Kinase Inhibitors and Nucleoside Analogues as Novel Therapies to Inhibit HIV-1 or ZEBOV Replication by Stephen D. S. McCarthy A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Laboratory Medicine and Pathobiology University of Toronto © Copyright by Stephen D. S. McCarthy (2017)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Kinase Inhibitors and Nucleoside Analogues as Novel
Therapies to Inhibit HIV-1 or ZEBOV Replication
by
Stephen D. S. McCarthy
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Laboratory Medicine and Pathobiology University of Toronto
© Copyright by Stephen D. S. McCarthy (2017)

ii
Kinase Inhibitors and Nucleoside Analogues as Novel
Therapies to Inhibit HIV-1 or ZEBOV Replication
Stephen D.S. McCarthy
Doctor of Philosophy
Graduate Department of Laboratory Medicine and Pathobiology
University of Toronto
2017
Abstract
Without a vaccine for Human Immunodeficiency Virus type 1 (HIV-1), or approved therapy for
treating Zaire Ebolavirus (ZEBOV) infection, new means to treat either virus during acute
infection are under intense investigation. Repurposing tyrosine kinase inhibitors of known
specificity may not only inhibit HIV-1 replication, but also treat associated inflammation or
neurocognitive disorders caused by chronic HIV-1 infection. Moreover, tyrosine kinase
inhibitors may effectively treat other infections, including ZEBOV. In addition, established
nucleoside/nucleotide analogues that effectively inhibit HIV-1 infection, could also be
repurposed to inhibit ZEBOV replication.
In this work the role of two host cell kinases, cellular protoncogene SRC (c-SRC) and Protein
Tyrosine Kinase 2 Beta (PTK2B), were found to have key roles during early HIV-1 replication in
primary activated CD4+ T-cells ex vivo. siRNA knockdown of either kinase increased
intracellular reverse transcripts and decreased nuclear proviral integration, suggesting they act at
the level of pre-integration complex (PIC) formation or PIC nuclear translocation. c-SRC siRNA
knockdown consistently reduced p24 levels of IIIB(X4) and Ba-L(R5) infection, or luciferase

iii
activity of HXB2(X4) or JR-FL(R5) recombinant viruses, prompting further drug inhibition
studies of this kinase. Four c-SRC kinase inhibitors (dasatinib, saracatinib, KX2-391 and SRC
Inhibitor-1) significantly reduced HXB2 and JR-FL infection in primary CD4+ T-cells. Thus,
these potent c-SRC inhibitors should be further evaluated in humanized mouse models of HIV-1
infection.
During 2014-16, the Ebola outbreak in West Africa prompted us to rapidly assess whether
conventional nucleoside analogs could inhibit in vitro ZEBOV replication. Employing a new
lifecycle model of ZEBOV infection in level 2 biocontainment, combinations of nucleoside
analogues and interferons were found to synergistically inhibit ZEBOV replication. These
included zidovudine, lamivudine and tenofovir, confirmed to show antiviral activity against fully
infectious ZEBOV-GFP in level 4 biocontainment. Findings from this thesis provided the
rationale for further preclinical development of nucleoside analogue combination treatments, and
a phase II EVD trial evaluating recombinant interferon in Guinea. Pre-clinical results using c-
SRC kinase inhibitors also suggest that this approach could also be effective in EVD.

iv
Acknowledgments
First and foremost, I will thank my mentor Dr. Donald R. Branch, for his inspiration, invaluable
advice and tremendous support throughout my time in his laboratory at Canadian Blood
Services. I also extend thanks to my Ph D advisory committee, Dr. Rupert Kaul and Dr. Dwayne
Barber, for their valuable advice, direction and input. I will next acknowledge the hard work of
Dr. Thomas Hoenen, Dr. Danila Leontyev, Dr. Anton Neschadim, Dr. Daniel Jung, Dr. Trina
Racine and Beata Majchrzak-Kita, who contributed immensely to the many joint projects related
to this work. As well, I am very grateful to Darinka Sakac, Yulia Petrenko and Amanda
Harrison Wong, who generously gave their time to train me, and Dr. Reed Siemieniuk for
insightful conversations. I also thank Dr. Eleanor Fish and Dr. Gary Kobinger for their
collaboration on a completely novel direction of this project, which has proven to be fruitful new
avenue of research.
I also thank my dear colleagues in the Branch laboratory, Evgenia Bloch, Carlyn Figueiredo,
Cindy Tong, Megan Blacquiere, Bonnie Lewis, Minji Kim, Eric Lai and Beth Binnington, for the
many opportunities to collaborate, abundant discussions, support of my ideas and work, and for
creating a joyful and lively lab environment. I also want to thank undergraduate summer
students Hannah Kozlowski, Shawn Goyal, Pauline Nicoletti, Ninon Guichard and Janette
Spears, whom I closely mentored and had good fortune to collaborate with over the years.
I also want to thank organizations that shaped my thinking around HIV and Ebola research. I am
eternally grateful for the support of my graduate department at the University of Toronto,
Laboratory Medicine and Pathobiology. In particular, I deeply appreciate the wisdom and
mentorship of Dr. Harry Elsholz, Dr. Maria Rozakis, Ferzeen Dharas-Sammy, Rama Ponda and
Katie Babcock, who encouraged my participation in the Three Minute Thesis competition, to
represent the department at the CIHR Canadian Student Health Research Forum in Winnipeg,
and to participate in our vibrant student union CLAMPS. Additionally, I am thankful to Sergio
Martinez, Duncan MacLachlan and Adam Busch, who at the AIDS Committee of Toronto taught
me valuable perspectives on people living with HIV-1, access to STI testing, and health care
inequality. I am also very thankful of Dr. Dana Devine and Don LaPierre at Canadian Blood

v
Services, who gave me a platform to advocate for blood donor equality at a national level. And I
am deeply thankful of Dr. Logan and Elizabeth Cohen, who through courageous actions,
reported potential alternative therapies to treat Liberian Ebola patients in 2014.
I will also take the opportunity to acknowledge the financial support of NSERC (CGS M
scholarship), Canadian Blood Services (GFP scholarship and support from the Centre for
Innovation to Dr. Branch), CIHR (Vanier CGS D scholarship, and support to Dr. Fish), the
Ontario HIV Treatment Network (Dr. Branch) and Health Canada (Dr. Branch), which have
financially supported me for the entirety of this project.
Finally, I would like to thank my dear parents, brothers and close friends. Your love and
unconditional support was the foundation of my success in graduate school.

vi
Table of Contents
Page Abstract ........................................................................................................................................... ii
Acknowledgments.......................................................................................................................... iv
Table of Contents ........................................................................................................................... vi
List of Tables ............................................................................................................................... viii
List of Figures ................................................................................................................................ ix
List of Abbreviations .................................................................................................................... xii
Chapter 1: Introduction ................................................................................................................... 1
1.1 HIV-1 and Ebola: Global Health Problems .......................................................................... 2
1.2 Human Immunodeficiency Virus (HIV) ............................................................................... 5
1.2.1 HIV-1 Epidemiology, Transmission and Replication Cycle .......................................... 5
1.2.2 HIV-1 Treatments, Potential Vaccines, and New Therapeutics ................................... 13
1.2.3 Host Kinases as Targets for HIV-1 Inhibition .............................................................. 17
1.2.4 The SRC Family of Non-Receptor Tyrosine Kinases in HIV-1 Infection ................... 22
1.2.5 Role of c-SRC in HIV-1 Infection ................................................................................ 37
1.2.6 Role of PTK2B in HIV-1 Infection .............................................................................. 42
1.3 Ebola Virus (EBOV) ........................................................................................................... 47
1.3.1 ZEBOV Epidemiology, Transmission and Replication Cycle ..................................... 47
1.3.2 ZEBOV Clinical Trials, Potential Vaccines and New Therapeutics ............................ 55
1.3.3 Repositioning Nucleoside Analogues for ZEBOV Inhibition ...................................... 59
1.3.4 Testing Other Potent Nucleoside/Nucleotide Analogues: Zidovudine, Lamivudine and
Tenofovir ............................................................................................................................... 69
1.4 Statement of the Problem, Rationale, Hypotheses and Objectives of this Work ................ 87
1.4.1 Statement of the Problem ............................................................................................. 87
1.4.2 Rationale ....................................................................................................................... 87
1.4.3 Hypotheses.................................................................................................................... 88
1.4.4 Objectives of this Work ................................................................................................ 89
1.4.5 Organization of the Thesis ............................................................................................ 89

vii
Chapter 2: c-SRC and PTK2B Protein Tyrosine Kinases Play Protective Roles in Early HIV-1
Infection of CD4+ T-Cell Lines. ................................................................................................... 91
2.1 Abstract ............................................................................................................................... 92
2.2 Introduction ......................................................................................................................... 93
2.3 Materials and Methods ........................................................................................................ 95
2.4 Results ............................................................................................................................... 101
2.5 Discussion ......................................................................................................................... 113
Chapter 3: c-SRC Protein Tyrosine Kinase Regulates Early HIV-1 Infection Post-Entry ......... 115
3.1 Abstract ............................................................................................................................. 116
3.2 Introduction ....................................................................................................................... 117
3.3 Materials and Methods ...................................................................................................... 119
3.4 Results ............................................................................................................................... 125
3.5 Discussion ......................................................................................................................... 140
Chapter 4: A Rapid Screening Assay Identifies Monotherapy with Interferon-ß and Combination
Therapies with Nucleoside Analogues as Effective Inhibitors of Ebola Virus .......................... 144
4.1 Abstract ............................................................................................................................. 145
4.2 Introduction ....................................................................................................................... 146
4.3 Materials and Methods ...................................................................................................... 148
4.4 Results ............................................................................................................................... 152
4.5 Discussion ......................................................................................................................... 169
Chapter 5: Key Findings, Future Perspectives, Conclusions and Broader Significance ............ 173
5.1 Key Findings, Limitations and Future Perspectives.......................................................... 174
5.2 Conclusions and Broader Significance ............................................................................. 189
References ................................................................................................................................... 195
Copyright Acknowledgments ..................................................................................................... 245

viii
List of Tables
Chapter 1
Table 1.1: Experimental drugs, therapeutics and vaccines fast-tracked for emergency II/III Ebola
clinical trials in 2014-2016. ............................................................................................................ 4
Chapter 4
Table 4.1: Fi and CI values for two-drug combination treatments. ............................................ 164
Table 4.2: Fi and CI values for three-drug combination treatments. .......................................... 165

ix
List of Figures
Chapter 1
Fig. 1.1: Global and national spread of HIV-1/AIDS. .................................................................... 6
Fig. 1.2: HIV-1 virion and genomic structure................................................................................. 8
Fig. 1.3: The HIV-1 replication cycle in T-cells. .......................................................................... 10
Fig. 1.4: FDA-approved kinase inhibitors. ................................................................................... 20
Fig. 1.5: Domain structure of chicken SRC-family kinases (SFKs). ............................................ 24
Fig. 1.6: Nef recycling of LCK and TCR impairs formation of the immunological synapse. ..... 27
Fig. 1.7: c-SRC domains, tertiary structure, and regulation. ........................................................ 38
Fig. 1.8: PTK2B domains, tertiary structure, and regulation. ....................................................... 43
Fig. 1.9: Spread of ZEBOV in West Africa during 2014-16. ....................................................... 48
Fig. 1.10: ZEBOV virion and genome structure. .......................................................................... 50
Fig. 1.11: The intracellular ZEBOV replication cycle. ................................................................. 52
Fig. 1.12: General structure of polymerases from the three main groups of RNA viruses and
retroviruses. ................................................................................................................................... 70
Fig. 1.13: Predicted structure of the RdRP L of ZEBOV. ............................................................ 72
Fig. 1.14: Model of ribonucleoside triphosphates in complex with RdRP L. .............................. 74
Fig. 1.15: Example nucleoside/nucleotide docking to the ZEBOV RdRP L. ............................... 76
Fig. 1.16: Nucleotide analogues binding ZEBOV RdRP L in the presence of NTPs................... 85
Chapter 2
Fig. 2.1: Western blot of phosphorylated proteins from serum-starved T-cells shortly after HIV-1
infection. ..................................................................................................................................... 102
Fig. 2.2: Jurkat T-cell lines inhibited with SRC-family kinase inhibitors. ................................. 104
Fig. 2.3: Cell survival was not significantly different between c-SRC drug or adenovirus vector
treatments. ................................................................................................................................... 105
Fig. 2.4: T-cell lines were transduced with adenovectors expressing c-SRC mutants. .............. 107
Fig. 2.5: siRNA titration and time-course optimization in Jurkat E6-1 cells. ............................ 109

x
Fig. 2.6: siRNA knockdown in Jurkat E6-1 cells infected with either VSV-G/HIV-1 or HXB2
virus............................................................................................................................................. 110
Fig. 2.7: qPCR of HXB2 cDNA extracted from Jurkat E6-1 following siRNA knockdown. .... 112
Chapter 3
Fig. 3.1: Cell Purity and Cell Viability of Enriched CD4+ T-Cells nucleofected with siRNA. . 126
Fig. 3.2: SRC-Family Drug Inhibitors do not Increase Necrosis or Apoptosis of Activated CD4+
T-Lymphocytes. .......................................................................................................................... 127
Fig. 3.3: Pre-treating CD4+ T-cells with c-SRC kinase inhibitors prior to HXB2 or JR-FL
infection. ..................................................................................................................................... 128
Fig. 3.4: c-SRC and PTK2B activation shortly after HIV-1 infection........................................ 130
Fig. 3.5: Luciferase reporter activity, viral integration and p24 production after c-SRC or PTK2B
siRNA knockdown. ..................................................................................................................... 132
Fig. 3.6: Cell surface receptor expression and RT activity. ........................................................ 134
Fig. 3.7: qPCR and PERT Assay Standard Curves. .................................................................... 136
Fig. 3.8: Time-Course and qPCR Melting Curves of HIV-1 DNA Targets. .............................. 137
Fig. 3.9: qPCR of early HXB2 or JR-FL infection after c-SRC or PTK2B siRNA knockdown.138
Fig. 3.10: qPCR of HXB2 or JR-FL Infection in CD4+ T-Cells from Three Separate Donors. . 139
Fig. 3.11: Summary schematic of the proposed mechanism for c-SRC involvement during early
HIV-1 infection. .......................................................................................................................... 140
Chapter 4
Fig. 4.1: Transcription and replication competent virus-like particle (trVLP) assay. ................ 149
Fig. 4.2: IFNs and nucleoside analogues inhibit Ebola mini-genome replication in vitro. ........ 153
Fig. 4.3: IFNs, toremifene, and nucleoside analogues inhibit trVLP replication. ...................... 154
Fig. 4.4: IFNs, toremifene and nucleoside analogues administered 24 hrs post-exposure inhibit
Ebola-mini-genome replication. ................................................................................................. 156
Fig. 4.5: No direct effect of drugs on Renilla luciferase reporter assay. .................................... 157
Fig. 4.6: CDV treatment increases trVLP replication. ................................................................ 158

xi
Fig. 4.7: IFNs, toremifene and nucleoside analogues reduce trVLP replication and transcription.
..................................................................................................................................................... 160
Fig. 4.8: 2 and 3 drug combinations synergistically inhibit Ebola trVLP infection. .................. 163
Fig. 4.9: IFNs, toremifene and nucleoside analogues administered 24 hrs post-exposure inhibit
ZEBOV-GFP............................................................................................................................... 167
Fig. 4.10: Synergistic 2 and 3 drug combinations against trVLP-LUC infection inhibit fully
infectious ZEBOV-GFP. ............................................................................................................. 168
Chapter 5
Fig. 5.1: Aberrant TCR signaling in Jurkat T-cells. ................................................................... 177
Fig. 5.2: Model of FPV-RTP and TFV-DP binding the RdRP L nucleotide pocket of ZEBOV.
..................................................................................................................................................... 187

xii
List of Abbreviations
(-)ssRNA negative-sense single-stranded RNA
(+)ssRNA positive-sense single-stranded RNA
1-LTR One Long Terminal Repeat
2ΔΔCT
Two delta delta Ct, comparative gene expression method
2-LTR Two Long Terminal Repeats
IIIB formerly HTLV-III B/H9, X4-tropic HIV-1
3P Triphosphate
3TC lamivudine, epivir
7-AAD 7-Aminoactinomycin D
α4β7 gut-specific homing integrin
Ab Antibody
ABL1 Abelson murine Leukemia viral oncogene homolog 1
ACK1 Activated CDC42 Kinase 1
ADV Adenovirus
AIDS Acquired Immunodeficiency Syndrome
ALT Alanine Transaminase
AMPK AMP-Activated Protein Kinase
ANP Acyclic Nucleotide Phosphonate
APC Antigen Presenting Cell
APC Allophycocyanin
APOBEC3G Apoliprotein B mRNA-editing Enzyme, Catalytic polypeptide-like 3G
ARG Abelson-Related Gene
ART Antiretroviral Therapy
ATCC American Type Culture Collection
ATP Adenosine Triphosphate
AZT Azidothymidine, zidovudine, retrovir
Ba-L Ba-Lung, R5-tropic HIV-1
BCR B-Cell Receptor
BCV Brincidofovir, CMX001
BCX4430 Immucillin-A
BDBV Bundibugyo Ebolavirus
BID Twice a Day
BLK B Lymphocyte Kinase
BMVEC Brain Microvascular Endothelial Cells
bNAB broadly Neutralizing Antibody
Bp Base Pair
BSL Biosafety Level
BVDV Bovine Diarrhea Virus
CA Capsid protein, p24
CADK Calcium-Dependent Tyrosine Kinase
cART combination Antiretroviral Therapy
CCL5 Chemokine (C-C motif) Ligand 5, RANTES

xiii
CCR5 Chemokine (C-C motif) Receptor 5
CD Cluster of Differentiation
CDC USA Centers for Disease Control and Prevention
CDV Cidofovir, CFV
cDNA complementary Deoxyribonucleic Acid
ChAd3-EBO-Z Chimpanzee Adenovirus type-3 vaccine vector expressing ZEBOV GP
CHAPS 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
ChIP Chromatin Immunoprecipitation
CHK C-terminal SRC kinase-Homologous Kinase
CI Combination Index
CML Chronic Myelogenous Leukemia
CMV Cytomegalovirus
CNS Central Nervous System
Co-IP Co-Immunoprecipitation
CR3 Complement Receptor 3
CRISPR/Cas9 Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR
associated protein 9
cRNA copy RNA
CRMP2 Collapsin Response Mediator Protein 2
CSF-1R Colony Stimulating Factor 1 Receptor
CSK C-terminal SRC Kinase
c-SRC cellular-SRC, proto-oncogene protein tyrosine kinase c-SRC, pp60c-SRC
CVID Common Variable Immunodeficiency
CXCR4 Chemokine (C-X-C motif) Receptor 4
c-YES cellular homolog of the Yamaguchi Sarcoma virus oncogene
DC Dendritic Cell
DC-SIGN Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-
integrin
DFP 4-amino substituted Diphenylfuropyrimidine
DISC Death-Inducing Signaling Complex
DMEM Dulbeco’s Modified Eagle’s Medium
DMSO Dimethyl sulfoxide
DN Dominant Negative
DNA Deoxyribonucleic Acid
DNAL4 DNA Ligase 4
dNTP deoxynucleoside Triphosphate
DP Dominant Positive
DP Diphosphate
ds DNA double-stranded Deoxyribonucleic Acid
DTT Dithiothreitol
DV Dengue Virus
DYRK1A Dual specificity tyrosine-phosphorylation-Regulated Kinase 1A
Early RT Early Reverse Transcripts
EBOV Ebolavirus

xiv
ECL Enhanced Chemiluminescence
EDTA Ethylenediaminetetraacetic Acid
EGFR Epidermal Growth Factor Receptor
ELISA Enzyme-Linked Immunosorbant Assay
EMD Emerin
Env Envelope
ERK Extracellular signal–Regulated Kinase
ESCRT Endosomal Sorting Complexes Required for Transport
ETC Ebola Treatment Center
EV Empty Vector
EV Enterovirus
EVD Ebola Virus Disease
EYFP Enhanced Yellow Fluorescent Protein
FACS Fluorescence-Activated Cell Sorting
FAK Focal Adhesion Kinase
FasR Fas Receptor
FAT Focal Adhesion Targeting domain
FBS Fetal Bovine Serum
FcγR Fc-gamma Receptor
FDA Food and Drug Administration (USA)
FERM Four point 1/Ezrin/Radixin/Moesin domain
FFU Focus-Forming Units
FGR Feline Gardner-Rasheed tyrosine kinase
FITC Fluorescein Isothiocyanate
FMDV Foot-and-Mouth Disease Virus
FPV Favipiravir, T-705
FSC Forward Scatter
FSV Fujinami Sarcoma Virus
FTC emtricitabine
FYB FYN Binding protein
FYN FGR- and c-YES-related protein kinase
Gag Group-specific antigen, polyprotein precursor Pr55
GAPDH Glyceraldehyde 3-Phosphate Dehydrogenase
GFP Green Fluorescent Protein
GM130 Golgi Matrix protein 130
gMFI geometric Mean Fluorescence Intensity
GORASP1 Golgi Reassembly-Stacking Protein 1
GP Glycoprotein
GP1,2 GP1 and GP2 glycoprotein
gp41 glycoprotein 41
gp120 glycoprotein 120
gp160 glycoprotein 160
GPCL cleaved Glycoprotein
GPCR G-Protein Coupled Receptor

xv
GRB2 Growth Factor Receptor-Bound protein 2
GS-5734 2-ethylbutyl l-alaninate phosphoramidate derivative
GTPase Guanosine Triphosphatase
HAART Highly Active Antiretroviral Therapy
HBV Hepatitis B Virus
HCK Hematopoietic Cell Kinase
HCV Hepatitis C Virus
HDAC Histone Deacetylase
HDP 3-hexadecyloxy-1-propanol
HEK 293T Human Embryonic Kidney cells 293 SV40 Large T-antigen
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer
HFV Human Foamy Virus
HIV-1 Human Immunodeficiency Virus type-1
HIV-2 Human Immunodeficiency Virus type-2
hnRNP-K heterogeneous nuclear Ribonucleoprotein K
HPC Hematopoietic Progenitor Cells
HRP Horseradish Peroxidase
HS Homo sapiens
HSV Herpes Simplex Virus
HTLV-1 Human T-cell Lymphotropic Virus type I
HXB2 HXB clone 2, X4-tropic HIV-1
IAV Influenza A Virus
ICAM-1 Intercellular Adhesion Molecule 1
IDR Intrinsically Disordered Region
IFN Interferon
IgG Immunoglobulin G
IL Interleukin
IL-2R Interleukin-2 Receptor
IN Integrase
IP Immunoprecipitation
IRF3 IFN Regulatory Factor 3
IS Immunological Synapse
ITAM Immunoreceptor Tyrosine-based Activation Motifs
JKC Jurkat C T-cells
JNK c-Jun N-terminal Kinase
JR-FL JR-Frontal Lobe, R5-tropic HIV-1
KNH1207 clinical isolate, R5-tropic HIV-1
KS Kaposi Sarcoma
Late RT Late Reverse Transcripts
LCK Lymphocyte-specific protein tyrosine Kinase
LDH Lactate Dehydrogenase
LEDGF Lens Epithelium-Derived Growth Factor, p75
LFA-1 Lymphocyte Function-associated Antigen-1
L-LEC Lung Lymphatic Endothelial Cells

xvi
LRA Latency Reversing Agents
LTR Long Terminal Repeat
LUC Luciferase
LYN LCK/Yes Novel protein tyrosine kinase
MA Matrix protein, p17
mAB monoclonal Antibody
MAPK Mitogen-Activated Protein Kinase
M-CSF Macrophage Colony-Stimulating Factor
MERS-CoV Middle East Respiratory Syndrome Coronavirus
MFI Median Fluorescent Intensity
MHC Major Histocompatibility Complex
MIP-1β Macrophage Inflammatory Protein 1 beta
MOI Multiplicity of Infection
MP Monophosphate
mRNA messenger Ribonucleic Acid
MS2 bacteriophage MS2
MSM Men who have Sex with Men
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
MVA-BN-Filo Modified Vaccinia Ankara expressing ZEBOV GP booster vaccine
MVC Maraviroc
NC Nucleocapsid, p7
Nef Negative effector
NF-κB Nuclear Factor and activator of transcription kappa B
NFAT Nuclear Factor of Activated T-cells
NHEJ Non-Homologous End-Join
NHP Non Human Primate
NIAID US National Institute of Allergy and Infectious Diseases
NNRTI Non-Nucleoside Reverse Transcriptase Inhibitor
NOD Non-Obese Diabetic
Nonidet P-40 octylphenoxypolyethoxyethanol
NoRT No Reverse Transcriptase
NP Nucleocapsid Protein
NPC1 Niemann-Pick C1
NRTI Nucleoside Reverse Transcriptase Inhibitor
N5SA Nonstructural protein 5A
N5SB Nonstructural protein 5B
NT Non-targeting siRNA
NTC No Template Control
NTP Nucleotide Triphosphate
P2Y2 P2Y purinoceptor 2
p38α mitogen-activated protein kinase 14
p53 tumor suppressor protein p53
PAK2 p21-Activated Kinase 2
PBMC Peripheral Blood Mononuclear Cell

xvii
PBS Phosphate Buffered Saline
PCR Polymerase Chain Reaction
PDGF-R Platelet-Derived Growth Factor Receptor
PE Phycoerythrin
PERT Product-Enhanced RT assay
PHA Phytohaemagglutinin
PHK Phosphorylase Kinase
PI3K Phosphoinositide 3-Kinase
PIC Pre-Integration Complex
PKC Protein Kinase C
PLC-γ1 Phospholipase C gamma 1
PMSF Phenylmethanesulfonyl Fluoride
Pol Polymerase
PP1 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo-d-3,4-pyrimidine PP2 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine PPE Personel Protective Equipment
PR Protease
PREVAIL Partnership for Research on Ebola Vaccines In Liberia
PTK Protein Tyrosine Kinase
PTK2B Protein Tyrosine Kinase 2 Beta, FAK2, Pyk2
PTP Protein Tyrosine Phosphatase
PTP-PEST Protein Tyrosine Phosphatase Non-receptor type 12, PTPN12
PTPRA Receptor-type Tyrosine-Protein Phosphatase alpha, PTP-α
Q151Mc A62V, V75I, F77L, F116Y and Q151M complex
qPCR quantitative Polymerase Chain Reaction, real-time PCR
qRT-PCR quantitative Reverse Transcription Polymerase Chain Reaction
R5 CCR5-tropic HIV-1, M-tropic
Rac Rac subfamily of Rho small GTPases
rAD recombinant Adenoviral Vector
Ras Ras family of small GTPases
RdRP L RNA-dependent RNA polymerase L
RESTV Reston Ebolavirus
Rev Regulator of expression of virion proteins
RIPA Radioimmunoprecipitation Assay
RISC RNA-Induced Silencing Complex
RLU Relative Luciferase Units
RNA Ribonucleic Acid
RNAse H Ribonuclease H, p15
RPMI Roswell Park Memorial Institute medium 1640
RSK1 Ribosomal S6 Kinase 1
RSV Rous Sarcoma Virus
RT Reverse Transcriptase
RTC Reverse Transcriptase Complex
RTP Ribofuranosyl Triphosphate

xviii
RVFV Rift Valley Fever Virus
rVSV-ZEBOV-GP recombinant VSV vaccine expressing ZEBOV GP
Sam68 SRC-Associated substrate in Mitosis of 68 kDa
SAMHD1 SAM domain and HD domain-containing protein 1
SARS-CoV Severe Acute Respiratory Syndrome Coronavirus
SCID Severe Combined Immunodeficiency
SDF-1α Stromal cell-Derived Factor 1 alpha, CXCL12
SDS-PAGE Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
SEM Standard Error of the Mean
SFK SRC Family of non-receptor tyrosine kinases
sGP soluble Glycoprotein
SH SRC Homology
SHP-1 Protein Tyrosine Phosphatase Non-receptor type 6, PTPN6
SHP-2 Protein Tyrosine Phosphatase Non-receptor type 11, PTPN11
siRNA small interfering RNA
SIV Simian Immunodeficiency Virus
SNALPS Stable Nucleic Acid Lipid Particles
SRC-I1 SRC Inhibitor-1
SSC Side Scatter
STAT1 Signal Transducer and Activator of Transcription protein 1
STI Sexually Transmitted Infection
SU6656 (3Z)-N,N-Dimethyl-2-oxo-3-(4,5,6,7-tetrahydro-1H-indol-2-
ylmethylidene)-2,3-dihydro-1H-indole-5-sulfonamide
SUDV Sudan Ebolavirus
SYBR Green nucleic acid staining dye
SYK Spleen-associated tyrosine Kinase
T-20 enfuvirtide
TAFV Taï Forest Ebolavirus
TAR Transactivation Response region
Tat Trans-activator of transcription, p16, p14
TBS Tris-Buffered Saline
TCID50 50% Tissue Culture Infective Dose
TCR T-Cell Receptor
TDF Tenofovir Disoproxil Fumarate
TEMED Tetramethylethylenediamine
Tev Tat, Env and Rev fusion protein
TFV Tenofovir, Viread
TGF-β Transforming Growth Factor beta
Th17 T helper 17
TIM-1 T-cell Immunoglobulin and Mucin Domain 1
TKM-Ebola Tekmira Pharmaceuticals siRNA treatment
TLR Toll-Like Receptor
TM DNA melting temperature
TNF-α Tumor Necrosis Factor alpha

xix
TOR Toremifene citrate
TP Triphosphate
Treg Regulatory T-cell
tRNA transfer Ribonucleic Acid
trVLP transcription and replication competent Virus-Like Particle
UCHT-1 anti-human CD3 antibody, clone UCHT-1
USAMRIID United States Army Medical Research Institute of Infectious Diseases
VACV Vaccinia Virus
VAV1 guanine nucleotide exchange factor 1
v-FPS viral FPS protein tyrosine kinase (avian)
Vif Viral infectivity factor
VMMC Voluntary Medical Male Circumcision
VP24 Viral Protein 24, matrix protein
VP30 Viral Protein 30, transcription activator
VP35 Viral Protein 35, polymerase cofactor
VP40 Viral Protein 40, matrix protein
Vpr Viral protein r
Vpu Viral protein u
Vpx Viral protein x
vRNA viral RNA
VS Virological Synapse
v-SRC viral-SRC
VSV Vesicular Stomatitis Virus
VSV-G Vesicular Stomatitis Virus Envelope G protein
VSV-G/HIV-1 VSV-G pseudoenveloped HIV virus
VZV Varicella Zoster Virus
WASp Wiskott-Aldrich Syndrome protein
WHO World Health Organization
WT Wild Type
X4 CXCR4-tropic HIV-1, T-tropic
ZEBOV Zaire Ebolavirus
ZMapp Mapp Biopharmaceutical monoclonal antibody cocktail

1
Chapter 1: Introduction

2
1.1 HIV-1 and Ebola: Global Health Problems
Notwithstanding decades of research in Human Immunodeficiency Virus type 1 (HIV-1) vaccine
design [1], prevention strategies to reduce transmission [2], or drug treatments to limit and
control infection [3], there are more than 36.7 million people living with HIV-1 worldwide as of
2015, and 1.1 million Acquired Immunodeficiency Syndrome (AIDS)-related deaths caused by
HIV-1 each year [4]. Another virus of global concern is Ebola virus (EBOV), which infected at
least 28,616 people and caused 11,310 deaths during the 2014-16 Zaire Ebola virus (ZEBOV)
outbreak in West Africa [5]. EBOV was discovered before HIV-1, yet there are no approved
antiviral agents to treat those infected [6]. The HIV-1 pandemic and EBOV outbreaks in Africa
in recent years have caused global shifts in how governments treat viral illnesses that
disproportionally affect the world’s poor, people who face stigma, and live in locations with
limited access to medications, trained medical staff or well-equipped hospitals. This has also
prompted changes in preclinical drug discovery research, to ultimately find effective treatments
that can prevent, cure or reduce chronic illnesses caused by either HIV-1 or ZEBOV, in novel
ways that can be implemented in resource limited settings or during a humanitarian crisis.
While treating HIV-1 infection in patients with combination antiretroviral therapy (cART) is
well tolerated and strongly suppresses patient viral load, these treatments are not without some
limitations [3, 7]. In the absence of a functional or sterilizing cure, the estimated 75,500
Canadians living with HIV-1 require life-time adherence to medications, which present new
challenges in reducing the rate of new HIV-1 infections [8]. Prolonged cART does not
significantly reduce the latent viral reservoir [3, 9], and there can be long-term toxicity
associated with sustained treatment with HIV-1 therapies [10-12]. In addition, some regimens
can cause unwanted drug-drug interactions in HIV-1 patients with comorbidities [13]. For
instance, the protease boosting agents ritonavir and cobicistat inhibit cytochrome P450-mediated
metabolism, altering the pharmacokinetics of co-medications [14, 15]. Thus there is a strong
incentive to discover novel therapeutics that can complement and improve current cART
medications.

3
Comparing to cART treatment of HIV-1, approved antiviral agents for treating Ebola Viral
Disease (EVD) are much further behind. Critical and supportive intensive care guidelines for
those suffering from EVD were still being developed during the 2014-16 ZEBOV outbreak in
West Africa, and continues to be an active area of research [16]. Critical care interventions
included: oral hydration therapy, intravenous fluid management to maintain blood electrolyte
balance, loperamide treatment for severe diarrhea, and pain relief [16]. Experimental drugs that
inhibit ZEBOV infection in vitro and in animal models of infection, which were safe and well-
tolerated in phase I clinical trials, became fast-tracked for emergency phase II/III trials that
began in 2014-15 (summarized in Table 1.1) [17-20]. These included trials assessing a cocktail
of three monoclonal antibodies called ZMapp [19], small-interfering RNA (siRNA) called TKM-
Ebola [21], transfusion of convalescent plasma [20] or recombinant interferon supplementation
[22]. Nucleoside analogues approved for treatment of other viruses that showed anti-ZEBOV
activity in vitro or in animal models of infection, such as favipiravir or brincidofovir, were also
considered for emergency phase II/III trials in West Africa [18, 23, 24]. Some of these clinical
trials are ongoing, in particular the PREVAIL vaccine trial in Liberia. Variability in non-
randomized trial design and limited patient enrollment produced modest support for any
particular treatment as the standard for treating EVD, while others were terminated before
efficacy could be assessed [25]. Success occurred with one vaccine trial, providing 100%
protection of those enrolled [26]. However treatment options for those with acute or chronic
EVD are much desired. Thus, continued preclinical research into novel ways to inhibit HIV-1
and ZEBOV replication is warranted.
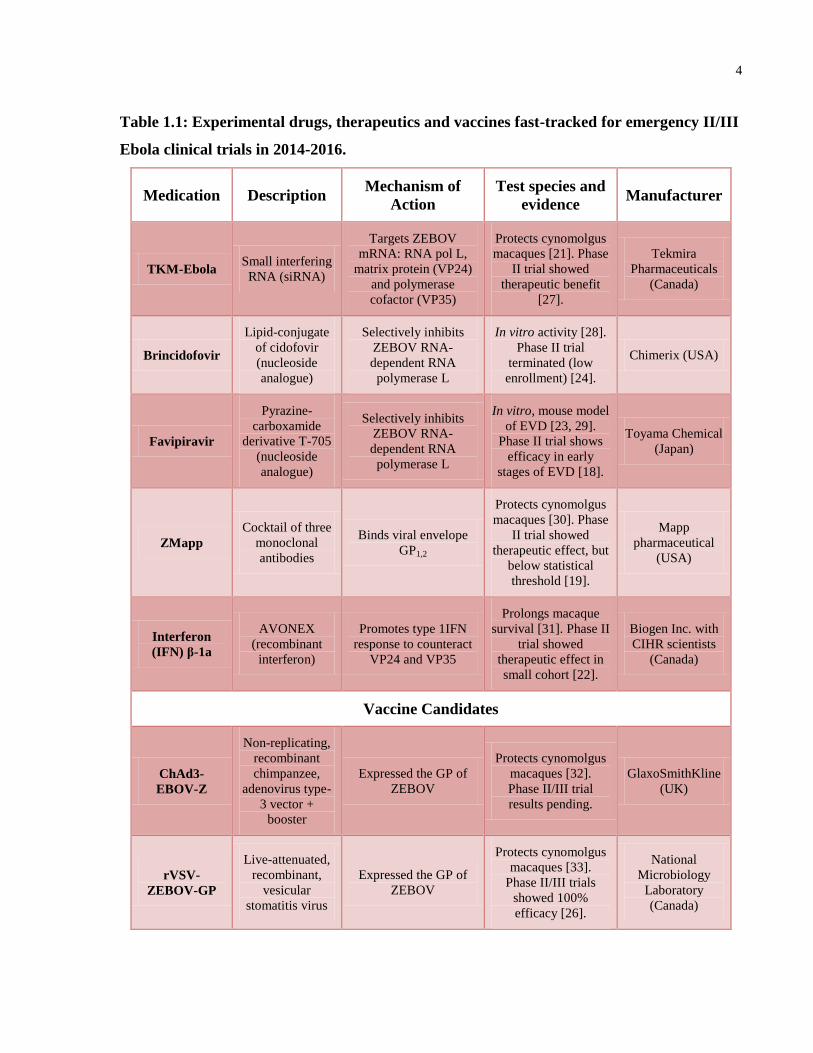
4
Table 1.1: Experimental drugs, therapeutics and vaccines fast-tracked for emergency II/III
Ebola clinical trials in 2014-2016.
Medication Description Mechanism of
Action
Test species and
evidence Manufacturer
TKM-Ebola Small interfering
RNA (siRNA)
Targets ZEBOV
mRNA: RNA pol L,
matrix protein (VP24)
and polymerase
cofactor (VP35)
Protects cynomolgus
macaques [21]. Phase
II trial showed
therapeutic benefit
[27].
Tekmira
Pharmaceuticals
(Canada)
Brincidofovir
Lipid-conjugate
of cidofovir
(nucleoside
analogue)
Selectively inhibits
ZEBOV RNA-
dependent RNA
polymerase L
In vitro activity [28].
Phase II trial
terminated (low
enrollment) [24].
Chimerix (USA)
Favipiravir
Pyrazine-
carboxamide
derivative T-705
(nucleoside
analogue)
Selectively inhibits
ZEBOV RNA-
dependent RNA
polymerase L
In vitro, mouse model
of EVD [23, 29].
Phase II trial shows
efficacy in early
stages of EVD [18].
Toyama Chemical
(Japan)
ZMapp
Cocktail of three
monoclonal
antibodies
Binds viral envelope
GP1,2
Protects cynomolgus
macaques [30]. Phase
II trial showed
therapeutic effect, but
below statistical
threshold [19].
Mapp
pharmaceutical
(USA)
Interferon
(IFN) β-1a
AVONEX
(recombinant
interferon)
Promotes type 1IFN
response to counteract
VP24 and VP35
Prolongs macaque
survival [31]. Phase II
trial showed
therapeutic effect in
small cohort [22].
Biogen Inc. with
CIHR scientists
(Canada)
Vaccine Candidates
ChAd3-
EBOV-Z
Non-replicating,
recombinant
chimpanzee,
adenovirus type-
3 vector +
booster
Expressed the GP of
ZEBOV
Protects cynomolgus
macaques [32].
Phase II/III trial
results pending.
GlaxoSmithKline
(UK)
rVSV-
ZEBOV-GP
Live-attenuated,
recombinant,
vesicular
stomatitis virus
Expressed the GP of
ZEBOV
Protects cynomolgus
macaques [33].
Phase II/III trials
showed 100%
efficacy [26].
National
Microbiology
Laboratory
(Canada)

5
1.2 Human Immunodeficiency Virus (HIV)
1.2.1 HIV-1 Epidemiology, Transmission and Replication Cycle
HIV-1, the primary cause of AIDS, was first clinically observed in the United States in men who
have sex with men (MSM) in June of 1981 [34]. Otherwise healthy men succumbed to
pneumonia caused by Pneumocystis carinii, and developed the rare skin cancer Kaposi’s
sarcoma (KS) [34]. It was soon discovered patients were vulnerable to a variety of opportunistic
infections caused by bacteria, viruses and fungi [35], which were recognized as AIDS-defining
illnesses, permitting early classification of disease progression. In 1983-84, Dr. Robert Gallo,
Dr. Luc Montagnier and Francoise Barré-Sinousi were the first to report a new human T-
lymphotrophic retrovirus from AIDS patients [36, 37]. Shortly thereafter, Dr. Robert Gallo
independently demonstrated this new retrovirus to be the causative agent of AIDS [38, 39].
It has since been determined the HIV-1 pandemic started many decades prior to the first AIDS
cases identified in North America. Human-to-human transmission in Africa occurred as early as
the 1920’s in Kinshasa, the capital city of the Democratic Republic of the Congo (see Figure
1.1A, originally published in [4]). Early spread of HIV-1 from that era shares genetic similarity
with simian immunodeficiency viruses (SIVs) that infect the common chimpanzee Pan
troglodytes, suggesting HIV-1 originated as a zoonose from at least three separate cross-species
transmissions to humans [40]. Another serotype, HIV-2, is found predominantly in West Africa
and is associated with weaker transmission and less likely to cause AIDS, however co-infection
with HIV-1 can complicate antiretroviral treatment [41].
HIV-1 is found in a variety of bodily fluids, but is primarily transmitted in semen, vaginal
secretions or rectal secretions during intercourse, through transfusions of untreated blood
products, by reusing needles without sterilization, and by mother-to-child (vertical) transmission
from mixing of maternal and child blood at birth, or from breastfeeding [42]. Infant exposure to
maternal blood and fluids during childbirth is the most common route of vertical transmission.
In Canada, of the estimated 75,500 people living with HIV-1, 21% are unaware of their diagnosis
[8]. The 2014 incidence of new infections was 2,570, where MSM accounted for 54.3% of new
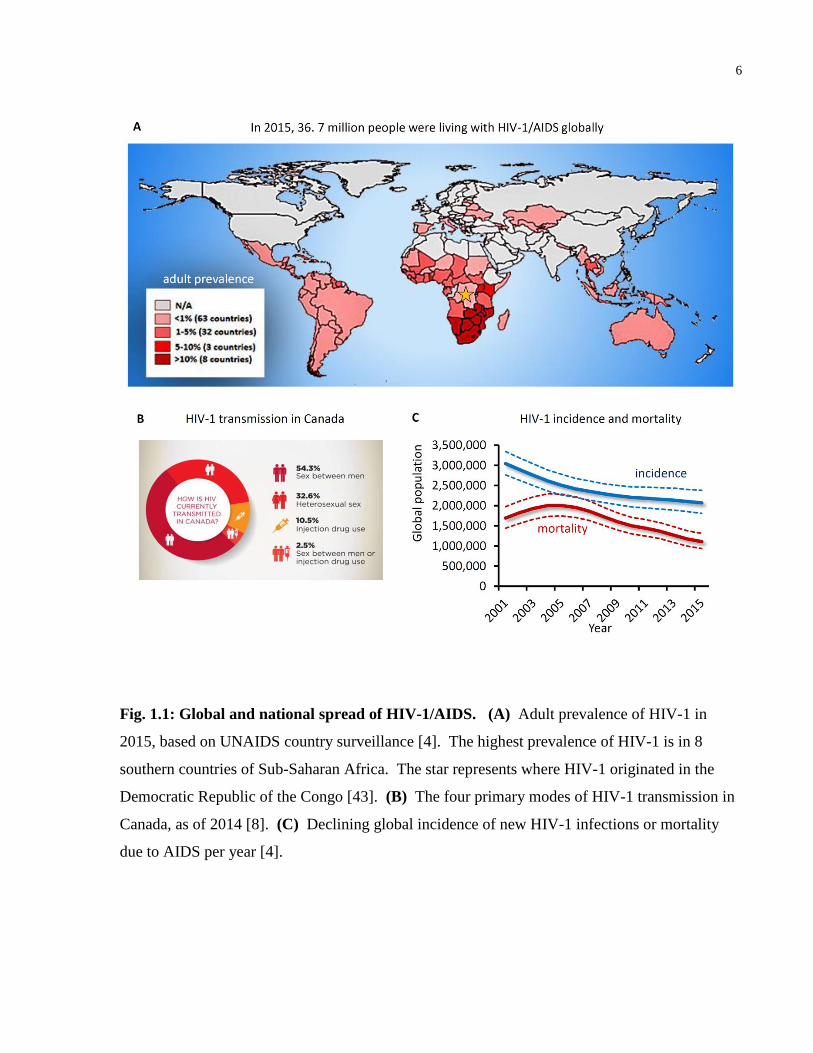
6
Fig. 1.1: Global and national spread of HIV-1/AIDS. (A) Adult prevalence of HIV-1 in
2015, based on UNAIDS country surveillance [4]. The highest prevalence of HIV-1 is in 8
southern countries of Sub-Saharan Africa. The star represents where HIV-1 originated in the
Democratic Republic of the Congo [43]. (B) The four primary modes of HIV-1 transmission in
Canada, as of 2014 [8]. (C) Declining global incidence of new HIV-1 infections or mortality
due to AIDS per year [4].

7
HIV-1 infections, followed by heterosexual intercourse (18.7%), people who emigrated from
HIV-endemic regions (13.9%), those of aboriginal ethnicity (10.8%) and injection drug users
(10.5%) (Figure 1.1B, originally published in [8]). Despite successes in reducing the global
levels of children born with HIV-1, HIV-1 testing of pregnant women, HIV-1 prevention
strategies, and increasing access to life-saving HIV-1 antiretroviral therapy, the global rate of
new infections remains persistently high at 2.1 million new infections in 2015, while HIV-1
mortality declines (Figure 1.1C, originally published in [4]).
HIV-1 is an enveloped retrovirus with a short 9.7 kb genome on positive sense, single-stranded
RNA ((+)ssRNA) (see Figure 1.2, originally published in [44] and [45]). The viral genome
contains three genomic regions (gag, pol and env) that encode genes that directly participate in
creating new virions, and 6 regulatory genes (tat, rev, nef, vif, vpr, and vpu). Together they
encode 19 distinct proteins [46]. The gag region encodes genes for the structural proteins p17
matrix (MA), p24 capsid (CA), p7 nucelocapsid (NC) and p6. The pol region encodes genes for
three viral enzymes: two subunits (p66 and p51) of reverse transcriptase (RT), integrase (IN) and
protease (PR), as well as p15 (RNase H). Finally, the env region encodes genes for the two
subunits of the external viral envelope protein, glycoprotein 120 (gp120) and glycoprotein 41
(gp41) [46]. The HIV-1 genome also encodes accessory proteins that regulate the viral lifecycle
and subvert immune responses to infection: negative regulatory factor (Nef), two splice variants
of trans-activator of transcription (p16 and p14 Tat), regulator of expression of virion proteins
(Rev), viral infectivity factor (Vif), viral protein u (Vpu), viral protein r (Vpr), and a fusion
protein encoded by tat, env and rev (Tev) [46].
During viral attachment and entry of a CD4+ T-cell, surface gp120, existing as a trimer of gp120-
gp41 heterodimers, binds to host CD4 [47]. Gp120 then binds a host chemokine co-receptor,
primarily α-chemokine CXCR4 or β-chemokine CCR5. CXCR4 is highly expressed on CD4+ T-
cells and CCR5 on the cell surface of macrophages, leading to the nomenclature of X4 viruses
(T-tropic) and R5 viruses (M-tropic), of which R5 viruses are more prevalent during early HIV-1
infection of a person and also more likely to be transmitted through intercourse [48, 49].
Following co-receptor engagement, virions that productively infect T-cells undergo
internalization by receptor-mediated endocytosis [50]. Within the endosome a conformational
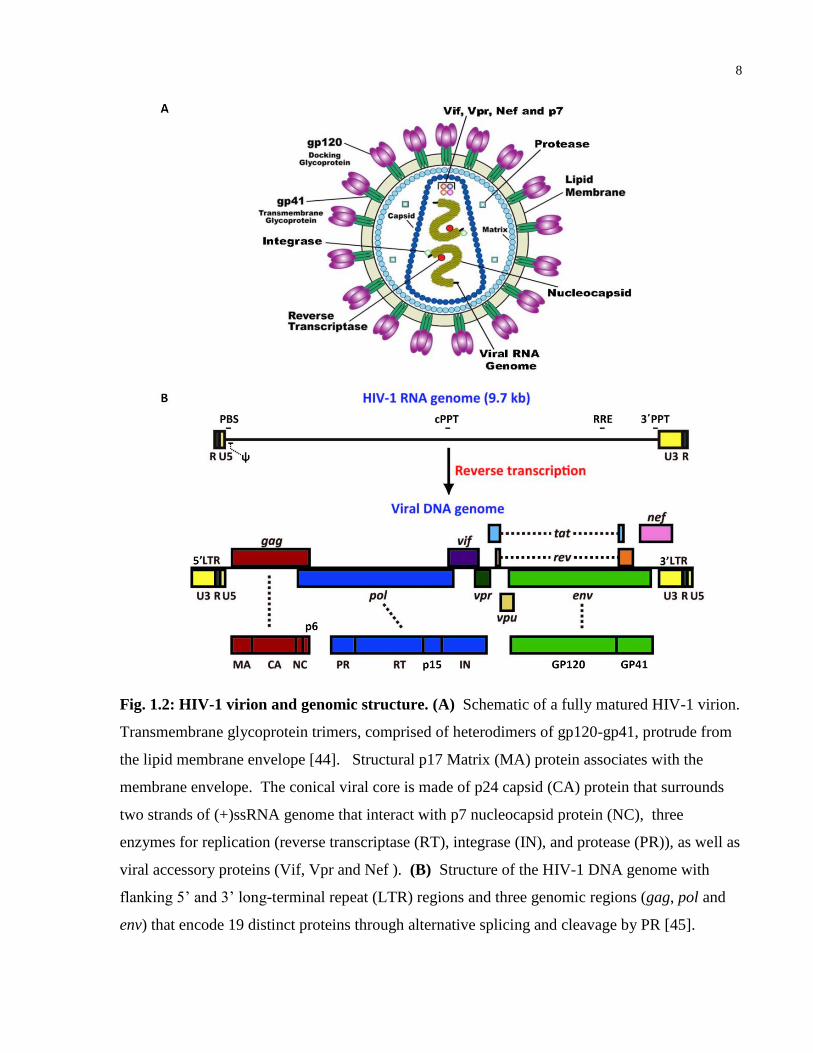
8
Fig. 1.2: HIV-1 virion and genomic structure. (A) Schematic of a fully matured HIV-1 virion.
Transmembrane glycoprotein trimers, comprised of heterodimers of gp120-gp41, protrude from
the lipid membrane envelope [44]. Structural p17 Matrix (MA) protein associates with the
membrane envelope. The conical viral core is made of p24 capsid (CA) protein that surrounds
two strands of (+)ssRNA genome that interact with p7 nucleocapsid protein (NC), three
enzymes for replication (reverse transcriptase (RT), integrase (IN), and protease (PR)), as well as
viral accessory proteins (Vif, Vpr and Nef ). (B) Structure of the HIV-1 DNA genome with
flanking 5’ and 3’ long-terminal repeat (LTR) regions and three genomic regions (gag, pol and
env) that encode 19 distinct proteins through alternative splicing and cleavage by PR [45].

9
change in the glycoprotein trimer exposes the fusion domain of gp41, causing insertion of the
fusion peptide into the target T-cell membrane [51]. Inside the cytoplasm, the virus core makes
use of fibrous actin remodeling to dock outside the nucleus and shed capsid protein [52], creating
the reverse transcriptase complex (RTC) containing both viral and host proteins (Figure 1.3,
originally published in [53]). Viral reverse transcriptase synthesizes linear, double-stranded
copy DNA (ds cDNA) from the viral RNA genome, which binds integrase and other host factors
to form the pre-integration complex (PIC) [54]. The PIC is actively transported through a
nuclear pore and integrated into a host chromosome by reactions catalyzed by integrase and host
DNA repair proteins [55]. Failed integration of the cDNA are circularized by non-homologous
end joining, homologous recombination, or autointegration, leading to closed circular forms with
one or two long-terminal repeats (LTR), called 1- or 2-LTR circles [56]. Integrated provirus
behaves as a set of genes while 1- or 2-LTR circles are episomal DNA that may lead to
preintegration latency, a potential source of latent viral infection [57]. Expression of the
integrated virus is regulated by several host transcription factors that bind enhancer and promoter
sequences in the viral 5’ LTR, as well as viral Tat that binds the Transactivation Response region
(TAR) of nascent HIV-1 RNA transcripts in the nucleus, considerably increasing transcription of
the viral genome [58]. Unspliced and singly-spliced viral mRNA transcripts are exported into
the cytosol by Rev, then translated into proteins by the ribosome [59]. Separate from other viral
proteins, Env glycoprotein precursor (gp160) undergoes glycosylation and proteolytic processing
in the endoplasmic reticulum and trans-Golgi apparatus [60, 61]. Mature gp120-gp41
glycoproteins are then inserted into the host plasma membrane as non-covalently bound trimers.
Virus assembly at the plasma membrane is directed predominantly by Gag polyprotein precursor
(Pr55) [62] and the host actin cytoskeleton [63], leading to virions budding with encapsidated
genomic (+)ssRNA [64], viral enzymes, and host proteins located on the viral membrane and in
the virion itself [65]. Viral protease cleaves Gag and Pol polyproteins during the release of the
virus particle, creating fully mature virions capable of infecting new cells [66].
Following sexual transmission, dendritic cells (DCs), the most potent antigen-presenting cells
(APCs) of the adaptive immune system, play a key role in acquiring HIV-1 at mucosal surfaces
and disseminating the virus during early infection [67, 68]. Binding of virus to C-type lectin
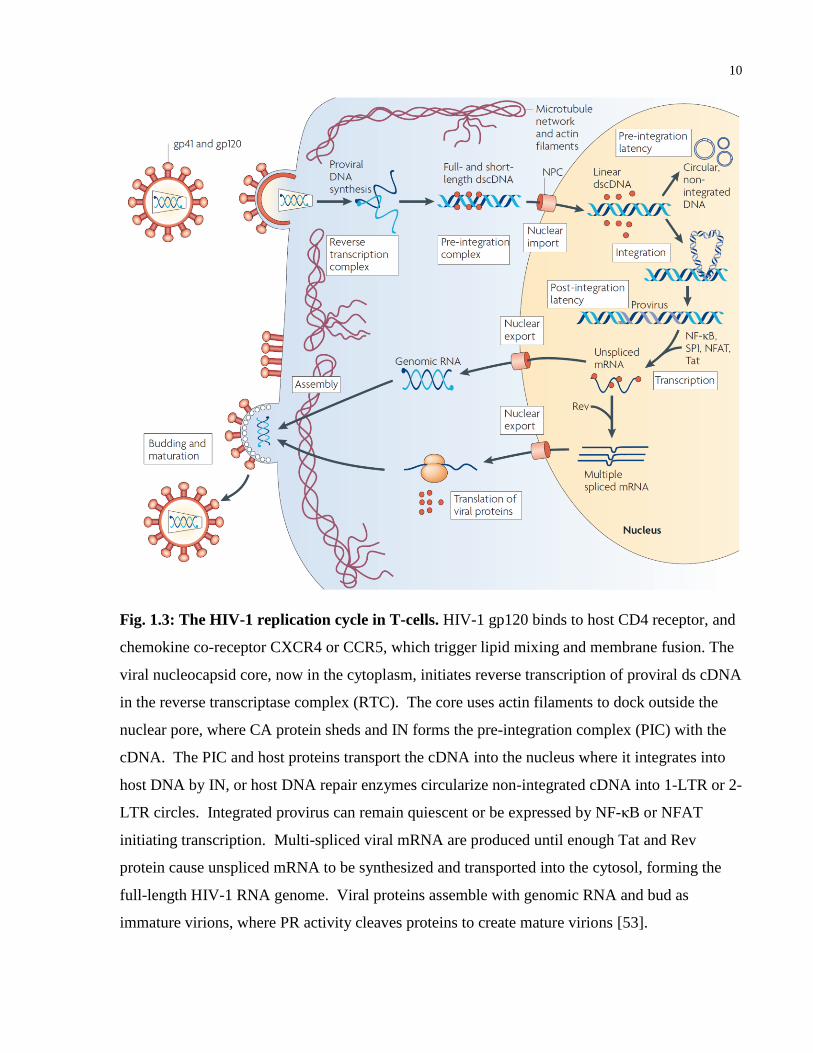
10
Fig. 1.3: The HIV-1 replication cycle in T-cells. HIV-1 gp120 binds to host CD4 receptor, and
chemokine co-receptor CXCR4 or CCR5, which trigger lipid mixing and membrane fusion. The
viral nucleocapsid core, now in the cytoplasm, initiates reverse transcription of proviral ds cDNA
in the reverse transcriptase complex (RTC). The core uses actin filaments to dock outside the
nuclear pore, where CA protein sheds and IN forms the pre-integration complex (PIC) with the
cDNA. The PIC and host proteins transport the cDNA into the nucleus where it integrates into
host DNA by IN, or host DNA repair enzymes circularize non-integrated cDNA into 1-LTR or 2-
LTR circles. Integrated provirus can remain quiescent or be expressed by NF-κB or NFAT
initiating transcription. Multi-spliced viral mRNA are produced until enough Tat and Rev
protein cause unspliced mRNA to be synthesized and transported into the cytosol, forming the
full-length HIV-1 RNA genome. Viral proteins assemble with genomic RNA and bud as
immature virions, where PR activity cleaves proteins to create mature virions [53].

11
receptors on immature DCs, such as mannose receptor, langerin or Dendritic Cell-Specific
Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), leads to protection from
degradation in endolysosomal compartments [69, 70]. There, HIV-1 can remain infectious for 1-
3 days, providing enough time for immature DCs to migrate from the submucosa to local
lymphoid tissues and efficiently infect CD4+ T-lymphocytes [71]. Transmission occurs via three
routes: trans-infection through a virological synapse (VS) [72], exocytosis of virions from
multivesicular bodies [73], or by cis-infection from budding progeny virus [74]. HIV-1 can also
directly infect CD4+ T-cells at mucosal surfaces. In addition to CD4
+ T-cells, HIV-1 also infects
monocytes, macrophages and microglia cells [75], but predominantly infects and depletes the
helper CD4+ T-cell population during the course of infection [76, 77]. In a matter of weeks,
CD8+ cytotoxic T-cell and B-cell responses to control infection inadvertently provide immune
pressure that selects for the evolution of HIV-1 quasispecies [78]. Given the extensive genetic
diversity of HIV-1 in a host, virus expressing gp120 variants with different glycan shields can be
selected to evade clearance by broadly neutralizing antibodies [79]. Escape from broad humoral
responses results in persistent infection, causing depletion of Th17 CD4+ T-cells in gut mucosa
[80], mucosal damage from pro-inflammatory cytokines released from activated T-cells, and loss
of immune protection at intestinal mucosa [81]. Dysfunction at the intestinal barrier allows
microbial translocation of bacteria may contribute to systemic immune activation [82], which is
characteristic of chronic HIV-1 infection. This continued immune stimulation disrupts the
dynamic regulation of CD4+ and CD8
+ T-cells [83], leading to a gradual loss of peripheral CD4
+
T-cells over time from persistent viral replication and apoptosis [84, 85].
Chronic immune activation and inflammation also causes lymphoid tissue fibrosis via regulatory
T-cells (Treg) depositing collagen [86], and persistent antigen stimulation produces defective
helper T-cells that are less responsive, leading to effector T-cell exhaustion over many years
[87]. In addition, the immune system is unable to eliminate latently infected, central memory
CD4+ T-cells that produce low copies of virus, despite a patient adhering to suppressive therapy
and having an undetectable viral load [3]. Unfortunately, combination antiretroviral therapy
(cART) is unable to prevent the homeostatic proliferation of latently infected CD4+ cells that
harbor integrated provirus that are transcriptionally silent, nor access certain compartments of the

12
human body, such as the central nervous system, testes or gut-associated lymphoid tissue
(GALT), where low levels of viruses are produced and HIV-1 RNA and DNA are detected [88-
90]. These have been called anatomical sanctuaries, hypothesized to maintain the latent viral
reservoir through low-level viral replication in resting CD4+ T-cells, and are established within
days of primary infection [91]. Integrase inhibitors have been demonstrated to reach viral
sanctuaries such as the GALT, however they are unable to decrease the latent viral reservoir.
Thus, designing new antiviral combination therapies to activate infected, quiescent T-cells, then
clear infected cells after reactivation, has become a prominent area of HIV-1 cure research [92].

13
1.2.2 HIV-1 Treatments, Potential Vaccines, and New Therapeutics
In 1986-87, the first antiretroviral therapy (ART) approved for AIDS treatment in the United
States was the nucleoside analogue zidovudine (AZT) [93-95]. AZT, an inhibitor of HIV-1
reverse transcriptase, was swiftly used to treat AIDS patients at high doses to increase the CD4+
T-cell population and reduce opportunistic infections, but was associated with acute and long-
term drug toxicity such as anemia and neutropenia [96]. Moreover, drug resistance, as measured
by HIV-1 antigen detected in serum, developed within 24 weeks of continuous AZT treatment
[97]. It was also determined that HIV-1 contains an error-prone reverse transcriptase enzyme
that introduces base substitutions, additions and deletions when it synthesizes double-stranded
cDNA from the viral RNA genome [98]. These provide genetic variation in integrated provirus
that are selected under pressure of drug monotherapy, leading to escape mutations and the
evolution of drug resistant HIV-1 quasispecies [99-101]. In 1995, it was discovered that
combining AZT with lamivudine (3TC) was superior at controlling viremia when compared to
either monotherapy treatment [102]. This was superseded by triple drug therapies in 1996,
which considerably suppressed patient viral loads [103, 104]. The drug strategy was termed
highly active antiretroviral therapy (HAART), now called cART.
With 25 different single drugs and 14 fixed-dose combination regimens currently FDA-approved
to treat HIV-1, the success of cART has turned HIV-1 infection into a chronic, manageable
illness [7]. This paradigm shift has averted millions of AIDS-related deaths and controlled the
rate of new infections in developed and developing nations with the highest HIV-1 burden [105].
First-line therapy for treatment-naïve adults include three drugs, consisting of two nucleoside
reverse transcriptase inhibitor (NRTI) and an inhibitor from another drug class [106]. These
include non-nucleoside reverse transcriptase inhibitors (NNRTI), inhibitors that target viral
entry, fusion, integration or viral protease, and the potential inclusion of pharmacokinetic
enhancers [106]. While cART regimens can be well-tolerated and have a low pill burden for
patients, they require lifetime adherence to control viremia and any interruption can lead to rapid
viral rebound in plasma [107]. Drug interactions, unwanted side-effects, and regiment
adjustments can also lead to poor adherence among patients, which increase the risk of drug

14
resistance mutations that may cause virological failure of first-line cART [7]. The emergence of
multi-drug resistant HIV-1 variants has prompted renewed focus into novel vaccine and
therapeutic strategies to improve or replace cART [108], and ultimately end the AIDS pandemic
by developing a functional or sterilizing cure [109, 110].
There have been numerous attempts over the last 30 years to design a safe, immunogenic, and
effective vaccine to prevent or treat HIV-1 infection [1, 111]. These include active
immunization strategies to elicit broadly neutralizing antibodies (bNAbs) [112], inducing CD8+
T-cell responses [113], priming with Adenovirus (ADV) or canarypox vectors [114], or passive
immunotherapy with a human monoclonal antibody (mAb) [115]. After two decades of failed
vaccine trials, and one trial that lead to an increase in HIV-1 acquisition [116], the 2004-2009
RV144 trial in Thailand became the first vaccine to demonstrate efficacy in preventing 60.5% of
new HIV-1 infections after 1 year, and 31.2% after 3.5 years [1]. The prime-boost vaccine given
to 16,402 participants was a combination of two strategies: a cannarypox vector (ALVAC-HIV)
expressing clade E env and clade B gag and pro, and recombinant gp120 from clade B/E
(AIDSVAX). Participants that expressed IgG antibodies to the V2 loop of HIV-1 gp120 were
the least likely to become infected [1]. Further immunogenicity testing of the vaccine was
tested in South Africa in the 2013-14 phase I/II HVTN097 trial [117], providing the groundwork
for the phase II/III HVTN702 trial recently initiated in 2016. Therapeutic vaccines to intensify
cART treatment and limit establishment of the viral reservoir have produced few promising
leads, with the exception of the VRC01 monoclonal antibody isolated from the B-cells of an elite
controller [118]. Continued challenges remain in eliciting broadly neutralizing antibodies,
maintaining durable cross-strain breadth, and improving B-cell and T-cell priming to develop an
effective HIV-1 vaccine [108].
Other innovative strategies to prevent HIV-1 infection include male circumcision, preventative
microbicides and pre-exposure prophylaxis (PrEP). Since 2008, Voluntary Medical Male
Circumcision (VMMC) has successfully led to 11 million adolescent boy and adult male
circumcisions in eastern and southern Africa, as part of the ongoing World Health Organization
(WHO) strategy to prevent female-to-male HIV-1 infection [2]. To prevent male-to-female
infection, a variety of HIV-1 microbicides are being developed (acidic buffers, surfactants,

15
polyanionic polymers, reverse transcriptase inhibitors, and other small molecule inhibitors), with
different modes of delivery (gels, vaginal rings, tablets and nanoparticles) [119]. However
microbicides derived from cART regimens raise concerns of potential selection of drug-resistant
HIV-1 strains in women who seroconvert in microbicide trials [120]. In addition, cultural
expectations, partner-related factors (ex. fear of disproval) and acceptability issues have so far
led to low microbicide treatment adherence in clinical trials [121, 122]. In Canada, the recent
expansion of PrEP is a promising new strategy to prevent new HIV-1 infections, complementing
other prevention strategies such as access to HIV-1/STI testing and free condom distribution.
PrEP involves administering two ART medications, often tenofovir disoproxil fumarate (TDF)
with emtricitabine (FTC) in the single pill Truvada, to seronegative people who are at high risk
of contracting HIV-1 from a sexual encounter or the sharing of needles [123]. This can inhibit
HIV-1 before it disseminates from the site of infection at mucosal tissues, preventing systemic
infection [124]. A recent randomized, double-blind trial called IPERGAY determined that when
MSM participants were optimally adherent to PrEP, high plasma levels of antiretrovirals could
prevent 86% of new HIV-1 transmissions [125].
In terms of pre-clinical HIV-1 cure research, two main approaches are gene-editing to remove
integrated provirus, and latency reversal followed by clearing the viral reservoir. CRISPR/Cas9
technology is the leading gene-editing strategy to excise integrated HIV-1 provirus from the
primary CD4+ T-cells of HIV-1 patients ex vivo [126]. However, insertions and deletions that
follow Cas9 cleavage, created by host Non-Homologous End-Join (NHEJ) repair proteins, can
either suppress HIV-1 replication or accelerate viral escape by selection of viral sequences
refractory to Cas9 recognition [127]. Although a recent in vivo study has demonstrated proof-
of-principle for removing HIV-1 viral DNA in transgenic mice with CRISPR/Cas9 [109],
further research is needed to produce safe and efficient delivery of a mildly immunogenic viral
vector amendable for clinical use. Lastly, a ‘shock and kill’ strategy to reactivate then clear
latent HIV-1 infection, potentially curing a patient of HIV-1, involves pairing small-molecule
Latency Reversing Agents (LRAs), such as Histone Deacetylase (HDAC) inhibitors, with
immunotherapies that promote clearance of persistently infected cells [92]. Challenges remain in
reversing latency in all cell types harboring integrated provirus [128], boosting HIV-1-specific

16
CD8+ T-cell responses in tissue sanctuaries [129], and combining multiple LRAs in a
coordinated fashion with approaches to clear infection in animal models and human clinical trials
[130]. These complimentary approaches to replace or improve conventional cART are
promising areas of research. Yet there is also considerable interest in studying host T-cell factors
that contribute to HIV-1 replication, as novel means to restrict infection and eliminate the viral
reservoir of latently infected cells [131].

17
1.2.3 Host Kinases as Targets for HIV-1 Inhibition
Decades of basic HIV-1 research have revealed many complex interactions between HIV-1 and
the host immune cells that it infects. The success of FDA-approved HIV-1 antivirals targeting
host T-cell factors demonstrate both the critical nature of host proteins participating in the HIV-1
lifecycle, and their therapeutic value as components of cART [132, 133]. For example,
Maraviroc (MVC) was developed as a selective CCR5 receptor antagonist to block binding of
envelope gp120 [134]. Thus, discovering antivirals that target host factors essential for HIV-1
replication is a compelling area of research, as they may pose higher barriers to drug resistance
by blocking multiple stages of infection, offer unique tissue distribution to purge the viral
reservoir, or alleviate symptoms not targeted by conventional cART [135, 136].
With a genome encoding only 19 proteins, HIV-1 requires many host T-cell protein interactions
in order to replicate. These interactions have been organized into protein networks in an HIV-1
human protein interaction database [137]. As many as 348 unique protein-protein interactions
have been found between HIV-1 and human proteins in the Jurkat T-cell line [138]. Moreover,
mutations caused by the error-prone HIV-1 reverse transcriptase are primarily selected to negate
cellular restriction factors, exploiting host defense mechanisms that then improve viral fitness
[139]. As with other enveloped viruses, HIV-1 promotes replication and avoids immune
responses by modifying the host plasma membrane, which it also uses for its lipid shell during
budding. For instance, viral Nef and Vpu modulate plasma membrane receptor expression and
localization, to increase viral fitness and evade immune detection [140]. They can cause CD4
downregulation, which promotes viral egress by allowing newly synthesized gp120 to traffic to
the cell membrane, and inhibit tetherin binding, which allows newly released particles to bud
from the plasma membrane [140]. Alongside Vpu and Nef, HIV-1 encodes two other accessory
proteins (Vif, and Vpr) to counteract host restriction factors, each playing unique roles in
different T-cell types and at different stages of infection [141]. For example, host apolipoprotein
B mRNA editing enzyme, catalytic polypeptide-like 3G (APOBEC3G) is a cytidine deaminase
packaged into newly synthesized virions, catalyzing the deaminiation of cytosine shortly after
reverse transcription of nascent single-stranded viral cDNA in a new cell, dramatically impairing

18
HIV-1 replication [142]. Another host protein, SAM domain and HD domain-containing protein
1 (SAMHD1), can also interfere with reverse transcription by hydrolysing cytosolic
deoxynucleoside triphosphates (dNTPs) [143]. Yet these host proteins are counteracted by Vif,
which prevents APOBEC3G packaging into virions, and Vpx (the HIV-2 equivalent of Vpr),
which mediates degradation of SAMHD1 [141]. Moreover, viral enzymes such as IN depend on
host-cofactors to such an extent that drugs that interfere with integrase interactions with Lens
Epithelium-Derived Growth Factor (LEDGF/p75), called LEDGIN’s, show pre-clinical promise
as a new class of allosteric integrase inhibitors [144, 145]. Thus cellular proteins hijacked during
the HIV-1 replication cycle continue to be attractive targets in developing new antiretroviral
therapies.
In particular, HIV-1 modifies host signal transduction pathways that disrupt virtually every
aspect of cellular metabolism [146]. When gp120 binds to the co-receptor CXCR4 or CCR5
during viral entry of a T-cell, the interaction also initiates signaling pathways downstream to
promote intracellular viral replication post-fusion [147]. Corroborating this finding, CD4+ T-
cells isolated from asymptomatic, HIV-1 infected patients show defective early tyrosine
phosphorylation downstream of T-cell receptor stimulation [148, 149]. General inhibition of
tyrosine kinase signaling with the broad-spectrum inhibitor genistein has demonstrated that
tyrosine kinase signaling is essential for HIV-1 entry and intracellular steps shortly thereafter
[150, 151]. It has been put forth that HIV-1 requires T-cell activation and reorganization of the
cytoskeleton for productive HIV-1 entry and replication [152], altering tyrosine kinase pathways
downstream of the T-cell Receptor (TCR) to facilitate its viral lifecycle. In support of this
hypothesis, the addition of mitogenic stimuli can activate host kinases to permit HIV-1
replication in quiescent T-cells that is otherwise inefficient, further suggesting a potential role of
host T-cell kinases in the HIV-1 lifecycle [153]. As mentioned earlier, chronic T-cell activation
also has an important role in maintaining persistent HIV-1 infection, helper T-cell disregulation,
and eventual exhaustion of effector T-cells [83, 87] further emphasizing the important roles of
tyrosine kinase signal transduction.
Small lentiviruses, such as HIV-1, appear to have evolved to become phosphorylated by host T-
cell kinases to facilitate infection in non-dividing cells, access the nuclear compartment, and

19
assemble proteins during viral egress [154]. All HIV-1 proteins are phosphorylated throughout
intracellular viral replication, however the functional purpose for these modifications, and the
kinases that regulate them, is an active area of research [155]. Thus far, phosphorylation events
have been found to regulate: HIV-1 viral entry [147], actin remodeling [52, 135], reverse
transcription [156], capsid shedding [157], nuclear translocation of HIV-1 ds cDNA [154, 155,
158], viral integration [159], post-integration repair and circularization of un-integrated cDNA
[160], proviral gene transcription [161], mRNA transport [162], viral assembly [63], and budding
from the host T-cell [163]. One of the most well studied HIV-1 proteins that modify cellular
tyrosine kinase activity is Nef, which has pleiotropic effects on cell signaling and strong binding
affinity to the SRC family of kinases (SFKs) [164-167]. Nef preferentially activates SFK
members LCK, HCK, LYN and c-SRC through allosteric displacement of intramolecular SRC
homology 3 (SH3)-linker interactions [164], however its functional role in activating SFKs
during HIV-1 replication is still under investigation, such as the SFK-Nef contribution to AIDS
progression [168].
Currently there are 28 small-molecule kinase inhibitors approved by the US Food and Drug
Administration for various cancer indications (Figure 1.4, originally published in [169] and
[170]). Their improved specificity from first generation inhibitors and known inhibition of host
kinases has made them ideal for other illnesses outside of cancer, such as pulmonary fibrosis
[171], arthritis [172], and potentially viral infections [173]. Unfortunately, HIV-1 patients taking
cART are at higher risk for non-AIDS defining cancers such as lung adenocarcinoma, Hodgkin’s
lymphoma and anal cancer, when compared with the average population [174]. It has been
suggested this increased risk in due to heightened immune activation and inflammation [175].
Repositioning safe and well-tolerated kinase inhibitors, with published safety and efficacy for
various cancer indications, offers the potential for a new class of antivirals to inhibit HIV-1. In
particular, tyrosine kinase inhibitors are attractive for these purposes because of

20
Fig. 1.4: FDA-approved kinase inhibitors. (A) The cancer and non-cancer indications of
small-molecule kinase inhibitors as of 2015. Dasatinib, a dual ABL/SRC inhibitor that has
shown antiviral activity, is highlighted in red [169]. (B) Chemical structure of dasatinib, with
emphasis of its mode of binding to the adenine pocket and hydrophobic pocket of ABL kinase
[170]. (C) Co-crystal structure of dasatinib (purple) in the Adenosine Triphosphate (ATP)
pocket of ABL, interacting with key amino acids residues (green) through hydrogen bonding (red
dashed lines) [170].

21
extensive research into their molecular interactions with host proteins, known off-targets, ease of
administration, biodistribution, and pharmacokinetics within humans [169, 170]. Some of these
inhibitors, such as imatinib, have been used successfully to treat Kaposi Sarcoma lesions and
Chronic Myeloid Leukemia (CML) in HIV-1 patients on cART [176, 177]. Others have shown
antiviral activity during virus replication, such as the dual SRC/ABL inhibitor dasatinib,
restricting Dengue Virus (DV) replication in vitro [178]. Moreover, extended use of drugs that
inhibit HIV-1 directly are prone to selection of drug-resistant variants, whereas small-molecule
inhibitors targeting host factors may pose a greater mutational barrier, reducing the chance of
selecting drug-resistant viral strains [152]. Thus novel kinase inhibitors that reduce
inflammation, target cancer, and restrict HIV-1 replication, would be ideal drug candidates to
improve cART regimens.
In the human genome there are 32 non-receptor tyrosine kinases (NRTKs) that catalyse the
phosphorylation of tyrosine residues on protein substrates, grouped into 10 families [146].
NRTKs regulate cell processes essential to life such as cell signaling, growth, differentiation,
motility, adhesion and cell death [179-183]. While NRTKs are generally appreciated for having
central roles in cancer and chronic inflammation [184], they are increasingly being recognized
for playing significant roles during viral infections. However, there are specific gaps of
knowledge stalling FDA-approved tyrosine kinase inhibitors from being repurposed to treat
HIV-1 in conjunction with cART. In the following three sections, the role of SRC family
kinases and focal adhesion kinases (FAK) during HIV-1 infection will be reviewed.

