485 ISSN 1479-6678 part of Future Cardiology 10.2217/FCA.11.24 © 2011 Future Medicine Ltd Future Cardiol. (2011) 7(4), 485–497 The epigenetic mechanisms are essentially DNA methylation, histone modifications and RNA interference. Epigenetics plays a crucial role in several pathological conditions, including immune dysfunction, inflammation, cancer and insulin resistance [1] . Recent studies have demon- strated that epigenetic alterations are associated with inflammation and cardiovascular disease (CVD) in patients with chronic kidney disease (CKD). The reversible nature of the epigenetic changes gives a unique opportunity to halt or even reverse the disease process through targeted therapeutic strategies [1] . Epigenetic change can occur as a real subversion of cell programming produced during normal ontogenetic develop- ment of organs and tissues, during differentia- tion and normal cell turnover. Investigating the role of epigenetic mechanisms in triggering/pro- gression of several common diseases has gained emphasis over the recent years. The study of such complex mechanisms has become very important in order to understand the etiopathology of these diseases, as well as to design appropriate targeted clinical therapies. In this article, we analyze the epigenetic-based mechanisms between heart and kidney in the clinical pathophysiology of cardiorenal syndrome (CRS) (FIGURE 1) . Cardiorenal axis dysfunction Coronary artery disease has emerged as the dominant etiologic factor in patients with heart failure (HF) and cardiac ischemia which are among the most common causes of death and hospitalization in most Western countries [2] . Patients with CKD are at higher risk of cardio- vascular disease than the general population, and demonstrate a higher rate of cardiovascu- lar mortality [2] . In the Acute Decompensated Heart Failure National Registry (ADHERE), 30% of hospitalized patients with acute HF had a history of chronic renal failure [2] . The early diagnosis and management of HF are important in patients with CKD. Moreover, early recogni- tion and management of CKD is also necessary in patients with HF [2] . Cardiac and renal bio- markers could facilitate and improve the clinical managements of HF and CKD patients [2] . It is also known that many patients with CKD have not achieved optimal treatment levels [3–4] . This phenomenon exists because CKD does not exist in isolation; the multiple risk factors commonly associated with the CKD patient make optimal management difficult, and the intricate relationship between cardiac and renal physiology demand that both organ systems be addressed [3] . Many years ago, the indexes of myocardial ischemia and vasoconstrictive hor- monal release were already evaluated in order to investigate the difference between essential hypertension and hypertension during chronic renal failure. Arterial hypertension induces several cardiovascular alterations that reflect Kidney and heart interactions during cardiorenal syndrome: a molecular and clinical pathogenic framework Claudio Napoli †1,2 , Amelia Casamassimi 1 , Valeria Crudele 1 , Teresa Infante 2 & Ciro Abbondanza 1 1 Dipartimento di Patologia Generale, Centro di Eccellenza sulle Malattie Cardiovascolari, Facoltà di Medicina e Chirurgia, Seconda Università di Napoli, Via Costantinopoli 16, 80138 Napoli, Italy 2 Istituto di Ricerca Diagnostica e Nucleare-SDN, IRCCS, Via E. Gianturco 113, 80143 Napoli, Italy † Author for correspondence: Tel.: +39 8156 6756 n Fax: +39 8145 0169 n [email protected] The heart and kidney are physiologically interconnected. Cardiorenal syndrome (CRS) is a pathological disorder where acute or chronic dysfunction in one organ may induce dysfunction in the other one. Although classical studies have proposed a role for hypertension, dyslipidemia and endothelial dysfunction, CRS should be considered as a complex molecular interplay of neurohumoral pathway activation including the sympathetic nervous system, the renin angiotensin aldosterone axis, the endothelin system and the arginine vasopressin system. This activation may induce vascular inflammation, oxidative stress, accelerated atherosclerosis, cardiac hypertrophy and both myocardial and intrarenal fibrosis with progression of CRS treatment. More recently, epigenetics has opened new pathogenic molecular routes for CRS. This will lead to a more rapid development of novel, safe and effective clinical therapies. Keywords n cardiac ischemia n cardiorenal syndrome n cardiovascular disease n chronic kidney disease n epigenetics n heart failure Review For reprint orders, please contact: [email protected]

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

485ISSN 1479-6678
part of
Futu
re C
ard
iolo
gy
10.2217/FCA.11.24 © 2011 Future Medicine Ltd Future Cardiol. (2011) 7(4), 485–497
The epigenetic mechanisms are essentially DNA methylation, histone modifications and RNA interference. Epigenetics plays a crucial role in several pathological conditions, including immune dysfunction, inflammation, cancer and insulin resistance [1]. Recent studies have demon-strated that epigenetic alterations are associated with inflammation and cardiovascular disease (CVD) in patients with chronic kidney disease (CKD). The reversible nature of the epigenetic changes gives a unique opportunity to halt or even reverse the disease process through targeted therapeutic strategies [1]. Epigenetic change can occur as a real subversion of cell programming produced during normal ontogenetic develop-ment of organs and tissues, during differentia-tion and normal cell turnover. Investigating the role of epigenetic mechanisms in triggering/pro-gression of several common diseases has gained emphasis over the recent years. The study of such complex mechanisms has become very important in order to understand the etiopathology of these diseases, as well as to design appropriate targeted clinical therapies. In this article, we analyze the epigenetic-based mechanisms between heart and kidney in the clinical pathophysiology of cardiorenal syndrome (CRS) (Figure 1).
Cardiorenal axis dysfunctionCoronary artery disease has emerged as the dominant etiologic factor in patients with heart
failure (HF) and cardiac ischemia which are among the most common causes of death and hospitalization in most Western countries [2]. Patients with CKD are at higher risk of cardio-vascular disease than the general population, and demonstrate a higher rate of cardiovascu-lar mortality [2]. In the Acute Decompensated Heart Failure National Registry (ADHERE), 30% of hospitalized patients with acute HF had a history of chronic renal failure [2]. The early diagnosis and management of HF are important in patients with CKD. Moreover, early recogni-tion and management of CKD is also necessary in patients with HF [2]. Cardiac and renal bio-markers could facilitate and improve the clinical managements of HF and CKD patients [2].
It is also known that many patients with CKD have not achieved optimal treatment levels [3–4]. This phenomenon exists because CKD does not exist in isolation; the multiple risk factors commonly associated with the CKD patient make optimal management difficult, and the intricate relationship between cardiac and renal physiology demand that both organ systems be addressed [3]. Many years ago, the indexes of myocardial ischemia and vasoconstrictive hor-monal release were already evaluated in order to investigate the difference between essential hypertension and hypertension during chronic renal failure. Arterial hypertension induces several cardiovascular alterations that reflect
Kidney and heart interactions during cardiorenal syndrome: a molecular and clinical pathogenic framework
Claudio Napoli†1,2, Amelia Casamassimi1, Valeria Crudele1, Teresa Infante2 & Ciro Abbondanza1
1Dipartimento di Patologia Generale, Centro di Eccellenza sulle Malattie Cardiovascolari, Facoltà di Medicina e Chirurgia, Seconda Università di Napoli, Via Costantinopoli 16, 80138 Napoli, Italy 2Istituto di Ricerca Diagnostica e Nucleare-SDN, IRCCS, Via E. Gianturco 113, 80143 Napoli, Italy †Author for correspondence: Tel.: +39 8156 6756 n Fax: +39 8145 0169 n [email protected]
The heart and kidney are physiologically interconnected. Cardiorenal syndrome (CRS) is a pathological disorder where acute or chronic dysfunction in one organ may induce dysfunction in the other one. Although classical studies have proposed a role for hypertension, dyslipidemia and endothelial dysfunction, CRS should be considered as a complex molecular interplay of neurohumoral pathway activation including the sympathetic nervous system, the renin angiotensin aldosterone axis, the endothelin system and the arginine vasopressin system. This activation may induce vascular inflammation, oxidative stress, accelerated atherosclerosis, cardiac hypertrophy and both myocardial and intrarenal fibrosis with progression of CRS treatment. More recently, epigenetics has opened new pathogenic molecular routes for CRS. This will lead to a more rapid development of novel, safe and effective clinical therapies.
