Mechanism of colon cancer cell apoptosis mediated by pyropheophorbide-a methylester photosensitization Jean-Yves Matroule 1 , Chris M Carthy 3 , David J Granville 3 , Olivier Jolois 2 , David WC Hunt 3 and Jacques Piette* ,1 1 Laboratory of Virology and Immunology, University of Liege, B-4000 Liege, Belgium; 2 Laboratory of Human Histology, University of Liege, B-4000 Liege, Belgium; 3 QLT Inc., 887 Great Northern Way, Vancouver, BC, Canada, V5T 4T5 Pyropheophorbide-a methylester (PPME) is a second generation of photosensitizers used in photodynamic therapy (PDT). We demonstrated that PPME photo- sensitization triggered apoptosis of colon cancer cells as measured by using several classical parameters such as DNA laddering, PARP cleavage, caspase activation and mitochondrial release of cytochrome c. Preincubation of cells with N-acetyl cysteine (NAC) or pyrolidine dithiocarbamate (PDTC) protected against apoptosis mediated by PPME photosensitization showing that reactive oxygen species (ROS) are involved as second messengers. On the other hand, photosensitization carried out in the presence of deuterium oxide (D 2 O) which enhances singlet oxygen ( 1 O 2 ) lifetime only increases necrosis without aecting apoptosis. Since PPME was localized in the endoplasmic reticulum (ER)/Golgi system and lysosomes, other messengers than ROS were tested such as calcium, Bid, Bap31, phosphorylated Bcl-2 and caspase-12 but none was clearly identified as being involved in triggering cytochrome c release from mitochondria. On the other hand, we demonstrated that the transduction pathways leading to NF-kB activation and apoptosis were clearly independent although NF-kB was shown to counteract apoptosis mediated by PPME photosensitization. Onco- gene (2001) 20, 4070 – 4084. Keywords: photosensitization; photodynamic therapy; pyropheophorbide; apoptosis; caspase; NF-kB Introduction Photodynamic therapy (PDT) is an emerging modality that shows considerable promise for the treatment of solid tumors and a range of non-oncologic disorders (Dougherty et al., 1998; Murphree et al., 1996; Pass, 1993). This approach is predicated on the use of a sensitizing molecule (photosensitizer) that is adminis- tered, usually via intravenous injection, and allowed to localize somewhat selectively in cancerous tissue or other sites of therapeutic interest (e.g. atheromatous plaque, neovascular regions). Visible light irradiation is used to activate the photosensitizer yielding primarily singlet oxygen, the reactive oxygen species (ROS) believed responsible for the cytotoxic action of PDT. The specificity achieved from drug uptake selectivity combined with light targeting makes PDT an appealing approach. The first photosensitizer approved for PDT is a porphyrin oligomer (Photofrin) which is highly eec- tive but exhibits the drawbacks of (i) a tendency to cause prolonged skin photosensitivity, (ii) an activation wavelength lower than that optimal for eective penetration through tissue, and (iii) a poorly defined chemical composition which makes a detailed under- standing of its mode of action and pharmacokinetics dicult. To address these issues, new photosensitizers are being developed and a number of new agents are now in clinical trials. Several groups have recently reported the antitumor ecacy of pheophorbide-and pyropheophorbide-based PDT (Bellnier et al., 1993; Henderson et al., 1997; Payne et al., 1996). These compounds are chemically well characterized, absorb light above 600 nm and produce less long-term skin photosensitivity than Photofrin. In the pyropheophor- bide-a series, either as methylesters or as carboxylic acids, photosensitizing activity increases with the length of the alkyl ether side-chain. These alkyl ether derivatives, although having similar photophysical properties (singlet oxygen and fluorescence yields), exhibit remarkable dierences in photosensitizing eciency in biological systems (Pandey et al., 1996). The cellular mechanisms of PDT are still under investigation. Singlet oxygen, the putative cytotoxic agent in PDT, is initially formed by the energy transfer from the excited triplet state of the photosensitizer to ground-state oxygen (Weishaupt et al., 1976). Several factors are thought to work together to achieve complete tumor eradication in vivo. These include direct cellular damage leading to necrosis or apoptosis, vascular destruction, inflammatory changes and sub- sequent immune responses (Korbelik, 1996). While the mechanism responsible for the eective destruction of the malignant tissues by PDT is not completely Oncogene (2001) 20, 4070 – 4084 ª 2001 Nature Publishing Group All rights reserved 0950 – 9232/01 $15.00 www.nature.com/onc *Correspondence: J Piette, Laboratory of Virology and Immunology, Institute of Pathology B23, University of Liege, B-4000 Liege, Belgium; E-mail: [email protected]. Received 2 January 2001; revised 28 March 2001; accepted 11 April 2001

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Mechanism of colon cancer cell apoptosis mediated by pyropheophorbide-amethylester photosensitization
Jean-Yves Matroule1, Chris M Carthy3, David J Granville3, Olivier Jolois2, David WC Hunt3 andJacques Piette*,1
1Laboratory of Virology and Immunology, University of Liege, B-4000 Liege, Belgium; 2Laboratory of Human Histology,University of Liege, B-4000 Liege, Belgium; 3QLT Inc., 887 Great Northern Way, Vancouver, BC, Canada, V5T 4T5
Pyropheophorbide-a methylester (PPME) is a secondgeneration of photosensitizers used in photodynamictherapy (PDT). We demonstrated that PPME photo-sensitization triggered apoptosis of colon cancer cells asmeasured by using several classical parameters such asDNA laddering, PARP cleavage, caspase activation andmitochondrial release of cytochrome c. Preincubation ofcells with N-acetyl cysteine (NAC) or pyrolidinedithiocarbamate (PDTC) protected against apoptosismediated by PPME photosensitization showing thatreactive oxygen species (ROS) are involved as secondmessengers. On the other hand, photosensitizationcarried out in the presence of deuterium oxide (D2O)which enhances singlet oxygen (1O2) lifetime onlyincreases necrosis without a�ecting apoptosis. SincePPME was localized in the endoplasmic reticulum(ER)/Golgi system and lysosomes, other messengersthan ROS were tested such as calcium, Bid, Bap31,phosphorylated Bcl-2 and caspase-12 but none wasclearly identi®ed as being involved in triggeringcytochrome c release from mitochondria. On the otherhand, we demonstrated that the transduction pathwaysleading to NF-kB activation and apoptosis were clearlyindependent although NF-kB was shown to counteractapoptosis mediated by PPME photosensitization. Onco-gene (2001) 20, 4070 ± 4084.
Keywords: photosensitization; photodynamic therapy;pyropheophorbide; apoptosis; caspase; NF-kB
Introduction
Photodynamic therapy (PDT) is an emerging modalitythat shows considerable promise for the treatment ofsolid tumors and a range of non-oncologic disorders(Dougherty et al., 1998; Murphree et al., 1996; Pass,1993). This approach is predicated on the use of asensitizing molecule (photosensitizer) that is adminis-
tered, usually via intravenous injection, and allowed tolocalize somewhat selectively in cancerous tissue orother sites of therapeutic interest (e.g. atheromatousplaque, neovascular regions). Visible light irradiation isused to activate the photosensitizer yielding primarilysinglet oxygen, the reactive oxygen species (ROS)believed responsible for the cytotoxic action of PDT.The speci®city achieved from drug uptake selectivitycombined with light targeting makes PDT an appealingapproach.
The ®rst photosensitizer approved for PDT is aporphyrin oligomer (Photofrin) which is highly e�ec-tive but exhibits the drawbacks of (i) a tendency tocause prolonged skin photosensitivity, (ii) an activationwavelength lower than that optimal for e�ectivepenetration through tissue, and (iii) a poorly de®nedchemical composition which makes a detailed under-standing of its mode of action and pharmacokineticsdi�cult. To address these issues, new photosensitizersare being developed and a number of new agents arenow in clinical trials. Several groups have recentlyreported the antitumor e�cacy of pheophorbide-andpyropheophorbide-based PDT (Bellnier et al., 1993;Henderson et al., 1997; Payne et al., 1996). Thesecompounds are chemically well characterized, absorblight above 600 nm and produce less long-term skinphotosensitivity than Photofrin. In the pyropheophor-bide-a series, either as methylesters or as carboxylicacids, photosensitizing activity increases with the lengthof the alkyl ether side-chain. These alkyl etherderivatives, although having similar photophysicalproperties (singlet oxygen and ¯uorescence yields),exhibit remarkable di�erences in photosensitizinge�ciency in biological systems (Pandey et al., 1996).
The cellular mechanisms of PDT are still underinvestigation. Singlet oxygen, the putative cytotoxicagent in PDT, is initially formed by the energy transferfrom the excited triplet state of the photosensitizer toground-state oxygen (Weishaupt et al., 1976). Severalfactors are thought to work together to achievecomplete tumor eradication in vivo. These includedirect cellular damage leading to necrosis or apoptosis,vascular destruction, in¯ammatory changes and sub-sequent immune responses (Korbelik, 1996). While themechanism responsible for the e�ective destruction ofthe malignant tissues by PDT is not completely
Oncogene (2001) 20, 4070 ± 4084ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00
www.nature.com/onc
*Correspondence: J Piette, Laboratory of Virology and Immunology,Institute of Pathology B23, University of Liege, B-4000 Liege,Belgium; E-mail: [email protected] 2 January 2001; revised 28 March 2001; accepted 11April 2001

elucidated, many reports show that tumor cell deathoccurred through apoptosis, at least for photosensiti-zers that distribute to mitochondria (Granville et al.,1998; Kessel and Luo, 1998). In recent years,considerable progress has been made towards amolecular understanding of the processes underlyingapoptotic cell death pathway(s) (Evan and Littlewood,1998; Granville and Hunt, 2000; Jacobson et al., 1997;Steller, 1995).
