ß 2007 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 143A:219–228 (2007) Rapid Publication Matthew-Wood Syndrome: Report of Two New Cases Supporting Autosomal Recessive Inheritance and Exclusion of FGF10 and FGFR2 Jelena Martinovic-Bouriel, 1 * Ce ´line Bernabe ´-Dupont, 2 Christelle Golzio, 4,5 Bettina Grattagliano-Bessie `res, 3 Vale ´rie Malan, 1,4 Maryse Bonnie `re, 1 Chantal Esculpavit, 4 Catherine Fallet-Bianco, 3 Ve ´ronique Mirlesse, 3 Jero ˆme Le Bidois, 3 Marie-Ce ´cile Aubry, 2 Michel Vekemans, 1,4,5 Nicole Morichon, 1,4 Heather Etchevers, 4,5 Tania Attie ´-Bitach, 1,4,5 Fe ´re ´chte ´ Encha-Razavi, 1,4 and Alexandra Benachi 2,4 1 Assistance Publique—Ho ˆpitaux de Paris; Ho ˆpital Necker—Enfants Malades, Department of Genetics, Embryo-Fetal Pathology Unit, Paris, France 2 Assistance Publique—Ho ˆpitaux de Paris; Ho ˆpital Necker—Enfants Malades, Department of Obstetrics, Paris, France 3 Institut de Pue ´riculture, Department of Fetal Pathology, Paris, France 4 Universite ´ Paris-Descartes; Ho ˆpital Necker—Enfants Malades, Paris, France 5 INSERM U781, Ho ˆpital Necker—Enfants Malades, Paris, France Received 28 July 2006; Accepted 27 October 2006 We describe two fetal cases of microphthalmia/anophthal- mia, pulmonary agenesis, and diaphragmatic defect. This rare association is known as Matthew-Wood syndrome (MWS; MIM 601186) or by the acronym ‘‘PMD’’ (Pulmonary agenesis, Microphthalmia, Diaphragmatic defect). Fewer than ten pre- and perinatal diagnoses of Matthew- Wood syndrome have been described to date. The cause is unknown, and the mode of transmission remains unclear. Most cases have been reported as isolated and sporadic, although recurrence among sibs has been observed once. Our two cases both occurred in consanguineous families, further supporting autosomal recessive transmission. In addition, in one family at least one of the elder sibs presented an evocatively similar phenotype. The spatiotem- poral expression pattern of the FGF10 and FGFR2 genes in human embryos and the reported phenotypes of knockout mice for these genes spurred us to examine their coding sequences in our two cases of MWS. While in our patients, no causative sequence variations were identified in FGF10 or FGFR2, this cognate ligand-receptor pair and its downstream effectors remain functional candidates for MWS and similar associations of congenital ocular, diaphragmatic and pulmonary malformations. ß 2007 Wiley-Liss, Inc. Key words: microphthalmia; pulmonary hypoplasia; con- genital diaphragmatic defect; polymalformative syndrome; association; growth retardation; facial dysmorphy; prenatal diagnosis; fibroblast growth factor; embryo How to cite this article: Martinovic-Bouriel J, Bernabe ´-Dupont C, Golzio C, Grattagliano-Bessie `res B, Malan V, Bonnie `re M, Esculpavit C, Fallet-Bianco C, Mirlesse V, Le Bidois J, Aubry M-C, Vekemans M, Morichon N, Etchevers H, Attie ´-Bitach T, Encha-Razavi F, Benachi A. 2007. Matthew-Wood syndrome: Report of two new cases supporting autosomal recessive inheritance and exclusion of FGF10 and FGFR2. Am J Med Genet Part A 143A:219 – 228. INTRODUCTION A distinct association of pulmonary, ocular, and diaphragmatic congenital malformations has been reported occasionally over the last 25 years. Spear et al. [1987] examined a patient with bilateral pulmonary agenesis, eventration of the left dia- phragm, bilateral microphthalmia, and a complex cardiac defect with a ventricular septal defect and absent pulmonary vessels. Engellenner et al. [1989] Jelena Martinovic-Bouriel, Ce ´line Bernabe ´-Dupont, and Christelle Golzio have contributed equally to this work. Grant sponsor: Association Franc ¸aise contre les Myopathies; Grant sponsor: Institut National pour la Sante ´ et la Recherche Me ´dicale (INSERM). *Correspondence to: Dr. Jelena Martinovic-Bouriel, Embryo-Fetal Pathology Unit, Department of Genetics, Hopital Necker-Enfants Malades, 149 rue de Sevres, 75015 Paris, France. E-mail: [email protected] DOI 10.1002/ajmg.a.31599

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

� 2007 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 143A:219–228 (2007)
Rapid Publication
Matthew-Wood Syndrome:Report of Two New Cases Supporting Autosomal Recessive
Inheritance and Exclusion of FGF10 and FGFR2
Jelena Martinovic-Bouriel,1* Celine Bernabe-Dupont,2 Christelle Golzio,4,5
Bettina Grattagliano-Bessieres,3 Valerie Malan,1,4 Maryse Bonniere,1 Chantal Esculpavit,4
Catherine Fallet-Bianco,3 Veronique Mirlesse,3 Jerome Le Bidois,3 Marie-Cecile Aubry,2
Michel Vekemans,1,4,5 Nicole Morichon,1,4 Heather Etchevers,4,5 Tania Attie-Bitach,1,4,5
Ferechte Encha-Razavi,1,4 and Alexandra Benachi2,41Assistance Publique—Hopitaux de Paris; Hopital Necker—Enfants Malades, Department of Genetics,
Embryo-Fetal Pathology Unit, Paris, France2Assistance Publique—Hopitaux de Paris; Hopital Necker—Enfants Malades, Department of Obstetrics, Paris, France
3Institut de Puericulture, Department of Fetal Pathology, Paris, France4Universite Paris-Descartes; Hopital Necker—Enfants Malades, Paris, France
5INSERM U781, Hopital Necker—Enfants Malades, Paris, France
Received 28 July 2006; Accepted 27 October 2006
We describe two fetal cases of microphthalmia/anophthal-mia, pulmonary agenesis, and diaphragmatic defect. Thisrare association is known as Matthew-Wood syndrome(MWS; MIM 601186) or by the acronym ‘‘PMD’’ (Pulmonaryagenesis, Microphthalmia, Diaphragmatic defect). Fewerthan ten pre- and perinatal diagnoses of Matthew-Wood syndrome have been described to date. The cause isunknown, and the mode of transmission remains unclear.Most cases have been reported as isolated and sporadic,although recurrence among sibs has been observed once.Our two cases both occurred in consanguineous families,further supporting autosomal recessive transmission. Inaddition, in one family at least one of the elder sibspresented an evocatively similar phenotype. The spatiotem-poral expression pattern of the FGF10 and FGFR2 genes in
human embryos and the reported phenotypes of knockoutmice for these genes spurred us to examine their codingsequences in our two cases of MWS. While in our patients, nocausative sequence variations were identified in FGF10 orFGFR2, this cognate ligand-receptor pair and its downstreameffectors remain functional candidates for MWS andsimilar associations of congenital ocular, diaphragmatic andpulmonary malformations. � 2007 Wiley-Liss, Inc.