22
1.2.4 The SRC Family of Non-Receptor Tyrosine Kinases in HIV-1 Infection
In 1911, Dr. Francis Peyton Rous observed that a virus in cell-free filtrate can cause
fibrosarcoma cancer in domestic chickens [185], latter named the Rous Sarcoma Virus (RSV).
For this discovery of tumor-inducing viruses, Dr. Rous was awarded the Nobel Prize in
Physiology or Medicine in 1966. This soon led to the search for the oncogene responsible for
transformation caused by retroviruses. Drs. John Michael Bishop and Harold Eliot Varmus
demonstrated in 1976 that the viral oncogene responsible for avian sarcomas was originally
acquired from normal avian cells [186]. The viral oncogene causing sarcomas was called v-
SRC, to distinguish it from the cellular homolog c-SRC. Drs. Bishop and Varmus received the
1989 Nobel Prize in Physiology or Medicine for their work showing the cellular origin of
retroviral oncogenes, and that a malignant tumor can originate from normal genes that regulate
growth within a cell. Then at the University of British Columbia in 1981, while investigating
Fujinami Sarcoma Virus (FSV) regulation of v-FPS signaling, Dr. Anthony James Pawson made
an important breakthrough as he uncovered the first modular domain that controls cellular signal
transduction [187]. He identified a phospho-tyrosine binding site similar to a non-catalytic
region of v-SRC, called the SRC homology 2 or SH2 domain, which became prototypic of other
non-catalytic modules that modify kinase activity allosterically, or through binding to other
signaling proteins [188]. Since these pioneering discoveries, hundreds of modular protein
domains have been identified that underpin multiprotein signaling complexes. Much has also
been learned of the structure, regulation, localization and function of v-SRC, c-SRC, and the
related SRC family of non-receptor tyrosine kinases [189].
In humans, there are eight SFK members: c-SRC, LCK, FYN, HCK, LYN, FGR, c-YES and
BLK [189]. The 52-62 kDa proteins play essential roles in the signaling of a variety of cell
processes, such as proliferation, differentiation, motility, adhesion, and proper functioning of
adaptive and innate immunity [189-191]. c-SRC, FYN and c-YES show ubiquitous expression
in many cell types and tissues, while the remaining five have time-specific and cell lineage-
specific expression, such as in hematopoietic cells [192-196]. SFKs are activated by the
stimulation of a variety of transmembrane G-protein coupled receptors (GPCRs), such as the C-

23
X-C chemokine receptor type 4 (CXCR4) and C-C chemokine receptor type 5 (CCR5), linking
surface receptors with downstream signaling pathways such as Mitogen-Activated Protein
Kinase (MAPK) activation [197]. SFKs have seven conserved domains that regulate their
function (see Fig. 1.5 for their domain structure, originally published in [198]). The N-terminus
contains a 15 amino-acid peptide sequence that forms the SH4 domain, involved in post-
translational lipid modifications of the SFK such as myristoylation or palmitoylation [199].
These modifications target SFKs towards membrane-associated signaling and adaptor protein
complexes in various cell compartments, or lipid rafts positioned at the inner leaflet of the
plasma membrane. The SH4 domain is followed by a unique region of 50-90 residues that is
characteristic to each SFK [199]. This intrinsically disordered region (IDR) can encode
sequences that promote cleavage of the SFK protein, phosphorylation of residues that add further
specificity to kinase activity, and localization signals. After the IDR is an SH3 module that
mediates intra- and intermolecular protein binding by recognizing the proline-rich PxxP
consensus sequence. Next there is an SH2 domain capable of binding proteins with
phosphorylated tyrosine in a pYEEI consensus sequence [199]. A short SH2-linker region
containing proline-rich sequences follows the SH2 domain, a target region for SH3 binding.
This linker region is followed by a large SH1 domain containing the catalytic site for substrate
and ATP binding, and an activation loop where the trans- or autophosphorylation of a specific
tyrosine residue fully activates the kinase [199]. Lastly, there is a short, negative regulatory C-
terminus tail with a terminal tyrosine residue that when phosphorylated, binds the SH2 domain.
While healthy cells tightly regulate the active and inactive conformations of each SFK, viruses
have been implicated in altering their expression, activity or cellular localization [178, 200].
These can impair innate or adaptive immune responses to clear viral infection, promote spread of
the virus within tissues or between cells, prevent apoptotic cell death of infected cells, or directly
assist in the viral replication cycle [201]. For instance, cellular SFKs facilitate the entry of the
coxsackievirus, the RNA replication of Hepatitis C Virus (HCV) and DV, as well as assembly of
West Nile Virus (WNV) [178, 201-203]. The multiple mechanisms by which the SRC-family of
kinases facilitate or impede the infectious cycle of HIV-1 will be examined in this section,
suggesting potential roles for c-SRC interactions with HIV-1, reviewed further in chapter 1.2.5.
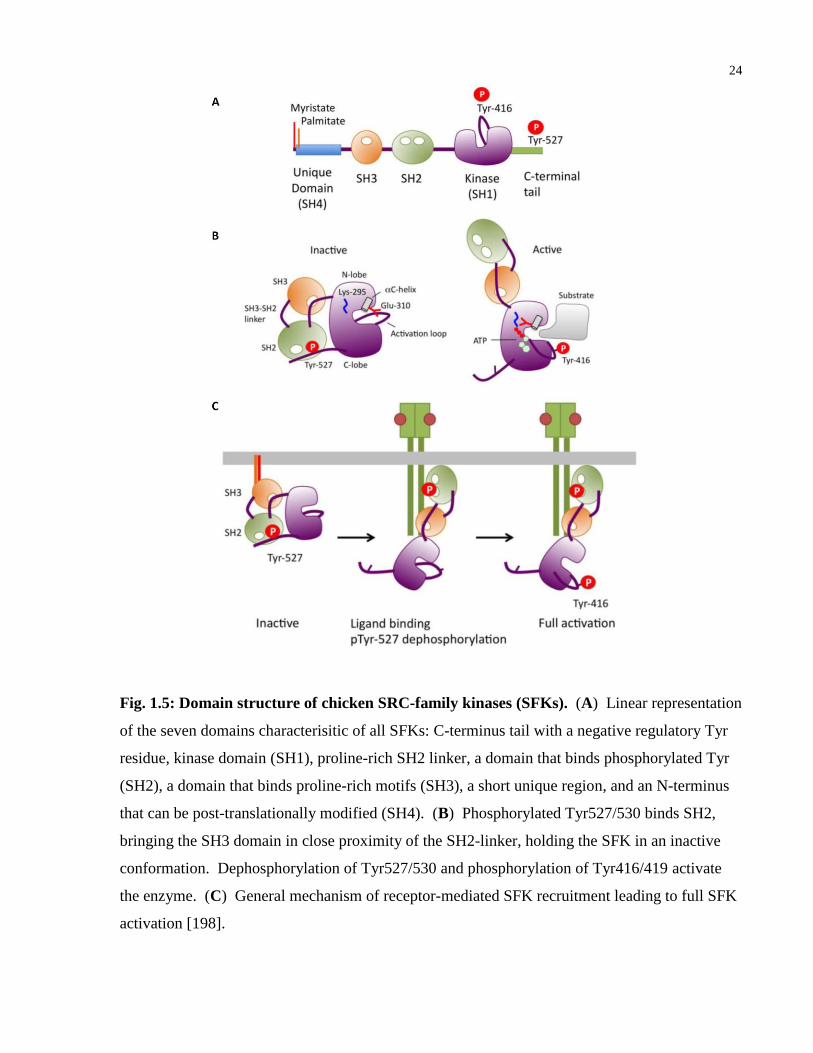
24
Fig. 1.5: Domain structure of chicken SRC-family kinases (SFKs). (A) Linear representation
of the seven domains characterisitic of all SFKs: C-terminus tail with a negative regulatory Tyr
residue, kinase domain (SH1), proline-rich SH2 linker, a domain that binds phosphorylated Tyr
(SH2), a domain that binds proline-rich motifs (SH3), a short unique region, and an N-terminus
that can be post-translationally modified (SH4). (B) Phosphorylated Tyr527/530 binds SH2,
bringing the SH3 domain in close proximity of the SH2-linker, holding the SFK in an inactive
conformation. Dephosphorylation of Tyr527/530 and phosphorylation of Tyr416/419 activate
the enzyme. (C) General mechanism of receptor-mediated SFK recruitment leading to full SFK
activation [198].

25
LCK
Lymphocyte-specific protein tyrosine kinase (LCK) is expressed in B-cells, T-cells, natural killer
cells, brain tissue and in the spleen [204-207]. It localizes to the plasma membrane and to
pericentrosomal vesicles in the cell, interacting with the cytoplasmic tails of transmembrane
receptors [208]. LCK non-covalently associates with CD4 or CD8, plays a major role in TCR-
CD3 mediated T-cell activation, and is a downstream signaling molecule of the interleukin-2 (IL-
2) receptor [209, 210]. In CD4-expressing T-cells, 50-96% of LCK is bound to the cytoplasmic
tail of CD4 receptors, through interactions between the N-terminus of LCK and cysteine residues
on CD4 [211]. Cross-linking of CD4 on the cell surface transiently increases the enzyme activity
of LCK, and it is believed autophosphorylation of Tyr394 participates in this increased LCK
activation [212]. In JCaM1 T-cells expressing a defective splice variant of LCK, cells are unable
to respond to TCR-CD3 stimulation [213]. However, expression of wild type LCK restored TCR
signaling, demonstrating the significance of LCK in normal TCR-CD3 signaling pathways [213].
In the CD4+ T-cells isolated from chronically infected HIV-1 patients, these cells show defective
responses to CD3 activation, reduced proximal TCR tyrosine phosphorylation, and decreased
levels of LCK expression [148]. Paradoxically, acute infection of Jurkat T-cells with HIV-1IIIB
show a global increase in tyrosine phosphorylation within 30 minutes, including activation and
phosphorylation of LCK [214]. Gp120/CD4/LCK complex signalling can induce NF-κB nuclear
translocation and gene transcription [215]. These studies suggest nuanced and different effects
of LCK throughout HIV-1 infection, which appears to be protective during early entry as LCK
associates with CD4. For example, point-mutations of LCK at Cys20 and Cys23, which prevent
LCK association with CD4, results in greater HIV-1 replication [209]. Moreover JCaM1.6 T-
cells, expressing truncated LCK, are far more infectable than Jurkat 45 cells, which lack CD45
and express much higher LCK activity [209]. It has also been demonstrated that short-term
exposure of gp120 can transiently enhance the autophosphorylation and activity of LCK, while
long-term incubation of gp120 with CD4+
T-cells decreases overall expression of LCK [148,
216]. This longer gp120 exposure also correlates with complete dissociation of LCK from CD4,
followed by downregulated surface expression of CD4 [217]. Furthermore, HIV-1 gp120 can
block the migration of CXCR4-expressing T-cells to stromal cell-derived factor 1 alpha (SDF-

26
1α), which is mediated by CD4/LCK signaling and associated with cofilin phosphorylation
[218]. The precursor protein, gp160, can also disrupt adhesion between CD4+ T-cells and B-
cells by donwregulating lymphocyte function-associated antigen-1 (LFA-1), in an LCK-
dependent manner [219].
The use of a variety of techniques to overexpress or knockdown LCK, coupled with
colocalization experiments, have revealed many of the mechanisms by which HIV-1 proteins, in
particular Nef, Vpu and Tat, interact with LCK to modify its initial protective function in order
to facilitate viral replication. In transgenic mice expressing HIV-1 Nef, the animals exhibit
profound thymocyte depletion and CD4+ T-cell lymphopenia [220]. It has been found that Nef
depletes the population of double-positive thymocytes and impairs lineage commitment towards
single-positive CD4+ thymocytes, which was dependent on reduced LCK activity [220].
Expressing constitutively active LCK in the mice rescued CD4+
T-cell maturation, potentially
through increased affinity of the TCR- Major Histocompatibility Complex (MHC) interaction
[220]. HIV-1 Nef disrupts the anterograde transport of LCK to the plasma membrane within T-
cells [167]. It does this by interfering with the vesicular transport of newly synthesized LCK to
membrane-microdomains, a mechanism dependent on the SH4 membrane-anchor domain of
LCK [167]. Nef also impairs the formation of the immunological synapse (IS) and early T-cell
signaling, by retargeting the TCR and LCK for recycling in endosomal compartments (see Figure
1.6, originally published in [221]). It induces the internalization and endocytosis of surface CD4
in a manner dependent on LCK in lymphoid cells [222]. In addition, Nef relocalizes LCK to the
trans-Golgi network, effectively decoupling downstream LCK kinase signaling from the surface
TCR-CD3 complex [223]. In this way, Nef diminishes TCR responses to antigenic stimulation
while rerouting LCK and other kinases to the trans-Golgi network, promoting T-cell survival
through IL-2 production and enhancing viral spread of infected cells [223]. Similar to Nef, the
viral accessory protein Vpu can also subvert the subcellular localization of LCK, implicating
LCK with Vpu-mediated downregulation of cell surface receptors [224]. Nef also recruits a
variety of proteins, including heterogeneous nuclear Ribonucleoprotein K( hnRNP-K) and LCK,
into a signaling complex that increases Tat-mediated HIV-1 transcription [225]. This
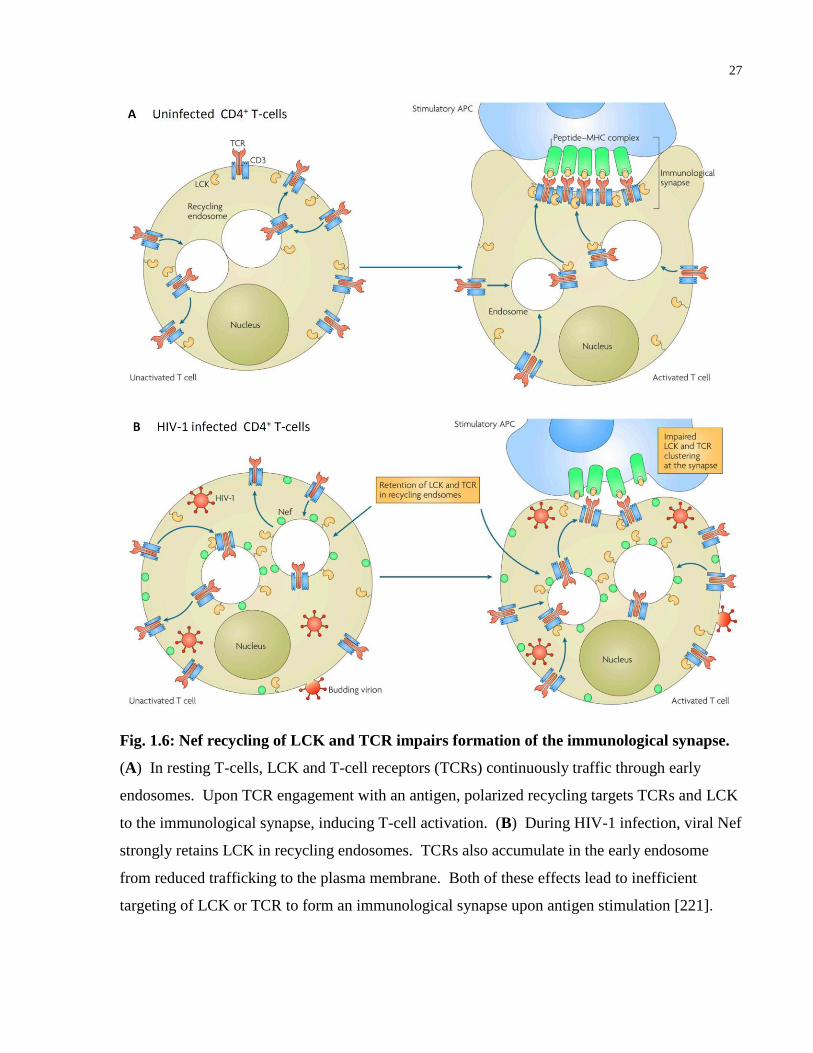
27
Fig. 1.6: Nef recycling of LCK and TCR impairs formation of the immunological synapse.
(A) In resting T-cells, LCK and T-cell receptors (TCRs) continuously traffic through early
endosomes. Upon TCR engagement with an antigen, polarized recycling targets TCRs and LCK
to the immunological synapse, inducing T-cell activation. (B) During HIV-1 infection, viral Nef
strongly retains LCK in recycling endosomes. TCRs also accumulate in the early endosome
from reduced trafficking to the plasma membrane. Both of these effects lead to inefficient
targeting of LCK or TCR to form an immunological synapse upon antigen stimulation [221].

28
interaction offers one potential explanation of how Nef enables high levels of viral replication in
HIV-1 infected people. In addition, LCK has been shown to participate in Tat-mediated
induction of NF-κB reporter gene expression in Jurkat T-cells [226].
LCK kinase activity also influences viral egress and cell-to-cell spread of HIV-1. Activated
LCK directly interacts with Gag protein through its unique domain in infected cells, and
facilitates viral assembly at the plasma membrane [227]. It was determined that palmitoylation of
the unique domain of LCK was required for the successful release of HIV-1 virus-like particles
[227]. In a phosphoproteome study of signaling pathways essential for virus cell-to-cell spread,
LCK and other distal pathways of the TCR were activated and found to be essential for viral
dissemination by the virological synapse [228]. Taken together, LCK initially promotes
protection against HIV-1 infection downstream of CD4 and TCR-CD3 engagement during acute
infection, becomes decoupled from CD4 and relocalized to the trans-Golgi complex where it
facilitates downregulation of cell surface receptors, assists with viral egress and viral spreading,
and becomes downregulated during chronic HIV-1 infection.
FYN
Similar to LCK, the FGR- and c-YES-related protein kinase (FYN) is involved in TCR-CD3
signaling in CD4+ and CD8
+ T-cells, as well as IL-2 production [149, 229]. FYN also
participates in Central Nervous System (CNS) myelination during neuronal development and the
formation of a variety of cancers [230, 231]. FYN can be found in many cell types, although a
unique isoform of FYN, called FYN-T, is expressed only in hematopoietic cells and contributes
to TCR-induced calcium fluxes of antigen-stimulated T-cells [232]. It participates in integrin
signalling, associates with FYN binding protein (FYB) to modulate IL-2 expression, and innate
immune signaling through Toll-Like Receptors (TLRs) expressed on T-cells [233-235].
In the CD4+ and CD8
+ T-cells from HIV-1 non-progressors, FYN is super-activated compared
with cells collected from uninfected controls or HIV-1 patients with AIDS symptoms [149].
This points towards a potential protective mechanism associated with FYN kinase activity.
Corroborating this finding, S1T cells, which lack LCK and FYN expression, are more infectable

29
by HIV-1 [209]. Similar to LCK, pre-treating cells with HIV-1 gp160 prevents CD3-mediated
activation of CD4+ T-cells and FYN phosphorylation, demonstrating that gp160/CD4 signaling
also deregulates the TCR-CD3 activation of FYN [236]. Also similar to LCK, acute HIV-1IIIB
infection of Jurkat T-cells activates FYN activity in the first 30 minutes of infection [214].
Furthermore, FYN can activate the transcription of the HIV-1 promoter by activating four NF-κB
DNA-binding proteins in Jurkat T-cells [237]. This activity required the SH2 domain of FYN
[237]. Unlike LCK however, the effect of FYN during HIV-1 infection is not through an
interaction with Nef. While Nef can bind the isolated SH3 domain of FYN in vitro, it does not
bind full-length FYN, nor has a direct effect on FYN kinase activity in vivo [164].
During the HIV-1 lifecycle, FYN has been shown to directly interact with viral Vif and promote
virion assembly and release [227, 238]. In the absence of Vif, activated FYN is able to
phosphorylate the cytosine deaminase APOBEC3G, increasing the level of phosphorylated
APOBEC3G becoming encapsidated into budding virus, which can restrict viral replication in
the cytosol in a subsequent target T-cell [238]. However in the presence of Vif, Vif protein
binds the SH3 domain of FYN, reducing FYN tyrosine kinase activity. Vif also reduces the
autophosphorylation of FYN [238]. This reduced FYN activity leads to less phosphorylation of
APOBEC3G packaged into virions, counteracting this natural HIV-1 resistance factor in
successive infection cycles and promoting viral infectivity [238]. Interestingly, palmitoylated
FYN can enhance the assembly and release of HIV-1 virus-like particles, in a similar manner as
LCK [227]. This was also dependent on the N-terminal sequence of FYN, localizing FYN to
sites of virus budding at the plasma membrane [227]. Thus FYN activity has multiple effects on
HIV-1 during the viral replication cycle, with different signaling functions as HIV-1 enters the
cell, initiates viral transcription, packages viral and host proteins into virions, and buds from an
infected cell.
HCK
Hematopoietic cell kinase (HCK) is primarily expressed in B-lymphoid and myeloid cell
lineages, such as promoncytic cells and monocyte-derived macrophages, and is not expressed in
CD4+ T-cells [239]. In phagocytic cells, HCK mediates proinflammatory cytokine production,

30
Fc-gamma receptor 1 (FcγRI) signaling, phagocytosis, migration and cell spreading [240-242].
It also has important functions in the migration and degranulation of neutrophils [243].
In primary, naive B-cells, it has been found that HIV-1 gp120 signals through integrin α4β7,
resulting in reduced proliferative responses that correlated with downregulated expression of
HCK [244]. This may contribute to the delayed and ineffective humoral response after acute
HIV-1 infection, as these primary B-cells treated with gp120 also increased expression of the
immunosuppressive cytokine TGF-β1 [244]. After B-cells, HCK is most strongly expressed in
monocytes/macrophages, which are important target T-cells of HIV-1 replication and
maintenance of the viral reservoir. In monocyte-derived macrophages stimulated by macrophage
colony-stimulating factor (M-CSF), high HCK expression correlated with increased HIV-1
infection, and reducing HCK expression with antisense oligonucleotides blocked viral replication
[245]. Of all the SFKs, viral Nef most strongly binds HCK, and this interaction has been
described in great detail [164]. Nef activates HCK in vitro and in primary macrophages, and the
Nef PxxP motif is essential for Nef binding to the SH3 domain of HCK [164]. This implies that
Nef activates SFKs through intramolecular displacement of SH3 binding to the SH2-linker.
Interestingly, dephosphorylation of the negative regulatory tyrosine on the C-terminal tail, or
displacement of the SH2 domain, are not required for Nef-mediated HCK activation, suggesting
this method of SFK activation of an otherwise inactive kinase is unique to Nef [164]. This Nef-
HCK interaction has spurred the structure-based design of novel HCK inhibitors that can reduce
HIV-1 infection without causing cell cytotoxicity [246, 247]. In transgenic mice expressing
mutant Nef lacking the PxxP motif, none of the mice developed an AIDS-like disease [168].
However, mice expressing wild type Nef and HCK knockout only showed delayed onset of the
AIDS-like phenotype [168]. This suggests the SH3 binding ability of Nef is critical for
developing severe AIDS-like disease, but that it depends on the interaction with a more essential
factor that binds PxxP on Nef, perhaps another SFK such as c-SRC.
Nef-mediated activation of HCK has multiple affects on intracellular HIV-1 replication, surface
receptor expression levels and cell motility. Similar to Nef-LCK interactions, HIV-1 Nef
inhibits the anterograde transport of HCK to plasma membrane microdomains, causing HCK to
accumulate at recycling endosomes and the trans-Golgi network [167]. Through HCK, Nef

31
perturbs the intracellular maturation and trafficking of receptors destined to the plasma
membrane, such as CSF-1 receptor (CSF-1R) expression on macrophages [248]. M-CSF is a
cytokine released during viral infection, and promotes the differentiation of macrophages.
However during HIV-1 infection, Nef activation of HCK in the trans-Golgi network
donwregulates surface expression of mature CSF-1R by causing accumulation of under-N-
glycosylated CSF-1R [248]. It was determined that Nef-activated HCK disrupts the distribution
of Golgi Matrix protein 130 (GM130), which is required for efficient protein glycosylation
[249]. Moreover, Nef-activated HCK induced ERK signaling that caused serine phosphorylation
of the GM130-interacting protein Golgi Reassembly-Stacking Protein 1 (GORASP1), causing
this structural protein to unstack Golgi cisternal membranes [249]. The Nef interaction with the
SH3 domain of HCK is also responsible for signalling that downregulates MHC I surface
expression, potentially enabling infected cells to evade killing by CD8+ cytotoxic T-cells [165].
In immature dendritic cells, HIV-1 Nef also induces the downregulation of CD1 surface
receptors, a class of non-MHC lipid antigen-presenting proteins [250]. This effect was mediated
by activated HCK and p21-activated kinase 2 (PAK2) [250]. The PxxP-SH3 interaction between
Nef and HCK can also enhance the incorporation of HCK into budding viral particles from 293T
cells [251]. In addition, Nef inhibits the amoeboid migration pattern of HIV-1 infected
monocytes-derived macrophages, promoting instead mesenchymal migration in vitro [252].
Mesenchymal motility requires podosome regulation and extracellular proteolysis of the matrix,
and it was found that Nef alters the stability, size and proteolytic function of podosomes through
increased HCK activity and Wiskott-Aldrich Syndrome protein (WASp) [252]. This Nef-HCK-
mediated migration reprogramming in macrophages could have important effects on the
dissemination and spread of HIV-1 infection in vivo, as transgenic mice expressing Nef show
increased tissue infiltration [252] and HIV-1 patient tissues accumulate macrophages during
infection [253].
While Nef-induced activation of HCK facilitates HIV-1 infection, HCK kinase activity impairs
later stages of virus assembly and release. This HCK activity is counteracted by viral Vif. Using
living cell fluorescence microscopy, it has been observed that Vif multimerization, an indication
of its role in Gag viral assembly and genomic RNA packaging into viral particles, is altered by

32
HCK expression in HeLa cells [254]. In HCK-expressing cells infected with Vif-deleted HIV-1,
both the release of viral particles and subsequent virion infectivity are strongly reduced [255].
Similar to FYN, Vif has been shown to bind the SH3 domain of HCK, reducing its activity and
preventing autophosphorylation of HCK tyrosine kinase [255]. Also like FYN, HCK
phosphorylates tyrosine on APOBEC3G, increasing the levels of this restriction factor being
packaged into budding virions [238]. As expected, HIV-1 expressing Vif can mitigate this by
reducing HCK activity [238]. Thus, the opposing effects of Nef and Vif on HCK activity
demonstrate the complexity of HIV-1 fine-tuning SFK activity, both in their location and timing,
during the virus replication cycle.
LYN
LCK/c-YES novel tyrosine kinase (LYN) is primarily expressed in hematopoietic cells, neural
cells, and liver tissue [256-258]. LYN kinase activity has important function in regulating
degranulation of mast cells, erythrocyte differentiation, and cell activation [259]. In particular,
LYN has been implicated downstream of B-Cell Receptor (BCR) engagement [260]. Activation
of the BCR receptor leads to LYN phosphorylating Immunoreceptor Tyrosine-based Activation
Motifs (ITAMs) on the cytosolic portions of receptor proteins, recruiting and activating Spleen-
associated tyrosine Kinase (SYK), Phospholipase C gamma 1 (PLC-γ1) and Phosphoinositide 3-
Kinase (PI3K), which further transduce activation signals, induce Ca2+
mobilization, and
promote B-cell proliferation and cell differentiation [194, 261, 262].
As with LCK, HCK and c-SRC, LYN has been shown to bind the Nef PxxP motif through its
SH3 domain, causing allosteric displacement of the SH2-linker, which increases LYN kinase
activity [164]. LYN and HCK share a key Ile residue in their SH3 domain, responsible for their
high affinity binding to a hydrophobic pocket within the core of Nef [164]. However, a
biological role for Nef activation of LYN during HIV-1 infection has yet to be described. Yet
the interactions between LYN, HCK or c-SRC with Nef are highly conserved, as shown with
different Nef proteins isolated from HIV-1 M-group subtypes [263]. In these experiments
testing infection of CEM-T4 lymphoblast cells with chimeric HIV-1 expressing each Nef
protein, Nef-SFK binding could be uniformly disrupted with the compound DFP [263]. This

33
demonstrates that Nef-dependent activation of SFKs are a conserved feature of M-group HIV-1
isolates.
Studies have also demonstrated other roles of LYN kinase activity during HIV-1 infection, with
particular focus on alterations to hepatocytes and dendritic cells. Coinfection with HCV and
HIV-1 has led to research into the effects of HIV-1 envelope proteins on hepatocytes, as patients
with both viruses are more likely to develop liver disease or liver cirrhosis [257]. HIV-1 gp120
has been found to cause bystander apoptosis of uninfected hepatocytes through a Signal
Transducer and Activator of Transcription 1 (STAT1) signaling pathway that involved activation
of LYN, mitogen-activated protein kinase 14 (p38α), and Protein Kinase C (PKC) [257]. In
immature monocyte-derived DCs, it has been demonstrated that inhibiting LYN with the SFK
inhibitor PP2, or reducing LYN expression with targeted siRNA, leads to greater HIV-1
production when co-cultured with primary CD4+ T-cells [264]. This finding suggests LYN may
play an important role in the transfer of HIV-1 from DCs, as LYN has previously been described
to associate with the cytoplasmic tail of DC-SIGN [265]. Furthermore, LYN kinase activity has
been implicated in immune evasion strategies of HIV-1 in immature DCs as they respond to free
HIV-1 virions and complement-opsonized HIV-1 [266]. While exposure of immature DCs to
free virus induced proinflammatory cytokines IL-1β, IL-6, and Tumor Necrosis Factor alpha
(TNF-α), these were dampened in cells treated with complement-opsonized HIV-1 [266].
Instead, these immature DCs showed a different signaling pattern that involved the activation of
IFN regulatory factor 3 (IRF3) and phosphorylated LYN, leading to enhanced infection of the
immature DCs [266]. It is hypothesized that C3-opsonized HIV-1 could be signaling through
TLRs and complement receptor 3 (CR3), which leads to a LYN/PI3K pathway that suppresses
inflammatory responses in immature DCs [266]. These findings suggest HIV-1 could be
subverting the host complement system in a LYN-dependent manner, which may have
implications in immature DCs of the genital tract and rectal mucosa, as these are one of the first
cell types to contact HIV-1 shortly after sexual transmission of the virus.
The effects of LYN kinase activity during HIV-1 infection have been most documented in
monocytes and macrophages, downstream of CCR5 and CXCR4 receptor stimulation. Treating
monocytes-derived macrophages with Macrophage Inflammatory Protein 1 Beta (MIP-1β) leads

34
to activating a CCR5/LYN/ERK-1/2 pathway, which can similarly be triggered by stimulation
with HIV-1 gp120 [267]. This inappropriate macrophage activation by gp120 can induce the
production of TNF-α and other proinflammatory cytokines, which could contribute to AIDS
pathologies and HIV-1 associated dementia [267]. A separate study on primary human,
monocytes-derived macrophages, also found that stimulation of CCR5 with gp120 or whole
virions triggers IL-1β release, which depended on coupling of the Gαi subunit to the CCR5
receptor [268]. This gp120/CCR5 stimulation also led to concomitant PTK2B and PI3K
activation, translocation of both of these signaling proteins from the cytoplasm to the plasma
membrane, and formation of a signaling complex with activated LYN tyrosine kinase [268].
Monocytes migration is also regulated by LYN activity downstream of CXCR4 receptor
engagement with its ligand, SDF-1α [269]. When Brain Microvascular Endothelial Cells
(BMVEC) are activated with TNF- α, IL-1β or treated with HIV-1 gp120, it has been observed
that SDF-1α-treated monocytes no longer adhere to Intercellular Adhesion Molecule 1 (ICAM-1)
ligands, but instead become migratory [269]. Conversly, targeted siRNA knockdown of LYN
prevented the SDF-1α-mediated migration of monocytes, promoting monocytes attachment to
ICAM-1 on activated BMVECs [269]. This data strongly suggests that LYN kinase regulation
of SDF-1α-activated monocytes migration could have an important role in the infiltration of
monocytes into the CNS and past the blood-brain barrier, which may contribute to the various
neurological disorders associated with chronic HIV-1 infection.
FGR, c-YES and BLK
For the final three SFK members, Feline Gardner-Rasheed tyrosine kinase (FGR), cellular
homolog of the Yamaguchi sarcoma virus oncogene (c-YES) and B lymphocyte kinase (BLK),
less is known about their roles during HIV-1 infection.
Expressed exclusively in hematopoietic cells, FGR is found in myeloid cells and B-cells, and
localizes to plasma membrane ruffles [195]. It acts a negative regulator of cell migration and
adhesion downstream of integrin beta-2 (ITGB2) signaling [270]. FGR overexpression has been
associated with a subset of myeloid leukemia and ovarian tumors [271, 272]. Interestingly, the
amino-acid sequence of FGR shares a limited but significant N-terminal sequence homology

35
with HIV-1 Nef [273]. Despite strong expression in macrophages and overlap in signaling
functions with HCK, FGR does not bind or become activated by Nef, likely because FGR lacks
the critical Ile residue in its SH3 domain [164]. In a study of the effects of HIV-1 gp120 on
downstream signaling in naïve B-cells, it was determined that gp120 binds integrin α4β7,
restricting proliferation [244]. This signaling was associated with increased production of the
immunosuppressive cytokine Transforming Growth Factor Beta 1 (TGF-β1), increased
expression of the inhibitory surface receptor FcRL4, and downregulation of FGR expression
[244]. Thus, HIV-1 may be subverting early HIV-1-specific humoral immune responses by
causing B-cell dysfunction through integrin signaling through FGR. Early HIV-1 infection also
appears to alter FGR expression in vaginal epithelial cells. Compared with untreated control
cells, vaginal epithelial cells treated with HIV-1 gp120 showed upregulation of FGR kinase
expression, which occurred in concert with genes that promote inflammation and proteases that
are capable of weakening the vaginal epithelium [274]. Accordingly, FGR may have unique
signaling functions that are tissue specific during HIV-1 infection, reminiscent of how LCK and
HCK exhibit up- and down-regulated activity at specific stages of the HIV-1 replication cycle.
In contrast with FGR, c-YES tyrosine kinase is ubiquitously expressed in many cells and tissues,
having an essential role in regulating cell growth, cell survival, cell adhesion, cytoskeletal
rearrangements, and cell differentiation [275-277] . c-YES signaling has important functions in
glucose activation of the cell cycle, translocation of Epidermal Growth Factor Receptor (EGFR)
into the nucleus during tumor progression, and Platelet-Derived Growth Factor Receptor (PDGF-
R) induced chemotaxis [278-280]. c-YES also has an important role in mediating chemokine-
directed T-cell motility [281]. Jurkat T-cells treated with SDF-1α stimulates CXCR4, which
induces c-YES to phosphorylate Collapsin Response Mediator Protein 2 (CRMP2) at Tyr479,
leading to T-cell polarization and lymphocyte migration [281]. As with FGR, the SH3 domain
of c-YES does not permit interactions with HIV-1 Nef [164]. In a study of HIV-1 Tat modifying
global gene expression in primary CD4+
T-cells, it was found after 24 hrs of Tat exposure that c-
YES showed a modest, yet significant reduction in gene expression [282]. This correlated with
the CD4+ T-cells increasing secretion of 11 different cytokines, with notable production of IL-
17, and proinflammatory gene expression reminiscent of the Th17 phenotype [282]. However,

36
this finding is incongruent with another study examining the role of c-YES and apoptosis-related
genes during HIV-1 infection of Jurkat T-cells [283]. In this work, Jurkat T-cells infected with
HIV-1 showed increased c-YES expression within 3 days, as confirmed by Western blot [283].
The authors suggest that c-YES may be signaling downstream of the Fas receptor (FasR), which
can oligomerize and trigger apoptosis through the Death-Inducing Signaling Complex (DISC)
[283].
Lastly, BLK is a tyrosine kinase expressed in pancreatic β cells and hematopoietic cells, with
essential function in B-cells in particular [284, 285]. It participates in pre-BCR signaling, B-cell
development, and B-cell differentiation [286, 287]. As with other SFKs, BLK transmits signals
from cell surface receptors, and has been implicated in B-cell associated autoimmune disorders
[288]. Little is known of changes to BLK activity in response to HIV-1 infection, although a
BLK gene variant has been found to disrupt B-cell proliferation and the ability to elicit antigen-
specific CD4+
T-cells in patients suffering from Common Variable Immunodeficiency (CVID)
[196]. A genome-wide study of host proteins involved in early HIV-1 infection found that
siRNA knockdown of BLK could reduce infection of VSV-g pseudoenveloped HIV-1 in 293T
cells [289]. However, confirmatory experiments exposing primary B-cells to extracellular HIV-
1 proteins such as gp120, Nef or Tat, have yet to ascribe a function for BLK during HIV-1
infection.

37
1.2.5 Role of c-SRC in HIV-1 Infection
c-SRC is the canonical member of the SFKs, often expressed as a cytosolic proto-oncogene
tyrosine kinase. In humans, it is expressed in nearly all cells and tissues, with high levels of
protein found in the brain (neuronal splice variant), testis, platelets and PBMCs [190, 290-292].
c-SRC is expressed in CD4+ T-cells 12 hours following TCR-CD3 activation [293]. Its primary
functions are thought to play a role in fetal organogenesis, differentiation, cell cycle progression,
proliferation, cell survival, cell adhesion, and cell migration via cytoskeletal rearrangement at
adhesion networks [294]. c-SRC is a key signaling molecule that integrates multiple signal
inputs that induce a variety of downstream signal cascades, as evidenced by the 132 c-SRC
mediated phosphorylation sites in the human proteome, 64 known substrates, and over 204
proteins that interact with c-SRC [294]. c-SRC is predominantly repressed in an inactive state,
and transiently activated during cellular processes such as mitosis [189]. Conversely, the kinase
is constitutively active in abnormal states associated with human cancers and viral infection of
chickens by RSV [295, 296]. c-SRC is often associated with cellular membranes, in particular
the plasma membrane, endosomal membranes and the nuclear envelope [297, 298]. The defined
subcellular locations of c-SRC are important for the regulation of specific cellular events, such as
cytoskeletal rearrangement, membrane trafficking and cell cycle progression [189].
Given the essential role of c-SRC in cellular processes, and its potential to cause aberrant tumor
growth or differentiation when constitutively active (ex. colon, breast or prostate cancers [296,
299, 300]), it is unsurprising that the cell regulates c-SRC activation with multiple post-
translational modifications (see Figure 1.7 for c-SRC protein structure and regulation, originally
published in [199]). Phosphorylation of the C-terminal tyrosine residue Tyr530 leads to
intermolecular folding and binding to the SH2 domain, suppressing c-SRC tyrosine kinase
activity [301]. Interestingly, the regulatory Tyr530 is absent in the truncated v-SRC protein,
accounting for its constitutive activity [302]. The conformational change caused by Tyr530
phosphorylation also allows intramolecular binding of the SH3 domain to a proline-rich motif,
further suppressing c-SRC activity [303]. A variety of c-SRC binding proteins regulate c-SRC
activity by disrupting its SH2 and SH3 intramolecular interactions. For example, PDGFR and
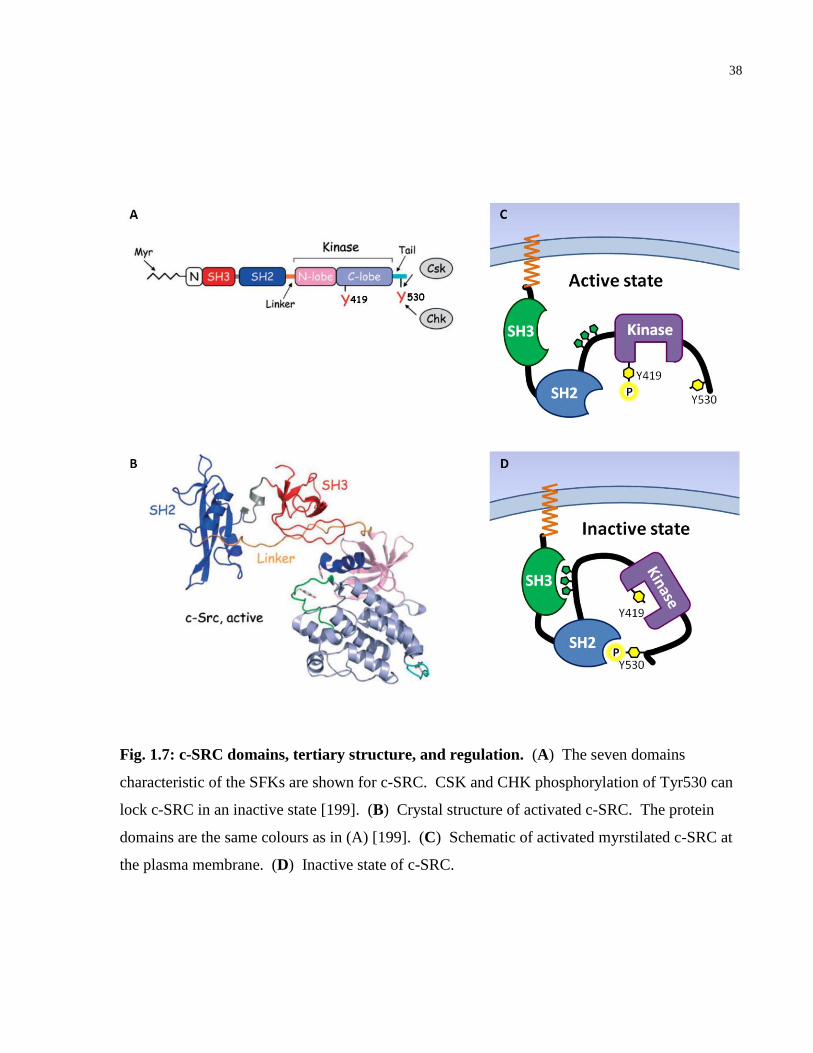
38
Fig. 1.7: c-SRC domains, tertiary structure, and regulation. (A) The seven domains
characteristic of the SFKs are shown for c-SRC. CSK and CHK phosphorylation of Tyr530 can
lock c-SRC in an inactive state [199]. (B) Crystal structure of activated c-SRC. The protein
domains are the same colours as in (A) [199]. (C) Schematic of activated myrstilated c-SRC at
the plasma membrane. (D) Inactive state of c-SRC.

39
Focal Adhesion Kinase (FAK) can bind the c-SRC SH2 domain and activate the enzyme [183,
304], while Sin is a protein that can bind the SH3 domain to activate c-SRC [305]. Moreover,
dephosphorylation of Tyr530 by cellular Protein Tyrosine Phosphatases (PTPs) relieves this
intramolecular inhibition of c-SRC kinase activity [306]. Full activation of the enzyme requires
a second major tyrosine residue, Tyr419, located in an activation loop in the catalytic domain
[302]. Phosphorylation of c-SRC at Tyr 419 displaces this residue from the substrate binding
pocket, permitting the kinase to phosphorylate substrate targets. Tyr419 can be phosphorylated
by other tyrosine kinases or through autophosphorylation [307]. Therefore, phosphorylation of
Tyr419 acts as a positive regulator of c-SRC activity, while phosphorylation of Tyr530 acts as a
negative regulator to suppress c-SRC activity [302].
c-SRC has multiple phosphatase and kinase binding partners that tightly control its kinase
activity. Phosphatases that dephosphorylate c-SRC Tyr530 to activate the enzyme include:
Receptor-type tyrosine-protein phosphatase alpha (PTPRA, PTP-α) and PTP non-receptor type 6
(PTPN6 or SHP-1), 11 (PTPN11 or SHP-2) and 12 (PTPN12 or PTP-PEST) [306, 308-310]. C-
terminal SRC Kinase (CSK) and C-terminal SRC kinase-Homologous Kinase (CHK) are two
kinases that counteract these effects at Tyr530 [191, 311]. In resting T-cells, CSK binding
protein (Cbp) and phosphoprotein associated with glycosphingolipid-enriched microdomains
(PAG) are constitutively phosphorylated, recruiting CSK which prevents the activation of c-SRC
[312]. In contrast, T-cell activation induces rapid dephosphorylation of Cbp/PAG, reducing
PAG-mediated inhibition of c-SRC by CSK [312].
As previously mentioned, the subcellular location of c-SRC has important function in the
signaling cascades that it regulates. Myristoylated c-SRC associated with the plasma membrane
regulates cell growth and cell proliferation downstream of growth factor receptors [304]. Plasma
membrane c-SRC also localizes at focal adhesion plaques and adheren junctions between cells
[313], which is dependent on an intact actin cytoskeleton [314]. c-SRC is also localized to the
perinuclear Golgi region of the cell, accounting for 30-40% of the total c-SRC cellular protein
[315]. Within the nucleus, c-SRC has been proposed to regulate cell cycle progression and entry
into mitosis by interacting with proteins that regulate cell division [298]. It has been shown that
the SH3 domain of c-SRC interacts with Sam68, a nuclear protein with 6 proline-rich regions

40
and a tyrosine-rich C-terminal domain [316]. Sam68 is a multifunctional RNA-binding protein
that is required for transactivation of Rev function and the cytoplasmic translation of HIV-1
RNA into proteins [317]. Cytoplasmic mutants of Sam68 have been shown to selectively inhibit
the translation of HIV-1 RNA transcripts [317]. This suggests Sam68 directly participates in the
nuclear exportation of unspliced and singly-spliced HIV-1 RNA. Moreover, overexpression of
Sam68 can enhance the expression of viral p24, through translational regulation of HIV-1 RNA
[317]. Beyond interactions with Sam68, c-SRC has also been found within the nucleolus [298].
Despite all this localization knowledge, little is known of the cytosolic effects of c-SRC signal
transduction away from the plasma membrane, or the specific substrates and binding proteins
that interact with cytosolic c-SRC. Moreover, the potential role of cytosolic c-SRC during the
lifecycle of HIV-1 infection has not been established or well defined.
Previous work has demonstrated that the Jurkat T-cell line exhibits robust c-SRC activity after 30
minutes of HIV-1IIIB infection [214]. Moreover, treatment of activated PBMCs from healthy
human donors with a synthetic peptide expressing the CD4-binding region of gp120 (Peptide T),
could induces an 11-fold increase in c-SRC kinase activity within 15 minutes [318]. As with
other SFK members, viral Nef binds to c-SRC through allosteric displacement of SH3 domain
interactions, requiring the PXXPXR motif on c-SRC [164]. Yet the direct effect of Nef on c-
SRC activity in vivo during infection is not clear [164, 319]. Relative to HCK, c-SRC lacks the
SH3 domain Ile residue that promotes high Nef binding affinity [164]. This creates an SH3
domain more constrained by hydrogen bonds, and may take a conformation less compatible for
Nef interactions [164]. In this study, Nef binding to c-SRC alone could not induce strong c-SRC
kinase activity in vitro [164]. Yet in another paper on immortalized podocytes, Nef was shown
to increase c-SRC activity and induce proliferation in this cell line by inducing activation of
STAT3, the Ras family of small GTPases (Ras) and MAPK1[320]. These Nef-c-SRC
interactions underpinning aberrant glomerular podocyte growth may contribute to HIV-1
associated nephropathy, which can lead to chronic renal failure in patients living with HIV-1
[320].
Kinase inhibitor experiments have also suggested a potential role for c-SRC during HIV-1
infection. A study examining the transfer of HIV-1 from primary DCs to CD4+ T-cells

41
demonstrated that CD4+ T-cells pre-treated with the broad SFK inhibitor PP2, reduced the p24
produced after 6 days of HIV-1JR-CSF or NL4-3balenv infection [264]. However, it is difficult to
ascertain the direct effects of c-SRC inhibition with pyrazolopyrimidines, such as PP1 or PP2, as
they inhibit LCK and FYN at similar IC50 values as c-SRC [184]. PP1 and PP2 also have many
off targets, such as CSK, which can counteract the effect of either drug on SFK activity in cells
[184]. Research into the effects of LCK, FYN and c-SRC in the budding of HIV-1 virus-like
particles suggests that LCK and FYN both facilitate this step of the viral lifecycle, where as c-
SRC had no effect on virus released from transfected 293T cells [227]. Thus c-SRC likely
affects HIV-1 replication at an earlier stage of the viral lifecycle. Another study investigating the
permeability of endothelial cells in the dissemination of HIV-1 found that pre-treating Lung
Lymphatic Endothelial Cells (L-LECs) with SRC inhibitor 1 (SRC-I1) induced
hyperpermeability of the lymphatic endothelial barrier [321]. Moreover HIV-1 gp120 could
induce similar L-LEC hyperpermeability that required SFK modulation of the cytoskeleton
[321]. However, the direct effects of c-SRC during the early entry events of HIV-1 in CD4+ T-
cells, and whether they modify the host cytoskeleton, has not yet been determined.
c-SRC involvement in cytoskeletal organization and membrane trafficking via its binding
partner, protein tyrosine kinase 2 beta (PTK2B), is a potential signaling pathway exploited by
HIV-1 during early infection [147, 197, 322]. PTK2B/c-SRC activity has also been implicated
in the migration and podosome formation of immature DCs exposed to soluble gp120, which
may enhance the mucosal transmission of HIV-1 as these cells migrate in response to HIV-1
envelope protein [323]. Immature DCs treated with gp120 enhanced the phosphorylation of c-
SRC and PTK2B, which activated Rac-1 subfamily of Rho small GTPases (Rac-1) signaling and
subsequent paxillin phosphorylation, an essential adhesion molecule that facilitates podosome
ring structure [323]. Whether PTK2B/c-SRC signalling plays a similar role in the migration of
CD4+ T-cells during HIV-1 infection, has yet to be evaluated.

42
1.2.6 Role of PTK2B in HIV-1 Infection
PTK2B, the second member of the FAK family, is a 112 kDa non-receptor tyrosine kinase highly
expressed in hematopoietic, neuronal and epithelial cell lineages [324-326]. Slightly smaller
than FAK, the two proteins share 45% sequence identity [327] (see Figure 1.8 for PTK2B
structure, originally published in [328] and [329]). An alternatively spliced form of PTK2B is
expressed in the spleen, with a 42 amino acid deletion within the C-terminal domain [324].
PTK2B contains tyrosine phosphorylation sites that recruit binding of SH2 domains of proteins
such as Growth Factor Receptor-Bound protein 2 (GRB2) and c-SRC [197]. In addition, the C-
terminal end of PTK2B contains two conserved proline-rich motifs that can further bind proteins
containing an SH3 domain [330]. The C-terminus also contains a Focal Adhesion Targeting
(FAT) sequence that allows for binding to paxillin or leupaxin [331]. Unique to the FAK family,
PTK2B has a central tyrosine kinase domain flanked by an N-terminal Four point
1/Ezrin/Radixin/Moesin (FERM) domain, which regulates PTK2B complex formation and
phosphorylation [332]. Like c-SRC, multiple phosphorylation sites tightly regulate PTK2B
function. Located in the linker region between the FERM domain and kinase domain is Tyr402,
a residue critical to the activation of PTK2B and the recruitment of SFKs [197]. The activation
loop in the kinase domain also contains Tyr579 and Tyr580, two amino acids that when
phosphorylated, induce maximal PTK2B kinase activity [333].
Various cell models have explored the interaction between PTK2B and c-SRC, which link Gi-
and Gq-coupled receptors with downstream effects within the cell [197]. C-terminal
dephosphorylation of c-SRC at Tyr530 by PTPs is one of the earliest steps to c-SRC activation
[306, 308-310]. The SH2 domain of c-SRC then becomes free to bind ligands such as PTK2B, at
the PTK2B autophosphorylation site Tyr402 [197]. The binding of c-SRC to PTK2B regulates
both the activation of c-SRC kinase activity and its cellular localization, bringing c-SRC in close
proximity of potential substrates. For instance, autophosphorylation of PTK2B creates a binding
site for the c-SRC SH2 domain, localizing c-SRC to focal adhesions where it can phosphorylate
paxillin to regulate cell migration [183].
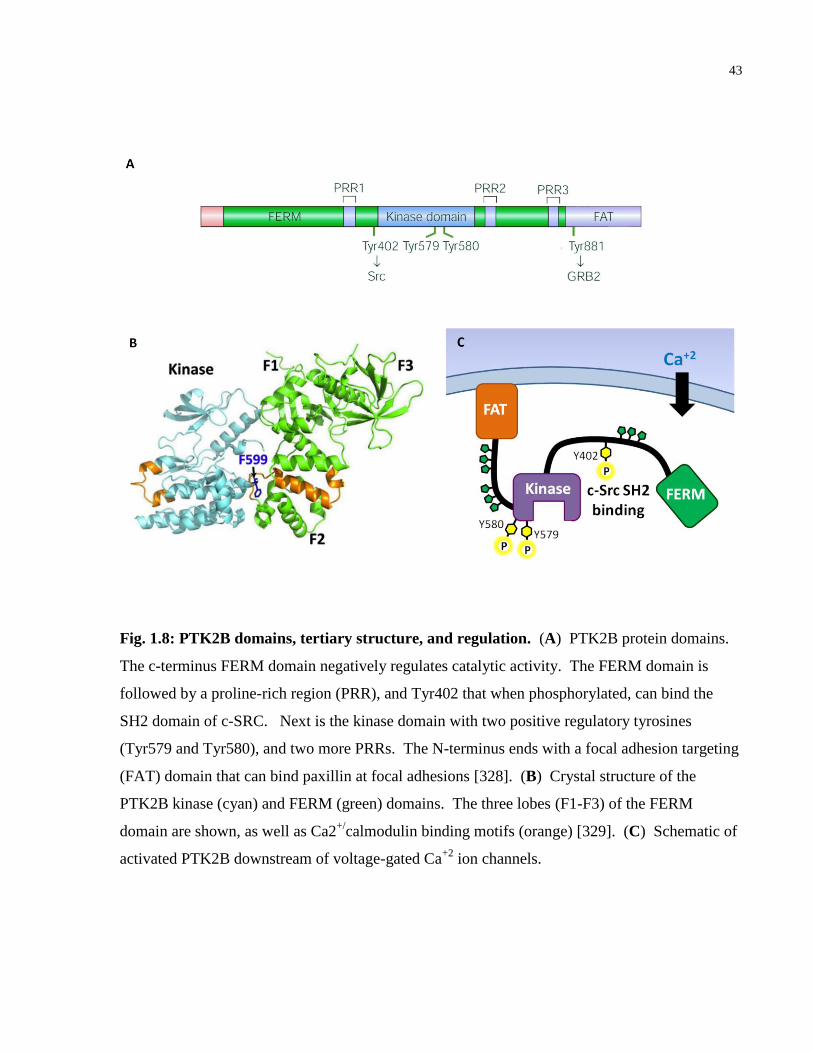
43
Fig. 1.8: PTK2B domains, tertiary structure, and regulation. (A) PTK2B protein domains.
The c-terminus FERM domain negatively regulates catalytic activity. The FERM domain is
followed by a proline-rich region (PRR), and Tyr402 that when phosphorylated, can bind the
SH2 domain of c-SRC. Next is the kinase domain with two positive regulatory tyrosines
(Tyr579 and Tyr580), and two more PRRs. The N-terminus ends with a focal adhesion targeting
(FAT) domain that can bind paxillin at focal adhesions [328]. (B) Crystal structure of the
PTK2B kinase (cyan) and FERM (green) domains. The three lobes (F1-F3) of the FERM
domain are shown, as well as Ca2+/
calmodulin binding motifs (orange) [329]. (C) Schematic of
activated PTK2B downstream of voltage-gated Ca+2
ion channels.

44
PTK2B is most well studied for its participation in integrin receptor signaling pathways at focal
adhesions in adherent cell types [334], signaling during apoptosis [335], cell migration [326], as
well as responding to elevated calcium signals [336]. Extracellular matrix proteins binding to
integrins are important for transducing signals that promote cell growth, cell survival, and cell
migration. In this integrin pathway, PTK2B colocalizes with integrin receptors at cell contact
sites, termed focal adhesions, to phosphorylate structural proteins such as paxillin, which directly
associate with the actin cytoskeleton [334]. PTK2B also recruits SFKs and other focal adhesion
proteins to activate signaling targets such as Rac1, a member of the Rho Guanosine
Triphosphatase (GTPase) family [337]. In the absence of integrin signaling that promotes
adhesion-dependent cell survival, PTK2B activation can induce early apoptotic signaling by
activating caspase-3, SFKs, and increase DNA fragmentation [335]. In conjunction with integrin
receptors, PTK2B transduces migratory signals downstream of growth factor, antigen,
chemokine and cytokine receptors [147, 326, 338]. PTK2B is increasingly being recognized for
its role in responding to elevated intracellular calcium signals as a Calcium-Dependent Tyrosine
Kinase (CADTK). An influx of extracellular calcium via voltage-gated calcium channels or
cytosolic release from intracellular stores can induce an increase in PTK2B tyrosine kinase
activity [339]. It has also been suggested that calpains, Ca2+
-dependent cytosolic cysteine
proteases, cleave PTK2B in order to regulate cell attachment and motility through assembly and
disassembly at focal adhesions [336].
Within T-cells, PTK2B has many important roles in T-cell stimulation and effector cell functions
[340, 341]. For instance, PTK2B is essential for LFA-1 dependent CD8+ T-cell activation,
migration and cell adhesion [341], forming micro-adhesion rings around the TCR to stabilize the
immunological synapse [342], establishing cell polarity [180], and donwregulating activated
surface receptors. In helper CD4+ and cytotoxic CD8
+ T-cells, TCR and integrin stimulation
lead to tyrosine phosphorylation of PTK2B, enhancing its catalytic activity, and causing PTK2B
translocation to sites of cell contact [343]. PTK2B also participates in downstream signaling of
TCR and CD28 costimulation that promote IL-2 production [344]. Unique in T-cells, PTK2B
exhibits a biphasic burst in activity shortly after TCR engagement that is dependent on SFK
activation [340]. Both Tyr402 and Tyr580 on PTK2B are phosphorylated early (5 min) and then

45
late (30-60 min) after TCR activation, causing two distinct phases of paxillin phosphorylation
and actin polymerization [340]. It has been hypothesized that the two periods of PTK2B
activation control the cytoskeletal assembly and then disassembly necessary for optimal T-
cell/APC contact, which occurs for approximately 1 hr [340]. In the first burst, PTK2B
facilitates cytoskeletal changes that may reinforce the T-cell and APC contact, while the second
burst of PTK2B activity disassembles the actin cytoskeleton, and may help disengage the T-cell
from the APC.
Past research suggests PTK2B signaling may influence HIV-1 binding and entry of CD4+ T-cells
through signaling downstream of G protein-coupled receptors engaged during viral co-receptor
binding [147], and responding to ATP that enhances viral membrane fusion to the target T-cell
[345]. After natural stimulation of surface CCR5 with Chemokine (C-C motif) Ligand 5 (CCL5)
or MIP-1β, or CXCR4 stimulation with SDF-1α, PTK2B becomes rapidly phosphorylated [147].
This suggests that HIV-1 binding to either surface receptor might mimic natural ligand
engagement and produce physiologically similar signals. Indeed, infecting the T-cell line DU6
(CCR5+) with JR-FL (R5) pseudoenveloped virus induced PTK2B activation, as did mixing
HL60 cells (CXCR4+) with 293T cells expressing HXB2 (X4) envelope protein [147]. Yet
whether PTK2B activation is simply a byproduct of HIV-1 envelope engagement, or has a
functional role in the intracellular lifecycle of HIV-1 post-fusion, has yet to be determined.
Binding of HIV-1 gp120 to CD4 and a chemokine receptor also induces mechanical membrane
stress, stimulating the release of ATP through pannexin-1 hemichannels [345]. In the
extracellular space, ATP then acts as an autocrine and paracrine signal, activating the purinergic
receptor P2Y2 on the cell surface, which are coupled to G-proteins that recruit PTK2B that
subsequently autophosphrylates at Tyr402 to become activated [345]. P2Y2 also has an SH3
binding domain, which can directly recruit PTK2B and induce its phosphorylation and c-SRC
activation [182]. These provide even more avenues by which PTK2B recruits signaling
complexes that contain SFKs or actin interacting proteins that may enhance HIV-1 fusion upon
entry.
Once HIV-1 enters a T-cell, studies suggest FAK apoptotic signaling is blocked [346], and that
PTK2B promotes cell migration during HIV-1 infection [323]. After gp120 engages CD4 and

46
CCR5, FAK localizes to the inner face of the plasma membrane at these receptors [346]. It then
becomes phosphorylated and cleaved by caspase-3 and caspase-6, promoting apoptosis and
inactivating FAK in uninfected cells [347, 348]. Interestingly, the interaction of the FAT domain
of FAK with the cytosolic portion of CD4 shares similarity with the interaction of HIV-1 Nef
with CD4 [346]. Both FAK and Nef bind CD4 in the same region and with similar affinity
[349], yet cause opposing signalling effects. The FAK-CD4 interaction with gp120 can trigger
apoptosis of a bystander T-cell, through FAK cleavage by caspases [348]. However, the Nef-
CD4 interaction can block this pro-apoptotic signal, permitting an infected T-cell to survive and
productively produce virions [346]. Whether PTK2B has a similar role as FAK, to induce
apoptosis signaling that is blocked by Nef in CD4+
T-cells during HIV-1 infection, has yet to be
assessed. In addition, the potential effect of PTK2B activation on CD4+ T-cell migration during
HIV-1 infection has not been determined. As mentioned earlier, gp120-induced transendothelial
migration of immature DCs relies on c-SRC signalling, as well as downstream signaling by
PTK2B, paxillin, and Rac1 [323]. In these cells, PTK2B activates the Rho GTPase Activated
CDC42 Kinase 1 (ACK1), promoting actin polymerization, ring formation, and nucleation of
actin filaments within the podosome core [323]. Taken together, there are multiple mechanisms
by which c-SRC and PTK2B signaling could be cooperating (or acting independently) to alter
intracellular HIV-1 infection post-entry in CD4+ T-cells.

47
1.3 Ebola Virus (EBOV)
1.3.1 ZEBOV Epidemiology, Transmission and Replication Cycle
In 1976, WHO medical staff and USA Center of Disease Control (CDC) researchers witnessed
two separate outbreaks of an unknown contagious disease in Nzara, southern Sudan (284 cases,
151 deaths), and in Yambuku, northern Zaire (318 cases, 280 deaths) [350, 351]. The disease
caused fever, joint and muscle pain, rash, abdominal pain, diarrhea with blood, and rapid
progression to death, leading to regional panic in both locations. A new virus was isolated by
Dr. Peter Piot in Belgium and further characterized by a CDC researcher, Dr. Karl Johnson, from
the blood sample of an infected Belgium nun in Zaire [352]. The virus was named after the Ebola
River located near the original Zaire outbreak, in what is now the Democratic Republic of the
Congo [352]. It was later discovered that both outbreaks were caused by two distinct species of
ebolavirus, now classified as Sudan ebolavirus (SUDV) and Zaire ebolavirus (ZEBOV) [353].
Three more have been isolated in the Ebolavirus genus: Bundibugyo ebolavirus (BDBV),
discovered in Uganda [354], Reston ebolavirus (RESTV) from the Philippines that infects
cynomolgus macaques and is non-pathogenic to humans [355], and Taï Forest ebolavirus
(TAFV), isolated from a chimpanzee in Côte d’Ivoire [356]. From 1976 to 2013, there have been
24 separate Ebola outbreaks primarily clustered in central Africa, with mortality rates ranging
between 25-90% [6, 357]. The difference in host fatality and disease severity attributed to each
ebolavirus strain is still under investigation [357]. None of these outbreaks compare with the
unprecedented 2014-16 ZEBOV epidemic in West Africa that led to 28,616 cases and 11,310
deaths in Liberia, Guinea and Sierra Leone (see Figure 1.9, originally published in [5]). This
spurred significant developments in Ebola virus therapeutics and vaccines that were tested for
safety and efficacy in human trials during the outbreak. While a licensed treatment has proven
elusive [25], a successful vaccine candidate has recently been announced [26]. Significant
challenges were faced with implementing clinical trials in resource limited settings, including
global support and preparedness, maintaining public trust, ethical concerns of implementing
randomized control trials during an outbreak, and a decline in Ebola cases once clinical trials
were initiated [25].
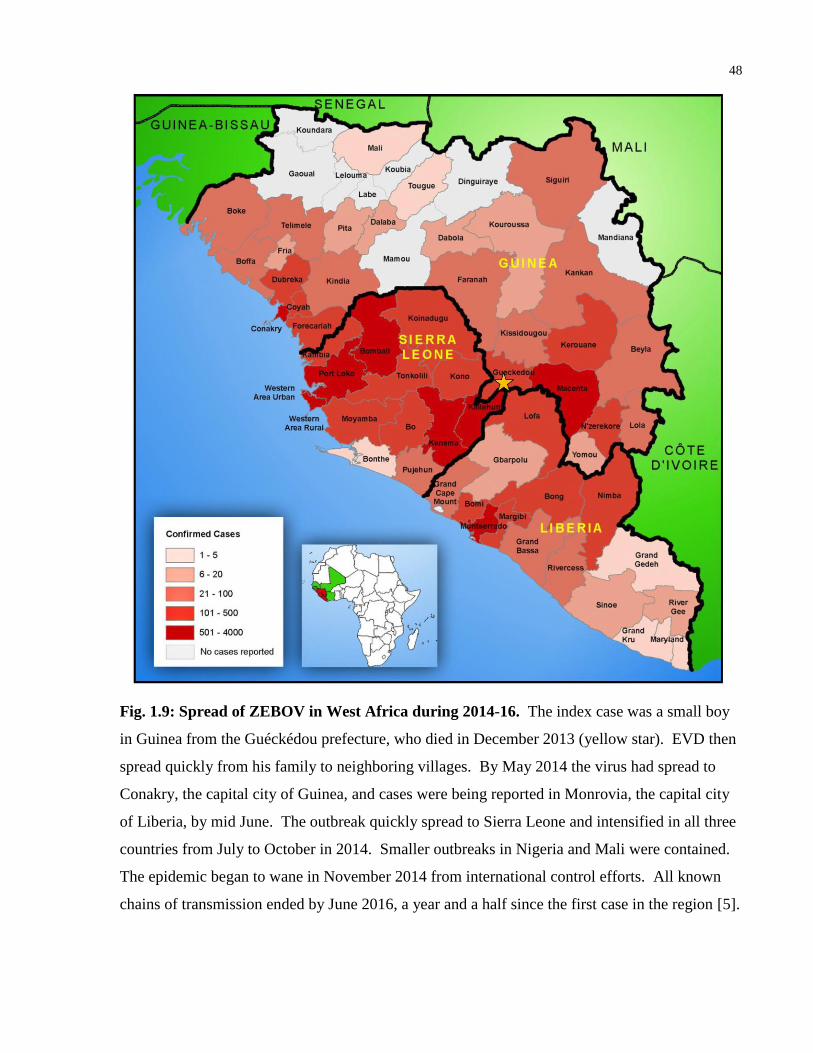
48
Fig. 1.9: Spread of ZEBOV in West Africa during 2014-16. The index case was a small boy
in Guinea from the Guéckédou prefecture, who died in December 2013 (yellow star). EVD then
spread quickly from his family to neighboring villages. By May 2014 the virus had spread to
Conakry, the capital city of Guinea, and cases were being reported in Monrovia, the capital city
of Liberia, by mid June. The outbreak quickly spread to Sierra Leone and intensified in all three
countries from July to October in 2014. Smaller outbreaks in Nigeria and Mali were contained.
The epidemic began to wane in November 2014 from international control efforts. All known
chains of transmission ended by June 2016, a year and a half since the first case in the region [5].

49
The West Africa ZEBOV outbreak began with cases reported in Guinea during December of
2013, then spread quickly to neighboring countries Liberia and Sierra Leone [358]. From a
single introduction into the human population, it caused an overall fatality rate of 40% over
several months [5]. The variant responsible for the epidemic was isolated and determined to be
genetically related to ZEBOV, classified as the EBOV-Makona reference strain [359]. In August
2014 an unrelated outbreak of ZEBOV, caused by the variant EBOV- Lomela, occurred in the
Democratic Republic of the Congo [359]. Transmission of this variant lead to a shorter ZEBOV
epidemic, with fewer fatalities (66 cases, 49 deaths) [359]. It was not until March 29, 2016 that
the WHO declared an end to the Public Health Emergency of International Concern regarding
the ZEBOV outbreak in West Africa, although sporadic cases continue to occur in Guinea and
Sierra Leone [5, 360]. It is not yet understood why some Ebola survivors are asymptomatic
seroconverters, while others succumb to life threatening conditions collectively called Ebola
Virus Disease (EVD) [361]. Studies of large patient cohorts demonstrate that overall survival
and disease severity could be predicted from initial viral load at time of admission to an Ebola
Treatment Center (ETC) [18, 19]. The unprecedented outbreak also produced the largest group
of Ebola survivors, with ~ 17,000 recovered patients in the three most affected countries, who
exhibit newly described chronic afflictions called post-Ebola disease syndrome [362]. Ebola
survivors are at greater risk for ocular problems (blurred vision, retro-orbital pain), loss of
hearing, difficulty swallowing, abdominal and back pain, fatigue, severe headaches, memory
problems, and confusion, among others [362-364]. After acute infection, ZEBOV has been
found to persist in semen, ocular fluid, cerebrospinal fluid, placenta, and amniotic fluid [365-
367]. A case of a woman contracting ZEBOV in Guinea after sexual intercourse with a male
Ebola survivor 470 days after the onset of his symptoms, who also shed ZEBOV in his semen up
to 531 days after disease onset, demonstrates that infectious virus can be transmitted sexually
from Ebola survivors [360].
Ebola viruses are in the family Filoviridae, and are lipid-enveloped, heavily glycosylated,
filamentous RNA viruses (Figure 1.10, originally published in [368]). The 19-kb genome
consists of a non-segmented, negative-sense single strand of RNA ((-)ssRNA) coding for 7
genes, each separated by short intergenic regions [369]. Flanking both ends of the genome
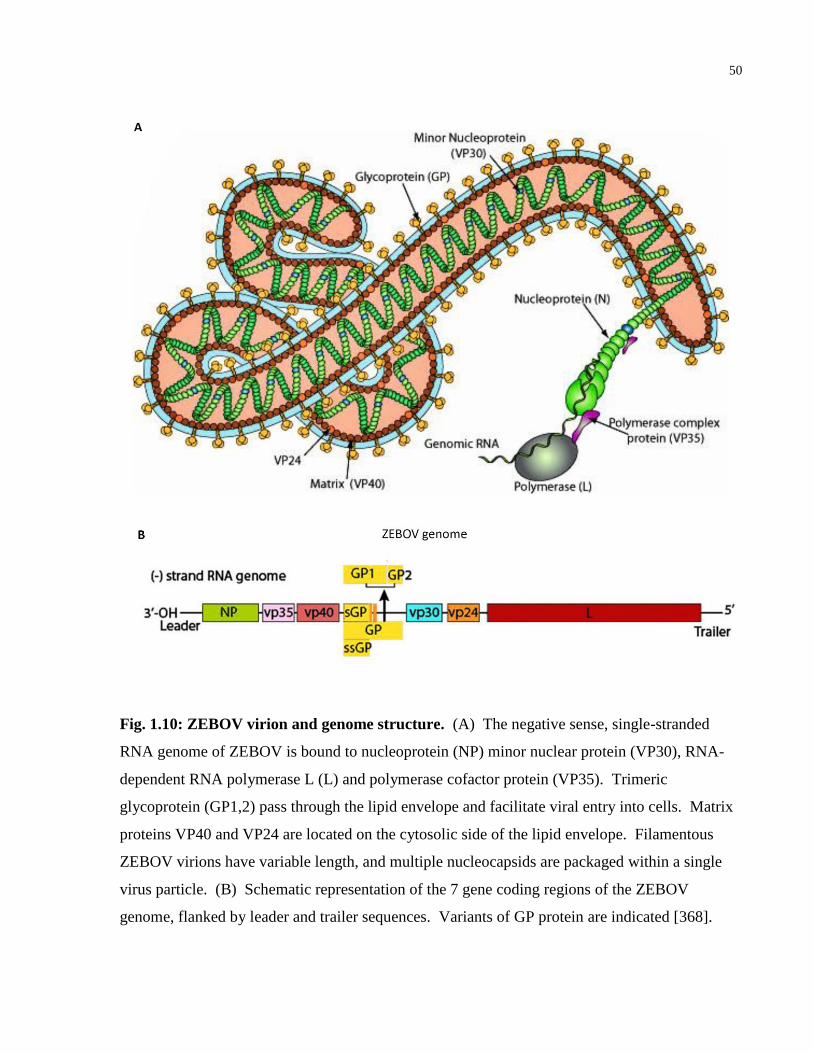
50
Fig. 1.10: ZEBOV virion and genome structure. (A) The negative sense, single-stranded
RNA genome of ZEBOV is bound to nucleoprotein (NP) minor nuclear protein (VP30), RNA-
dependent RNA polymerase L (L) and polymerase cofactor protein (VP35). Trimeric
glycoprotein (GP1,2) pass through the lipid envelope and facilitate viral entry into cells. Matrix
proteins VP40 and VP24 are located on the cytosolic side of the lipid envelope. Filamentous
ZEBOV virions have variable length, and multiple nucleocapsids are packaged within a single
virus particle. (B) Schematic representation of the 7 gene coding regions of the ZEBOV
genome, flanked by leader and trailer sequences. Variants of GP protein are indicated [368].

