Research Report Lithium modulates cortical excitability in vitro Charlotte Butler-Munro, Emma J. Coddington, Cristina H. Shirley, Philip M. Heyward ⁎ Department of Physiology, University of Otago, Dunedin, New Zealand ARTICLE INFO ABSTRACT Article history: Accepted 9 July 2010 Available online 15 July 2010 The sometimes devastating mood swings of bipolar disorder are prevented by treatment with selected antiepileptic drugs, or with lithium. Abnormal membrane ion channel expression and excitability in brain neurons likely underlie bipolar disorder, but explaining therapeutic effects in these terms has faced an unresolved paradox: the antiepileptic drugs effective in bipolar disorder reduce Na + entry through voltage-gated channels, but lithium freely enters neurons through them. Here we show that lithium increases the excitability of output neurons in brain slices of the mouse olfactory bulb, an archetypical cortical structure. Treatment in vitro with lithium (1 to 10 mM) depolarizes mitral cells, blocks action potential hyperpolarization, and modulates their responses to synaptic input. We suggest that Na + entry through voltage-gated channels normally directly activates K + channels regulating neuron excitability, but that at therapeutic concentrations, lithium entry and accumulation reduces this K + channel activation. The antiepileptic drugs effective in bipolar disorder and lithium may thus share a membrane target consisting of functionally coupled Na + and K + channels that together control brain neuron excitability. © 2010 Elsevier B.V. All rights reserved. Keywords: Lithium Bipolar disorder Membrane excitability Neuron Potassium current 1. Introduction Li + is the primary treatment for bipolar disorder, but after more than 50 years since its first reported use (Cade, 1949), its mechanism of action remains enigmatic. Reported actions of Li + include effects on monoamine metabolites, neurotrans- mitter receptors, intracellular second messengers (the phos- photidylinositol cycle in particular), immediate early genes and response elements, membrane ion pumps, neuroprotec- tion and neurogenesis, but which of these, if any, might be the primary therapeutic action of Li + remains unclear (reviewed in Atack et al., 1995; Belmaker, 2004; Gurvich and Klein, 2002; Askland, 2006). Bipolar disorder is a heritable, chronic, periodic distur- bance of function in an electrically excitable tissue, and as such it has the characteristics of an ion channel disease (Gargus, 2006; Askland, 2006; Askland and Parsons, 2006). Extensive genetic association studies (Askland and Parsons, 2006; Askland, 2006; The Welcome Trust Case Control Consortium, 2007; Askland et al., 2009) conclude that bipolar disorder may result from altered expression of membrane ion channels in brain neurons. Antiepileptic drugs acting on ion channels are commonly used to treat bipolar disorder (Gargus, 2006, Rogawski and Loscher, 2004b), for which they are the only established alternative to Li + . The primary shared mode of action of the antiepileptic drugs is to block voltage-gated Na + channels (Rogawski and Loscher, 2004a; Errington et al., 2005; Askland, 2006), but Li + readily passes through these same channels (reviewed in Hille, 2001). Thus although Li + and antiepileptic drugs each stabilize mood, it has not been BRAIN RESEARCH 1352 (2010) 50 – 60 ⁎ Corresponding author. Department of Physiology, University of Otago, Dunedin, New Zealand 9054. Fax: +64 4 479 7323. E-mail address: [email protected] (P.M. Heyward). 0006-8993/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.brainres.2010.07.021 available at www.sciencedirect.com www.elsevier.com/locate/brainres

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res
Research Report
Lithium modulates cortical excitability in vitro
Charlotte Butler-Munro, Emma J. Coddington, Cristina H. Shirley, Philip M. Heyward⁎
Department of Physiology, University of Otago, Dunedin, New Zealand
A R T I C L E I N F O
⁎ Corresponding author. Department of PhysiE-mail address: phil.heyward@stonebow.
0006-8993/$ – see front matter © 2010 Elsevidoi:10.1016/j.brainres.2010.07.021
A B S T R A C T
Article history:Accepted 9 July 2010Available online 15 July 2010
The sometimes devastating mood swings of bipolar disorder are prevented by treatmentwith selected antiepileptic drugs, or with lithium. Abnormal membrane ion channelexpression and excitability in brain neurons likely underlie bipolar disorder, but explainingtherapeutic effects in these terms has faced an unresolved paradox: the antiepileptic drugseffective in bipolar disorder reduce Na+ entry through voltage-gated channels, but lithiumfreely enters neurons through them. Here we show that lithium increases the excitability ofoutput neurons in brain slices of themouse olfactory bulb, an archetypical cortical structure.Treatment in vitro with lithium (1 to 10 mM) depolarizes mitral cells, blocks action potentialhyperpolarization, and modulates their responses to synaptic input. We suggest that Na+
entry through voltage-gated channels normally directly activates K+ channels regulatingneuron excitability, but that at therapeutic concentrations, lithium entry and accumulationreduces this K+ channel activation. The antiepileptic drugs effective in bipolar disorder andlithium may thus share a membrane target consisting of functionally coupled Na+ and K+
channels that together control brain neuron excitability.© 2010 Elsevier B.V. All rights reserved.
Keywords:LithiumBipolar disorderMembrane excitabilityNeuronPotassium current
1. Introduction
Li+ is the primary treatment for bipolar disorder, but aftermore than 50 years since its first reported use (Cade, 1949), itsmechanism of action remains enigmatic. Reported actions ofLi+ include effects on monoamine metabolites, neurotrans-mitter receptors, intracellular second messengers (the phos-photidylinositol cycle in particular), immediate early genesand response elements, membrane ion pumps, neuroprotec-tion and neurogenesis, but which of these, if any, might be theprimary therapeutic action of Li+ remains unclear (reviewed inAtack et al., 1995; Belmaker, 2004; Gurvich and Klein, 2002;Askland, 2006).
Bipolar disorder is a heritable, chronic, periodic distur-bance of function in an electrically excitable tissue, and as
ology, University of Otagootago.ac.nz (P.M. Heyward
er B.V. All rights reserved
such it has the characteristics of an ion channel disease(Gargus, 2006; Askland, 2006; Askland and Parsons, 2006).Extensive genetic association studies (Askland and Parsons,2006; Askland, 2006; The Welcome Trust Case ControlConsortium, 2007; Askland et al., 2009) conclude that bipolardisorder may result from altered expression of membrane ionchannels in brain neurons. Antiepileptic drugs acting on ionchannels are commonly used to treat bipolar disorder (Gargus,2006, Rogawski and Loscher, 2004b), for which they are theonly established alternative to Li+. The primary shared modeof action of the antiepileptic drugs is to block voltage-gatedNa+ channels (Rogawski and Loscher, 2004a; Errington et al.,2005; Askland, 2006), but Li+ readily passes through these samechannels (reviewed in Hille, 2001). Thus although Li+ andantiepileptic drugs each stabilize mood, it has not been
, Dunedin, New Zealand 9054. Fax: +64 4 479 7323.).
.
51B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
apparent how ion channels controlling membrane excitabilitycould be their common target of action.
