La carga mutacional del tumor es poligénica y está genéticamente asociada con rasgos y enfermedades complejas Xiwei Sun, Angli Xue, Ting Qi, Dan Chen, Dandan Shi, Yang Wu, Zhili Zheng, Jian Zeng y Jian Yang Cancer Resaerch. Marzo 2021. Volumen 81, Número 5 https://cancerres.aacrjournals.org/content/81/5/1230.long Resumen La carga mutacional tumoral (TMB) es un biomarcador emergente de respuesta a la inmunoterapia en tumores sólidos. Sin embargo, la medida en que la variación en TMB entre pacientes es atribuible a la variación genética de la línea germinal sigue siendo difícil de determinar. Aquí, utilizando 7004 pacientes no emparentados de ascendencia europea en 33 tipos de cáncer de The Cancer Genome Atlas, mostramos que la TMB pancancerosa es poligénica con aproximadamente el 13 % de su variación explicada por aproximadamente 1,1 millones de variantes comunes en total. Identificamos variantes de línea germinal que afectan a TMB en adenocarcinoma de estómago mediante la alteración de los niveles de expresión de BAG5 y KLC1. Análisis adicionales proporcionan evidencia de que la TMB está genéticamente asociada con rasgos y enfermedades complejos, como fumar, artritis reumatoide, altura y cánceres, y algunas de las asociaciones son probablemente causales. En general, estos resultados brindan nuevos conocimientos sobre la base genética de las mutaciones somáticas en los tumores y pueden informar los esfuerzos futuros para usar variantes genéticas para estratificar a los pacientes para la inmunoterapia. Importancia: este estudio proporciona evidencia de una arquitectura poligénica de la carga mutacional del tumor y abre una vía para el uso de variaciones genéticas de la línea germinal del genoma completo para estratificar a los pacientes con cáncer para la inmunoterapia. Introducción Las mutaciones somáticas, en particular aquellas que confieren una ventaja de crecimiento selectivo a las células, contribuyen al desarrollo del cáncer ( 1 ). Los patrones y funciones de las mutaciones somáticas en el genoma del cáncer humano se han analizado ampliamente en una amplia variedad de cánceres, lo que ha dado lugar a numerosos conocimientos biológicos ( 2– 4 ). En particular, se ha demostrado que un evento somático importante denominado carga mutacional tumoral (TMB), determinado por el número total de mutaciones somáticas no sinónimas en un genoma de cáncer, tiene una gran relevancia práctica. En particular, las líneas de evidencia sugirieron que TMB es un biomarcador predictivo para identificar a los pacientes con cáncer que tienen más probabilidades de responder a las terapias con inhibidores del punto de control inmunitario (ICI) ( 5– 9 ). Varios estudios han revelado la asociación de variantes genéticas de línea germinal con eventos somáticos en tumores, incluidas firmas mutacionales ( 10–12 ) y mutaciones somáticas en genes específicos relacionados con el cáncer ( 13, 14 ). Además, Zhu y sus colegas descubrieron una asociación negativa entre los alelos de riesgo de cáncer de línea germinal y la carga de mutaciones somáticas en el cáncer de mama ( 15 ). Robles-Espinoza y colegas encontraron que las variantes de MC1R de la línea germinal estaban asociadas con la carga de mutación somática del melanoma ( 16). A pesar de estos avances, nuestra comprensión de la genética de la línea germinal de TMB aún es muy limitada. Una pieza clave que falta es la aclaración de la arquitectura genética de TMB (p. ej., la contribución general de las variantes genéticas comunes a TMB y la distribución del tamaño del efecto de las variantes comunes) y las relaciones genéticas y causales de rasgos complejos (incluyendo enfermedades) con TMB. Responder a estas preguntas será fundamental para orientar los futuros estudios de asociación del genoma completo (GWAS) para la TMB, identificar los factores de riesgo de la TMB y evaluar la posible utilidad clínica de las características genéticas de la TMB en las terapias de ICI. Aquí, estudiamos la arquitectura genética de TMB a partir de un análisis pancanceroso de 7004 pacientes de ascendencia europea en 33 tipos de cáncer en The Cancer Genome Atlas (TCGA). Aunque es probable que existan diferencias en la arquitectura genética de TMB entre los tipos de cáncer, los tamaños de muestra en TCGA son insuficientes para los métodos que usan variantes comunes en todo el genoma para representar la arquitectura genética de TMB para tipos de cáncer individuales. Por lo tanto, enfocamos este estudio en la TMB pancancerosa al mismo tiempo que reconocemos que es probable que exista una heterogeneidad genética en la TMB entre los tipos de cáncer (consulte más adelante para obtener más información). Estimamos que aproximadamente el 13 % de la variación en la TMB pancancerosa puede explicarse por todas las variantes genéticas comunes juntas. Identificamos loci de línea germinal asociados con TMB en tipos de cáncer específicos, así como los genes causales putativos subyacentes que impulsan las asociaciones. Además, nuestros hallazgos muestran un componente genético compartido entre la TMB pancancerosa y rasgos complejos (incluidas enfermedades), como el tabaquismo, la altura, la artritis reumatoide y los cánceres. Investigamos más a fondo las relaciones causales entre la TMB y los rasgos complejos mediante análisis de aleatorización (MR) mendeliana, que proporcionan evidencia de los efectos causales putativos del tabaquismo en la TMB pancancerosa.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

La carga mutacional del tumor es poligénica y está genéticamente asociada con rasgos y enfermedades complejas
Xiwei Sun, Angli Xue, Ting Qi, Dan Chen, Dandan Shi, Yang Wu, Zhili Zheng, Jian Zeng y Jian Yang
Cancer Resaerch. Marzo 2021. Volumen 81, Número 5
https://cancerres.aacrjournals.org/content/81/5/1230.long
Resumen
La carga mutacional tumoral (TMB) es un biomarcador emergente de respuesta a la inmunoterapia en tumores sólidos. Sin embargo, la medida en que la variación en TMB entre pacientes es atribuible a la variación genética de la línea germinal sigue siendo difícil de determinar. Aquí, utilizando 7004 pacientes no emparentados de ascendencia europea en 33 tipos de cáncer de The Cancer Genome Atlas, mostramos que la TMB pancancerosa es poligénica con aproximadamente el 13 % de su variación explicada por aproximadamente 1,1 millones de variantes comunes en total. Identificamos variantes de línea germinal que afectan a TMB en adenocarcinoma de estómago mediante la alteración de los niveles de expresión de BAG5 y KLC1. Análisis adicionales proporcionan evidencia de que la TMB está genéticamente asociada con rasgos y enfermedades complejos, como fumar, artritis reumatoide, altura y cánceres, y algunas de las asociaciones son probablemente causales. En general, estos resultados brindan nuevos conocimientos sobre la base genética de las mutaciones somáticas en los tumores y pueden informar los esfuerzos futuros para usar variantes genéticas para estratificar a los pacientes para la inmunoterapia.
Importancia: este estudio proporciona evidencia de una arquitectura poligénica de la carga mutacional del tumor y abre una vía para el uso de variaciones genéticas de la línea germinal del genoma completo para estratificar a los pacientes con cáncer para la inmunoterapia.
Introducción
Las mutaciones somáticas, en particular aquellas que confieren una ventaja de crecimiento selectivo a las células, contribuyen al desarrollo del cáncer ( 1 ). Los patrones y funciones de las mutaciones somáticas en el genoma del cáncer humano se han analizado ampliamente en una amplia variedad de cánceres, lo que ha dado lugar a numerosos conocimientos biológicos ( 2–4 ). En particular, se ha demostrado que un evento somático importante denominado carga mutacional tumoral (TMB), determinado por el número total de mutaciones somáticas no sinónimas en un genoma de cáncer, tiene una gran relevancia práctica. En particular, las líneas de evidencia sugirieron que TMB es un biomarcador predictivo para identificar a los pacientes con cáncer que tienen más probabilidades de responder a las terapias con inhibidores del punto de control inmunitario (ICI) ( 5–9 ).
Varios estudios han revelado la asociación de variantes genéticas de línea germinal con eventos somáticos en tumores, incluidas firmas mutacionales ( 10–12 ) y mutaciones somáticas en genes específicos relacionados con el cáncer ( 13, 14 ). Además, Zhu y sus colegas descubrieron una asociación negativa entre los alelos de riesgo de cáncer de línea germinal y la carga de mutaciones somáticas en el cáncer de mama ( 15 ). Robles-Espinoza y colegas encontraron que las variantes de MC1R de la línea germinal estaban asociadas con la carga de mutación somática del melanoma ( 16). A pesar de estos avances, nuestra comprensión de la genética de la línea germinal de TMB aún es muy limitada. Una pieza clave que falta es la aclaración de la arquitectura genética de TMB (p. ej., la contribución general de las variantes genéticas comunes a TMB y la distribución del tamaño del efecto de las variantes comunes) y las relaciones genéticas y causales de rasgos complejos (incluyendo enfermedades) con TMB. Responder a estas preguntas será fundamental para orientar los futuros estudios de asociación del genoma completo (GWAS) para la TMB, identificar los factores de riesgo de la TMB y evaluar la posible utilidad clínica de las características genéticas de la TMB en las terapias de ICI.
