and metastasis byinducing angiogenesis, invasion and migration.11,12
2. I
n addition to the interaction with the tumour stroma,primary melanoma progression as well as cutaneousmelanoma metastases impact on the epidermal tumourmicroenvironment: the multilayered epithelium of theskin.133. D
ifferent microenvironmental conditions within thetumour itself are created by differential access to nutrientsand oxygen.14–16The microenvironment is not only important for the primarytumour, but also for colonisation of a secondary organ.The ‘seed and soil’ hypothesis implies that the metastaticprocess depends on the tumorigenic capacity of the cellsand, again, on their interactions with the microenvironment.17
Connexins have been identified as key molecules for directcell–cell communication and are also thought to be important
authorized reproduction of this article is prohibited.
ogists of Australasia

Copy
Melanocyte
Keratinocyte
Fibroblast
Oxygen andnutrient-deprivedmelanoma cell
Melanoma cell
Endothelial cell
Immune cell
Basement membrane
1 Tumour-stroma interactions
2 Tumour-ETM interactions
3 Tumour subcompartments
1
1
1
2
2?
?
3
Fig. 1 Melanoma tumour microenvironment. Under normal conditions, keratinocytes control the localisation and proliferative behaviour of melanocytes (green one-way arrow). Once this balance is disturbed and a melanoma has developed, melanoma cells may take over the control of their epidermal tumour microenvironment (ETM;red one-way arrows, 2). It is likely that keratinocytes controlled by melanoma cells, in turn, impact positively on melanoma cells favouring progression (red one-wayarrow, ?). There is bilateral communication between melanoma cells and the stroma, which includes fibroblasts, endothelial cells, immune cells, soluble molecules, andthe extracellular matrix (red two-way arrows, 1). Finally, there are subcompartments within tumours with different microenvironmental milieu defined by their access tooxygen and nutrients (3).
444 BRANDNER and HAASS Pathology (2013), 45(5), August
for the release of signalling molecules from cells to themicroenvironment. Here we will give an update on changesof expression patterns of connexins in cancer, particularly inmelanoma, and in the tumour microenvironment.
CONNEXINS, GAP JUNCTIONS ANDHEMICHANNELS
This review assumes a familiarity with the general biology ofgap junctions (GJs), which has been reviewed pre-viously.6,18,19 Briefly, connexins belong to a family of gapjunction forming transmembrane proteins, which are essentialfor GJIC by exchange of small molecules (<1.2 kDa) such asions (Ca2þ, Hþ), signalling molecules (cAMP, ATP) andmetabolic products (amino acids). In addition to molecularweight and size, the ability of a solute to transverse thesechannels depends on its net charge, shape, and interactionswith specific connexins that constitute gap junctions inparticular cells.20 Twenty-one connexins have been identified;eleven of those are expressed in the skin.21–23 In the epidermis,GJIC is involved in maintenance of homeostasis, regulation ofproliferation and differentiation, barrier function and recruit-ment of inflammatory cells. GJIC is thus a critical factor in thelife and death balance of cells.24–29 Furthermore, GJIC hasbeen shown to be critical in keratinocyte-melanocyte inter-action.30,31 In addition, connexins can form hemichannels,which allow release (e.g., ATP, NADþ) or putatively uptake ofmolecules and ions to and from the cellular environment.32,33
Finally, connexins, especially Cx43, interact with structural
right © Royal College of pathologists of Australasia
and signalling molecules, which may add further functions tothese molecules.34
CONNEXINS ARE CONDITIONAL TUMOURSUPPRESSORS
Almost half a century ago Loewenstein and Kanno firstdescribed the lack of communication between cancer cells.35
Since then, loss of GJIC and/or down-regulation of connexinshas been reported both in cell lines as well as in tissues of manytumour types,36–38 such as hepatocellular carcinoma,39–42
gastric carcinoma,43 colon cancer,44 prostate cancer,45 bladdercarcinoma,46 cervical carcinoma,47 ovarian carcinoma,48,49
lung cancer,50,51 glioblastoma,52 mammary carcinoma,53 andvarious skin cancers including basal cell carcinoma, squamouscell carcinoma and melanoma.54–56 For example, electronmicroscopy investigations have shown that basal and squamouscell carcinomas do not have fully developed GJs, and that Cx43is not restricted to these poorly developed GJs but is present inthe cytoplasm.55 The connexin species lost during tumourprogression can vary between tumour entities; for examplewhile in mammary carcinoma cells both Cx43 and Cx26 aredown-regulated,53 in basal cell carcinoma Cx43 is down- butCx26 up-regulated.54,56 Loss of GJIC might help the tumourcells to survive, as GJIC has been shown to spread cell-killingsignals, most likely Ca2þ ions.57 In addition, it was shown thatdown-regulation of Cx43 expression or function resulted inincreased proliferation and migration in primary keratinocytes,implying a contribution of Cx43 to controlling early stages of
. Unauthorized reproduction of this article is prohibited.

Copy
MELANOMA’S CONNECTIONS TO THE TUMOUR MICROENVIRONMENT 445
tumourigenesis.58–60 Xu and Nicholson reviewed the evidencethat increased opening of hemichannels formed by connexinsresulted in cell death in cochlear supporting cells of the ear andin keratinocytes of the epidermis.61 Several oncogenes andhormone and growth regulators (such as epidermal growthfactor, transforming growth factor-beta and peptide hormones),known to promote tumour onset or progression frequentlyinhibit GJIC or down-regulate connexin expression.62–64
This observation was confirmed experimentally, as functionalabrogation of connexins (Cx26, Cx32 or Cx43), using geneknock-down studies, antisense or dominant negativemutant approaches, have demonstrated an enhancement ofthe malignant phenotype in several tumour types.65–73 Further,Cx32 knock-out mice have an increased incidence of tumouronset when challenged with carcinogens.74–77 Corres-pondingly, the reverse experiment—ectopic expression ofconnexins in cancer cells—resulted in partial differentiationof transformed cells and restored functional communicationand reduced tumour proliferation and growth both in vitro andin vivo.38,53,78–80
This large body of evidence may lead to the assumption thatconnexins are general tumour suppressors, but it appears thatthis is only the case in the earlier steps of cancerogenesis. Therole of connexins in invasion and metastasis is very complex,and several reports suggest that connexins might facilitateinvasion, intravasation, extravasation and metastasis.41,81–94
Cronier and colleagues support both the tumour suppressorand the tumour driver theories by proposing the followingmodel.36 For the step from primary to invasive tumours thereis need for disruption of intercellular junctions including GJs,consistent with the model that connexins are tumour suppres-sors. In contrast, for the tumour cell dissemination and metas-tasis steps, increased cell contacts and communication areneeded in order to enable interaction with the tumour stroma,especially between cancer cells and endothelial cells. There-fore, connexins might be better classified as conditional tumoursuppressors that modulate cell proliferation, as well as adhesionand migration.38
CONNEXINS IN MELANOMA
Reflecting the situation in many other cancer types as discussedabove, the role of connexins and GJIC is still highly contro-versial also in melanoma and its tumour microenvironment.