51
are extragenic regions called the leader and trailer sequences, which signal for encapsidation and
contain promoters for replication and transcription [370]. The genome encodes for 4 structural
proteins (nucleocapsid protein (NP), glycoprotein (GP1,2), viral protein 24 (VP24) and viral
protein 40 (VP40)), and 3 replication proteins (RNA-dependent RNA polymerase L (RdRP L),
viral protein 35 (VP35) and viral protein 30 (VP30)) [369]. GP1,2 is the only surface
glycoprotein that mediates virus fusion and entry, and the mature trimer of GP1-GP2
heterodimers shares structural similarity with retroviruses such as HIV-1 [371]. Transcriptional
stuttering of the polymerase also produces two soluble GP variants called sGP and small sGP.
In the EBOV cellular replication cycle, a variety of host surface receptors act as cofactors prior
to viral entry (see Figure 1.11, originally published in [368]). These include T-cell
Immunoglobulin and Mucin Domain 1 (TIM-1), integrins, asialoglycoprotein expressed on
hepatocytes, and C-type lectins such as DC-SIGN on dendritic cells and macrophages [372-374].
Upon GP1,2 binding a surface cofactor, the virus enters the early endosome by macropinocytosis
[375]. The endosome vesicle acidifies, and host cysteine cathepsins cleave GP1, which unmask
GP2 [376]. This allows host Niemann-Pick C1 (NPC1) to bind the cleaved GP (GPCL) as the
bonafide entry receptor, triggering a conformational change in GP2 that promotes fusion of the
late endosome and viral membranes, releasing the virus into the cytoplasm [376, 377]. Within 6-
12 hours, the RdRP L forms a complex with NP in the form of cytoplasmic inclusion bodies
outside the nucleus [378]. Polymerase L performs primary transcription of individual mRNAs
from the negative-sense RNA genome, with assistance of VP30 (transcription activator) and
VP35 (polymerase cofactor), and the mRNAs are capped at the 5’ end and polyadenylated at the
3’ tail [379, 380]. Each gene is transcribed sequentially from 3’ to 5’, producing abundant
transcripts of genes at the 3’ end relative to the 5’ end [370]. Thus NP transcripts are the most
abundant, while RdRP L transcripts are the least transcribed. Phosphorylation of VP35 triggers
RdRP L, NP and VP30 to switch to genomic replication, producing full-length positive-sense,
anti-genomic RNA that serve as templates for negative-sense genomic synthesis (secondary
transcription) [381]. Newly translated NP, VP35, VP30 and RdRP L proteins associate with the
progeny negative-sense RNA strands, while GP and sGP mature separately in the endoplasmic
reticulum and Golgi bodies to be
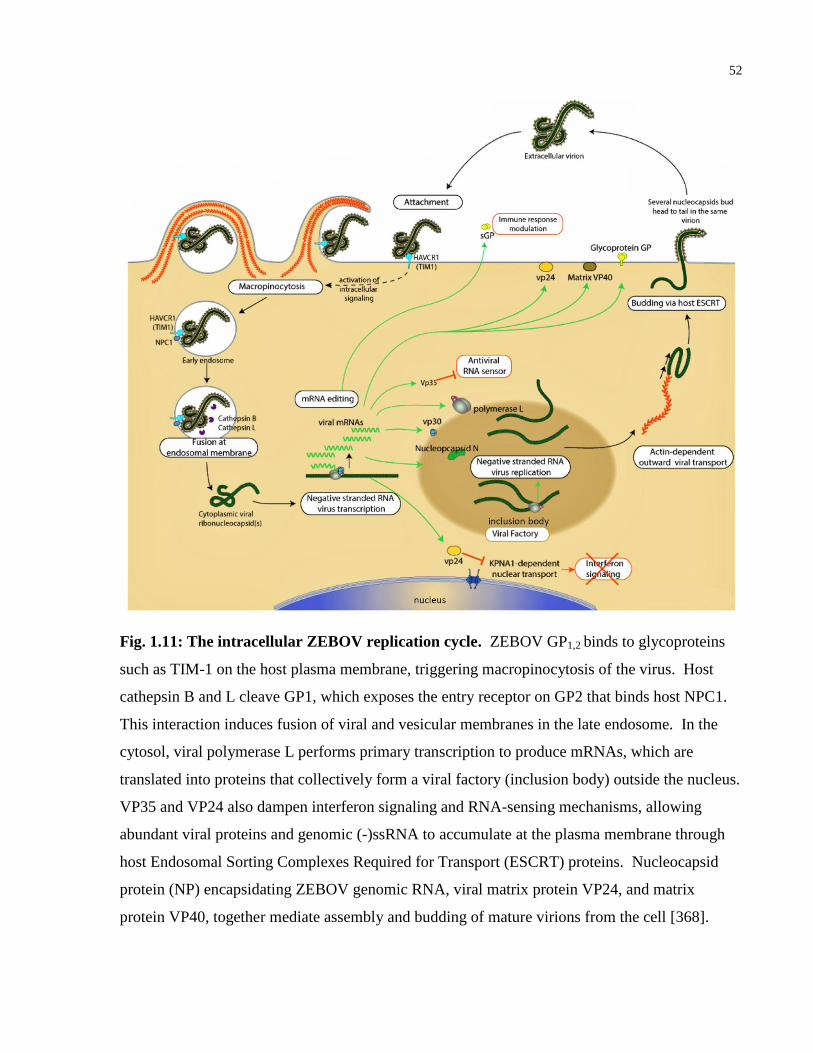
52
Fig. 1.11: The intracellular ZEBOV replication cycle. ZEBOV GP1,2 binds to glycoproteins
such as TIM-1 on the host plasma membrane, triggering macropinocytosis of the virus. Host
cathepsin B and L cleave GP1, which exposes the entry receptor on GP2 that binds host NPC1.
This interaction induces fusion of viral and vesicular membranes in the late endosome. In the
cytosol, viral polymerase L performs primary transcription to produce mRNAs, which are
translated into proteins that collectively form a viral factory (inclusion body) outside the nucleus.
VP35 and VP24 also dampen interferon signaling and RNA-sensing mechanisms, allowing
abundant viral proteins and genomic (-)ssRNA to accumulate at the plasma membrane through
host Endosomal Sorting Complexes Required for Transport (ESCRT) proteins. Nucleocapsid
protein (NP) encapsidating ZEBOV genomic RNA, viral matrix protein VP24, and matrix
protein VP40, together mediate assembly and budding of mature virions from the cell [368].

53
post-transcriptionally modified [378]. Concurrently, membrane-associated matrix proteins VP24
and VP40 are translated and localize to the plasma membrane [382]. When sufficient levels of
NP and negative-sense RNA genomes are produced, VP24 helps assemble viruses at the plasma
membrane, then VP40 induces budding of the newly formed virions [382]. Outside of
replication, VP24 and VP35 have additional roles that help EBOV evade host innate immune
responses, chiefly by antagonizing type I IFN pathways [383].
ZEBOV is transmitted between humans by direct contact with mucosal membranes or bodily
fluids (saliva, sweat, vomit, diarrhea, blood or semen) of an infected or deceased individual
[367]. Following a 6-12 day incubation period, a symptomatic patient will be highly contagious
during the acute phase of EVD. Early symptoms consist of fever, chills, malaise and muscle
pain, which are non-specific and can be confused with other hemorrhagic fevers or viral
infections [357, 367]. When the virus enters broken skin or mucosal surface, antigen-presenting
cells are the preferred initial cellular targets, such as monocytes, macrophages or dendritic cells
[384]. In vitro infection of these cells demonstrates robust expression of inflammatory mediators
IL-1β, IL-6, IL-8, MIP-1α, MIP-1β, MCP-1, and TNF-α [384-386]. Infected cells also release
chemokines that recruit additional monocytes and macrophages to the site of infection, which in
turn become infected [384]. Symptoms at this stage are gastrointestinal (vomiting, diarrhea
abdominal pain, nausea, anorexia), respiratory (nasal discharge, cough, shortness of breath, chest
pain) and vascular (postural hypotension, edema), demonstrating multisystem involvement [387].
As the host immune system attempts to mobilize an adaptive response, ZEBOV may be
impairing the essential role of APCs to stimulate T-cell and B-cell responses required to clear
infection [388]. Moreover, infected APCs secrete soluble GP, which may act as decoys that
counteract neutralizing antibodies [389]. Infection of APCs, coupled with elevated
proinflammatory cytokines and a weak adaptive immune response, are likely why ZEBOV can
disseminate systemically to infect hepatocytes, adrenal cortical cells, and endothelial cells [390].
This results in hepatocellular necrosis that decreases secretion of coagulation proteins, and
abnormal adrenal cortical cells that can no longer maintain blood pressure homeostasis.
In the late stages of illness, symptoms include intravascular volume depletion, hypoperfusion,
electrolyte perturbations and renal impairment [387]. Severe lymphopenia and lymphoid tissue

54
destruction occur, with apoptosis causing the greatest loss of peripheral blood CD4+ and CD8
+ T-
cells [391]. In concert, the large release of TNF-α from infected monocytes and macrophages
induces endothelial permeability, causing vascular leakage and hemorrhage [384]. Together the
blood leakage, extensive cytokine release and viral replication in endothelial cells lead to
hemorrhage syndrome, often characterized by gastrointestinal bleeding [387]. At the terminal
stage of disease, multi-organ failure and shock are the main causes of death.

55
1.3.2 ZEBOV Clinical Trials, Potential Vaccines and New Therapeutics
Despite international efforts to accelerate research into ZEBOV therapeutics and conduct clinical
trials during the 2014-16 West Africa outbreak, there remains no licensed treatment for EVD
[25]. However, there was success with a clinical trial testing a preventative vaccine [26]. A
variety of vaccine platforms have been used to treat EBOV infection in Non-Human Primate
(NHP) models of infection, including: virus-like particle antigen-based vaccines, replication-
deficient DNA and recombinant Adenoviral Vector (rAD) vaccines, and replication-competent
viral vectors that employ recombinant parainfluenza, rabies, Cytomegalovirus (CMV) or
Vesicular Stomatitis Virus (VSV) [392]. However the two most promising vaccine candidates
that reached phase II/III clinical testing during the West Africa ZEBOV outbreak were ChAd3-
EBO-Z and rVSV-ZEBOV-GP [26].
ChAd3-EBO-Z is a non-replicating, recombinant, chimpanzee adenovirus type-3 vector-based
vaccine expressing ZEBOV GP [393]. Earlier studies on cynomolgus macaques demonstrated
that single immunization 5 weeks prior to ZEBOV challenge led to complete protection, but this
dropped to 50% protection when the animals were challenged 10 weeks post-immunization [32].
Pre-existing immunity to ADVs, which also occurs in humans, likely explains the lower than
desired GP-specific humoral response. Thus a booster vaccine with modified vaccinia Ankara
expressing ZEBOV GP (MVA-BN-Filo) was administered 8 weeks after ChAd3-EBO-Z
immunization, leading to complete ZEBOV protection in macaques challenged as late as 10
months post-immunization [32]. In September 2014, the US National Institute of Allergy and
Infectious Diseases (NIAID) and UK Welcome Trust promptly tested the safety, tolerability, and
immunogenicity of ChAd3-EBO-Z in 91 healthy participants in two, single-blind, dose-
escalation phase I trials in Mali and the USA [393]. Another phase I trial was performed on 60
patients in the United Kingdom, and it was found that the vaccine boosted with MVA elicited
strong B-cell and T-cell responses, with no safety concerns reported [394]. The vaccine platform
then entered a phase II/III trial called Partnership for Research on Ebola Vaccines in Liberia
(PREVAIL), which enrolled 27,000 healthy adult participants, and is estimated to complete by

56
June 2020. PREVAIL is a randomized, double-blind, controlled, 3-arm study that is also
evaluating the other most promising vaccine platform, rVSV-ZEBOV-GP .
rVSV-ZEBOV-GP is a live attenuated, recombinant, replication-competent vaccine consisting of
VSV expressing ZEBOV GP [26]. It has the advantage over non-replicating vaccines by
providing longer durability, but possesses the potential risk of reversion to wild type (WT), and
could cause complications in people with compromised immunity [357]. No adverse events
were observed in 80 NHPs immunized with single dose rVSV-ZEBOV-GP [395], or cause
disease in immunocompromised Non-Obese Diabetic (NOD) Severe Combined
Immunodeficiency (SCID) mice [396]. The vaccine could protect 100% of macaques from lethal
ZEBOV challenge up to 6 months after immunization with a single vaccine dose, and confer
50% protection when administered 30 min post-exposure of ZEBOV challenge [33, 397]. rVSV-
ZEBOV-GP safety and immunogenicity in humans was also determined in multiple phase I trials
across Europe and Africa in 2014 [398, 399]. Recent findings from a phase III, cluster-
randomized ring trial in Guinea and Sierra Leone showed this vaccine to be 100% efficacious in
the 7,651 people enrolled [26]. When a case of EVD was confirmed, contacts and contacts of
contacts were randomized to either immediate vaccination or delayed vaccination (21 days later).
None of the participants that were vaccinated immediately developed EVD within 10 days,
compared with 23 contacts in the delayed vaccination clusters [26]. As a potential post-exposure
vaccine, rVSV-ZEBOV-GP has been administered twice after ZEBOV needle stick injuries, and
neither subject displayed EVD, although they developed fevers [400, 401]. While both ChAd3-
EBO-Z and rVSV-ZEBOV-GP are very promising vaccine candidates, individual-level
correlates of protection and durability in humans remain to be determined.
In parallel with vaccine research, a variety of experimental treatments and therapeutics were
prioritized by the WHO for further evaluation in September 2014, based on in vitro and animal
models of ZEBOV infection [6]. These were also considered for emergency phase I or phase
II/III trials during the ZEBOV outbreak. In addition to intensive supportive care, treatments
included: convalescent plasma [20], antisense siRNA drugs [27], monoclonal antibodies [19],
and type I interferon regimens [22]. Without an approved specific therapy, intensive supportive
care became the benchmark treatment by providing intravenous fluids, electrolyte solutions, and

57
oral rehydration to help maintain blood volume [16]. Convalescent plasma, whereby plasma
from ZEBOV survivors was transfused twice into patients during acute infection, was
administered in a non-randomized comparative study in Guinea [20]. However this treatment
required ABO-compatibility and optimal collection time from donors. It was determined after
the trial completed that the 84 patients received low levels of neutralizing anti-ZEBOV IgG
antibodies from the transfusions [402]. Moreover, convalescent plasma transfusions were not
easily amendable to sparsely equipped ETCs, and have yet to demonstrate clinical efficacy [20].
Stable Nucleic Acid Lipid Particles (SNALPS) were developed by Tekmira to deliver siRNA in
vivo, to target three ZEBOV mRNAs encoding for RdRP L, VP24 and VP35 proteins [403]. The
treatment, TKM-Ebola, could protect 100% of rhesus macaques three days post-exposure to a
lethal challenge of EBOV-Makona [21]. However, TKM-Ebola failed to demonstrate efficacy in
humans in a phase II, single-arm trial, despite infusions being well tolerated [27]. In addition,
siRNA strategies are viral strain and variant-specific, which may not be effective in future
outbreaks of EBOV [25]. As another strategy, the Public Health Agency of Canada and Mapp
Biopharmaceuticals jointly created a cocktail of three monoclonal antibodies called ZMapp,
which was administered to ZEBOV patients in a randomized, controlled phase I trial of 71
patients in Liberia, Guinea, Sierra Leone and the USA [19]. ZMapp plus standard care did not
show improved efficacy over standard care alone. The antibody cocktail has also been
administered for emergency use in seven ZEBOV patients, of which five have survived [404].
However, the slow production of antibodies from tobacco plants limits the usefulness of ZMapp
in future EBOV outbreaks. Recombinant interferon supplementation was also considered for
treatment because ZEBOV infection is associated with strong downregulation of type I
interferons α and β [405, 406]. IFN- α and IFN- β normally clear infection by activating
apoptosis in infected cells and recruiting cytotoxic cells [407]. The single-arm, historically
controlled phase II trial testing recombinant interferon β recently completed in Guinea. Nine
ZEBOV patients received daily subcutaneous injections of IFN- β-1a, and showed higher
survival (67%) when compared with 21 patients who received supportive care alone (19%) [22].
While these results are encouraging, enrollment was limited due to potential risk of interferon
treatment exacerbating EVD symptoms [22]. Taken together, these therapeutic approaches
demonstrate the logistical hurdles of amending an efficacious and well-tolerated strategy that

58
works well in small animal and NHP models of EBOV infection, with practical challenges of
implementing a clinical trial during an ongoing EBOV outbreak: patient recruitment changes
rapidly with local transmission rates, resources can be limited, electricity is often intermittent,
and limited personnel who are qualified and trained to safely administer therapies with needles in
an ETC. For all of these reasons, oral drugs that directly inhibit ZEBOV replication that are safe,
stable in warm climates, affordable, and in abundant supply, were strongly considered for
prioritization by the WHO during the ZEBOV outbreak in West Africa [6].

59
1.3.3 Repositioning Nucleoside Analogues for ZEBOV Inhibition
Nucleoside and nucleotide analogues are one of the most successful classes of antivirals for
treating HIV-1, Hepatitis B Virus (HBV), Herpes Simplex Virus (HSV), or Varicella Zoster
Virus (VZV) in patients [408-412], and are under clinical evaluation for treating HCV, CMV or
influenza virus infections [413, 414]. Pharmacological advantages include: direct inhibition of
viral polymerases that transcribe either DNA or RNA, long cellular half-lives due to sequential
phosphorylation of nucleoside prodrug to an active nucleotide triphosphate by cellular kinases,
and broad tissue distribution [415-417]. While viral and human polymerases typically have
selectivity to either Nucleotide Triphosphates (NTPs) or dNTPs, based on the ribose or
deoxyribose sugar bound to a nitrogenous base, the ability of synthetic nucleotide analogues to
disrupt nascent DNA or RNA chain synthesis is empirically determined for each viral
polymerase. Unfortunately the broad-spectrum antiviral ribavirin, active against many RNA
viruses that cause hemorrhagic fever, is a weak inhibitor of ZEBOV in vitro. Thus the ZEBOV
outbreak in West Africa accelerated the development of many nucleoside/nucleotide analogues
for the potential treatment of EVD, namely brincidofovir, favipiravir, BCX4430 and GS-5734 [6,
18, 24, 25, 418, 419].
Brincidofovir (BCV)
Brincidofovir, a broad-spectrum antiviral developed by Chimerix, is a covalent lipid conjugate of
the nucleotide cidofovir (CDV), a cytosine monophosphate analogue [25, 420]. Cidofovir is an
Acyclic Nucleotide Phosphonate (ANP), cautiously administered intravenously to treat CMV
infections of the eye in HIV-1 patients [421]. To enhance bioavailability by imitating the
digestion of monoacyl phospholipids, CDV has been conjugated to the lipid 3-hexadecyloxy-1-
propanol (HDP), making it BCV [422]. This has been found to increase the cellular uptake and
tissue distribution of orally administered BCV, with improved safety profile over CDV [423].
BCV associates with phospholipids at the cell membrane where HDP becomes cleaved, releasing
CDV into the cytosol [424]. Intracellular CDV then becomes phosphorylated twice by cell
enzymes to become CDV diphosphate (CDV-DP), the active form of the drug [425]. CDV-DP
competes with the DNA polymerase substrates of CMV and vaccinia viruses, inhibiting DNA

60
elongation by causing chain termination from its incorporation into the nascent DNA strand
[426]. BCV was originally developed to inhibit the replication of double-stranded DNA
(dsDNA) viruses, such as CMV and ADV [357]. BCV has broad antiviral activity against many
dsDNA viruses, including viruses from the families Herpesviridae, Poxviridae, Polyomaviridae,
and Adenoviridae [28]. However, long-term treatment with BCV can lead to escape mutations in
the viral DNA polymerases, selecting for BCV-resistant poxvirus or CMV [427]. In 2014, it was
discovered that BCV showed activity against ZEBOV replication in vitro, the first example of
this drug inhibiting the replication of an RNA virus. An in silico screen identifying drugs that
could be repurposed to inhibit ZEBOV replication predicted CDV may inhibit the RdRP L of
ZEBOV, suggesting a potential mechanism [428]. Yet an in vitro study on the effects of BCV
and BCV-derivatives on ZEBOV replication in cell lines only demonstrated antiviral activity of
CDV when conjugated to HDP [28]. This could suggest CDV proper has no ZEBOV antiviral
effect, or rather, that higher drug concentrations are needed in comparison with BCV. However
these two possibilities have yet to be tested, and the mechanism of inhibition of RdRP L,
potentially through RNA chain termination, has yet to be determined for either BCV or CDV.
In vivo animal studies have well documented the antiviral activity of BCV in ADV, poxvirus or
Vaccinia Virus (VACV) infections, identifying tolerated doses of BCV that protect against lethal
viral challenge and reduce disease symptoms [420, 429, 430]. However, the rapid metabolism of
BCV in non-human primates precluded the testing of BCV in gold standard models of ZEBOV
infection, complicating the design of human efficacy studies evaluating BCV for treating EVD
[25]. Nonetheless in October 2014, the FDA approved BCV for emergency use after ZEBOV
exposure. Two of three American EVD patients treated with BCV have recovered after
treatment; although their survival cannot be directly linked with BCV due to other interventions
conducted during treatment [25, 431]. Both patients were treated with a single 200 mg oral BCV
dose, and blood samples were drawn before and after 2 days of treatment [427]. Neither patient
showed genetic mutations to the ZEBOV RdRP L sequence, although the viral genome from one
patient developed a silent point mutation relative to the EBOV-Makona reference strain [427].
The third EVD patient, a man from Liberia visiting Texas and in the late stages of EVD, died
four days after initiating BCV treatment.

61
Because of advanced clinical trials testing the safety and efficacy of BCV in preventing CMV
infection [413, 432], and in vitro data of its antiviral effect against ZEBOV that has not been
made public, BCV was prioritized by the WHO in late 2014 as a compound to be evaluated in
patients with EVD [24]. The drug became fast-tracked for a single-arm, phase II ZEBOV
clinical trial that started in Liberia on January 1st, 2015 [24]. Four ZEBOV patients were
enrolled, receiving 200 mg oral BCV on day 0, followed by 100 mg oral BCV on days 3, 7, 10
and 14. Unfortunately all four patients died of illness consistent with EVD, despite no serious or
unexpected serious adverse reactions reported from BCV treatment [24]. The single-arm study
was terminated on January 31st, 2015 by the manufacturer Chimerix, before BCV efficacy could
be determined. This was due to insufficient enrollment and fewer new cases of EVD in Liberia
during the trial period [24].
Favipiravir (FPV)
Favipiravir (T-705) is another broad-spectrum antiviral, developed by Toyama Chemical Co
Ltd., to treat influenza strains that are resistant to conventional antivirals [433]. It is a purine
nucleoside analogue currently under investigation in phase III trials of uncomplicated influenza
in adults [6]. Within the cell, FPV is ribosylated and phosphorylated by cellular enzymes,
becoming the active drug favipiravir-RTP (FPV-RTP) [434]. The active drug inhibits the RdRP
of Influenza A Virus (IAV) when two molecules of FPV-RMP are incorporated consecutively
during primer extension [434]. FPV-RTP outcompetes both GTP and ATP, preventing further
primer extension and terminating RNA chain synthesis [434]. FPV has demonstrated in vitro
and in vivo activity against many RNA virus families that infect humans, including negative-
sense single-stranded RNA (-)ssRNA viruses (Orthomyxoviridae, Arenaviridae and
Bunyaviridae) and (+)ssRNA viruses (Caliciviridae, Togaviridae, Picornaviridae and
Flaviviridae) [434-440].
Two separate studies have reported activity of FPV against ZEBOV replication in vitro [23, 29].
The first study administered FPV to Vero E6 cells (African green monkey kidney epithelial) 1
hour prior to infection with ZEBOV-Mayinga, at a Multiplicity of Infection (MOI) of 0.01 [23].
Five days thereafter, infectious particles were quantified by an immunofocus assay using anti-

62
EBOV polyclonal antibodies, and cell viability measured by the MTT assay. In cell culture, FPV
was able to suppress ZEBOV infection with an IC50 of 67 μM and an IC90 of 110 μM, with no
affects on cell viability [23]. In the second report, Vero C1008 cells were infected with ZEBOV-
E718 or ZEBOV-Kikwit at a higher MOI of 0.1, and then treated immediately with FPV [29].
Infection was visualized by plaque layer formation, and FPV could inhibit ZEBOV plaque
formation with an EC50 of ~200 μM and an EC90 of ~400 μM [29], higher doses than the
previous report, which assessed FPV pretreatment and a lower ZEBOV MOI [23].
To plan for potential human studies, both reports then examined the efficacy and
pharmacokinetics FPV in small animal models of ZEBOV infection [23, 29]. This was followed
by a third study investigating the pharmokinetics of FPV in non-human primates [441]. In the
first publication, 18 IFNα/βR−/−
(A 129) knockout mice, which are immunodeficient and
susceptible to EVD, were challenged with aerosolized ZEBOV-E718, at a lethal dose equivalent
to 1 TCID50 [29]. All 6 mice given twice-daily (BID) oral FPV at 50 mg/kg starting 1 hr post-
exposure, survived for 4 weeks-post challenge, exhibiting transient weight loss but were
otherwise normal after 30 days. Conversely, the 12 mice receiving no treatment showed clinical
signs of weight loss, severe ruffling, hunched posture and blindness, and all of them died 7-8
days post-ZEBOV exposure [29]. In the second study, which used IFNα/βR−/−
(C57BL/6)
knockout mice, FPV was administered either 6 or 8 days after ZEBOV exposure [23]. Fifteen
mice were infected intranasally with 1,000 Focus-Forming Units (FFU) of ZEBOV-Mayinga,
and treated with 150 mg/kg of FPV BID, starting at 6 (N = 5) or 8 (N = 5) days post-infection
[23]. Prior to treatment, all mice lost weight rapidly, showed increasing viremia, exhibited
elevated symptoms of EVD, and a decrease in body temperature consistent with shock. In the
mice starting FPV treatment at 6 days post-infection, all 5 were able to clear infection from
blood within 4 days, and all 5 mice recovered during the 3 weeks of post-infection observation
[23]. These mice developed anti-EBOV specific antibodies, and CD8+ T-cells responding to
viral nucleoprotein, suggesting a virus-specific adaptive immune response was mounted in the
absence of type I IFN signaling [23]. In contrast, all 5 of the untreated mice died within 10 days
of ZEBOV infection [23]. Of the 5 mice starting FPV treatment 8 days-post infection, treatment
delayed death in 1/5 animals, but all of them eventually died of infection by day 15. This

63
provides evidence of a critical treatment window period where once overt symptoms and high
viremia are present, FPV may no longer be beneficial at terminal stages of EVD [23]. Lastly, a
pharmacokinetics study of daily maintenance doses of FPV (60 to 180 mg/kg BID) in uninfected
Chinese or Mauritian cynomolgus macaques, determined that a 20% dose increase would be
needed by day 7 to treat EVD, to compensate for higher drug clearance over time in larger
animals [441]. This study helped inform dosing regimens that were useful in planning EVD
clinical trials testing the efficacy of FPV.
With strong anti-ZEBOV effects shown in animal models [23, 29], acceptable safety data from
human dose-escalation trials in Japan [442], and large stocks of drug readily available, by
September 2014 FPV quickly became short-listed by the WHO for emergency use after ZEBOV
exposure. A case study of a 43-year old doctor who contracted ZEBOV in Sierra Leone, and
was treated with a combination of FPV and ZMapp in Switzerland, demonstrates the complexity
in attributing drug efficacy in non-controlled, emergency treatment settings [443]. While the
patient’s viral load decreased rapidly and the patient fully recovered, it is not clear whether
immune responses cleared infection, whether either treatment had an effect, or both [443]. To
answer such questions, international collaborations in the fall of 2014 led to the rapid planning of
the JIKI trial: a multicenter, historically controlled, single-arm phase II clinical trial in Guinea
[18]. It recruited EVD patients beginning in December 2014 and completed by April 2015.
Patients (N = 126) were offered standardized care and optional oral FPV treatment, with an
initial adult loading does of 6,000 mg FPV on the first day, and maintenance doses of 2,400 mg
FPV per day for the next 9 days [18]. Of 99 adults and adolescent analyzed, the mortality rate
was marginally reduced during the clinical trial (52.6%) compared with the historical mortality
rate at the ETC prior to the study (55%), leading to no firm conclusion of the benefits of FPV
treatment [18]. FPV was well tolerated by all patients who received it, but the study was unable
to determine the efficacy of FPV on EVD mortality or relationship with RNA viral load,
suggesting further FPV dose optimization is required [18]. Most importantly, it was observed
that baseline viremia was a strong prognostic of patient survival, regardless of treatment: the
mortality rate of those with low baseline viremia (< 107.7
genome copies/mL) was 20%,

64
compared with the 91% mortality rate of patients with high baseline viremia (> 107.7
genome
copies/mL) [18].
After the trial ended, analysis of plasma FPV concentrations from 66 of the treated patients
revealed that by day 4 of treatment, the mean plasma concentration dropped and was much lower
than anticipated by modeling: 64.4 μg/mL predicted versus 25.9 μg/mL observed [444]. Thus,
the JIKI trial did not achieve the target plasma drug exposure level defined before the trial began.
Previous pharmacokinetic research in healthy Japanese volunteers shows that single-dose FPV
has a short half-life of 2-5.5 hrs [442]. Nevertheless, a retrospective clinical trial case series
provides optimism for further FPV efficacy research in treating EVD [445]. In a Sierra Leone
hospital, 85 EVD patients were enrolled into supportive therapy (control group), while 39
patients were given oral FPV in addition to supportive therapy [445]. Relative to the control
group, patients adminstered FPV showed greater symptom improvement, reduced viral load,
longer average survival time (46.9 days vs. 28.9 days) and a higher overall survival rate (56.4%
vs. 35.3%) [445]. These are compelling reasons to design future randomized controlled trials to
test the efficacy of FPV in treating EVD, with careful optimization of drug doses to achieve
target FPV plasma levels. Currently a phase II, dose-escalation trial (FORCE) is recruiting male
Ebola survivors in Guinea, to evaluate whether high doses of FPV can reduce ZEBOV RNA
shedding in semen.
BCX4430
BCX4430 is an experimental adenosine analogue created by BioCryst Pharmaceuticals, studied
for its inhibition of RNA viruses, such as HCV, Middle East Respiratory Syndrome Coronavirus
(MERS-CoV) and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) [6]. Like
FPV, BCX4430 can broadly inhibit (-)ssRNA viruses (Orthomyxoviridae, Arenaviridae,
Paramyxoviridae and Bunyaviridae) and (+)ssRNA viruses (Filoviridae, Togaviridae,
Picornaviridae Flaviviridae and Coronaviridae) [418, 446, 447]. It is highly selective for viral
RNA polymerases, and has not been shown to incorporate into human RNA or DNA, when
human Huh-7 cells are treated with BCX4430 [418].

65
BCX4430 was originally identified from a small-molecule library of inhibitors designed to target
RNA polymerase and reverse transcriptase activity. Inside a cell it is converted to BCX4430-
triphosphate (BCX4430-TP), where after pyrophosphate cleavage, becomes incorporated into the
nascent viral RNA strand [418]. Its mechanism of action is through non-obligate RNA chain
termination, causing premature termination of viral replication or transcription. This has been
demonstrated for the HCV RNA polymerase in cell-free experiments, where BCX4430-TP
caused premature termination of RNA chain synthesis two bases after BCX3330-MP
incorporation into the growing RNA chain [418]. Within various cell lines and primary human
hepatocytes, BCX4430 becomes rapidly phosphorylated within hours [418]. In HeLa cells
transfected with an EBOV minigenome, which creates transcription and replication competent
virus-like particles (trVLPs), viral glycoprotein-GFP expressed on the cell surface was inhibited
by BCX4430 adminstered 5 hrs post-transfection, with an EC50 of ~ 12.5 μM. No adverse
effects on cell viability were detected by the lactate dehydrogenase (LDH) release assay [418].
In Hela cells infected with EBOV-Kikwit, surface expression of viral GP1,2 was completely
inhibited by BCX4430 at concentrations greater than 100 μM, with an EC50 of ~ 11.8 μM. This
was repeated in primary human monocyte-derived macrophages infected with EBOV-Kikwit and
treated with BCX4430 (EC50 of ~ 32 μM) [418]. Taken together, these in vitro findings suggest
BCX4430 inhibits ZEBOV replication; however, they only come from a single report, and have
yet to be reproduced independently.
The pharmacokinetics and efficacy of BCX4430 have been tested in both a small animal model
and non-human primate models of ZEBOV infection [418, 448]. While the prodrug has been
found to rapidly decrease in the plasma of rodents after intramuscular injection (half-life of 5
minutes), the active metabolite BCX4430-TP has an intracellular half-life of 6.2 hours in the
liver of rats [418]. In an experiment with immunocompromised C57Bl/6 mice, 30 mice were
injected daily with 0.9% saline vehicle (N = 10) or administered 150 mg/kg BCX4430 BID,
either by intramuscular injection (N = 10) or orally (N = 10), starting 4 hours prior to infection.
The mice were then infected with a mouse-adapted strain of ZEBOV-Mayinga at 1,000 PFU
[418]. All mice administered saline vehicle died within 8 days. Meanwhile, intramuscular
injections of BCX4430 provided 100% protection against lethal ZEBOV challenge, and all mice

66
lived for the 14 days of follow-up [418]. Oral administration of BCX4430 was also protective,
with 80% of the mice surviving until day 14. In a separate dose-range study in cynomolgus
macaques, 3.4 to 16 mg/kg of BCX4430 BID was injected intramuscularly into animals 48 hours
post-ZEBOV exposure [448]. Despite the 16 mg/kg dose group showing a significantly
prolonged lifespan relative to the control group (13.2 days vs 7.2 days), none of the animals
survived [448]. Thus, a second study was performed with higher doses, administered closer to
the time of infection. Rhesus macaques were injected with 16 mg/kg BID (N = 6) or 25 mg/kg
BID (N = 6) of BCX4430, or injected with saline vehicle (N = 3) after 30-60 minutes of lethal
ZEBOV challenge [448]. Following 14 days of follow-up, all animals administered 25 mg/kg
BID of BCX4430 survived, while 4/6 animals in the 16 mg/kg BID group survived, and all 3
animals in the control group died by day 9 [448]. The mean peak viral load on day 8 also
decreased in both groups of BCX4430-treated animals. Clinical trial data has yet to be reported
for BCX4430 in humans, however, a phase I study of the safety, tolerability and
pharmacokinetics of daily intramuscular injection of BCX4430 in healthy participants is ongoing
[448].
GS-5734
Recently, Gilead has reported that a novel adenosine analogue, GS-5734, inhibits ZEBOV
replication with high in-vitro efficacy [6]. GS-5734 is a monophosphoramidate prodrug with
broad-spectrum activity against (+)ssRNA viruses (Filoviridae and Coronaviridae), (-)ssRNA
viruses (Paramyxoviridae and Arenaviridae), but not against (+)ssRNA Togaviridae or
retroviruses such as HIV-1 [417, 449]. It has been shown to have low toxicity in primary human
cells and cell lines, with selectivity against viral polymerases [417].
GS-5734 was discovered from a library of nucleoside and nucleoside phosphonate analogues, in
a ZEBOV collaboration between the CDC and United States Army Medical Research Institute of
Infectious Diseases (USAMRIID) [419]. Focused screening of the ~ 1,000 compounds
identified a 2-ethylbutyl l-alaninate phosphoramidate parent drug, which through further
optimization by structure activity relationships, led to identifying an isomer that inhibited
ZEBOV in human macrophages with an EC50 = 86 nM, called GS-5734 [419]. Nucleoside

67
analogue drugs typically have slow kinetics during the first phosphorylation event, whereas GS-
5734 was designed to have a monophosphate promoiety, to bypass this rate-limiting step and
greatly enhance intracellular triphosphate concentrations of the compound [417]. GS-5734-TP
has shown to have a long half-life of 24 hrs in primary human monocyte-derived macrophages,
30-times greater when compared to its parent drug [417]. When GS-5734 was added to HeLa or
HFF-1 cells 2 hrs prior to infection with ZEBOV-Makona (5 PFU) or ZEBOV-Kikwit (0.5 PFU),
it could markedly inhibit infection measured by immune-staining 2 days later, with low EC50
values: ~0.2 μM for inhibiting ZEBOV-Makona and ~ 0.16 μM for inhibiting ZEBOV-Kikwit
[417]. In similar experiments, GS-5734 could inhibit ZEBOV-Makona infection in pretreated
primary human macrophages (EC50 = 0.086 μM) and in the hepatocyte cell line Huh-7 (EC50 =
0.07 μM) [417]. At doses greater than 0.1 μM, pretreated Huh-7 cells infected with ZEBOV-
Makona for 3 days demonstrated a GS-5734 dose-dependent reduction in viral RNA produced in
infected cells. Consistent with the mechanism of action of FPV and BCX4430, GS-5734 causes
premature chain termination during RNA synthesis of a viral RdRP, as demonstrated by its cell-
free inhibition of the RNA polymerase isolated from Respiratory Syncytial Virus (RSV) [417].
GS-5734 was also found not to inhibit human RNA Pol II or human mitochondrial RNA
polymerase, bolstering its selectivity to viral RNA polymerases [417].
In non-human primate models of ZEBOV infection, GS-5734 has exhibited unique properties
that make it a strong candidate for further development [417]. Intravenous administration of
10 mg/kg of GS-5734 to rhesus macaques lead to rapid elimination of the prodrug (half-life of
0.39 hours), where as intracellular GS-5734-TP was persistent in PBMCs (half-life of 14 hours)
[417]. Tissue distribution of the drug administered to cynomolgus macaques shows that it can
reach viral sanctuary sites (testes, epididymis, eyes, and brain) within 4 hours [417], suggesting
GS-5734 could be useful for Ebola survivors, who continue to shed virus or suffer from post-
Ebola virus syndrome [362]. Efficacy studies of GS-5734 in rhesus macaques found that daily
intramuscular injection of 3 mg/kg or 6 mg/kg of drug, starting 30-90 minutes following ZEBOV
challenge, were suboptimal in reducing systemic viremia or improving animal survival [417].
However, animals given a 10 mg/kg loading dose of GS-5734 three days-post ZEBOV exposure,
followed by daily GS-5734 injections of either 3 mg/kg (N = 6) or 10 mg/kg (N = 6), showed

68
100% survival after 28 days [417]. All animals administered saline vehicle died by day 9, and
the rhesus macaques given 10 mg/kg of GS-5734 daily showed the lowest levels of plasma viral
RNA, with no evidence of genetic variants that would indicate selection of GS-5734-resistent
virus [417].
Similar to BCV and FPV, GS-5734 is also reserved for emergency use after high-risk exposure
to ZEBOV. During the 2014-16 West Africa ZEBOV outbreak, there were two instances of the
compassionate use of GS-5734 [450, 451]. The first case was a female nurse from Scotland who
contracted ZEBOV in Sierra Leone, and was readmitted to hospital 9 months after suffering
acute meningitis and EVD relapse, with detectable virus found in her cerebrospinal fluid [450].
She was treated with corticosteroids and GS-5734 for 14 days, until her viral RNA load became
undetectable [450]. The second case was a newborn girl in Guinea, who was diagnosed with
EVD the day she was born from a ZEBOV-positive mother diagnosed with EVD [451]. The
newborn was promptly administered ZMapp, plasma from an Ebola survivor and GS-5734.
After 20 days of treatment, she showed no detectable ZEBOV RNA in her blood, and was
discharged by day 33 in good health [451]. This newborn girl is the first documented case of a
patient surviving vertical transmission of ZEBOV.
Given the propensity for GS-5734-TP to accumulate in mononuclear cells, the primary target
cells of ZEBOV infection [417], this drug may be valuable for post-exposure prophylaxis. The
large-scale manufacturing of GS-5734 permitted for phase I clinical trials evaluating the safety
and pharmacokinetics of single and multiple doses of GS-5734, administered by intravenous
infusion [419]. No serious adverse effects to GS-5734 have yet to be observed. In addition, the
distribution of the drug to viral sanctuary sites prompted interest in evaluating GS-5734 for
efficacy in treating post-Ebola syndrome, and to potentially reduce persistent viral shedding in
the male genital tract [419]. Adult male Ebola survivors are currently being enrolled in a double-
blind, randomized, placebo-controlled phase II study (PREVAIL IV), to assess whether 100 mg
of daily GS-5734 treatment can cause long-term clearance of ZEBOV and reduce viral shedding
in semen [419].

69
1.3.4 Testing Other Potent Nucleoside/Nucleotide Analogues: Zidovudine,
Lamivudine and Tenofovir
The rapid development of preclinical research and EVD clinical trials to test the efficacy of
brincidofovir, favipiravir, BCX4430 and GS-5734, demonstrate the escalated interest in
nucleoside and nucleotide analogue inhibitors of the ZEBOV RNA-dependent RNA polymerase
L. Since the West Africa ZEBOV outbreak began in 2014, several compounds approved for
other indications have shown in vitro activity against ZEBOV replication [6, 452-454]. In this
section, the tertiarty structure of RdRP L will be compared with other related viral polymerases,
allowing for comparisons to be drawn between nucleoside/nucleotide analogues and NTP
binding to RdRP L [455, 456]. This will provide the rationale for considering potent nucleoside
analogue inhibitors of HIV-1 and HBV, such as zidovudine (AZT), lamivudine (3TC) and
tenofovir (TFV), for testing of antiviral activity against ZEBOV replication. The case for
investigating various combinations of nucleoside analogues targeting the RdRP L of ZEBOV
will also be made.
Modeling ZEBOV RdRP L and nucleotide binding
Given the lack of a defined crystal structure of the EBOV polymerase, two separate modeling
studies have used the tertiary structures of related viral RNA polymerases and reverse
transcriptases [455], as well as combined homology and ab initio modeling [456], to describe
putative domains of the RdRP L of ZEBOV. The crystal structures of nucleotide binding sites
on RNA and DNA polymerases are of particular interest, as no polymerase has been shown to
have absolute template or substrate specificity [457-459]. Moreover, available structures of
polymerases, bound either to natural nucleotides or nucleotide analogues, demonstrate similar
conserved binding mechanisms [460-462].
In the first study, the ZEBOV protein L sequence together with the crystal structures of 20 RTs
from retroviruses and monomeric RdRPs from (+)ssRNA, (-)ssRNA and dsRNA viruses, led to
a model of the tertiary protein structure of RdRP L [455]. The general structure of viral RNA
polymerases and retroviral reverse transcriptases in Figure 1.12 are from [455]. The crystal
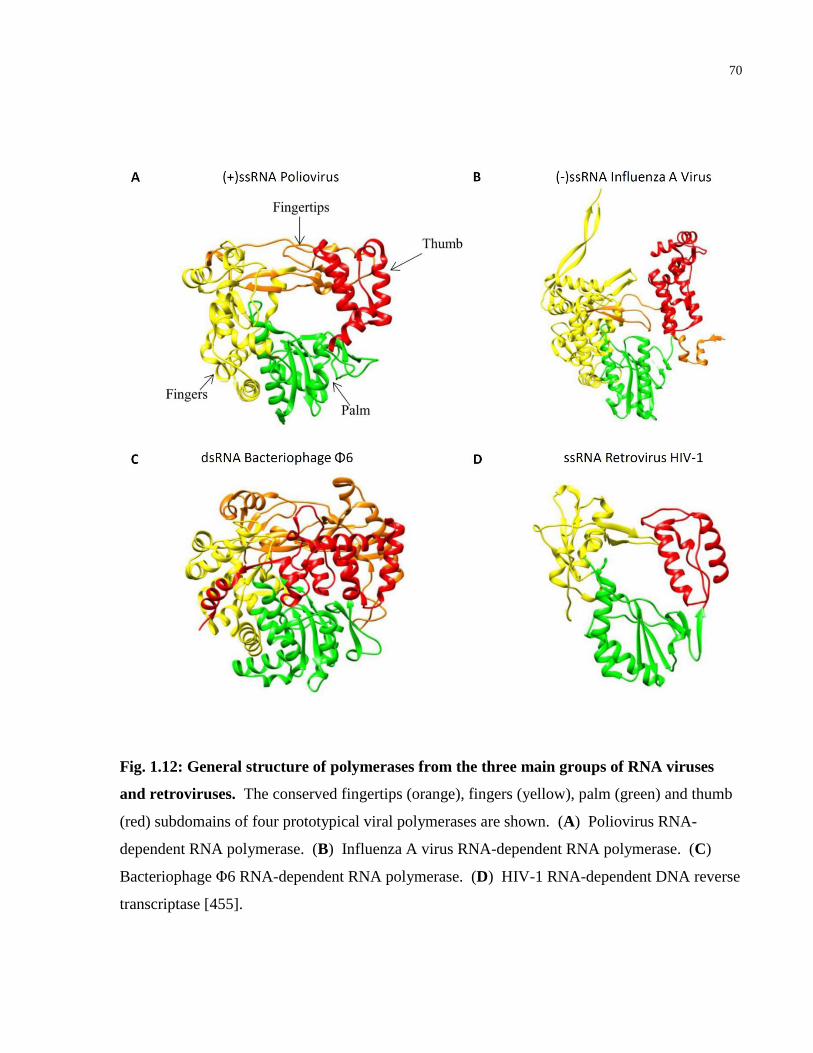
70
Fig. 1.12: General structure of polymerases from the three main groups of RNA viruses
and retroviruses. The conserved fingertips (orange), fingers (yellow), palm (green) and thumb
(red) subdomains of four prototypical viral polymerases are shown. (A) Poliovirus RNA-
dependent RNA polymerase. (B) Influenza A virus RNA-dependent RNA polymerase. (C)
Bacteriophage Φ6 RNA-dependent RNA polymerase. (D) HIV-1 RNA-dependent DNA reverse
transcriptase [455].

71
structures of 20 polymerases from four of the main viral classes share a right-hand appearance,
with three characteristic subdomains called the fingers, palm and thumb domains [463]. The
thumb domain participates in non-specific interactions with the primer strand, while the palm
domain coordinates the phosphoryl transfer reaction of an incoming ribonucleotide to the
growing RNA chain [464]. A common mechanism of nucleotidyl transfer is used by RdRP
enzymes within the palm subdomain. In this region, a conserved aspartate residue coordinates
with two divalent metal ions to catalyze the polymerization reaction of the growing RNA chain
[465, 466]. Consequently, this area of the palm subdomain is the most conserved sequence of all
viral RdRP. In many cases, a fourth fingertips domain can be present, creating a more closed
right-hand structure [455]. This fingertips domain is known to influence the interaction between
an incoming nucleotide and the template strand [467].
Specifically within the family of RdRPs, six conserved structural motifs have been identified
within the palm subdomain (labeled A-F), containing key residues for proper ribonucleotide
binding and protein conformational changes [468-470]. Key residues within the active site have
several interactions with an incoming nucleotide. Conserved aspartic acid residues within motifs
A and C of the palm subdomain, along with basic residues in motif F of the fingers subdomain,
specifically interact with the triphosphate moiety of the incoming nucleotide [468, 471]. An
aspartic residue in motif A and an asparagine in motif B of the palm subdomain also interact
with the 2’OH moiety of ribonucleotides, providing selection for ribonucleotides in the
nucleotide binding pocket of RdRPs [471, 472]. Residues in motifs A and B of the palm and
fingers subdomains of RdRPs also interact with the sugar moiety of the nucleotide, playing an
additional role in substrate discrimination [468]. Unique to RTs and DNA-dependent DNA
polymerases is a bulky side chain that creates a steric barrier preventing ribonucleotides from
entering the nucleotide binding pocket [473]. Lastly, the RNA template and primer bases
provide many interactions with the incoming nucleotide base moiety in RdRPs, strongly
influencing ribonucleiotide specificity.
Considered together, the four conserved subdomains of viral RNA and DNA polymerases, and
six highly conserved structural motifs within the palm subdomain, allowed for homology-based
construction of a general three-dimensional model for ZEBOV L (see Figure 1.13, from[455]).
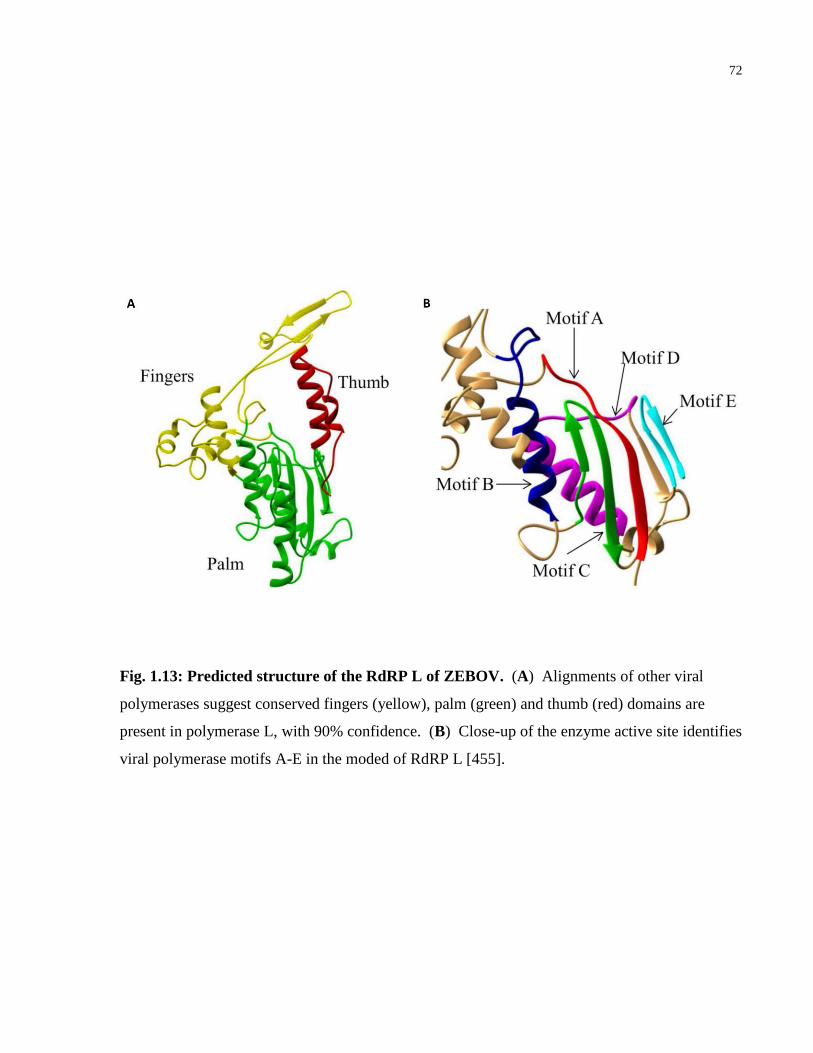
72
Fig. 1.13: Predicted structure of the RdRP L of ZEBOV. (A) Alignments of other viral
polymerases suggest conserved fingers (yellow), palm (green) and thumb (red) domains are
present in polymerase L, with 90% confidence. (B) Close-up of the enzyme active site identifies
viral polymerase motifs A-E in the moded of RdRP L [455].

73
The protein L model was identified to contain the finger, palm and thumb subdomains, with
conserved structure in the palm subdomain, consistent with other viral RNA and DNA
polymerases [467, 474]. It also showed similar structural motifs with the crystal structures of
DNA and RNA polymerases bound to nucleotides or nucleotide analogues, suggesting similar
binding mechanisms may be involved [455]. These conserved regions, in particular the palm
subdomain which contains the residues for nucleotide binding, could explain why FPV and
BCX4430 exhibit broad-spectrum inhibition of many (+)ssRNA and (-)ssRNA viruses, and why
BCV was recently found to have activity against RNA viruses [455]. Thus, nucleotide analogues
designed to inhibit the active site of polymerases from different viruses, such as 3TC inhibiting
HIV-1 RT, may also inhibit ZEBOV RdRP catalytic activity [455].
In the second study, a 3-D model of the middle polymerase domain of ZEBOV protein L (AAs
648-1457) was generated by homology-mediated modelling, based on the known X-ray
structures of the viral RNA polymerases of human Enterovirus (EV), Bovine Diarrhea Virus
(BVDV) and Foot-and-Mouth Disease Virus (FMDV) [456, 475-477]. This model of the middle
domain of protein L shared significant architectural similarities with the polymerases of other
RNA viruses, namely Poliovirus, Rotavirus, HCV and Rhinovirus [456]. To determine the
putative ribonucleotide binding pocket, the crystal structures of EV and BVDV polymerases
complexed with GTP substrate were superimposed on the predicted protein L structure [456]
Complementing this approach, the solved structure of FMDV polymerase complex with the
nucleotide analogue ribavirin and template-primer RNA, also helped to identify a helical motif at
AAs 983-990 as the probable ribonucleotide binding domain: FLRQIVRR [456].
Next, with the predicted structure of this nucleotide binding domain, and conformational
information of nucleotides (ATP, CTP, GTP and UTP) and select nucleotide analogues, in silico
molecular docking studies of polymerase-ligand complexes and their associated interaction
energies could be modeled [456, 478]. The putative nucleotide binding domain appears to have
two entry sites, one on the “left” and one on the “right”. The lowest total interaction energy for
each natural nucleotide suggests that all four NTPs bind the FLRQIVRR motif from the “left”
(ATP = -8.83 kcal/mol, CTP = -11.59 kcal/mol, GTP = -9.23 kcal/mol and UTP = -8.96
kcal/mol) (Figure 1.14, from [456]). Interestingly, triphosphorylated nucleotide analogues were
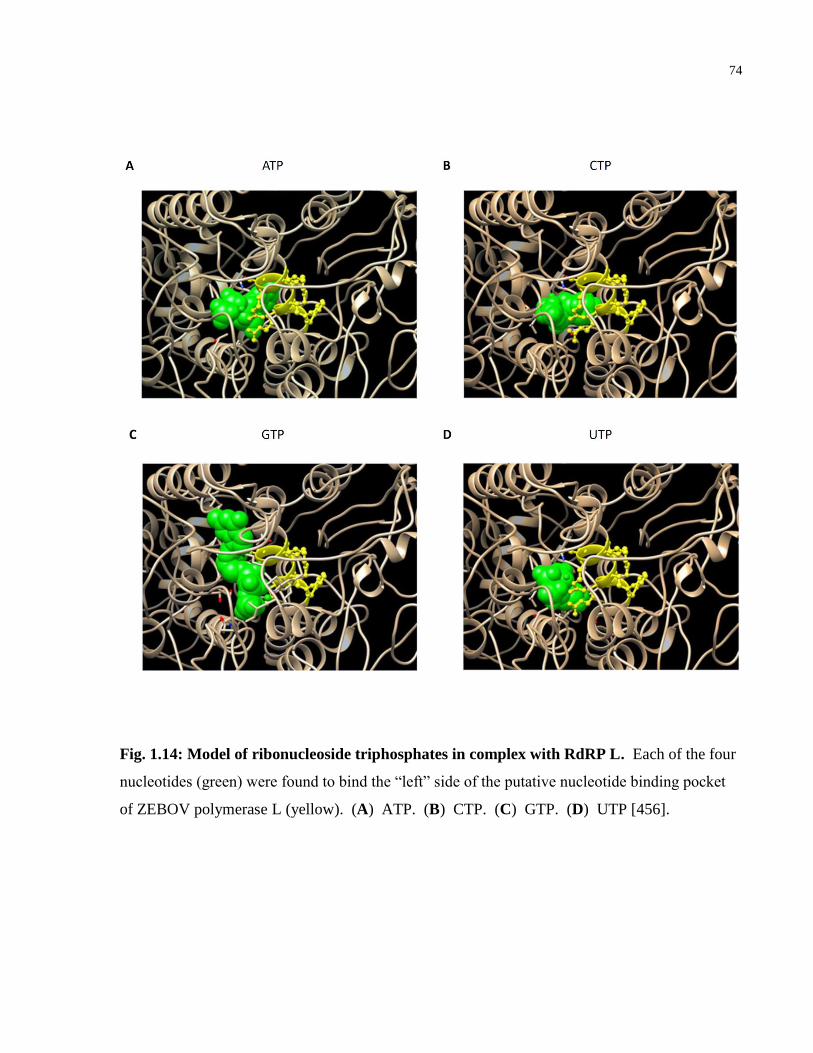
74
Fig. 1.14: Model of ribonucleoside triphosphates in complex with RdRP L. Each of the four
nucleotides (green) were found to bind the “left” side of the putative nucleotide binding pocket
of ZEBOV polymerase L (yellow). (A) ATP. (B) CTP. (C) GTP. (D) UTP [456].

75
found to be either “left-handed” or “right-handed” in terms of their lowest interaction energy
with RdRP L. This separated nucleotide analogues into two distinct docking groups. For
instance, abacavir-TP (-10.42 kcal/mol), favipiravir-RTP (-11.51 kcal/mol) and BCX4430-TP (-
8.23 kcal/mol) were predicted to bind from the “left”, while tenofovir-DP (-12.51 kcal/mol) is
modeled to bind from the “right” (see Figure 1.15 for an example of nucleotide analogues
binding differently, from [456]). Moreover, these total energy interaction values of
triphosphorylated nucleotide analogues were very similar to the natural NTPs, suggesting they
could compete for substrate binding to RdRP L [456]. As an example, the model predicted FPV-
RTP would outcompete binding of CTP or UTP to the putative nucleotide binding pocket, while
BCX4430-3P would outcompete CTP binding [456]. Potential interactions between AZT, 3TC
and TFV with the nucleotide binding pocket were also described [456]. In the following three
sections, the established interactions of AZT, 3TC and TFV nucleoside/nucleotide analogues
binding and inhibiting viral RdRPs and RTs, their clinical indications for treating other viral
infections, and potential for inhibiting the RdRP L of ZEBOV, will be discussed.
Zidovudine (AZT)
In 1985, it was discovered that the thymidine analogue zidovudine, also known as
azidothymidine or retrovir, could inhibit HIV-1 replication in vitro [479]. Originally designed to
target DNA synthesis in breast cancer, AZT became the first antiviral FDA-approved for the
antiretroviral treatment of HIV-1/AIDS in 1986 [480]. AZT shows strong synergy with 3TC in
inhibiting HIV-1, and was an important component of first-line cART regimens [481, 482].
As with most other nucleoside analogues, intracellular AZT becomes phosphorylated to its active
form (AZT-TP) by host cell enzymes [483]. AZT-TP inhibits HIV-1 reverse transcriptase when
it is incorporated into the nascent DNA chain, causing early chain termination [483]. It is a weak
inhibitor of cellular DNA polymerases α and β, but can inhibit mitochondrial DNA polymerase γ
[484]. This can impair mitochondrial DNA synthesis at high AZT concentrations, causing
apoptosis of cardiac and skeletal muscle cells, which can lead to toxic mitochondrial myopathy
when used long-term in patients [485, 486]. Research into the mechanism of AZT-TP inhibition

76
Fig. 1.15: Example nucleoside/nucleotide docking to the ZEBOV RdRP L. (A) Abacavir-TP
(red) binds the FLRQIVRR motif (yellow) from the “left” side, similar to favipiravir-RTP and
BCX4430-TP. (B) Tenofovir-DP (red) binds the nucleotide binding pocket from the “right”
side [456].

77
of HIV-1 RT revealed that the enzyme undergoes isomerisation in a two-step binding reaction
with template-primer, before binding free dNTP substrate [487]. Incorporated AZT-MP into the
DNA strand inhibits the ribonuclease H (RNase H) cleavage activity of HIV-1 RT, acting as a
competitive substrate inhibitor when RT is in the presence of divalent cation activator Mn2+
, or
as an uncompetitive substrate inhibitor in the presence of Mg2+
[488]. This suggests the
conformation of RNase H is dependent on the divalent cation present, altering its mode of
hydrolysis between endonucleolytic and exonucleolytic cleavage [488].
Research into HIV-1 RT resistance mutations towards AZT treatment provides valuable insight
into the mechanisms of this drug, suggesting potential means of how AZT might inhibit the
RdRP L of ZEBOV. Mutations in the RNase H primer grip region have shown to enhance
resistance to AZT, by decreasing template switching, altering the balance between template RNA
degradation and nucleotide excision, and changing RT interactions with the template-primer
complex [489]. While other RT mutations also permit HIV-1 to overcome AZT monotherapy
and increase viral replication, substitutions at amino acids D67N, K70R, T215Y, and K219Q
come at the cost of processive DNA synthesis [490]. HIV-1 RT resistant to AZT inhibition also
show mutations that enhance the excision of AZT-MP from the end of the primer strand at the
nucleotide binding site [491]. These mutations increase AZT-MP excision because of a new
binding site that can recruit ATP, which acts as the pyrophosphate donor for AZT-MP-mediated
excision [492].
In addition to HIV-1 antiviral activity, AZT has been found to inhibit the replication of a variety
of retroviruses, DNA viruses and RNA viruses [493-496]. AZT can inhibit the replication of
HBV in HepG2 cells, a virus encoding a DNA-dependent DNA polymerase [493]. The
polymerase of HBV can transcribe from RNA as well as DNA templates, and AZT can inhibit in
vivo HBV replication in chronically infected patients [494]. Furthermore, in vitro AZT treatment
of uninfected PBMCs can prevent infection from Human T-cell Lymphotropic Virus type I
(HTLV-1) [495]. The HTLV-1 genome is ssRNA, similar to HIV-1, and encodes a reverse
transcriptase enzyme. Human Foamy Virus (HFV), another retrovirus that occasionally infects
humans, can also be efficiently inhibited with AZT treatment [496].

78
With respect to RNA viruses that cause hemorrhagic fever, AZT has also shown strong antiviral
activity [497, 498]. Hantaan Virus (HTN) is a (-)ssRNA virus that causes hemorrhagic fever
with renal syndrome (HFRS) in humans [499]. The virus is endemic to China and the Korean
Peninsula [500, 501]. An in vitro study of inhibitors of HTN found that cells treated with AZT
24 hrs post-infection, showed no detectable HTN RNA by qRT-PCR [497]. In addition, a study
of an aryl phosphate derivative of AZT, called zidampidine, showed this compound could protect
mice from lethal Lassa virus challenge [498]. Lassa virus has a bisegmented, ambisense ssRNA
genome [502]. In the zidampidine pilot study, CBA mice were injected with 250 mg/kg of
zidampidine prior (-24 hr), during (1 hr) or post-challenge (24, 48, 72 or 96 hr) of intracerbral
injections of Lassa virus (Josiah strain) [498]. The zidampidine-treated mice showed
significantly higher survival compared to mice administered vehicle [498].
From the model of nucleotide analogue docking to the putative nucleotide binding pocket of
ZEBOV RdRP L described in the previous section, it was predicted AZT-TP would have a
similar interaction energy to the viral polymerase (-11.47 kcal/mol) as natural NTPs (-8.83 to -
11.59 kcal/mol) [456]. Moreover, AZT-TP belongs to the group of nucleotide analogues that
would dock at the “right” side of the nucleotide binding motif, opposite of FPV and BCX4430
[456]. AZT-TP was also predicted to outcompete CTP binding directly, providing a potential
mechanism of this drug during in vitro ZEBOV infection [456].
Lamivudine (3TC)
Lamivudine is a nucleoside analogue of cytidine that can inhibit HIV-1 and HBV replication,
used for salvage therapy when treating HBV [503-505]. Similar to AZT-TP, the active
metabolite 3TC-TP inhibits HIV-1 reverse transcriptase when 3TC-MP becomes incorporated
into the growing DNA chain, causing early chain termination [506]. Also similar to AZT, 3TC
is a weak inhibitor of human DNA polymerases α and β [506]. 3TC is orally administered, has
high bioavailability, and may cross the blood-brain barrier [507]. 3TC is a component of cART
regimens to treat HIV-1 in patients, and is highly synergistic in inhibiting HIV-1 when combined
with AZT [408, 481].

79
In 1992, 3TC was first reported to be a potent and highly selective inhibitor of HIV-1 and HIV-2
replication in CD4+ T-cell lines and human PBMCs ex vivo [505]. It was then determined in
HeLa cells that 3TC-TP was a direct competitor of dCTP in the RNA-dependent DNA
polymerase activity of HIV-1 RT [506]. 3TC-TP can also outcompete dCTP in the presence of
human DNA polymerase γ, with 3TC-MP becoming incorporated into mitochondrial DNA
[508]. However, this enzyme has substrate exonuclease activity that can remove 3TC-MP from
the dsDNA, likely explaining the low levels of mitochondrial toxicity associated with 3TC when
compared with AZT [508].
Interestingly, HIV-1 variants that become resistant to 3TC also show reduced processivity
similar to AZT-resistant variants; however, it is at a cost to viral replication in infected primary
cells [509]. Often these mutations occur at the catalytic core of RT in a conserved YMDD motif,
where the methionine residue becomes replaced with isoleucine (M184I), and then valine
(M18V4) in subsequent HIV-1 quasispecies [509]. Under limiting dNTP concentrations, HIV-1
RT enzymes carrying either mutation produce shorter cDNA transcripts when compared with
wild type RT [509]. The decreased processivity of M184I and M184V RT mutants, slower
primer extension rate, and increased strand transfer activity can all be attributed to defective
dNTP utilization [510]. These alterations are not caused by changes in binding of the DNA
primer-RNA template or RNAse H ribonuclease activity. Thus slower cDNA synthesis, coupled
with normal dissociation rate of the primer-template, accounts for the lower processivity and
shorter HIV-1 cDNA transcripts exhibited in 3TC-resistant HIV-1 strains [510]. HIV-1 RT
enzymes carrying the M184I and M184V substitutions also demonstrate increased fidelity, and
are more accurate than wild type RT in assays testing misinsertion and mispair extension
efficiency [511]. This suggests a trade-off in 3TC-resistent RT enzymes: increased polymerase
fidelity comes at the cost of slower cDNA synthesis and reduced synthesis of full-length viral
cDNA [511]. Indeed, HIV-1 carrying the RT M184V mutation, which produces more accurately
transcribed viral cDNA, is less capable of compensatory mutagenesis that allows reversion back
to wild type replication kinetics, impairing viral fitness [512]. The effects of substitutions at
Met184 have been further clarified with crystal structures of 3TC-TP bound to M184I mutant
HIV-1 RT, and compared with 3TC-TP bound to wild type HIV-1 RT [513]. As a beta-branched

80
amino acid, the isoleucine mutation creates steric hindrance at position 184, allowing entry of
dNTPs to the nucleotide binding pocket but not 3TC-TP [513]. The valine mutation at the same
position likely has the same effect, as it is also a beta-branched amino acid. Moreover, the
crystal structure of the M184I mutant HIV-1 RT revealed repositioning of the template-primer,
suggesting how the 3TC-resistence mutation could be interfering with optimal enzyme
polymerase activity [513].
3TC has also shown to effectively inhibit the polymerase activity of the dsDNA virus HBV and
retrovirus HTLV-1, similar to AZT [514, 515]. In experiments with duck HBV, 3TC-TP was
shown to cause DNA chain termination as a competitive inhibitor of the viral DNA polymerase,
with respect to dCTP [514]. However, development of HBV resistance mutations such as
M552V, M552I or L528M, precludes 3TC as a first-line therapy for HBV infection in patients
[516]. It appears that mutations to HBV DNA polymerase do not confer 3TC resistance through
removal of 3TC-MP by an exonuclease mechanism [517]. These mutations likely cause steric
hindrance with their side-chains, preventing 3TC-TP access to the nucleotide binding pocket,
similar to HIV-1 RT resistance to 3TC [516]. Homology modeling of the catalytic core of HBV
polymerase revealed that the mutation at Met522 was most likely to directly contribute to this
steric hindrance, where as mutation at Leu528 induced broader rearrangement of nucleotide
pocket residues [516]. 3TC treatment has also been administered to reduce HTLV-1 infection
and lessen the associated myelopathy exhibited by HTLV-1 infected patients [515]. For
instance, in a pilot study of 5 HTLV-1 patients administered 3TC, a 10-fold reduction in viral
DNA was observed [515].
The potential use of 3TC to inhibit ZEBOV replication or treat EVD patients in an emergency
context remains controversial. During the 2014-16 West Africa ZEBOV outbreak, there were
anecdotal reports of 3TC being used to treat patients with EVD in Liberia. For instance Dr.
Logan, the Chief Health Officer of Bomi County in Liberia, administered AZT or 3TC to 15
ZEBOV patients in his care, of which 13 survived [518]. However, these results have not been
published as peer-reviewed clinical case studies. For a potential mechanism of action for 3TC,
3TC-TP is predicted to interact with the putative nucleotide binding site of ZEBOV RdRP L,
comparable to FPV-RTP, BCX4430-TP and AZT-TP [456]. Modeling of 3TC-TP docking

81
suggests it would bind this region from the “left” side, resembling FPV-RTP and BCX4430-TP
binding [456]. The interaction energy of 3TC-TP with the polymerase was comparatively higher
(-7.26 kcal/mol) than other nucleotide analogues modeled in silico, suggesting weaker
competition with natural NTP binding would be predicted [456]. Indeed, the effects of 3TC in a
preliminary cell-based assay did not show activity in reducing ZEBOV-Kikwit infection in Vero
E6 cells, Hep G2 cells, or in human monocyte-derived macrophages [519]. However, these
experiments tested 3TC treatment 1 hr prior to infection, which may not be enough time for
cellular enzymes to convert the prodrug to active 3TC-TP. It has been reported in human
subjects that at 3TC oral doses of 150 or 300 mg, peak intracellular concentration of 3TC-TP in
PBMCs occured at 2.95 and 4.10 hours, respectively [415]. Moreover the in vitro ZEBOV study
of 3TC did not include a nucleoside analogue prodrug as a +ve control for ZEBOV inhibition,
but rather the drug toremifene (TOR), which destabilizes the ZEBOV GP1,2 trimer and triggers
early release of GP2, [519, 520]. Already in its active form, TOR would be expected to show
antiviral activity 1 hr prior to ZEBOV infection [519]. Accordingly, these disparate findings that
predict 3TC interacting with the ZEBOV nucleotide binding pocket [456], weak in vitro
inhibition of ZEBOV replication [519], and use to treat some EVD patients in Liberia [518],
warrant further in vitro testing of 3TC during ZEBOV replication, to clarify potential antiviral
effects of this drug.
Tenofovir (TFV)
Tenofovir disoproxil fumarate (TFV-DF) is an adenosine monophosptate nucleotide analogue,
belonging to the same class of phosphonates as BCV and CDV [521]. While TFV cannot cure
HIV-1 or HBV infection, it is used in combination with other drugs to prevent or manage HIV-1,
and ameliorate chronic HBV symptoms [522, 523]. After oral administration, TFV disoproxil
fumarate becomes absorbed in the gut and cleaved, releasing TFV into the bloodstream [521].
The phosphonate moiety of TFV allows it to be rapidly mono- and diphosphorylated to the active
drug TFV-DP in two steps, rather than three. TFV-DP inhibits HIV-1 RT activity through
premature cDNA chain termination, and shows limited inhibition of human DNA polymerase α,
β, γ [521, 524]. While viral RNA and DNA polymerases discriminate between NTPs and dNTPs

82
during RNA or DNA chain elongation, this does not occur with TFV-DP because of its absence
of a ribose or deoxyribose moiety in its bioactive structure [456].
In 1998, it was discovered that TFV selectively inhibits HIV-1 reverse transcriptase [521]. TFV
alone, or in combination with rilpivirine (RPV) and emtricitabine (FTV), can form dead-end
complexes with the RT polymerase of HIV-1, producing chain-terminated DNA primer/template
[525]. This effect was synergistically enhanced by the presence of the other two NRTI drugs,
leading to more stable chain termination and inhibition of HIV-1 replication [525]. TDF
treatment in HIV-1 patients has been successful when they harbor HIV-1 quasispecies with
substitution mutations that provide resistance to multiple NRTIs, described as the Q151M
complex [526]. This multidrug-resistant HIV-1 genome carries five characterisitic mutations in
the RT gene: A62V, V75I, F77L, F116Y and Q151M [526]. However, it has recently been
found in an HIV-1 clinical isolate that an additional RT mutation, K70Q, can add a high level of
HIV-1 resistance to TFV treatment, and only in viruses that already have the Q151M complex
background [526]. The additional K70Q mutation does not confer increased excision of TFV
from the cDNA chain, but changes hydrogen bonding patterns in the polymerase [526]. This
selectively reduces the affinity of TFV-DP to the nucleotide binding pocket of HIV-1 RT,
reducing TFV incorporation into the newly synthesized cDNA chain [526]. Another important
HIV-1 RT resistance mutation to TFV is K65R, however there is a low prevalence of sexual
transmission of various HIV-1 subtypes carrying this substitution mutation [527]. This finding
has important implications for the inclusion of TDF in PrEP regimens, as TDF reduces the
transmission of HIV-1 to uninfected people [527].
The interactions of TFV with the RT of HBV have also been extensively studied. In an open-
label study of twenty HIV-1 and HBV coinfected patients, a significant decrease in HBV viral
load was measured after 52 weeks of daily TFV treatment, as part of their cART [522]. This was
true even for 11 coinfected patients with HBV carrying the 3TC resistance sequence YMDD or
YIDD [522]. Unique to HBV DNA polymerase, its own protein sequence can initiate viral
cDNA synthesis as a protein primer [528]. It has been shown that TFV-DP strongly inhibits the
second stage of protein priming, becoming incorporated into the viral DNA primer instead of
dAMP [528]. From 2008 until 2014, no TFV drug resistance mutations have been detected in

83
clinical samples of HBV from patients treated with TFV-DF monotherapy [529]. To explain this
phenomenon, in silico modeling of the HBV polymerase suggests TFV has significantly strong
binding affinity to the nucleotide pocket of this enzyme [529]. In the HBV genome, the
polymerase gene overlaps another gene coding for surface protein. This creates an additional
genetic constraint that limits the mutational freedom that would allow for TFV drug resistance to
develop during HBV infection [529].
Little preclinical research has evaluated the potential antiviral activity of TFV or TFV-DF
against RNA viruses, including ZEBOV. Of the 20 nucleotide analogues modeled to dock to the
putative nucleotide binding domain of ZEBOV RdRP L, TFV-DP had the lowest total interaction
energy (-12.51 kcal/mol) [456]. TFV-DP was the best nucleotide/nucleoside candidate
predicted to outcompete natural NTPs binding the FLRQIVRR motif [456]. Similar to AZT-TP,
TFV-DP was modeled to bind this motif from the “right” side [456]. Given the absence of a
ribose moiety in the structure of TFV, whether TFV is capable of inhibiting the RdRP L of
ZEBOV or ZEBOV replication in living cells, deserves further attention [456].
Combination regimens to inhibit ZEBOV
When infected healthcare workers were airlifted from sites of ZEBOV exposure in West Africa
to be treated in Western hospitals, these patients had a collectively low mortality rate [25]. This
lower rate might be attributed to the multiple therapeutics administered to these patients, in
addition to intensive supportive care [431, 443, 451]. Thus, research into therapy combinations
that inhibit ZEBOV replication, such as ZMapp or TKM-Ebola formulations, are a high priority
in developing an effective ZEBOV treatment [21, 30]. In vitro and small animal model research
into combinations of IFNs with ZMapp, which may synergistically inhibit ZEBOV replication,
are underway [6]. Furthermore the JIKI trial authors surmised that in addition to optimization of
the FPV dosing regimen, the possibility of treatment combinations with FPV should also be
evaluated in future EVD clinical trials [6, 18]. This led to oral FPV and ZMapp combinations
being administered in the randomized, controlled trial PREVAIL II ,which had sites in Libera,
Guinea, Sierra Leone and the United States [19]. In addition, low doses of the nucleoside
analogue ribavirin have shown to potentiate the activity of FPV in guinea pigs challenged with

84
Junin virus, the (-)ssRNA arenavirus responsible for Argentine hemorrhagic fever [435]. FPV
and ribavirin synergistically inhibit a variety of other (-)ssRNA viruses that cause hemorrhagic
fevers: Pichinde virus in hamsters [435], Lassa virus in cell culture and in lethal mouse model of
infection [530], and mice infected with Rift Valley Fever Virus (RVFV) [436]. Hence, drug
combinations that improve the efficacy of FPV are of great interest in ZEBOV preclinical and
clinical research.
The molecular docking model of the ZEBOV nucleotide binding pocket provides additional
rationale for considering two-drug combinations for inhibition of ZEBOV replication in vitro
[456]. When nucleoside analogues preferentially dock on the “left” side of the nucleotide
binding motif (FPV-RTP, BCX4430-TP and 3TC-TP), it leaves the “right” side open for binding
of ATP, CTP, GTP, and UTP can still bind from the “left” side (see Figure 1.16, from [456]).
Likewise, nucleotide analogues predicted to bind the “right” side of the nucleotide binding motif
(AZT-TP and TFV-DP), leave entry on the “left” side for ATP, CTP, GTP and UTP binding
[456]. Thus, optimal inhibition of the nucleotide binding pocket predicts a combination of two
drugs that flank both sides of the motif, where one preferentially occupies the “left” and the other
inhibitor occupies the “right” side, to sufficiently outcompete incoming NTPs [456].
Recalculating the interaction energy in the scenario of two different nucleotide analogues
binding the enzyme, such as FPV-RTP + TFV-DP, considerably lowers their total interaction
energy [456]. It becomes -12.28 kcal/mol for FPV-RTP and -10.80 kcal/mol for TFV-DP, much
lower than the values for ATP (-4.84 kcal/mol), CTP (-6.83kcal/mol), GTP (-7.55 kcal/mol) or
UTP (-8.11 kacal/mol) [456]. A reduction of 1 kcal/mol, indicates a 10-fold higher affinity for
binding, in a simple E + S⇔ES model [456]. Thus, the interaction energies of natural NTPs for
RdRP L are significantly surpassed by specific combinations of two nucleotide analogues, which
specifically occupy both flanking sites of the nucleotide binding pocket [456]. Administration of
FPV + TFV for example, may inhibit ZEBOV polymerase activity and subsequent transcription
and replication of the viral (-)ssRNA genome. This strategy has two additional benefits over
monotherapy: 1) It could impede the rate of selection for drug resistant mutations against both
drugs, and 2) Inhibiting viral replication in vivo during the window period of high viremia days

85
Fig. 1.16: Nucleotide analogues binding ZEBOV RdRP L in the presence of NTPs. (A)
Abacavir-TP (red) docks to the nucleotide binding motif from the “left”, allowing most NTPs
(green) access to the “right” side. (B) The opposite trend is found for tenofovir-DP (red), where
it docks from the “right”, permitting NTPs to bind at the “left” [456].

