This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Cardiovascular Pharmacology
Levosimendan preserves the contractile responsiveness of hypoxic humanmyocardium via mitochondrial KATP channel and potential pERK 1/2 activation
Paul F. Soeding a,b,⁎, Peter J. Crack c, Christine E. Wright a, James A. Angus a, Colin F. Royse a
a Cardiovascular Therapeutics Unit, Department of Pharmacology, University of Melbourne, Parkville, Australiab Department of Anaesthesia and Pain Medicine, Royal Melbourne Hospital, Parkville, Australiac Department of Pharmacology, University of Melbourne, Parkville, Australia
a b s t r a c ta r t i c l e i n f o
Article history:Received 17 October 2010Received in revised form 10 December 2010Accepted 15 December 2010Available online 13 January 2011
Keywords:Human right atrium levosimendanHypoxia–reoxygenationProsurvival mediatorApoptosis
This study investigated the role of levosimendan, a mitochondrial KATP channel opener, during hypoxia–reoxygenation injury in human isolated tissue. The activation of preconditioning pathways, and the release ofmitochondrial cytochrome cwere determined. Human right atrial trabeculae were mounted in an organ bath,electrically paced and contractile force measured. Tissue was subjected to hypoxia–reoxygenation, andisoproterenol concentration–response experiments were performed as an index of contractile viability. Theintracellular activities of Akt, ERK 1/2, P38, caspase 3, and cytochrome c were assayed by western blot.Following hypoxia–reoxygenation, the maximal contractile response of trabeculae to isoproterenol wassignificantly increased with levosimendan pretreatment compared to the hypoxia–reoxygenation control(0.88±0.02 versus 0.60±0.01 g, Pb0.01). This enhanced response was blocked by 5-hydroxydecoanate(0.54±0.09 g, Pb0.01). A significant increase in both phosphorylated and total ERK 1/2 and P38 occurred at60 min reoxygenation, compared to control tissue. No difference was observed in phosphorylated or total Akt,though there was a trend for increased levels in hypoxic tissue. Cytochrome c was detected at 60 min postreoxygenation, in both levosimendan treated and untreated tissue. No increase in cleaved-caspase 3 activitywas observed. Our findings suggest that levosimendan preserves the contractile force to isoproterenol afterhypoxia–reoxygenation, a response mediated via mKATP channel activation. The significant increase in theactivity of prosurvival mediators ERK 1/2 and P38 following hypoxia indicates a potential mechanism of actionfor levosimendan-induced cardioprotection.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
In cardiac surgery, cardiopulmonary bypass subjects the myocar-dium to a varying degree of ischemia–reperfusion injury, and isresponsible for the transient myocardial dysfunction observed onbypass separation. Restoration of normal myocardial perfusionfollowing cardioplegic arrest, or similarly in patients with acutecoronary syndrome, can paradoxically lead to myocyte death, aphenomenon termed lethal reperfusion-induced injury (Kloner, 2004).Activation of apoptotic or necrotic cell death pathways during theearly phase of reperfusion is seen as an important contributor to lethalreperfusion-induced injury (Gottlieb et al., 1994; Maulik et al., 1998;Stephanou et al., 2001)
Mitochondria play a central role in the triggeringof apoptosis (Greenand Reed, 1998). Hypoxic stress alters outer membrane permeability,
leading to mitochondrial matrix calcium overload, severe organelledysfunction and disruption. The release of mitochondrial cytochrome cinto the cytosol is postulated to activate apoptosis (Brookes et al., 2004;Halestrap et al., 2004; Hausenloy et al., 2004). Secondly the activation ofimportant prosurvival mediators may influence mitochondrial perme-ability and function; and include themitoxgen-activated protein (MAP)kinases, extracellular signal-regulated kinase (ERK1/2), P38MAPkinase(P38), phosphoinositide 3-kinase (PI-3), serine/threonine-specifickinase (Akt), and c-Jun N-terminal kinase (JNK 1/2) (Engelbrechtet al., 2004; Ma et al., 1999; Saurin et al., 2000; Yue et al., 1998).
Levosimendan, a known mitochondrial KATP channel opener(Kopustinskiene et al., 2001), has been demonstrated to have apreconditioning-like effect on myocardial function (Kersten et al.,2000) and thereby has the potential to protect the heart duringischemia–reperfusion. During cardiac surgery levosimendan ad-ministration is reported to reduce myocardial damage (Tritapepeet al., 2006) and improve cardiac function following cardiopulmo-nary bypass (De Hert et al., 2007). Whether levosimendan actspredominantly as an inotrope via calcium sensitization, vasodilatoror cardioprotective agent at clinically used concentrations isunknown.
European Journal of Pharmacology 655 (2011) 59–66
⁎ Correspondingauthor. Cardiovascular TherapeuticsUnit,Departmentof Pharmacology,University of Melbourne, Victoria 3010, Australia. Tel.: +61 3 8344 5673; fax: +61 3 83445193.
E-mail address: [email protected] (P.F. Soeding).
0014-2999/$ – see front matter © 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.ejphar.2010.12.035
Contents lists available at ScienceDirect
European Journal of Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /e jphar

Author's personal copy
In this study we investigated the effect of levosimendan oncontractile function in human isolated atrial tissue, followinghypoxia–reoxygenation, and tested whether mKATP channels areinvolved. We measured the response to the β-adrenoceptor agonistisoproterenol as a ‘contractility viability index’ and the effects thereonafter various pretreatments. Secondly the study evaluated whetherpretreatment with levosimendan had an effect on preconditioningpathways including MAP kinases ERK 1/2, P38, MAPKa/h, PI-3 kinase/Akt, the release of mitochondrial cytochrome c and the activation ofapoptosis.