Keywords
n cardiac ischemia n cardiorenal syndrome n cardiovascular disease n chronic kidney disease n epigenetics n heart failure
Revie
wFor reprint orders, please contact: [email protected]

Future Cardiol. (2011) 7(4)486 future science group
themselves either on the heart and/or on the cor-onary blood flow enhancing the cardiovascular risk. Since chronic renal failure can influence the neuroendocrine response, various mechanisms involved in hypertension during chronic renal failure are still unclear [4].
Several pathologies such as metabolic syn-drome, increased arterial stiffness, anemia, pro-teinuria, chronic inflammatory state and uremic toxins are described as nontraditional risk factors for HF in CKD patients [4].
Endothelial progenitor cells (EPCs) are very relevant in vascular biology. These progenitors have been studied as potential therapeutic cells
for vascular regeneration and as biomarkers to assess the risk of several diseases including CKD and CVD. Particularly, their role as mediators of chronic ischemia and myocardial infarction has been assessed. Indeed, transplantation of EPCs into patients induces blood flow recov-ery in ischemic limbs and improves myocardial viability after infarction [4,5]. Circulating EPC levels are reduced in CRS patients and, short-term erythropoietin (EPO) treatment has no effect on EPC levels in these patients, while long term EPO treatment prevents a decline in these cells [6]. Thus, it would be interesting to further investigate the role of EPCs in CRS.
Causes
• ncDNA• Defective DNA repair• Uremic insult?• Exogenous insult
Epigenetic modifications
• DNA methylation• Histone modification
Consequences
• Aberrant gene expression• Loss of imprinting• Chromosomal instability• Microsatellite instability
Histonemodifications
Chromosome
DNA
↑ H3acetyation
Kidney failure
Heart failure
Kidney failure
Aberrant methylation
CH3
CH3
Figure 1. Epigenetic modifications between heart and kidney in cardiorenal syndrome causing both cardiac and renal dysfunction. Epigenetic effects correlate with covalent modifications of the genome resulting from changes in promoter methylation and histone modifications. Thus, epigenetic phenomena are central to the induction of heritable changes in gene expression occurring without alteration of DNA sequence. The H3 acetylation is the only epigenetic direct correlation between kidney and heart during cardiorenal syndrome.
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 487future science group
Hence, dysfunctional cardiac and renal effects can coexist through a complex combination of neurohormonal mechanisms. Drug therapies utilized in the management of renal disease and associated complications may influence cardiac performance. Approximately one-third of the patients with HF have reduced kidney func-tion that is associated with diuretic resistance and increased mortality [7]. Another cardiorenal connection is observed in patients with CKD, who can have an increased risk of cardiovascular complications and death [8,9]. For many years CRS had been generally defined as an acute or chronic renal dysfunction resulting from pri-mary changes in cardiac function. A consensus definition of CRS, stressing the bidirectional nature of heart-kidney interactions, has recently been proposed [10] that describes CRS as a patho-logical disorder of the heart and the kidney in which acute or chronic dysfunction in one organ may induce acute or chronic dysfunction in the other organ [10]. It includes the five subtypes described below.
Cardiorenal syndrome type I, or acute CRS, describes the acute kidney injury as a consequence of a sudden worsening of cardiac function (acute cardiogenic shock or acute decompensation of congestive HF) [11–15].
Cardiorenal syndrome type II, or chronic CRS, describes the progressive and CKD caused by chronic cardiac dysfunction [16–23].
Cardiorenal syndrome type III, or acute reno-cardiac syndrome, describes a sudden cardiac disorder as a consequence of an impairment of kidney function [10].
Cardiorenal syndrome type IV, or chronic renocardiac syndrome, is attributed to the decreased cardiac function, accelerated ath-erosclerosis, left ventricular hypertrophy and increased risk of cardiovascular events in patients with CKD [24–25].
Cardiorenal syndrome type V describes a situ-ation in which a systemic condition causes both cardiac and renal dysfunction [26,27].
Epigenetic mechanismsEpigenetic inheritance is responsible for the huge number of phenotypic differences between cell types in multicellular organisms [28]. This may explain for example why subjects having similar genetic background and both environmental and classical risk factors for CVD and/or CKD could have a very different outcome in clinical manifestation of these diseases. Interestingly, it is still unclear if the altered gene expression pat-terns can be passed to the progeny upon cell
division or even transgenerationally [29]. The main mechanisms of epigenetic modifications in mammals include DNA methylation, his-tone modifications, which result in changes of chromatin structure and RNA-based silencing (miRNAs). miRNAs are important for kidney development and homeostasis, and are known to play a pathogenic role in renal diseases. Thus, a better understanding of their mechanisms in this context could revolutionize both the diagnosis and treatment of major renal diseases [30–32].
Indeed epigenetics is a dynamic process, which regulates gene expression patterns in normal and diseased state. These mechanisms alter the physical accessibility to the genome through gene expression and molecular complexes can then alter the function of genes [28].
The mechanism of gene silencing by DNA methylation may be related to stearic obstacle of the transcriptional machinery, recruitment of repressors, or alteration in chromatin con-figuration [28]. The induction and maintenance of DNA methylation are catalyzed by DNA methyl transferase-1 and DNA methyltrans-ferase-3b and are responsible for maintaining abnormal promoter methylation in diseased cells [28]. Furthermore, DNA methyltransfer-ases interact directly with histone deacetylases (HDACs) to recruit them to gene promoters.
Histone acetyltransferases and HDACs act in an opposing manner to control the acetylation state of proteins. The most well characterized role for protein acetylation is in the control of gene transcription. Acetylation of lysine residues in nucleosome histone tails by histone acetyltrans-ferases makes chromatin structure relaxed by weakening the interaction of histone tails with DNA and accessible to transcriptional activa-tors. On the other side, deacetylation of histones by HDACs alters the electrostatic properties of chromatin thereby facilitating gene repression. A recent study has revealed HDAC activity linked to induction of certain genes, suggesting that HDACs do not only turn genes off, but rather dynamically regulate gene expression levels [31]. Moreover, acetylation additionally provides a mechanism to control the activity of nonhistone proteins [31]. The first mammalian HDAC was isolated in 1996 [33]. Distinct genes encoding 18 mammalian HDACs have now been identified and they are grouped into four classes on the basis of their similarity with yeast. Emerging evidence suggests that aberrant epigenetic modifications have a potential role in several conditions includ-ing atherosclerosis, autoimmune diseases and kid-ney disease among others [33,34]. Thus, we assume
Kidney & heart interactions during cardiorenal syndrome Review

Future Cardiol. (2011) 7(4)488 future science group
that epigenetic mechanisms can also be essential in kidney and heart diseases, especially in their correlation (Figure 1).
Pathophysiology & epigenetics of the cardiorenal axis
Clinical studies have not yet found a direct rela-tionship between kidney dysfunction and cardiac efficiency, with the exception of general hemody-namic parameters, such as intrarenal hemodynam-ics, transrenal perfusion pressure, central venous pressure [34,35]. Several studies have also proposed a role for hypertension, dyslipidemia and endothelial dysfunction in this context [2]. However, CRS is not limited to hemodynamic changes and should be considered as a complex interplay of neuro-humoral pathways activation [2,36]. The involved neurohumoral systems include the sympathetic nervous system, the renin–angiotensin–aldoste-rone system (RAAS), the endothelin system, and the arginine vasopressin system. Their activation induces inflammation and oxidative stress that lead to vasoconstriction, salt and water retention, accelerated atherosclerosis, cardiac remodeling and hypertrophy, myocardial and intrarenal fibrosis, and progression of renal disease (Figure 2) [36,37]. Pathogenic contribution of increased oxidative stress suggested a role for antioxidant strategies in preserving the atherosclerotic and ischemic kidney in these pathological conditions [38]. It is impor-tant to underline the role of BNP that provides some beneficial effects by counteracting many of the negative adaptations. Particularly, BNP inhib-its the RAAS and vasoconstriction. Moreover, it promotes diuresis, enhances sodium excretion and may even increase glomerular filtration rate.
