KERNFORSCHUNGSANLAGE JÜLICH GmbH Institut für Chemie 4 : Angewandte Physikalische Chemie Beiträge zur Umweltprobenbank Herausgeber : M . Stoeppler und H . W. Dürbeck 3 . Aufstellung von Elementkonzentrations- katastern in unterschiedlichen Pflanzenarten und Bodentypen in Deutschland, Österreich und Schweden von Bernd Markert Jül - Spez- 360 Juli 1986 ISSN 0343 - 7639

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

KERNFORSCHUNGSANLAGE JÜLICH GmbHInstitut für Chemie 4 : Angewandte Physikalische Chemie
Beiträge zur Umweltprobenbank
Herausgeber :M. Stoeppler und H. W. Dürbeck
3. Aufstellung von Elementkonzentrations-katastern in unterschiedlichen Pflanzenartenund Bodentypen in Deutschland, Österreichund Schweden
von
Bernd Markert
Jül - Spez- 360Juli 1986
ISSN 0343 - 7639
Autobahn
MotonvayE E E °-E Autobahn im Bau
Motonvay in ConstructionBundesstraße
Main Road Schnellzugstrecke
Main Railway LineNebenstrecke
Brauch-LinieO
Flughafen
AirportKernforschungsanlage
Jülich Nuclear ResearchJülich
Centre
Als Manuskript gedruckt
Spezielle Berichte der Kernforschungsanlage Jülich - Nr. 360Institut für Chemie 4 : Angewandte Physikalische Chemie Jül-Spez-360
Zu beziehen durch : ZENTRALBIBLIOTHEK der Kernforschungsanlage Jülich GmbH
Postfach 1913 • D-5170 Jülich (Bundesrepublik Deutschland)Telefon : 02461/610 • Telex : 833556-0 kf d

Beiträge zur Umweltprobenbank
Herausgeber :M. Stoeppler und H. W. Dürbeck
3. Aufstellung von Elementkonzentrations-katastern in unterschiedlichen Pflanzenartenund Bodentypen in Deutschland, Österreichund Schweden
von
* Bernd Markert
*D 700 (Diss. Uni . Osnabrück)


Zusammenfassung
Diese Arbeit demonstriert die Durchführbarkeit, Methoden,
Ergebnisse und Grenzen der quantitativen Erfassung sämtlicher
Elemente des periodischen Systems in unterschiedlichen Öko-
systemen . Es werden die Analysetechniken AAS, ICP-AES, AES,
EA, NAA, MS und PIXE auf ihre Nachweisgrenzen und Brauch-
barkeit für Ökosystemanalysen in bezug auf Pflanzen- und
Bodenmatrix miteinander verglichen.
Es wird die Erstellung von Elementkonzentrationskatastern
vorgestellt . Elementkonzentrationskataster werden für Öko-
systeme in Schweden, Österreich und Deutschland erstellt.
Die Verteilungsmuster sämtlicher nachgewiesener Elemente
werden diskutiert.
Evaluation of element concentration cadasters in different
plants and soll types in Germany
Summary
This work demonstrates the feasibility, methods, results
and limitations for a quantitative determination of all
elements of the periodic table in different ecosystems.
A comparison is made for the analytical methods AAS,
ICP-AES, AES, EA, NAA, MS and PIXE from the view of
detection limits and applicability to the analysis of
plants and soils in ecosystems.
An element concentration cadaster scheme is presented.
Those cadasters have been evaluated for ecosystems in
Sweden, Austria and Germany . The obtained element pat-
terns are discussed .


Gliederung:
1 .
Einleitung und Fragestellung
1 -
4
II . Ombrotrophe und minerotrophe Systeme
5 - 8
III . Ökosystembeschreibung
1 . Österreich
a. die Lage
b. das Klima
c. die Vegetation
2 . Deutschland
a. die Lage
b. das Klima
c. die Vegetation
3 . Schweden
a. die Lage
b. das Klima
c. die Vegetation
9 - 19
9 - 12
9
9 - 11
11 - 12
12 - 16
12 - 15
15
16
16 - 19
16 - 17
18
19
IV . Probenaufbereitung
20 - 35
1. Probennahme
22 - 24
2. Probenhomogenisierung
24 - 26
3. Veraschung des Probenmaterials
26 - 35
a . Trockene Veraschunga 1 Tieftemperaturveraschung im an-
geregten Sauerstoff
27a2 Hochtemperaturveraschung
27 - 28
b. Nasse Veraschung
28 - 35b 1 Nassveraschung im offenen
Systemb2 Nassveraschung im geschlossenen
Systemb3 Aufschluß der Pflanzen- und
Bodenproben an der Univer-sität Osnabrück
31b4 Test der Aufschlußapperatur
31 - 33b5 Bemerkungen zum Referenzmaterial 34 - 35
26 - 28
29
29 - 30

V. Analytische Bestimmungsverfahren
36 - 51
1 . Atomabsorptionsspektroskopie
36 - 42
a. MARKERT, Osnabrück, Deutschland
36
b. STOEPPLER, Jülich, Deutschland
36 - 40
c. WILSON, Experiment, Georgia, USA
40
d. YUAN, Gainesville, Florida, USA
41
e. PAWLUK and DUDAS, Edmonton, Alberta,Canada
41
f. DAVID, Canberra, Australien
41 - 42
g. MACALALAD, Manila, Phillipinen
42
2 . Atomemissionsspektroskopie mit Hilfeeines induktiv gekoppelten Plasmas(AES-ICP)
42 - 43
a. SCHRAMEL, München, Deutschland
42
b. HOFFMANN, München, Deutschland
43
c. SUGIMAE, Osaka, Japan
43
3 . Massenspektroskopie (MAS)
43 - 45
a. YLIRUOKANEN, Helsinki, Finnland
44
b. URE, Aberdeen, Schottland
45
4 . Neutronenaktivierungsanalyse (NAA)
45 - 48
a. EHMANN und TIAN, Lexington,Kentucky, USA
46
b. HEYDORN und DAMSGAARD, Roskilde,Dänemark
46
c. OHNO, Chiba, Japan
47
d. DE BRUIN, Delft, Niederlande
48
5 . Quantitative Elementaranalyse
48 - 49
a. Kohlenstoff und Wasserstoff
48 - 49
b. Stickstoff
49
6 . Partikel induzierte Röntgenfluoreszenz-spektroskopie (PIXE)
VI . Vergleich der unterschiedlichen Messme-thoden
1. Nachweisgrenze
2. Vergleich der einzelnen Messdatenuntereinander
50
50 - 55
50
51 - 55

VII . Gesamtbetrachtung mit Hilfe der Elementkon-zentrationskataster 56 -
62
1 . Boden 57 -
592 . Pflanze 59 -
62
VIII .Literatur 63 -
72
Anhang 1 Elementkonzentrationskataster 73 -
95
Anhang 2 Vergleich der Einzelmessdaten 96 - 158
Anhang 3 Mitarbeitende Institute 159 - 166
Lebenslauf 167
Eidesstattliche Erklärung 168


1
1 . Einleitung und Fragestellung
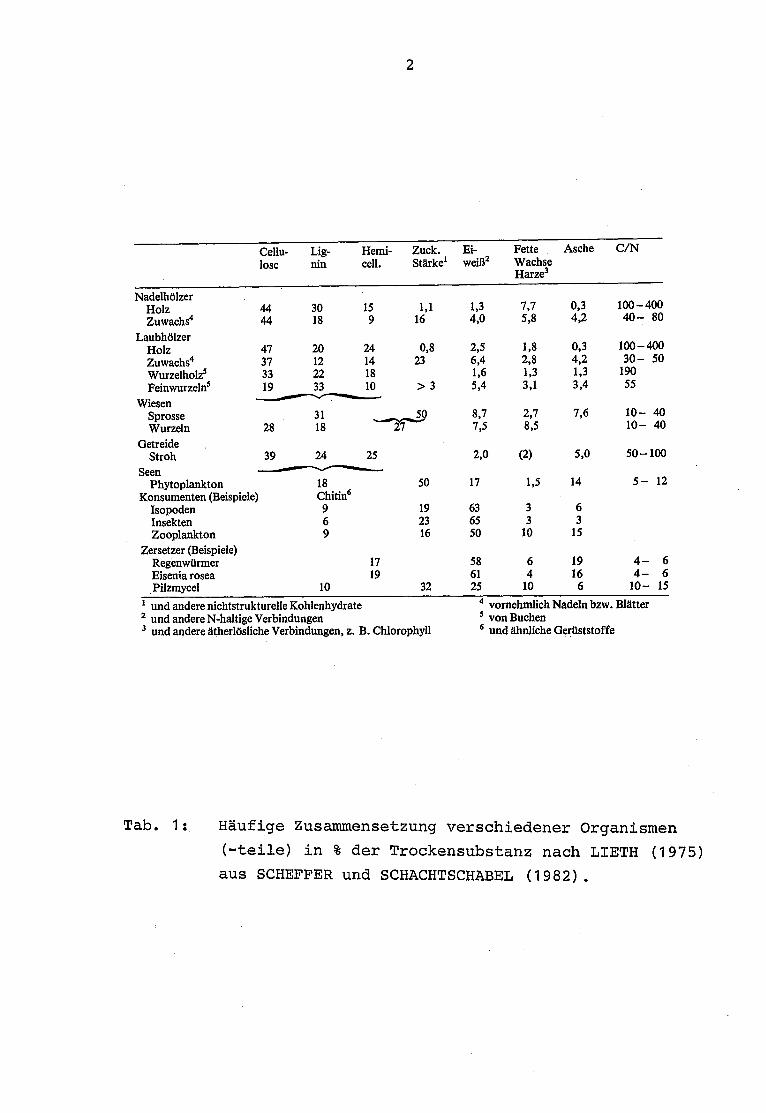
Die heutigen Forschungsaufgaben in der Systemökologie verlangeneine weitgehende Kenntnis der stofflichen Zusammensetzung der
Ökosysteme . Im Prinzip scheint es notwendig, die chemische Zu-
sammensetzung der organischen Substanz in allen Einzelheitenzu kennen, ehe man das Funktionieren der Organismen in denÖkosystemen verstehen kann . Über die Bedeutung und prozentualeVerteilung organischer Stoffklassen in verschiedenen Ökosyste-
men hat LIETH (1975) berichtet (Tab . 1).
Versuche, sämtliche Elemente des periodischen Systems in unter-schiedlichen Matrizes zu bestimmen, wurden bisher ausschließ-lich an Referenzmaterialien durchgeführt, etwa BOWEN's "kale
powder" (1979) . Ein komplettes Elementkataster für Ökosystemeexistiert in dieser Vollkommenheit bisher nicht.
Für die moderne Ökosystemanalyse ist die Beschränkung auf dieklassischen Mineralstoffe nicht mehr ausreichend . Wie aus unter-schiedlichen Untersuchungsergebnissen hervorgeht (etwa BOWEN,1979), kommen scheinbar alle Elemente in der Erdkruste, im Bo-den, im Wasser und in tierischen und pflanzlichen Organismen
vor. Jedes Element, jede Stoffklasse kann an einer Stelle desÖkosystems von Bedeutung sein und an anderer Stelle wirkungs-los . Dabei ist es gleichgültig, ob die Elemente chemische Ver-bindungen eingehen oder durch ihre Gegenwart anderen reagieren-den Stoffen den Platz wegnehmen. Viele Elemente zeigen bereitsbei sehr geringen Konzentrationen eine Wirkung.
Der gegenwärtige Stand der Umweltverschmutzung durch anthropo-gene Aktivitäten und deren Veränderung im Laufe der Zeit sowiewachsende Beherrschung der analytischen Techniken für alle in
den Ökosystemen vorkommenden chemischen Elemente eröffnen dieNotwendigkeit und Möglichkeit neuartiger Elementkonzentrations-
untersuchungen im Ökosystem . Es ist heute bereits möglich zu
2
Cellu-lose
Lig-nin
Hemi
Zuck.Gell .
Stärke"Ei-weiß 2
FetteWachseHarzei
Asche C/N
NadelhölzerHolz 30 15
1,1 1,3 7,7 0,3 100 - 400Zuwachs° 18 9
16 4,0 5,8 4,2 40 - 80
LaubhölzerHolz 47 20 24
0,8 2,5 1,8 0,3 100-400Zuwachs4 37 12 14
23 6,4 2,8 4,2 30- 50Wurzelholz' 33 22 18 1,6 1,3 1,3 190
Feinwurzeln' 19 33 10
>3 5,4 3,1 3,4 55
WiesenSprosse 31 50 8,7 2,7 7,6 10 - 40Wurzeln 28 18 7,5 8,5 10- 40
Getreide
..-ne-''--50
50-100Stroh 39 24 25 2,0 (2) 5,0
SeenPhytoplankton 18 17 1,5 14 5- 12
Konsumenten (Beispiele)Isopoden
Chitin9 19 63 3 6
Insekten 6 23 65 3 3Zooplankton 9 16 50 10 15
Zersetzer (Beispiele)Regenwürmer 17 58 6 19 4-
6Eisenia rosea 19 61 4 16 4 -
6Pilzmycel 10 32 25 10 6 10 - 15
' und andere nichtstrukturelle Kohlenhydrate
4 vornehmlich Nadeln bzw. Blätter2 und andere N-haltige Verbindungen
' von Buchenund andere ätherlösliche Verbindungen, z. B . Chlorophyll
6 und ähnliche Gerüststoffe
Tab . 1 : Häufige Zusammensetzung verschiedener Organismen
(-teile) in % der Trockensubstanz nach LIETH (1975)
aus SCHEFFER und SCHACHTSCHABEL (1982).

3
untersuchen, welche Elemente in welchen Pflanzen angereichert
werden, welche davon aus der unmittelbaren physikalischen Um-
gebung (Erde, Wasser, Luft, Staub etc .) stammen, und welche
Elemente aus größeren Entfernungen in die Ökosysteme eingeführt
werden. Dabei ist es möglich, Schwellenwerte für die einzelnen
Elemente in den unterschiedlichen biologischen Systemen zu
finden, in denen diese in aktiver Form vorliegen.
Es ist erforderlich, mehr über die globale Zirkulation der
Elemente zu erfahren, da das gegenwärtige Interesse auf die-
jenigen Elemente gerichtet ist, die in größeren Konzentrationen
in der Biosphäre vorkommen . Soweit es die analytischen Techni-
ken erlauben, sollte das Interesse mit gleicher Intensität
auf Elemente gerichtet sein, die in geringen Mengen vorkommen.
Das unmittelbare Anliegen dieser Arbeit liegt in der Frage,
in welchem Maße verschiedene Elemente in der Nähe von mensch-
lichen Ortschaften bzw . industrieller Aktivität akkumulieren.
Dies erfordert die Analyse derjenigen Elemente, die durch die
unmittelbare Nachbarschaft in ein Ökosystem gelangen und
welche durch den Transport über große geographische Räume
hinweg (MARKERT und LIETH, 1983).
Unter den oben erwähnten Gesichtspunkten wurde an der Univer-
sität Osnabrück ein Arbeitsprogramm mit folgender Zielsetzung
entwickelt:
1. Komplette Elementkataster für vergleichbare Pflanzenarten
in mehreren kontrastierenden Ökosystemen sollen erstellt
werden.
2. Ein Sammelverfahren soll entwickelt werden, das für die
Erfassung der Elemente die besten Ergebnisse liefert.
3. Die geeignetsten analytischen Methoden sollen gefunden
werden (Aufstellung eines Methodenrasters) .

4
4. Die unterschiedlichen Untersuchungs- und Anreicherungs-
methoden sollen auf ihre Brauchbarkeit hin untersucht
werden.
5. Basisdaten der Elementkonzentrationen in unterschiedlichen
Ökosystemen sollen geschaffen werden.
6. Frequenzmuster einzelner Elemente für unterschiedliche
Pflanzenarten auf unterschiedlichem Substrat sollen er-
arbeitet werden.
7. Es soll untersucht werden, welche Pflanzen welche Elemente
unter welchen Umweltbedingungen anreichern .oder ausschlie-
ßen und damit einen Indikatorwert besitzen.
Um diese zahlreichen Ziele zu verfolgen, wurden Ökosysteme ge-
wählt, die es ermöglichen, einen Einblick in den Nah- und Fern-
transport der Elemente zu gewähren . Für die ersten Vorunter-suchungen eines solchen Programms haben wir Hochmoore gewählt,
in deren Umgebung die gleichen Pflanzenarten vorkommen, die
auch auf dem Hochmoor stehen .

5
II . Ombrotrophe und minerotrophe Systeme
Der für diese Untersuchung ausschlaggebende Unterschied der
untersuchten Ökosysteme wurde bereits 1823 von DAUE erkannt.
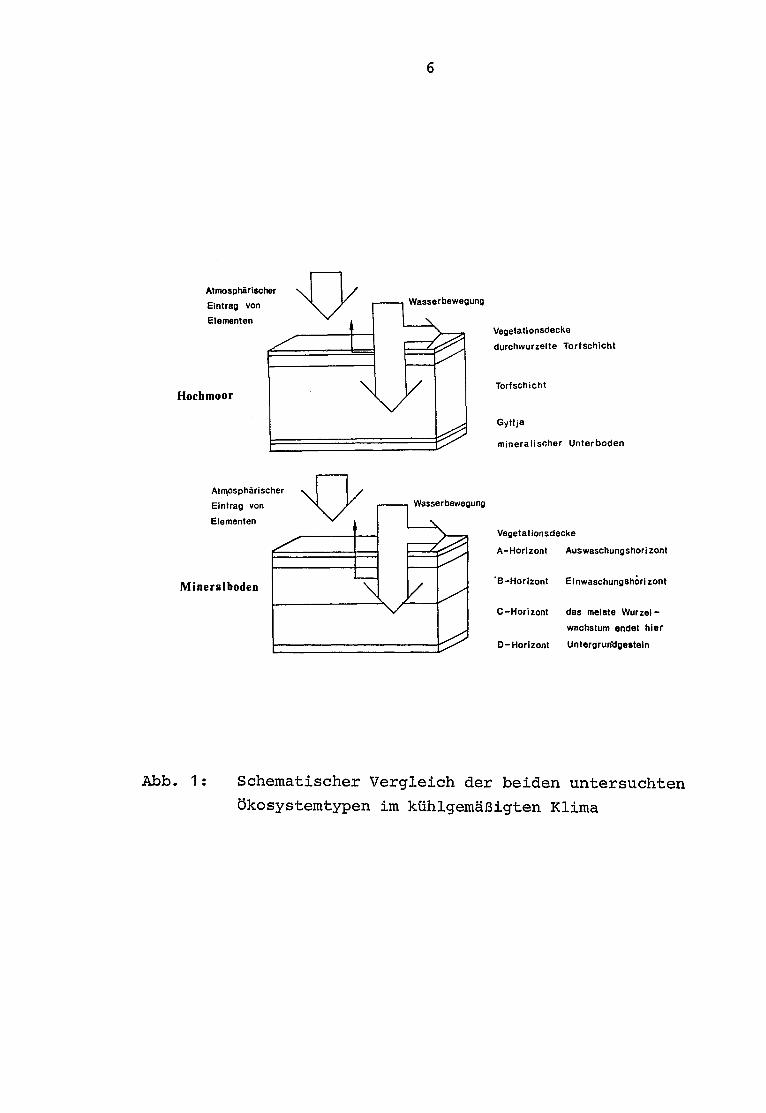
Hochmoorpflanzen decken ihren Nährstoffbedarf ausschließlich
aus der Atmosphäre, da durch die torfogene Schicht eine Ver-
bindung ihrer Wurzeln bzw . Rhizoide zum mineralischen Boden
verhindert wird . Dieser Unterschied soll durch den schemati-
schen Vergleich der unterschiedlichen Ökosysteme in Abb . 1verdeutlicht werden.
Eine regelrechte Stoffbilanz für die Hochmoorernährung versucht
FIRBAS (1952) aufzustellen . Als mögliche Stoffquellen sind da-nach anzusehen:
1. Absorption von Gasen (speziell NH3) durch den sauren Torf
oder Bindung durch lebende Organismen.
2.
Im Regenwasser gelöste oder suspendierte Stoffe.
3. Die minerotrophen und organotrophen Bestandteile des Flug-
staubes.
4. Gelegentliche Zufuhr von größeren toten Pflanzenteilen,
von Exkrementen und Leichen von Tieren, die sich vorwiegend
außerhalb der Moore ernähren.
5.
Diffusion von Stoffen aus stoffreicheren Torfen oder
dem mineralischen Unterboden.
6. Umlagerung der Stoffe aus basalen in höhere Schichten durch
die Aufnahme seitens tiefwurzelnder Pflanzen (Eriophorum,
Scirpus caespitosus) und deren spätere Zersetzung.
Was die unter 5 . genannte Stoffdiffusion aus dem Untergrund be-
trifft, so kann diese kaum wesentlich ins Gewicht fallen ange-
sichts der durch viele Beobachtungen belegten, im allgemeinen
6
Vegetationsdecke
durchwurzelte Torfschicht
Torfschicht
Gyttja
mineralischer Unterboden
Vegetationsdecke
A-Horizont
Auswaschungshorizont
' B-Horizont
Einwaschungshörizont
C-Horizont
das meiste Wurzel -
wachstum endet hier
D-Horizont
Untergrundgestein
Abb . 1 : Schematischer Vergleich der beiden untersuchten
ökosystemtypen im kühlgemäßigten Klima

7
abwärtsgerichteten Wasserbewegung im Hochmoor (OVERBECK, 1975).
Innerhalb eines rein ombrogenen, gleichmäßig zusammengesetzten
jüngeren Hochmoortorfs ist daher auch keine regelmäßige Abnahme
der Mineralstoffe von unten nach oben zu erkennen, die auf
einen Transport derselben durch aufsteigendes Wasser oder Dif-
fusion von der Basis her schließen ließe . Nichtsdestoweniger
konnten im Gesamtprofil des ombrogenen Hochmoortorfs schicht-
weise mehr oder weniger bedeutende Schwankungen im Aschenge-halt nachgewiesen werden, die aber andere Ursachen haben . Sie
stehen im Zusammenhang mit Veränderungen der Wachstumsgeschwin-
digkeit und Vegetationsdecke der Moore oder mit anthropogen
bedingten Veränderungen in der Umgebung . So kann man als Fol-
gen eines zu Stillstand gekommenen oder verlangsamten Moor-
wachstums, wie unter dem Grenzhorizont oder anderen Rekurrenz-
flächen, ebenso aber auch infolge stärkerer Flugstaubbildung in
Kulturlandschaften, eine Anreicherung von Mineralstoffen in den
zeitlich entsprechenden Schichten finden . Sehr eingehend wurde
dieser Aspekt etwa von ERNST et al . (1974) zur Beurteilung der
großflächigen Belastung in Westfalen untersucht . Die Unter-
suchung der Torfprofile eines westfälischen ombrogenen Hoch-
moores erbrachte Hinweise über den zeitlichen Verlauf durch
Blei- und Zinkemissionen . Bei 90 bis 100 cm Moortiefe steigt
der Bleigehalt um mehr als 100 %, der Zinkgehalt um das Vier-
fache . Diese Schicht fällt etwa in die Zeit um Chr . Geb . und
dürfte die Schwermetallkontamination durch den römischen Berg-
bau anzeigen . Bis auf 60 cm nehmen der Blei- und Zinkwert wieder
ab, um von dort an auf das Fünffache für Blei und das Dreifache
für Zink zu steigen . Dieser Anstieg der Schwermetallbelastung
des Moores fällt etwa in das 12 ./13 . Jahrhundert, in dem aus
vielen Teilen Westfalens eine hohe Aktivität des Blei- und
Zinkbergbaus urkundlich nachgewiesen ist . Das Maximum der
Blei- und Zinkwerte fällt in die zweite Industrialisierungs-
phase des 18 . und 19 . Jahrhunderts.
Weitere Untersuchungsergebnisse in bezug auf Nah- und Fern-
transport von Nährstoffen liegen etwa vor bei ROHLING und
TYLER (1971), STEINNES (1980), MARKERT und MEER (1985) .
8
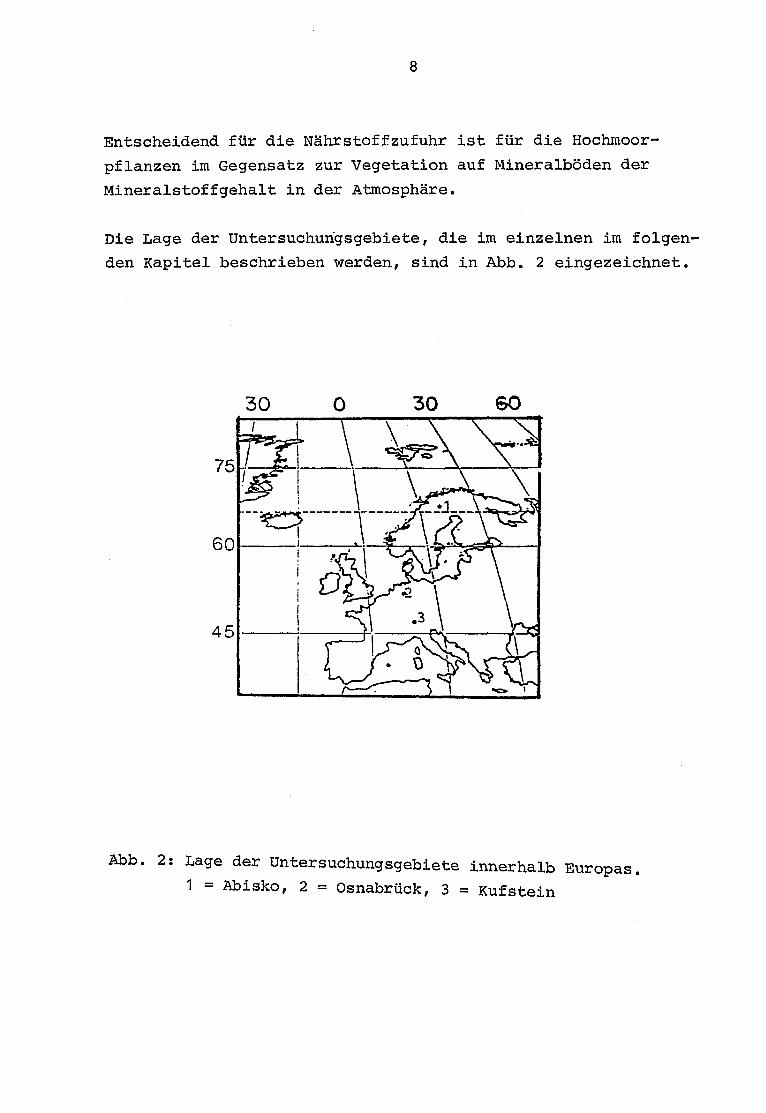
Entscheidend für die Nährstoffzufuhr ist für die Hochmoor-
pflanzen im Gegensatz zur Vegetation auf Mineralböden der
Mineralstoffgehalt in der Atmosphäre.
Die Lage der Untersuchungsgebiete, die im einzelnen im folgen-
den Kapitel beschrieben werden, sind in Abb . 2 eingezeichnet.
30
0
30
---------------
----
.~
3
Abb . 2 : Lage der Untersuchungsgebiete innerhalb Europas.1 = Abisko, 2 = Osnabrück, 3 = Kufstein
75
60
45

9
III . Ökosystembeschreibung
1 . Österreich
a. Die Lage
Die beiden untersuchten Biotope liegen im Bezirk Kufstein in
der nordöstlichen Landesecke Nordtirols am Fuße des Kaiserge-
birges, einem Teil der nördlichen Kalkalpen (Abb . 3).
Abb . 3 . Lage der österreichischen Ökosysteme (')
Der Kaisergebirgsstock ist aus Wettersteinkalk auf einem Sockel-
gebirge aufgebaut und besteht aus zwei parallelen west-östlich
streichenden Hauptkämmen von 20 km Länge und 14 km Gesamtbreite
(LEHMANN, 1980) . Die höchste Erhebung erreicht 2344 NN . Am
Nordfuß liegt das Moorgebiet "Schwemm" bei Walchsee auf 644 NN.
Nördlich davon liegt ein Steilhang aus Kalk.
b. Das Klima
Das Klima zeigt durch die Lage am Nordalpenrand sowohl ozea-
nische als auch kontinentale Züge (LEHMANN, 1980) .
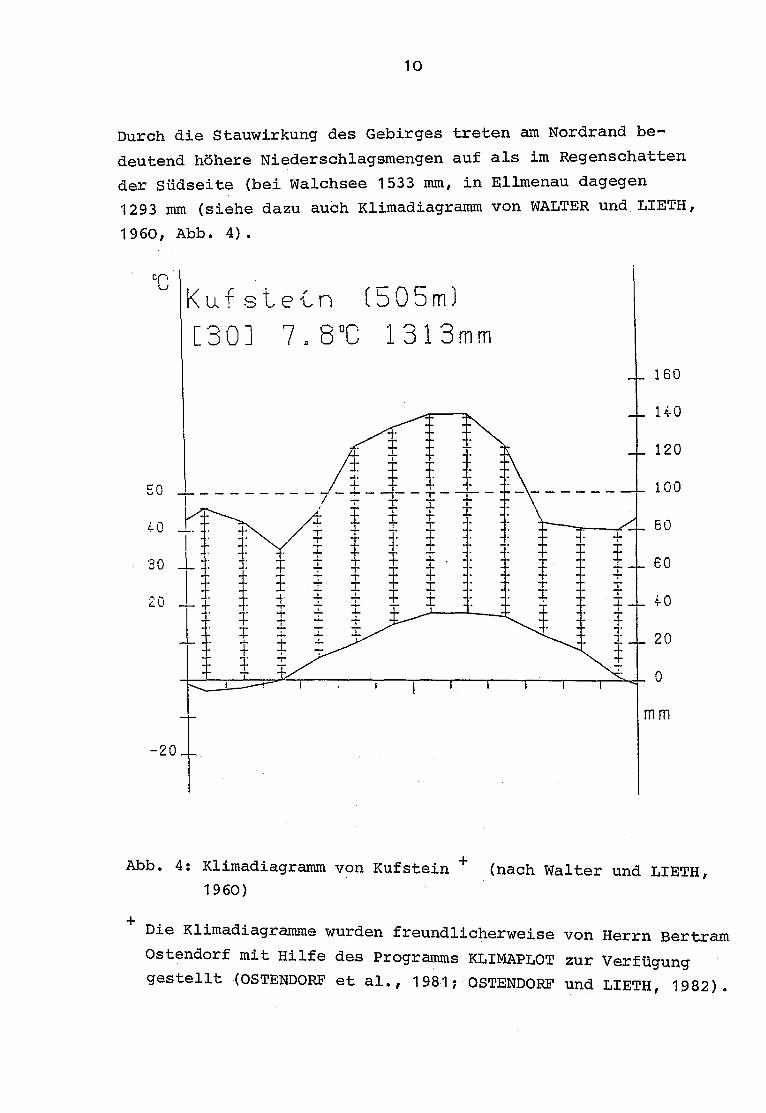
10
Durch die Stauwirkung des Gebirges treten am Nordrand be-
deutend höhere Niederschlagsmengen auf als im Regenschatten
der Südseite (bei Walchsee 1533 mm, in Ellmenau dagegen
1293 mm (siehe dazu auch Klimadiagramm von WALTER und LIETH,
1960, Abb . 4).
Abb. 4 : Klimadiagramm von Kufstein
(nach Walter und LIETH,1960)
+ Die Klimadiagramme wurden freundlicherweise von Herrn Bertram
Ostendorf mit Hilfe des Programms KLIMAPLOT zur Verfügunggestellt (OSTENDORF et al ., 1981 ; OSTENDORF und LIETH, 1982) .

11
Etwa die Hälfte des Niederschlags fällt in den Monaten Mai
bis August, mit Maximum im Juli . Für Kufstein beträgt die
mittlere Zahl der heiteren Tage (Bewölkungsgrad unter 20 %)
im Sommer 12 .2, sowie das Jahresmittel der Temperatur 7 .8 °C
(LEHMANN, 1980).
c. Die Vegetation
Die "Schwemm" bei Walchsee ist die größte noch ungestörte
Moorlandschaft Nordtirols (LEHMANN, 1980) . Der von Wiesen
und Weiden umrandete Moorkomplex ist 7 .7 km lang und 0 .5 km
breit und nimmt dabei eine Gesamtfläche von 63 ha ein . Der
Entstehung nach ein Verlandungsmoor, ist das Untersuchungs-
gebiet im heutigen Zustand im wesentlichen dem Übergangsmoor-
typus zuzuordnen . Es läßt folgende Zonierung erkennen:
Der zentrale Bereich (ca . 25 ha = 40 %) zeigt deutliche Ten-
denz zur Hochmoorbildung . Man findet mit flutenden Sphagnen
gefüllte Schlenken und auch unbewachsene mit freier Wasser-
fläche, deren nackter Torfgrund sich beim sommerlichen Trocken-
fallen mit Drosera anglica und Lycopodium inundata überzieht.
Die Bülten, auf denen randlich Carex inflata und Carex limosa
die Verlandung einleiten, sind mit den typischen Hochmoor-
pflanzen besiedelt . Der sehr verstreute Baumbestand besteht
aus Latschen (von 12 cm bis 2 m Höhe), einigen Birken, Schwarz-
erlen und Faulbaumgebüsch . über prunkvollen Schwingrasen mit
Trichophorum caespitosum, Scheuchzeria palustris und Rhynchos-
pora fusca geht der Zentralbereich in ein schilfreiches meso-
trophes Übergangsmoor über (ca . 20 ha = 32 %), wobei der Phrag-
mites-Bestand gegen den Rand mit sinkender Oligotrophie so-
wohl an Höhe als auch an Dichte zunimmt. Die Zonierung schließt
nach außen (mit Ausnahme des Südrandes) mit Grosseggenwiesen
(ca . 18 ha = 28 %) ab, die teilweise zur Streugewinnung bewirt-
schaftet werden . Am Südrand der "Schwemm" befindet sich an
Stelle der Magno-Cariceten eine Kette eutropher Weiher, in
denen Pflanzengesellschaften des Myriophyllo-Nupharetum ge-
deihen .

