HUMAN MUTATION DATABASE IN BRIEF HUMAN MUTATION Database in Brief #____ (2015) Online © 2015 WILEY-LISS, INC. Received 23 April 2015; accepted revised manuscript 26 June 2015. Mediterranean Founder Mutation Database (MFMD): Taking Advantage from Founder Mutations in Genetics Diagnosis, Genetic Diversity and Migration History of the Mediterranean Population Hicham Charoute 1,2 , Amina Bakhchane 1 , Houda Benrahma 1 , Lilia Romdhane 3 , Khalid Gabi 1 , Hassan Rouba 1 , Malika Fakiri 2 , Sonia Abdelhak 3 , Guy Lenaers 4 and Abdelhamid Barakat 1,* 1 Laboratoire de Génétique Moléculaire Humaine, Institut Pasteur du Maroc, 1 Place Louis Pasteur, 20360 Casablanca, Morocco; 2 Laboratory of Agri-food and Health, Faculty of Sciences and Techniques, Hassan 1st University, BP 577, 26000, Settat, Morocco; 3 Laboratory of Biomedical Genomics and Oncogenetics LR11IPT05, Institut Pasteur de Tunis, 1002 Tunis, Tunisia; 4 Pôle de Recherche et d’Enseignement en Médecine Mitochondriale (PREMMi), Université d'Angers, CHU Bât IRIS/IBS, Rue des Capucins, 49933 Angers cedex 9, France *Correspondence to Abdelhamid Barakat, Laboratoire de Génétique Moléculaire Humaine, Département de Recherche Scientifique, Institut Pasteur du Maroc. 1 Place Louis Pasteur, 20360 Casablanca, Morocco. E-mail: [email protected]. Communicated by Alastair F. Brown ABSTRACT: The Mediterranean basin has been the theater of migration crossroads followed by settlement of several societies and cultures in prehistoric and historical times, with important consequences on genetic and genomic determinisms. Here, we present the Mediterranean Founder Mutation Database (MFMD), established to offer web-based access to founder mutation information in the Mediterranean population. Mutation data were collected from the literature and other online resources and systematically reviewed and assembled into this database. The information provided for each founder mutation includes DNA change, amino-acid change, mutation type and mutation effect, as well as mutation frequency and coalescence time when available. Currently, the database contains 383 founder mutations found in 210 genes related to 219 diseases. We believe that MFMD will help scientists and physicians to design more rapid and less expensive genetic diagnostic tests. Moreover, the coalescence time of founder mutations gives an overview about the migration history of the Mediterranean population. MFMD can be publicly accessed from http://mfmd.pasteur.ma. ©2015 Wiley Periodicals, Inc. KEY WORDS: Database, Founder Mutation, Mediterranean population, human genetics, mutation screening INTRODUCTION The Mediterranean Basin is the region of lands that surround the Mediterranean Sea at the intersection between three different continents: Africa, Asia and Europe. Since the original diaspora of peoples from the African OFFICIAL JOURNAL www.hgvs.org

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

HUMAN MUTATION
DATABASE IN BRIEF
HUMAN MUTATION Database in Brief #____ (2015) Online
© 2015 WILEY-LISS, INC.
Received 23 April 2015; accepted revised manuscript 26 June 2015.
Mediterranean Founder Mutation Database (MFMD):
Taking Advantage from Founder Mutations in
Genetics Diagnosis, Genetic Diversity and Migration
History of the Mediterranean Population
Hicham Charoute1,2, Amina Bakhchane1, Houda Benrahma1, Lilia Romdhane3, Khalid Gabi1, Hassan Rouba1, Malika Fakiri2, Sonia Abdelhak3, Guy Lenaers4 and Abdelhamid Barakat1,*
1 Laboratoire de Génétique Moléculaire Humaine, Institut Pasteur du Maroc, 1 Place Louis Pasteur, 20360 Casablanca, Morocco; 2 Laboratory of Agri-food and Health, Faculty of Sciences and Techniques, Hassan 1st University, BP 577, 26000, Settat, Morocco; 3 Laboratory of Biomedical Genomics and Oncogenetics LR11IPT05, Institut Pasteur de Tunis, 1002 Tunis, Tunisia; 4 Pôle de Recherche et d’Enseignement en Médecine Mitochondriale (PREMMi), Université d'Angers, CHU Bât IRIS/IBS, Rue des Capucins, 49933 Angers cedex 9, France
*Correspondence to Abdelhamid Barakat, Laboratoire de Génétique Moléculaire Humaine, Département de Recherche Scientifique, Institut Pasteur du Maroc. 1 Place Louis Pasteur, 20360 Casablanca, Morocco. E-mail: [email protected]. Communicated by Alastair F. Brown
ABSTRACT: The Mediterranean basin has been the theater of migration crossroads followed by settlement of several societies and cultures in prehistoric and historical times, with important consequences on genetic and genomic determinisms. Here, we present the Mediterranean Founder Mutation Database (MFMD), established to offer web-based access to founder mutation information in the Mediterranean population. Mutation data were collected from the literature and other online resources and systematically reviewed and assembled into this database. The information provided for each founder mutation includes DNA change, amino-acid change, mutation type and mutation effect, as well as mutation frequency and coalescence time when available. Currently, the database contains 383 founder mutations found in 210 genes related to 219 diseases. We believe that MFMD will help scientists and physicians to design more rapid and less expensive genetic diagnostic tests. Moreover, the coalescence time of founder mutations gives an overview about the migration history of the Mediterranean population. MFMD can be publicly accessed from http://mfmd.pasteur.ma. ©2015 Wiley Periodicals, Inc.