86
after infection [18, 23], could provide the immune system enough relief to mount an effective
adaptive response to clears ZEBOV infection [456].
Treating retrovirus or RNA virus infections with specific combinations of nucleoside analogues
was a paradigm shift in 1996, ushering in the era of combination antiretroviral therapy for
treating HIV-1/AIDS [531]. Drug synergy can help lower plasma viremia by inhibiting multiple
viral targets, reduce drug toxicity by requiring lower drug doses, and provide a higher barrier to
selecting resistance mutations that occur with monotherapy, as previously described for HIV-1,
HBV and HCV [178, 481, 532, 533]. While routinely used to treat HIV-1, HBV or co-infection
of both viruses [410], cART has not yet been explored in preclinical ZEBOV research. This is
partly due to constraints caused by limited access to BSL4 facilities for in vitro or animal testing.
In 2014, a sophisticated new trVLP model of the ZEBOV replication lifecycle was developed,
allowing for rapid testing of therapeutics targeting any stage of the virus lifecycle in a BSL2
laboratory [534, 535]. This permitted efficient testing of drug combinations that was not
previously feasible. Furthermore, the potential mechanism of action of AZT, 3TC or TFV
during ZEBOV replication remained untested in living cells. Nucleoside analogues can disrupt
viral replication by interfering with polymerase NTP binding [506], induce RNA chain
termination [417, 426], or increase lethal mutagenesis [536], among other mechanisms [488].
Additionally, it has not been tested whether 3TC, AZT or TFV potentially synergize with leading
ZEBOV drugs candidates such as FPV, fast-tracked by the WHO for phase II/III EVD clinical
trials.

87
1.4 Statement of the Problem, Rationale, Hypotheses and
Objectives of this Work
1.4.1 Statement of the Problem
Discovering host non-receptor tyrosine kinases that facilitate HIV-1 infection, yet lessen
replication when inhibited with small-molecule inhibitors, is an underexplored area of research
that could improve conventional cART. Moreover, with no approved treatments for EVD,
repositioning drugs that directly inhibit ZEBOV replication, such as nucleoside analogues that
inhibit DNA or RNA synthesis of other viruses, may inhibit ZEBOV replication in vitro.
1.4.2 Rationale
Both c-SRC and PTK2B have been implicated during HIV-1 infection from various means of
investigation. Shortly after TCR-CD3 activation of a CD4+ T-cell, or G protein-coupled receptor
stimulation, both c-SRC and PTK2B interact and become activated [197, 293, 340]. In addition,
c-SRC and PTK2B also become activated after treating immature DCs with HIV-1 gp120 [323],
or infection of Jurkat T-cells with intact virus [214]. HIV-1 also rearranges the cytoskeleton
upon fusion and entry, and PTK2B has defined roles in T–cell cytoskeletal rearrangements at
focal adhesions [340]. Moreover JCaM1.6 T-cells, which lack functional LCK and FYN
expression but expresses c-SRC, are more infectable with HIV-1 [209]. Inhibition of the SFKs
in CD4+
T-cells with PP2 inhibited viral p24 production after 6 days [264], while shRNA
knockdown of PTK2B in T-cell lines reduced HIV-1 infection after 6 days [345]. HIV-1 Nef
binds and activates c-SRC kinase activity [320], and may be the cause of Nef-induced AIDS
phenotype in transgenic mice [164, 168]. In addition, proteins downstream of c-SRC signaling
(SAMHD1 in the cytosol, Sam68 in the nucleus) already have defined roles during HIV-1
infection [317, 537]. Furthermore, c-SRC does not contribute to Gag-mediated viral assembly
during budding [227], suggesting the effects of c-SRC signalling may occur at earlier stages of
the viral lifecycle. My thesis will attempt to answer the following questions: Are c-SRC and
PTK2B natural restriction factors of HIV-1 during early T-cell infection, or do they facilitate

88
early HIV-1 infection in T-cell lines and primary CD4+ T-lymphocytes? At which stages of the
viral replication cycle do c-SRC and PTK2B have an effect? And if one or both facilitate
infection, can FDA-approved small-molecule kinase inhibitors that target either of these NRTKs,
be used to inhibit HIV-1 infection in primary CD4+ T-cells?
My thesis will also make use of a new trVLP model of ZEBOV replication, permitting rapid
assessment of new therapies and combinations of therapies that can be tested in a BSL2 lab
environment [534, 535]. The nucleoside analogues BCV, FPV, BCX4430 and GS5734
demonstrate broad-spectrum inhibition of other RNA viruses, DNA viruses and retroviruses [6,
357, 417, 455], becoming fast-tracked by the WHO for additional ZEBOV preclinical research
[28, 418, 441], emergency phase II/III efficacy trials in West Africa [18, 24], and ZEBOV post-
exposure treatment [427, 443, 450]. Other nucleoside analogues AZT, 3TC and TFV, which
show potent inhibition of HIV-1, HBV and other (-)ssRNA viruses that cause hemorrhagic fever
[479, 493, 497, 505, 514, 521], have been predicted to bind the ZEBOV nucleotide binding motif
of RdRP L, with similar or greater affinity than natural NTPs or other nucleotide analogues
modeled [456]. Moreover, combinations of nucleotide analogues that preferentially occupy the
“left” side of the nucleotide binding pocket (FPV-RTP, BCX4430-TP and 3TC-TP) are predicted
to synergize with drugs that enter the “right” side of the binding pocket (AZT-TP and TFV-DP)
[456]. AZT, 3TC and TFV show limited inhibition of human DNA polymerases, and broad
biodistribution to immune cells and tissues where ZEBOV replication persists during acute and
chronic infection [416, 484, 508, 538]. Furthermore, anecdotal reports of patients treated for
EVD with 3TC and AZT in Liberia warrant further testing of these drugs in cell culture infection
of ZEBOV [518]. Combination regimens with FPV, the most promising drug candidate in
ZEBOV clinical trials to date [18, 19], and other experimental therapies such as IFN treatment,
should also be compared to one another to prioritize which combinations are tested against fully
infectious ZEBOV in BSL4.
1.4.3 Hypotheses
1) Inhibiting or reducing host non-receptor tyrosine kinases c-SRC or PTK2B will restrict early
HIV-1 infection in T-cell lines or primary human CD4+ T-cells.

89
2) Nucleoside/nucleotide analogue inhibitors AZT, 3TC and TFV, which demonstrate potent
antiviral activity against other RNA viruses and retroviruses, will inhibit an in vitro model of
ZEBOV replication alone or in combination with other therapies.
1.4.4 Objectives of this Work
1) Assess whether c-SRC and PTK2B facilitate HIV-1 infection in T-cell lines, by inhibiting or
reducing their expression with ADV vectors expressing dominant-negative kinase mutants, tool-
drug inhibitors or siRNA knockdown.
2) Determine whether c-SRC and PTK2B have similar roles in primary CD4+ T-cells collected
from healthy human donors, and investigate whether FDA-approved SFK inhibitors, or SFK
inhibitors in advanced clinical trials, can inhibit HIV-1 replication ex vivo.
3) Evaluate whether antivirals with established safety profiles and efficacy in inhibiting other
RNA viruses or retroviruses, can inhibit ZEBOV replication alone or in combination, in an in
vitro trVLP model of infection or with fully-infectious ZEBOV.
1.4.5 Organization of the Thesis
The strong evidence of the SFK roles during the HIV-1 replication cycle, coupled with new
kinase inhibitors with enhanced target specificity being evaluated in clinical trials outside of
cancer research, motivated the investigations contained in this thesis. Moreover drug
repurposing and combination therapy with nucleoside analogues, successful at treating HIV-1 or
HBV infection, have not been adequately investigated in ZEBOV replication in vitro.
In chapter 2, “c-SRC and PTK2B Protein Tyrosine Kinases Play Protective Roles in Early HIV-1
Infection of CD4+ T-Cell Lines,” kinase inhibitors, protein overexpression ADV vectors and
siRNA knockdown are used to determine the roles of c-SRC and PTK2B during in vitro HIV-1
infection in BSL3 containment. These T-cell line experiments laid the groundwork towards a
more comprehensive study of both kinases during HIV-1 replication in primary human CD4+
T-
cells in chapter 3; “c-SRC Protein Tyrosine Kinase Regulates Early HIV-1 Infection Post-Entry.”

90
This chapter investigates four c-SRC inhibitors (FDA-approved or under clinical investigation)
for their ability to inhibit HIV-1 infection ex vivo, using X4 and R5 lab-adapted strains and
clinical isolates of HIV-1. It was also determined at which early stage during the viral lifecycle
that is most dependent on c-SRC or PTK2B kinase expression.
In chapter 4, “A Rapid Screening Assay Identifies Monotherapy with Interferon-ß and
Combination Therapies with Nucleoside Analogues as Effective Inhibitors of Ebola Virus,”
nucleoside/nucleotide analogue inhibitors were assessed in a new model system of ZEBOV
replication in vitro. Timely during the 2014-16 West African ZEBOV outbreak, nucleoside
analogues and therapeutics fast-tracked for EVD clinical trials were compared or combined in
vitro, to assess whether they inhibit ZEBOV transcription and replication competent virus-like
particles, or fully-infectious virus tested under BSL4 conditions at the National Microbiology
Laboratory in Winnipeg.
Lastly in chapter 5, “Key Findings, Future Perspectives, Conclusions and Broader Significance,”
limitations of the present work are discussed, as well as how the findings of this thesis relate to
preclinical HIV-1 and ZEBOV drug discovery. This chapter also suggests future studies that
could investigate promising therapeutics evaluated in this thesis, to potentially inhibit viral
replication in animal models of HIV-1 or ZEBOV infection.

91
Chapter 2: c-SRC and PTK2B Protein Tyrosine Kinases
Play Protective Roles in Early HIV-1 Infection of CD4+ T-
Cell Lines.
Role of author McCarthy S.D.: Performed the experiments for Figure 2.1B, and all the
experiments for Figures 2.2-2.7. I analyzed all the data, wrote the manuscript draft, and
performed experiments to address reviewer’s comments.
A version of this chapter was published in the Journal of Acquired Immune Deficiency
Syndromes:
McCarthy, S.D., Jung, D., Sakac, D., and Branch, D.R. 2014. c-SRC and Pyk2 protein tyrosine
kinases play protective roles in early HIV-1 infection of CD4+ T-cell lines. JAIDS, 66(2): 118-
26.

92
2.1 Abstract
Background: During early HIV-1 infection of CD4+ T-lymphocytes, many host protein tyrosine
kinases (PTK) become activated within minutes, including phosphoprotein pp60c-SRC
(c-SRC)
and the focal adhesion kinase family member, protein tyrosine kinase 2 beta (PTK2B, Pyk2).
Whether their activation facilitates or impedes infection remains to be determined.
Methods: c-SRC kinase inhibitors (SU6656, PP1 and PP2), adenovectors (wild type (WT) and
dominant-negative (DN) c-SRC) or siRNA (targeting c-SRC or PTK2B) were used to inhibit,
compete with or knockdown c-SRC in Jurkat C, Jurkat E6-1, Hut 78 or Kit225 T-cell lines.
Cells were then infected with HIV-1 luciferase reporter virus expressing VSV-G or HXB2(X4)
envelope and luciferase activity was measured after 2 days. Reverse transcriptase activity and
viral cDNA were measured 1 hr post-infection, while integrated virus was measured 12 hr post-
infection.
Results: Pre-treating Jurkat T-cells with SU6656 led to increased VSV-G luciferase activity. In
the adenovector experiments, T-cells overexpressing DN c-SRC, but not WT c-SRC, showed
increased luciferase activity following VSV-G infection. siRNA knockdown of c-SRC or
PTK2B, followed by HXB2 infection in Jurkat T-cells, lead to increased reverse transcriptase
activity, viral cDNA, integrated virus, as well as increased luciferase activity.
Conclusions: PTK2B is known to interact with c-SRC. Thus PTK2B activation, which
coincides with increased c-SRC activity during HIV-1 infection, could be responsible for c-SRC
activation. Reduced c-SRC activation increases HIV-1 reverse transcription, integration and/or
transcription, suggesting the high c-SRC activity seen early in HIV-1 infection may be a cellular
response to slow or prevent early infection in CD4+ T-cells.

93
2.2 Introduction
Chronic immune activation and T-cell dysregulation persists during asymptomatic HIV-1
infection, and yet we still do not have a clear understanding of the early stages of these events
[83]. From the initial binding of viral glycoprotein 120 (gp120) to T lymphocyte receptor CD4
and chemokine coreceptors CXCR4 or CCR5, signaling cascades are induced that promote viral
infection [147]. It is known that tyrosine kinases are major regulators of HIV-1 infection
because the pan-tyrosine kinase inhibitor, genistein, inhibits HIV-1 infection in macrophages
[151]. Moreover, HIV-1-infected patients show defective early protein tyrosine phosphorylation
in peripheral blood mononuclear cells [149] and in CD4+ T-cells specifically [148]. Indeed,
much research has been done on the proximal signaling cascades induced upon viral binding and
fusion, which interfere with normal T-cell activation and cortical actin rearrangement [539].
However, a functional role for many kinases hijacked immediately following viral entry remains
to be discovered.
Non-receptor tyrosine kinases play critical roles throughout the HIV-1 lifecycle in T-cells.
Changes in tyrosine phosphorylation signaling are necessary for viral entry [209], actin
remodeling [135, 539], viral RNA reverse transcription [135], translocation of the viral pre-
integration complex (PIC) to the nucleus, viral integration [155], viral DNA transcription and
viral egress [152]. In particular, HIV-1 infected T-cells show striking changes in the activity of
the SRC-family of tyrosine kinases [214]. They become activated within minutes of HIV-1
infection; however their roles at this early time point are only partially understood. There are
eight SRC family members, four of which are expressed in T-cells: LCK, c-SRC, FYN and c-
YES. LCK has been the most studied of these four in terms of early HIV-1 infection [148, 209,
217], as it is a proximal signaling molecule directly associated with CD4 and is critical for T-cell
activation and growth [213]. Increased LCK activity was found to reduce viral replication in
various T-cell lines [209]. In addition, increased FYN activity correlates with greater
recruitment of tyrosine-phosphorylated APOBEC3G into HIV-1 particles [238], corroborating
our previous observation of higher FYN activity in patients with asymptomatic HIV-1 infection

94
[149]. Clearly the SRC family of protein kinases is implicated in HIV-1 infection, but the
functional role of activated c-SRC during early HIV-1 entry remains unknown.
Previously we have shown that negligible c-SRC protein exists in resting peripheral blood T
lymphocytes, but that it is produced within 12 hours of T-cell stimulation [293]. c-SRC
phosphorylates more than 64 known cellular substrates, activating signaling cascades involved in
cell migration, cytoskeletal rearrangement, cell proliferation and cell survival [294]. The
enzyme often associates with cell membranes, in particular at focal adhesions of the plasma
membrane and the perinuclear golgi region [189]. When HIV-1 initially attaches to the host
membrane, viral gp120 is able to bind the G protein-coupled receptor CXCR4 or CCR5,
inducing the autophosphorylation of protein tyrosine kinase 2 beta (PTK2B, Pyk2) within the
cell [147]. PTK2B plays an important role at focal adhesions at the cell periphery and in
cytoskeleton remodeling. Activated PTK2B can then recruit the binding of the c-SRC SH2
domain, which allows c-SRC to be autophosphorylated at Tyr419, activating the enzyme [197,
322]. Both of these kinases become phosphorylated within minutes of HIV-1 infection. Recent
work has shown that PTK2B shRNA knockdown in T CEM cells reduced viral p24 production
within six days of HIV-1NDK infection [345]. Moreover Gilbert et al. showed that inhibiting c-
SRC with the kinase inhibitor PP2 caused a dramatic decrease in p24 production after six days of
infection [264]. However, the roles of PTK2B and c-SRC in the first few hours of early HIV-1
infection, when these enzymes are first activated, are not well defined.
We hypothesize that c-SRC and PTK2B non-receptor tyrosine kinases play an important role in
facilitating early HIV-1 infection of CD4+ T-cells. By using c-SRC drug inhibitors, a dominant
negative c-SRC construct and siRNA knockdown, we were surprised to find that reducing c-SRC
protein levels or its activity caused an increase in viral infection in three laboratory T-cell lines.
This research is significant because small molecular kinase inhibitors targeting c-SRC may not
be efficacious in reducing initial HIV-1 infection, but may be valuable in studying the formation
and/or stability of the HIV-1 reverse transcriptase complex and pre-integration complex.

95
2.3 Materials and Methods
Cells and Cell Culture Conditions: In these in vitro experiments, we used the human T-cell
leukemia cell lines Jurkat C (from Jurkat-FHCRC, a gift from Dr. G. B. Mills, MD Anderson
Cancer Center, Houston, TX, USA), Jurkat E6-1 and a cutaneous T lymphocyte cell line Hut 78
from the American Type Culture Collection (ATCC, Rockville, USA). We also used a chronic
lymphocytic leukemia T-cell line, Kit 225, also a gift from Dr. G. B. Mills. These T-cell lines
were grown in complete medium consisting of RPMI-1640 with L-glutamine and NaHCO3
(SIGMA, St. Louis, USA), supplemented with 10% (vol/vol) heat-inactivated Fetal Bovine
Serum (FBS; Wisent, Saint-Jean-Baptiste, Canada), 100 units/mL penicillin (Gibco, Burlington,
Canada), 100 μg/mL streptomycin (Gibco), 10 μg/mL gentamycin (Gibco), and 10 units/mL
human recombinant Il-2 (Kit 225 cells only; SIGMA). Human embryonic kidney cells (HEK
293T, ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) with the
same proportion of FBS and antibiotics as above. MT-4 cells from the NIH AIDS Research and
Reference Reagent Program (Germantown, USA) were used to determine viral multiplicity of
infection (MOI). All cells were incubated at 37°C in a 5% CO2 atmosphere.
Generation of infectious HIV-1, VSV-G pseudoenveloped and HXB2 enveloped virus: In
brief, X4 HIV-1IIIB from the NIH AIDS Research and Reference Reagent Program (Rockville)
was grown in Jurkat C cells (1 x 106
cells/mL) in a biosafety level 3 (BSL3) lab [540]. The cell
supernatant was collected, viral stocks were titrated and assessed by p24 Antigen ELISA
(ZeptoMetrix, Buffalo, USA), and aliquots were frozen at -80°C. MOI was determined using
MT-4 cells [540].
We produced recombinant, replication deficient VSV-G pseudoenveloped or HXB2 enveloped
(X4) HIV-1 containing a luciferase gene, as described previously [541]. Briefly, 2.5 x 106 HEK
293T cells were plated with 10 mL of DMEM 24 hours prior to transfection. We co-transfected
15 μg of HIV-1 NL4-3luc (luciferase gene inserted into viral nef gene and env deficient; a kind
gift from Dr. Veneet KewalRamani, New York Medical University, NY) with 10 μg of VSV-G
env plasmid (amphotropic envelope of the vesicular stomatitis virus; a generous gift from Dr. M.
Tremblay, Quebec City, PQ) into each 10 cm plate of HEK 293T cells with the CalPhosTM

96
Mammalian Transfection Kit (Clontech, Mountain View, USA), following the manufacturer’s
instructions. The same was performed for the HXB2 plasmid (HIV-1 NL4-3luc lacking nef and
containing HXB2 (X4) env; another generous gift from Dr. M. Tremblay) at 15 μg per plate.
DMEM media was replaced after 25 hours and viral supernatant was collected on days 2 and 3.
The supernatant was syringed through 0.45 μm pore filters (Millipore, Billerica, USA) , then
concentrated by underlaying with a 20% sucrose gradient and centrifuging at 16,000 x g for 90
minutes at 4 °C. Virus pellet were resuspended in TNE buffer (20 mM Tris-HCl, pH 7.5, 1 mM
EDTA and 100 mM NaCl) and stored in -80°C aliquots. p24 levels were measured by ELISA.
All viruses underwent one freeze-thaw cycle prior to infection studies.
c-SRC Drug Inhibition: Prior to infection, 2 x 106 T-cells were inhibited with 5-20 μM of
SU6656 (In SolutionTM
), 10 μM PP1 (Analogue) or 10 μM PP2 (Calbiochem, La Jolla, USA) for
1 hr, washed with Dulbecco’s Phosphate Buffered Saline (PBS) lacking calcium chloride and
magnesium chloride (SIGMA), resuspended in RPMI media, and then counted again. The
solvent Dimethyl Sulfoxide (DMSO, Bioshop, Burlington, Canada) was used as a -ve control for
SU6656.
Kinase Assay: Jurkat C cells treated for 1 hr with SU6656 were lysed with
radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM HEPES, pH 7.3, 1% Nonidet P-40,
0.1% SDS, 0.1% sodium deoxycholate, 150 mM NaCl, 1 mM Na3VO4, 50 mM ZnCl2, 2 mM
EDTA and 2 mM PMSF), and immunoprecipitated with anti-c-SRC clone 327 (Oncogene,
Cambridge, USA). Lysates and beads were washed then re-suspended in kinase buffer,
including 5 μCi γ32
P-ATP (MP Biomedicals, Santa Ana, USA) and heat inactivated enolase (MP
Biomedicals) as substrate. Protein samples were boiled for 10 minutes and then separated on a
12% reducing SDS-PAGE gel and transferred to an Immobilon-P membrane (Millipore).
Enolase phosphorylation by c-SRC was determined by autoradiography. In vitro kinase activity
was measured as the densitometry of enolase activity divided by the amount of c-SRC per lane,
and normalized to the DMSO control treatment.
c-SRC Adenovirus Gene Transduction: Recombinant Ad5/F35 adenovirus vectors were
created by Daniel Jung. These bicistronic vectors contained enhanced yellow fluorescent protein

97
(EYFP), and expressed wild type human c-SRC (WT), a dominant negative, kinase inactive c-
SRC (DN) or no c-SRC as a control (empty vector; EV) [190]. Cells were titrated with virus to
determine the highest multiplicity of infection that permitted 70-80% gene transduction, while
minimizing cell death 48 hours post-infection. EYFP detection and viability staining (7-AAD,
which stains both apoptotic cells and necrotic bodies, BD Sciences, Franklin Lakes, USA) were
performed on fixed cells using FACS (see flow cytometry and data acquisition).
siRNA Knockdown: Pools of four siRNA duplexes, specific for human c-SRC (cat # M-
003175-03) or PTK2B (cat # M-003164-02) mRNA, and a control pool of non-targeting siRNA
(cat # D-001206-13-05) were ordered from Dharmacon RNAi technologies (siGENOME
SMART Pool, Thermo Scientific, Lafayette, USA). In Jurkat E6-1 cells, siRNA knockdown
was achieved using the siRNA cationic lipid transfection reagent GeneSilencer® (Genlantis, San
Diego, USA), following the manufacturer’s instructions. Two μL of siGuard RNase inhibitor®
(Genlantis) was added to each 1 mL reaction to prevent RNAse degradation of the RNA
duplexes. Upon siRNA titration, 900 nM was determined to give the optimal level of protein
knockdown for both c-SRC and PTK2B, via Western blot densitometry at 48 hours.
HIV-1 Infection and Luciferase Assay: 1 x 106 T-cells pre-treated with drug, adenovector or
siRNA were infected with 1.4 ng of VSV-G/HIV-1 or 7.5 ng of HXB2 at 37°C in 2 mL of
growth media in 12-well plates for 2 days. Cells were counted, while others were lysed with the
Luciferase Assay System (Promega, Madison, USA) or kept for Western blots.
Antibodies/Immunoblots: Jurkat C cells were serum-starved and then immediately following
15 minutes of HIV-IIIB infection, were washed in PBS and lysed with RIPA buffer. Monoclonal
anti-phosphotyrosine (anti-ptyr) clone 4G10 (Millipore) and protein G-Sepharose beads (Santa
Cruz Biotechnology, Dallas, USA) were used to immunopreciptate phosphoproteins from 4 x 106
Jurkat C cell lysates.
Following VSV-G/HIV-1 or HXB2 infection, 5-10 x 105 T-cells were washed in PBS and lysed
with RIPA buffer. After boiling samples, proteins were resolved on an 8%, reducing SDS-
PAGE gel, and transferred to an Immobilon-P membrane for Western Blot detection. The blots

98
were blocked with 5% skimmed-milk powder and 0.5% Nonidet P-40 (BioShop, Burlington,
Canada). Primary antibodies diluted in 5% milk and 0.5% Nonidet P-40, anti-β-actin (Sigma),
anti-α-tubulin (Sigma) anti-PTK2B (Transduction Laboratories) and anti-c-SRC GD11
(Millipore) were used to probe the blots. After washing, secondary goat anti-mouse HRP
antibody (Bio-Rad, Mississauga, Canada) was added, followed by enhanced chemiluminescent
(ECL) detection (GE Healthcare, Buckinghamshire, UK). PTK2B and c-SRC protein levels were
determined by comparing their densitometry to normalizers β-actin or α-tubulin with the
Molecular Imager GelDoc XR+ Imagin System (Bio-Rad).
DNA isolation and quantitative PCR: Genomic and viral cDNA was isolated from 5 x 105
Jurkat E6-1 cells, pre-treated by siRNA transfection and infected with HXB2 virus for 1 or 12
hours, using the QIAamp DNA Blood Mini Kit (Qiagen, Toronto, Canada). Samples were
heated at 56°C for 2 hours in QIAamp DNA Blood Mini Kit buffer AL to deactivate viable viral
particles.
The primer sets used to detect human or viral DNA (synthesized by the Center for Applied
Genomics, The Hospital for Sick Children, Toronto, Canada) were as follows [159, 542, 543]: β-
globin forward, 5′-CCCTTGGACCCAGAGGTTCT -3′; β-globin reverse, 5′-CGAGCACTTTCT
TGCCATGA-3′; early reverse transcripts (early RT) forward, 5′-GTAACTAGAGATCCCTCAG
ACCCTTTTAG-3′; early RT reverse, 5′- TAGCAGTGGCGCCCGA-3′; late reverse transcripts
(late RT) forward, 5′-CCGTCTGTTGTGTGACTCTGG-3′; late RT reverse, 5′-GAGTCCTGCG
TCGAGAGATCT-3′; genomic Alu forward, 5′-GCCTCCCAAAGTGCTGGGATTACAG-3′;
HIV-1 gag reverse, 5′-GCTCTCGCACCCATCTCTCTCC-3′; HIV-1 LTR forward, 5′-GCCTCA
ATAAAGCTTGCCTTGA-3′; HIV-1 LTR reverse, 5′-TCCACACTGACTAAAAGGGTCTGA-
3′. For each qPCR run, 50-100 ng template DNA (with the exception of the no-template control,
NTC) was added to PCR tubes containing: 12.5 μL 2x SYBR Green PCR Master Mix (Applied
Biosystems), 300-900 nM of each forward and reverse primers, and PCR grade H2O (Roche
Diagnostics, Indianapolis, USA) up to a final volume of 25 μL. PCR cycling parameters were as
follows: initial denaturation at 95°C for 10 min; 40-50 cycles of amplification of 95°C for 15
sec, 58°C for 30 sec (β-globin or late RT, 54°C for early RT) and 60°C for 30 sec. To measure
integrated virus by Alu-gag PCR [543], tubes were first pre-amplified with 100 nM Alu forward

99
and 600 nM gag reverse primers, with 2x SYBR Green PCR Master Mix as above, for 10 cycles
of 93°C for 30 sec, 50°C for 60 sec, and 60°C for 100 sec. Nested PCR was performed on these
samples by adding 300 nM each of HIV-1 LTR forward and reverse primers, with 2x SYBR
Green PCR Master Mix, for 40 more cycles using the above parameters. Duplicate reactions
were analyzed using the Rotor-Gene RG-3000 thermocycler (Corbett Research, Montreal,
Canada). c-SRC and PTK2B siRNA-treated groups were compared to the non-targeting siRNA-
treated group by the 2-ΔΔCT
comparative CT method [544].
Flow Cytometry and data acquisition: Cell surface expression of CD4 and CXCR4 were
determined on 2.5 x 105 Jurkat E6-1 cells per FACS tube, 48 hours after siRNA knockdown.
Cell surface staining was performed as previously described [545]. In brief, cells were stained
with 5 μL of FITC mouse anti-human CD4, 5 μL of PE mouse anti-human CXCR4 and 2.5 μL of
7-AAD, in 100 μL of FACS buffer (PBS + 2.5% FBS). Isotype control antibodies were FITC-
conjugated mouse IgG1κ and PE-conjugated mouse IgG2aκ. All of these antibodies were
purchased from BD Pharmingen, Mississauga, Canada. Data on 20,000 events per tube was
collected using Becton Dickenson FACSCalibur (BD Calibrite; BD Biosciences). Signals were
acquired for the forward scatter (FSC), side scatter (SSC), the green (FITC, EYFP), yellow (PE)
and red (7-AAD) channels.
Reverse Transcriptase Activity Assay: Quantification of reverse transcriptase activity from
infected cell lysates was performed using the SYBR Green I qPCR-based product-enhanced RT
(PERT) assay [546]. Briefly, 5 x 105 JE6-1 cells were infected with HXB2 virus for 1 hr,
washed twice with PBS, lysed, diluted 10-fold with 10x sample dilution buffer, and given 1 μg
of MS2 RNA template (Roche Diagnostics) to reverse transcribe in vitro. The cDNA product
was measured by qPCR, and a standard curve of recombinant HIV-1 RT (Calbiochem) was used
to assign enzyme activity units to reverse transcriptase from the infected cell lysate samples.
Statistical Analysis: Means were compared using a two-tailed, unpaired Student’s t test and
corrected for multiple comparisons when more than two means were considered in an
experiment, to minimize the probability of type 1 errors. FACS data shows the Median
Fluorescent Intensity (MFI) and percent positive cells collected with CellQuest software (BD

100
Biosciences) and analyzed by FlowJo version X software. Comparisons of histogram EYFP
expression was performed with the two sample Kolmogorov-Smirnov test. For all figures, an
asterisk (*) denotes a p value < 0.05, two asterisks (**) denotes a p value < 0.01 and three
asterisks (***) denotes a p value < 0.005. Error bars shown are the standard error around the
mean (SEM).

101
2.4 Results
Tyrosine kinases become activated within minutes of HIV-1IIIB infection
To confirm previous reports that non-receptor kinases become activated within minutes of HIV-1
infection, we infected serum-starved Jurkat C T-cells with HIV-1IIIB, HXB2 (X4 strains), or with
VSV-G pseudoenveloped HIV-1 virus. We then immunoprecipitated protein substrates from T-
cell lysates with anti-ptyr clone 4G10 (Fig. 2.1). Probing the blot of HIV-1IIIB infected cell lysate
with anti-ptyr revealed a robust phosphorylation of substrates in the 55-130 kDa range, and one
low molecular band at ~40-42 kDa (Fig. 2.1A). Re-probing the Western blot for PTK2B and c-
SRC (Fig. 2.1B), demonstrated phosphorylation of these kinases within 15 minutes of HIV-1
infection, indicative of their activation and recruitment during early HIV-1 entry events.
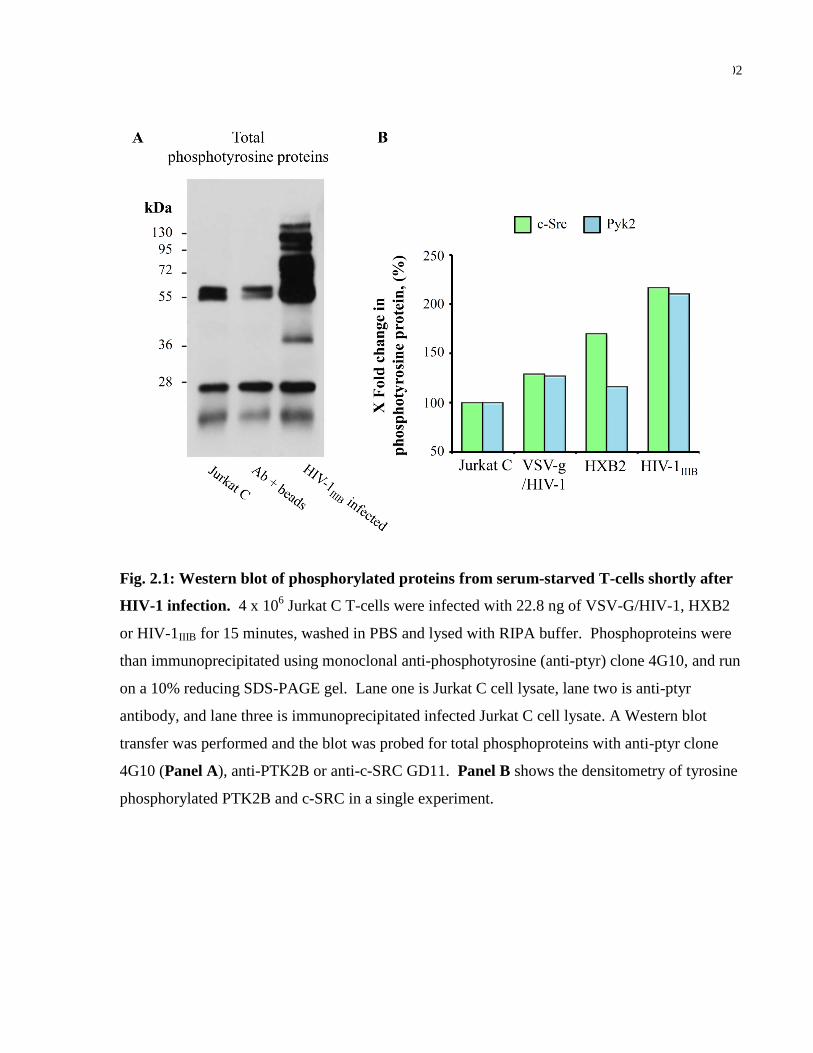
102
Fig. 2.1: Western blot of phosphorylated proteins from serum-starved T-cells shortly after
HIV-1 infection. 4 x 106 Jurkat C T-cells were infected with 22.8 ng of VSV-G/HIV-1, HXB2
or HIV-1IIIB for 15 minutes, washed in PBS and lysed with RIPA buffer. Phosphoproteins were
than immunoprecipitated using monoclonal anti-phosphotyrosine (anti-ptyr) clone 4G10, and run
on a 10% reducing SDS-PAGE gel. Lane one is Jurkat C cell lysate, lane two is anti-ptyr
antibody, and lane three is immunoprecipitated infected Jurkat C cell lysate. A Western blot
transfer was performed and the blot was probed for total phosphoproteins with anti-ptyr clone
4G10 (Panel A), anti-PTK2B or anti-c-SRC GD11. Panel B shows the densitometry of tyrosine
phosphorylated PTK2B and c-SRC in a single experiment.

103
SU6656 consistently inhibited c-SRC activity, and increased VSV-G/HIV-1 infection
To directly implicate a role for c-SRC activity during early HIV-1 infection, we used well known
c-SRC inhibitors (PP1, PP2 and SU6656) [184] to inhibit c-SRC activity prior to HIV-1
infection. For these experiments a replication deficient, pseudotyped nef-deficient VSV-G/HIV-
1 virus carrying a luciferase reporter gene was used, so as not to produce viral protein products
which are known to directly bind and activate the SRC family of kinases, such as viral protein
Nef [164]. After pre-treating Jurkat E6-1 cells with 10 μM of drug for 1 hour, only the SU6656
treated cells showed a significant increase in VSV-G/HIV-1 infection after two days, compared
with DMSO control (Fig. 2.2A). Cell growth was not adversely affected during this experiment
(Figure 2.3A). The non-specific effects of SU6656 differ from PP1 and PP2 [184], which is why
we decided to move forward using SU6656 as our c-SRC inhibitor. Our in-vitro kinase assay
showed that SU6656 reduces c-SRC activity by 50% at 10 μM (Fig. 2.2B). We repeated our
VSV-G/HIV-1 infection experiment at two different SU6656 drug concentrations (Fig. 2.2C) and
in another Jurkat T-cell line (Fig. 2.2D) to confirm our finding that c-SRC drug inhibition leads
to increased VSV-G/HIV-1 infection in T-cell lines.
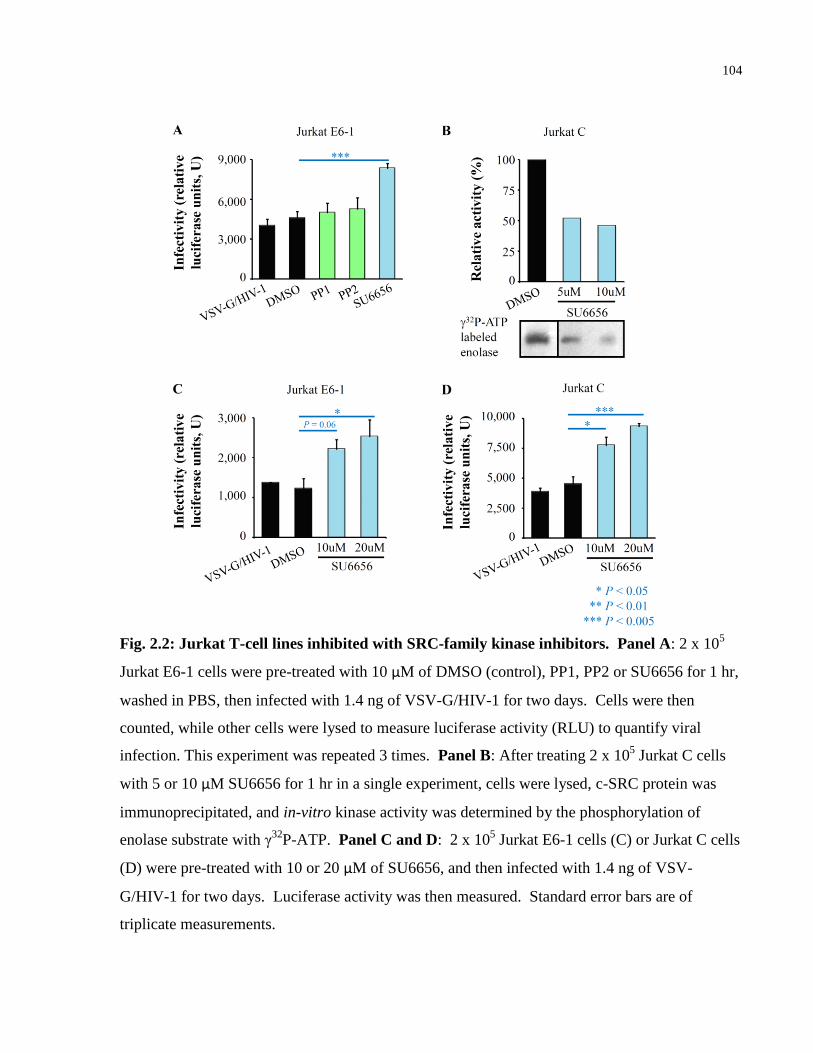
104
Fig. 2.2: Jurkat T-cell lines inhibited with SRC-family kinase inhibitors. Panel A: 2 x 105
Jurkat E6-1 cells were pre-treated with 10 μM of DMSO (control), PP1, PP2 or SU6656 for 1 hr,
washed in PBS, then infected with 1.4 ng of VSV-G/HIV-1 for two days. Cells were then
counted, while other cells were lysed to measure luciferase activity (RLU) to quantify viral
infection. This experiment was repeated 3 times. Panel B: After treating 2 x 105 Jurkat C cells
with 5 or 10 μM SU6656 for 1 hr in a single experiment, cells were lysed, c-SRC protein was
immunoprecipitated, and in-vitro kinase activity was determined by the phosphorylation of
enolase substrate with γ32
P-ATP. Panel C and D: 2 x 105 Jurkat E6-1 cells (C) or Jurkat C cells
(D) were pre-treated with 10 or 20 μM of SU6656, and then infected with 1.4 ng of VSV-
G/HIV-1 for two days. Luciferase activity was then measured. Standard error bars are of
triplicate measurements.
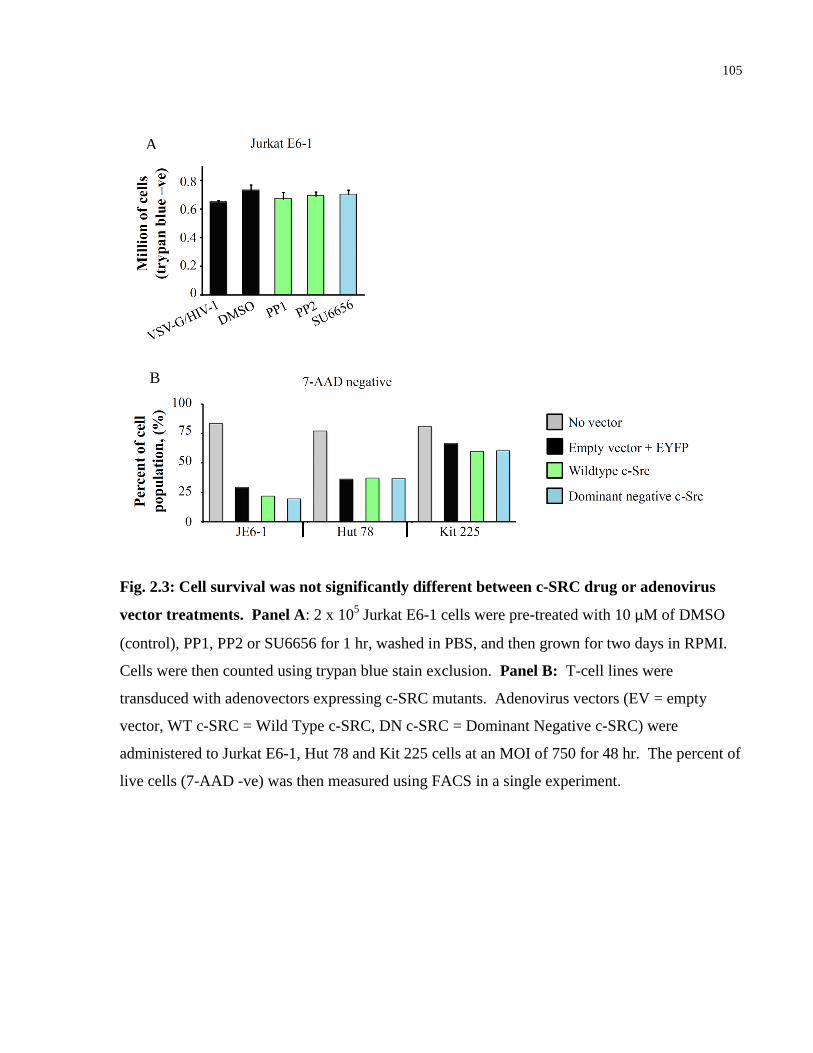
105
Fig. 2.3: Cell survival was not significantly different between c-SRC drug or adenovirus
vector treatments. Panel A: 2 x 105 Jurkat E6-1 cells were pre-treated with 10 μM of DMSO
(control), PP1, PP2 or SU6656 for 1 hr, washed in PBS, and then grown for two days in RPMI.
Cells were then counted using trypan blue stain exclusion. Panel B: T-cell lines were
transduced with adenovectors expressing c-SRC mutants. Adenovirus vectors (EV = empty
vector, WT c-SRC = Wild Type c-SRC, DN c-SRC = Dominant Negative c-SRC) were
administered to Jurkat E6-1, Hut 78 and Kit 225 cells at an MOI of 750 for 48 hr. The percent of
live cells (7-AAD -ve) was then measured using FACS in a single experiment.
A
B

106
Adenovector expressing dominant negative c-SRC increased VSV-G/HIV-1 infection in
CD4+ T-cell lines
As with most kinase inhibitors, we cannot ignore that SU6656 inhibits related SRC family
members, such as LCK and FYN, in conjunction with c-SRC. Thus, we used adenovirus gene
transduction to alter the pool of functional c-SRC in three T-cell lines, prior to VSV-G/HIV-1
infection. Jurkat E6-1, Hut 78 and Kit 225 T-cells were transduced with adenoviruses containing
an Empty Vector control (EV) Wild Type (WT) c-SRC or a Dominant Negative (DN) c-SRC
that can bind but not phosphorylate substrate (Figure 2.4). Administering adenovector
expressing WT c-SRC or DN c-SRC did not alter cell survival when compared to EV, within
each cell type (Figure 2.3B). Once we optimized multiplicity of infection (MOI) to reduce cell
death and maintain high gene transduction, we achieved 62-84% EYFP expression in Jurkat E6-1
and Hut 78 cells after 2 days (Fig. 2.4B-C). Kit 225 cells were less transducible (36-47%, Fig.
2.4D), but showed higher overall survival. No statistical difference was found between EV, WT
c-SRC or DN c-SRC EYFP expression, by the Kolmogorov-Smirnov test.
Viral infection was normalized to the total cells in each well after two days of VSV-G/HIV-1
infection. At this time point, no significant difference in VSV-G/HIV-1 infectivity was found
between EV and WT c-SRC treatments (Fig. 2.4E-G). However, administering the DN c-SRC
adenovector caused a significant increase in VSV-G/HIV-1 infection in two of the three T-cell
lines tested. This finding complements our previous c-SRC drug experiments. Competing
endogenous c-SRC enzyme with the dominant negative mutant increases the integration or
transcription of VSV-G/HIV-1.
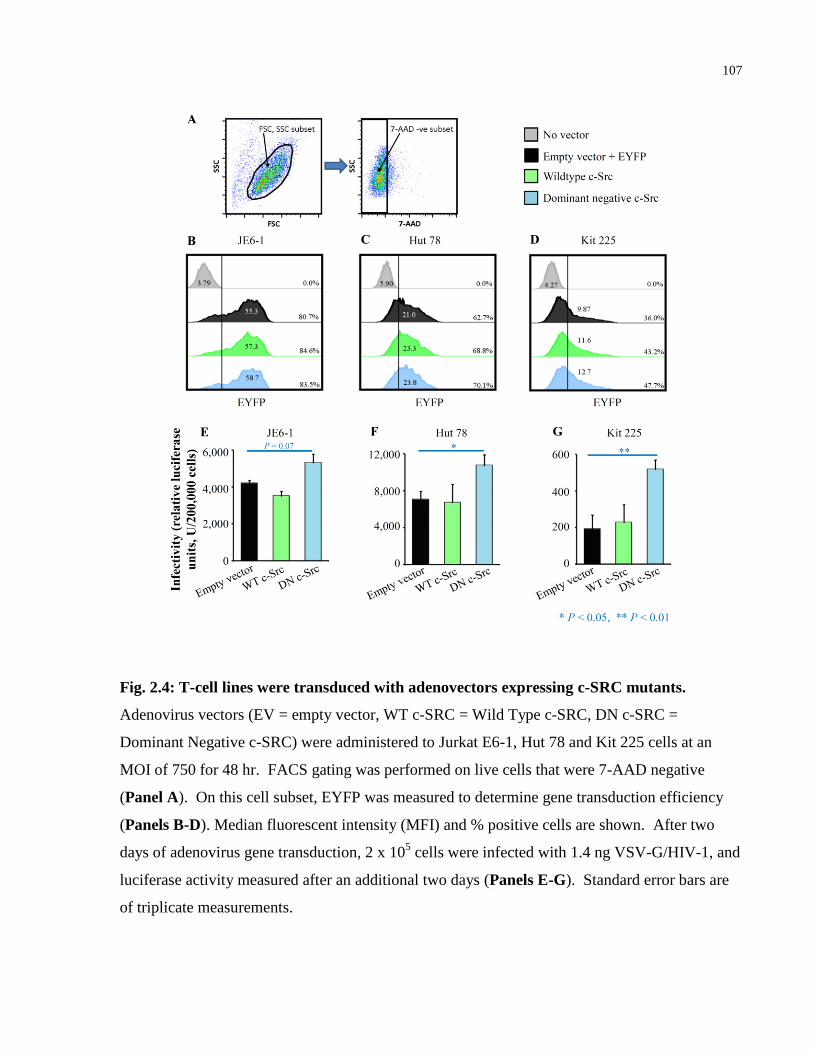
107
Fig. 2.4: T-cell lines were transduced with adenovectors expressing c-SRC mutants.
Adenovirus vectors (EV = empty vector, WT c-SRC = Wild Type c-SRC, DN c-SRC =
Dominant Negative c-SRC) were administered to Jurkat E6-1, Hut 78 and Kit 225 cells at an
MOI of 750 for 48 hr. FACS gating was performed on live cells that were 7-AAD negative
(Panel A). On this cell subset, EYFP was measured to determine gene transduction efficiency
(Panels B-D). Median fluorescent intensity (MFI) and % positive cells are shown. After two
days of adenovirus gene transduction, 2 x 105 cells were infected with 1.4 ng VSV-G/HIV-1, and
luciferase activity measured after an additional two days (Panels E-G). Standard error bars are
of triplicate measurements.

108
Both c-SRC and PTK2B knockdown caused an increase in either VSV-G/HIV-1 or HXB2
infection.
To assess whether the upstream binding partner PTK2B mediates the effects of c-SRC during
HIV-1 infection, we used siRNA to knockdown mRNA expression of either kinase in Jurkat E6-
1 cells. Relative to a non-targeting siRNA control, cell proliferation was not affected by PTK2B
or c-SRC knockdown (Figure 2.5A). T-cells are a challenging non-adherent T-cell line to
transfect for siRNA knockdown, hence we optimized the siRNA concentration delivered and the
time-point to assess protein knockdown by Western blot (Fig. 2.5B-G). Qualitatively, we found
900 nM of siRNA transfected by lipofection could reliably knockdown either c-SRC or PTK2B
by 40-50%, 48 hours post-transfection (Fig. 2.6A-D).
At this time point, we then infected the Jurkat E6-1 cells with VSV-G/HIV-1 and measured
infection two days later. Interestingly, both c-SRC and PTK2B knockdown in Jurkat E6-1 cells
lead to an increase in luciferase activity upon infection with the VSV-G/HIV-1 reporter virus,
when compared with the non-targeting siRNA control group (Fig. 2.6E). This implicates both
kinases as having important catalytic activity during early HIV-1 infection of T-cells. To
determine whether this phenomenon can be attributed to the differences in vesicular stomatitis
entry into a host T-cell (clathrin-dependent endocytosis) [547] and that of HIV-1 (receptor-
mediated internalization), we performed the siRNA knockdown experiment once more with an
HXB2 (X4) luciferase reporter virus that is also replication-deficient. Similar to the VSV-
G/HIV-1 infection, when either c-SRC or PTK2B protein expression is reduced by siRNA
knockdown, we saw a concomitant increase in HXB2 viral infection after two days (Fig. 2.6F).
This suggests that PTK2B and c-SRC may play similar roles in the early HIV-1 entry events of
both viruses downstream of HIV-1 receptor binding and fusion.
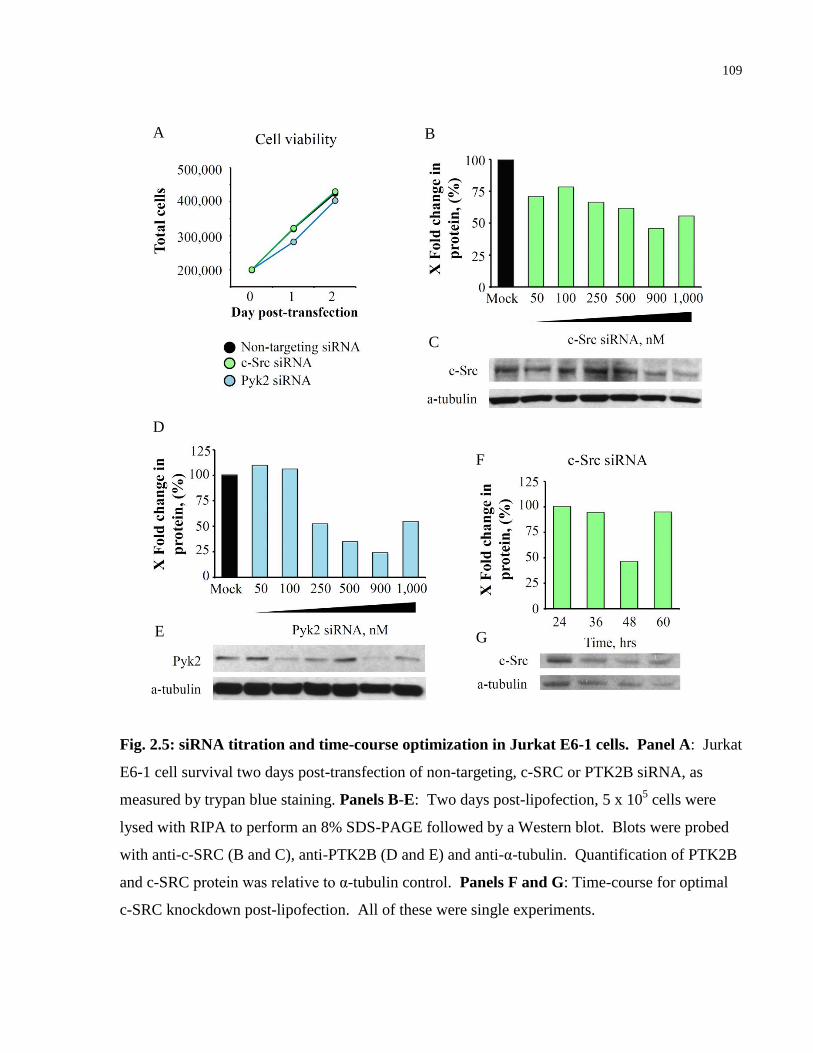
109
Fig. 2.5: siRNA titration and time-course optimization in Jurkat E6-1 cells. Panel A: Jurkat
E6-1 cell survival two days post-transfection of non-targeting, c-SRC or PTK2B siRNA, as
measured by trypan blue staining. Panels B-E: Two days post-lipofection, 5 x 105 cells were
lysed with RIPA to perform an 8% SDS-PAGE followed by a Western blot. Blots were probed
with anti-c-SRC (B and C), anti-PTK2B (D and E) and anti-α-tubulin. Quantification of PTK2B
and c-SRC protein was relative to α-tubulin control. Panels F and G: Time-course for optimal
c-SRC knockdown post-lipofection. All of these were single experiments.
B
C
D
F
G E
A
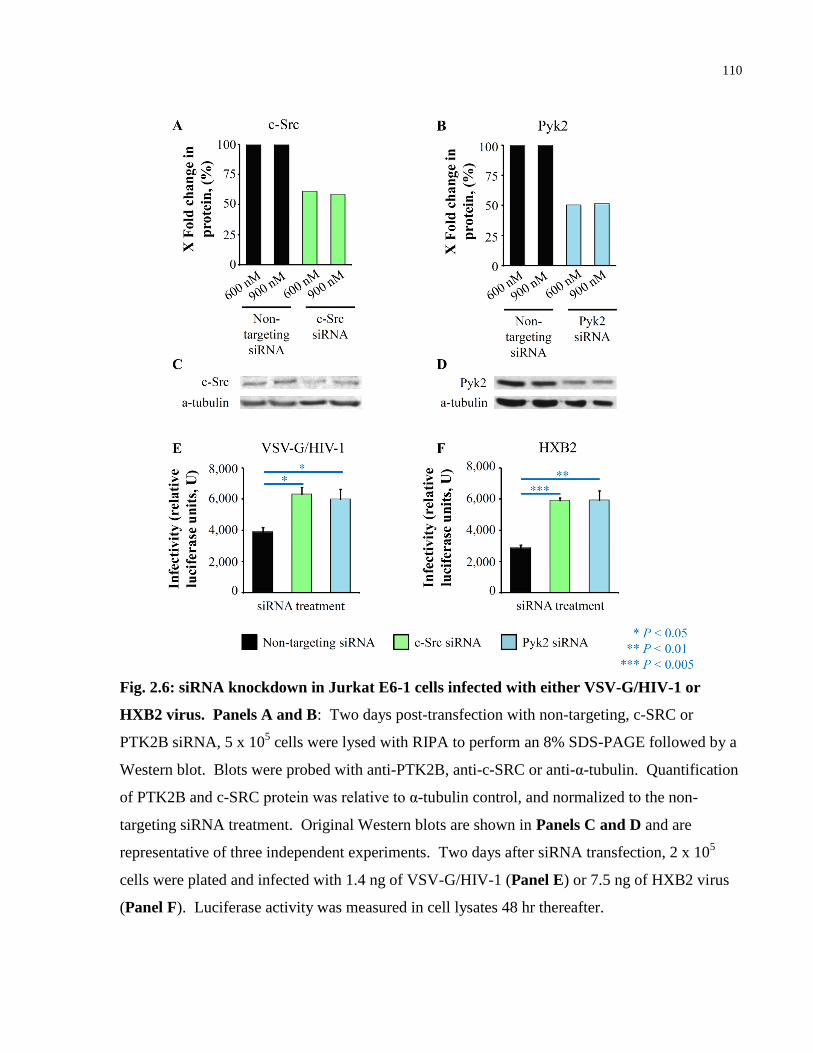
110
Fig. 2.6: siRNA knockdown in Jurkat E6-1 cells infected with either VSV-G/HIV-1 or
HXB2 virus. Panels A and B: Two days post-transfection with non-targeting, c-SRC or
PTK2B siRNA, 5 x 105 cells were lysed with RIPA to perform an 8% SDS-PAGE followed by a
Western blot. Blots were probed with anti-PTK2B, anti-c-SRC or anti-α-tubulin. Quantification
of PTK2B and c-SRC protein was relative to α-tubulin control, and normalized to the non-
targeting siRNA treatment. Original Western blots are shown in Panels C and D and are
representative of three independent experiments. Two days after siRNA transfection, 2 x 105
cells were plated and infected with 1.4 ng of VSV-G/HIV-1 (Panel E) or 7.5 ng of HXB2 virus
(Panel F). Luciferase activity was measured in cell lysates 48 hr thereafter.

111
c-SRC and PTK2B siRNA knockdown increased the reverse transcription of HXB2 cDNA
To assess at which early stage c-SRC or PTK2B were having their effect, we measured the
production of HIV-1 early reverse transcripts (early RT) late reverse transcripts (late RT), as well
as integrated virus, shortly after HXB2 infection by real-time qPCR (Fig. 2.7A). One hour post-
infection, both c-SRC and PTK2B siRNA knockdown led to a 4-10 fold increase in early and
late reverse transcripts, relative to non-targeting siRNA treated cells. Knocking down c-SRC or
PTK2B also led to a 3-4 fold increase in integrated virus measured after 12 hours of infection.
We detected no increase in CD4 or CXCR4 cell surface expression after c-SRC or PTK2B
knockdown (Fig. 2.7B), suggesting the increase in HIV-1 early RT, late RT and integrated virus
is not due to increased receptor binding upon entry. Measuring reverse transcriptase activity 1
hour post-infection revealed increased activity from lysates of Jurkat T-cells administered c-SRC
or PTK2B siRNA (Fig. 2.7C), suggesting that they act, at least in part, at the level of HIV-1
reverse transcription.
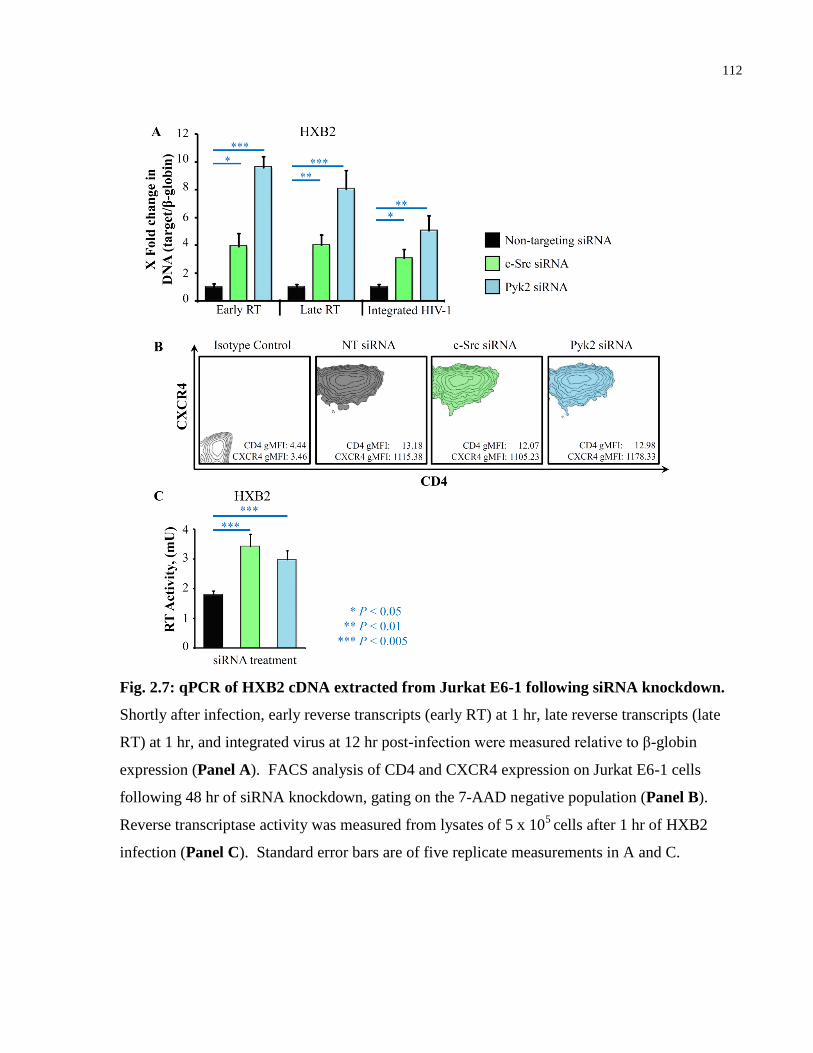
112
Fig. 2.7: qPCR of HXB2 cDNA extracted from Jurkat E6-1 following siRNA knockdown.
Shortly after infection, early reverse transcripts (early RT) at 1 hr, late reverse transcripts (late
RT) at 1 hr, and integrated virus at 12 hr post-infection were measured relative to β-globin
expression (Panel A). FACS analysis of CD4 and CXCR4 expression on Jurkat E6-1 cells
following 48 hr of siRNA knockdown, gating on the 7-AAD negative population (Panel B).
Reverse transcriptase activity was measured from lysates of 5 x 105
cells after 1 hr of HXB2
infection (Panel C). Standard error bars are of five replicate measurements in A and C.

113
2.5 Discussion
The present study demonstrates that reducing c-SRC enzyme activity, outcompeting endogenous
c-SRC with a dominant negative SRC mutant, or reducing c-SRC protein levels with siRNA
knockdown caused an increase in VSV-G/HIV-1 infection in various T-cell lines. We first
demonstrated that c-SRC becomes activated within 15 minutes of HIV-1IIIB infection, which
agrees with our previous findings [214]. We also found that PTK2B becomes phosphorylated in
the same 15 minute time span, faster than Seror et al. reported using HIV-1NDK virus [345].
Reducing c-SRC activity with SU6656 consistently caused an increase in VSV-G/HIV-1
infection in Jurkat T-cells. Initially, this finding appears paradoxical considering that CD4+ T-
lymphocytes pre-treated with PP2 were previously reported to show a decrease in p24 levels six
days after NL4-3balenv or JR-CSF (R5) infection [264]. This discrepancy may be because our
viral read-out measured transcription of luciferase to detect successful viral integration, instead
of p24 to monitor viral production and egress. The initial activation of non-receptor tyrosine
kinases is rapid [214], and kinases that play a role during early entry may have different roles
during viral egress and p24 release. Thus inhibiting c-SRC activity may promote early entry of
HIV-1, as we have found, and yet reduce viral p24 production at later stages of the viral
replication cycle. This difference in function could perhaps be attributed to the downstream
substrate, the 68 kDa SRC-associated protein in mitosis (Sam 68), which is known to assist in the
nuclear export and translation of HIV-1 RNA [317].
We also observed that the addition of adenovector producing dominant negative c-SRC in three
T-cell lines caused an increase in VSV-G/HIV-1 infection, yet adding adenovector expressing
wild type c-SRC did not reduce infection. Perhaps this can be attributed to the dynamic
regulation of c-SRC by cellular phosphatases (SHP-1, CD45) and kinases (CSK), explaining why
the addition of wild type c-SRC could not reduce viral infection. Nonetheless, the ability of
dominant negative c-SRC to compete with endogenous c-SRC for substrates and cause an
increase in VSV-G/HIV-1 infection agrees well with our drug experiments. Whether c-SRC
directly phosphorylates HIV-1 proteins, or triggers signaling pathways that hinder viral entry and
integration, warrants further investigation.

114
In terms of our siRNA knockdown findings, both c-SRC and PTK2B knockdown caused an
increase in VSV-G/HIV-1 infection in Jurkat T-cells. This agrees well with the previous finding
that activated PTK2B links G protein-coupled receptors to downstream signaling by complexing
with c-SRC and subsequently causing c-SRC activation [197]. It is interesting to note that while
VSV-G/HIV-1 enters cells via clathrin-dependent endocytosis [547], using a virus with HXB2
(X4) envelope gave a similar result, suggesting both kinases play important roles in HIV-1
infection regardless of initial entry route. We also found increased reverse transcriptase activity,
early reverse transcripts, late reverse transcripts and integrated virus after c-SRC or PTK2B
siRNA knockdown. PTK2B, being upstream of c-SRC, had a stronger effect on viral DNA
produced in cells. It will be of interest to determine the mechanism by which these kinases affect
early infection, perhaps stabilizing the reverse transcriptase complex or pre-integration complex
through phosphorylation or allosteric interactions.
Our research is the first study to explore the impact of c-SRC and PTK2B activation on early
HIV-1 infection in CD4+ T-cell lines. Discovering novel mechanisms in the entry of HIV-1 and
their impact on T-cell signal transduction may lead to targeting these pathways with drugs and
improving the treatment options available to people with HIV-1. Tyrosine kinase inhibitors with
low toxicity profiles exist for cancer therapies, and could be repositioned as viable options for
treating HIV-1 infection. For instance cyclin-dependent kinase inhibitors, such as r-roscovitine
and alsterpaullone, are showing promising anti-HIV-1 properties in PBMCs by preventing viral
Tat transactivation [548]. We also know that tyrosine kinase inhibitors can be safe in HIV-1
patients, as a case study of patients with AIDS-related Kaposi Sarcoma given the ABL kinase
inhibitor imatinib showed no increase in viral plasma load [176]. Our results show increased
HIV-1 early reverse transcripts, late reverse transcripts and integration when c-SRC or PTK2B
are inhibited, which may preclude the use of SRC inhibitors during early HIV-1 infection.
However, the potential use of c-SRC kinase inhibitors to study the stability of the reverse
transcriptase complex and pre-integration complex warrants further investigation.

115
Chapter 3: c-SRC Protein Tyrosine Kinase Regulates
Early HIV-1 Infection Post-Entry
Role of author McCarthy S.D.: Performed all the experiments for Figures 3.1-3.10 and
illustrated Figure 3.11. I analyzed all the data, wrote the manuscript draft, and performed
experiments to address reviewer’s comments.
A version of this chapter was published in the Journal AIDS:
McCarthy, S.D., Sakac, D., Neschadim, A., and Branch, D.R. 2016. c-SRC protein tyrosine
kinase regulates early HIV-1 infection post-entry. AIDS, 30(6): 849-58.

116
3.1 Abstract
Objective: We investigated whether HIV-1 inhibition by SRC-family kinase inhibitors is
through the non-receptor tyrosine kinase phosphoprotein pp60c-SRC
(c-SRC) and its binding
partner, protein tyrosine kinase 2 beta (PTK2B).
Design: CD4+ T-lymphocytes were infected with R5 (JR-FL) or X4 (HXB2) HIV-1. SRC-
family kinase inhibitors or targeted siRNA knockdown of c-SRC and PTK2B were used and
various stages of the early HIV-1 lifecycle were examined.
Methods: Four SRC-family kinase inhibitors or targeted siRNA knockdown were used to reduce
c-SRC or PTK2B protein expression. Activated CD4+ T-lymphocytes were infected with
recombinant, nef-deficient, or replication-competent infectious viruses. Knockdown experiments
examined multiple stages of early infection by monitoring: luciferase activity, expression of host
surface receptors, reverse transcriptase activity, p24 levels and qPCR of reverse transcripts,
integrated HIV-1 and 2-LTR circles.
Results: All SRC-family kinase inhibitors inhibited R5 and X4 HIV-1 infection. Neither c-
SRC nor PTK2B siRNA knockdown had an effect on cell surface receptors (CD4, CXCR4,
CCR5) nor on reverse transcriptase activity. However, using JR-FL both decreased luciferase
activity while increasing late reverse transcripts (16-fold) and 2-LTR circles (8-fold) while also
decreasing viral integration (4-fold). With HXB2, c-SRC but not PTK2B siRNA knockdown
produced similar results.
Conclusions: Our results suggest c-SRC tyrosine kinase is a major regulator of HIV-1 infection,
participating in multiple stages of infection post-viral entry: Reduced proviral integration with
increased 2-LTR circles is reminiscent of integrase inhibitors used in combination antiretroviral
therapies. Decreasing c-SRC expression and/or activity provides a new target for antiviral
intervention and the potential for repurposing existing FDA-approved kinase inhibitors.

117
3.2 Introduction
To overcome HIV-1 drug resistance, targeting host T-cell factors critical to HIV-1 infection has
become a promising field of research [146]. Therapeutics designed to target host proteins are
being explored in conjunction with combination antiretroviral therapy (cART) to reduce the size
of the initial HIV-1 reservoir [549] or reactivate then purge HIV-1 in the cells of infected people
[550]. For example, some drugs disrupt allosteric interactions between viral integrase and host
proteins LEDGF/p75 [551], and other therapeutics can inhibit histone deacetylases to reactivate
provirus in latently infected cells [552]. However, inhibition of tyrosine kinases activated during
early HIV-1 infection remains an underexplored area of antiretroviral drug discovery. From
initial binding of viral glycoprotein 120 (gp120) to the T-lymphocyte receptor CD4 and
chemokine co-receptor CXCR4 (X4 viruses) or CCR5 (R5 viruses), tyrosine kinases are induced
that promote viral infection [147]. Moreover, non-receptor tyrosine kinases play critical roles
during the early stages of the HIV-1 lifecycle in T-cells (reviewed in [146]). Tyrosine
phosphorylation signaling is necessary for viral entry [135], actin remodeling [539] and
translocation of the viral pre-integration complex (PIC) into the nucleus [154, 158]. In
particular, HIV-1 infected CD4+ T-cells show striking changes in the activity of the SRC-family
of tyrosine kinases, which become activated within minutes of HIV-1 infection [214]. Previous
work suggests that dasatinib, a dual SRC/ABL ATP competitive inhibitor, approved for the
treatment of imatinib-resistant chronic myeloid leukemia [553], effectively inhibits HIV-1
production in activated CD4+ T-cells isolated from chronically infected HIV-1 patients [554].
However, a specific role for c-SRC at earlier time points, post initial infection up through
integration, has not been examined.
When HIV-1 initially attaches to the host T-cell membrane through its interaction with cell-
surface CD4, viral gp120 then binds the G protein-coupled receptor CXCR4/CCR5, inducing the
autophosphorylation of protein tyrosine kinase 2 beta (PTK2B, Pyk2 or FAK2) within the cell
[147]. PTK2B plays a key role in focal adhesions at the cell periphery and in cytoskeleton
remodeling [340]. Phosphorylation of PTK2B at Tyr402 can then recruit c-SRC (SRC, pp60c-
SRC) to its SRC-homology-2 (SH2) binding domain, permitting c-SRC to autophosphorylate at

118
Tyr419, activating c-SRC [197, 322]. Both of these kinases become phosphorylated within
minutes of HIV-1 infection [147, 214].
Previously, we have reported that inhibiting or reducing PTK2B or c-SRC in various T-cell lines
increased HIV-1 reverse transcription and integrated provirus [555]. However, another study has
shown that PTK2B shRNA knockdown in the T CEM cell line reduced viral p24 production after
six days of HIV-1NDK infection [345]. In addition, Gilbert et al. have demonstrated that
inhibiting c-SRC with the kinase inhibitor PP2 decreased p24 production in primary CD4+ T-
cells after six days of infection [264]. The discrepancy of PTK2B and c-SRC inhibition that
promotes early HIV-1 entry within hours, yet restricts virion maturation and p24 release within
days, remains unresolved. One possibility is that PTK2B and c-SRC function differently during
early HIV-1 entry in activated primary CD4+ T-lymphocytes than in immortalized T-cell lines.
To test this possibility, we hypothesized that inhibiting the non-receptor tyrosine kinases PTK2B
and c-SRC will restrict HIV-1 early entry in activated primary CD4+ T-cells.
We demonstrate that reducing c-SRC expression with siRNA restricted the infection of X4-tropic
HXB2 or R5-tropic JR-FL, Ba-L or KNH1207, within 12 hrs of infection. Interestingly, PTK2B
siRNA knockdown only reduced early infection of R5-tropic viruses, suggesting different
dependence on downstream signaling contingent on the co-receptor engaged during virus
binding and fusion. Our findings not only provide the rationale for exploring the use of drugs to
inhibit these two kinases as novel means to inhibit HIV-1 replication, but they may also suggest
a unique method for arresting HIV-1 at the stage of pre-integration complex (PIC) formation
and/or integration.

119
3.3 Materials and Methods
Cell Isolation and Culture Conditions: Peripheral blood mononuclear cells (PBMCs) were
isolated from the whole blood of healthy human donors (Canadian Blood Services Research
Ethics Board Committee Protocol Reference #2005-003) by the Ficoll gradient method,
according to manufacturer’s instructions (GE healthcare, Buckinghamshire, UK). Informed
written consent was obtained from all subjects. CD4+ T-cells were then isolated from PBMCs
using the EasySep negative selection human CD4+ T-cell enrichment kit, following the protocol
by StemCell Technologies (Vancouver, Canada). For all experiments, isolated CD4+ T-cells
were grown in complete RPMI-1640 media (Sigma-Aldrich, Oakville, Canada) containing 10%
fetal bovine serum, 1% penicillin, 1% streptomycin and 0.1% gentamycin. Human embryonic
kidney cells (HEK 293T, ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM,
Gibco, Burlington, Canada) with the same proportion of FBS and antibiotics as above. Cell
cultures were incubated at 37°C in 5% CO2 atmosphere. CD4+ T-cells were activated by the
addition of the following: 10 μg/mL anti-CD3 (UCHT-1, Antibody Core Facility, Sunnybrook
Research Institute, Toronto, Canada), 2 μg/mL anti-CD28 (10R-CD28bHU, Fitzgerald Industries
International, Acton, USA) and 10 units/mL recombinant human Interleukin-2 (IL-2, Sigma-
Aldrich).
Virus Production: We produced recombinant, replication deficient JR-FL enveloped (R5) or
HXB2 enveloped (X4) HIV-1 containing a luciferase gene, as described previously [555].
Briefly, 2.5 x 106 HEK 293T cells were plated with 10 mL of DMEM 24 hours prior to
transfection. We co-transfected 10 μg of HIV-1 NL4-3luc (luciferase gene inserted into viral nef
gene and env deficient; a kind gift from Dr. Veneet KewalRamani, New York Medical
University, NY) with 10 μg of JR-FL env plasmid (encodes the R5-tropic JR-FL glycoproteins; a
generous gift from Dr. M. Tremblay, Quebec City, PQ) into each 10 cm plate of HEK 293T cells
with the CalPhosTM
Mammalian Transfection Kit (Clontech, Mountain View, USA), following
the manufacturer’s instructions. This occurred in a biosafety level 3 (BSL3) lab. The same was
performed for the HXB2 plasmid (HIV-1 NL4-3luc lacking nef and containing HXB2 (X4) env;
another generous gift from Dr. M. Tremblay) at 15 μg per plate. DMEM media was replaced

120
after 24 hours and viral supernatant was collected on days 2 and 3. The supernatant was
syringed through 0.45 μm pore filters (Millipore, Billerica, USA) , then concentrated by
underlaying with a 20% sucrose gradient and centrifuging at 16,000 x g for 90 minutes at 4 °C.
Virus pellet were resuspended in TNE buffer (20 mM Tris-HCl, pH 7.5, 1 mM EDTA and 100
mM NaCl) and stored in -80°C aliquots. p24 levels were measured by ELISA (ZeptoMetrix,
Buffalo, USA). Replication competent IIIB (formerly HTLV-III B/H9), Ba-L (Reference ID:
85US_Ba-L) and KNH1207 (Reference ID: 00KE_KNH1207) viruses were acquired from the
NIH AIDS Research & Reference Reagent Program’s International panel of 60 HIV-1 isolates
(Germantown, USA). All viruses underwent one freeze-thaw cycle prior to infection studies.
HIV-1 Infection and Luciferase Activity: 2 x 105 CD4
+ T-cells pretreated with siRNA or drugs
were infected with 1.0 ng of HXB2, 26 ng of JR-FL, 1.1 ng IIIB, 43 ng of Ba-L or 16 ng of
KNH1207 at 37°C in 200 μL of clear growth media including activators in 96-well plates for 2
days. Cells were counted, while others were lysed with the Luciferase Assay System (Promega,
Madison, USA). Other CD4+ T-cells were infected with replication competent IIIB (X4), Ba-L
(R5) or KNH1207 (R5), and integrated copies of provirus were quantified by qPCR after 12 hrs
of infection.
HIV-1 p24 ELISA: 2 x 105 CD4
+ T-cells pretreated with siRNA were infected with IIIB or Ba-
L for 1 hr at 37°C, washed in PBS three times, and incubated in 200 μL complete RPMI media
supplemented with activators. Cell-free supernatant (180 μL) was collected on day 3 and assayed
for p24gag
antigen following the manufacturer’s instructions (ZeptoMetrix, Franklin, USA).
Drug Experiments: Enriched CD4+ T-cells were administered potent small molecule c-SRC
inhibitors. Cells were given complete media containing activators, and one of the four inhibitors:
dasatinib (BMS-354825), saracatinib (AZD0530), KX2-391 (Selleckchem, Houston, USA) or
SRC Inhibitor-1 (Sigma-Aldrich). DMSO solvent served as a negative control. Twenty-four
hours thereafter, cells were prepared for HIV-1 infection or FACS viability staining (Annexin V-
FITC Apoptosis Detection Kit I, BD Pharmingen, Mississauga, Canada) to measure apoptosis
and/or necrosis. In other experiments, enriched CD4+ T-cells were given 20 μM of raltegravir 24
hours prior to HIV-1 infection.