Aquí, estudiamos la arquitectura genética de TMB a partir de un análisis pancanceroso de 7004 pacientes de ascendencia europea en 33 tipos de cáncer en The Cancer Genome Atlas (TCGA). Aunque es probable que existan diferencias en la arquitectura genética de TMB entre los tipos de cáncer, los tamaños de muestra en TCGA son insuficientes para los métodos que usan variantes comunes en todo el genoma para representar la arquitectura genética de TMB para tipos de cáncer individuales. Por lo tanto, enfocamos este estudio en la TMB pancancerosa al mismo tiempo que reconocemos que es probable que exista una heterogeneidad genética en la TMB entre los tipos de cáncer (consulte más adelante para obtener más información). Estimamos que aproximadamente el 13 % de la variación en la TMB pancancerosa puede explicarse por todas las variantes genéticas comunes juntas. Identificamos loci de línea germinal asociados con TMB en tipos de cáncer específicos, así como los genes causales putativos subyacentes que impulsan las asociaciones. Además, nuestros hallazgos muestran un componente genético compartido entre la TMB pancancerosa y rasgos complejos (incluidas enfermedades), como el tabaquismo, la altura, la artritis reumatoide y los cánceres. Investigamos más a fondo las relaciones causales entre la TMB y los rasgos complejos mediante análisis de aleatorización (MR) mendeliana, que proporcionan evidencia de los efectos causales putativos del tabaquismo en la TMB pancancerosa.

Materiales y métodos
cohorte TCGA
TCGA incluyó bioespecímenes tumorales y normales emparejados de 10 224 muestras humanas con consentimiento informado por escrito bajo la autorización de las juntas de revisión institucionales locales (IRB; https://cancergenome.nih.gov/abouttcga/policies/informedconsent ). Excluimos 344 muestras altamente mutadas que probablemente reflejaban artefactos técnicos detectados por un estudio anterior ( 4 ). Las muestras que fueron marcadas por el Grupo de Trabajo de Análisis sobre la base de la patología también se descartaron, pero aquellas con el indicador de "degradación de ARN" se mantuvieron porque no se utilizaron datos de ARN en este estudio ( 4 ). Todas las muestras en la base de datos pública de TCGA se recopilaron y utilizaron siguiendo estrictas pautas de protección de sujetos humanos, consentimiento informado por escrito y revisión de protocolos por parte del IRB (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/history/policies ). Nuestro estudio fue aprobado por los comités de ética de la Universidad de Queensland (Brisbane, Queensland, Australia, ID: 2011001173), la Universidad Médica de Wenzhou (Wenzhou, Zhejiang, PR China, ID: 2019094) y la Universidad de Westlake (Hangzhou, Zhejiang, PR China, identificación: 20200722YJ001).
Cuantificación de la carga mutacional tumoral
TMB se define como el número total de variantes no sinónimas (incluidas variantes de un solo nucleótido e indeles) por Mb de un exoma examinado sobre la base de llamadas MC3 de PanCanAtlas (mc3.v0.2.8.PUBLIC.maf; https://www. synapse.org/#!Synapse:syn7214402/files/ ).
datos de matriz SNP
Utilizamos datos de genotipo de 522 606 SNP derivados de matrices Affymetrix 6.0 SNP. Los archivos de genotipo de alpiste ( n = 11 459, en 33 tipos de cáncer) se descargaron del archivo heredado de Genome Data Commons (GRCh37/hg19) ( https://portal.gdc.cancer.gov/legacy-archive ). Birdseed2vcf ( https://github.com/ding-lab/birdseed2vcf ) se utilizó para transformar archivos de genotipo de alpiste en archivos VCF individuales, que luego se fusionaron en un solo archivo VCF combinado. Elegimos un solo archivo de acuerdo con los siguientes criterios, si 1 paciente tenía códigos de barras de alícuotas múltiples, como se describe en detalle en otra parte ( 17): (i) tipo de muestra: sangre > sólido (código de tipo de muestra, 10 > 11), (ii) tipo molecular de analito: se prefirieron los analitos D (ADN nativo) sobre G, W o X (genoma completo amplificado) ; (iii) orden de porción: se seleccionó la porción con mayor número, (iv) orden de placa: se seleccionó la placa con mayor número, y (v) centro de secuenciación o análisis: se seleccionó el centro con mayor número. Finalmente, se obtuvo un total de 10 406 muestras no redundantes para 33 tipos de cáncer para el análisis posterior.
Análisis de componentes principales
Para eliminar los valores atípicos ancestrales de la muestra de TCGA, realizamos un análisis de componentes principales (PCA) en un conjunto de datos de genotipo combinado de TCGA y 1000 Genomes Project (1000GP, RRID:SCR_008801; ref. 18 ). Solo se incluyeron en el PCA los SNP autosómicos con una tasa de ausencia < 5 % y una frecuencia de alelos menores (MAF) > 1 % y los individuos con una tasa de ausencia < 5 % (446 018 SNP en 12 909 individuos en total). Después del PCA, eliminamos a los individuos TCGA cuyo componente principal 1 (PC1) o PC2 se desviaba más de seis DE de la media del PC correspondiente de los 1000 GP de ascendencia europea. Finalmente, se retuvo un total de 8.179 individuos TCGA de ascendencia europea para su posterior análisis.
Imputación de genotipo
Imputamos un conjunto de datos de genotipos limpios que consta de 423 239 SNP autosómicos (tasa de ausencia < 5 %, MAF > 1 % y equilibrio de Hardy Weinberg P > 10 −6 ) en 8179 individuos de ascendencia europea al Haplotype Reference Consortium ( 19 ) usando el servidor de imputación de Sanger ( https://imputation.sanger.ac.uk/ ; ref. 20 ). Las probabilidades posteriores del genotipo de la imputación se convirtieron en genotipos de llamada dura utilizando PLINK2 (-hard-call-thresh 0.1) ( 21 ). Filtramos los SNP con una tasa de ausencia > 5 %, MAF < 1 %, equilibrio de Hardy Weinberg P < 10 −6, o puntuación INFO de imputación < 0,3. Tenga en cuenta que debido a que el tamaño de la muestra de la cohorte TCGA era demasiado pequeño para analizar variantes raras, incluimos solo las variantes comunes (es decir, MAF> 1%) en este estudio.
Análisis de asociación de todo el genoma
Primero eliminamos a los individuos a los que les faltaba información de TMB o de covariables (p. ej., edad, sexo o pureza del tumor). Luego explotamos el análisis de rasgos complejos de todo el genoma (GCTA; ref. 22) para calcular las relaciones genéticas entre todos los pares de los sujetos restantes utilizando variantes en común con las del HapMap 3 (denominados SNP HM3 en lo sucesivo) y excluyó uno de cada par de individuos con una relación genética estimada > 0,05, lo que resultó en un conjunto de 7.004 individuos no relacionados. Realizamos una regresión de TMB contra la edad y estandarizamos los residuos a puntajes z dentro de cada grupo de sexo de cada tipo de cáncer. Para la TMB pancancerosa, primero combinamos los residuos estandarizados de todos los tipos de cáncer y luego aplicamos una transformación normal inversa basada en rangos (RINT) a los residuos combinados. También aplicamos RINT a TMB en cada tipo de cáncer.
https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/history/policies

Para los datos de genotipo imputados, realizamos pasos de control de calidad adicionales en los individuos no relacionados: excluyendo variantes con tasa de ausencia > 5 %, MAF < 1 % o equilibrio de Hardy Weinberg P < 10 −6 y muestras con tasa de ausencia > 5 %. Finalmente, se utilizó un total de 7 567 484 variantes comunes, con 1 139 665 en común con los SNP HM3 y 7 004 individuos no relacionados para los análisis a continuación.
Se realizaron pruebas de asociación para TMB pancáncer y TMB de cada tipo de cáncer con base en un modelo de regresión lineal implementado en fastGWA ( 23 ). Incluimos las primeras 20 PC y la pureza del tumor como covariables en el modelo de regresión. Las variantes con P < 5 × 10-8 se consideraron significativas en todo el genoma.
Estimación de heredabilidad basada en SNP
Usamos 1,139,665 HM3 SNP y 7,004 muestras no relacionadas para estimar la heredabilidad basada en SNP ( ) para pan-cáncer TMB. Optamos por usar los SNP de HM3 porque son un conjunto recortado de variantes optimizadas para capturar la variación genética común del genoma humano ( 24 ) y para mejorar la comparabilidad de este estudio con estudios anteriores.
análisis de estimación. Utilizamos los siguientes métodos para Estimacion.
Análisis de máxima verosimilitud restringido por matriz de relación genómica
La máxima verosimilitud restringida por matriz de relación genómica (GREML; también conocida como GERML de un solo componente o GREML-SC) es un método de modelo lineal mixto que aprovecha una matriz de relación genética (GRM) para estimar la proporción de variación fenotípica en un rasgo que puede ser explicado por todos los SNP en individuos no relacionados. GREML se implementa en el paquete de software GCTA ( 22 ). Los detalles de la metodología GREML se han descrito en otra parte ( 25 ). Construimos un GRM de los 7,004 individuos no relacionados usando los SNP HM3 y luego
estimamos con las primeras 20 PC y la pureza del tumor ajustadas como covariables en el modelo GREML.
Análisis GREML estratificado por MAF
Se ha demostrado en estudios previos que obtenidos de GREML-SC pueden estar sesgados si la distribución de MAF de las variantes causales es diferente de la de los SNP utilizados en el análisis ( 26, 27 ). Por lo tanto, GREML estratificado por MAF (GREML-MS) se desarrolló para abordar esta brecha al estratificar los SNP por MAF en múltiples GRM y ajustar los GRM conjuntamente en un modelo. Estratificamos los SNP HM3 utilizados en cinco contenedores MAF (es decir, 0.01 < MAF < 0.1, 0.1 < MAF < 0.2, 0.2 < MAF < 0.3, 0.3 < MAF < 0.4 y 0.4 < MAF < 0.5) y generamos los GRM correspondientes. Luego se usó GCTA
para realizar un análisis GREML-MS con ajuste para las primeras 20 PC y la pureza del tumor para estimar para pan-cáncer TMB.