It is becoming clear that di�erent cytotoxic stimuliacting through various mechanisms converge on theactivation of caspases which instigate the well-conservedexecution and terminal stages of apoptosis. However,during the induction phase of apoptosis, multiplesignaling pathways in¯uence the central control of thecell death machinery. Mitochondria are situated at thecrossroad between the activation and execution phasesand their role as orchestrators of apoptosis is now®rmly established in many systems (Green and Reed,1998; Halestrap et al., 2000; Kroemer and Reed, 2000).Thus, a variety of apoptosis-related events involvemitochondria, including the release of caspase activa-tors (such as cytochrome c), changes in electrontransport, loss of mitochondrial transmembrane poten-tial and altered cellular redox state. Mitochondria areconsidered the site where photosensitizers are bestlocalized for e�cient cell killing with PDT. Under-standing the signals that converge on mitochondria andin¯uence these events and their downstream e�ects areof great signi®cance in biology.
We have previously shown that photosensitizationwith pyropheophorbide-a methylester (PPME) ofcolon cancer cells activates transcription factor NF-kB by mobilization of the IL-1 receptor transductionmachinery. NF-kB has been shown to have either anti-or pro-apoptotic activity depending on the cell type orthe nature of the stimulus (Van Antwerp et al., 1996;Wang et al., 1998; Ward et al., 1999). We sought toinvestigate the mechanism of colon cancer cellapoptosis mediated by PPME photosensitization andto clarify the role of NF-kB in this process. BecausePPME mainly localizes in membranes of the endo-plasmic reticulum (ER) and in lysosomes, it makes thisphotosensitizer an interesting compound to studymechanisms of apoptosis triggered by damage gener-ated in organelles other than mitochondria.
Results
PPME photosensitization triggers colon cancer cell deaththrough an apoptotic mechanism
Incubation of HCT-116 colon cancer cells with PPMEat 6 mM followed by irradiation with red light produceda signi®cant level of cytotoxicity as indicated by theTrypan blue exclusion method. Approximatively 10%of the cells survived treament at a 96 kJ/m2 light dose(Figure 1a). Cell killing was clearly in¯uenced by thelight dose used as well as the concentration of thephotosensitizer applied to the cells. HCT-116 cells
unirradiated or incubated with PPME but notirradiated displayed a 100% survival.
In order to determine the nature of cell death,photosensitized cells were assessed for DNA fragmen-tation by agarose gel electrophoresis. PPME photo-sensitization produced extensive dose-dependent DNAfragmentation to a degree comparable to that inducedby daunomycin (Figure 1b). In a time-course study,DNA laddering was detectable by 3 h with a 96 kJ/m2
light dose (Figure 1b, lower panel). Untreated controlsdid not exhibit DNA fragmentation. In situ labeling ofDNA cleavage products by TUNEL method reinforcedthese observations. PPME photosensitization gave riseto appearance of TUNEL positive cells as compared tothe negative control (Figure 1c). A separate means tomeasure apoptotic cell death consisted of visualizingDNA condensation along the nuclear membrane byDAPI staining (Figure 1d). This condensation wasevident by 3 h after photosensitization and wasaccompanied by nuclear disruption and the appearanceof apoptotic bodies. Since caspase-3 is known to playan executioner role in apoptosis, we evaluated itsinvolvement in PPME-mediated apoptosis. Figure 1eshows a dose-dependent stimulation of caspase-3activity. This activity was completely abrogated bypre-incubation of HCT-116 cells with a speci®ccaspase-3 inhibitor (Z-DEVD-fmk).
Although HCT-116 cells expresses a functional p53protein, stabilization of this protein was not observedin response to PPME photosensitization (data notshown). Further, PPME treatment did not induceexpression of p21, a p53-encoded protein involved inthe control of cell cycle (data not shown). These resultsindicate that PPME-mediated cell death is unrelated top53 activation or to cell cycle arrest.
In order to assess the importance of the apoptoticprocess in PPME-mediated cytotoxicity, HCT-116 cellswere analysed by AnnexinV/PI staining method whichis commonly used to di�erentiate necrotic and apoptoticcell populations. As soon as 4 h after a 64 kJ/m2 or a96 kJ/m2 irradiation, 4.8% (Figure 2a) and 7.8%(Figure 2b) of cells undergo apoptosis, respectively toreach 19.3% (Figure 2c) and 1.1% (Figure 2d) after24 h. The ratio necrosis/apoptosis turns out to bedependent on light dose and post-irradiation time.Moreover, di�erent PPME concentration used in thetreatment turned out also to modulate the ratio betweennecrosis and apoptosis (data not shown).
Mitochondria play a central role in PPME-mediatedapoptosis
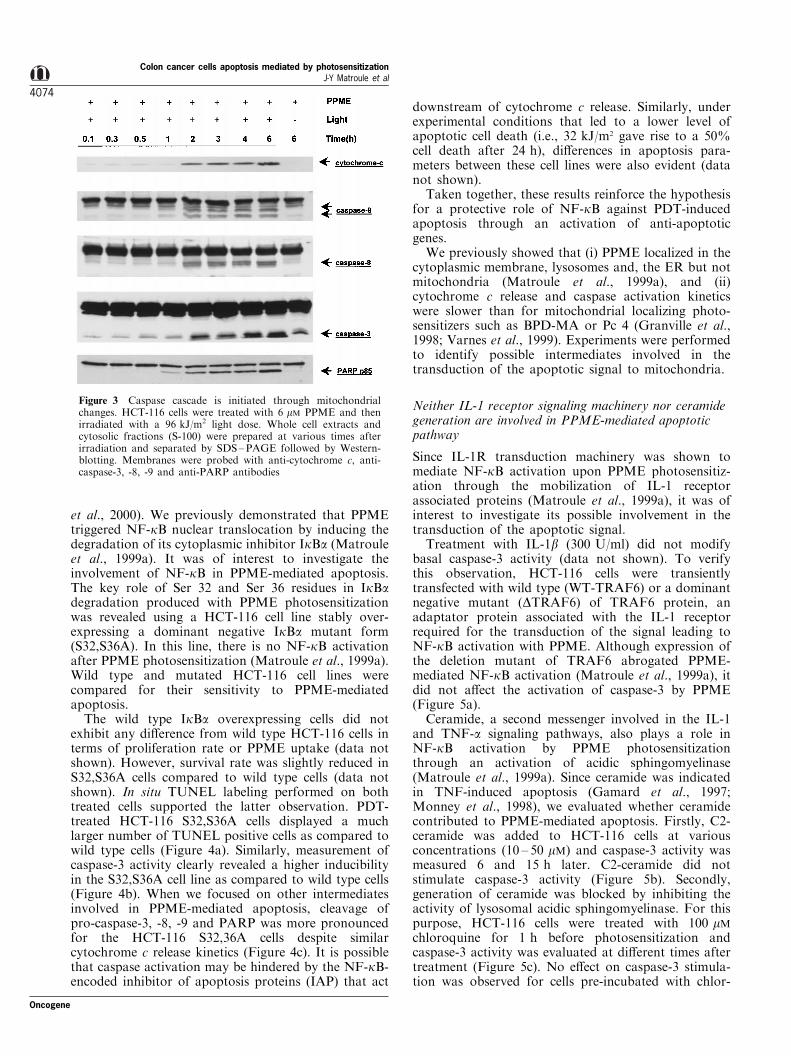
To further identify the di�erent intermediates trans-ducing the apoptotic signal, it was of interest to assessthe contribution of some well-characterized apoptosisfactors. Firstly, PPME photosensitization gave rise tothe appearance of cytochrome c in the cytosol (Figure3). This event occurred by 2 h after PDT and wasaccompanied by pro-caspase-9 cleavage into activecaspase-9. Activation of caspase-3, PARP cleavageand DNA fragmentation occurred in temporal proxi-
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4071
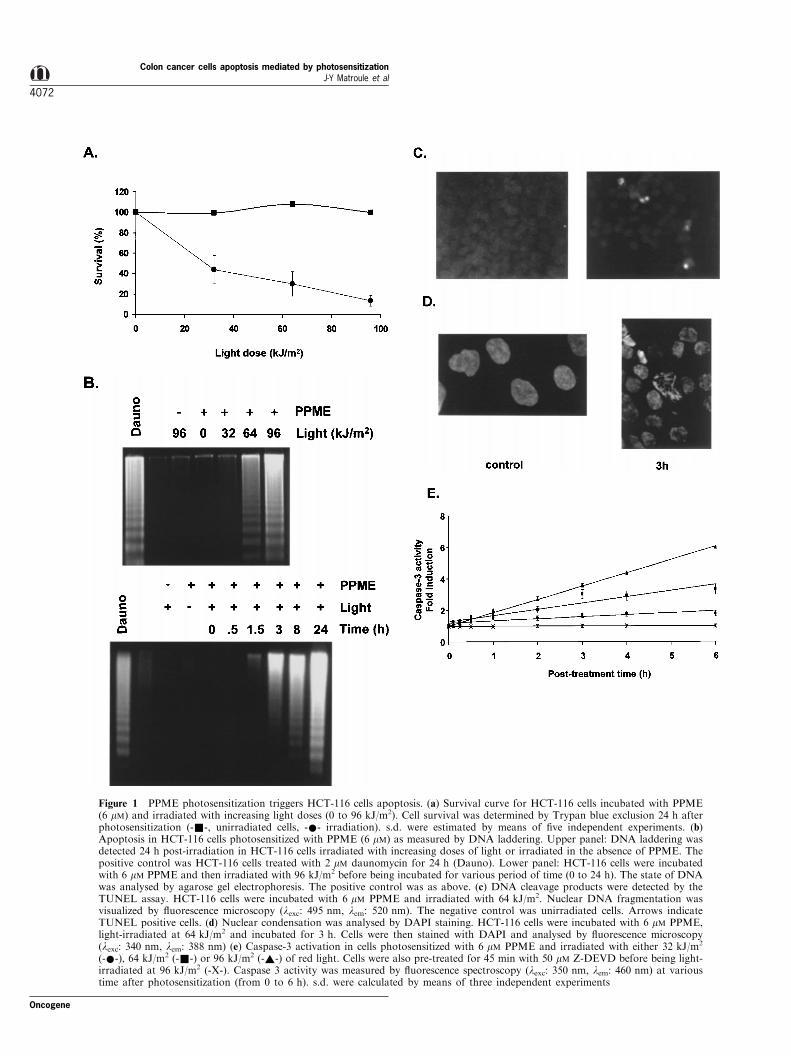
Figure 1 PPME photosensitization triggers HCT-116 cells apoptosis. (a) Survival curve for HCT-116 cells incubated with PPME(6 mM) and irradiated with increasing light doses (0 to 96 kJ/m2). Cell survival was determined by Trypan blue exclusion 24 h afterphotosensitization (-&-, unirradiated cells, -*- irradiation). s.d. were estimated by means of ®ve independent experiments. (b)Apoptosis in HCT-116 cells photosensitized with PPME (6 mM) as measured by DNA laddering. Upper panel: DNA laddering wasdetected 24 h post-irradiation in HCT-116 cells irradiated with increasing doses of light or irradiated in the absence of PPME. Thepositive control was HCT-116 cells treated with 2 mM daunomycin for 24 h (Dauno). Lower panel: HCT-116 cells were incubatedwith 6 mM PPME and then irradiated with 96 kJ/m2 before being incubated for various period of time (0 to 24 h). The state of DNAwas analysed by agarose gel electrophoresis. The positive control was as above. (c) DNA cleavage products were detected by theTUNEL assay. HCT-116 cells were incubated with 6 mM PPME and irradiated with 64 kJ/m2. Nuclear DNA fragmentation wasvisualized by ¯uorescence microscopy (lexc: 495 nm, lem: 520 nm). The negative control was unirradiated cells. Arrows indicateTUNEL positive cells. (d) Nuclear condensation was analysed by DAPI staining. HCT-116 cells were incubated with 6 mM PPME,light-irradiated at 64 kJ/m2 and incubated for 3 h. Cells were then stained with DAPI and analysed by ¯uorescence microscopy(lexc: 340 nm, lem: 388 nm) (e) Caspase-3 activation in cells photosensitized with 6 mM PPME and irradiated with either 32 kJ/m2