Cx43
This is certainly the most studied connexin in melanoma.Hsu and colleagues showed Cx43 protein expression in allhuman melanocytic cell lines they investigated by Westernblotting (in foreskin-derived melanocytes and several mela-noma cell lines).30 This was confirmed by Lin and colleagueswho detected Cx43 expression in human melanoma celllines using immunofluorescence.95 However, neither studyquantified the Cx43 protein expression levels. Schiffner andcolleagues showed by qRT-PCR and immunofluorescence thatCx43 in human melanoma cell lines had lower expressionlevels than in human melanocytes.96 Su and colleagues reportedthat Cx43 was expressed at low levels in human melanoma celllines and, importantly, that its overexpression suppressedanchorage-independent growth in colony-forming efficiencyassays, suggesting a tumour-suppressor role of Cx43 inmelanoma.97 Zucker and colleagues showed by qRT-PCR inhuman melanoma cell lines no detectable expression for
right © Royal College of pathologists of Australasia
Cx26, Cx30, Cx31.1, Cx36, and Cx37, low expressionfor Cx30.3 and Cx31 and higher expression levels for Cx32,Cx40, Cx43 and Cx45.23 Surprisingly, they discovered muchhigher Cx43 expression levels in migrating versus non-migrating cells (WB).23 Also Villares and colleagues foundhigh levels of Cx43 protein expression in human metastaticmelanoma cell lines.98 Loss of protease-activated receptor-1(PAR-1) expression resulted in the loss of Cx43 and, corre-spondingly, overexpression of PAR-1 contributed to melanomametastasis via up-regulation of Cx43.98,99 Interestingly,Saccheri and colleagues found that, while initial levels ofCx43 were low in B16 mouse melanoma cells, Cx43 proteinlevels increased after infection with bacteria or treatmentwith interferon-g.100 This was followed by the transfer ofpreprocessed antigenic peptides from melanoma cells todendritic cells, which then presented those peptides on theirsurface and consequently activated cytotoxic T cells against thetumour antigen. Correspondingly, melanoma cells in whichCx43 had been silenced, failed to elicit a cytotoxic antitumourresponse after infection with bacteria.100
In addition to the above-listed in vitro data, there are also anumber of studies on human melanoma tissue. Using immuno-fluorescence on human tissue samples we did not detectCx43 (nor Cx26 and Cx30) in naevi, primary melanomas orcutaneous melanoma metastases, while the internal controls(adjacent epidermis) were positive in the expected layers.13,54
In contrast, using immunohistochemistry, Rezze as well asSargen and colleagues reported Cx43 expression in humanmelanoma tissue, higher than in human naevi.101,102 However,neither of these studies provided high magnification imagesto confirm the subcellular localisation nor did they showappropriate positive and negative controls. Indeed, in bothstudies Cx43 expression in melanoma cells appeared to becytoplasmic and hence would argue for a cell–cell or cell–matrix communication-independent role of these connexins.This would not support the mechanism for melanoma survivalin brain metastasis proposed by Lin and colleagues, whoshowed that reactive astrocytes protect metastatic melanomacells in the brain from chemotherapy by sequestering intra-cellular calcium through direct cell–cell communication.95
Moreover, in the Rezze and Sargen studies the expressionpattern of Cx43 in naevi and different melanoma stagesappeared very variable and the typical Cx43 staining inthe epidermis was missing.101,102 Analysing the on-line databank of human tissue Oncomine, Stoletov and colleaguesshowed that increased Cx43 (and Cx26) gene expression inprimary lesions correlated with metastasis and poor patientsurvival.103
Cx26 and Cx30
These connexins are much less studied. Ito and colleaguesfound that Cx26 was up-regulated in the highly aggressive BL6sub-line of B16 mouse melanoma cells compared to the lessaggressive F10 sub-line.83 F10 cells transfected with wild-typeCx26 exhibited similar metastatic behaviour to the BL6 cells.Correspondingly, BL6 cells transfected with a dominant-negative Cx26 mutant showed the less aggressive behaviourcharacteristic of F10 cells. They did not find Cx26 in humanmelanoma in situ but found an up-regulation of Cx26 ininvasive melanomas.83 However, in their study Cx26 stainingin both melanoma cells and epidermal keratinocytes wascytoplasmic. Moreover, they did not distinguish between
. Unauthorized reproduction of this article is prohibited.

Copy
446 BRANDNER and HAASS Pathology (2013), 45(5), August
Cx26 and Cx30. In contrast, we showed in immuno-fluorescence studies on human melanoma tissue samples, thatall areas of melanocytic naevi, primary melanomas andcutaneous melanoma metastases lacked Cx26 and Cx30 expres-sion,13,54 similar to our findings in Merkel cell carcinoma.104
This was confirmed by Sargen and colleagues who did notdetect Cx26 in melanoma using immunohistochemistry onhuman tissue samples,102 as well as by Zucker and colleagueswho did not find Cx26 and Cx30 expression in human mela-noma cell lines using qRT-PCR.23 Contrastingly, Stoletov andcolleagues reported a positive correlation between Cx26expression and metastatic potential using Cx26 shRNA inB16 mouse melanoma cells.103 This was supported by analysisof the on-line data bank of human tissue Oncomine, whichshowed that increased Cx26 expression in primary lesionscorrelated with metastasis and poor patient survival.103
Pannexin 1
Interestingly, Penuela and colleagues showed recently that lossof Pannexin 1, a channel-forming glycoprotein remotely relatedto connexins, attenuated melanoma progression by reversion toa melanocyte-like phenotype.105
The Oncomine data103 do not seem to match the data onprimary melanomas in other studies; however, it would beinteresting to re-analyse these data more in detail. As thereappears to be a correlation to tumour thickness, is there no orlittle expression on thin tumours and a differential expressionpattern in different areas of thick melanomas?
Of course, one is surprised about the differences betweensimilar studies investigating Cx43 as well as Cx26 (also sum-marised in Table 1). These may be due to the following reasons:
1. S
rig
everal studies investigated the molecules on mRNA levelonly. The presence of mRNA does not necessarily meanthat the respective protein is present.
2. I
n tissues it is difficult to separate between connexinspresent in melanoma cells and those present inepidermal, mesenchymal or endothelial tissues enclosedby the tumour.3. I
mmunohistochemistry is often dependent on stainingconditions and can result in false positive and false negativeresults. Appropriate positive and negative controls show-ing the sensitivity and specificity of the antibody areindispensable for the interpretation of these results. Forexample, the Cx26 antibody used in some of the discussedstudies shows cross-reactivity with Cx30.Importantly, most of the apparent discrepancies in thisparagraph can be explained by the model proposed by Cronierand colleagues,36 which implies that connexins are tumoursuppressors during early melanomagenesis but tumourdrivers during metastasis. During early melanomagenesis therespective connexins are typically located in the cell mem-branes indicating that they are functioning through GJIC.In contrast, in advanced stages connexins are typically locatedin the cytoplasm, indicating a different function, possiblythrough interaction with signalling molecules.