KEY WORDS: Database, Founder Mutation, Mediterranean population, human genetics, mutation screening
INTRODUCTION
The Mediterranean Basin is the region of lands that surround the Mediterranean Sea at the intersection between
three different continents: Africa, Asia and Europe. Since the original diaspora of peoples from the African
OFFICIAL JOURNAL
www.hgvs.org

e2 Charoute et al.
continent (Capelli et al. 2006), its population history witnessed many civilization movements both in prehistorical
and historical times, as it was and still is an important route of transport, trade and interaction. The Neolithic
period is marked by the transition from hunter-gathering to the spread of agriculture, that led to a demographic
transition, by increasing the population growth rate (Bocquet-Appel and Bar-Yosef 2008). In addition, the
Neolithic period is marked by the development of trading between different civilizations and by several migratory
movements. Ancient civilizations were located around the Basin, including, the Phoenician, the Greek, the Roman
and the Arab ones. The Phoenicians used their maritime expertise to become the principal traders in the
Mediterranean Sea, as they spread throughout the Mediterranean basin and establish important colonies and
centers of trade (Zalloua et al. 2008). Indeed, starting from Carthage, the most important Phoenician colony, this
population spread to settle throughout the western Mediterranean regions of the North African coast and the
Iberian Peninsula (Abulafia 2011). The Greeks had multiple colonies along the western Mediterranean and Black
Sea coasts, many of these being in Italy. They contested with the Phoenicians over control of colonies and trading
posts in Sicily and the western Mediterranean (Sacks and Murray 2009). After the Third Punic War between the
Romans and Phoenicians, the Roman Empire became the most powerful force in the Mediterranean region
(Abulafia 2011). Then, in the early part of the middle ages, the Arabs conquered a large region of the
Mediterranean Sea, unifying the Arabian Peninsula with a vast part of the Byzantine Empire, the whole Persian
lands, North Africa and the Iberian Peninsula (Hitti 1948).
All these demographic events, population interactions and migratory movements that occurred along the
Mediterranean Sea, had a crucial impact on the genetic structure of the Mediterranean population, which have
already been studied using Mitochondrial DNA (mtDNA), Y chromosome and Autosomal molecular markers and
sequencing. The population history and diversity have important medical genetic implications (Laberge et al.
2005). In particular populations, many disease-causing mutations can be traced to a founder individual, in whom
the mutation first appeared. Founder effect occurs when a new group is established by a small number of
individuals that are separated from a larger population. The new population represents only a restricted sample of
the genetic variability that was present in the original population (Neuhausen 2000) and further endogamy led to
modifications in DNA variation frequencies. Therefore, founder effects explain the high prevalence of some
disease-causing mutations in a particular population, and haplotype analysis can demonstrate that a common
mutation is due to a founder effect with linkage disequilibrium to the adjacent genetic markers. The size of the
linkage disequilibrium intervals helps to estimate the age of the original mutation spreading into a given
population (Zeegers et al. 2004). Identification of founder mutations in a particular population has been useful for
the improvement of molecular diagnosis and genetic counseling. Screening for few prevalent founder mutations is
quicker and cheaper than testing many rare mutations (Ferla et al. 2007). Moreover, determining founder
mutations and their regional distribution in different ethnic groups can help scientists to study the genetic diversity
and migration history of these populations (Ostrer 2001).
There are a lot of published findings that highlight the importance of founder mutations in medical genetics. For
instance, various ancient founder mutations transmitted across tens or hundreds of generations, tend to migrate
over long distances and spread widely around the Mediterranean Sea. This case is illustrated by the c.35delG
common founder mutation in the GJB2 gene that causes deafness, and appears to have arisen in the Middle East
about 500 generations ago (10000 years) and spread across Europe and around the Mediterranean basin (Van Laer
et al. 2001). Furthermore, several genetic disorders have a wide geographical presence along the Mediterranean
region, with multiple disease-causing founder mutations. This situation is observed for the familial Mediterranean
fever disease; multiple founder mutations affecting the MEFV gene were shared between different Mediterranean
populations. Phylogenetic analyses based on allele frequencies of the most common MEFV founder mutations
(M694V, V726A, M680I, M694I, and E148Q) in 14 Mediterranean populations, suggested three major possible
gene flows between populations (Papadopoulos et al. 2008). The current distribution pattern of MEFV mutations
may be influenced by several historical events: the Byzantine Empire, the Arab conquests, the Ottoman
dominance, the dispersal of the Armenian nation and the Jewish Diaspora (Papadopoulos et al. 2008). Screening
frequent founder mutations in a particular population as the first step in molecular genetic diagnosis is a cost-
effective methodology before considering alternative approaches such as Next Generation Sequencing (Ponti et al.
2015).
In this paper, we describe the Mediterranean Founder Mutation Database (MFMD), a comprehensive online
database established to collect and document human founder mutations reported in the Mediterranean population.
