Large-Scale Phylogenetic Large-Scale Phylogenetic Analysis Analysis Tandy Warnow Associate Professor Department of Computer Sciences Graduate Program in Evolution and Ecology Co-Director The Center for Computational Biology and Bioinformatics The University of Texas at Austin

Large-Scale Phylogenetic Analysis Tandy Warnow Associate Professor Department of Computer Sciences Graduate Program in Evolution and Ecology Co-Director.
Dec 17, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Large-Scale Phylogenetic Large-Scale Phylogenetic AnalysisAnalysis
Tandy WarnowAssociate Professor
Department of Computer SciencesGraduate Program in Evolution and Ecology
Co-DirectorThe Center for Computational Biology and Bioinformatics
The University of Texas at Austin

Outline of TalkOutline of Talk
• Phylogenetic reconstruction from DNA sequences – the problems, and the progress
• Phylogenetic reconstruction from gene order and content in whole genomes – initial work
• The future of large-scale phylogeny, and the possibilities of inferring the “Tree of Life”
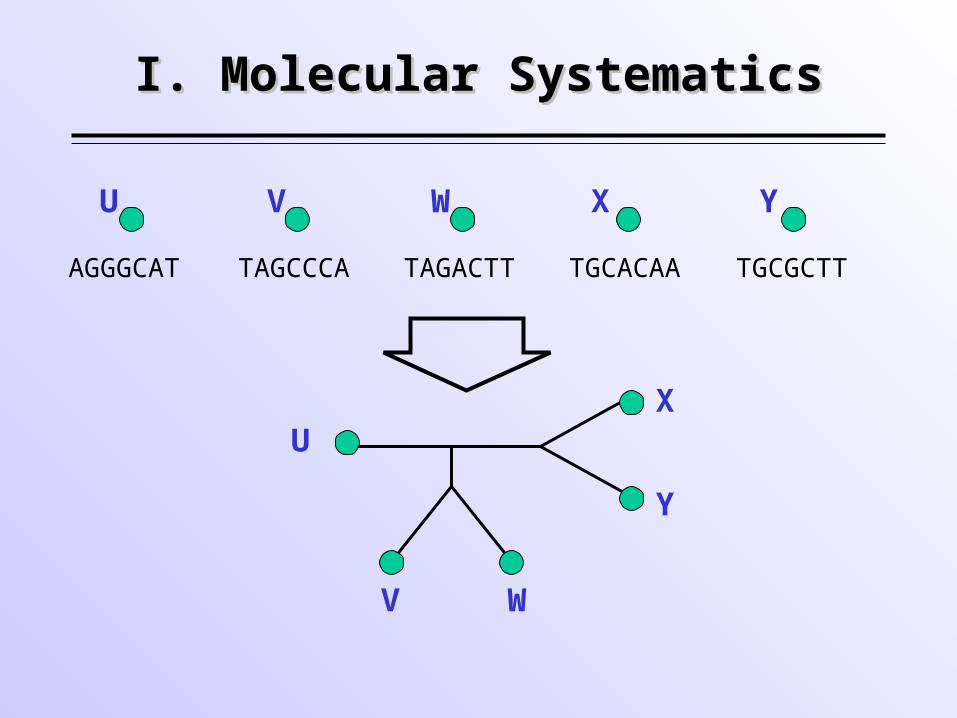
I. Molecular SystematicsI. Molecular Systematics
TAGCCCA TAGACTT TGCACAA TGCGCTTAGGGCAT
U V W X Y
U
V W
X
Y

DNA Sequence EvolutionDNA Sequence Evolution
AAGACTT
TGGACTTAAGGCCT
-3 mil yrs
-2 mil yrs
-1 mil yrs
today
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
TAGCCCA TAGACTT AGCGCTTAGCACAAAGGGCAT
AGGGCAT TAGCCCT AGCACTT
AAGACTT
TGGACTTAAGGCCT
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT

Major Phylogenetic Reconstruction Major Phylogenetic Reconstruction MethodsMethods
• Polynomial-time distance-based methods (neighbor joining the most popular)
• NP-hard sequence-based methods– Maximum Parsimony– Maximum Likelihood
• Heated debates over the relative performance of these methods
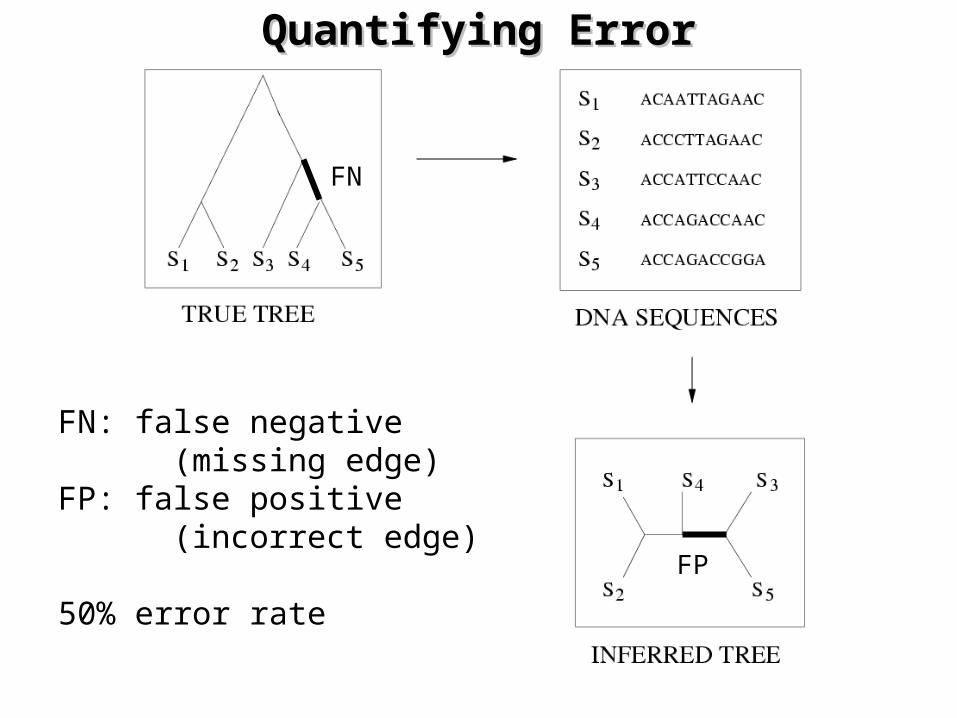
Quantifying ErrorQuantifying Error
FN: false negative (missing edge)FP: false positive (incorrect edge)
50% error rate
FN
FP

Main Result: DCM-Boosting and Main Result: DCM-Boosting and DCMDCMNJNJ+ML +ML
We have developed the first polynomial time methods that improveupon NJ (with respectto topological accuracy)and are never worsethan NJ.
The method is obtainedthrough DCM-boosting.

Basis of Distance-Based Methods: Basis of Distance-Based Methods: AdditivityAdditivity
• A distance matrix is additive if there exists a tree and such that .
• Waterman et al. (1977) showed that:
D),( EVT RE:
ijPeij eD )(
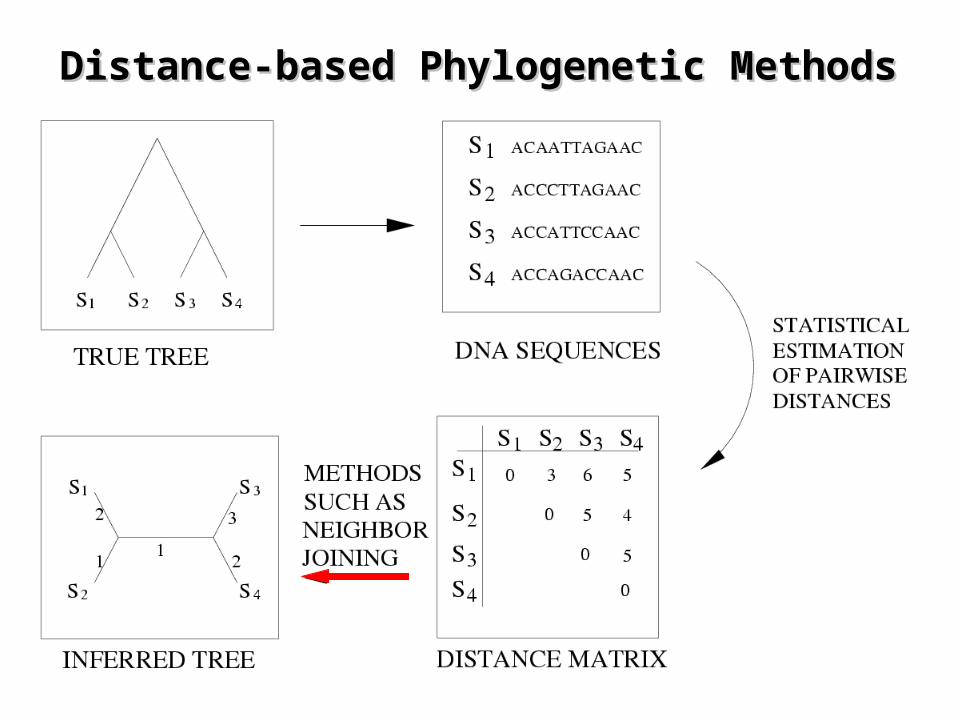
Distance-based Phylogenetic Distance-based Phylogenetic MethodsMethods