CONNEXINS IN THE TUMOUR ENVIRONMENTOF MELANOMA
Hsu and colleagues showed that keratinocytes communicatewith melanocytes but not with melanoma cells via GJIC.
ht © Royal College of pathologists of Australasia
Instead, melanoma cells communicate amongst themselvesand with fibroblasts and endothelial cells.30 This switch incommunication partners coincides with the E- to N-cadherinswitch, suggesting that the gain of N-cadherin with theconcurrent loss of E-cadherin facilitates GJ formation withfibroblasts and endothelial cells.30 Additionally, GJ formationin human melanoma cell lines appears to require MCAM.31
This switch will allow melanoma cells to de-couple from theepidermal microenvironment and to communicate with celltypes important for their metastatic spread. Several studies havesuggested that connexins may promote metastasis in melanomaand other tumours by forming intercellular connectionsbetween cancer cells and vascular endothelium that are ableto initiate tumour cell diapedesis.30,81,82,87,98,106 Melanomacells expressing higher levels of Cx43 show increased couplingto vascular endothelial cells81 and the ability of tumour cells tometastasise appears to correlate with the ability of tumour cellsto communicate with endothelial cells.87 Also Cx26 maycontribute to the metastasis of melanoma by facilitatingcommunication between melanoma cells and their surroundingendothelial cells.98 Cx26 expression is associated with lym-phatic vessel invasion and poor prognosis in human breastcancer.107
Lin and colleagues recently showed that melanoma brainmetastases are surrounded and infiltrated by astrocytes, and thatthese astrocytes can play a role similar to their establishedability to protect neurons from apoptosis.95 In co-cultureexperiments, astrocytes reduced apoptosis in human melanomacells treated with various chemotherapeutic drugs. Thischemoprotective effect was dependent on physical contactand GJIC between astrocytes, which express high levels ofCx43, and tumour cells. Moreover, the protective effect ofastrocytes resulted from their sequestering calcium from thecytoplasm of tumour cells. These data suggest that brainmetastases can harness the neuroprotective effects of reactiveastrocytes for their own survival.95 Using a chick embryomodel, Stoletov and colleagues showed that B16 mousemelanoma cells, which express Cx26, but not Cx43, colonisedthe chicken brain forming numerous microtumours invadingalong the pre-existing vasculature.103 In contrast, Cx26 knock-down B16 cells formed significantly fewer and less invasivetumours, suggesting that in metastatic melanoma cells Cx26expression enhances microtumour formation in the brain inassociation with the existing vasculature.103
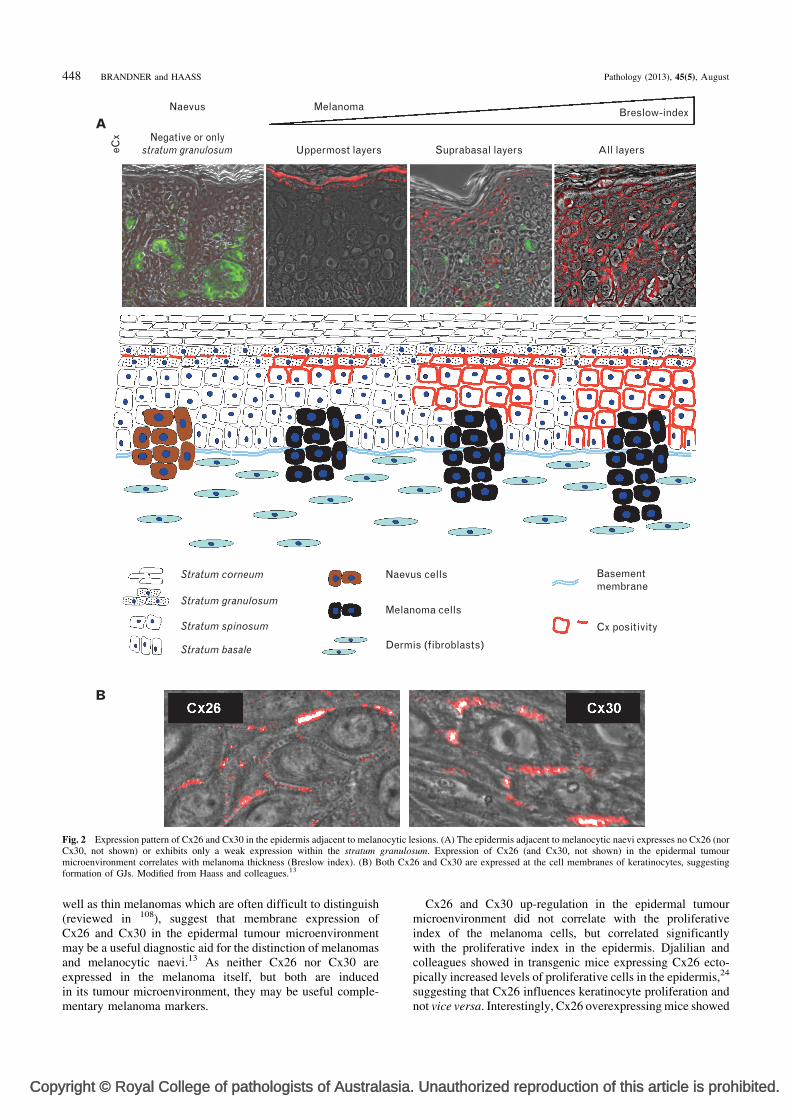
While these studies demonstrate the interaction of melanomacells with the stroma and the role of connexins and/or GJIC inthe early and late steps of melanomagenesis, interactionsbetween melanoma and the epidermal tumour microenviron-ment (ETM)—the multilayered epithelium of the skin—arepoorly understood. In this regard, we have demonstrated theinduction of Cx26 and Cx30 in the epidermis adjacentto malignant tumours (e.g., melanoma and Merkel cellcarcinoma), but not in the epidermis adjacent to benign tumours(e.g., melanocytic naevi and angiomas)54,104 (Fig. 2). Morerecently, we found correlation between (1) tumour thickness(Breslow index) and vertical Cx26 and Cx30 expression inthe ETM (Fig. 2), (2) tumour thickness and horizontal Cx26dissemination in the ETM, (3) metastasis and horizontal Cx26expression in the ETM, and (4) vertical epidermal expressionpatterns of Cx26 and Cx30 and the proliferative index inthe ETM. Thus we provided evidence for the association ofETM alteration with tumour malignancy and progression.13
The results of this study, which included dysplastic naevi as
. Unauthorized reproduction of this article is prohibited.