Desequilibrio de ligamiento y análisis GREML estratificado por MAF
El desequilibrio de ligamiento y GREML estratificado por MAF (GREML-LDMS) es otra variante del método GREML desarrollado para dar cuenta de la distribución no aleatoria de variantes causales con respecto tanto a MAF como al desequilibrio de ligamiento (LD; refs. 27–29 ). En este análisis, cada uno de los cinco contenedores MAF en el análisis GREML-MS anterior se dividió en un contenedor LD bajo y uno alto en función de las puntuaciones LD de los SNP, lo que resultó en 10 contenedores MAF y LD en total. Luego ejecutamos GCTA para realizar un análisis GREML-LDMS ( 27 ) para TMB pancanceroso con las primeras 20 PC y la pureza del tumor incluidas como covariables.
Modelo lineal mixto bayesiano con un análisis de parámetros S
El modelo lineal mixto bayesiano con un método de parámetro S (denominado BayesS), implementado en el software GCTB ( http://cnsgenomics.com/software/gctb/ ), se empleó para estimar tres parámetros de arquitectura genética simultáneamente,
es decir, , poligenicidad (proporción de SNP con efectos distintos de cero) y relación entre el tamaño del efecto de SNP y MAF ( 30 ). Realizamos un análisis BayesS para pan-cáncer TMB con una corrección para las primeras 20 PC y la pureza del tumor.
Análisis de regresión de puntuación de LD
La regresión de puntuación de LD (LDSC) es un método basado en datos resumidos que estima por regresión GWAS estadísticas de todos los SNP en sus puntuaciones de LD ( https://github.com/bulik/ldsc ; ref. 31 ). LDSC es computacionalmente
más eficiente que el convencional métodos de estimación que se basan en datos a nivel individual, especialmente para conjuntos de datos con muestras de gran tamaño. Realizamos un análisis LDSC utilizando las estadísticas resumidas de GWAS para las puntuaciones de TMB y LD pancancerosas proporcionadas por el paquete LDSC (calculadas a partir de los 1000GP individuos de ascendencia europea basados en HM3 SNP). Excluimos variantes en la región MHC debido a la complejidad de esta región.
Estimación de correlación genética usando LDSC bivariado
Similar a la estimación de por LDSC para un solo rasgo, el LDSC bivariante estima la covarianza genética mediante la regresión del producto de las puntuaciones z para dos rasgos en la puntuación LD a través de SNP ( 32 ). Usamos el LDSC bivariado para estimar la correlación genética (rg ) entre el TMB pancanceroso y un rasgo complejo. Los datos resumidos de GWAS para 46 rasgos complejos (incluidas las enfermedades) se recopilaron de estudios publicados (Tabla complementaria S3).

Análisis de puntaje de riesgo poligénico
SBayesR es un método de regresión múltiple bayesiano basado en datos resumidos, que se desarrolló principalmente para el análisis de puntuación de riesgo poligénico (PRS) y se ha demostrado que mejora la precisión de la predicción en comparación con otros enfoques de PRS ampliamente utilizados ( 21, 33, 34 ). SBayesR está disponible en el paquete de software GCTB ( 30). Ejecutamos SBayesR para estimar los efectos conjuntos de todos los SNP de HM3 utilizando datos de resumen de GWAS para un rasgo complejo e información de LD de una muestra de referencia con genotipos de nivel individual. La cadena de MCMC se realizó con 50 000 iteraciones, 20 000 de inicio y otros parámetros predeterminados. La reducción de los efectos SNP hacia cero proviene de dos componentes, uno de la probabilidad de una masa puntual en cero y el otro de la distribución previa (una mezcla de distribuciones normales centradas en cero) para los efectos no nulos. Utilizamos un conjunto de 50 000 personas no emparentadas de ascendencia europea seleccionadas al azar del Biobanco del Reino Unido (UKB) como referencia de LD para los datos resumidos de UKB GWAS y otro conjunto de 50 000 personas no emparentadas de ascendencia europea muestreadas del GERA (Recurso para la Investigación en Epidemiología Genética). sobre el envejecimiento) cohorte (35 ) como referencia de LD para los otros conjuntos de datos de resumen de GWAS. De manera similar a lo anterior, se excluyeron los SNP en las regiones del MHC.
Después de obtener las estimaciones de SBayesR de los efectos SNP, calculamos la PRS del rasgo en la cohorte TCGA usando la siguiente ecuación:
donde m es el número de todos los HM3 SNP sin selección, es el efecto estimado de la th SNP de SBayesR, y , codificado como 0, 1 o 2, es el número de alelos de efecto del el SNP. Luego correlacionamos la PRS de cada uno de los 46 rasgos con la TMB pancancerosa, así como la TMB de tipos de cáncer individuales.
análisis de RM
La RM basada en datos resumidos generalizados (GSMR) es un método de RM que utiliza múltiples variantes genéticas como instrumentos para inferir la relación causal entre dos fenotipos utilizando estadísticas resumidas GWAS ( 36 ). Aquí, empleamos GSMR para explorar las asociaciones causales entre 11 rasgos, que mostraron asociaciones nominalmente significativas con TMB pancanceroso en el análisis PRS o r g, y TMB pancanceroso. Siguiendo el estudio de Zhu y colegas ( 36 ), seleccionamos SNP casi independientes (LD r 2 < 0.05) con P < 5 × 10 −8como instrumentos genéticos y solo analizó rasgos con al menos 10 instrumentos genéticos. Ejecutamos el análisis GSMR con el paso de filtrado de valores atípicos del instrumento dependiente de heterogeneidad (HEIDI) (configuración predeterminada) para excluir los instrumentos genéticos que están asociados con los fenotipos de exposición y resultado debido a la pleiotropía horizontal. También repetimos los análisis de MR utilizando el enfoque ponderado de varianza inversa (MR-IVW) implementado en el paquete R "MR-base" ( 37 ). Al igual que en los análisis de GSMR, se eligieron como instrumentos SNP casi independientes asociados con una exposición con P < 5 × 10-8, que luego se podaron para LD siguiendo la configuración predeterminada en MR-base.
Resumen de análisis de RM basado en datos
La MR basada en datos resumidos (SMR) es un método que integra estadísticas resumidas de un GWAS para un rasgo con las de un estudio de locus de rasgos cuantitativos de expresión (eQTL) para identificar genes cuyos niveles de expresión son los mediadores de los efectos SNP en el rasgo ( 38 ). Usamos SMR para identificar genes causales putativos para TMB en carcinoma invasivo de mama, cromófobo de riñón y adenocarcinoma de estómago. Utilizamos datos resumidos de cis-eQTL del consorcio eQTLGen, un metanálisis de eQTL de 31 684 individuos en sangre ( 39 ). Incluimos en el análisis SMR solo los genes con al menos un cis-eQTL con P < 5 × 10 −8(la configuración predeterminada en SMR) y genes excluidos en las regiones MHC, lo que da como resultado 15.487 genes en total. Utilizamos un umbral de significación de todo el genoma de P < 3,23 × 10 −6 (es decir, 0,05/15 487) para reclamar asociaciones significativas de SMR y la prueba HEIDI para distinguir la causalidad o la pleiotropía vertical de la vinculación ( PHEIDI > 0,05).