12
Der gesamte Hochmoor-Zwischenmoorkomplex wird zum größten Teil
von einem für Hochmoor oft typischen Randgehänge (Laag) umge-
ben, auf dem die untersuchten Pflanzen Molinia, Frangula und
Alnus vorkommen . Auf der Steilwand nahe der Waldgrenze wurde
Pinus mugo auf kalkigem Boden entnommen.
2 . Deutschland
a . Die Lage
Das "Venner Moor" liegt am Südrand des "Großen Moores" und
gehört damit zu einem umfangreichen Hochmoorkomplex, der sich
einst vom Fuße der Mittelgebirge bei Kalkriese bis zu den
Ausläufern der Dammer Berge im Norden und in west-östlicher
Richtung von Vörden bis Schwege Hunteburg erstreckte (WEBER,
1980), (Abb . 5).
Abb . 5 : Lage des Venner Moores und,Achmer in Nordwest -deutschland

13
Von den Rändern aus wurden schon im vorigen Jahrhundert
bäuerliche Torfstiche in das Moor vorangetrieben . Während
die eigentliche Hochfläche damals noch unberührt blieb,
war der Bereich des heutigen NSG "Venner Moor", der sehr
am Rande liegt, schon mit Torfstichen durchsetzt und schon
so stark entwässert, daß einzelne Birken in die vorher baum-
lose Fläche eindringen konnten . Zur Entwässerung trug ins-
besondere der in nächster Nähe südlich am gesamten heutigen
Naturschutzgebiet entlanglaufende "Venner Moorkanal" bei,
der damals schon angelegt war . Der großflächige maschinelle
Torfabbau ist jedoch in dem früher von Hand verstochenen
Randbereich uninteressant, auch die Kultivierung in einzu-
ebnende Grünländereien wäre sehr kostenaufwendig . Daher
blieb rund um die eigentliche Hochmoorfläche des Großen
Moores ein breiter Kranz derartig zerstochener, inzwischen
von Birkenaufwuchs geprägter Moorreste übrig . Das NSG "VennerMoor" ist ein Teil dieser Randzone (WEBER, 1980) . Die Torf-
mächtigkeit ist wegen dieser Randlage gering . Sie beträgt im
mehr oder wenig entwässerten Zustand am Südrande des Natur-
schutzgebietes im Westteil nur 50 cm und befindet sich damit
an der untersten Grenze eines Hochmoorprofils . Im Osten liegendie Werte stellenweise etwas höher (bis ca . 1 .5 m) . Im Norden
des Gebiets, das heißt, nur 200 - 500 m näher zum 2 .5 km ent-
fernten Moorzentrum hin, werden meist 1 .5 m und maximal 2 .5 m
Torfmächtigkeit erreicht, die gegen das Zentrum hin wesentlich
weiter ansteigen dürfte.
Die Untersuchungsfläche bei Achmer liegt etwa 2 km südlichvon Achmer (Abb . 6).
Zur Zeit der größten Ausdehnung des Inlandeises im Norden
Europas herrschte im Blattgebiet ein besonders intensives
Periglazialklima (THIERMANN, 1983) . Aus der damals von pflanz-
lichem Bewuchs fast freien Niederterrassenebene, aber auch
aus älteren pleistozänen Ablagerungen wie der Grundmoräne,
den Schmelzwassersanden und dem Verwitterungsschutt des

14
uhlullmor Lehm, Ioh.8 bn10stark
dmlehmpnr Smsl
oderschluliperSand oder -Sand
»
1 - - 1Nredermomtor13- 10 dm
Sand, sloeonwnrse schlulhp 000 lehmig4km
atemper kalkNltp .r sandgar Lehm.atallenw .ae tonq,Y-5dm
Kalkstein
IIIIIIiIIP;;iii! I
schlulfgibNmger Sand.viellaoh lehmp. S.d e- »20 dm
lehmgar Sand, Sandstein und Tollstem
schwach al.nper Nhmger Sand.tT stempor sandq. Lehm 0- 6dm
oder Inhnuger Schlull 18- 20 dmstempor s .dper Lehm oderIomp. Lehm bis lehmgar Ton
TodundSand0-8dmSand atellenw.se schlullp oder lehmp
humoser Form ds Mdlelsand.elal»mwenelehmpat Sand.S- t0dm
Sand . T sl .nq oder Nhmp. 0 - 15 dmsandyer Lehm
Schwach .hnugr Iss Iehmger ScNu116- 20 dm.endgar Lehm oder Sandstarn
lani b F.msand 6->20denSand, sl&100w.se scNullp 0Nr lohmp
Abb . 6 : Bodenkundliche Karte des Untersuchungsgebietes (aus
THIERMANN, 1983) "Achmer " , Ä= Probennahmestelle
Gebirges wurde das feine Material ausgeblasen . Dies setzte
sich in geringerem Maße wegen der zunehmend stärker hervor-
tretenden Vegetation bis in das Holozän hinein fort . Der fein-
körnige Staub wurde als Löß abgelagert, der weniger feinkör-
nige als Sandlöß . Der Feinsand wurde als Flugsand und Dünenwieder sedimentiert (THIERMANN, 1983).
Erst in dieser Zeit entstand der so ausgeprägte Podsol-Hori-zont als Abschluß.
Entstehung des Podsol : Sinkt auf basenarmen Sanden, meist unterEinfluß von Sauerhumus, der pH-Wert in den stark sauren Be-reich (unter pH 4 .5) ab, so werden die Tonminerale zerstört.
Durch die Niederschläge werden die im Oberboden gelösten
Stoffe in den Unterboden gewaschen und dort wieder ausgefällt.
Durch diesen Prozeß entwickeln sich die aus einem graufarbenen
15
Bleich- oder Verarmungshorizont und darunterliegenden schwarz-,
braun- oder rostfarbenen Anreicherungshorizont bestehenden
Podsole . Im Untersuchungsgebiet sind sie aus basenarmen Flug-
sanden hervorgegangen.
b . Das Klima
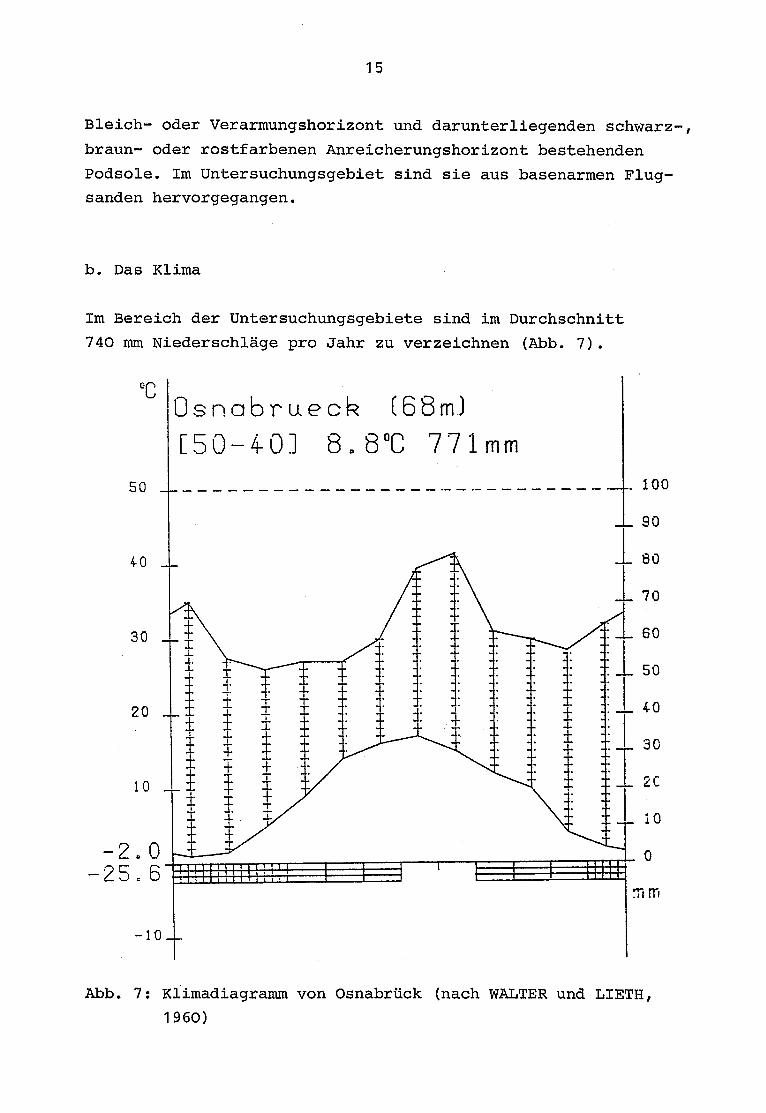
Im Bereich der Untersuchungsgebiete sind im Durchschnitt
740 mm Niederschläge pro Jahr zu verzeichnen (Abb . 7).
oc
Osnabruec
(68)
C50-4-01 8 .8°C 771 mm
mi mi
-10_
Abb . 7 : Klimadiagramm von Osnabrück (nach WALTER und LIETH,
1960)

16
c . Die Vegetation
Die natürliche Hochmoorflora ist insbesondere durch die Ent-
wässerung verarmt (WEBER, 1980) . Empfindlichere Arten sind
ausgestorben, so Drosera anglica, die früher im Großen Moor
vorkam (WEBER, 1980) . Vor allem sind zahlreiche der typischen
Hochmoorsphagnen wie Sphagnum magellanicum, Sph . rubellum
u .a . ebenso wie andere Kryptogamen ganz verschwunden oder
nur in spärlichen Resten vorhanden . Andererseits haben sich
das gesamte ursprüngliche Inventar an Zwergsträuchern und
andere weniger gegen Entwässerung empfindliche Arten halten
können (WEBER, 1980) . Folgende Arten sind heute noch im
Schutzgebiet anzutreffen:
Andromeda polifolia, Calluna vulgaris, Drosera rotundifolia,
Empetrum nigrum, Erica tetralix, Eriophorum angustifolium,
Eriophorum vaginatum, Rhynchospora alba, Vaccinium oxycoccus.
Alle übrigen, heute dort zum Teil häufigen und oft dominie-
renden Arten, fehlten der ursprünglichen Vegetation vollständig
und sind erst durch den anthropogenen Einfluß in das Gebiet
eingewandert oder verschleppt worden, so vor allem das Pfei-
fengras (Molinia coerulea) und der gesamte Strauch- und Baum-bewuchs (besonders mit Betula pubescens, WEBER, 1980).
Auf der Flugsanddüne in Achmer hat sich auf dem stark sauren
Boden (Podsol) als Endstufe die Vaccinio-Piceeta Nadelwald-
gesellschaft entwickelt, die am Boden von Vaccinium vitis-idaea rasenartig überzogen wird.
3 . Schweden
a . Die Lage
Das Hochmoor Stordalen liegt etwa 10 km östlich von Abisko,
200 km nördlich des Polarkreises, am Südrand des Torneträsk(Abb . 8) .

17
Abb . 8 : Lage des Moores Stordalen in der Nähe von Abisko
Die Gesamtfläche beträgt etwa 25 ha, wovon der zentrale Teil
von ca . 15 ha als ombrotroph anzusehen ist . Das Moor ist von
hohen Bulten gekennzeichnet, die an der Spitze durch Wind oft
erodiert sind . Das starke Höhenwachstum der Bulte wird wahr-
scheinlich durch Permafrost verursacht (SONESSON, 1983) . In
Schweden haben diese Moore den Namen Pals-Moore (nach einem
lappischen Wort mit der Bedeutung : trockene Gebirgskämme).
Das schwedische Vergleichsökosystem "Mineralboden" liegt etwa
1 km südlich von Abisko . Der Boden weist sich als schwach
entwickelter Podsol aus, da die Podsolierung durch das nur
etwa 40 cm tief liegende Untergrundgestein durch Verwitterung
ständig zurückgedrängt wird+ . Vaccinium vitis idaea überzieht
großflächig den Boden.
+ persönliche Mitteilung von Prof . Bailey, Fachhochschule
Osnabrück
18
b . Das Klima
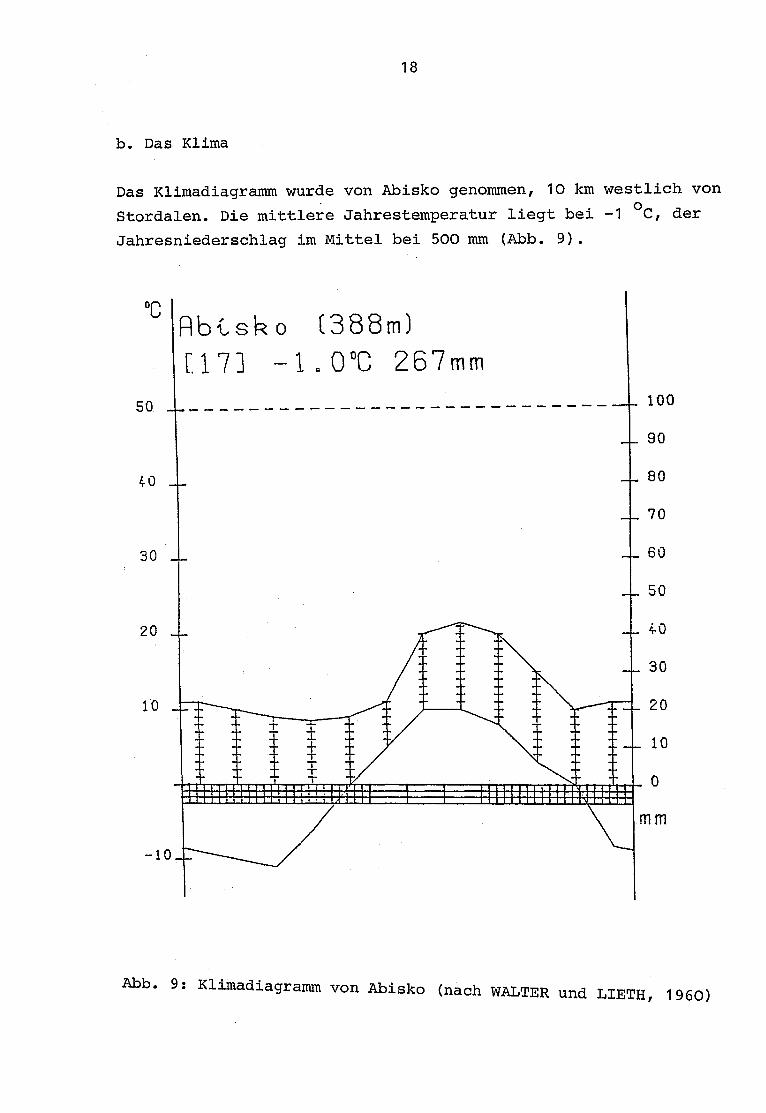
Das Klimadiagramm wurde von Abisko genommen, 10 km westlich von
Stordalen . Die mittlere Jahrestemperatur liegt bei -1 °C, der
Jahresniederschlag im Mittel bei 500 mm (Abb . 9).
Rbtsko (388m)[173 -1 .0°C 267mmMMM MMMMMMMMMMMMMMMMMMMMMMMMM
DC
50 100
4-0
30
90
80
70
60
50
20
4-0
30
rrrrrnrrrrrrCrrrrrrr~rrrr~rr~r~rrrrrrrrr.
10
-10
IMIMMrrarrrrrrr.rmaorrnnrrrrrrr.r•r rrrrrelmrrmrrr
Wrrrrrrrr•rn
'
MIM.EIrrrrrl nrrrrrrrrrrrrvrorr•n
20
10
mm
Abb . 9 : Klimadiagramm von Abisko (nach WALTER und LIETH, 1960)

19
c . Die Vegetation
Die wesentlichen Pflanzenarten von Stordalen sind in Tab . 2wiedergegeben:
Andromeda poZifoZia L.
BetuZa nana L.
Carex rotundata Wg
Cetraria cucuZZata (Bell .) Ach.
C. delisei (Borg) Th . Fr.
C. nivaZis (L.) Ach.
CZadonia sect . Cenomycae
C. sect. CZadina
C.rangiferina (L .) Web.
C. svZvatica toll.
Dicranum eZongatum Schleich.
DrepanocZadus schulzei Roth.
Empetrum hermaphroditum Hagerup
Eriophorum angustifoZium Honck.
E. vaginatum L .
Gymnocolea infZata Bude.
IcmadophiZa ericetorum L.
Orchrolechia frigida (5w .) Lynge
PinguicoZa viZZosa L.
Polytrichum jensenii Hag.
P. strictum Banks
PtiZidium ciZiare L.
Rubus chamaemorus L.
Sphagnum baZticum (Sw .) Lynge
5. fuscum KZinggr.
S. Zindbergii Schimp.
S. russowii Warnst.
Vaccinium microcarpum (Turcz .) Hook.
V. uliginosum L.
V. vitis-idaea L.
Tab . 2 : Hauptsächliche Vegetation auf dem Moor Stordalen

20
IV . Probenaufbereitung
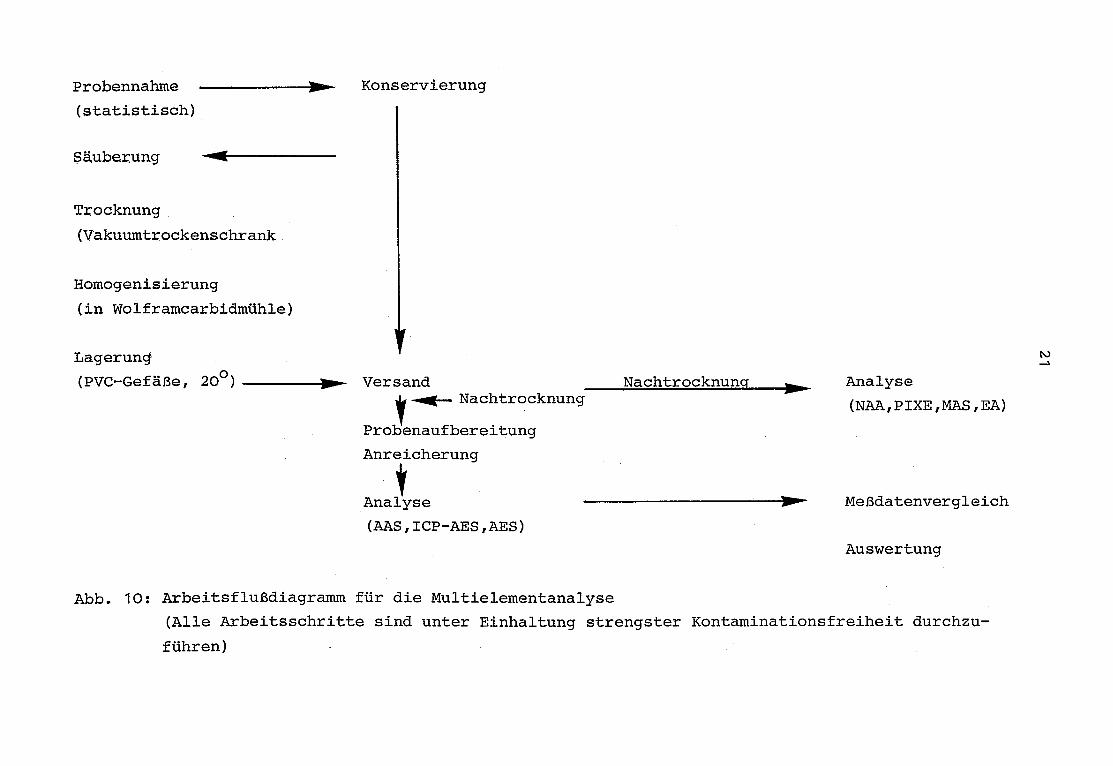
In Abb . 10 ist ein Arbeitsflußdiagramm dargestellt, dessen
einzelne Schritte im folgenden besprochen werden sollen . Das
dargestellte Arbeitsflußdiagramm stellt den idealen Arbeits-
gang dar, der für zukünftige Arbeiten auf dem Gebiet der anor-
ganisch-analytischen Ökologie eingehalten werden sollte.
Im Rahmen dieser Arbeit mußte häufig vom analytischen Ideal-
pfad abgewichen werden . Sei es, daß die für die geplanten
Untersuchungen benötigten Geräte nicht vorhanden waren, bzw.
ihre Anschaffung den finanziellen Rahmen gesprengt hätte
(z .B . Wolf ramcarbidmühle, Vakuumtrockenschrank), oder daß
gewisse Arbeitsschritte bewußt gekürzt werden müßten, damit
die Anzahl der Einzelproben nicht zu groß wurde, was eine
Untersuchung auf sämtliche Elemente des periodischen Systemsunmöglich gemacht hätte . Beispielsweise wurde auf eine sta-tistische Probennahme verzichtet . Im folgenden sei auf dieeinzelnen Punkte des Arbeitsflußschemas näher eingegangen .
Probennahme ie- Konservierung
(statistisch)
Säuberung
Trocknung
(Vakuumtrockenschrank
Homogenisierung
(in Wolframcarbidmühle)
VersandIrme-. Nachtrocknung
Probenaufbereitung
Anreicherung
Analyse
Meßdatenvergleich
(AAS,ICP-AES,AES)Auswertung
Abb . 10 : Arbeitsflußdiagramm für die Multielementanalyse
(Alle Arbeitsschritte sind unter Einhaltung strengster Kontaminationsfreiheit durchzu-
führen)
Lagerung
(PVC-Gefäße, 200 ) Analyse
(NAA,PIXE,MAS,EA)
Nachtrocknung

22
1 . Probennahme
Tab . 3 gibt diejenigen Pflanzenarten und Entnahmestellen wieder,
die in die Untersuchungen im Rahmen dieser Arbeit eingingen.
Schweden
Deutschland
Österreich
Torf Podsol Torf Podsol Torf Laag Kalk
Vaccinium vitis idaea
Pinus mugo (sylvestris)
Alnus glutinosaFrangula alnus
Molinia coerulea
x
x
x
x
X
X
X
X
X
X
x
x
x
Tab . 3 : Pflanzenarten, die aus den unterschiedlichen Systemen
gezogen wurden
Die Proben wurde im Juli 1983 und teilweise nochmal im Juni
1984 in Schweden (Abisko, Stordalen), Deutschland (Venne,
Achmer) und in Österreich (Kufstein) entnommen . Zur Proben-
nahme wurden in den Mooren nach Möglichkeit Bulte ausgewählt,
in denen alle Arten in wüchsiger Form auftraten . Bei der
Auswahl der Bulte wurden solche bevorzugt, die sich auf den
offenen Hochmoorflächen befanden und nicht durch erdwüchsige
Vegetation geschützt waren . Eine mögliche Interzeption der
Spurenelementemission durch Gebüsch oder Baumkronen war
somit ausgeschlossen . Die beblätterten Sprosse von Vaccinium
vitis idaea wurden dabei auf quadratischen Flächen von etwa10 x 10 m2 an 20 Stellen 2 cm oberhalb der Bultoberflächeabgerupft . Die gesammelten Proben wurden auf einer Kunst-
stoffolie ausgebreitet und innerhalb von drei Tagen amStandort getrocknet (Nachtrocknung im Labor bei 105 oC imTrockenschrank) . In gleicher Weise wurde mit Molinia coeruleaverfahren . Die nadeltragenden Zweige von Pinus mugo und die
beblätterten Äste von Frangula alnus und Alnus glutinosa

23
wurden etwa einen Meter vor dem Zweigende abgebrochen und eben-
falls der Lufttrocknung überlassen . Die für die Quecksilberbe-stimmung vorgesehenen Proben wurden nicht getrocknet (Ver-
flüchtigung (MAY und STOEPPLER, 1978)), sondern im Frischzu-stand analysiert . Dazu wurden die Proben in ultrareinen Duran-
Schottgläsern aufbewahrt, denen zur Konservierung des Pflanzen-
materials jeweils 1 g Thymol (3-Hydroxy-4-iso-propyl-toluol)pro 250 ml Glas zugesetzt wurden . Thymol wurde getrennt ana-
lysiert.
In den Hochmooren Schwedens, Deutschlands und Österreichs
wurden Torfproben bis zu einer Tiefe von 60 cm ab Oberfläche
der Vegetationsdecke mit Hilfe eines metallfreien Spatens
(Hart-PVC) entnommen. Bei den Mineralbodenproben lag in
Schweden das Untergrundgestein sehr hoch, so daß oberhalb
der 60 cm Marke abgebrochen werden mußte . Anschließend wurden
die Proben wie schon das Pflanzenmaterial der Lufttrocknung
überlassen.
Bei den gesammelten Pflanzenproben wurden die artfremden Be-
standteile aussortiert (z .B . Blätter und Wurzeln von Erica-
ceen, Sphagnumarten, Lebermoose, Birkenblätter) . Bei Vacci-
nium vitis idaea wurden die Blättchen von den Sproßachsen
getrennt, bei Pinus die Nadeln von den Zweigen, bei Alnus
glutinosa und Fragula alnus die Blätter von den Zweigen.
Die verholzten Teile wurden getrennt gelagert.
Die Mineralbodenproben wurden durch ein Plastiksieb (Maschen-
weite : 0 .4 mm) geschickt, um zu grobes silikates Gestein
abzutrennen.
Während der laufenden Untersuchungen wurde Probenmaterial
von folgenden Wissenschaftlern von ihren Reisen mitgebracht,
bzw. zugesandt:
Dr . Esser (Schweden), Prof . Lieth (Österreich), Prof . Sonesson
(Schweden) und Prof . Bortenschlager (Österreich) .

24
Ihnen sei an dieser Stelle gedankt . Die Probennahme dieser
Proben weicht in einigen Punkten, insbesondere in der Proben-
trocknung (Trockenschrank) und Probenanzahl, von dem oben
Gesagten ab.
Die Proben wurden in Polyethylenbehältern bzw . -tüten bei
Raumtemperatur kontaminationsfrei gelagert . Ein Element-
verlust konnte sich während der Lagerung nach einem Jahr
nicht feststellen lassen.
Soweit nicht anders erwähnt, sind die Proben im getrockneten,
nicht homogenisierten Zustand an die mitarbeitenden Institute
(meist in 10 g Portionen), verpackt in Polyethylenbehältern,
versandt worden.
2 . Probenhomogenisierung
Die getrockneten Proben wurden soweit wie möglich weiter
manuell zerkleinert . Anschließend wurden sie in einem
elektrischen Achatmörser der Universität Münster (Institut
für angewandte Botanik) innerhalb einer viertel Stunde
zu einem feinen Pulver homogenisiert, bzw . im unhomogeni-
sierten Zustand aufgeschlossen . Der Abrieb des Achatmörsers
ist aufgrund des insgesamt resultierenden Verdünnungsfaktors
verschwindend klein und beeinflußt somit nicht die Analysen-
ergebnisse (BLOOM und BARNETT, 1955, THOMPSON und BANKSTON,
1979) . Von einer Waschung des Pflanzenmaterials wurde ab-
gesehen . Zum einen wird durch die Behandlung der Probenmit Wasser (bzw . anderen Lösungsmitteln) ein Teil der
Elemente (insbesondere natürlich der von leicht wasserlös-lichen) in Abhängigkeit von der Waschdauer mitgerissen
und geht dadurch für die quantitative Erfassung verloren.
Andererseits gelangt ein Teil dieser Aerosole auch kurz-fristig in die Pflanze . Eine Aufnahme durch die Spaltöff-nungen soll . zwar aufgrund der hohen Oberflächenspannung

25
nicht möglich sein (HULL, 1970, ROBERTS, 1975), aller-
dings konnte STEENKEN (1973) in Gewächshausexperimenten
mit bleifreien und bleihaltigen Autoabgasen zeigen, daß
Blei durch die Cuticula bis in die Zellwände der äußeren
Epidermis gelangt . Zwar wird es vom Stoffwechsel der Pflanze
ferngehalten, allerdings ist es als Bestandteil der Gesamt-
trockensubstanz der Pflanze zu betrachten . Sehr eindrucks-
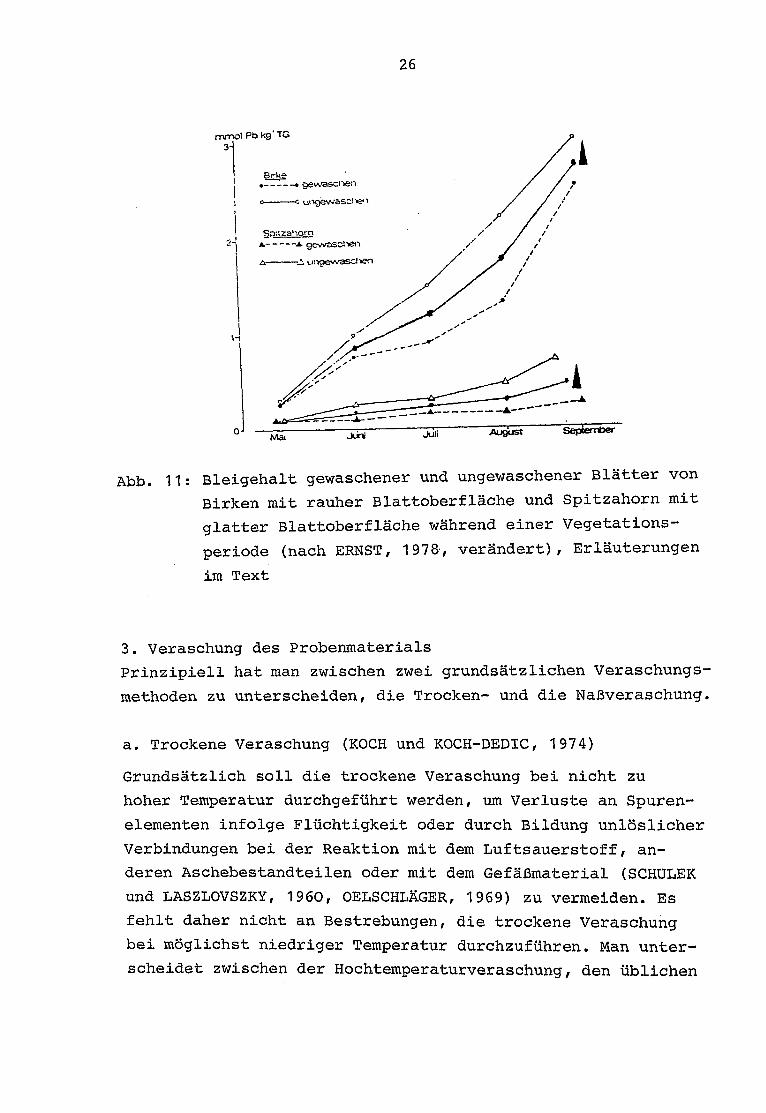
voll beschreibt ERNST (1978) den Bleigehalt gewaschener
und ungewaschener Blätter von Birken mit rauher Blattober-
fläche und Spitzahorn mit glatter Blattoberfläche während
einer Vegetationsperiode (Abb . 11).
Unter Berücksichtigung des Gesamtfehlers der chemischen Pro-
benaufbereitung wäre es vermutlich am günstigsten, einen
Mittelwert aus gewaschenen und ungewaschenen Meßergebnissen
zu ziehen (Abb . 11) . Aufgrund der großen Blätter- und Nadel-
anzahl dieses Projektes wurde auf eine Waschung verzichtet.
Dieses Problem soll in weiteren Arbeiten eingehend diskutiert
werden .
26
Abb . 11 : Bleigehalt gewaschener und ungewaschener Blätter von
Birken mit rauher Blattoberfläche und Spitzahorn mit
glatter Blattoberfläche während einer Vegetations -
periode (nach ERNST, 197&, verändert), Erläuterungen
im Text
3 . Veraschung des Probenmaterials
Prinzipiell hat man zwischen zwei grundsätzlichen Veraschungs -
methoden zu unterscheiden, die Trocken- und die Naßveraschung.
a . Trockene Veraschung (KOCH und KOCH-DEDIC, 1974)
Grundsätzlich soll die trockene Veraschung bei nicht zu
hoher Temperatur durchgeführt werden, um Verluste an Spuren-
elementen infolge Flüchtigkeit oder durch Bildung unlöslicher
Verbindungen bei der Reaktion mit dem Luftsauerstoff, an-
deren Aschebestandteilen oder mit dem Gefäßmaterial (SCHULEK
und LASZLOVSZKY, 1960, OELSCHLÄGER, 1969) zu vermeiden . Esfehlt daher nicht an Bestrebungen, die trockene Veraschung
bei möglichst niedriger Temperatur durchzuführen . Man unter-scheidet zwischen der Hochtemperaturveraschung, den üblichen

27
Verfahren der trockenen Veraschung, den Bereich von 400 °C um-
fassend, und der Tief temperaturveraschung im Bereich von1000 - 150°C unter Anwendung von angeregtem Sauerstoff.
a1. Tieftemperaturveraschung im angeregten Sauerstoff
Die Kalt-Plasma-Veraschung eignet sich für Untersuchungen
im untersten Konzentrationsbereich, d .h . pg/g . Das Prinzipdieser Methode läßt sich in wenigen Worten zusammenfassen:
Bei geringem Druck (einige mbar) wird Sauerstoff in ein
Hochfrequenz- oder Mikrowellenfeld geleitet . Es entstehtein Plasma (DORNER, 1982) . Kommt der angeregte Sauerstoff
mit dem Probenmaterial in Kontakt, wird das organische
Material abgebaut (GLEIT und HOLLAND, 1962, KAISER et al .,1971) . Im Gegensatz zu anderen Verfahren können auf diese
Weise auch sehr widerstandsfähige Materialien wie PFTE oder
Graphit verascht werden . Nach beendeter Veraschung wird die
gesamte Probe im geschlossenen Gefäß in den Heizblock eines
Rückflußkühlers eingesetzt, durch den wenig konzentrierte
Säure eingefüllt wird . Die veraschten Probenrückstände sam-
meln und lösen sich in der Säure durch Kochen unter Rück-fluß . Vorteil dieses Verfahrens ist weitgehende Kontamina-
tionsfreiheit, nachteilig wirkt sich allerdings der geringe
Probendurchsatz aus.
a2. Hochtemperaturveraschung
Bei der Hochtemperaturveraschung wird das getrocknete Proben-
material meist in einem Tiegel (unterschiedlichster Zusammen-
setzung) im Muffelofen zwischen 400° - 600 °C verascht, der
meist weiße Rückstand mit wenig Säure aufgenommen und auf
das gewünschte Endvolumen mit bidest . Wasser aufgefüllt.
Bei dieser Art der Veraschung sind die Fehlerquellen recht
komplex und vielzahlig:
1 . besteht die Möglichkeit der Einschleppung von Spuren-
elementen durch den Staub während der Veraschung (ge-
gebenenfalls muß in staubfreien Räumen gearbeitet werden)

28
oder durch Bestandteile des Gefäßmaterials (die Mindestfor -
derung ist ein mit Quarz ausgekleideter Muffelofen) . Es ist
ein gewisser Nachteil der trockenen Veraschung, daß man noch
verhältnismäßig wenig über die während der Veraschung statt-
findenden Reaktionen zwischen Spurenelementen, Bestandteilen
des Probenmaterials bzw . dem Gefäßmaterial und den damit ver-
bundenen Spurenverlusten weiß (KOCH und KOCH-DEDIC, 1974).
2 . besteht die Möglichkeit des Verlustes an Spurenelementen
infolge ihrer Flüchtigkeit oder durch Adsorption oder
Bindung mit anderen Aschebestandteilen bzw . dem Gefäßma-
terial.
Die Ursache für das Auftreten von Verlusten bei der
trockenen Veraschung.ist komplexer Natur, da mehrere
ineinandergreifende Faktoren einen Einfluß ausüben:
Temperatur, chemische und physikalische Natur des orga-
nischen Materials und seiner Bestandteile, Tiegelmaterial,
Vorgeschichte (Alter und Zustand) des Tiegels, Veraschungs-
hilfe und chemische Bindungsform des Elements (KOCH undKOCH-DEDIC, 1974) . Ein Beispiel, das die Komplexität
dieses Veraschungsmodi darstellen soll:
Verluste durch Verflüchtigung treten bei Blei nicht auf,
wenn dieses als Nitrat oder Sulfat vorliegt, sie tretenaber bei PbC12 auf (AGAZZI et al ., 1963) . Mit Pb-Ver-lusten ist daher auch zu rechnen, wenn Chlor ionogen
oder organisch gebunden im Probenmaterial vorkommt . EinZusatz von H2SO4 kann günstig sein, hat aber bei organischgebundenem Chlor (z .B . Polyvinylchlorid) keinen Einfluß(KOCH und KOCH-DEDIC, 1974).
b. Nasse Veraschung
Die nasse Veraschung zeichnet sich besonders dadurch aus,daß sie niedrige Veraschungstemperaturen aufweist . Da sichhier die Veraschung in flüssiger Phase vollzieht, entfallenbzw. verringern sich die Verluste durch Adsorption oderReaktion mit dem Gefäßmaterial, Aschebestandteilen und dem

29
Luftsauerstoff . Prinzipiell haben wir die Naßveraschung
im offenen und geschlossenen System zu unterscheiden.
b 1 . Naßveraschung im offenen System
Hierbei wird meist in offenen Bechergläsern (meist be-
stehend aus Glas, Quarz oder Teflon) 1 - 2 g der Probe
mit Salpetersäure oder einer anderen Säure bzw . Säure-
gemisch gekocht und anschließend die Säure bis fast zur
Trockne eingedampft . Dieser Vorgang kann sich im Ver-
hältnis zur Widerstandsfähigkeit des aufzuschließenden
Materials mehrere Male wiederholen . Salpetersäure hat
sich für diesen Aufschluß besonders bewährt, da diese
Säure leicht zu reinigen ist (Erniedrigung der Konta-
minationsgefahr) und außerdem der Gefahr der Bildung
von explosiven Perchloraten in Verbindung mit Perchlor-
säure beim unvorsichtigen Abrauchen aus dem Weg gegangen
wird.
Der offene Naßaufschluß weist neben anderen drei prin-
zipielle Schwächen auf.
1. Die Veraschungstemperatur muß dem Siedepunkt der Säure
angepaßt werden, was oft zu unvollständigen Aufschlüssen
führt.
2. Im offenen Naßveraschungssystem ist die Verflüchtigung
gewisser Elemente durch Bildung leichtflüchtiger Halo-
genide besonders hoch (Dorner, 1982).
3. Wie bei jedem offenen Aufschluß findet beim Arbeiten
in nicht staubgefilterten Räumen Kontamination durch
Bestandteile der Luft statt.
b2 . Naßveraschung im geschlossenen System
Zur Naßveraschung in geschlossenen Systemen wurden ver-
schiedene Druckbombem konstruiert, die sich in Bau und
Größe voneinander unterscheiden . Der augenscheinlichste
Vorteil dieser Methode ist das Unterdrücken jeglicher Ver-

30
flüchtigung . Es können somit Proben ohne Elementverlust
aufgeschlossen werden . Weitere Vorteile geschlossener
Systeme liegen auf der Hand : Es werden geringere Proben-
mengen benötigt . Es wird in der Regel mit einer Einwaage
von 200 bis 500 mg (BERNAS et al ., 1986, KOTZ et al ., 1972,
SCHRAMEL et al ., 1982) gearbeitet . Zudem beschleunigen
erhöhte Temperaturen und Druck den Aufschluß . Damit
läßt sich die Veraschung in der Bombe meist in kürzerer
Zeit durchführen als im offenen System.
Hier muß jedoch auch bemerkt werden, daß gerade die im
offenen System vorteilhafte - weil leicht handhabbare -
Salpetersäure keine optimalen Ergebnisse liefert, falls
die Proben für andere Bestimmungsmethoden als AAS und
AES-ICP aufbereitet werden (DÖRNER, 1982) . Bei einer Ver-
aschungstemperatur von 170 °C bleiben oft noch Reste der
biologischen Matrix übrig und stören elektrochemische
Nachweisverfahren . Anders sieht es aus, wenn eine AAS-
Untersuchung geplant ist . In•diesem Fall bleiben die
organischen Reste ohne Einfluß, da sie ebenfalls in
der Flamme bzw . im Graphitofen atomisiert werden . Einweiteres Problem der Druckbomben liegt in den verwen-
deten PFTE-Einsätzen, die sich nach längerem Gebrauch
zu verformen beginnen, Dampf durchlassen und damit zu
einer Verfälschung der Analysenergebnisse führen . Außer-dem haben sich in letzter Zeit Meinungen gehäuft, die
eine ausgesprochen hohe Oberflächenadsorption für ge-
wisse Elemente bei den verwandten Drücken aufweisen.
Zudem sind aufgrund der hohen Anschaffungskosten wenigparallele Aufschlüsse möglich.
Aus den oben genannten Gründen wurde an der Universität
Osnabrück eine Aufschlußapparatur nach Jülicher Vorbild(MAY und STOEPPLER, 1978) konstruiert.
Auf eine Heizplatte wird ein Aluminiumblock mit 24 Löchernmontiert . In diese Löcher passen 24 mit Klammern und Deckelverschließbare Quarzgläser, die uns von der KFA Jülichzur Verfügung gestellt wurden .