Copyright © Royal College of pathologists of Australasia. Unauthorized reproduction of this article is prohibited.
Tab
le1
Expre
ssio
nof
connex
ins
Cx26,
Cx30
and
Cx43
inm
elan
om
aan
din
its
tum
our
mic
roen
vir
onm
ent
Mel
anom
aM
elan
om
am
icro
envir
onm
ent
Cx26
Cx30
Cx43
Cx26
Cx30
Cx43
Hsu
etal.
2000
30
Hum
anm
elan
om
ace
llli
nes
N/A
N/A
Expre
ssed
(WB
),but
loss
of
com
munic
atio
nw
ith
ker
atin
ocy
tes,
com
munic
atio
nw
ith
oth
erm
elan
om
ace
lls
or
fibro
bla
sts
inst
ead
N/A
N/A
N/A
Ito
etal.
2000
83
Hum
anti
ssue
(pri
mar
ym
elan
om
a)P
osi
tive
(IF
;lo
win
mel
anom
ain
situ
,in
crea
sing
inin
vas
ive
mel
anom
a)
N/A
N/A
N/A
N/A
N/A
Ito
etal.
2000
83
B16
cell
s(m
ouse
)C
x26
up-r
egula
ted
inhig
hly
aggre
ssiv
eB
16-B
L6
cell
s,but
not
inth
ele
ssag
gre
ssiv
eB
16-F
10
cell
sS
uet
al.
2000
97
Hum
anm
elan
om
ace
llli
nes
N/A
N/A
Low
(DN
Am
icro
arra
yan
alysi
s),
over
expre
ssio
nsu
ppre
ssed
anch
ora
ge-
indep
enden
tgro
wth
N/A
N/A
N/A
Haa
sset
al.
2006
54
and
Haa
sset
al.
2010
13
Hum
anti
ssue
(pri
mar
ym
elan
om
a)N
egat
ive
(IF
)N
egat
ive
(IF
)N
egat
ive
(IF
)U
p(I
F;
com
par
edto
norm
alep
ider
mis
;posi
tive
corr
elat
ion
wit
hB
resl
ow
index
)
Up
(IF
;co
mpar
edto
norm
alep
ider
mis
;posi
tive
corr
elat
ion
wit
hB
resl
ow
index
)
Unch
anged
(IF
;co
mpar
edto
norm
alep
ider
mis
)
Haa
sset
al.
2006
54
Hum
anti
ssue
(mel
anom
ask
inm
etas
tasi
s)N
egat
ive
(IF
)N
egat
ive
(IF
)N
egat
ive
(IF
)U
p(I
F;
com
par
edto
norm
alep
ider
mis
)U
p(I
F;
com
par
edto
norm
alep
ider
mis
)U
nch
anged
(IF
;co
mpar
edto
norm
alep
ider
mis
)H
aass
etal.
2006
54
Hum
anti
ssue
(nae
vus)
Neg
ativ
e(I
F)
Neg
ativ
e(I
F)
Neg
ativ
e(I
F)
Unch
anged
(IF
;co
mpar
edto
norm
alep
ider
mis
)U
nch
anged
(IF
;co
mpar
edto
norm
alep
ider
mis
)U
nch
anged
(IF
;co
mpar
edto
norm
alep
ider
mis
)V
illa
res
etal.
2009
98
Hum
anm
elan
om
ace
llli
nes
N/A
N/A
Up-r
egula
ted
by
PA
R-1
inm
etas
tati
cm
elan
om
aN
/AN
/AN
/A
Lin
etal.
2010
95
Hum
anm
elan
om
ace
llli
nes
N/A
N/A
Expre
ssed
(IF
)N
/AN
/AE
xpre
ssed
(IF
)in
astr
ocy
tes;
dir
ect
inte
ract
ion
wit
hm
elan
om
ace
lls
Rez
zeet
al.
2010
101
Hum
anm
elan
om
ati
ssue
N/A
N/A
Up
(IH
C,
com
par
edto
nae
vi)
N/A
N/A
N/A
Sac
cher
iet
al.
2010
100
B16
cell
s(m
ouse
)N
/AN
/AL
ow
(WB
,IF
).B
acte
ria
and
IFN
-gup-r
egula
teC
x43
expre
ssio
n
N/A
N/A
N/A
Sch
iffn
eret
al.
2011
96
Hum
anm
elan
om
ace
llli
nes
N/A
N/A
Dow
n(q
RT
-PC
R,
IF;
com
par
edto
mel
anocy
tes)
N/A
N/A
N/A
Zuck
eret
al.
2012
23
Hum
anm
elan
om
ace
llli
nes
Neg
ativ
e(q
RT
-PC
R)
Neg
ativ
e(q
RT
-PC
R)
Expre
ssed
(qR
T-P
CR
).M
uch
hig
her
expre
ssio
nin
mig
rati
ng
ver
sus
non-
mig
rati
ng
cell
s(W
B)
N/A
N/A
N/A
Sto
leto
vet
al.
2013
103
Hum
anm
elan
om
ati
ssue
(Onco
min
e)C
orr
elat
esw
ith
met
asta
sis
and
poor
pat
ient
surv
ival
N/A
Corr
elat
esw
ith
met
asta
sis
and
poor
pat
ient
surv
ival
N/A
N/A
N/A
Sto
leto
vet
al.
2013
103
B16
cell
s(m
ouse
)C
orr
elat
esw
ith
met
asta
sis
(shR
NA
study)
N/A
N/A
N/A
N/A
N/A
Sar
gen
etal.
2013
102
Hum
anm
elan
om
ati
ssue
Neg
ativ
e(I
HC
)N
/AU
p(I
HC
,co
mpar
edto
nae
vi)
N/A
N/A
N/A
IF,
imm
unofl
uore
scen
ce;
IHC
,im
munohis
toch
emis
try;
qR
T-P
CR
,quan
tita
tive
rever
se-t
ransc
ripta
sepoly
mer
ase
chai
nre
acti
on;
WB
,W
este
rnblo
t.