Key words: microphthalmia; pulmonary hypoplasia; con-genital diaphragmatic defect; polymalformative syndrome;association; growth retardation; facial dysmorphy; prenataldiagnosis; fibroblast growth factor; embryo
How to cite this article: Martinovic-Bouriel J, Bernabe-Dupont C, Golzio C, Grattagliano-Bessieres B,Malan V, Bonniere M, Esculpavit C, Fallet-Bianco C, Mirlesse V, Le Bidois J, Aubry M-C, Vekemans M,Morichon N, Etchevers H, Attie-Bitach T, Encha-Razavi F, Benachi A. 2007. Matthew-Wood syndrome:Report of two new cases supporting autosomal recessive inheritance and exclusion of FGF10 and
FGFR2. Am J Med Genet Part A 143A:219–228.
INTRODUCTION
A distinct association of pulmonary, ocular, anddiaphragmatic congenital malformations has beenreported occasionally over the last 25 years. Spearet al. [1987] examined a patient with bilateralpulmonary agenesis, eventration of the left dia-phragm, bilateral microphthalmia, and a complexcardiac defect with a ventricular septal defect andabsent pulmonary vessels. Engellenner et al. [1989]
Jelena Martinovic-Bouriel, Celine Bernabe-Dupont, and ChristelleGolzio have contributed equally to this work.
Grant sponsor: Association Francaise contre les Myopathies; Grantsponsor: Institut National pour la Sante et la Recherche Medicale(INSERM).
*Correspondence to: Dr. Jelena Martinovic-Bouriel, Embryo-FetalPathology Unit, Department of Genetics, Hopital Necker-EnfantsMalades, 149 rue de Sevres, 75015 Paris, France.E-mail: [email protected]
DOI 10.1002/ajmg.a.31599

described another patient with bilateral pulmonaryagenesis, an inverted right diaphragm, right micro-phthalmia and a small heart with absent pulmonaryveins. Seller et al. [1996] coined the term Matthew-Wood syndrome (MWS) in reference to an associa-tion of microphthalmia and pulmonary hypoplasia,after the name of a firstborn sib. Berkenstadtet al. [1999] reported the coincidence of unilateralpulmonary agenesis, microphthalmia, diaphrag-matic hernia, and intrauterine growth retardationunder the acronym ‘‘PMD.’’ The authors proposedthat PMD might be a new entity, and that MWS,reported as anophthalmia and pulmonary hypo-plasia, might represent an incomplete form of PMD.
A few cases have been observed in the peri- andpostnatal period [Spear et al., 1987; Enns et al., 1998;Priolo et al., 2004; Li and Wei, 2006; Robert Lee et al.,2006] as well as three cases of prenatal diagnosis ofthe syndrome at the respective ages of 18, 22, and36 weeks’ gestation [Engellenner et al., 1989; Selleret al., 1996; Berkenstadt et al., 1999]. While mostcases were apparently sporadic and isolated, MWSseems to have a genetic basis because of one reportof familial recurrence [Seller et al., 1996]. A similarphenotype, presenting in addition with tetralogy ofFallot, was observed in a patient with a balancedreciprocal translocation de novo 46,XY,t(1;15) (q41;21.2) [Smith et al., 1994]. No one animal modelrecapitulates this particular human association.However, knock-outs of the murine Fgf10 (fibroblastgrowth factor 10) [Min et al., 1998; Sekine et al., 1999]or its binding-specific receptor isoform, Fgfr2(IIIb)[De Moerlooze et al., 2000], have multiple congenitaldefects including pulmonary agenesis. Other organsystems are affected including ablepharon forFgfr2b, atretic or stenotic colon [Fairbanks et al.,2005] or imperforate anus, hypoplasic pituitary,lacrimal and salivary glands, pancreas and spleen,abnormal limbs and, inconsistently, kidneys [Ohuchiet al., 1997; Min et al., 1998]. Cardiac outflow tractmalformations have also been noted.
Given a certain number of similarities betweenthese animal models and the clinical signs ofMatthew-Wood cases described here and elsewherein the literature, we therefore undertook a molecularanalysis of the FGF10 and FGFR2 genes in our twopatients and were able to exclude both genes aspathogenic candidates in these individuals.
METHODS
Standard Karyotype and FISH (Fluorescence InSitu Hybridization) Analysis on Chromosomes
Standard karyotyping using GTG and RHG band-ing analysis was carried out on cultured amnioticfluid cells according to standard procedures. FISHwas performed using BACs, RP11-297K5, RP11-
1149B18, RP11-239E10 spanning the GATA4(8p23.1) and CHD2 (15q26.1) genes and the 1q41region, respectively, to rule out any possibledeletions. BACs were selected from several data-bases accessible through the Internet (UCSC, Uni-versity of California, Santa Cruz http://www.genome.ucsc.edu/ and NCBI, National centre forBiotechnology Information http://www.ncbi.nlm.nih.gov/). FISH experiments were performed onchromosome preparations as described previously[Romana et al., 1993].