2. Material and methods
2.1. Tissue preparation, dissection and mounting
Human discarded right atrial appendages were obtained frompatients undergoing routine cardiac surgery at the Royal MelbourneHospital (Ethics approval 2006/006). Patients gave informed consentbut were excluded from this study if they were on oral hypoglycemicagents. At surgery, the discarded tip of the right atrial appendage wasexcised and immediately placed in pre-oxygenated ice-cold Krebs'solution (mM): NaCl 119; KCl 4.7; KH2PO4 1.18; MgSO4 1.17; NaHCO3
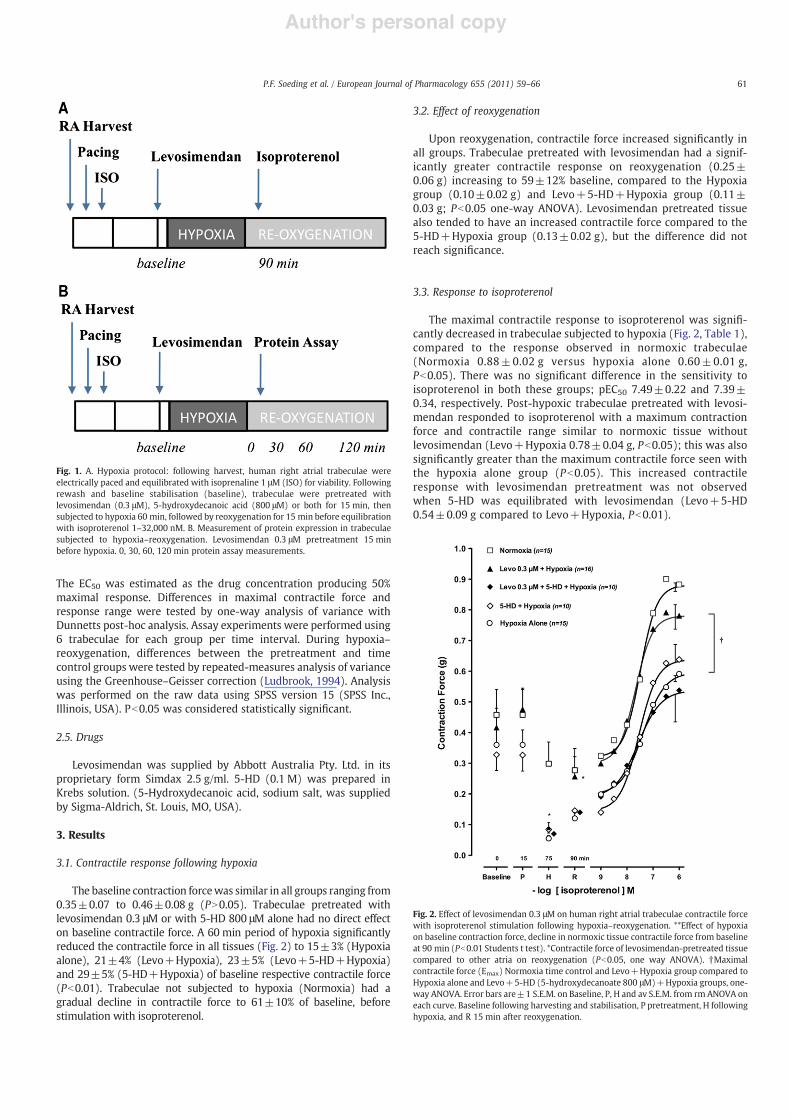
25; CaCl2 2.0; Na EDTA 0.026; and glucose 5.5 and transported to thelaboratory within 5–10 min. Individual trabeculae (≤1 mm thick)were dissected free, threaded with stainless steel hooks at each endand mounted vertically in 15 ml organ baths. The upper hook wasattached to a strain gauge transducer. Experiments were conducted inan isolated bio-cabinet (Gelman class 2, GHA 180). Baths containedKrebs solution (as above), saturated with carbogen at 34 °C. Eachappendage provided an average of 4–6 trabeculae. Following a 60 minstabilisation period the tissue was then stimulated with square wavepulses (1 Hz, 5 ms duration, 20% above threshold voltage) viaplatinum wire field electrodes. The resting passive force was adjustedto 0.5 g, ensuring muscle contraction was measured at the peak of thelength–tension curve, Lmax (Serone and Angus, 1999). A positivecontractile response to isoproterenol 1 μM indicated tissue viabilityand tissues that failed to contract with a force≥0.2 g (2 mN) wererejected. The tissue bath solution was replaced with fresh Krebs'solution and resting force of stimulated trabeculae continuouslymeasured. Experiments then proceeded after a stable period, usually30–60 min, of baseline force (Baseline). Responseswere recorded on aPowerLab 8SP data acquisition system (AD Instruments, Bella VistaNSW, Australia) and Chart v5.5 software.
2.2. Hypoxia–reoxygenation protocol
Five types of experiments were performed, including three withdrug pretreatment: levosimendan 0.3 μM, levosimendan 0.3 μM and5-hydroxydecoanate (5-HD) 800 μM and 5-HD 800 μM alone; andtwo without drug pretreatment (normoxia and hypoxia alone). 5-HDis regarded as a specific mKATP antagonist, known to blockmitochondrial channels at concentrations of 50–500 μM (Wanget al., 2001) and has been used in organ bath preparations atconcentrations of 800 μM (Hanouz et al., 2002; Yvon et al., 2003).Drug pretreatment occurred 15 min before the onset of hypoxia andeach drug remained within the organ bath without further washingfor the remainder of the experimental protocol. In the Hypoxia alonegroup trabeculae were subjected to hypoxia without pretreatment,and a normoxia time control group (Normoxia) did not receivehypoxia exposure.
Trabeculae were subjected to a 60 min period of hypoxia, byreplacement of carbogen with a N2/CO2 (95:5%) gas mixture. Inbaths saturated with carbogen, solution pH was 7.47±0.02, pCO2
28±2 mm Hg, pO2 497±27 mm Hg, compared to the hypoxic N2/CO2 saturated solution, with pH 7.51±0.01, pCO2 26±1 mm Hg,pO2 65±8 mm Hg. Following the hypoxic period, tissue was
reoxygenated with carbogen for 15 min, and then stimulated withisoprenaline (Fig. 1). A concentration–contractile response curvewas constructed by equilibrating trabeculae with cumulativeconcentrations of isoprenaline 1–32,000 nM. At each concentration,the maximal contractile response was measured when contractileforce plateaued at 5–8 min. Increasing concentration was notassociated with episodes of arrhythmia.
2.3. Intracellular mediator protocol
In another series of experiments trabeculae (n=4) were removedat different time intervals following the hypoxia–reoxygenation phase(time 0 at the end of hypoxia, then 15, 60 and 120 min duringreoxygenation), weighed and immediately frozen in liquid nitrogen at−80 °C, and assayed later for intracellular markers (phosphorylatedAkt, ERK and P38, cleaved-caspase 3, and cytochrome c). Thesetrabeculae were not subjected to isoproterenol stimulation. Analysisof protein activation, from 0–120 min following reoxygenation, wasaimed to provide a temporal picture of the response to hypoxia–reoxygenation. At assay, samples were ground and homogenised inlysis buffer 15 μl mg−1 (Bio-Rad cytoplasmic protein extraction buffer4307616, Bio-Rad Laboratories, Hercules CA, USA). Following 5 minincubation on ice, homogenates underwent centrifugation for 10 min(1000×g at 4 °C) and the supernatant decanted.
The protein content of each homogenate was determined byspectrophotometry. In view of the small volumes used, proteindetermination was made by measuring the absorbance of a 1.5 μlsample aliquot at 280 nm wavelength (NanoDrop 280, NanodropTechnologies, Wilmington, DE, USA). In addition a small number ofsamples was taken for protein estimation by the Bradford method.Absorbance of 300 μl samples were made at 590 nM (FLUOstarOPTIMA, BMG Labtechnologies Pty. Ltd., Offenburg, Germany). Samplemeasurements are referred to a standard absorbance curve based onknown concentrations of bovine serum albumin (Bradford, 1976).