31
b3. Aufschluß der Pflanzen- und Bodenproben an der Universität
Osnabrück
0 .5 g Probenmaterial wird mit 10 ml HNO 3 suprapur (Merck)
versetzt und innerhalb von 1 h in den geschlossenen Quarz-
gefäßen auf 170 °C erhitzt . Der Druck liegt zwischen 2
und 4 bar+ . Die Temperatur wird 2 h auf 170 °C gehalten
und anschließend durch Ausschalten des Heizgerätes ab-
gekühlt . Anschließend werden die Proben auf das gewünschte
Endvolumen von 50 ml mit bidestilliertem Wasser aufge-
füllt.
b4. Test der Auf schlußapperatur
Die Brauchbarkeit und Zuverlässigkeit wurde durch ver-
schiedene Tests geprüft:
1. Referenzmaterial (Apfelblätter, 1978) wurde zunächst
in der oben beschriebenen Apperatur aufgeschlossen
und unabhängig davon in einer Aufschlußapparatur
von der GSF München . Anschließend wurden sowohl die
Münchner als auch die Osnabrücker Proben von der GSF
München mit Hilfe der AES-ICP gemessen (Abb . 12).
Tabelle 4 zeigt die ausgezeichnete Übereinstimmung
der erhaltenen Meßergebnisse.
2. Parallelaufschluß jeweils von drei Einzelproben:
a. Referenzmaterial (Apfelblätter 1978)
b. Vaccinium vitis idaea
c. Torfboden
Tabelle 5 zeigt die gute Übereinstimmung der Meßer-
gebnisse am Beispiel Mangan, das mit der Flamme be-
stimmt wurde.
Diese Untersuchungen der Aufschlußapparatur gab Auf-
schluß darüber, zufriedenstellende Ergebnisse im Spuren-
elementbereich zu erhalten.
+MAY, Kernforschungsanlage Jülich, mündliche Mitteilung

32
Messung. AES-ICP an der GSF-München
Aufschluß GSFAufschluß OS
Probe 51Probe 51
Abb . 12 : Flußschema zur Testung der Aufschlußapperatur für
die Spurenelementanalytik (Erläuterungen im Text)
Element Aufschluß Univer-
sität Osnabrück
Aufschluß GSF
München
Ca 5350 6309
Cd 0 .10 0 .09Cr 1 .22 1 .60Cu 9 .0 8 .5Fe 160 150Mg 1802 1735Mn 29 .1 32 .8Ni 3 .79 4 .3P 1917 1900Pb 2 .75 4 .7Zn 22 .2 25 .2
Tab . 4 : Messung des Elementgehaltes im Referenzmaterial
Apfelblätter mit Hilfe der AES-ICP und der GSF
München nach unabhängigem Aufschluß der Proben
an der Universität Osnabrück und der GSF München(Angaben in mg/kg)

33
3 Einzelmessungen (digits)
Referenzmaterial Probe 70
Referenzmaterial Probe 71
Referenzmaterial Probe 72
Vaccinium vitis idaea Probe 76
Vaccinium vitis idaea Probe 77
Vaccinium vitis idaea Probe 78
Torf Probe 73
Torf Probe 74
Torf Probe 75
0 .038
+ 0 .001
0 .035
+ 0 .001
0 .035
+ 0 .001
0 .029
+ 0 .001
0 .030
+ 0 .001
0 .029
+ 0 .001
0 .027
+ 0 .001
0 .026
+ 0 .001
0 .027
+ 0 .001
0 .474
+ 0 .002
0 .463
+ 0 .002
0 .477
+ 0 .002
0 .488
+ 0 .002
0 .488
+ 0 .002
0 .475
+ 0 .002
0 .472
+ 0 .002
0 .472
+ 0 .002
0.481
+ 0 .002
0 .012
+ 0 .001
0 .012
+ 0 .001
0 .014
+ 0 .001
0 .011
+ 0 .001
0 .010
+ 0 .001
0 .011
+ 0 .001
0 .010
+ 0 .001
0 .009
+ 0 .0010 .010
+ 0 .001
Tab . 5 : Vergleich der Meßdaten für Mangan paralleler Auf-schlüsse von Referenzmaterial (Apfelblätter, 1978),Vaccinium vitis idaea und Torfboden . (3 Einzelproben
ä 3 Einzelmessungen, Angaben in digits)

34
b 5 . Bemerkungen zum Referenzmaterial (SCHRAMEL et al ., 1982)
Die Entwicklung einer neuen analytischen Technik für be -
stimmte Elemente - besonders natürlich für Spurenelemente
- und deren Überprüfung auf Richtigkeit und Reproduzier -
barkeit setzt die Verfügbarkeit von geeignetem Standard-
Referenz-Material (SRM) voraus . Dabei ist natürlich die
Notwendigkeit zu beachten, daß die SRM's hinsichtlich der
Matrixzusammensetzung und der Konzentration der zu bestim -
menden Elemente möglichst gut an die zu untersuchenden
Proben angeglichen sind, was eine relativ große Anzahl
SRM's voraussetzt . Leider ist diese Forderung gerade auf
dem medizinisch-biologischen Sektor und für die Umweltpro -
ben nicht erfüllt . Ausreichend zertifizierte Materialien
stehen praktisch nur vom NBS (National Bureau of Standards,
USA) zur Verfügung, aber leider hinsichtlich der Matrix-
verschiedenheit nur in einem sehr beschränkten Maße . Aus
diesem Grund müssen die Aktivitäten des BCR (Community
Bureau of Reference) der EG (Europäische Gemeinschaft)
hinsichtlich der Anfertigung und Zertifizierung einer
breiten Palette solcher SRM's für Umwelt- und medizinisch-
biologische Proben unterstrichen und nachdrücklich von
allen Analytikern unterstützt werden . Diese Palette sollvon Böden, Klärschlämmen, Flugasche, Pflanzen, Geweben,Körperflüssigkeiten, Milchpulver usw . einen möglichst um-fassenden Bogen über die verschiedenen Matrizes spannen,
so daß jeder Analytiker in der Lage ist, eine interne
und externe Qualitätskontrolle vorzunehmen.
Auch die Kosten größerer Mengen eines Referenzmaterialssind immens . Aus diesem Grunde wurde in dieser Arbeit auf
nicht zertifiziertes Referenzmaterial der FachhochschuleOsnabrück zurückgegriffen, das mir von Prof . Alt und FrauPeters freundlicherweise zur Verfügung gestellt wurde . Ihnensei an dieser Stelle gedankt . Bei diesem Referenzmaterialhandelt es sich um Apfelblätter aus dem Jahre 1978, die von
den Versuchsanlagen der Fachhochschule Osnabrück stammen.
Tabelle 6 zeigt einige Ergebnisse, die aus diesem Materialgewonnen werden konnten .
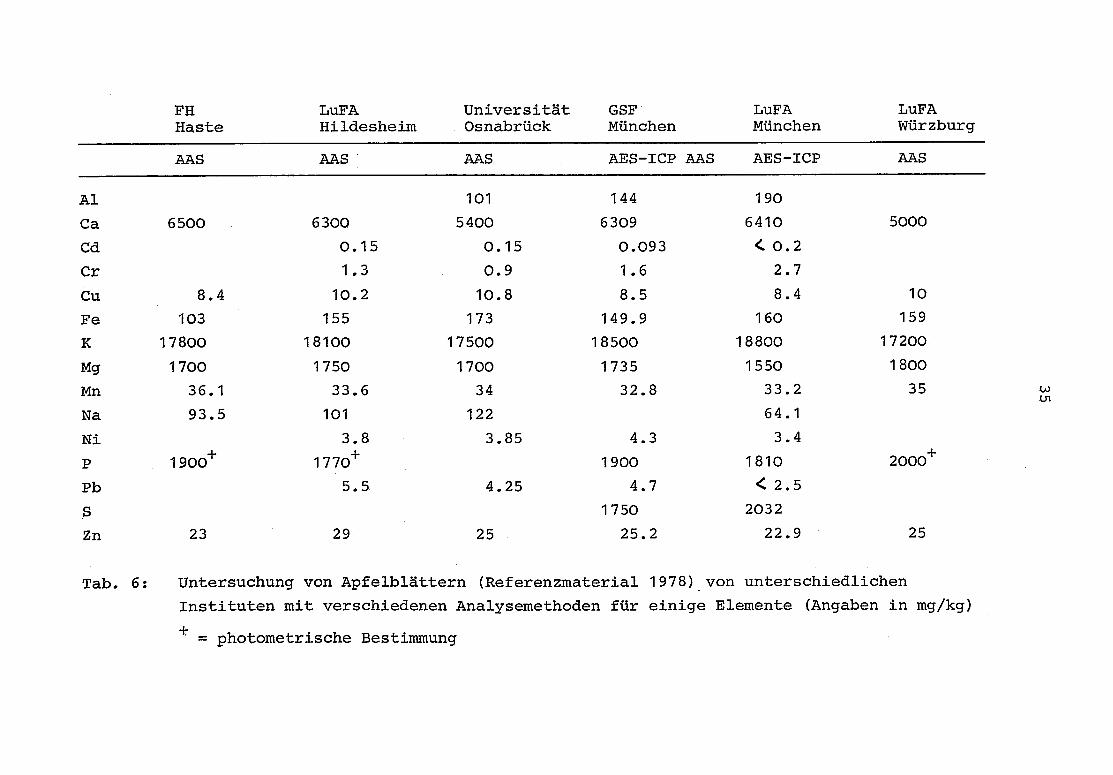
FHHaste
LuFAHildesheim
UniversitätOsnabrück
GSFMünchen
LuFAMünchen
LuFAWürzburg
AAS AAS AAS. AES-ICP AAS AES-ICP AAS
Al 101 144 190
Ca 6500 6300 5400 6309 6410 5000
Cd 0 .15 0 .15 0 .093 <0.2
Cr 1 .3 0 .9 1 .6 2 .7
Cu 8 .4 10 .2 10 .8 8 .5 8 .4 10
Fe 103 155 173 149 .9 160 159
K 17800 18100 17500 18500 18800 17200
Mg 1700 1750 1700 1735 1550 1800
Mn 36 .1 33 .6 34 32 .8 33 .2 35
Na 93 .5 101 122 64 .1
Ni 3 .8 3 .85 4 .3 3 .4
P 1900+ 1770+ 1900 1810 2000+
Pb 5 .5 4 .25 4 .7 < 2 .5
S 1750 2032
Zn 23 29 25 25 .2 22 .9 25
Tab .
6 : Untersuchung von Apfelblättern (Referenzmaterial 1978) von unterschiedlichen
Instituten mit verschiedenen Analysemethoden für einige Elemente (Angaben in mg/kg)
= photometrische Bestimmung

36
V. Analytische Bestimmungsverfahren
1 . Atomabsorptionsspektroskopie (AAS), (HEIN und SCHRADER,
1976 ; WELZ, 1983 ; INSTRUMENTATION LABORATORY, 1981)
Bei der Atomabsorptionsspektroskopie wird die zu untersuchende
(meist flüssige) Probe in atomaren Dampf überführt . Das von
einer Hohikathodenlampe emittierte Licht einer bestimmten
Wellenlänge wird in Abhängigkeit von der Anzahl der in der
Lösung vorhandenen Atome mehr oder weniger stark absorbiert.
Die Atomisierung kann entweder mit Hilfe einer Flamme (Bren-
nergemisch Luft/Acetylen bzw . Lachgas/Acetylen) oder flammen-
los mit Hilfe eines Graphitofens erfolgen . Nachgeschaltete
Monochromatoren dienen zur Selektierung der gewünschten
Wellenlänge aus dem vom Atomreservoir (Flamme oder Ofen)
kommenden, polychromatischen Lichts . Die Intensität der Pho-
tonenstrahlung wird innerhalb des Arbeitsbereichs des Empfän-
gers linear proportional in elektrischen Strom umgewandelt.
a . MARKERT, Universität Osnabrück, Deutschland
Ag, Al, Ba, Ca, Cd, Co, Cr, Cu, Fe, Li, Mg, Mn, Mo, Na, Ni,
Pb, Sr, Ti, Zn
Die Proben wurden, wie in Kapitel IV .3 .b2 beschrieben, auf-geschlossen und mit Hilfe eines AAS der Firma IL (357) mit
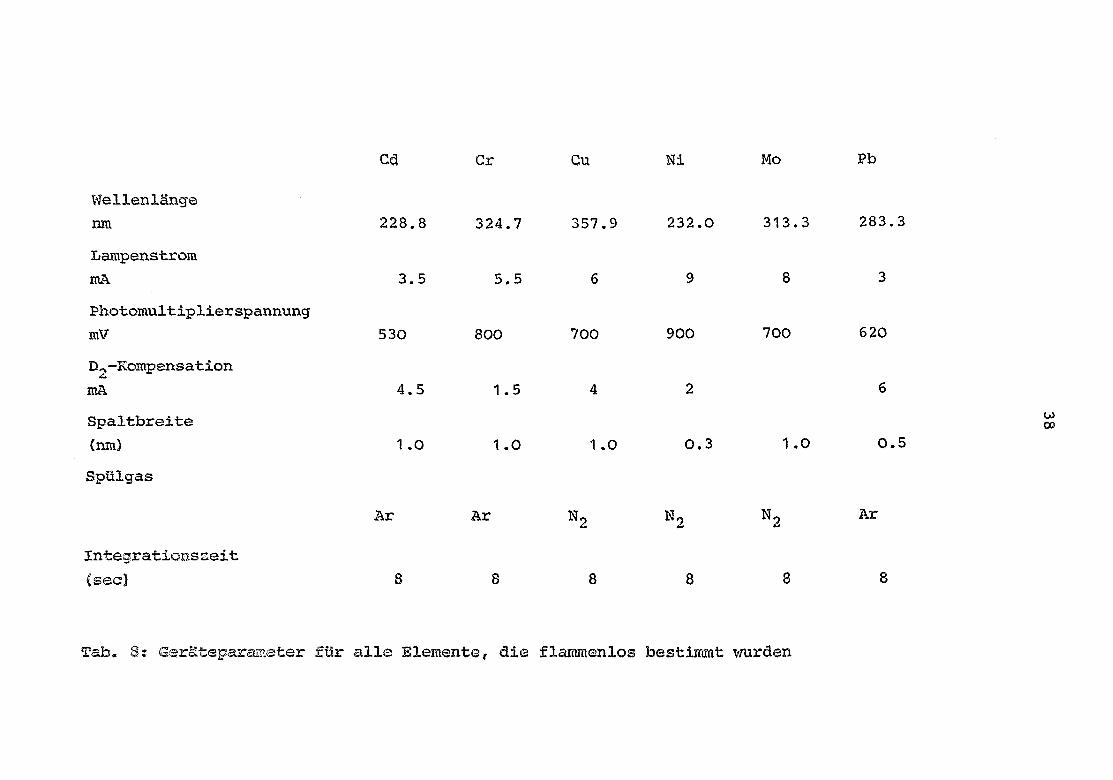
der Flamme und des Graphitofens (IL 655) obige Elemente be -stimmt . Die dabei benutzten Geräteparameter (Wellenlänge,
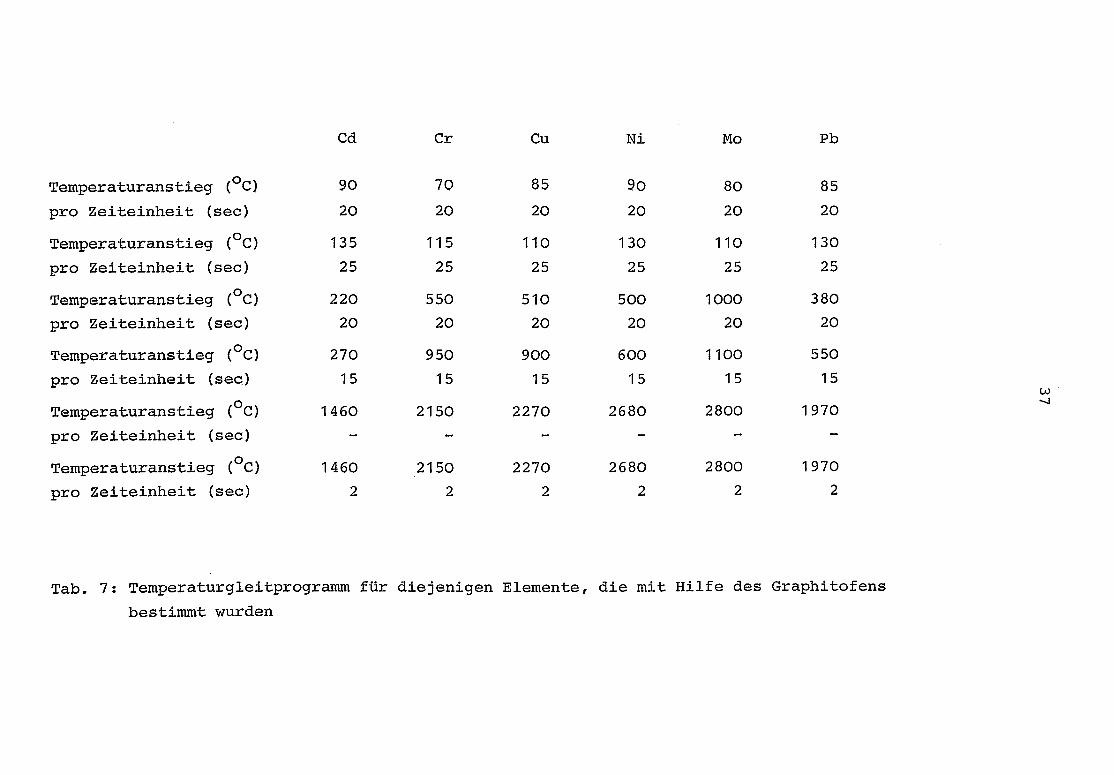
Lampenstrom, Photomultiplierspannung, Untergrundkompensation,Spaltbreite und Integrationszeit) sind in Tab . 8 und 9 ange-geben . Die verwandten Temperatürgleitprogramme (Temperatur-anstieg pro Zeiteinheit) sind in Tab . 7 dargestellt.
As, Hg, Se
Unter Verwendung eines vorhandenen Auto-Analyzer-Ii-Systems(STOEPPLER, 1980) wurde in Kombination mit verschiedenen Per-
kin-Elmer-Geräten ein weitgehend automatisiertes Hg-Bestim-
mungssystem von der KFA Jülich entwickelt, dessen Probendurch-
satz bei maximal 120 Proben pro Arbeitstag (10 h in automati-siertem Betrieb) liegt.
b . STOEPPLER, Jülich, Deutschland (STOEPPLER, 1980 ; MATTHES etal ., 1978)
Cd Cr Cu Ni Mo Pb
90 70 85 90 80 85
20 20 20 20 20 20
135 115 110 130 110 130
25 25 25 25 25 25
220 550 510 500 1000 380
20 20 20 20 20 20
270 950 900 600 1100 550
15 15 15 15 15 15
1460 2150 2270 2680 2800 1970
- - - - - -
1460 2150 2270 2680 2800 1970
2 2 2 2 2 2
Tab . 7 : Temperaturgleitprogramm für diejenigen Elemente, die mit Hilfe des Graphitofens
bestimmt wurden
Temperaturanstieg (°C)
pro Zeiteinheit (sec)
Temperaturanstieg (°C)
pro Zeiteinheit (sec)
Temperaturanstieg (°C)pro Zeiteinheit (sec)
Temperaturanstieg (°C)
pro Zeiteinheit (sec)
Temperaturanstieg (°C)
pro Zeiteinheit (sec)
Temperaturanstieg (°C)
pro Zeiteinheit (sec)
Cd
Cr
Cu
Ni
Mo
Pb
Wellenlängenm
228 .8
324 .7
357 .9
232 .0
313 .3
283 .3
LampenstrommA 3 .5
5 .5
6
9
8
3
PhotomultiplierspannungmV
530
800
700
900
700
620
D2-KompensationmA
4 .5
1 .5
4
2
6
Spaltbreite(nm)
1 .0
1 .0
1 .0
0 .3
1 .0
0 .5
Spülgas
Ar
Ar
N 2
Integrationszeit
(sec)
8
8
8
8
8
8
2 ArN N 2
Tab. 8 : Geräteparameter für alle Elemente, die flammenlos bestimmt wurden

Al
Al+ Ba
Ca
Co
Fe
Li
Mg
Mn
Na
Sr
Ti
Zn
Wellenlängenm
328 .1 309 .3 553 .5 422 .7 240 .7 248 .3 670 .8 285 .2 279 .5 589 .0 460 .7 364 .3 214 .0
LampenstrommA
5
6
10
3
8
8
9
4
5
4
10
8
Photomultiplier-spannung mV
320 460 620 530 620 700 530
620 700 530 620 530 620
D 2-KompensationmA
2
2
2
Spaltbreite(nm)
1 .0
1 .0
0 .5
1 .0
0 .3
1 .0
0 .5
1 .0
0 .5 1 .0
0 .5
0 .3
1 .0
Integrationszeit(sec)
1
1
1
1
1
1
1
1
1
1
1
1
1
Tab . 9 : Geräteparameter für alle Elemente, die mit der Flamme bestimmt wurden.
+Lachgas-Acetylen

40
Durch die Zwischenschaltung eines Silberwolle-Absorptions -
röhrchens - das gebildete Silberamalgam wird rasch ausge-
heizt, was zu scharfen Hg-Peaks in der Quarz-Meßküvette
führt - ist es mit diesem System möglich, auch relativ
niedrige Hg-Konzentrationen zu erfassen . Bei sorgfältiger
Blindwertkontrolle können z .B . 30 ng Hg/g (30 ppb) noch mit
einer relativen Standardabweichung von 10 bestimmt werden,
was einer Hg-Konzentration von 1 ng/ml Probenlösung ent-
spricht (MATTHES et al ., 1978) . Das Vorhandensein verschie-
dener As-Verbindungen in terrestrischen und marinen Umwelt-
material erfordert zur Bestimmung des Gesamt-Arsengehaltes
einen sehr wirkungsvollen Aufschluß, da vor allem die in
den Proben vorhandenen höhermolekularen As-Verbindungen
chemisch sehr stabil sind, wenn anschließend das Hydrid-
verfahren (mit NaBH 4 ) angewandt werden soll (STOEPPLER, 1980).
Ein Naßaufschluß mit HNO 3/HC10 4/H2S04 mit Maximaltempera-
turen um 300 °C führt zu quantitativen As-Ausbeuten, wie
durch instrumentelle Aktivierungsanalyse nachgewiesen
werden konnte.
c . WILSON, Experiment, Georgia, USA
(WILSON, 1979;
WILSON, pers . Mitt .)Mo
Für die Pflanzenproben wurde ein Salpetersäure/Perchlorsäure-
Aufschluß gewählt.
Die Bodenproben wurden mit Hilfe einer Ammoniumoxalatlösung(2 .5 g Boden und 25 ml Lösung) innerhalb von einer Stunde
extrahiert, gefiltert und das Filtrat mit der AAS aufMolybdän untersucht.
Die Ammoniumoxalatlösung enthielt 12 .6 g Oxalsäuredihydratgelöst in einem Liter Wasser . Der extrem niedrige Molybdän-gehalt im österreichischen Kalkboden mit 0 .004 ppm muß alsangenähert betrachtet werden, da keine Erfahrungen fürMolybdängehalte unter 0 .01 ppm vorliegen . Die Messung wurdeausgeführt an einem AAS der Firma Perkin Eimer (403) mitder Graphitofenküvette HGA 2100 .

41
d . YUAN, Gainesville, Florida, USA
Al
a . Pflanzenproben und Torf
(YUAN und BRELAND,
YUAN, pers . Mitt .)
1969 ;
Die gesamte Probe wurde zerkleinert und ordentlich gemischt.
Jeweils zwei 1 g-Portionen der getrockneten Probe wurden in-
nerhalb von 3 Stunden bei 450 °C verascht . Nach Erkalten
wurde die Asche mit einigen Tropfen Salpetersäure aufgenommen
und unter dem Abzug getrocknet . Die Asche wurde nochmals für
10 Minuten auf 400 °C erhitzt und anschließend in 25 ml 0 .1 M
Salzsäure aufgenommen . Anschließend wurde auf 50 ml Endvo-
lumen aufgefüllt und die Probe der Determination mit Hilfe
der AAS unterzogen.
b . Mineralboden und Torf
Jeweils zwei 5 g Mineralbodenproben und 1 .25 g Torfbodenpro-
ben wurden mit 25 m1 1 M Ammoniumacetatlösung für 30 min.
bei einem konstanten pH-Wert von 4 .8 geschüttelt . Die Proben
wurden anschließend zentrifugiert und die Extrakte mit Hilfe
der AAS auf Aluminium untersucht . Die Messungen wurden an
einem AAS der Firma Perkin Elmer (303) in der Lachgas/Ace-
tylen-Flamme durchgeführt.
e. PAWLUK und DUDAS, Edmonton, Alberta, .Canada (PAWLUK, 1967;
DUDAS und PAWLUK, 1976;
Al
PAWLUK, pers . Mitt .)
Die Pflanzenproben wurden trocken verascht.
Bevor die Bodenproben homogenisiert wurden, wurden sie mit
Hilfe eines 1 mm 0 Siebes gesiebt.
Anschließend wurden die Bodenproben einem Flußsäure-Salzsäure-
Aufschluß unterworfen . Die quantitative Erfassung wurde mit
einem AAS der Firma IL durchgeführt (IL 751).
f. DAVID, Canberra, Australien (DAVID, 1962 ; DAVID 1964, DAVID,
1969 ; DAVID, pers . Mitt .)Sr
2 g der pulverisierten Probe wurde in einem Muffelofen ver-
ascht, die Asche, in Salzsäure aufgenommen, an einer Anion-Aus-

42
tauschersäule extrahiert und der Strontiumgehalt mit einem
AAS der Firma Perkin Elmer bestimmt.
g . MACALALAD, Manila, Philippinen (RUBESKA et al ., 1977;
MACALALAD, pers . Mitt .)
Au, Pd
Gold und Palladium wurden nach Extraktion mit Dibutylsulfat
und Toluol bestimmt (Perkin Eimer 503 und HGA 74).
Auf eine exaktere Beschreibung des Analyseverfahrens wird an
dieser Stelle verzichtet, da die Proben scheinbar während der
Probenaufbereitung kontaminierten und somit keine zuverlässi -
gen Ergebnisse erzielt werden konnten.
2 . Atomemissionspektroskopie mit Hilfe eines induktiv gekoppel -
ten Plasmas (AES-ICP)
(SCHRAMEL et al ., 1982)
Bei der Anregung durch ein induktiv gekoppeltes Plasma wird
das Probenmaterial - meist nach geeigneter Zerstäubung derProbenlösung - in den Kern eines Plasmas injiziert . Dieses
Plasma ist einer chemischen Flamme sehr ähnlich . . Während der
Verweilzeit im Plasma wird das Aerosol getrocknet und die
Probenpartikelchen atomisiert und angeregt, so daß die ver-
schiedenen Atome durch ihre Strahlungsemission in einem
nachgeschalteten Spektrometer möglichst hoher spektraler
Auflösung quantitativ nachgewiesen werden können.
a, SCHRAMEL, München, Deutschland (SCHRAMEL und KLOSE, 1981)
Al, B, La, Cd, Cr, Cu, Fe, Mg, Mn, Ni, P, Pb, Ti, Zn
Alle Messungen wurden nach nasser Veraschung (Kap .IV .3 .b 2 ) aneinem ICP - Emissionsspektrometer JY 38 P (Sequenzspektro-meter der Firma Instruments S .A . (Jobin-Yvon)) durchgeführt .

43
b . HOFFMANN, München, Deutschland (HOFFMANN, 1980 ; HOFFMANN,pers . Mitt .)
Ag, Al, B, Ba, Be, Ca, Cd, Co, Cr, Cu, Fe, K, Mg, Mn, Mo, Na,
Ni, P, Pb, S, Si, Sn, Sr, V, Zn
Alle Messungen wurden nach nasser Veraschung (HF/HN0 3 -Gemisch)ebenfalls an einem ICP - Emissionsspektrometer JY 38 P der
Firma Instruments S .A . durchgeführt.
c . SUGIMAE, Osaka, Japan
(SUGIMAE, 1980 ; SUGIMAE,
pers . Mitt .)
Lanthaniden
Die Messungen wurden nach vorheriger Anreicherung der Lantha -
niden an einer Ionen-Austauschersäule vorgenommen . Alle
Messungen wurden zur überprüfung der Einzelergebnisse
gegen Referenzmaterial durchgeführt (NBS SRM 1571 und 1575,
USGS G-2 und BCR-1).
3 . Massenspektroskopie (MAS)
(BINGHAM und FLLIOT, 1974)
Die zu analysierende Substanz wird einer Ionenquelle zuge-
führt und dort ionisiert . Die Ionen werden in einem elek-
trischen Feld beschleunigt . Es entsteht ein gebündelter
Ionenstrahl, der durch Ablenkung im magnetischen Feld in
seine verschiedenen Massenanteile zerlegt wird . Die meist
auf einer Bildschirmplatte ankommenden Ionen werden auf
einer photographischen Platte registriert .

44
a . YLIRUOKANEN, Helsinki, Finnland (NIEMINEN und YLIRUOKANEN;YLIRUOKANEN, 1980 ; YLIRUO-
KANEN, pers. Mitt .)
Lanthaniden, Bi, Hf, Nb, Th, U, W, Y
YLIRUOKANEN benutzte die konventionelle Scanning-Methode (BING-
HAM und ELLIOT, 1971) . Das magnetische Feld wurde ständig
exponentiell abgebaut, so daß die einzelnen Isotope am Nach-
weisgitterschlitz einzeln vorbeiglitten und mit einem Elektro-
nen-Multiplier gemessen wurden. Die Probenhomogenität ist sehr
ausschlaggebend, weil jedes Isotop nur für eine kurze Zeit ge-
messen wurde . Die Outputs des Elektronenmultipliers und des
gesamten Ionenstrahl-Monitors wurden über einen konventionellen
doppelt logarithmierten Verstärker gesteuert, um mögliche
Fehler, die durch Fluktuation des gesamten Ionenstroms auf-treten können, zu verhindern.
Die Geräteparameter, die von YLIRUOKANEN bei diesen Unter-
suchungen verwendet wurden, sind in Tab . 10 zusammengefaßt.
Instrument
Spannung des Ionenstrahis:
Pulsfrequenz:
Pulslänge:
Beschleunigerspannung:
Bereich der magnetischen Scannung :
Doppeltfokussiertes Massen-spektrometer mit eingebautemVerstärker- und Autospark-Ein-heit (Associated ElectricalIndustries)
30 kV
1000 Herz
25 „us
19 .5 kV
220 - 20 mA (U-Li)Zeitkonstante der Scannung : 600 secDruck in der Ionenquelle : 10
-4Torr
Druck im Analysator : 3 x 10-9 Torr
Tab. 10 : Geräte und Arbeitsparameter des MS 702 (YLIRUOKANEN,1980)

45
b . URE, Aberdeen, Schottland (URE und BACON, 1978 ; URE,pers . Mitt .)
Ag, Bi, Ga, Ge, Hf, Nb, Sb, Sn, Th, U, Zr
Die Bodenproben wurden luftgetrocknet (30 °C), gesiebt (2 mm),
schließlich zerkleinert (150 b um und abschließend über Nachtbei 450 °C verascht.
Instrument:
Pulslänge:
Eintrittsspalt:
Beschleunigerspannung:
Druck in der Ionenquelle:
Druck im Analysator :
Doppelt fokussiertes Massen-spektrometer mit elektro-nischem Nachweissystem
25 aus
0 .051 mm
21 kV
10-7 Torr
10-6 Torr
Tab . 11 : Arbeitsparameter des von URE verwandten Massen-
spektrometers
4 . Neutronenaktivierungsanalyse (NAA) (SCHNIER und SCHNUG,
1981 ; KRUGER, 1971 ; DE SOETE, 1972)
Mit Hilfe von Neutronen wird die Probe in einem Kernreaktor
bestrahlt . Bei dieser Bestrahlung wandeln sich die stabilen
Kerne der zu bestimmenden Elemente in radioaktive Kerne des
gleichen Elements oder benachbarter Elemente um . Das durch
die Kernreaktion entstandene Radionuklid zerfällt mit be-
kannter Halbwertszeit und sendet dabei charakteristische
radioaktive Strahlen aus . Über diese beiden Eigenschaften4
werden die Indikator-Radionuklide identifiziert und unter
Berücksichtigung der dazugehörigen Reaktionsarten, die der
Aktivierung zugrundeliegen, den stabilen Ausgangsnukliden
der entsprechenden Elemente zugeordnet . Die quantitative
Bestimmung basiert auf der Proportionalität zwischen der
Elementmenge und der Aktivität des gebildeten Radionuklides .

46
a . EHMANN und TIAN, Lexington, Kentucky, USA
(NADKARNI et
al ., 1969 ; EHMANN und TIAN,
pers . Mitt .)
0, Si
5 - 10 mg der Proben, die bei 90 °C nachgetrocknet wurden,
werden in Quarzampullen gefüllt und während einer Woche
mit einem Neutronenfluß von 5 x 10 12 bis 1 x 1013 n/sec cm2
im Argonne National-Labor-Reaktor CP 5 bestrahlt . Nach der
Bestrahlung werden die Proben in frische Zählampullen über-
führt.
Die Aktivitäten wurden mit Hilfe eines Ge(Li)-Detektors
und eines Vielkanalanalysators bestimmt.
b. HEYDORN und DAMSGAARD, Roskilde, Dänemark (HEYDORN et al .,
1980 ; HEYDORN und DAMSGAARD,
pers . Mitt .)
V
Die Proben wurden am dänischen Reaktor DR 2 mit einem Neutro-
nenfluß von 7 x 1012 n/sec cm2 innerhalb einer Stunde be-
strahlt.
250 - 300 mg an Probenmaterial werden in einem Porzellan-
tiegel innerhalb von 2 h getrocknet . Die Temperatur wird
zunächst auf 200 °C erhöht und 15 h gehalten und anschließendauf 475 °C erhöht und nochmals 15 h gehalten . Eine Total-veraschung wurde durch 3 h Erhitzen auf 700 °C erzielt.
Nach der Lösung der Asche in Salpetersäure und anschließender
Neutralisation mit Ammoniumhydroxid wird der pH-Wert mit
Kalium-Hydrogen-Phtalat auf 4 .5 eingestellt . Vanadium wirdmit 2 x 10 ml 1
8-Hydroxyquinaldin in Chloroform extra-hiert. Die Lösung wird zur Trockne eingedampft und der ver-
bleibende Rückstand in 0 .5 m1 Pyridin gelöst .