(-*-), 64 kJ/m2 (-&-) or 96 kJ/m2 (-~-) of red light. Cells were also pre-treated for 45 min with 50 mM Z-DEVD before being light-irradiated at 96 kJ/m2 (-X-). Caspase 3 activity was measured by ¯uorescence spectroscopy (lexc: 350 nm, lem: 460 nm) at varioustime after photosensitization (from 0 to 6 h). s.d. were calculated by means of three independent experiments
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4072
Oncogene

mity. Pro-caspase-8 cleavage into its active form wasobserved concomitant with the activation of the othercaspases. However, caspase-8 did not appear to have aprimary role in the initiation of the apoptotic signal.Caspase-8 activation was completely blocked by thecaspase-3 speci®c inhibitor Z-DEVD (data not shown).Caspase-8 is therefore likely activated downstream ofcaspase-3. This suggests that PPME photosensitizationdoes not lead to apoptosis through the recruitment ofthe Fas, TNF-a or related death receptors, cell surfaceproteins known to induce apoptosis through mobiliza-tion of caspase-8 (Schneider and Tschopp, 2000).Moreover, pre-treatment of cells with 10 mM speci®c
caspase-9 inhibitor (LEHD-CHO) completely abro-gated caspase-3-like cleavage activity thereby indicatingthat caspase-9 is likely an initiator caspase for PDT-induced apoptosis (data not shown).
Taken together, these results reveal mitochondrialevents as key components in the transmission of theapoptotic signal upon PPME photosensitization.
NF-kB partially protects HCT-116 cells againstPPME-mediated apoptosis
The role of NF-kB in apoptosis remains controversial(Kuhnel et al., 2000; Plumpe et al., 2000; Rivera-Walsh
Figure 2 Determination of necrotic vs apoptotic cells ratio by Annexin V/PI. HCT-116 cells were incubated with 6 mM PPME andthen irradiated with either a 96 kJ/m2 (b and d) or a 64 kJ/m2 (a and c) light dose. Cells were harvested 4 h (a and b) or 24 h (c andd) after photosensitization and stained with Annexin V/PI for FACS analysis. Lower left panel represents living cells, lower rightpanel represents apoptotic cells, upper right panel represents necrotic cells and upper left panel represents pre-necrotic cells. Stainingof control cells after 24 h displayed 85% living cells, 10% necrotic cells and 5% apoptotic cells
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4073
et al., 2000). We previously demonstrated that PPMEtriggered NF-kB nuclear translocation by inducing thedegradation of its cytoplasmic inhibitor IkBa (Matrouleet al., 1999a). It was of interest to investigate theinvolvement of NF-kB in PPME-mediated apoptosis.The key role of Ser 32 and Ser 36 residues in IkBadegradation produced with PPME photosensitizationwas revealed using a HCT-116 cell line stably over-expressing a dominant negative IkBa mutant form(S32,S36A). In this line, there is no NF-kB activationafter PPME photosensitization (Matroule et al., 1999a).Wild type and mutated HCT-116 cell lines werecompared for their sensitivity to PPME-mediatedapoptosis.
The wild type IkBa overexpressing cells did notexhibit any di�erence from wild type HCT-116 cells interms of proliferation rate or PPME uptake (data notshown). However, survival rate was slightly reduced inS32,S36A cells compared to wild type cells (data notshown). In situ TUNEL labeling performed on bothtreated cells supported the latter observation. PDT-treated HCT-116 S32,S36A cells displayed a muchlarger number of TUNEL positive cells as compared towild type cells (Figure 4a). Similarly, measurement ofcaspase-3 activity clearly revealed a higher inducibilityin the S32,S36A cell line as compared to wild type cells(Figure 4b). When we focused on other intermediatesinvolved in PPME-mediated apoptosis, cleavage ofpro-caspase-3, -8, -9 and PARP was more pronouncedfor the HCT-116 S32,36A cells despite similarcytochrome c release kinetics (Figure 4c). It is possiblethat caspase activation may be hindered by the NF-kB-encoded inhibitor of apoptosis proteins (IAP) that act
downstream of cytochrome c release. Similarly, underexperimental conditions that led to a lower level ofapoptotic cell death (i.e., 32 kJ/m2 gave rise to a 50%cell death after 24 h), di�erences in apoptosis para-meters between these cell lines were also evident (datanot shown).
Taken together, these results reinforce the hypothesisfor a protective role of NF-kB against PDT-inducedapoptosis through an activation of anti-apoptoticgenes.
We previously showed that (i) PPME localized in thecytoplasmic membrane, lysosomes and, the ER but notmitochondria (Matroule et al., 1999a), and (ii)cytochrome c release and caspase activation kineticswere slower than for mitochondrial localizing photo-sensitizers such as BPD-MA or Pc 4 (Granville et al.,1998; Varnes et al., 1999). Experiments were performedto identify possible intermediates involved in thetransduction of the apoptotic signal to mitochondria.
Neither IL-1 receptor signaling machinery nor ceramidegeneration are involved in PPME-mediated apoptoticpathway
Since IL-1R transduction machinery was shown tomediate NF-kB activation upon PPME photosensitiz-ation through the mobilization of IL-1 receptorassociated proteins (Matroule et al., 1999a), it was ofinterest to investigate its possible involvement in thetransduction of the apoptotic signal.
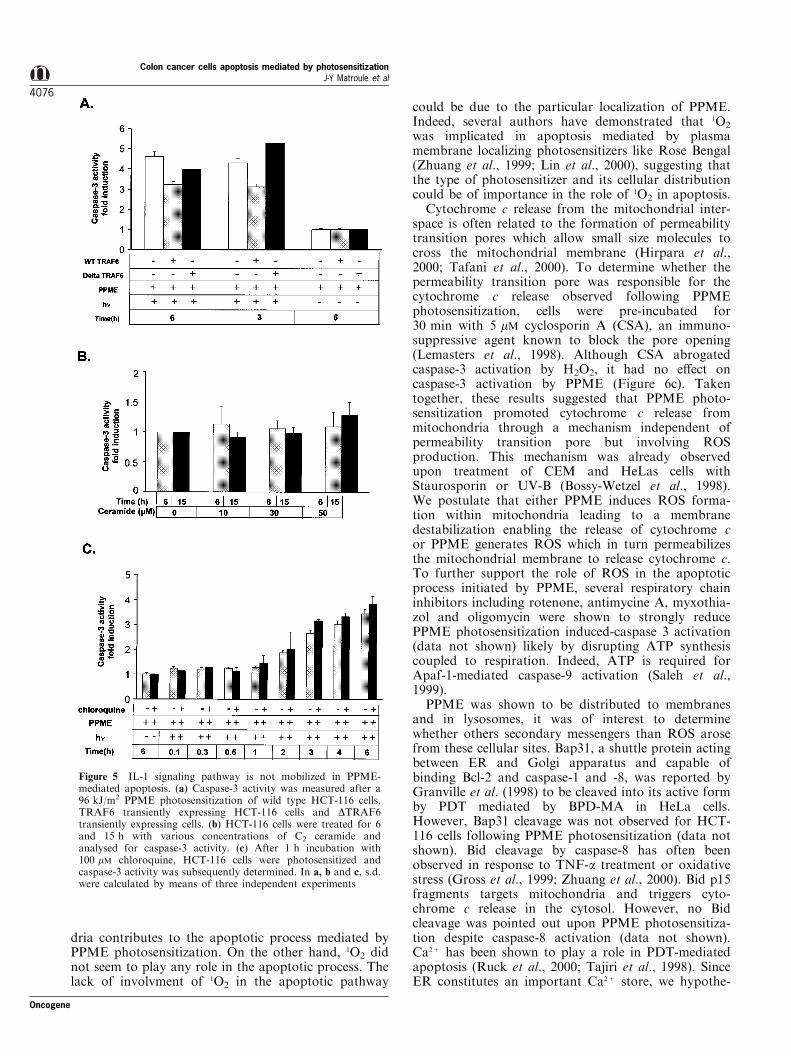
Treatment with IL-1b (300 U/ml) did not modifybasal caspase-3 activity (data not shown). To verifythis observation, HCT-116 cells were transientlytransfected with wild type (WT-TRAF6) or a dominantnegative mutant (DTRAF6) of TRAF6 protein, anadaptator protein associated with the IL-1 receptorrequired for the transduction of the signal leading toNF-kB activation with PPME. Although expression ofthe deletion mutant of TRAF6 abrogated PPME-mediated NF-kB activation (Matroule et al., 1999a), itdid not a�ect the activation of caspase-3 by PPME(Figure 5a).