Neuronal membrane potential is controlled mainly byrelative membrane permeabilities to Na+ and K+ ions. Over-lapping ranges of voltage-sensitivity allow voltage-gated Na+
and K+ channels to functionally interact, and in addition, Na+
entry directly activates K+ channels, generating a Na+-activated K+ current (IKNa) (Bhattachargee and Kaczmarek,2005). Li+ entry readily substitutes for Na+ in generating inwardcurrent, but reduces outward current, affecting the restingmembrane potentials or action potentials in many types ofbrain neuron (Mayer et al., 1984; Schwindt et al., 1989;Safronov and Vogel, 1996; Bischoff et al., 1998; Colino et al.,1998, Franceschetti et al., 2003; Liu and Leung, 2004). Ifclinically relevant extracellular concentrations of Li+
(∼1 mM) result in sufficient Li+ entry to block outward current,IKNa in particular, this could contribute to the effectiveness ofLi+ in bipolar disorder. We therefore investigated electrophys-iological effects of Li+ on a representative cortical principalneuron.We chosemitral cells of the olfactory bulb of the brain,as these neurons are the principal output neuron within anarchetypical cortical modular circuit (Chen and Shepherd,2005), with well-characterized membrane properties andresponses to synaptic inputs in vitro (Carlson et al., 2000;Heyward et al., 2001; Aungst et al., 2003). Similar tomany othertypes of central neuron, they generate voltage-gated Na+
current at resting potentials (Heyward et al., 2001; Balu andStrowbridge, 2007) providing a route of Li+ entry independent
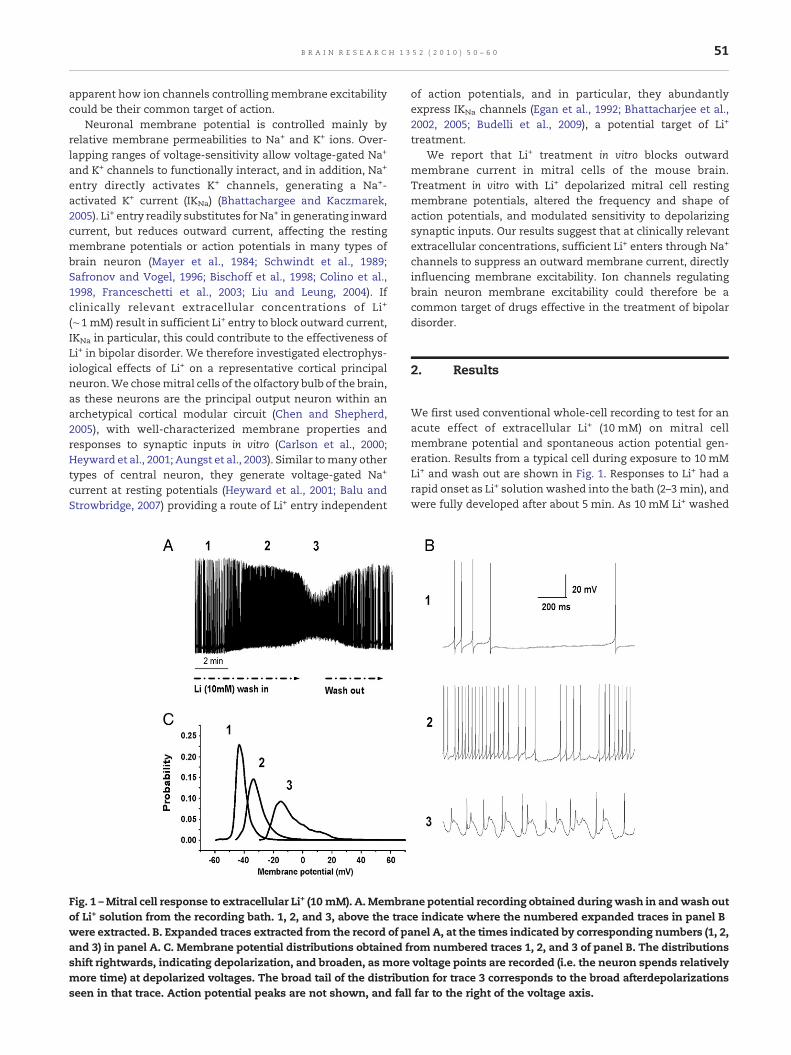
Fig. 1 –Mitral cell response to extracellular Li+ (10 mM). A.Membraof Li+ solution from the recording bath. 1, 2, and 3, above the tracwere extracted. B. Expanded traces extracted from the record of pand 3) in panel A. C. Membrane potential distributions obtained fshift rightwards, indicating depolarization, and broaden, as moremore time) at depolarized voltages. The broad tail of the distribuseen in that trace. Action potential peaks are not shown, and fal
of action potentials, and in particular, they abundantlyexpress IKNa channels (Egan et al., 1992; Bhattacharjee et al.,2002, 2005; Budelli et al., 2009), a potential target of Li+
treatment.We report that Li+ treatment in vitro blocks outward
membrane current in mitral cells of the mouse brain.Treatment in vitro with Li+ depolarized mitral cell restingmembrane potentials, altered the frequency and shape ofaction potentials, and modulated sensitivity to depolarizingsynaptic inputs. Our results suggest that at clinically relevantextracellular concentrations, sufficient Li+ enters through Na+
channels to suppress an outward membrane current, directlyinfluencing membrane excitability. Ion channels regulatingbrain neuron membrane excitability could therefore be acommon target of drugs effective in the treatment of bipolardisorder.
2. Results
We first used conventional whole-cell recording to test for anacute effect of extracellular Li+ (10 mM) on mitral cellmembrane potential and spontaneous action potential gen-eration. Results from a typical cell during exposure to 10 mMLi+ and wash out are shown in Fig. 1. Responses to Li+ had arapid onset as Li+ solution washed into the bath (2–3 min), andwere fully developed after about 5 min. As 10 mM Li+ washed
ne potential recording obtained duringwash in andwash oute indicate where the numbered expanded traces in panel Banel A, at the times indicated by corresponding numbers (1, 2,rom numbered traces 1, 2, and 3 of panel B. The distributionsvoltage points are recorded (i.e. the neuron spends relativelytion for trace 3 corresponds to the broad afterdepolarizationsl far to the right of the voltage axis.
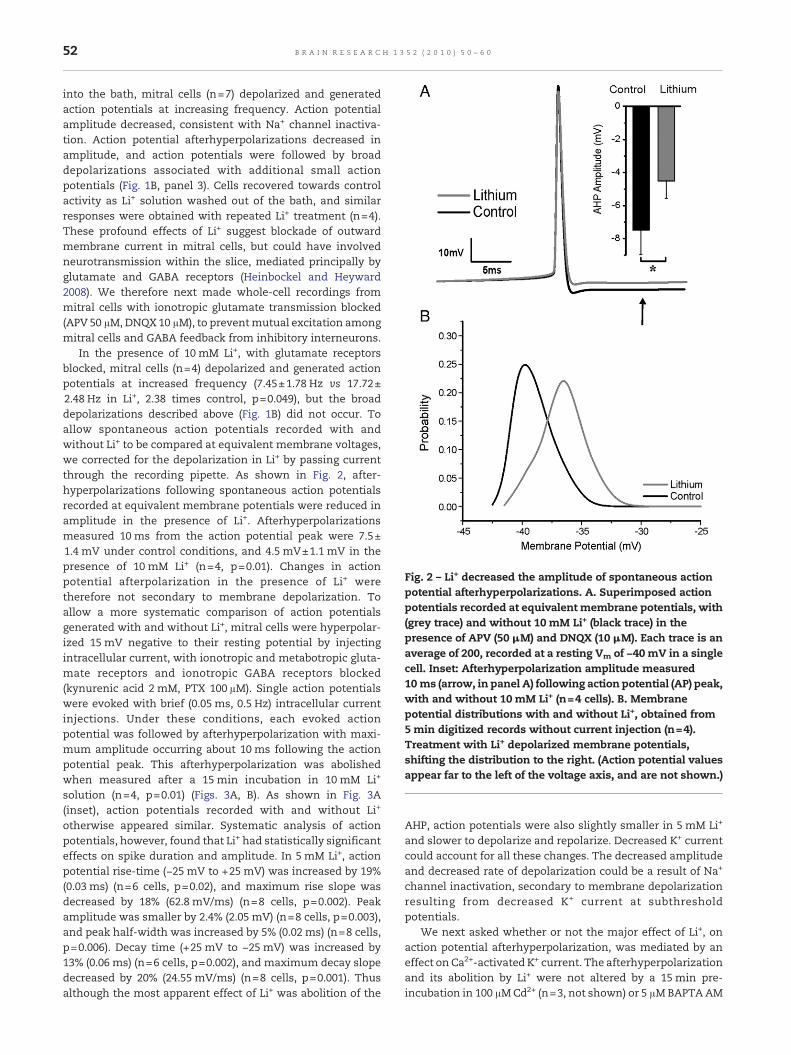
Fig. 2 – Li+ decreased the amplitude of spontaneous actionpotential afterhyperpolarizations. A. Superimposed actionpotentials recorded at equivalent membrane potentials, with(grey trace) and without 10 mM Li+ (black trace) in thepresence of APV (50 μM) and DNQX (10 μM). Each trace is anaverage of 200, recorded at a resting Vm of −40 mV in a singlecell. Inset: Afterhyperpolarization amplitude measured10 ms (arrow, in panel A) following action potential (AP) peak,with and without 10 mM Li+ (n=4 cells). B. Membranepotential distributions with and without Li+, obtained from5min digitized records without current injection (n=4).Treatment with Li+ depolarized membrane potentials,shifting the distribution to the right. (Action potential valuesappear far to the left of the voltage axis, and are not shown.)
52 B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
into the bath, mitral cells (n=7) depolarized and generatedaction potentials at increasing frequency. Action potentialamplitude decreased, consistent with Na+ channel inactiva-tion. Action potential afterhyperpolarizations decreased inamplitude, and action potentials were followed by broaddepolarizations associated with additional small actionpotentials (Fig. 1B, panel 3). Cells recovered towards controlactivity as Li+ solution washed out of the bath, and similarresponses were obtained with repeated Li+ treatment (n=4).These profound effects of Li+ suggest blockade of outwardmembrane current in mitral cells, but could have involvedneurotransmission within the slice, mediated principally byglutamate and GABA receptors (Heinbockel and Heyward2008). We therefore next made whole-cell recordings frommitral cells with ionotropic glutamate transmission blocked(APV 50 μM, DNQX 10 μM), to preventmutual excitation amongmitral cells and GABA feedback from inhibitory interneurons.
In the presence of 10 mM Li+, with glutamate receptorsblocked, mitral cells (n=4) depolarized and generated actionpotentials at increased frequency (7.45±1.78 Hz vs 17.72±2.48 Hz in Li+, 2.38 times control, p=0.049), but the broaddepolarizations described above (Fig. 1B) did not occur. Toallow spontaneous action potentials recorded with andwithout Li+ to be compared at equivalent membrane voltages,we corrected for the depolarization in Li+ by passing currentthrough the recording pipette. As shown in Fig. 2, after-hyperpolarizations following spontaneous action potentialsrecorded at equivalent membrane potentials were reduced inamplitude in the presence of Li+. Afterhyperpolarizationsmeasured 10 ms from the action potential peak were 7.5±1.4 mV under control conditions, and 4.5 mV±1.1 mV in thepresence of 10 mM Li+ (n=4, p=0.01). Changes in actionpotential afterpolarization in the presence of Li+ weretherefore not secondary to membrane depolarization. Toallow a more systematic comparison of action potentialsgenerated with and without Li+, mitral cells were hyperpolar-ized 15 mV negative to their resting potential by injectingintracellular current, with ionotropic and metabotropic gluta-mate receptors and ionotropic GABA receptors blocked(kynurenic acid 2 mM, PTX 100 μM). Single action potentialswere evoked with brief (0.05 ms, 0.5 Hz) intracellular currentinjections. Under these conditions, each evoked actionpotential was followed by afterhyperpolarization with maxi-mum amplitude occurring about 10 ms following the actionpotential peak. This afterhyperpolarization was abolishedwhen measured after a 15 min incubation in 10 mM Li+
solution (n=4, p=0.01) (Figs. 3A, B). As shown in Fig. 3A(inset), action potentials recorded with and without Li+
otherwise appeared similar. Systematic analysis of actionpotentials, however, found that Li+ had statistically significanteffects on spike duration and amplitude. In 5 mM Li+, actionpotential rise-time (−25 mV to +25 mV) was increased by 19%(0.03 ms) (n=6 cells, p=0.02), and maximum rise slope wasdecreased by 18% (62.8 mV/ms) (n=8 cells, p=0.002). Peakamplitude was smaller by 2.4% (2.05 mV) (n=8 cells, p=0.003),and peak half-width was increased by 5% (0.02 ms) (n=8 cells,p=0.006). Decay time (+25 mV to −25 mV) was increased by13% (0.06 ms) (n=6 cells, p=0.002), and maximum decay slopedecreased by 20% (24.55 mV/ms) (n=8 cells, p=0.001). Thusalthough the most apparent effect of Li+ was abolition of the
AHP, action potentials were also slightly smaller in 5 mM Li+
and slower to depolarize and repolarize. Decreased K+ currentcould account for all these changes. The decreased amplitudeand decreased rate of depolarization could be a result of Na+
channel inactivation, secondary to membrane depolarizationresulting from decreased K+ current at subthresholdpotentials.