We believe that this is the first database for founder mutations in the Mediterranean region. The database will help

Mediterranean Founder Mutation Database e3
scientists and physicians in having an overview about the spectrum of founder mutations found in the
Mediterranean population, and will be beneficial to understand the history, demography and migration flows of the
Mediterranean populations. Furthermore, MFMD provides useful information for the diagnosis and prevention of
genetic diseases. The MFMD can be publicly accessed from http://mfmd.pasteur.ma.
METHODS
Software design and implementation
The MFMD was implemented using a three-tier model (client, application server and database). The web
interface was designed following the Model-View-Controller (MVC) design pattern, and developed using HTML
(HyperText Markup Language), CSS (Cascading Style Sheets) and Javascript programming languages. The PHP
(version 5.4) scripting language was used for all data retrieval and output. The data were managed with MySQL
(version 5.5) database management system. The PHPlot 6.1.0 tool (http://phplot.sourceforge.net/) was used to
generate graphs in the statistics page of the MFMD web interface. The R statistical programming language was
used to draw Bar charts and to perform Heatmap Analysis.
Literature search and data extraction
We conducted a literature search using the PubMed database for articles, published before March, 1st 2015. The
search strategy to identify all relevant studies was based on a combination of the following keywords: (“founder”
OR “common”) AND (“effect” OR “mutation” OR “allele” OR “variation” OR “variant” OR “loci” OR “locus”
OR “haplotype” OR “microsatellite” OR “polymorphism”), without any restriction on language. In addition, to
determine articles conducted in subjects of Mediterranean origin, we used a list of Mediterranean country names to
filter out non relevant studies. Remaining abstracts were downloaded in XML-format from PubMed and stored in
the database. Data were extracted from abstracts by independent reviewers and validated by the database editor in
chief. Genes, diseases and mutations were identified and highlighted in abstracts using the PubTator text mining
tool, to facilitate data extraction. Eligible studies were selected if they were conducted in subjects of Mediterranean
origin, and reported a founder mutation. We collected both founder mutations proved by haplotype analysis and
mutations with suggested founder effect. Moreover, we also checked all references cited in the identified articles
for any additional literature. The following data were extracted from the eligible studies: publication information,
subject information (population of origin, geographical region and ethnic group) and mutation information (DNA
change, amino acid change, prevalence of mutation among affected individuals and coalescence time) (Figure 1A).
The Mutalyzer 2.0.6 software (Wildeman et al. 2008) was used to check mutation nomenclature according to the
HGVS recommendations. Gene symbol follows the HUGO gene nomenclature (Gray et al. 2014). Genetic diseases
registered in the database have been classified using the World Health Organization (WHO) International
Statistical Classification of Diseases and Related Health Problems 10th Revision (ICD-10) Version 2010
(http://apps.who.int/classifications/icd10/browse/2010/en).
The current version of the database registered founder mutation data from the following Mediterranean
countries: Albania, Algeria, Bosnia and Herzegovina, Croatia, Cyprus, Egypt, France, Greece, Israel, Italy,
Lebanon, Libya, Malta, Monaco, Morocco, Palestine, Slovenia, Spain, Syria, Tunisia and Turkey. All patients are
classified according to their population of origin before emigration.

e4 Charoute et al.
RESULTS
Data included in the database
The flowchart of the study selection is shown in Figure 1B. The literature search yielded 1397 publications. A
total of 1006 studies did not meet the inclusion criteria and were rejected based on the title, abstract or the full text
screening. Three hundred and eighty-one studies met the inclusion criteria and were included in the database.
Moreover, 28 additional articles were obtained from reviewing references from the eligible studies. Finally, a total
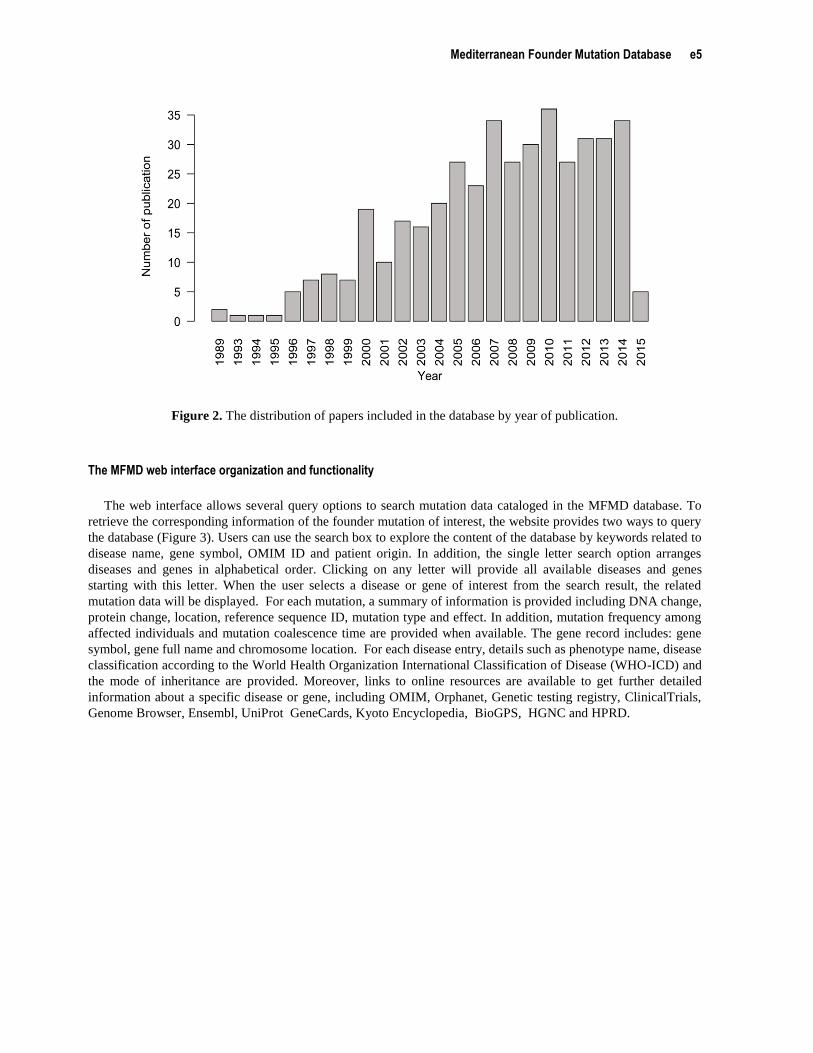
of 419 studies were included in the database. These relevant articles dated from 1989 to 2015 and the distribution
of number of articles per year showed an increasing trend (Figure 2).