Ceramide, a second messenger involved in the IL-1and TNF-a signaling pathways, also plays a role inNF-kB activation by PPME photosensitizationthrough an activation of acidic sphingomyelinase(Matroule et al., 1999a). Since ceramide was indicatedin TNF-induced apoptosis (Gamard et al., 1997;Monney et al., 1998), we evaluated whether ceramidecontributed to PPME-mediated apoptosis. Firstly, C2-ceramide was added to HCT-116 cells at variousconcentrations (10 ± 50 mM) and caspase-3 activity wasmeasured 6 and 15 h later. C2-ceramide did notstimulate caspase-3 activity (Figure 5b). Secondly,generation of ceramide was blocked by inhibiting theactivity of lysosomal acidic sphingomyelinase. For thispurpose, HCT-116 cells were treated with 100 mMchloroquine for 1 h before photosensitization andcaspase-3 activity was evaluated at di�erent times aftertreatment (Figure 5c). No e�ect on caspase-3 stimula-tion was observed for cells pre-incubated with chlor-
Figure 3 Caspase cascade is initiated through mitochondrialchanges. HCT-116 cells were treated with 6 mM PPME and thenirradiated with a 96 kJ/m2 light dose. Whole cell extracts andcytosolic fractions (S-100) were prepared at various times afterirradiation and separated by SDS±PAGE followed by Western-blotting. Membranes were probed with anti-cytochrome c, anti-caspase-3, -8, -9 and anti-PARP antibodies
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4074
Oncogene

oquine. NH4Cl, a separate inhibitor of acidic sphingo-myelinase, also did not a�ect PPME-mediated caspase-3 activation (data not shown).
Reactive oxygen species (ROS) instigate the apoptoticprocess
Since singlet oxygen (1O2) is one of the reactive oxygenspecies (ROS) generated by PDT and because ROS[superoxide anion (O2
.7), hydrogen peroxide (H2O2),hydroxyl radical (OH.)] can also be released bymitochondria as second messenger during the apoptosisprocess, we decided to investigate the role of ROS inthe cell death process induced by PPME photosensi-tization (Dumont et al., 1999; Katschinski et al., 2000;Shimura et al., 2000; Stridh et al., 1998; Zorov et al.,2000). To test their respective roles, two kinds ofexperiments were carried out. Firstly, HCT-116 cellswere irradiated in PBS where H2O was substituted byD2O since singlet oxygen lifetime (1O2) was known tobe speci®cally increased by this isotopic substitution.Cell killing was reproductibly slightly exaggeratedwhen cells were irradiated in the presence of D2O(data not shown). This suggests that 1O2 generatedduring PPME photosensitization may be involvedeither in the necrotic or in the apoptotic process. Todetermine whether apoptosis could be initiated through1O2 generation, caspase-3 activity was measured undersimilar experimental conditions as above (Figure 6a).Unexpectedly, caspase-3 activation was unmodi®ed bythe isotopic substitution suggesting that 1O2 generatedby PPME photosensitization may control necrosis butnot apoptosis. Second, PPME photosensitization wascarried out in HCT-116 cells either pre-incubated with300 mM PDTC or with 20 mM NAC as antioxidants.Survival curves obtained for cells treated with PDTCand NAC indicated a reduced sensitivity to photo-dynamic killing (data not shown) reinforcing the ideathat other ROS than 1O2 are also involved in the celldeath process mediated by PPME photosensitization.Importantly, PDTC and NAC completely or partiallyinhibited caspase-3 activation by PPME, suggestingthat ROS others than 1O2 were involved in theapoptosis process (Figure 6b). These data werecon®rmed by AnnexinV/PI staining of HCT-116irradiated in the presence of 300 mM PDTC. Weobserved a 43% reduction of apoptotic cells as soonas 4 h after a 64 kJ/m2 irradiation and a 61.2%reduction 24 h after irradiation (data not shown).These results indicate that ROS (sensitive to NACand PDTC) very likely generated through mitochon-
Figure 4 Protective role of NF-kB against PPME-mediatedapoptosis. (a) TUNEL assay after a 64 kJ/m2 irradiation with6 mM PPME in wild type HCT-116 cells (left panel) and S32,S36AIkB-a overexpressing HCT116 cells (right panel). Nuclear DNAfragmentation was visualized by ¯uorescence microscopy (lexc:495 nm, lem: 520 nm). Arrows indicate TUNEL positive cells.(b) Comparative caspase-3 activation following a 64 kJ/m2 PPME
photosensitization in wild type cells (-&-) and S32,S36A IkB-aoverexpressing cells (-*-). s.d. were calculated by means of threeindependent experiments. (c) Both wild type cells and S32,S36AIkB-a overexpressing cells were treated by PPME and thenirradiated with a 64 kJ/m2 light dose. Whole cell lysates wereseparated by SDS±PAGE and Western-blotting was performedwith anti-cytochrome c, anti-caspase-3, -8, -9 and anti-PARPantibodies
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4075
dria contributes to the apoptotic process mediated byPPME photosensitization. On the other hand, 1O2 didnot seem to play any role in the apoptotic process. Thelack of involvment of 1O2 in the apoptotic pathway
could be due to the particular localization of PPME.Indeed, several authors have demonstrated that 1O2
was implicated in apoptosis mediated by plasmamembrane localizing photosensitizers like Rose Bengal(Zhuang et al., 1999; Lin et al., 2000), suggesting thatthe type of photosensitizer and its cellular distributioncould be of importance in the role of 1O2 in apoptosis.
Cytochrome c release from the mitochondrial inter-space is often related to the formation of permeabilitytransition pores which allow small size molecules tocross the mitochondrial membrane (Hirpara et al.,2000; Tafani et al., 2000). To determine whether thepermeability transition pore was responsible for thecytochrome c release observed following PPMEphotosensitization, cells were pre-incubated for30 min with 5 mM cyclosporin A (CSA), an immuno-suppressive agent known to block the pore opening(Lemasters et al., 1998). Although CSA abrogatedcaspase-3 activation by H2O2, it had no e�ect oncaspase-3 activation by PPME (Figure 6c). Takentogether, these results suggested that PPME photo-sensitization promoted cytochrome c release frommitochondria through a mechanism independent ofpermeability transition pore but involving ROSproduction. This mechanism was already observedupon treatment of CEM and HeLas cells withStaurosporin or UV-B (Bossy-Wetzel et al., 1998).We postulate that either PPME induces ROS forma-tion within mitochondria leading to a membranedestabilization enabling the release of cytochrome cor PPME generates ROS which in turn permeabilizesthe mitochondrial membrane to release cytochrome c.To further support the role of ROS in the apoptoticprocess initiated by PPME, several respiratory chaininhibitors including rotenone, antimycine A, myxothia-zol and oligomycin were shown to strongly reducePPME photosensitization induced-caspase 3 activation(data not shown) likely by disrupting ATP synthesiscoupled to respiration. Indeed, ATP is required forApaf-1-mediated caspase-9 activation (Saleh et al.,1999).
PPME was shown to be distributed to membranesand in lysosomes, it was of interest to determinewhether others secondary messengers than ROS arosefrom these cellular sites. Bap31, a shuttle protein actingbetween ER and Golgi apparatus and capable ofbinding Bcl-2 and caspase-1 and -8, was reported byGranville et al. (1998) to be cleaved into its active formby PDT mediated by BPD-MA in HeLa cells.However, Bap31 cleavage was not observed for HCT-116 cells following PPME photosensitization (data notshown). Bid cleavage by caspase-8 has often beenobserved in response to TNF-a treatment or oxidativestress (Gross et al., 1999; Zhuang et al., 2000). Bid p15fragments targets mitochondria and triggers cyto-chrome c release in the cytosol. However, no Bidcleavage was pointed out upon PPME photosensitiza-tion despite caspase-8 activation (data not shown).Ca2+ has been shown to play a role in PDT-mediatedapoptosis (Ruck et al., 2000; Tajiri et al., 1998). SinceER constitutes an important Ca2+ store, we hypothe-
Figure 5 IL-1 signaling pathway is not mobilized in PPME-mediated apoptosis. (a) Caspase-3 activity was measured after a96 kJ/m2 PPME photosensitization of wild type HCT-116 cells,TRAF6 transiently expressing HCT-116 cells and DTRAF6transiently expressing cells. (b) HCT-116 cells were treated for 6and 15 h with various concentrations of C2 ceramide andanalysed for caspase-3 activity. (c) After 1 h incubation with100 mM chloroquine, HCT-116 cells were photosensitized andcaspase-3 activity was subsequently determined. In a, b and c, s.d.were calculated by means of three independent experiments
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4076
Oncogene

sized that PPME might trigger Ca2+ release to thecytosol. Therefore, cells were pre-incubated with theCa2+ chelator, BAPTA-AM, before photosensitizationand assessed for subsequent caspase-3 activity. Noalteration in caspase-3 activity was observed forBAPTA-AM treated cells (data not shown) indicatingthat either Ca2+ was not released from ER store orCa2+ is not a messenger in the apoptotic processmediated by PPME photosensitization. The lastintermediate tested was caspase-12 which was recentlyshown to be involved in ER-speci®c apoptosis andcytotoxicity by amyloid-b in neuronal cells (Nakagawaet al., 2000). In HCT-116 cells photosensitized byPPME, no procaspase-12 cleavage could be visualizedand no inhibition of caspase-3 activation or PARPcleavage was visualized after transfecting photosensi-tized HCT-116 cells with antisense oligonucleotides(data not shown).