Western blotting was done based on a previously establishedprotocol (Taylor et al., 2005) with slight modifications. Proteinextracts (approximately 3–10 μg) were separated on 12% SDS-PAGEgels and subsequently transferred to polyvinylidene difluoridemembranes (Millipore Co., MA, USA). Membranes were blockedwith 4% w/v skim milk in tris buffered saline containing 0.05% v/vTween 20 (TBST; pH 7.6) for 1 h at room temperature andsubsequently incubated with 1:1000 dilution of rabbit polyclonalantibodies specific for either Phospho-ERK1/2, Phospho-Akt, Phos-pho-P38–MAPK, ERK1/2, or P38–MAPK, cleaved caspase 3 andcytochrome c (Cell Signaling) in TBST containing 1% w/v skim milk(skim milk–TBST buffer) overnight at 4 °C. Following that, mem-branes were incubated with 1:1000 dilution of anti-rabbit polyclonalantibodies conjugated to horseradish peroxidase in skim milk–TBSTbuffer for 1 h at room temperature. β-tubulin was used as loadingcontrol and detected by incubating membranes with 1:1000 dilutionof mouse monoclonal anti-β-tubulin antibodies in skim milk–TBSTbuffer, followed by 1:1000 dilution of anti-mouse polyclonalantibodies conjugated to horseradish peroxidase (HRP) in skimmilk–TBST buffer, both antibody incubations 1 h each at roomtemperature. Membranes were washed in TBST 3 times after antibodyincubation. Proteins were revealed using SuperSignal Chemilumines-cence Substrate system (Pierce Biotechnology Inc., IL, USA), visualisedby exposure to X-ray film and protein bands quantitated using NIHImageJ densitometry analysis.
2.4. Statistical methods
Force was expressed in grams, as mean±S.E.M. Concentration–response (CR) curves were constructed from themaximum total forcegenerated in response to drug addition. CR curves were fitted usingnon-linear regression analysis (Prism 5 Graphpad software 2008, Inc).
60 P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66
Author's personal copy
The EC50 was estimated as the drug concentration producing 50%maximal response. Differences in maximal contractile force andresponse range were tested by one-way analysis of variance withDunnetts post-hoc analysis. Assay experiments were performed using6 trabeculae for each group per time interval. During hypoxia–reoxygenation, differences between the pretreatment and timecontrol groups were tested by repeated-measures analysis of varianceusing the Greenhouse–Geisser correction (Ludbrook, 1994). Analysiswas performed on the raw data using SPSS version 15 (SPSS Inc.,Illinois, USA). Pb0.05 was considered statistically significant.
2.5. Drugs
Levosimendan was supplied by Abbott Australia Pty. Ltd. in itsproprietary form Simdax 2.5 g/ml. 5-HD (0.1 M) was prepared inKrebs solution. (5-Hydroxydecanoic acid, sodium salt, was suppliedby Sigma-Aldrich, St. Louis, MO, USA).
3. Results
3.1. Contractile response following hypoxia
The baseline contraction forcewas similar in all groups ranging from0.35±0.07 to 0.46±0.08 g (PN0.05). Trabeculae pretreated withlevosimendan 0.3 μM or with 5-HD 800 μM alone had no direct effecton baseline contractile force. A 60 min period of hypoxia significantlyreduced the contractile force in all tissues (Fig. 2) to 15±3% (Hypoxiaalone), 21±4% (Levo+Hypoxia), 23±5% (Levo+5-HD+Hypoxia)and 29±5% (5-HD+Hypoxia) of baseline respective contractile force(Pb0.01). Trabeculae not subjected to hypoxia (Normoxia) had agradual decline in contractile force to 61±10% of baseline, beforestimulation with isoproterenol.
3.2. Effect of reoxygenation
Upon reoxygenation, contractile force increased significantly inall groups. Trabeculae pretreated with levosimendan had a signif-icantly greater contractile response on reoxygenation (0.25±0.06 g) increasing to 59±12% baseline, compared to the Hypoxiagroup (0.10±0.02 g) and Levo+5-HD+Hypoxia group (0.11±0.03 g; Pb0.05 one-way ANOVA). Levosimendan pretreated tissuealso tended to have an increased contractile force compared to the5-HD+Hypoxia group (0.13±0.02 g), but the difference did notreach significance.
3.3. Response to isoproterenol
The maximal contractile response to isoproterenol was signifi-cantly decreased in trabeculae subjected to hypoxia (Fig. 2, Table 1),compared to the response observed in normoxic trabeculae(Normoxia 0.88±0.02 g versus hypoxia alone 0.60±0.01 g,Pb0.05). There was no significant difference in the sensitivity toisoproterenol in both these groups; pEC50 7.49±0.22 and 7.39±0.34, respectively. Post-hypoxic trabeculae pretreated with levosi-mendan responded to isoproterenol with a maximum contractionforce and contractile range similar to normoxic tissue withoutlevosimendan (Levo+Hypoxia 0.78±0.04 g, Pb0.05); this was alsosignificantly greater than the maximum contractile force seen withthe hypoxia alone group (Pb0.05). This increased contractileresponse with levosimendan pretreatment was not observedwhen 5-HD was equilibrated with levosimendan (Levo+5-HD0.54±0.09 g compared to Levo+Hypoxia, Pb0.01).
Fig. 1. A. Hypoxia protocol: following harvest, human right atrial trabeculae wereelectrically paced and equilibrated with isoprenaline 1 μM (ISO) for viability. Followingrewash and baseline stabilisation (baseline), trabeculae were pretreated withlevosimendan (0.3 μM), 5-hydroxydecanoic acid (800 μM) or both for 15 min, thensubjected to hypoxia 60 min, followed by reoxygenation for 15 min before equilibrationwith isoproterenol 1–32,000 nM. B. Measurement of protein expression in trabeculaesubjected to hypoxia–reoxygenation. Levosimendan 0.3 μM pretreatment 15 minbefore hypoxia. 0, 30, 60, 120 min protein assay measurements.
Fig. 2. Effect of levosimendan 0.3 μM on human right atrial trabeculae contractile forcewith isoproterenol stimulation following hypoxia–reoxygenation. **Effect of hypoxiaon baseline contraction force, decline in normoxic tissue contractile force from baselineat 90 min (Pb0.01 Students t test). *Contractile force of levosimendan-pretreated tissuecompared to other atria on reoxygenation (Pb0.05, one way ANOVA). †Maximalcontractile force (Emax) Normoxia time control and Levo+Hypoxia group compared toHypoxia alone and Levo+5-HD (5-hydroxydecanoate 800 μM)+Hypoxia groups, one-way ANOVA. Error bars are±1 S.E.M. on Baseline, P, H and av S.E.M. from rm ANOVA oneach curve. Baseline following harvesting and stabilisation, P pretreatment, H followinghypoxia, and R 15 min after reoxygenation.