Figure 1. Database curation steps and data extraction form literature. (A) Workflow for the retrieval of articles and
extraction of founder mutation information from literature. (B) Flowchart of study identification, inclusion, and
exclusion.
Mediterranean Founder Mutation Database e5
Figure 2. The distribution of papers included in the database by year of publication.
The MFMD web interface organization and functionality
The web interface allows several query options to search mutation data cataloged in the MFMD database. To
retrieve the corresponding information of the founder mutation of interest, the website provides two ways to query
the database (Figure 3). Users can use the search box to explore the content of the database by keywords related to
disease name, gene symbol, OMIM ID and patient origin. In addition, the single letter search option arranges
diseases and genes in alphabetical order. Clicking on any letter will provide all available diseases and genes
starting with this letter. When the user selects a disease or gene of interest from the search result, the related
mutation data will be displayed. For each mutation, a summary of information is provided including DNA change,
protein change, location, reference sequence ID, mutation type and effect. In addition, mutation frequency among
affected individuals and mutation coalescence time are provided when available. The gene record includes: gene
symbol, gene full name and chromosome location. For each disease entry, details such as phenotype name, disease
classification according to the World Health Organization International Classification of Disease (WHO-ICD) and
the mode of inheritance are provided. Moreover, links to online resources are available to get further detailed
information about a specific disease or gene, including OMIM, Orphanet, Genetic testing registry, ClinicalTrials,
Genome Browser, Ensembl, UniProt GeneCards, Kyoto Encyclopedia, BioGPS, HGNC and HPRD.
e6 Charoute et al.
Figure 3. The home page of the MFMD website.
Characteristics of disease and gene data
A total of 219 Mendelian diseases are registered in MFMD. Data analysis reveals that 67.1% of all the inherited
diseases follow autosomal recessive mode of inheritance, while autosomal dominant transmission represents
21.9% of all cases. In addition, 2.3% are X-linked and 8.7% of genetic diseases have multiple modes of
transmission (Figure 4A). According to the WHO-ICD-10 disease classification, the main disease categories in the
database include endocrine, nutritional and metabolic diseases (26.5%), followed by nervous system diseases
(20.5%) and congenital malformations and chromosomal abnormalities (17.4%). Diseases of blood and immune
mechanism (11%), neoplasms (7.3%), diseases of the eye and adnexa (5.5%) are also prevalent (Figure 4B).

Mediterranean Founder Mutation Database e7
Figure 4. Summary statistics about genetic diseases registered in the database. (A) Distribution of genetic diseases
according to the mode of inheritance. (B) Classification of genetic diseases using the WHO ICD-10 system.
Comparison of founder mutation spectrum between Mediterranean populations
At the time of its first online release, the MFMD database contained 383 founder mutations. At present, the
disease with the highest number of founder mutations is Breast-ovarian cancer, with 21 mutations in the BRCA1
gene and 13 mutations in the BRCA2 gene, the majority of them being found in Spanish, Italian and Greek
populations. Founder mutations are also prevalent in Ataxia-telangiectasia disease, with 15 out of 18 mutations in
the ATM gene reported in the Italian population. The 35delG mutation in GJB2 gene is the most common
mutation, detected in 8 Mediterranean countries from Europe (Greece, Italy and Spain), Asia (Lebanon and
Turkey), and Africa (Egypt, Morocco and Tunisia). More than one founder mutation is responsible for the Beta
thalassemia in the Mediterranean population. The most recurrent mutation c.118C>T (also known as Codon 39,
C>T) in HBB gene has been reported in 3 populations. Five founder mutations responsible for Familial
Mediterranean fever widely spread around the Mediterranean Sea, were described in 4 populations (Egypt,
Lebanon, Syria and Tunisia). On the other hand, current data showed that 270 mutations were reported in only one
population.