1O2 mainly generated from the ER and Golgi membranesis involved in necrosis and not in apoptosis
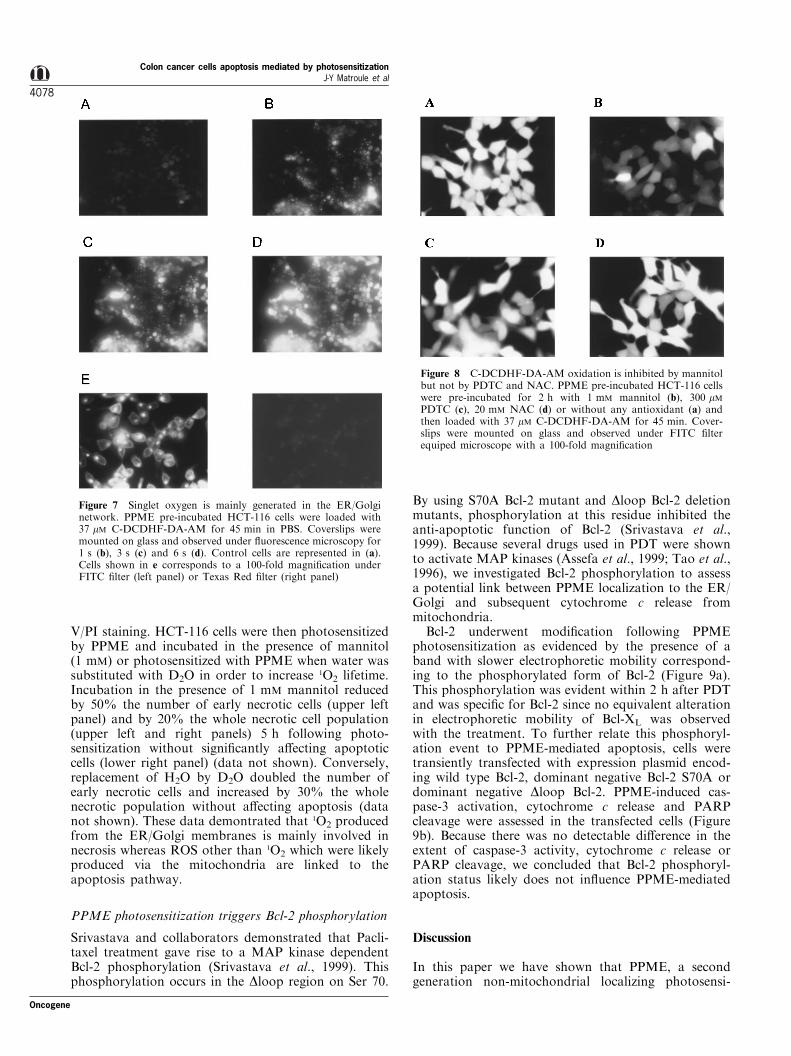
Since 1O2 lifetime and range of di�usion are quite shortin cells, it was of interest to identify its production sitein order to better correlate the molecular mechanismsto oxidative status. For that purpose, we useddichloro¯uorescein staining method which is commonlyused to monitor intracellular ROS production (Hock-berger et al., 1999). In the present study, cells pre-incubated with PPME were subsequently loaded with6-carboxy-2',7'-dichlorodihydro¯uorescein diacetatedi(acetoxymethyl ester) (C-DCDHF-DA-AM) andobserved by ¯uorescence microscopy with a FITC®lter. As shown in Figure 7b ± d, PPME photosensiti-zation under the ®ltered light of the microscopeimmediately triggered C-DCDHF-DA-AM oxidationas visualized by yellow ¯uorescence. In Figure 7e, the100-fold magni®cation allowed us to visualize, at earlyirradiation times, a perfectly similar ¯uorescencepattern between oxidized C-DCDHF-DA-AM (leftpanel) and PPME (right panel). These data demon-strate that PPME photosensitization triggered theproduction of 1O2 by photoexcited PPME localized inthe ER and the Golgi apparatus. We could not detectany colocalization between C-DCDHF-DA-AM andmitochondria (Figure 7e) reinforcing the idea that theprobe detected 1O2 generated by PPME photosensitiza-tion and not ROS released by mitochondria. Im-portantly, the C-DCDHF-DA-AM ¯uorescence wasquenched by a pre-incubation with 1 mM mannitol(Figure 8a,b), a well-known 1O2 inhibitor(k1O24108 M71 s71), but not by PDTC and NAC(k1O25105 M71 s71) (Figure 8c,d). This suggests thatPDTC and NAC could not e�ciently quench the 1O2
production by PPME photosensitization and acteddownstream in the oxidative process. From theseexperiments we can conclude that PPME photosensi-tization led to 1O2 generation from the ER/Golgimembranes. In order to determine whether or not this1O2 production was linked to the apoptosis or to thenecrosis process, we used the FACS coupled Annexin
Figure 6 ROS participate in PPME-mediated apoptosis. (a)Caspase-3 activity levels were measured after a 96 kJ/m2
irradiation in either PBS-H2O or PBS-D2O. Caspase-3 wasdetermined on whole cell extracts. (b) Caspase-3 activity inHCT-116 cells were pretreated for 1 h with either PDTC (300 mM)or NAC (20 mM) before photosensitization with a 96 kJ/m2 lightdose. Caspase-3 activity was measured for whole cell extracts. (c)Caspase-3 activity measurements when 5 mM cyclosporin A (CSA)was added to HCT-116 cells 30 min prior 96 kJ/m2 irradiation.Positive control was made by treating the cells for 24 h with500 mM H2O2. Caspase-3 activity was measured for whole cellextracts. s.d. were calculated by means of three independentexperiments
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4077
V/PI staining. HCT-116 cells were then photosensitizedby PPME and incubated in the presence of mannitol(1 mM) or photosensitized with PPME when water wassubstituted with D2O in order to increase 1O2 lifetime.Incubation in the presence of 1 mM mannitol reducedby 50% the number of early necrotic cells (upper leftpanel) and by 20% the whole necrotic cell population(upper left and right panels) 5 h following photo-sensitization without signi®cantly a�ecting apoptoticcells (lower right panel) (data not shown). Conversely,replacement of H2O by D2O doubled the number ofearly necrotic cells and increased by 30% the wholenecrotic population without a�ecting apoptosis (datanot shown). These data demontrated that 1O2 producedfrom the ER/Golgi membranes is mainly involved innecrosis whereas ROS other than 1O2 which were likelyproduced via the mitochondria are linked to theapoptosis pathway.
PPME photosensitization triggers Bcl-2 phosphorylation
Srivastava and collaborators demonstrated that Pacli-taxel treatment gave rise to a MAP kinase dependentBcl-2 phosphorylation (Srivastava et al., 1999). Thisphosphorylation occurs in the Dloop region on Ser 70.
By using S70A Bcl-2 mutant and Dloop Bcl-2 deletionmutants, phosphorylation at this residue inhibited theanti-apoptotic function of Bcl-2 (Srivastava et al.,1999). Because several drugs used in PDT were shownto activate MAP kinases (Assefa et al., 1999; Tao et al.,1996), we investigated Bcl-2 phosphorylation to assessa potential link between PPME localization to the ER/Golgi and subsequent cytochrome c release frommitochondria.
Bcl-2 underwent modi®cation following PPMEphotosensitization as evidenced by the presence of aband with slower electrophoretic mobility correspond-ing to the phosphorylated form of Bcl-2 (Figure 9a).This phosphorylation was evident within 2 h after PDTand was speci®c for Bcl-2 since no equivalent alterationin electrophoretic mobility of Bcl-XL was observedwith the treatment. To further relate this phosphoryl-ation event to PPME-mediated apoptosis, cells weretransiently transfected with expression plasmid encod-ing wild type Bcl-2, dominant negative Bcl-2 S70A ordominant negative Dloop Bcl-2. PPME-induced cas-pase-3 activation, cytochrome c release and PARPcleavage were assessed in the transfected cells (Figure9b). Because there was no detectable di�erence in theextent of caspase-3 activity, cytochrome c release orPARP cleavage, we concluded that Bcl-2 phosphoryl-ation status likely does not in¯uence PPME-mediatedapoptosis.