121
Flow Cytometry and Data Acquisition: All antibodies were purchased from BD Pharmingen.
Cell surface expression of CD3 and CD4 were determined on 1 x 106 CD4
+ T-cells after using
the EasySep negative selection human CD4+ T-cell enrichment kit (StemCell). In other
experiments, cell surface expression of CD4, CXCR4 and CCR5 were determined on 2 x 105
enriched CD4+ T-cells per tube, 48 hours after siRNA knockdown. In brief, cells were stained
with 5 μL of PE mouse anti-human CD3, 5 μL of APC mouse anti-human CD4, 5 μL of FITC
mouse anti-human CD4, 5 μL of PE mouse anti-human CXCR4, or 10 μL of Alexa Fluor®647
rat anti-human CCR5 in a 100 μL of FACS buffer (PBS + 2.5% FBS). Isotype control
antibodies were FITC-conjugated mouse IgG1κ, PE-conjugated mouse IgG1κ or IgG2aκ, APC-
conjugated mouse IgG1κ and Alexa Fluor®647-conjugated rat IgG2a. Data on 20,000 events per
sample was collected using the Becton Dickenson FACSCalibur flow cytometer (BD
Biosciences, Mississauga, Canada).
Antibodies and Western Blot: Activated CD4+ T-cells (1 x 10
6) infected with HXB2 or JR-FL
virus for 1 hr were washed in PBS and lysed with radioimmunoprecipitation assay (RIPA) lysis
buffer (50 mM HEPES, pH 7.3, 1% Nonidet P-40, 0.1% SDS, 0.1% sodium deoxycholate, 150
mM NaCl, 1 mM Na3VO4, 50 M ZnCl2, 2 mM EDTA and 2 mM PMSF). In other co-
immunoprecipitation experiments, 3x106 activated CD4
+ T-cells were infected with HXB2 or
JR-FL for 1 hr, lysed, and rotated at 4°C overnight with protein A/G PLUS-Agarose (Santa Cruz,
Dallas, USA) and primary anti-v-SRC clone 327 (Millipore). After boiling samples, proteins
from cell lysates were resolved on an 8%, reducing SDS-PAGE gel, and transferred to an
Immobilon-P membrane for Western Blot detection. The blots were blocked with 1x Tris-buffer
saline (TBS), 5% skim-milk powder and 0.5% Nonidet P-40 (BioShop, Burlington, Canada).
Primary antibodies were diluted in 5% milk and 0.5% Nonidet P-40. Anti-GAPDH (Millipore),
anti-PTK2B (BD Transduction Laboratories, Mississauga, Canada) and anti-c-SRC GD11
(Millipore) were used to probe the blots. To measure phospho-c-SRC (Tyr419, BD Transduction
Laboratories) or phospho-PTK2B (Tyr402, New England Biolabs, Whitby, Canada), blots were
stripped and then blocked with 1x TBS, 5% w/v BSA and 0.1% Tween-20. After washing,
secondary goat anti-mouse HRP (Bio-Rad, Mississauga, Canada) or goat anti-rabbit HRP (Bio-
Rad) antibody was added, followed by enhanced chemiluminescent (ECL) detection (GE

122
Healthcare). PTK2B and c-SRC protein levels were determined by comparing their densitometry
to normalizer GAPDH, with the Molecular Imager GelDoc XR+ Imagin System (Bio-Rad).
siRNA Knockdown Experiments: Pools of four siRNA duplexes, specific for human c-SRC
(cat # M-003175-03) or PTK2B (cat # M-003164-02) mRNA, and a control pool of non-
targeting siRNA (cat # D-001206-13-05) were ordered from Dharmacon RNAi technologies
(siGENOME SMART Pool, Thermo Scientific, Lafayette, USA). With enriched CD4+ T-cells,
siRNA knockdown was performed using the Amaxa® Human T-cell Nucleofector
® Kit (Lonza,
Cologne, Germany), following the manufacturer’s instructions. Enriched T-cells (1.0 x 107)
were resuspended with 100 μL of Nucleofector solution, 900 nM of siRNA and 2 μL of siGuard
RNase inhibitor® (Genlantis, San Diego, USA), then electroporated with program V-024. Cells
were grown in 2 mL media lacking activators in a 37°C incubator for 12 hr, spun down, and
resuspended in 2 mL complete media containing activators. Forty-eight hours post-transfection,
cells were prepared for HIV-1 infection, harvested to measure protein knockdown of either c-
SRC or PTK2B by Western blot densitometry, or prepared for FACS staining.
qPCR Amplification of HIV-1 cDNA: Genomic and viral cDNA was isolated from 5 x 105
CD4+ T-cells, pre-treated by siRNA transfection and infected with HXB2 or JR-FL virus for 1 or
12 hours, using the QIAamp DNA Blood Mini Kit (Qiagen, Toronto, Canada). Samples were
heated at 56°C for 2 hours in QIAamp DNA Blood Mini Kit buffer AL to deactivate viable viral
particles.
The primer sets used to detect human or viral DNA (synthesized by the Center for Applied
Genomics, The Hospital for Sick Children, Toronto, Canada) were as follows: β-globin forward,
5′-CCCTTGGACCCAGAGGTTCT -3′; β-globin reverse, 5′-CGAGCACTTTCT TGCCATGA-
3′; early reverse transcripts (early RT) forward, 5′-GTAACTAGAGATCCCTCAG
ACCCTTTTAG-3′; early RT reverse, 5′-TAGCAGTGGCGCCCGA-3′; late reverse transcripts
(late RT) forward, 5′-CCGTCTGTTGTGTGACTCTGG-3′; late RT reverse, 5′-GAGTCCTGCG
TCGAGAGATCT-3′; 2-LTR circles forward, 5′-CCCTCAGACCCTTTTAGTCAGTG-3′; 2-
LTR circles reverse, 5′-TGGTGTGTAGTTCTGCCAATCA-3′; genomic Alu forward, 5′-GCCT
CCCAAAG TGCTGGGATTACAG-3′; HIV-1 gag reverse, 5′-GCTCTCGCACCCATCTCTCT

123
CC-3′; HIV-1 LTR forward, 5′-GCCTCA ATAAAGCTTGCCTTGA-3′; HIV-1 LTR reverse, 5′-
TCCACACT GACTAAAAGGGTCTGA-3’ [159, 542, 543]. For each qPCR run, 50-100 ng
template DNA (with the exception of the no-template control, NTC) was added to PCR tubes
containing: 12.5 μL 2x SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK),
300-900 nM of each forward and reverse primers, and PCR grade H2O (Roche Diagnostics,
Indianapolis, USA) up to a final volume of 25 μL. PCR cycling parameters were as follows:
initial denaturation at 95°C for 10 min; 40-50 cycles of amplification of 95°C for 15 sec, 58°C
for 30 sec (β-globin or late RT, 54°C for early RT or 2-LTR) and 60°C for 30 sec. To measure
integrated virus by Alu-gag PCR [542], tubes were first pre-amplified with 100 nM Alu forward
and 600 nM gag reverse primers, with 2x SYBR Green PCR Master Mix as above, for 10 cycles
of 93°C for 30 sec, 50°C for 60 sec, and 60°C for 100 sec. Nested PCR was performed on these
samples by adding 300 nM each of HIV-1 LTR forward and reverse primers, with 2x SYBR
Green PCR Master Mix, for 40 more cycles using the above parameters. Duplicate reactions
were analyzed using the Rotor-Gene RG-3000 thermocycler (Corbett Research, Montreal,
Canada). Five biological replicates in the c-SRC and PTK2B siRNA-treated groups were
compared to the non-targeting siRNA-treated group by the 2-ΔΔCT
comparative CT method [544].
Absolute quantification was performed for PCR amplicons of integrated virus, relative to the
chronically infected 8E5 cell line carrying one integrated proviral copy (NIH AIDS Reagent
Program, Dr. Thomas Folks).
In-Vitro Reverse Transcriptase Assay: Quantification of reverse transcriptase activity from
infected cell lysates was performed using the SYBR Green I qPCR-based product-enhanced RT
(PERT) assay [556]. Briefly, 5 x 105 CD4
+ T-cells were infected with HXB2 or JR-FL virus for
1 hr, washed twice with PBS, lysed, diluted 10-fold with 10x sample dilution buffer, and given 1
μg of MS2 RNA template (Roche Diagnostics) to reverse transcribe in vitro. The cDNA product
was measured by qPCR, and a standard curve of recombinant HIV-1 RT (Calbiochem) was used
to assign enzyme activity units to reverse transcriptase from the infected cell lysate samples.
Statistical Analysis: Means were compared using a two-tailed, unpaired Student’s t test and
corrected for multiple comparisons when more than two means were considered in an
experiment, to minimize the probability of type 1 errors. FACS data shows the percent positive

124
cells collected with CellQuest software (BD Biosciences) and analyzed by FlowJo version X
software. For all figures, an asterisk (*) denotes a p value < 0.05, two asterisks (**) denotes a p
value < 0.01 and three asterisks (***) denotes a p value < 0.001. Error bars shown are the
standard error around the mean (SEM).

125
3.4 Results
Kinase inhibitors targeting c-SRC significantly reduce HXB2 or JR-FL infection in
activated CD4+ T-lymphocytes
To confirm and extend the finding that dasatinib inhibits HIV-1 production [554], we
investigated whether pharmacological inhibitors of the SRC-family kinases, approved or under
clinical investigation for the treatment of various cancers, could inhibit early HIV-1 infection of
X4 or R5 luciferase-reporter viruses. For this purpose we tested dasatinib, saracatinib, a
SRC/ABL ATP reversible inhibitor in a phase II clinical trial for the treatment of advanced
ovarian cancer (http://clinicaltrials.gov/ct2/show/NCT00610714); KX2-391, a SRC substrate
competitive inhibitor in a phase II clinical trial evaluating efficacy in gastric and breast cancers
(http://clinicaltrials.gov/ct2/show/NCT01764087); and SRC Inhibitor-1, a dual ATP and
substrate competitive inhibitor in pre-clinical studies [557].
We first isolated peripheral blood mononuclear cells (PBMCs) from the whole blood of healthy
human donors, and could enrich a CD3+CD4
+ cell population to 98% purity with a negative
selection human CD4+ T-cell enrichment kit (Fig. 3.1a). No increase in apoptosis or necrosis
was observed after treating activated CD4+ T-cells with the four SRC inhibitors at the
concentrations tested (Fig. 3.2). Following drug-treatment and activation of primary CD4+ T-
cells for 24 hrs, we measured a strong decrease in luciferase activity after two days of HXB2
(X4) or JR-FL (R5) viral infection (Fig. 3.3). The dasatinib treatment exhibited the greatest
potency at doses in the 10-100 nM range.
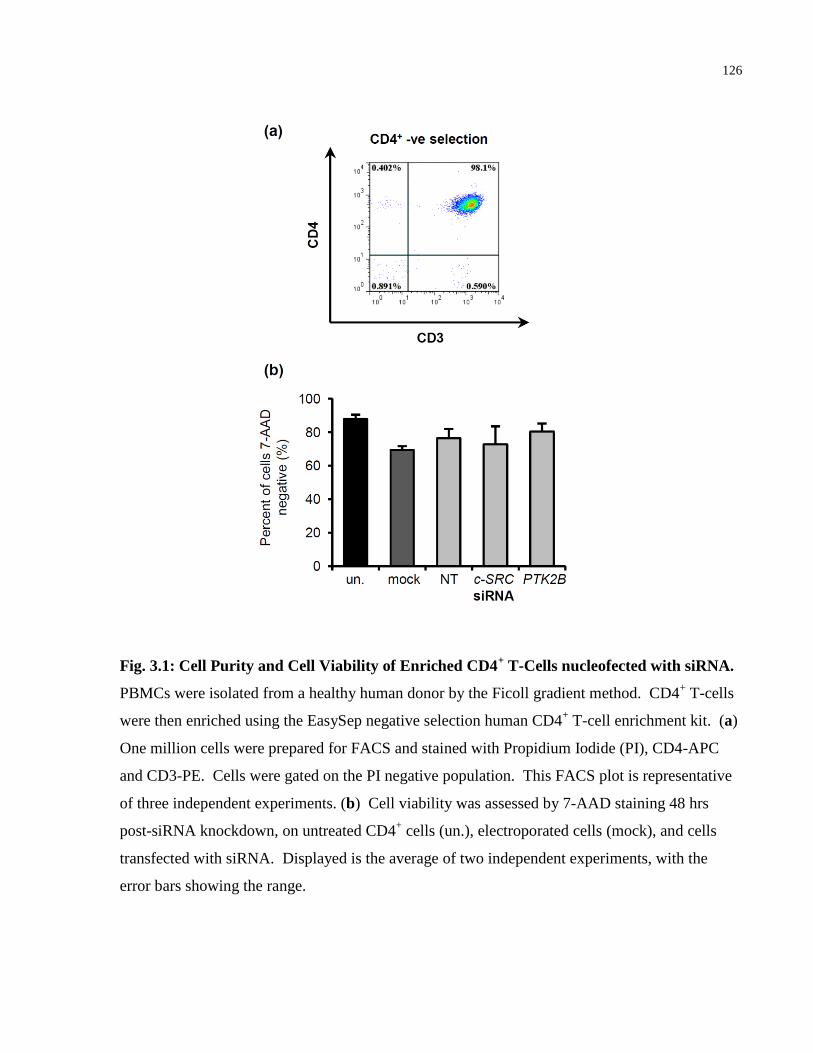
126
Fig. 3.1: Cell Purity and Cell Viability of Enriched CD4+ T-Cells nucleofected with siRNA.
PBMCs were isolated from a healthy human donor by the Ficoll gradient method. CD4+ T-cells
were then enriched using the EasySep negative selection human CD4+ T-cell enrichment kit. (a)
One million cells were prepared for FACS and stained with Propidium Iodide (PI), CD4-APC
and CD3-PE. Cells were gated on the PI negative population. This FACS plot is representative
of three independent experiments. (b) Cell viability was assessed by 7-AAD staining 48 hrs
post-siRNA knockdown, on untreated CD4+ cells (un.), electroporated cells (mock), and cells
transfected with siRNA. Displayed is the average of two independent experiments, with the
error bars showing the range.
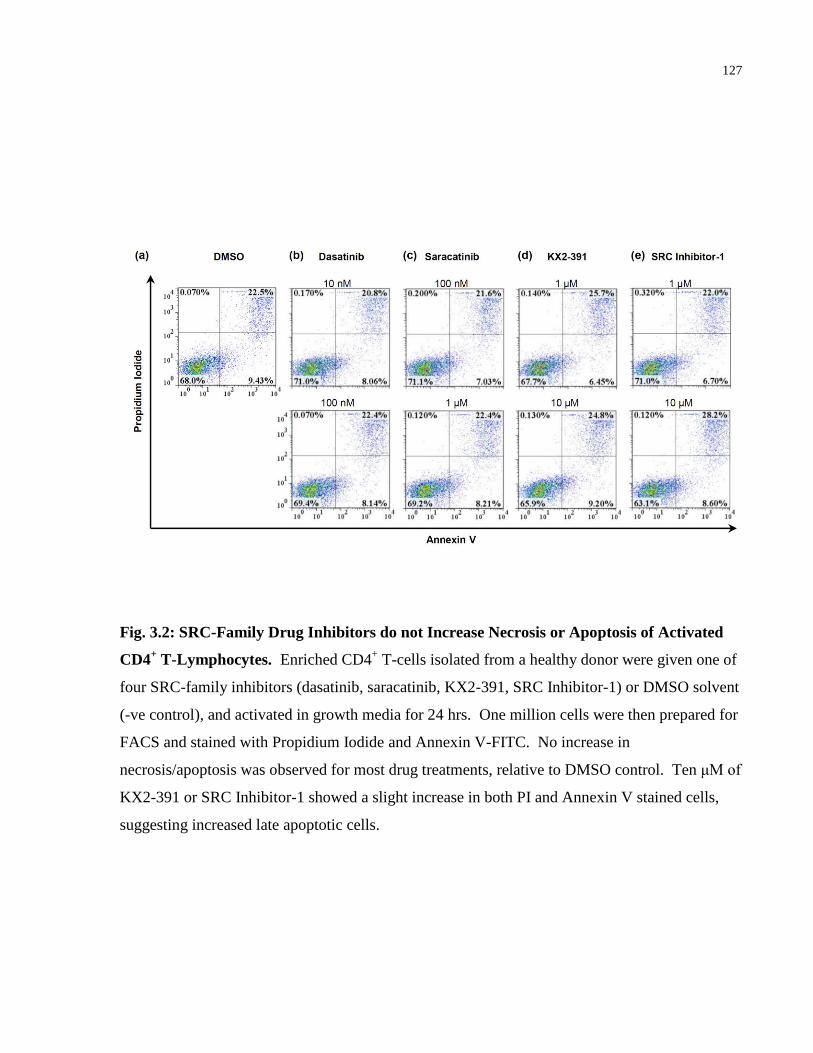
127
Fig. 3.2: SRC-Family Drug Inhibitors do not Increase Necrosis or Apoptosis of Activated
CD4+ T-Lymphocytes. Enriched CD4
+ T-cells isolated from a healthy donor were given one of
four SRC-family inhibitors (dasatinib, saracatinib, KX2-391, SRC Inhibitor-1) or DMSO solvent
(-ve control), and activated in growth media for 24 hrs. One million cells were then prepared for
FACS and stained with Propidium Iodide and Annexin V-FITC. No increase in
necrosis/apoptosis was observed for most drug treatments, relative to DMSO control. Ten μM of
KX2-391 or SRC Inhibitor-1 showed a slight increase in both PI and Annexin V stained cells,
suggesting increased late apoptotic cells.
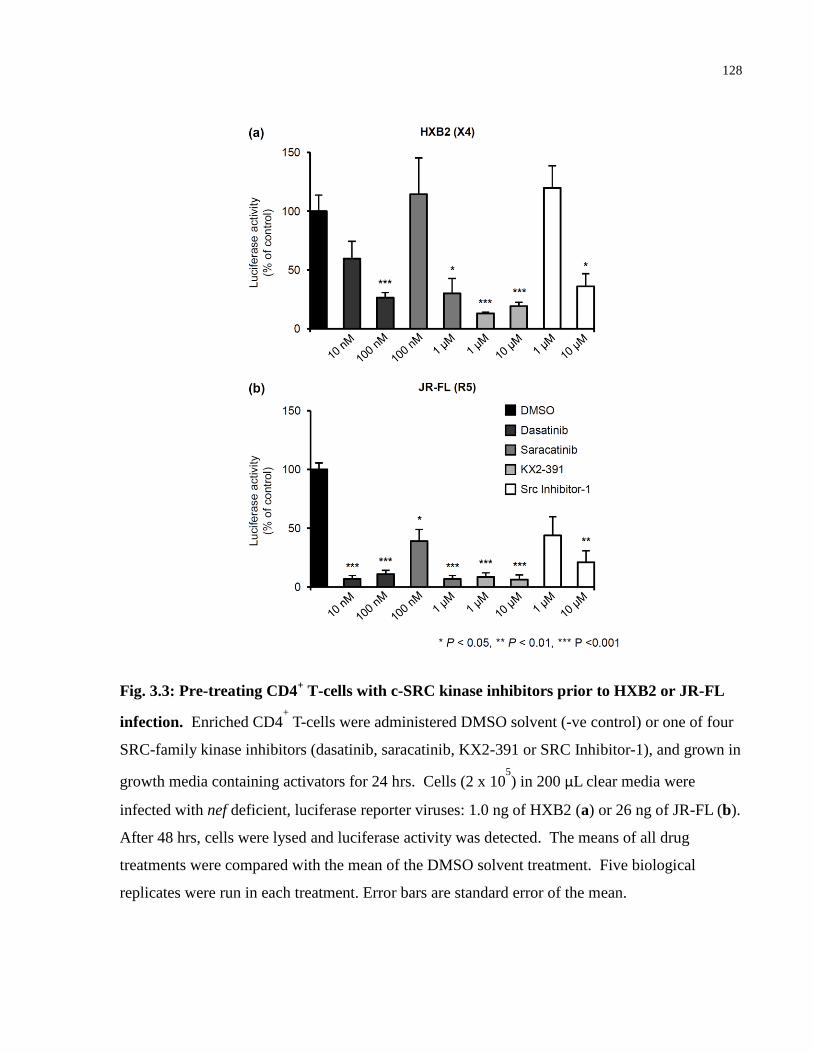
128
Fig. 3.3: Pre-treating CD4+ T-cells with c-SRC kinase inhibitors prior to HXB2 or JR-FL
infection. Enriched CD4+ T-cells were administered DMSO solvent (-ve control) or one of four
SRC-family kinase inhibitors (dasatinib, saracatinib, KX2-391 or SRC Inhibitor-1), and grown in
growth media containing activators for 24 hrs. Cells (2 x 105) in 200 μL clear media were
infected with nef deficient, luciferase reporter viruses: 1.0 ng of HXB2 (a) or 26 ng of JR-FL (b).
After 48 hrs, cells were lysed and luciferase activity was detected. The means of all drug
treatments were compared with the mean of the DMSO solvent treatment. Five biological
replicates were run in each treatment. Error bars are standard error of the mean.

129
c-SRC and PTK2B become activated and co-immunoprecipitate shortly after HXB2 or JR-
FL infection
Given the SRC-family kinase inhibitor findings, we asked whether c-SRC signaling via PTK2B
plays an important role during early viral entry. c-SRC protein expression was strongly induced
in CD4+ T-cells 48 hrs after stimulating with anti-CD28, anti-CD3 and IL-2 (Fig. 3.4a). PTK2B
expression remained unaffected during T-cell activation. Meanwhile c-SRC became
phosphorylated at Tyr419 1 hr following HXB2 (X4) or JR-FL (R5) infection, a key
phosphorylation step that leads towards high c-SRC activity [558]. PTK2B also became
phosphorylated at Tyr402 after 1 hr of infection, suggesting SRC-family phosphorylation of
PTK2B [559]. Immunoprecipitation of c-SRC followed by probing for phosphorylated PTK2B
revealed the two kinases strongly co-associate after 1 hr of HXB2 or JR-FL infection, and that
PTK2B has been cleaved into a smaller fragment (Fig. 3.4b).
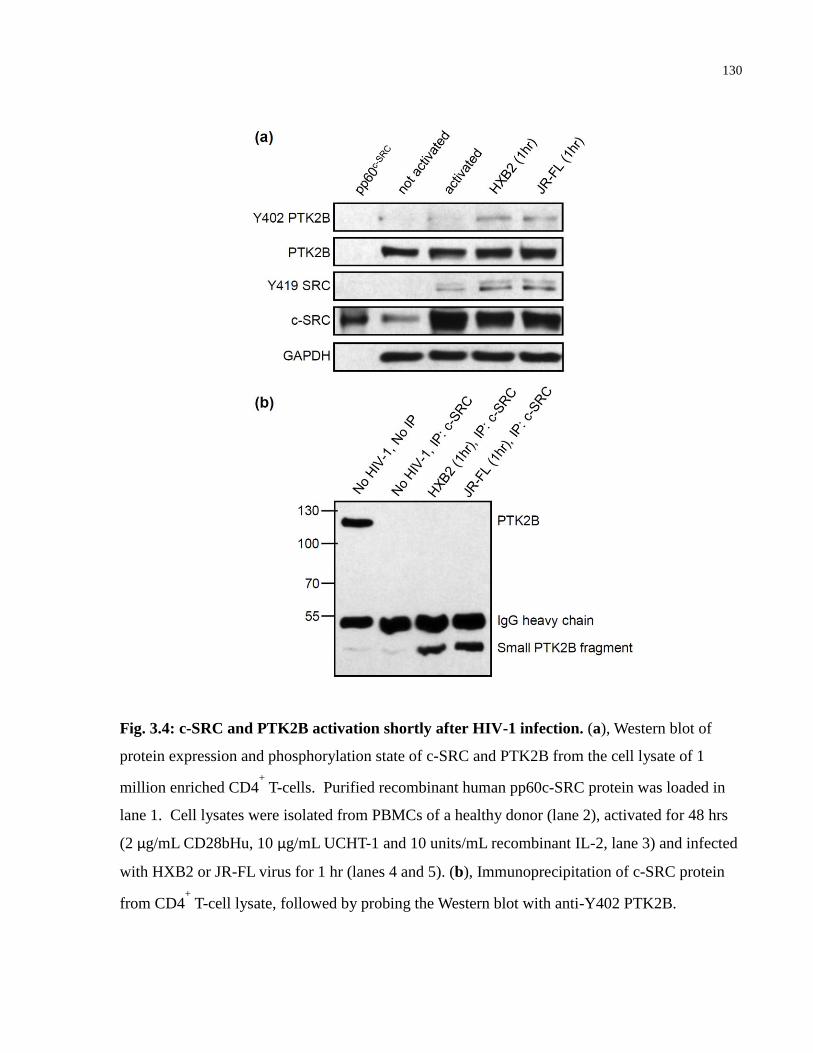
130
Fig. 3.4: c-SRC and PTK2B activation shortly after HIV-1 infection. (a), Western blot of
protein expression and phosphorylation state of c-SRC and PTK2B from the cell lysate of 1
million enriched CD4+ T-cells. Purified recombinant human pp60c-SRC protein was loaded in
lane 1. Cell lysates were isolated from PBMCs of a healthy donor (lane 2), activated for 48 hrs
(2 μg/mL CD28bHu, 10 μg/mL UCHT-1 and 10 units/mL recombinant IL-2, lane 3) and infected
with HXB2 or JR-FL virus for 1 hr (lanes 4 and 5). (b), Immunoprecipitation of c-SRC protein
from CD4+ T-cell lysate, followed by probing the Western blot with anti-Y402 PTK2B.

131
c-SRC and PTK2B siRNA knockdown reduce the infection of luciferase reporter viruses
and replication competent viruses
To test whether siRNA knockdown of either kinase reduces viral infection, we electroporated
siRNA into primary CD4+ T-cells, performed activation, and measured protein knockdown
48 hrs thereafter (Fig. 3.5a-b). We could consistently reduce c-SRC expression by 40% and
PTK2B expression by 80%. Cell viability at this time-point ranged from 70-80% (Fig. 3.1b). We
then infected the siRNA-transfected, activated CD4+ T-cells with HXB2 virus, and observed
decreased luciferase activity in c-SRC siRNA-treated, but not in PTK2B siRNA-treated cells,
relative to the non-targeting siRNA control (Fig. 3.5c). In contrast, siRNA knockdown of either
c-SRC or PTK2B reduced the luciferase activity following JR-FL viral infection (Fig. 3.5d).
This suggests that the co-receptor engaged upon entry (CXCR4 or CCR5) affects downstream
signaling events, with c-SRC common to the entry of both viruses tested and PTK2B playing a
role primarily during JR-FL (R5) infection. Further supporting this evidence, we also found that
knockdown of only c-SRC reduced viral integration of replication competent X4 IIIB, while
knockdown of either kinase reduced integration of R5 viruses Ba-L and clinical isolate
KNH1207 (Fig. 3.5e). Indeed, the unique response of X4 and R5 viruses to c-SRC or PTK2B
siRNA also affected the later stages of the replication cycle of IIIB or Ba-L, as determined by
p24 detected in cell-free supernatant three days post-infection (Fig. 3.5f-g).
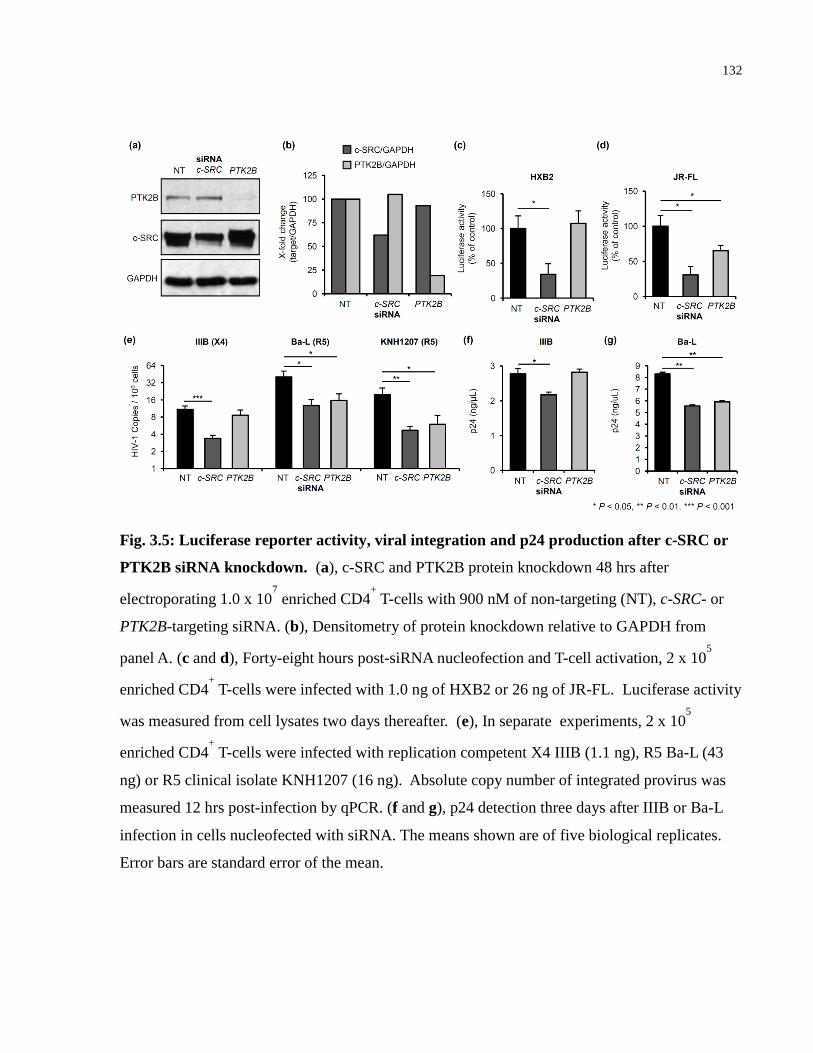
132
Fig. 3.5: Luciferase reporter activity, viral integration and p24 production after c-SRC or
PTK2B siRNA knockdown. (a), c-SRC and PTK2B protein knockdown 48 hrs after
electroporating 1.0 x 107 enriched CD4
+ T-cells with 900 nM of non-targeting (NT), c-SRC- or
PTK2B-targeting siRNA. (b), Densitometry of protein knockdown relative to GAPDH from
panel A. (c and d), Forty-eight hours post-siRNA nucleofection and T-cell activation, 2 x 105
enriched CD4+ T-cells were infected with 1.0 ng of HXB2 or 26 ng of JR-FL. Luciferase activity
was measured from cell lysates two days thereafter. (e), In separate experiments, 2 x 105
enriched CD4+ T-cells were infected with replication competent X4 IIIB (1.1 ng), R5 Ba-L (43
ng) or R5 clinical isolate KNH1207 (16 ng). Absolute copy number of integrated provirus was
measured 12 hrs post-infection by qPCR. (f and g), p24 detection three days after IIIB or Ba-L
infection in cells nucleofected with siRNA. The means shown are of five biological replicates.
Error bars are standard error of the mean.

133
siRNA knockdown of c-SRC or PTK2B did not reduce surface expression of receptors
required for viral entry or reverse transcriptase activity
We then determined whether c-SRC or PTK2B siRNA knockdown altered the surface expression
of CD4 or the chemokine co-receptors, CXCR4 or CCR5, which are necessary for HIV-1
binding and fusion. Surface expression of CD4, CXCR4 and CCR5 was not affected by siRNA
knockdown of either kinase (Fig.3.6a-b). We then infected the siRNA-transfected, activated
CD4+ T-cells with HXB2 or JR-FL virus. After measuring reverse transcriptase (RT) activity
1 hr post-viral infection, we found no significant changes in RT activity following c-SRC or
PTK2B siRNA knockdown, relative to the non-targeting siRNA control (Fig. 3.6c-d).

134
Fig. 3.6: Cell surface receptor expression and RT activity. (a and b), T-cell surface expression
of CD4, CXCR4 and CCR5 48 hrs post-siRNA nucleofection, gating on 7-AAD- cells. (c and d),
5 x 105 siRNA-treated and activated CD4
+ T-cells were infected with 1.0 ng of HXB2 or 26 ng of
JR-FL in 200 μL of growth media containing activators. Cell lysates were collected after 1 hr to
measure in vitro reverse transcriptase activity. The means shown are of five biological replicates.
Error bars are standard error of the mean.

135
Suppressing c-SRC or PTK2B with siRNA reduced viral integration and increased late
reverse transcripts and 2-LTR circles
Next we explored at which stage following reverse transcription c-SRC and PTK2B are exerting
their effects, by measuring viral synthesis of early reverse transcripts (Early RT), late reverse
transcripts (Late RT), integrated virus in the host genome, and two-long terminal repeat (2-LTR)
circular DNA, which are indicative of failed integration [56]. Primer efficiency curves, time-
course experiments to optimally measure viral cDNA, and qPCR product melting curves are
presented in Figs. 3.7 and 3.8. We observed a 4-fold reduction in integration of either HXB2 or
JR-FL viruses in the c-SRC siRNA-treated cells (Fig. 3.9a-b). Surprisingly, this was associated
with increased early reverse transcripts, late reverse transcripts, as well as 2-LTR circles. These
experiments are the average of three separate donors (see Fig. 3.10 for the three independent
experiments). PTK2B siRNA knockdown also showed a similar trend as c-SRC siRNA
knockdown during JR-FL infection, but not with HXB2 infection. This is consistent with the
luciferase findings in Fig. 3.5c-d. The qPCR data suggest multiple interactions of both kinases
during HIV-1 entry, as we observed decreased integration that coincides with increased early
transcripts, late reverse transcripts and 2-LTR circles. The increase in late reverse transcripts
caused by c-SRC or PTK2B siRNA knockdown was not due to increased reverse transcription
(Fig. 3.6c-d), but rather, taken together with the observed increase in 2-LTR circles and decrease
in integrated provirus, that both kinases likely have a role in pre-integration complex (PIC)
formation or nuclear import of viral cDNA. Their nuclear affects on 2-LTR circles and
integration are akin to the integrase inhibitor raltegravir in HXB2 or Ba-L infection (Fig. 3.9c).
Interestingly, JR-FL virus appears resistant to high-dose raltegravir treatment, despite being
susceptible to c-SRC or PTK2B siRNA knockdown and SRC-family kinase inhibitors (Fig.
3.1b).
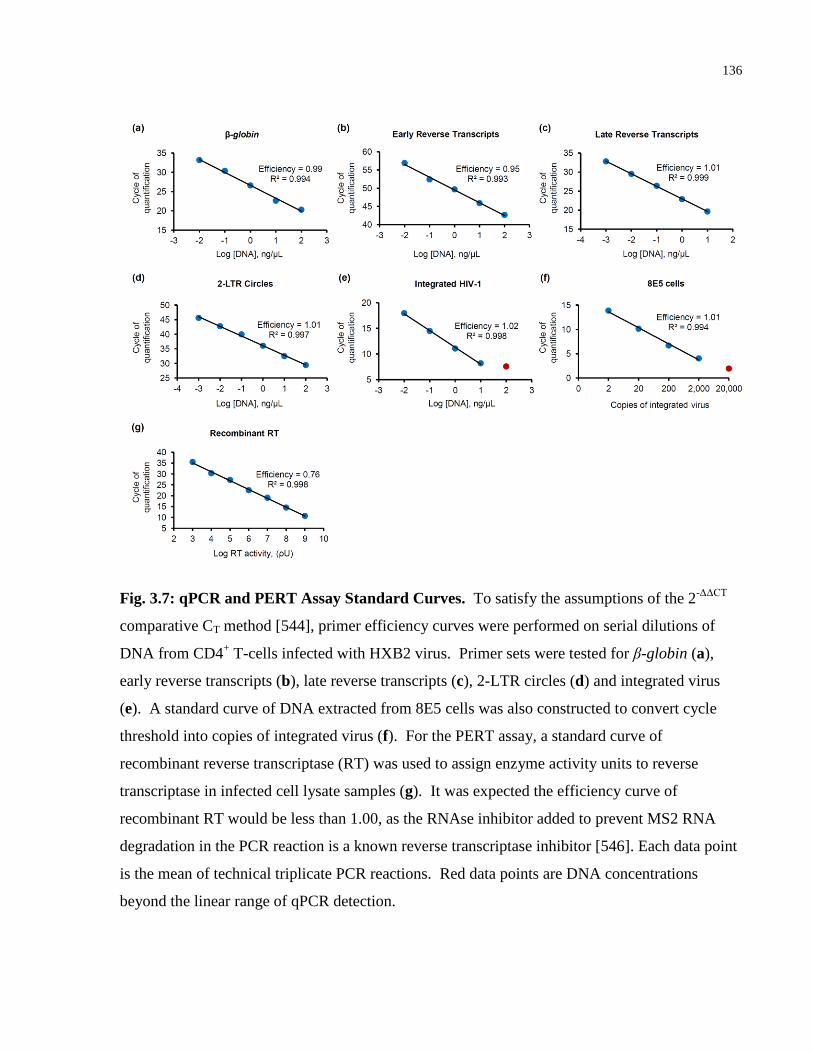
136
Fig. 3.7: qPCR and PERT Assay Standard Curves. To satisfy the assumptions of the 2-ΔΔCT
comparative CT method [544], primer efficiency curves were performed on serial dilutions of
DNA from CD4+ T-cells infected with HXB2 virus. Primer sets were tested for β-globin (a),
early reverse transcripts (b), late reverse transcripts (c), 2-LTR circles (d) and integrated virus
(e). A standard curve of DNA extracted from 8E5 cells was also constructed to convert cycle
threshold into copies of integrated virus (f). For the PERT assay, a standard curve of
recombinant reverse transcriptase (RT) was used to assign enzyme activity units to reverse
transcriptase in infected cell lysate samples (g). It was expected the efficiency curve of
recombinant RT would be less than 1.00, as the RNAse inhibitor added to prevent MS2 RNA
degradation in the PCR reaction is a known reverse transcriptase inhibitor [546]. Each data point
is the mean of technical triplicate PCR reactions. Red data points are DNA concentrations
beyond the linear range of qPCR detection.
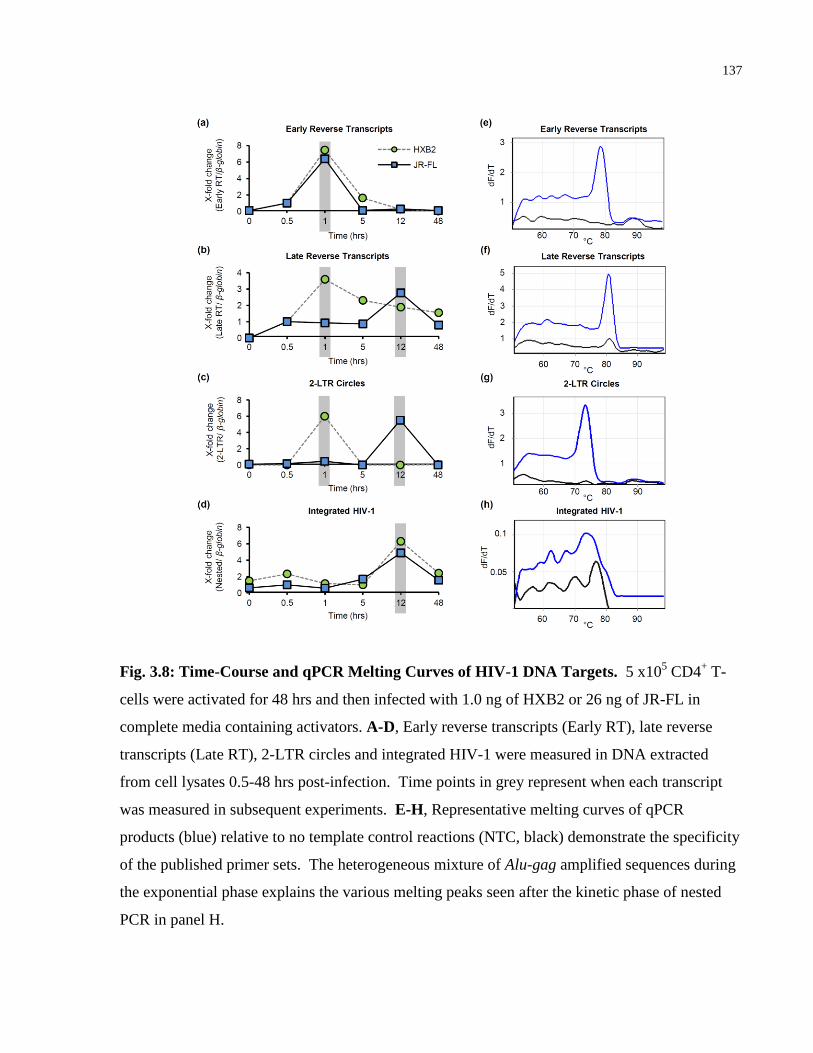
137
Fig. 3.8: Time-Course and qPCR Melting Curves of HIV-1 DNA Targets. 5 x105 CD4
+ T-
cells were activated for 48 hrs and then infected with 1.0 ng of HXB2 or 26 ng of JR-FL in
complete media containing activators. A-D, Early reverse transcripts (Early RT), late reverse
transcripts (Late RT), 2-LTR circles and integrated HIV-1 were measured in DNA extracted
from cell lysates 0.5-48 hrs post-infection. Time points in grey represent when each transcript
was measured in subsequent experiments. E-H, Representative melting curves of qPCR
products (blue) relative to no template control reactions (NTC, black) demonstrate the specificity
of the published primer sets. The heterogeneous mixture of Alu-gag amplified sequences during
the exponential phase explains the various melting peaks seen after the kinetic phase of nested
PCR in panel H.
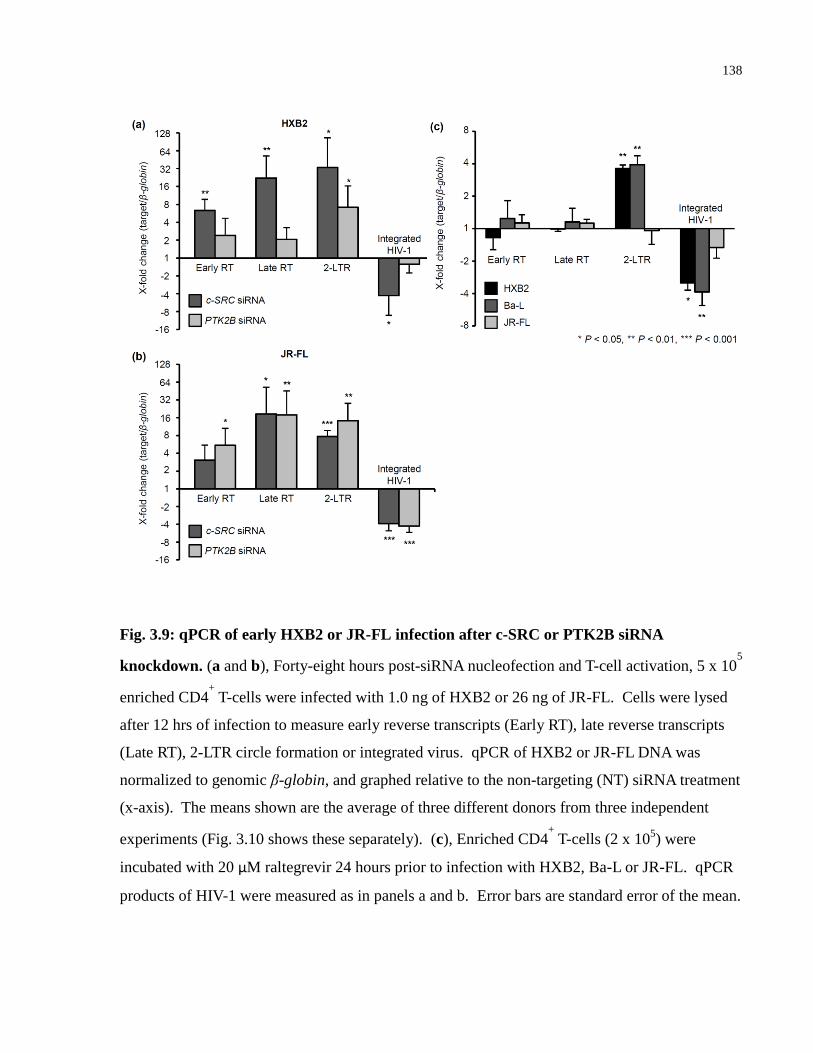
138
Fig. 3.9: qPCR of early HXB2 or JR-FL infection after c-SRC or PTK2B siRNA
knockdown. (a and b), Forty-eight hours post-siRNA nucleofection and T-cell activation, 5 x 105
enriched CD4+ T-cells were infected with 1.0 ng of HXB2 or 26 ng of JR-FL. Cells were lysed
after 12 hrs of infection to measure early reverse transcripts (Early RT), late reverse transcripts
(Late RT), 2-LTR circle formation or integrated virus. qPCR of HXB2 or JR-FL DNA was
normalized to genomic β-globin, and graphed relative to the non-targeting (NT) siRNA treatment
(x-axis). The means shown are the average of three different donors from three independent
experiments (Fig. 3.10 shows these separately). (c), Enriched CD4+ T-cells (2 x 10
5) were
incubated with 20 μM raltegrevir 24 hours prior to infection with HXB2, Ba-L or JR-FL. qPCR
products of HIV-1 were measured as in panels a and b. Error bars are standard error of the mean.
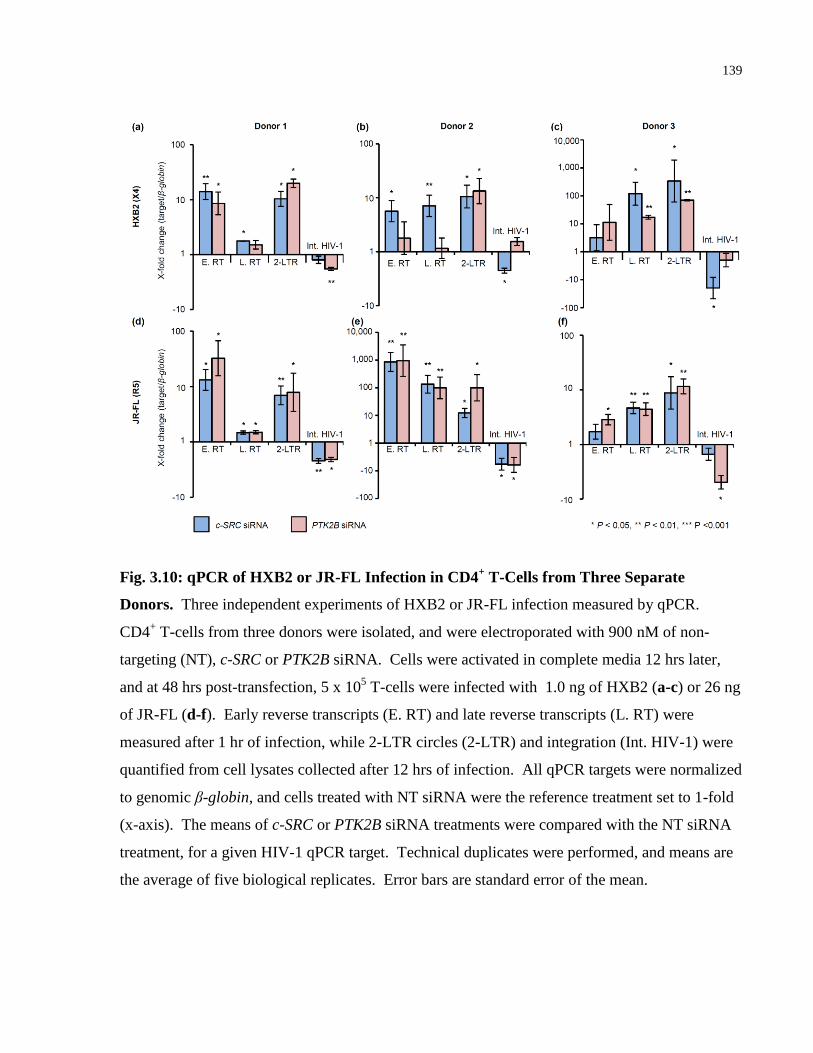
139
Fig. 3.10: qPCR of HXB2 or JR-FL Infection in CD4+ T-Cells from Three Separate
Donors. Three independent experiments of HXB2 or JR-FL infection measured by qPCR.
CD4+ T-cells from three donors were isolated, and were electroporated with 900 nM of non-
targeting (NT), c-SRC or PTK2B siRNA. Cells were activated in complete media 12 hrs later,
and at 48 hrs post-transfection, 5 x 105 T-cells were infected with 1.0 ng of HXB2 (a-c) or 26 ng
of JR-FL (d-f). Early reverse transcripts (E. RT) and late reverse transcripts (L. RT) were
measured after 1 hr of infection, while 2-LTR circles (2-LTR) and integration (Int. HIV-1) were
quantified from cell lysates collected after 12 hrs of infection. All qPCR targets were normalized
to genomic β-globin, and cells treated with NT siRNA were the reference treatment set to 1-fold
(x-axis). The means of c-SRC or PTK2B siRNA treatments were compared with the NT siRNA
treatment, for a given HIV-1 qPCR target. Technical duplicates were performed, and means are
the average of five biological replicates. Error bars are standard error of the mean.
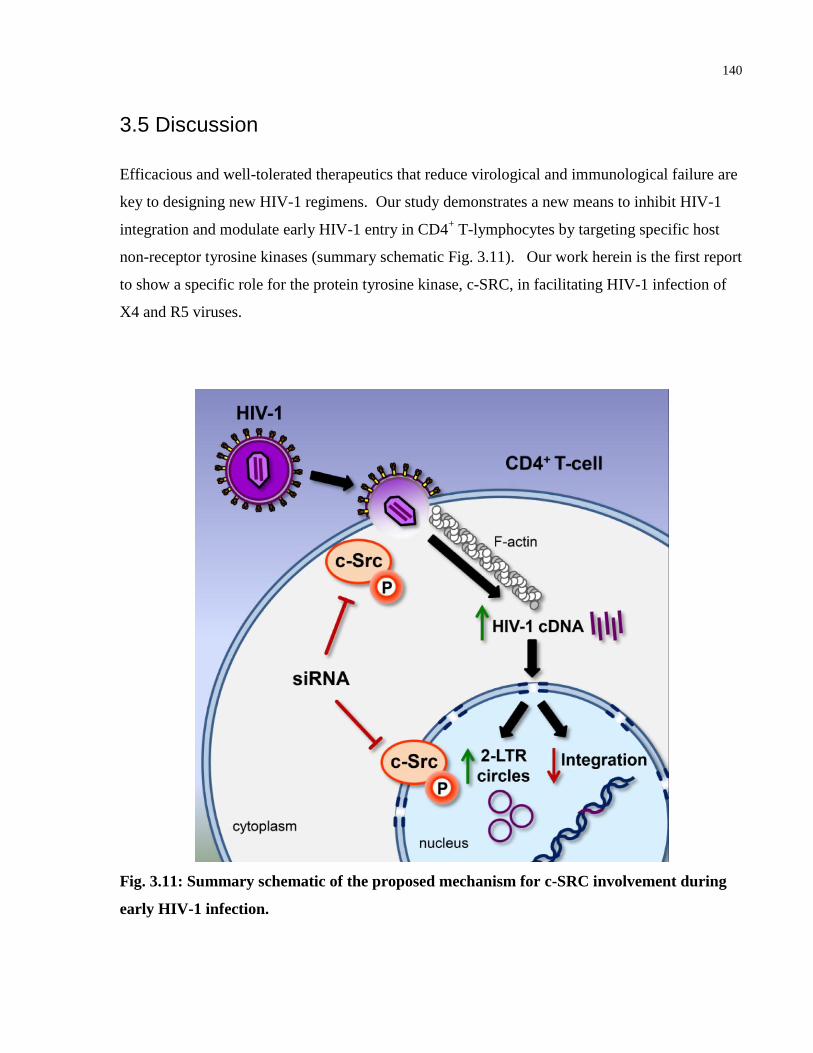
140
3.5 Discussion
Efficacious and well-tolerated therapeutics that reduce virological and immunological failure are
key to designing new HIV-1 regimens. Our study demonstrates a new means to inhibit HIV-1
integration and modulate early HIV-1 entry in CD4+ T-lymphocytes by targeting specific host
non-receptor tyrosine kinases (summary schematic Fig. 3.11). Our work herein is the first report
to show a specific role for the protein tyrosine kinase, c-SRC, in facilitating HIV-1 infection of
X4 and R5 viruses.
Fig. 3.11: Summary schematic of the proposed mechanism for c-SRC involvement during
early HIV-1 infection.

141
We first demonstrate that four SRC-family kinase inhibitors reduced HXB2 or JR-FL infection
by as much as 90% in activated CD4+ T-cells (Fig. 3.3). The diversity of mechanisms by which
these four drugs inhibit c-SRC, and differential on- and off-target efficacy, provide a strong
rationale for testing these inhibitors for their ability to reduce in vivo HIV-1 infection in a
humanized mouse model. While preclinical-stage inhibitors targeting other host kinases have
been found to influence viral infection (c-Jun N-terminal Kinase (JNK) inhibitors that reduce
integrase stability, protein kinase C agonists that reactivate latent HIV-1 provirus to purge latent
reservoirs [560]), our study is the first to describe mechanisms by which clinically approved (in
the case of dasatinib), safe, and well-tolerated SRC-family inhibitors act on early HIV-1
infection, suggesting these inhibitors could be promptly repurposed for potential treatment of
HIV-1 infection. For instance, a 75 nM dose of dasatinib has recently been shown to strongly
inhibit p24 production in activated CD4+ T-cells cultured from HIV
+, treatment-naïve patients
[554]. This dasatinib concentration was below the well-tolerated in vivo level of 180 nM [561],
and Pogliaghi et al. reported no increase in cell toxicity in cultured CD4+ T-cells [554].
Moreover, dasatinib has recently been shown to synergistically enhance the antiviral properties
of sofosbuvir, further inhibiting in vitro hepatitis C infection [532]. It also has potency in
inhibiting dengue virus 2 RNA replication by inhibiting the SRC-family kinase member FYN
[178].
To investigate potential targets of these four SRC kinase inhibitors, we demonstrated c-SRC and
its binding partner PTK2B become activated within 1 hr of HXB2 or JR-FL infection, and that
phosphorylated PTK2B co-immunoprecipitates with c-SRC after infection (Fig. 3.4). Activity of
calpains, which are Ca2+-dependent proteases known to cleave PTK2B at focal adhesions [336],
explains why a smaller PTK2B fragment immunoprecipitated with c-SRC in Fig. 3.4B. The
activation or autophosphorylation of cleaved PTK2B at tyrosine 402 may be induced in response
to chemokine engagement [147] or activation of the purinergic receptor P2Y2 during early HIV-
1 infection [345]. This may form a signaling complex that contains c-SRC and actin interacting
proteins that facilitate early HIV-1 entry and subsequent formation of the reverse transcriptase
complex in the cytosol. We then showed that reducing c-SRC protein with targeted-siRNA
reduced the luciferase activity of HXB2 and JR-FL viruses, and decreased the copy number of

142
integrated IIIB, Ba-L or KNH1207 provirus (Fig. 3.5). c-SRC siRNA knockdown affects HIV-1
infection at a stage following reverse transcription and prior to genomic integration (Fig. 3.6 and
3.9). We also show a similar finding with PTK2B siRNA knockdown, reducing JR-FL luciferase
reporter activity and viral integration (Fig. 3.5d and 3.9b). However, PTK2B siRNA knockdown
had little effect on the entry of the X4 viruses HXB2 or IIIB (Fig. 3.5c, 3.5e and 3.9a). While
PTK2B activation has been reported to be downstream of both CXCR4 and CCR5 co-receptor
engagement with gp120/gp41 in a T-cell line [147], our finding suggests that PTK2B signaling is
more dispensable during HXB2 (X4) infection in primary CD4+ T-cells. It is possible that
engagement of CXCR4, abundantly expressed on activated CD4+ T-cells compared to CCR5
(Fig. 3.6a-b), may provide a more robust signal that causes increased activation of c-SRC
independently of PTK2B activation.
It is intriguing that reducing either kinase had no effect on JR-FL reverse transcription (Fig.
3.6d) yet inhibited viral integration (Fig. 3.9b). Meanwhile, the same siRNA knockdown
conditions increased reverse transcription and viral integration in our previous report on
transformed T-cell lines [555]. Jurkat T-cells show constitutive c-SRC and PTK2B expression
and activation, as well as other signaling abnormalities downstream of the T-cell receptor [562],
suggesting a very different intracellular environment compared with activated primary CD4+ T-
cells. Indeed, our work is adding to a growing body of evidence that the involvement of receptor
and non-receptor tyrosine kinases during HIV-1 infection is highly dependent on the cell model
investigated [289, 563, 564], and that transformed cell lines may not be physiologically relevant
models of signal transduction during in vivo HIV-1 infection.
Our findings also suggest c-SRC signaling is a common feature of the early HIV-1 life cycle, and
that its interaction with viral and host proteins during PIC formation, viral cDNA nuclear
transport and genomic integration warrants further research. In light of the 20-fold increase in
late reverse transcripts following c-SRC siRNA knockdown (Fig. 3.9a), our findings offer the
potential to advance the study of host proteins integral to the PIC. Cellular kinases have been
reported to directly phosphorylate viral PIC proteins (reviewed in [155]) and assist with
integration into the host genome [154, 159]. Moreover, the identities of tyrosine kinases that
phosphorylate known viral constituents of the PIC remain unknown. For instance, the tyrosine

143
kinases that phosphorylate matrix protein p17 at C-terminal Y132 and preferentially target
matrix protein to the nucleus, have yet to be identified [158]. Future experiments using
knockdown of c-SRC or PTK2B could reveal whether these tyrosine kinases directly
phosphorylate host or viral PIC proteins, and could also uncover novel protein interactions for
pharmacological intervention. Whether nuclear c-SRC directly participates in HIV-1 integration
warrants further research. Moreover, c-SRC siRNA knockdown produced a viral qPCR signal
reminiscent of integrase inhibitors (Fig. 3.9c): reduced viral integration and increased 2-LTR
circles [565-567]. siRNA targeting PTK2B has also been shown to inhibit mutated HIV-1
strains that are resistant to reverse transcriptase or integrase inhibitors [345]. The unexpected
finding that JR-FL was resistant to raltegravir treatment opens up the possibility of c-SRC or
PTK2B inhibitors substituting for integrase inhibitors when combination therapies fail due to
integrase mutations. This warrants further experiments in humanized animal models to explore
how these drugs can improve existing HIV-1 treatment regimens.
In conclusion, our findings demonstrate the importance of host non-receptor tyrosine kinases in
the early stages of HIV-1 entry, specifically c-SRC and to a lesser extent PTK2B. Furthermore,
we demonstrate that these kinases can have multifunctional roles when interacting with the virus
at various stages of viral entry. The mechanism by which reducing or inhibiting c-SRC or
PTK2B restricts viral integration remains to be explored. Nonetheless, the availability of well-
studied SRC-family kinase inhibitors of known efficacy, specificity, pharmacokinetics and
toxicity profile, opens up new avenues for repurposing these drugs in combination with known
antivirals as novel means to inhibit HIV-1 and other viral infections. In particular, these may
prove useful for patients with contraindications for current HIV-1 drug combinations.

144
Chapter 4: A Rapid Screening Assay Identifies
Monotherapy with Interferon-ß and Combination Therapies
with Nucleoside Analogues as Effective Inhibitors of Ebola
Virus
Role of author McCarthy S.D.: Illustrated Figure 4.1, and performed all experiments for Figures
4.2, 4.3 and Figures 4.5-4.8. I measured % inhibition for the majority of Figure 4.4. I also
performed all experiments for Tables 4.1 and 4.2. I analyzed all the data, co-wrote the
manuscript draft, and performed experiments to address reviewer’s comments.
A version of this chapter was published in the Journal PLoS Neglected Tropical Diseases:
McCarthy, S.D., Majchrzak-Kita, B., Racine, T., Kozlowski, H.N., Baker, D.P., Hoenen, T.,
Kobinger, G.P., Fish, E.N., and Branch, D.R. 2016. A Rapid Screening Assay Identifies
Monotherapy with Interferon-ß and Combination Therapies with Nucleoside Analogues as
Effective Inhibitors of Ebola Virus. PLoS NTD, 10(1):e0004364.

145
4.1 Abstract
To date there are no approved antiviral drugs for the treatment of Ebola virus disease (EVD).
While a number of candidate drugs have shown limited efficacy in vitro and/or in non-human
primate studies, differences in experimental methodologies make it difficult to compare their
therapeutic effectiveness. Using an in vitro model of Ebola Zaire replication with transcription-
competent virus like particles (trVLPs), requiring only level 2 biosafety containment, we
compared the activities of the type I interferons (IFNs) IFN-α and IFN-ß, a panel of viral
polymerase inhibitors (lamivudine (3TC), zidovudine (AZT) tenofovir (TFV), favipiravir (FPV),
the active metabolite of brincidofovir, cidofovir (CDV)), and the estrogen receptor modulator,
toremifene (TOR), in inhibiting viral replication in dose-response and time course studies. We
also tested 28 two- and 56 three-drug combinations against Ebola replication. IFN-α and IFN-ß
inhibited viral replication 24 hours post-infection (IC50 0.038 µM and 0.016 µM, respectively).
3TC, AZT and TFV inhibited Ebola replication when used alone (50-62%) or in combination
(87%). They exhibited lower IC50 (0.98-6.2 μM) compared with FPV (36.8 μM), when
administered 24 hours post-infection. Unexpectedly, CDV had a narrow therapeutic window
(6.25-25 μM). When dosed > 50 µM, CDV treatment enhanced viral infection. IFN-ß exhibited
strong synergy with 3TC (97.3% inhibition) or in triple combination with 3TC and AZT (95.8%
inhibition). This study demonstrates that IFNs and viral polymerase inhibitors may have utility in
EVD. We identified several 2 and 3 drug combinations with strong anti-Ebola activity,
confirmed in studies using fully infectious ZEBOV, providing a rationale for testing combination
therapies in animal models of lethal Ebola challenge. These studies open up new possibilities for
novel therapeutic options, in particular combination therapies, which could prevent and treat
Ebola infection and potentially reduce drug resistance.

146
4.2 Introduction
As of December 13, 2015, the current outbreak of Ebola virus disease (EVD) in West Africa
resulted in 28,633 cumulative cases and 11,314 deaths [568]. Two potential vaccine candidates,
rVSV∆G-ZEBOV and ChAd3-EBO Z, have shown durable protection from lethal Ebola
challenge in mice [569] and macaques [32] respectively, and are part of the phase II/III
PREVAIL trial in Liberia and Guinea (https://clinicaltrials.gov/ct2/show/NCT02344407). Other
potential therapeutics, such as convalescent plasma and the antibody cocktail ZMapp [30] have
been approved for an emergency phase II/III trial in Guinea
(https://clinicaltrials.gov/ct2/show/NCT02342171) and a phase I trial in Liberia
(https://clinicaltrials.gov/ct2/show/NCT02363322), respectively. However, to date there is no
licensed vaccine or treatment for EVD, although improvements in supportive care are increasing
survival rates [570].
Repurposing antivirals used for other viral infections, based on knowledge of mechanisms of
action, has prompted accumulating interest in the application of different nucleoside/nucleotide
analogues and type I interferons (IFNs) for the treatment of Ebola virus disease (EVD).
Experimental nucleoside analogues may have therapeutic efficacy for EVD, given the evidence
of protection in primate and rodent disease models, 2-6 days after lethal Ebola or the related
hemorrhagic Marburg virus challenges [23, 418]. Favipiravir (FPV), a viral polymerase inhibitor,
provides 100% protection when administered 6 days after challenge with a lethal dose of Ebola
virus [23] and has been evaluated in the phase II/III JIKI trial in Guinea
(https://clinicaltrials.gov/ct2/show/NCT02329054). TKM-Ebola, a cocktail of siRNAs targeting
VP35 and L polymerase and brincidofovir (BCV), a viral polymerase inhibitor that has activity
against dsDNA viruses such as adenovirus and cytomegalovirus [571], were also considered for
treatment against EVD. The brincidofovir trial was halted, ostensibly because of projections of
low recruitment.
Despite infecting different target cells, Ebola and HIV-1 share many similar features early in
their replication cycle. Both are RNA viruses that package a viral polymerase (L for Ebola, RT
for HIV-1) required for early replication in the cytosol of the host cell [572]. Homology-based

147
structural prediction of the RNA-dependent RNA polymerase of Ebola indicates the polymerase
contains conserved structural motifs in the catalytic palm subdomain similar to viral DNA
polymerases [455], supportive of nucleoside analogues potentially inhibiting Ebola replication.
Inhibiting HIV-1 reverse transcription with nucleoside analogues such as lamivudine (3TC,
cytidine analogue), zidovudine (AZT, thymidine analogue) or tenofovir (TFV, adenosine
monophosphate analogue) is the basis for highly active antiretroviral treatment (HAART) [523,
573]. Nucleoside analogues are on the WHO list of essential medicines and can be deployed in
limited resource settings [574]. Moreover, AZT binds RNA through G-C and A-U bases [575],
prompting us to evaluate whether these nucleoside analogues might also inhibit Ebola
replication.
Type I IFNs mediate diverse biological effects, including cell type-independent antiviral
responses and cell type-restricted responses of immunological relevance. IFNs inhibit viral
infection by preventing viral entry into target cells and by blocking different stages of the viral
replication cycle for different viruses. Moreover, type I IFNs have a critical role in linking the
innate and adaptive immune responses to viral challenge. IFN-α/β expression occurs as the
earliest non-specific response to viral infection. Indeed, viruses have evolved immune evasion
strategies specifically targeted against an IFN response, confirming the importance of IFNs as
antivirals. This immune evasion strategy is relevant when one considers the IFN response to
Ebola infection [576]. Ebola proteins VP24 and VP35 inhibit host cell systems that lead to IFN
production and also inhibit events associated with an IFN response [577-579]. VP24 blocks the
binding of importins to phosphorylated STAT1, preventing STAT1 nuclear translocation
required for transcription of interferon simulated genes [577]. VP35 binds viral dsRNA,
preventing dsRNA degradation [578] and inhibits the phosphorylation of IRF-3 and the
SUMOylation of IRF-3 and IRF-7, thereby limiting IFN production [579]. Despite these virally-
encoded mechanisms to limit an IFN response to infection, different rodent and non-human
primate studies provide evidence for IFN-induced partial protection: the effects of IFN-α/β
treatment in lethal Ebola virus infection reduced viremia and prolonged survival [31, 580, 581].
Thus, a potential therapeutic effect for IFNs as monotherapy in EVD, or in combination with
other anti-Ebola therapies, has not been resolved.

148
4.3 Materials and Methods
Cell culture and trVLP infection: We employed an established mini-genome system to rapidly
evaluate candidate drugs that could inhibit Ebola Zaire replication under BSL2 conditions (see
Fig. 4.1 for an an illustration of the reporter system) [534]. The mini-genome encodes 3 of the 7
ZEBOV proteins (VP24, VP40 and GP1,2) and a luciferase reporter gene. Expression plasmids
for the remaining four Ebola nucleocapsid proteins (L, NP, VP30 and VP35) were also included
during transfection. Cell culture conditions and virus infections were performed as previously
described [534]. Briefly, 80,000 producer 293T cells (American Type Culture Collection;
ATCC, Rockville, USA) were seeded in individual wells of 24-well plates in 400 μL Dulbecco’s
Modified Eagle Medium (DMEM) containing 10% FBS, 1% penicillin and 1% streptomycin,
and grown in 5% C02 atmosphere at 37°C. Cells were transfected with the viral replication
protein plasmids (L, NP, VP30, VP35), a tetracistronic Ebola mini-genome and the T7
polymerase, using the CalPhos Mammalian Transfection Kit (Clontech Laboratories). Twenty-
four hours later, medium was replaced with 800 μL DMEM with 5% FBS. The replication and
transcription-competent virus like particles (trVLPs) were harvested 3 days later. Virus stock
was frozen at -80°C.
For infection, 293T target cells were seeded at 80,000 cells in 400 μL of DMEM supplemented
with 10% FBS. Target cells were then transfected with the four viral replication protein
plasmids, as well as Tim-1, to allow efficient virus binding and entry. Twenty-four hours post-
transfection, 25 μL of trVLP stock was diluted in 600 μL of DMEM with 5% FBS, warmed to
37°C for 30 min, then added to target cells. Medium was removed the following day and
replaced with 800 μL DMEM with 5% FBS. Four days post-infection, the medium was
aspirated and cells were re-suspended in 200 μL of 1x Renila Luciferase Assay Lysis Buffer
(Renilla Luciferase Assay System, Promega). Lysates were assayed for luciferase activity.
ZEBOV-eGFP infection: We generated recombinant ZEBOV expressing enhanced green
fluorescent protein (eGFP) from cDNA clones of full-length infectious ZEBOV, as previously
described [582]. The eGFP reporter protein was expressed as an eighth gene, and the virus
exhibited an in vitro phenotype similar to wild type ZEBOV. Notably, in vivo, incorporation of
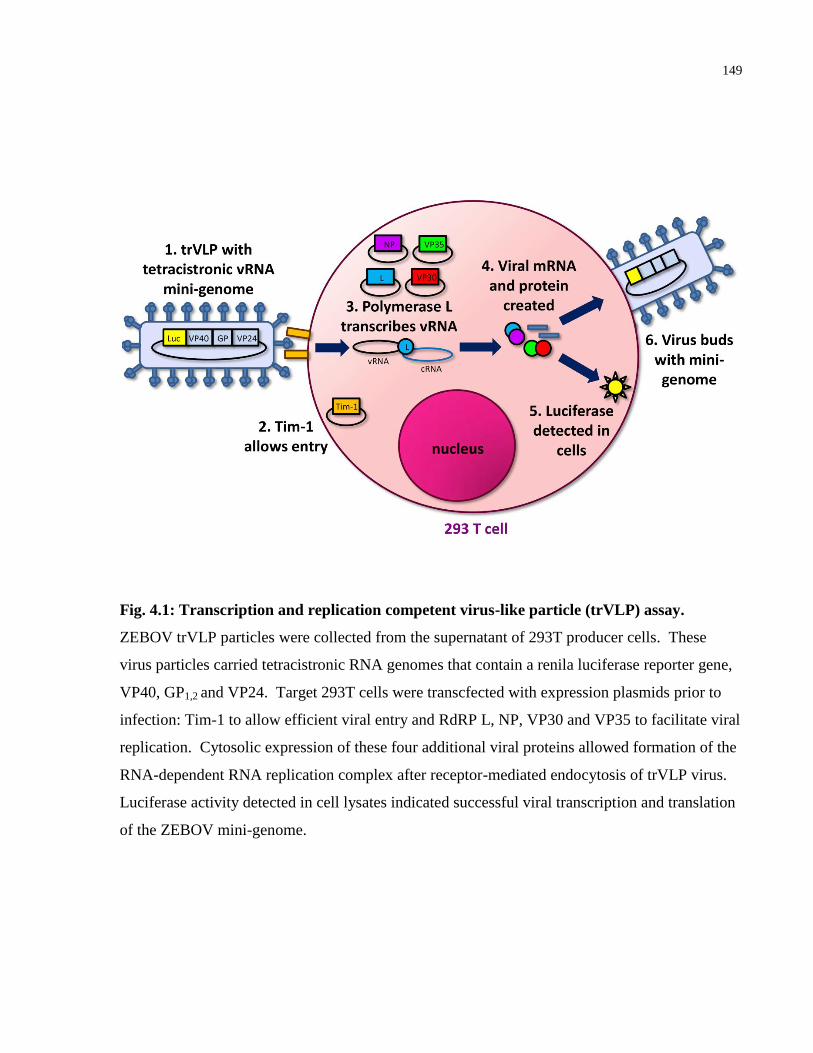
149
Fig. 4.1: Transcription and replication competent virus-like particle (trVLP) assay.
ZEBOV trVLP particles were collected from the supernatant of 293T producer cells. These
virus particles carried tetracistronic RNA genomes that contain a renila luciferase reporter gene,
VP40, GP1,2 and VP24. Target 293T cells were transcfected with expression plasmids prior to
infection: Tim-1 to allow efficient viral entry and RdRP L, NP, VP30 and VP35 to facilitate viral
replication. Cytosolic expression of these four additional viral proteins allowed formation of the
RNA-dependent RNA replication complex after receptor-mediated endocytosis of trVLP virus.
Luciferase activity detected in cell lysates indicated successful viral transcription and translation
of the ZEBOV mini-genome.

150
GFP into wild type ZEBOV results in some attenuation of disease [582]. All work with
infectious ZEBOV was performed in biosafety level 4 (BSL4), at the National Microbiology
Laboratory of the Public Health Agency of Canada in Winnipeg, Manitoba. 293T cells (30,000)
were seeded in 96-well plates in 100 μL DMEM with 10% FBS. Twenty-four hours thereafter,
the medium was replaced with 100 μL DMEM with 10% FBS containing ZEBOV-GFP at an
MOI of 0.1. Twenty-four hours post-infection, the medium was removed and replaced with 200
µL of DMEM with 5% FBS, or 190 μL DMEM with 5% FBS and 10 μL of single or
combinations of drugs. eGFP fluorescence was measured 3 days post-infection using a Synergy
HTX Multi-Mode Microplate Reader (BioTek).
Drugs: For these experiments, we used toremifiene citrate (TOR; Sigma), cidofovir hydrate
(CDV; Sigma) favipiravir (FPV, T-705; Cellagen Technology), lamivudine (3TC; Sigma)
zidovudine (AZT), tenofovir (TFV) maraviroc (MVC; NIH AIDS Reagent Program), Infergen
(IFN alfacon-1, Pharmunion Bsv Development Ltd.) or human interferon beta-1a (IFN-β,
Avonex; Biogen).
Mini-genome RNA extraction and qRT-PCR quantification of viral RNA: Forty-eight hours
after trVLP infection, medium was aspirated from 293T cells that had either been left untreated
or treated with the various drugs and total RNA extracted from cell lysates with 500 μL of
TRIzol (Thermo Fisher Scientific). cDNA synthesis was performed on 5 µg of total RNA, using
the First-Strand cDNA Synthesis Kit (GE Healthcare Life Sciences), according to the
manufacturer’s instructions. A 20 µl reaction also contained bulk first-strand cDNA reaction
mix, DTT solution and 40 pmol of one of two trVLP specific primers [583]: vRNA forward (5’-
GGC CTC TTC TTA TTT ATG GCG A -3’), or cRNA/mRNA reverse (5’-AGA ACC ATT
ACC AGA TTT GCC TGA-3’). Both primers were synthesized by the Center for Applied
Genomics (The Hospital for Sick Children, Toronto, Canada). Real-time qPCR reactions (25 µl)
were conducted in duplicate, using the Rotor-Gene RG-3000 thermocycler (Corbett Research,
Montreal, Canada). Each reaction contained 100 ng template cDNA, 12.5 µL 2 x SYBR Green
PCR Master Mix (Applied Biosystems, Warrington, UK), 300 nM of both the forward (vRNA)
and reverse (cRNA/mRNA) primers, and PCR grade H2O (Roche Diagnostics, Indianapolis,
USA). Samples lacking reverse transcriptase (No RT) during first-strand cDNA synthesis served

151
as negative controls. Cycling parameters were as follows: initial denaturation at 95°C for 10
min, followed by 40 cycles of amplification with 95°C for 15 seconds, 56°C for 30 seconds, and
60°C for 30 seconds. Biological triplicates in the drug-treated groups were normalized to the
average Ct of infected cells given DMSO solvent alone, by the 2-ΔCT
comparative CT method.
Cell viability assay: Dose-response cytotoxicity/viability assays were conducted in 293T cells 4
days post-infection for each of the drugs examined, either alone or in the various combinations
indicated, using the MTT assay as previously described [584].
Statistics: Means were compared using a two-tailed, unpaired Student’s t test and corrected for
multiple comparisons. For all figures, (*) denotes a p value <0.05, (**) denotes a p value <0.01
and (***) denotes a p value <0.001. Error bars shown are the standard error around the mean
(SEM). Synergy between two and three-drug combinations and combination index (CI) were
calculated with CompuSyn Version 1.0 [533]. The coefficient of determination (R2) was
determined for simple linear regressions.