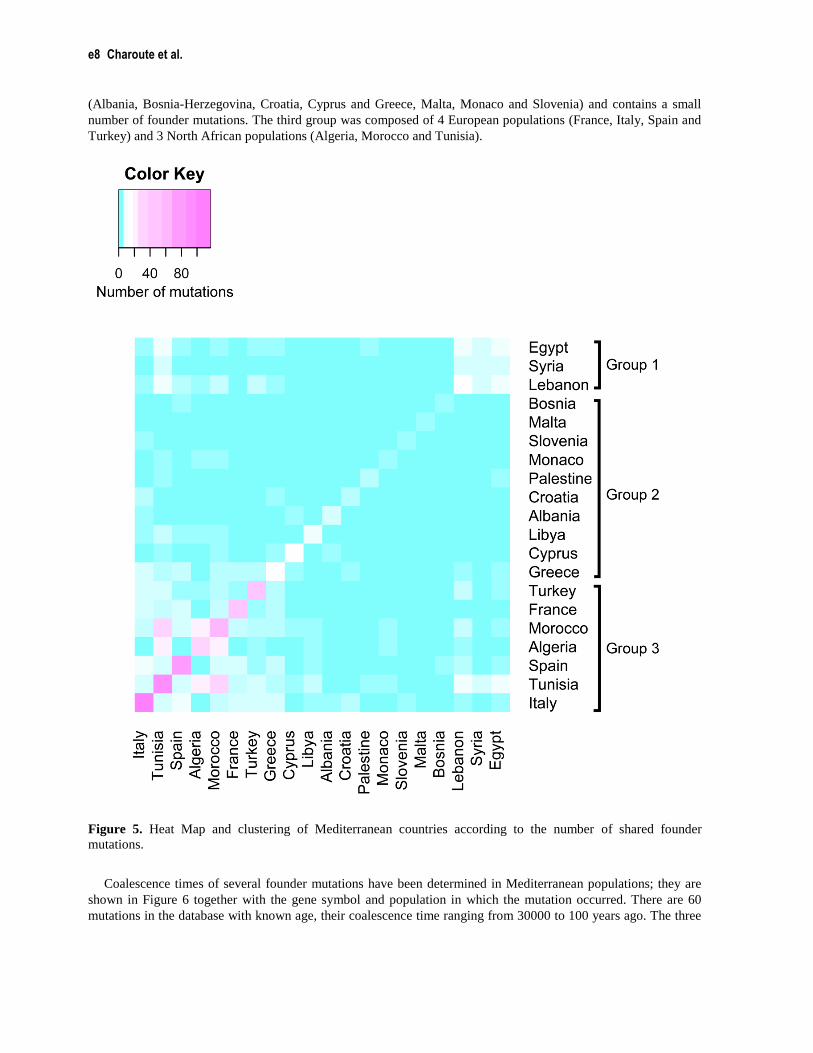
We clustered Mediterranean populations, using the number of shared mutations as a linkage criterion which
indicates the similarity between populations. The results are presented as a heatmap in Figure 5. Each colored cell
of the Heat Map indicates the number of founder mutations shared between two countries, and the diagonal cells
show the total number of mutations in a particular country. Current data indicate the presence of more than 117
founder mutations in the Italian population, which represents 30.55 % of all mutations included in the database.
Italy is followed by Tunisia, Spain and Morocco with 89, 71 and 54 mutations, respectively. Based on the resulting
pair wise comparisons between populations, we can define three clusters: the first is composed of 3 Arab
populations, including Egypt, Lebanon and Syria. The second is mainly composed of European populations
e8 Charoute et al.
(Albania, Bosnia-Herzegovina, Croatia, Cyprus and Greece, Malta, Monaco and Slovenia) and contains a small
number of founder mutations. The third group was composed of 4 European populations (France, Italy, Spain and
Turkey) and 3 North African populations (Algeria, Morocco and Tunisia).
Figure 5. Heat Map and clustering of Mediterranean countries according to the number of shared founder
mutations.
Coalescence times of several founder mutations have been determined in Mediterranean populations; they are
shown in Figure 6 together with the gene symbol and population in which the mutation occurred. There are 60
mutations in the database with known age, their coalescence time ranging from 30000 to 100 years ago. The three

Mediterranean Founder Mutation Database e9
oldest mutations were found in the MEFV gene with the following coalescence time: 30000, 23000 and 15000
years ago. The most recent founder effect was reported in the ACVRL1 gene and occurred 100 years ago.
Figure 6. Timing of founder mutations in Mediterranean populations.
DISCUSSION
The Mediterranean basin has been the point of intersection between several civilizations, and religions that
influenced each other (Abulafia 2011), thus providing an exceptional context to examine the impact of
demographic events and migration on the actual genetic landscape. The study of the genetic structure of
Mediterranean populations using Y-chromosome microsatellites (Quintana-Murci et al. 2003) and mtDNA
variations (Plaza et al. 2003), revealed a low genetic structure between the northern and southern shores of the
Mediterranean sea. In addition, Y-chromosome analysis failed to find any significant barrier to genetic exchange
between populations from the eastern part of the Mediterranean (Manni et al. 2002). On the basis of X-
chromosome polymorphisms, high overall genetic population homogeneity was found in the Mediterranean area,
except in north-west African populations (Morocco) (Tomas et al. 2008). In the western part of the Mediterranean
area, the Strait of Gibraltar has been suggested by several studies as a geographic barrier to gene flow between
north-west Africa and the Iberian Peninsula (Tomas et al. 2008)(Ennafaa et al. 2009). The Mediterranean Sea
might also act as a partial barrier to gene flow between the northern and southern shores (Athanasiadis et al. 2010).