In Situ Hybridization
Human embryos were collected from pregnancieslegally terminated using the mefiprestone protocol,in concordance with French law 00-800 and hospitalethics committee recommendations. Primers wereselected for PCR amplification (FGF10: [F] 50-CTGGATGGCTTGTATCAAATG-30 [R] 50-TTGGCA-AAAGAGCCATTGGT-30 corresponding to exon 3;FGFR2(IIIb): [F] 50-CTTTAATGCCGCTGTTTAG-30 [R] 50-TCTTTTCAGCTTCTATATCCAG-30 corre-sponding to alternatively spliced exon 9, includedas the 8th exon in the IIIb RNA isoform). A T7promotor sequence extension (TAATACGACTCAC-TATAGGGAGA) was added at the 50 end of eachprimer. T7F/R and F/T7R primer pairs allowedthe amplification of sense and antisense templatesrespectively, specific to the FGF10 or theFGFR2(IIIb) transcripts. Riboprobe labeling, tissuefixation, hybridization, and developing were carriedout according to standard protocols, as previouslydescribed [Wilkinson, 1992; Trueba et al., 2005].
DNA Analysis
Both cases had normal chromosomes according tokaryotype. DNA was extracted from thymus samplesafter informed consent and autopsy using standardprotocols. PCRs were carried out using intronicprimers for the 3 exons of the FGF10 gene and the18 coding exons (including the alternativelyincluded exons 8 and 9) for the FGFR2 gene;sequences and conditions presented in Table II.
CLINICAL REPORT
Patient 1
A consanguineous healthy couple of Romanianorigin presented with a history of neonatal demise intheir two first-born children. The first child, a boydelivered vaginally at 33 weeks’ gestation (birth-weight: 800 g, <3rd centile), died on the first daypostpartum with no autopsy. The second child, agirl delivered at 43 weeks’ gestation (birthweight:2,800 g, 3rd centile), died within the first hour of life.The parents brought to our attention her respiratory
220 MARTINOVIC-BOURIEL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

problems associated with a bilateral absence ofocular globes. However, the parents had no medicalrecords and autopsy was not performed. Overthe following years, the mother had four healthychildren. Her seventh pregnancy ended in a volun-tary interruption.
Their eighth child is a boy who developed oculartroubles but he has had neither medical follow-upnor an obvious diagnosis.
In the ninth pregnancy, at a maternal age of34 years, the first sonogram, performed at 12 weeks’gestation showed normal nuchal translucency andno anomalies. A second sonogram at 23 weeks’gestation showed bilateral anophthalmia (Fig. 1) andgastro-duodenal dilatation in a male fetus. Thecontrol ultrasound at 29 weeks’ gestation confirmedthe absence of ocular globes and jejunal distension.In addition, polyhydramnios, a left diaphragmaticdefect (Fig. 2), and a short femoral length, measuredat the 3rd centile, were noted. A TORCH study wasnegative. Amniocentesis showed a normal malekaryotype (46,XY). After genetic counseling, theparents opted for termination of pregnancy at31 weeks, in the light of a poor prognosis.
The fetus (birth weight: 1,250 g, <3rd centile;length: 38 cm, 3rd centile; head circumference: 28cm, 10th–25th centile) presented with bilateralmicrophthalmia with recessed orbits, hypotelorism,narrow palpebral fissures, short nose, large ears,and retrognathia (Fig. 3A,B). Autopsy showed thebilateral absence of bronchial and pulmonaryanlagebelow blind-ended tracheae. The heart was normal,except for the absence of pulmonary artery branchesand pulmonary veins. Bilateral diaphragmatic even-tration, as well as stomach and duodenal dilatationupstream of stenosis at the duodeno-jejunal junctionwere confirmed (Fig. 4). In addition, a commonmesentery was noted. Microscopic ocular examina-tion showed bilateral cataracts and the presence ofretinal tissue in severely hypoplastic globes. The
cerebral examination was normal, except for aslightly hypoplastic lateral geniculate body. Thevermis displayed a few heterotopic Purkinje cells(not shown).
Patient 2
A 32-year-old G2P2 (Tanner scale) womanpresented on her second pregnancy with noparticular medical history and a previous, healthy
FIG. 1. Ultrasonography at 29 SA in Patient 1 showing a left diaphragmaticdefect with an ascended stomach. [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
FIG. 2. 3D ultrasonography at 29 SA in Patient 1 showing bilateralmicrophthalmia. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
FIG. 3. Facial gestalt in Patient 1 (A: front, B: profile) and Patient 2 (C: front,D: profile) showing common features: narrow palpebral fissures withmicrophthalmia, high forehead, short nose with anteverted nostrils. [Colorfigure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
MATTHEW-WOOD SYNDROME 221
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

son. The couple is consanguineous, of Portugueseorigin.
The second sonogram, performed at 23 weeks’gestation, showed a left diaphragmatic defect withmediastinal shift, hypoechogenic digestive tract,bilateral anophthalmia, long philtrum, abnormalears, and moderate renal dysplasia. Amniocentesisyielded a normal female karyotype (46,XX).Echocardiography showed atresia of the pulmonaryartery with ventricular septal defect. Given a verypoor prognosis, the parents opted for termination ofthe pregnancy at 29 weeks.
The fetus (birthweight: 1,160g, 10th centile; length:37 cm, 10th centile; HC: 27.5 cm, 25th–50th centile)presented bilateral microphthalmia, a high forehead,a flat nosewith antevertednares, a bifid uvula, a largeneck, and camptodactyly (Fig. 3C,D). The autopsyshowed bilateral pulmonary agenesis with no mainbronchi, bilateral diaphragmatic eventration, a hor-izontalized heart with pulmonary artery agenesis andperimembraneous septal defect (Fig. 5). In addition,duodenal stenosis (Fig. 6), pancreatic agenesis, and amultilobulated spleen were noted. Microscopicocular examination confirmed bilateral microphthal-mia with retinal dysplasia and cataracts (Fig. 7). No
anomalies were found on neuropathologic exam-ination.
MOLECULAR STUDIES
Standard Karyotype and FISH Analysis
In all 20 metaphases analyzed, chromosomalanalysis of the fetus 1 and 2 were normal. FISHanalysis with BAC clones RP11-297K5, RP11-1149B18, RP11-239E10 showed two hybridizationsignals on chromosomes 8, 15, and 1, respectively.According to these results, a submicroscopicdeletionwas excluded.
