Лизозомна болест на натрупување Lysosomal Storage Disease Клиничка биохемија МАРТИН ЈОВАНОВСКИ 1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Лизозомна болест нанатрупување
Lysosomal Storage Disease
Клиничка биохемија
МАРТИН ЈОВАНОВСКИ
1

Скопје, 2014
Содржина:
Вовед.........................................................
..............................................................
...................3
Лизозоми и лизозомна болест на натрупување
(LSD)...............................................................
........4
Клинички
испитувања..........................................................
.................................................................6
Заклучок............................................................
....................................................................
................10
2

Литература..........................................................
....................................................................
..............12
ВоведLysosomal storage diseases (LSD) или лизозомна болест на
натрупување претставува група од приближно 50 ретко наследни
метаболни нарушувања кои се резултат на дефети во
лизозомалната функција. Овие заболувања се јавуваат кога
3

лизозомите – специфични органели во клетките, не функционираат
правилно. Дисфункционирањето се однесува на дефицит на
единечен ензим кој е потребен за метаболизам на липидите,
гликопротеините и мукополисахаридите.
Индивидуално, LSD се јавива со инциденца помала од 1:100.000;
сепак, како група на болести, инциденцата е околу 1:5.000 –
1:10.000. повеќето од нарушувањата кои се јавуваат
претставуваат автозомно рецесивни наследни болести, како
Niemann-Pick заболувањето, тип С, потоа Х-поврзани рецесивно
наследени болести, како Fabry-ва болест, Hunter-ова болест и
така натаму.
Како и сите други генетски заболувања, индивидуите ја
наследуваат оваа болест од своите родители. Иако секое
нарушување резултира со различна генска мустација тоја се
транслира во дефицит на ензимска активност, сите тие делат
исти биохемиски карактеристики – сите лизозомални нарушувања
потекнуваат од абнормална акумулација на супстанци во
лизозомите.
4

Лизозоми и лизозомна болест на натрупување (LSD)
Лизозомите претставуваат субцелуларни органели одговорни за
физиолошкиот turnover на клеточните конституенти. Тие содржат
катаболни ензими, кои бараат ниска рН средина со цел да
функционираат оптимално. LSD како поим, опишува хетерогена
група на 50тина ретки, наследни болести кои се карактеризираат
со акумулација на недигестирани или парцијално дигестирани
макромолекули, што на крајот резултира со целуларна
дисфункција и клинички абнормалности. Како најчести
абнормалности се јавуваат органомегалија, дисфункција на
конективно ткиво, окуларна патологија и дисфункција на
централниот нервен систем.
Првото заболување од оваа група бил Tay-Sachs-ово заболување
кое било опишано во 1881 година, а потоа следел и описот на
Gaucher-овото заболување во 1882 година. Во доцните `50ти
години и раните `60ти години од минатиот век, de Duve и
неговите соработници употребиле техника за фракционирање на
клетката, цитолошки студии и биохемиски анализи, со што
успеале да ги идентификуваат и карактеризираат лизозомите како
целуларни органели одговорни за интрацелуланата дигестија и
рециклирање на макромолекулите. Ова било научно откритие кое
ќе доведе до разбирање на физиолошките основи на LSD. Pompe-
овото заболување било првото заболување кое било
5
Ензимски комплекс
Мембрана
СТРУКТУРА НА ЛИЗОЗОМ

идентификувано како LSD vo 1963
година, дефинирано како
заболување поради недостаток на
α-глукозидаза. Покрај ова
заболување, било сугерирано дека
постојат и други заболувања,
како мукополисахаридозата, кои
може да бидат резултат на
ензимски дефицит.
LSD опфаќа само дефицит на ензимите хидролази. Поновите
истражувања сугерираат дека концептот на оваа болест опфаќа и
дефицит или дефект на протеините потребни за нормална пост-
транслациски модификации на лизозомалните ензими, (кои самите
по себе се гликопротеини), активаторните протеини, или
протеините важни за соодветен, важен „промет“ на
интрацелуларни материи помеѓу лизозомите и другите целуларни
компартменти (Winchester et al., 2000).
6