Resultados
La arquitectura poligénica del pan-cáncer TMB
Definimos TMB como el número total de variantes no sinónimas por Mb de un exoma examinado (Materiales y Métodos). Primero estimamos la proporción de varianza en la TMB pancancerosa explicada por todos los SNP comunes (es decir,
) en 7004 pacientes europeos no relacionados en 33 tipos de cáncer en TCGA (en lo sucesivo denominado TMB pancanceroso; figuras complementarias S1 y S2). El fenotipo TMB se ajustó por covariables, como la edad, el género y el tipo de
cáncer, y se estandarizó a la puntuación z mediante RINT (Materiales y Métodos). Empleamos cinco de uso común métodos de estimación, con covariables adicionales ajustadas para controlar los efectos debido a la estratificación de la población y la pureza del tumor (Materiales y Métodos). Los cinco métodos utilizados fueron: (i) análisis GREML-SC ( 25 ), (ii) GREML-MS (cinco GRM; ref. 28 ), (iii) GREML-LDMS (10 GRM; ref. 28 ), (iv) BayesS ( 30 ), y (v) LDSC ( 31 ). GREML-MS y GREML-LDMS pueden
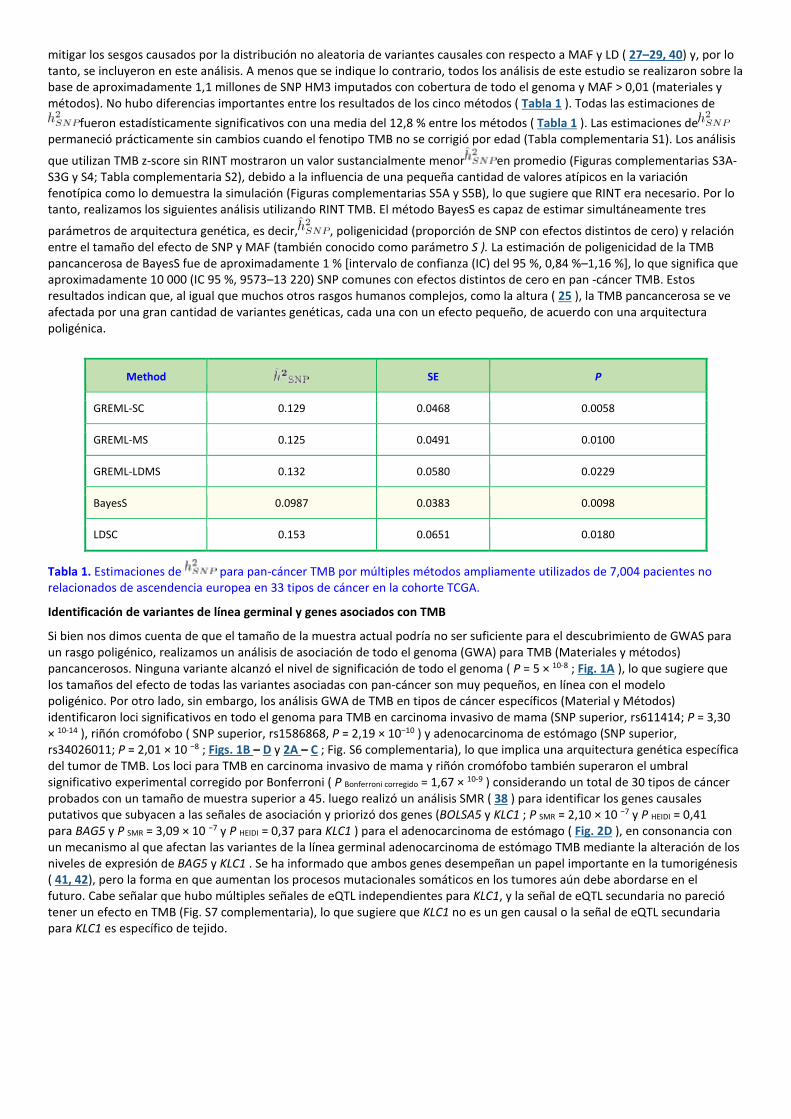
mitigar los sesgos causados por la distribución no aleatoria de variantes causales con respecto a MAF y LD ( 27–29, 40) y, por lo tanto, se incluyeron en este análisis. A menos que se indique lo contrario, todos los análisis de este estudio se realizaron sobre la base de aproximadamente 1,1 millones de SNP HM3 imputados con cobertura de todo el genoma y MAF > 0,01 (materiales y métodos). No hubo diferencias importantes entre los resultados de los cinco métodos ( Tabla 1 ). Todas las estimaciones de
fueron estadísticamente significativos con una media del 12,8 % entre los métodos ( Tabla 1 ). Las estimaciones depermaneció prácticamente sin cambios cuando el fenotipo TMB no se corrigió por edad (Tabla complementaria S1). Los análisis
que utilizan TMB z-score sin RINT mostraron un valor sustancialmente menor en promedio (Figuras complementarias S3A-S3G y S4; Tabla complementaria S2), debido a la influencia de una pequeña cantidad de valores atípicos en la variación fenotípica como lo demuestra la simulación (Figuras complementarias S5A y S5B), lo que sugiere que RINT era necesario. Por lo tanto, realizamos los siguientes análisis utilizando RINT TMB. El método BayesS es capaz de estimar simultáneamente tres
parámetros de arquitectura genética, es decir, , poligenicidad (proporción de SNP con efectos distintos de cero) y relación entre el tamaño del efecto de SNP y MAF (también conocido como parámetro S ). La estimación de poligenicidad de la TMB pancancerosa de BayesS fue de aproximadamente 1 % [intervalo de confianza (IC) del 95 %, 0,84 %–1,16 %], lo que significa que aproximadamente 10 000 (IC 95 %, 9573–13 220) SNP comunes con efectos distintos de cero en pan -cáncer TMB. Estos resultados indican que, al igual que muchos otros rasgos humanos complejos, como la altura ( 25 ), la TMB pancancerosa se ve afectada por una gran cantidad de variantes genéticas, cada una con un efecto pequeño, de acuerdo con una arquitectura poligénica.
Method
SE P
GREML-SC 0.129 0.0468 0.0058
GREML-MS 0.125 0.0491 0.0100
GREML-LDMS 0.132 0.0580 0.0229
BayesS 0.0987 0.0383 0.0098
LDSC 0.153 0.0651 0.0180
Tabla 1. Estimaciones de para pan-cáncer TMB por múltiples métodos ampliamente utilizados de 7,004 pacientes no relacionados de ascendencia europea en 33 tipos de cáncer en la cohorte TCGA.
Identificación de variantes de línea germinal y genes asociados con TMB
Si bien nos dimos cuenta de que el tamaño de la muestra actual podría no ser suficiente para el descubrimiento de GWAS para un rasgo poligénico, realizamos un análisis de asociación de todo el genoma (GWA) para TMB (Materiales y métodos) pancancerosos. Ninguna variante alcanzó el nivel de significación de todo el genoma ( P = 5 × 10-8 ; Fig. 1A ), lo que sugiere que los tamaños del efecto de todas las variantes asociadas con pan-cáncer son muy pequeños, en línea con el modelo poligénico. Por otro lado, sin embargo, los análisis GWA de TMB en tipos de cáncer específicos (Material y Métodos) identificaron loci significativos en todo el genoma para TMB en carcinoma invasivo de mama (SNP superior, rs611414; P = 3,30 × 10-14 ), riñón cromófobo ( SNP superior, rs1586868, P = 2,19 × 10−10 ) y adenocarcinoma de estómago (SNP superior, rs34026011; P = 2,01 × 10 −8 ; Figs. 1B – D y 2A – C ; Fig. S6 complementaria), lo que implica una arquitectura genética específica del tumor de TMB. Los loci para TMB en carcinoma invasivo de mama y riñón cromófobo también superaron el umbral significativo experimental corregido por Bonferroni ( P Bonferroni corregido = 1,67 × 10-9 ) considerando un total de 30 tipos de cáncer probados con un tamaño de muestra superior a 45. luego realizó un análisis SMR ( 38 ) para identificar los genes causales putativos que subyacen a las señales de asociación y priorizó dos genes (BOLSA5 y KLC1 ; P SMR = 2,10 × 10 −7 y P HEIDI = 0,41 para BAG5 y P SMR = 3,09 × 10 −7 y P HEIDI = 0,37 para KLC1 ) para el adenocarcinoma de estómago ( Fig. 2D ), en consonancia con un mecanismo al que afectan las variantes de la línea germinal adenocarcinoma de estómago TMB mediante la alteración de los niveles de expresión de BAG5 y KLC1 . Se ha informado que ambos genes desempeñan un papel importante en la tumorigénesis ( 41, 42), pero la forma en que aumentan los procesos mutacionales somáticos en los tumores aún debe abordarse en el futuro. Cabe señalar que hubo múltiples señales de eQTL independientes para KLC1, y la señal de eQTL secundaria no pareció tener un efecto en TMB (Fig. S7 complementaria), lo que sugiere que KLC1 no es un gen causal o la señal de eQTL secundaria para KLC1 es específico de tejido.

Figura 1. Diagrama de Manhattan de GWAS para pan-cáncer TMB ( A ) y TMB en carcinoma invasivo de mama (BRCA; B ), riñón cromófobo (KICH; C ) y adenocarcinoma de estómago (STAD; D ). Las posiciones de los cromosomas se muestran en el eje x, y los valores de −log 10 P se muestran en el eje y . La línea discontinua corresponde a P = 5 × 10 −8 .
Figura 2. Gráficos de locus GWAS y SMR. Se muestran los resultados de GWAS en los loci asociados con TMB en carcinoma invasivo de mama (BRCA; A ), riñón cromófobo (KICH; B ) y adenocarcinoma de estómago (STAD; C ), y resultados de SMR en los locus BAG5 y KLC1 para TMB en adenocarcinoma de estómago ( D ). A – C, las posiciones de los cromosomas se muestran en el

eje x junto con los nombres y tamaños de los genes, y los valores −log 10 P se muestran en el eje y izquierdo. El punto morado muestra el SNP asociado superior y los otros colores punteados indican LD por pares ( r 2) con este SNP. El eje y derecho representa la tasa de recombinación regional (cM/Mb). D, Los puntos grises de la gráfica superior muestran los valores de -log 10 P de los SNP del análisis GWAS para TMB en el adenocarcinoma de estómago, y los diamantes indican los valores de -log 10 P de los genes del análisis SMR. Los valores de −log 10 P de eQTL de eQTLGen para BAG5 y KLC1 se muestran en el gráfico inferior. Resaltados en rojo están los genes ( BAG5 y KLC1 ) que pasaron las pruebas SMR ( P < 3.23 × 10 −6 ) y HEIDI ( P > 0.05).
Efectos poligénicos compartidos entre pan-cáncer TMB y 46 rasgos y enfermedades complejos
Habiendo sabido que la TMB pancancerosa tiene un componente poligénico sustancial, buscamos investigar si existían factores genéticos compartidos entre la TMB pancancerosa y los rasgos complejos (incluidas las enfermedades). Para hacer esto, seleccionamos 46 rasgos complejos para los cuales las estadísticas resumidas de GWAS a gran escala estaban disponibles a partir de trabajos anteriores (Tabla complementaria S3), y usamos dos enfoques analíticos, el PRS ( 43 ) y el LDSC bivariado ( 32 ), para cuantificar el medida en que los efectos poligénicos se comparten entre los rasgos (Materiales y Métodos). Ambos métodos utilizan todos los SNP comunes en todo el genoma y son robustos en presencia de un gran número de SNP no asociados.