On the other hand, the Arab region played an important role in shaping the current genetic structure of the
Mediterranean population. It was an important area for human population interactions and movements, as well as
an interesting bridge for gene flow between Africa, Asia, and Europe (Tadmouri et al. 2014).
MFMD is a carefully curated database extracted from the literature for all described founder mutations reported
in the Mediterranean population, providing detailed information for each one. A user-friendly web interface was

e10 Charoute et al.
designed to provide a flexible access to the database content. MFMD could be a useful resource for research
purposes in medical and population genetics studies on the Mediterranean population.
More than 30% of founder mutations registered in the database were reported in the Italian population; several
of them have been found in Sardinia and Sicily islands. Due to geographical and cultural barriers to gene flow,
some populations can be considered as genetic isolates which lack significant genetic sub-structures such as
Sardinian (Pardo et al. 2012; Di Gaetano et al. 2014) and Sicilian populations (Rickards et al. 1998; Sarno et al.
2014). The Italian Peninsula hosted various human migration events and cultural exchanges, due to its central
geographical location in the Mediterranean basin. Furthermore, numerous ethno-linguistic minorities inhabit Italy
and represent 5% of the total population (Capocasa et al. 2014). Consequently, the combination of linguistic and
geographic factors may have influenced the genetic isolation of Italian populations (Capocasa et al. 2014) in
combination with endogamy and genetic drift phenomena.
The database registered 60 mutations with known coalescence time, 24 older than 2000 years ago, and 36
mutations more recent. We observed that 13 out of 24 old mutations were reported in more than two populations;
in contrast only 6 out of 36 recent mutations were reported in different populations. Thus, we can hypothesize that
founder mutations reported in only one population, which is the case of 270 mutations, can be due to recent
founder effects.
The Heat Map constructed based on the number of shared mutations between Mediterranean populations,
revealed three main groups (labelled as Group 1, Group 2 and Group 3). The number of founder mutations
reported in populations belonging to Group 1 and Group 2 was relatively low. Both groups were mainly composed
of Eastern Mediterranean countries, for which insufficient amount of molecular data on genetic diseases has been
published. The interest for human genetics research in this part of the Mediterranean region is relatively recent and
there remains a clear need for more investments to build capacities in medical genetics. In contrast, remarkable
efforts in exploring the molecular bases of genetic disorders in western Mediterranean countries forming Group 3,
has led to a wide characterization of disease-causing mutations. Clinicians and researchers from North Africa
(Morocco, Tunisia and Algeria) benefit from collaboration with northern Mediterranean partners to bridge the gap
in healthcare infrastructures and to overcome the lack of molecular genetic technologies. The Heat Map showed
that western Mediterranean populations (Morocco, Algeria, Tunisia, France, Italy and Spain) hold the majority of
founder mutations and have several shared founder mutations. In particular, North African populations (Morocco,
Algeria and Tunisia) shared 15 founder mutations. This is in agreement with the analysis of mitochondrial DNA
(mtDNA) sequences performed in Western Mediterranean populations that showed multidirectional gene flow in
this region (Plaza et al. 2003). Moreover, recent migration events from North Africa have a significant contribution
to the genetic diversity in the southern European region, consequently North African disease risk alleles should be
taken in consideration when developing molecular diagnostic tests for genetic disorders in European populations
(Botigué et al. 2013).
Differences in the prevalence of many diseases and mutations have been observed in different populations and
ethnic groups, as a result of founder effects (Neuhausen 2000). For example, more than 30 inherited diseases are
more prevalent in the Finland than in other populations (Polvi et al. 2013). On the other hand, many mutations in
MFMD database are unique to a specific ethnicity, such as Jewish groups. Thus, patient ancestry can be used as a
criterion for diagnosis (Ostrer 2001).
To date, the database contains 219 genetic diseases; the majority of them follow an autosomal recessive mode
of inheritance (67.1%). The high rate of consanguinity in this region and especially in Arab countries may
contribute to this observation (Tadmouri et al. 2009). The disease classification reveals that the majority of
diseases in MFMD belong to three categories: endocrine, nutritional and metabolic diseases (26%),
neurodegenerative diseases (21%) and congenital malformations and chromosomal abnormalities (17%).
Similarly, endocrine and metabolic diseases are also prevalent in Morocco; they represent 24% of all genetic
disorders reported in the Moroccan Genetics Disease Database (MGDD), then followed by congenital
malformations and chromosomal abnormalities (22%) and nervous system diseases (19%) (Charoute et al. 2014).
In contrast, the classification of genetics disease registered in the “Catalogue of Transmission Genetics in Arabs"
(CTGA) database, showed that congenital malformations and chromosomal abnormalities are the most prevalent
category of genetic disorders in the Arab population (the United Arab Emirates, Bahrain, Oman and Qatar). They
represent 34% of all recorded diseases, followed by endocrine and metabolic disorders (19%) and nervous system
disorders (11%) (Tadmouri 2012). Moreover, the classification of genetic diseases reported in Tunisia is in
accordance with the data observed in the Arab population. Congenital malformations and chromosomal

Mediterranean Founder Mutation Database e11
abnormalities constitute 30% of all genetic disorders found in the Tunisian population, followed by endocrine and
metabolic disorders (19%) and nervous system disorders (11%) (Romdhane et al. 2011). Differences in the
classification of genetic disorders may be explained by the fact that genetic disorders are not equally distributed in
the Arab region, where some genetic diseases are presents in many countries, whereas almost half of diseases were
reported only in a single population (Tadmouri et al. 2014). In addition, the majority of MFMD data were collected
from European countries (Italy, France and Spain), consequently the heterogeneity between populations registered
in both databases (MFMD and CTGA) may be responsible for these differences in the classification diseases.