47
c . OHNO, Chiba, Japan
(OHNO und YATAZAWA, 1970 ; ICHIKAWAund OHNO, 1974 ; OHNO, pers . Mitt .)
Co, Cs
Die getrockneten Proben wurden bei 450 0C in einem Muffel-ofen verascht . Die veraschte Probe wurde in Polyethylenbeutelnverpackt und hitzeversiegelt.
Die Bestrahlungszeit dauerte 5 h im Triga Mark II Reaktor
am Muzashi Institut für Technologie mit einem durchschnitt-
lichen Neutronenfluß von 4 x 10 12 n/sec cm2 .
Die bestrahlten Proben wurden in einen Schmelztiegel über-
führt und mit 100 /ug Co und Cs als Träger versetzt . Darauf-hin wurden 1 g Natriumhydroxid und Natriumperoxid hinzuge -fügt und das Gemisch 5 h bei 450 °C in einem Muffelofen ge-
schmolzen. Der entstandene Schmelzkuchen wurde in 1N Salz-
säure gelöst und gefiltert . Verdünntes Ammoniakwasser wird
dem Filtrat solange hinzugefügt, bis ein Hydroxidnieder-
schlag ausfällt . Der Niederschlag wird filtriert und das
Filtrat auf einem Sandbad bis zur Trockne eingedampft.
Abtrennung von Co : Der Rückstand wird mit verdünnter Salz-
säure aufgenommen und der pH-Wert mit verdünntem Ammoniak-wasser auf pH 5 eingestellt . Anschließend wird 1 % Natrium-
diethyldithiocarbamatlösung hinzugefügt, bis sich ein voll-
ständiger Niederschlag gebildet hat . Anschließend wird die
organische Phase mit 2 x 5 m1 10 % Kaliumcyanidlösung ver-
setzt . Die verbleibende organische Phase wird auf einem Sand-
bad bis zur Trockne eingedampft . Der Rückstand wird in 5 ml
Salpetersäure aufgelöst und in eine Zählampulle zur y-Spek-
trometrie auf 6OCo überführt.
Abtrennung von Cs : Nach der Extraktion von Cobalt wird die
verbleibende wäßrige Phase auf einem Sandbad eingedampft
und der Rückstand in 30 ml 1 N Salzsäure gelöst . Anschließend
wird 134Cs an einem Kupfer-Eisen-Cyanid-Anionaustauscherharz
extrahiert und das Harz anschließend in eine Zählampulle
zur y-Spektrometrie auf134
Cs überführt .

48
d . DE BRUIN, Delft, Niederlande
(TJIOE et al ., 1973;
TJIOE et al ., 1977;
DE BRUIN, pers . Mitt .)
A1, As, Au, Ba, Br, Ca, Cd, Ce, Cl, Co, Cr, Cs, Cu, Eu,
F, Fe, Ge, Hf, Hg, K, La, Lu, Mg, Mn, Mo, Na, Nd, Ni, Rb,
Ru, Sb, Sc, Sn, Se, Sr, Ta, Th, Ti, U, V, W, Zn
Die Probenbestrahlungen wurden am Interuniversitair-Reaktor-
Zentrum in Deift mit einem Neutronenfluß von 10 13 x cm-2 x
sec-1 durchgeführt . Die Proben befanden sich während der
Bestrahlung in Quarzbehältern von 3 ml Inhalt, die zuvor
mit dampfender HNO3 gereinigt wurden.
Nach der Bestrahlung wurden die Quarzbehälter an der Außen-
seite gereinigt, um eine mögliche Kontamination zu vermeiden.
Anschließend wurden sie im flüssigen Stickstoff gekühlt, um
einem möglichen Innendruck, der bei der Bestrahlung ent-
standen sein könnte, entgegenzuwirken (TJIOE et al ., 1973;
TJIOE et al ., 1977).
5 . Quantitative Elementaranalyse (BAYER, 1978 ; KRONER, pers.
Mitt .)
a . Kohlenstoff und Wasserstoff
Die C-H-Analyse entspricht dem qualitativen Nachweis beider
Elemente, und zwar verbrennt man eine genau abgewogene Menge
der zu analysierenden Substanz in einer geeigneten Apparatur
im Sauerstoffstrom durch eine glühende Kupfer (II) -oxid-Schjcht,
wobei der Kohlenstoff in Kohlendioxid und der Wasserstoffin Wasser übergeht .

49
Das Wasser wird in einem mit Magnesiumperchlorat gefüllten
Röhrchen absorbiert und das Kohlendioxid in einem mit Na-
tronasbest (NAOH + Asbest) gefüllten Absorptionsgefäß
chemisch gebunden . Aus der Gewichtsdifferenz der beiden
Absorptionsgefäße vor und nach der Verbrennung kann der
Prozentgehalt an Kohlenstoff und Wasserstoff berechnetwerden.
Da viele organische Substanzen neben Kohlenstoff und Wasser-
stoff noch Stickstoff, Schwefel und Halogene enthalten, die
bei der Verbrennung Stickoxide, Schwefeloxide und freie
Halogene bilden, muß das Rohr mit einer "Universalfüllung"
versehen werden . Hinter dem Kupfer(II)-oxid, das durch einen
Langbrenner auf einer Temperatur von 750° - 800°C gehalten
wird, folgt eine Schicht Silberwolle, die die störend wir-
kenden Halogene und Schwefeloxide bindet . Die Stickoxidewerden an Blei(IV)-oxid zerlegt, das, auf Bimstein oder
Asbest aufgezogen, konstant auf 180 °C gehalten wird.
b . Stickstoff
Bei der quantitativen Bestimmung des Stickstoffgehalts einer
Verbindung verbrennt man die abgewogene Substanzmenge in
einem Verbrennungsrohr durch glühendes Kupfer(II)-oxid in
einem luftfreien Kohlendioxidstrom, wobei neben Kohlendioxid
und Wasser elementarer Stickstoff entsteht . Die bei der
Analyse zuweilen auftretenden Stickoxide werden durch eine
der Kupfer(II)-oxid-Füllung folgende glühende Kupferschicht
zu Stickstoff reduziert . Der entweichende Stickstoff wird
in einer graduierten Meßbürette (Azotometer) über starker
Kalilauge, die zugleich Kohlendioxid und Wasser absorbiert,
volumetrisch bestimmt .

50
6 . Partikel induzierte Röntgenfluoreszenzspektroskopie (PIXE)
(GARTEN, 1984)
Neben der herkömmlichen Röntgenfluoreszenzanalyse und der
Röntgenemissionsspektroskopie mit Anregung durch Elektronen
wird seit 1970 auch die ioneninduzierte Röntgenemission
(Proton Induced X-Ray Emission (PIXE)) für die Elementana -
lytik genutzt . Die Entwicklung der hochauflösenden Si (Li)-
Halbleiterdetektoren und die zunehmende Verfügbarkeit kleiner
Beschleuniger und Beschleunigerspannungen im MV-Bereich
waren wichtige technische Voraussetzungen . Von diesen Be-schleunigern existieren etwa 200 . In etwa 10 Beschleuniger-
laboratorien der Bundesrepublik Deutschland werden derzeit
Untersuchungsprogramme mit PIXE durchgeführt . Das physika-
lische Prinzip von PIXE ist die Emission charakteristischerRöntgenstrahlung der Probenatome als Folge von Ionisation
innerer Schalen durch Ionenbeschuß . PIXE ermöglicht eineverbrauchs- und häufig zerstörungsfreie Analyse geringerElementgehalte im unteren ppm-Bereich.
Im Rahmen dieser Arbeit wurden die PIXE nur testmäßig im Be-
schleunigerreaktor in Gainesville Florida mit wenigen Probengefahren . Letztendlich konnten in ökologischen Matrizes auchnur die herkömmlichen Elemente bestimmt werden . Für weiter-reichende Untersuchungen, über den Rahmen dieser Arbeit hinaus,
kann auf dieses Bestimmungsverfahren verzichtet werden .

.51
Vl . Vergleich der unterschiedlichen Meßmethoden
1. Nachweisgrenze:
Tab . 12 gibt für die einzelnen Elemente des periodischen
Systems die mit den verwandten Methoden (AAS, AES-ICP, AES,
NAA, MAS, PIXE, EA) erzielten Nachweisgrenzen wieder . Folgende
Elemente konnten nicht quantitativ erfaßt werden:
Die Actiniden (mit Ausnahme von U und Th), die Edelgase, die
Platinmetalle und die Elemente Be, F, I, In, Li, Pm, Ru, Re,
Ta, Tc und T1.
In weiterführenden Arbeiten (LIETH und MARKERT, 1986) sollen
diese Elemente mit Hilfe von verfeinerten Techniken nachge-
wiesen werden.
2. Vergleich der einzelnen Meßdaten untereinander:
Alle Einzelmeßdaten der Elemente sind im Anhang (2) zusammen-
gefaßt . Die erste Spalte gibt dabei die Probennummer an, die
im einzelnen folgender Probenart zukommt:
33 = Torfboden aus Schweden
62 = Torfboden aus Deutschland
64 = Torfboden aus Österreich
34 = Mineralboden aus Schweden
63 = Mineralboden aus Deutschland
69 = Mineralboden aus Österreich
35 = Vaccinium vitis idaea auf Torfboden aus Schweden
31 = Vaccinium vitis idaea auf Mineralboden in Schweden
60 = Vaccinium vitis idaea auf Torfboden in Deutschland
39 = Vaccinium vitis idaea auf Mineralboden in Deutschland
61 = Pinus sylvestris auf Torfboden in Deutschland
43 = Pinus sylvestris auf Mineralboden in Deutschland
65 = Pinus mugo auf Torfboden in Österreich
66 = Pinus mugo auf Mineralboden in Österreich
47 = Molinia coerulea auf Torfboden in Deutschland
28 = Molinia coerulea auf Torfboden in Österreich
29 = Molinia coerulea auf Laag in Österreich

52
Tab . 12 : Methodenliste für die Multielementanalyse in ökolo-gischen Matrizes (Pflanze und Boden).
X = Das Element wurde mit dieser Methode quantitativerfaßt.
= Nachweisgrenze für die angewandte Analysenmethode.
- = Das Element konnte bisher mit den angewandten
Analysenmethoden nicht quantitativ erfaßt werden.
AAS ICP-AES AES NAA MAS PIXE EA
Ag <2 X X <20Al X X < 200 XAr < 30As X X <
5Au X <
5B X
Ba X <
10 < 20Be < 0 .5Bi X <
5Br X XC X
Ca X X X XCd X <0 .2 <
0 .5 < 20Ce X <
0 .15 < 0 .1 < 10Cl X XCo < 0 .5 X <
2 .5Cr X X <
0 . 5 XCs X < 20Cu X X X X
Dy X < 0 .3 < 20Er X <0 .3 <10Eu X <
0 .01 <0 .1 < 25F <
500Fe X X X XGa < 0 .02 XGd X < 0 .3Ge < 1 < 10000 XH
Hf~ <
5X
<
0 .05 X
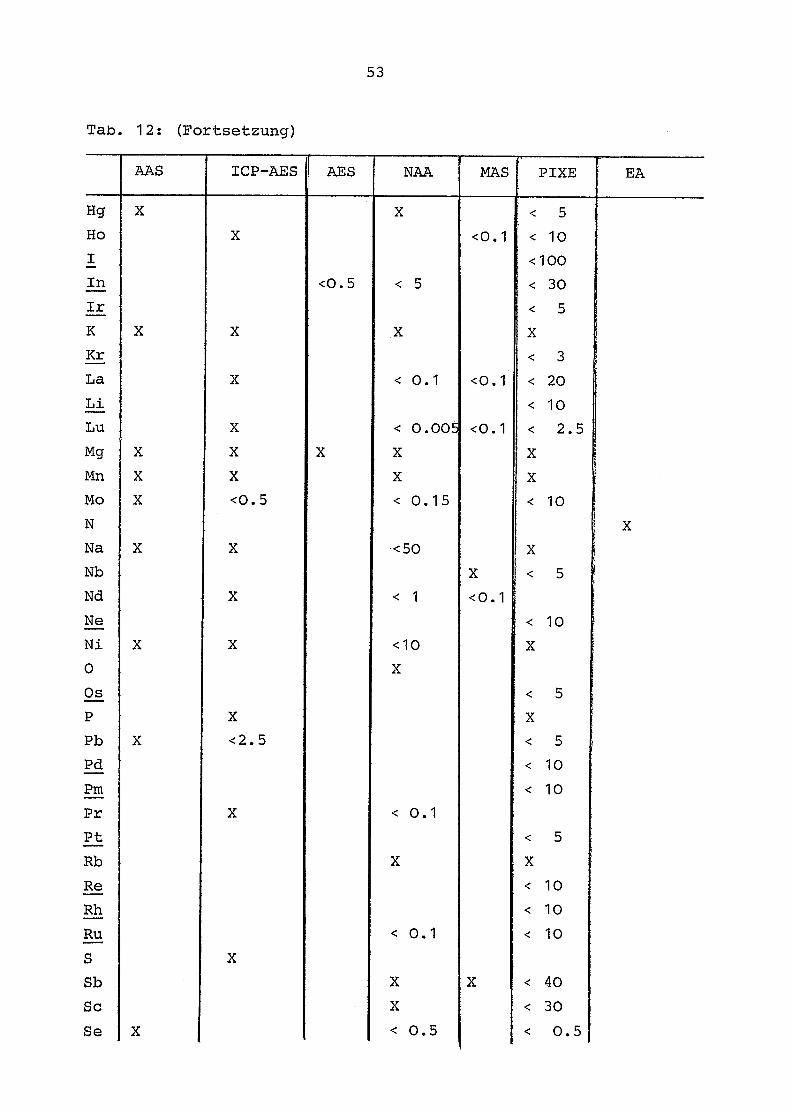
53
Tab . 12 : (Fortsetzung)
AAS ICP-AES AES NAA MAS PIXE EA
Hg X X <
5
Ho X <0 .1 <
10
I 1
<100
In <0 .5 < 5 < 30
Ir 5
K X X X X
Kr <
3
La X <
0 .1 <0 .1 < 20
Li <
10
Lu X < 0 .005 <0 .1 <
2 .5
Mg X X X X X
Mn X X X X
Mo X <0 .5 < 0 .15 <
10
N X
Na X X •<50 X
Nb X <
5
Nd
Ne
X <
1 <0 .1
<
10
Ni
1X X <10 X
0 X
Os <
5
P X X
Pb X <2 .5 <
5
,
Pd <
10
Pm <
10
Pr X <
0 .1
Pt <
5
Rb X X
Re <
10
Rh <
10
Ru <
0 .1 <
10
S X
Sb X X <
40
Sc X <
30
Se X < 0 .5 <
0 .5
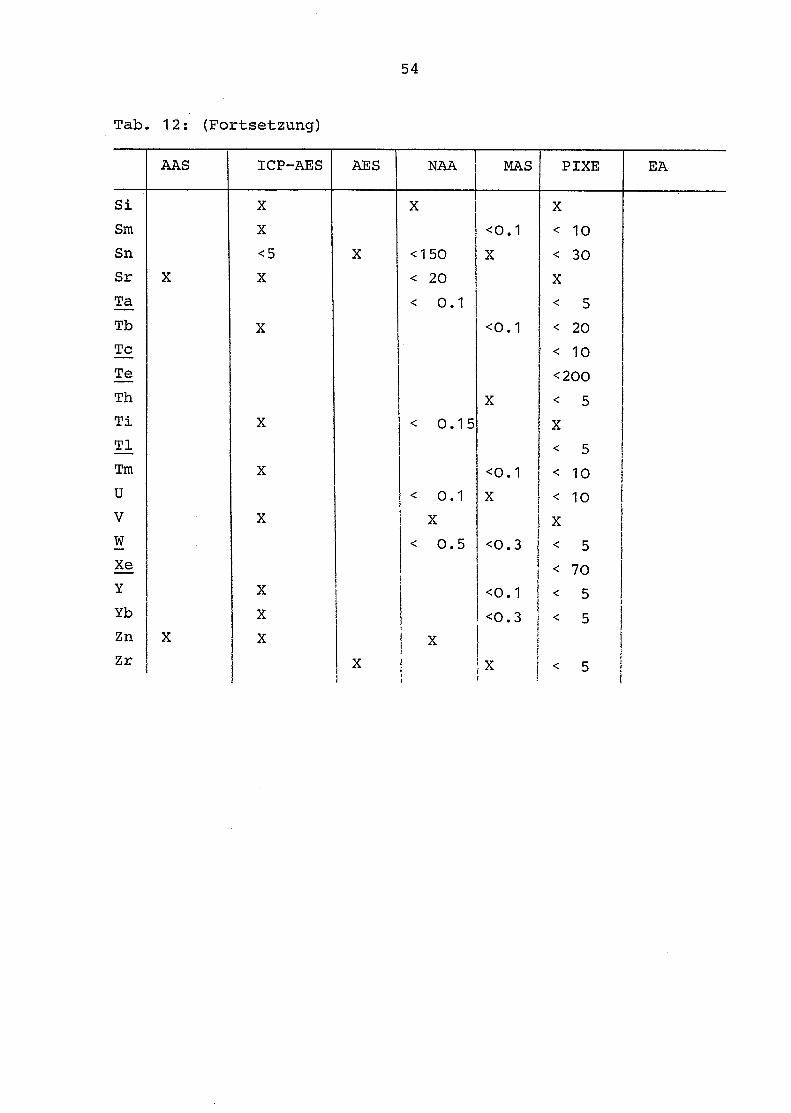
54
Tab . 12 : (Fortsetzung)
AAS ICP-AES AES NAA
1 MAS PIXE EA
Si X X X
Sm X <0 .1 <
10
Sn <5 X <150 X < 30
Sr X X < 20 X
Ta <
0 .1 <
5
Tb X <0 .1 < 20
Tc <10
Te <200
Th X <
5
Ti X <
0 .15 XTl 1 <
5Tm X <0 .1 <
10U <
0 .1 X <
10
V x X ! X
W , <
0 .5 <0 .3 <
5Xe < 70
Y X <0 .1
{
<
5Yb X <0 .3
<
5Zn X XZr
1X : X
j
<
5

55
67 = Frangula alnus auf Torfboden in Österreich
68 = Frangula alnus auf Laag in Österreich
7 = Alnus glutinosa auf Torfboden in Österreich
11 = Alnus glutinosa auf Laag in Österreich
Die Analysendaten der einzelnen Analytiker wurden zunächst
nacheinander aufgetragen, anschließend der Mittelwert x
gebildet und dann die Standardabweichung berechnet . DerVariationskoeffizient ist in % angegeben.
Bei manchen Elementen lag der Variationskoeffizient ineinigen Proben über 100 % . Diese Elemente sind im folgenden
aufgeführt, in Klammern dahinter die Anzahl der Proben, in
denen dieser hohe Variationskoeffizient gefunden wurde:
Ag (1), Al (1), Bi (1), Ca (1), Ce (1), Er (2), Mo (1), P (1)
Pb (1), Sc (1), Th (1), Yb (1).
Betont werden muß, daß sich die zunächst sehr hoch erscheinen-
den Variationskoeffizienten dadurch ergeben, daß das be-
treffende Element von unterschiedlichen Analytikern, in unter-
schiedlichen Instituten mit unterschiedlichen Analysenver-
fahren untersucht wurde .

56
VII . Gesamtbetrachtung mit Hilfe von Elementkonzentrations-
katastern
Sämtliche Daten, die im Rahmen dieser Arbeit erschienen sind,
werden im folgenden in sogenannten Elementkonzentrationska -
tastern zusammengefaßt . Dazu wird jedes Element aufgrund
seines durchschnittlichen Gehaltes in den unterschiedlichen
Proben einer bestimmten Elementkonzentrationsklasse zuge-
ordnet . Erscheint beispielsweise das Element in Probe X für
das Element Eisen mit 457 ppm, so wird das Elementsymbol für
Eisen in die Konzentrationsklasse zwischen 10 2 - 103 ppm
eingeordnet.
Im Rahmen dieser Arbeit sollen in Abweichung von früheren Ver-
öffentlichungen (MARKERT und LIETH, 1985 ; LIETH und MARKERT,
1985) folgende Änderungen vorgenommen werden:
1. Die einzelnen Konzentrationsklassen der Elemente stehen
zwischen den eigentlichen ppm-Reihen, da der Bereich, in
dem sich das Element befindet, in der Realität auchzwischen 102 - 103 ppm liegt.
2. Elemente, die nicht in allen Proben nachgewiesen werdenkonnten, erscheinen mit der. entsprechenden Nachweisgrenzerechts neben dem Elementkonzentrationskataster.
3. Elemente, die aufgrund analytischer Unzulänglichkeiten in
keiner Probe nachgewiesen werden konnten, werden nicht
mehr in die Elementkonzentrationskataster aufgenommen.
Darunter fallen, wie aus Tabelle 12 zu ersehen ist, die
Edelgase, die Platinmetalle, die Actiniden (mit Ausnahme
von Th und U), Be, F, Fr, 1, In, Li, Pm, Ra, Re, Ta, ?c, Teund Ti.
Die Abänderung der Darstellung der Elementkonzentrationska-taster, insbesondere durch die Punkte 2 und 3, ergab sich
daraus, daß ein oder mehrere Elemente, die nicht nachge -wiesen werden konnten, in eine bestimmte Konzentrationsklasse

57
gebracht wurden, die sich aufgrund ihrer Nachweisgrenze ergab.
Beispielsweise wurde Tantal stets mit der Nachweisgrenze von
0 .1 ppm analysiert und wurde somit dem Konzentrationsbereich
zwischen 10-1 und 100 zugeordnet, obwohl es theoretisch auch
dem Konzentrationsbereich 10 -2 bis 10-1 zugeordnet werden
konnte . Falls diese Zuordnung mit mehreren Elementen geschieht,
kann das äußere Bild der Elementkonzentrationskataster sehr
verzerrt werden und entspricht nicht mehr der Realität.
Diese Änderung der Elementkonzentrationskataster führt uns in
bezug auf das periodische System zu unvollständigen Klassen,
jedoch dürfte diese Lücke durch die Weiterentwicklung und
Intensivierung der analytischen Techniken bald geschlossen
werden . Die Änderung der Elementkonzentrationskataster war
für die Vergleichbarkeit der Kataster untereinander unab-
dingbar.
1 . Boden
Bei der Betrachtung des Elementkonzentrationskatasters des
schwedischen Hochmoorbodens fällt zunächst die gleichmäßige
Normalverteilungskurve mit Verteilungsmaximum zwischen 0 .01
ppm - 10 ppm auf . Es sind keine außergewöhnlichen Elementver-
schiebungen festzustellen, daß heißt, keine unerwarteten
Verteilungsmaxima treten auf . Scheinbar ist der schwedische
Hochmoorboden noch durch keine anthropogenen Einflüsse sei-
tens der Industrie, des Autoverkehrs oder privater Haushalte
beeinflußt . Aus diesem Grunde wird vorgeschlagen, den schwe-
dischen Hochmoorboden als Referenzstandard für unbelastete
Ökosysteme zu verwenden, um die Entwicklung dieses Moores
in bezug auf Elementeintrag im Lauf der nächsten Jahrzehnte
näher zu untersuchen (MARKERT und LIETH, 1986).
Vergleichen wir das Elementkonzentrationskataster des schwe-
dischen Hochmoorbodens mit dem des deutschen Hochmoorbodens,
so fällt auf, daß 1 . ein neues Verteilungsmaximum zwischen

58
103 und 10 4 ppm aufgetreten ist, und 2 ., daß sich das zweite
Verteilungsmaximum, das beim schwedischen Hochmoorboden zwi-
schen 0 .01 ppm - 10 ppm lag, um eine Zehnerpotenz weiter nach
rechts verschoben hat (0 .1 ppm - 100 ppm) . Gründe hierfür
dürften die im nordwestdeutschen Hochmoor wesentlich stär-
keren anthropogenen Beeinflußungen des Hochmoores sein.
Besonders erwähnt sei an dieser Stelle, wie schon bei der
Einzelelementbetrachtung, daß hoch toxische Elemente, wie
beispielsweise Cadmium, um 2 Zehnerpotenzen in den höheren
Bereich verschoben sind . Letztendlich werden dadurch Elemente
wie Stickstoff, die in Hochmooren häufig sowieso schon den
wachstumsbegrenzenden Faktor darstellen, weiter in den linken
Bereich des Elementkonzentrationskataster verschoben, werden
also im Gesamtgehalt zurückgedrängt . Dies dürften auch die
Gründe für das neue Verteilungsmaximum des deutschen Torfes
zwischen 1000 ppm - 10000 ppm sein.
Betrachten wir als drittes Verteilungssystem den öster-
reichischen Torfboden, so fällt auf, daß wir uns innerhalb
unseres Elementkonzentrationskatasters einer Form der Nor-
malverteilungskurve nähern, mit Hauptmaximum zwischen 0 .01ppm und 10 ppm, wie es uns schon vom schwedischen Hochmoorbekannt ist . Das zweite Verteilungsmaximum zwischen 1000 ppm
und 10000 ppm, wie es im deutschen Hochmoor aufgetreten ist,fällt weg . Aufgrund dieser Beobachtungen darf davon ausge-
gangen werden, daß sich das österreichische Moor in einer
Umgebung befindet, in der noch wenig anthropogene Einflüsse
zu finden sind.
Die erhöhten Werte im deutschen Torf reflektieren somit mög-
licherweise die Geschlossenheit der dichten Population und
würden einen Eintrag von Elementen aus Nah- und Ferndeposi-tion nahelegen.
Es bliebe noch die Möglichkeit zu diskutieren, ob der Eintragder Elemente aus den umgebenden Mineralböden stammt, deren
Elementkonzentrationskataster im folgenden diskutiert werden .

59
Allerdings fällt bei Betrachtung der Elementkonzentrations-
kataster der Mineralböden sofort auf, daß diese eine voll-
kommen andere Zusammensetzung aufweisen als die Hochmoorböden
und somit ein Eintrag von diesen Systemen ausgeschlossen
werden kann.
Der schwedische Mineralboden . dürfte mit einem Verteilungs-
maximum zwischen 1 ppm - 100 ppm im Vergleich zu den anderen
beiden Mineralböden bei weitem der nährstoffreichste sein.
Er besteht in der Hauptsache aus Siliciumdioxid, das einen
Großteil an Aluminium- und Eisenoxiden mit sich führt . Auchder Anteil an phosphatischen Mineralien dürfte einen Groß-teil ausmachen . Aufgrund des nur 60 cm tief liegenden Unter-grundgesteins dürfte eine Podsolisierung ständig zurückge-
drängt werden, so daß nicht das Verteilungsmuster erreicht
werden konnte, wie wir es beim deutschen Podsolboden finden.
Das Elementkonzentrationskataster des deutschen Podsolbodens
ist deutlich geprägt durch den extrem hohen Si0 2 -Anteil unddurch die Verarmung von vielen Elementen durch Auswaschung,was dazu führt, daß das Hauptmaximum um eine Zehnerpotenz
weiter nach rechts in den niedrigeren ppm-Bereich gedrängtwird.
Der österreichische Kalkboden weist sich mit seinem Element-
konzentrationskataster als sehr nährstoffarm aus . Erscheintdas erste Verteilungsmaximum zwischen 100000 - 1000000 ppm,
hervorgerufen durch den hohen Magnesiumcarbonat und Calcium-
carbonatanteil, werden sämtliche anderen Elemente auf ein
tieferes Konzentrationsniveau zurückgedrängt, unter denen
sich auch bekanntermaßen essentielle Elemente wie etwa
Molybdän befinden .

60
2 . Pflanze
Betrachten wir die Elementkonzentrationskataster von Vaccinium
vitis idaea in Schweden und Deutschland jeweils auf Torf und
Mineralboden, so stellen wir zunächst mehr Gemeinsamkeiten als
Unterschiede fest . Ganz im Gegensatz zu den untersuchten Böden
scheinen sich die Elemente in den einzelnen Konzentrations-
klassen immer wiederzufinden.
In der Tat scheint sich im Laufe der Evolution ein spezifischer
Selektions- bzw . Akkumulationsmechanismus innerhalb der unter-
schiedlichen Pflanzenarten ausgebildet zu haben, der die Pflanze
befähigt, diejenigen Stoffe aufzunehmen, die für ihr existenti-
elles Dasein lebensnotwendig sind . Dieser Mechanismus der selek-
tiven Ionenaufnahme soll an nur zwei Beispielen der Untersuchungs-
reihen verdeutlicht werden:
Zunächst sei auf den Mangangehalt von Vaccinium vitis idaea
verwiesen . Wie seit langem bekannt ist, akkumulieren alle
Ericaceen Mangan (zwischen etwa 1000 ppm bis 2000 ppm) . Diesesscheinbar für Ericaceen essentielle Element liegt in den
Hochmoorböden in äußerst geringer Menge vor . Trotz des sehr
unterschiedlichen Mangangehaltes aller untersuchten Bodentypen
ist Vaccinium vitis idaea in der Lage, sich das notwendige
Mangan in der hohen ppm-Menge einzuverleiben . Die Pflanzensind scheinbar durch eine genetische Vorgabe dazu befähigt,
die für sie lebensnotwendigen Stoffe zu akkumulieren.
Andererseits ist Vaccinium vitis idaea auch in der Lage, einen
Stoff, wie beispielsweise das für viele Pflanzen toxische
Aluminium, zu verschmähen . Aluminium wird der Pflanze imschwedischen Mineralboden in sehr hohen Konzentrationen
geboten, trotzdem findet sich Aluminium in Vaccinium vitis
idaea in allen vier untersuchten Ökosystemen nur mit Gehal-ten zwischen 100 ppm - 1000 ppm wieder .

61
Innerhalb der schwedischen Preiselbeeren erscheint unabhängig
vom Standort ein Verteilungsmaximum zwischen 1000 ppm -
10000 ppm, das in der Regel die Makroelemente beinhaltet.Dieses erste Verteilungsmaximum findet sich bei den deut-
schen Vaccinium vitis idaeen nicht mehr wieder . Scheinbar
ist bei Vaccinium vitis idaea weniger das Nährsubstrat (Hoch-
moorboden oder Mineralboden), sondern vielmehr seine geo-
graphische Verteilung (Deutschland oder Schweden) für die
Elementverteilung und somit seine anthropogen beeinflußte
Umwelt ausschlaggebend . Die Elementkonzentrationskatastervon Vaccinium vitis idaea ähneln sich bei gleicher geogra-
phischer Lage mehr als sich bei den großen Unterschieden
der Nährsubstrate vermuten ließ.
Betrachten wir das Verteilungsmuster der unterschiedlichen
Pinusarten, so stellen wir fest, daß ein zunächst für
Pflanzen typisches Verteilungsmuster mit 1 . und 2 . Maximum
auftritt . Auffällig ist allerdings, daß sich im Verteilungs-
maximum Nr . 1 nicht nur essentielle Makroelemente befinden,
sondern auch toxische Elemente wie beispielsweise Aluminium.
Wie schon bei Vaccinium vitis idaea ähneln sich die Pflanzen
gleicher geographischer Herkunft mehr, als die sehr unter-
schiedlichen Nährsubstrate hätten vermuten lassen . Bei den
österreichischen Pinusarten erscheint das erste Verteilungs-
maximum zwischen 100 ppm - 1000 ppm mit steilem Abfall zwi-
schen 10 ppm - 100 ppm . Bei den deutschen Pinusarten ist
das Verteilungsmaximum um eine Zehnerpotenz nach rechts
verschoben.
Werfen wir einen Blick auf die untersuchten Laubbäume (Frangula
alnus und Alnus glutinosa), die jeweils auf dem österreichi-
schen Hochmoor und der Laagzone gezogen wurden, so stellen
wir fest, daß die beiden Konzentrationskataster für die je-
weiligen Pflanzenarten fast identisch sind . Scheinbar konnte
der leicht versetzte Entnahmeort (Hochmoor bzw . Laag) nicht
zu einer Änderung des Elementkonzentrationskatasters führen .

62
Bei leichter Verschiebung des einen oder anderen Elements
dürfte es sich um die allgmein vorhandene biologische Varianz
handeln oder durchaus auch einmal die Schwäche von Element-
konzentrationskatastern aufzeigen . Vergleichen wir die Vertei-
lungsmuster der untersuchten Laubbäume mit denen der unter-
suchten Nadelbäume (Pinus mugo bzw. sylvestris), so beob-achtet man insbesondere bei Alnus glutinosa, aber auch bei
Frangula alnus, eine Verschiebung bzw . Verbreiterung desersten Verteilungsmusters zwischen 1 ppm bis 10000 ppm.
Scheinbar brauchen Laubbäume mehr essentielle Elemente als
bisher angenommen wurde.
Bei der Betrachtung der Elementkonzentrationskataster von
Molinia coerulea fällt auf, daß bei allen drei Standorten
das erste Verteilungsmaximum zwischen 1000 ppm - 10000 ppmliegt . Innerhalb des essentiellen Bereiches fällt wie beiallen Gräsern das Element Silicium auf . Trotz der unter-schiedlichen Standorte und einzelner Elementverschiebungenweisen doch alle drei Elementkonzentrationskataster sehrgroße Ähnlichkeit auf . Scheinbar zeigt die Laagzone, wie
schon bei den Laubbäumen andiskutiert, keine großen Unter-schiede in ihrer elementaren Zusammensetzung im Vergleichzum untersuchten Hochmoorboden .

63
VIII . Literatur
AMBERGER, A ., 1979:Pflanzenernährung, 237 S ., Ulmer, Stuttgart.
BAUMEISTER, W . und ERNST, W ., 1978:Mineralstoffe und Pflanzenwachstum, 416 S ., Gustav FischerVerlag, Stuttgart, New York.
BERNAS, B ., 1968:A novel method for decomposition and comprehensive analysisof silicates by atomic absorption spectroscopy, Anal . Chem .,40, 1682-1684.
BEYER, H ., 1978:Lehrbuch der organischen Chemie, S . Hirzel-Verlag,Stuttgart, 860 S.
BINGHAM, R .A . und ELLIOT, R .M ., 1971:Accuracy of analysis by electrical detection in sparksource mass spectrometry, Anal . Chem ., 43, 43-45.
BLOOM, H . und BARNETT, P .R ., 1955:A new ceramic buckboard and muller, Anal . Chem ., 27,1037-1038.
BOWEN, H .J .M ., 1979:Environmental chemistry of the elements, 333 S ., AcademicPress, London, New York.
BUTTGEREIT, G ., 1973:Atomabsorptions-, Flammenemissions- und Atomfluoreszens-spektroskopie, in : Methodicum Chimicum, Band 1, Analytik,Teil 2, Spurenanalyse, Biologische Methoden, Substanz-klassen, Automatisierung, Friedhelm Korte (Herausgeber),S . 736-752, Georg Thieme Verlag, Stuttgart und AcademicPress, New York, London.
CAROLI, S ., ALIMONTI, A ., VIOLANTE, N ., 1980:Determination of gallium in biological samples by meansof the hollow cathode discharge, Spectroscopy Letters,13 (5), 313-319.
CAROLI, S ., DELLE FEMMINE, P ., ALIMONTE, A ., PETRUCCI, F .,VIOLANTE, N ., 1982:Applicability of spectroscopic methods to the determina-tion of Aluminium in biological samples, SpectroscopyLetters, 15 (6), 211-215.
CAROLI, S ., ALIMONTI, A ., DELLE FEMMINE, P ., SHUKLA, S .K .,1982 : Determination of gallium in tumor-affected tissuesby means of spectroscopic techniques, Analytica ChimicaActa, 136, 225-231 .

64
CHAO, S .S . und PICKETT, E .E ., 1980:Trace chromium determination by furnace atomic absorptionspectrometry following enrichment by extraction, Anal.Chem ., 52, 335-339
CHAPMAN, H .D ., 1972:Diagnostic criteria for plants and soils, 793 S ., Uni-versity of California, Riverside, Calif.
DAUE, J .H ., 1823:Neues Handbuch über den Torf, 240 S ., Hinrichs Buchhand-lung, Leipzig.
DAVID, D .J ., 1962:Determination of strontium in biological materials andexchangeable strontium in soils by atomic absorptionspectrophotometry, The Analyst, Vol . 87, No . 1036,576-587.
DAVID, D .J ., 1964:An ion-exchange column for use with atomic-absorptionanalysis, The Analyst, Vol . 89, No . 1064, 747-748.
DAVID, D .J ., 1969:Atomic-absorption determination of strontium in a standardplant material : comment an results of interlaboratorycomparison, Analyst, Vol . 94, 884-885.
DE SOETE, GIJBELS, R ., HOSTE, J ., 1972:Neutron activation analysis, 380 S ., Wiley, New York.
DORNER, W .G ., 1982:Aufschlußmethoden in der Spurenanalyse, zu beziehen durch:Hans Kürner, Analysentechnik, Herderstraße 2, 8200 Rosen-heim.
DUDAS, M .J . und PAWLUK, S ., 1976:The nature of mercury in chernozemic and luvisolic soilsin Alberta, Can . J . Soil . Sci ., 56, 413-423.
ERÄMETSÄ, 0., YLIRUOKANEN, I ., 1971:Niobium, molybdenium, hafnium, tungsten, thorium, anduranium in lichens and mosses, Suomen Kemistilekti,B . 44, 372-378.
ERÄMETSÄ, 0 ., HAARALA, A .R ., YLIRUOKANEN, I ., 1973:Lanthanoid content three species of Equisetum, SuomenKemistilekti, B . 46, 234-236.
ERNST, W ., 1978:Ökologische Risiken, verursacht durch emittierte Fein-stäube aus Verbrennungsmotoren : Folgerungen für dieanalytische Chemie, Chemische Rundschau, 32, 31-55 .