152
4.4 Results
We employed an established mini-genome system to rapidly evaluate candidate drugs that could
inhibit Ebola Zaire replication under BSL 2 conditions [534, 535, 585]. At the outset, we
established the experimental conditions for infection with replication and transcription-
competent virus like particles (trVLPs), by examining luciferase activity under various
transfection and drug treatment conditions, which included transfection with viral support protein
plasmids (Fig. 4.2). We included treatment with maraviroc, a CCR5 inhibitor, that would have
no effect on trVLP entry and infection, thereby serving as a negative control for subsequent
treatment regimens. The triple combination of 3TC + AZT + TFV significantly inhibited trVLP
infection, and further inhibited luciferase activity of a second passage of 293T cells when
administered supernatant of a previous passage of cells.
In a second series of experiments, we examined the inhibitory effects of IFN-α (0.5 µM/10,000
U/mL), IFN-ß (0.2 µM/1,000 U/ml), TOR (5 µM), CDV (100 µM), FPV (100 µM), and a
combination of 3TC, AZT and TFV (5 µM each) on trVLP infection of 293T cells (Fig. 4.3).
Specifically, the 293T cells were treated with the different drugs at four different times relative
to infection with trVLP, as indicated. We provide evidence that for each of the individual drugs
and for the triple drug combination, at the doses indicated, trVLP infection of 293T cells is
inhibited when treatment is initiated at +2, +6 or +24 hours post-infection. Interestingly, TOR,
an estrogen receptor modulator discovered in a high throughput screen as a potent inhibitor of
Ebola [25], significantly reduced viral luciferase activity at all time-points tested. For IFN-α,
IFN-ß, TOR and FPV treatments, maximal inhibition of trVLP infection was achieved when the
cells were treated prior to challenge with trVLP. By contrast, pre-treatment with CDV at 100
µM, 24 hours prior to infection with trVLP, resulted in enhanced infection.
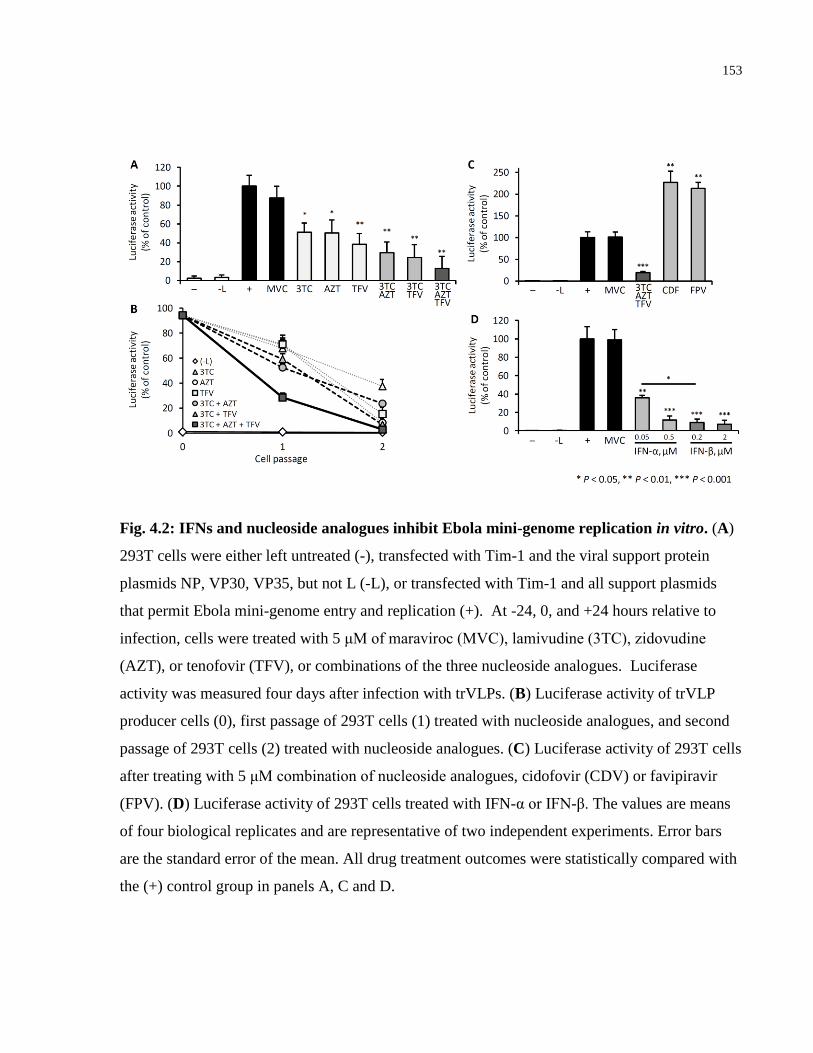
153
Fig. 4.2: IFNs and nucleoside analogues inhibit Ebola mini-genome replication in vitro. (A)
293T cells were either left untreated (-), transfected with Tim-1 and the viral support protein
plasmids NP, VP30, VP35, but not L (-L), or transfected with Tim-1 and all support plasmids
that permit Ebola mini-genome entry and replication (+). At -24, 0, and +24 hours relative to
infection, cells were treated with 5 μM of maraviroc (MVC), lamivudine (3TC), zidovudine
(AZT), or tenofovir (TFV), or combinations of the three nucleoside analogues. Luciferase
activity was measured four days after infection with trVLPs. (B) Luciferase activity of trVLP
producer cells (0), first passage of 293T cells (1) treated with nucleoside analogues, and second
passage of 293T cells (2) treated with nucleoside analogues. (C) Luciferase activity of 293T cells
after treating with 5 μM combination of nucleoside analogues, cidofovir (CDV) or favipiravir
(FPV). (D) Luciferase activity of 293T cells treated with IFN-α or IFN-β. The values are means
of four biological replicates and are representative of two independent experiments. Error bars
are the standard error of the mean. All drug treatment outcomes were statistically compared with
the (+) control group in panels A, C and D.
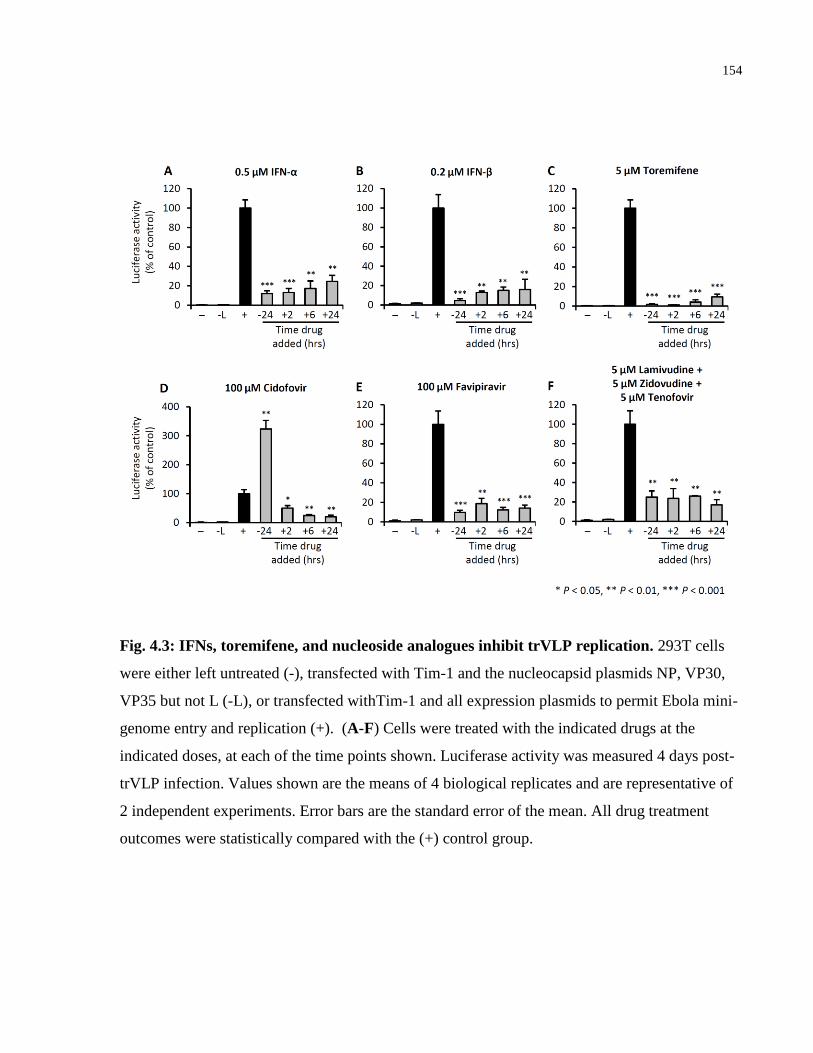
154
Fig. 4.3: IFNs, toremifene, and nucleoside analogues inhibit trVLP replication. 293T cells
were either left untreated (-), transfected with Tim-1 and the nucleocapsid plasmids NP, VP30,
VP35 but not L (-L), or transfected withTim-1 and all expression plasmids to permit Ebola mini-
genome entry and replication (+). (A-F) Cells were treated with the indicated drugs at the
indicated doses, at each of the time points shown. Luciferase activity was measured 4 days post-
trVLP infection. Values shown are the means of 4 biological replicates and are representative of
2 independent experiments. Error bars are the standard error of the mean. All drug treatment
outcomes were statistically compared with the (+) control group.

155
In subsequent dose-response studies, we compared the inhibitory effects of IFN-α, IFN-ß, TOR,
CDV, FPV, 3TC, AZT or TFV when administered 24 hours post trVLP infection (Fig. 4.4). The
data in Figure 4.4I summarize the IC50 dose for each drug. The IFNs exhibited the lowest IC50
values at 0.016 µM for IFN-ß and 0.038 µM for IFN-α. The data show a log-fold difference in
IC50 values for IFN-α and IFN-ß when compared in terms of U/ml, the norm for antiviral activity
measurements (Figure 4.4 A, B). TOR had the next lowest IC50 (0.36 µM) and completely
inhibited infection at doses > 5 µM (Figure 4.4C). TFV had an IC50 at 0.98 µM. CDV, 3TC and
AZT all exhibited similar IC50 values in the dose range 4.2-7.8 µM, while FPV had the highest
IC50 of the nucleoside analogues at 36.8 µM. At their IC50 concentration, none of these drugs
directly inhibited luciferase reporter activity (Fig. 4.5). We observed a relatively small antiviral
dose range for CDV (1.5-25 µM) (Figure 4.4D), beyond which the drug appeared to enhance
viral infection (Fig. 4.6). In cell viability assays we observe that at doses >10 µM CDV affects
cell viability, confounding the interpretation of the effects of CDV on viral replication.
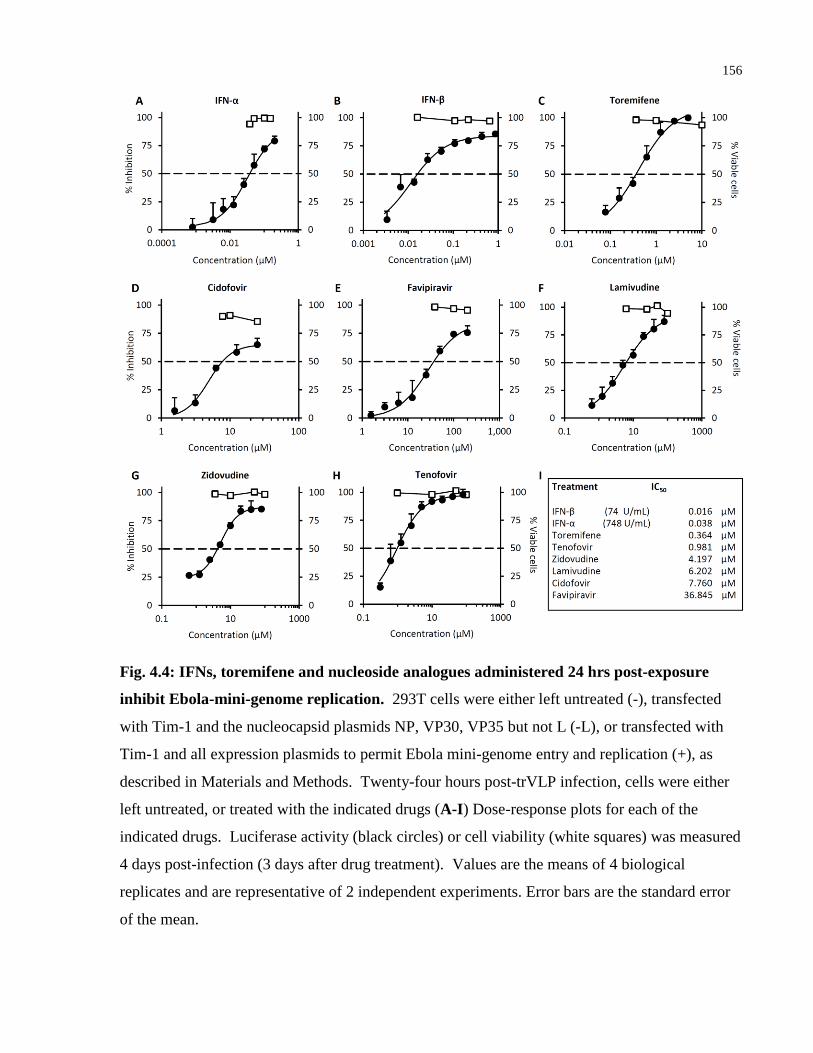
156
Fig. 4.4: IFNs, toremifene and nucleoside analogues administered 24 hrs post-exposure
inhibit Ebola-mini-genome replication. 293T cells were either left untreated (-), transfected
with Tim-1 and the nucleocapsid plasmids NP, VP30, VP35 but not L (-L), or transfected with
Tim-1 and all expression plasmids to permit Ebola mini-genome entry and replication (+), as
described in Materials and Methods. Twenty-four hours post-trVLP infection, cells were either
left untreated, or treated with the indicated drugs (A-I) Dose-response plots for each of the
indicated drugs. Luciferase activity (black circles) or cell viability (white squares) was measured
4 days post-infection (3 days after drug treatment). Values are the means of 4 biological
replicates and are representative of 2 independent experiments. Error bars are the standard error
of the mean.
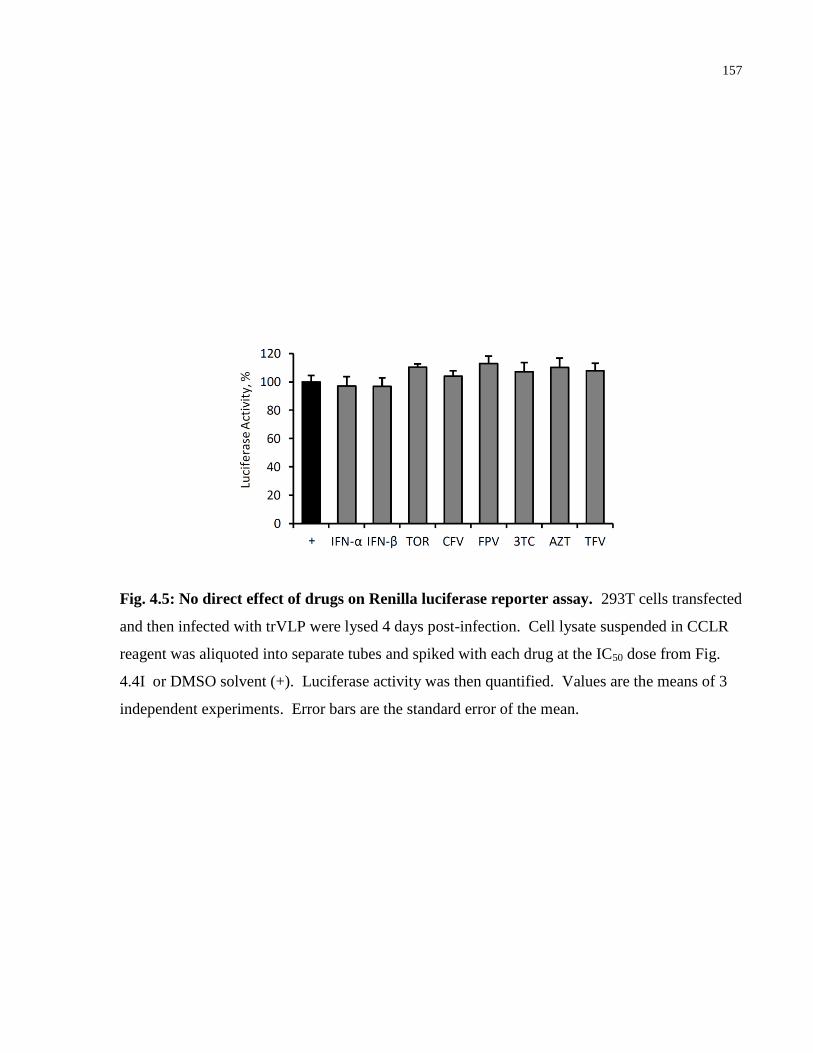
157
Fig. 4.5: No direct effect of drugs on Renilla luciferase reporter assay. 293T cells transfected
and then infected with trVLP were lysed 4 days post-infection. Cell lysate suspended in CCLR
reagent was aliquoted into separate tubes and spiked with each drug at the IC50 dose from Fig.
4.4I or DMSO solvent (+). Luciferase activity was then quantified. Values are the means of 3
independent experiments. Error bars are the standard error of the mean.
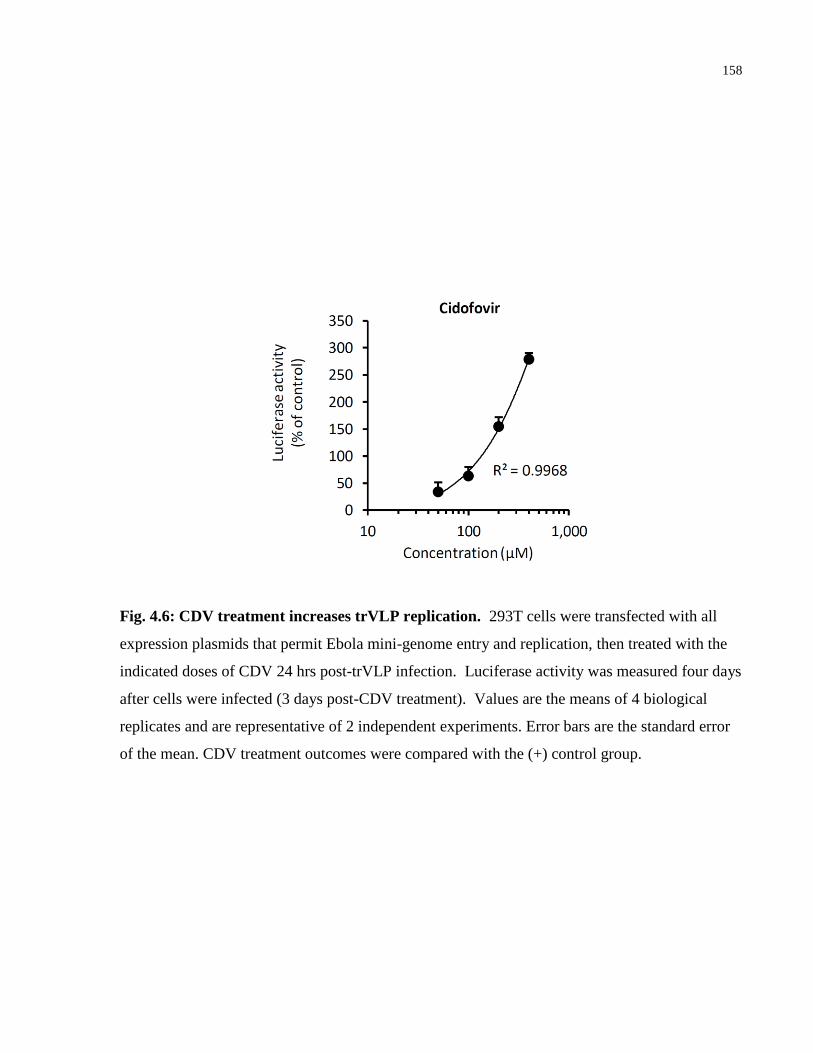
158
Fig. 4.6: CDV treatment increases trVLP replication. 293T cells were transfected with all
expression plasmids that permit Ebola mini-genome entry and replication, then treated with the
indicated doses of CDV 24 hrs post-trVLP infection. Luciferase activity was measured four days
after cells were infected (3 days post-CDV treatment). Values are the means of 4 biological
replicates and are representative of 2 independent experiments. Error bars are the standard error
of the mean. CDV treatment outcomes were compared with the (+) control group.

159
In an orthogonal assay to confirm these findings, we next measured viral replication and
transcription by qRT-PCR, following trVLP infection. trVLP-infected cells were either left
untreated, or treated with the different drugs 24 hours post-infection, then viral replication and
transcription evaluated 24 hours later (Fig. 4.7). All treatments, with the exception of TOR,
significantly reduced the amount of genomic vRNA detected within cells (Figure 4.7A) and all
treatments significantly reduced the synthesis of cRNA and mRNA isolated from infected cells
(Figure 4.7B). Notably, IFN-ß treatment of trVLP-infected cells resulted in the greatest
reduction in viral replication and transcription.
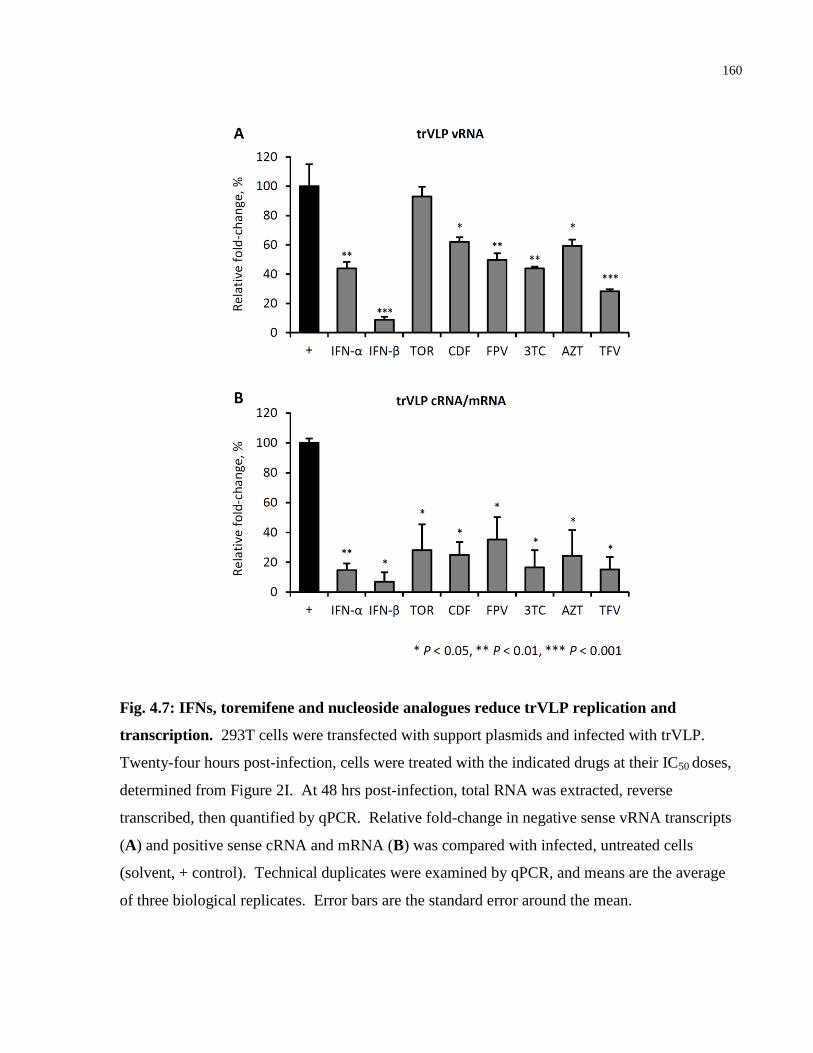
160
Fig. 4.7: IFNs, toremifene and nucleoside analogues reduce trVLP replication and
transcription. 293T cells were transfected with support plasmids and infected with trVLP.
Twenty-four hours post-infection, cells were treated with the indicated drugs at their IC50 doses,
determined from Figure 2I. At 48 hrs post-infection, total RNA was extracted, reverse
transcribed, then quantified by qPCR. Relative fold-change in negative sense vRNA transcripts
(A) and positive sense cRNA and mRNA (B) was compared with infected, untreated cells
(solvent, + control). Technical duplicates were examined by qPCR, and means are the average
of three biological replicates. Error bars are the standard error around the mean.

161
Next we examined the effectiveness of two and three drug combinations on trVLP infection. We
first examined 28 two-drug combinations, using each drug at its luciferase IC50 value, and used
the median-effect equation and combination index theorem [533] to determine drug synergy,
additive or sub-additive effects (Figure 4.8A). Synergy is defined as greater than additive effect
when drugs were combined (CI<1), additive as the effect expected when combining each drug
(CI=1) and sub-additive as a smaller than expected additive effect (CI>1). Fractional inhibition
(Fi) is defined as the percent reduction in luciferase activity. When administered 24 hours post-
infection, many of the two-drug combinations showed strong synergism in inhibiting trVLP
replication (Figure 4.8J), with IFN-β + 3TC demonstrating the greatest synergism (97.3%
inhibition, CI = 0.028). 3TC was synergistic with all seven other drugs tested. Notably, when
CDV was used in combination with FPV, AZT, TFV or IFN-α, it produced a sub-additive effect.
Next we tested all possible 56 three-drug combinations, using each drug at its IC50 value, to
assess whether adding a third drug enhanced efficacy compared with two-drug combinations
(Figure 4.8B-I) . This series of experiments served to validate our two-drug findings, as
synergistic two-drug combinations such as IFN-β + 3TC and IFN-β + AZT, predicted strong
synergy for the triple drug combination of IFN-ß + 3TC + AZT. As anticipated from the two-
drug polygonogram, CDV was sub-additive when combined in three-drug combinations (Figure
4.8E). This was most evident even when CDV was administered in conjunction with two-drug
combinations that had shown strong synergy, such as IFN-β + 3TC or FPV + TFV, further
indicating that CDV diminishes the antiviral effects of other drugs. IFN-ß, 3TC, AZT and TFV
all promoted strong synergism when included in triple drug combinations, with IFN-β + AZT
specifically providing strong synergism when combined in three unique triple therapies.
From these two-drug and three-drug screens, we calculated the combination index (CI) and
fractional inhibition (Fi) (Figure 4.8J-K). Many of the synergistic drug combinations (i.e. low
CI) included one nucleoside analogue and an IFN, while those drug combinations that were sub-
additive all included CDV. IFN-β was predominant in the most efficacious two- and three-drug
combinations. In particular, IFN-β + 3TC and IFN-β + 3TC + AZT consistently exhibited the
strongest synergism and highest Fi when administered 24 hours post-infection. Refer also to
Tables 4.1 and 4.2.
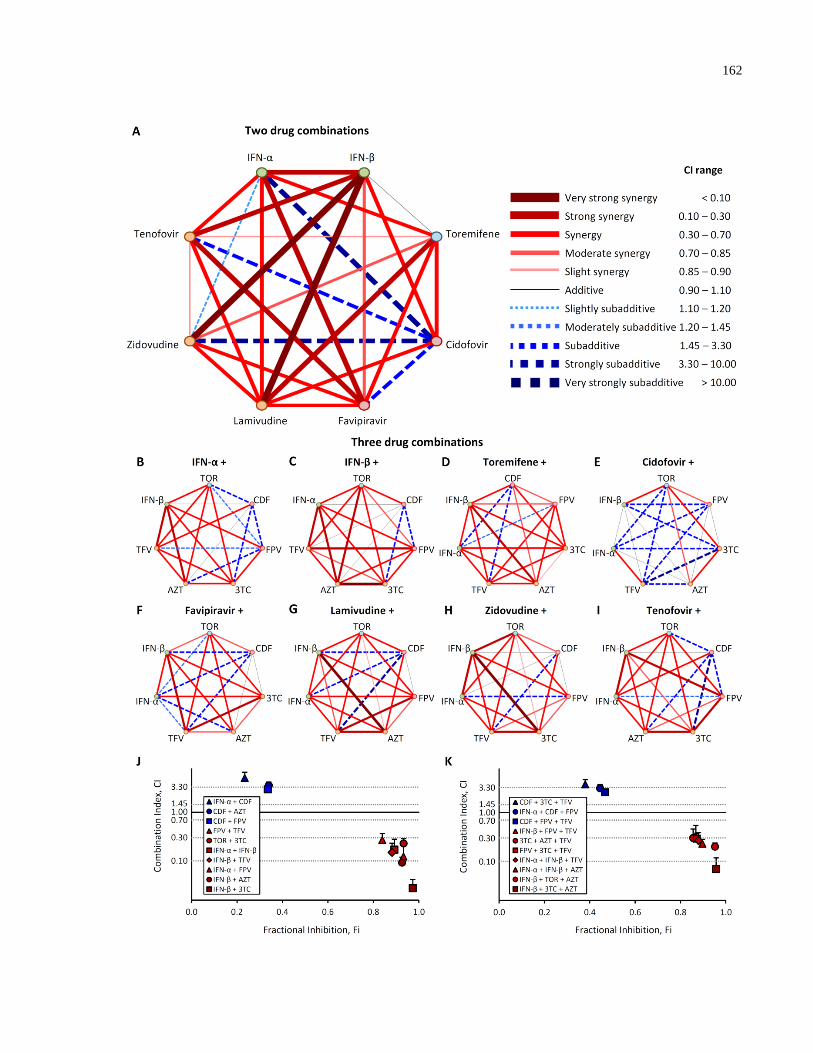
162

163
Fig. 4.8: 2 and 3 drug combinations synergistically inhibit Ebola trVLP infection. 293T
cells were either left untreated, transfected with Tim-1 and the support plasmids NP, VP30,
VP35, but not L, or the mini-genome plus all the support plasmids. (A) Polygonogram of 28 two-
drug combinations (at monotherapy IC50 doses) administered at 24 hrs post-trVLP infection, then
luciferase activity evaluated 3 days later. A thick red line represents strong synergy between two
drugs (CI<1), a thin black line represents additive effects (CI=1), and a thick blue line represents
strong sub-additive (less than additive) between two drugs (CI>1). (B-I) Polygonograms of 56
three-drug combinations. The backbone drug in each triple combination is listed above the
heptagon, and the synergism/sub-additive effect of the additional two drugs is represented within
each heptagon. (J-K) Combination index (CI) vs. fractional inhibition (Fi, percent luciferase
inhibition) plots of the most synergistic and sub-additive double (J) and triple (K) drug
combinations on trVLP luciferase activity. Dotted lines identify thresholds of synergy/additive
effects. Values shown are the means of 2-4 biological replicates in 2 independent experiments.
Error bars are the standard error of the mean.
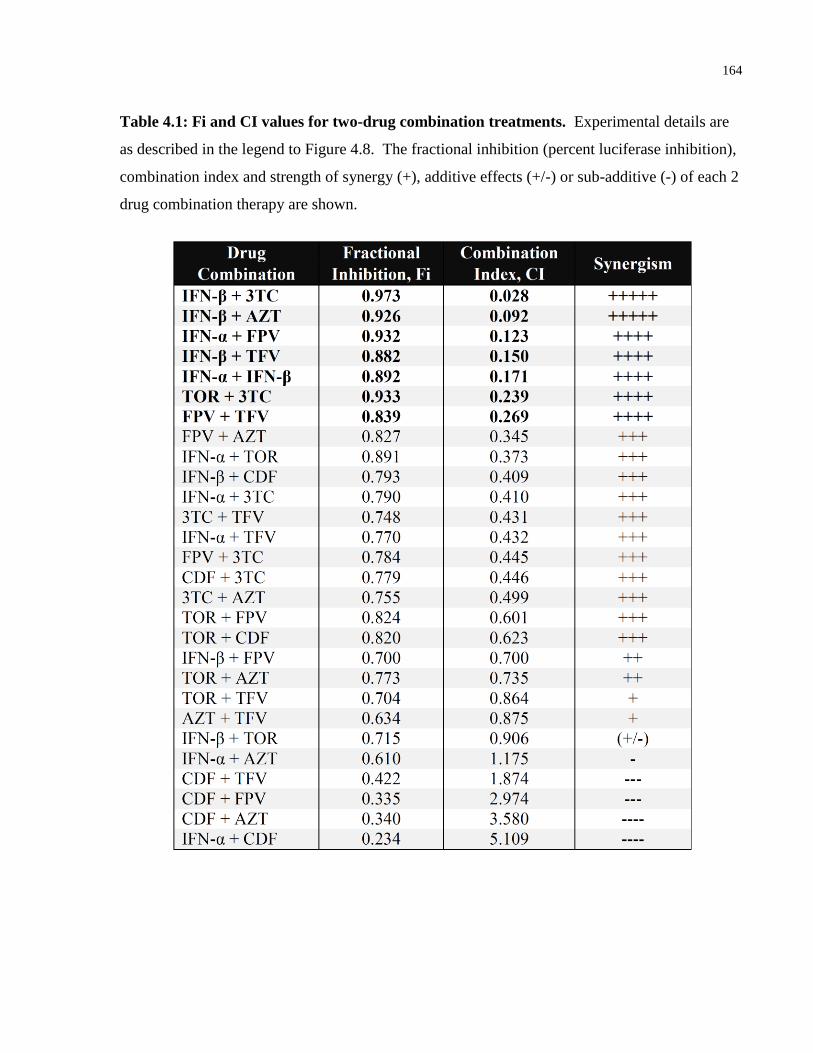
164
Table 4.1: Fi and CI values for two-drug combination treatments. Experimental details are
as described in the legend to Figure 4.8. The fractional inhibition (percent luciferase inhibition),
combination index and strength of synergy (+), additive effects (+/-) or sub-additive (-) of each 2
drug combination therapy are shown.
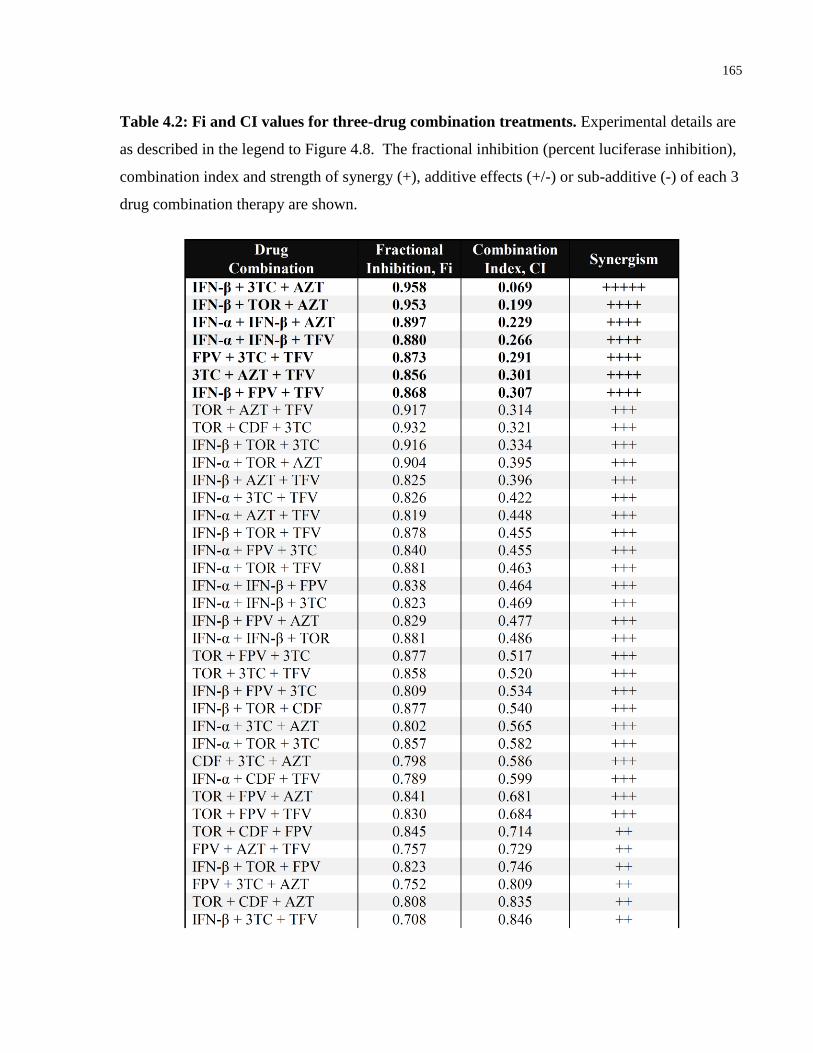
165
Table 4.2: Fi and CI values for three-drug combination treatments. Experimental details are
as described in the legend to Figure 4.8. The fractional inhibition (percent luciferase inhibition),
combination index and strength of synergy (+), additive effects (+/-) or sub-additive (-) of each 3
drug combination therapy are shown.
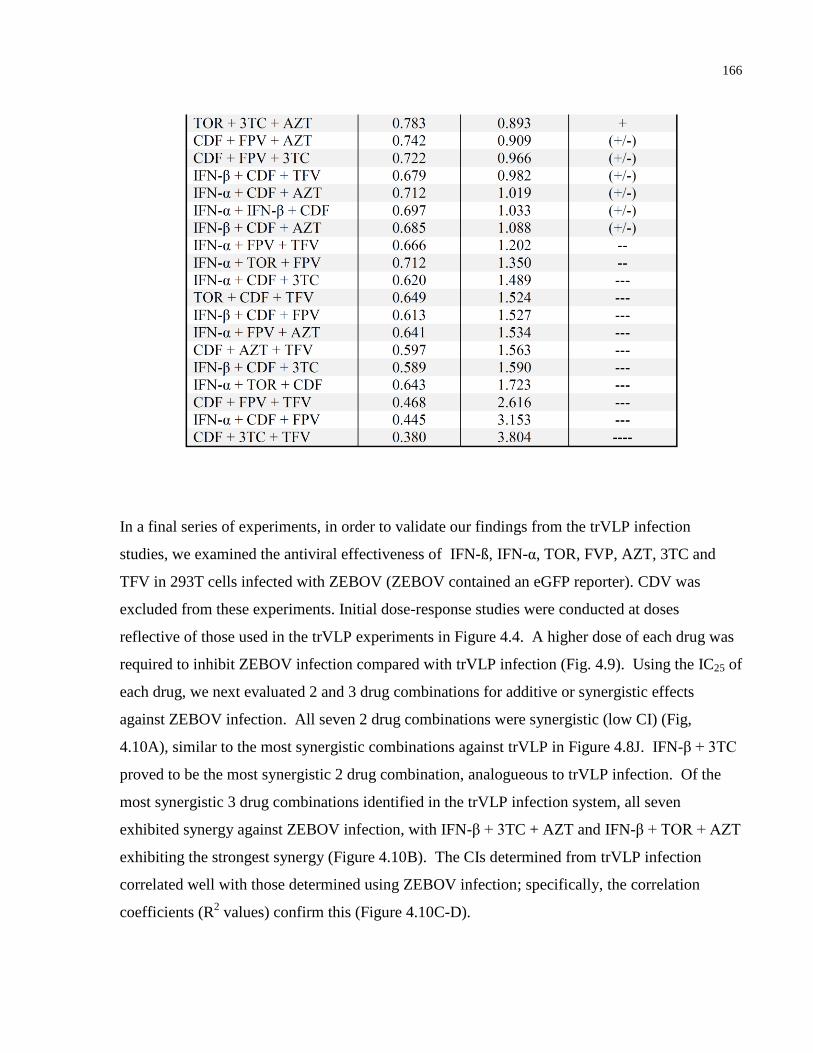
166
In a final series of experiments, in order to validate our findings from the trVLP infection
studies, we examined the antiviral effectiveness of IFN-ß, IFN-α, TOR, FVP, AZT, 3TC and
TFV in 293T cells infected with ZEBOV (ZEBOV contained an eGFP reporter). CDV was
excluded from these experiments. Initial dose-response studies were conducted at doses
reflective of those used in the trVLP experiments in Figure 4.4. A higher dose of each drug was
required to inhibit ZEBOV infection compared with trVLP infection (Fig. 4.9). Using the IC25 of
each drug, we next evaluated 2 and 3 drug combinations for additive or synergistic effects
against ZEBOV infection. All seven 2 drug combinations were synergistic (low CI) (Fig,
4.10A), similar to the most synergistic combinations against trVLP in Figure 4.8J. IFN-β + 3TC
proved to be the most synergistic 2 drug combination, analogueous to trVLP infection. Of the
most synergistic 3 drug combinations identified in the trVLP infection system, all seven
exhibited synergy against ZEBOV infection, with IFN-β + 3TC + AZT and IFN-β + TOR + AZT
exhibiting the strongest synergy (Figure 4.10B). The CIs determined from trVLP infection
correlated well with those determined using ZEBOV infection; specifically, the correlation
coefficients (R2 values) confirm this (Figure 4.10C-D).
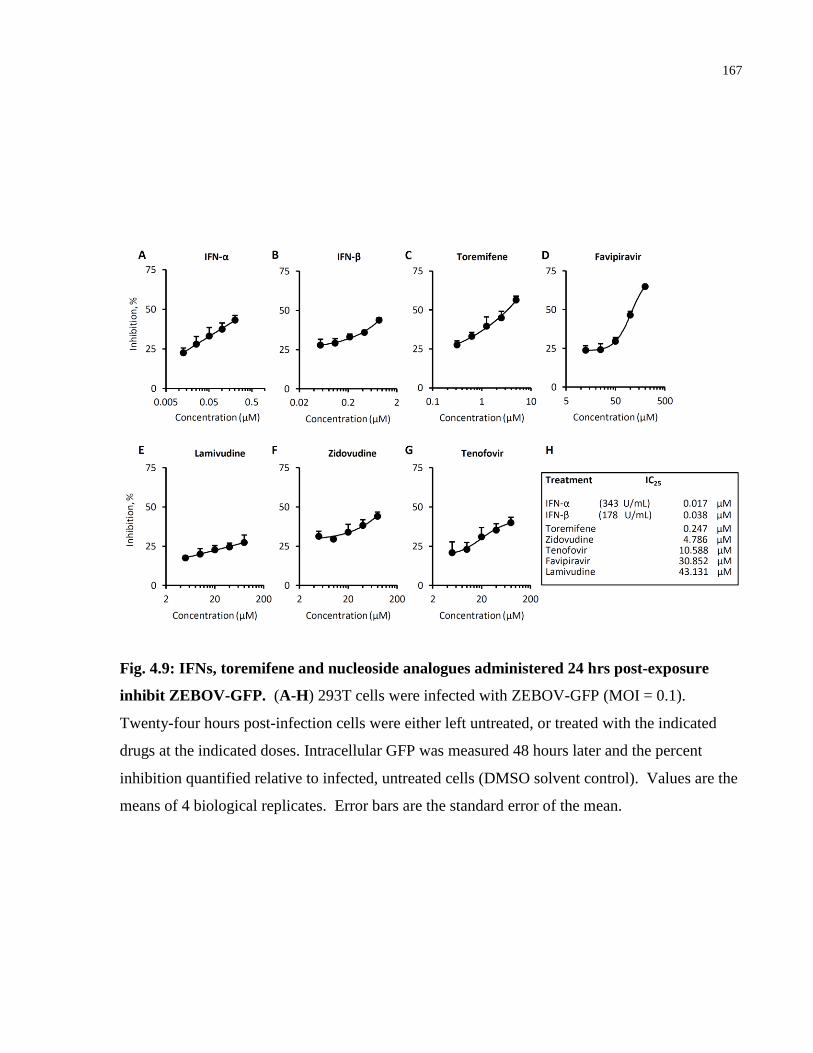
167
Fig. 4.9: IFNs, toremifene and nucleoside analogues administered 24 hrs post-exposure
inhibit ZEBOV-GFP. (A-H) 293T cells were infected with ZEBOV-GFP (MOI = 0.1).
Twenty-four hours post-infection cells were either left untreated, or treated with the indicated
drugs at the indicated doses. Intracellular GFP was measured 48 hours later and the percent
inhibition quantified relative to infected, untreated cells (DMSO solvent control). Values are the
means of 4 biological replicates. Error bars are the standard error of the mean.
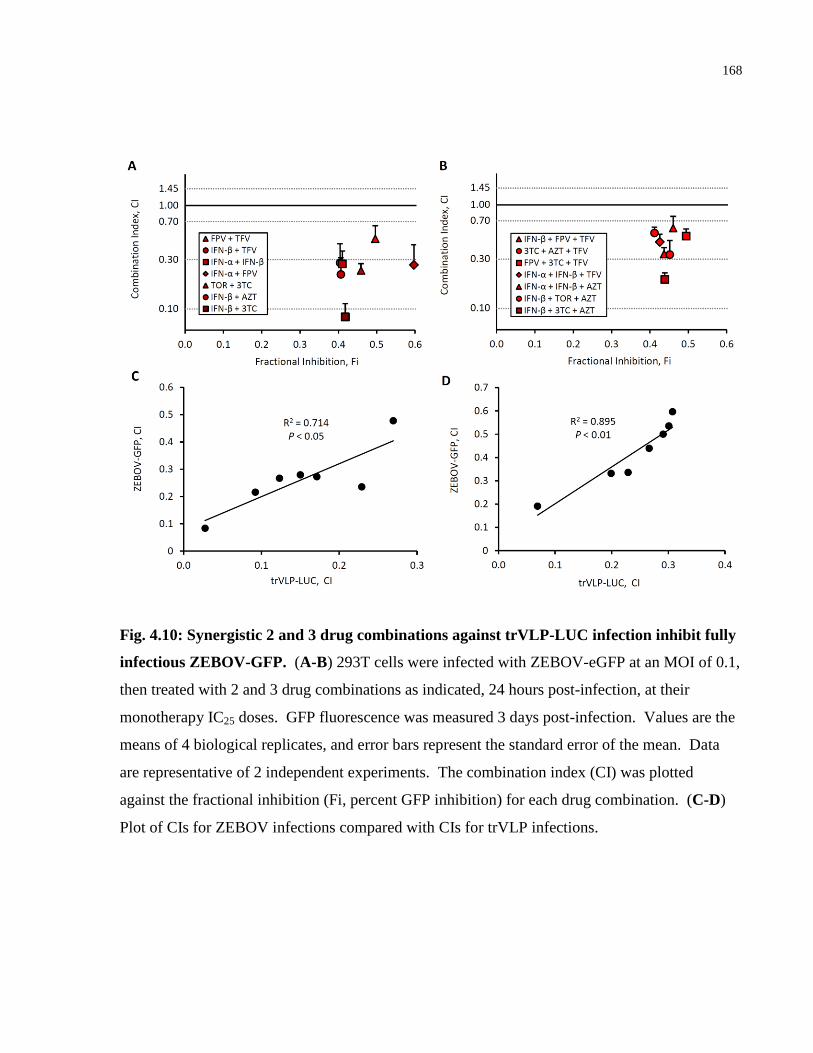
168
Fig. 4.10: Synergistic 2 and 3 drug combinations against trVLP-LUC infection inhibit fully
infectious ZEBOV-GFP. (A-B) 293T cells were infected with ZEBOV-eGFP at an MOI of 0.1,
then treated with 2 and 3 drug combinations as indicated, 24 hours post-infection, at their
monotherapy IC25 doses. GFP fluorescence was measured 3 days post-infection. Values are the
means of 4 biological replicates, and error bars represent the standard error of the mean. Data
are representative of 2 independent experiments. The combination index (CI) was plotted
against the fractional inhibition (Fi, percent GFP inhibition) for each drug combination. (C-D)
Plot of CIs for ZEBOV infections compared with CIs for trVLP infections.

169
4.5 Discussion
In September 2014, the WHO hosted a conference to facilitate development of a global action
plan to deal with the Ebola outbreak in West Africa. Delegates from affected West African
countries, ethicists, scientists, health care providers, logisticians and representatives from
different funding agencies were in attendance. A committee had been struck to evaluate the
different vaccine candidates and therapeutic interventions being developed, which subsequently
received an overwhelming number of submissions for consideration, and was hampered by an
inability to compare antiviral effectiveness, since in vitro and pre-clinical in vivo model systems
vary, treatment regimens vary from prophylaxis to post-exposure administration, and direct
readouts of antiviral efficacy differ. Moreover, given the virulence and high mortality associated
with EVD, all of these studies have been conducted under BSL 4 conditions, limiting the number
of laboratories that can engage in these antiviral studies. Cognizant of these limitations, we
employed the trVLP model system to compare the antiviral effectiveness of eight antiviral
candidates from three drug classes. We evaluated their antiviral activities in the context of
inhibition of Ebola replication, using this mini-genome model that allows for rapid comparisons
among compounds under BSL 2 conditions. The tetracistronic minigenome represents the most
sophisticated in vitro replication model of Ebola virus to date. trVLPs proceed through every
replication step as wild type Ebola virus, and have been tested in multiple cell lines. Using TOR,
there has been some validation of the trVLP assay. Specifically, TOR has been evaluated in
limiting Ebola virus infection of VeroE6 and HepG2 cells, and exhibited IC50 values of 0.2 µM
and 0.03 µM, respectively [586], in line with the IC50 dose for TOR (0.36 µM) observed with
trVLP infection. Likewise, the IC50 identified in the trVLP system for FPV (36.8 µM), is
consistent with that of 67 µM recorded using Ebola virus infection [23], suggesting that this
Ebola mini-genome system has relevance for screening potential antiviral compounds. Indeed,
our validation studies using ZEBOV (ZEBOV-eGFP) suggest that the trVLP infection model has
utility as an in vitro screening assay when comparing different drugs as monotherapies or in 2
and 3 drug combinations.

170
As mentioned, the Ebola virus encodes in its genome factors that limit a type I IFN response to
infection [577-579]. Yet, both rodent and non-human primate studies suggest that IFN-α and
IFN-ß treatment can confer partial protection from infection, reducing viremia and prolonging
survival [31, 580, 581], suggesting that it may be possible to override the inhibitory effects of the
virus by treatment with IFN. At the outset, we conducted a series of experiments to compare the
antiviral activities of IFN-α and IFN-ß in the trVLP infection system, and our findings suggest
that whether treatment is administered prior to or post-infection, both IFN-α and IFN-ß exhibit
antiviral activity. These findings only have relevance for the direct antiviral activities of these
IFNs, since the effects of IFN-α or IFN-ß on immune modulation for viral clearance cannot be
determined using this system. Nevertheless, these data contributed to the decision to conduct a
clinical trial of IFN-ß treatment for EVD in Guinea.
We provide evidence that the nucleoside/nucleotide analogues 3TC, AZT, TFV, FPV and CDV
inhibit Ebola trVLP replication in vitro. The results with 3TC are in contrast to published data
that show no evidence for 3TC inhibiting Ebola virus infection in vitro [519]. These studies
examined the antiviral effectiveness of 3TC when administered one hour prior to infection, in
contrast to our studies that have focused on post-exposure protection. In cells, the kinetics of
3TC phosphorylation are such that a minimum of four hours are required for optimal activity,
perhaps distinguishing why our 24 hour pre-treatment, specifically a combination treatment,
offered protection. Post-exposure treatment with 3TC and the other nucleoside/nucleotide
analogues we examined, would more likely reveal activity against viral RNA synthesis than pre-
treatment. When comparing the IC50 values of each of the nucleoside analogues that we tested,
TFV exhibited the lowest IC50 at ~1 µM. Whether this reflects the fact that this adenosine
monophosphate analogue only requires two phosphorylation events to become an active drug
versus three for the other nucleoside analogues, remains undetermined. Extensive published data
reveal both the safety profiles [523, 587, 588] and the biodistribution of 3TC, AZT and TFV in
the circulation and liver [416, 538], the same compartments where Ebola infects monocytes,
macrophages, dendritic cells, endothelial cells and hepatocytes. Moreover, drug interactions
with other nucleoside analogues have been well studied: e.g. tenofovir disoproxil fumarate, when

171
used alone or in combination with emtricitabine effectively prevents HIV-1 infection in
antiretroviral pre-exposure prophylaxis (PrEP) [588].
Our studies also revealed that the active metabolite of brincidofovir, CDV, has a narrow
therapeutic window of efficacy (6.25-25 µM) when assessed in the trVLP assay, enhancing viral
replication at higher doses when added either prior to or post-infection. In cell viability assays,
CDV exhibits cytotoxicity at doses >10 µM. These findings suggest that caution is required if
CDV is to be considered further for the treatment of EVD, specifically that phase I/II trials
define the safety profile of this drug for EVD.
Another advantage of this in vitro system is that it allowed us to evaluate various 2 and 3 drug
combinations and demonstrates that combination treatments limit viral replication up to 97.3%.
A benefit of combination treatment is the potential to limit/avoid the emergence of drug
resistance. Interestingly, IFN-ß was predominant among all the 8 antivirals considered in terms
of contributing very strong synergism in combination treatments: e.g. IFN-ß + 3TC; IFN-ß +
3TC + AZT. Using this system, we observe that FPV, when administered 24 hours post-
infection, has an IC50 of ~ 37 µM. To date, the phase II/III JIKI trial examining the efficacy of
FPV against EVD has reported only modestly encouraging results. In our 2 drug combination
treatment studies we show that, with the exception of CDV, whenever FPV is included, synergy
occurs, effectively reducing the CI. It may transpire that for treating EVD, FPV is most effective
in a drug combination regimen.
Viewed altogether, we present an in vitro Ebola trVLP screening system, that requires only level
2 biocontainment, which allowed us to compare the antiviral activities of 8 compounds, either
alone or in combination. We provide evidence that IFNs are effective inhibitors of Ebola
replication, with IFN-ß exhibiting greater efficacy over IFN-α, or when used in combination with
nucleoside analogues. We infer from our data that whether IFN-ß treatment is administered 24
hours prior to, or up to 24 hours post-infection, reduced Ebola replication is achieved. As
additional antiviral therapeutic candidates become available, we now have the capability to
measure and compare their direct antiviral activities with the existing panel. This allows for rapid

172
in vitro evaluation and the opportunity to prioritize antiviral candidates for further pre-clinical
and clinical trial studies.

173
Chapter 5: Key Findings, Future Perspectives,
Conclusions and Broader Significance

174
5.1 Key Findings, Limitations and Future Perspectives
HIV-1
As new indications for small-molecule kinase inhibitors expand beyond the treatment of various
cancers [169, 170], the essential role of non-receptor tyrosine kinases facilitating viral replication
are increasingly being recognized, along with their associated disease pathologies that can be
targeted with small-molecule inhibitors [146, 178, 201, 554]. At the outset, this work
investigated the role of the SFK member c-SRC during early HIV-1 infection, to determine
whether it has particular signaling functions during early HIV-1 replication in diverse CD4+
T-
cell lines and primary human CD4+ T-cells. A variety of techniques were used, including SFK
tool-drug compounds PP1, PP2 and SU6656, ADV vector overexpressing DN c-SRC, and
targeted siRNA knockdown (chapter 2). It was also determined whether the focal adhesion
kinase binding partner PTK2B exhibited similar affects as c-SRC during early HIV-1 infection,
and a variety of HIV-1 viruses were considered, including: X4- or R5-tropic viruses, nef-
deficient or fully infectious viruses, and laboratory strains versus primary clinical isolates. The
essential role of c-SRC during early time-points of HIV-1 infection led to further exploration of
four potent c-SRC kinase inhibitors, providing evidence to support further testing of these
preclinical or FDA-approved compounds for further studies of HIV-1 inhibition (chapter 3).
Lastly, the work presented in this thesis expanded on the use of FDA-approved drugs, used to
treat HIV-1, for repurposing for use in EVD. Monotherapy and combination antiviral therapy of
8 antiviral candidates for inhibition of a ZEBOV mini-genome model of replication or in vitro
ZEBOV infection were used (chapter 4). Nucleoside analogues that were used for
compassionate EVD treatment during the 2014-16 ZEBOV outbreak in West Africa were a
particular focus [519]. The trVLP model of ZEBOV replication permitted rapid assessment of
drugs and combinations of therapies to prioritize testing of compounds, leading to confirmatory
BLS4 testing of the most synergistic drug combinations, and providing rationale for future drug
development in small animal models of EVD.
Prior to this work, the roles of LCK and FYN signaling had been the most studied in HIV-1
infection of CD4+ T-cells [227, 238], but an understanding of a role for c-SRC signaling had not

175
been clearly defined. Early LCK activation downstream of CD4 can be decoupled from the
TCR-CD3 complex by Nef and impair formation of the immunological synapse [589]. LCK also
becomes recycled with CD4 at endosomes, contributes to Tat-mediated HIV-1 transcription,
facilitates Gag-assembly during viral egress, and promotes virological synapse transmission of
HIV-1 [222, 226-228, 589]. Activated FYN signaling promotes HIV-1 transcription through
NF-κB, enhances assembly and release of virions, and yet becomes inhibited by viral Vif to
reduce phosphorylated APOBEC3G packaged into budding virions [227, 237, 238]. Similar to
both LCK and FYN, this work demonstrates that c-SRC becomes activated and phosphorylated
at Tyr419 within the first hour of HIV-1 IIIB (X4) or HXB2 (X4) infection of Jurkat C T-cells
(chapter 2), or HXB2 (X4) or JR-FL (R5) infection of activated primary CD4+ T-cells (chapter
3). This was strongest for virus expressing Nef, but not completely absent in pseudo-enveloped
viruses lacking Nef (HXB2 or JR-FL), which may suggest c-SRC activation is partially
independent of Nef in the first hour of CD4+ T-cell infection. Similar results were found for the
binding partner PTK2B, where its phosphorylation at Tyr402 increased after 1 hr of HIV-1 IIIB
infection in Jurkat C T-cells, or HXB2 or JR-FL infection in activated CD4+ T-cells. However,
PTK2B was not phosphorylated at Tyr402 after 1 hr of HXB2 infection in Jurkat C cells. This
was the first of many indications that signaling within T-cell lines may not be the best models to
recapitulate physiological signaling cascades of c-SRC or PTK2B in primary CD4+ T-cells.
In chapter 2, it was demonstrated that pan-inhibition of SFKs with 10-20 μM of the tool
compound SU6656 significantly enhanced HIV-1 luciferase activity of nef-deficient VSV-
G/HIV-1 in Jurkat C or Jurkat E6-1 infection. This observation correlated with decreased in
vitro c-SRC kinase activity and no change in cell viability. The SU6656 experiments were
corroborated with follow-up tests of DN c-SRC ADV vector overexpression, which increased
VSV-G/HIV-1 infection in Hut 78 and Kit 225 cells. However, these ADV results should be
interpreted cautiously, as ADV treatment alone caused high cell death in Jurkat E6-1 and Hut 78
cells (70-80% of cells were 7-AAD+), and EYFP transfection efficiency was not equivalent
between cell lines. These findings suggest further optimization of ADV vector MOI is needed in
the three T-cell lines. Future experiments should also evaluate whether a dominant positive (DP)
mutant of c-SRC decreases VSV-G/HIV-1 infection in T-cell lines, as would be predicted. This

176
work from chapter 2 also revealed that siRNA reduction of c-SRC or PTK2B expression in
Jurkat E6-1 cells increased HXB2 reverse transcription. The pools of 4 siRNAs were kinase-
specific, had no affect on cell viability when administered by lipofection, nor change in surface
expression of CD4 or CXCR4 (chapter 2). The increased HXB2 luciferase activity from c-SRC
or PTK2B siRNA knockdown also correlated with increased early and late reverse transcript
cDNA, as well as 3-4-fold higher integrated virus after 24 hrs of infection.
However, these drug and siRNA findings are in contrast with the results of primary CD4+ T-cells
studied in chapter 3, and the published research of others describing a protective effect of c-SRC
inhibition or PTK2B siRNA knockdown 6 days post-infection [264, 345]. Different SFK
inhibitors were employed in chapters 2 and 3 that inhibit different off-target kinases. SRC
Inhibitor-1 is reported to be the most selective for SFKs at 1 μM in an in vitro study of 73 human
kinases (4 off-targets), followed by PP1 (5 off-targets), PP2 (5 off-targets) and SU6656 (9 off-
targets) [557]. In comparison, dasatinib, saracatinib and KX2 391 are less selective inhibitors of
SFKs, targeting several other kinases such as ABL1, ARG and serine/threonine kinases [590,
591]. Yet employing the same pool of siRNA to knockdown c-SRC in chapters 2 and 3 caused
opposite changes to luciferase activity in HIV-1 infected Jurkat cells compared with primary
CD4+ T-cells, suggesting the observed differences are primarly caused by the cell type
investigated.
Taken together, these findings cast doubt on the generalizability of c-SRC and PTK2B signaling
in transformed T-cell lines, with their corresponding signaling events in primary human CD4+ T-
cells during HIV-1 infection. Indeed, a comprehensive study on the proximal TCR signaling in
Jurkat E6-1 and Hut 78 cells, relative to activated primary CD4+
T-cells, showed that upon TCR
stimulation the cell lines exhibited greater Ca2+
flux, uncharacteristic expression of costimulatory
receptors, atypical cytokine release, and hyperphosphorylation of PTK2B and c-SRC, among
other downstream signal transduction abnormalities (see Figure 5.1, originally published in
[562]). Thus, while mutant T-cell sublines can be useful in other contexts [592], the work in this
thesis shifted instead to focus on the role of early c-SRC and PTK2B signaling in HIV-1
infection of primary CD4+ T-cells, isolated from healthy human donors (chapter 3).
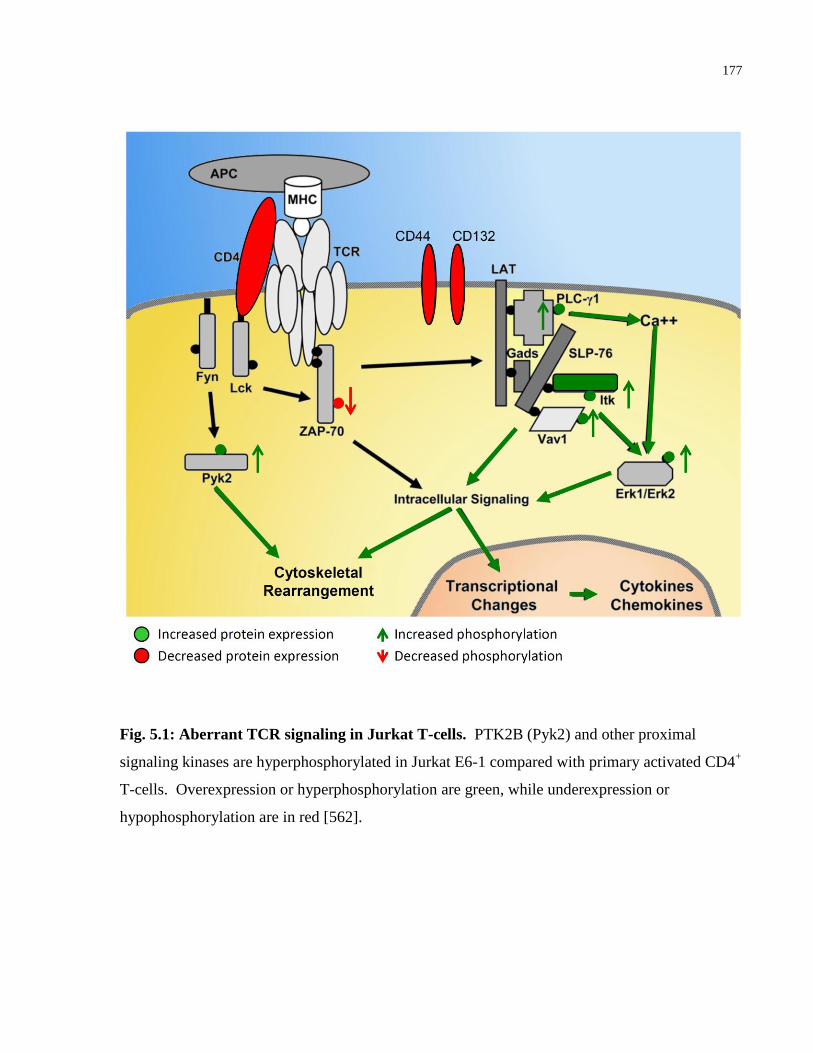
177
Fig. 5.1: Aberrant TCR signaling in Jurkat T-cells. PTK2B (Pyk2) and other proximal
signaling kinases are hyperphosphorylated in Jurkat E6-1 compared with primary activated CD4+
T-cells. Overexpression or hyperphosphorylation are green, while underexpression or
hypophosphorylation are in red [562].

178
In activated primary CD4+
T-cells, this work demonstrates that c-SRC co-immunoprecipitates
with a truncated PTK2B fragment phosphorylated at Tyr402, 1 hr following infection of nef-
deficient HXB2 or JR-FL virus (chapter 3). To explain this finding, HIV-1 chemokine receptor
binding may induce cleavage of PTK2B, potentially through CXCR4, CCR5, or P2Y2 activation
and signaling [147, 345]. This highlights a potential PTK2B similarity with closely related FAK
during HIV-1 infection. Downstream of gp120/CD4 signaling, phosphorylated FAK becomes
cleaved by caspase-3 and caspase-6 into a 90 kDa fragment containing the FAT domain, and a
35 kDa fragment containing the kinase domain of FAK [322, 346]. Separating the FAT domain
from focal adhesions in this manner has been shown to promote p53-dependent apoptosis [347,
348]. Thus, it should be explored in future experiments whether during HIV-1 infection similar
signaling from the larger PTK2B fragment containing the FAT domain occurs, as the smaller,
phosphorylated kinase fragment of PTK2B binds to c-SRC.
To investigate the effects of activated c-SRC and PTK2B on the early stages of HIV-1
replication, careful optimization of siRNA electroporation was performed, as CD4+ T-cells are
challenging non-adherent cells to transfect (chapter 3). Moreover, activated primary CD4+ T-
cells degrade argonaute, an enzyme essential to the RNA-Induced Silencing Complex (RISC)
[593]. Without argonaute, many mRNAs become de-repressed, which promotes rapid protein
translation as part of normal T-cell activation and T-cell differentiation [593]. This natural
phenomenon of argonaute degradation upon activation therefore precluded testing of c-SRC or
PTK2B siRNA knockdown after T-cell activation. Primary CD4+
T-cells administered siRNA,
and then activated, showed no changes to CD4, CXCR4 or CCR5 expression, or reduction in cell
viability (chapter 3).
The findings in chapter 3 reveal that viral reverse transcription of either HXB2 or JR-FL was
unaffected by c-SRC or PTK2B siRNA knockdown. However, reducing c-SRC expression in
CD4+ T-cells prior to infection with nef-deficient HXB2 or JR-FL viruses, increased the
detection of early reverse transcripts, late reverse transcripts, and 2-LTR circles, while reducing
viral integration and luciferase reporter activity. Similar results were found after PTK2B siRNA
knockdown, although only during JR-FL (R5) infection. This suggests PTK2B activation is a
byproduct of HXB2 (X4) and IIIB (X4) entry, perhaps through CXCR4 stimulation [147], and is

179
not essential in the early stages of HXB2 or IIIB replication in an activated CD4+ T-cell. While
both c-SRC and PTK2B are activated upon HIV-1 binding and fusion, they could be transmitting
signals that prepare the cell for later stages of the intracellular virus replication cycle [147]. To
generalize these c-SRC and PTK2B findings to multiple strains of HIV-1, testing of more X4 and
R5 HIV-1 clinical isolates are needed. Nonetheless, infection with HIV-1 strains expressing Nef
(IIIB (X4), Ba-L (R5) or KNH1207 (R5)) could not rescue infection after c-SRC siRNA
knockdown, as quantified by qPCR integration and p24 ELISA measurements, suggesting the
function of c-SRC may be independent of Nef during early infection. This outcome was not
entirely surprising, given the low affinity of in vitro c-SRC SH3 binding to Nef PxxP, relative to
the stronger binding of HCK or LYN to Nef that is mediated by a key Ile residue in their SH3
domain [164]. Collectively, these results from chapter 3 suggest changing roles for c-SRC and
PTK2B signaling at various time-points and stages of the early HIV-1 replication cycle, similar
to SFKs LCK and HCK during HIV-1 infection [223, 227, 238, 249]. In future experiments, it
will be of interest to test whether c-SRC or PTK2B siRNA knockdown increase HIV-1 reverse
transcripts and reduce viral integration in a similar fashion in T-cell sub populations. These
experiments could be performed in T-cells that maintain the viral reservoir in lymph nodes
(central memory peripheral T-follicular cells), prevent microbial translocation at the gut and
control inflammation at mucosal barriers (Th17 cells), or play a role in suppressing immune
hyperactivation (Tregs) [80, 594, 595].
The increase in observed early and late reverse transcripts of JR-FL virus after c-SRC or PTK2B
siRNA knockdown, together with no effect on RT activity, strongly suggest these kinases act
after reverse transcription and prior to viral integration (chapter 3). Other kinases have shown
similar effects on HIV-1, such as JNK inhibition increasing reverse transcripts and impairing
viral integration [159]. Unlike LCK and FYN, c-SRC interacts with membrane proteins of
multiple compartments in addition to microdomains at the plasma membrane [189], suggesting
c-SRC could have roles in the cytosolic or nuclear stages of HIV-1 replication. Moreover, the
SH3 domain of c-SRC regulates its localization from actin-associated focal adhesions to the
perinuclear region in a microtubule-dependent manner [189]. The function of host and viral
proteins that constitute the pre-integration complex of HIV-1 are still under investigation [155].

180
It also remains to be determined whether c-SRC or PTK2B directly phosphorylate host and viral
proteins in the PIC, or bind PIC proteins through SH2 or SH3 interactions to stabilize the
complex. In support of these hypotheses, c-SRC has been predicted by NetPhos 3.1 to
phosphorylate HIV-1 RT at eight different sites in its protein sequence (Tyr56, Tyr115, Tyr188,
Tyr342, Tyr354, Tyr405, Tyr441 and Tyr457) with 31-52% confidence, and three sites in the IN
sequence (Tyr15, Tyr83 and Tyr99) with 37-48% confidence [596]. Comparing with dengue
virus infection, it has been proposed the related family member FYN phosphorylates host factors
in that RNA replication complex, or may act as an adaptor protein that scaffolds interactions
between components of the dengue virus replication complex [178]. In addition, c-SRC has been
shown to directly interact with the HCV RdRP complex during HCV infection, facilitating
intracellular replication [202]. In the context of HCV replication, the SH3 domain of c-SRC is
exploited to bind the viral polymerase NS5B, linking c-SRC to the non-structural phosphoprotein
NS5A through its SH2 domain [202]. These c-SRC interactions were also found to be essential
for NS5B and NS5A binding each other and HCV replication, and were not mediated by related
SFK members FYN or c-YES [202]. Thus, c-SRC could have a novel and essential interaction
within the PIC of HIV-1. This line of inquiry is part of future experiments that involve isolating
the HIV-1 PIC, to determine a potential scaffolding role for c-SRC.
The buildup of 2-LTR circles and reduced proviral integration after c-SRC or PTK2B
knockdown further implicate these kinases in the nuclear translocation of the HIV-1 PIC (chapter
3). Indeed, this work showed that in the presence of the integrase inhibitor raltegravir, the
synthesis of HXB2 or Ba-L cDNA was similar to the nuclear effects of c-SRC or PTK2B siRNA
knockdown. Others have reported similar increases in 2-LTR circles after raltegravir
intensification in HIV-1 patients taking cART [565]. There is precedent for non-receptor kinases
affecting the nuclear biology of HIV-1, such as MAPK phosphorylation of the inner nuclear
lamina protein emerin (EMD) [154]. If EMD phosphorylation is blocked, or EMD expression
silenced by siRNA, proviral integration decreases and 1- and 2-LTR circles accumulate [154].
The phosphorylation of nuclear proteins may very well be a common lentivirus mechanism for
accessing the nuclear compartment [154], and may include c-SRC kinase activity during HIV-1
infection. c-SRC has been found to localize to the nucleus and nucleolus, and may regulate

181
proteins involved in cell cycle progression and entry into mitosis [189]. Tyr419 phosphorylation
on nuclear c-SRC has been shown to induce c-SRC interactions with Sam68 in other cell types
[597]. Thus nuclear c-SRC may already have a role in HIV-1 replication through Sam68, as
Sam68 regulates HIV-1 RNA processing, the transactivation of Rev, viral mRNA nuclear
transport, and mRNA translation in the cytosol [317]. Research into c-SRC protein interactions
with perinuclear membrane and nuclear protein substrates is ongoing [189]. One hypothesis is
that nuclear c-SRC could be regulating DNA ligase 4 (DNAL4), an essential DNA repair protein
in the NHEJ pathway that specifically catalyzes the ligation of unintegrated HIV-1 cDNA into 2-
LTR circles [155]. It has been put forth that the free ends of unintegrated HIV-1 cDNA are
sensed as DNA double-strand breaks, which activate nuclear kinases that regulate DNA repair
mechanisms to circularize viral cDNA, thus pre-empting apoptosis signaling by the T-cell [155].
Our lab is now investigating the potential role of c-SRC signaling in this NHEJ repair pathway
that circularizes HIV-1 cDNA into 2-LTR circles during HIV-1 replication [598].
The findings in chapter 3 also demonstrate the novelty of four specific c-SRC inhibitors that
strongly reduce JR-FL or HXB2 infection in activated CD4+ T-cells. Dasatinib, saracatinib,
KX2-391 and SRC inhibitor-1 all inhibited the luciferase activity of these recombinant viruses,
and caused little to no increase in apoptosis or necrosis. Dasatinib was the most potent of the
four inhibitors examined, reducing HXB2 or JR-FL infection in the 10-100 nM range. This drug
has recently been shown to inhibit the phosphorylation of SAMHD1, a c-SRC substrate [537].
Without phosphorylation of negative regulatory tyrosine residue Tyr592, SAMHD1 becomes
activated and reduces the cytosolic pool of free dNTPs, impairing HIV-1 reverse transcription in
dasatinib-pretreated primary CD4+ T-cells [537]. Dasatinib is a safe and well tolerated kinase
inhibitor, approved for second-line treatment of CML [553]. Moreover, the related SRC/ABL
inhibitor imatinib has been used to safely treat KS or CML cancers in HIV-1 patients on fully
suppressive cART, improving long-term survival [176, 599, 600]. These clinical examples of
kinase inhibitor provokes the question: Can dasatinib be used safely in conjunction with cART to
further suppress HIV-1 infection? At doses within well-tolerated levels in humans, dasatinib has
also been shown to inhibit the ex vivo reactivation of HIV-1 from the CD4+
T-cells of treatment
naïve, HIV-1 donors [554]. To investigate the mechanism of action of dasatinib, which targets

182
Abelson murine Leukemia viral oncogene homolog 1 (ABL1) and Abelson-Related Gene (ARG)
kinases as well as c-SRC, the role of these two ABL family kinases during HIV-1 infection is
another active area of future research. Furthermore, current investigations in our lab are
expanding on the findings presented in chapter 3, to test whether dasatinib treatment can reduce
in vivo Ba-L infection in a humanized mouse model of HIV-1 infection. This model will be an
excellent means of studying HIV-1 resistance mutations that may develop from weeks of
dasatinib monotherapy, which could be pre-empted with combination therapy if they occur. We
will also explore whether dasatinib can be included in cART as an alternative integration
inhibitor in this mouse model of HIV-1 infection. Furthermore, CRISPR/Cas9 may be used to
knockout c-SRC or PTK2B in human CD34+
Hematopoietic Progenitor Cells (HPCs) prior to
their transplantation into mice, to ascribe more definitive in vivo roles of c-SRC and PTK2B
signaling during HIV-1 infection.
ZEBOV
In September of 2014, during the height of the ZEBOV outbreak in West Africa and when the
first documented case of ZEBOV transmission occurred in the United States [601], the need to
rapidly assess drugs for preclinical ZEBOV antiviral activity was of paramount concern to the
National Microbiology Laboratory of Canada, US CDC, USAMRIID, and the WHO Working
Group on Ebola Therapeutics. By establishing a new trVLP model of ZEBOV replication in the
lab [534], the four c-SRC inhibitors from chapter 3 were initially tested for activity against
ZEBOV trVLP replication. While this SFK research was a novel avenue of its own, and
preliminary results were promising, the urgent need to test and compare experimental therapies,
either fast-tracked for upcoming phase II/III clinical trials in West Africa (BCV, FPV and IFN-
β), or already being used for compassionate EVD treatment (AZT and 3TC), took precedent for
in vitro testing [29, 31, 431, 519]. Thus, 8 compounds from 3 drug classes were evaluated alone
or in combination, to provide additional rationale for further preclinical development or their
potential use in emergency ZEBOV clinical trials (chapter 4).