Molecular mechanismsIn order to understand the pathophysiology of the kidney and heart inter-relationship, it would be interesting to analyze the molecular mechanisms of these complex pathways and highlight their epigenetic correlations. For example, it is still unclear how CRS risk fac-tors such as arterial hypertension, hypergly-cemia, hyperinsulinemia, obesity, proteinuria and uremic toxins individually affect histone modifying enzymes. Furthermore, it would be noteworthy to understand whether drugs mod-ulating epigenetic modifications could prevent CRSs such as diabetic nephropathy-associated cardiomyopathy [39].
A recent study by Gaikwad et al has dem-onstrated that renal failure increased cardiac mRNA expression of most of these genes together with cardiomyocyte size in diabetic mice. This increase might be a direct result of a specific epigenetic histone modification pat-tern [40]. This currently is the only study which has investigated the mechanism directly link-ing renal failure with the induction of cardio-myopathy-related genes. However, this link in the cardiorenal axis can be indirectly suggested by studies that have examined epigenetic modi-fications in the single organ (heart or kidney) diseases. Moreover, it would be interesting to explore in more depth those pathways that are involved in the cardiorenal axis. These studies are discussed below.
For instance, it is known that aberrant DNA methylation may contribute to accelerated ath-erosclerosis by upregulation of atherosclerosis-susceptible genes and downregulation of ath-erosclerosis-protective genes [41]. Since several features of uremia may contribute to aberrant DNA methylation, epigenetic changes can also be involved in CRS. The homocysteine pre-cursor S-adenosylmethionine is a competitive inhibitor of S-adenosylmethionine methyltrans-ferases [42]. Thus, because protein car\1–\2yl methylation and repair is inhibited, this may lead to a state of unbalanced methylation in clinical states with hyperhomocysteinemia. In vascular disease patients increased homocyste-ine and S-adenosylmethionine concentrations were associated with DNA hypomethylation [41]. Moreover, the presence of inflammation may account for the association between low homo-cysteine levels and cardiovascular outcome in CKD. Indeed, It is interesting the consider-ation that inflammation may also cause aber-rant DNA methylation [43]. In accordance, another study demonstrated that global DNA
CRS
PathwaysSNPRAASEndothelin systemAVP
ConsequencesInflammationOxidative stressVasoconstrictionSalt and water retentionAtherosclerosisCardiac remodelingCardiac hypertrophyMyocardial fibrosisIntrarenal fibrosis
Renal disease
Figure 2. Molecular pathways linking renal failure and progression of cardiac disease.AVP: Arginine vasopressin system; CRS: Cardiorenal syndrome; RAAS: Renin–angiotensin–aldosterone system.
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 489future science group
hypermethylation was associated with inflam-mation and increased mortality in CVD [44]. However, there are also discordant recent data that do not support the role of DNA hypometh-ylation in CKD-associated vascular disease in patients with advanced CKD [45].
Most evidence regarding histone modifica-tions have been primarily obtained through the use of HDAC inhibitors (HDACis) especially in the control of cardiac hypertrophy. Indeed, the heart responds to stress signals by hypertrophic growth and an important role for HDACs in the regulation of cardiac growth was initially revealed by the discovery that some HDACs act as signal-responsive repressors of pathological cardiac hypertrophy through the regulation of a specific transcription factor and reprogramming of cardiac gene expression [46]. Several studies also suggested that pharmacological inhibitors of HDACs have antihypertrophic action through neutralization of HDACs [47–52].
With regards to the molecular basis for the cardioprotective effects of HDACis in the heart, studies indicate that they de-repress the expres-sion of protective cardiac genes mainly coding for endogenous antioxidant enzymes. Moreover, another example involves the so called ‘myosin isoform switch’ that is predicted to have adverse functional consequences for the heart failure [53] with the a-myosin heavy chain found to be cardio protective [54]. HDACis have been found to reverse the myosin isoform switch [55].
A further cardiac protective gene that is derepressed by HDACis is the Kruppel-like factor-4 (Klf4) transcription factor. Its expres-sion is downregulated by hypertrophic agonists and induced in cardiomyocytes exposed to HDACis [56]. It has been demonstrated that many genes are important during develop-ment of the fetal heart are re-expressed in the adult tissue, resulting in pathophysiologi-cal changes leading to arrhythmias, cardiac failure and sudden death. One transcription factor thought to be important in repressing the expression of fetal genes in the adult heart is the transcriptional repressor repressor ele-ment 1-silencing transcription factor (REST) [57]. Indeed, it has been demonstrated that con-tinued REST expression prevents increases in the levels of the BNP (Nppb) and ANP (Nppa) genes, encoding brain and atrial natriuretic peptides respectively, in adult rat ventricular myocytes in response to endothelin-1 (ET-1), the most powerful endothelium-derived vaso-constrictor. Likewise, REST inhibition results in increased expression of these genes, which
correlates with increased histone H4 acetyla-tion and histone H3 lysine 4 methylation of their gene promoters [57].
The peptide hormone ET-1 plays multiple, complex roles in both cardiovascular and renal physiology [56,57]. Arterial hypertension is a sig-nificant risk factor for the high rate of cardiovas-cular disease in chronic uremic patients [58,59]. Indeed, the elevation in ET-1 levels was corre-lated with diastolic dysfunction in man, thus suggesting the possibility of an early therapeu-tic approach in these patients [60]. High ET-1 levels have been found both in arterial hyper-tension and during chronic renal failure [61,62]. Studies targeting the ET-1 pathway for patients suffering from a range of disorders, including the progression of CKD and CVDs, are under investigation. Transcriptional activity of the ET-1 encoding gene (edn1) is also modulated by DNA methylation and histone modification patterns [63].
Endothelial dysfunction has been shown to participate in postischemic vasoconstriction fol-lowing reperfusion, with the nitric oxide (NO) pathway as a main player. NO is well known for its role in vascular tone regulation and is gener-ated through a reaction catalyzed by nitric oxide synthase (NOS), which exists in three different isoforms, all of which functioning in heart and kidney. NOS inhibitors have highlighted the role of NO in maintaining also regional renal blood flow. Importantly, the inability of hypoxic endo-thelial cells to produce NO has been suggested to have detrimental effects and participate in renal damage [64]. Epigenetic mechanisms involved in the expression of endothelial, and inducible NOS isoforms have been recognized [65–67].
In addition to promoting expression of pro-tective genes, HDACis also appear to directly block the expression of pathological genes, which could be considered a paradox since HDAC action is usually associated with gene repression. Two recent studies have clarified the mechanism by which HDACis repress genes in the heart. Expression of the gene encoding BNP is dramatically enhanced in ventricular myo-cytes during pathological cardiac hypertrophy. In a study on cultured rat cardiac myocytes, it has been demonstrated that upregulation of BNP in response to endothelin signaling is dependent on association of HDAC2 with the yin-yang 1 (YY1) transcription factor on the BNP gene promoter [68]. YY1 was found in the acetylated form in cardiac myocytes and, when deacetylated by HDAC2, its capability to stimulate BNP gene transcription increased.
Kidney & heart interactions during cardiorenal syndrome Review

Future Cardiol. (2011) 7(4)490 future science group
TSA treatment disrupted YY1/HDAC2 com-plexes and suppressed endothelin-induced BNP expression. Similarly, in a model of cultured adult feline cardiac myocytes, other authors demonstrated that HDAC1 activity stimulates sodium/calcium exchanger (NCX1) gene expression during cardiac hypertrophy (Table 1) [69].