Founder mutation data were collected from 419 articles, the distribution of these studies against time shows an
increasing trend. The number of founder mutations discovered in the Mediterranean population is expected to keep
increasing in the coming years, due to the use of next-generation sequencing technology. In this respect,
identification of founder mutations facilitates the development of efficient molecular diagnostic assays. Testing
prevalent founder mutations would be cheaper and quicker than screening for various rare mutations. In addition,
the number of carriers of a given founder mutation, allows scientists to perform penetrance analyses of this
mutation among populations (Neuhausen 2000; Zeegers et al. 2004; Ferla et al. 2007; Romdhane et al. 2012).
In conclusion, we developed an online database of founder mutations in the Mediterranean population. It
provides relevant information to the research community, through a user-friendly interface. All efforts have been
made to construct a comprehensive database by conducting an exhaustive search for relevant studies reporting
founder mutations. However, the current version of the MFMD database might miss some mutation data. To
ensure the collection of the complete set of founder mutation data in the Mediterranean population, we will
develop new research strategies including more biomedical literature databases and text-mining tools. We will
continue collecting new founder mutation data from published literature, to keep MFMD updated. In addition, the
database supports new data submission from public users, using an online submission form. The MFMD database
will be beneficial to the improvement of disease diagnosis and better understanding the gene flow in the
Mediterranean population.
ACKNOWLEDGMENTS
This work was supported by Pasteur Institute of Morocco (IPM) and a collaborative project between the French
National Institute of Health and Medical Research (INSERM) and the Moroccan National Centre for Scientific and
Technical Research (CNRST).
REFERENCES
Abulafia D. 2011. The Great Sea: A Human History of the Mediterranean. Oxford University Press.
Athanasiadis G, González-Pérez E, Esteban E, Dugoujon J-M, Stoneking M, Moral P. 2010. The Mediterranean Sea as a barrier
to gene flow: evidence from variation in and around the F7 and F12 genomic regions. BMC Evol. Biol. 10: 84.
Bocquet-Appel J-P, Bar-Yosef O. 2008. The Neolithic Demographic Transition and its Consequences. New York: Springer.
Botigué LR, Henn BM, Gravel S, Maples BK, Gignoux CR, Corona E, Atzmon G, Burns E, Ostrer H, Flores C, Bertranpetit J,
Comas D, et al. 2013. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe.
Proc. Natl. Acad. Sci. U. S. A. 110: 11791–11796.
Capelli C, Redhead N, Romano V, Calì F, Lefranc G, Delague V, Megarbane A, Felice AE, Pascali VL, Neophytou PI, Poulli
Z, Novelletto A, et al. 2006. Population structure in the Mediterranean basin: a Y chromosome perspective. Ann. Hum.
Genet. 70: 207–225.
Capocasa M, Anagnostou P, Bachis V, Battaggia C, Bertoncini S, Biondi G, Boattini A, Boschi I, Brisighelli F, Caló CM, Carta
M, Coia V, et al. 2014. Linguistic, geographic and genetic isolation: a collaborative study of Italian populations. J.
Anthropol. Sci. Riv. Antropol. JASS Ist. Ital. Antropol. 92: 201–231.
Charoute H, Nahili H, Abidi O, Gabi K, Rouba H, Fakiri M, Barakat A. 2014. The Moroccan Genetic Disease Database
(MGDD): a database for DNA variations related to inherited disorders and disease susceptibility. Eur. J. Hum. Genet. EJHG
22: 322–326.
Ennafaa H, Cabrera VM, Abu-Amero KK, González AM, Amor MB, Bouhaha R, Dzimiri N, Elgaaïed AB, Larruga JM. 2009.
Mitochondrial DNA haplogroup H structure in North Africa. BMC Genet. 10: 8.

e12 Charoute et al.
Ferla R, Calò V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, Surmacz E, Colucci G, Bazan V, Russo A. 2007. Founder
mutations in BRCA1 and BRCA2 genes. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 18 Suppl 6: vi93–98.
Gaetano C Di, Fiorito G, Ortu MF, Rosa F, Guarrera S, Pardini B, Cusi D, Frau F, Barlassina C, Troffa C, Argiolas G,
Zaninello R, et al. 2014. Sardinians genetic background explained by runs of homozygosity and genomic regions under
positive selection. PloS One 9: e91237.
Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. 2014. Genenames.org: the HGNC resources in 2015. Nucleic Acids
Res.
Hitti PK. 1948. The Arabs: A Short History. MacMillan.
Laberge A-M, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, Mitchell GA. 2005. Population history and its impact on
medical genetics in Quebec. Clin. Genet. 68: 287–301.
Laer L Van, Coucke P, Mueller RF, Caethoven G, Flothmann K, Prasad SD, Chamberlin GP, Houseman M, Taylor GR,
Heyning CM Van de, Fransen E, Rowland J, et al. 2001. A common founder for the 35delG GJB2 gene mutation in
connexin 26 hearing impairment. J. Med. Genet. 38: 515–518.