Statistical ConsistencyStatistical Consistency
Atteson (1990) showed that if is small enough.
TdNJ )( ),( dL
Sequence length
Hence NJ is statistically consistent for many modelsof evolution.
But what about performance on finite sequence lengths?

We focus on performance on finite We focus on performance on finite sequence lengthssequence lengths

Absolute fast convergence vs. Absolute fast convergence vs. exponential convergenceexponential convergence

General Markov (GM) ModelGeneral Markov (GM) Model
• A GM model tree is a pair where
– is a rooted binary tree.
– , and is a
stochastic substitution matrix with
.– The sequence at the root of is drawn
from a uniform distribution.– the rates of evolution across the sites can
be drawn from a fixed distribution• GM contains models like Jukes-Cantor (JC) and
Kimura 2-Parameter (K2P) models.
)( MT,
)}(:)({ TEeeM M
T
)(eM
1,0))(det( eM
T

Absolute Fast ConvergenceAbsolute Fast Convergence
• Let . Define . We parameterize the GM model:
• A phylogenetic reconstruction method is absolute fast-converging (AFC) for the GM model if for all positive there is a polynomial such that for all on set of sequences of length at least generated on , we have
0, gf |)det(|log)( eMe
})(),(:),{(, gefTEeGMTGM gf M
,, gfp gfGMT ,),( M
S n )(npT
1])(Pr[ TS

Theoretical Comparison of Early AFC Theoretical Comparison of Early AFC Methods to NJMethods to NJ
• Theorem 1 [Warnow et al. 2001]DCMNJ+SQS is absolute fast converging for the GM model.
• Theorem 2 [Csűrös 2001]HGT+FP is absolute fast converging for the GM model.
• Theorem 3 [Atteson 1999]NJ is exponentially converging for the GM model (but is not known to be AFC).

DCM-Boosting DCM-Boosting [Warnow et al. 2001][Warnow et al. 2001]
• DCM+SQS is a two-phase procedure which reduces the sequence length requirement of methods.
DCM SQSExponentiallyconvergingmethod
Absolute fast convergingmethod
• DCMNJ+SQS is the result of DCM-boosting NJ.

Experimental Comparison of Early Experimental Comparison of Early AFC Methods to NJAFC Methods to NJ
•rbcL 500-taxon tree•Jukes-Cantor model•Avg. branch length = 0.264

Improving upon early AFC Improving upon early AFC methodsmethods
• These early AFC methods outperform NJ only on long enough sequences and on large enough trees with high enough rates of evolution.
• Hence we need new fast converging methods which improve upon NJ on more of the parameter space, and are never worse than NJ.
• We modify the second phase to improve the empirical performance, replacing SQS with ML (maximum likelihood) or MP (maximum parsimony).