Kidney & heart associationsIn the development of a targeted therapy for CKD an important issue is the multifactorial nature of the renal fibrosis pathogenesis [70]. Tissue fibrosis is a common pathway involved in the response to chronic stress and injury and is the result of epithelial to mesenchymal transition (EMT), the activation of fibroblasts to produce extracellular matrix, recruitment of inflamma-tory cells, and cellular regeneration at sites of damage. In the kidney chronic hypertension damages glomerular cells, resulting in cytokine release and inflammation. For example, TGF-b has been demonstrated to induce tubular EMT and it is thought to be one of the major causes of renal fibrosis [71,72]. An in vitro study demon-strated that an HDACi (TSA) blocked TGF-b-driven EMT [64]. TSA also increased expression of a renal-protective factor, the bone morphoge-netic protein-7, which is an endogenous inhibitor of TGF-b signaling known to suppress EMT and reverse renal fibrosis [73,74]. More recently, TSA was also demonstrated to reduce fibrosis, sup-press EMT and improve renal function in a rat model of diabetic nephropathy [75]. In the kidney, tubulointerstitial fibrosis is also associated with hypoxia and the activation of hypoxia-inducible
factor (HIF) signaling. HIF activation has been shown to promote EMT and renal fibrogenesis [76]. Moreover, it is important to consider that a variety of human disorders, including isch-emic heart disease, stroke and kidney disease, all share the deleterious consequences of a com-mon, hypoxic and oxidative stress pathway with HIF as the key player. Epigenetics has been demonstrated to play a crucial role in the cel-lular response to hypoxia with the involvement of HIF family and its transcriptional targets [77]. Moreover, hypoxia is closely related to oxidative stress and these two phenomena are also strictly linked in both organs. Thus, it would be inter-esting to analyze their related epigenetic mecha-nisms to potentially gain a greater understanding of CRS.
Chronic inflammation triggers pathological fibrosis in the heart and kidney. Potent anti-inflammatory effects of HDACis on multiple immune cell types have been observed [78], thus explaining the broad efficacy observed with HDACis in preclinical models of cardiorenal dis-ease. The main suggested mechanisms of action seem to be diverse, since also several nonhistone proteins have been demonstrated as targets of HDACs, including the transcription factors STAT1 [79,80] and nuclear factor kB [81]. For inflammatory diseases with no currently effec-tive treatments HDACis may represent a new therapeutic approach and it would be interest-ing to determine whether induction of regulatory T cells also contributes to the efficacy of HDACs in the settings of heart and renal failure [82].
Recent findings strongly support an impor-tant role of angiotensin II in the pathophysi ology of renal water and sodium retention associated with HF [83]. Renal water retention in HF is in part mediated through the release of arginine vasopressin system, which acts, through bind-ing to vasopressin-2 receptor, on the water channel aquaporin-2 (AQP2) in the collecting duct. Moreover, in HF the increase of angioten-sin II and aldosterone levels upregulated type 2 11b-hydroxysteroid dehydrogenase and epithe-lial sodium channel subunits [83]. However, no studies have investigated the involvement of pos-sible epigenetic mechanisms in this important molecular pathway (Table 1).
Fetal programmingSeveral genetic factors as well as lifelong envi-ronmental factors, such as salt and fat intake, obesity, diabetes, smoke and alcohol consump-tion, amongst others, clearly contribute to the development of hypertension and/or other risk
Table 1. Relevant epigenetic mechanisms for the heart and kidney.
Epigenetic mechanism Heart Kidney
DNA methylation Yes YesHDAC inhibitors Yes YesHistone H4 acetylation Yes NoHistone H3 lysine methylation Yes NoNO pathway on NOS isoforms Yes YesHIF pathways Yes YesRAAS and AVP pathways No YesFetal programming Yes YesImplications of miRNA in CRS: cardiomyocyte hypertrophy: miR-1, miR-21, miR-23, miR-133, miR-208a; Cardiomyocyte apoptosis and regeneration: miR-195, miR-199a, miR-320; fibrosis and heart failure: miR-133, miR-21, miR-29; restenosis: miR-21, miR-145, miR-221; angiogenesis: miR-221, miR-222, miR-210, miR-126, miR-17~92 cluster; diabetic nephropathy: miR-192, miR-216a, miR-217, miR-382, miR-129, miR-377, miR-93; acute kidney injury: miR-132, miR-362, miR-379, miR-668, miR-687, miR-34a; polycystic kidney disease: miR-17, miR-15a; renal allograft rejection: miR-142-5p, miR-155; kidney cancer: miR-210, miR-21, miR-155, miR-562, miR-185, miR-483-3p; other renal diseases: miR-371-5P, miR-423-5P, miR-638, miR-1224-3P, miR-663, miR-200c, miR-141, miR-205, miR-192, miR-200a, miR-200b, miR-429, miR-205, miR-29b. AVP: Arginine vasopressin system; HDAC: Histone deacetylase; HIF: Hypoxia-inducible factor; NO: Nitric oxide; NOS: Nitric oxide synthase; RAAS: Renin–angiotensin–aldosterone system.
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 491future science group
factors for these disorders, but it has also been established that stress in utero may program the later development of these diseases [84,85]. This phenomenon, known as fetal programming can be modeled in a range of experimental animal models and is related to epigenetic mechanisms.
Interestingly, recent studies have underlined the importance of epigenetic alterations in the renin–angiotensin system following maternal high salt intake or low-protein diet during pregnancy in the fetal programming of adult hypertension [85–88]. These data suggest a link between epigenetic modification of genes dur-ing fetal life and the consequent alteration of gene expression in adult life leading ulti-mately to the development of hypertension. Although these results have been obtained in
animal models, similar mechanisms may also be involved in the development of hypertension and other pathologic conditions in humans. Indeed, animal models have established cause-effect relationships consistent with epidemio-logical findings in humans and have demon-strated, in principle, that interventions before or during pregnancy can reduce or prevent pathogenic in utero programming of CVD (Table 1) [89,90].
In the context of the fetal programming of renal disease, metabolic responses play an important role by metabolic responses. The best-investigated conditions are nutri-ent deficiency leading to low birth weight, as well as maternal obesity or maternal dia-betes mellitus during pregnancy, resulting in
Figure 3. Pathophysiological interactions between heart and kidney during cardiorenal syndrome leading to kidney injury. HDACis target multiple pathological mechanisms of chronic cardiac and renal disease. The chronic stresses of hypertension and diabetes produce cardiomyocyte hypertrophy and inflammation leading to cardiorenal fibrosis, reduced organ function, cell death and, ultimately, organ failure. In the pathological state, the YY1 transcription factor is bound to HDAC. HDAC deacetylates YY1, enhancing its ability to stimulate BNP gene expression. HDAC inhibition may reduce development of CRS and acetylation of YY1 occurring during heart failure. CRS: Cardiorenal syndrome; HDAC: Histone deacetylase; HDACi: Histone deacetylase inhibitor.
HypertensionMetabolic disease
InflammationInflammation
Organ damage/dysfunction
HDACi HDACi
BNP Hormonal factors
Systemic diseaseDiabetes
AmyloidosisVasculitis and sepsis
HDACi
ACYY1 YY1BNP gene
Low expression of BNP High expression of BNP
HDAC2BNP gene
Kidney & heart interactions during cardiorenal syndrome Review
Future Cardiol. (2011) 7(4)492 future science group
high-birthweight infants. These birth condi-tions can cause adverse renal outcomes which can become evident during early childhood. Exposure to corticosteroids, particularly gluco-corticoids, during pregnancy has also been sug-gested to have an effect on the fetal program-ming of these diseases. However, to date only animal studies clearly demonstrate an adverse effect of glucocorticoids on renal development. Potential underlying mechanisms of fetal pro-gramming can include a marked reduction of nephron number through reduced nephrogen-esis, inhibition of the intrarenal RAAS, dimin-ished circulating insulin-like growth factor 1, and increased apoptosis. Moreover, early-life nutrition can alter the metabolic responses and influence renal outcome postnatally [91,92].
ConclusionIt is clear that inflammation and metabolic stress encountered in CKD could promote epigenetic
changes, leading to altered gene expression and abnormal cellular function (Figure 1). As regard to the epigenetic effects in CKD an important example are the HDACis, as described in this article, with some studies that indicate they are able to de-repress the expression of important pro-tective cardiac genes. Thus, we can also assume that many genes with important functions during development of the fetal heart can be re-expressed in the adult tissue. This expression modification can result in pathophysiological changes leading to cardiac ischemia and HF.