In Situ Hybridization
The expression patterns of FGF10 and theFGFR2(IIIb) isoform, corresponding to the onlyreceptor form to which FGF10 specifically binds,
FIG. 4. Diaphragmatic eventration and pulmonary agenesis in Patient 1.[Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
FIG. 5. Pulmonary artery agenesis in Patient 2. [Color figure can be viewed inthe online issue, which is available at www.interscience.wiley.com.]
FIG. 6. Duodenal dilatation in Patient 1 (A) and Patient 2 (B). [Color figurecan be viewed in the online issue, which is available at www.interscience.wiley.com.]
222 MARTINOVIC-BOURIEL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
were analyzed at multiple stages of development ofnormal human embryos.
At Carnegie stage (C) 11 (24 days’ development,not shown) FGF10was restricted to themesenchymeof the secondary heart field (future outflow tract)andweak expression began in the anlage of theadenohypophysis; diffuse central nervous system(CNS) expression was seen at C12 (26 days).FGFR2(IIIb) was transcribed in both cardiac inflowand outflow segments, throughout the CNS, andstrongly expressed in the pharyngeal endoderm andlateral plate mesoderm. FGF10 transcripts wereobserved at C13–C14 (28–32 days) in restrictedportions of the otic vesicle and pharyngeal archmesenchyme, as well as uniformly in fore- andhindlimb bud mesenchyme in similar domains tothose already noted in vertebrate animal models.FGFR2(IIIb)began to be expressed at higher levels inboth dorsal and ventral posterior diencephalon, inthe ventral CNS elsewhere, in the ectodermal/endodermal epithelia of the pharyngeal arches, inthe gut endoderm, and in the otic vesicle. At C15 (34days, not shown) and C19 (47 days), FGF10expression was present in the nasal pit ectoderm aswell as in the neural retina (Fig. 8A, inset). Anequivalent retinal expression domain had nothitherto been noted in the reports of spatiotemporaltranscript distribution in the mouse or chick.
Expression was maintained in the germinal layersof the CNS for both FGF10 and FGFR2(IIIb) at C19.Particularly intenseFGF10 signalwas observed in the
future hypothalamus (mirroring the strong receptorexpression visible between C12 and C15), while bothFGF10 and its receptor were expressed in thedeveloping adenohypophysis (ah; Fig. 8A–C). Com-plementary patterns were observed in the facialprimordia,with strongFGF10 expression in the toothmesenchyme and tongue muscle (tb, to; Fig. 8C), andmorediscretely in thenasal andpalatialmesenchyme(fm), while FGFR2(IIIb) was transcribed within thebuccal ectoderm (be) and pharyngeal endodermalepithelia, within the thymic and thyroid primordia(thry, tm), and around the condensing mesenchymeof Meckel’s cartilage (MC; Fig. 8D). Intense FGF10expression was found in the muscular layer ofthe stomach (not shown), intestine and rectum(int; Fig. 8A), and lower levels were observed inthe mesenchyme of the urogenital folds (uf);FGFR2(IIIb) transcripts were localized to the uro-genital fold epithelium (Fig. 8B) and their signal wasonly abovebackgroundwithin the intestinalmucosalepithelium. However, both ligand and receptor wereexpressed in the muscular layer of the physiological(at this stage) intestinal hernia into the umbilical cord(Fig. 8A,B). Lung (lu) expression patterns were alsocomplementary, with FGF10 transcripts observed inthe interstitial mesenchyme between the developinglobes (Fig. 8E), and FGFR2(IIIb) highly expressed inthe tracheal and bronchial epithelia (Fig. 8F). Bothgenes were expressed in the pericartilaginouscondensations of the developing digits at this stage(Fig. 8A,B).
FGF10 and FGFR2 Gene Analyses
Direct sequencing of both cases was carried out forthe FGF10 and FGFR2 genes. Case 1 had a hetero-zygous, conservative substitution in FGFR2 at V534V,inherited from his non-affected mother. FGF10 hadno sequence variations from the published sequence(RefSeq NM_004465). Genescan analysis showedthat unaffected parents and affected fetus shared onecommon allele encompassing the entire geneand flanking regions (data not shown), renderingan interstitial chromosomal deletion unlikely in theface of probable familial recurrence.
Case 2 had no coding variations in FGF10but demonstrated heterozygosity at IVS2-15g> c,thereby excluding a heterozygous deletion of theentire gene. For FGFR2, while no coding variationswere seen, a known SNP was identified at V232V(dbSNP: rs17859273), allowing the same conclusionto be drawn.