En el análisis de PRS, utilizamos SBayesR ( 33 ), un método de regresión múltiple bayesiano basado en datos resumidos, para estimar los pesos de todos los SNP HM3 para construir un PRS (es decir, una suma ponderada de alelos de efecto en todos los SNP) para cada uno. de los 46 rasgos en TCGA y probó la correlación entre PRS y pan-cancer TMB. Optamos por usar SBayesR para el análisis de PRS porque solo requiere estadísticas de resumen del GWAS de descubrimiento y muestra una precisión de predicción mejorada en comparación con los enfoques de la competencia ( 44, 45 ). Descubrimos que la TMB pancancerosa se correlacionó significativamente de manera positiva con la PRS de la artritis reumatoide y se correlacionó negativamente con la PRS para dejar de fumar, con un FDR < 0,05 ( Fig. 3 ; Tabla complementaria S3).
Figura 3. Efectos poligénicos compartidos entre pan-cáncer TMB y 46 rasgos complejos. El eje x muestra la estimación de r g entre TMB pancanceroso y un rasgo complejo, y el eje y indica la correlación entre TMB pancanceroso y el rasgo PRS. Las estimaciones con FDR < 0,05 en el análisis PRS o r g se resaltan en azul oscuro y las que tienen P < 0,05 se resaltan en azul claro.
Luego empleamos el método bivariado LDSC para estimar entre pan-cáncer TMB y cada uno de los 46 rasgos, donde puede interpretarse como la correlación de efectos poligénicos entre TMB y un rasgo. Los análisis de LDSC se realizaron utilizando todos los SNP de HM3 de los datos de resumen de GWAS para TMB de pancáncer y 46 rasgos mencionados
anteriormente. Encontramos que el TMB pancanceroso mostró una r g significativa con la altura ( = 0,28; SE, 0.081) con FDR <0.05 ( Fig. 3 ; Tabla complementaria S3). Observamos buenas concordancias entre las estimaciones de PRS y LDSC tanto en la magnitud (correlación de Pearson = 0,83 y P = 1,39 × 10 −12 ) como en el nivel de significancia (correlación de Pearson = 0,71 y P = 3,53 × 10 −8 ; Fig. 3 ; Figura complementaria S8), con ocho estimaciones nominalmente significativas (es decir, P < 0,05) de la primera y nueve de la última, incluida la iniciación al tabaquismo y la artritis reumatoide (Tabla complementaria S3).
Asociaciones causales putativas del tabaquismo y la altura con pan-cáncer TMB: un análisis de RM
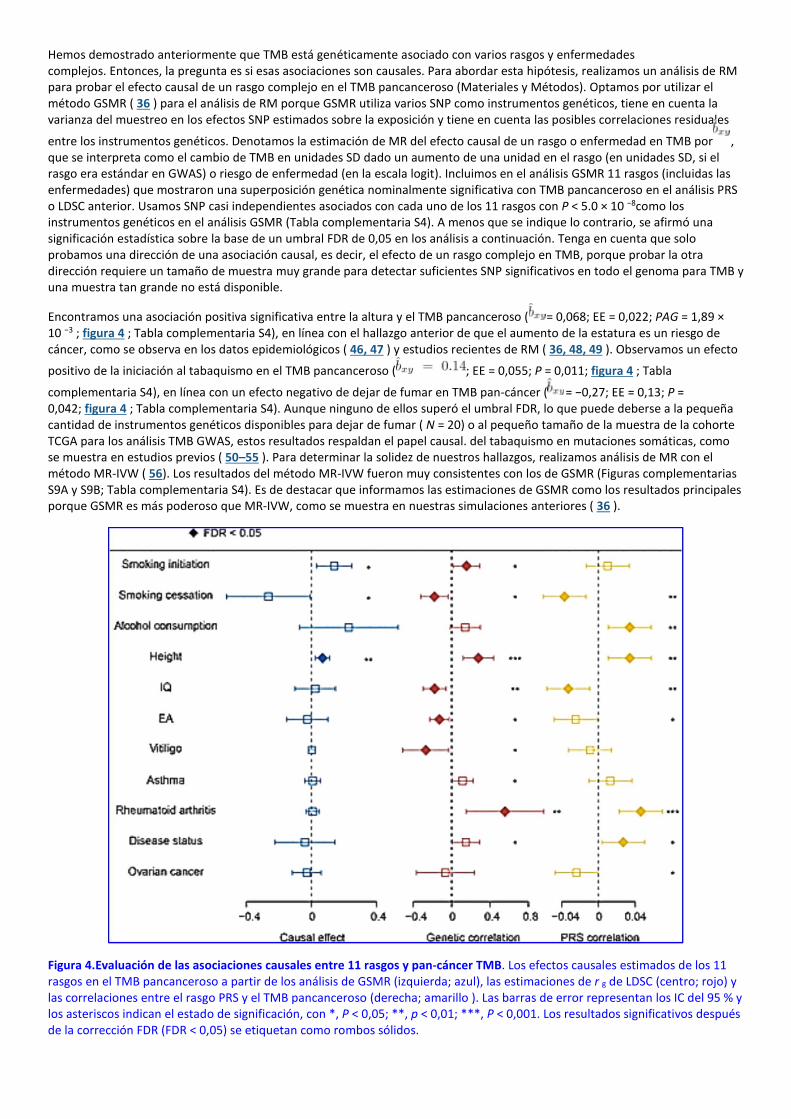
Hemos demostrado anteriormente que TMB está genéticamente asociado con varios rasgos y enfermedades complejos. Entonces, la pregunta es si esas asociaciones son causales. Para abordar esta hipótesis, realizamos un análisis de RM para probar el efecto causal de un rasgo complejo en el TMB pancanceroso (Materiales y Métodos). Optamos por utilizar el método GSMR ( 36 ) para el análisis de RM porque GSMR utiliza varios SNP como instrumentos genéticos, tiene en cuenta la varianza del muestreo en los efectos SNP estimados sobre la exposición y tiene en cuenta las posibles correlaciones residuales
entre los instrumentos genéticos. Denotamos la estimación de MR del efecto causal de un rasgo o enfermedad en TMB por , que se interpreta como el cambio de TMB en unidades SD dado un aumento de una unidad en el rasgo (en unidades SD, si el rasgo era estándar en GWAS) o riesgo de enfermedad (en la escala logit). Incluimos en el análisis GSMR 11 rasgos (incluidas las enfermedades) que mostraron una superposición genética nominalmente significativa con TMB pancanceroso en el análisis PRS o LDSC anterior. Usamos SNP casi independientes asociados con cada uno de los 11 rasgos con P < 5.0 × 10 −8como los instrumentos genéticos en el análisis GSMR (Tabla complementaria S4). A menos que se indique lo contrario, se afirmó una significación estadística sobre la base de un umbral FDR de 0,05 en los análisis a continuación. Tenga en cuenta que solo probamos una dirección de una asociación causal, es decir, el efecto de un rasgo complejo en TMB, porque probar la otra dirección requiere un tamaño de muestra muy grande para detectar suficientes SNP significativos en todo el genoma para TMB y una muestra tan grande no está disponible.
Encontramos una asociación positiva significativa entre la altura y el TMB pancanceroso ( = 0,068; EE = 0,022; PAG = 1,89 × 10 −3 ; figura 4 ; Tabla complementaria S4), en línea con el hallazgo anterior de que el aumento de la estatura es un riesgo de cáncer, como se observa en los datos epidemiológicos ( 46, 47 ) y estudios recientes de RM ( 36, 48, 49 ). Observamos un efecto
positivo de la iniciación al tabaquismo en el TMB pancanceroso ( ; EE = 0,055; P = 0,011; figura 4 ; Tabla
complementaria S4), en línea con un efecto negativo de dejar de fumar en TMB pan-cáncer ( = −0,27; EE = 0,13; P = 0,042; figura 4 ; Tabla complementaria S4). Aunque ninguno de ellos superó el umbral FDR, lo que puede deberse a la pequeña cantidad de instrumentos genéticos disponibles para dejar de fumar ( N = 20) o al pequeño tamaño de la muestra de la cohorte TCGA para los análisis TMB GWAS, estos resultados respaldan el papel causal. del tabaquismo en mutaciones somáticas, como se muestra en estudios previos ( 50–55 ). Para determinar la solidez de nuestros hallazgos, realizamos análisis de MR con el método MR-IVW ( 56). Los resultados del método MR-IVW fueron muy consistentes con los de GSMR (Figuras complementarias S9A y S9B; Tabla complementaria S4). Es de destacar que informamos las estimaciones de GSMR como los resultados principales porque GSMR es más poderoso que MR-IVW, como se muestra en nuestras simulaciones anteriores ( 36 ).
Figura 4.Evaluación de las asociaciones causales entre 11 rasgos y pan-cáncer TMB. Los efectos causales estimados de los 11 rasgos en el TMB pancanceroso a partir de los análisis de GSMR (izquierda; azul), las estimaciones de r g de LDSC (centro; rojo) y las correlaciones entre el rasgo PRS y el TMB pancanceroso (derecha; amarillo ). Las barras de error representan los IC del 95 % y los asteriscos indican el estado de significación, con *, P < 0,05; **, p < 0,01; ***, P < 0,001. Los resultados significativos después de la corrección FDR (FDR < 0,05) se etiquetan como rombos sólidos.