Manni F, Leonardi P, Barakat A, Rouba H, Heyer E, Klintschar M, McElreavey K, Quintana-Murci L. 2002. Y-chromosome
analysis in Egypt suggests a genetic regional continuity in Northeastern Africa. Hum. Biol. 74: 645–658.
Neuhausen SL. 2000. Founder populations and their uses for breast cancer genetics. Breast Cancer Res. BCR 2: 77–81.
Ostrer H. 2001. A genetic profile of contemporary Jewish populations. Nat. Rev. Genet. 2: 891–898.
Papadopoulos VP, Giaglis S, Mitroulis I, Ritis K. 2008. The population genetics of familial mediterranean fever: a meta-
analysis study. Ann. Hum. Genet. 72: 752–761.
Pardo LM, Piras G, Asproni R, Gaag KJ van der, Gabbas A, Ruiz-Linares A, Knijff P de, Monne M, Rizzu P, Heutink P. 2012.
Dissecting the genetic make-up of North-East Sardinia using a large set of haploid and autosomal markers. Eur. J. Hum.
Genet. EJHG 20: 956–964.
Plaza S, Calafell F, Helal A, Bouzerna N, Lefranc G, Bertranpetit J, Comas D. 2003. Joining the pillars of Hercules: mtDNA
sequences show multidirectional gene flow in the western Mediterranean. Ann. Hum. Genet. 67: 312–328.
Polvi A, Linturi H, Varilo T, Anttonen A-K, Byrne M, Fokkema IFAC, Almusa H, Metzidis A, Avela K, Aula P, Kestilä M,
Muilu J. 2013. The Finnish disease heritage database (FinDis) update-a database for the genes mutated in the Finnish
disease heritage brought to the next-generation sequencing era. Hum. Mutat. 34: 1458–1466.
Ponti G, Castellsagué E, Ruini C, Percesepe A, Tomasi A. 2015. Mismatch repair genes founder mutations and cancer
susceptibility in Lynch syndrome. Clin. Genet. 87: 507–516.
Quintana-Murci L, Veitia R, Fellous M, Semino O, Poloni ES. 2003. Genetic structure of Mediterranean populations revealed
by Y-chromosome haplotype analysis. Am. J. Phys. Anthropol. 121: 157–171.
Rickards O, Martinez-Labarga C, Scano G, Stefano GF De, Biondi G, Pacaci M, Walter H. 1998. Genetic history of the
population of Sicily. Hum. Biol. 70: 699–714.
Romdhane L, Abdelhak S, Research Unit on Molecular Investigation of Genetic Orphan Diseases, Collaborators. 2011. Genetic
diseases in the Tunisian population. Am. J. Med. Genet. A. 155A: 238–267.
Romdhane L, Kefi R, Azaiez H, Halim N Ben, Dellagi K, Abdelhak S. 2012. Founder mutations in Tunisia: implications for
diagnosis in North Africa and Middle East. Orphanet J. Rare Dis. 7: 52.
Sacks D, Murray O. 2009. Encyclopedia of the Ancient Greek World. Infobase Publishing.
Sarno S, Boattini A, Carta M, Ferri G, Alù M, Yao DY, Ciani G, Pettener D, Luiselli D. 2014. An ancient Mediterranean
melting pot: investigating the uniparental genetic structure and population history of sicily and southern Italy. PloS One 9:
e96074.
Tadmouri GO. 2012. Genetic disorders in Arabs. In : Tadmouri GO, Taleb Al Ali M, and Al Khaja N, editors. Genetic
Disorders in the Arab World: Qatar. Centre for Arab Genomic Studies, Dubai, United Arab Emirates, p 10–29.

Mediterranean Founder Mutation Database e13
Tadmouri GO, Nair P, Obeid T, Ali MT Al, Khaja N Al, Hamamy HA. 2009. Consanguinity and reproductive health among
Arabs. Reprod. Health 6: 17.
Tadmouri GO, Sastry KS, Chouchane L. 2014. Arab gene geography: From population diversities to personalized medical
genomics. Glob. Cardiol. Sci. Pract. 2014: 394–408.
Tomas C, Sanchez JJ, Barbaro A, Brandt-Casadevall C, Hernandez A, Dhiab M Ben, Ramon M, Morling N. 2008. X-
chromosome SNP analyses in 11 human Mediterranean populations show a high overall genetic homogeneity except in
North-west Africans (Moroccans). BMC Evol. Biol. 8: 75.
Wildeman M, Ophuizen E van, Dunnen JT den, Taschner PEM. 2008. Improving sequence variant descriptions in mutation
databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 29: 6–13.
Zalloua PA, Platt DE, El Sibai M, Khalife J, Makhoul N, Haber M, Xue Y, Izaabel H, Bosch E, Adams SM, Arroyo E, López-
Parra AM, et al. 2008. Identifying Genetic Traces of Historical Expansions: Phoenician Footprints in the Mediterranean.
Am. J. Hum. Genet. 83: 633–642.
Zeegers MPA, Poppel F van, Vlietinck R, Spruijt L, Ostrer H. 2004. Founder mutations among the Dutch. Eur. J. Hum. Genet.
EJHG 12: 591–600.
Related Documents