DCMDCMNJNJ+ML vs. other methods on a +ML vs. other methods on a fixed treefixed tree
•500-taxon rbcL tree•K2P+ model (=2, =1)•Avg. branch length = 0.278•Typical performance
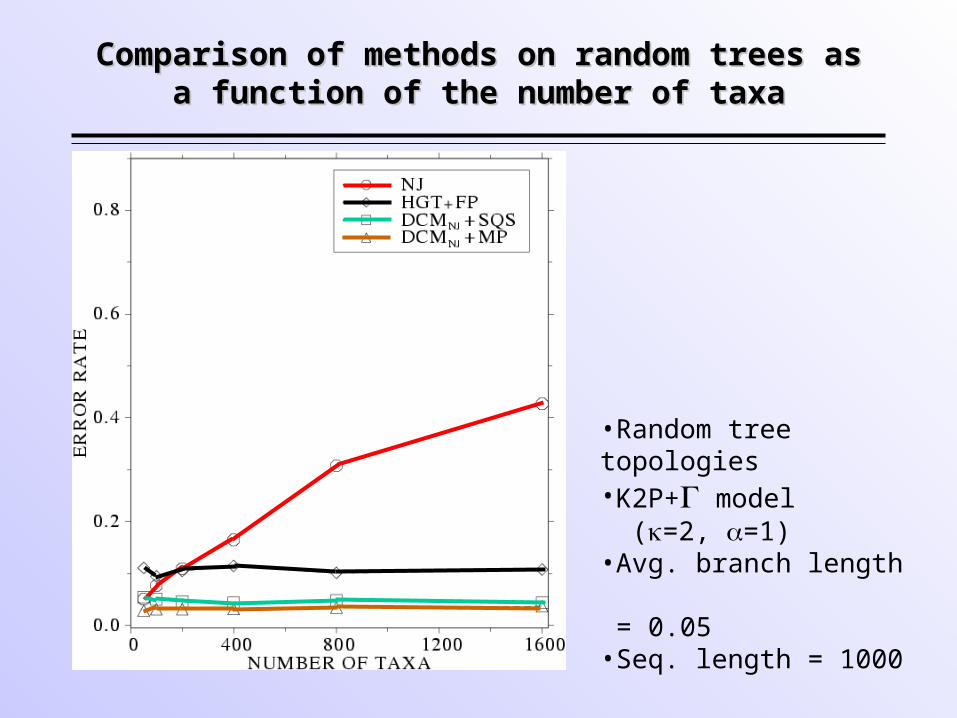
Comparison of methods on random trees as Comparison of methods on random trees as a function of the number of taxaa function of the number of taxa
•Random tree topologies•K2P+ model (=2, =1)•Avg. branch length = 0.05•Seq. length = 1000

SummarySummary
• These are the first polynomial time methods that improve upon NJ (with respect to topological accuracy) and are never worse than NJ.
• The advantage obtained with DCMNJ+MP and
DCMNJ+ML increases with number of taxa.
• In practice these new methods are slower than NJ (minutes vs. seconds), but still much faster than MP and ML (which can take days).
• Conjecture: DCMNJ+ML is AFC.

II. Whole-Genome PhylogenyII. Whole-Genome Phylogeny
A
B
C
D
E
F
X
Y
ZW
A
B
C
D
E
F
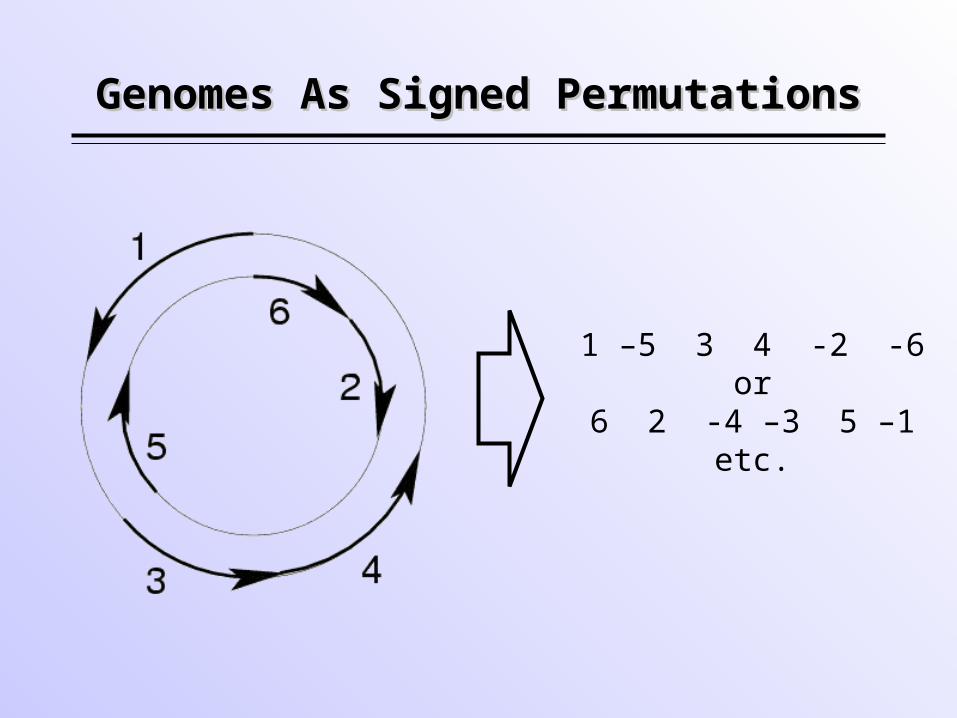
Genomes As Signed PermutationsGenomes As Signed Permutations
1 –5 3 4 -2 -6or
6 2 -4 –3 5 –1etc.