Discussion
In this paper we have shown that PPME, a secondgeneration non-mitochondrial localizing photosensi-
Figure 7 Singlet oxygen is mainly generated in the ER/Golginetwork. PPME pre-incubated HCT-116 cells were loaded with37 mM C-DCDHF-DA-AM for 45 min in PBS. Coverslips weremounted on glass and observed under ¯uorescence microscopy for1 s (b), 3 s (c) and 6 s (d). Control cells are represented in (a).Cells shown in e corresponds to a 100-fold magni®cation underFITC ®lter (left panel) or Texas Red ®lter (right panel)
Figure 8 C-DCDHF-DA-AM oxidation is inhibited by mannitolbut not by PDTC and NAC. PPME pre-incubated HCT-116 cellswere pre-incubated for 2 h with 1 mM mannitol (b), 300 mMPDTC (c), 20 mM NAC (d) or without any antioxidant (a) andthen loaded with 37 mM C-DCDHF-DA-AM for 45 min. Cover-slips were mounted on glass and observed under FITC ®lterequiped microscope with a 100-fold magni®cation
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4078
Oncogene

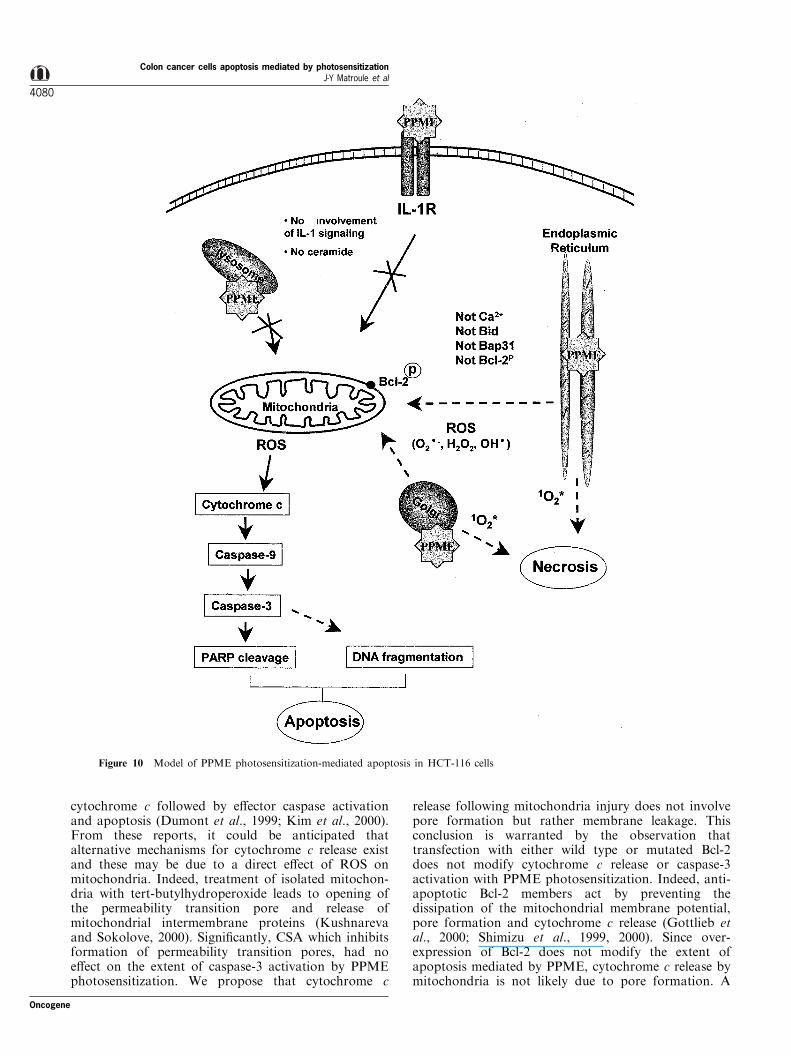
tizer, can rapidly trigger apoptosis despite having anER/Golgi and lysosomal distribution in cells. Apopto-sis generated in colon cancer cells by PPME photo-sensitization does not depend on p53 activation/stabilisation but leads to the release of cytochrome cfrom mitochondria and activation of caspase-9 and -3.We have also demonstrated that apoptosis does notoriginate from neither mobilization of IL-1 receptortransduction machinery nor ceramide generation byacidic sphingomyelinase as shown for NF-kB activa-tion by this compound (Matroule et al., 1999a). Theapoptosis initiation mechanism appears classic with theexception of the existence of a critical link between themain localization sites of PPME and mitochondria.This connection is likely based on ROS generation butnot on Bid cleavage, Ca2+ release from stores or
cleavage of Bap-31, a pro-apoptotic ER molecule(Figure 10) (Granville et al., 1998).
Cellular localization studies with PPME or amino-pyropheophorbide (APP) (Matroule et al., 1999a,b)with ¯uorescence microscopy using organelle speci®cprobes, revealed that PPME mainly localized in theER/Golgi apparatus and lysosomes. Lysosomes arebelieved to quench photodynamic activity by acting asa sink which lowers the active drugs concentrations(MacDonald et al., 1999). It is thus likely that theactive cellular site for triggering apoptosis mediated byPPME photosensitization is the ER/Golgi system. Theabsence of co-localization with the mitochondrialprobe rhodamine-123 indicates the likelihood that theER/Golgi is the site from which the apoptotic signaloriginates (Matroule et al., 1999b). Others have foundthat photosensitizers localize to a variety of cellularsites including the outer membrane, lysosomes, mito-chondria and the nucleus resulting in damage to thesestructures upon light exposure (Morgan et al., 2000;Oleinick and Evans, 1998). Although mitochondria areprimary targets for some photosensitizers, otherorganelles were also considered as sites of action(Granville and Hunt, 2000; Lee et al., 1995; Miller etal., 1995). The present data provides evidence thatinternal membranes, particularly, the ER/Golgi appa-ratus is a distinct site from which an apoptotic signalcan emerge.
ROS turns out to be intermediates in the apoptoticprocess following PPME photosensitization. ROSgenerated from mitochondria by a perturbation ofthe electron transport chain may occur subsequent tothe primary photodynamic events in the ER/Golgi. Themechanism that causes mitochondria to produce ROSin response to apoptotic stimuli is still unclear butseveral mitochondrial regulators have been identi®edsuch as Bax-like proteins, nitric oxide, ceramide, ATP/ADP depletion, NADH oxidation (Kroemer and Reed,2000). Among them, none were clearly identi®ed asemerging from the ER/Golgi where the photosensitiz-ation by PPME is triggered. Recently, caspase-12 wasshown to mediate ER-speci®c apoptosis and cytotox-icity of neuronal cells by amyloid-b (Nakagawa et al.,2000). Using antibodies directed against caspase-12 andantisense oligonucleotides we were unable to demon-strate processing of pro-caspase-12 and an inhibition ofapoptosis mediated by PPME photosensitization rulingout the role of caspase-12 as mediator in our system.
The most likely possibility is that other ROS than1O2 (O2
.7, H2O2 or OH.) produced at the primary siteof the photosensitization reaction (in the ER/Golinetwork) act as second messengers causing mitochon-dria to release cytochrome c and, via caspase-3activation, apoptosis. This hypothesis was recentlysupported by the observation that ROS accumulationin individual mitochondria in isolated cardiac myocytesreproductibly triggered abrupt mitochondrial depolar-ization which coincided with a burst of mitochondrialROS generation (Zorov et al., 2000). On the otherhand, Jurkat cells treated with oxidants such as H2O2
or tributylin, rapidly caused mitochondria to release
Figure 9 Bcl-2 phosphorylation does not contribute to PPME-induced apoptosis. (a) Whole cell extracts from photosensitizedcells were separated by SDS±PAGE (12%) and analysed byWestern-blotting with either Bcl-2 antibody (upper panel) or Bcl-XL antibody (lower panel). (b) Wild type HCT-116 cells, S70Aor Dloop Bcl-2 transiently overexpressing cells were photosensi-tized at a 96 kJ/m2 light dose. Whole cell extracts and cytosolicfractions (S-100) were prepared 5 h following irradiation andassessed for caspase-3 activity (upper panel), cytochrome crelease (middle panel) and PARP cleavage (lower panel). HSindicates herring sperm DNA used as negative control fortransfection
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4079
cytochrome c followed by e�ector caspase activationand apoptosis (Dumont et al., 1999; Kim et al., 2000).From these reports, it could be anticipated thatalternative mechanisms for cytochrome c release existand these may be due to a direct e�ect of ROS onmitochondria. Indeed, treatment of isolated mitochon-dria with tert-butylhydroperoxide leads to opening ofthe permeability transition pore and release ofmitochondrial intermembrane proteins (Kushnarevaand Sokolove, 2000). Signi®cantly, CSA which inhibitsformation of permeability transition pores, had noe�ect on the extent of caspase-3 activation by PPMEphotosensitization. We propose that cytochrome c
release following mitochondria injury does not involvepore formation but rather membrane leakage. Thisconclusion is warranted by the observation thattransfection with either wild type or mutated Bcl-2does not modify cytochrome c release or caspase-3activation with PPME photosensitization. Indeed, anti-apoptotic Bcl-2 members act by preventing thedissipation of the mitochondrial membrane potential,pore formation and cytochrome c release (Gottlieb etal., 2000; Shimizu et al., 1999, 2000). Since over-expression of Bcl-2 does not modify the extent ofapoptosis mediated by PPME, cytochrome c release bymitochondria is not likely due to pore formation. A
Figure 10 Model of PPME photosensitization-mediated apoptosis in HCT-116 cells
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4080
Oncogene

similar circumstance was described for apoptosisinduced either by UV or staurosporine (Bossy-Wetzelet al., 1998). In these situations, the release ofcytochrome c to the cytosol preeceded mitochondrialmembrane depolarization which was dependent oncaspase activity.
Another important feature demonstrated in thispaper is a clear separation between the transductionpathways initiated by PPME photosensitization leadingto NF-kB activation and apoptosis. NF-kB activationis ROS-independent but requires IL-1 receptor internal-ization and recruitment of its signal transductionmachinery whereas apoptosis is ROS-dependent anddoes not involve any components of the IL-1Rtransduction machinery. This is the ®rst descriptionthat two very important cellular phenomena initiatedby a photosensitization reaction can be clearlyseparated in terms of the mechanisms involved.However, these two distinct pathways are inter-relatedbecause apoptosis is more extensive when NF-kBactivation is prohibited by over-expression of adominant negative form of IkBa. All apoptosisparameters examined except cytochrome c release, weremore pronounced for the IkBa mutated cell line.Several reports show that in di�erent cell types, NF-kB controls genes encoding proteins that exert anti-apoptotic e�ects. For example, binding of TNF-a to itsreceptor may initiate apoptosis and concomitantlyactivate NF-kB, which suppresses apoptosis. TRAF1(TNFR-associated factor 1), TRAF2, and the inhibitorof apoptosis (IAP) proteins c-IAP1, c-IAP2, A1, A20,Bcl-XL have been identi®ed as target genes of NF-kBtranscriptional activity (Green, 2000; Hu et al., 1998;Wang et al., 1998, 1999). In cells in which NF-kB isinactive, expression of all the above mentioned proteinshave been shown to be required to suppress TNF-induced apoptosis, whereas c-IAP1 and c-IAP2 aresu�cient to suppress etoposide-induced apoptosis(Wang et al., 1998). One important issue of the workpresented above will be to identify the anti-apoptoticgenes in¯uenced by NF-kB following PPME-mediatedphotosensitization.
In conclusion, we have shown for the ®rst time thatan ER/Golgi-localized photosensitizer can trigger animportant apoptosis in colon cancer cells. The e�ectorof apoptosis are very likely to be ROS other than 1O2
causing mitochondria to release cytochrome c initiatingthe subsequent late stages of apoptosis. This mechan-ism is clearly separated from the one leading to NF-kBwhich was shown to be activated by the recruitment ofthe IL-1 transduction machinery (Matroule et al.,1999a) and demonstrated in the present manuscriptto counteract apoptosis.