.65
FILBY, R .H ., NGUYEN, S ., GRIMM, C .A ., MARKOWSKI, G .R ., 1985:Evaluation of geochemical standard reference materialsfor micröanalysis, Analytical Chemistry, 57, 551-568.
FIRBAS, F ., 1952:Einige Berechnungen zur Ernährung der Hochmoore, Ver-öffentl . des Geobotanischen Instituts der ETH, StiftungRubel, 25, 177-182.
FUSBAN, H .U ., SEGEBADE, C ., SCHMITT, B .F ., 1981:Instrumental multielement analysis of soll samples,Journal of Radioanalytical Chemistry, 67, 101-117.
GLEIT, C .E .W . und HOLLAND, W .D ., 1962:Use of electrically excited oxygen for the low tempera-ture decomposition of organic substances . Anal . Chem .,34, 1454-1457.
HEIN, H . und SCHRADER, W ., 1976:Anwendungstechnische Grundlagen für das Arbeiten mit derGraphitrohrküvette HGA-76, Angewandte Atom-Absorptions-spektroskopie, 3, 1-57.
HEYDORN, K ., DAMSGAARD, E ., RIETZ, B ., 1980:Systematic differences in the determination of vanadiumin standard reference material 1571 Orchard Leaves, Anal.Chem ., 52, 1045-1049.
HOFFMANN, H .J ., 1980:Der Einsatz der simultanen ICP-AES in der Wasseranalytik,Laborpraxis, Juli/August, 18-24.
HOROWITZ, L .T ., SCHOCK, H .H . und HOROWITZ-KISIMOVA, L .A ., 1974:The content of scandium, thorium, silver and other traceelements in different plant species, Plant Soil, 40,397-408.
HUDSON, G .M ., KAUFMANN, H .C ., NELSON, J .W ., BONACCI, M .A .,1980:Advances in the use of PIXE and PESA for air pollutionsampling, Nuclear Instruments and Methods, 168, 259-263.
ICHIKAWA, R . und OHNO, S ., 1974:Levels of cobalt, cesium and zinc in some marine orga-nisms in Japan, Bulletin of the Japanese Society ofScientific Fisheries, 40, 501-508.
INSTRUMENTATION LABORATORY GMBH, 1981:Bedienungsanleitung für das Graphitofen-Modell IL 655 CTF,73 S ., Bornheim.
JACOBS, F .S ., EKAMBARAM, V ., FILBY, R .H ., 1982:Determination of trace elements in 13 organic solvents byinstrumental neutron activation analysis, AnalyticalChemistry, 54, 1240-1243 .

66
JOHANSSON, T .B ., VAN GRIEKEN, R .E ., NELSON, J .W ., WINCHESTER,J .W ., 1975:Elemental trace analysis of small samples by proton in-duced x-ray emission, Analytical Chemistry, 47, 855-860.
KABATA-PENDIAS, A . und PENDIAS, H ., 1984:Trace elements in soils and plants, 315 S ., CRC Press,Boca Raton, Florida.
KAISER, G ., TSCHÖPEL, P ., TÖLG, G ., 1971:Aufschluß im aktivierten Sauerstoff bei Bestimmung extremniedriger Spurenelementgehalte in organischem Material,Fresenius Z . Anal . Chem ., 253, 177-198.
KOCH, O .G . und KOCH-DEDIC, G .A ., 1974:Handbuch der Spurenanalyse, Teil 1, 750 S ., Springer Ver-lag, Berlin, Heidelberg, New York.
KOONS, R .D . und HELMKE, P .A ., 1978:Neutron activation analysis of standard soils, SoilSci . Soc . Am . J., 42, 237-248.
KOTZ, L ., KAISER, G ., TSCHÖPEL, P ., TÖLG,,G ., 1972:Aufschluß biologischer Matrizes für die Bestimmung sehrniedriger Spurenelementgehalte bei begrenzter Einwaagemit Salpetersäure unter Druck in einem Teflongefäß,Fresenius Z . Anal . Chem ., 260, 207-215.
KRUGER, P ., 1971:Principles of activation analysis, 612 S ., Wiley andSons, New York, London, Sydney, Toronto.
LABREQUE, J .J ., ROSALES, P .A ., QUIERO, A ., 1982:Application of "direct" beta particle excitation for thedetermination of noble metals in catalysts by radioiso-tope excited x-ray fluorescence ., Journal of Radioana-lytical Chemistry, 75, 205-211.
LABREQUE, J .J ., ROSALES, P .A ., PARKER, W .C ., 1983:Sensitivities for eight medium z-elements with elevendifferent radionuclides using a radiometrie x-ray ana-lysis technique, Journal of Radioanalytical Chemistry,78, 87-99.
LAG, J . und BOLVINKEN, B ., 1974:Some naturally heavy metal poisoned areas of interestin prospecting, soll chemistry, and geomedicine, Norgesgeoliske undersokels, 304, 73-85.
LAG, J . und STEINNES, E ., 1976:Regional distribution of halogens in Norwegian forestsoils, Geoderma, 16, 317-332.
LAG, J . und STEINNES, E ., 1978:Regional distribution of mercury in humus layers of Nor-wegian forest soils, Acta Agriculture Scandinavica, 28,393-396 .

67
LEHMANN, G ., 1980:Die "Schemm" bei Walchsee - Nordtirols größte erhalteneMoorlandschaft, 73 . Jahresbericht des BundesgymnasiumsKufstein, S . 16-18, Kufstein.
LIETH, H . und WHITTAKER, R .H ., 1975:Ecological Studies 14, Springer Verlag, New York, Heidel-berg, Berlin, 339 S.
LIETH, H . und MARKERT, B ., 1985:Concentration cadasters of chemical elements in contra-sting ecosystems, Naturwissenschaften, 72, 322-324.
LIETH, H . und MARKERT, B ., 1986 a:Elementkonzentrationskataster für Böden und einigePflanzen in Walchsee/Österreich, Veröfftl . der Eidg.Techn . Hochschule Zürich, im Druck.
LIETH, H . und MARKERT, B ., 1986 b:The establishment of element concentration cadastersfor ecosystems (ECCE) in the different vegetationzones of the earth, Research concept for an Interna-tional Geoecological program to be discussed by theIUBS Committee, 25 S.
MAOLIANG, L . und FILBY, R .H ., 1983:Determination of sulfur in fly ash and fuel oil Standardreference materials by radiochemical neutron activationanalysis and liquid scintillation counting, AnalyticalChemistry, 55, 2336-2340.
MARKERT, B . und LIETH, H ., 1983:Vergleichende Elementbestimmung in einem ombrogenen undminerogenen System, Symp . anorg . Analytik in Umweltfor-schung und Umweltschutz, 13 .-16 . Juni 1983, Jülich.
MARKERT, B . und LIETH, H ., 1984:Vergleichende Elementbestimmung in einem ombrogenen undminerogenen System, Fresenius Z . f . anal . Chem ., 317,412.
MARKERT, B . und LIETH, H ., 1985:Elementkonzentrationskataster für einige Pflanzen in kon-trastierenden Ökosystemen, Veröffentlichungen der Natur-forschenden Gesellschaft zu Emden, Band 5, Jahresbericht1985, 27-56.
MARKERT, B . und MEER, G ., 1985:Biomonitoring mittels Hypnum cupressiforme (Hedw .) imGroßraum Osnabrück, Veröffentl . der NaturforschendenGesellschaft zu Emden, Serie 1, 65-76 .

68
MARKERT, B . und LIETH, H ., 1986:Element Concentration Cadasters in a Swedish Biotope/Reference standard for inorganic environmental chemistry,Fresenius, Z . f . anal . Chemie, im Druck.
MATTHES, W ., FLUCHT, R ., STOEPPLER, M ., 1978:Beiträge zur automatisierten Spurenanalyse, III : Eineempfindliche automatisierte Methode zur Quecksilber-bestimmung in biologischen und Umweltproben, Fresenius,Z . Anal . Chem ., 291, 20-23.
MAY, K . und STOEPPLER, M ., 1978:Pretreatment studies with biological and environmentalmaterials by perchloric acid mixtures, Fresenius Z.Anal . Chem ., 293, 127-130.
MERIAN, E ., 1984:Metalle in der Umwelt, 722 S ., Verlag Chemie, Weinheim.
NADKARNI, R .A ., FLIEDER, D .E ., EHMANN, W .D ., 1969:Instrumental neutron activation analysis of biologicalmaterials, Radiochimica Acta, 11, 97-100.
NIEMINEN, K . und YLIRUOKANEN, I ., 1974:Trace element analysis of granitic and radioactive rocksby spark source mass spectrometry with electrical de-tection, Bull . Geol . Soc . Finland, 46, 167-176.
OHNO, S . und YATAZAWA, M ., 1970:Simultaneous determination of arsenic and antimony insoil by neutron activation analysis, Radioisotopes, 19,565-570.
OSTENDORF, B ., LIETH, H ., LEHKER, H ., 1981:Klimaplot, a Computer routine to plot climate diagrams,Veröffentl . des Geobotanischen Instituts der ETH,Stiftung Rübel, Zürich, Heft 77, 149-171.
OSTENDORF, B . und LIETH, H ., 1982:The Computer drawn climate diagrams, S . XIX-XXIV in:MÜLLER, M .J ., Selected climatic data for a global setof standard stations for vegetation science, T :VS, Vo-lume 5, Dr . W . Junk, Den Haag.
OVERBECK, F ., 1975:Botanisch-geologische Moorkunde, 380 S ., Karl Wach-holtz Verlag, Neumünster.
PALMER, C .A . und FILBY, R .H ., 1984:Distribution of trace elements in coal from the Pow-hatan No . 6 mine, Ohio, Fuel, 63, 318-328 .

69
PARRY, S .J ., 1980:Simultaneous determination of the noble metals in geolo-gical materials by radiochemical neutron activationanalysis, Analyst, 105, 1157-1162.
PAU, J .C .M ., PICKETT, E .E ., KOIRTYOHANN, S .R ., 1972:Determination of boron in plants by emission spectros-copy with the nitrous oxide-hydrogen flame, Analyst, 97,860-865.
PAWLUK, S ., 1967:Soll analysis by atomic absorption spectrophotometry,Atomic Absorption Newsletter, 6, 53-56.
PICKETT, E .E . und PAU, J .C .M ., 1973:Emission photometric determination of boron in unboro-nated fertilizers, using the nitrous oxidehydrogen flame,Journal of the AOAC, 56, 151-153.
POTTS, P .J ., 1983:Gamma-ray photopeak interferences found in the instru-mental activation analysis of silicate rocks, Journalof Radioanalytical Chemistry, 79, 363-370.
POTTS, P .J . und HUSSEY, R ., 1983:Effect of sample to setector geometry an the accuracy ofinstrumental neutron activation analysis and implicationsfor the design of a simple automatic samplechangingwheel suitable for the routine counting of low activitygeological Samples, Journal of Radioanalytical Chemistry,78, 339-346.
ROBERTS, T .M ., 1975:A review of some biological effects of lead emissionsfrom primary and secondary smelters, Int . Conf . HeavyMetals in the Environment, . Toronto, Vol . 2, 503-532.
RUBESKA, I ., KORELKOVA, J ., WEISS, D ., 1977:The determination of gold and palladium in geologicalmaterials by atomic absorption after extraction withdibuthylsulfide, Atomic Absorption Newsletter, 16, 1-3.
RÜHLING, A . und TYLER, G ., 1971:Regional differences in the deposition of heavy metalsover Skandinavia, J . Appl . Ecol ., 8, 497-507.
SCHEFFER, F . und SCHACHTSCHABEL, P ., 1982:Lehrbuch der Bodenkunde, 442 S ., Ferdinand Enke Verlag,Stuttgart.
SCHMITT, B .F ., SEGEBADE, C ., FUSBAN, H .U ., 1980:Waste incineration ash - a versatile environmental re-ference material-, Journal of Radioanalytical Chemistry,60, 99-109 .

70
SCHNIER, C . und SCHNUG, E ., 1981:Spurenelementbestimmung in Pflanzenmaterial mit instru-menteller Neutronenaktivierungsanalyse (NAA) unter be-sonderer Berücksichtigung des Molybdäns, Landwirtschaft-liche Forschung, 38, 736-750.
SCHORIN, H ., 1982:Quantitative determination of Ga, Zn, Cu, Ni, Mn, and Crby x-ray fluorescence in laterites and bauxites usingtwo evaluation methods, aus : Advances in x-ray analysis,Vol . 25, Herausgeber : J .C . Russ, C .S . Barrett, P .K.Predecki and D .E . Leyden, Plenum, Publishing Corporation.
SCHORIN, H . und PICCIONI, L ., 1984:X-ray fluorescence spectrometric analysis of uncontami-nated and contaminated tropical plant materials fortraces of heavy metals, aus : Advances in x-ray analysis,Vol . 27, herausgegeben von Cohen, Russ, Leyden, Barettund Predecki, 187 S ., Plenum, Publishing Corporation.
SCHRAMEL, P ., 1973:Determination of eight metals in the international biolo-gical standard by flameless atomic absorption spectro-metry, Analytica Chimica Acta, 67, 69-77.
SCHRAMEL, P ., WOLF, A ., SEIF, R ., KLOSE, B .J ., 1980:Eine neue Apparatur zur Druckveraschung von biologischemMaterial, Fresenius Z . Anal . Chem ., 302, 62-77.
SCHRAMEL, P . und KLOSE, B .J ., 1981:Direktbestimmung von Cu, Fe, Zn, Ca, Mg und Na im Serummittels ICP-Emissionsspektralanalyse, Fresenius Z . Anal.Chem ., 307, 26-30.
SCHRAMEL, P ., KLOSE, B .J ., HASSE, S ., 1982:Die Leistungsfähigkeit der ICP-Emissionsspektroskopiezur Bestimmung von Spurenelementen in biologisch-medizi-nischen und in Umweltproben, Fresenius Z . Anal . Chem .,310, 209-216.
SCHULEK, E . und LASZLOVSKY, J ., 1960:Zur Frage der Zerstörung und Anreicherung in der Mikro-analyse, Mikrochim . Acta, 4, 485-501.
SEGEBADE, C ., FUSBAN, H .U ., WEISE, H .P ., 1980:Analysis of some toxic components of environmental samplesby high energy photon activation, Journal of Radioanaly-tical Chemistry, 59, 399-405.
SONESSON, M ., 1983:Ecology of a subarctic mixe, Ecol . Bull ., 30, 250 S .,Stockholm.
STEENKEN, F ., 1973:Begasungen von Nutzpflanzen mit bleihaltigen und blei-freien Autoabgasen, Z . Pflanzenkrankh . Pflanzenschutz,80, 513-527 .

71
STEINNES, E ., 1980:Atmospheric deposition of heavy metals in Norway studiedby the analysis of moss samples using neutron activationanalysis and atomic absorption spectrometry, Journal ofRadioanal . Chem ., 58, 387-391.
STOEPPLER, M ., 1980:Beiträge zur Umweltforschung und Umweltüberwachung, III:Optimierung und Einsatz automatisierter Methoden beiBilanzierungsstudien mit toxischen Elementen, 79 S .,KFA Jülich.
SUGIMAE, A ., 1980:Atmospheric concentrations and sources of rare earth ele-ments in the Osaka area, Japan, Atmospheric Environment,14, 1171-1175.
THIERMANN, A ., 1983:Geologische Karte von Nordrhein-Westfalen, Nr . 3613Westerkappeln (1 :25000), Geologisches Landesamt Nord-rhein-Westfalen, Krefeld.
THOMPSON, G. und BANKSTON, D .C ., 1970:Sample contamination from grinding and silving determinedby emission spectrometry, Applied Spectrometry, 24,310-319.
TJIOE, P .S ., DE GOEIJ, J .J .M ., HOUTMAN, J .P .W ., 1973:Automated chemical separations in routine activationanalysis, J . of Radioanal . Chem ., 16, 153-164.
TJIOE, P .S ., DE GOEIJ, J .J .M ., HOUTMAN, J .P .W ., 1977:Extended automated separation techniques in destructiveneutron activation analysis ; application to various bio-logical materials, including human tissues and blood,Journal of Radioanal . Chem ., 37, 511-522.
URE, A .M ., BACON, J .R ., 1978:Comprehensive analysis of soils and rocks by sparksource mass spectrometry, Analyst, 103, 807-822.
URE, A .M ., BACON, J .R ., BERROW, M .L ., WATT, J .J ., 1979:The total trace element content of some Scottish soilsby spark source mass spectrometry, Geoderma, 22, 1-17.
WALTER, H . und LIETH, H ., 1960:Klimadiagramm-Weltatlas, Gustav Fischer Verlag, Jena.
WEBER, E ., 1980:Venner-Moor-Schutzbericht, unveröffentlicht.
WELZ, B ., 1983:Atomabsorptionsspektroskopie, 527 S ., Verlag Chemie,Weinheim .

72
WILSON, W .O ., 1979:Determination of molybdenum in wet ashed digests ofplant material using flameless atomic absorption,Communication in Soil Science and Plant Analysis, 10,1319-1330.
WINOGRADOW, A .P ., 1954:Geochemie seltener und nur in Spuren vorhandener chemischerElemente im Boden, 454 S ., Akademie-Verlag, Berlin.
WYTTENBACH, A ., BAJO, S ., TOBLER, L ., 1983:Group separation of rare earth elements by liquid-liquidextraction for the neutron activation analysis of Sili-cate rocks, Journal of the Radioanalytical Chemistry,78,, 283-294.
YLIRUOKANEN, 1 ., 1975:Uranium, thorium, lead, lanthanoids and yttrium in someplants growing an granitic and radioactive rocks, Bull.Geol . Soc . Finland, 47, 71-77.
YLIRUOKANEN, I ., 1980:The occurrence of uranium in some Finnish peat bogs,Kemina-Kemi, 7, 213-217.
YUAN, T .L . und BRELAND, H .L ., 1969:Evaluation of atomic absorption methods for determi-nations of aluminium, iron and silicon in clay andsoll extracts . Soll Sci . Soc . Amer . Proc ., 33, 868-872 .