Genomes Evolve by RearrangementsGenomes Evolve by Rearrangements
• Inverted Transposition
1 2 3 9 -8 –7 –6 –5 –4 10
1 2 3 4 5 6 7 8 9 10
• Inversion (Reversal)
1 2 3 –8 –7 –6 –5 -4 9 10
• Transposition
1 2 3 9 4 5 6 7 8 10

Genome Rearrangement Has Genome Rearrangement Has A Huge State SpaceA Huge State Space
• DNA sequences : 4 states per site• Signed circular genomes with n genes:
states, 1 site
• Circular genomes (1 site)
– with 37 genes: states
– with 120 genes: states
)!1(2 1 nn
521056.2 2321070.3

Distance-based Phylogenetic Distance-based Phylogenetic Methods for GenomesMethods for Genomes

Genomic Distance EstimatorsGenomic Distance Estimators
• Standard: – Breakpoint distance– (Minimum) Inversion distance
• Our estimators: We attempt to estimate the actual number of events (the ``true
evolutionary distance”):– EDE [Moret et al, ISMB’01]
– Approx-IEBP [Wang and Warnow, STOC’01]
– Exact-IEBP [Wang, WABI’01]

Breakpoint DistanceBreakpoint Distance
• Breakpoint distance=5
1 2 3 4 5 6 7 8 9 10
1 –3 –2 4 5 9 6 7 8 10
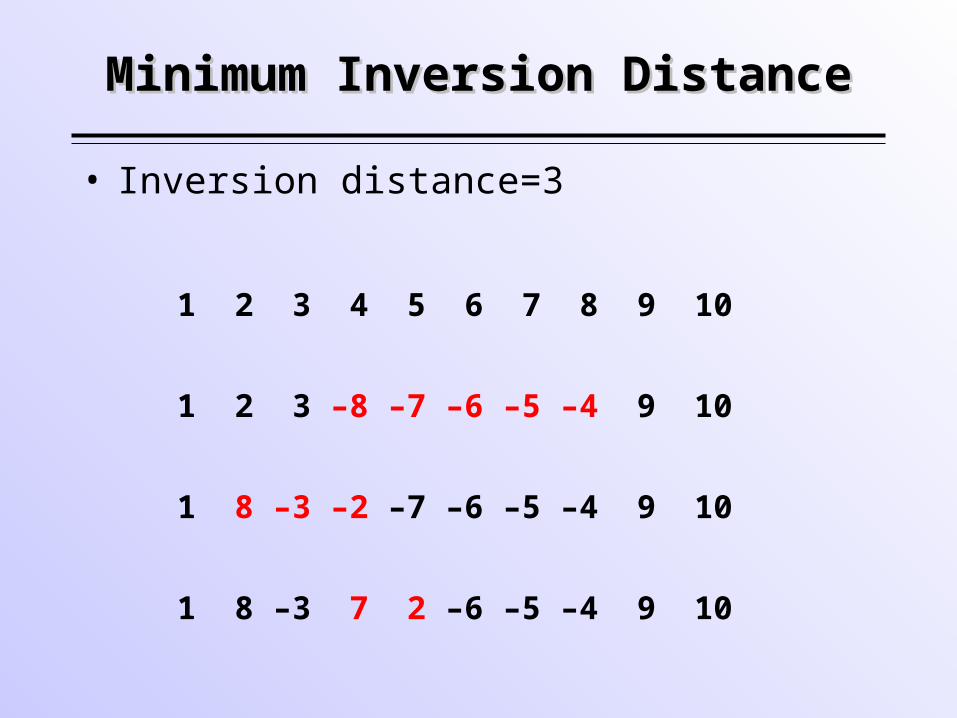
Minimum Inversion DistanceMinimum Inversion Distance
1 2 3 4 5 6 7 8 9 10
1 2 3 –8 –7 –6 –5 –4 9 10
1 8 –3 –2 –7 –6 –5 –4 9 10
1 8 –3 7 2 –6 –5 –4 9 10
• Inversion distance=3
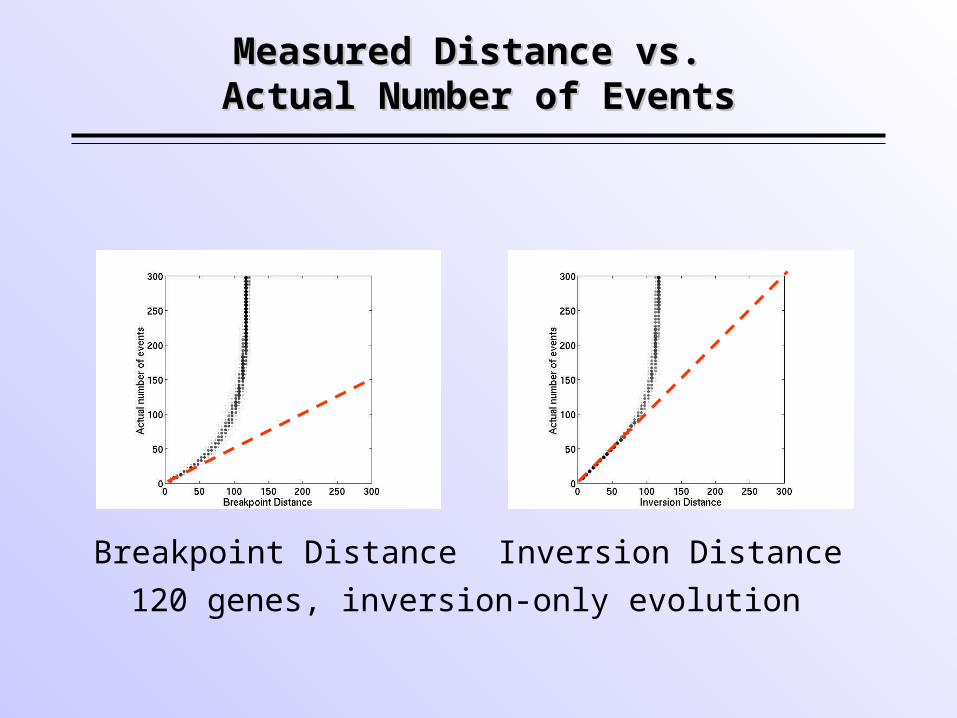
Measured Distance vs. Measured Distance vs. Actual Number of EventsActual Number of Events
Breakpoint Distance Inversion Distance
120 genes, inversion-only evolution