We next asked whether or not the major effect of Li+, onaction potential afterhyperpolarization, was mediated by aneffect on Ca2+-activated K+ current. The afterhyperpolarizationand its abolition by Li+ were not altered by a 15 min pre-incubation in 100 μMCd2+ (n=3, not shown) or 5 μMBAPTAAM
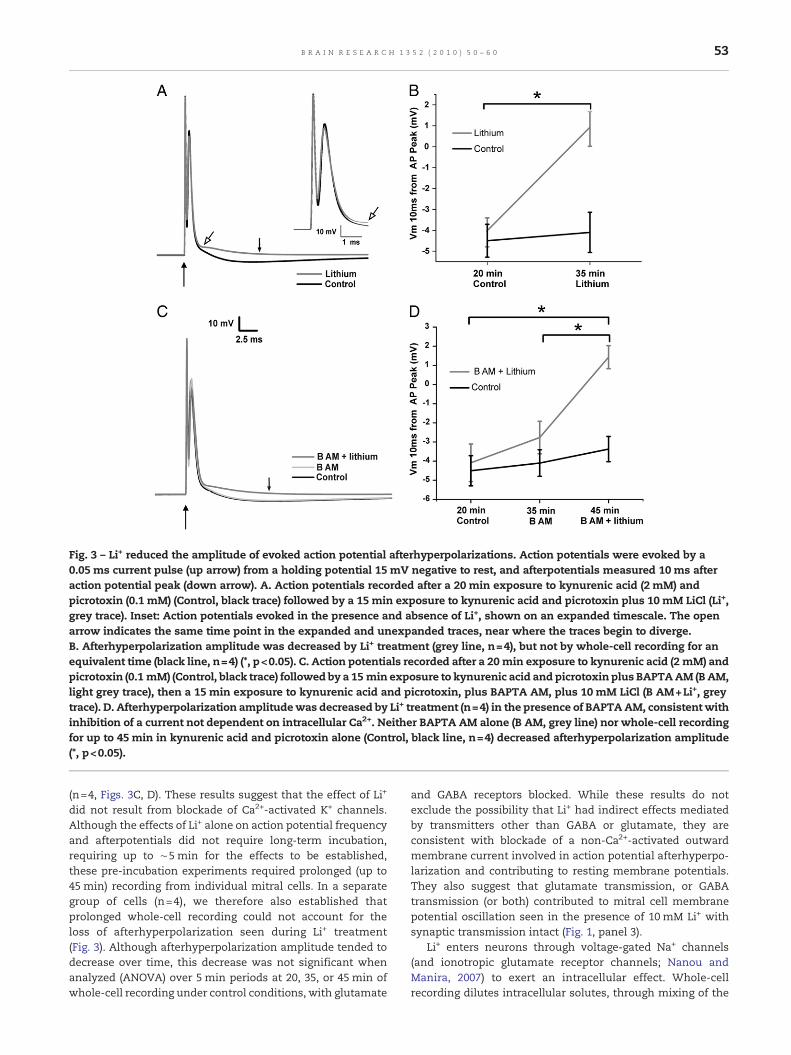
Fig. 3 – Li+ reduced the amplitude of evoked action potential afterhyperpolarizations. Action potentials were evoked by a0.05 ms current pulse (up arrow) from a holding potential 15 mV negative to rest, and afterpotentials measured 10 ms afteraction potential peak (down arrow). A. Action potentials recorded after a 20 min exposure to kynurenic acid (2 mM) andpicrotoxin (0.1 mM) (Control, black trace) followed by a 15 min exposure to kynurenic acid and picrotoxin plus 10 mM LiCl (Li+,grey trace). Inset: Action potentials evoked in the presence and absence of Li+, shown on an expanded timescale. The openarrow indicates the same time point in the expanded and unexpanded traces, near where the traces begin to diverge.B. Afterhyperpolarization amplitude was decreased by Li+ treatment (grey line, n=4), but not by whole-cell recording for anequivalent time (black line, n=4) (*, p<0.05). C. Action potentials recorded after a 20 min exposure to kynurenic acid (2 mM) andpicrotoxin (0.1 mM) (Control, black trace) followed by a 15 min exposure to kynurenic acid and picrotoxin plus BAPTAAM (BAM,light grey trace), then a 15 min exposure to kynurenic acid and picrotoxin, plus BAPTA AM, plus 10 mM LiCl (B AM+Li+, greytrace). D. Afterhyperpolarization amplitudewas decreased by Li+ treatment (n=4) in the presence of BAPTAAM, consistent withinhibition of a current not dependent on intracellular Ca2+. Neither BAPTA AM alone (B AM, grey line) nor whole-cell recordingfor up to 45 min in kynurenic acid and picrotoxin alone (Control, black line, n=4) decreased afterhyperpolarization amplitude(*, p<0.05).
53B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
(n=4, Figs. 3C, D). These results suggest that the effect of Li+
did not result from blockade of Ca2+-activated K+ channels.Although the effects of Li+ alone on action potential frequencyand afterpotentials did not require long-term incubation,requiring up to ∼5 min for the effects to be established,these pre-incubation experiments required prolonged (up to45 min) recording from individual mitral cells. In a separategroup of cells (n=4), we therefore also established thatprolonged whole-cell recording could not account for theloss of afterhyperpolarization seen during Li+ treatment(Fig. 3). Although afterhyperpolarization amplitude tended todecrease over time, this decrease was not significant whenanalyzed (ANOVA) over 5 min periods at 20, 35, or 45 min ofwhole-cell recording under control conditions, with glutamate
and GABA receptors blocked. While these results do notexclude the possibility that Li+ had indirect effects mediatedby transmitters other than GABA or glutamate, they areconsistent with blockade of a non-Ca2+-activated outwardmembrane current involved in action potential afterhyperpo-larization and contributing to resting membrane potentials.They also suggest that glutamate transmission, or GABAtransmission (or both) contributed to mitral cell membranepotential oscillation seen in the presence of 10 mM Li+ withsynaptic transmission intact (Fig. 1, panel 3).
Li+ enters neurons through voltage-gated Na+ channels(and ionotropic glutamate receptor channels; Nanou andManira, 2007) to exert an intracellular effect. Whole-cellrecording dilutes intracellular solutes, through mixing of the
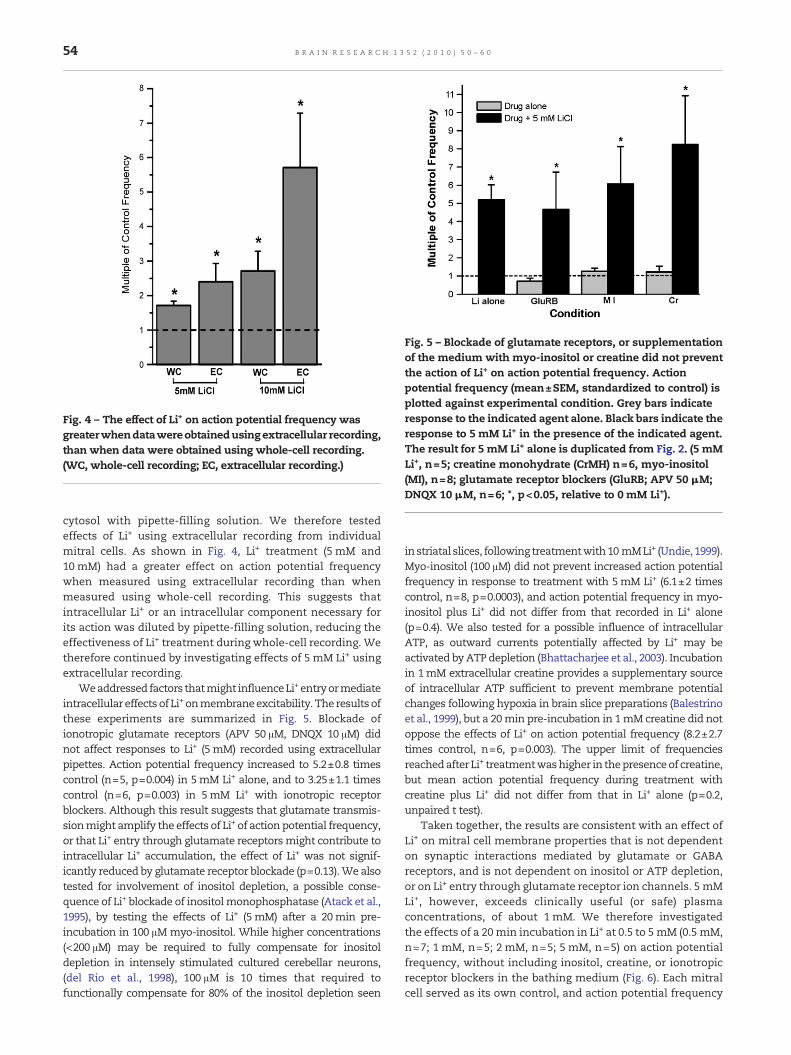
Fig. 4 – The effect of Li+ on action potential frequency wasgreaterwhendatawereobtainedusingextracellular recording,than when data were obtained using whole-cell recording.(WC, whole-cell recording; EC, extracellular recording.)