MELANOMA’S CONNECTIONS TO THE TUMOUR MICROENVIRONMENT 447
Copy
Melanoma cells
Dermis (fibroblasts)
Naevus cells
B
A
Stratum basale
Stratum spinosum
Stratum granulosum
Stratum corneum Basementmembrane
Cx positivity
MelanomaNaevuseC
x Negative or onlystratum granulosum
Breslow-index
All layersSuprabasal layersUppermost layers
Fig. 2 Expression pattern of Cx26 and Cx30 in the epidermis adjacent to melanocytic lesions. (A) The epidermis adjacent to melanocytic naevi expresses no Cx26 (norCx30, not shown) or exhibits only a weak expression within the stratum granulosum. Expression of Cx26 (and Cx30, not shown) in the epidermal tumourmicroenvironment correlates with melanoma thickness (Breslow index). (B) Both Cx26 and Cx30 are expressed at the cell membranes of keratinocytes, suggestingformation of GJs. Modified from Haass and colleagues.13
448 BRANDNER and HAASS Pathology (2013), 45(5), August
well as thin melanomas which are often difficult to distinguish(reviewed in 108), suggest that membrane expression ofCx26 and Cx30 in the epidermal tumour microenvironmentmay be a useful diagnostic aid for the distinction of melanomasand melanocytic naevi.13 As neither Cx26 nor Cx30 areexpressed in the melanoma itself, but both are inducedin its tumour microenvironment, they may be useful comple-mentary melanoma markers.
right © Royal College of pathologists of Australasia
Cx26 and Cx30 up-regulation in the epidermal tumourmicroenvironment did not correlate with the proliferativeindex of the melanoma cells, but correlated significantlywith the proliferative index in the epidermis. Djalilian andcolleagues showed in transgenic mice expressing Cx26 ecto-pically increased levels of proliferative cells in the epidermis,24
suggesting that Cx26 influences keratinocyte proliferation andnot vice versa. Interestingly, Cx26 overexpressing mice showed
. Unauthorized reproduction of this article is prohibited.

Copy
Keratinocyte
Fibroblast
Melanoma cell
Biomarker-positivemelanoma cell
Biomarker-positivekeratinocyteNaevus cell
Naevus Melanoma Melanoma
Basement membrane
Fig. 3 Proposed biomarker assay. Complementary diagnostic biomarkers in melanoma cells (e.g., Cx43, red biomarker-positive melanoma cells) and in the epidermaltumour microenvironment (e.g., Cx26 and/or Cx30, red-bordered biomarker-positive keratinocytes).
MELANOMA’S CONNECTIONS TO THE TUMOUR MICROENVIRONMENT 449
a delay in wound healing, which needs to be explored withregards to ulceration, a biomarker associated with very poorprognosis for melanoma patients.109 In our study, all melano-mas with ulceration showed Cx26 (and Cx30) expression in alllayers of the epidermal tumour microenvironment.13 McCartyand colleagues hypothesised that induction of angiogenesisby the hyperplastic epithelium could stimulate growth andprogression of melanoma.110 This suggests a positive feedbackmechanism: tumour cells induce alterations in keratinocytes,which results in the production of growth factors which, inturn, stimulates tumour survival via endothelial cells. Theinduction of Cx26 and Cx30 in the epidermis adjacent tomelanoma putatively leading to GJIC or signalling via hemi-channels may play a role in this feedback mechanism byinducing proliferation and other functions. An interruptionof this vicious circle may provide a novel therapeuticapproach.
right © Royal College of pathologists of Australasia
OUTLOOKFurther work is needed to understand the complex role andtime and stage-dependent expression patterns of connexins inmelanoma and its microenvironment. However, the publishedevidence summarised in this review suggests that expressionlevels of Cx43 in melanocytic lesions could be optimisedas markers for ‘early’ melanomas (Cx43 negative/low) andmelanomas that are likely to metastasise (Cx43 high) (Fig. 3).Levels and distribution of Cx26 and Cx30 expression inthe epidermal tumour microenvironment may provide acomplementary diagnostic marker for the distinction of mel-anomas and melanocytic naevi (Fig. 3). Similarly, Bijnsdorpand colleagues proposed to measure Cx26 expression in theadjacent non-cancerous tissues (rather than cancer tissues) ofprostatectomy sections to identify high-risk patients whomay benefit from adjuvant therapy to decrease the risk ofmetastasis.111 Furthermore, it is not surprising that connexins
. Unauthorized reproduction of this article is prohibited.

Copy
450 BRANDNER and HAASS Pathology (2013), 45(5), August
have been viewed as potential therapeutic targets in cancer, atopic that has been extensively discussed by Kandouz andBatist recently.112
Conflicts of interest and sources of funding: NKH is arecipient of the Cameron Fellowship from the Melanomaand Skin Cancer Research Institute/Melanoma Foundation/Dermatology Foundation, Australia. NKH is CIA on projectgrants RG 09-08 and RG 13-06 (Cancer Council New SouthWales), project grant 570778 (Priority-driven collaborativecancer research scheme/Cancer Australia/Cure Cancer Austra-lia Foundation), Research Innovation Grant 08/RFG/1-27(Cancer Institute New South Wales) and project grant1003637 (National Health and Medical Research Council).JMB was supported by the Johann and Anny Thomas Stiftung(grant 01/07). The authors state that there are no conflicts ofinterest to disclose.
Address for correspondence: Dr N. K. Haass, The University of Queensland
Diamantina Institute, Translational Research Institute, 37 Kent St, Woolloon-
gabba, Qld 4102, Australia. E-mail: [email protected]
References1. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001;
1: 46–54.2. Fitzpatrick TB, Breathnach AS. The epidermal melanin unit system.
(German.) Dermatol Wochenschr 1963; 147: 481–9.3. Jimbow K, Quevedo WC Jr, Fitzpatrick TB, et al. Some aspects of
melanin biology: 1950–1975. J Invest Dermatol 1976; 67: 72–89.4. Fitzpatrick TB, Szabo G, Seiji M, et al. Biology of the melanin
pigmentary system. In: Fitzpatrick TB, Eisen A, Wolff K, et al., editors.Dermatology in General Medicine. New York: McGraw-Hill, 1979; 131–45.
5. Haass NK, Herlyn M. Normal human melanocyte homeostasis as aparadigm for understanding melanoma. J Investig Dermatol Symp Proc2005; 10: 153–63.
6. Haass NK, Smalley KS, Herlyn M. The role of altered cell-cell com-munication in melanoma progression. J Mol Histol 2004; 35: 309–18.
7. Haass NK, Smalley KS, Li L, et al. Adhesion, migration andcommunication in melanocytes and melanoma. Pigment Cell Res 2005;18: 150–9.
8. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57–70.
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation.Cell 2011; 144: 646–74.
10. Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the micro-environment on the malignant phenotype. Mol Med Today 2000; 6: 324–9.
11. Villanueva J, Herlyn M. Melanoma and the tumor microenvironment.Curr Oncol Rep 2008; 10: 439–46.
12. Zigler M, Kamiya T, Brantley EC, et al. PAR-1 and thrombin: the ties thatbind the microenvironment to melanoma metastasis. Cancer Res 2011; 71:6561–6.
13. Haass NK, Ripperger D, Wladykowski E, et al. Melanoma progressionexhibits a significant impact on connexin expression patterns in theepidermal tumor microenvironment. Histochem Cell Biol 2010; 133:113–24.
14. Groebe K, Mueller-Klieser W. Distributions of oxygen, nutrient,and metabolic waste concentrations in multicellular spheroids and theirdependence on spheroid parameters. Eur Biophys J 1991; 19: 169–81.
15. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat RevCancer 2006; 6: 583–92.