Discusión
En este estudio, nuestros resultados proporcionaron la primera evidencia genética molecular de una arquitectura poligénica de la TMB pancancerosa mediante análisis de predicción y estimación del genoma completo utilizando datos de TCGA. Junto con varias asociaciones emergentes de variantes de la línea germinal con eventos somáticos ( 11–16 ), nuestros hallazgos respaldan la hipótesis de que la TMB no solo es hereditaria, sino también poligénica. Usando genotipos de línea germinal y fenotipos de mutaciones somáticas en TCGA, obtuvimos una proporción moderada y significativa de varianza explicada por todos los SNP
comunes (es decir, ) de aproximadamente 13% para pan-cáncer TMB. Debido a los tamaños de muestra pequeños de los
tipos de cáncer individuales, enfocamos el análisis de estimación solo en pan-cáncer TMB. La varianza genética para la TMB pancancerosa puede considerarse como la media de una matriz de varianza genética y covarianza para la TMB de los tipos de
cáncer individuales. En teoría, la estimación de en la muestra agrupada es menor que la media de las estimaciones de las submuestras, a menos que el r g entre las submuestras sea 1. Por lo tanto, si existe heterogeneidad genética en la TMB entre los
tipos de cáncer individuales, que es muy probable que sea el caso, el para pan-cáncer TMB proporciona un límite inferior
de la media a través de TMB de tipos de cáncer individuales. Nuestro análisis GWA para TMB pancanceroso no identificó ningún loci significativo en todo el genoma para TMB pancanceroso ( Fig. 1A), en consonancia con lo que se predice a partir de un modelo poligénico de que los tamaños del efecto de los SNP individuales son demasiado pequeños para alcanzar el nivel de significación de todo el genoma cuando el tamaño de la muestra no es lo suficientemente grande. La inferencia de poligenicidad del análisis de BayesS apoyó aún más la arquitectura poligénica de la TMB pancancerosa con una estimación de aproximadamente 10 000 (IC del 95 %, 9573–13 220) variantes comunes de la línea germinal con efectos distintos de cero. Se requerirán GWAS con tamaños de muestra mucho más grandes en el futuro para el descubrimiento de variantes genéticas individuales asociadas con TMB pancanceroso. Sin embargo, por otro lado, identificamos algunos loci significativos en todo el genoma para TMB en tipos de cáncer específicos, como el carcinoma invasivo de mama, el riñón cromófobo y el adenocarcinoma de estómago ( Fig. 2B – D; Figura complementaria S6), lo que sugiere heterogeneidad genética entre los tipos de cáncer. Un ejemplo notable son las variantes en el locus BAG5 / KLC1, donde encontramos evidencia de que los efectos de las variantes de la línea germinal en el adenocarcinoma de estómago TMB están mediados por el nivel de expresión de BAG5 o KLC1 . Estos resultados indican que es probable que la TMB específica del tipo de cáncer sea menos poligénica que la TMB pancancerosa porque la última es un rasgo compuesto de la primera.
A pesar de la falla en la detección de SNP individuales asociados con el TMB pancanceroso en un nivel de significancia de todo el genoma, la estimación del genoma completo y los análisis de predicción que prueban el intercambio general de efectos poligénicos entre el TMB pancanceroso y una amplia gama de rasgos y enfermedades complejos reveló varias ideas importantes. En primer lugar, encontramos una r g positiva de TMB pancáncer con el inicio del tabaquismo (aunque solo fue nominalmente significativa) y una correlación negativa con el cese del tabaquismo. Nuestros análisis de RM encontraron evidencia sugestiva de un vínculo causal entre el tabaquismo y la TMB pancancerosa. Estos resultados sugieren que fumar tabaco puede causar mutaciones somáticas, en línea con hallazgos previos ( 50–55). Por ejemplo, la asociación del tabaquismo con el riesgo de cáncer humano se ha establecido durante más de 60 años mediante estudios epidemiológicos y clínicos ( 57–59 ). Las firmas mutacionales asociadas con el tabaquismo se detectaron y caracterizaron bien en tumores humanos a partir de estudios de secuenciación a gran escala ( 53 ). Recientemente, estas firmas mutacionales se indujeron experimentalmente en líneas de células madre pluripotentes inducidas por humanos expuestas al hidrocarburo aromático policíclico, uno de los componentes mutagénicos más importantes del tabaquismo ( 54 ).). Un estudio más reciente evaluó el panorama de mutaciones somáticas de 632 colonias derivadas de una sola célula en el epitelio bronquial normal mediante la secuenciación del genoma completo y encontró que las células de los fumadores actuales y exfumadores revelaron una carga mutacional sustancialmente mayor ( 55 ). Todos estos descubrimientos respaldan un supuesto efecto causal del tabaquismo en la generación de mutaciones somáticas.
Nuestro trabajo también tiene varias limitaciones. En primer lugar, como se mencionó anteriormente, no intentamos estimar la
para TMB de tipos de cáncer individuales debido a sus pequeños tamaños de muestra; tenga en cuenta que el SE de
es aproximadamente 318/ n, siendo n el tamaño de la muestra ( 60 ). El para pan-cáncer TMB sirve un límite inferior de la
estimación media de para TMB de los tipos de cáncer individuales. Es probable que el para TMB de cada tipo de cáncer varía, siendo algunos sustancialmente más grandes que otros. Se requieren tamaños de muestra grandes en estudios
futuros para estimar la para TMB de tipos de cáncer específicos, especialmente aquellos para los cuales el es pequeño. En segundo lugar, la diferencia en la varianza de TMB antes y después de RINT puede dar lugar a una diferencia en la
. Por lo tanto, la debe interpretarse específicamente para el fenotipo utilizado en el análisis de estimación. Sin
embargo, independientemente del problema con respecto a RINT, la magnitud de junto con su SE es significativo porque al
menos sugiere que el para pan-cáncer TMB es muy poco probable que sea grande. En tercer lugar, los loci significativos en todo el genoma para TMB en carcinoma invasivo de mama, cromófobo de riñón y adenocarcinoma de estómago justifican la replicación en conjuntos de datos independientes cuando las muestras apropiadas estén disponibles en el futuro. En cuarto lugar, aunque hemos recopilado los datos de GWAS más grandes disponibles para los rasgos complejos hasta el momento, el poder para detectar algunos de los r gs, correlaciones PRS o asociaciones MR todavía es limitada. Por ejemplo, las PRS de la

iniciación al tabaquismo se asociaron con la TMB pancancerosa solo en el nivel de significancia nominal; estas asociaciones deben confirmarse en conjuntos de datos más grandes en el futuro. Por último, pero no menos importante, observamos una correlación negativa significativa entre la TMB pancancerosa y la PRS por cáncer de ovario. Una de las posibles explicaciones es la hipótesis de los dos aciertos de Knudsen ( 61), que establece que las personas con un riesgo genético de línea germinal más bajo de cáncer de ovario pueden necesitar acumular más mutaciones somáticas para desarrollar cáncer. Sin embargo, esta hipótesis no explica por qué no tuvimos una observación similar para otros tipos de cáncer, como el de mama y el de próstata. Se justifican más estudios para comprender la causa de la correlación negativa entre la TMB pancancerosa y la PRS del cáncer de ovario.
En resumen, nuestros análisis pancancerosos muestran que la TMB es un rasgo hereditario con una arquitectura poligénica. Dado que TMB es un sustituto del resultado de ICI, estos conocimientos ofrecen la posibilidad de que las variantes de la línea germinal asociadas con TMB puedan usarse para estratificar a los pacientes para terapias de ICI mediante un ensayo simple basado en sangre o saliva. Si bien el poder para identificar variantes individuales asociadas con un rasgo poligénico, como TMB pancanceroso, es limitado dado el tamaño de la muestra actual, nuestros análisis GWA para TMB específico del tipo de cáncer identificaron tres loci significativos en todo el genoma, y el seguimiento funcional el análisis de integración destacó un mecanismo putativo por el cual las variantes en el locus BAG5/KLC1 se asocian con el adenocarcinoma estomacal TMB a través de sus efectos sobre BAG5/KLC1expresión, proporcionando una pista importante para diseñar futuros estudios funcionales para comprender los mecanismos moleculares que sustentan el control genético de las mutaciones somáticas. Utilizando los datos resumidos de GWAS, identificamos asociaciones causales putativas de tabaquismo y altura con TMB pancanceroso. Estos hallazgos confirman los vínculos entre el tabaquismo y la altura con el riesgo de cáncer y respaldan la teoría de la mutación somática de la carcinogénesis. Nuestros resultados no solo contribuyen a la comprensión de los mecanismos subyacentes de las mutaciones somáticas, sino que también pueden estimular los esfuerzos sistemáticos para evaluar las variantes genéticas como biomarcadores no invasivos putativos para estratificar a los pacientes con cáncer para las terapias de ICI.
Divulgaciones de los autores
No se informaron revelaciones.
Contribuciones de los autores
X. Sun: Conceptualización, curación de datos, análisis formal, investigación, visualización, metodología, redacción-borrador original, redacción-revisión y edición. A. Xue: Curación de datos, software, análisis formal. T. Qi: Curación de datos, análisis formal. D. Chen: Curación de datos, análisis formal. D. Shi: Curación de datos, análisis formal. Y. Wu: Curación de datos, análisis formal. Z. Zheng: Software, análisis formal. J. Zeng: Curación de datos, software, análisis formal. J. Yang: Conceptualización, supervisión, captación de fondos, metodología, redacción-borrador original, administración del proyecto, redacción-revisión y edición.
Expresiones de gratitud
Esta investigación fue financiada en parte por Sylvia & Charles Viertel Charitable Foundation, el Australian Research Council (FT180100186 para J. Yang) y la Westlake Education Foundation. Los resultados publicados aquí se basan, en parte, en datos generados por TCGA Research Network: https://www.cancer.gov/tcga (número de acceso de dbGaP phs000178). Este proyecto también utiliza datos del Biobanco del Reino Unido (aplicación n.º 54336). Agradecemos a tres revisores anónimos por sus comentarios constructivos sobre una primera versión del artículo.