61P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66
Author's personal copy
3.4. Activation of pERK 1/2
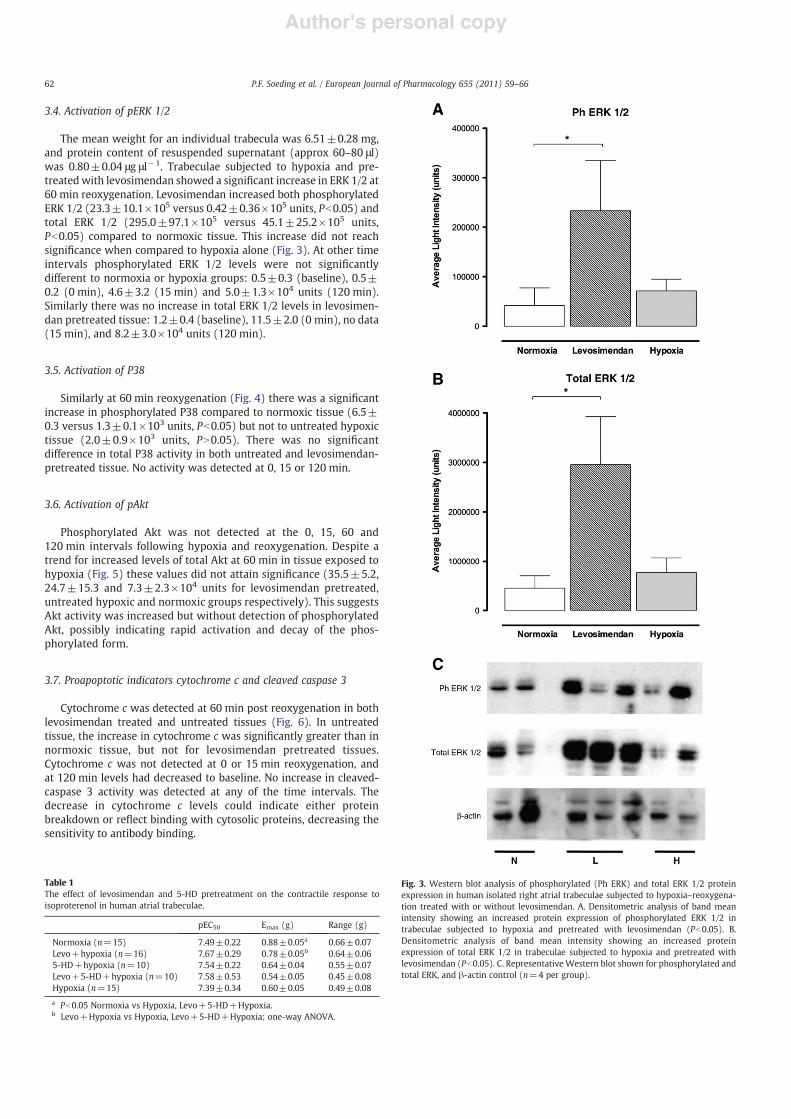
The mean weight for an individual trabecula was 6.51±0.28 mg,and protein content of resuspended supernatant (approx 60–80 μl)was 0.80±0.04 μg μl−1. Trabeculae subjected to hypoxia and pre-treated with levosimendan showed a significant increase in ERK 1/2 at60 min reoxygenation. Levosimendan increased both phosphorylatedERK 1/2 (23.3±10.1×105 versus 0.42±0.36×105 units, Pb0.05) andtotal ERK 1/2 (295.0±97.1×105 versus 45.1±25.2×105 units,Pb0.05) compared to normoxic tissue. This increase did not reachsignificance when compared to hypoxia alone (Fig. 3). At other timeintervals phosphorylated ERK 1/2 levels were not significantlydifferent to normoxia or hypoxia groups: 0.5±0.3 (baseline), 0.5±0.2 (0 min), 4.6±3.2 (15 min) and 5.0±1.3×104 units (120 min).Similarly there was no increase in total ERK 1/2 levels in levosimen-dan pretreated tissue: 1.2±0.4 (baseline), 11.5±2.0 (0 min), no data(15 min), and 8.2±3.0×104 units (120 min).
3.5. Activation of P38
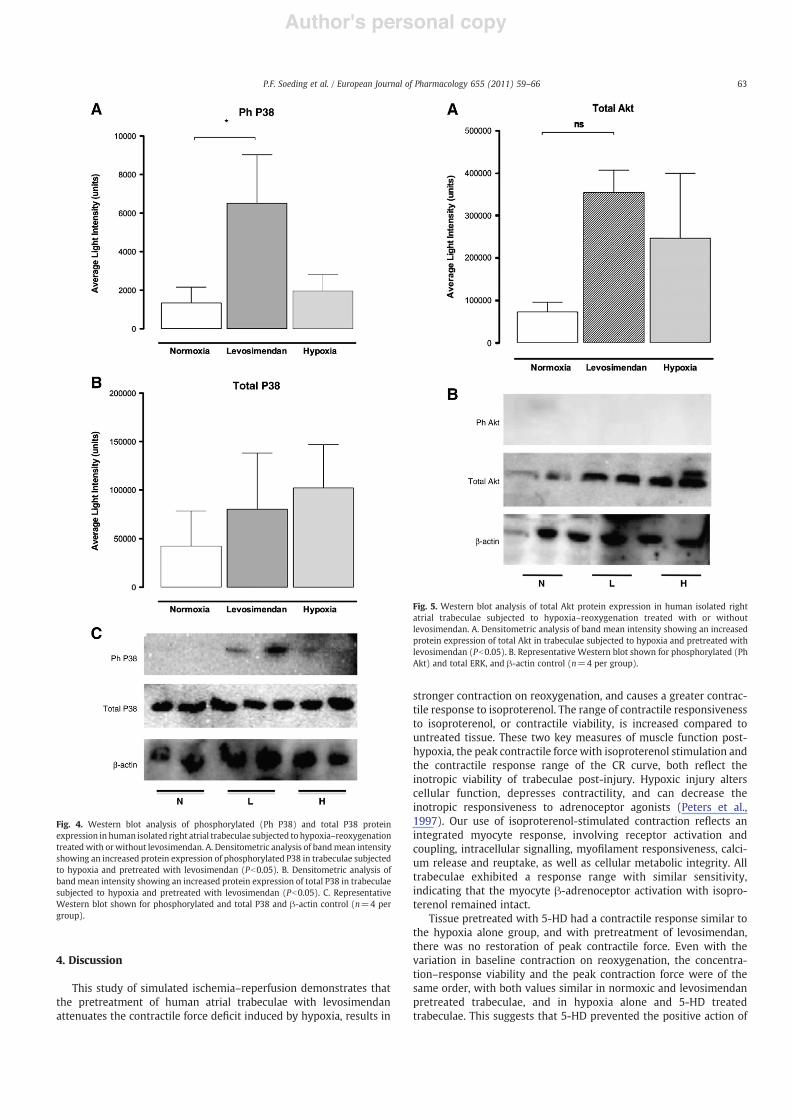
Similarly at 60 min reoxygenation (Fig. 4) there was a significantincrease in phosphorylated P38 compared to normoxic tissue (6.5±0.3 versus 1.3±0.1×103 units, Pb0.05) but not to untreated hypoxictissue (2.0±0.9×103 units, PN0.05). There was no significantdifference in total P38 activity in both untreated and levosimendan-pretreated tissue. No activity was detected at 0, 15 or 120 min.