Materials and methods
Chemicals and reagents
Pyropheophorbide methylester (PPME) was from Sigma(Sigma Chemical Co., St. Louis, MO, USA) and usedwithout modi®cation. A stock solution was made in ethanol
(1 mM) and kept in the dark at 7208C. PPME was diluted inthe culture medium just before use and added to exponen-tially growing cells. Deuterium oxide (99.8% purity) wasfrom Merck (Darmstadt, Germany). Antibodies were ob-tained from the following sources: rabbit anti-caspase-3 andmouse anti-caspase-8 (Upstate Biotechnology Inc., LakePlacid, NY, USA), mouse anti-caspase-9 and anti-cyto-chrome c (Pharmingen, Mississauga, Ont., Canada), ratanti-human caspase-12 was a generous gift from J Yuan(Harvard Medical School, USA), mouse anti-poly(ADP-ribose) polymerase (PARP) (Biomol Research Laboratories,Plymouth Meeting, PA, USA), rabbit anti-PARP p85fragment (Promega, NL, Canada), mouse anti-Bcl-2 andanti-Bcl-XL (Santa Cruz Biotechnology, USA). Caspase-3and caspase-9 peptide inhibitors were from Calbiochem (SanDiego, USA). Chicken anti Bap-31 antibody was a generousgift from Dr Gordon Shore (McGill University, Quebec,Canada). All other chemicals were of reagent grade.
Cell culture
The human colon carcinoma cell line HCT-116 was grown inMcCoy's 5A medium (Biowhittaker, Belgium) supplementedwith 10% fetal calf serum (FCS, Gibco-BRL, UK). HCT-116cells overexpressing IkBa mutated at serines 32 and 36(S32,36A) were generated as described (Matroule et al.,1999a) and cultivated in the presence of neomycin (250 mg/mL).
Plasmids
pTRAF6 and pDTRAF6 (289 ± 522) constructs were giftsfrom D Goeddel (Tularik, San Francisco, USA). pS70A Bcl-2and pDloop Bcl-2 were generous gifts from D Longo(National Institute on Aging, Baltimore, USA). Wild typeBcl-2 was kindly provided by C Borner (Fribourg University,Switzerland). All plasmids were puri®ed using Qiagen columnchromatography (Qiagen, NL, Canada) and their integritywas checked by agarose gel electrophoresis.
Exposure of HCT-116 cells to PPME photosensitization
Before photosensitization with PPME, HCT-116 cells werecultivated in 25 cm2 ¯asks for three days in McCoys 5Amedium with 10% FCS and incubated with 6 mM PPMEduring the last 20 h in McCoys 5A medium with 2% FCS.Prior to irradiation, HCT-116 cells were washed once withPBS and then irradiated with red light (l4600 nm) atvarious ¯uence rate covered with PBS. After irradiation,HCT-116 cells were returned in culture at 378C in McCoys5A medium supplemented with 10% FCS. Cell survival wasdetermined after 24 h using Trypan blue exclusion.
Transient transfection assays
HCT-116 cells were grown in 25 cm2 ¯ask for 2 days inMcCoys5A medium supplemented with 10% FCS and transfected with5 mg expression plasmids. Plasmids were mixed in McCoys 5Amedium, added to Fugene liposomes (7 ml) (Boehringer-Mannheim, Germany) for 15 min at room temperature andloaded on cells in 2 ml of McCoys 5A containing 10% FCS for24 h. Then, HCT-116 cells were incubated with PPME (6 mM)for 24 h and irradiated with red light.
Whole cell lysate extraction
After photodynamic treatment, cells were collected byscraping, washed once in ice-cold PBS and treated with lysis
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4081

bu�er (1% Nonidet P-40, 20 mM Tris pH 8.0, 137 mM NaCl,10% glycerol) supplemented with 1 mM phenyl-methylsulfo-nyl ¯oride, 1 mM sodium orthovanadate and anti-proteasescomplex (Complete, Boehringer Mannheim, Germany) for15 min on ice and further centrifuged at 10 000 g for 15 min.Protein concentrations were determined using the bisinconinicacid method (micro BCA, Pierce, USA). For the phospho-Bcl-2 assay, a phosphatase inhibitor mix was added to lysisbu�er as described by Kroll et al. (1999).
Preparation of S-100 fraction (cytosolic extracts)
Harvested cells were washed once with cold PBS andresuspended in ice-cold bu�er (20 mM Tris-HCl pH 8.0,137 mM NaCl, 10% glycerol) supplemented with 1 mM
phenyl-methylsulfonyl ¯oride, 1 mM sodium orthovanadateand an anti-protease cocktail (Boehringer Mannheim,Germany). Cells were disrupted using a Kontes douncehomogenizer. Lysates were centrifuged at 10 000 g for 10 minand the supernatant was further ultracentrifuged at 100 000 gfor 1 h in a Beckman Airfuge (Analis, Belgium). Proteinamounts were measured using Bradford method (Bio-Radprotein assay, Bio-Rad, Germany).
DNA laddering
After photosensitization,HCT-116 cellswere returned to culturefor various times as described above and washed once in PBSbefore being trypsinized, pooled with cells recovered from thesupernatant and centrifuged at 5000 g for 2 min. Cells werewashed again in PBS and treated with two cycles of lysis bu�er(1%NP-40 in 20 mMEDTA,50 mMTris-HCl, pH 7.5; 10 ml per106 cells, minimum 50 ml). After centrifugation for 5 min at1600 g the supernatant was collected and treated with RNAse Afor 120 min at 568C and brought to 1% (w/v) SDS, followed bydigestionwith proteinaseK (®nal concentration 2.5 mg/ml) for atleast 120 min at 378C. DNAwas then precipitated and analysedby gel electrophoresis in 1% agarose gels.
DAPI and TUNEL assays
Various times after photosensitization, HCT-116 cells werewashed with PBS and then washed once with 4', 6-Diamine-2'-phenylindole (DAPI) (1 mg/ml, Boehringer, Mannheim,Germany) in methanol and incubated for 15 min at 378C.After staining cells washed with methanol and placed oncoverslip to be observed by ¯uorescence microscopy (lexc:340 nm, lem: 388 nm). TUNEL (terminal deoxyuridine nick-end-labeling) assays were performed by using TUNEL labelmix and tunel enzyme (Boehringer, Mannheim, Germany)according to the procedure described by the manufacturer.Brie¯y, after photosensitization cells were cultured inMcCoy's medium supplemented with 10% FCS for di�erentperiods of time until ®xation. Cells were then washed twice inPBS, ®xed in paraformaldehyde (4%) and permeabilized in70% ethanol. After two washes in PBS, cells were labeled by¯uorescein-dUTP in the presence of terminal deoxynucleo-tidyl transferase (in situ cell death detection kit, BoehringerMannheim, Germany) and incubated for 60 min at 378C in ahumidi®ed atmosphere in the dark. After several washes inPBS, cells were analysed by ¯uorescence microscopy (NikonEclipse E800, Japan) using ®lters for ¯uorescein.
Immunoblot analysis
Detergent soluble proteins (30 ± 50 mg) were loaded andseparated by SDS±PAGE (10 ± 12% gels) under reducing
conditions followed by Western blotting. Membranes wereincubated for 45 ± 120 min with primary antibody at roomtemperature. Membranes were then probed with horseradishperoxidase-labeled anti-mouse IgG (1/2000 ± 1/5000), anti-rabbit IgG (1/2000 ± 1/5000) and anti-rat IgG (1/1000) inPBS, 0.05% Tween 20, 5% (w/v) milk powder for 30 ± 60 minat room temperature. Proteins were detected using achemiluminescence detection system (Amersham, ArlingtonHeights, IL, USA) and bands visualized by autoradiography.
AnnexinV ±PI staining
HCT-116 cells were cultivated in six-well plates for 24 h inMcCoy's medium supplemented with 10% FCS and thenincubated for another 24 h with 6 mM PPME in McCoy'smedium supplemented with 2% FCS before photosensitiza-tion. Photosensitized cells were gently scraped and pooledwith medium ¯oating cells. Cells were individualized in PBS-EDTA (10 mM) and stained as described by the manufacturer(Roche, Germany). Brie¯y, cell pellet was resuspended in500 mL AnnexinV-HEPES solution (10 mM HEPES-NaOH,pH 7.4, 140 mM NaCl, 5 mM CaCl2) and incubated on ice for30 min in the dark. Cells were then washed once in ice-coldHEPES bu�er and PI was added just before FACS analysis.Cells were analysed with a FacsCalibur (Becton-Dickinson,Sunnyvale, CA, USA).
Protease assay
A cell-free caspase-3 assay was performed as described byGranville et al. (1998). Brie¯y, 50 mg of whole cell lysate wasincubated at 378C for 4 h with 20 mM Ac-DEVD-AMC assubstrate (Calbiochem, San Diego, USA). Fluorescence wasmeasured using a VictorTM 1420 multilabel counter (Wallac,Sweden) or CytoFluor 2350 (PerSeptive Biosystems, Canada)set at 360 and 460 nm for excitation and emissionrespectively. Z-DEVD-fmk was provided by Enzyme SystemsProducts (Livermore, USA).
Fluorescence detection of ROS generation
HCT-116 cells were grown for 24 h on coverslips in McCoy'smedium supplemented with 10% FCS. After a 24 hincubation with 6 mM PPME, cells were loaded at RT inthe dark with 37 mM membrane permeable 6-carboxy-2',7'-dichlorodihydro¯uorescein diacetate, di(acetoxymethyl ester)(C-DCDHF-DA-AM) (Molecular Probe, USA) for 45 min.Coverslips were rinsed once in PBS to remove the excess ofprobe and then mounted on glass for microscopic observa-tions. Photosensitization reaction was iniated with the bluelight emitting microscope FITC ®lter and C-DCDHF-DA-AM ¯uorescence was analysed under the same conditions.