Chronic kidney disease has been associated with increased cardio vascular morbidity and mortality and this risk has been demonstrated in several clinical studies. Indeed, as described in this article, all risk factors in patients with kidney disease, such as modifiable, nonmodifi-able and uremia related risk factors, contribute to generate coronary artery disease, peripheral artery disease and left ventricular hypertrophy
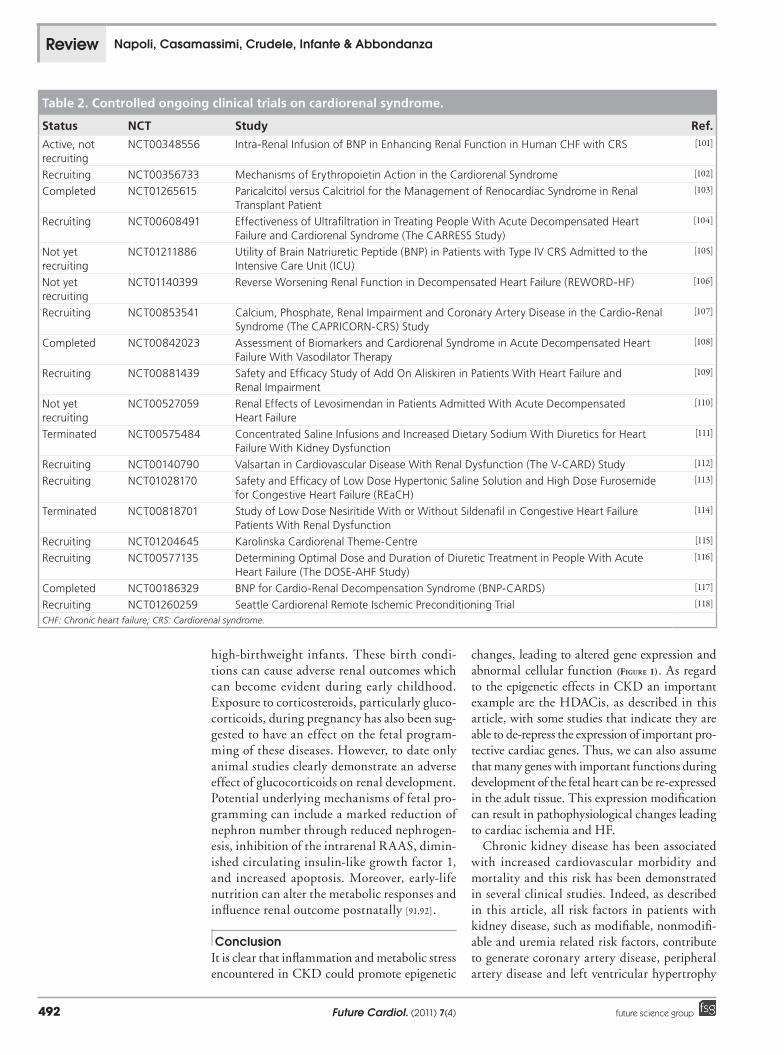
Table 2. Controlled ongoing clinical trials on cardiorenal syndrome.
Status NCT Study Ref.
Active, not recruiting
NCT00348556 Intra-Renal Infusion of BNP in Enhancing Renal Function in Human CHF with CRS [101]
Recruiting NCT00356733 Mechanisms of Erythropoietin Action in the Cardiorenal Syndrome [102]
Completed NCT01265615 Paricalcitol versus Calcitriol for the Management of Renocardiac Syndrome in Renal Transplant Patient
[103]
Recruiting NCT00608491 Effectiveness of Ultrafiltration in Treating People With Acute Decompensated Heart Failure and Cardiorenal Syndrome (The CARRESS Study)
[104]
Not yet recruiting
NCT01211886 Utility of Brain Natriuretic Peptide (BNP) in Patients with Type IV CRS Admitted to the Intensive Care Unit (ICU)
[105]
Not yet recruiting
NCT01140399 Reverse Worsening Renal Function in Decompensated Heart Failure (REWORD-HF) [106]
Recruiting NCT00853541 Calcium, Phosphate, Renal Impairment and Coronary Artery Disease in the Cardio-Renal Syndrome (The CAPRICORN-CRS) Study
[107]
Completed NCT00842023 Assessment of Biomarkers and Cardiorenal Syndrome in Acute Decompensated Heart Failure With Vasodilator Therapy
[108]
Recruiting NCT00881439 Safety and Efficacy Study of Add On Aliskiren in Patients With Heart Failure and Renal Impairment
[109]
Not yet recruiting
NCT00527059 Renal Effects of Levosimendan in Patients Admitted With Acute Decompensated Heart Failure
[110]
Terminated NCT00575484 Concentrated Saline Infusions and Increased Dietary Sodium With Diuretics for Heart Failure With Kidney Dysfunction
[111]
Recruiting NCT00140790 Valsartan in Cardiovascular Disease With Renal Dysfunction (The V-CARD) Study [112]
Recruiting NCT01028170 Safety and Efficacy of Low Dose Hypertonic Saline Solution and High Dose Furosemide for Congestive Heart Failure (REaCH)
[113]
Terminated NCT00818701 Study of Low Dose Nesiritide With or Without Sildenafil in Congestive Heart Failure Patients With Renal Dysfunction
[114]
Recruiting NCT01204645 Karolinska Cardiorenal Theme-Centre [115]
Recruiting NCT00577135 Determining Optimal Dose and Duration of Diuretic Treatment in People With Acute Heart Failure (The DOSE-AHF Study)
[116]
Completed NCT00186329 BNP for Cardio-Renal Decompensation Syndrome (BNP-CARDS) [117]
Recruiting NCT01260259 Seattle Cardiorenal Remote Ischemic Preconditioning Trial [118]
CHF: Chronic heart failure; CRS: Cardiorenal syndrome.
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 493future science group
finally leading to myocardial infarction and congestive heart failure. Patients with moder-ate to severe CKD have an increase in mortality from cardiovascular disease, even long before they develop kidney failure. Emerging evidence suggests that endothelial dysfunction, oxidative stress, vascular calcification, systemic diseases, infections and inflammation are strongly inter-related and together play a major role in the initiation and progression of vascular disease in CKD (Figure 3).
Future perspectiveCurrently, controlled clinical studies involving CRS are based on molecules such as homocyste-ine, EPO and Natrecor drug (nasiritide), which is a recombinant form of BNP (Table 2). Other clinical trials in cardiovascular disease with renal dysfunction are based on valsartan, a spe-cific drug with an indication of an active sub-stance against hypertension. From the clinical
perspective it is clear that it is necessary a better understanding of the underlying mechanisms of kidney and heart diseases, to achieve better diagnostic, preventive and therapeutic mea-sures. Overall, there is an emerging interest in the possible pathogenic role of endoplasmic reticulum stress in the framework of cardiac hypertrophy, HF, and CKD [93]. Similarly, epoxyeicosatrienoic acids [94] or statins [90] could have a therapeutic value in the treatment of chronic CRS.
Epigenetic studies may be helpful in the identif ication of new vascular markers or biofactors and facilitate a more rapid devel-opment of novel, safe and effective thera-pies [95,96]. Future perspectives could be based on the study of those genes playing an essen-tial role during development of the fetal heart and vasculature [90] that are re-expressed in the adult tissue with later pathophysiological consequences. These findings could be useful
Executive summary
Kidney & heart interactionnHeart and kidney function are closely interconnected.nCardiorenal syndrome (CRS) is a pathological disorder where acute or chronic dysfunction in one organ may induce dysfunction in the
other organ.