3.6. Activation of pAkt
Phosphorylated Akt was not detected at the 0, 15, 60 and120 min intervals following hypoxia and reoxygenation. Despite atrend for increased levels of total Akt at 60 min in tissue exposed tohypoxia (Fig. 5) these values did not attain significance (35.5±5.2,24.7±15.3 and 7.3±2.3×104 units for levosimendan pretreated,untreated hypoxic and normoxic groups respectively). This suggestsAkt activity was increased but without detection of phosphorylatedAkt, possibly indicating rapid activation and decay of the phos-phorylated form.
3.7. Proapoptotic indicators cytochrome c and cleaved caspase 3
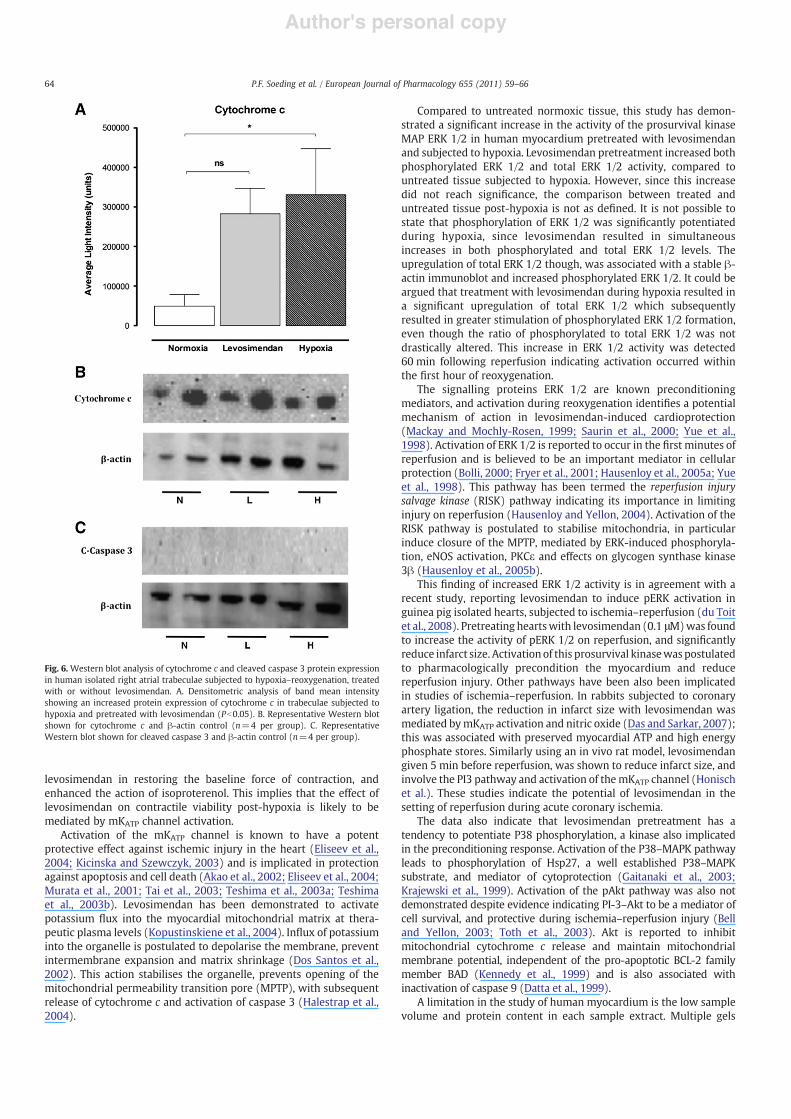
Cytochrome c was detected at 60 min post reoxygenation in bothlevosimendan treated and untreated tissues (Fig. 6). In untreatedtissue, the increase in cytochrome c was significantly greater than innormoxic tissue, but not for levosimendan pretreated tissues.Cytochrome c was not detected at 0 or 15 min reoxygenation, andat 120 min levels had decreased to baseline. No increase in cleaved-caspase 3 activity was detected at any of the time intervals. Thedecrease in cytochrome c levels could indicate either proteinbreakdown or reflect binding with cytosolic proteins, decreasing thesensitivity to antibody binding.
Table 1The effect of levosimendan and 5-HD pretreatment on the contractile response toisoproterenol in human atrial trabeculae.
pEC50 Emax (g) Range (g)
Normoxia (n=15) 7.49±0.22 0.88±0.05a 0.66±0.07Levo+hypoxia (n=16) 7.67±0.29 0.78±0.05b 0.64±0.065-HD+hypoxia (n=10) 7.54±0.22 0.64±0.04 0.55±0.07Levo+5-HD+hypoxia (n=10) 7.58±0.53 0.54±0.05 0.45±0.08Hypoxia (n=15) 7.39±0.34 0.60±0.05 0.49±0.08a Pb0.05 Normoxia vs Hypoxia, Levo+5-HD+Hypoxia.b Levo+Hypoxia vs Hypoxia, Levo+5-HD+Hypoxia; one-way ANOVA.
Fig. 3. Western blot analysis of phosphorylated (Ph ERK) and total ERK 1/2 proteinexpression in human isolated right atrial trabeculae subjected to hypoxia–reoxygena-tion treated with or without levosimendan. A. Densitometric analysis of band meanintensity showing an increased protein expression of phosphorylated ERK 1/2 intrabeculae subjected to hypoxia and pretreated with levosimendan (Pb0.05). B.Densitometric analysis of band mean intensity showing an increased proteinexpression of total ERK 1/2 in trabeculae subjected to hypoxia and pretreated withlevosimendan (Pb0.05). C. Representative Western blot shown for phosphorylated andtotal ERK, and β-actin control (n=4 per group).
62 P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66
Author's personal copy
4. Discussion
This study of simulated ischemia–reperfusion demonstrates thatthe pretreatment of human atrial trabeculae with levosimendanattenuates the contractile force deficit induced by hypoxia, results in
stronger contraction on reoxygenation, and causes a greater contrac-tile response to isoproterenol. The range of contractile responsivenessto isoproterenol, or contractile viability, is increased compared tountreated tissue. These two key measures of muscle function post-hypoxia, the peak contractile force with isoproterenol stimulation andthe contractile response range of the CR curve, both reflect theinotropic viability of trabeculae post-injury. Hypoxic injury alterscellular function, depresses contractility, and can decrease theinotropic responsiveness to adrenoceptor agonists (Peters et al.,1997). Our use of isoproterenol-stimulated contraction reflects anintegrated myocyte response, involving receptor activation andcoupling, intracellular signalling, myofilament responsiveness, calci-um release and reuptake, as well as cellular metabolic integrity. Alltrabeculae exhibited a response range with similar sensitivity,indicating that the myocyte β-adrenoceptor activation with isopro-terenol remained intact.