Fig. 5 – Blockade of glutamate receptors, or supplementationof the medium with myo-inositol or creatine did not preventthe action of Li+ on action potential frequency. Actionpotential frequency (mean±SEM, standardized to control) isplotted against experimental condition. Grey bars indicateresponse to the indicated agent alone. Black bars indicate theresponse to 5 mM Li+ in the presence of the indicated agent.The result for 5 mM Li+ alone is duplicated from Fig. 2. (5 mMLi+, n=5; creatine monohydrate (CrMH) n=6, myo-inositol(MI), n=8; glutamate receptor blockers (GluRB; APV 50 μM;DNQX 10 μM, n=6; *, p<0.05, relative to 0 mM Li+).
54 B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
cytosol with pipette-filling solution. We therefore testedeffects of Li+ using extracellular recording from individualmitral cells. As shown in Fig. 4, Li+ treatment (5 mM and10 mM) had a greater effect on action potential frequencywhen measured using extracellular recording than whenmeasured using whole-cell recording. This suggests thatintracellular Li+ or an intracellular component necessary forits action was diluted by pipette-filling solution, reducing theeffectiveness of Li+ treatment during whole-cell recording. Wetherefore continued by investigating effects of 5 mM Li+ usingextracellular recording.
Weaddressed factors thatmight influenceLi+entryormediateintracellular effects of Li+ onmembraneexcitability. The results ofthese experiments are summarized in Fig. 5. Blockade ofionotropic glutamate receptors (APV 50 μM, DNQX 10 μM) didnot affect responses to Li+ (5 mM) recorded using extracellularpipettes. Action potential frequency increased to 5.2±0.8 timescontrol (n=5, p=0.004) in 5mM Li+ alone, and to 3.25±1.1 timescontrol (n=6, p=0.003) in 5mM Li+ with ionotropic receptorblockers. Although this result suggests that glutamate transmis-sionmight amplify the effects of Li+ of action potential frequency,or that Li+ entry through glutamate receptorsmight contribute tointracellular Li+ accumulation, the effect of Li+ was not signif-icantly reduced by glutamate receptor blockade (p=0.13).We alsotested for involvement of inositol depletion, a possible conse-quence of Li+ blockade of inositol monophosphatase (Atack et al.,1995), by testing the effects of Li+ (5 mM) after a 20min pre-incubation in 100 μM myo-inositol. While higher concentrations(<200 μM) may be required to fully compensate for inositoldepletion in intensely stimulated cultured cerebellar neurons,(del Rio et al., 1998), 100 μM is 10 times that required tofunctionally compensate for 80% of the inositol depletion seen
in striatal slices, following treatmentwith10mMLi+ (Undie, 1999).Myo-inositol (100 μM) did not prevent increased action potentialfrequency in response to treatment with 5mM Li+ (6.1±2 timescontrol, n=8, p=0.0003), and action potential frequency in myo-inositol plus Li+ did not differ from that recorded in Li+ alone(p=0.4). We also tested for a possible influence of intracellularATP, as outward currents potentially affected by Li+ may beactivated byATP depletion (Bhattacharjee et al., 2003). Incubationin 1mM extracellular creatine provides a supplementary sourceof intracellular ATP sufficient to prevent membrane potentialchanges following hypoxia in brain slice preparations (Balestrinoet al., 1999), but a 20min pre-incubation in 1mM creatine did notoppose the effects of Li+ on action potential frequency (8.2±2.7times control, n=6, p=0.003). The upper limit of frequenciesreachedafter Li+ treatmentwashigher in thepresenceof creatine,but mean action potential frequency during treatment withcreatine plus Li+ did not differ from that in Li+ alone (p=0.2,unpaired t test).
Taken together, the results are consistent with an effect ofLi+ on mitral cell membrane properties that is not dependenton synaptic interactions mediated by glutamate or GABAreceptors, and is not dependent on inositol or ATP depletion,or on Li+ entry through glutamate receptor ion channels. 5 mMLi+, however, exceeds clinically useful (or safe) plasmaconcentrations, of about 1 mM. We therefore investigatedthe effects of a 20 min incubation in Li+ at 0.5 to 5 mM (0.5 mM,n=7; 1 mM, n=5; 2 mM, n=5; 5 mM, n=5) on action potentialfrequency, without including inositol, creatine, or ionotropicreceptor blockers in the bathing medium (Fig. 6). Each mitralcell served as its own control, and action potential frequency
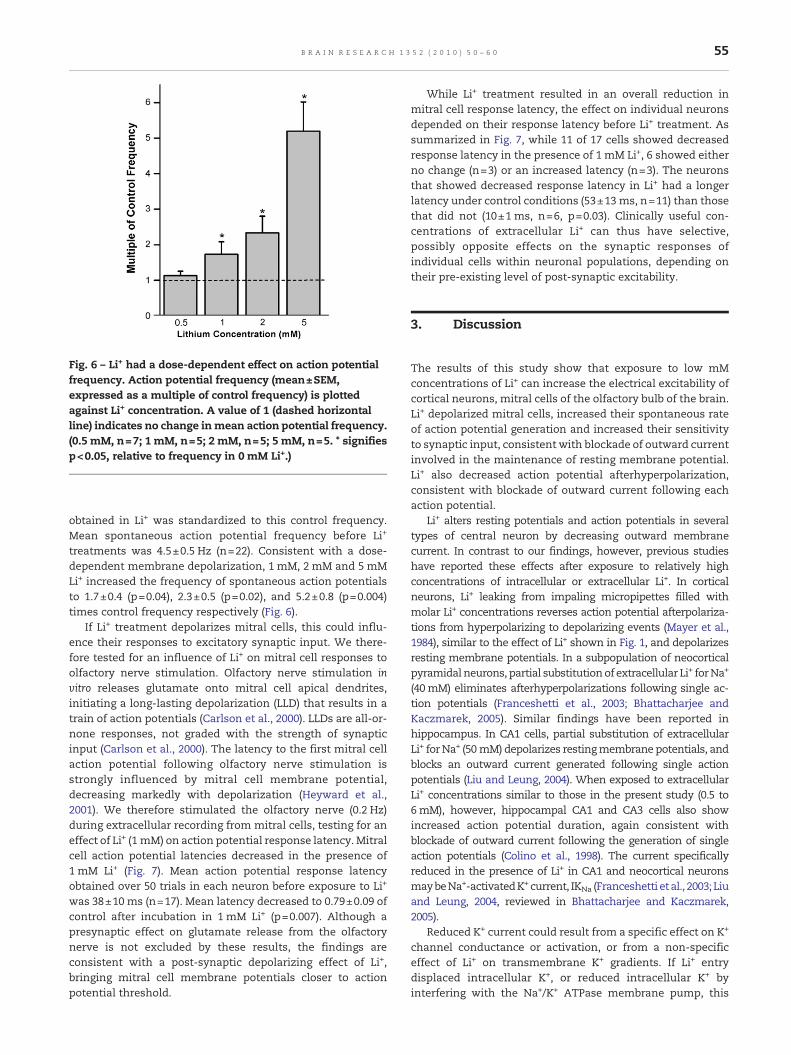
Fig. 6 – Li+ had a dose-dependent effect on action potentialfrequency. Action potential frequency (mean±SEM,expressed as a multiple of control frequency) is plottedagainst Li+ concentration. A value of 1 (dashed horizontalline) indicates no change inmean action potential frequency.(0.5 mM, n=7; 1 mM, n=5; 2 mM, n=5; 5 mM, n=5. * signifiesp<0.05, relative to frequency in 0 mM Li+.)
55B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
obtained in Li+ was standardized to this control frequency.Mean spontaneous action potential frequency before Li+
treatments was 4.5±0.5 Hz (n=22). Consistent with a dose-dependent membrane depolarization, 1 mM, 2 mM and 5 mMLi+ increased the frequency of spontaneous action potentialsto 1.7±0.4 (p=0.04), 2.3±0.5 (p=0.02), and 5.2±0.8 (p=0.004)times control frequency respectively (Fig. 6).