Cardiorenal axis dysfunctionnThe early diagnosis and management of heart failure are important in patients with chronic kidney disease (CKD). nCardiac and renal biomarkers facilitate and improve the clinical managements of heart failure and CKD patients.nCRS type I describes the acute kidney injury as a consequence of a sudden worsening of cardiac function.nCRS type II describes the progressive and permanent kidney disease caused by chronic cardiac dysfunction.nCRS type III describes a sudden cardiac disorder as a consequence of an impairment of kidney function.nCRS type IV is attributed to the decreased cardiac function and increased risk of cardiovascular events in patients with CKD.nCRS type V describes a situation in which a systemic condition causes both cardiac and renal dysfunction.
Epigenetic mechanismsnThe main epigenetic mechanisms include DNA methylation, histone modification and RNA interference, which result in changes of
chromatin structure and miRNA alterations.nRecent studies have demonstrated that epigenetic alterations are also associated with inflammation and cardiovascular disease in
patients with CKD and CRS.nHypertension, dyslipidemia and endothelial dysfunction are important in this context. nCRS is not limited to hemodynamic changes and should be considered as a complex interplay of neurohumoral pathways activation.
Pathophysiology & epigenetics in the cardiorenal axisnCRS involves neurohumoral systems that include the sympathetic nervous system, the renin–angiotensin–aldosterone system, the
endothelin system, and the arginine vasopressin system. nThe activation of the sympathetic nervous system, renin–angiotensin–aldosterone system, endothelin system and arginine vasopressin
system induces inflammation and oxidative stress that lead to vasoconstriction, salt and water retention, accelerated atherosclerosis, cardiac remodeling and hypertrophy, myocardial and intrarenal fibrosis and progression of CKD.
ConclusionnHistone deacetylase inhibitor studies indicate they are able to de-repress the expression of important protective cardiac genes.nGenes with important functions during heart development can be re-expressed in the adult, thereby leading to pathophysiological
modifications ultimately responsible of cardiac ischemia, heart failure and CRS.
Future perspectivenEpigenetic studies may be helpful in the identification of new vascular markers or factors that ultimately may facilitate a more rapid
development of novel, safe and effective therapies in CRS patients.
Kidney & heart interactions during cardiorenal syndrome Review

Future Cardiol. (2011) 7(4)494 future science group
Financial & competing interests disclosureThe authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the sub-ject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
BibliographyPapers of special note have been highlighted as:n of interestnn of considerable interest
1. Dwivedi RS, Herman JG, McCaffrey et al. Beyond genetics: epigenetic code in CKD. Kidney Int. 79(1), 23–32 (2011).
2. Iwanaga Y, Miyazaki S. Heart failure, chronic kidney disease, and biomarkers--an integrated viewpoint. Circ. J. 74(7), 1274–1282 (2010).
3. Balestrieri ML, Giovane A, Milone L et al. Modification of the detrimental effect of TNF-a on human endothelial progenitor cells by fasudil and Y27632. J. Biochem. Mol. Toxicol. 24(6), 351–360 (2010).
n First evidence that some drugs modify detrimental effect of TNF-a on endothelial progenitor cells.
4. Smith GL, Masoudi FA, Shlipak MG et al. Renal impairment predicts long-term mortality risk after acute myocardial infarction. J. Am. Soc. Nephrol. 19(1), 141–150 (2008).
5. Napoli C, Balestrieri A, Ignarro LJ. Therapeutic approaches in vascular repair induced by adult bone marrow cells and circulating progenitor endothelial cells. Curr. Pharm. Des. 13(31), 3245–3251 (2007).
6. Jie KE, van der Putten K, Bergevoet MW et al. Short- and long-term effects of erythropoietin treatment on endothelial progenitor cell levels in patients with cardiorenal syndrome. Heart 97(1), 60–65 (2011).
n Data presented in this paper show that circulating endothelial progenitor cell levels are reduced in cardiorenal syndrome patients also after erythropoietin treatment.
7. Smith GL, Lichtman JH, Bracken MB et al.: Renal impairment and outcomes in heart failure: systematic review and meta-ana lysis. J. Am. Coll. Cardiol. 47(10), 1987–1996 (2006).
8. Levin A. Clinical epidemiology of cardiovascular disease in CKD prior to dialysis. Semin. Dial. 16(2), 101–105 (2003).
9. Kendrick J, Chonchol MB. Nontraditional risk factors for cardiovascular disease in patients with CKD. Nat. Clin. Pract. Nephrol. 4(12), 672–681 (2008).
10. Ronco C, Haapio M, House AA et al. Cardiorenal syndrome. J. Am. Coll. Cardiol. 52(19), 1527–1539 (2008).
nn In this paper a consensus definition of cardiorenal syndrome, that emphasizes the bidirectional nature of heart–kidney interactions, has been proposed for the first time.
11. Han WK, Bonventre JV. Biologic markers for the early detection of acute kidney injury. Curr. Opin. Crit. Care 10(6), 476–482 (2004).
12. Devarajan P, Mishra J, Supavekin S et al. Gene expression in early ischemic renal injury: clues towards pathogenesis, biomarker discovery, and novel therapeutics. Mol. Genet. Metab. 80(4), 365–376 (2003).
13. Nguyen MT, Ross GF, Dent CL et al. Early prediction of acute renal injury using urinary proteomics. Am. J. Nephrol. 25(4), 318–326 (2005).
14. Ronco C. NGAL: an emerging biomarker of acute kidney injury. Int. J. Artif. Organs 31(3), 199–200 (2008).
15. Nickolas TL, Barasch J, Devarajan P. Biomarkers in acute and CKD. Curr. Opin. Nephrol. Hypertens. 17(2), 127–132 (2008).
16. Dharnidharka VR, Kwon C, Stevens G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta-ana lysis. Am. J. Kidney Dis. 40(2), 221–226 (2002).
17. Han WK, Bailly V, Abichandani R et al. Kidney injury molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 62(1), 237–244 (2002).
18. Vaidya VS, Ramirez V, Ichimura T et al. Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am. J. Physiol. Renal. Physiol. 290(2), F517–F529 (2005).
19. Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and
predicts mortality in the intensive care unit. J. Am. Soc. Nephrol. 16(10), 3046–3052 (2005).
20. Liang KV, Williams AW, Greene EL et al. Acute decompensated heart failure and the cardiorenal syndrome. Crit. Care Med. 36(Suppl. 1), S75–S88 (2008).
21. Jie KE, Verhaar MC, Cramer MJ et al. Erythropoietin and the CRS: Cellular mechanisms on the cardiorenal connectors. Am. J. Physiol. Renal Physiol. 291(5), F932–F944 (2006).
22. Riksen NP, Hausenloy DJ, Yellon DM. Erythropoietin: ready for prime-time cardioprotection. Trends Pharmacol. Sci. 29(5), 258–267 (2008).
23. Palazzuoli A, Silverberg DS, Iovine F et al. Effects of b-erythropoietin treatment on left ventricular remodeling, systolic function, and B-type natriuretic peptide levels in patients with the cardiorenal anemia syndrome. Am. Heart J. 154(4), 645e9–645e15 (2007).
24. Rattazzi M, Puato M, Faggin E et al. New markers of accelerated atherosclerosis in end-stage renal disease. J. Nephrol. 16(1), 11–20 (2003).
25. Tonelli M, Wiebe N, Culleton B et al. Chronic kidney disease and mortality risk: a systematic review. J. Am. Soc. Nephrol. 17(7), 2034–2047 (2006).
26. Cunningham PN, Dyanov HM, Park P et al. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J. Immunol. 168(11), 5817–5823 (2002).
27. Kumar A, Paladugu B, Mensing J et al. Nitric oxide-dependent and -independent mechanisms are involved in TNF-a-induced depression of cardiac myocyte contractility. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292(5), R1900–R1906 (2007).
28. Portela A, Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 28(10), 1057–1068 (2010).
29. Kaufman PD, Rando OJ. Chromatin as a potential carrier of heritable information. Curr. Opin. Cell Biol. 22(3), 284–290 (2010).
to utilize compounds that act on epigenetic mechanisms as novel therapeutic strategies. To date, the available molecules targeting epigenetic modifications have been studied mainly in tumors [91–98], while studies involv-ing the cardiovascular and kidney diseases, as well as CRS, are still at a preliminary stage. For example, with regard to atherosclerosis and HF, the administration of curcumin (an inhibitor KAT) has been tested [99].