DISCUSSION
The principal clinical signs of hitherto reportedcases of MWS or PMD syndrome in comparison toour two cases, are presented in Table I. In both ourcases, bilateral pulmonary agenesis is associated
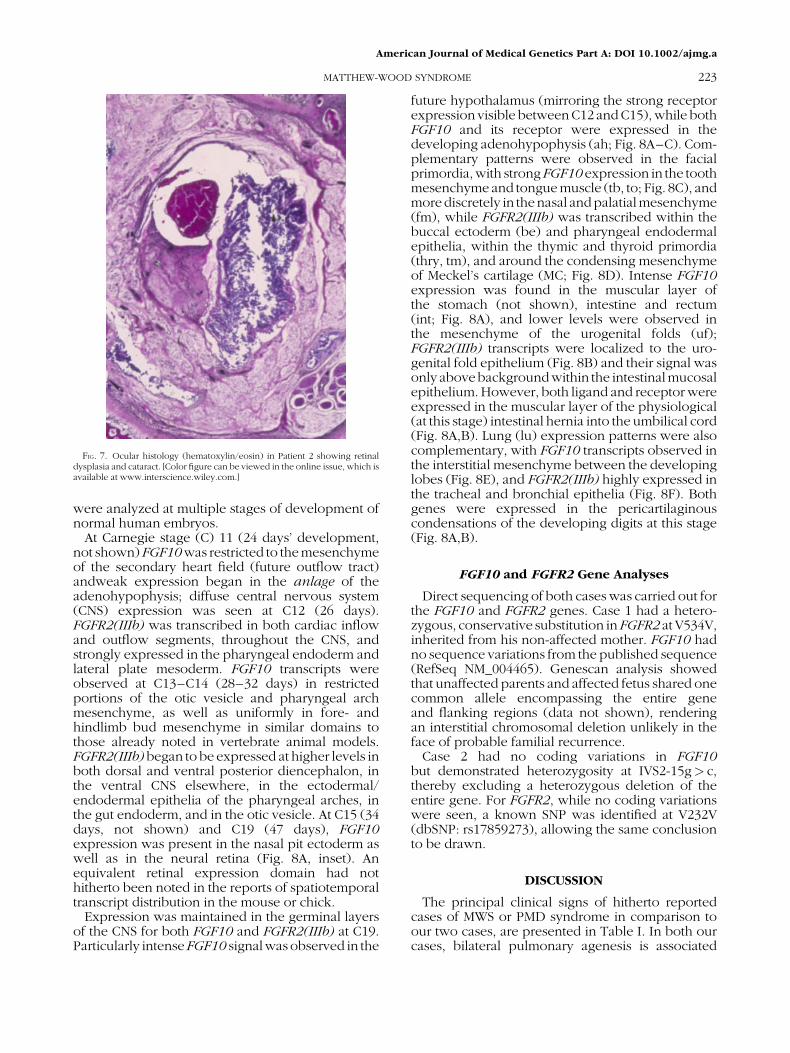
FIG. 7. Ocular histology (hematoxylin/eosin) in Patient 2 showing retinaldysplasia and cataract. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
MATTHEW-WOOD SYNDROME 223
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

with diaphragmatic eventration. In one case, thepulmonary arterywas absent, in theother pulmonaryartery branches were missing. Two other casesdescribed previously had bilateral pulmonary agen-esis associated with unilateral diaphragmatic even-tration [Spear et al., 1987; Engellenner et al., 1989].Similar facial gestalt exists in our cases, and includeda high forehead, short palpebral fissures and micro-phthalmia, short nose with anteverted nostrils and asmall chin. Similar features such as flat face, longphiltrum, narrow palpebral fissures, prominent nosehave been observed by other groups [Seller et al.,1996; Steiner et al., 2002].
It is noteworthy that duodenal stenosis was presentin both of our cases, though not previously reported.Moreover, we have observed pancreatic agenesisand a multilobulated spleen in the second case.Seller et al. [1996] had observed a case with a
hypoplastic spleen. Two other cases presented withrenal dysplasia and malrotation [Engellenner et al.,1989; Priolo et al., 2004]. In our second case, thesonogram noted a moderate renal dysplasia whichwas not confirmed at the autopsy.
Microphthalmia/anophthalmia in association withdiaphragmatic defect and pulmonary hypoplasia hasbeen reported in multiple syndromes, includingFryns syndrome [Lubinsky et al., 1983], Frasersyndrome, Goldenhar syndrome, and Goltz–Gorlinsyndrome [Kunze et al., 1979; Warburg et al., 1997].Fryns syndrome is the best-characterized syndromeof diaphragmatic defects with eye abnormalities[reviewed in Fryns, 1987; Cunniff et al., 1998].The combination of features in Fryns syndromewas described as follows: hydramnios, coarseface, cleft palate, distal limb hypoplasia, diaphrag-matic defect, lung hypoplasia, cloudy cornea,
FIG. 8. In situ hybridization on human parasagittal embryo sections at Carnegie stage 19 (47 days) using ribosondes against FGF10 (A,C,E) or FGFR2(IIIb) (B,D,F).Signal in white, aside from refringent red blood cells. Rostral left, ventral top. A: FGF10 transcripts are observed in this near-sagittal section in the germinal zone of theCNS, in particular in the ventral diencephalon; in themuscle of the tongue, esophagus and intestine, with strong expression in the rectum, and in themesenchyme of theurogenital folds. Inset: distinct expression is observed in the neural retina. B: An adjacent section hybridized with the FGFR2(IIIb) antisense probe. Transcripts areobserved in the endodermal and ectodermal epithelia, in the adenohypophysis, the urogenital fold and hindlimb ectoderm. C: A close-up of the craniofacial regionshows low FGF10 expression in the adenohypophysis, more intense signal in the tooth buds and forming salivary glands, and in the facial mesenchyme andoesophageal muscles. D: FGFR2(IIIb) is expressed complementarily in the buccal ectoderm and pharyngeal endodermal epithelium; in the thyroid and thymicprimordia, and around but not within Meckel’s cartilage. E: FGF10 transcripts are seen at low levels in the muscular walls of the pulmonary artery and aorta, and in theinterlobar mesenchyme of the lung. F: Its receptor is transcribed within the tracheal and bronchial epithelia, but not in the great vessels. Non-specific, flocular signal isseen at the sites of red blood cell accumulation.
224 MARTINOVIC-BOURIEL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

TABLE
I.Com
par
ison
ofClinic
alFin
din
gs
inM
atth
ew
-Wood
Syndro
me
Reported
inth
eLi
tera
ture
Fin
din
gPat
ient1
Pat
ient2
Berk
enst
adtetal
.Sp
ear
etal
.Engelleneretal
.Se
ller
etal
.(t
wo
sibs)
Stei
neretal
.(t
wo
spora
dic
case
s)Priolo
etal
.