Generalized Nadeau-Taylor ModelGeneralized Nadeau-Taylor Model
• Three types of events: – Inversions – Transpositions– Inverted Transpositions
• Events of the same type are equiprobable• Probability of the three types have fixed
ratio: Inv : Trp : Inv.Trp = (1--)::

Estimating True Evolutionary Estimating True Evolutionary Distances for GenomesDistances for Genomes
Given fixed probabilities for each type of event, we estimate the expected breakpoint distance after k random events:
• Approx-IEBP [Wang, Warnow 2001]
– Polynomial-time closed-form approximation to the expected breakpoint distance
– Proven error bound• Exact-IEBP [Wang 2001]
– Exact, recursive solution for the expected breakpoint distance
– Polynomial-time but slower than Approx-IEBP

Estimating True Evolutionary Estimating True Evolutionary Distances for Genomes (cont.)Distances for Genomes (cont.)
Estimating the expected Inversion distance:
EDE [Moret, Wang, Warnow, Wyman 2001]
– Closed-form formula based upon an empirical estimation of the expected inversion distance after k random events (based upon 120 genes and inversion only, but robust to errors in the model) .
– Polynomial time, fastest of the three.
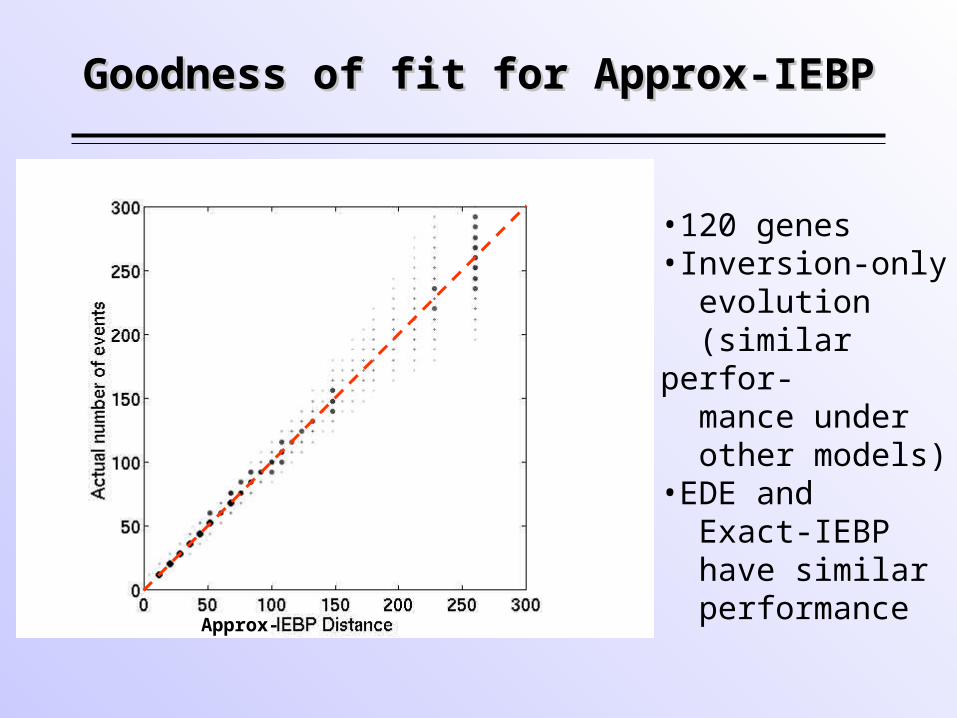
Goodness of fit for Approx-IEBPGoodness of fit for Approx-IEBP
•120 genes•Inversion-only evolution (similar perfor- mance under other models)•EDE and Exact-IEBP have similar performance
Approx-