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 495future science group
nn Illustrates the evidence found in the literature that some chromatin states are indeed heritable.
30. Bhatt K, Mi QS, Dong Z. Invited review- microRNAs in kidneys: biogenesis, regulation and pathophysiological roles. Am. J. Physiol. Renal Physiol. (2011) (Epub ahead of print).
31. Wang Z, Zang C, Cui K et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138(5), 1019 –1031 (2009).
32. Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation 121(8), 1022–1032 (2010).
33. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272(5260), 408–411 (1996).
34. Wierda RJ, Geutskens SB, Jukema JW et al. Epigenetics in atherosclerosis and inflammation. J. Cell. Mol. Med. 14(6A), 1225–1240 (2010).
35. Mullens W, Abrahams Z, Francis GS et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 53(7), 589–596 (2009).
36. Schetz M. Cardiorenal syndrome. F1000 Medicine Reports 1, 78 (2009).
37. Kociol R, Rogers J, Shaw A. Organ cross talk in the critically ill: the heart and the kidney. Blood Purif. 27(4), 311–320 (2009).
38. Chade AR, Rodriguez-Porcel M, Herrmann J et al. Antioxidant intervention blunts renal injury in experimental renovascular disease. J. Am. Soc. Nephrol. 15(4), 958–966 (2004).
n First evidence that antioxidant intervention blunts renal failure.
39. Berry JM, Cao DJ, Rothermel BA et al. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin. Drug Saf. 7(1), 53–67 (2008).
40. Gaikwad AB, Sayyed SG, Lichtnekert J et al. Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am. J. Pathol. 176(3), 1079–1083 (2010).
nn Investigates for the first time the epigenetic mechanism directly linking renal failure with the induction of cardiomyopathy-related genes.
41. Turunen MP, Aavik E, Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim. Biophys. Acta 1790(9), 886–891 (2009).
42. Castro R, Rivera I, Struys EA et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin. Chem. 49(8), 1292–1296 (2003).
43. Teitell M, Richardson B. DNA methylation in the immune system. Clin. Immunol. 109(1), 2–5 (2003).
44. Stenvinkel P, Karimi M, Johansson S et al. Impact of inflammation on epigenetic DNA methylation: A novel risk factor for cardiovascular disease? J. Intern. Med. 261(5), 488–499 (2007).
45. Nanayakkara PW, Kiefte-de Jong JC, Stehouwer CD et al. Association between global leukocyte DNA methylation, renal function, carotid intima-media thickness and plasma homocysteine in patients with stage 2–4 chronic kidney disease. Nephrol. Dial. Transplant. 23(8), 2586–2592 (2008).
46. Zhang CL, McKinsey TA, Chang S et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110(4), 479–488 (2002).
47. Antos CL, McKinsey TA, Dreitz M et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 278(31), 28930–28937 (2003).
48. Kook H, Lepore JJ, Gitler AD et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. Invest. 112(6), 863–871 (2003).
49. Kee HJ, Sohn IS, Nam KI et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113(1), 51–59 (2006).
50. Kee HJ, Eom GH, Joung H et al. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 103(11), 1259–1269 (2008).
51. Cardinale JP, Sriramula S, Pariaut R et al. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 56(3), 437–444 (2010).
52. Trivedi CM, Luo Y, Yin Z et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 b activity. Nat. Med. 13(3), 324 –331 (2007).
53. Gupta MP. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol. 43(4), 388–403 (2007).
54. James J, Martin L, Krenz M et al. Forced expression of a-myosin heavy chain in the rabbit ventricle results in cardioprotection under cardiomyopathic conditions. Circulation 111(18), 2339–2346 (2005).
55. Davis FJ, Pillai JB, Gupta M et al. Concurrent opposite effects of trichostatin A, an inhibitor of histone deacetylases, on expression of a-MHC and cardiac tubulins: implication for gain in cardiac muscle contractility. Am. J. Physiol. Heart Circ. Physiol. 288(3), H1477–H1490 (2005).
56. Kee HJ, Kook H. Kruppel-like factor 4 mediates histone deacetylase inhibitor-induced prevention of cardiac hypertrophy. J. Mol. Cell. Cardiol. 47(6), 770 –780 (2009).
57. Bingham AJ, Ooi L, Kozera L et al. The repressor element 1-silencing transcription factor regulates heart-specific gene expression using multiple chromatin-modifying complexes. Mol. Cell. Biol. 27(11), 4082–4092 (2007).
58. Wada A, Tsutamoto T, Ohnishi M et al. Effects of a specific endothelin-converting enzyme inhibitor on cardiac, renal, and neurohumoral functions in congestive HF: comparison of effects with those of endothelin A receptor antagonism. Circulation 99(4), 570–577 (1999).
59. Chade AR, Best PJ, Rodriguez-Porcel M et al. Endothelin-1 receptor blockade prevents renal injury in experimental hypercholesterolemia. Kidney Int. 64(3), 962–969 (2003).
60. Napoli C, Liguori A, Sorice P et al. Relations between vasoactive hormones and diastolic function in hypertensive uraemic patients. J. Intern. Med. 240(6), 389–394 (1996).
61. Napoli C, Di Gregorio F, Sorice P et al. High prevalence of myocardial ischemia and vasoconstrictive hormonal release in hypertension during chronic renal failure. Nephron 76(4), 434–444 (1997).
n Seminal study establishing that cardiac ischemia is more severe in hypertension during chronic kidney disease.
62. Napoli C, Di Gregorio F, Leccese M et al. Evidence of exercise-induced myocardial ischemia in patients with primary aldosteronism: the cross-sectional primary aldosteronism and heart Italian multicenter study. J. Investig. Med. 47(5), 212–221 (1999).
63. Stow LR, Jacobs ME, Wingo CS et al. Endothelin-1 gene regulation. FASEB J. 25(1), 16–28 (2010).
64. Legrand M, Mik EG, Johannes T et al. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol. Med. 14(7–8), 502–516 (2008).
65. Ignarro LJ, Cirino G, Casini A et al. Nitric oxide as a signaling molecule in the vascular system: an overview. J. Cardiovasc. Pharmacol. 34(6), 879–886 (1999).
Kidney & heart interactions during cardiorenal syndrome Review

Future Cardiol. (2011) 7(4)496 future science group
66. Chan Y, Fish JE, D’Abreo C et al. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J. Biol. Chem. 279(33), 35087–35100 (2004).
67. Chan GC, Fish JE, Mawji IA et al. Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J. Immunol. 175(6), 3846–3861 (2005).
68. Glenn DJ, Wang F, Chen S et al. Endothelin stimulated human B-type natriuretic peptide gene expression is mediated by Yin Yang 1 in association with histone deacetylase 2. Hypertension 53(3), 549–555 (2009).
nn Demonstrates that upregulation of BNP in response to endothelin signaling is dependent on the association of histone deacetylase 2 with YY1 transcription factor on its promoter.
69. Chandrasekaran S, Peterson RE, Mani SK et al. Histone deacetylases facilitate sodium/calcium exchanger up-regulation in adult cardiomyocytes. FASEB J. 23(11), 3851–3864 (2009).
70. Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 69(2), 213–217 (2006).
71. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119(6), 1420 –1428 (2009).
72. Yoshikawa M, Hishikawa K, Marumo T et al. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-b1 in human renal epithelial cells. J. Am. Soc. Nephrol. 18(1), 58–65 (2007).
73. Zeisberg M, Hanai J, Sugimoto H et al. BMP-7 counteracts TGF-b1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9(7), 964–968 (2003).
74. Zeisberg M, Kalluri R. Reversal of experimental renal fibrosis by BMP7 provides insights into novel therapeutic strategies for chronic kidney disease. Pediatr. Nephrol. 23(9), 1395–1398 (2008).