Los costos de publicación de este artículo fueron sufragados en parte por el pago de los cargos por página. Por lo tanto, este artículo debe marcarse como publicidad de acuerdo con 18 USC Sección 1734 únicamente para indicar este hecho.
notas al pie
Nota: Los datos complementarios de este artículo están disponibles en Cancer Research Online (http://cancerres.aacrjournals.org/).
Cáncer Res 2021;81:1230–9
Recibido el 12 de octubre de 2020.
Revisión recibida el 14 de diciembre de 2020.
Aceptado el 4 de enero de 2021.
Publicado primero el 8 de enero de 2021.
©2021 Asociación Americana para la Investigación del Cáncer.
Referencias

1.↵ Sr. Stratton, Campbell PJ, Futréal PA. El genoma del cáncer . Naturaleza 2009 ; 458 : 719 - 24 .Referencia cruzadaPubMedGoogle Académico
2.↵ Greenman c, Esteban P, Smith R, Dalgliesh GL, cazador c, Bignell G, et al. Patrones de mutación somática en genomas de cáncer humano . Naturaleza 2007 ; 446 : 153 - 8 .Referencia cruzadaPubMedGoogle Académico
3.↵de PK, LiJ, _Jeong KJ, Shao S, Chen H, Tsang YH, et al. Anotación funcional sistemática de mutaciones somáticas en cáncer . Célula de cáncer 2018 ; 33 : 450 - 62 .Referencia cruzadaPubMedGoogle Académico
4.↵Bailey M. H., Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Caracterización integral de genes y mutaciones impulsoras del cáncer . Célula 2018 ; 174 : 1034 - 5 .Referencia cruzadaPubMedGoogle Académico
5.↵SamsteinRM, Lee CH, Shoushtari AN, Dr. Hellmann, ShenR, Janjigian YY, et al. La carga mutacional del tumor predice la supervivencia después de la inmunoterapia en múltiples tipos de cáncer . Nat Genet 2019 ; 51 : 202 - 6 .Referencia cruzadaPubMedGoogle Académico
6.↵ Dr. Hellmann, Callahan MK, Awad MM, Calvo E, Ascierto PA, Halcón A, et al. Carga tumoral mutacional y eficacia de la monoterapia con nivolumab y en combinación con ipilimumab en el cáncer de pulmón de células pequeñas . Célula de cáncer 2019 ; 35 : 329 .Google Académico
7.↵ Dr. Hellmann, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab más ipilimumab en cáncer de pulmón con alta carga tumoral mutacional . N Engl J Med 2018 ; 378 : 2093 - 104 .Referencia cruzadaPubMedGoogle Académico
8.↵Robert C, Schachter J, VG largo, Arance A, Grob JJ, Mortero L, et al. Pembrolizumab versus ipilimumab en melanoma avanzado . N Engl J Med 2015 ; 372 : 2521 - 32 .Referencia cruzadaPubMedGoogle Académico
9.↵Borghaei H, Paz-Ares L, Cuerno L, Dr. Spigel, Steins M, Listo NE, et al. Nivolumab versus docetaxel en el cáncer de pulmón no microcítico no escamoso avanzado . N Engl J Med 2015 ; 373 : 1627 - 39 .Referencia cruzadaPubMedGoogle Académico
10.↵ Disco de Middlebrook, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquín A, et al. Asociación de variantes de línea germinal en la región APOBEC3 con riesgo de cáncer y enriquecimiento con mutaciones de la firma APOBEC en tumores . Nat Genet 2016 ; 48 : 1330 - 8 .Referencia cruzadaPubMedGoogle Académico
11.↵Chen ZS, Wen WQ, Beeghly-Fadiel A, Shu XO, Diez-Obrero V, Largo JR, et al. Identificar genes de susceptibilidad putativa y evaluar sus asociaciones con mutaciones somáticas en cánceres humanos . Soy J Hum Genet 2019 ; 105 : 477 - 92 .Google Académico
12.↵ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium . Pan-análisis de cáncer de genomas completos . Naturaleza 2020 ; 578 : 82 - 93 .Referencia cruzadaPubMedGoogle Académico
13.↵esta X, Gu JC, MS Huang, Tannir Nuevo México, Mañana SF, Karam JA, et al. Las variantes genéticas de la línea germinal en genes con mutaciones somáticas significativas en tumores están asociadas con el riesgo y el resultado del carcinoma de células renales . Carcinogénesis 2018 ; 39 : 752 - 7 .Google Académico
14.↵Carter H, marty r, Hofree M, AM bruto, JensenJ, pescado km, et al. Paisaje de interacción de polimorfismos heredados con eventos somáticos en el cáncer . Cáncer Descubrimiento 2017 ; 7 : 410 – 23 .Resumen / Texto Completo GRATISGoogle Académico
15.↵Zhu B, Mukherjee A, Machiela MJ, Canción L, Huá X, Shi JX, et al. Una investigación de la asociación del riesgo de susceptibilidad genética con la carga de mutaciones somáticas en el cáncer de mama . Br J Cáncer 2016 ; 115 : 752 - 60 .Google Académico
16.↵Robles-Espinoza CD, Roberts ND, Chen SY, Leacy FP, Alexandrov LB, Pornputtapong N, et al. El estado de la línea germinal MC1R influye en la carga de mutaciones somáticas en el melanoma . Nat Comun 2016 ; 7 : 12064 .Referencia cruzadaPubMedGoogle Académico
17.↵YuanJ, HuZY, Estimado BA, Zhao SHD, Kensler KH, PiJJ, et al. Análisis integrado de la ascendencia genética y las alteraciones genómicas en los cánceres . Célula de cáncer 2018 ; 34 : 549 .Referencia cruzadaGoogle Académico
18.↵Altshuler DM, DurbinRM, Abecasis GR, Bentley República Dominicana, Chakravarti A, Clark AG, et al. Una referencia global para la variación genética humana . Naturaleza 2015 ; 526 : 68 .Referencia cruzadaPubMedGoogle Académico
19.↵la S, Anterior L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Servicio y métodos de imputación de genotipos de última generación . Nat Genet 2016 ; 48 : 1284-7 . Referencia cruzadaPubMedGoogle Académico
20.↵McCarthy S, la S, Kretzschmar W. Delaneau O, RA de madera, Teumero A, et al. Un panel de referencia de 64.976 haplotipos para la imputación de genotipos . Nat Genet 2016 ; 48 : 1279 - 83 .Referencia cruzadaPubMedGoogle Académico
21.↵ChangCC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee J.J.. PLINK de segunda generación: a la altura del desafío de conjuntos de datos más grandes y ricos . Gigaciencia 2015 ; 4 : 7 .Referencia cruzadaPubMedGoogle Académico

22.↵Yang JA, Lee SH, Goddard YO, pescador PM. GCTA: una herramienta para el análisis de rasgos complejos de todo el genoma . Soy J Hum Genet 2011 ; 88 : 76 – 82 .Referencia cruzadaPubMedGoogle Académico
23.↵JiangLD, Zheng ZL, QiT, Kemper KE, Wray NR, Pescador PM, et al. Una herramienta eficiente en recursos para el análisis de asociación de modelos mixtos de datos a gran escala . Nat Genet 2019 ; 51 : 1749 .Google Académico
24.↵Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, conductor sf, Yo FL, et al. Integración de variaciones genéticas comunes y raras en diversas poblaciones humanas . Naturaleza 2010 ; 467 : 52 - 8 .Referencia cruzadaPubMedGoogle Académico
25.↵yang j, Benjamín B, McEvoy BP, gordon s, Henders AK, Nyholt DR, et al. Los SNP comunes explican una gran proporción de la heredabilidad de la altura humana . Nat Genet 2010 ; 42 : 565 - 9 .Referencia cruzadaPubMedGoogle Académico
26.↵Lee SH, yang j, Chen GB, Ripke S, acero ea, HultmanCM, et al. Estimación de la heredabilidad de SNP a partir de datos de genotipos densos . Soy J Hum Genet 2013 ; 93 : 1151 - 5 .Referencia cruzadaPubMedGoogle Académico
27.↵yang j, Bakshi A ZhuZ, Hemani G, Vanabayzen AAE, Lee SH, et al. La estimación de la varianza genética con variantes imputadas encuentra que la heredabilidad faltante es insignificante para la altura humana y el índice de masa corporal . Nat Genet 2015 ; 47 : 1114 - 20 .Referencia cruzadaPubMedGoogle Académico
28.↵Evans LM, Tahmasbi R, Congelar SI, Abecasis GR, la S, Gazal S, et al. Comparación de métodos que utilizan datos del genoma completo para estimar la heredabilidad y la arquitectura genética de rasgos complejos . Nat Genet 2018 ; 50 : 737 .Referencia cruzadaGoogle Académico
29.↵yang j, Zeng J, Goddard YO, Wray NR, pescador PM. Conceptos, estimación e interpretación de la heredabilidad basada en SNP . Nat Genet 2017 ; 49 : 1304 – U243 .Referencia cruzadaPubMedGoogle Académico
30.↵Zeng J, la R flamenca, WuY, Robinson Sr., Lloyd-Jones LR, yengo l, et al. Firmas de selección negativa en la arquitectura genética de los rasgos complejos humanos . Nat Genet 2018 ; 50 : 746 - 53 .Referencia cruzadaGoogle Académico