Absolute DifferenceAbsolute Difference
•120 genes•Inversion only evolution (Similar relative performance under other models)
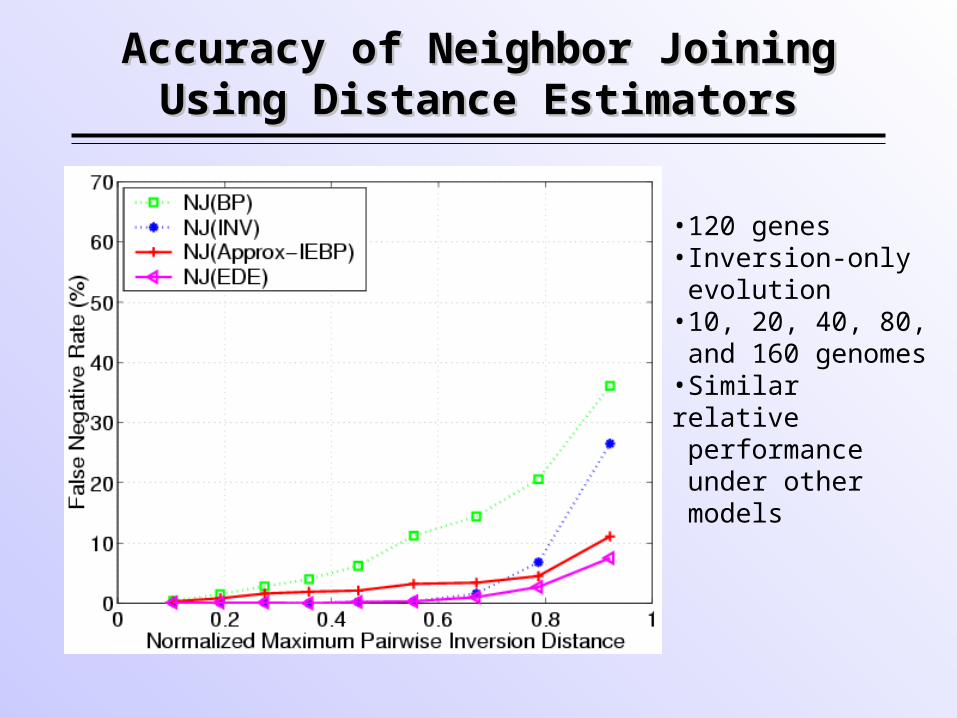
Accuracy of Neighbor Joining Accuracy of Neighbor Joining Using Distance EstimatorsUsing Distance Estimators
•120 genes•Inversion-only evolution •10, 20, 40, 80, and 160 genomes•Similar relative performance under other models
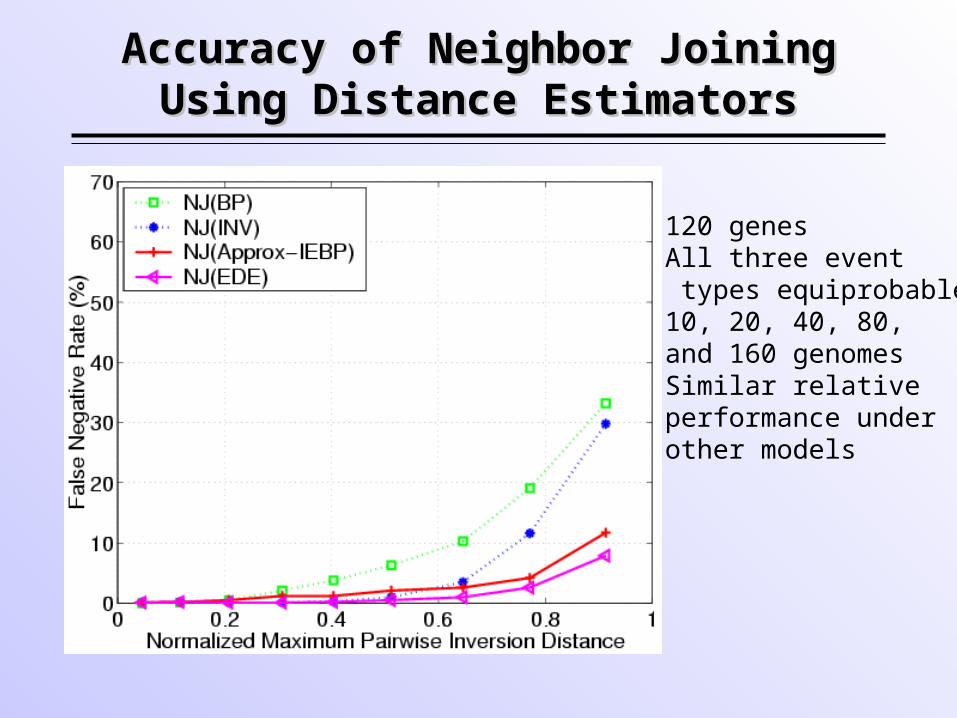
Accuracy of Neighbor Joining Accuracy of Neighbor Joining Using Distance EstimatorsUsing Distance Estimators
•120 genes•All three event types equiprobable•10, 20, 40, 80, and 160 genomes•Similar relative performance under other models