16. Santiago-Walker A, Li L, Haass NK, et al. Melanocytes: from morphologyto application. Skin Pharmacol Physiol 2009; 22: 114–21.
17. Fidler IJ. The pathogenesis of cancer metastasis: the ’seed and soil’hypothesis revisited. Nat Rev Cancer 2003; 3: 453–8.
18. Richard G. Connexins: a connection with the skin. Exp Dermatol 2000; 9:77–96.
19. Maeda S, Tsukihara T. Structure of the gap junction channel and itsimplications for its biological functions. Cell Mol Life Sci 2011; 68:1115–29.
20. Goldberg GS, Valiunas V, Brink PR. Selective permeability of gapjunction channels. Biochim Biophys Acta 2004; 1662: 96–101.
right © Royal College of pathologists of Australasia
21. Di WL, Rugg EL, Leigh IM, et al. Multiple epidermal connexins areexpressed in different keratinocyte subpopulations including connexin 31.J Invest Dermatol 2001; 117: 958–64.
22. Willecke K, Eiberger J, Degen J, et al. Structural and functional diversityof connexin genes in the mouse and human genome. Biol Chem 2002; 383:725–37.
23. Zucker SN, Bancroft TA, Place DE, et al. A dominant negative Cx43mutant differentially affects tumorigenic and invasive properties in humanmetastatic melanoma cells. J Cell Physiol 2013; 228: 853–9.
24. Djalilian AR, McGaughey D, Patel S, et al. Connexin 26 regulatesepidermal barrier and wound remodeling and promotes psoriasiformresponse. J Clin Invest 2006; 116: 1243–53.
25. Langlois S, Maher AC, Manias JL, et al. Connexin levels regulatekeratinocyte differentiation in the epidermis. J Biol Chem 2007; 282:30171–80.
26. Maass K, Ghanem A, Kim JS, et al. Defective epidermal barrier inneonatal mice lacking the C-terminal region of connexin 43. Mol BiolCell 2004; 15: 4597–608.
27. Man YK, Trolove C, Tattersall D, et al. A deafness-associated mutanthuman connexin 26 improves the epithelial barrier in vitro. J Membr Biol2007; 218: 29–37.
28. Kretz M, Maass K, Willecke K. Expression and function of connexins inthe epidermis, analyzed with transgenic mouse mutants. Eur J Cell Biol2004; 83: 647–54.
29. Mese G, Richard G, White TW. Gap junctions: basic structure andfunction. J Invest Dermatol 2007; 127: 2516–24.
30. Hsu M, Andl T, Li G, et al. Cadherin repertoire determines partner-specificgap junctional communication during melanoma progression. J Cell Sci2000; 113: 1535–42.
31. Satyamoorthy K, Muyrers J, Meier F, et al. Mel-CAM-specificgenetic suppressor elements inhibit melanoma growth and invasionthrough loss of gap junctional communication. Oncogene 2001; 20:4676–84.
32. Barr TP, Albrecht PJ, Hou Q, et al. Air-stimulated ATP release fromkeratinocytes occurs through connexin hemichannels. PLoS One 2013; 8:e56744.
33. Chandrasekhar A, Bera AK. Hemichannels: permeants and their effecton development, physiology and death. Cell Biochem Funct 2012; 30: 89–100.
34. Herve JC, Bourmeyster N, Sarrouilhe D, et al. Gap junctional complexes:from partners to functions. Prog Biophys Mol Biol 2007; 94: 29–65.
35. Loewenstein WR, Kanno Y. Intercellular communication and thecontrol of tissue growth: lack of communication between cancer cells.Nature 1966; 209: 1248–9.
36. Cronier L, Crespin S, Strale PO, et al. Gap junctions and cancer: newfunctions for an old story. Antioxid Redox Signal 2009; 11: 323–38.
37. Mesnil M, Crespin S, Avanzo JL, et al. Defective gap junctionalintercellular communication in the carcinogenic process. Biochim BiophysActa 2005; 1719: 125–45.
38. Naus CC, Laird DW. Implications and challenges of connexin connectionsto cancer. Nat Rev Cancer 2010; 10: 435–41.
39. Eghbali B, Kessler JA, Reid LM, et al. Involvement of gap junctions intumorigenesis: transfection of tumor cells with connexin 32 cDNA retardsgrowth in vivo. Proc Natl Acad Sci USA 1991; 88: 10701–5.
40. Loewenstein WR, Rose B. The cell-cell channel in the control of growth.Semin Cell Biol 1992; 3: 59–79.
41. Krutovskikh V, Mazzoleni G, Mironov N, et al. Altered homologousand heterologous gap-junctional intercellular communication in primaryhuman liver tumors associated with aberrant protein localization but notgene mutation of connexin 32. Int J Cancer 1994; 56: 87–94.
42. Yamaoka K, Nouchi T, Tazawa J, et al. Expression of gap junctionprotein connexin 32 and E-cadherin in human hepatocellular carcinoma.J Hepatol 1995; 22: 536–9.
43. Uchida Y, Matsuda K, Sasahara K, et al. Immunohistochemistry ofgap junctions in normal and diseased gastric mucosa of humans.Gastroenterology 1995; 109: 1492–6.
44. Friedman EA, Steinberg M. Disrupted communication between late-stage premalignant human colon epithelial cells by 12-O tetradecanoyl-phorbol-13-acetate. Cancer Res 1982; 42: 5096–105.
45. Tsai H, Werber J, Davia MO, et al. Reduced connexin 43 expression inhigh grade, human prostatic adenocarcinoma cells. Biochem Biophys ResCommun 1996; 227: 64–9.
46. Krutovskikh VA, Troyanovsky SM, Piccoli C, et al. Differential effect ofsubcellular localization of communication impairing gap junction proteinconnexin43 on tumor cell growth in vivo. Oncogene 2000; 19: 505–13.
47. King TJ, Fukushima LH, Hieber AD, et al. Reduced levels of connexin43in cervical dysplasia: inducible expression in a cervical carcinoma cell linedecreases neoplastic potential with implications for tumor progression.Carcinogenesis 2000; 21: 1097–109.
48. Hanna EA, Umhauer S, Roshong SL, et al. Gap junctional intercellularcommunication and connexin43 expression in human ovarian surface
. Unauthorized reproduction of this article is prohibited.

Copy
MELANOMA’S CONNECTIONS TO THE TUMOUR MICROENVIRONMENT 451
epithelial cells and ovarian carcinomas in vivo and in vitro. Carcino-genesis 1999; 20: 1369–73.
49. Umhauer S, Ruch RJ, Fanning J. Gap junctional intercellularcommunication and connexin 43 expression in ovarian carcinoma.Am J Obstet Gynecol 2000; 182: 999–1000.
50. Jinn Y, Ichioka M, Marumo F. Expression of connexin32 and connexin43gap junction proteins and E-cadherin in human lung cancer. Cancer Lett1998; 127: 161–9.