73
Anhang 1
Elementkonzentrationskataster
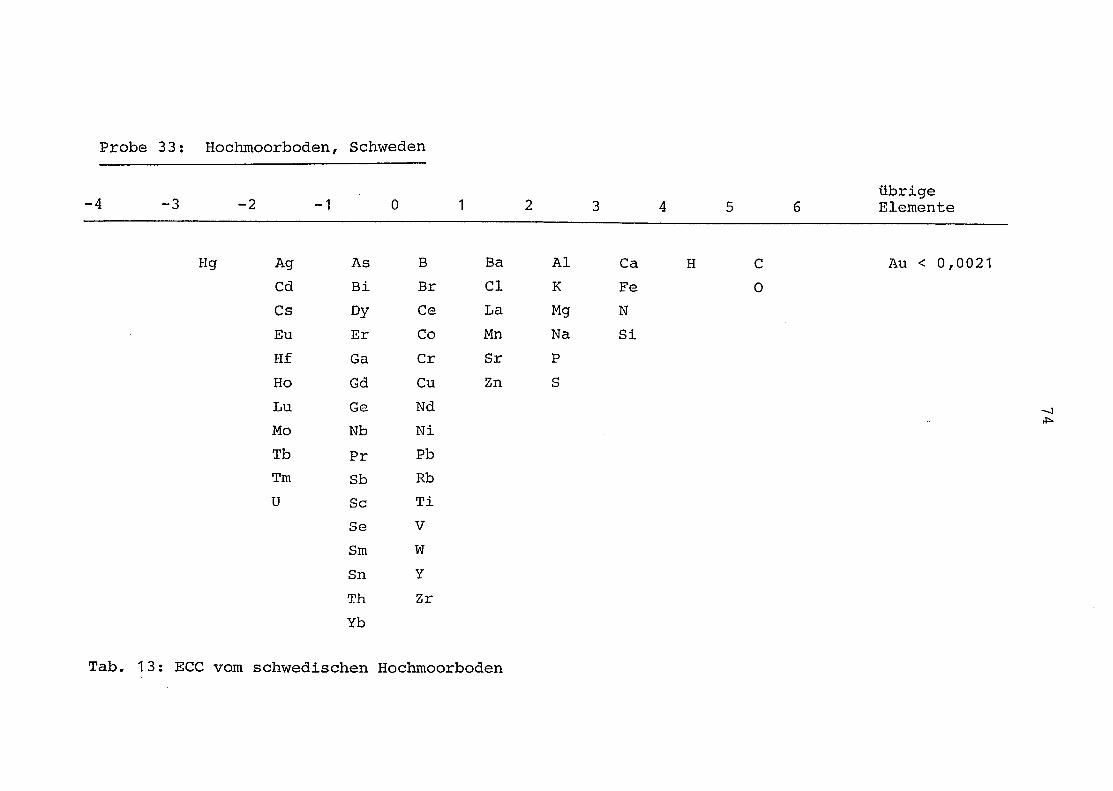
Probe 33 : Hochmoorboden, Schweden
-4
-3 -2
-1 0 1 2 3 4 5 6übrigeElemente
Hg Ag As B Ba Al Ca H C Au < 0,0021
Cd Bi Br Cl K Fe 0
Cs
Eu
Hf
Ho
Lu
Mo
TbTm
U
Dy
Er
Ga
Gd
Ge
Nb
PrSb
Sc
Se
Sm
Sn
Th
Yb
Ce
Co
Cr
Cu
Nd
Ni
PbRb
Ti
V
W
Y
Zr
La
Mn
Sr
Zn
Mg
Na
P
S
N
Si
Tab . 13 : ECC vom schwedischen Hochmoorboden
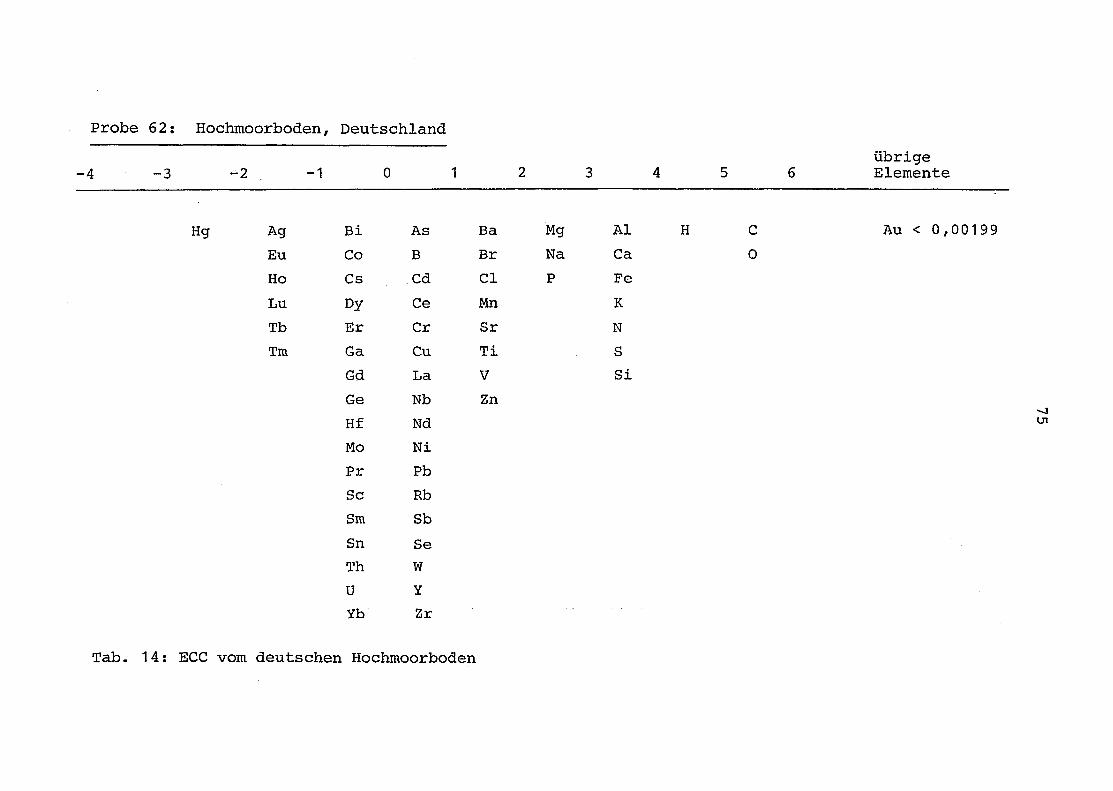
Probe 62 : Hochmoorboden, Deutschland
übrige-4
-3
-2
-1
0
1
2
3
4
5
6
Elemente
Hg Ag Bi As Ba Mg Al H C Au < 0,00199
Eu Co B Br Na Ca 0
Ho
Lu
Cs
Dy
Cd
Ce
Cl
Mn
P Fe
K
Tb Er Cr Sr N
Tm Ga Cu Ti S
Gd La V Si
Ge
HfMo
PrSc
Sm
SnTh
U
Yb
Nb
NdNi
Pb
Rb
Sb
SeW
YZr
Zn
Tab . 14 : ECC vom deutschen Hochmoorboden
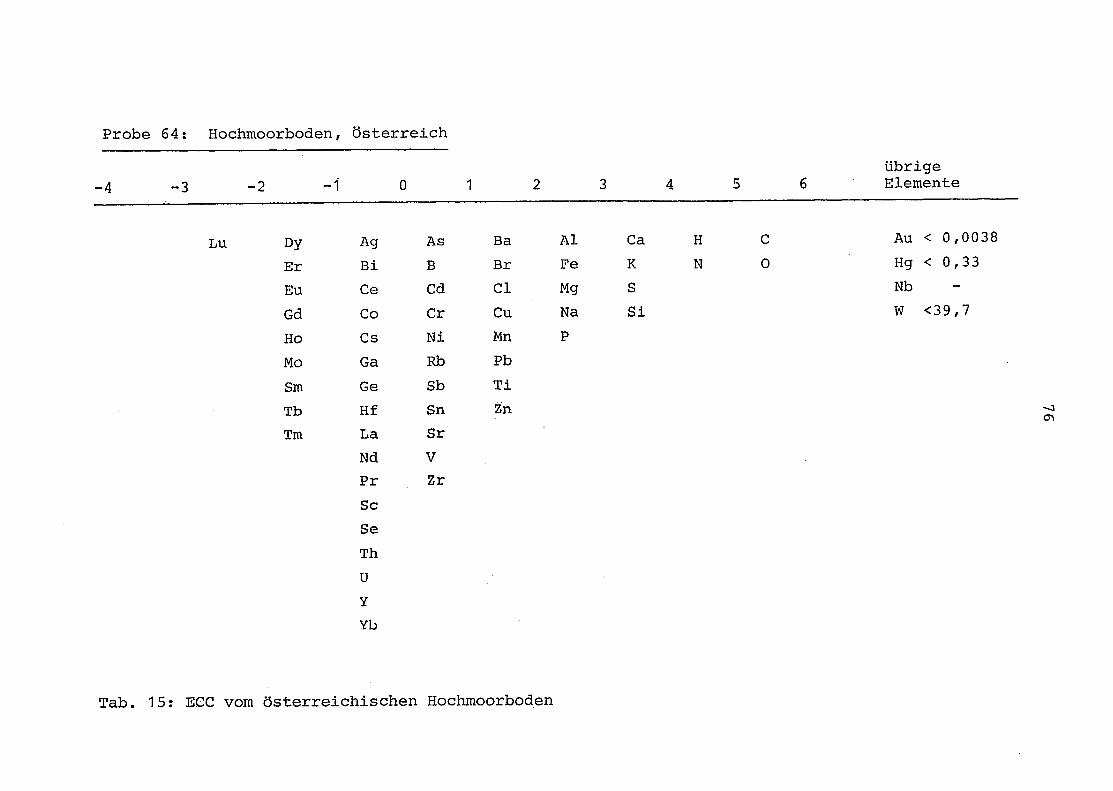
Probe 64 : Hochmoorboden, Österreich
übrige-4
-3
-2
-1
0
1
2
3
4
5
6
Elemente
Lu Dy Ag As Ba Al Ca H C Au < 0,0038
Er Bi B Br Fe K N 0 Hg < 0,33
Eu Ce Cd C1 Mg S Nb
-
Gd Co Cr Cu Na Si W
<39,7
Ho
Mo
Sm
Tb
Tm
Cs
Ga
GeHf
La
Nd
Pr
Sc
Se
Th
U
Y
Yb
Ni
Rb
Sb
Sn
Sr
VZr
Mn
Pb
Ti
Zn
P
rn
Tab . 15 : ECC vom österreichischen Hochmoorboden
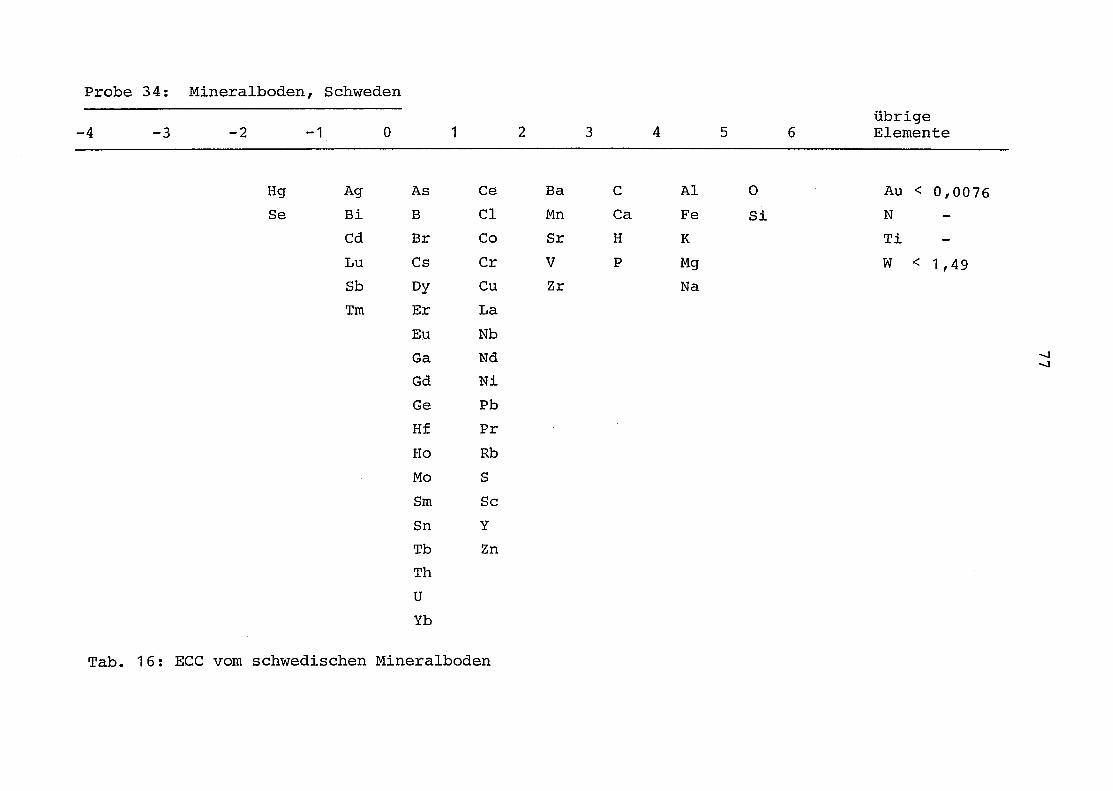
Probe 34 :
Mineralboden, Schweden
0 1 2 3 4 5 6übrigeElemente
Hg Ag As Ce Ba C Al 0 Au < 0,0076
Se Bi B Cl Mn Ca Fe Si N
-
Cd Br Co Sr H K Ti
-Lu
Sb
Tm
Cs
Dy
Er
Eu
Ga
Gd
Ge
Hf
Ho
Mo
Sm
Sn
Tb
ThU
Yb
Cr
Cu
La
Nb
Nd
Ni
PbPr
Rb
S
Sc
Y
Zn
V
ZrMg
Na
W
<
1,49
Tab . 16 : ECC vom schwedischen Mineralboden
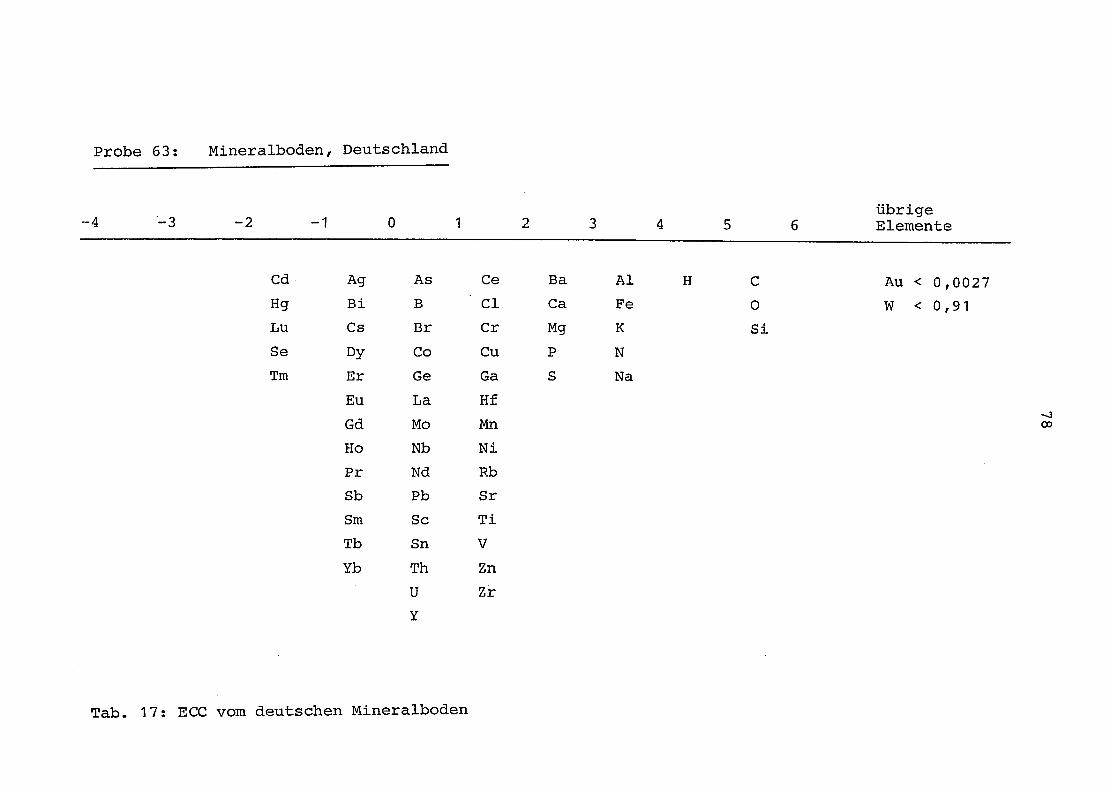
Probe 63 : Mineralboden, Deutschland
-4
-3 -2
-1 0
Cd Ag As
Hg Bi B
Lu Cs Br
Se Dy Co
Tm Er
Eu
GdHo
Pr
Sb
Sm
Tb
Yb
Ge
La
Mo
Nb
Nd
Pb
Sc
Sn
Th
UY
1 2 3 4 5 6übrigeElemente
Ce Ba Al H C Au < 0,0027C1
Cr
Cu
Ga
Hf
Mn
Ni
Rb
Sr
Ti
V
Zn
Zr
Ca
Mg
P
S
FeK
N
Na
0
Si
W
< 0,91
Tab . 17 : ECC vom deutschen Mineralboden
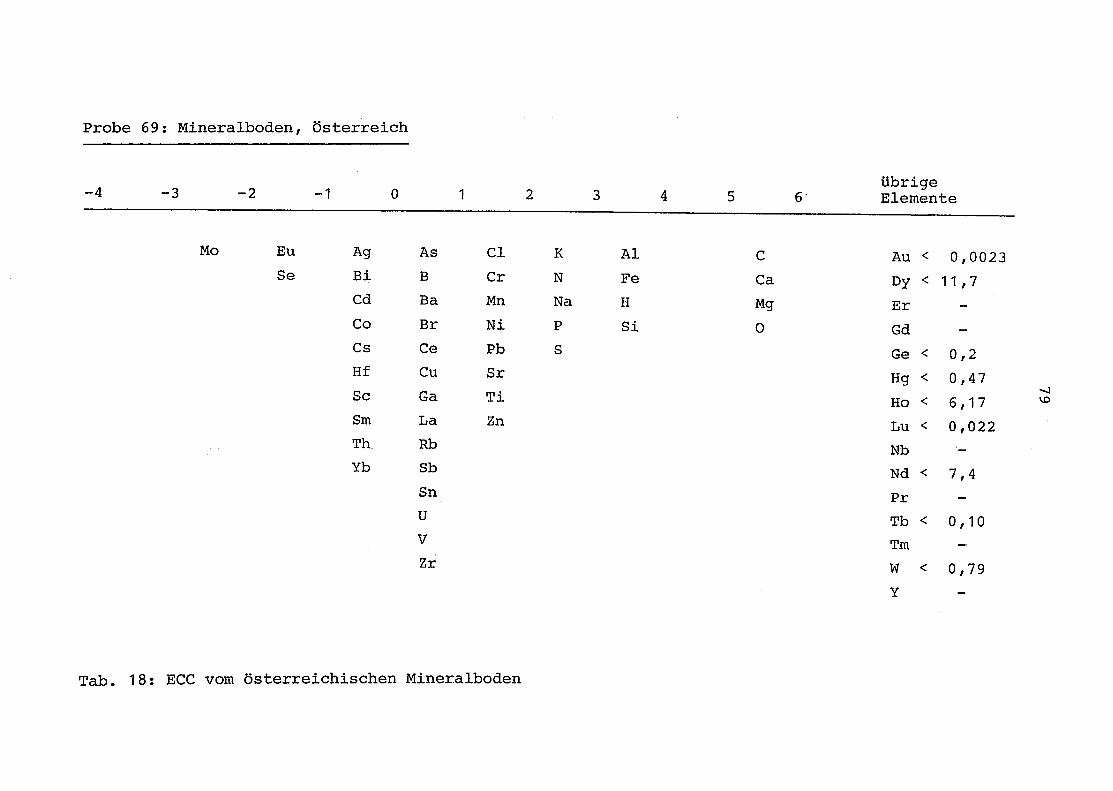
Probe 69 : Mineralboden, Österreich
-4
-3
-2 -1
0
Mo Eu
SeAgBi
Cd
CoCs
Hf
Sc
Sm
ThYb
1 2 3 4
5 6 .übrigeElemente
As Cl K Al C Au < 0,0023B Cr N Fe Ca Dy < 11,7Ba Mn Na H Mg Er -Br Ni P Si 0 Gd -Ce Pb S Ge < 0,2Cu Sr Hg < 0,47Ga Ti Ho < 6,17La Zn Lu < 0,022Rb Nb -Sb
SnNd <
Pr
7,4
U Tb < 0,10V Tm -
W
< 0,79Y -
Tab . 18 : ECC vom österreichischen Mineralboden
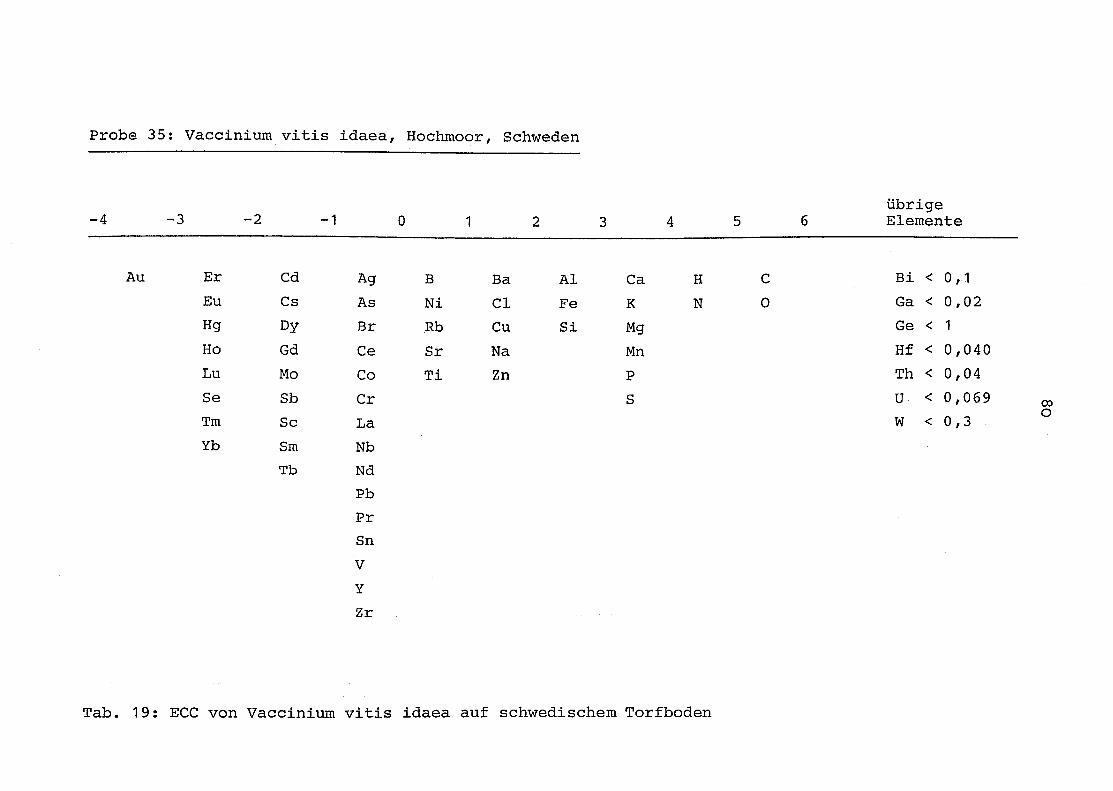
Probe 35 : Vaccinium vitis idaea, Hochmoor, Schweden
-4 -3 -2 -1 0 1 2
Au Er
EuHg
Ho
Lu
Se
Tm
Yb
Cd
CsDy
Gd
Mo
Sb
Sc
SmTb
Ag
As
Br
Ce
Co
Cr
La
Nb
NdPb
PrSn
V
Y
Zr
B
Ni
Rb
Sr
Ti
Ba
Cl
Cu
Na
Zn
Al
Fe
Si
3 4 5 6übrigeElemente
Ca H C Bi < 0,1
K N 0 Ga < 0,02
Mg Ge < 1
Mn Hf < 0,040
P Th < 0,04
S U
< 0,069
W
< 0,3
Tab . 19 : ECC von Vaccinium vitis idaea auf schwedischem Torfboden
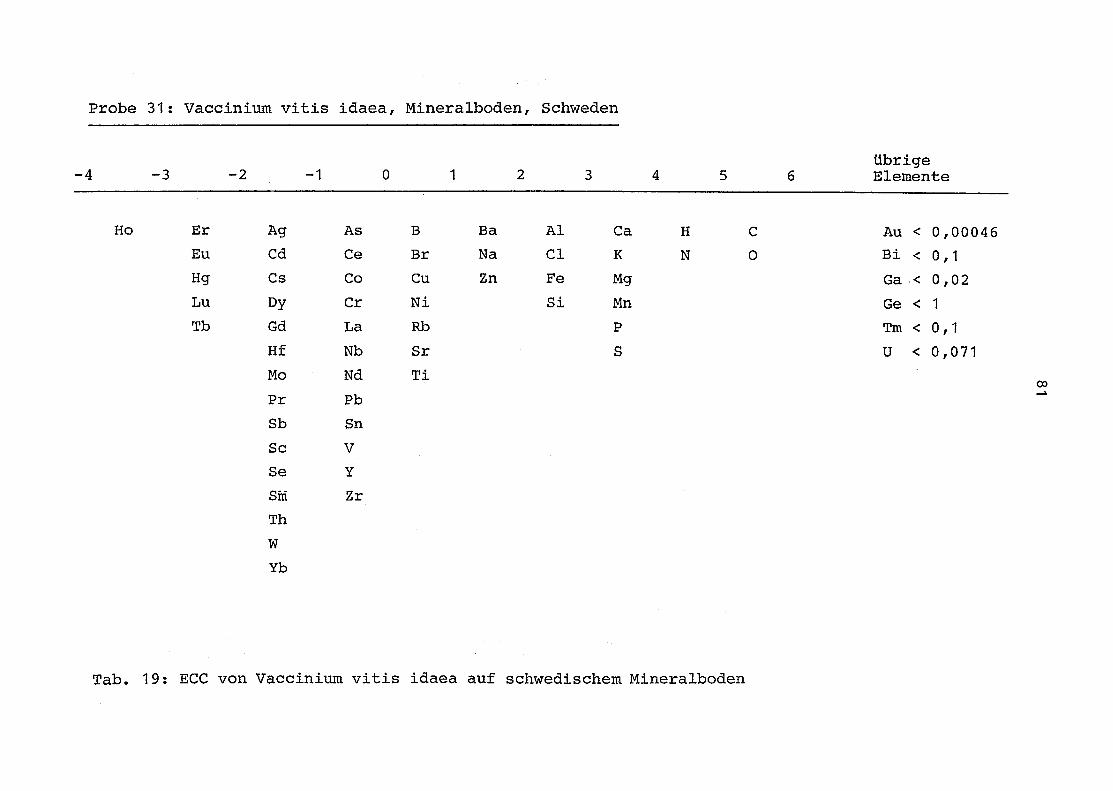
Probe 31 : Vaccinium vitis idaea, Mineralboden, Schweden
-4 -3 -2
-1 0 1 2 3 4 5 6übrigeElemente
Ho Er Ag As B Ba Al Ca H C Au < 0,00046Eu Cd Ce Br Na Cl K N 0 Bi < 0,1Hg Cs Co Cu Zn Fe Mg Ga < 0,02Lu Dy Cr Ni Si Mn Ge < 1Tb Gd La Rb P Tm < 0,1
Hf Nb Sr S U
< 0,071Mo
Pr
Sb
Sc
Se
Sm
Th
W
Yb
Nd
Pb
Sn
V
Y
Zr
Ti
Tab. 19 : ECC von Vaccinium vitis idaea auf schwedischem Mineralboden
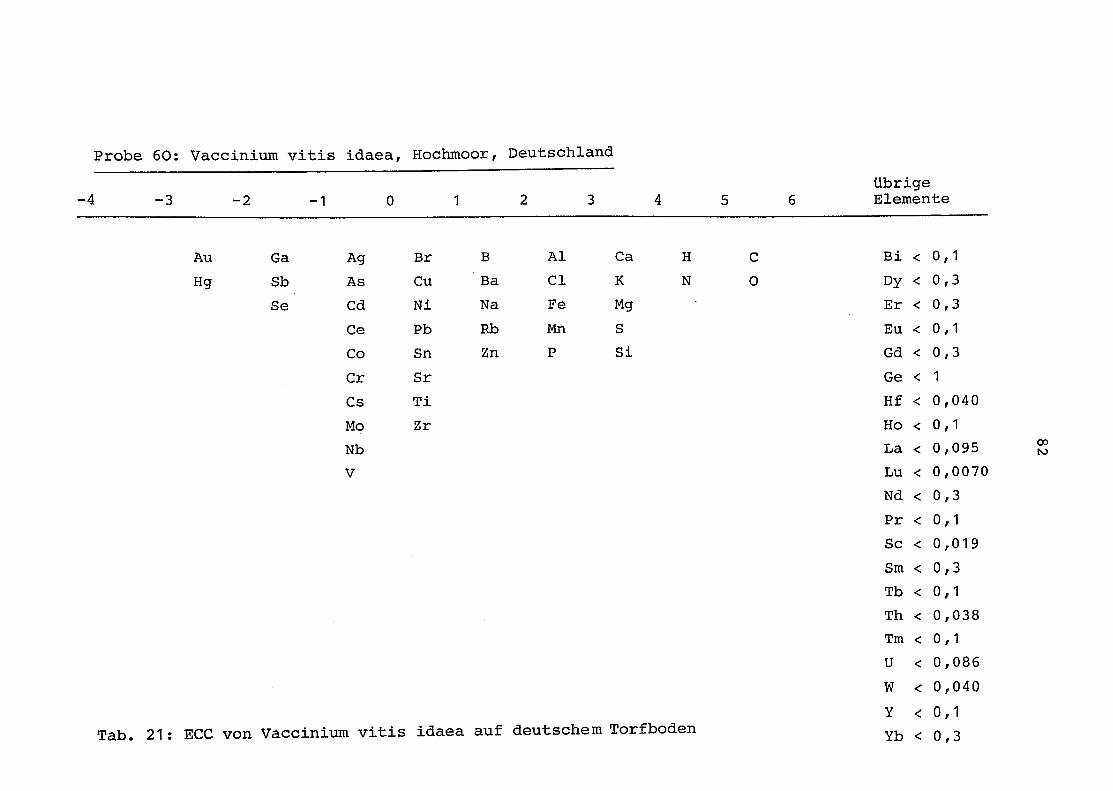
Probe 60 : Vaccinium vitis idaea, Hochmoor, Deutschland
-2
-1
0
1
2
3
4
5 6übrigeElemente
Au
Ga
Ag
Br
B
Al
Ca
H C Bi < 0,1
Hg
Sb
As
Cu
Ba
Cl
K
N 0 Dy < 0,3
Se
Cd
Ni
Na
Fe
Mg Er < 0,3
Ce
Pb
Rb
Mn
S Eu < 0,1
Co
Sn
Zn
P
Si Gd < 0,3
Cr
Sr Ge < 1
Cs
Ti Hf < 0,040
Mo
Zr Ho < 0,1
Nb La < 0,095
V Lu < 0,0070
Nd < 0,3
Pr < 0,1
Sc < 0,019
Sm < 0,3
Tb < 0,1
Th < 0,038Tm < 0,1
U
< 0,086
W
< 0,040
Y
< 0,1Tab . 21 : ECC von Vaccinium vitis idaea auf deutschem Torfboden Yb < 0,3
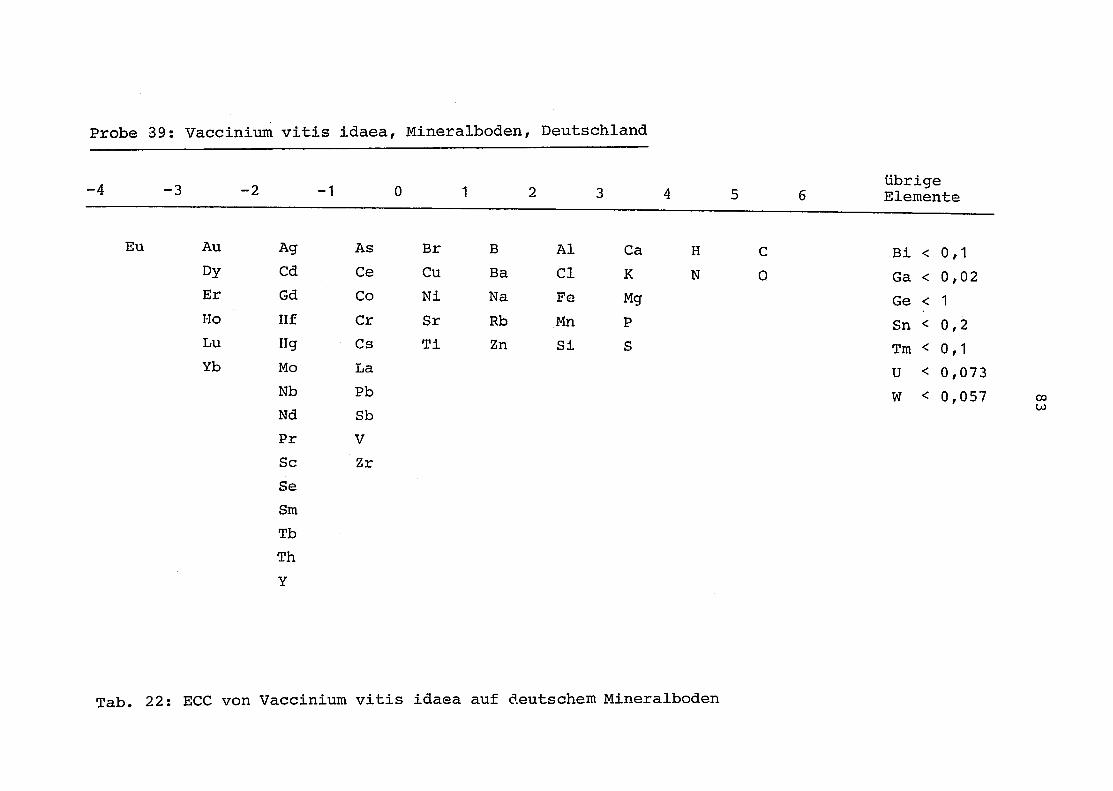
Probe 39 : Vaccinium vitis idaea, Mineralboden, Deutschland
-4 -3 -2
-1 0 1 2 3
Eu Au Ag As Br B Al CaDy
Er
I1o
Lu
Yb
Cd
Gd
IIf
IIg
Mo
Nb
Nd
Pr
Sc
SeSm
Tb
Th
Y
Ce
Co
Cr
Cs
La
Pb
SbV
Zr
Cu
Ni
Sr
Ti
Ba
Na
RbZn
C1
Fe
Mn
Si
K
Mg
P
S
4 5 6übrigeElemente
H C Bi < 0,1N 0 Ga < 0,02
Ge < 1
Sn < 0,2Tm < 0,1
U
< 0,073
W
< 0,057
Tab. 22 : ECC von Vaccinium vitis idaea auf deutschem Mineralboden
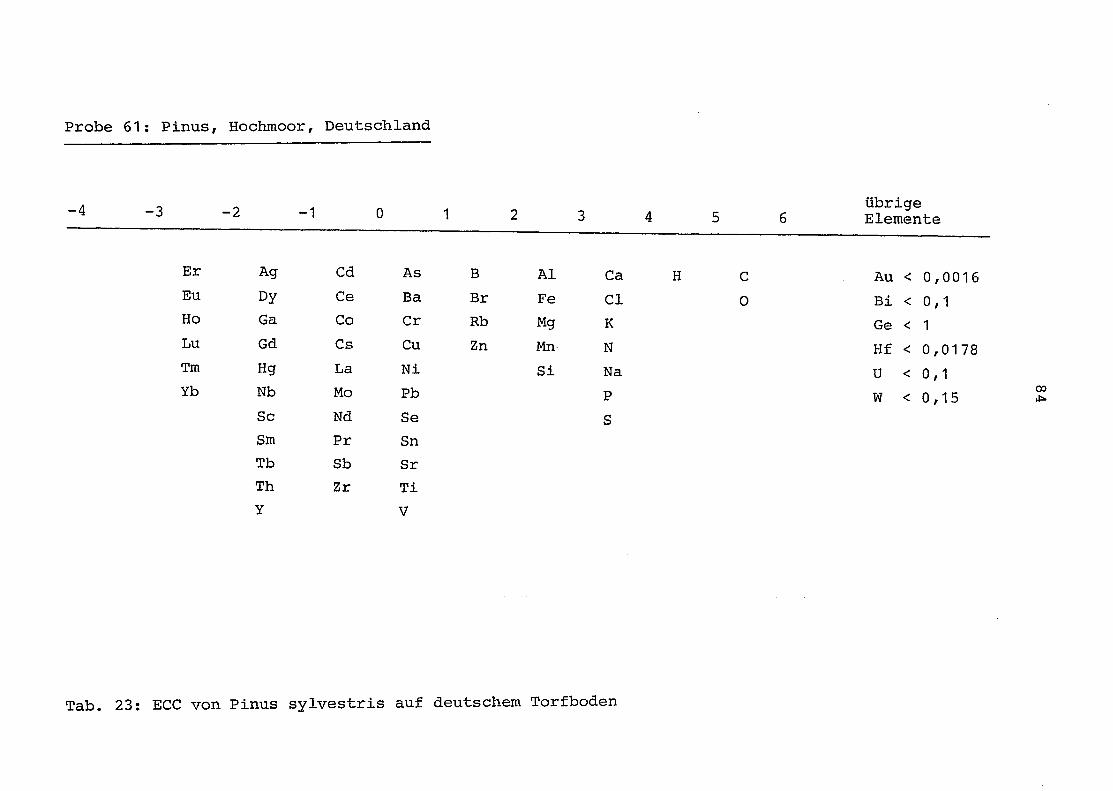
Probe 61 : Pinus, Hochmoor, Deutschland
-4
-3 -2 -1 0
Er Ag Cd AsEu Dy Ce BaHo Ga Co CrLu Gd Cs CuTm Hg La NiYb Nb
Sc
SmTb
ThY
Mo
Nd
Pr
SbZr
Pb
Se
SnSr
Ti
V
1 2 3 4 5 6übrigeElemente
B Al Ca H C Au < 0,0016Br Fe C1 0 Bi < 0,1Rb Mg K Ge < 1Zn Mn N Hf < 0,0178
Si Na U
< 0,1P
SW
< 0,15
Tab . 23 : ECC von Pinus sylvestris auf deutschem Torfboden
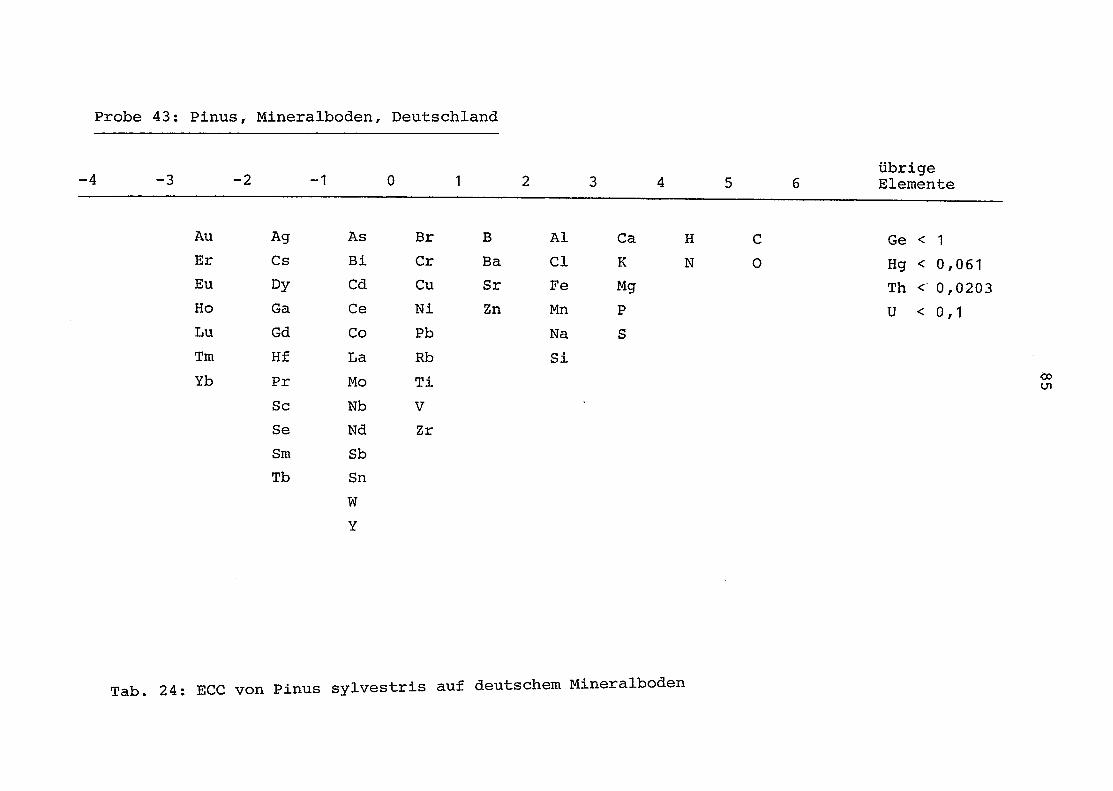
Probe 43 : Pinus, Mineralboden, Deutschland
-4
-3 -2 -1 0 1 2 3 4 5 6übrigeElemente