Настанување на LSD
Денес опишани се преку 50 лизозомални болести на натрупување.
Годините на појава на ова заболување и клиничките манифестации
може да варираат во широк опсег помеѓу пациентите со дадено
лизозомално нарушување и постои значителна фенотипска
хетерогеност помеѓу членовите од семејството кои носат
идентични мутации. LSD се генерално класифицирани преку
акумулираниот супстрат и вклучуваат:
- Сфинголипидози,
- Олисахаридози,
- Муколипидози,
- Липопротеинска болест на натрупување,
- Дефекти во лизозомалниот транспорт,
- Невронска цероидна липофусциноза и други.
7
Комплекс на
супстра
Нормалнализозомнадеградациј
а
Лизозомнаензимскаактивност
Натрупани недигестирани метаболити
Мали дифузабилни крајни продукти

Покрај ваквата класификација, LSD може да биде генерално
класифицирана според природата на примарниот акумулиран
материјал и може да биде грубо поделена на следниве категории
(според интернационалната статистичка класификација на
болестите-ICD:
- (Е75) нарушување со липидна акумулација, главно
сфинголипидоза (вклучувајќи ги и Gaucher-ова болест,
Niemann-Pick-ова болест) (Е75.0-Е75.1) ганглиозидоза
(Tay-Sach-ова болест) и (Е75.2) леукодистрофија
- (Е76.0) мукополисахаридоза (вклучувајќи ја и Hunter-овата
болест)
- (Е77) нарушување со акумулација на гликопротеини
- (Е77.0-77.1) муколипидози
Исто така, гликоген акумулирачката болест тип II (Pompe-ова
болест) исто така е дефект на лизозомниот метаболизам, иако е
поинаку класифицирана во групата на Е74.0.
И покрај што се откриени повеќе од 50 видови на LSD, денес се
откриваат и некои сосема нови. Иако се јавуваат различни
типови на LSD кај индивидуи, се проценува дека се јавуваат кај
1 од 7.700 раѓања, што го прави ова заболување релативно често
и претставува значителен здравствен проблем.
Сите форми на LSD се прогресивни, со брзина на прогресија,
сериозност на симптоми и зафатениоргански системи кои варираат
од болест до болест, дури и помеѓу секој тип за заболување
(Clarke et al., 2005).
8