51. Zhang ZQ, Zhang W, Wang NQ, et al. Suppression of tumorigenicityof human lung carcinoma cells after transfection with connexin43.Carcinogenesis 1998; 19: 1889–94.
52. Huang RP, Hossain MZ, Sehgal A, et al. Reduced connexin43expression in high-grade human brain glioma cells. J Surg Oncol1999; 70: 21–4.
53. Hirschi KK, Xu CE, Tsukamoto T, et al. Gap junction genes Cx26 andCx43 individually suppress the cancer phenotype of human mammarycarcinoma cells and restore differentiation potential. Cell Growth Differ1996; 7: 861–70.
54. Haass NK, Wladykowski E, Kief S, et al. Differential inductionof connexins 26 and 30 in skin tumors and their adjacent epidermis.J Histochem Cytochem 2006; 54: 171–82.
55. Tada J, Hashimoto K. Ultrastructural localization of gap junction proteinconnexin 43 in normal human skin, basal cell carcinoma, and squamouscell carcinoma. J Cutan Pathol 1997; 24: 628–35.
56. Wilgenbus KK, Kirkpatrick CJ, Knuechel R, et al. Expression of Cx26,Cx32 and Cx43 gap junction proteins in normal and neoplastic humantissues. Int J Cancer 1992; 51: 522–9.
57. Krutovskikh VA, Piccoli C, Yamasaki H. Gap junction intercellularcommunication propagates cell death in cancerous cells. Oncogene2002; 21: 1989–99.
58. Mori R, Power KT, Wang CM, et al. Acute downregulation of connexin43at wound sites leads to a reduced inflammatory response, enhancedkeratinocyte proliferation and wound fibroblast migration. J Cell Sci2006; 119: 5193–203.
59. Wright CS, van Steensel MA, Hodgins MB, et al. Connexin mimeticpeptides improve cell migration rates of human epidermal keratinocytesand dermal fibroblasts in vitro. Wound Repair Regen 2009; 17: 240–9.
60. Pollok S, Pfeiffer AC, Lobmann R, et al. Connexin 43 mimetic peptideGap27 reveals potential differences in the role of Cx43 in wound repairbetween diabetic and non-diabetic cells. J Cell Mol Med 2011; 15:861–73.
61. Xu J, Nicholson BJ. The role of connexins in ear and skin physiology –functional insights from disease-associated mutations. Biochim BiophysActa 2013; 1828: 167–78.
62. Lampe PD. Analyzing phorbol ester effects on gap junctionalcommunication: a dramatic inhibition of assembly. J Cell Biol 1994;127: 1895–905.
63. Trosko JE, Chang CC, Madhukar BV, et al. Chemical, oncogene andgrowth factor inhibition gap junctional intercellular communication:an integrative hypothesis of carcinogenesis. Pathobiology 1990; 58:265–78.
64. Atkinson MM, Menko AS, Johnson RG, et al. Rapid and reversiblereduction of junctional permeability in cells infected with a tempera-ture-sensitive mutant of avian sarcoma virus. J Cell Biol 1981; 91: 573–8.
65. Duflot-Dancer A, Mesnil M, Yamasaki H. Dominant-negative abrogationof connexin-mediated cell growth control by mutant connexin genes.Oncogene 1997; 15: 2151–8.
66. Dagli ML, Yamasaki H, Krutovskikh V, et al. Delayed liver regenerationand increased susceptibility to chemical hepatocarcinogenesis in trans-genic mice expressing a dominant-negative mutant of connexin32 only inthe liver. Carcinogenesis 2004; 25: 483–92.
67. Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, et al. Increased suscept-ibility to urethane-induced lung tumors in mice with decreased expressionof connexin43. Carcinogenesis 2004; 25: 1973–82.
68. Omori Y, Yamasaki H. Mutated connexin43 proteins inhibit rat glioma cellgrowth suppression mediated by wild-type connexin43 in a dominant-negative manner. Int J Cancer 1998; 78: 446–53.
69. Krutovskikh VA, Yamasaki H, Tsuda H, et al. Inhibition of intrinsic gap-junction intercellular communication and enhancement of tumorigenicityof the rat bladder carcinoma cell line BC31 by a dominant-negativeconnexin 43 mutant. Mol Carcinog 1998; 23: 254–61.
70. Shao Q, Wang H, McLachlan E, et al. Down-regulation of Cx43 byretroviral delivery of small interfering RNA promotes an aggressive breastcancer cell phenotype. Cancer Res 2005; 65: 2705–11.
71. Czyz J. The stage-specific function of gap junctions during tumourigen-esis. Cell Mol Biol Lett 2008; 13: 92–102.
72. Gershon E, Plaks V, Dekel N. Gap junctions in the ovary: expression,localization and function. Mol Cell Endocrinol 2008; 282: 18–25.
73. Shen Y, Khusial PR, Li X, et al. SRC utilizes Cas to block gapjunctional communication mediated by connexin43. J Biol Chem 2007;282: 18914–21.
right © Royal College of pathologists of Australasia
74. Temme A, Buchmann A, Gabriel HD, et al. High incidence of spontaneousand chemically induced liver tumors in mice deficient for connexin 32.Curr Biol 1997; 7: 713–6.
75. King TJ, Lampe PD. The gap junction protein connexin32 is a mouse lungtumor suppressor. Cancer Res 2004; 64: 7191–6.
76. King TJ, Lampe PD. Mice deficient for the gap junction proteinConnexin32 exhibit increased radiation-induced tumorigenesis associatedwith elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2)activation. Carcinogenesis 2004; 25: 669–80.
77. Moennikes O, Buchmann A, Willecke K, et al. Hepatocarcinogenesis infemale mice with mosaic expression of connexin32. Hepatology 2000; 32:501–6.
78. Zhu D, Caveney S, Kidder GM, et al. Transfection of C6 glioma cells withconnexin 43 cDNA: analysis of expression, intercellular coupling, and cellproliferation. Proc Natl Acad Sci USA 1991; 88: 1883–7.
79. McLachlan E, Shao Q, Wang HL, et al. Connexins act as tumorsuppressors in three-dimensional mammary cell organoids by regu-lating differentiation and angiogenesis. Cancer Res 2006; 66:9886–94.
80. Hellmann P, Grummer R, Schirrmacher K, et al. Transfection withdifferent connexin genes alters growth and differentiation of humanchoriocarcinoma cells. Exp Cell Res 1999; 246: 480–90.
81. el-Sabban ME, Pauli BU. Cytoplasmic dye transfer between metastatictumor cells and vascular endothelium. J Cell Biol 1991; 115: 1375–82.
82. el-Sabban ME, Pauli BU. Adhesion-mediated gap junctional communica-tion between lung-metastatatic cancer cells and endothelium. InvasionMetastasis 1994; 14: 164–76.
83. Ito A, Katoh F, Kataoka TR, et al. A role for heterologous gap junctionsbetween melanoma and endothelial cells in metastasis. J Clin Invest 2000;105: 1189–97.