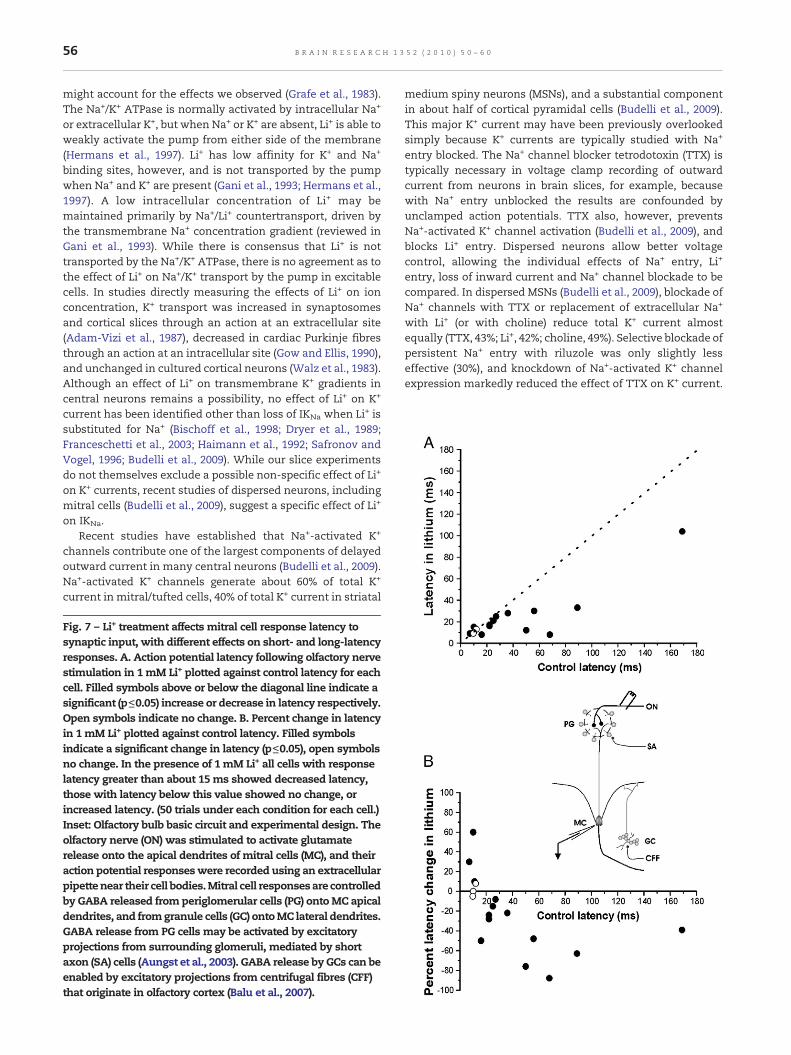
If Li+ treatment depolarizes mitral cells, this could influ-ence their responses to excitatory synaptic input. We there-fore tested for an influence of Li+ on mitral cell responses toolfactory nerve stimulation. Olfactory nerve stimulation invitro releases glutamate onto mitral cell apical dendrites,initiating a long-lasting depolarization (LLD) that results in atrain of action potentials (Carlson et al., 2000). LLDs are all-or-none responses, not graded with the strength of synapticinput (Carlson et al., 2000). The latency to the first mitral cellaction potential following olfactory nerve stimulation isstrongly influenced by mitral cell membrane potential,decreasing markedly with depolarization (Heyward et al.,2001). We therefore stimulated the olfactory nerve (0.2 Hz)during extracellular recording from mitral cells, testing for aneffect of Li+ (1 mM) on action potential response latency. Mitralcell action potential latencies decreased in the presence of1 mM Li+ (Fig. 7). Mean action potential response latencyobtained over 50 trials in each neuron before exposure to Li+
was 38±10 ms (n=17). Mean latency decreased to 0.79±0.09 ofcontrol after incubation in 1 mM Li+ (p=0.007). Although apresynaptic effect on glutamate release from the olfactorynerve is not excluded by these results, the findings areconsistent with a post-synaptic depolarizing effect of Li+,bringing mitral cell membrane potentials closer to actionpotential threshold.
While Li+ treatment resulted in an overall reduction inmitral cell response latency, the effect on individual neuronsdepended on their response latency before Li+ treatment. Assummarized in Fig. 7, while 11 of 17 cells showed decreasedresponse latency in the presence of 1 mM Li+, 6 showed eitherno change (n=3) or an increased latency (n=3). The neuronsthat showed decreased response latency in Li+ had a longerlatency under control conditions (53±13 ms, n=11) than thosethat did not (10±1 ms, n=6, p=0.03). Clinically useful con-centrations of extracellular Li+ can thus have selective,possibly opposite effects on the synaptic responses ofindividual cells within neuronal populations, depending ontheir pre-existing level of post-synaptic excitability.
3. Discussion
The results of this study show that exposure to low mMconcentrations of Li+ can increase the electrical excitability ofcortical neurons, mitral cells of the olfactory bulb of the brain.Li+ depolarized mitral cells, increased their spontaneous rateof action potential generation and increased their sensitivityto synaptic input, consistent with blockade of outward currentinvolved in the maintenance of resting membrane potential.Li+ also decreased action potential afterhyperpolarization,consistent with blockade of outward current following eachaction potential.
Li+ alters resting potentials and action potentials in severaltypes of central neuron by decreasing outward membranecurrent. In contrast to our findings, however, previous studieshave reported these effects after exposure to relatively highconcentrations of intracellular or extracellular Li+. In corticalneurons, Li+ leaking from impaling micropipettes filled withmolar Li+ concentrations reverses action potential afterpolariza-tions from hyperpolarizing to depolarizing events (Mayer et al.,1984), similar to the effect of Li+ shown in Fig. 1, and depolarizesresting membrane potentials. In a subpopulation of neocorticalpyramidal neurons, partial substitutionof extracellular Li+ forNa+
(40mM) eliminates afterhyperpolarizations following single ac-tion potentials (Franceshetti et al., 2003; Bhattacharjee andKaczmarek, 2005). Similar findings have been reported inhippocampus. In CA1 cells, partial substitution of extracellularLi+ for Na+ (50mM) depolarizes restingmembrane potentials, andblocks an outward current generated following single actionpotentials (Liu and Leung, 2004). When exposed to extracellularLi+ concentrations similar to those in the present study (0.5 to6mM), however, hippocampal CA1 and CA3 cells also showincreased action potential duration, again consistent withblockade of outward current following the generation of singleaction potentials (Colino et al., 1998). The current specificallyreduced in the presence of Li+ in CA1 and neocortical neuronsmaybeNa+-activatedK+current, IKNa (Franceshetti etal., 2003; Liuand Leung, 2004, reviewed in Bhattacharjee and Kaczmarek,2005).
Reduced K+ current could result from a specific effect on K+
channel conductance or activation, or from a non-specificeffect of Li+ on transmembrane K+ gradients. If Li+ entrydisplaced intracellular K+, or reduced intracellular K+ byinterfering with the Na+/K+ ATPase membrane pump, this
56 B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
might account for the effects we observed (Grafe et al., 1983).The Na+/K+ ATPase is normally activated by intracellular Na+
or extracellular K+, but when Na+ or K+ are absent, Li+ is able toweakly activate the pump from either side of the membrane(Hermans et al., 1997). Li+ has low affinity for K+ and Na+
binding sites, however, and is not transported by the pumpwhen Na+ and K+ are present (Gani et al., 1993; Hermans et al.,1997). A low intracellular concentration of Li+ may bemaintained primarily by Na+/Li+ countertransport, driven bythe transmembrane Na+ concentration gradient (reviewed inGani et al., 1993). While there is consensus that Li+ is nottransported by the Na+/K+ ATPase, there is no agreement as tothe effect of Li+ on Na+/K+ transport by the pump in excitablecells. In studies directly measuring the effects of Li+ on ionconcentration, K+ transport was increased in synaptosomesand cortical slices through an action at an extracellular site(Adam-Vizi et al., 1987), decreased in cardiac Purkinje fibresthrough an action at an intracellular site (Gow and Ellis, 1990),and unchanged in cultured cortical neurons (Walz et al., 1983).Although an effect of Li+ on transmembrane K+ gradients incentral neurons remains a possibility, no effect of Li+ on K+
current has been identified other than loss of IKNa when Li+ issubstituted for Na+ (Bischoff et al., 1998; Dryer et al., 1989;Franceschetti et al., 2003; Haimann et al., 1992; Safronov andVogel, 1996; Budelli et al., 2009). While our slice experimentsdo not themselves exclude a possible non-specific effect of Li+
on K+ currents, recent studies of dispersed neurons, includingmitral cells (Budelli et al., 2009), suggest a specific effect of Li+
on IKNa.Recent studies have established that Na+-activated K+
channels contribute one of the largest components of delayedoutward current in many central neurons (Budelli et al., 2009).Na+-activated K+ channels generate about 60% of total K+
current in mitral/tufted cells, 40% of total K+ current in striatal
Fig. 7 – Li+ treatment affects mitral cell response latency tosynaptic input, with different effects on short- and long-latencyresponses. A. Action potential latency following olfactory nervestimulation in 1mM Li+ plotted against control latency for eachcell. Filled symbols above or below the diagonal line indicate asignificant (p≤0.05) increase or decrease in latency respectively.Open symbols indicate no change. B. Percent change in latencyin 1mM Li+ plotted against control latency. Filled symbolsindicate a significant change in latency (p≤0.05), open symbolsno change. In the presence of 1mM Li+ all cells with responselatency greater than about 15ms showed decreased latency,those with latency below this value showed no change, orincreased latency. (50 trials under each condition for each cell.)Inset: Olfactory bulb basic circuit and experimental design. Theolfactory nerve (ON) was stimulated to activate glutamaterelease onto the apical dendrites of mitral cells (MC), and theiraction potential responseswere recorded using an extracellularpipettenear their cell bodies.Mitral cell responsesare controlledby GABA released from periglomerular cells (PG) ontoMC apicaldendrites, and fromgranule cells (GC) ontoMC lateral dendrites.GABA release from PG cells may be activated by excitatoryprojections from surrounding glomeruli, mediated by shortaxon (SA) cells (Aungst et al., 2003). GABA release by GCs can beenabled by excitatory projections from centrifugal fibres (CFF)that originate in olfactory cortex (Balu et al., 2007).
medium spiny neurons (MSNs), and a substantial componentin about half of cortical pyramidal cells (Budelli et al., 2009).This major K+ current may have been previously overlookedsimply because K+ currents are typically studied with Na+
entry blocked. The Na+ channel blocker tetrodotoxin (TTX) istypically necessary in voltage clamp recording of outwardcurrent from neurons in brain slices, for example, becausewith Na+ entry unblocked the results are confounded byunclamped action potentials. TTX also, however, preventsNa+-activated K+ channel activation (Budelli et al., 2009), andblocks Li+ entry. Dispersed neurons allow better voltagecontrol, allowing the individual effects of Na+ entry, Li+
entry, loss of inward current and Na+ channel blockade to becompared. In dispersed MSNs (Budelli et al., 2009), blockade ofNa+ channels with TTX or replacement of extracellular Na+
with Li+ (or with choline) reduce total K+ current almostequally (TTX, 43%; Li+, 42%; choline, 49%). Selective blockade ofpersistent Na+ entry with riluzole was only slightly lesseffective (30%), and knockdown of Na+-activated K+ channelexpression markedly reduced the effect of TTX on K+ current.