Au Ag As Br B Al Ca H C Ge < 1Er Cs Bi Cr Ba Cl K N 0 Hg < 0,061Eu Dy Cd Cu Sr Fe Mg Th < - 0,0203Ho
Lu
Tm
Yb
Ga
Gd
Hf
Pr
Sc
Se
Sm
Tb
Ce
Co
La
Mo
Nb
Nd
Sb
Sn
W
Y
Ni
Pb
Rb
Ti
V
Zr
Zn Mn
Na
Si
P
SU
<
0,1
Tab . 24 : ECC von Pinus sylvestris auf deutschem Mineralboden
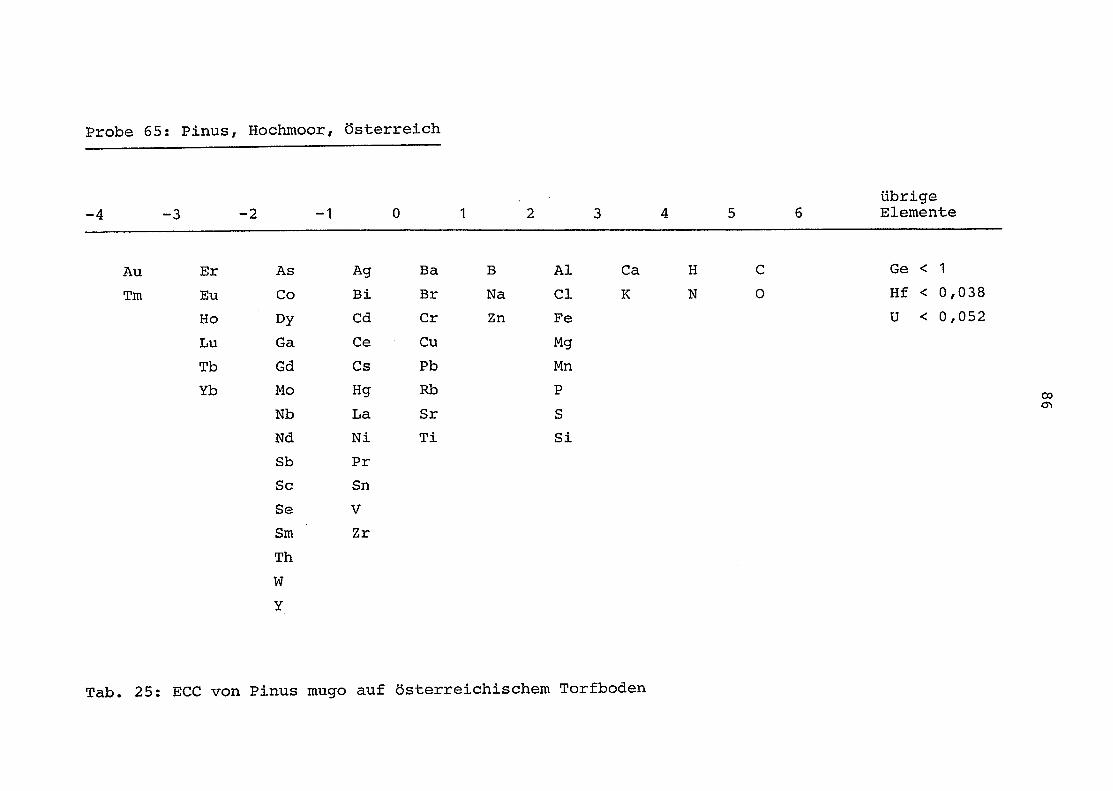
Probe 65 : Pinus, Hochmoor, Österreich
-4 -3 -2
-1 0
Au Er As Ag Ba
Tm Eu
Ho
Lu
Tb
Yb
CoDy
Ga
Gd
Mo
NbNd
Sb
Sc
Se
Sm
Th
W
Bi
Cd
Ce
Cs
Hg
La
Ni
Pr
Sn
V
Zr
Br
Cr
Cu
Pb
Rb
Sr
Ti
1 3 4 5 6übrigeElemente
B Al Ca H C Ge < 1
Na C1 K N 0 Hf < 0,038
Zn Fe
Mg
Mn
U
< 0,052
P co
S
Si
rn
Tab . 25 : ECC von Pinus mugo auf österreichischem Torfboden
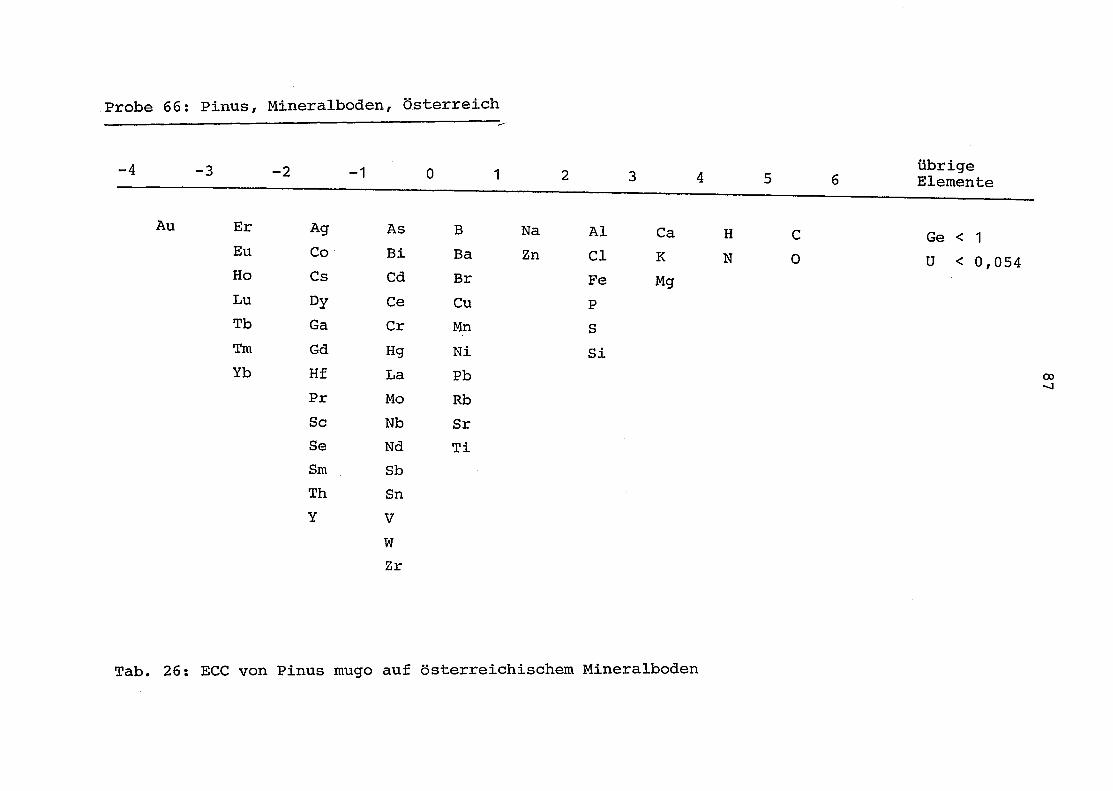
Probe 66 : Pinus, Mineralboden, Österreich
-4 -3 -2
-1 0
1
Au Er
Eu
Ho
Lu
Tb
Tm
Yb
Ag
Co
Cs
DyGa
Gd
Hf
Pr
ScSe
Sm
Th
Y
As
Bi
Cd
Ce
Cr
Hg
La
Mo
Nb
Nd
Sb
Sn
V
WZr
B
Ba
Br
Cu
Mn
Ni
Pb
Rb
Sr
Ti
Tab . 26 : ECC von Pinus mugo auf österreichischem Mineralboden
übrige4
5
6
Elemente
Ca
H
C
Ge < 1K
N
0
U < 0,054Mg
2
3
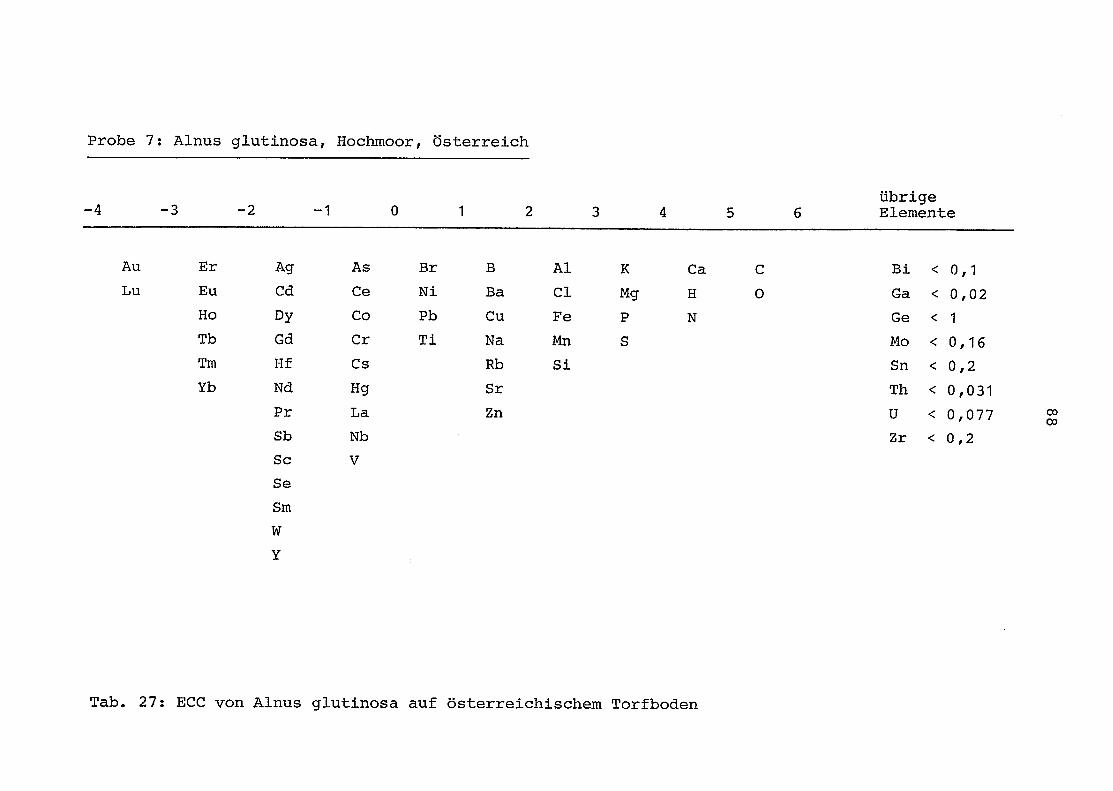
Probe 7 : Alnus glutinosa, Hochmoor, Österreich
-4 -3 -2 -1 0 1
2 3 4 5 6übrigeElemente
Au Er Ag As Br B Al K Ca C Bi
< 0,1Lu Eu Cd Ce Ni Ba Cl Mg H 0 Ga
< 0,02Ho Dy Co Pb Cu Fe P N Ge
< 1Tb Gd Cr Ti Na Mn S Mo
< 0,16Tm Hf Cs Rb Si Sn
< 0,2Yb Nd Hg Sr Th
< 0,031Pr La Zn U
< 0,077Sb Nb Zr
< 0,2ScSe
Sm
W
Y
V
Tab . 27 : ECC von Alnus glutinosa auf österreichischem Torfboden
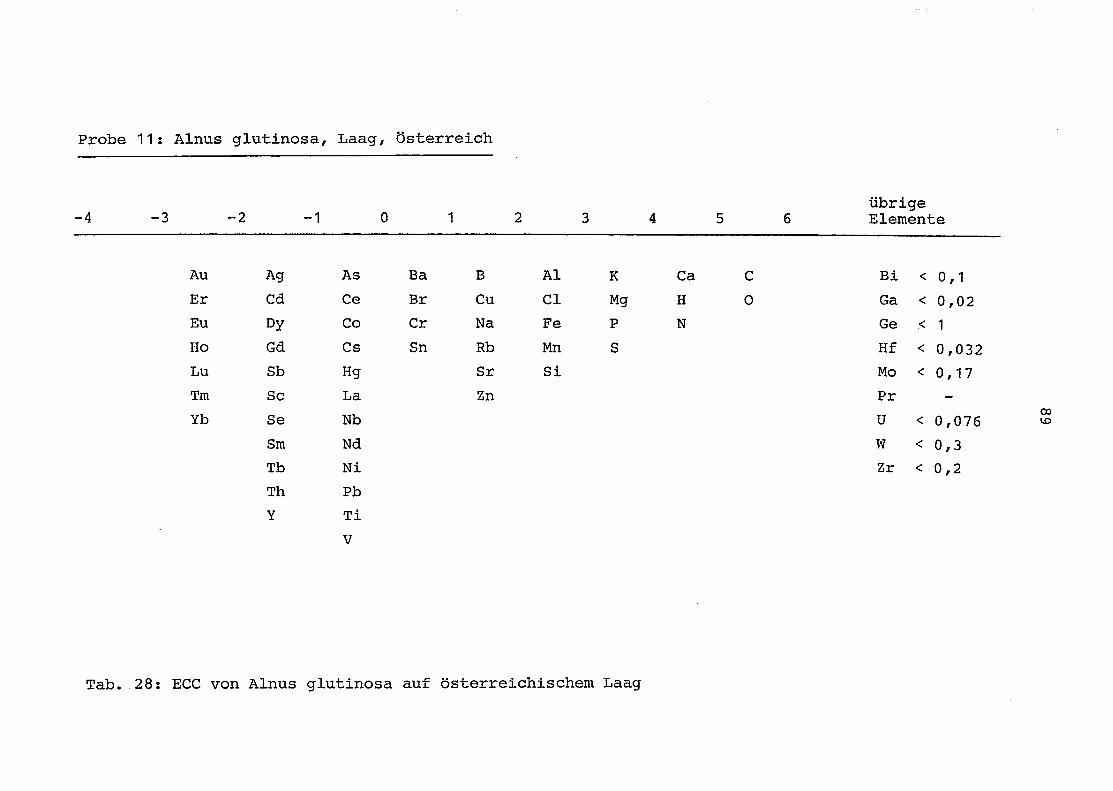
Probe 11 : Alnus glutinosa, Laag, Österreich
-4
-3 -2 -1 0 1 2 3 4 5 6übrigeElemente
Au Ag As Ba B Al K Ca C Bi
< 0,1Er Cd Ce Br Cu Cl Mg H 0 Ga
< 0,02Eu Dy Co Cr Na Fe P N Ge
< 1Ho Gd Cs Sn Rb Mn S Hf
< 0,032Lu Sb Hg Sr Si Mo
< 0,17Tm Sc La Zn Pr -
Yb Se Nb U
< 0,076Sm Nd W
< 0,3Tb Ni Zr
< 0,2
Th
Y
Pb
Ti
V
Tab . 28 : ECC von Alnus glutinosa auf österreichischem Laag
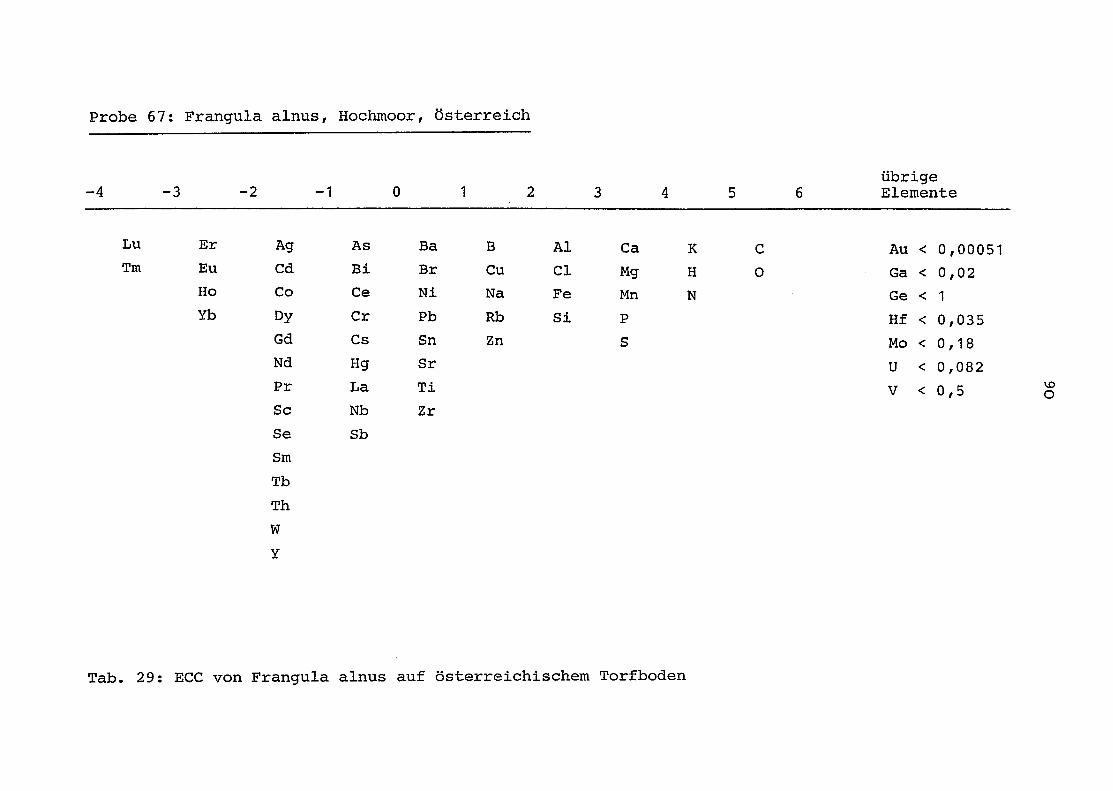
Probe 67 : Frangula alnus, Hochmoor, Österreich
übrige-4
-3
-2
-1
0
1
2
3
4
5
6
Elemente
Lu
Er
Ag
As
Ba
B
Al
Ca
K
CTm
Eu
Cd
Bi
Br
Cu
Cl
Mg
H
0Ho
Co
Ce
Ni
Na
Fe
Mn
NYb
Dy
Cr
Pb
Rb
Si
PGd
Cs
Sn
Zn
SNd
Hg
SrPr
La
TiSc
Nb
ZrSe
SbSm
Tb
Th
W
Y
Au < 0,00051Ga < 0,02
Ge < 1
Hf < 0,035Mo < 0,18
U < 0,082
V < 0,5
Tab . 29 : ECC von Frangula alnus auf österreichischem Torfboden
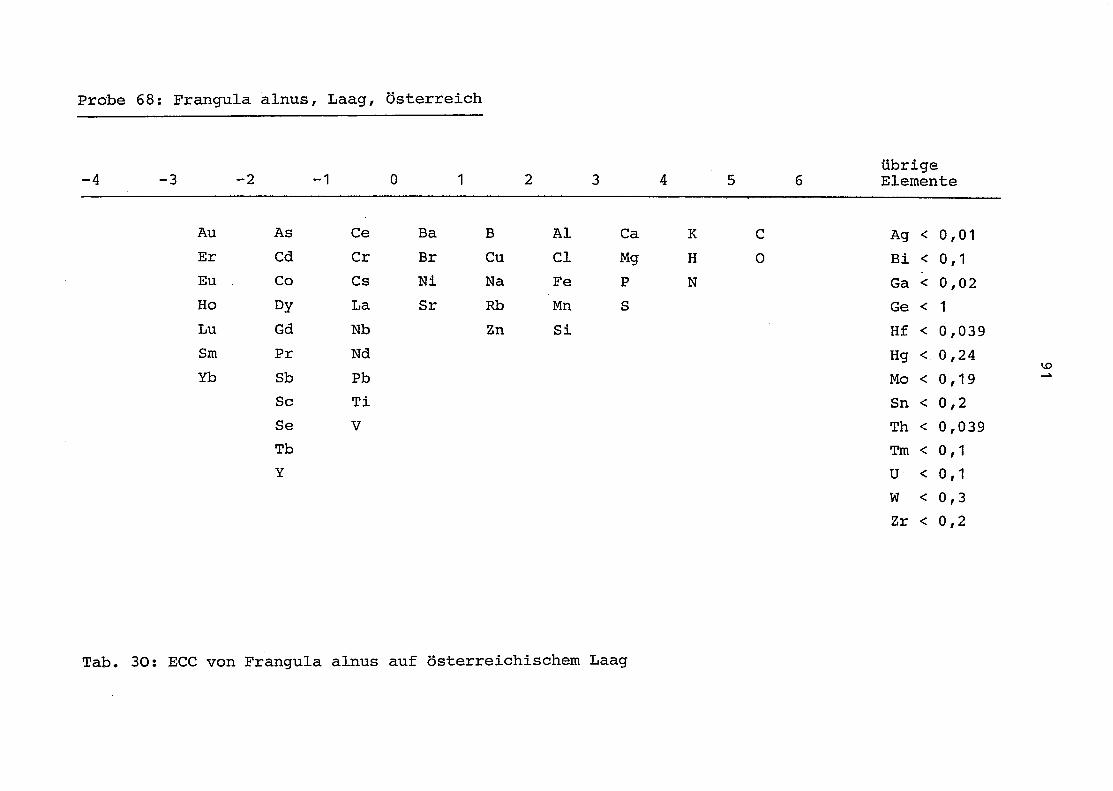
Probe 68 : Frangula alnus, Laag, Österreich
-4 -3
-2 -1 0 1
Au As Ce Ba
Er Cd Cr Br
Eu Co Cs Ni
Ho Dy La Sr
Lu
Sm
Yb
Gd
Pr
Sb
Sc
Se
TbY
Nb
Nd
Pb
Ti
V
2 3 4 6übrigeElemente
B Al Ca K C Ag < 0,01Cu Cl Mg H 0 Bi < 0,1Na Fe P N Ga < 0,02Rb Mn S Ge < 1Zn Si Hf < 0,039
Hg < .0,24
Mo < 0,19
Sn < 0,2
Th < 0,039Tm < 0,1U
< 0,1
W
< 0,3
Zr < 0,2
Tab. 30 : ECC von Frangula alnus auf österreichischem Laag
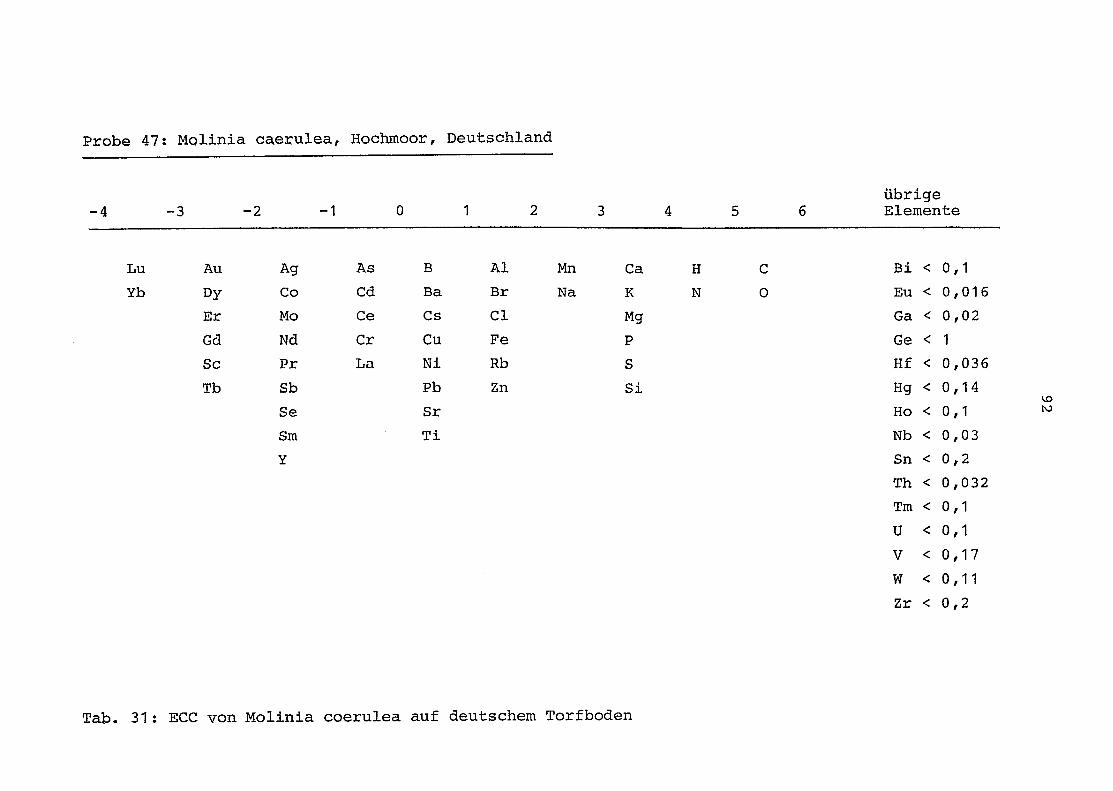
Probe 47 : Molinia caerulea, Hochmoor, Deutschland
-4 -3
-2 -1 0 1
2
Lu Au Ag As B Al
Yb Dy
Er
Gd
Sc
Co
Mo
Nd
Pr
Cd
Ce
Cr
La
Ba
Cs
CuNi
Br
Cl
FeRb
Tb Sb Pb Zn
Se Sr
Sm
Y
Ti
3 4 5 6übrigeElemente
Mn Ca H C Bi < 0,1
Na K N 0 Eu < 0,016
Mg Ga < 0,02
P Ge < 1
S Hf < 0,036
Si Hg < 0,14
Ho < 0,1
Nb < 0,03
Sn < 0,2
Th < 0,032
Tm < 0,1
U
< 0,1
V
< 0,17
W
< 0,11
Zr < 0,2
Tab. 31 : ECC von Molinia coerulea auf deutschem Torfboden
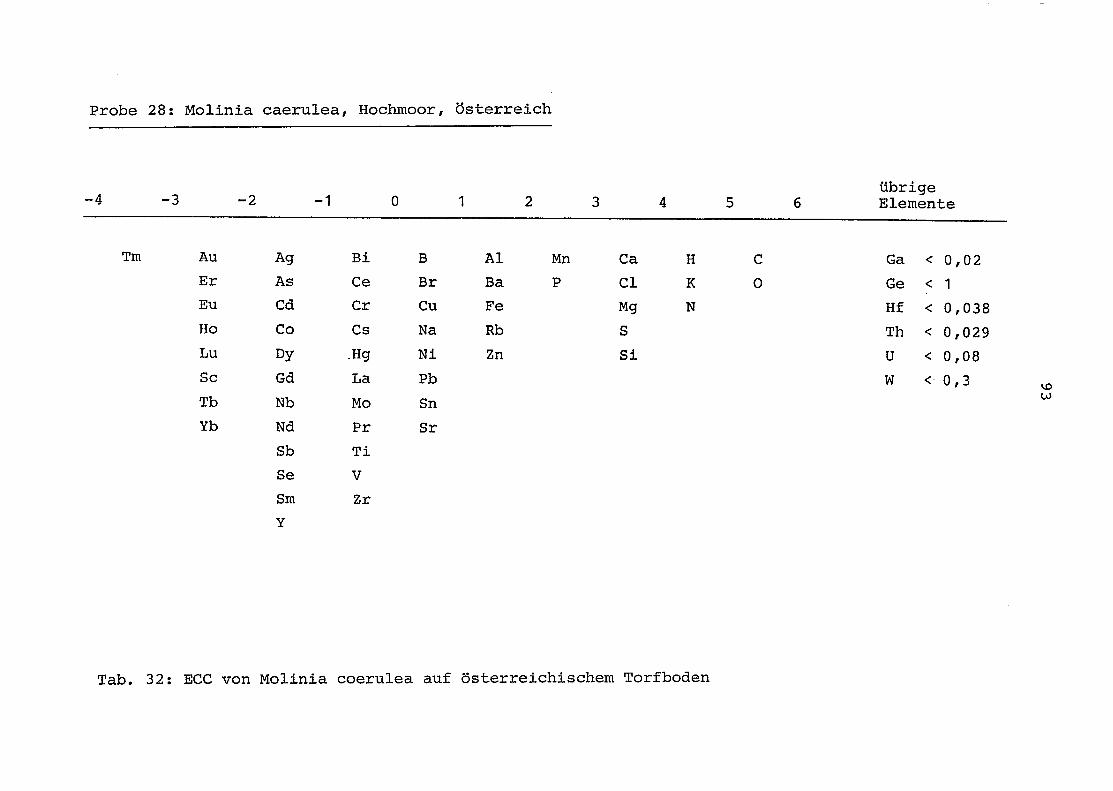
Probe 28 : Molinia caerulea, Hochmoor, Österreich
-4 -3 -2
-1 0 1
2 3 4 5 6übrigeElemente
Tm Au Ag Bi B Al Mn Ca H C Ga
< 0,02Er As Ce Br Ba P Cl K 0 Ge
< 1Eu Cd Cr Cu Fe Mg N Hf
< 0,038Ho Co Cs Na Rb S Th
< 0,029Lu Dy .Hg Ni Zn Si U
< 0,08Sc Gd La Pb W
< 0,3Tb
YbNb
Nd
Sb
Se
Sm
Y
Mo
Pr
TiV
Zr
Sn
Sr
Tab . 32 : ECC von Molinia coerulea auf österreichischem Torfboden
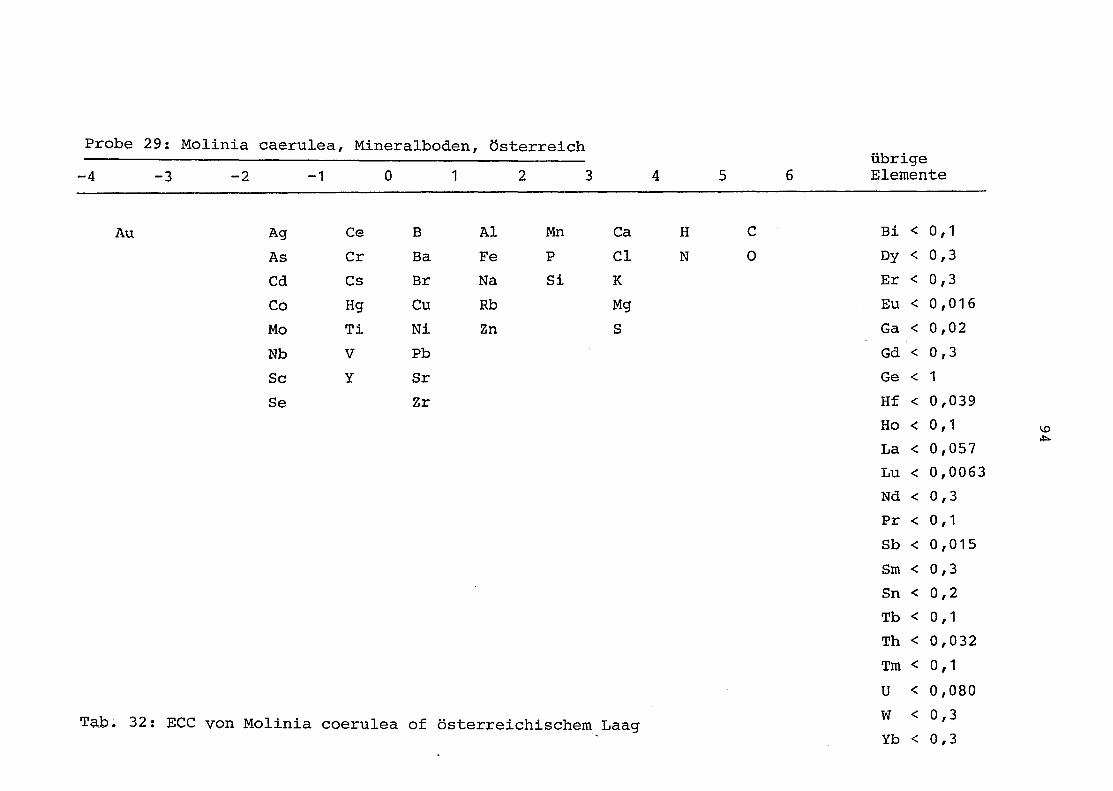
Probe 29 : Molinia caerulea, Mineralboden, Österreich
5 6übrigeElemente-4
-3
-2
-1
0
1
2
3
4
Au
Ag
Ce
B
Al
Mn
Ca H C Bi < 0,1
As
Cr
Ba
Fe
P
Cl N 0 Dy < 0,3
Cd
Cs
Br
Na
Si
K Er < 0,3
Co
Hg
Cu
Rb
Mg Eu < 0,016
Mo
Ti
Ni
Zn
S Ga < 0,02
Nb
V
Pb Gd < 0,3
Sc
Y
Sr Ge < 1
Se
Zr Hf < 0,039
Ho < 0,1
La < 0,057
Lu < 0,0063
Nd < 0,3
Pr < 0,1
Sb < 0,015
Sm < 0,3
Sn < 0,2
Tb < 0,1
Th < 0,032
Tm < 0,1
U
< 0,080
W
< 0,3Tab . 32 : ECC von Molinia coerulea of österreichischem LaagYb < 0,3
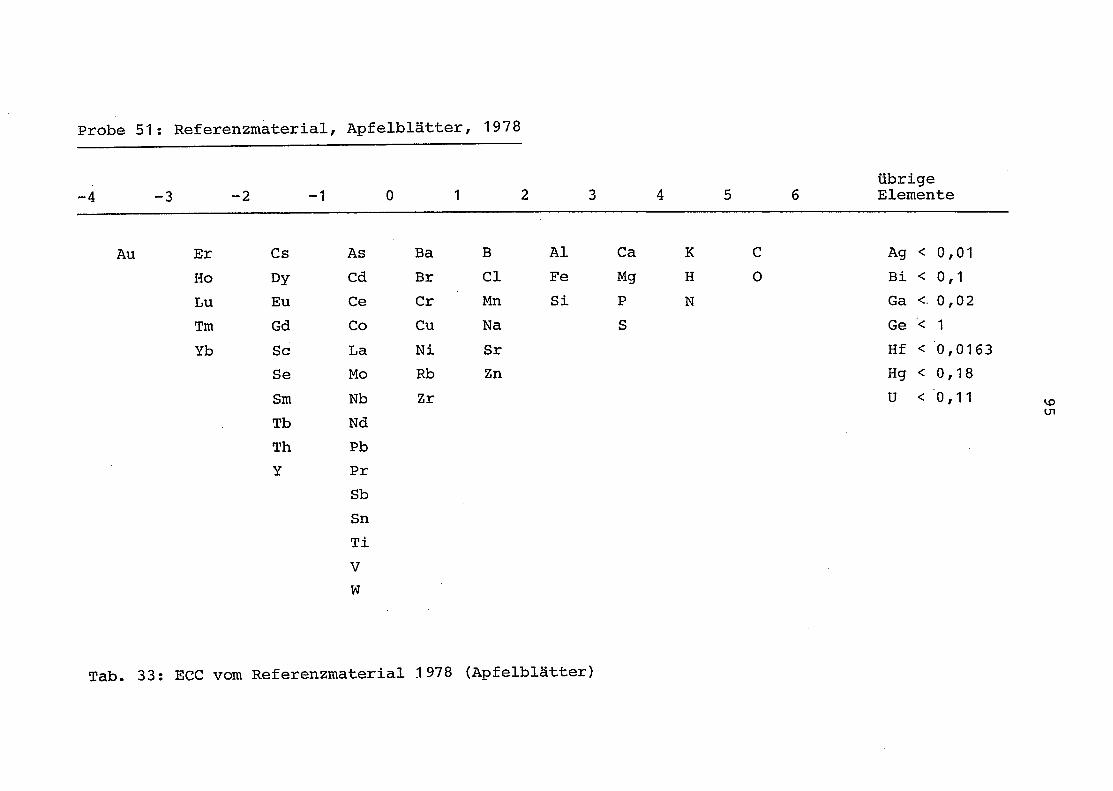
Probe 51 : Referenzmaterial, Apfelblätter, 1978
übrige-3
-2
-1
0
1
2
3
4
5
6
Elemente
Au
Er
Cs
As
Ba
B
Al
Ca
K
C
Ho
Dy
Cd
Br
C1
Fe
Mg
H
0
Lu
Eu
Ce
Cr
Mn
Si
P
N
Tm
Gd
Co
Cu
Na
S
Yb
Sc
La
Ni
Sr
Se
Mo
Rb
Zn
Sm
Nb
Zr
Tb
Nd
Th
Pb
Y Pr
Sb
Sn
Ti
V
W
Tab . 33 : ECC vom Referenzmaterial 1978 (Apfelblätter)
Ag < 0,01
Bi < 0,1
Ga <. 0,02
Ge < 1
Hf < 0,0163Hg < 0,18
U < 0,11