Mic
rophth
alm
ia/
anophth
alm
iaBilat
era
lBilat
era
lBilat
era
lBilat
era
lU
nilat
era
lBilat
era
l(2
/2)
Bilat
era
l(2
/2)
Bilat
era
l
Pulm
onar
ym
alfo
rmat
ion
Bilat
era
lag
enesi
sof
pulm
onar
yar
tery
bra
nch
es
Bilat
era
lag
enes
isU
nilat
era
lag
enes
isBilat
era
lag
enes
isBilat
era
lag
enesi
sBilat
era
lag
enes
isofupperlo
bes
(1/2
),bilat
era
lhypopla
sia
(1/2
)
No
lung
anom
aly
(1/2
),bilat
era
lbro
nch
o-p
ulm
onar
ydysp
lasi
a(1
/2)
Bilat
era
lhypopla
sia
Dia
phra
gm
atic
defe
ctBilat
era
l(e
ventrat
ion)
Bilat
era
l(e
ven
trat
ion)
Unilat
era
l(h
ern
ia)
Unilat
era
l(e
ventrat
ion)
Unilat
era
l(inverted)
No
(2/2
)Bilat
era
l(e
ven
trat
ion)
(2/2
)U
nilat
era
l(h
ern
ia)
Car
dia
cm
alfo
rmat
ion
No
Pulm
onar
yar
tery
agenes
is,
ventric
ula
rse
pta
ldefe
ct
No
Pulm
onar
yar
tery
agenes
is,
ventric
ula
rse
pta
ldefe
ct
Modera
tedilat
atio
nof
rightat
rium
,cl
ose
dnar
row
duct
us
Single
ventric
le,
hypopla
stic
left
atrium
,enla
rged
pulm
onar
ytrunk
Ost
ium
prim
um
ASD
,rightventric
ula
rhypertro
phy
(1/2
)
No
Fac
ialan
om
alie
sSh
ort
pal
peb
ral
fiss
ure
s,sh
ort
nose
,re
trognat
hia
,la
rge
ear
s
Short
pal
pebra
lfiss
ure
s,lo
ng
philtrum
,la
rge
ear
s
Notdocu
mente
dN
otdocu
mente
dN
otdocu
mente
dPro
min
entnose
,sh
ort
upperlip,
mic
rognat
hia
with
cleft
pal
ate,lo
w-s
et
ear
s(1
/2)
Short
pal
pebra
lfiss
ure
s,pro
min
ent
nose
with
ante
verted
nar
es
Oth
er
Duodenal
stenosi
s,hypopla
stic
pan
creat
icst
ruct
ure
s
Duodenal
stenosi
s,poly
-lo
bed
sple
en,
agenes
isof
pan
creas
��
Renal
dysp
lasi
aH
ypopla
stic
sple
en
and
ute
rus
Cry
pto
rchid
ism
(1/2
)M
alro
tation
of
the
left
kid
ney
Gro
wth
reta
rdat
ion
þþ
þþ
þþ
þ(1
/2)
þ
Poly
hydra
mnio
sþ
þþ
�þ
�þ
(1/2
)þ
Kar
yoty
pe
Norm
alN
orm
alN
orm
alN
orm
alN
orm
alN
orm
alN
orm
al(2
/2)
Norm
alConsa
nguin
ity
Yes,
thre
esi
bs
Yes
No
No
No
No,tw
osi
bs
No
(2/2
)N
o
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

microphthalmia, renal dysplasia, and cerebralmalformations. It is interesting that primary pulmon-ary hypoplasia without a diaphragmatic defecthas also been described in Fryns syndrome [Willemset al., 1991; Wilgenbus et al., 1994]. Furthermore,chromosomal abnormalities, such as deletionsof 15q26.2, 8p23.1, and 1q41-q42.12, have beenassociated with the congenital diaphragmatic her-niaof Fryns syndrome [Slavotinek et al., 2005;Kantarci et al., 2006]. We excluded submicroscopicdeletions in these regions of both present patientsusing FISH.
The hallmarks of Fraser syndrome are cryptoph-talmos or anophthalmia (93%), laryngeal atresia withenlarged lungs, cutaneous syndactyly of digits (54%),genital or renal anomalies with frequent renalagenesis (90%), abnormal ears and an anomaly ofcord implantation on the abdominal wall [Tibboeland Gaag, 1996]. Almost half of the affected infantsare stillborn or die in infancy, and mental retardation
is common. In humans, this autosomal recessivedisorder is genetically heterogeneous. The FRAS1gene maps to 4q21 and encodes a large extracellularmatrix protein highly homologous to the murineequivalent [McGregor et al., 2003]. In two familieswith Fraser syndrome unlinked to the FRAS1 gene,Jadeja et al. [2005] found a missense mutation in theFREM2 gene. Both proteins are involved in ectoder-mal adhesion to underlying basal mesenchymeduring development [reviewed in Smyth and Scam-bler, 2005]. The absence of characteristic signs ofFraser syndrome, in particular, digital and renalanomalies, support a different condition in our cases,although they may be functionally related throughimpaired epithelial-mesenchymal signaling duringfetal life.
The mode of transmission of the Matthew-Woodsyndrome has been a subject of debate. Most casesreported in the literature appear to be isolated.However, Seller et al. [1996] reported two sibs of
TABLE II. PCR Primers for Direct Sequencing of Human FGF10 and FGFR2
Name Temperature Amplicon size
TCCAGTATGTTCCTTCTGATG FGF10-1F 54.58C 424 bpTGGGGGTGGATAATTGGAA FGF10-1R 54.58CTTGCCGGGTTTTAAGACACA FGF10-2F 558C 332 bpGGTAATGGTTTACTGGAGTGG FGF10-2R 558CCTGGATGGCTTGTATCAAATG FGF10-3F 54.58C 319 bpTTGGCAAAAGAGCCATTGGT FGF10-3R 54.58CTCCCTGACTCGCCAATCTCTTTC FGFR2-EX2-F 558C 343 bpTGCCCCCAGACAAATCCCAAAAC FGFR2-EX2-R 558CCACTGACCTTTGTTGGACGTTC FGFR2-EX3-F 558C 380 bpGAGAAGAGAGAGCATAGTGCTGG FGFR2-EX3-R 558CTGGAGAAGGTCTCAGTTGTAGAT FGFR2-EX4-F 558C 232 bpAGACAGGTGACAGGCAGAACT FGFR2-EX4-R 558CCAAAGCGAAATGATCTTACCTG FGFR2-EX5-F 558C 291 bpAGAAATGTGATGTTCTGAAAGC FGFR2-EX5-R 558CGCTAGGATTGTTAAATAACCGCC FGFR2-EX6-F 558C 226 bpAAACGAGTCAAGCAAGAATGGG FGFR2-EX6-R 558CACAGCCCTCTGGACAACACA FGFR2-EX7-F 558C 393 bpCTGGCTAGTCAAAAAAGAGAA FGFR2-EX7-R 558CCTTTAATGCCGCTGTTTAG FGFR2-EXIIIB-F 548C 333 bpTCTTTTCAGCTTCTATATCCAG FGFR2-EXIIIB-R 548CATCATTCCTGTGTCGTCTAG FGFR2-EXIIIC-F 548C 224 bpAAAAACCCAGAGAGAAAGAACAGTATA FGFR2-EXIIIC-R 548CTGCGTCAGTCTGGTGTGCTAAC FGFR2-EX9-F 558C 341 bpAGGACAAGATCCACAAGCTGGC FGFR2-EX9-R 558CTGACTTCCAGCCTTCTCAGATG FGFR2-EX10-F 558C 252 bpAGTCTCCATCCTGGGACATGG FGFR2-EX10-R 558CCCCCATCACCAGATGCTATGTG FGFR2-EX11-F 558C 221 bpTTGATAAGACTCTCCACCCAGCC FGFR2-EX11-R 558CGAGGAAATGAACTGATTTGTG FGFR2-EX12-F 558C 192 bpGCAGAGTATTTGGGCGAATG FGFR2-EX12-R 558CCTGGATTCTCTCTTTAGGGAG FGFR2-EX13-F 558C 263 bpCACCCAGCCAAGTAGAATG FGFR2-EX13-R 558CACATATTTCCTTTTTGTTCTGG FGFR2-EX14-F 558C 256 bpTCTTCCTGGAACATTCTGAG FGFR2-EX14-R 558CGAGCCTGCTAAGATAAATTCTT FGFR2-EX15-F 558C 180 bpAGCTCAAGCCCAGGAAAAAG FGFR2-EX15-R 558CGGTTTTGGCAACGTGGATGGG FGFR2-EX16-F 558C 254 bpGAGAGGTATTACTGGTGTGGCAAG FGFR2-EX16-R 558CACACCACGTCCCCATATTGCC FGFR2-EX17-F 558C 243 bpCTCACAAGACAACCAAGGACAAG FGFR2-EX17-R 558CTCCCACGTCCAATACCCACAT FGFR2-EX18-F 558C 368 bpTTCCCAGTGCTGTCCTGTTTGG FGFR2-EX18-R 558C