Tissue pretreated with 5-HD had a contractile response similar tothe hypoxia alone group, and with pretreatment of levosimendan,there was no restoration of peak contractile force. Even with thevariation in baseline contraction on reoxygenation, the concentra-tion–response viability and the peak contraction force were of thesame order, with both values similar in normoxic and levosimendanpretreated trabeculae, and in hypoxia alone and 5-HD treatedtrabeculae. This suggests that 5-HD prevented the positive action of
Fig. 4. Western blot analysis of phosphorylated (Ph P38) and total P38 proteinexpression in human isolated right atrial trabeculae subjected to hypoxia–reoxygenationtreated with or without levosimendan. A. Densitometric analysis of bandmean intensityshowing an increased protein expression of phosphorylated P38 in trabeculae subjectedto hypoxia and pretreated with levosimendan (Pb0.05). B. Densitometric analysis ofband mean intensity showing an increased protein expression of total P38 in trabeculaesubjected to hypoxia and pretreated with levosimendan (Pb0.05). C. RepresentativeWestern blot shown for phosphorylated and total P38 and β-actin control (n=4 pergroup).
Fig. 5. Western blot analysis of total Akt protein expression in human isolated rightatrial trabeculae subjected to hypoxia–reoxygenation treated with or withoutlevosimendan. A. Densitometric analysis of band mean intensity showing an increasedprotein expression of total Akt in trabeculae subjected to hypoxia and pretreated withlevosimendan (Pb0.05). B. Representative Western blot shown for phosphorylated (PhAkt) and total ERK, and β-actin control (n=4 per group).
63P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66
Author's personal copy
levosimendan in restoring the baseline force of contraction, andenhanced the action of isoproterenol. This implies that the effect oflevosimendan on contractile viability post-hypoxia is likely to bemediated by mKATP channel activation.
Activation of the mKATP channel is known to have a potentprotective effect against ischemic injury in the heart (Eliseev et al.,2004; Kicinska and Szewczyk, 2003) and is implicated in protectionagainst apoptosis and cell death (Akao et al., 2002; Eliseev et al., 2004;Murata et al., 2001; Tai et al., 2003; Teshima et al., 2003a; Teshimaet al., 2003b). Levosimendan has been demonstrated to activatepotassium flux into the myocardial mitochondrial matrix at thera-peutic plasma levels (Kopustinskiene et al., 2004). Influx of potassiuminto the organelle is postulated to depolarise the membrane, preventintermembrane expansion and matrix shrinkage (Dos Santos et al.,2002). This action stabilises the organelle, prevents opening of themitochondrial permeability transition pore (MPTP), with subsequentrelease of cytochrome c and activation of caspase 3 (Halestrap et al.,2004).
Compared to untreated normoxic tissue, this study has demon-strated a significant increase in the activity of the prosurvival kinaseMAP ERK 1/2 in human myocardium pretreated with levosimendanand subjected to hypoxia. Levosimendan pretreatment increased bothphosphorylated ERK 1/2 and total ERK 1/2 activity, compared tountreated tissue subjected to hypoxia. However, since this increasedid not reach significance, the comparison between treated anduntreated tissue post-hypoxia is not as defined. It is not possible tostate that phosphorylation of ERK 1/2 was significantly potentiatedduring hypoxia, since levosimendan resulted in simultaneousincreases in both phosphorylated and total ERK 1/2 levels. Theupregulation of total ERK 1/2 though, was associated with a stable β-actin immunoblot and increased phosphorylated ERK 1/2. It could beargued that treatment with levosimendan during hypoxia resulted ina significant upregulation of total ERK 1/2 which subsequentlyresulted in greater stimulation of phosphorylated ERK 1/2 formation,even though the ratio of phosphorylated to total ERK 1/2 was notdrastically altered. This increase in ERK 1/2 activity was detected60 min following reperfusion indicating activation occurred withinthe first hour of reoxygenation.
The signalling proteins ERK 1/2 are known preconditioningmediators, and activation during reoxygenation identifies a potentialmechanism of action in levosimendan-induced cardioprotection(Mackay and Mochly-Rosen, 1999; Saurin et al., 2000; Yue et al.,1998). Activation of ERK 1/2 is reported to occur in the first minutes ofreperfusion and is believed to be an important mediator in cellularprotection (Bolli, 2000; Fryer et al., 2001; Hausenloy et al., 2005a; Yueet al., 1998). This pathway has been termed the reperfusion injurysalvage kinase (RISK) pathway indicating its importance in limitinginjury on reperfusion (Hausenloy and Yellon, 2004). Activation of theRISK pathway is postulated to stabilise mitochondria, in particularinduce closure of the MPTP, mediated by ERK-induced phosphoryla-tion, eNOS activation, PKCε and effects on glycogen synthase kinase3β (Hausenloy et al., 2005b).
This finding of increased ERK 1/2 activity is in agreement with arecent study, reporting levosimendan to induce pERK activation inguinea pig isolated hearts, subjected to ischemia–reperfusion (du Toitet al., 2008). Pretreating heartswith levosimendan (0.1 μM)was foundto increase the activity of pERK 1/2 on reperfusion, and significantlyreduce infarct size. Activation of this prosurvival kinasewas postulatedto pharmacologically precondition the myocardium and reducereperfusion injury. Other pathways have been also been implicatedin studies of ischemia–reperfusion. In rabbits subjected to coronaryartery ligation, the reduction in infarct size with levosimendan wasmediated bymKATP activation and nitric oxide (Das and Sarkar, 2007);this was associated with preserved myocardial ATP and high energyphosphate stores. Similarly using an in vivo rat model, levosimendangiven 5 min before reperfusion, was shown to reduce infarct size, andinvolve the PI3 pathway and activation of themKATP channel (Honischet al.). These studies indicate the potential of levosimendan in thesetting of reperfusion during acute coronary ischemia.
The data also indicate that levosimendan pretreatment has atendency to potentiate P38 phosphorylation, a kinase also implicatedin the preconditioning response. Activation of the P38–MAPK pathwayleads to phosphorylation of Hsp27, a well established P38–MAPKsubstrate, and mediator of cytoprotection (Gaitanaki et al., 2003;Krajewski et al., 1999). Activation of the pAkt pathway was also notdemonstrated despite evidence indicating PI-3–Akt to be a mediator ofcell survival, and protective during ischemia–reperfusion injury (Belland Yellon, 2003; Toth et al., 2003). Akt is reported to inhibitmitochondrial cytochrome c release and maintain mitochondrialmembrane potential, independent of the pro-apoptotic BCL-2 familymember BAD (Kennedy et al., 1999) and is also associated withinactivation of caspase 9 (Datta et al., 1999).
A limitation in the study of human myocardium is the low samplevolume and protein content in each sample extract. Multiple gels
Fig. 6. Western blot analysis of cytochrome c and cleaved caspase 3 protein expressionin human isolated right atrial trabeculae subjected to hypoxia–reoxygenation, treatedwith or without levosimendan. A. Densitometric analysis of band mean intensityshowing an increased protein expression of cytochrome c in trabeculae subjected tohypoxia and pretreated with levosimendan (Pb0.05). B. Representative Western blotshown for cytochrome c and β-actin control (n=4 per group). C. RepresentativeWestern blot shown for cleaved caspase 3 and β-actin control (n=4 per group).
64 P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66

Author's personal copy
were plated for five antibody probes, with each well receiving only4–6 μg of protein sample. The efficacy of protein transfer with lowmass samples onto the membrane ultimately determines the successof immunodetection, and may explain failure to detect pAkt. For thisreason the measurement of total p38, Akt and ERK 1/2, as well asβ-actin were used as internal controls. The data suggest that total Akthowever, may be upregulated by hypoxia and further study couldexplore this.