Клинички испитувањаСимптоми Збирот од дисморфични карактерисики (остри линии на лицето,
макроглосија), коскени абнормалности (дисостоза мултиплекс),
кардијални нарушувања (аритмија или кардиомегалија),
хепатоспленомегалија, офталмолошки знаци (корнеално
замаглување или макуларни црвени точки) и невролошки знаци
може да доведат до клиничко сомневање за лизозомна болест на
натрупување. Симптомите се типично градуално прогресивни
наместо епизодични, како што се појавуваат кај другите
неурометаболни нарушувања. Невролошките симптоми може да
вклучуваат одложување на развојот, хипотонија, епилепсија
(комплексна парцијална или миоклонична), периферна
неуропатија, инетелактуални нарушувања, атаксија и/или
спастичност. Другите симптоми кои може да се јават се напади,
деменција, глувост и/или слепило. Некои пациенти со LSD може
да покажуваат и хепатомегалија, без спленомегалија, и обратно,
како и пулмонални и кардијални проблеми, како и абнормалности
на коските, како што е кажано на почетокот.
9
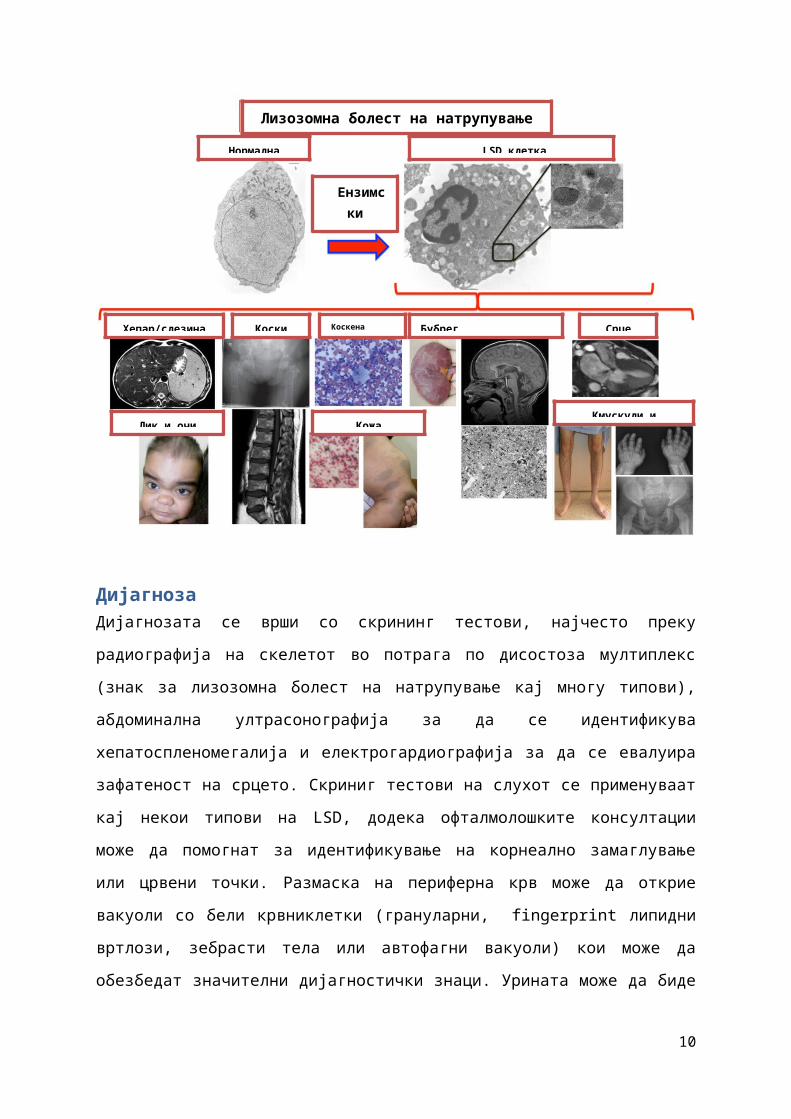
ДијагнозаДијагнозата се врши со скрининг тестови, најчесто преку
радиографија на скелетот во потрага по дисостоза мултиплекс
(знак за лизозомна болест на натрупување кај многу типови),
абдоминална ултрасонографија за да се идентификува
хепатоспленомегалија и електрогардиографија за да се евалуира
зафатеност на срцето. Скриниг тестови на слухот се применуваат
кај некои типови на LSD, додека офталмолошките консултации
може да помогнат за идентификување на корнеално замаглување
или црвени точки. Размаска на периферна крв може да открие
вакуоли со бели крвниклетки (грануларни, fingerprint липидни
вртлози, зебрасти тела или автофагни вакуоли) кои може да
обезбедат значителни дијагностички знаци. Урината може да биде
10
Лизозомна болест на натрупување
Нормална LSD клетка
Ензимски
дефицит
Хепар/слезина Коски Коскена Бубрег Срце
Лик и очи Кожа Кмускули и

тестирана за евалуација на екскрецијата на олигосахариди
(олигосахаридоза) и глукозаминогликани (мукополисахариди). Кај
резултатите, вредноста на крвната хитотриозидаза (ензимски
маркер за макрофагна активност) може да биде покачена.
Дефинитивен тест е најефикасно изведен со мерење на ензимската
активност во референтни лаборатории, типично одредувани кај
префиерни бели крвни клетки (иако кожните фибробласти може
исто така, да се употребат дури се и есенцијално важни во
некои случаи). За некои нарушувања, ензимската активност може
да се одредува на сува филтерна хартија, и понекогаш,
ензимската активност може да се мери од други ткива, како
мускулно. Одредување на ензимската активност во урина е
понекогаш од помош, иако екскрецијата на супстрат преку урина
може да обезбеди корисни информации. Но некои случаи, се прави
и конформативна (потврдна) DNA мутациска анализа (Bruni et
al., 2007).
МанифестацијаИако еден дефектен ген типично резултира со акумулација на
супстрат, прецизниот механизам по кој делува патофизиологијата
и води до појава на клинички симптоми не е целосно јасен.
Дистрибуцијата на акумулираниот материјал корелира со органот
кој е афектиран. Клетките на мононуклеарниот фагоцитен систем
се посебно богати со лизозоми, па според ова, пофрекфентно се
нападнати од болеста.
Невроните и глиа клетките се често пати нападнати од болеста,
веројатно поради релативната оскудност на клеточен turnover во
централниот нервен систем, иако остојат и други не-
11