226 MARTINOVIC-BOURIEL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

a non-consanguineous Caucasian couple whopresented MWS. Both children had pulmonaryhypoplasia and anophthalmia. One also had anumber of other malformations, as micrognathia, acleft palate reminiscent of the bifid uvula in oursecond case, a short upper lip and low-set ears. Theautopsy showeda single ventricle, anhypoplastic leftatrium, an hypoplastic spleen and a bicornuateuterus.
Both our cases occurred in consanguineouscouples, as may have been the case for one of theoriginal reports comprising bilateral colobomatousmicrophthalmia and diaphragmatic eventration[Radhakrishnan, 1981], highly supporting a recessiveautosomal inheritance. Moreover, in our first case anadditional sibling presented with similar features(respiratory anomalies and anophthalmia).
Our cases, together with previously publishedcases with similar features, strongly supportthe hypothesis that this combination of defects isnon-random [Steiner et al., 2002]. The spectrum ofmalformations seems to correspond to organsdeveloping simultaneously from the fourth week ofgestation on [Priolo et al., 2004]. The expressionpatterns in both animal models and humans ofFGF10 and FGFR2(IIIb) were evocative of the organsystems affected in MWS, although coding anomaliesin these genes were excluded in our cases. Thepresence of FGF10 transcripts in the human neural,non-pigmented retina was novel relative to reportsmade in animal models to date, demonstrating therelevance of performing expression analysis inhuman embryos. Further studies will be needed torule out mutations in the promoter regions of thesegenes spread over large genomic territories, as wellas modified functional interactions with heparinsulfate or intracellular effector gene candidates.
In summary, rare cases of microphthalmia/anophthalmia associated with pulmonary hypopla-sia/agenesis have been hitherto reported in theliterature. We report on two patients presentingmicrophthalmia and pulmonary agenesis associatedwith bilateral diaphragmatic eventration. In addition,not previously reported, both cases presentedduodenal stenosis. Facial gestalt is rather similar tohitherto reported cases. Most cases of Matthew-Wood syndrome are described as sporadic. Ourcases occurred in consanguineous families withrecurrence among sibs in the first family. As in asimilar, published case [Seller et al., 1996], theseobservations strongly support autosomal recessiveinheritance of the syndrome. However, additionalcases will be necessary to further delineate thissyndrome, as well as to provide some informationon its natural history. Further molecular studiesmay help us understand these pleiotropic fielddefects. Meanwhile, careful sonogram examinationin further pregnancies should be offered to thefamilies.
ACKNOWLEDGMENTS
We thank the medical staff of the Centre d’Ortho-genie at the Hopital Broussais for their collaborationand G. Goudefroye, C. Ozilou, and M. Alcaraz fortheir excellent technical assistance. We offer ourappreciation to the families for participating inthis study. The support of A. Munnich is alsogratefully acknowledged. This work was supportedby the Association Francaise contre les Myopathies,the Institut National pour la Sante et la RechercheMedicale (INSERM) ‘‘Avenir’’ program and theAssistance Publique—Hopitaux de Paris (AP-HP).
REFERENCES
Berkenstadt M, Lev D, Achiron R, Rosner M, Barkai G. 1999.Pulmonary agenesis, microphthalmia, and diaphragmaticdefect: New syndrome or association? Am J Med Genet86:6–8.
Cunniff C, Curtis M, Hassed SJ, Hoyme HE. 1998. Blepharophi-mosis: A causally heterogeneous malformation frequentlyassociated with developmental disabilities. Am J Med Genet75:52–54.
De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M,Rosewell I, Dickson C. 2000. An important role for the IIIbisoform of fibroblast growth factor receptor 2 (FGFR2) inmesenchymal-epithelial signalling during mouse organogen-esis. Development 127:483–492.
Engellenner W, Kaplan C, Van de Vegte GL. 1989. Pulmonaryagenesis association with nonimmune hydrops. Pediat Path9:725–730.
Enns GM, Cox VA, Goldstein RB, Gibbs DL, Harrison ML, GolabiM. 1998. Congenital diaphragmatic defects and associatedsyndromes, malformations, and chromosome anomalies: Aretrospective study of 60 patients and literature review. Am JMed Genet 79:215–225.
Fairbanks TJ, Kanard RC, Del Moral PM, Sala FG, De Langhe SP,Lopez CA, Veltmaat JM, Warburton D, Anderson KD, BellusciS, Burns RC. 2005. Colonic atresia without mesenteric vascularocclusion. The role of the fibroblast growth factor 10 signalingpathway. J Pediatr Surg 40:390–396.