84. Saunders MM, Seraj MJ, Li Z, et al. Breast cancer metastatic potentialcorrelates with a breakdown in homospecific and heterospecific gapjunctional intercellular communication. Cancer Res 2001; 61: 1765–7.
85. Lin JH, Takano T, Cotrina ML, et al. Connexin 43 enhances the adhesivityand mediates the invasion of malignant glioma cells. J Neurosci 2002; 22:4302–11.
86. Miekus K, Czernik M, Sroka J, et al. Contact stimulation of prostatecancer cell migration: the role of gap junctional coupling and migrationstimulated by heterotypic cell-to-cell contacts in determination of themetastatic phenotype of Dunning rat prostate cancer cells. Biol Cell 2005;97: 893–903.
87. Pollmann MA, Shao Q, Laird DW, et al. Connexin 43 mediatedgap junctional communication enhances breast tumor cell diapedesis inculture. Breast Cancer Res 2005; 7: R522–34.
88. Kanczuga-Koda L, Sulkowski S, Lenczewski A, et al. Increased expres-sion of connexins 26 and 43 in lymph node metastases of breast cancer.J Clin Pathol 2006; 59: 429–33.
89. Bates DC, Sin WC, Aftab Q, et al. Connexin43 enhances glioma invasionby a mechanism involving the carboxy terminus. Glia 2007; 55: 1554–64.
90. Li Q, Omori Y, Nishikawa Y, et al. Cytoplasmic accumulation of con-nexin32 protein enhances motility and metastatic ability of human he-patoma cells in vitro and in vivo. Int J Cancer 2007; 121: 536–46.
91. Dobrowolski R, Sasse P, Schrickel JW, et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculoden-todigital dysplasia in humans. Hum Mol Genet 2008; 17: 539–54.
92. Cotrina ML, Lin JH, Nedergaard M. Adhesive properties of connexinhemichannels. Glia 2008; 56: 1791–8.
93. Elzarrad MK, Haroon A, Willecke K, et al. Connexin-43 upregulation inmicrometastases and tumor vasculature and its role in tumor cell attach-ment to pulmonary endothelium. BMC Med 2008; 6: 20.
94. Ezumi K, Yamamoto H, Murata K, et al. Aberrant expression of connexin26 is associated with lung metastasis of colorectal cancer. Clin Cancer Res2008; 14: 677–84.
95. Lin Q, Balasubramanian K, Fan D, et al. Reactive astrocytes protectmelanoma cells from chemotherapy by sequestering intracellular calciumthrough gap junction communication channels. Neoplasia 2010; 12: 748–54.
96. Schiffner S, Zimara N, Schmid R, et al. p54nrb is a new regulator ofprogression of malignant melanoma. Carcinogenesis 2011; 32: 1176–82.
97. Su YA, Bittner ML, Chen Y, et al. Identification of tumor-suppressor genesusing human melanoma cell lines UACC903, UACC903(þ6), and SRS3by comparison of expression profiles. Mol Carcinog 2000; 28: 119–27.
98. Villares GJ, Dobroff AS, Wang H, et al. Overexpression of protease-activated receptor-1 contributes to melanoma metastasis via regulation ofconnexin 43. Cancer Res 2009; 69: 6730–7.
99. Villares GJ, Zigler M, Bar-Eli M. The emerging role of the thrombinreceptor (PAR-1) in melanoma metastasis–a possible therapeutic target.Oncotarget 2011; 2: 8–17.
100. Saccheri F, Pozzi C, Avogadri F, et al. Bacteria-induced gap junctions intumors favor antigen cross-presentation and antitumor immunity. SciTransl Med 2010; 2: 44–57.
. Unauthorized reproduction of this article is prohibited.

Copy
452 BRANDNER and HAASS Pathology (2013), 45(5), August
101. Rezze GG, Fregnani JH, Duprat J, et al. Cell adhesion and communicationproteins are differentially expressed in melanoma progression model.Hum Pathol 2011; 42: 409–18.
102. Sargen MR, Gormley RH, Pasha TL, et al. Melanocytic tumors expressconnexin 43 but not 26: immunohistochemical analysis with potentialsignificance in melanocytic oncogenesis. Am J Dermatopathol 2013; Jan22: (Epub ahead of print).
103. Stoletov K, Strnadel J, Zardouzian E, et al. Role of connexins in metastaticbreast cancer and melanoma brain colonization. J Cell Sci 2013; 126:904–13.
104. Haass NK, Houdek P, Brandner JM, et al. Expression patterns of con-nexins in merkel cell carcinoma and adjacent epidermis. In: Baumann KI,Moll I, Halata Z, editors. The Merkel Cell: Structure-Development-Function-Cancerogenesis. Berlin: Springer-Verlag, 2003; 219–22.
105. Penuela S, Gehi R, Laird D W. The biochemistry and function of pannexinchannels. Biochim Biophys Acta 2013; 1828: 15–22.
106. Saito-Katsuragi M, Asada H, Niizeki H, et al. Role for connexin 26 inmetastasis of human malignant melanoma: communication between mel-anoma and endothelial cells via connexin 26. Cancer 2007; 110: 1162–72.
right © Royal College of pathologists of Australasia
107. Naoi Y, Miyoshi Y, Taguchi T, et al. Connexin 26 expression is associatedwith lymphatic vessel invasion and poor prognosis in human breast cancer.Breast Cancer Res Treat 2007; 106: 11–7.
108. Haass NK, Smalley KS. Melanoma biomarkers: current status and utilityin diagnosis, prognosis, and response to therapy. Mol Diagn Ther 2009;13: 283–96.
109. Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysisof 17,600 melanoma patients: validation of the American JointCommittee on Cancer melanoma staging system. J Clin Oncol 2001;19: 3622–34.
110. McCarty MF, Bielenberg DR, Nilsson MB, et al. Epidermal hyperplasiaoverlying human melanoma correlates with tumour depth and angiogen-esis. Melanoma Res 2003; 13: 379–87.
111. Bijnsdorp IV, Rozendaal L, van Moorselaar RJ, et al. A predictive role fornoncancerous prostate cells: low connexin-26 expression in radicalprostatectomy tissues predicts metastasis. Br J Cancer 2012; 107:1963–8.
112. Kandouz M, Batist G. Gap junctions and connexins as therapeutic targetsin cancer. Expert Opin Ther Targets 2010; 14: 681–92.
. Unauthorized reproduction of this article is prohibited.
Related Documents











![A three-gene signature based on tumour microenvironment ......incidence is higher at age 15 to 19 years old emerging therapies such[1]. OSs has a high potential to metastasize immunotherapy](https://static.cupdf.com/doc/110x72/60e482f838222c48a42a973b/a-three-gene-signature-based-on-tumour-microenvironment-incidence-is-higher.jpg)