Hypoxia resulted in increased cytochrome c levels, evident at60 min reoxygenation. Since cytochrome c was not elevated innormoxic tissue, the detection of an increase in hypoxic tissue reflectscell damage in both treated and untreated groups. Since these werenot significantly different, a limitation of injury with levosimendanpretreatment, on the basis of cytochrome c levels, could not bedemonstrated. The release of cytochrome c is associated with theinitiation of cell death primarily through the activation of caspase 3. Inthis study there was no detection of cleaved caspase 3, and the reasonfor this could be explained by a longer time frame of 6–8 h beingrequired for caspase activation.
This study indicates that levosimendan has the potential to up-regulate the RISK pathway, attenuate ischemic injury, and limitmyocardial infarction in patients undergoing cardiac surgery. Inpatients with acute coronary syndromes, levosimendanmay also limitmyocardial damage during the reperfusion phase of injury (Staatet al., 2005). During cardiac surgery, therapeutic inhibition ofapoptosis may protect against ischemia–reperfusion injury, improvemyocyte recovery and maintain contractile function.
5. Conclusions
Our findings have shown that in human tissue exposed to hypoxia,levosimendan preserves both the contractile responsiveness and peakcontraction force to isoproterenol. This action on contractile functionindicates that myocytes pretreated with levosimendan withstandhypoxia more favourably than untreated tissue, an effect mediated bymKATP channel activation and potential upregulation of the prosurvi-val pathway ERK 1/2.
Acknowledgements
We thank Mark Ross-Smith and Dr. Moses Zhang for theirassistance. Levosimendan was supplied by Abbott Australia.
References
Akao, M., Teshima, Y., Marban, E., 2002. Antiapoptotic effect of nicorandil mediated bymitochondrial ATP-sensitive potassium channels in cultured cardiac myocytes. J.Am. Coll. Cardiol. 40, 803–810.
Bell, R.M., Yellon, D.M., 2003. Bradykinin limits infarction when administered as anadjunct to reperfusion in mouse heart: the role of PI3K. Akt and eNOS. J. Mol. Cell.Cardiol. 35, 185–193.
Bolli, R., 2000. The late phase of preconditioning. Circ. Res. 87, 972–983.Bradford, M.M., 1976. A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem.72, 248–254.
Brookes, P.S., Yoon, Y., Robotham, J.L., Anders, M.W., Sheu, S.S., 2004. Calcium, ATP, andROS: a mitochondrial love–hate triangle. Am. J. Physiol. Cell Physiol. 287,C817–C833.
Das, B., Sarkar, C., 2007. Pharmacological preconditioning by levosimendan is mediatedby inducible nitric oxide synthase and mitochondrial KATP channel activation inthe in vivo anesthetized rabbit heart model. Vascul. Pharmacol. 47, 248–256.
Datta, S.R., Brunet, A., Greenberg, M.E., 1999. Cellular survival: a play in three Akts.Genes Dev. 13, 2905–2927.
De Hert, S.G., Lorsomradee, S., Cromheecke, S., Van der Linden, P.J., 2007. The effects oflevosimendan in cardiac surgery patients with poor left ventricular function.Anesth. Analg. 104, 766–773.
Dos Santos, P., Kowaltowski, A.J., Laclau, M.N., Seetharaman, S., Paucek, P., Boudina, S.,Thambo, J.B., Tariosse, L., Garlid, K.D., 2002. Mechanisms by which opening themitochondrial ATP-sensitive K(+) channel protects the ischemic heart. Am. J.Physiol. Heart Circ. Physiol. 283, H284–H295.
du Toit, E.F., Genis, A., Opie, L.H., Pollesello, P., Lochner, A., 2008. A role for the RISKpathway and K(ATP) channels in pre- and post-conditioning induced bylevosimendan in the isolated guinea pig heart. Br. J. Pharmacol. 154, 41–50.
Eliseev, R.A., Vanwinkle, B., Rosier, R.N., Gunter, T.E., 2004. Diazoxide-mediatedpreconditioning against apoptosis involves activation of cAMP-response element-binding protein (CREB) and NFkappaB. J. Biol. Chem. 279, 46748–46754.
Engelbrecht, A.M., Niesler, C., Page, C., Lochner, A., 2004. p38 and JNK have distinctregulatory functions on the development of apoptosis during simulatedischaemia and reperfusion in neonatal cardiomyocytes. Basic Res. Cardiol. 99,338–350.
Fryer, R.M., Pratt, P.F., Hsu, A.K., Gross, G.J., 2001. Differential activation of extracellularsignal regulated kinase isoforms in preconditioning and opioid-induced cardio-protection. J. Pharmacol. Exp. Ther. 296, 642–649.
Gaitanaki, C., Konstantina, S., Chrysa, S., Beis, I., 2003. Oxidative stress stimulatesmultiple MAPK signalling pathways and phosphorylation of the small HSP27 in theperfused amphibian heart. J. Exp. Biol. 206, 2759–2769.
Gottlieb, R.A., Burleson, K.O., Kloner, R.A., Babior, B.M., Engler, R.L., 1994. Reperfusioninjury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 94, 1621–1628.
Green, D.R., Reed, J.C., 1998. Mitochondria and apoptosis. Science 281, 1309–1312.Halestrap, A.P., Clarke, S.J., Javadov, S.A., 2004. Mitochondrial permeability transition
pore opening during myocardial reperfusion—a target for cardioprotection.Cardiovasc. Res. 61, 372–385.
Hanouz, J.L., Yvon, A., Massetti, M., Lepage, O., Babatasi, G., Khayat, A., Bricard, H.,Gerard, J.L., 2002. Mechanisms of desflurane-induced preconditioning in isolatedhuman right atria in vitro. Anesthesiology 97, 33–41.
Hausenloy, D.J., Yellon, D.M., 2004. New directions for protecting the heart againstischaemia–reperfusion injury: targeting the Reperfusion Injury Salvage Kinase(RISK)-pathway. Cardiovasc. Res. 61, 448–460.
Hausenloy, D., Wynne, A., Duchen, M., Yellon, D., 2004. Transient mitochondrialpermeability transition pore opening mediates preconditioning-induced protec-tion. Circulation 109, 1714–1717.
Hausenloy, D.J., Tsang, A., Mocanu, M.M., Yellon, D.M., 2005a. Ischemic preconditioningprotects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Heart Circ.Physiol. 288, H971–H976.
Hausenloy, D.J., Tsang, A., Yellon, D.M., 2005b. The reperfusion injury salvage kinasepathway: a common target for both ischemic preconditioning and postcondition-ing. Trends Cardiovasc. Med. 15, 69–75.
Honisch, A., Theuring, N., Ebner, B., Wagner, C., Strasser, R.H., Weinbrenner, C., 2010.Postconditioning with levosimendan reduces the infarct size involving the PI3Kpathway and KATP-channel activation but is independent of PDE-III inhibition.Basic Res. Cardiol. 105, 155–167.