57B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
The absence of Na+ entry rather than the absence of inwardcurrent (or presence of TTX or Li+) therefore accounts for thereduction of K+ current (Budelli et al., 2009). If Li+ reduced K+
gradients (or had other pan-specific effects on K+ channels) inaddition to blocking IKNa, replacing extracellular Na+ with Li+
would be expected to cause a greater decrease of total K+
current than blocking Na+ entry with TTX, or replacing Na+
with choline. Although effects on K+ gradients were notdirectly excluded in those experiments, the equivalent effectsof Li+ and TTX are consistent with a requirement for Na+ entryfor channel activation, and suggest that at least in striatalMSNs, failure to activate Na+-activated K+ channels can fullyaccount for the effect of Li+ on K+ current. Na+-activated K+
channels are widely distributed in brain, and function over awide range of time scales in different neuron types (Bhatta-
et al., 2002, 2005; Bhattachargee and Kazmarek,2005). Expression of IKNa channels is particularly strong inmitral cells (Egan et al., 1992; Bhattacharjee et al., 2002, 2005,Budelli et al., 2009). IKNa is activated by Na+ entry during singleor multiple action potentials (reviewed in Bhattachargee andKaczmarek, 2005), and in neurons generating persistent Na+
currents at subthreshold potentials (such as mitral cells;Heyward et al., 2001; Balu and Strowbridge, 2007), IKNa
contributes to long-term regulation of membrane potential.Consistent with our findings, partial blockade of IKNa in suchneurons depolarizes resting membrane potentials, increasingmembrane excitability (Schwindt et al., 1989).
Li+ enters neurons through voltage-gated Na+ channels,readily substituting for Na+ in the generation of action potentials(Hille, 2001), but fails to activate IKNa. Our findings are consistentwith reduced activation of IKNa as a result of Li+ entry into mitralcells, but the concentrations of intracellular Na+ and Li+ in ourexperiments are unknown. Specific blockade when extracellularLi+ is substituted forNa+ isan identifyingpropertyof IKNa (Bischoffet al., 1998; Dryer et al., 1989; Franceschetti et al., 2003; Haimannet al., 1992; Safronov and Vogel, 1996; Budelli et al., 2009), but theconcentrations of intracellular Na+ required for activation of IKNa,
have been unclear. Recent studies (Budelli et al., 2009) haveclarified the apparent requirement for high intracellular Na+ foractivation of IKNa. Voltage-gated Na+ entry activates IKNa even ifbulk cytoplasmic Na+ has been manipulated using whole-cellpatch pipette-filling solutions containing 0 to 70mMNa+ (Budelliet al., 2009). This is explained by a highly localized accumulationof Na+ that has entered through voltage-gated Na+ channels,persistent Na+ channels in particular. Although Li+ may notaccumulate in the cytoplasm of neurons to concentrationsapproaching extracellular levels (Birch, 1999), this does notexclude local accumulation of Li+. Na+-activated K+ channelsfound in mitral cells and many other central neurons (SLACKchannels, ‘Sequence like a Ca-activated K channel’) have astructure like that of BK channels, with a ring of negative chargessurrounding the channel inner vestibule (Brelidze et al., 2003;Budelli et al., 2009; Nimigean et al., 2003). This ring of charge (andpossibly other charged cytoplasmic domains) may create a Na+-richmicrodomain involved in channel gating (Budelli et al., 2009).Accumulation of ions by the ring of charge is purely electrostatic,and therefore non-selective (Brelidze et al., 2003; Budelli et al.,2009). In BK channels, the ring of charge creates a very higheffective local concentration of K+ (0.5 M), doubling K+ conduc-tance (Brelidze et al., 2003). In pituitary melanotrophs, 20mM
charjee
intracellular Li+ does not decrease BK channel conductance (Kehl,1996), suggesting that Li+ doesnot interferewithK+ accumulation.Much higher local concentrations of Li+ could, however, beachieved by the accumulation of Li+ entering through persistentNa+ channels (Budelli et al., 2009). Li+mayhowever bemore likelyto interfere with the electrostatic accumulation of Na+ proposedto endow Na+-activated K+ channels with their sensitivity topersistent Na+ entry (Budelli et al., 2009). If a fraction of Na+ entryis replaced by Li+ entry, Li+ may accumulate and displace Na+ atsites of electrostatic attraction responsible for channel activation,reducing the normal coupling of Na+ entry to K+ current. Thishypothesis could in principle be tested in single-channel record-ings from inside-out membrane patches. If Li+ displaces Na+
accumulating near Na+-activated K+ channels, but does notdisplace K+, then Li+ would decrease their sensitivity to Na+
without changing the K+ conductance.When outward membrane current is reduced in mitral cells,
their resting membrane potentials depolarize, decreasing theirsynaptic response latency to glutamate released by olfactorynerve stimulation (Hayar et al., 2001). The effect of Li+ (1 mM) onsynaptic response latencywe report here, however, depended onthe response latency before Li+ treatment. Long responselatencies (>15ms) were decreased during Li+ treatment, butshort response latencies (<15ms) were unaffected, or wereincreased. These differential effects are not readily explained bya post-synaptic effect on outward current inmitral cells alone, orby a presynaptic effect on glutamate release from the olfactorynerve. Inhibitory interneurons in the olfactory bulb, periglomer-ular and granule cells, also abundantly express IKNa channels(Bhattacharjee et al., 2002, 2005). Periglomerular cells releaseGABA to inhibit mitral cell responses to olfactory nerve stimula-tion in brain slices (Aungst et al., 2003). Granule cells perform asimilar function, andmay bemost effective when depolarized byinput extrinsic to the olfactory bulb (Balu et al., 2007). Li+ couldtherefore increase the excitability of periglomerular and granulecells, increasing their inhibitory output tomitral cells. The overalleffect of Li+ will depend on the relative strengths of the indirecteffectmediated throughdecreased IKNa in interneurons, acting toincrease mitral cell response latency, and the direct effectmediated through decreased IKNa in mitral cells, acting todecrease response latency. IKNa is uniformly strong in dispersedmitral/tufted cells (Budelli et al., 2009), but could show morevariability in situ. Li+ will have the strongest direct effect onmitralcells in which IKNa is relatively strong, and mitral cells in whichthis outward current is relatively strongmay be those respondingtoolfactorynervestimulationat longer latency (Hayaret al., 2001).Conversely, Li+ may have relatively little direct effect on mitralcells responding at shorter latency, in which IKNa may berelatively weak. The results thus suggest that the overall effectof Li+ could be to stabilize the responsiveness of these corticalprincipal neurons to excitatory synaptic input, through opposingdirect and indirect effects on their post-synaptic excitability.
Ion channels regulating neuron excitability are the commontarget of the antiepileptic drugs effective in the treatment ofbipolar disorder (El-Mallakh and Huff, 2001; Askland 2006;Rogawski and Loscher, 2004a). It has not been apparent, however,how this target might be shared with Li+ (see Rogawski andLoscher, 2004b). The antiepileptics effective in bipolar disorderbindpreferentially toandstabilize inactivatedNa+ channel states,resulting in use-dependent blockade of Na+ entry (Taverna et al.,

58 B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
1998; Errington et al., 2005). They therefore act preferentially ondepolarized or rapidly firing neurons. Blockade of channelsgenerating persistent Na+ current at subthreshold potentialsmay be a major contributor to their ability to reduce neuronalhyperexcitability (Taverna et al., 1998; Johanessen 2000; Rogawskiand Loscher, 2004a; Errington et al., 2005), and persistent Na+
current may be the major activator of IKNa (Budelli et al., 2009).Moreover, sodium channels and IKNa channels can cluster inneuronal membranes, allowing them to interact in the control ofmembrane potential (Koh et al., 1994). The anticonvulsants andLi+ might thus target structurally and functionally coupled ionchannels that interact in the long-term control of neuronalmembranepotentialandexcitability (Schwindtetal., 1989;Taylor,1993; Koh et al., 1994; Bhattacharjee andKaczmarek, 2005, Budelliet al., 2009). The anticonvulsants reduce Na+ entry, and maythereby also decrease K+ channel activation, while Li+ maydecrease the functional coupling of Na+ entry to K+ channelactivation. Consistent with such distinct but overlapping modesof action, treatment of bipolar disorder using Li+ and anantiepileptic drug together can be more effective than usingeither agent alone (Gajwani et al., 2005).