невропатични типови на болеста. LSD може да резултира со
сериозен неуродегенеративен фенотип. Поблаг (типично касно
појавување или појавување кај адулти) фенотип е идентификуван
и генерално поврзан со степенот на резидуална ензимска
активност (ponder et al., 2007).
ПатофизиологијаИако, генерално, акумулацијата на недеградираните супстрати се
смета за причина на целуларната дисфункција и смртта која ја
придружува оваа болест, прецизниот механизам под кој подлежи
оваа дегенерација е некомплетно дефиниран. Шемата на
невронската деградација кај некои субтипови на LSD може да
биде изненадувачки клеточно-тип специфична (специфична за
одредени клеточни типови). Во некои случаи, акумулацијата на
супстратот, е исто така, асоцирана со секвестрација на важни
компонентни молекули, што води до релативна состојба на
дефицит.
Лизозомите служат како централни компоненти на ендозомалниот-
лизозомен систем. Овој систем е важен за одржување на
нормалниот целуларен метаболизам, делува на конјукцијата со
каперон-посредувана автофагија и убиквитин-протеазомалниот
систем. Абнормалната функција на овој систем може да води до
ектопична дендрична поддршка (карактеристика релативно
уникатна за LSD) и нарушен рецикличен пат на глутамерните AMPA
рецептори. Неуроаксијалната сфероидна формација е
карактеристика на ганглиозидната болест на натрупување,
Niemann-Pick тип А и С и а-манозидоза, имплицирајќи заедничка
12

Акумулација на супстрат и интервенција во ефектите (секундарни и биохемиски патишта)
Терапија за редукција на супстрат
Супстрат
Продукт
Генерален третман
Дефектни лизозомни хидролазиГенска терапија
Терапија со замена на ензимиДизајн на мали молекули за реактивација на дефектните ензими
патологија која води до продукција на овие компактни
акумулации на митрохондриите и тубовезикуларните тела.
ТретманМоментално не постои лек за LSD и третманот воглавно е
симптоматски, иако пресадувањето на коскена срцевина и
терапија на замена на ензимите биле пробувани и се покажале
како доволно успешни третмани. Како нешто ново, се применува
трансплантација на крв од папочна врвца, третман кој се
изведува во специлаизирани центри, но претставува можен
третман само за неколку типови на заболувања од групата на
LSD. Терапија за редукција на супстратот, е исто така метод
употребен за намалување на акумулираниот материјал, но овој
метод е во фаза на евалуација. Понатаму, каперон терапија,
техника употребена за стабилизирање на дефектните ензими
продуцирани кај пациентите, е во почеток на истражувањата за
одредени типови на нарушувања. Во иднина, се смета дека
генската терапија би била решение за оваа болест.
Заклучок 13

Лизозомната болест на натрупување претставува заболување кое
ги погодува лизозомните ензими – хидролази, при што доаѓа до
дефицит на ензимите или до продукција на дефицитни ензими.
Дефицитот или дефектноста на ензимите влијае на многу функции
во организмот, поради тоа што лизозомите се места за обработка
на интраклеточните продукти.
Ова заболување не е единечно заболување, туку претставува
група на заболувња, група која вбројува преку 50 заболувања,
класифицирани според неколку категории. Треба да се напомене
дека ова нарушување е вродено генетско нарушување и се
пренесува автозомно рецесивно.
LSD е релативно ретко нарушување, но сепак погодува значителен
број на лица од светската популација.
Се јавува со различни симптоми, со силина на симптомите
рзлична за секој тип на LSD. Дијагнозата може да се изведе
според одредени скриниг тестови или директно преку одредување
на ензимска активност, што е значајно, едноставено и
најефикасно откривање на ова заболување.
Што се однесува на третманот, тој не постои, се третираат
симптомите кои ја пратат болеста од соодветниот тип на
заболување. Единствената терапија, која, теоретски, е возможна
– генската терапија, е сеуште во развој, со надеж за идна
примена.
14

Литература
1. Winchester B, Vellodi A, Young E (2000). "The molecular
basis of lysosomal storage diseases and their
treatment". Biochem. Soc. Trans. 28 (2): 150–4
2. Clarke JT, Iwanochko RM (2005). "Enzyme replacement
therapy of Fabry disease". Mol. Neurobiol. 32 (1): 043–
050.
3. Bruni S, Loschi L, Incerti C, Gabrielli O, Coppa GV
(2007). "Update on treatment of lysosomal storage
diseases". Acta Myol 26 (1): 87–92.
4. Ponder KP, Haskins ME (2007). "Gene therapy for
mucopolysaccharidosis".Expert Opin Biol Ther 7 (9): 1333–
1345.
15
Related Documents