AbbreviationsPDT, photodynamic therapy; PPME, pyropheophorbide-amethylester; FCS, foetal calf serum; PARP, poly (ADP-ribose) polymerase; DAPI, 4',6-diamidine-2'-phenylindoledihydrochloride; TUNEL, terminal deoxyuridine nick-end-labeling; ROS, reactive oxygen species; 1O2, singlet oxygen;H2O2, hydrogen peroxide; IL, interleukin; TNF-a, tumornecrosis factor a; ER, endoplasmic reticulum; CSA,cyclosporin A; PDTC, pyrrolidine dithiocarbamate; NAC,N-acetyl cysteine; D2O, deuterium oxide, C-DCDHF-DA-AM, 6-carboxy-2',7'-dichlorodihydro¯uorescein diacetatedi(acetoxymethyl ester)
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4082
Oncogene

AcknowledgmentsThis work was supported by a grant from the BelgianNational Fund for Scienti®c Research (NFSR) (Brussels,
Belgium) and Te le vie (Brussels, Belgium). J-Y Matroule isResearch fellow from the FRIA and J Piette is ResearchDirector from the NFSR (Brussels, Belgium).
References
Assefa Z, Vantieghem A, Declercq W, Vandenabeele P,Vandenheede JR, Merlevede W, de Witte P and AgostinisP. (1999). J. Biol. Chem., 274, 8788 ± 8796.
Bellnier DA, Henderson BW, Pandey RK, Potter WR andDougherty TJ. (1993). J. Photochem. Photobiol. B, 20,55 ± 61.
Bossy-Wetzel E, Newmeyer DD and Green DR. (1998).EMBO J., 17, 37 ± 49.
Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D,Korbelik M, Moan J and Peng Q. (1998). J. Natl. CancerInst., 90, 889 ± 905.
Dumont A, Hehner SP, Hofmann TG, Ue�ng M, Droge Wand Schmitz ML. (1999). Oncogene, 18, 747 ± 757.
Evan G and Littlewood T. (1998). Science, 281, 1317 ± 1322.Gamard CJ, Dbaibo GS, Liu B, Obeid LM and Hannun YA.
(1997). J. Biol. Chem., 272, 16474 ± 16481.Gottlieb E, Vander Heiden MG and Thompson CB. (2000).
Mol. Cell. Biol., 20, 5680 ± 5689.Granville DJ, Carthy CM, Jiang H, Shore GC, McManus
BM and Hunt DW. (1998). FEBS Lett., 437, 5 ± 10.Granville DJ and Hunt DWC. (2000). Curr. Opin. Drug Disc.
Dev., 3, 232 ± 243.Green DR. (2000). Cell, 102, 1 ± 4.Green DR and Reed JC. (1998). Science, 281, 1309 ± 1312.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C,
Erdjument-Bromage H, Tempst P and Korsmeyer SJ.(1999). J. Biol. Chem., 274, 1156 ± 1163.
Halestrap AP, Doran E, Gillespie JP and O'Toole A. (2000).Biochem. Soc. Trans., 28, 170 ± 177.
Henderson BW, Bellnier DA, Greco WR, Sharma A, PandeyRK, Vaughan LA, Weishaupt KR and Dougherty TJ.(1997). Cancer Res., 57, 4000 ± 4007.
Hirpara JL, Seyed MA, Loh KW, Dong H, Kini RM andPervaiz S. (2000). Blood, 95, 1773 ± 1780.
Hockberger PE, Skimina TA, Centonze VE, Lavin C, Chu S,Dadras S, Reddy JK and White JG. (1999). Proc. Natl.Acad. Sci. USA, 96, 6255 ± 6260.
Hu X, Yee E, Harlan JM, Wong F and Karsan A. (1998).Blood, 92, 2759 ± 2765.
Jacobson MD, Weil M and Ra� MC. (1997). Cell, 88, 347 ±354.
Katschinski DM, Boos K, Schindler SG and Fandrey J.(2000). J. Biol. Chem., 275, 21094 ± 21098.
Kessel D and Luo Y. (1998). J. Photochem. Photobiol. B, 42,89 ± 95.
Kim DK, Cho ES and Um HD. (2000). Exp. Cell. Res., 257,82 ± 88.
Korbelik M. (1996). J. Clin. Laser Med. Surg., 14, 329 ± 334.Kroemer G and Reed JC. (2000). Nat. Med., 6, 513 ± 519.Kroll M, Margottin F, Kohl A, Renard P, Durand H,
Concordet JP, Bachelerie F, Arenzana-Seisdedos F andBenarous R. (1999). J. Biol. Chem., 274, 7941 ± 7945.
Kuhnel F, Zender L, Paul Y, Tietze MK, Trautwein C,Manns M and Kubicka S. (2000). J. Biol. Chem., 275,6421 ± 6427.
Kushnareva YE and Sokolove PM. (2000). Arch. Biochem.Biophys., 376, 377 ± 388.
Lee C, Wu SS and Chen LB. (1995). Cancer Res., 55, 2063 ±2069.
Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP,Nishimura Y, Crowe RA, Cascio WE, Bradham CA,Brenner DA and Herman B. (1998). Biochim. Biophys.Acta, 1366, 177 ± 196.
Lin CP, Lynch MC and Kochevar IE. (2000). Exp. Cell. Res.,259, 351 ± 359.
MacDonald IJ, Morgan J, Bellnier DA, Paszkiewicz GM,Whitaker JE, Litch®eld DJ and Dougherty TJ. (1999).Photochem. Photobiol., 70, 789 ± 797.
Matroule JY, Bonizzi G, Morliere P, Paillous N, Santus R,Bours V and Piette J. (1999a). J. Biol. Chem., 274, 2988 ±3000.
Matroule JY, Hellin AC, Morliere P, Fabiano AS, Santus R,Merville MP and Piette J. (1999b). Photochem. Photobiol.,70, 540 ± 548.
Miller GG, Brown K, Moore RB, Diwu ZJ, Liu J, Huang L,Lown JW, Begg DA, Chlumecky V and Tulip J. (1995).Photochem. Photobiol., 61, 632 ± 638.
Monney L, Olivier R, Otter I, Jansen B, Poirier GG andBorner C. (1998). Eur. J. Biochem., 251, 295 ± 303.
Morgan J, Potter WR and Osero� AR. (2000). Photochem.Photobiol., 71, 747 ± 757.
Murphree AL, Villablanca JG, Deegan WF, III, Sato JK,Malogolowkin M, Fisher A, Parker R, Reed E and GomerCJ. (1996). Arch. Ophthalmol., 114, 1348 ± 1356.
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BAand Yuan J. (2000). Nature, 403, 98 ± 103.
Oleinick NL and Evans HH. (1998). Radiat. Res., 150,S146 ± S156.
Pandey RK, Sumlin AB, Constantine S, Aoudla M, PotterWR, Bellnier DA, Henderson BW, Rodgers MA, SmithKM and Dougherty TJ. (1996). Photochem. Photobiol., 64,194 ± 204.
Pass HI. (1993). J. Natl. Cancer Inst., 85, 443 ± 456.Payne JT, McCaw DL, Casteel SW, Frazier D, Rogers K and
Tompson RV. (1996). Lasers Surg. Med., 18, 406 ± 409.Plumpe J, Malek NP, Bock CT, Rakemann T, Manns MP
and Trautwein C. (2000). Am. J. Physiol. Gastrointest.Liver Physiol., 278, G173 ±G183.
Rivera-Walsh I, Cvijic ME, Xiao G and Sun SC. (2000). J.Biol. Chem., 275, 25222 ± 25230.
Ruck A, Heckelsmiller K, Kaufmann R, Grossman N,Haseroth E and Akgun N. (2000). Photochem. Photobiol.,72, 210 ± 216.
Saleh A, Srinivasula SM, Acharya S, Fishel R and AlnemriES. (1999). J. Biol. Chem., 274, 17941 ± 17945.
Schneider P and Tschopp J. (2000). Pharm. Acta Helv., 74,281 ± 286.
Shimizu S, Konishi A, Kodama T and Tsujimoto Y. (2000).Proc. Natl. Acad. Sci. USA, 97, 3100 ± 3105.
Shimizu S, Narita M and Tsujimoto Y. (1999). Nature, 399,483 ± 487.
Shimura M, Osawa Y, Yuo A, Hatake K, Takaku F andIshizaka Y. (2000). J. Leukoc. Biol., 68, 87 ± 96.
Srivastava RK, Mi QS, Hardwick JM and Longo DL. (1999).Proc. Natl. Acad. Sci. USA, 96, 3775 ± 3780.
Steller H. (1995). Science, 267, 1445 ± 1449.Stridh H, Kimland M, Jones DP, Orrenius S and Hampton
MB. (1998). FEBS Lett., 429, 351 ± 355.
Oncogene
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4083

Tafani M, Schneider TG, Pastorino JG and Farber JL.(2000). Am. J. Pathol., 156, 2111 ± 2121.
Tajiri H, Hayakawa A, Matsumoto Y, Yokoyama I andYoshida S. (1998). Cancer Lett., 128, 205 ± 210.
Tao J, Sanghera JS, Pelech SL, Wong G and Levy JG. (1996).J. Biol. Chem., 271, 27107 ± 27115.
Van Antwerp DJ, Martin SJ, Kafri T, Green DR and VermaIM. (1996). Science, 274, 787 ± 789.
Varnes ME, Chiu SM, Xue LY and Oleinick NL. (1999).Biochem. Biophys. Res. Commun., 255, 673 ± 679.
Wang CY, Guttridge DC, Mayo MW and Baldwin ASJ.(1999). Mol. Cell. Biol., 19, 5923 ± 5929.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV andBaldwin Jr AS. (1998). Science, 281, 1680 ± 1683.
Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S,Farrow SN, Haslett C and Rossi AG. (1999). J. Biol.Chem., 274, 4309 ± 4318.
Weishaupt KR, Gomer CJ and Dougherty TJ. (1976).Cancer Res., 36, 2326 ± 2329.
Zhuang S, Lynch MC and Kochevar IE. (1999). Exp. CellRes., 250, 203 ± 212.
Zhuang S, Demirs JT and Kochevar IE. (2000). J. Biol.Chem., 275, 25939 ± 25948.
Zorov DB, Filburn CR, Klotz LO, Zweier JL and Sollott SJ.(2000). J. Exp. Med., 192, 1001 ± 1014.
Colon cancer cells apoptosis mediated by photosensitizationJ-Y Matroule et al
4084
Oncogene
Related Documents