Bipolar disorder may result from altered expression ofmembrane ion channels in brain neurons (Askland et al.,2009). IKNa channels, for example, are genetically encoded atidentified affective disorder susceptibility loci (chromosomes9q34.3, 1q31.3) (Bhattacharjee and Kaczmarek, 2005; Askland2006). In addition to ion channel deficits, recent studies (TheWelcome Trust Case Control Consortium, 2007) highlight thepotential importance of genes encoding neurotransmitterreceptors in bipolar disorder, GABAA receptors in particular.In addition to their role in inhibitory neurotransmission,GABAA receptors tonically regulate membrane excitability,and genetic deficiency in GABAA receptors can result inadaptive changes in neuronal ion channel expression (Brickleyet al., 2001). In mutant mice lacking crucial subunits of
GABAA receptors, for example, increased K+
current compensates for lack of tonic GABA-dependentchloride current, maintaining normal levels of post-synapticexcitability (Brickley et al., 2001). The drugs acting onmembrane ion channels may thus be effective in bipolardisorder whether abnormal ion channel function and mem-brane excitability results directly from deficits in ion channelgene expression, or from adaptive (or maladaptive), possiblycyclical, responses to altered neurotransmitter function inspecific brain regions.
extrasynaptic
4. Experimental procedures
Experiments were conducted with the approval of the Universityof Otago Animal Ethics Committee. The data were obtained frommale Swiss out bredmice aged 18–30 days. Slices of the olfactorybulb of the brainwere prepared as previously described (Heywardet al., 2001; Aungst et al., 2003). In brief, mice were killed bydecapitation, a single olfactory bulb was dissected out andmounted on the stage of a vibratingmicrotome (VibratomeSeries1000, The VibratomeCompany, USA, or Leica VT1000S, Germany)with cyanoacrylate glue. The tissue was submerged in chilled(∼4 °C) bathing medium (see below) and 350 μm thick horizontalslices were cut parallel to the long axis of the bulb. Slices were
transferred into a bath through which medium flowed at 2.5–3ml/min past both sides of the tissue, and maintained at 30 °C(TC2BIP, Cell Microcontrol, USA). The bath formed the fixed stageof a modified Nikon compound videomicroscope. Slices wereviewed using a Dage MTI CCD100 monochrome camera withonboardcontrast enhancement. Forextracellular recording, sliceswere visualized using a long-working distance 10× dry objective(Nikon), and oblique incident white light illumination wasprovided through a 1mm diameter fibre optic light pipe with itstip submerged in the bathing medium above the slice. Thisallowed illumination of the tissue at a shallow angle of incidence,giving good visualization of cell layers in the slicewithout surfacereflection from the bathing medium. For whole-cell recording,mitral cells were visualized using a 40× 0.75NA water immersionobjective (Zeiss) and transilluminated using Asymmetric Illumi-nation Contrast (Kachar, 1985) with near-infra-red light (850±10 nM). Slices were allowed to recover for about 1.5 h beforerecording, and each experiment continued for up to 6 h.
4.1. Solutions and drugs
The bathing medium used to prepare and maintain slicesconsisted of (inmM)NaCl 120, KCl 3, CaCl2 1.3, MgSO4 1.3, glucose10, NaHCO3 25, and BES (N,N-Bis(2-hydroxyethyl)-2-aminoetha-nesulfonic acid) 5. This mediumwas continuously gassed withinthe reservoirwith 5%CO2/95%O2, pH7.2, andwasdelivered to therecording bath by peristaltic pump through tubing of low CO2/O2
permeability (Sanipure-60, Cole Parmer). Control and Li+-contain-ing solutions were made equimolar by substitution of LiCl forNaCl or addition of NaCl.
In some experiments, mitral cells were isolated fromexcitatory glutamate synaptic inputs by using the broadspectrum excitatory amino acid receptor antagonist 4-hydro-xyquinoline-2-carboxylic acid (kynurenic acid, 2 mM), or thespecific NMDA receptor antagonist DL-2-amino-5-phosphono-pentanoic acid (APV, 50 μM) and non-NMDA antagonist 6-7-dinitroquinoxaline-2-3-dione (DNQX, 10 μM), and isolatedfrom inhibitory GABAA and glycine inputs using picrotoxin(PTX, 100 μM). Myo-inositol (100 μM) or creatine monohydrate(1 mM) was included in the bathing medium to test for effectsof Li+ possibly mediated through myo-inositol or ATP deple-tion. The cell permeable Ca2+ chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)(BAPTA AM) was used to test for Ca-activated K+ currents.DNQX and BAPTA AM were from Tocris (UK), other chemicalswere from Sigma Aldrich.
4.2. Electrophysiological recording
Conventional patch-clamp micropipettes with an outside tipdiameter of about 3 μm, 6 to 8 MΩ, were used for extracellularsingle-unit or whole-cell recording. For extracellular recording,pipettes were filled with bathing medium. For whole-cellrecording, pipettes were filled with a solution containing (inmM) K gluconate 125, MgCl2 2, HEPES 10, Mg2ATP 2, Na3GTP 0.2,NaCl 1, and EGTA 0.2. Extracellular recordings were made usingan Axoprobe amplifier (Axon Instruments USA) or AM Systems(USA) model 2400 amplifier. Whole-cell recordings were madeusing an AM Systems model 2400 amplifier. Seal resistance wasroutinely >1 GΩ. Vm under control conditions was 46±1mV

59B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
(n=16), access resistance 10.4 MΩ. Voltage measurements werenot corrected for a liquid junction potential of 9–10mV. Record-ings were digitized on personal computer and analyzed usingPClamp 9 software (Axon Instruments). Unless otherwise noted,the effects of each experimental manipulation were assessed byanalyzing and comparing 5min epochs of recording obtainedimmediately before and after pre-incubation in experimentalsolutions for20min.Unlessotherwisenoted in the text, statisticalanalysis was by Student's paired t test, or for multiple compar-isons, ANOVA. Statistical significance was accepted at p≤0.05.
In some experiments, intracellular current injection wasused to activate single action potentials from hyperpolarizedholding potentials. Mitral cells were held 15 mV negative totheir resting membrane potential using current generated bythe recording amplifier, and a Grass Instruments SD9 stimu-lator used to command superimposed monophasic squarewave depolarizing current pulses (0.05 ms, 0.5 Hz), with anamplitude (2 to 6 nA) just sufficient to evoke an actionpotential in 100% of trials.
Electrical stimulation of the olfactory nerve was used toinvestigate the effects of Li+ on mitral cell responses todepolarizing synaptic input. Bipolar stimulating electrodeswere constructed in the laboratory from twisted pairs of 70 μmdiameter silver wire (AM Systems USA), insulated with Teflonexcept at squarely cut tips. Stimuli were monophasic squarewave pulses (0.1 ms) delivered at 0.5 Hz, generated using aPulsar 4i isolated constant-current stimulator (Frederick Haer& Co, USA) synchronized with the acquisition system, andwere applied to the olfactory nerve opposite the recordedmitral cell. Stimulus amplitude (80 to 200 μA) was adjusted tobe just sufficient to synaptically activate amitral cell responseon 100% of trials.
Acknowledgments
These studies were supported by the Otago Medical ResearchFoundation (PMH), the Department of Physiology University ofOtago (CHS and CB-M), and the Marsden Fund (PMH and EJC).
R E F E R E N C E S
Adamvizi, V., Banayschwartz, M., Wajda, I., Lajtha, A., 1987.Depolarization of brain cortex slices and synaptosomes bylithium — determination of K+ equilibrium potential in cortexslices. Brain Res. 410, 257–263.
Askland, K., Read, C., Moore, J., 2009. Pathways-based analyses ofwhole-genome association study data in bipolar disorderreveal genes mediating ion channel activity and synapticneurotransmission. Hum. Genet. 125, 63–79.
Askland, K., 2006. Toward a biaxial model of “bipolar” affectivedisorders: further exploration of genetic, molecular andcellular substrates. J. Affect. Disord. 94, 35–66.
Askland, K., Parsons, M., 2006. Towards a biaxial model of“bipolar” affective disorders: spectrum phenotypes as theproducts of neuroelectrical and neurochemical alterations.J. Affect. Disord. 94, 15–33.
Atack, J.R., Broughton, H.B., Pollack, S.J., 1995. Inositolmonophosphatase — a putative target for Li+ in the treatmentof bipolar disorder. Trends Neurosci. 18, 343–349.
Aungst, J.L., Heyward, P.M., Puche, A.C., Karnup, S.V., Hayar, A.,Szabo, G., Shipley, M.T., 2003. Centre-surround inhibitionamong olfactory bulb glomeruli. Nature 426, 623–629.
Balestrino, M., Rebaudo, R., Lunardi, G., 1999. Exogenous creatinedelays anoxic depolarization and protects from hypoxicdamage: dose–effects relationship. Brain Res. 816, 124–130.
Balu, R., Pressler, R.T., Strowbridge, B.W., 2007. Multiple modes ofsynaptic excitation of olfactory bulb granule cells. J. Neurosci.27, 5621–5632.
Balu, R., Strowbridge, B.W., 2007. Opposing inward and outwardconductances regulate rebound discharges in olfactory mitralcells. J. Neurophysiol. 97, 1959–1968.