183
HEK 293T cells are an ideal cell line for the ZEBOV trVLP model because their high
transfection rate produces many virions [534], and the cells can quickly convert nucleoside
analogs to their active triphosphate form analagous to primary hepatocytes [418]. Indeed,
VeroE6 show slower phosphorylation of nucleoside analogs and may under represent drug
potency [418]. As 293T cells are not the natural target cell infected by ZEBOV, it was important
to first validate the in vitro model with therapeutic treatments published to inhibit fully infectious
ZEBOV in other cell lines [23, 586]. The ZEBOV GP1,2 entry inhibitor TOR showed very
similar IC50 results in the trVLP model (0.36 μM) as with publications of TOR inhibiting
ZEBOV in VeroE6 (0.2 μM) or HepG2 cells (0.03 μM) [586]. Similarly, FPV has been reported
to inhibit ZEBOV in VeroE6 cells with an IC50 of 67 μM [23], comparable to the IC50 of
36.8 μM found for ZEBOV trVLP in chapter 4. Together, this data suggested the trVLP model
of ZEBOV infection in 293T cells could be used to screen potential antiviral compounds.
As predicted from in silico simulations of RdRP L nucleotide analogue docking [456], 3TC,
AZT and TFV each showed dose-dependent inhibition of ZEBOV trVLP luciferase activity,
which was independent of whether the drug was administered a day before, during or one day
after infection (chapter 4). TFV, which was predicted out of 20 nucleoside and nucleotide
analogs to have the strongest interaction with the putative RdRP L nucleotide binding pocket
[456], showed the lowest IC50 of the 5 nucleoside analogs tested (0.98 μM). This was followed
by AZT (4.2 μM), 3TC (6.2 μM) and CDV (7.7 μM) giving similar doses for 50% inhibition.
Interestingly FPV had the highest IC50 (36.8 μM). FPV is a drug that is required in high doses to
protect small animals from lethal ZEBOV exposure [29]. It is also required in high doses in non-
human primates to achieve target plasma levels [441]. Furthermore, FPV showed sub-optimal
dosing in the blood samples of EVD patients in the recently completed phase II JIKI trial in
Guinea [444]. From these multiple lines of investigation, it would appear FPV is a mild inhibitor
of ZEBOV RdRP that could benefit from synergistic drug combinations to reduce the high doses
of drug required in monotherapy.
The work presented in chapter 4 is the first account of 3TC, AZT or TFV having antiviral
activity against ZEBOV trVLP replication or against ZEBOV-GFP in vitro infection in BSL4.
As a potential mechanism of action, these three nucleoside/nucleotide analogues were then

184
shown to inhibit viral synthesis of negative-sense genomic vRNA and positive-sense cRNA and
mRNA, as measured by qRT-PCR. TFV showed the strongest antiviral activity of the 5
nucleoside analogues tested in this qRT-PCR assay. At first, these findings appear to be in
contrast with recent published work suggesting 3TC and AZT exhibit weak in vitro activity
against ZEBOV replication [518]. However, the publication showed similarly mild
dose/response curves of 3TC and AZT against ZEBOV-GFP, as reported in chapter 4. For
instance, 25 μM of 3TC caused 18% inhibition of ZEBOV-Makona in their report [518], while
the same 25 μM dose of 3TC caused 22% inhibition of ZEBOV-GFP in chapter 4. It was
anticipated that the required drug doses would be higher when moving from the ZEBOV trVLP
model to fully intact ZEBOV-GFP infection at an MOI of 0.1. Nevertheless, the primary value
in testing 3TC and AZT came not from monotherapy alone, but their potential to synergize in the
trVLP model of ZEBOV infection. In the top 7 two-drug and top 7 three-drug combinations
demonstrating the strongest synergy against ZEBOV-GFP, 3TC was included in 5 of these
combinations, and AZT was in 5 combinations as well (chapter 4). Likewise, the recent ZEBOV
study testing 3TC and AZT also reported drug synergy when combining 12.5 μM of 3TC with 25
μM of AZT, markedly reducing ZEBOV-Makona infection by 70% [518]. To further clarify the
mechanisms of 3TC and AZT ZEBOV inhibition, future work demonstrating RNA chain
termination of RdRP L in the presence of either nucleoside analogue, and crystal structures of the
RdRP L enzyme bound with either compound, would be desirable. In addition, the antiviral
activity of 3TC and AZT in ZEBOV infection should be confirmed in primary monocyte-derived
macrophages, to strengthen the preclinical evidence for evaluating these two drugs further in
small animal models of ZEBOV infection.
Treating 293T cells with the metabolite of BCV (CDV) at or above 100 μM prior to or post-
ZEBOV trVLP infection, unexpectedly and consistently increased viral luciferase activity 4 days
post-infection (chapter 4). Although CDV inhibited trVLP luciferase activity in a small range of
concentrations (1.25-25 μM), it reduced the antiviral activity of many of the other 7 drugs tested
in two- or three-drug combinations. Others have reported BCV to enhance ZEBOV-GFP
infection in Huh7 cells, at doses less than 0.2 μM [28]. In two recent phase III trials, BCV has
failed to show efficacy in treating ADV infections or preventing CMV infection during

185
hematopoietic cell transplantation in patients. Additionally, a third phase III trial of BCV to
prevent CMV infection after kidney transplant was recently terminated. BCV was originally
created to be a less toxic alternative to CDV, and shows no associated nephrotoxicity in patients
[602]. However, an oral BCV dose of 200 mg twice a week in a study of 30 participants was
shown to elevate liver enzymes detected in blood samples, such as Alanine Transaminase (ALT)
(40% of participants were > 3x upper limit of normal range) [413]. Elevated bloodstream levels
of liver enzymes can indicate potential inflammation or injury to the liver [603]. Two hundred
mg of BCV twice a week also caused diarrhea for 70% of the participants, leading to 60% of
those taking the drug to withdraw from the study from adverse effects [413]. It is possible that in
the BCV phase II ZEBOV trial, which only recruited 4 EVD patients of which none survived
[24], BCV may have unintentionally exacerbated liver dysfunction and diarrhea symptoms.
Evidence supporting this are: 1) Patient #2 in the BCV ZEBOV trial had hepatic injury (high
ALT levels) and persistent diarrhea, leading to suspension of BCV treatment by day 7 [24]. 2) It
was determined in the JIKI trial, which measured baseline viremia in 58 EVD patients and serum
ALT over 25 days, that patients nearing death had elevated ALT levels compared with those
who survived [18]. Collectively, these BCV findings and CDV results presented in chapter 4
highlight the necessity of establishing clear preclinical in vitro antiviral activity and in vivo
animal efficacy in preventing or treating lethal ZEBOV challenge, prior to initiating a phase II
trial during an epidemic setting [24].
The last key finding from chapter 4 was that specific combinations of the 8 drugs tested
exhibited strong synergy against ZEBOV trVLP, which were confirmed with fully infectious
ZEBOV-GFP in BSL4. FPV showed synergy with all other drugs tested, suggesting
recombinant IFNs, TOR or nucleoside analogs could be used with FPV to reduce its
monotherapy dose and maintain the same level of inhibition. This is in line with FPV
combinations with ribavirin that synergistically inhibit other RNA viral infections, such as
(-)ssRNA Lassa virus, Pichinde virus and RVFV, and (+)ssRNA Junin arenavirus [435, 436,
530]. Moreover, synergistic two drug combinations, such as IFN-β + 3TC or IFN-β + AZT,
were predictive of the synergy when testing the three drugs together (IFN-β + 3TC + AZT). Of
combinations only comprising nucleoside/nucleotide analogues, FPV + TFV showed the highest

186
synergy. This result was predicted in silico from their combined interaction energy displacing
NTPs at the putative nucleotide binding motif of RdRP L, with FPV binding from the “left” and
TFV binding from the “right” of the polymerase nucleotide binding pocket (see Figure 5.2 for
binding of TFV + FPV outcompeting NTP binding, originally published in [456]). In addition,
the in vitro trVLP model in chapter 4 permitted rapid testing of multiple drug combinations, with
hits inhibiting luciferase activity as high as 97.3% and with no appreciable affect on cell
viability. This data allowed for 14 combinations to then be prioritized for BSL4 testing at the
National Microbiology Laboratory in Winnipeg. As in ZEBOV trVLP infection, the
combinations of IFN-β + 3TC or IFN-β + 3TC + AZT showed the strongest drug synergy against
ZEBOV-GFP in vitro. Moreover, the consistency in which low doses of recombinant IFN- β
inhibited ZEBOV alone or in combination added to the strong preclinical evidence supporting
this treatment to be considered in a phase II ZEBOV clinical trial [22, 31]. On March 26th
2015,
a phase II IFN- β-1a proof-of-concept, historically controlled, single-arm trial started in the
western town of Coyah, Guinea, and completed by June 12th
2015 [22]. While the results in
chapter 4 are but one of many sources of data that contributed to the planning of this trial, it is an
example of real-world impact this thesis has already made.
Presently, there are no active cases of acute EVD reported by the WHO in West Africa [5]. Yet
from May to June 2017, a small outbreak of EVD (5 confirmed cases, 3 probable cases) occurred
in the North-East province of Likati in the Democratic Republic of the Congo [604]. While the
WHO is confident this new outbreak will be contained, this should not engender research
complacency between larger EBOV outbreaks. On the contrary, this time provides an
opportunity for clinicians and scientists to produce rigorous and sound preclinical evidence that
better inform policy makers, who may again priorotize EBOV clinical trials during an active
EBOV outbreak. In our lab, future work has begun on screening antimalarial derivatives of
quinoline, to determine antiviral activity in the ZEBOV trVLP model of infection in 293T cells.
Moreover, earlier work on SFK inhibitors that inhibit trVLP replication has been

187
Fig. 5.2: Model of FPV-RTP and TFV-DP binding the RdRP L nucleotide pocket of
ZEBOV. Preferential binding of favipiravir-RTP (red) to the “left” side and tenofovir-DP
(orange) to the “right” side of the nucleotide binding motif (yellow). The total interaction energy
of both nucleotide analogues with the polymerase is significantly lower than each NTP (green),
displacing them to locations with decreased affinity. (A) ATP. (B) CTP. (C) GTP. (D) UTP
[456].

188
reinitiated. This investigation of host tyrosine kinases will likely be a promising new area of
research, as 53 host-protein interactions during ZEBOV infection of VeroE6 cells have recently
been described, and little is known of the role of NRTK signaling during ZEBOV replication
[357]. Only two such studies have been published [605, 606]. The first showed that ABL1
phosphorylation of VP40 matrix protein at Tyr13 was essential for budding of newly assembled
ZEBOV virions, by employing siRNA or a specific inhibitor (nilotinib) targeting ABL1 activity
[605]. The second publication also used FDA-approved kinase inhibitors (sunitinib and
erlotinib) to inhibit ZEBOV entry [606], providing additional rationale to explore SFK signaling
and the repurposing of kinase inhibitors in future ZEBOV lifecycle research.

189
5.2 Conclusions and Broader Significance
HIV-1
In summary, this work has specifically shown that c-SRC and PTK2B non-receptor tyrosine
kinases are key mediators of early HIV-1 replication in various T-cell lines and in activated
primary CD4+ T-cells from healthy donors. Using multiple experimental methods (including
tool-drug inhibitors, adenovirus vectors overexpressing DN c-SRC mutant, and targeted siRNA
knockdown) and testing of various strains of HIV-1 viruses (X4- or R5-tropic, fully infectious or
nef-deleted luciferase reporter viruses, VSV-G pseudo-enveloped viruses, laboratory strains and
primary clinical isolates), it was determined that reducing the activity or expression of c-SRC
consistently increased viral infection in transformed T-cell lines, but reduced viral infection in
primary CD4+ T-cells. Results with primary CD4
+ T-cells were taken as the more physiological
relevant results and further examined. Inhibition of viral infection of CD4+ T-cells was
associated with no change in RT activity, increased early and late reverse transcripts, reduced
proviral integration, and higher accumulation of 2-LTR circles, suggesting two novel
mechanisms by which this kinase may be altering intracellular HIV-1 replication: 1) At the level
of the pre-integration complex formation in the cytosol, and 2) During PIC nuclear translocation
and viral integration. Similar results were discovered for the c-SRC binding partner PTK2B.
However reduced PTK2B kinase expression only appeared to decrease HIV-1 infection when R5
viruses were tested. These findings offer the potential for better characterization of the PIC and
its associated host proteins, as arresting the virus at this stage from c-SRC siRNA knockdown
could allow for better isolation of this elusive protein/cDNA complex that is hard to characterize
in primary CD4+ T-cells [138, 607, 608].
The dependency of early HIV-1 infection on c-SRC signaling was a novel and complex finding.
This was unlike other SFKs FYN and LCK, which transmit signals as HIV-1 binds and fuses at
the plasma membrane, interact with viral Nef (LCK), change the recycling of surface proteins at
the trans-Golgi network (LCK), phosphorylate APOBEC3G (FYN) or facilitate viral egress
(LCK) [208, 217, 218, 223, 227, 238]. The work presented here strongly supports that c-SRC
has a unique cytosolic and nuclear role in the cell biology of HIV-1 infection of CD4+ T-cells. It

190
demonstrates that c-SRC regulates HIV-1 infection at multiple stages post-entry, reminiscent of
the integrase inhibitor raltegravir with respect to the nuclear effects of c-SRC [565, 566].
Furthermore, this research shows that four specific inhibitors of c-SRC (dasatinib, saracatinib,
KX2-391 and SRC Inhibitor-1), each with different off-target activity, can significantly reduce
HIV-1 infection of CD4+ T-cells. These four inhibitors, FDA-approved or under clinical
evaluation as potential cancer treatments [553, 561, 609], showed excellent cell viability data at
doses that significantly inhibit HIV-1 replication ex vivo. These results lay the foundation for
further preclinical testing of dasatinib, alone or concurrent with cART treatment, in a humanized
mouse model of HIV-1 infection.
The findings in this work make a significant advance in the fields of HIV-1 cell signalling and
novel HIV-1 treatment strategies, by demonstrating the roles of two non-receptor tyrosine
kinases during early HIV-1 entry of CD4+ T-cells, and evaluating kinase inhibitors that can
specifically inhibit one of these two targets. The fundamental roles of host kinases in the HIV-1
lifecycle are increasing being recognized, allowing for rapid discovery of kinase inhibitors as
new potential therapies of HIV-1 infection [146, 156, 160, 610]. Small-molecule kinase
inhibitors that can be repurposed for new indications in virology offer many significant
advantages [169, 170]. They often have low toxicity profiles, and broad tissue distribution. In
vitro studies suggests they may offer unique ways to inhibit HIV-1 replication in viral
sanctuaries [560, 611, 612], potentially altering the dynamics of the latent viral reservoir. They
can also be paired with latency reversal agents to prevent unwanted inflammation during “shock
and kill” methods [173]. Moreover, kinase inhibitors have the potential for ameliorating
underlining inflammation and neural cognitive dysfunction that occur during HIV-1 infection
[173, 613], which are not addressed adequately with current cART regimens. Kinase inhibitors
could also reduce the risk of cancers in HIV-1 patients, as demonstrated by their ability to treat
KS or CML without causing additional complications, extending the life expectancy of these
HIV-1 patients [176, 600]. Additionally, targeting host kinases essential for early intracellular
infection may pose higher barriers towards selecting drug-resistant mutations, as they would not
directly inhibit viral proteins [135, 159, 539]. Thus, tyrosine kinase inhibitors could be a useful
part of a multidrug regimen for those living with HIV-1. There is a strong need to design not

191
only intensifying therapies, but therapies that improve immune function in HIV-1 patients, such
as re-establishing CD4+ T-cell populations at gut mucosa [80]. The clinical application of SRC-
family kinase inhibitors of known specificity, biodistribution, pharmacokinetics, toxicity profile
and ease of administration, suggest these drugs could be repositioned as novel inhibitors of HIV-
1 infection, and should be explored further in small animal models of HIV-1 disease progression.
ZEBOV
The 2014-16 outbreak of ZEBOV in West Africa challenged global healthcare workers and
virology researchers from different backgrounds to come together and quickly assess which
preclinical drugs, treatments and vaccines should be prioritized for emergency evaluation in
phase II/III ZEBOV clinical trials. Many new molecules were discovered to have in vitro
antiviral activity [586], or improved the survival in animal models when treated after an
otherwise lethal ZEBOV exposure [6, 25]. The phase II/III ZEBOV clinical trials were the first
of their kind in an outbreak setting during a humanitarian crisis, which led to many lessons for
future trials, such as: ethical considerations of consent and the use of placebos; standardization of
intensive supportive care; the fostering of local, regional and global partnerships; establishing
multiple treatment sites as the spread of disease rapidly shifted; and new trial formats such as the
cluster-randomized ring design [16, 25, 26]. In particular, multiple approaches from HIV-1 drug
research, spanning from preclinical drug screening to treating patients who suffer not only from
EVD, but associated disease stigmatization, were very relevant in Liberia, Guinea and Sierra
Leone during the ZEBOV outbreak.
The creation of a sophisticated in vitro lifecycle model of ZEBOV in 2014 [534] allowed for
rapid assessment of 8 different drugs from three drug classes, including toremifene,
nucleoside/nucleotide analogues, and recombinant interferons. This thesis made significant
contributions by comparing and contrasting each of these compounds, testing various time-
points, doses, and 84 unique combinations, while assessing cell viability and antiviral activity
against a ZEBOV trVLP luciferase reporter virus. It was demonstrated for the first time that safe
and well tolerated nucleoside analogues 3TC, AZT and TFV, often used to treat HIV-1 or HBV
infection [408, 410], can inhibit ZEBOV trVLP replication in vitro. This was confirmed with

192
qRT-PCR measurements of ZEBOV vRNA and cRNA/mRNA synthesis 2 days post-infection.
The antiviral activity of 3TC, AZT and TFV were comparable or lower than the IC50 of
nucleoside analogues fast-tracked for phase II trials in Guinea and Liberia: FPV and the
metabolite of BCV (CDV) [18, 24]. While neither FPV nor BCV demonstrated significant
therapeutic benefit in these clinical trials, lessons were learned of unwanted side effects from
BCV administration [24], and lower than anticipated plasma levels from FPV monotherapy [18].
Retrospective analysis of compassionate FPV use suggest this drug should continue to be
developed for experimental treatment of EVD in a future EBOV outbreak, in addition to
intensive supportive care [445].
Testing 5 nucleoside/nucleotide analogs in the trVLP replication model confirmed many of the in
silico predictions of how these drugs might dock to the nucleotide binding motif of RdRP L and
outcompete NTP substrates [456]. TFV was predicted to have the strongest interaction energy
with the enzyme [456], and it was confirmed to have the lowest IC50 of the 5 nucleoside analogs
tested. Moreover, simulations of two nucleoside analogs flanking the nucleotide binding motif
from the “left” and “right” predicted strong synergy when combining TFV with FPV [456], a
drug combination that experimentally was confirmed to be highly synergistic in vitro. In
addition, testing of two-drug combinations rationally predicted synergistic three drug
combinations, adding more strength to the methodology used [533]. Lastly, confirmation of
antiviral activity of the 14 most synergistic drug combinations occurred with fully infectious
ZEBOV expressing a GFP reporter, demonstrating multiple efficacious combinations that
included FPV or IFN-β. Thus both of these drugs, which were evaluated in phase II ZEBOV
clinical trials as monotherapies [18, 22], could very well be improved by combinations with
other nucleoside analogues. These hypotheses should be investigated further in small animal or
non-human primate models of lethal ZEBOV challenge.
Combinations of monoclonal antibodies and siRNAs were behind the early success of ZMapp
and TKM-Ebola; however, they had shortcomings in drug production or treating people with
EVD in controlled clinical trials [27, 404]. Combinations with nucleoside analogues are now on
the leading edge of EBOV drug discovery, as ZMapp is actively being studied in combination
with either FPV or recombinant IFN-β. Thus, the nucleoside analogues 3TC, AZT and TFV

193
could fill the gap where monotherapy has so far failed to demonstrate therapeutic benefit, by
increasing the efficacy of other administered treatments as combination regimens. Moreover,
nucleoside analogues may have the additional advantage of exploiting the spontaneous mutation
rate of ZEBOV [614]. While the frequency of ZEBOV mutations are high, as other RNA
viruses, they are not represented in the genetic diversity of viable strains, suggesting nucleoside
analogues that increase the error rate of RdRP L cause harmful mutagenesis, reducing the fitness
of ZEBOV during infection [614]. Nucleoside analogs are also on the WHO list of essential
medicines, and can be deployed in resource limited settings, often without need of cold storage
[574]. Taken together, the findings presented in this thesis demonstrate the value of an in vitro
BSL2 trVLP screening system prior to BSL4 ZEBOV testing, allowing for direct comparisons of
8 compounds with suggested or known antiviral activity against ZEBOV replication [29, 31].
This work also contributes to the growing body of evidence advocating for combination therapy
as standard treatment of RNA viruses and retroviruses, to improve efficacy and theoretically
reduce the risk of drug resistance [456, 523, 530, 532, 615]. In future studies, this ZEBOV
trVLP model will continue to allow rapid assessment of new compounds of interest, such as SFK
inhibitors and quinoline derivatives, to directly compare their antiviral activity with leading
compounds in ongoing or planned ZEBOV clinical trials.
As of June 2016, there are approximately 17,300 Ebola survivors in West Africa, the largest such
Ebola cohort ever recorded [5]. The management of chronic disease that is becoming increasing
recognized as post-Ebola syndrome, is under active investigation [6, 450]. Moreover the
mechanisms of viral persistence, and the risk of ZEBOV reemergence and shedding, are also
being actively studied [357, 360]. Ebola survivors with persistent symptoms and low-level viral
replication in immune-privileged organs may benefit from antiviral drugs with appropriate
permeability, such as the nucleoside analog GS-5734 [417]. ZEBOV virions have been found in
the semen of male Ebola survivors, which can cause acute EVD if transmitted to their partner,
suggesting ZEBOV virus transmission can be similar to other sexually transmitted viruses [360].
Hence clinical trials testing the efficacy of nucleoside analogues to reduce the shedding of
virions in the semen of male Ebola survivors are currently underway: a phase II FPV trial in
Guinea and a phase II GS-5734 trial in Liberia [419].

194
The successful rVSV-ZEBOV-GP cluster-randomized ring vaccine trial in Guinea, which
protected 100% of those immeditaly vaccinated (N = 2,119) compared with those receiving
delayed vaccination (N = 2,041) [26], may provide protection in future ZEBOV outbreaks for
health care workers and quarantined contacts of EVD patients. Until the vaccine demonstrates
durable protection and broad neutralization of the five Ebolavirus species (ZEBOV, SUDV,
BDBV, RESTV and TAFV) a future outbreak emerging from a new EBOV strain may be
resistant to the current rVSV-ZEBOV-GP formulation. Thus, the rVSV-ZEBOV-GP vaccine is
one of many components in the EBOV toolkit needed to reduce the spread of future infections.
Government cooperation, effective border screening of EVD symptoms, robust healthcare
infrastructure, extensive training of healthcare personnel, proper Personel Protective Equipment
(PPE), rapid onsite EBOV antigen testing, contact tracing, community engagement, public health
education, and access to the basic necessities of life, all contribute to the epidemiological spread
and control of an EBOV epidemic. Thus, an effective treatment or combination of treatments
could have far reaching consequences in the transmission of EBOV in addition to immediate
patient care. An effective treatment could provide psychological reassurance that improves trust
between healthcare staff and ill patients, encouraging people to seek treatment at an ETC and
reduce EBOV spread among family caregivers at home. An emergent EBOV zoonose may also
show higher lethality compared to the ZEBOV-Makona and ZEBOV-Kikwit strains of the most
recent EBOV outbreak. For all of these reasons, future research into effective nucleoside analog
drug combinations should continue to proceed in preclinical in vivo models of EBOV infection,
until there is an effective treatment for acute and chronic EBOV infection.

195
References

196
1. de Souza MS, Ratto-Kim S, Chuenarom W, et al. The Thai phase III trial (RV144)
vaccine regimen induces T cell responses that preferentially target epitopes within the V2
region of HIV-1 envelope. J Immunol 2012; 188:5166-5176.
2. UNAIDS, World Health Organization. A framework for voluntary medical male
circumcision: Effective HIV prevention and a gateway to improved adolescent boys’ &
men’s health in eastern and southern Africa by 2021. 2016.
http://www.who.int/hiv/pub/malecircumcision/vmmc-policy-2016/en/.
3. Zhang L, Ramratnam B, Tenner-Racz K, et al. Quantifying residual HIV-1 replication in
patients receiving combination antiretroviral therapy. N Engl J Med 1999; 340:1605-
1613.
4. GARP, UNAIDS. Global AIDS Update. 2016.
http://www.unaids.org/en/resources/documents/2016/Global-AIDS-update-2016
5. World Health Organization. Ebola Situation Report, 10 June 2016. 2016.
http://apps.who.int/ebola/ebola-situation-reports
6. Madelain V, Nguyen TH, Olivo A, et al. Ebola Virus Infection: Review of the
Pharmacokinetic and Pharmacodynamic Properties of Drugs Considered for Testing in
Human Efficacy Trials. Clin Pharmacokinet 2016; 55:907-923.
7. Solomon DA, Sax PE. Current state and limitations of daily oral therapy for treatment.
Curr Opin HIV AIDS 2015; 10:219-225.
8. Public Health Agency of Canada. Summary: Estimates of HIV Incidence, Prevalence and
Proportion Undiagnosed in Canada, 2014. 2015.
http://www.catie.ca/sites/default/files/2014-HIV-Estimates-in-Canada-EN.pdf
9. Archin NM, Vaidya NK, Kuruc JD, et al. Immediate antiviral therapy appears to restrict
resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection.
Proc Natl Acad Sci U S A 2012; 109:9523-9528.
10. Ma Q, Vaida F, Wong J, et al. Long-term efavirenz use is associated with worse
neurocognitive functioning in HIV-infected patients. J Neurovirol 2015; 22:170-178.
11. Gibb DM, Kizito H, Russell EC, et al. Pregnancy and infant outcomes among HIV-
infected women taking long-term ART with and without tenofovir in the DART trial.
PLoS Med 2012; 9:e1001217.
12. Scherzer R, Estrella M, Li Y, et al. Association of tenofovir exposure with kidney disease
risk in HIV infection. Aids 2012; 26:867-875.
13. El-Sherif O, Khoo S, Solas C. Key drug-drug interactions with direct-acting antiviral in
HIV-HCV coinfection. Curr Opin HIV AIDS 2015; 10:348-354.

197
14. von Moltke LL, Greenblatt DJ, Grassi JM, et al. Protease inhibitors as inhibitors of
human cytochromes P450: high risk associated with ritonavir. J Clin Pharmacol 1998;
38:106-111.
15. Marzolini C, Gibbons S, Khoo S, et al. Cobicistat versus ritonavir boosting and
differences in the drug-drug interaction profiles with co-medications. J Antimicrob
Chemother 2016; 71:1755-1758.
16. Leligdowicz A, Fischer WA, 2nd, Uyeki TM, et al. Ebola virus disease and critical
illness. Crit Care 2016; 20:10.1186.
17. Mendoza EJ, Qiu X, Kobinger GP. Progression of Ebola Therapeutics During the 2014-
2015 Outbreak. Trends Mol Med 2016; 22:164-173.
18. Sissoko D, Laouenan C, Folkesson E, et al. Experimental Treatment with Favipiravir for
Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-
Concept Trial in Guinea. PLoS Med 2016; 13:e1001967.
19. Davey RT, Jr., Dodd L, Proschan MA, et al. A Randomized, Controlled Trial of ZMapp
for Ebola Virus Infection. N Engl J Med 2016; 375:1448-1456.
20. van Griensven J, Edwards T, de Lamballerie X, et al. Evaluation of Convalescent Plasma
for Ebola Virus Disease in Guinea. N Engl J Med 2016; 374:33-42.
21. Thi EP, Mire CE, Lee AC, et al. Lipid nanoparticle siRNA treatment of Ebola-virus-
Makona-infected nonhuman primates. Nature 2015; 521:362-365.
22. Konde MK, Baker DP, Traore FA, et al. Interferon beta-1a for the treatment of Ebola
virus disease: A historically controlled, single-arm proof-of-concept trial. PLoS One
2017; 12:e0169255.
23. Oestereich L, Ludtke A, Wurr S, et al. Successful treatment of advanced Ebola virus
infection with T-705 (favipiravir) in a small animal model. Antiviral Res 2014; 105:17-
21.
24. Dunning J, Kennedy SB, Antierens A, et al. Experimental Treatment of Ebola Virus
Disease with Brincidofovir. PLoS One 2016; 11:e0162199.
25. Cardile AP, Downey LG, Wiseman PD, et al. Antiviral therapeutics for the treatment of
Ebola virus infection. Curr Opin Pharmacol 2016; 30:138-143.
26. Henao-Restrepo AM, Camacho A, Longini IM, et al. Efficacy and effectiveness of an
rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea
ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 2016;
389:505-518.

198
27. Dunning J, Sahr F, Rojek A, et al. Experimental Treatment of Ebola Virus Disease with
TKM-130803: A Single-Arm Phase 2 Clinical Trial. PLoS Med 2016; 13:e1001997.
28. McMullan LK, Flint M, Dyall J, et al. The lipid moiety of brincidofovir is required for in
vitro antiviral activity against Ebola virus. Antiviral Res 2016; 125:71-78.
29. Smither SJ, Eastaugh LS, Steward JA, et al. Post-exposure efficacy of oral T-705
(Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res
2014; 104:153-155.
30. Qiu X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman
primates with ZMapp. Nature 2014; 514:47-53.
31. Smith LM, Hensley LE, Geisbert TW, et al. Interferon-beta therapy prolongs survival in
rhesus macaque models of Ebola and Marburg hemorrhagic fever. J Infect Dis 2013;
208:310-318.
32. Stanley DA, Honko AN, Asiedu C, et al. Chimpanzee adenovirus vaccine generates acute
and durable protective immunity against ebolavirus challenge. Nat Med 2014; 20:1126-
1129.
33. Qiu X, Fernando L, Alimonti JB, et al. Mucosal immunization of cynomolgus macaques
with the VSVDeltaG/ZEBOVGP vaccine stimulates strong ebola GP-specific immune
responses. PLoS One 2009; 4:e5547.
34. Curran JW. Pneumocystis pneumonia--Los Angeles. MMWR Morb Mortal Wkly Rep
1981; 30:250-252.
35. Quagliarello V. The Acquired Immunodeficiency Syndrome: current status. Yale J Biol
Med 1982; 55:443-452.
36. Barre-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus
from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983;
220:868-871.
37. Vilmer E, Barre-Sinoussi F, Rouzioux C, et al. Isolation of new lymphotropic retrovirus
from two siblings with haemophilia B, one with AIDS. Lancet 1984; 1:753-757.
38. Gallo RC, Salahuddin SZ, Popovic M, et al. Frequent detection and isolation of
cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS.
Science 1984; 224:500-503.
39. Sarngadharan MG, Popovic M, Bruch L, et al. Antibodies reactive with human T-
lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science 1984;
224:506-508.

199
40. Keele BF, Van Heuverswyn F, Li Y, et al. Chimpanzee reservoirs of pandemic and
nonpandemic HIV-1. Science 2006; 313:523-526.
41. Jespersen S, Tolstrup M, Honge BL, et al. High level of HIV-1 drug resistance among
patients with HIV-1 and HIV-1/2 dual infections in Guinea-Bissau. J Virol 2015;
12:10.1186.
42. Vermund SH, Hayes RJ. Combination prevention: new hope for stopping the epidemic.
Curr HIV/AIDS Rep 2013; 10:169-186.
43. Faria NR, Rambaut A, Suchard MA, et al. HIV epidemiology. The early spread and
epidemic ignition of HIV-1 in human populations. Science 2014; 346:56-61.
44. Disease NIoAaI, National Institutes Health. How HIV Causes AIDS. 2004.
45. Suzuki Y, Suzuki Y. Gene Regulatable Lentiviral Vector System. In: Viral Gene
Therapy. Edited by Xu K. Rijeka: InTech; 2011. pp. 10.5772.
46. Brian Foley TL, Cristian Apetrei, Beatrice Hahn, Ilene Mizrachi, James Mullins, Andrew
Rambaut, Steven Wolinsky, and Bette Korber. HIV Sequence Compendium 2016. In. Los
Alamos, New Mexico.: Los Alamos National Laboratory; 2016.
47. Kwong PD, Wyatt R, Robinson J, et al. Structure of an HIV gp120 envelope glycoprotein
in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998;
393:648-659.
48. Coakley E, Petropoulos CJ, Whitcomb JM. Assessing chemokine co-receptor usage in
HIV. Curr Opin Infect Dis 2005; 18:9-15.
49. Lawson VA, Oelrichs R, Guillon C, et al. Adaptive changes after human
immunodeficiency virus type 1 transmission. AIDS Res Hum Retroviruses 2002; 18:545-
556.
50. Miyauchi K, Kim Y, Latinovic O, et al. HIV enters cells via endocytosis and dynamin-
dependent fusion with endosomes. Cell 2009; 137:433-444.
51. Furuta RA, Wild CT, Weng Y, et al. Capture of an early fusion-active conformation of
HIV-1 gp41. Nat Struct Biol 1998; 5:276-279.
52. Spear M, Guo J, Turner A, et al. HIV-1 triggers WAVE2 phosphorylation in primary
CD4 T cells and macrophages, mediating Arp2/3-dependent nuclear migration. J Biol
Chem 2014; 289:6949-6959.
53. Coiras M, Lopez-Huertas MR, Perez-Olmeda M, et al. Understanding HIV-1 latency
provides clues for the eradication of long-term reservoirs. Nat Rev Microbiol 2009;
7:798-812.

200
54. Jayappa KD, Ao Z, Yao X. The HIV-1 passage from cytoplasm to nucleus: the process
involving a complex exchange between the components of HIV-1 and cellular machinery
to access nucleus and successful integration. Int J Biochem Mol Biol 2012; 3:70-85.
55. Daniel R, Katz RA, Skalka AM. A role for DNA-PK in retroviral DNA integration.
Science 1999; 284:644-647.
56. Bukrinsky M, Sharova N, Stevenson M. Human immunodeficiency virus type 1 2-LTR
circles reside in a nucleoprotein complex which is different from the preintegration
complex. J Virol 1993; 67:6863-6865.
57. Bukrinsky MI, Stanwick TL, Dempsey MP, et al. Quiescent T lymphocytes as an
inducible virus reservoir in HIV-1 infection. Science 1991; 254:423-427.
58. Sweet T, Sawaya BE, Khalili K, et al. Interplay between NFBP and NF-kappaB
modulates tat activation of the LTR. J Cell Physiol 2005; 204:375-380.
59. Jayaraman B, Crosby DC, Homer C, et al. RNA-directed remodeling of the HIV-1
protein Rev orchestrates assembly of the Rev-Rev response element complex. Elife 2014;
3:e04120.
60. Fenouillet E, Jones IM. The glycosylation of human immunodeficiency virus type 1
transmembrane glycoprotein (gp41) is important for the efficient intracellular transport of
the envelope precursor gp160. J Gen Virol 1995; 76:1509-1514.
61. Pfeiffer T, Zentgraf H, Freyaldenhoven B, et al. Transfer of endoplasmic reticulum and
Golgi retention signals to human immunodeficiency virus type 1 gp160 inhibits
intracellular transport and proteolytic processing of viral glycoprotein but does not
influence the cellular site of virus particle budding. J Gen Virol 1997; 78:1745-1753.
62. Borsetti A, Ohagen A, Gottlinger HG. The C-terminal half of the human
immunodeficiency virus type 1 Gag precursor is sufficient for efficient particle assembly.
J Virol 1998; 72:9313-9317.
63. Wen X, Ding L, Wang JJ, et al. ROCK1 and LIM kinase modulate retrovirus particle
release and cell-cell transmission events. J Virol 2014; 88:6906-6921.
64. Liang C, Hu J, Russell RS, et al. Translation of Pr55(gag) augments packaging of human
immunodeficiency virus type 1 RNA in a cis-acting manner. AIDS Res Hum Retroviruses
2002; 18:1117-1126.
65. Linde ME, Colquhoun DR, Ubaida Mohien C, et al. The conserved set of host proteins
incorporated into HIV-1 virions suggests a common egress pathway in multiple cell
types. J Proteome Res 2013; 12:2045-2054.

201
66. Tessmer U, Krausslich HG. Cleavage of human immunodeficiency virus type 1
proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag
polyprotein processing and viral infectivity. J Virol 1998; 72:3459-3463.
67. Ayehunie S, Groves RW, Bruzzese AM, et al. Acutely infected Langerhans cells are
more efficient than T cells in disseminating HIV type 1 to activated T cells following a
short cell-cell contact. AIDS Res Hum Retroviruses 1995; 11:877-884.
68. Gurney KB, Elliott J, Nassanian H, et al. Binding and transfer of human
immunodeficiency virus by DC-SIGN+ cells in human rectal mucosa. J Virol 2005;
79:5762-5773.
69. Turville SG, Cameron PU, Handley A, et al. Diversity of receptors binding HIV on
dendritic cell subsets. Nat Immunol 2002; 3:975-983.
70. Kwon DS, Gregorio G, Bitton N, et al. DC-SIGN-mediated internalization of HIV is
required for trans-enhancement of T cell infection. Immunity 2002; 16:135-144.
71. Cameron PU, Freudenthal PS, Barker JM, et al. Dendritic cells exposed to human
immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells.
Science 1992; 257:383-387.
72. McDonald D, Wu L, Bohks SM, et al. Recruitment of HIV and its receptors to dendritic
cell-T cell junctions. Science 2003; 300:1295-1297.
73. Wiley RD, Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1
trans infection. Proc Natl Acad Sci U S A 2006; 103:738-743.
74. Nobile C, Petit C, Moris A, et al. Covert human immunodeficiency virus replication in
dendritic cells and in DC-SIGN-expressing cells promotes long-term transmission to
lymphocytes. J Virol 2005; 79:5386-5399.
75. Sharpless NE, O'Brien WA, Verdin E, et al. Human immunodeficiency virus type 1
tropism for brain microglial cells is determined by a region of the env glycoprotein that
also controls macrophage tropism. J Virol 1992; 66:2588-2593.
76. Alouf JE, Geoffroy C, Klatzmann D, et al. High production of the acquired
immunodeficiency syndrome virus (lymphadenopathy-associated virus) by human T
lymphocytes stimulated by streptococcal mitogenic toxins. J Clin Microbiol 1986;
24:639-641.
77. Gerstoft J, Petersen CS, Kroon S, et al. The immunological and clinical outcome of HIV
infection: 31 months of follow-up in a cohort of homosexual men. Scand J Infect Dis
1987; 19:503-509.

202
78. Jones NA, Wei X, Flower DR, et al. Determinants of human immunodeficiency virus
type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J Exp Med
2004; 200:1243-1256.
79. Wei X, Decker JM, Wang S, et al. Antibody neutralization and escape by HIV-1. Nature
2003; 422:307-312.
80. Kim CJ, McKinnon LR, Kovacs C, et al. Mucosal Th17 cell function is altered during
HIV infection and is an independent predictor of systemic immune activation. J Immunol
2013; 191:2164-2173.
81. Nazli A, Chan O, Dobson-Belaire WN, et al. Exposure to HIV-1 directly impairs
mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog 2010;
6:e1000852.
82. Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of
systemic immune activation in chronic HIV infection. Nat Med 2006; 12:1365-1371.
83. Catalfamo M, Wilhelm C, Tcheung L, et al. CD4 and CD8 T cell immune activation
during chronic HIV infection: roles of homeostasis, HIV, type I IFN, and IL-7. J
Immunol 2011; 186:2106-2116.
84. Rothen M, Gratzl S, Hirsch HH, et al. Apoptosis in HIV-infected individuals is an early
marker occurring independently of high viremia. AIDS Res Hum Retroviruses 1997;
13:771-779.
85. Nardelli B, Gonzalez CJ, Schechter M, et al. CD4+ blood lymphocytes are rapidly killed
in vitro by contact with autologous human immunodeficiency virus-infected cells. Proc
Natl Acad Sci U S A 1995; 92:7312-7316.
86. Zeng M, Smith AJ, Wietgrefe SW, et al. Cumulative mechanisms of lymphoid tissue
fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest 2011; 121:998-
1008.
87. Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is
associated with T-cell exhaustion and disease progression. Nature 2006; 443:350-354.
88. Chomont N, El-Far M, Ancuta P, et al. HIV reservoir size and persistence are driven by T
cell survival and homeostatic proliferation. Nat Med 2009; 15:893-900.
89. Langford D, Marquie-Beck J, de Almeida S, et al. Relationship of antiretroviral treatment
to postmortem brain tissue viral load in human immunodeficiency virus-infected patients.
J Neurovirol 2006; 12:100-107.

203
90. Lamers SL, Rose R, Maidji E, et al. HIV DNA Is Frequently Present within Pathologic
Tissues Evaluated at Autopsy from Combined Antiretroviral Therapy-Treated Patients
with Undetectable Viral Loads. J Virol 2016; 90:8968-8983.
91. Chun TW, Engel D, Berrey MM, et al. Early establishment of a pool of latently infected,
resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A 1998;
95:8869-8873.
92. Margolis DM, Garcia JV, Hazuda DJ, et al. Latency reversal and viral clearance to cure
HIV-1. Science 2016; 353:aaf6517.
93. Parks WP, Parks ES, Fischl MA, et al. HIV-1 inhibition by azidothymidine in a
concurrently randomized placebo-controlled trail. J Acquir Immune Defic Syndr 1988;
1:125-130.
94. Chaisson RE, Allain JP, Leuther M, et al. Significant changes in HIV antigen level in the
serum of patients treated with azidothymidine. N Engl J Med 1986; 315:1610-1611.
95. Fischl MA, Richman DD, Grieco MH, et al. The efficacy of azidothymidine (AZT) in the
treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-
controlled trial. N Engl J Med 1987; 317:185-191.
96. Fischl MA, Parker CB, Pettinelli C, et al. A randomized controlled trial of a reduced
daily dose of zidovudine in patients with the acquired immunodeficiency syndrome. The
AIDS Clinical Trials Group. N Engl J Med 1990; 323:1009-1014.
97. Reiss P, Lange JM, Boucher CA, et al. Resumption of HIV antigen production during
continuous zidovudine treatment. Lancet 1988; 1:421.
98. Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1.
Science 1988; 242:1171-1173.
99. Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-
level resistance to zidovudine (AZT). Science 1989; 246:1155-1158.
100. Maxeiner HG, Keulen W, Schuurman R, et al. Selection of zidovudine resistance
mutations and escape of human immunodeficiency virus type 1 from antiretroviral
pressure in stavudine-treated pediatric patients. J Infect Dis 2002; 185:1070-1076.
101. La Seta Catamancio S, De Pasquale MP, Citterio P, et al. In vitro evolution of the human
immunodeficiency virus type 1 gag-protease region and maintenance of reverse
transcriptase resistance following prolonged drug exposure. J Clin Microbiol 2001;
39:1124-1129.

204
102. Eron JJ, Benoit SL, Jemsek J, et al. Treatment with lamivudine, zidovudine, or both in
HIV-positive patients with 200 to 500 CD4+ cells per cubic millimeter. North American
HIV Working Party. N Engl J Med 1995; 333:1662-1669.
103. D'Aquila RT, Hughes MD, Johnson VA, et al. Nevirapine, zidovudine, and didanosine
compared with zidovudine and didanosine in patients with HIV-1 infection. A
randomized, double-blind, placebo-controlled trial. National Institute of Allergy and
Infectious Diseases AIDS Clinical Trials Group Protocol 241 Investigators. Ann Intern
Med 1996; 124:1019-1030.
104. Collier AC, Coombs RW, Schoenfeld DA, et al. Combination therapy with zidovudine,
didanosine and saquinavir. Antiviral Res 1996; 29:99.
105. Granich R, Gupta S, Hersh B, et al. Trends in AIDS Deaths, New Infections and ART
Coverage in the Top 30 Countries with the Highest AIDS Mortality Burden; 1990-2013.
PLoS One 2015; 10:e0131353.
106. World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for
treating and preventing HIV infection: Recommendations for a public health approach.
2016. http://www.who.int/hiv/pub/arv/arv-2016/en/
107. Kearney MF, Wiegand A, Shao W, et al. Origin of Rebound Plasma HIV Includes Cells
with Identical Proviruses That Are Transcriptionally Active before Stopping of
Antiretroviral Therapy. J Virol 2015; 90:1369-1376.
108. Gray GE, Laher F, Lazarus E, et al. Approaches to preventative and therapeutic HIV
vaccines. Curr Opin Virol 2016; 17:104-109.
109. Kaminski R, Bella R, Yin C, et al. Excision of HIV-1 DNA by gene editing: a proof-of-
concept in vivo study. Gene Ther 2016; 23:690-695.
110. Wang CX, Cannon PM. Clinical Applications of Genome Editing to HIV Cure. AIDS
Patient Care STDS 2016; 30:539-544.
111. Flynn NM, Forthal DN, Harro CD, et al. Placebo-controlled phase 3 trial of a
recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J Infect Dis 2005;
191:654-665.
112. Spearman P, Lally MA, Elizaga M, et al. A trimeric, V2-deleted HIV-1 envelope
glycoprotein vaccine elicits potent neutralizing antibodies but limited breadth of
neutralization in human volunteers. J Infect Dis 2011; 203:1165-1173.
113. De Rosa SC, Thomas EP, Bui J, et al. HIV-DNA priming alters T cell responses to HIV-
adenovirus vaccine even when responses to DNA are undetectable. J Immunol 2011;
187:3391-3401.

205
114. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and
AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 2009; 361:2209-2220.
115. Caskey M, Klein F, Lorenzi JC, et al. Viraemia suppressed in HIV-1-infected humans by
broadly neutralizing antibody 3BNC117. Nature 2015; 522:487-491.
116. Moodie Z, Metch B, Bekker LG, et al. Continued Follow-Up of Phambili Phase 2b
Randomized HIV-1 Vaccine Trial Participants Supports Increased HIV-1 Acquisition
among Vaccinated Men. PLoS One 2015; 10:e0137666.
117. Lazarus EM, Otwombe K, Adonis T, et al. Uptake of genital mucosal sampling in HVTN
097, a phase 1b HIV vaccine trial in South Africa. PLoS One 2014; 9:e112303.
118. Chun TW, Murray D, Justement JS, et al. Broadly neutralizing antibodies suppress HIV
in the persistent viral reservoir. Proc Natl Acad Sci U S A 2014; 111:13151-13156.
119. Alexandre KB, Mufhandu HT, London GM, et al. Progress and Perspectives on HIV-1
microbicide development. Virology 2016; 497:69-80.
120. Riddler SA, Husnik M, Gorbach PM, et al. Long-term follow-up of HIV seroconverters
in microbicide trials - rationale, study design, and challenges in MTN-015. HIV Clin
Trials 2016; 17:204-211.
121. Montgomery CM, Gafos M, Lees S, et al. Re-framing microbicide acceptability: findings
from the MDP301 trial. Cult Health Sex 2010; 12:649-662.
122. Greene E, Batona G, Hallad J, et al. Acceptability and adherence of a candidate
microbicide gel among high-risk women in Africa and India. Cult Health Sex 2010;
12:739-754.
123. Martin M, Vanichseni S, Suntharasamai P, et al. The impact of adherence to preexposure
prophylaxis on the risk of HIV infection among people who inject drugs. AIDS 2015;
29:819-824.
124. Denton PW, Othieno F, Martinez-Torres F, et al. One percent tenofovir applied topically
to humanized BLT mice and used according to the CAPRISA 004 experimental design
demonstrates partial protection from vaginal HIV infection, validating the BLT model for
evaluation of new microbicide candidates. J Virol 2011; 85:7582-7593.
125. Molina JM, Capitant C, Spire B, et al. On-Demand Preexposure Prophylaxis in Men at
High Risk for HIV-1 Infection. N Engl J Med 2015; 373:2237-2246.
126. Kaminski R, Chen Y, Fischer T, et al. Elimination of HIV-1 Genomes from Human T-
lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep 2016; 6:10.1038.

206
127. Wang Z, Pan Q, Gendron P, et al. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1
Replication and Accelerate Viral Escape. Cell Rep 2016; 15:481-489.
128. Tsunetsugu-Yokota Y, Kobayahi-Ishihara M, Wada Y, et al. Homeostatically Maintained
Resting Naive CD4+ T Cells Resist Latent HIV Reactivation. Front Microbiol 2016;
7:10.3389.
129. Deng K, Pertea M, Rongvaux A, et al. Broad CTL response is required to clear latent
HIV-1 due to dominance of escape mutations. Nature 2015; 517:381-385.
130. Laird GM, Bullen CK, Rosenbloom DI, et al. Ex vivo analysis identifies effective HIV-1
latency-reversing drug combinations. J Clin Invest 2015; 125:1901-1912.
131. Weydert C, Rijck JD, Christ F, et al. Targeting Virus-host Interactions of HIV
Replication. Curr Top Med Chem 2016; 16:1167-1190.
132. Cooper DA, Heera J, Ive P, et al. Efficacy and safety of maraviroc vs. efavirenz in
treatment-naive patients with HIV-1: 5-year findings. AIDS 2014; 28:717-725.
133. Reynes J, Arasteh K, Clotet B, et al. TORO: ninety-six-week virologic and immunologic
response and safety evaluation of enfuvirtide with an optimized background of
antiretrovirals. AIDS Patient Care STDS 2007; 21:533-543.
134. Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally
bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with
broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents
Chemother 2005; 49:4721-4732.
135. Readinger JA, Schiralli GM, Jiang JK, et al. Selective targeting of ITK blocks multiple
steps of HIV replication. Proc Natl Acad Sci U S A 2008; 105:6684-6689.
136. Wrasidlo W, Crews LA, Tsigelny IF, et al. Neuroprotective effects of the anti-cancer
drug sunitinib in models of HIV neurotoxicity suggests potential for the treatment of
neurodegenerative disorders. Br J Pharmacol 2014; 171:5757-5773.
137. van Dijk D, Ertaylan G, Boucher CA, et al. Identifying potential survival strategies of
HIV-1 through virus-host protein interaction networks. BMC Syst Biol 2010; 4:10.1186.
138. Jager S, Cimermancic P, Gulbahce N, et al. Global landscape of HIV-human protein
complexes. Nature 2012; 481:365-370.
139. Smyth RP, Negroni M. A step forward understanding HIV-1 diversity. Retrovirology
2016; 13:27.

207
140. Sugden SM, Bego MG, Pham TN, et al. Remodeling of the Host Cell Plasma Membrane
by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence. Viruses
2016; 8.
141. Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of
viral escape. Nat Immunol 2015; 16:546-553.
142. Mangeat B, Turelli P, Caron G, et al. Broad antiretroviral defence by human
APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003; 424:99-
103.
143. Goldstone DC, Ennis-Adeniran V, Hedden JJ, et al. HIV-1 restriction factor SAMHD1 is
a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011; 480:379-382.
144. Vranckx LS, Demeulemeester J, Saleh S, et al. LEDGIN-mediated Inhibition of
Integrase-LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV.
EBioMedicine 2016; 8:248-264.
145. Christ F, Voet A, Marchand A, et al. Rational design of small-molecule inhibitors of the
LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol 2010; 6:442-448.
146. Bertoletti F, Crespan E, Maga G. Tyrosine kinases as essential cellular cofactors and
potential therapeutic targets for human immunodeficiency virus infection. Cell Mol Biol
(Noisy-le-grand) 2012; 58:31-43.
147. Davis CB, Dikic I, Unutmaz D, et al. Signal transduction due to HIV-1 envelope
interactions with chemokine receptors CXCR4 or CCR5. J Exp Med 1997; 186:1793-
1798.
148. Cayota A, Vuillier F, Siciliano J, et al. Defective protein tyrosine phosphorylation and
altered levels of p59fyn and p56lck in CD4 T cells from HIV-1 infected patients. Int
Immunol 1994; 6:611-621.
149. Phipps DJ, Yousefi S, Branch DR. Increased enzymatic activity of the T-cell antigen
receptor-associated fyn protein tyrosine kinase in asymptomatic patients infected with the
human immunodeficiency virus. Blood 1997; 90:3603-3612.
150. Guo J, Xu X, Rasheed TK, et al. Genistein interferes with SDF-1- and HIV-mediated
actin dynamics and inhibits HIV infection of resting CD4 T cells. Retrovirology 2013;
10:10.1186.
151. Stantchev TS, Markovic I, Telford WG, et al. The tyrosine kinase inhibitor genistein
blocks HIV-1 infection in primary human macrophages. Virus Res 2007; 123:178-189.

208
152. Schiralli Lester GM, Akiyama H, Evans E, et al. Interleukin 2-inducible T cell kinase
(ITK) facilitates efficient egress of HIV-1 by coordinating Gag distribution and actin
organization. Virology 2012.
153. Zack JA, Arrigo SJ, Weitsman SR, et al. HIV-1 entry into quiescent primary
lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 1990; 61:213-
222.
154. Bukong TN, Hall WW, Jacque JM. Lentivirus-associated MAPK/ERK2 phosphorylates
EMD and regulates infectivity. J Gen Virol 2010; 91:2381-2392.
155. Francis AC, Di Primio C, Allouch A, et al. Role of phosphorylation in the nuclear
biology of HIV-1. Curr Med Chem 2011; 18:2904-2912.
156. Pauls E, Badia R, Torres-Torronteras J, et al. Palbociclib, a selective inhibitor of cyclin-
dependent kinase4/6, blocks HIV-1 reverse transcription through the control of sterile
alpha motif and HD domain-containing protein-1 (SAMHD1) activity. AIDS 2014;
28:2213-2222.
157. Dochi T, Nakano T, Inoue M, et al. Phosphorylation of human immunodeficiency virus
type 1 capsid protein at serine 16, required for peptidyl-prolyl isomerase-dependent
uncoating, is mediated by virion-incorporated extracellular signal-regulated kinase 2. J
Gen Virol 2014; 95:1156-1166.
158. Gallay P, Swingler S, Aiken C, et al. HIV-1 infection of nondividing cells: C-terminal
tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 1995; 80:379-
388.
159. Manganaro L, Lusic M, Gutierrez MI, et al. Concerted action of cellular JNK and Pin1
restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat Med 2010;
16:329-333.
160. Lau A, Swinbank KM, Ahmed PS, et al. Suppression of HIV-1 infection by a small
molecule inhibitor of the ATM kinase. Nat Cell Biol 2005; 7:493-500.
161. Flory E, Weber CK, Chen P, et al. Plasma membrane-targeted Raf kinase activates NF-
kappaB and human immunodeficiency virus type 1 replication in T lymphocytes. J Virol
1998; 72:2788-2794.
162. Meggio F, D'Agostino DM, Ciminale V, et al. Phosphorylation of HIV-1 Rev protein:
implication of protein kinase CK2 and pro-directed kinases. Biochem Biophys Res
Commun 1996; 226:547-554.
163. Schubert U, Strebel K. Differential activities of the human immunodeficiency virus type
1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular
compartments. J Virol 1994; 68:2260-2271.

209
164. Trible RP, Emert-Sedlak L, Smithgall TE. HIV-1 Nef selectively activates Src family
kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J Biol Chem 2006;
281:27029-27038.
165. Chang AH, O'Shaughnessy MV, Jirik FR. Hck SH3 domain-dependent abrogation of
Nef-induced class 1 MHC down-regulation. Eur J Immunol 2001; 31:2382-2387.
166. Hung CH, Thomas L, Ruby CE, et al. HIV-1 Nef assembles a Src family kinase-ZAP-
70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 2007;
1:121-133.
167. Pan X, Geist MM, Rudolph JM, et al. HIV-1 Nef disrupts membrane-microdomain-
associated anterograde transport for plasma membrane delivery of selected Src family
kinases. Cell Microbiol 2013; 15:1605-1621.
168. Hanna Z, Weng X, Kay DG, et al. The pathogenicity of human immunodeficiency virus
(HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-
binding domain, and disease development is delayed in the absence of Hck. J Virol 2001;
75:9378-9392.
169. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-
approved drugs. Drug Discov Today 2016; 21:5-10.
170. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends
Pharmacol Sci 2015; 36:422-439.
171. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic
pulmonary fibrosis. N Engl J Med 2014; 370:2071-2082.
172. Asahina A, Etoh T, Igarashi A, et al. Oral tofacitinib efficacy, safety and tolerability in
Japanese patients with moderate to severe plaque psoriasis and psoriatic arthritis: A
randomized, double-blind, phase 3 study. J Dermatol 2016.
173. Spivak AM, Larragoite ET, Coletti ML, et al. Janus kinase inhibition suppresses PKC-
induced cytokine release without affecting HIV-1 latency reversal ex vivo. Retrovirology
2016; 13:10.1186.
174. Worm SW, Bower M, Reiss P, et al. Non-AIDS defining cancers in the D:A:D Study--
time trends and predictors of survival: a cohort study. BMC Infect Dis 2013; 13:471.
175. Kowalkowski MA, Day RS, Du XL, et al. Cumulative HIV viremia and non-AIDS-
defining malignancies among a sample of HIV-infected male veterans. J Acquir Immune
Defic Syndr 2014; 67:204-211.
176. Koon HB, Bubley GJ, Pantanowitz L, et al. Imatinib-induced regression of AIDS-related
Kaposi's sarcoma. J Clin Oncol 2005; 23:982-989.

210
177. Hagiwara S, Yotsumoto M, Odawara T, et al. Non-AIDS-defining hematological
malignancies in HIV-infected patients: an epidemiological study in Japan. Aids 2013;
27:279-283.
178. de Wispelaere M, LaCroix AJ, Yang PL. The small molecules AZD0530 and dasatinib
inhibit dengue virus RNA replication via Fyn kinase. J Virol 2013; 87:7367-7381.
179. Guntermann C, Zheng R, Nye KE. The effects of CD3, CD4 and CD28 signaling on
lymphocytes during human immunodeficiency virus-1 infection. Eur J Immunol 1997;
27:1966-1972.
180. Maqueda A, Moyano JV, Gutierrez-Lopez MD, et al. Activation pathways of
alpha4beta1 integrin leading to distinct T-cell cytoskeleton reorganization, Rac1
regulation and Pyk2 phosphorylation. J Cell Physiol 2006; 207:746-756.
181. Sol-Foulon N, Sourisseau M, Porrot F, et al. ZAP-70 kinase regulates HIV cell-to-cell
spread and virological synapse formation. Embo j 2007; 26:516-526.
182. Liu J, Liao Z, Camden J, et al. Src homology 3 binding sites in the P2Y2 nucleotide
receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2,
and growth factor receptors. J Biol Chem 2004; 279:8212-8218.
183. Wu JC, Chen YC, Kuo CT, et al. Focal adhesion kinase-dependent focal adhesion
recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion
and migration. Sci Rep 2015; 5:10.1038.
184. Bain J, McLauchlan H, Elliott M, et al. The specificities of protein kinase inhibitors: an
update. Biochem J 2003; 371:199-204.
185. Rous P. A SARCOMA OF THE FOWL TRANSMISSIBLE BY AN AGENT
SEPARABLE FROM THE TUMOR CELLS. J Exp Med 1911; 13:397-411.
186. Stehelin D, Varmus HE, Bishop JM, et al. DNA related to the transforming gene(s) of
avian sarcoma viruses is present in normal avian DNA. Nature 1976; 260:170-173.
187. Pawson T, Kung TH, Martin GS. Structure and phosphorylation of the Fujinami sarcoma
virus gene product. J Virol 1981; 40:665-672.
188. DeClue JE, Sadowski I, Martin GS, et al. A conserved domain regulates interactions of
the v-fps protein-tyrosine kinase with the host cell. Proc Natl Acad Sci U S A 1987;
84:9064-9068.
189. Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of
Src kinase. Oncogene 2000; 19:5620-5635.

211
190. Cayer MP, Proulx M, Ma XZ, et al. c-Src tyrosine kinase co-associates with and
phosphorylates signal transducer and activator of transcription 5b which mediates the
proliferation of normal human B lymphocytes. Clin Exp Immunol 2009; 156:419-427.
191. Kaplan KB, Bibbins KB, Swedlow JR, et al. Association of the amino-terminal half of c-
Src with focal adhesions alters their properties and is regulated by phosphorylation of
tyrosine 527. Embo j 1994; 13:4745-4756.
192. Bergman M, Mustelin T, Oetken C, et al. The human p50csk tyrosine kinase
phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. Embo j 1992;
11:2919-2924.
193. Kedzierska K, Vardaxis NJ, Jaworowski A, et al. FcgammaR-mediated phagocytosis by
human macrophages involves Hck, Syk, and Pyk2 and is augmented by GM-CSF. J
Leukoc Biol 2001; 70:322-328.
194. Takata M, Sabe H, Hata A, et al. Tyrosine kinases Lyn and Syk regulate B cell receptor-
coupled Ca2+ mobilization through distinct pathways. Embo j 1994; 13:1341-1349.
195. Tronick SR, Popescu NC, Cheah MS, et al. Isolation and chromosomal localization of the
human fgr protooncogene, a distinct member of the tyrosine kinase gene family. Proc
Natl Acad Sci U S A 1985; 82:6595-6599.
196. Compeer EB, Janssen W, van Royen-Kerkhof A, et al. Dysfunctional BLK in common
variable immunodeficiency perturbs B-cell proliferation and ability to elicit antigen-
specific CD4+ T-cell help. Oncotarget 2015; 6:10759-10771.
197. Dikic I, Tokiwa G, Lev S, et al. A role for Pyk2 and Src in linking G-protein-coupled
receptors with MAP kinase activation. Nature 1996; 383:547-550.
198. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci 2012; 8:1385-
1397.
199. Engen JR, Wales TE, Hochrein JM, et al. Structure and dynamic regulation of Src-family
kinases. Cell Mol Life Sci 2008; 65:3058-3073.
200. Nakashima K, Takeuchi K, Chihara K, et al. HCV NS5A protein containing potential
ligands for both Src homology 2 and 3 domains enhances autophosphorylation of Src
family kinase Fyn in B cells. PLoS One 2012; 7:e46634.
201. Hirsch AJ, Medigeshi GR, Meyers HL, et al. The Src family kinase c-Yes is required for
maturation of West Nile virus particles. J Virol 2005; 79:11943-11951.
202. Pfannkuche A, Buther K, Karthe J, et al. c-Src is required for complex formation between
the hepatitis C virus-encoded proteins NS5A and NS5B: a prerequisite for replication.
Hepatology 2011; 53:1127-1136.

212
203. Delorme-Axford E, Sadovsky Y, Coyne CB. Lipid raft- and SRC family kinase-
dependent entry of coxsackievirus B into human placental trophoblasts. J Virol 2013;
87:8569-8581.
204. Majolini MB, D'Elios MM, Galieni P, et al. Expression of the T-cell-specific tyrosine
kinase Lck in normal B-1 cells and in chronic lymphocytic leukemia B cells. Blood 1998;
91:3390-3396.
205. Moroi Y, Koga Y, Nakamura K, et al. Induction of interleukin 2-responsiveness in
thymocytes of the transgenic mice carrying lck-transgene. Microbiol Immunol 1993;
37:369-381.
206. Einspahr KJ, Abraham RT, Dick CJ, et al. Protein tyrosine phosphorylation and p56lck
modification in IL-2 or phorbol ester-activated human natural killer cells. J Immunol
1990; 145:1490-1497.
207. Omri B, Crisanti P, Marty MC, et al. The Lck tyrosine kinase is expressed in brain
neurons. J Neurochem 1996; 67:1360-1364.
208. Ley SC, Marsh M, Bebbington CR, et al. Distinct intracellular localization of Lck and
Fyn protein tyrosine kinases in human T lymphocytes. J Cell Biol 1994; 125:639-649.
209. Yousefi S, Ma XZ, Singla R, et al. HIV-1 infection is facilitated in T cells by decreasing
p56lck protein tyrosine kinase activity. Clin Exp Immunol 2003; 133:78-90.
210. Barber EK, Dasgupta JD, Schlossman SF, et al. The CD4 and CD8 antigens are coupled
to a protein-tyrosine kinase (p56lck) that phosphorylates the CD3 complex. Proc Natl
Acad Sci U S A 1989; 86:3277-3281.
211. Veillette A, Bookman MA, Horak EM, et al. The CD4 and CD8 T cell surface antigens
are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 1988;
55:301-308.
212. Luo KX, Sefton BM. Cross-linking of T-cell surface molecules CD4 and CD8 stimulates
phosphorylation of the lck tyrosine protein kinase at the autophosphorylation site. Mol
Cell Biol 1990; 10:5305-5313.
213. Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in
signal transduction through the T cell antigen receptor. Cell 1992; 70:585-593.
214. Phipps DJ, Read SE, Piovesan JP, et al. HIV infection in vitro enhances the activity of
src-family protein tyrosine kinases. AIDS 1996; 10:1191-1198.
215. Briant L, Robert-Hebmann V, Acquaviva C, et al. The protein tyrosine kinase p56lck is
required for triggering NF-kappaB activation upon interaction of human

213
immunodeficiency virus type 1 envelope glycoprotein gp120 with cell surface CD4. J
Virol 1998; 72:6207-6214.
216. Hivroz C, Mazerolles F, Soula M, et al. Human immunodeficiency virus gp120 and
derived peptides activate protein tyrosine kinase p56lck in human CD4 T lymphocytes.
Eur J Immunol 1993; 23:600-607.
217. Juszczak RJ, Turchin H, Truneh A, et al. Effect of human immunodeficiency virus gp120
glycoprotein on the association of the protein tyrosine kinase p56lck with CD4 in human
T lymphocytes. J Biol Chem 1991; 266:11176-11183.
218. Trushin SA, Bren GD, Badley AD. CXCR4 Tropic HIV-1 gp120 Inhibition of SDF-
1alpha-Induced Chemotaxis Requires Lck and is Associated with Cofilin
Phosphorylation. Open Virol J 2010; 4:157-162.
219. Mazerolles F, Barbat C, Fischer A. Down-regulation of LFA-1-mediated T cell adhesion
induced by the HIV envelope glycoprotein gp160 requires phosphatidylinositol-3-kinase
activity. Eur J Immunol 1997; 27:2457-2465.
220. Chrobak P, Simard MC, Bouchard N, et al. HIV-1 Nef disrupts maturation of CD4+ T
cells through CD4/Lck modulation. J Immunol 2010; 185:3948-3959.
221. Fackler OT, Alcover A, Schwartz O. Modulation of the immunological synapse: a key to
HIV-1 pathogenesis? Nat Rev Immunol 2007; 7:310-317.
222. Laguette N, Bregnard C, Bouchet J, et al. Nef-induced CD4 endocytosis in human
immunodeficiency virus type 1 host cells: role of p56lck kinase. J Virol 2009; 83:7117-
7128.
223. Pan X, Rudolph JM, Abraham L, et al. HIV-1 Nef compensates for disorganization of the
immunological synapse by inducing trans-Golgi network-associated Lck signaling. Blood
2012; 119:786-797.
224. Haller C, Muller B, Fritz JV, et al. HIV-1 Nef and Vpu are functionally redundant broad-
spectrum modulators of cell surface receptors, including tetraspanins. J Virol 2014;
88:14241-14257.
225. Wolf D, Witte V, Clark P, et al. HIV Nef enhances Tat-mediated viral transcription
through a hnRNP-K-nucleated signaling complex. Cell Host Microbe 2008; 4:398-408.
226. Manna SK, Aggarwal BB. Differential requirement for p56lck in HIV-tat versus TNF-
induced cellular responses: effects on NF-kappa B, activator protein-1, c-Jun N-terminal
kinase, and apoptosis. J Immunol 2000; 164:5156-5166.
227. Strasner AB, Natarajan M, Doman T, et al. The Src kinase Lck facilitates assembly of
HIV-1 at the plasma membrane. J Immunol 2008; 181:3706-3713.

214
228. Len AC, Starling S, Shivkumar M, et al. HIV-1 Activates T Cell Signaling Independently
of Antigen to Drive Viral Spread. Cell Rep 2017; 18:1062-1074.
229. Samelson LE, Phillips AF, Luong ET, et al. Association of the fyn protein-tyrosine
kinase with the T-cell antigen receptor. Proc Natl Acad Sci U S A 1990; 87:4358-4362.
230. Xie YG, Yu Y, Hou LK, et al. FYN promotes breast cancer progression through
epithelial-mesenchymal transition. Oncol Rep 2016; 36:1000-1006.
231. Peckham H, Giuffrida L, Wood R, et al. Fyn is an intermediate kinase that BDNF utilizes
to promote oligodendrocyte myelination. Glia 2016; 64:255-269.
232. Davidson D, Viallet J, Veillette A. Unique catalytic properties dictate the enhanced
function of p59fynT, the hemopoietic cell-specific isoform of the Fyn tyrosine protein
kinase, in T cells. Mol Cell Biol 1994; 14:4554-4564.
233. Hunter AJ, Shimizu Y. Alpha 4 beta 1 integrin-mediated tyrosine phosphorylation in
human T cells: characterization of Crk- and Fyn-associated substrates (pp105, pp115, and
human enhancer of filamentation-1) and integrin-dependent activation of p59fyn1. J
Immunol 1997; 159:4806-4814.
234. da Silva AJ, Li Z, de Vera C, et al. Cloning of a novel T-cell protein FYB that binds FYN
and SH2-domain-containing leukocyte protein 76 and modulates interleukin 2
production. Proc Natl Acad Sci U S A 1997; 94:7493-7498.
235. Sharma N, Akhade AS, Qadri A. Src kinases central to T-cell receptor signaling regulate
TLR-activated innate immune responses from human T cells. Innate Immun 2016;
22:238-244.
236. Tamma SM, Chirmule N, McCloskey TW, et al. Signals transduced through the CD4
molecule interfere with TCR/CD3-mediated ras activation leading to T cell
anergy/apoptosis. Clin Immunol Immunopathol 1997; 85:195-201.
237. Hohashi N, Hayashi T, Fusaki N, et al. The protein tyrosine kinase Fyn activates
transcription from the HIV promoter via activation of NF kappa B-like DNA-binding
proteins. Int Immunol 1995; 7:1851-1859.
238. Douaisi M, Dussart S, Courcoul M, et al. The tyrosine kinases Fyn and Hck favor the
recruitment of tyrosine-phosphorylated APOBEC3G into vif-defective HIV-1 particles.
Biochem Biophys Res Commun 2005; 329:917-924.
239. Quintrell N, Lebo R, Varmus H, et al. Identification of a human gene (HCK) that encodes
a protein-tyrosine kinase and is expressed in hemopoietic cells. Mol Cell Biol 1987;
7:2267-2275.

215
240. Dale BM, Traum D, Erdjument-Bromage H, et al. Phagocytosis in macrophages lacking
Cbl reveals an unsuspected role for Fc gamma receptor signaling and actin assembly in
target binding. J Immunol 2009; 182:5654-5662.
241. Dwyer AR, Mouchemore KA, Steer JH, et al. Src family kinase expression and
subcellular localization in macrophages: implications for their role in CSF-1-induced
macrophage migration. J Leukoc Biol 2016; 100:163-175.
242. Smolinska MJ, Page TH, Urbaniak AM, et al. Hck tyrosine kinase regulates TLR4-
induced TNF and IL-6 production via AP-1. J Immunol 2011; 187:6043-6051.
243. Mohn H, Le Cabec V, Fischer S, et al. The src-family protein-tyrosine kinase p59hck is
located on the secretory granules in human neutrophils and translocates towards the
phagosome during cell activation. Biochem J 1995; 309:657-665.
244. Jelicic K, Cimbro R, Nawaz F, et al. The HIV-1 envelope protein gp120 impairs B cell
proliferation by inducing TGF-beta1 production and FcRL4 expression. Nat Immunol
2013; 14:1256-1265.
245. Komuro I, Yokota Y, Yasuda S, et al. CSF-induced and HIV-1-mediated distinct
regulation of Hck and C/EBPbeta represent a heterogeneous susceptibility of monocyte-
derived macrophages to M-tropic HIV-1 infection. J Exp Med 2003; 198:443-453.
246. Tintori C, Laurenzana I, La Rocca F, et al. Identification of Hck inhibitors as hits for the
development of antileukemia and anti-HIV agents. ChemMedChem 2013; 8:1353-1360.
247. Breuer S, Espinola S, Morelli X, et al. A Biochemical/Biophysical Assay Dyad for HTS-
Compatible Triaging of Inhibitors of the HIV-1 Nef/Hck SH3 Interaction. Curr Chem
Genom Transl Med 2013; 7:16-20.
248. Hiyoshi M, Suzu S, Yoshidomi Y, et al. Interaction between Hck and HIV-1 Nef
negatively regulates cell surface expression of M-CSF receptor. Blood 2008; 111:243-
250.
249. Hiyoshi M, Takahashi-Makise N, Yoshidomi Y, et al. HIV-1 Nef perturbs the function,
structure, and signaling of the Golgi through the Src kinase Hck. J Cell Physiol 2012;
227:1090-1097.
250. Shinya E, Shimizu M, Owaki A, et al. Hemopoietic cell kinase (Hck) and p21-activated
kinase 2 (PAK2) are involved in the down-regulation of CD1a lipid antigen presentation
by HIV-1 Nef in dendritic cells. Virology 2016; 487:285-295.
251. Cornall A, Mak J, Greenway A, et al. HIV-1 infection of T cells and macrophages are
differentially modulated by virion-associated Hck: a Nef-dependent phenomenon.
Viruses 2013; 5:2235-2252.