Summary of Genomic Summary of Genomic Distance EstimatorsDistance Estimators
• Statistically based estimation of genomic distances improves NJ analyses
• Our IEBP estimators assume knowledge of the probabilities of each type of event, but are robust to model violations
• NJ(EDE) outperforms NJ on other estimators, under all models studied
• Accuracy is very good, except when very close to saturation

Maximum Parsimony on Maximum Parsimony on Rearranged Genomes (MPRG)Rearranged Genomes (MPRG)
• The leaves are rearranged genomes.• Find the tree that minimizes the total number of rearrangement events
A
B
C
D
3 6
2
3
4
A
B
C
D
EF
Total length= 18

GRAPPA GRAPPA [Bader et al., PSB’01][Bader et al., PSB’01]
(Genome Rearrangements Analysis under Parsimony and other Phylogenetic Algorithms)
Reimplementation of BPAnalysis [Blanchette et al. 1997] for the Breakpoint Phylogeny problem.
• Uses algorithm engineering to improve performance.
• Improves the algorithm by reducing the number of tree length evaluations. (Evaluating the length of a fixed tree is NP-hard)

CampanulaceaeCampanulaceae

Analysis of Analysis of CampanulaceaeCampanulaceae
• 12 genomes + 1 outgroup (Tobacco)• 105 gene segments• BPAnalysis [Blanchette et al. 1997]
over 200 years [Cosner et al. 2000]
Using GRAPPA v1.1 on the 512-processor Los Lobos Supercluster machine:
2 minutes = 100 million-fold speedup(200,000-fold speedup per processor)

Consensus of 216 MP TreesConsensus of 216 MP Trees
Strict Consensus of 216 trees;6 out of 10 internal edges recovered.
Trachelium
Campanula
Adenophora
Symphandra
Legousia
Asyneuma
Triodanus
Wahlenbergia
Merciera
Codonopsis
Cyananthus
Platycodon
Tobacco

Future WorkFuture Work
• New focus on Rare Genomic Changes– New data– New models– New methods
• New techniques for large scale analyses– Divide-and-conquer methods– Non-tree models– Visualization of large trees and large sets of
trees

AcknowledgementsAcknowledgements
• Funding: The David and Lucile Packard Foundation, The National Science Foundation, and Paul Angello• Collaborators: Robert Jansen (U. Texas) Bernard Moret, David Bader, Mi-Yan
(U. New Mexico) Daniel Huson (Celera) Katherine St. John (CUNY) Linda Raubeson (Central Washington U.) Luay Nakhleh, Usman Roshan, Jerry Sun,
Li-San Wang, Stacia Wyman (Phylolab, U. Texas)
Phylolab, U. TexasPhylolab, U. Texas
Please visit us athttp://www.cs.utexas.edu/users/phylo/
Related Documents