Belmaker, R.H., 2004. Bipolar disorder. N Engl J. Med. 351, 476–486.Bhattacharjee, A., Kaczmarek, L.K., 2005. For K+ channels, Na+ is
the new Ca2+. Trends Neurosci. 28, 422–428.Bhattacharjee, A., Gan, L., Kaczmarek, L.K., 2002. Localization of
the Slack potassium channel in the rat central nervous system.J. Comp. Neurol. 454, 241–254.
Bhattacharjee, A., Joiner, W.J., Wu, M., Yang, Y., Sigworth, F.J.,Kaczmarek, L.K., 2003. Slick (Slo2.1), a rapidly-gatingsodium-activated potassium channel inhibited by ATP. J.Neurosci. 23, 11681–11691.
Bhattacharjee, A., von Hehn, C.A., Mei, X., Kaczmarek, L.K., 2005.Localization of the Na+-activated K+ channel Slick in the ratcentral nervous system. J. Comp. Neurol. 484, 80–92.
Birch, N.J., 1999. Inorganic pharmacology of lithium. Chem. Rev.99, 2659–2682.
Bischoff, U., Vogel, W., Safronov, B.V., 1998. Na+-activated K+
channels in small dorsal root ganglion neurones of rat. J.Physiol. 510 (Pt 3), 743–754.
Brelidze, T.I., Niu, X.W., Magleby, K.L., 2003. A ring of eight conservednegatively charged amino acids doubles the conductance of BKchannels and prevents inward rectification. Proc. Natl Acad. Sci.USA 100, 9017–9022.
Brickley, S.G., Revilla, V., Cull-Candy, S.G., Wisden, W., Farant, M.,2001. Adaptive regulation of neuronal excitability by avoltage-independent potassium conductance. Nature 409, 88–92.
Budelli, G., Hage, T.A., Wei, A.A., Rojas, P., Jong, Y.J.I., O'Malley, K.,Salkoff, L., 2009. Na+-activated K+ channels express a largedelayed outward current in neurons during normal physiology.Nat. Neurosci. 12, 745–750.
Cade, J., 1949. Lithium salts in the treatment of psychoticexcitement. Med. J. Aust. 36, 349–352.
Carlson, G.C., Shipley, M.T., Keller, A., 2000. Long-lastingdepolarizations in mitral cells of the rat olfactory bulb. J.Neurosci. 20, 2011–2021.
Chen,W.R., Shepherd,G.M., 2005.Theolfactoryglomerulus:a corticalmodule with specific functions. J. Neurocytol. 34, 353–360.
Colino, A., Garcia-Seoane, J.J., Valentin, A., 1998. Action potentialbroadening induced by lithium may cause a presynapticenhancement of excitatory synaptic transmission in neonatalrat hippocampus. Eur. J. Neurosci. 10, 2433–2443.
del Rio, E., Shinomura, T., van der Kaay, J., Nicholls, D.G., Downes,C.P., 1998. Disruption by lithium of phosphoinositide signallingin cerebellar granule cells in primary culture. J. Neurochem. 70,1662–1669.
Dryer, S.E., Fujii, J.T., Martin, A.R., 1989. A Na+-activated K+ currentin cultured brain stem neurones from chicks. J. Physiol. 410,283–296.
Egan, T.M., Dagan, D., Kupper, J., Levitan, I.B., 1992. Properties andrundown of sodium-activated potassium channels in ratolfactory bulb neurons. J. Neurosci. 5, 1964–1978.
El-Mallakh, R.S., Huff, M.O., 2001. Mood stabilizers and ionregulation. Harv. Rev. Psychiatry 9, 23–32.
Errington, A.C., Stor, T., Lees, G., 2005. Voltage gated ion channels:targets for anticonvulsant drugs. Curr. Top.Med. Chem. 5, 15–30.
Franceschetti, S., Lavazza, T., Curia, G., Aracri, P., Panzica, F.,Sancini, G., Avanzini, G., Magistretti, J., 2003. Na+-activated K+
current contributes to postexcitatory hyperpolarization in

60 B R A I N R E S E A R C H 1 3 5 2 ( 2 0 1 0 ) 5 0 – 6 0
neocortical intrinsically bursting neurons. J. Neurophysiol. 89,2101–2111.
Gajwani, P., Forsthoff, A., Muzina, D., Amann, B., Gao, K., Elhaj, O.,Calabrese, J.R., Grunze, H., 2005. Antiepileptic drugs inmood-disordered patients. Epilepsia 46, 38–44.
Gani, D., Downes, C.P., Batty, I., Bramham, J., 1993. Lithium andmyoinositol homeostasis. Biochim. Biophys. Acta 1177,253–269.
Gargus, J.J., 2006. Ion channel functional candidate genes inmultigenic neuropsychiatric disease. Biol. Psychiatry 60,177–185.
Gow, I.F., Ellis, D., 1990. Effect of lithium on the intracellularpotassium concentration of sheep heart Purkinje-fibers. Exp.Physiol. 75, 427–430.
Grafe, P., Reddy, M.M., Emmert, H., Tenbruggencate, G., 1983.Effects of lithium on electrical activity and potassium-iondistribution in the vertebrate central nervous system. BrainRes. 279, 65–76.
Gurvich, N., Klein, P.S., 2002. Lithium and valproic acid: parallelsand contrasts in diverse signaling contexts. Pharmacol. Ther. 96,45–66.
Haimann, C., Magistretti, J., Pozzi, B., 1992. Sodium-activatedpotassium current in sensory neurons: a comparison ofcell-attached and cell-free single-channel activities. PflugersArch 422, 287–294.
Hayar, A., Heyward, P.M., Heinbockel, T., Ennis,M., Shipley,M.T., 2001.Direct excitation of mitral cells via activation of α1-noradrenergicreceptors in rat olfactory bulb slices. J. Neurophysiol. 86,2173–2182.
Heinbockel T., Heyward P.M., 2008. Glutamate synapses inolfactory neural circuits. In Amino Acid ReceptorResearch, Paley F., Warfield T.E. ed., Nova Science Publisherspp. 391–425.
Hermans, A.N., Glitsch, H.G., Verdonck, F., 1997. Activation of theNa+/K+ pump current by intra- and extracellular Li ions insingle guinea-pig cardiac cells. Biochim. Biophys. Acta Bio-membr. 1330, 83–93.
Heyward, P.M., Ennis, M., Keller, A., Shipley, M.T., 2001. Membranebistability in olfactory bulb mitral cells. J. Neurosci. 21, 5311–5320.
Hille, B., 2001. Ion Channels of Excitable Membranes, 3rd edition.Sinauer, MA USA.
Johanessen, C.U., 2000. Mechanisms of action of valproate: acommentary. Neurochem. Int. 37, 103–110.
Kachar, B., 1985. Asymmetric illumination contrast: a method ofimage formation for video light microscopy. Science 227, 766–768.
Kehl, S.J., 1996. Block of BK (maxi K) channels of rat pituitarymelanotrophs by Na+ and other alkali metal ions. PflügersArch. Eur. J. Physiol. 432, 623–629.
Koh, D.-S., Jonas, P., Vogel, W., 1994. Na+-activated K+ channelslocalized in the nodal region of myelinated axons of Xenopus.J. Physiol. 479, 183–197.
Liu, X., Leung, L.S., 2004. Sodium-activated potassium conductanceparticipates in the depolarizing afterpotential following a singleactionpotential in rathippocampalCA1pyramidal cells. BrainRes.1023, 185–192.
Mayer, M.L., Crunelli, V., Kemp, J.A., 1984. Lithium ions increaseaction potential duration of mammalian neurons. Brain Res.293, 173–177.
Nanou, E., El, Manira A., 2007. A post-synaptic negative feedbackmediatedbycouplingbetweenAMPAreceptors andNa+-activatedK+ channels in spinal cord neurons. Eur. J. Neurosci. 25, 445–450.
Nimigean, C.M., Chappie, J.S., Miller, C., 2003. Electrostatic tuningof ion conductance in potassium channels. Biochemistry 42,9263–9268.
Rogawski, M.A., Loscher, W., 2004a. The neurobiology of antiepilepticdrugs. Nat Rev Neurosci 5, 553–564.
Rogawski, M.A., Loscher, W., 2004b. The neurobiology ofantiepileptic drugs for the treatment of nonepilepticconditions. Nat. Med. 10, 685–692.
Safronov, B.V., Vogel, W., 1996. Properties and functionsof Na+-activated K+ channels in the soma of rat motoneurones.J. Physiol. 497, 727–734.
Schwindt, P.C., Spain, W.J., Crill, W.E., 1989. Long-lasting reductionof excitability by a sodium-dependent potassium current in catneocortical neurons. J. Neurophysiol. 61, 233–244.
Taverna, S., Mantegazza, M., Franceschetti, S., Avanzini, G., 1998.Valproate selectively reduces the persistent fraction of Na+
current in neocortical neurons. Epilepsy Res. 32, 304–308.Taylor, C.P., 1993. Na+ currents that fail to inactivate. TINS 16,
455–460.TheWelcome Trust Case Control Consortium, 2007. Genome-wide
association study of 14,000 cases of seven common diseasesand 3,000 shared controls. Nature 447, 61–678.
Undie, A.S., 1999. Relationship between dopamine agoniststimulation of inositol phosphate formation and cytidinediphosphate-diacylglycerol accumulation in brain slices.Brain Res. 816, 286–294.
Walz,W., Hertz, E., Hertz, L., 1983. Lithium–potassium interactionsin acutely treated cortical-neurons and astrocytes. Prog.Neuro-Psychopharmacolog. Biol. Psychiatry 7, 697–702.
Related Documents