31.↵Bulik-Sullivan BK, Hola PR, Finucane Hong Kong, Ripke S, yang j, Grupo de Trabajo de Esquizofrenia del Consorcio de Genómica Psiquiátricaet al. La regresión de la puntuación de LD distingue la confusión de la poligenicidad en los estudios de asociación de todo el genoma . Nat Genet 2015 ; 47 : 291 - 5 .Referencia cruzadaPubMedGoogle Académico
32.↵Bulik-Sullivan B, Finucane Hong Kong, Antila V, Gusev A, Día FR, Hola PR, et al. Un atlas de correlaciones genéticas entre enfermedades y rasgos humanos . Nat Genet 2015 ; 47 : 1236-41 . Referencia cruzadaPubMedGoogle Académico
33.↵Lloyd-Jones LR, Zeng J, Sidorenko J, yengo l, moser g, Kemper KE, et al. Predicción poligénica mejorada mediante regresión múltiple bayesiana en estadísticas de resumen . Nacional Comun 2019 ; 10 : 5086 .Google Académico
34.↵Vilhjalmsson BJ, yang j, Finucane Hong Kong, Gusev A, lindstrom s, Ripke S, et al. Modelar el desequilibrio de ligamiento aumenta la precisión de las puntuaciones de riesgo poligénico . Soy J Hum Genet 2015 ; 97 : 576 - 92 .Referencia cruzadaPubMedGoogle Académico
35.↵Kvale MN, hesselson s, Hoffman TJ, cao y Chan D, Connell S, et al. Informática de genotipado y control de calidad para 100 000 sujetos en la cohorte de Investigación de epidemiología genética sobre la salud y el envejecimiento de adultos (GERA) . Genética 2015 ; 200 : 1051 – 60 .Resumen / Texto Completo GRATISGoogle Académico
36.↵Zhu ZH, Zheng ZL, Zhang FT, WuY, Trzaskowski M, Maier R, et al. Asociaciones causales entre factores de riesgo y enfermedades comunes deducidas de los datos resumidos de GWAS . Nat Comun 2018 ; 9 : 224 .Google Académico
37.↵Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V. Baird D, et al. La plataforma basada en MR admite la inferencia causal sistemática en todo el fenómeno humano . Elife 2018 ; 7 : e34408 .Referencia cruzadaPubMedGoogle Académico
38.↵ZhuZ, Zhang F, Hu H, Bakshi A Robinson Sr., Powell JE, et al. La integración de datos resumidos de estudios GWAS y eQTL predice objetivos de genes de rasgos complejos . Nat Genet 2016 ; 48 : 481 - 7 .Referencia cruzadaPubMedGoogle Académico
39.↵Cepillo U, Claringbould A, Westra HJ, Bonder MJ, Compartir P, Zeng B, et al. Desentrañar la arquitectura poligénica de rasgos complejos mediante el metanálisis de eQTL en sangre . bioRxiv 2018 : 447367 . DOI: https://doi.org/10.1101/447367 .Google Académico
40.↵yang j, Manolio T.A. Pascual LR, Boerwinkle E, caporaso n, Cunningham JM, et al. Partición del genoma de la variación genética para rasgos complejos utilizando SNP comunes . Nat Genet 2011 ; 43 : 519 – U44 .Referencia cruzadaPubMedGoogle Académico
41.↵yue x, Zhao Y, Huang G, LiJ, Zhu J, feng z, et al. Un nuevo compañero de unión a p53 mutante BAG5 estabiliza p53 mutante y promueve los GOF de p53 mutantes en la tumorigénesis . Descubrimiento celular 2016 ; 2 : 16039 .Google Académico
42.↵Moamer A, Hajim IY, binothman n, wang n, LebrunJJ, ali s. Un papel para las subunidades de kinesina-1 KIF5B/KLC1 en la regulación de la plasticidad mesenquimatosa epitelial en la tumorigénesis mamaria . EBioMedicine 2019 ; 45 : 92 - 107 .Google Académico

43.↵Clarke TK, Lupton MK, Fernandez-Pujals AM, estrella j, davies g, cox s, et al. El riesgo poligénico común para el trastorno del espectro autista (TEA) está asociado con la capacidad cognitiva en la población general . Mol Psiquiatría 2016 ; 21 : 419 - 25 .Google Académico
44.↵moser g, Lee SH, Hayes BJ, Goddard YO, Wray NR, Pescador PMJPG. Análisis simultáneo de descubrimiento, estimación y predicción de rasgos complejos utilizando un modelo de mezcla bayesiano . PLoS Genet 2015 ; 11 : e1004969 .Referencia cruzadaPubMedGoogle Académico
45.↵NiG, Zeng J, Revez JR, Wang Y, Ge T, Restaudi R, et al. Una evaluación integral de los métodos de puntuación poligénica en cohortes en trastornos psiquiátricos . medRxiv 2020 . DOI: https://doi.org/10.1101/2020.09.10.20192310 .Google Académico
46.↵ J verde, Cairns B.J., Casabonne D, Wright Florida, reeves g, Beral V, et al. Altura e incidencia de cáncer en el Million Women Study: cohorte prospectiva y metanálisis de estudios prospectivos de altura y riesgo total de cáncer . Lancet Oncol 2011 ; 12 : 785 - 94 .Referencia cruzadaPubMedGoogle Académico
47.↵Walter R.B., BraskyTM, Buckley SA, alfarero JD, Blanco E. La altura como factor explicativo de las diferencias sexuales en el cáncer humano . Instituto Nacional del Cáncer J 2013 ; 105 : 860-8 . Referencia cruzadaPubMedGoogle Académico
48.↵qian f, Wang SF, mitchell j, McGuffog L, Barrowdale D, leslie g, et al. La altura y el índice de masa corporal como modificadores del riesgo de cáncer de mama en portadoras de mutación BRCA1/2: un estudio de aleatorización mendeliana . Instituto Nacional del Cáncer J 2019 ; 111 : 350 - 64 .Google Académico
49.↵Ong JS, Un JY, Ley MH, hombre blanco dc, Neale RE, Gharahkhani P, et al. Altura y riesgo general de cáncer y mortalidad: evidencia de un estudio de aleatorización mendeliana en 310 000 participantes del Biobanco del Reino Unido . Brit J Cáncer 2018 ; 118 : 1262-7 . Google Académico
50.↵Rozek LS, Herrón CM, Greenson JK, Moreno V, capella g, Rennert G, et al. El tabaquismo, el género y el origen étnico predicen mutaciones BRAF somáticas en el cáncer colorrectal . Biomarcadores del Epidemiol del Cáncer Prev 2010 ; 19 : 838 - 43 .Resumen / Texto Completo GRATISGoogle Académico
51.↵Brennan JA, Boyle JO, Koch WM, Goodman SN, Hruban RH, por YJ, et al. Asociación entre el tabaquismo y la mutación del gen P53 en el carcinoma de células escamosas de cabeza y cuello . New Engl J Med 1995 ; 332 : 712 - 7 .Referencia cruzadaPubMedGoogle Académico
52.↵Blackford A, Parmigiani G, Kensler TW, Wolfgang C, jones s, Zhang XS, et al. Mutaciones genéticas asociadas con el tabaquismo en el cáncer de páncreas . Cáncer Res 2009 ; 69 : 3681 - 8 .Resumen / Texto Completo GRATISGoogle Académico
53.↵Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Firmas mutacionales asociadas con el tabaquismo en el cáncer humano . Ciencia 2016 ; 354 : 618 - 22 .Resumen / Texto Completo GRATISGoogle Académico
54.↵Kucab JE, Zou XQ, morganella s, joel m, nanda as, E grande, et al. Un compendio de firmas mutacionales de agentes ambientales . Célula 2019 ; 177 : 821 .Google Académico
55.↵Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Cooren T, Maughan EF, et al. Tabaquismo y mutaciones somáticas en el epitelio bronquial humano . Naturaleza 2020 ; 578 : 266 - 72 .Referencia cruzadaPubMedGoogle Académico
56.↵burgess s, Butterworth A, Thompson SGJGe. Análisis de aleatorización mendeliana con múltiples variantes genéticas utilizando datos resumidos . Genet Epidemiol 2013 ; 37 : 658 - 65 .Referencia cruzadaPubMedGoogle Académico
57.↵Pesch B, Kendzia B, Gustavsson P, Jöckel KH, Juan G, Pohlabeln H, et al. Tabaquismo y cáncer de pulmón: estimaciones del riesgo relativo para los principales tipos histológicos a partir de un análisis combinado de estudios de casos y controles . Int J Cáncer 2012 ; 131 : 1210 - 9 .Referencia cruzadaPubMedGoogle Académico
58.↵Secretan B, Straif K, BaanR, Gran Y, El Ghissassi F, Bouvard V, et al. Una revisión de carcinógenos humanos-parte E: tabaco, nuez de areca, alcohol, humo de carbón y pescado salado . Lancet Oncol 2009 ; 10 : 1033 - 4 .Referencia cruzadaPubMedGoogle Académico
59.↵ A aguda, Bonet C, Travier N, Gonzalez CA, Viñedos Bueno-de-Mesquita HB, et al. Impacto del consumo de cigarrillos en el riesgo de cáncer en la investigación prospectiva europea sobre el estudio del cáncer y la nutrición . J Clin Oncol 2012 ; 30 : 4550 – 7 .Resumen / Texto Completo GRATISGoogle Académico
60.↵Pescador PM, Hemani G, Vanabayzen AA, Chen G-B, Lee SH, Wray NR, et al. Poder estadístico para detectar la (co)varianza genética de rasgos complejos utilizando datos de SNP en muestras no relacionadas . PLoS Genet 2014 ; 10 : e1004269 .Referencia cruzadaPubMedGoogle Académico
61.↵Knudson AG Jr. . Mutación y cáncer: estudio estadístico del retinoblastoma . Proc Natl Acad Sci USA 1971 ; 68 : 820 - 3 .Resumen / Texto Completo GRATISGoogle Académico Anteriorpróximo
Related Documents