Fryns JP. 1987. Fryns syndrome: A variable MCA syndrome withdiaphragmatic defects, coarse face, and distal limb hypopla-sia. J Med Gen 24:271–274.
Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E,McGregor L, Hopkins J, Chalepakis G, Philip N, Perez Aytes A,Watt FM, Darling SM, Jackson I, Woolf A, Scambler PJ. 2005.Identification of a new gene mutated in Fraser syndrome andmouse myelencephalic blebs. Nat Genet 37:520–525.
Kantarci S, Casavant D, Prada C, Russell M, Byrne J, Haug LW,Jennings R, Manning S, Blaise F, Boyd TK, Fryns JP, HolmesLB, Donahoe PK, Lee C, Kimonis V, Pober BR. 2006. Findingsfrom aCGH in patients with congenital diaphragmatic hernia(CDH): A possible locus for Fryns syndrome. Am J Med GenetPart A 140A:17–23.
Kunze J, Heyne K, Wiedemann HR. 1979. Diaphragmatic herniain a female newborn with focal dermal hypoplasia andmarked asymmetric malformations (Goltz-Gorlin syndrome).Eur J Pediatr 28:213–218.
Li L, Wei J. 2006. A newborn with anophthalmia and pulmonaryhypoplasia (the Matthew-Wood syndrome). Clin Dysmorphol15:43–44.
LubinskyM, SevernC, Rapoport JM. 1983. Fryns syndrome:Anewvariable multiple congenital anomaly (MCA) syndrome. Am JMed Genet 14:461–466.
McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G,Roberts C, Smart N, Rutland P, Prescott N, Hopkins J, BentleyE, Shaw A, Roberts E, Mueller R, Jadeja S, Philip N, Nelson
MATTHEW-WOOD SYNDROME 227
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

J, Francannet C, Perez-Aytes A, Megarbane A, Kerr B,Wainwright B, Woolf AS, Winter RM, Scambler PJ. 2003.Fraser syndrome and mouse blebbed phenotype caused bymutations in FRAS1/Fras1 encoding a putative extracellularmatrix protein. Nat Genet 34:203–208.
Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE,DeRose M, Simonet WS. 1998. Fgf-10 is required for both limband lung development and exhibits striking functionalsimilarity to Drosophila brancheless. Genes Dev 12:3156–3161.
Ohuchi H,NakagawaT, Yamamoto A,Araga A, Ohata T, IshimaruY, Yoshioka H, Kuwana T, Nohno T, Yamasaki M, Itoh N, NijoS. 1997. The mesenchymal factor, FGF10, initiates andmaintains the outgrowth of the chick limb bud throughinteraction with FGF8, an apical ectodermal factor. Develop-ment 124:2235–2244.
Priolo M, Casile G, Lagana C. 2004. Pulmonary agenesis/hypoplasia, microphthalmia, and diaphragmatic defects:Report of an additional case. Clin Dysmorphol 13:45–46.
Radhakrishnan N. 1981. Colobomatous microphthalmia withdiaphragmatic eventration (a case report). Indian J Ophthal-mol 28:221–222.
Robert Lee SYR, Shiu YK, Ng WF, Chow CB. 2006. Another patientwith pulmonary hypoplasia, microphthalmia and diaphrag-matic hernia. Clin Dysmorphol 15:43–44.
Romana SP, Cherif D, Le Coniat M, Derre J, Flexor MA, Berger R.1993. In situ hybridization to interphase nuclei in acuteleukemia. Genes Chromosomes Cancer 8:98–103.
Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, SatoT, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. 1999. Fgf10is essential for limb and lung formation. Nat Genet 21:138–141.
Seller MJ, Davis TB, Fear CN, Flinter FA, Ellis I, Gibson AG.1996. Two sibs with anophthalmia and pulmonary hypoplasia(the Matthew-Wood syndrome). Am J Med Genet 62:227–229.
Slavotinek A, Lee SS, Davis R, Shrit A, Leppig KA, Rhim J, JasnoszK, Albertson D, Pinkel D. 2005. Fryns syndrome phenotypecaused by chromosome microdeletions at 15q26.2 and8p23.1. J Med Genet 42:730–736.
Smith SA, Martin KE, Dodd KL, Young ID. 1994. Severemicrophthalmia, diaphragmatic hernia and Fallot’s tetralogyassociated with a chromosome 1;15 translocation. ClinDysmorphol 3:287–291.
Smyth I, Scambler P. 2005. The genetics of Fraser syndrome andthe blebs mouse mutants. Hum Mol Genet 14:R269–R274.
Spear GS, Yetur P, Beyerlein RA. 1987. Bilateral pulmonaryagenesis and microphthalmia. Am J Med Genet 3:379–382.
Steiner RD, Dignan PSJ, Hopkin RJ, Kozielski R, Bove KE. 2002.Combination of diaphragmatic eventration and microphthal-mia/anophthalmia is probably non-random. Am J Med Genet108:45–50.
Tibboel D, Gaag AV. 1996. Etiologic and genetic factors incongenital diaphragmatic hernia. Clin Perinatol 23:689–699.
Trueba SS, Auge J, Mattei G, Etchevers H, Martinovic J,Czernichow P, Vekemans M, Polak M, Attie-Bitach T. 2005.PAX8, TITF1, and FOXE1 gene expression patterns duringhuman development: New insights into human thyroiddevelopment and thyroid dysgenesis-associated malforma-tions. J Clin Endocrinol Metab 90:455–462.
WarburgM, JensenH, Prause JU, Bolund S, Skovby F,Miranda MJ.1997. Anophthalmia-microphthalmia-oblique clefting syn-drome: Confirmation of the Fryns anophthalmia syndrome.Am J Med Genet 73:36–40.
Wilgenbus KK, Engers R, Crombach G, Majewski F. 1994. Twofetuses with Fryns syndrome without diaphragmatic defect.J Med Genet 31:962–964.
Wilkinson DG. 1992. In situ hybridization: A practical approach.UK: IRL Press.
WillemsPJ, KeersmaekersGHA,DomKE,Colpaert C, SchattemanE, Vergote IBP, Dumon JE. 1991. Fryns syndrome withoutdiaphragmatic hernia? Am J Med Genet 41:255–257.
228 MARTINOVIC-BOURIEL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
Related Documents