216
252. Verollet C, Souriant S, Bonnaud E, et al. HIV-1 reprograms the migration of
macrophages. Blood 2015; 125:1611-1622.
253. Cassol E, Rossouw T, Malfeld S, et al. CD14(+) macrophages that accumulate in the
colon of African AIDS patients express pro-inflammatory cytokines and are responsive to
lipopolysaccharide. BMC Infect Dis 2015; 15:10.1186.
254. Batisse J, Guerrero SX, Bernacchi S, et al. APOBEC3G impairs the multimerization of
the HIV-1 Vif protein in living cells. J Virol 2013; 87:6492-6506.
255. Hassaine G, Courcoul M, Bessou G, et al. The tyrosine kinase Hck is an inhibitor of
HIV-1 replication counteracted by the viral vif protein. J Biol Chem 2001; 276:16885-
16893.
256. Yamanashi Y, Mori S, Yoshida M, et al. Selective expression of a protein-tyrosine
kinase, p56lyn, in hematopoietic cells and association with production of human T-cell
lymphotropic virus type I. Proc Natl Acad Sci U S A 1989; 86:6538-6542.
257. Balasubramanian A, Ganju RK, Groopman JE. Signal transducer and activator of
transcription factor 1 mediates apoptosis induced by hepatitis C virus and HIV envelope
proteins in hepatocytes. J Infect Dis 2006; 194:670-681.
258. Hernandez-Rapp J, Martin-Lanneree S, Hirsch TZ, et al. A PrP(C)-caveolin-Lyn complex
negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci Rep 2014;
4:10.1038.
259. Kawakami Y, Kitaura J, Satterthwaite AB, et al. Redundant and opposing functions of
two tyrosine kinases, Btk and Lyn, in mast cell activation. J Immunol 2000; 165:1210-
1219.
260. Yamanashi Y, Kakiuchi T, Mizuguchi J, et al. Association of B cell antigen receptor with
protein tyrosine kinase Lyn. Science 1991; 251:192-194.
261. Nishizumi H, Taniuchi I, Yamanashi Y, et al. Impaired proliferation of peripheral B cells
and indication of autoimmune disease in lyn-deficient mice. Immunity 1995; 3:549-560.
262. Rolli V, Gallwitz M, Wossning T, et al. Amplification of B cell antigen receptor
signaling by a Syk/ITAM positive feedback loop. Mol Cell 2002; 10:1057-1069.
263. Narute PS, Smithgall TE. Nef alleles from all major HIV-1 clades activate Src-family
kinases and enhance HIV-1 replication in an inhibitor-sensitive manner. PLoS One 2012;
7:e32561.
264. Gilbert C, Barat C, Cantin R, et al. Involvement of Src and Syk tyrosine kinases in HIV-1
transfer from dendritic cells to CD4+ T lymphocytes. J Immunol 2007; 178:2862-2871.

217
265. Caparros E, Munoz P, Sierra-Filardi E, et al. DC-SIGN ligation on dendritic cells results
in ERK and PI3K activation and modulates cytokine production. Blood 2006; 107:3950-
3958.
266. Ellegard R, Crisci E, Burgener A, et al. Complement opsonization of HIV-1 results in
decreased antiviral and inflammatory responses in immature dendritic cells via CR3. J
Immunol 2014; 193:4590-4601.
267. Tomkowicz B, Lee C, Ravyn V, et al. The Src kinase Lyn is required for CCR5 signaling
in response to MIP-1beta and R5 HIV-1 gp120 in human macrophages. Blood 2006;
108:1145-1150.
268. Cheung R, Ravyn V, Wang L, et al. Signaling mechanism of HIV-1 gp120 and virion-
induced IL-1beta release in primary human macrophages. J Immunol 2008; 180:6675-
6684.
269. Malik M, Chen YY, Kienzle MF, et al. Monocyte migration and LFA-1-mediated
attachment to brain microvascular endothelia is regulated by SDF-1 alpha through Lyn
kinase. J Immunol 2008; 181:4632-4637.
270. Berton G, Fumagalli L, Laudanna C, et al. Beta 2 integrin-dependent protein tyrosine
phosphorylation and activation of the FGR protein tyrosine kinase in human neutrophils.
J Cell Biol 1994; 126:1111-1121.
271. Kim HS, Han HD, Armaiz-Pena GN, et al. Functional roles of Src and Fgr in ovarian
carcinoma. Clin Cancer Res 2011; 17:1713-1721.
272. Willman CL, Stewart CC, Longacre TL, et al. Expression of the c-fgr and hck protein-
tyrosine kinases in acute myeloid leukemic blasts is associated with early commitment
and differentiation events in the monocytic and granulocytic lineages. Blood 1991;
77:726-734.
273. King FJ, Cole MD. Molecular cloning and sequencing of the murine c-fgr gene.
Oncogene 1990; 5:337-344.
274. Fanibunda SE, Modi DN, Bandivdekar AH. HIV gp120 induced gene expression
signatures in vaginal epithelial cells. Microbes Infect 2013; 15:806-815.
275. Sugawara K, Sugawara I, Sukegawa J, et al. Distribution of c-yes-1 gene product in
various cells and tissues. Br J Cancer 1991; 63:508-513.
276. Semba K, Yamanashi Y, Nishizawa M, et al. Location of the c-yes gene on the human
chromosome and its expression in various tissues. Science 1985; 227:1038-1040.

218
277. Fuhrer DK, Yang YC. Complex formation of JAK2 with PP2A, P13K, and Yes in
response to the hematopoietic cytokine interleukin-11. Biochem Biophys Res Commun
1996; 224:289-296.
278. Yoder SM, Dineen SL, Wang Z, et al. YES, a Src family kinase, is a proximal glucose-
specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet beta
cells. J Biol Chem 2014; 289:11476-11487.
279. Iida M, Brand TM, Campbell DA, et al. Yes and Lyn play a role in nuclear translocation
of the epidermal growth factor receptor. Oncogene 2013; 32:759-767.
280. Siegbahn A, Johnell M, Nordin A, et al. TF/FVIIa transactivate PDGFRbeta to regulate
PDGF-BB-induced chemotaxis in different cell types: involvement of Src and PLC.
Arterioscler Thromb Vasc Biol 2008; 28:135-141.
281. Varrin-Doyer M, Vincent P, Cavagna S, et al. Phosphorylation of collapsin response
mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. J
Biol Chem 2009; 284:13265-13276.
282. Johnson TP, Patel K, Johnson KR, et al. Induction of IL-17 and nonclassical T-cell
activation by HIV-Tat protein. Proc Natl Acad Sci U S A 2013; 110:13588-13593.
283. Wang X, Viswanath R, Zhao J, et al. Changes in the level of apoptosis-related proteins in
Jurkat cells infected with HIV-1 versus HIV-2. Mol Cell Biochem 2010; 337:175-183.
284. Dymecki SM, Niederhuber JE, Desiderio SV. Specific expression of a tyrosine kinase
gene, blk, in B lymphoid cells. Science 1990; 247:332-336.
285. Borowiec M, Liew CW, Thompson R, et al. Mutations at the BLK locus linked to
maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci U S A
2009; 106:14460-14465.
286. Saijo K, Schmedt C, Su IH, et al. Essential role of Src-family protein tyrosine kinases in
NF-kappaB activation during B cell development. Nat Immunol 2003; 4:274-279.
287. Tretter T, Ross AE, Dordai DI, et al. Mimicry of pre-B cell receptor signaling by
activation of the tyrosine kinase Blk. J Exp Med 2003; 198:1863-1873.
288. Simpfendorfer KR, Armstead BE, Shih A, et al. Autoimmune disease-associated
haplotypes of BLK exhibit lowered thresholds for B cell activation and expansion of Ig
class-switched B cells. Arthritis Rheumatol 2015; 67:2866-2876.
289. Konig R, Zhou Y, Elleder D, et al. Global analysis of host-pathogen interactions that
regulate early-stage HIV-1 replication. Cell 2008; 135:49-60.

219
290. Pyper JM, Bolen JB. Neuron-specific splicing of C-SRC RNA in human brain. J
Neurosci Res 1989; 24:89-96.
291. Golden A, Nemeth SP, Brugge JS. Blood platelets express high levels of the pp60c-src-
specific tyrosine kinase activity. Proc Natl Acad Sci U S A 1986; 83:852-856.
292. Nishio H, Tokuda M, Itano T, et al. pp60c-src expression in rat spermatogenesis.
Biochem Biophys Res Commun 1995; 206:502-510.
293. Branch DR, Mills GB. pp60c-src expression is induced by activation of normal human T
lymphocytes. J Immunol 1995; 154:3678-3685.
294. Amanchy R, Zhong J, Molina H, et al. Identification of c-Src tyrosine kinase substrates
using mass spectrometry and peptide microarrays. J Proteome Res 2008; 7:3900-3910.
295. Smart JE, Oppermann H, Czernilofsky AP, et al. Characterization of sites for tyrosine
phosphorylation in the transforming protein of Rous sarcoma virus (pp60v-src) and its
normal cellular homologue (pp60c-src). Proc Natl Acad Sci U S A 1981; 78:6013-6017.
296. Yang CC, Fazli L, Loguercio S, et al. Downregulation of c-SRC kinase CSK promotes
castration resistant prostate cancer and pinpoints a novel disease subclass. Oncotarget
2015; 6:22060-22071.
297. Sandilands E, Cans C, Fincham VJ, et al. RhoB and actin polymerization coordinate Src
activation with endosome-mediated delivery to the membrane. Dev Cell 2004; 7:855-869.
298. David-Pfeuty T, Nouvian-Dooghe Y. Highly specific antibody to Rous sarcoma virus src
gene product recognizes nuclear and nucleolar antigens in human cells. J Virol 1995;
69:1699-1713.
299. Weber TK, Steele G, Summerhayes IC. Differential pp60c-src activity in well and poorly
differentiated human colon carcinomas and cell lines. J Clin Invest 1992; 90:815-821.
300. Qian XL, Zhang J, Li PZ, et al. Dasatinib inhibits c-src phosphorylation and prevents the
proliferation of Triple-Negative Breast Cancer (TNBC) cells which overexpress
Syndecan-Binding Protein (SDCBP). PLoS One 2017; 12:e0171169.
301. Roussel RR, Brodeur SR, Shalloway D, et al. Selective binding of activated pp60c-src by
an immobilized synthetic phosphopeptide modeled on the carboxyl terminus of pp60c-
src. Proc Natl Acad Sci U S A 1991; 88:10696-10700.
302. Piwnica-Worms H, Saunders KB, Roberts TM, et al. Tyrosine phosphorylation regulates
the biochemical and biological properties of pp60c-src. Cell 1987; 49:75-82.
303. Ren R, Mayer BJ, Cicchetti P, et al. Identification of a ten-amino acid proline-rich SH3
binding site. Science 1993; 259:1157-1161.

220
304. Anderson D, Koch CA, Grey L, et al. Binding of SH2 domains of phospholipase C
gamma 1, GAP, and Src to activated growth factor receptors. Science 1990; 250:979-982.
305. Alexandropoulos K, Baltimore D. Coordinate activation of c-Src by SH3- and SH2-
binding sites on a novel p130Cas-related protein, Sin. Genes Dev 1996; 10:1341-1355.
306. Somani AK, Bignon JS, Mills GB, et al. Src kinase activity is regulated by the SHP-1
protein-tyrosine phosphatase. J Biol Chem 1997; 272:21113-21119.
307. Cooper JA, MacAuley A. Potential positive and negative autoregulation of p60c-src by
intermolecular autophosphorylation. Proc Natl Acad Sci U S A 1988; 85:4232-4236.
308. Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by
overexpression of a protein tyrosine phosphatase. Nature 1992; 359:336-339.
309. Chellaiah MA, Schaller MD. Activation of Src kinase by protein-tyrosine phosphatase-
PEST in osteoclasts: comparative analysis of the effects of bisphosphonate and protein-
tyrosine phosphatase inhibitor on Src activation in vitro. J Cell Physiol 2009; 220:382-
393.
310. Oh ES, Gu H, Saxton TM, et al. Regulation of early events in integrin signaling by
protein tyrosine phosphatase SHP-2. Mol Cell Biol 1999; 19:3205-3215.
311. Chong YP, Mulhern TD, Cheng HC. C-terminal Src kinase (CSK) and CSK-homologous
kinase (CHK)--endogenous negative regulators of Src-family protein kinases. Growth
Factors 2005; 23:233-244.
312. Davidson D, Bakinowski M, Thomas ML, et al. Phosphorylation-dependent regulation of
T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol Cell
Biol 2003; 23:2017-2028.
313. Tsukita S, Oishi K, Akiyama T, et al. Specific proto-oncogenic tyrosine kinases of src
family are enriched in cell-to-cell adherens junctions where the level of tyrosine
phosphorylation is elevated. J Cell Biol 1991; 113:867-879.
314. Fincham VJ, Unlu M, Brunton VG, et al. Translocation of Src kinase to the cell periphery
is mediated by the actin cytoskeleton under the control of the Rho family of small G
proteins. J Cell Biol 1996; 135:1551-1564.
315. Bard F, Patel U, Levy JB, et al. Molecular complexes that contain both c-Cbl and c-Src
associate with Golgi membranes. Eur J Cell Biol 2002; 81:26-35.
316. Finan PM, Hall A, Kellie S. Sam68 from an immortalised B-cell line associates with a
subset of SH3 domains. FEBS Lett 1996; 389:141-144.

221
317. He JJ, Henao-Mejia J, Liu Y. Sam68 functions in nuclear export and translation of HIV-1
RNA. RNA Biol 2009; 6:384-386.
318. Phipps DJ, Reed-Doob P, MacFadden DK, et al. An octapeptide analogue of HIV gp120
modulates protein tyrosine kinase activity in activated peripheral blood T lymphocytes.
Clin Exp Immunol 1995; 100:412-418.
319. Briggs SD, Lerner EC, Smithgall TE. Affinity of Src family kinase SH3 domains for HIV
Nef in vitro does not predict kinase activation by Nef in vivo. Biochemistry 2000;
39:489-495.
320. He JC, Husain M, Sunamoto M, et al. Nef stimulates proliferation of glomerular
podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin
Invest 2004; 114:643-651.
321. Zhang X, Yu J, Kuzontkoski PM, et al. Slit2/Robo4 Signaling Modulates HIV-1 gp120-
Induced Lymphatic Hyperpermeability. PLoS Pathogens 2012; 8:e1002461.
322. Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog
Biophys Mol Biol 1999; 71:435-478.
323. Prasad A, Kuzontkoski PM, Shrivastava A, et al. Slit2N/Robo1 Inhibit HIV-gp120-
Induced Migration and Podosome Formation in Immature Dendritic Cells by
Sequestering LSP1 and WASp. PLoS One 2012; 7:e48854.
324. Xiong WC, Macklem M, Parsons JT. Expression and characterization of splice variants
of PYK2, a focal adhesion kinase-related protein. J Cell Sci 1998; 111:1981-1991.
325. Dikic I, Dikic I, Schlessinger J. Identification of a new Pyk2 isoform implicated in
chemokine and antigen receptor signaling. J Biol Chem 1998; 273:14301-14308.
326. Block ER, Tolino MA, Klarlund JK. Pyk2 activation triggers epidermal growth factor
receptor signaling and cell motility after wounding sheets of epithelial cells. J Biol Chem
2010; 285:13372-13379.
327. Sasaki H, Nagura K, Ishino M, et al. Cloning and characterization of cell adhesion kinase
beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem
1995; 270:21206-21219.
328. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control
of cell motility. Nat Rev Mol Cell Biol 2005; 6:56-68.
329. Walkiewicz KW, Girault JA, Arold ST. How to awaken your nanomachines: Site-specific
activation of focal adhesion kinases through ligand interactions. Prog Biophys Mol Biol
2015; 119:60-71.

222
330. Avraham S, London R, Fu Y, et al. Identification and characterization of a novel related
adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem
1995; 270:27742-27751.
331. Vanarotti MS, Finkelstein DB, Guibao CD, et al. Structural basis for the interaction
between Pyk2-FAT domain and Leupaxin LD repeats. Biochemistry 2016; 55:1332-1345.
332. Riggs D, Yang Z, Kloss J, et al. The Pyk2 FERM regulates Pyk2 complex formation and
phosphorylation. Cell Signal 2011; 23:288-296.
333. Han S, Mistry A, Chang JS, et al. Structural characterization of proline-rich tyrosine
kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design.
J Biol Chem 2009; 284:13193-13201.
334. Litvak V, Tian D, Shaul YD, et al. Targeting of PYK2 to focal adhesions as a cellular
mechanism for convergence between integrins and G protein-coupled receptor signaling
cascades. J Biol Chem 2000; 275:32736-32746.
335. Melendez J, Turner C, Avraham H, et al. Cardiomyocyte apoptosis triggered by
RAFTK/pyk2 via Src kinase is antagonized by paxillin. J Biol Chem 2004; 279:53516-
53523.
336. Marzia M, Chiusaroli R, Neff L, et al. Calpain is required for normal osteoclast function
and is down-regulated by calcitonin. J Biol Chem 2006; 281:9745-9754.
337. Miao L, Xin X, Xin H, et al. Hydrogen Sulfide Recruits Macrophage Migration by
Integrin β1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction. Sci Rep 2016;
6:10.1038.
338. Genua M, Xu S-Q, Buraschi S, et al. Proline-Rich Tyrosine Kinase 2 (Pyk2) Regulates
IGF-I-Induced Cell Motility and Invasion of Urothelial Carcinoma Cells. PLoS One
2012; 7:e40148.
339. Corvol JC, Valjent E, Toutant M, et al. Depolarization activates ERK and proline-rich
tyrosine kinase 2 (PYK2) independently in different cellular compartments in
hippocampal slices. J Biol Chem 2005; 280:660-668.
340. Collins M, Bartelt RR, Houtman JC. T cell receptor activation leads to two distinct
phases of Pyk2 activation and actin cytoskeletal rearrangement in human T cells. Mol
Immunol 2010; 47:1665-1674.
341. Beinke S, Phee H, Clingan JM, et al. Proline-rich tyrosine kinase-2 is critical for CD8 T-
cell short-lived effector fate. Proc Natl Acad Sci U S A 2010; 107:16234-16239.
342. Hashimoto-Tane A, Sakuma M, Ike H. Micro-adhesion rings surrounding TCR
microclusters are essential for T cell activation. J Exp Med 2016; 213:1609-1625.

223
343. Sancho D, Montoya MC, Monjas A, et al. TCR engagement induces proline-rich tyrosine
kinase-2 (Pyk2) translocation to the T cell-APC interface independently of Pyk2 activity
and in an immunoreceptor tyrosine-based activation motif-mediated fashion. J Immunol
2002; 169:292-300.
344. Tsuchida M, Knechtle SJ, Hamawy MM. CD28 ligation induces tyrosine phosphorylation
of Pyk2 but not Fak in Jurkat T cells. J Biol Chem 1999; 274:6735-6740.
345. Seror C, Melki MT, Subra F, et al. Extracellular ATP acts on P2Y2 purinergic receptors
to facilitate HIV-1 infection. J Exp Med 2011; 208:1823-1834.
346. Garron ML, Arthos J, Guichou JF, et al. Structural basis for the interaction between focal
adhesion kinase and CD4. J Mol Biol 2008; 375:1320-1328.
347. Cicala C, Arthos J, Ruiz M, et al. Induction of phosphorylation and intracellular
association of CC chemokine receptor 5 and focal adhesion kinase in primary human
CD4+ T cells by macrophage-tropic HIV envelope. J Immunol 1999; 163:420-426.
348. Cicala C, Arthos J, Rubbert A, et al. HIV-1 envelope induces activation of caspase-3 and
cleavage of focal adhesion kinase in primary human CD4(+) T cells. Proc Natl Acad Sci
U S A 2000; 97:1178-1183.
349. Preusser A, Briese L, Willbold D. Presence of a helix in human CD4 cytoplasmic domain
promotes binding to HIV-1 Nef protein. Biochem Biophys Res Commun 2002; 292:734-
740.
350. Simpson DIH. Ebola haemorrhagic fever in Sudan, 1976. Report of a WHO/International
Study Team. Bull World Health Organ 1978; 56:247-270.
351. Johnson KM, Breman JG. Ebola haemorrhagic fever in Zaire, 1976. Bull World Health
Organ 1978; 56:271-293.
352. Johnson KM, Lange JV, Webb PA, et al. Isolation and partial characterisation of a new
virus causing acute haemorrhagic fever in Zaire. Lancet 1977; 1:569-571.
353. Richman DD, Cleveland PH, McCormick JB, et al. Antigenic analysis of strains of Ebola
virus: identification of two Ebola virus serotypes. J Infect Dis 1983; 147:268-271.
354. Towner JS, Sealy TK, Khristova ML, et al. Newly discovered ebola virus associated with
hemorrhagic fever outbreak in Uganda. PLoS Pathog 2008; 4:e1000212.
355. Hayes CG, Burans JP, Ksiazek TG, et al. Outbreak of fatal illness among captive
macaques in the Philippines caused by an Ebola-related filovirus. Am J Trop Med Hyg
1992; 46:664-671.

224
356. Formenty P, Boesch C, Wyers M, et al. Ebola virus outbreak among wild chimpanzees
living in a rain forest of Cote d'Ivoire. J Infect Dis 1999; 179:S120-S126.
357. Rivera A, Messaoudi I. Molecular mechanisms of Ebola pathogenesis. J Leukoc Biol
2016; 100:889-904.
358. Baize S, Pannetier D, Oestereich L, et al. Emergence of Zaire Ebola virus disease in
Guinea. N Engl J Med 2014; 371:1418-1425.
359. Kuhn JH, Andersen KG, Baize S, et al. Nomenclature- and database-compatible names
for the two Ebola virus variants that emerged in Guinea and the Democratic Republic of
the Congo in 2014. Viruses 2014; 6:4760-4799.
360. Diallo B, Sissoko D, Loman NJ, et al. Resurgence of Ebola Virus Disease in Guinea
Linked to a Survivor With Virus Persistence in Seminal Fluid for More Than 500 Days.
Clin Infect Dis 2016; 63:1353-1356.
361. Leroy EM, Baize S, Volchkov VE, et al. Human asymptomatic Ebola infection and
strong inflammatory response. Lancet 2000; 355:2210-2215.
362. Tiffany A, Vetter P, Mattia J, et al. Ebola Virus Disease Complications as Experienced
by Survivors in Sierra Leone. Clin Infect Dis 2016; 62:1360-1366.
363. Hereth He'bert E, Oury Bah M, E'Tard J F, et al. Ocular complications in survivors of the
Ebola outbreak in Guinea. Am J Ophthalmol 2016; 175:114-121.
364. Clark DV, Kibuuka H, Millard M, et al. Long-term sequelae after Ebola virus disease in
Bundibugyo, Uganda: a retrospective cohort study. Lancet Infect Dis 2015; 15:905-912.
365. Sissoko D, Duraffour S, Kerber R, et al. Persistence and clearance of Ebola virus RNA
from seminal fluid of Ebola virus disease survivors: a longitudinal analysis and modelling
study. Lancet Glob Health 2017; 5:e80-e88.
366. Caluwaerts S, Fautsch T, Lagrou D, et al. Dilemmas in Managing Pregnant Women With
Ebola: 2 Case Reports. Clin Infect Dis 2016; 62:903-905.
367. Vetter P, Fischer WA, 2nd, Schibler M, et al. Ebola Virus Shedding and Transmission:
Review of Current Evidence. J Infect Dis 2016; 214:S177-S184.
368. Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB bioinformatics resource portal.
Nucleic Acids Res 2012; 40:W597-603.
369. Sanchez A, Kiley MP, Holloway BP, et al. Sequence analysis of the Ebola virus genome:
organization, genetic elements, and comparison with the genome of Marburg virus. Virus
Res 1993; 29:215-240.

225
370. Neumann G, Watanabe S, Kawaoka Y. Characterization of Ebolavirus regulatory
genomic regions. Virus Res 2009; 144:1-7.
371. Sanchez A, Yang ZY, Xu L, et al. Biochemical analysis of the secreted and virion
glycoproteins of Ebola virus. J Virol 1998; 72:6442-6447.
372. Dahlmann F, Biedenkopf N, Babler A, et al. Analysis of Ebola Virus Entry Into
Macrophages. J Infect Dis 2015; 212:S247-257.
373. Takada A, Fujioka K, Tsuiji M, et al. Human macrophage C-type lectin specific for
galactose and N-acetylgalactosamine promotes filovirus entry. J Virol 2004; 78:2943-
2947.
374. Chan SY, Empig CJ, Welte FJ, et al. Folate receptor-alpha is a cofactor for cellular entry
by Marburg and Ebola viruses. Cell 2001; 106:117-126.
375. Aleksandrowicz P, Marzi A, Biedenkopf N, et al. Ebola virus enters host cells by
macropinocytosis and clathrin-mediated endocytosis. J Infect Dis 2011; 204:S957-967.
376. Aman MJ. Chasing Ebola through the Endosomal Labyrinth. MBio 2016; 7:e00346.
377. Wang H, Shi Y, Song J, et al. Ebola Viral Glycoprotein Bound to Its Endosomal
Receptor Niemann-Pick C1. Cell 2016; 164:258-268.
378. Nanbo A, Watanabe S, Halfmann P, et al. The spatio-temporal distribution dynamics of
Ebola virus proteins and RNA in infected cells. Sci Rep 2013; 3:10.1038.
379. Weik M, Modrof J, Klenk HD, et al. Ebola virus VP30-mediated transcription is
regulated by RNA secondary structure formation. J Virol 2002; 76:8532-8539.
380. Zinzula L, Esposito F, Pala D, et al. dsRNA binding characterization of full length
recombinant wild type and mutants Zaire ebolavirus VP35. Antiviral Res 2012; 93:354-
363.
381. Biedenkopf N, Lier C, Becker S. Dynamic Phosphorylation of VP30 Is Essential for
Ebola Virus Life Cycle. J Virol 2016; 90:4914-4925.
382. Licata JM, Johnson RF, Han Z, et al. Contribution of ebola virus glycoprotein,
nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol 2004; 78:7344-
7351.
383. Yen BC, Basler CF. Effects of Filovirus Interferon Antagonists on Responses of Human
Monocyte-Derived Dendritic Cells to RNA Virus Infection. J Virol 2016; 90:5108-5118.

226
384. Hensley LE, Young HA, Jahrling PB, et al. Proinflammatory response during Ebola virus
infection of primate models: possible involvement of the tumor necrosis factor receptor
superfamily. Immunol Lett 2002; 80:169-179.
385. Stroher U, West E, Bugany H, et al. Infection and activation of monocytes by Marburg
and Ebola viruses. J Virol 2001; 75:11025-11033.
386. Gupta M, Mahanty S, Ahmed R, et al. Monocyte-derived human macrophages and
peripheral blood mononuclear cells infected with ebola virus secrete MIP-1alpha and
TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 2001; 284:20-25.
387. Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet 2011; 377:849-862.
388. Lubaki NM, Ilinykh P, Pietzsch C, et al. The lack of maturation of Ebola virus-infected
dendritic cells results from the cooperative effect of at least two viral domains. J Virol
2013; 87:7471-7485.
389. Basler CF. A novel mechanism of immune evasion mediated by Ebola virus soluble
glycoprotein. Expert Rev Anti Infect Ther 2013; 11:475-478.
390. Geisbert TW, Hensley LE, Larsen T, et al. Pathogenesis of Ebola hemorrhagic fever in
cynomolgus macaques: evidence that dendritic cells are early and sustained targets of
infection. Am J Pathol 2003; 163:2347-2370.
391. Gupta M, Spiropoulou C, Rollin PE. Ebola virus infection of human PBMCs causes
massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology
2007; 364:45-54.
392. Friedrich BM, Trefry JC, Biggins JE, et al. Potential vaccines and post-exposure
treatments for filovirus infections. Viruses 2012; 4:1619-1650.
393. Tapia MD, Sow SO, Lyke KE, et al. Use of ChAd3-EBO-Z Ebola virus vaccine in
Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1,
single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation
trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis
2016; 16:31-42.
394. Ewer K, Rampling T, Venkatraman N, et al. A Monovalent Chimpanzee Adenovirus
Ebola Vaccine Boosted with MVA. N Engl J Med 2016; 374:1635-1646.
395. Jones SM, Feldmann H, Stroher U, et al. Live attenuated recombinant vaccine protects
nonhuman primates against Ebola and Marburg viruses. Nat Med 2005; 11:786-790.
396. Jones SM, Stroher U, Fernando L, et al. Assessment of a vesicular stomatitis virus-based
vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J Infect Dis 2007;
196:S404-412.

227
397. Feldmann H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment
of Ebola infection. PLoS Pathog 2007; 3:e2.
398. Agnandji ST, Huttner A, Zinser ME, et al. Phase 1 Trials of rVSV Ebola Vaccine in
Africa and Europe. N Engl J Med 2016; 374:1647-1660.
399. Regules JA, Beigel JH, Paolino KM, et al. A Recombinant Vesicular Stomatitis Virus
Ebola Vaccine. N Engl J Med 2015; 376:330-331.
400. Gunther S, Feldmann H, Geisbert TW, et al. Management of accidental exposure to
Ebola virus in the biosafety level 4 laboratory, Hamburg, Germany. J Infect Dis 2011;
204:S785-790.
401. Lai L, Davey R, Beck A, et al. Emergency postexposure vaccination with vesicular
stomatitis virus-vectored Ebola vaccine after needlestick. JAMA 2015; 313:1249-1255.
402. van Griensven J, Edwards T, Baize S. Efficacy of Convalescent Plasma in Relation to
Dose of Ebola Virus Antibodies. N Engl J Med 2016; 375:2307-2309.
403. Geisbert TW, Lee AC, Robbins M, et al. Postexposure protection of non-human primates
against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study.
Lancet 2010; 375:1896-1905.
404. McCarthy M. US signs contract with ZMapp maker to accelerate development of the
Ebola drug. BMJ 2014; 349:g5488.
405. Basler CF, Amarasinghe GK. Evasion of interferon responses by Ebola and Marburg
viruses. J Interferon Cytokine Res 2009; 29:511-520.
406. Wong G, Kobinger GP, Qiu X. Characterization of host immune responses in Ebola virus
infections. Expert Rev Clin Immunol 2014; 10:781-790.
407. Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth
Factor Rev 2014; 25:369-376.
408. Eron JJ, Jr. The treatment of antiretroviral-naive subjects with the 3TC/zidovudine
combination: a review of North American (NUCA 3001) and European (NUCB 3001)
trials. AIDS 1996; 10:S11-19.
409. Cooper DA, Katlama C, Montaner J, et al. Randomised trial of addition of lamivudine or
lamivudine plus loviride to zidovudine-containing regimens for patients with HIV-1
infection: the CAESAR trial. Lancet 1997; 349:1413-1421.
410. Thio CL, Smeaton L, Hollabaugh K, et al. Comparison of HBV-active HAART regimens
in an HIV-HBV multinational cohort: outcomes through 144 weeks. AIDS 2015;
29:1173-1182.

228
411. Strand A, Bottiger D, Gever LN, et al. Safety and tolerability of combination acyclovir
5% and hydrocortisone 1% cream in adolescents with recurrent herpes simplex labialis.
Pediatr Dermatol 2012; 29:105-110.
412. Madkan VK, Arora A, Babb-Tarbox M, et al. Open-label study of valacyclovir 1.5 g
twice daily for the treatment of uncomplicated herpes zoster in immunocompetent
patients 18 years of age or older. J Cutan Med Surg 2007; 11:89-98.
413. Marty FM, Winston DJ, Rowley SD, et al. CMX001 to prevent cytomegalovirus disease
in hematopoietic-cell transplantation. N Engl J Med 2013; 369:1227-1236.
414. Wedemeyer H, Forns X, Hezode C, et al. Mericitabine and Either Boceprevir or
Telaprevir in Combination with Peginterferon Alfa-2a plus Ribavirin for Patients with
Chronic Hepatitis C Genotype 1 Infection and Prior Null Response: The Randomized
DYNAMO 1 and DYNAMO 2 Studies. PLoS One 2016; 11:e0145409.
415. Else LJ, Jackson A, Puls R, et al. Pharmacokinetics of lamivudine and lamivudine-
triphosphate after administration of 300 milligrams and 150 milligrams once daily to
healthy volunteers: results of the ENCORE 2 study. Antimicrob Agents Chemother 2012;
56:1427-1433.
416. Parang K, Wiebe LI, Knaus EE. Pharmacokinetics and tissue distribution of (+/-)-3'-
azido-2',3'-dideoxy-5'-O-(2-bromomyristoyl)thymidine, a prodrug of 3'-azido-2',3'-
dideoxythymidine (AZT) in mice. J Pharm Pharmacol 1998; 50:989-996.
417. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734
against Ebola virus in rhesus monkeys. Nature 2016; 531:381-385.
418. Warren TK, Wells J, Panchal RG, et al. Protection against filovirus diseases by a novel
broad-spectrum nucleoside analogue BCX4430. Nature 2014; 508:402-405.
419. Siegel D, Hui HC, Doerffler E, et al. Discovery and Synthesis of a Phosphoramidate
Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the
Treatment of Ebola and Emerging Viruses. J Med Chem 2017; 60:1648-1661.
420. Parker S, Touchette E, Oberle C, et al. Efficacy of therapeutic intervention with an oral
ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res
2008; 77:39-49.
421. Bainbridge JW, Raina J, Shah SM, et al. Ocular complications of intravenous cidofovir
for cytomegalovirus retinitis in patients with AIDS. Eye 1999; 13:353-356.
422. Kern ER, Hartline C, Harden E, et al. Enhanced inhibition of orthopoxvirus replication in
vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents
Chemother 2002; 46:991-995.

229
423. Ciesla SL, Trahan J, Wan WB, et al. Esterification of cidofovir with alkoxyalkanols
increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res
2003; 59:163-171.
424. Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral
antiviral activity and reduce toxicity: current state of the art. Antiviral Res 2009; 82:A84-
A98.
425. Aldern KA, Ciesla SL, Winegarden KL, et al. Increased antiviral activity of 1-O-
hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained
by unique cellular uptake and metabolism. Mol Pharmacol 2003; 63:678-681.
426. Xiong X, Smith JL, Chen MS. Effect of incorporation of cidofovir into DNA by human
cytomegalovirus DNA polymerase on DNA elongation. Antimicrob Agents Chemother
1997; 41:594-599.
427. Whitmer SL, Albarino C, Shepard SS, et al. Preliminary Evaluation of the Effect of
Investigational Ebola Virus Disease Treatments on Viral Genome Sequences. J Infect Dis
2016; 214:S333-S341.
428. Zhao Z, Martin C, Fan R, et al. Drug repurposing to target Ebola virus replication and
virulence using structural systems pharmacology. BMC Bioinformatics 2016; 17:10.1186.
429. Toth K, Spencer JF, Dhar D, et al. Hexadecyloxypropyl-cidofovir, CMX001, prevents
adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc
Natl Acad Sci U S A 2008; 105:7293-7297.
430. Zaitseva M, McCullough KT, Cruz S, et al. Postchallenge administration of brincidofovir
protects healthy and immune-deficient mice reconstituted with limited numbers of T cells
from lethal challenge with IHD-J-Luc vaccinia virus. J Virol 2015; 89:3295-3307.
431. Florescu DF, Kalil AC, Hewlett AL, et al. Administration of Brincidofovir and
Convalescent Plasma in a Patient With Ebola Virus Disease. Clin Infect Dis 2015;
61:969-973.
432. Painter W, Robertson A, Trost LC, et al. First pharmacokinetic and safety study in
humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active
against double-stranded DNA viruses. Antimicrob Agents Chemother 2012; 56:2726-
2734.
433. Yen HL. Current and novel antiviral strategies for influenza infection. Curr Opin Virol
2016; 18:126-134.
434. Jin Z, Smith LK, Rajwanshi VK, et al. The ambiguous base-pairing and high substrate
efficiency of T-705 (Favipiravir) Ribofuranosyl 5'-triphosphate towards influenza A virus
polymerase. PLoS One 2013; 8:e68347.

230
435. Westover JB, Sefing EJ, Bailey KW, et al. Low-dose ribavirin potentiates the antiviral
activity of favipiravir against hemorrhagic fever viruses. Antiviral Res 2016; 126:62-68.
436. Scharton D, Bailey KW, Vest Z, et al. Favipiravir (T-705) protects against peracute Rift
Valley fever virus infection and reduces delayed-onset neurologic disease observed with
ribavirin treatment. Antiviral Res 2014; 104:84-92.
437. Jin Z, Tucker K, Lin X, et al. Biochemical Evaluation of the Inhibition Properties of
Favipiravir and 2'-C-Methyl-Cytidine Triphosphates against Human and Mouse
Norovirus RNA Polymerases. Antimicrob Agents Chemother 2015; 59:7504-7516.
438. Delang L, Segura Guerrero N, Tas A, et al. Mutations in the chikungunya virus non-
structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J
Antimicrob Chemother 2014; 69:2770-2784.
439. Wang Y, Li G, Yuan S, et al. In Vitro Assessment of Combinations of Enterovirus
Inhibitors against Enterovirus 71. Antimicrob Agents Chemother 2016; 60:5357-5367.
440. Morrey JD, Taro BS, Siddharthan V, et al. Efficacy of orally administered T-705
pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 2008;
80:377-379.
441. Madelain V, Guedj J, Mentre F, et al. Favipiravir Pharmacokinetics in Nonhuman
Primates and Insights for Future Efficacy Studies of Hemorrhagic Fever Viruses.
Antimicrob Agents Chemother 2017; 61: e01305-01316.
442. Kumagai Y, Murakawa Y, Hasunuma T, et al. Lack of effect of favipiravir, a novel
antiviral agent, on QT interval in healthy Japanese adults. Int J Clin Pharmacol Ther
2015; 53:866-874.
443. Schibler M, Vetter P, Cherpillod P, et al. Clinical features and viral kinetics in a rapidly
cured patient with Ebola virus disease: a case report. Lancet Infect Dis 2015; 15:1034-
1040.
444. Nguyen TH, Guedj J. Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI
trial reveals concentrations lower than targeted. PLoS Negl Trop Dis 2017; 11:e0005389.
445. Bai CQ, Mu JS, Kargbo D, et al. Clinical and Virological Characteristics of Ebola Virus
Disease Patients Treated With Favipiravir (T-705)-Sierra Leone, 2014. Clin Infect Dis
2016; 63:1288-1294.
446. Julander JG, Bantia S, Taubenheim BR, et al. BCX4430, a novel nucleoside analog,
effectively treats yellow fever in a Hamster model. Antimicrob Agents Chemother 2014;
58:6607-6614.

231
447. Julander JG, Siddharthan V, Evans J, et al. Efficacy of the broad-spectrum antiviral
compound BCX4430 against Zika virus in cell culture and in a mouse model. Antiviral
Res 2017; 137:14-22.
448. Taylor R, Kotian P, Warren T, et al. BCX4430 - A broad-spectrum antiviral adenosine
nucleoside analog under development for the treatment of Ebola virus disease. J Infect
Public Health 2016; 9:220-226.
449. Lo MK, Jordan R, Arvey A, et al. GS-5734 and its parent nucleoside analog inhibit Filo-,
Pneumo-, and Paramyxoviruses. Sci Rep 2017; 7:10.1038.
450. Jacobs M, Rodger A, Bell DJ, et al. Late Ebola virus relapse causing
meningoencephalitis: a case report. Lancet 2016; 388:498-503.
451. Dornemann J, Burzio C, Ronsse A, et al. First Newborn Baby to Receive Experimental
Therapies Survives Ebola Virus Disease. J Infect Dis 2017; 215:171-174.
452. Dowall SD, Bewley K, Watson RJ, et al. Antiviral Screening of Multiple Compounds
against Ebola Virus. Viruses 2016; 8:10.3390.
453. Liu G, Nash PJ, Johnson B, et al. A Sensitive in Vitro High-Throughput Screen To
Identify Pan-filoviral Replication Inhibitors Targeting the VP35-NP Interface. ACS Infect
Dis 2017; 3:190-198.
454. Madrid PB, Panchal RG, Warren TK, et al. Evaluation of Ebola Virus Inhibitors for Drug
Repurposing. ACS Infect Dis 2015; 1:317-326.
455. Jacome R, Becerra A, Ponce de Leon S, et al. Structural Analysis of Monomeric RNA-
Dependent Polymerases: Evolutionary and Therapeutic Implications. PLoS One 2015;
10:e0139001.
456. van Hemert FJ, Zaaijer HL, Berkhout B. In silico prediction of ebolavirus RNA
polymerase inhibition by specific combinations of approved nucleotide analogues. J Clin
Virol 2015; 73:89-94.
457. Lazcano A, Valverde V, Hernandez G, et al. On the early emergence of reverse
transcription: theoretical basis and experimental evidence. J Mol Evol 1992; 35:524-536.
458. Ricchetti M, Buc H. E. coli DNA polymerase I as a reverse transcriptase. Embo j 1993;
12:387-396.
459. Yang X, Smidansky ED, Maksimchuk KR, et al. Motif D of viral RNA-dependent RNA
polymerases determines efficiency and fidelity of nucleotide addition. Structure 2012;
20:1519-1527.

232
460. Doublie S, Tabor S, Long AM, et al. Crystal structure of a bacteriophage T7 DNA
replication complex at 2.2 A resolution. Nature 1998; 391:251-258.
461. Huang H, Chopra R, Verdine GL, et al. Structure of a covalently trapped catalytic
complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998;
282:1669-1675.
462. Alam I, Lee JH, Cho KJ, et al. Crystal structures of murine norovirus-1 RNA-dependent
RNA polymerase in complex with 2-thiouridine or ribavirin. Virology 2012; 426:143-
151.
463. Thompson AA, Albertini RA, Peersen OB. Stabilization of poliovirus polymerase by
NTP binding and fingers-thumb interactions. J Mol Biol 2007; 366:1459-1474.
464. Biswal BK, Cherney MM, Wang M, et al. Crystal structures of the RNA-dependent RNA
polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest
mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem 2005; 280:18202-
18210.
465. Lohmann V, Korner F, Herian U, et al. Biochemical properties of hepatitis C virus NS5B
RNA-dependent RNA polymerase and identification of amino acid sequence motifs
essential for enzymatic activity. J Virol 1997; 71:8416-8428.
466. Vazquez AL, Alonso JM, Parra F. Mutation analysis of the GDD sequence motif of a
calicivirus RNA-dependent RNA polymerase. J Virol 2000; 74:3888-3891.
467. Bressanelli S, Tomei L, Roussel A, et al. Crystal structure of the RNA-dependent RNA
polymerase of hepatitis C virus. Proc Natl Acad Sci U S A 1999; 96:13034-13039.
468. Ng KK, Arnold JJ, Cameron CE. Structure-function relationships among RNA-dependent
RNA polymerases. Curr Top Microbiol Immunol 2008; 320:137-156.
469. Cerny J, Cerna Bolfikova B, Valdes JJ, et al. Evolution of tertiary structure of viral RNA
dependent polymerases. PLoS One 2014; 9:e96070.
470. Ferrer-Orta C, Arias A, Escarmis C, et al. A comparison of viral RNA-dependent RNA
polymerases. Curr Opin Struct Biol 2006; 16:27-34.
471. Gohara DW, Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase
(3Dpol): kinetic, thermodynamic, and structural analysis of ribonucleotide selection.
Biochemistry 2004; 43:5149-5158.
472. Garriga D, Ferrer-Orta C, Querol-Audi J, et al. Role of motif B loop in allosteric
regulation of RNA-dependent RNA polymerization activity. J Mol Biol 2013; 425:2279-
2287.

233
473. Joyce CM. Choosing the right sugar: how polymerases select a nucleotide substrate. Proc
Natl Acad Sci U S A 1997; 94:1619-1622.
474. Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem
1999; 274:17395-17398.
475. Wu Y, Lou Z, Miao Y, et al. Structures of EV71 RNA-dependent RNA polymerase in
complex with substrate and analogue provide a drug target against the hand-foot-and-
mouth disease pandemic in China. Protein Cell 2010; 1:491-500.
476. Choi KH, Groarke JM, Young DC, et al. The structure of the RNA-dependent RNA
polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo
initiation. Proc Natl Acad Sci U S A 2004; 101:4425-4430.
477. Ferrer-Orta C, Arias A, Perez-Luque R, et al. Sequential structures provide insights into
the fidelity of RNA replication. Proc Natl Acad Sci U S A 2007; 104:9463-9468.
478. Schneidman-Duhovny D, Inbar Y, Nussinov R, et al. PatchDock and SymmDock: servers
for rigid and symmetric docking. Nucleic Acids Res 2005; 33:W363-367.
479. Mitsuya H, Weinhold KJ, Furman PA, et al. 3'-Azido-3'-deoxythymidine (BW A509U):
an antiviral agent that inhibits the infectivity and cytopathic effect of human T-
lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad
Sci U S A 1985; 82:7096-7100.
480. Furman PA, Fyfe JA, St Clair MH, et al. Phosphorylation of 3'-azido-3'-deoxythymidine
and selective interaction of the 5'-triphosphate with human immunodeficiency virus
reverse transcriptase. Proc Natl Acad Sci U S A 1986; 83:8333-8337.
481. Larder BA, Kemp SD, Harrigan PR. Potential mechanism for sustained antiretroviral
efficacy of AZT-3TC combination therapy. Science 1995; 269:696-699.
482. WHO Guidelines Approved by the Guidelines Review Committee. In: Antiretroviral
Therapy for HIV Infection in Adults and Adolescents: Recommendations for a Public
Health Approach: 2010 Revision. Geneva: World Health Organization; 2010.
483. Parker WB, White EL, Shaddix SC, et al. Mechanism of inhibition of human
immunodeficiency virus type 1 reverse transcriptase and human DNA polymerases alpha,
beta, and gamma by the 5'-triphosphates of carbovir, 3'-azido-3'-deoxythymidine, 2',3'-
dideoxyguanosine and 3'-deoxythymidine. A novel RNA template for the evaluation of
antiretroviral drugs. J Biol Chem 1991; 266:1754-1762.
484. Konig H, Behr E, Lower J, et al. Azidothymidine triphosphate is an inhibitor of both
human immunodeficiency virus type 1 reverse transcriptase and DNA polymerase
gamma. Antimicrob Agents Chemother 1989; 33:2109-2114.

234
485. Desai VG, Lee T, Moland CL, et al. Effect of short-term exposure to zidovudine (AZT)
on the expression of mitochondria-related genes in skeletal muscle of neonatal mice.
Mitochondrion 2009; 9:9-16.
486. Dalakas MC, Illa I, Pezeshkpour GH, et al. Mitochondrial myopathy caused by long-term
zidovudine therapy. N Engl J Med 1990; 322:1098-1105.
487. Jaju M, Beard WA, Wilson SH. Human immunodeficiency virus type 1 reverse
transcriptase. 3'-Azidodeoxythymidine 5'-triphosphate inhibition indicates two-step
binding for template-primer. J Biol Chem 1995; 270:9740-9747.
488. Zhan X, Tan CK, Scott WA, et al. Catalytically distinct conformations of the
ribonuclease H of HIV-1 reverse transcriptase by substrate cleavage patterns and
inhibition by azidothymidylate and N-ethylmaleimide. Biochemistry 1994; 33:1366-1372.
489. Delviks-Frankenberry KA, Nikolenko GN, Barr R, et al. Mutations in human
immunodeficiency virus type 1 RNase H primer grip enhance 3'-azido-3'-deoxythymidine
resistance. J Virol 2007; 81:6837-6845.
490. Caliendo AM, Savara A, An D, et al. Effects of zidovudine-selected human
immunodeficiency virus type 1 reverse transcriptase amino acid substitutions on
processive DNA synthesis and viral replication. J Virol 1996; 70:2146-2153.
491. Sarafianos SG, Clark AD, Jr., Das K, et al. Structures of HIV-1 reverse transcriptase with
pre- and post-translocation AZTMP-terminated DNA. Embo j 2002; 21:6614-6624.
492. Boyer PL, Sarafianos SG, Clark PK, et al. Why do HIV-1 and HIV-2 use different
pathways to develop AZT resistance? PLoS Pathog 2006; 2:e10.
493. Galle PR, Theilmann L. Inhibition of hepatitis B virus polymerase-activity by various
agents. Transient expression of hepatitis B virus DNA in hepatoma cells as novel system
for evaluation of antiviral drugs. Drug Res 1990; 40:1380-1382.
494. Berk L, Schalm SW, Heijtink RA. Zidovudine inhibits hepatitis B virus replication.
Antiviral Res 1992; 19:111-118.
495. Macchi B, Faraoni I, Zhang J, et al. AZT inhibits the transmission of human T cell
leukaemia/lymphoma virus type I to adult peripheral blood mononuclear cells in vitro. J
Gen Virol 1997; 78:1007-1016.
496. Yvon-Groussin A, Mugnier P, Bertin P, et al. Efficacy of dideoxynucleosides against
human foamy virus and relationship to its reverse transcriptase amino acid sequence and
structure. J Virol 2001; 75:7184-7187.
497. Nam JH, Yu CH, Hwang KA, et al. Real-time RT-PCR of Hantaan virus RNA used for
the detection of virus response to antiviral drugs. Acta Virol 2008; 52:67-70.

235
498. Uckun FM, Venkatachalam TK, Erbeck D, et al. Zidampidine, an aryl phosphate
derivative of AZT: in vivo pharmacokinetics, metabolism, toxicity, and anti-viral efficacy
against hemorrhagic fever caused by Lassa virus. Bioorg Med Chem 2005; 13:3279-3288.
499. McCormick JB, Sasso DR, Palmer EL, et al. Morphological identification of the agent of
Korean haemorrhagic fever (Hantaan virus)as a member of the Bunyaviridae. Lancet
1982; 1:765-768.
500. Xiao SY, Liang M, Schmaljohn CS. Molecular and antigenic characterization of HV114,
a hantavirus isolated from a patient with haemorrhagic fever with renal syndrome in
China. J Gen Virol 1993; 74:1657-1659.
501. Kim JA, Kim WK, No JS, et al. Genetic Diversity and Reassortment of Hantaan Virus
Tripartite RNA Genomes in Nature, the Republic of Korea. PLoS Negl Trop Dis 2016;
10:e0004650.
502. Auperin DD, Sasso DR, McCormick JB. Nucleotide sequence of the glycoprotein gene
and intergenic region of the Lassa virus S genome RNA. Virology 1986; 154:155-167.
503. Lin MT, Chou YP, Hu TH, et al. Telbivudine and adefovir combination therapy for
patients with chronic lamivudine-resistant hepatitis B virus infections. Arch Virol 2014;
159:29-37.
504. Xie H, Voronkov M, Liotta DC, et al. Phosphatidyl-2',3'-dideoxy-3'-thiacytidine:
synthesis and antiviral activity in hepatitis B-and HIV-1-infected cells. Antiviral Res
1995; 28:113-120.
505. Coates JA, Cammack N, Jenkinson HJ, et al. (-)-2'-deoxy-3'-thiacytidine is a potent,
highly selective inhibitor of human immunodeficiency virus type 1 and type 2 replication
in vitro. Antimicrob Agents Chemother 1992; 36:733-739.
506. Hart GJ, Orr DC, Penn CR, et al. Effects of (-)-2'-deoxy-3'-thiacytidine (3TC) 5'-
triphosphate on human immunodeficiency virus reverse transcriptase and mammalian
DNA polymerases alpha, beta, and gamma. Antimicrob Agents Chemother 1992;
36:1688-1694.
507. Zhang Y, Song F, Gao Z, et al. Long-term exposure of mice to nucleoside analogues
disrupts mitochondrial DNA maintenance in cortical neurons. PLoS One 2014; 9:e85637.
508. Gray NM, Marr CL, Penn CR, et al. The intracellular phosphorylation of (-)-2'-deoxy-3'-
thiacytidine (3TC) and the incorporation of 3TC 5'-monophosphate into DNA by HIV-1
reverse transcriptase and human DNA polymerase gamma. Biochem Pharmacol 1995;
50:1043-1051.

236
509. Back NK, Nijhuis M, Keulen W, et al. Reduced replication of 3TC-resistant HIV-1
variants in primary cells due to a processivity defect of the reverse transcriptase enzyme.
Embo j 1996; 15:4040-4049.
510. Gao L, Hanson MN, Balakrishnan M, et al. Apparent defects in processive DNA
synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse
transcriptase derive solely from a dNTP utilization defect. J Biol Chem 2008; 283:9196-
9205.
511. Oude Essink BB, Back NK, Berkhout B. Increased polymerase fidelity of the 3TC-
resistant variants of HIV-1 reverse transcriptase. Nucleic Acids Res 1997; 25:3212-3217.
512. Wei X, Liang C, Gotte M, et al. The M184V mutation in HIV-1 reverse transcriptase
reduces the restoration of wild-type replication by attenuated viruses. AIDS 2002;
16:2391-2398.
513. Sarafianos SG, Das K, Clark AD, Jr., et al. Lamivudine (3TC) resistance in HIV-1
reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc Natl
Acad Sci U S A 1999; 96:10027-10032.
514. Severini A, Liu XY, Wilson JS, et al. Mechanism of inhibition of duck hepatitis B virus
polymerase by (-)-beta-L-2',3'-dideoxy-3'-thiacytidine. Antimicrob Agents Chemother
1995; 39:1430-1435.
515. Taylor GP, Hall SE, Navarrete S, et al. Effect of lamivudine on human T-cell leukemia
virus type 1 (HTLV-1) DNA copy number, T-cell phenotype, and anti-tax cytotoxic T-
cell frequency in patients with HTLV-1-associated myelopathy. J Virol 1999; 73:10289-
10295.
516. Das K, Xiong X, Yang H, et al. Molecular modeling and biochemical characterization
reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and
emtricitabine (FTC). J Virol 2001; 75:4771-4779.
517. Urban S, Urban S, Fischer KP, et al. Efficient pyrophosphorolysis by a hepatitis B virus
polymerase may be a primer-unblocking mechanism. Proc Natl Acad Sci U S A 2001;
98:4984-4989.
518. Cong Y, Dyall J, Hart BJ. Evaluation of the Activity of Lamivudine and Zidovudine
against Ebola Virus. PLoS One 2016; 11:e0166318.
519. Hensley LE, Dyall J, Olinger GG, Jr., et al. Lack of effect of Lamivudine on ebola virus
replication. Emerg Infect Dis 2015; 21:550-552.
520. Zhao Y, Ren J, Harlos K, et al. Toremifene interacts with and destabilizes the Ebola virus
glycoprotein. Nature 2016; 535:169-172.

237
521. Suo Z, Johnson KA. Selective inhibition of HIV-1 reverse transcriptase by an antiviral
inhibitor, (R)-9-(2-Phosphonylmethoxypropyl)adenine. J Biol Chem 1998; 273:27250-
27258.
522. Nelson M, Portsmouth S, Stebbing J, et al. An open-label study of tenofovir in HIV-1
and Hepatitis B virus co-infected individuals. AIDS 2003; 17:F7-10.
523. Cassetti I, Madruga JV, Suleiman JM, et al. The safety and efficacy of tenofovir DF in
combination with lamivudine and efavirenz through 6 years in antiretroviral-naive HIV-
1-infected patients. HIV Clin Trials 2007; 8:164-172.
524. Johnson AA, Ray AS, Hanes J, et al. Toxicity of antiviral nucleoside analogs and the
human mitochondrial DNA polymerase. J Biol Chem 2001; 276:40847-40857.
525. Kulkarni R, Feng JY, Miller MD, et al. Dead-end complexes contribute to the synergistic
inhibition of HIV-1 RT by the combination of rilpivirine, emtricitabine, and tenofovir.
Antiviral Res 2014; 101:131-135.
526. Hachiya A, Kodama EN, Schuckmann MM, et al. K70Q adds high-level tenofovir
resistance to "Q151M complex" HIV reverse transcriptase through the enhanced
discrimination mechanism. PLoS One 2011; 6:e16242.
527. Chan PA, Huang A, Kantor R. Low prevalence of transmitted K65R and other tenofovir
resistance mutations across different HIV-1 subtypes: implications for pre-exposure
prophylaxis. J Int AIDS Soc 2012; 15:10.7448.
528. Jones SA, Murakami E, Delaney W, et al. Noncompetitive inhibition of hepatitis B virus
reverse transcriptase protein priming and DNA synthesis by the nucleoside analog
clevudine. Antimicrob Agents Chemother 2013; 57:4181-4189.
529. van Hemert FJ, Berkhout B, Zaaijer HL. Differential binding of tenofovir and adefovir to
reverse transcriptase of hepatitis B virus. PLoS One 2014; 9:e106324.
530. Oestereich L, Rieger T, Ludtke A, et al. Efficacy of Favipiravir Alone and in
Combination With Ribavirin in a Lethal, Immunocompetent Mouse Model of Lassa
Fever. J Infect Dis 2016; 213:934-938.
531. Staszewski S, Loveday C, Picazo JJ, et al. Safety and efficacy of lamivudine-zidovudine
combination therapy in zidovudine-experienced patients. A randomized controlled
comparison with zidovudine monotherapy. Lamivudine European HIV Working Group.
JAMA 1996; 276:111-117.
532. Xiao F, Fofana I, Thumann C, et al. Synergy of entry inhibitors with direct-acting
antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut
2014; 64:483-494.

238
533. Chou TC. Theoretical basis, experimental design, and computerized simulation of
synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621-
681.
534. Hoenen T, Watt A, Mora A, et al. Modeling The Lifecycle Of Ebola Virus Under
Biosafety Level 2 Conditions With Virus-like Particles Containing Tetracistronic
Minigenomes. J Vis Exp 2014; 91:52381.
535. Hoenen T, Feldmann H. Reverse genetics systems as tools for the development of novel
therapies against filoviruses. Expert Rev Anti Infect Ther 2014; 12:1253-1263.
536. de Avila AI, Gallego I, Soria ME, et al. Lethal Mutagenesis of Hepatitis C Virus Induced
by Favipiravir. PLoS One 2016; 11:e0164691.
537. Bermejo M, Lopez-Huertas MR, Garcia-Perez J, et al. Dasatinib inhibits HIV-1
replication through the interference of SAMHD1 phosphorylation in CD4+ T cells.
Biochem Pharmacol 2016; 106:30-45.
538. Di Mascio M, Srinivasula S, Bhattacharjee A, et al. Antiretroviral tissue kinetics: in vivo
imaging using positron emission tomography. Antimicrob Agents Chemother 2009;
53:4086-4095.
539. Harmon B, Campbell N, Ratner L. Role of Abl kinase and the Wave2 signaling complex
in HIV-1 entry at a post-hemifusion step. PLoS Pathog 2010; 6:e1000956.
540. Lund N, Olsson ML, Ramkumar S, et al. The human P(k) histo-blood group antigen
provides protection against HIV-1 infection. Blood 2009; 113:4980-4991.
541. Bokaei PB, Ma XZ, Sakac D, et al. HIV-1 integration is inhibited by stimulation of the
VPAC2 neuroendocrine receptor. Virology 2007; 362:38-49.
542. Yamamoto N, Tanaka C, Wu Y, et al. Analysis of human immunodeficiency virus type 1
integration by using a specific, sensitive and quantitative assay based on real-time
polymerase chain reaction. Virus Genes 2006; 32:105-113.
543. O'Doherty U, Swiggard WJ, Jeyakumar D, et al. A sensitive, quantitative assay for
human immunodeficiency virus type 1 integration. J Virol 2002; 76:10942-10950.
544. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T)
method. Nat Protoc 2008; 3:1101-1108.
545. Kim M, Binnington B, Sakac D, et al. Comparison of detection methods for cell surface
globotriaosylceramide. J Immunol Methods 2011; 371:48-60.

239
546. Vermeire J, Naessens E, Vanderstraeten H, et al. Quantification of reverse transcriptase
activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and
retroviral vectors. PLoS One 2012; 7:e50859.
547. Akari H, Uchiyama T, Fukumori T, et al. Pseudotyping human immunodeficiency virus
type 1 by vesicular stomatitis virus G protein does not reduce the cell-dependent
requirement of vif for optimal infectivity: functional difference between Vif and Nef. J
Gen Virol 1999; 80 ( Pt 11):2945-2949.
548. Guendel I, Agbottah ET, Kehn-Hall K, et al. Inhibition of human immunodeficiency
virus type-1 by cdk inhibitors. AIDS Res Ther 2010; 7:7.
549. Puertas MC, Massanella M, Llibre JM, et al. Intensification of a raltegravir-based
regimen with maraviroc in early HIV-1 infection. Aids 2014; 28:325-334.
550. Wei DG, Chiang V, Fyne E, et al. Histone deacetylase inhibitor romidepsin induces HIV
expression in CD4 T cells from patients on suppressive antiretroviral therapy at
concentrations achieved by clinical dosing. PLoS Pathog 2014; 10:e1004071.
551. Desimmie BA, Schrijvers R, Demeulemeester J, et al. LEDGINs inhibit late stage HIV-1
replication by modulating integrase multimerization in the virions. Retrovirology 2013;
10:57.
552. Shan L, Xing S, Yang HC, et al. Unique characteristics of histone deacetylase inhibitors
in reactivation of latent HIV-1 in Bcl-2-transduced primary resting CD4+ T cells. J
Antimicrob Chemother 2014; 69:28-33.
553. Shah NP, Guilhot F, Cortes JE, et al. Long-term outcome with dasatinib after imatinib
failure in chronic-phase chronic myeloid leukemia: follow-up of phase 3 study. Blood
2014.
554. Pogliaghi M, Papagno L, Lambert S, et al. The tyrosine kinase inhibitor Dasatinib blocks
in-vitro HIV-1 production by primary CD4+ T cells from HIV-1 infected patients. AIDS
2014; 28:278-281.
555. McCarthy SD, Jung D, Sakac D, et al. c-Src and Pyk2 Protein Tyrosine Kinases Play
Protective Roles in Early HIV-1 Infection of CD4+ T-Cell Lines. J Acquir Immune Defic
Syndr 2014; 66:118-126.
556. Pizzato M, Erlwein O, Bonsall D, et al. A one-step SYBR Green I-based product-
enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture
supernatants. J Virol Methods 2009; 156:1-7.
557. Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further
update. Biochem J 2007; 408:297-315.

240
558. Harvey R, Hehir KM, Smith AE, et al. pp60c-src variants containing lesions that affect
phosphorylation at tyrosines 416 and 527. Mol Cell Biol 1989; 9:3647-3656.
559. Collins M, Tremblay M, Chapman N, et al. The T cell receptor-mediated phosphorylation
of Pyk2 tyrosines 402 and 580 occurs via a distinct mechanism than other receptor
systems. J Leukoc Biol 2010; 87:691-701.
560. Mehla R, Bivalkar-Mehla S, Zhang R, et al. Bryostatin modulates latent HIV-1 infection
via PKC and AMPK signaling but inhibits acute infection in a receptor independent
manner. PLoS One 2010; 5:e11160.
561. Tyler T. Once-daily dasatinib for treatment of patients with chronic myeloid leukemia.
Ann Pharmacother 2009; 43:920-927.
562. Bartelt RR, Cruz-Orcutt N, Collins M, et al. Comparison of T cell receptor-induced
proximal signaling and downstream functions in immortalized and primary T cells. PLoS
One 2009; 4:e5430.
563. Rato S, Maia S, Brito PM, et al. Novel HIV-1 knockdown targets identified by an
enriched kinases/phosphatases shRNA library using a long-term iterative screen in Jurkat
T-cells. PLoS One 2010; 5:e9276.
564. Zhou H, Xu M, Huang Q, et al. Genome-scale RNAi screen for host factors required for
HIV replication. Cell Host Microbe 2008; 4:495-504.
565. Hatano H, Strain MC, Scherzer R, et al. Increase in 2-long terminal repeat circles and
decrease in D-dimer after raltegravir intensification in patients with treated HIV
infection: a randomized, placebo-controlled trial. J Infect Dis 2013; 208:1436-1442.
566. Buzon MJ, Massanella M, Llibre JM, et al. HIV-1 replication and immune dynamics are
affected by raltegravir intensification of HAART-suppressed subjects. Nat Med 2010;
16:460-465.
567. Hazuda DJ, Felock P, Witmer M, et al. Inhibitors of strand transfer that prevent
integration and inhibit HIV-1 replication in cells. Science 2000; 287:646-650.
568. World Health Organization. Ebola Situation Report – 16 December 2015. 2015.
http://apps.who.int/ebola/en/current-situation/ebola-situation-report-13-december-2015
569. Wong G, Audet J, Fernando L, et al. Immunization with vesicular stomatitis virus
vaccine expressing the Ebola glycoprotein provides sustained long-term protection in
rodents. Vaccine 2014; 32:5722-5729.
570. Wong KK, Perdue CL, Malia J, et al. Supportive Care of the First 2 Ebola Virus Disease
Patients at the Monrovia Medical Unit. Clin Infect Dis 2015; 61:e47-51.

241
571. Florescu DF, Keck MA. Development of CMX001 (Brincidofovir) for the treatment of
serious diseases or conditions caused by dsDNA viruses. Expert Rev Anti Infect Ther
2014; 12:1171-1178.
572. Muhlberger E, Weik M, Volchkov VE, et al. Comparison of the transcription and
replication strategies of marburg virus and Ebola virus by using artificial replication
systems. J Virol 1999; 73:2333-2342.
573. Phanuphak N, Ananworanich J, Teeratakulpisarn N, et al. A 72-week randomized study
of the safety and efficacy of a stavudine to zidovudine switch at 24 weeks compared to
zidovudine or tenofovir disoproxil fumarate when given with lamivudine and nevirapine.
Antivir Ther 2012; 17:1521-1531.
574. World Health Organization. 18th WHO Model List of Essential Medicines (April 2013).
2013. http://www.who.int/medicines/publications/essentialmedicines/en/
575. Ahmed Ouameur A, Marty R, Neault JF, et al. AZT binds RNA at multiple sites. DNA
Cell Biol 2004; 23:783-788.
576. Cardenas WB. Evasion of the interferon-mediated antiviral response by filoviruses.
Viruses 2010; 2:262-282.
577. Xu W, Edwards MR, Borek DM, et al. Ebola virus VP24 targets a unique NLS binding
site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated
STAT1. Cell Host Microbe 2014; 16:187-200.
578. Fabozzi G, Nabel CS, Dolan MA, et al. Ebolavirus proteins suppress the effects of small
interfering RNA by direct interaction with the mammalian RNA interference pathway. J
Virol 2011; 85:2512-2523.
579. Chang TH, Kubota T, Matsuoka M, et al. Ebola Zaire virus blocks type I interferon
production by exploiting the host SUMO modification machinery. PLoS Pathog 2009;
5:e1000493.
580. Qiu X, Wong G, Fernando L, et al. mAbs and Ad-vectored IFN-alpha therapy rescue
Ebola-infected nonhuman primates when administered after the detection of viremia and
symptoms. Sci Transl Med 2013; 5:207ra143.
581. Qiu X, Wong G, Fernando L, et al. Monoclonal antibodies combined with adenovirus-
vectored interferon significantly extend the treatment window in Ebola virus-infected
guinea pigs. J Virol 2013; 87:7754-7757.
582. Ebihara H, Theriault S, Neumann G, et al. In vitro and in vivo characterization of
recombinant Ebola viruses expressing enhanced green fluorescent protein. J Infect Dis
2007; 196:S313-322.

242
583. Hoenen T, Jung S, Herwig A, et al. Both matrix proteins of Ebola virus contribute to the
regulation of viral genome replication and transcription. Virology 2010; 403:56-66.
584. Mehrotra S, Sharma B, Joshi S, et al. Essential role for the Mnk pathway in the inhibitory
effects of type I interferons on myeloproliferative neoplasm (MPN) precursors. J Biol
Chem 2013; 288:23814-23822.
585. Watt A, Moukambi F, Banadyga L, et al. A novel life cycle modeling system for Ebola
virus shows a genome length-dependent role of VP24 in virus infectivity. J Virol 2014;
88:10511-10524.
586. Johansen LM, DeWald LE, Shoemaker CJ, et al. A screen of approved drugs and
molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med
2015; 7:290ra289.
587. Lok AS, Lai CL, Leung N, et al. Long-term safety of lamivudine treatment in patients
with chronic hepatitis B. Gastroenterol 2003; 125:1714-1722.
588. Baeten JM, Donnell D, Mugo NR, et al. Single-agent tenofovir versus combination
emtricitabine plus tenofovir for pre-exposure prophylaxis for HIV-1 acquisition: an
update of data from a randomised, double-blind, phase 3 trial. Lancet Infect Dis 2014;
14:1055-1064.
589. Thoulouze MI, Sol-Foulon N, Blanchet F, et al. Human immunodeficiency virus type-1
infection impairs the formation of the immunological synapse. Immunity 2006; 24:547-
561.
590. Uitdehaag JCM, Verkaar F, Alwan H, et al. A guide to picking the most selective kinase
inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol
2012; 166:858-876.
591. Shi H, Zhang CJ, Chen GY, et al. Cell-based proteome profiling of potential dasatinib
targets by use of affinity-based probes. J Am Chem Soc 2012; 134:3001-3014.
592. Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling
paradigm. Nat Rev Immunol 2004; 4:301-308.
593. Bronevetsky Y, Villarino AV, Eisley CJ, et al. T cell activation induces proteasomal
degradation of Argonaute and rapid remodeling of the microRNA repertoire. J Exp Med
2013; 210:417-432.
594. Kinter A, McNally J, Riggin L, et al. Suppression of HIV-specific T cell activity by
lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc Natl Acad Sci
U S A 2007; 104:3390-3395.

243
595. Pallikkuth S, Sharkey M, Babic DZ, et al. Peripheral T Follicular Helper Cells Are the
Major HIV Reservoir within Central Memory CD4 T Cells in Peripheral Blood from
Chronically HIV-Infected Individuals on Combination Antiretroviral Therapy. J Virol
2015; 90:2718-2728.
596. Blom N, Sicheritz-Ponten T, Gupta R, et al. Prediction of post-translational glycosylation
and phosphorylation of proteins from the amino acid sequence. Proteomics 2004; 4:1633-
1649.
597. Chen P, Li F, Xu Z, et al. Expression and distribution of Src in the nucleus of myocytes
in cardiac hypertrophy. Int J Mol Med 2013; 32:165-173.
598. Cooper A, Garcia M, Petrovas C, et al. HIV-1 causes CD4 cell death through DNA-
dependent protein kinase during viral integration. Nature 2013; 498:376-379.
599. Patel M, Philip V, Fazel F, et al. Human immunodeficiency virus infection and chronic
myeloid leukemia. Leuk Res 2012; 36:1334-1338.
600. Schlaberg R, Fisher JG, Flamm MJ, et al. Chronic myeloid leukemia and HIV-infection.
Leuk Lymphoma 2008; 49:1155-1160.
601. Chung WM, Smith JC, Weil LM, et al. Active Tracing and Monitoring of Contacts
Associated With the First Cluster of Ebola in the United States. Ann Intern Med 2015;
163:164-173.
602. Tippin TK, Morrison ME, Brundage TM, et al. Brincidofovir Is Not a Substrate for the
Human Organic Anion Transporter 1: A Mechanistic Explanation for the Lack of
Nephrotoxicity Observed in Clinical Studies. Ther Drug Monit 2016; 38:777-786.
603. M'Kada H, Munteanu M, Perazzo H, et al. What are the best reference values for a
normal serum alanine transaminase activity (ALT)? Impact on the presumed prevalence
of drug induced liver injury (DILI). Regul Toxicol Pharmacol 2011; 60:290-295.
604. World Health Organization. Ebola Situation Report, 20 June 2017 2017.
http://apps.who.int/ebola/ebola-situation-reports
605. García M, Cooper A, Shi W, et al. Productive Replication of Ebola Virus Is Regulated by
the c-Abl1 Tyrosine Kinase. Sci Transl Med 2012; 4:123ra124.
606. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular
viral trafficking and exert broad-spectrum antiviral effects. J Clin Invest 2017; 127:1338-
1352.
607. Schweitzer CJ, Jagadish T, Haverland N, et al. Proteomic analysis of early HIV-1
nucleoprotein complexes. J Proteome Res 2013; 12:559-572.

244
608. Raghavendra NK, Shkriabai N, Graham R, et al. Identification of host proteins associated
with HIV-1 preintegration complexes isolated from infected CD4+ cells. Retrovirology
2010; 7:10.1186.
609. Fujisaka Y, Onozawa Y, Kurata T, et al. First report of the safety, tolerability, and
pharmacokinetics of the Src kinase inhibitor saracatinib (AZD0530) in Japanese patients
with advanced solid tumours. Invest New Drugs 2013; 31:108-114.
610. Haile WB, Gavegnano C, Tao S, et al. The Janus kinase inhibitor ruxolitinib reduces HIV
replication in human macrophages and ameliorates HIV encephalitis in a murine model.
Neurobiol Dis 2016.
611. Bermejo M, Lopez-Huertas MR, Garcia-Perez J, et al. Dasatinib inhibits HIV-1
replication through the interference of SAMHD1 phosphorylation in CD4+ T cells.
Biochem Pharmacol 2016.
612. Gavegnano C, Detorio M, Montero C, et al. Ruxolitinib and tofacitinib are potent and
selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob
Agents Chemother 2014; 58:1977-1986.
613. Haile WB, Gavegnano C, Tao S, et al. The Janus kinase inhibitor ruxolitinib reduces HIV
replication in human macrophages and ameliorates HIV encephalitis in a murine model.
Neurobiol Dis 2016; 92:137-143.
614. Alfson KJ, Worwa G, Carrion R, Jr., et al. Determination and Therapeutic Exploitation of
Ebola Virus Spontaneous Mutation Frequency. J Virol 2015; 90:2345-2355.
615. Sun W, He S, Martinez-Romero C, et al. Synergistic drug combination effectively blocks
Ebola virus infection. Antiviral Res 2017; 137:165-172.

245
Copyright Acknowledgments
The author acknowledges the following publications, from which previously published work has
been reproduced in this thesis, with permission:
McCarthy, S.D., Jung, D., Sakac, D., and Branch, D.R. 2014. c-SRC and Pyk2 protein tyrosine
kinases play protective roles in early HIV-1 infection of CD4+ T-cell lines. JAIDS, 66(2):
118-26.
McCarthy, S.D., Sakac, D., Neschadim, A., and Branch, D.R. 2016. c-SRC protein tyrosine
kinase regulates early HIV-1 infection post-entry. AIDS, 30(6): 849-58.
McCarthy, S.D., Majchrzak-Kita, B., Racine, T., Kozlowski, H.N., Baker, D.P., Hoenen, T.,
Kobinger, G.P., Fish, E.N., and Branch, D.R. 2016. A Rapid Screening Assay Identifies
Monotherapy with Interferon-ß and Combination Therapies with Nucleoside Analogues
as Effective Inhibitors of Ebola Virus. PLoS NTD, 10(1):e0004364.
4. GARP, UNAIDS. Global AIDS Update. 2016.
http://www.unaids.org/en/resources/documents/2016/Global-AIDS-update-2016
5. World Health Organization. Ebola Situation Report, 10 June 2016. 2016.
http://apps.who.int/ebola/ebola-situation-reports
43. Faria NR, Rambaut A, Suchard MA, et al. HIV epidemiology. The early spread and
epidemic ignition of HIV-1 in human populations. Science 2014; 346:56-61.
44. Disease NIoAaI, National Institutes Health. How HIV Causes AIDS. 2004.
45. Suzuki Y, Suzuki Y. Gene Regulatable Lentiviral Vector System. In: Viral Gene
Therapy. Edited by Xu K. Rijeka: InTech; 2011. pp. 10.5772.
53. Coiras M, Lopez-Huertas MR, Perez-Olmeda M, et al. Understanding HIV-1 latency
provides clues for the eradication of long-term reservoirs. Nat Rev Microbiol 2009;
7:798-812.
169. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-
approved drugs. Drug Discov Today 2016; 21:5-10.

246
170. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends
Pharmacol Sci 2015; 36:422-439.
198. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci 2012; 8:1385-
1397.
199. Engen JR, Wales TE, Hochrein JM, et al. Structure and dynamic regulation of Src-family
kinases. Cell Mol Life Sci 2008; 65:3058-3073.
221. Fackler OT, Alcover A, Schwartz O. Modulation of the immunological synapse: a key to
HIV-1 pathogenesis? Nat Rev Immunol 2007; 7:310-317.
328. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control
of cell motility. Nat Rev Mol Cell Biol 2005; 6:56-68.
329. Walkiewicz KW, Girault JA, Arold ST. How to awaken your nanomachines: Site-specific
activation of focal adhesion kinases through ligand interactions. Prog Biophys Mol Biol
2015; 119:60-71.
368. Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB bioinformatics resource portal.
Nucleic Acids Res 2012; 40:W597-603.
455. Jacome R, Becerra A, Ponce de Leon S, et al. Structural Analysis of Monomeric RNA-
Dependent Polymerases: Evolutionary and Therapeutic Implications. PLoS One 2015;
10:e0139001.
456. van Hemert FJ, Zaaijer HL, Berkhout B. In silico prediction of ebolavirus RNA
polymerase inhibition by specific combinations of approved nucleotide analogues. J Clin
Virol 2015; 73:89-94.
562. Bartelt RR, Cruz-Orcutt N, Collins M, et al. Comparison of T cell receptor-induced
proximal signaling and downstream functions in immortalized and primary T cells. PLoS
One 2009; 4:e5430.
Related Documents