75. Noh H, Oh EY, Seo JY et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-b1-induced renal injury. Am. J. Physiol. Renal Physiol. 297(3), F729–F739 (2009).
76. Higgins DF, Kimura K, Iwano M et al. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 7(9), 1128–1132 (2008).
77. Watson JA, Watson CJ, McCann A et al. Epigenetics, the epicenter of the hypoxic response. Epigenetics 5(4), 293–296 (2010).
78. Halili MA, Andrews MR, Sweet MJ et al. Histone deacetylase inhibitors in inflammatory disease. Curr. Top. Med. Chem. 9(3), 309–319 (2009).
79. Klampfer L, Huang J, Swaby LA et al. Requirement of histone deacetylase activity for signaling by STAT1. J. Biol. Chem. 279(29), 30358–30368 (2004).
80. Kramer OH, Knauer SK, Greiner G et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 23(2), 223–235 (2009).
81. Calao M, Burny A, Quivy V et al. A pervasive role of histone acetyltransferases and deacetylases in an NF-kB signaling code. Trends Biochem. Sci. 33(7), 339–349 (2008).
82. Bush EW, McKinsey TA. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ. Res. 106(2), 272–284 (2010).
83. Lütken SC, Kim SW, Jonassen T et al. Changes of renal AQP2, ENaC, and NHE3 in experimentally induced HF: response to angiotensin II AT1 receptor blockade. Am. J. Physiol. Renal Physiol. 297(6), F1678-F1688 (2009).
84. Napoli C, Lerman LO, de Nigris F et al. Rethinking primary prevention of atherosclerosis-related diseases. Circulation 114(23), 2517–2527 (2006).
85. Napoli C, Cacciatore F: Novel pathogenic insights in the primary prevention of cardiovascular disease. Prog. Cardiovasc. Dis. 51(6), 503–523 (2009).
n An updated review regarding novel pathogenic insights in primary prevention of cardiovascular disease.
86. Bogdarina I, Welham S, King PJ et al. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 100(4), 520–526 (2007).
nn Suggests a link between epigenetic modification during fetal life of genes belonging to the renin–angiotensin system and the development of hypertension.
87. Ding Y, Lv J, Mao C et al. High-salt diet during pregnancy and angiotensin-related cardiac changes. J. Hypertens. 28(6), 1290–1297 (2010).
88. Gilbert JS and Nijland MJ. Sex differences in the developmental origins of hypertension and cardiorenal disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295(6), R1941–R1952 (2008).
89. Palinski W, Nicolaides E, Liguori A et al. Influence of maternal dysmetabolic conditions during pregnancy on cardiovascular disease. J. Cardiovasc. Transl. Res. 2(3), 277–285 (2009).
90. Napoli C. Developmental mechanisms involved in the primary prevention of atherosclerosis and cardiovascular disease. Curr. Atheroscler. Rep. 13(2), 170–175 (2011).
nn An update review on developmental mechanisms in the prevention of cardiovascular disease.
91. Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr. Nephrol. 26(5), 705–711 (2011).
92. Dötsch J, Plank C, Amann K. Fetal programming of renal function. Pediatr. Nephrol. (2011) (Epub ahead of print).
93. Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ. Res. 108(5), 629–642 (2011).
94. Huang H, Chen J, Lin T et al. Epoxyeicosatrienoic acids–novel mechanism and pharmacological therapy of chronic renocardiac syndrome. Med. Hypotheses 76(4), 550–552 (2011).
95. Stenvinkel P, Carrero JJ, Axelsson J et al. Emerging biomarkers for evaluating cardiovascular risk in the CKD patient: how do new pieces f it into the uremic puzzle? Clin. J. Am. Soc. Nephrol. 3(2), 505–521 (2008).
96. Ingelman-Sundberg M, Gomez A. The past, present and future of pharmacoepigenomics. Pharmacogenomics 11(5), 625–627 (2010).
97. Mann BS, Johnson JR, He K, Sridhara R et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 13(8), 2318–2322 (2007).
98. Gerstner T, Bell N, König S. Oral valproic acid for epilepsy--long-term experience in therapy and side effects. Expert Opin. Pharmacother. 9(2), 285–292 (2008).
99. Wongcharoen W, Phrommintikul A. The protective role of curcumin in cardiovascular diseases. Int. J. Cardiol. 133(2), 145–151 (2009).
Websites101. Intra-Renal Infusion of BNP in Enhancing
Renal Function in Human CHF with CRS http://clinicaltrials.gov/ct2/show/NCT00348556
102. Mechanisms of Erythropoietin Action in the Cardiorenal Syndrome http://clinicaltrials.gov/ct2/show/NCT00356733
Review Napoli, Casamassimi, Crudele, Infante & Abbondanza

www.futuremedicine.com 497future science group
103. Paricalcitol versus Calcitriol for the management of Renocardiac Syndrome in Renal Transplant Patient http://clinicaltrials.gov/ct2/show/NCT01265615
104. Effectiveness of Ultrafiltration in Treating People With Acute Decompensated Heart Failure and Cardiorenal Syndrome (The CARRESS Study) http://clinicaltrials.gov/ct2/show/NCT00608491
105. Utility of Brain Natriuretic Peptide (BNP) in Patients with Type IV CRS Admitted to the Intensive Care Unit (ICU) http://clinicaltrials.gov/ct2/show/NCT01211886
106. Reverse Worsening Renal Function in Decompensated Heart Failure (REWORD-HF) http://clinicaltrials.gov/ct2/show/NCT01140399
107. Calcium, Phosphate, Renal Impairment and Coronary Artery Disease in the Cardio-Renal Syndrome (The CAPRICORN-CRS) Study http://clinicaltrials.gov/ct2/show/NCT00853541
108. Assessment of Biomarkers and Cardiorenal Syndrome in Acute Decompensated Heart Failure With Vasodilator Therapy http://clinicaltrials.gov/ct2/show/NCT00842023
109. Safety and Efficacy Study of Add On Aliskiren in Patients With Heart Failure and Renal Impairment http://clinicaltrials.gov/ct2/show/NCT00881439
110. Renal Effects of Levosimendan in Patients Admitted With Acute Decompensated Heart Failure http://clinicaltrials.gov/ct2/show/NCT00527059
111. Concentrated Saline Infusions and Increased Dietary Sodium With Diuretics for Heart Failure With Kidney Dysfunction http://clinicaltrials.gov/ct2/show/NCT00575484
112. Valsartan in Cardiovascular Disease With Renal Dysfunction (The V-CARD) Study http://clinicaltrials.gov/ct2/show/NCT00140790
113. Safety and Efficacy of Low Dose Hypertonic Saline Solution and High Dose Furosemide for Congestive Heart Failure (REaCH) http://clinicaltrials.gov/ct2/show/NCT01028170
114. Study of Low Dose Nesiritide With or Without Sildenafil in Congestive Heart Failure Patients With Renal Dysfunction http://clinicaltrials.gov/ct2/show/NCT00818701
115. Karolinska Cardiorenal Theme-Centre http://clinicaltrials.gov/ct2/show/NCT01204645
116. Determining Optimal Dose and Duration of Diuretic Treatment in People With Acute Heart Failure (The DOSE-AHF Study) http://clinicaltrials.gov/ct2/show/NCT00577135
117. BNP for Cardio-Renal Decompensation Syndrome (BNP-CARDS) http://clinicaltrials.gov/ct/show/NCT00186329?order=1
118. Seattle Cardiorenal Remote Ischemic Preconditioning Trial http://clinicaltrials.gov/ct2/show/NCT01260259
Kidney & heart interactions during cardiorenal syndrome Review
Related Documents





![Cardiorenal biomarkers in acute heart failure · characterized as cardiorenal syndrome.[10,11] Ronco et al.,[11] proposed the definition for cardiorenal syndrome as, “disorders](https://static.cupdf.com/doc/110x72/5f0c081e7e708231d43369b5/cardiorenal-biomarkers-in-acute-heart-failure-characterized-as-cardiorenal-syndrome1011.jpg)