Kennedy, S.G., Kandel, E.S., Cross, T.K., Hay, N., 1999. Akt/Protein kinase B inhibits celldeath by preventing the release of cytochrome c frommitochondria. Mol. Cell. Biol.19, 5800–5810.
Kersten, J.R., Montgomery, M.W., Pagel, P.S., Warltier, D.C., 2000. Levosimendan, a newpositive inotropic drug, decreases myocardial infarct size via activation of K(ATP)channels. Anesth. Analg. 90, 5–11.
Kicinska, A., Szewczyk, A., 2003. Protective effects of the potassium channel opener-diazoxide against injury in neonatal rat ventricular myocytes. Gen. Physiol.Biophys. 22, 383–395.
Kloner, R.a.R.S., 2004. Cardiac protection during acute myocardial infarction: where dowe stand in 2004? J. Am. Coll. Cardiol. 44, 276–286.
Kopustinskiene, D.M., Pollesello, P., Saris, N.E., 2001. Levosimendan is a mitochondrial K(ATP) channel opener. Eur. J. Pharmacol. 428, 311–314.
Kopustinskiene, D.M., Pollesello, P., Saris, N.E., 2004. Potassium-specific effects oflevosimendan on heart mitochondria. Biochem. Pharmacol. 68, 807–812.
Krajewski, S., Krajewska,M., Ellerby, L.M.,Welsh, K., Xie, Z., Deveraux, Q.L., Salvesen, G.S.,Bredesen, D.E., Rosenthal, R.E., Fiskum, G., Reed, J.C., 1999. Release of caspase-9 frommitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl Acad. Sci.USA 96, 5752–5757.
Ludbrook, J., 1994. Repeated measurements and multiple comparisons in cardiovas-cular research. Cardiovasc. Res. 28, 303–311.
Ma, X.L., Kumar, S., Gao, F., Louden, C.S., Lopez, B.L., Christopher, T.A., Wang, C., Lee, J.C.,Feuerstein, G.Z., Yue, T.L., 1999. Inhibition of p38 mitogen-activated protein kinasedecreases cardiomyocyte apoptosis and improves cardiac function after myocardialischemia and reperfusion. Circulation 99, 1685–1691.
Mackay, K., Mochly-Rosen, D., 1999. An inhibitor of p38 mitogen-activated proteinkinase protects neonatal cardiac myocytes from ischemia. J. Biol. Chem. 274,6272–6279.
Maulik, N., Yoshida, T., Das, D.K., 1998. Oxidative stress developed during thereperfusion of ischemic myocardium induces apoptosis. Free Radic. Biol. Med. 24,869–875.
Murata, M., Akao, M., O'Rourke, B., Marban, E., 2001. Mitochondrial ATP-sensitivepotassium channels attenuate matrix Ca(2+) overload during simulated ischemiaand reperfusion: possible mechanism of cardioprotection. Circ. Res. 89, 891–898.
Peters, S.L., Pfaffendorf, M., van Zwieten, P.A., 1997. The influence of oxidative stress onvarious inotropic responses in isolated rat left atria. Naunyn-Schmiedeberg's Arch.Pharmacol. 355, 390–397.
Saurin, A.T., Martin, J.L., Heads, R.J., Foley, C., Mockridge, J.W., Wright, M.J., Wang, Y.,Marber, M.S., 2000. The role of differential activation of p38-mitogen-activatedprotein kinase in preconditioned ventricular myocytes. FASEB J. 14, 2237–2246.
Serone, A.P., Angus, J.A., 1999. Role of N-type calcium channels in autonomicneurotransmission in guinea-pig isolated left atria. Br. J. Pharmacol. 127, 927–934.
Staat, P., Rioufol, G., Piot, C., Cottin, Y., Cung, T.T., L'Huillier, I., Aupetit, J.F., Bonnefoy, E.,Finet, G., Andre-Fouet, X., Ovize, M., 2005. Postconditioning the human heart.Circulation 112, 2143–2148.
65P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66

Author's personal copy
Stephanou, A., Brar, B., Liao, Z., Scarabelli, T., Knight, R.A., Latchman, D.S., 2001. Distinctinitiator caspases are required for the induction of apoptosis in cardiac myocytesduring ischaemia versus reperfusion injury. Cell Death Differ. 8, 434–435.
Tai, K.K., McCrossan, Z.A., Abbott, G.W., 2003. Activation of mitochondrial ATP-sensitivepotassium channels increases cell viability against rotenone-induced cell death. J.Neurochem. 84, 1193–1200.
Taylor, J.M., Ali, U., Iannello, R.C., Hertzog, P., Crack, P.J., 2005. Diminished Aktphosphorylation in neurons lacking glutathione peroxidase-1 (Gpx1) leads toincreased susceptibility to oxidative stress-induced cell death. J. Neurochem. 92,283–293.
Teshima, Y., Akao, M., Baumgartner, W.A., Marban, E., 2003a. Nicorandil preventsoxidative stress-induced apoptosis in neurons by activating mitochondrial ATP-sensitive potassium channels. Brain Res. 990, 45–50.
Teshima, Y., Akao, M., Li, R.A., Chong, T.H., Baumgartner, W.A., Johnston, M.V., Marban, E.,2003b. Mitochondrial ATP-sensitive potassium channel activation protects cerebellargranule neurons from apoptosis induced by oxidative stress. Stroke 34, 1796–1802.
Toth, A., Halmosi, R., Kovacs, K., Deres, P., Kalai, T., Hideg, K., Toth, K., Sumegi, B., 2003.Akt activation induced by an antioxidant compound during ischemia–reperfusion.Free Radic. Biol. Med. 35, 1051–1063.
Tritapepe, L., De Santis, V., Vitale, D., Santulli, M., Morelli, A., Nofroni, I., Puddu, P.E.,Singer, M., Pietropaoli, P., 2006. Preconditioning effects of levosimendan incoronary artery bypass grafting—a pilot study. Br. J. Anaesth. 96, 694–700.
Wang, S., Cone, J., Liu, Y., 2001. Dual roles of mitochondrial K(ATP) channels indiazoxide-mediated protection in isolated rabbit hearts. Am. J. Physiol. Heart Circ.Physiol. 280, H246–H255.
Yue, T.L., Ma, X.L., Gu, J.L., Ruffolo Jr., R.R., Feuerstein, G.Z., 1998. Carvedilol inhibitsactivation of stress-activated protein kinase and reduces reperfusion injury inperfused rabbit heart. Eur. J. Pharmacol. 345, 61–65.
Yvon, A., Hanouz, J.L., Haelewyn, B., Terrien, X., Massetti, M., Babatasi, G., Khayat, A.,Ducouret, P., Bricard, H., Gerard, J.L., 2003. Mechanisms of sevoflurane-inducedmyocardial preconditioning in isolated human right atria in vitro. Anesthesiology99, 27–33.
66 P.F. Soeding et al. / European Journal of Pharmacology 655 (2011) 59–66
Related Documents