96
Anhang 2
Vergleich der Einzelmeßdaten
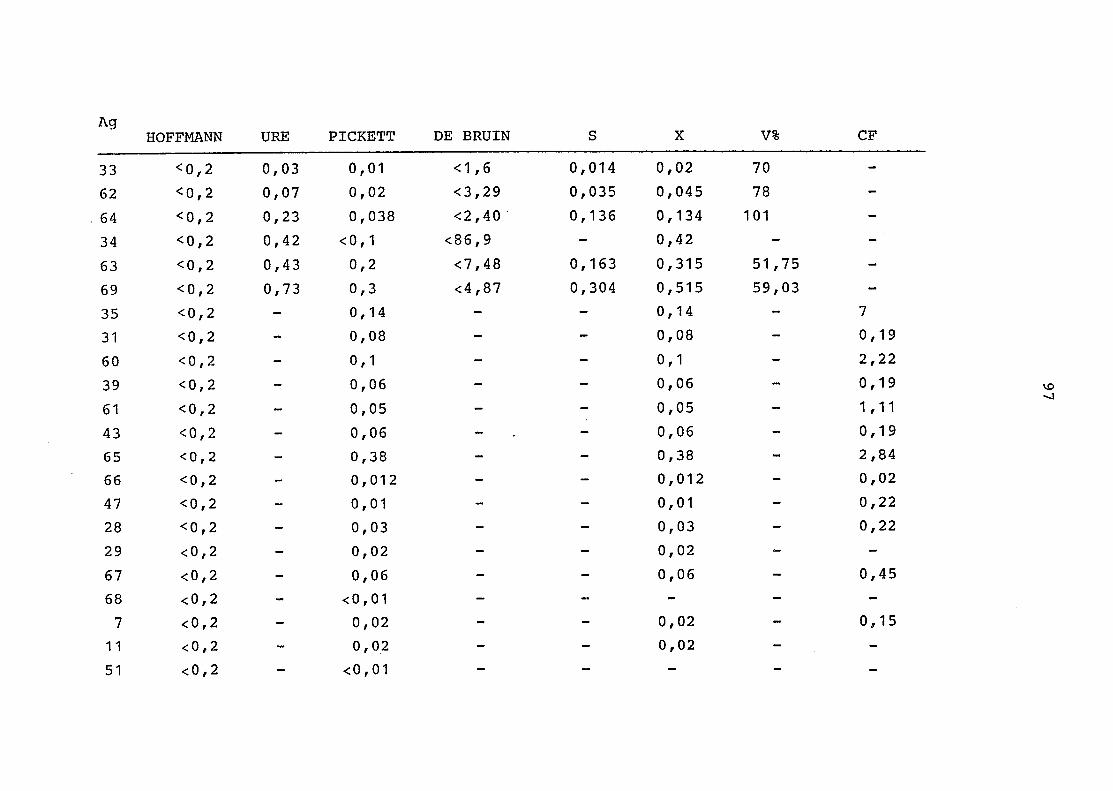
AgHOFFMANN
URE PICKETT DE BRUIN
S X
V% CF
33 < 0,2
0,03 0,01 <1,6
0,014 0,02
70 -
62 <0,2
0,07 0,02 <3,29
0,035 0,045
78 -
.64 <0,2
0,23 0,038 <2,40'
0,136 0,134
101 -
34 <0,2
0,42 <0,1 <86,9
- 0,42
-
63 <0,2
0,43 0,2 <7,48
0,163 0,315
51,75 -
69 <0,2
0,73 0,3 <4,87
0,304 0,515
59,03 -
35 <0,2
- 0,14 -
- 0,14
- 7
31 <0,2
- 0,08 -
- 0,08
- 0,19
60 <0,2
- 0,1 -
- 0,1
- 2,22
39 <0,2
- 0,06 -
- 0,06
- 0,19
t.o
61 <0,2
- 0,05 -
- 0,05
- 1,11
43 <0,2
- 0,06 -
- 0,06
- 0,19
65 <0,2
- 0,38 -
- 0,38
- 2,84
66 <0,2
- 0,012 -
- 0,012
- 0,02
47 <0,2
- 0,01 -
- 0,01
- 0,22
28 <0,2
- 0,03 -
- 0,03
- 0,22
29 <0,2
- 0,02 -
- 0,02
- -
67 <0,2
- 0,06 -
- 0,06
- 0,45
68 <0,2
- <0,01 -
- -
- -
7 <0,2
- 0,02 -
- 0,02
- 0,15
11 <0,2
- 0,02 -
- 0,02
-
51 <0,2
- <0,01 -
- -
- -
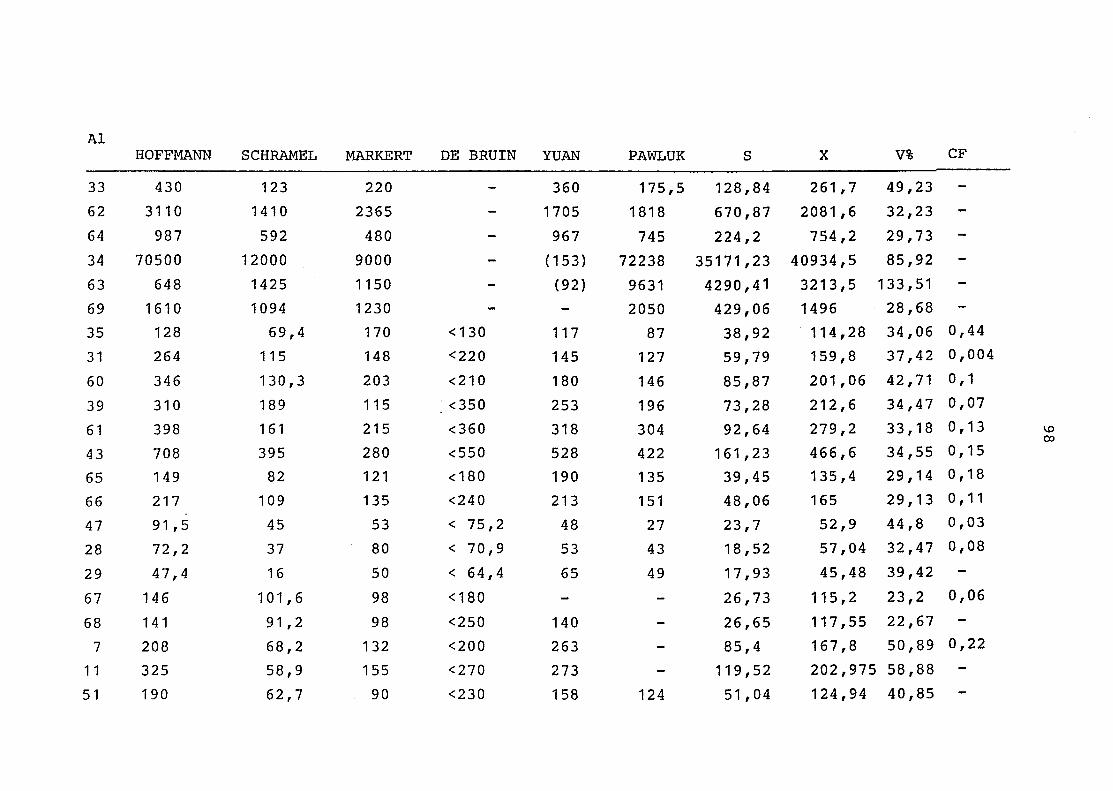
AlHOFFMANN SCHRAMEL MARKERT DE BRUIN YUAN PAWLUK S X V% CF
33 430 123 220 360 175,5 128,84 261,7 49,23 -
62 3110 1410 2365 1705 1818 670,87 2081,6 32,23 -
64 987 592 480 967 745 224,2 754,2 29,73 -
34 70500 12000 9000 - (153) 72238 35171,23 40934,5 85,92 -
63 648 1425 1150 - (92) 9631 4290,41 3213,5 133,51 -
69 1610 1094 1230 - 2050 429,06 1496 28,68 -
35 128 69,4 170 <13-0 117 87 38,92 114,28 34,06 0,44
31 264 115 148 <220 145 127 59,79 159,8 37,42 0,004
60 346 130,3 203 <210 180 146 85,87 201,06 42,71 0,1
39 310 189 115 :<350 253 196 73,28 212,6 34,47 0,07
61 398 161 215 <360 318 304 92,64 279,2 33,18 0,13
43 708 395 280 <550 528 422 161,23 466,6 34,55 0,15
65 149 82 121 <180 190 135 39,45 135,4 29,14 0,18
66 217 109 135 <240 213 151 48,06 165 29,13 0,11
47 91,5 45 53 <
75,2 48 27 23,7 52,9 44,8 0,03
28 72,2 37 80 <
70,9 53 43 18,52 57,04 32,47 0,08
29 47,4 16 50 <
64,4 65 49 17,93 45,48 39,42 -
67 146 101,6 98 <180 26,73 115,2 23,2 0,06
68 141 91,2 98 <250 140 - 26,65 117,55 22,67 -
7 208 68,2 132 <200 263 - 85,4 167,8 50,89 0,22
11 325 58,9 155 <270 273 - 119,52 202,975 58,88 -
51 190 62,7 90 <230 158 124 51,04 124,94 40,85 -
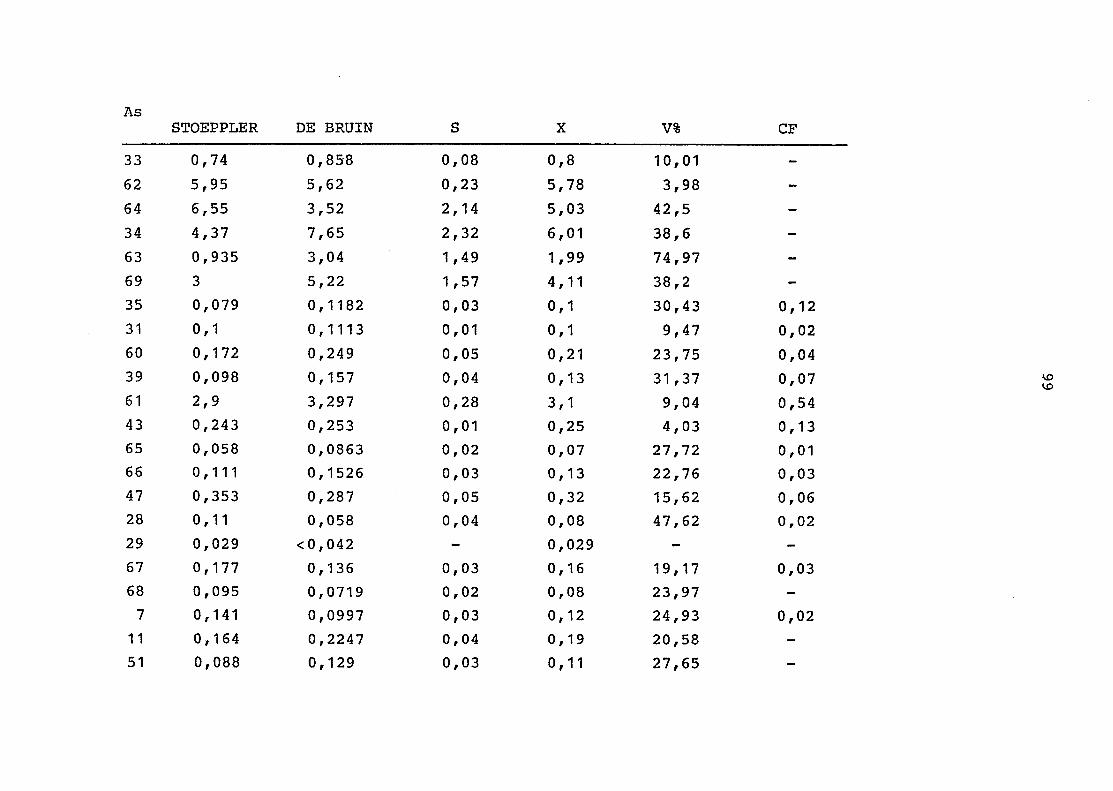
AsSTOEPPLER DE BRUIN S X V% CF
33 0,74 0,858 0,08 0,8 10,01 -
62 5,95 5,62 0,23 5,78 3,98 -
64 6,55 3,52 2,14 5,03 42,5 -
34 4,37 7,65 2,32 6,01 38,6 -
63 0,935 3,04 1,49 1,99 74,97 -
69 3 5,22 1,57 4,11 38,2 -
35 0,079 0,1182 0,03 0,1 30,43 0,12
31 0,1 0,1113 0,01 0,1 9,47 0,02
60 0,172 0,249 0,05 0,21 23,75 0,04
39 0,098 0,157 0,04 0,13 31,37 0,07
61 2,9 3,297 0,28 3,1 9,04 0,54
43 0,243 0,253 0,01 0,25 4,03 0,13
65 0,058 0,0863 0,02 0,07 27,72 0,01
66 0,111 0,1526 0,03 0,13 22,76 0,03
47 0,353 0,287 0,05 0,32 15,62 0,06
28 0,11 0,058 0,04 0,08 47,62 0,02
29 0,029 <0,042 - 0,029 - -
67 0,177 0,136 0,03 0,16 19,17 0,03
68 0,095 0,0719 0,02 0,08 23,97 -
7 0,141 0,0997 0,03 0,12 24,93 0,02
11 0,164 0,2247 0,04 0,19 20,58 -
51 0,088 0,129 0,03 0,11 27,65 -
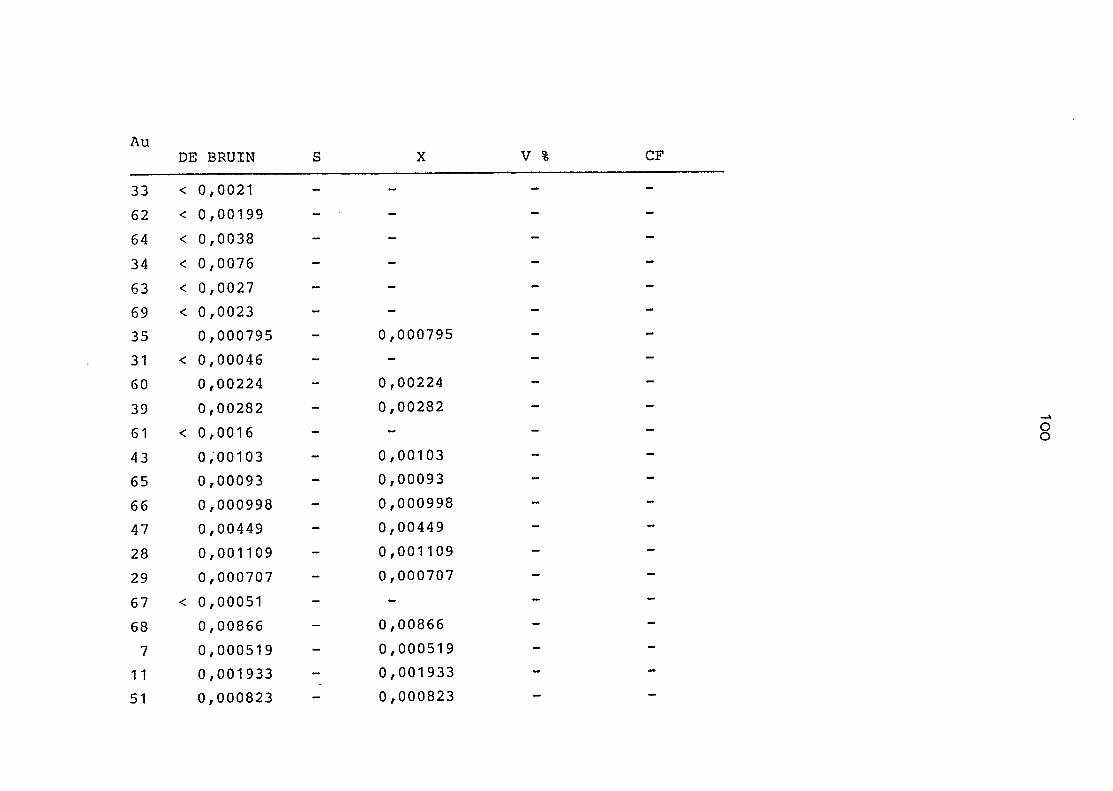
AuDE BRUIN
S
X
V %
CF
33
< 0,0021
-
-
62
< 0,00199
-
-
64
< 0,0038
-
-
34
< 0,0076
-
-
63
< 0,0027
-
-
69
< 0,0023
-
-
35 0,000795
-
0,000795
31
< 0,00046
-
-
60 0,00224
-
0,00224
39 0,00282
-
0,00282
61
< 0,0016
-
-
43 0,00103
-
0,00103
65 0,00093
-
0,00093
66 0,000998
-
0,000998
47 0,00449
-
0,00449
28 0,001109
-
0,001109
29 0,000707
-
0,000707
67
< 0,00051
-
-
68 0,00866
-
0,00866
7 0,000519
-
0,000519
11 0,001933
-
0,001933
51 0,000823
-
0,000823
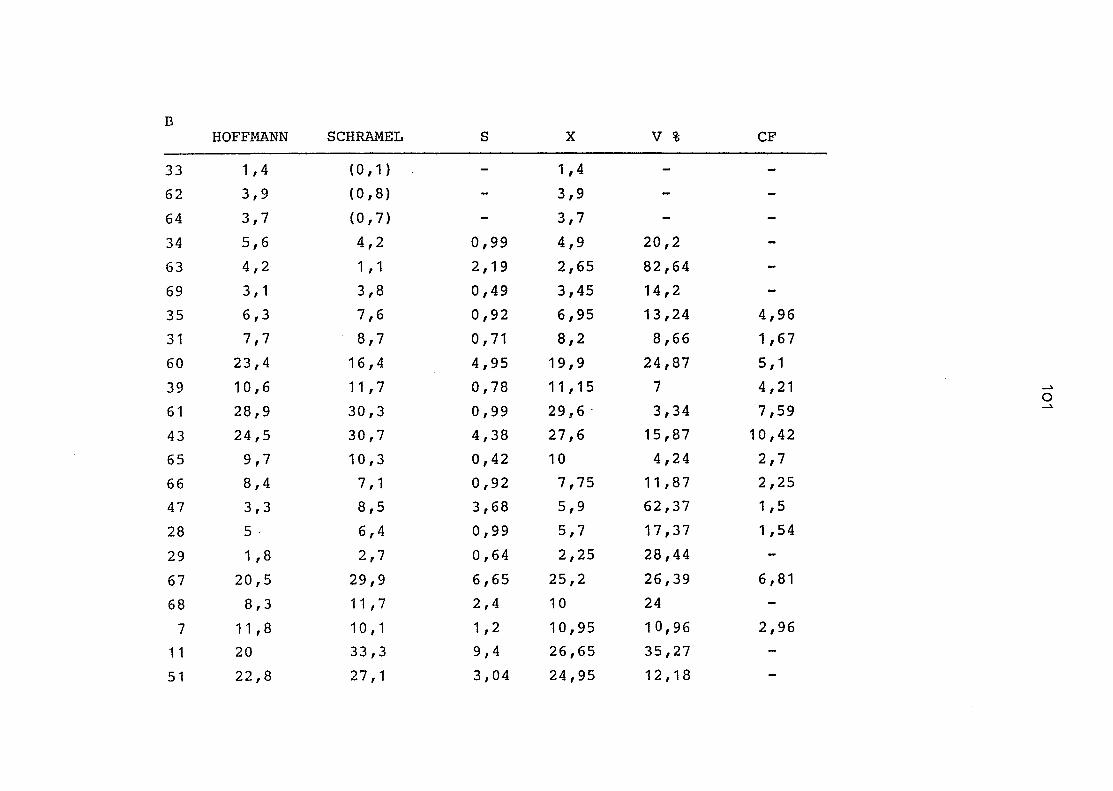
BHOFFMANN SCHRAMEL S X V % CF
33 1,4 (0,1) - 1,4 - -
62 3,9 (0,8) - 3,9 - -
64 3,7 (0,7) - 3,7 - -
34 5,6 4,2 0,99 4,9 20,2 -
63 4,2 1,1 2,19 2,65 82,64 -
69 3,1 3,8 0,49 3,45 14,2 -
35 6,3 7,6 0,92 6,95 13,24 4,96
31 7,7 8,7 0,71 8,2 8,66 1,67
60 23,4 16,4 4,95 19,9 24,87 5,1
39 10,6 11,7 0,78 11,15 7 4,21
61 28,9 30,3 0,99 29,6- 3,34 7,59
43 24,5 30,7 4,38 27,6 15,87 10,42
65 9,7 10,3 0,42 10 4,24 2,7
66 8,4 7,1 0,92 7,75 11,87 2,25
47 3,3 8,5 3,68 5,9 62,37 1,5
28 5• 6,4 0,99 5,7 17,37 1,54
29 1,8 2,7 0,64 2,25 28,44 -
67 20,5 29,9 6,65 25,2 26,39 6,81
68 8,3 11,7 2,4 10 24 -
7 11,8 10,1 1,2 10,95 10,96 2,96
11 20 33,3 9,4 26,65 35,27 -
51 22,8 27,1 3,04 24,95 12,18 -
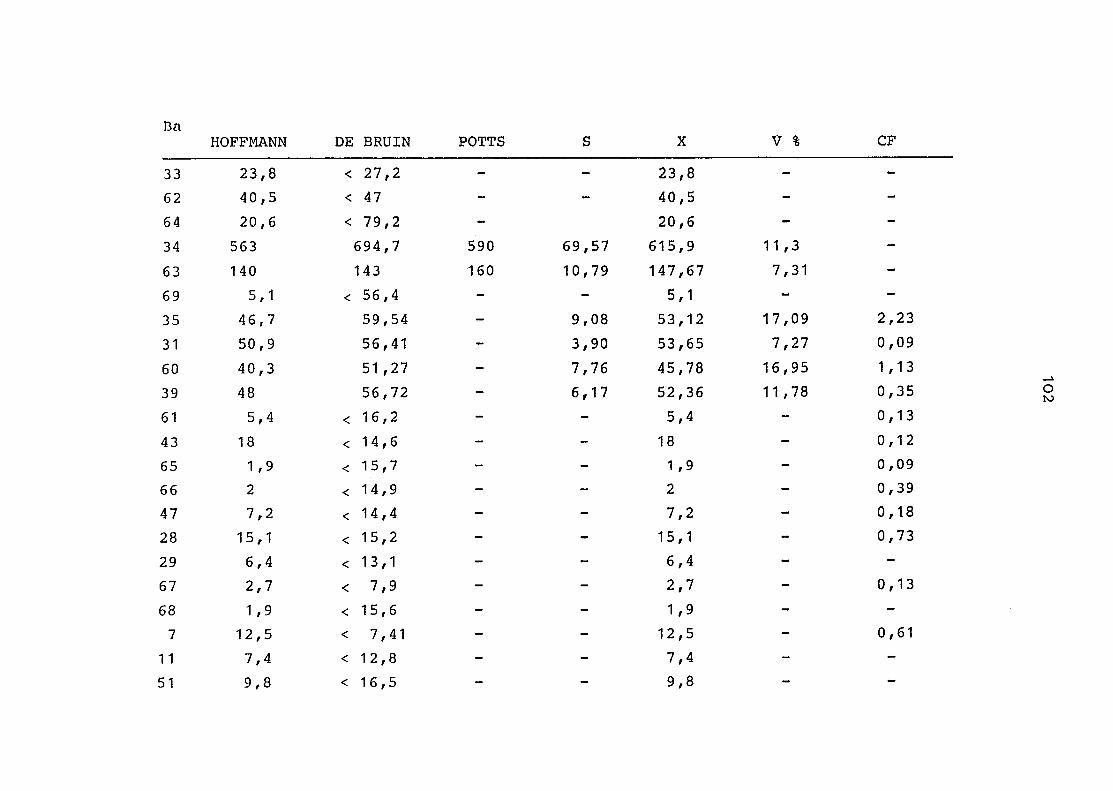
13aHOFFMANN
DE BRUSN
POTTS
S
X
V %
CP
33
23,8
< 27,2
-
-
23,8
-
-
62
40,5
< 47
-
-
40,5
-
-
64
20,6
< 79,2
-
20,6
-
-
34
563
694,7
590
69,57
615,9
11,3
-
63
140
143
160
10,79
147,67
7,31
-
69
5,1
< 56,4
-
-
5,1
-
-
35
46,7
59,54
-
9,08
53,12
17,09
2,23
31
50,9
56,41
-
3,90
53,65
7,27
0,09
60
40,3
51,27
-
7,76
45,78
16,95
1,13
39
48
56,72
-
6,17
52,36
11,78
0,35
61
5,4
< 16,2
-
-
5,4
-
0,13
43
18
< 14,6
-
-
18
-
0,12
65
1,9
< 15,7
-
-
1,9
-
0,09
66
2
< 14,9
-
-
2
-
0,39
47
7,2
< 14,4
-
-
7,2
-
0,18
28
15,1
< 15,2
-
-
15,1
-
0,73
29
6,4
< 13,1
-
-
6,4
-
-
67
2,7
< 7,9
-
-
2,7
-
0,13
68
1,9
< 15,6
-
-
1,9
-
-
7
12,5
< 7,41
-
-
12,5
-
0,61
11
7,4
< 12,8
-
-
7,4
-
-
51
9,8
< 16,5
-
-
9,8
-
-
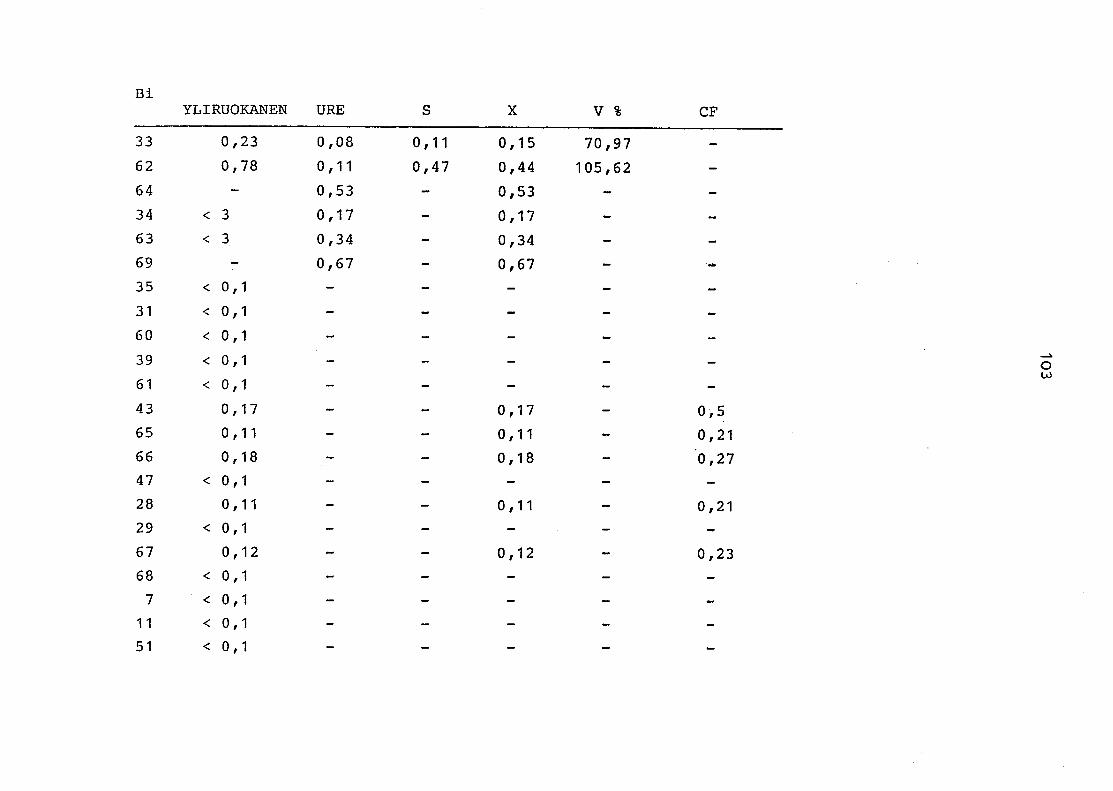
BiYLIRUOKANEN URE
S
X
V %
C'
33
0,23
0,08
0,11
0,15
70,97
62
0,78
0,11
0,47
0,44
105,62
64
-
0,53
-
0,53
-
34
< 3
0,17
0,17
-
63
< 3
0,34
-
0,34
-
69
-
0,67
-
0,67
-
35
< 0,1
-
-
-
-
31
< 0,1
-
-
-
60
< 0,1
-
-
-
-
39
< 0,1
-
-
-
-
61
< 0,1
-
-
-
-
43
0,17
-
-
0,17
-
65
0,11
-
-
0,11
-
66
0,18
-
-
0,18
-
47
< 0,1
-
-
-
-
28
0,11
-
-
0,11
-
29
< 0,1
-
-
-
-
67
0,12
-
-
0,12
-
68
< 0,1
-
-
-
-
7
< 0,1
-
-
-
-
11
< 0,1
-
-
-
-
51
< 0,1
-
-
-
-
0,5
0,21
0,27
0,21
0,23
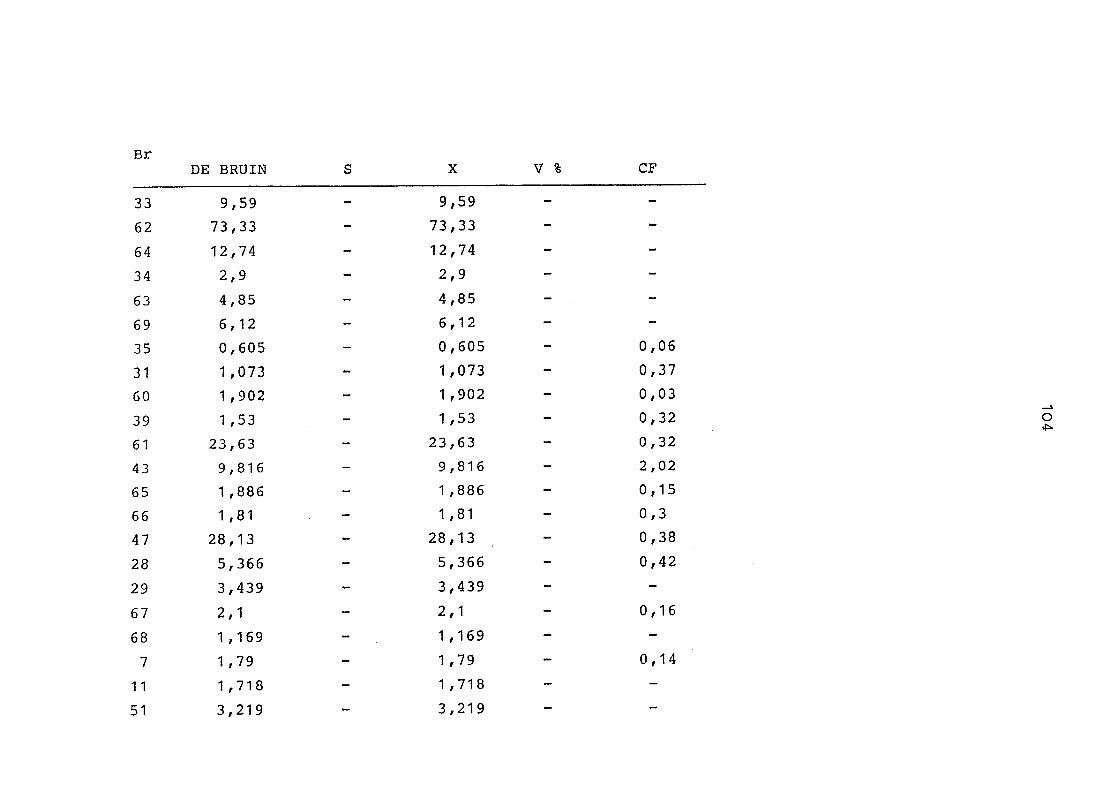
BrDE BRUIN
S X
V % CF
33 9,59
- 9,59
- -
62 73,33
- 73,33
- -
64 12,74
- 12,74
- -
34 2,9
- 2,9
- -
63 4,85
- 4,85
- -
69 6,12
- 6,12
- -
35 0,605
- 0,605
- 0,06
31 1,073
- 1,073
- 0,37
60 1,902
- 1,902
- 0,03
39 1,53
- 1,53
- 0,32
61 23,63
- 23,63
- 0,32
43 9,816
- 9,816
- 2,02
65 1,886
- 1,886
- 0,15
66 1,81
- 1,81
- 0,3
47 28,13
- 28,13
- 0,38
28 5,366
- 5,366
- 0,42
29 3,439
- 3,439
- -
67 2,1
- 2,1
- 0,16
68 1,169
- 1,169
- -
7 1,79
- 1,79
- 0,14
11 1,718
- 1,718
- -
51 3,219
- 3,219
- -
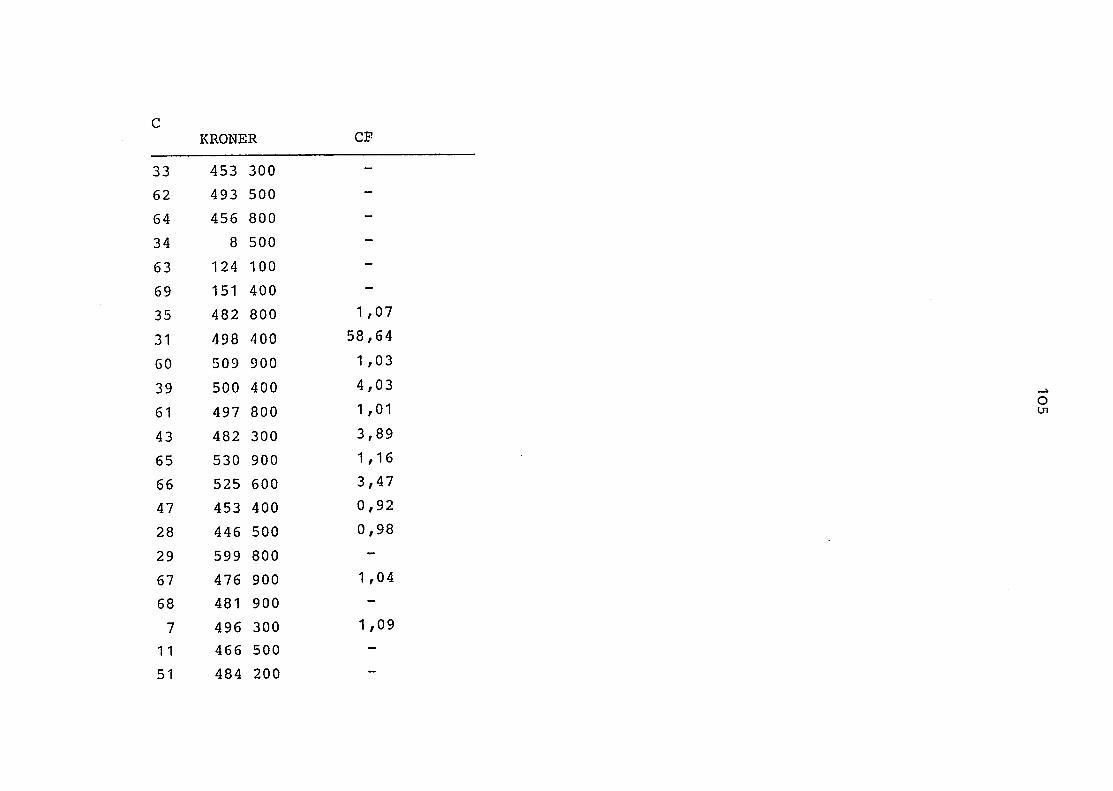
CKRONER
CF
33 453 300
62 493 500
64 456 800
34 8 500
63 124 100
69 151 400
35 482 800 1,07
31 498 400 58,64
60 509 900 1,03
39 500 400 4,03
61 497 800 1,01
43 482 300 3,89
65 530 900 1,16
66 525 600 3,47
47 453 400 0,92
28 446 500 0,98
29 599 800
67 476 900 1,04
68 481 900
7 496 300 1,09
11 466 500
51 484 200
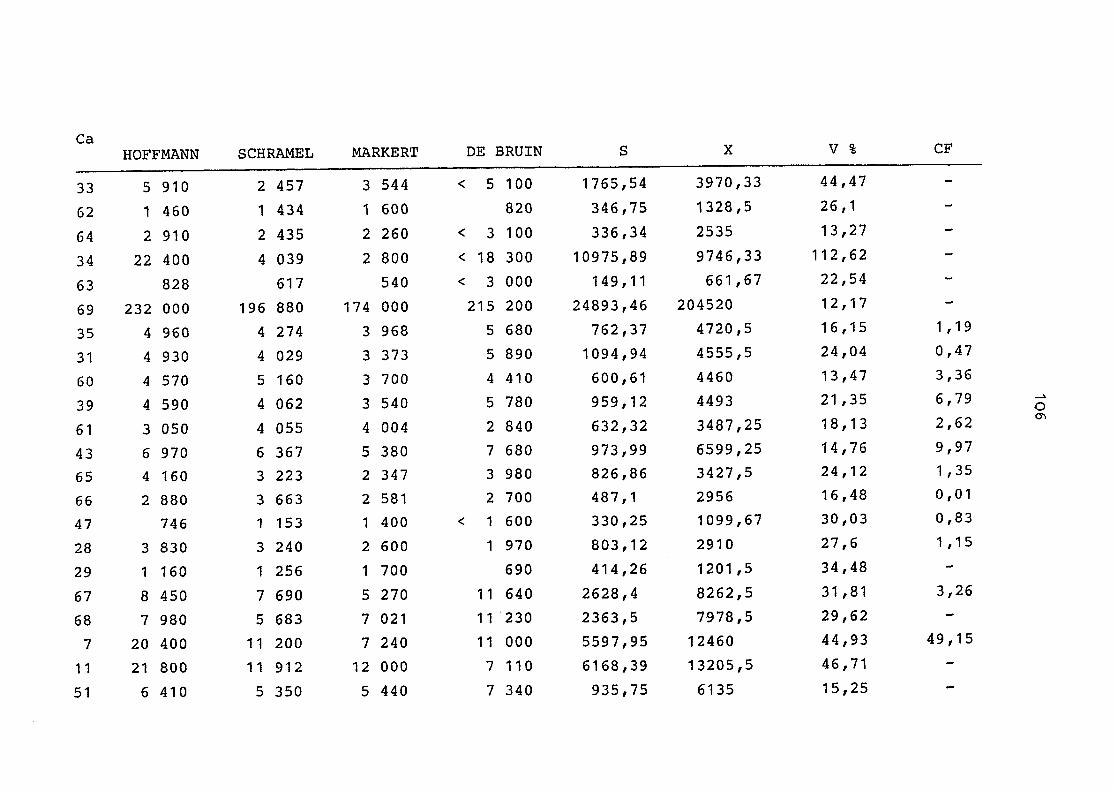
Ca HOFFMANN
SCHRAMEL
MARKE RT
DE BRUIN
S
X
V %
CF
33 5 910 2 457 3 544
< 5 100 1765,54 3970,33 44,47
62 1 460 1 434 1 600 820 346,75 1328,5 26,1
64 2 910 2 435 2 260
< 3 100 336,34 2535 13,27
34 22 400 4 039 2 800
< 18 300 10975,89 9746,33 112,62
63 828 617 540
< 3 000 149,11 661,67 22,54
69 232 000 196 880 174 000 215 200 24893,46 204520 12,17
35 4 960 4 274 3 968 5 680 762,37 4720,5 16,15 1,19
31 4 930 4 029 3 373 5 890 1094,94 4555,5 24,04 0,47
60 4 570 5 160 3 700 4 410 600,61 4460 13,47 3,36
39 4 590 4 062 3 540 5 780 959,12 4493 21,35 6,79
61 3 050 4 055 4 004 2 840 632,32 3487,25 18,13 2,62
43 6 970 6 367 5 380 7 680 973,99 6599,25 14,76 9,97
65 4 160 3 223 2 347 3 980 826,86 3427,5 24,12 1,35
66 2 880 3 663 2 581 2 700 487,1 2956 16,48 0,01
47 746 1 153 1 400
< 1 600 330,25 1099,67 30,03 0,83
28 3 830 3 240 2 600 1 970 803,12 2910 27,6 1,15
29 1 160 1 256 1 700 690 414,26 1201,5 34,48
67 8 450 7 690 5 270 11 640 2628,4 8262,5 31,81 3,26
68 7 980 5 683 7 021 11 230 2363,5 7978,5 29,62
7 20 400 11 200 7 240 11 000 5597,95 12460 44,93 49,15
11 21 800 11 912 12 000 7 110 6168,39 13205,5 46,71
51 6 410 5 350 5 440 7 340 935,75 6135 15,25
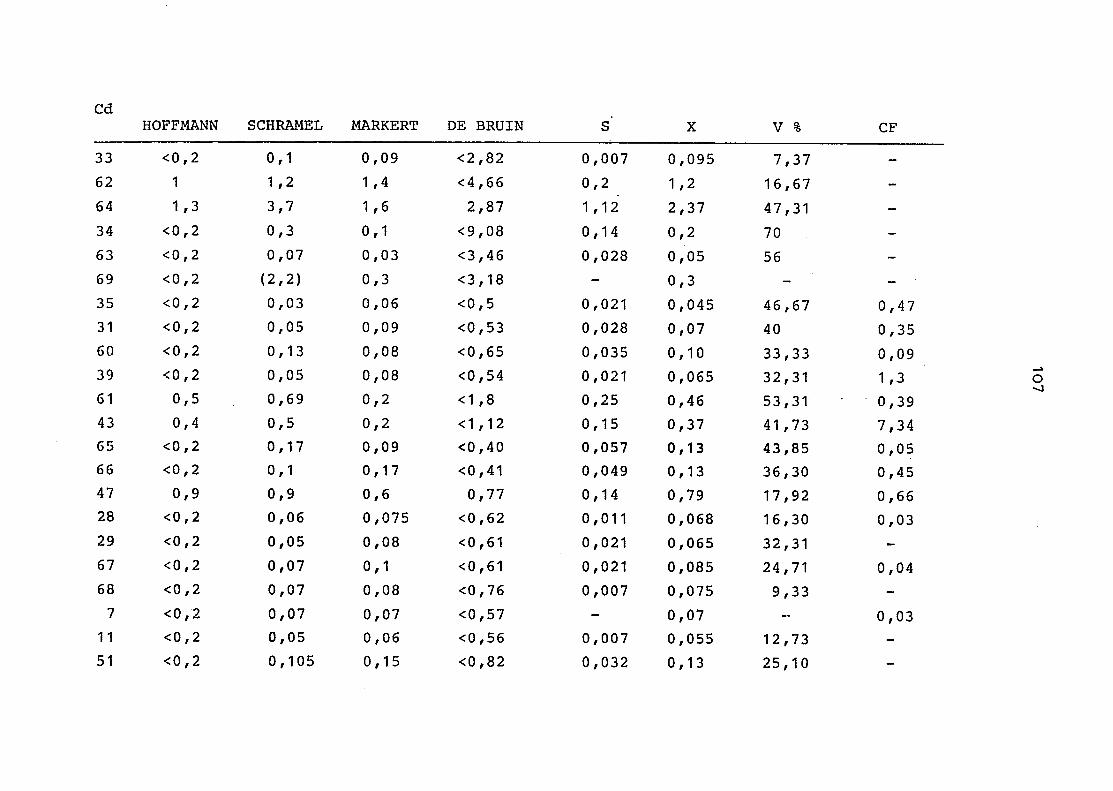
CdHOFFMANN SCHRAMEL MARKERT DE BRUIN S X V % CF
33 <0,2 0,1 0,09 <2,82 0,007 0,095 7,37 -
62 1 1,2 1,4 <4,66 0,2 1,2 16,67 -
64 1,3 3,7 1,6 2,87 1,12 2,37 47,31 -
34 <0,2 0,3 0,1 <9,08 0,14 0,2 70 -
63 <0,2 0,07 0,03 <3,46 0,028 0,05 56 -
69 <0,2 (2,2) 0,3 <3,18 - 0,3 - -
35 <0,2 0,03 0,06 <0,5 0,021 0,045 46,67 0,47
31 <0,2 0,05 0,09 <0,53 0,028 0,07 40 0,35
60 <0,2 0,13 0,08 <0,65 0,035 0,10 33,33 0,09
39 <0,2 0,05 0,08 <0,54 0,021 0,065 32,31 1,3
61 0,5 0,69 0,2 <1,8 0,25 0,46 53,31 0,39
43 0,4 0,5 0,2 <1,12 0,15 0,37 41,73 7,34
65 <0,2 0,17 0,09 <0,40 0,057 0,13 43,85 0,05
66 <0,2 0,1 0,17 <0,41 0,049 0,13 36,30 0,45
47 0,9 0,9 0,6 0,77 0,14 0,79 17,92 0,66
28 <0,2 0,06 0,075 <0,62 0,011 0,068 16,30 0,03
29 <0,2 0,05 0,08 <0,61 0,021 0,065 32,31 -
67 <0,2 0,07 0,1 <0,61 0,021 0,085 24,71 0,04
68 <0,2 0,07 0,08 <0,76 0,007 0,075 9,33 -
7 <0,2 0,07 0,07 <0,57 - 0,07 0,03
11 <0,2 0,05 0,06 <0,56 0,007 0,055 12,73 -
51 <0,2 0,105 0,15 <0,82 0,032 0,13 25,10 -
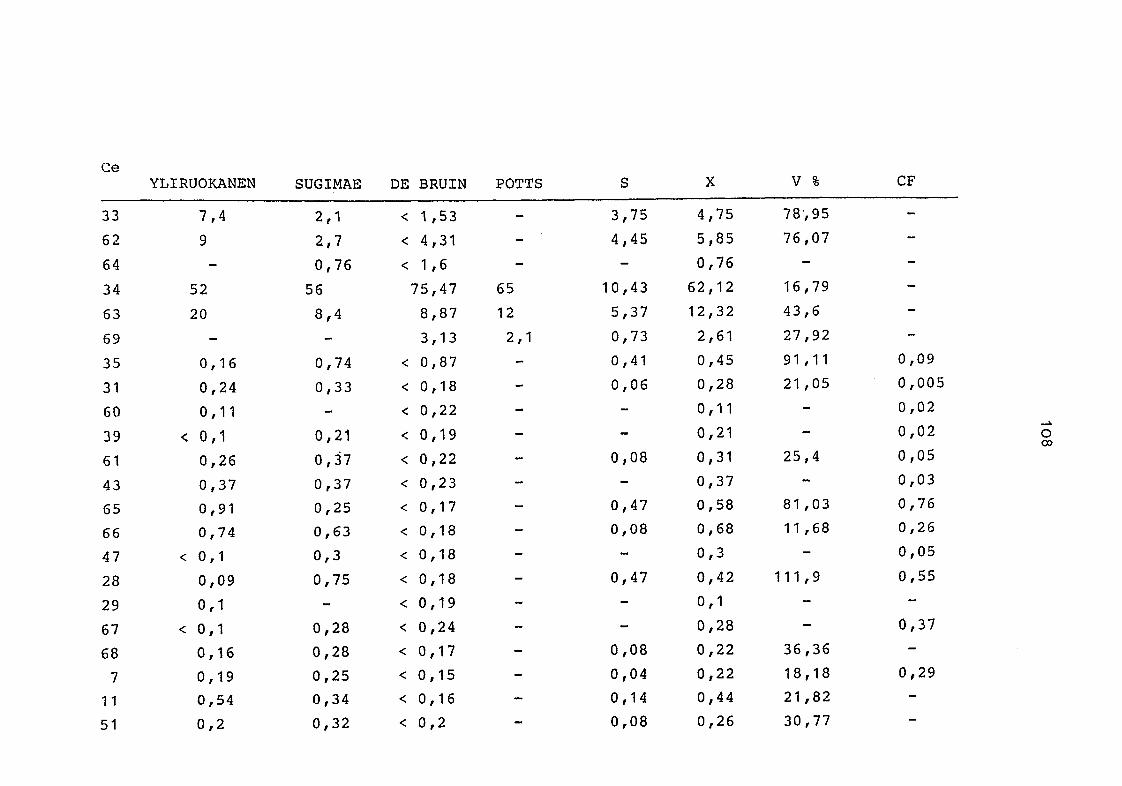
CeS X V % CFYLIRUOKANEN
SUGIMAE
DE BRUIN
POTTS
33
7,4
2,1
< 1,53
- 3,75 4,75 78,95
62
9
2,7
< 4,31
- 4,45 5,85 76,07
64
-
0,76
< 1,6
- - 0,76 -
34
52
56 75,47
65 10,43 62,12 16,79
63
20
8,4 8,87
12 5,37 12,32 43,6
69
-
- 3,13
2,1 0,73 2,61 27,92
35
0,16
0,74
< 0,87
- 0,41 0,45 91,11 0,09
31
0,24
0,33
< 0,18
- 0,06 0,28 21,05 0,005
60
0,11
-
< 0,22
- - 0,11 - 0,02
39
<
0,1
0,21
< 0,19
- - 0,21 - 0,02
61
0,26
0,37
< 0,22
- 0,08 0,31 25,4 0,05
43
0,37
0,37
< 0,23
- - 0,37 - 0,03
65
0,91
0,25
< 0,17
- 0,47 0,58 81,03 0,76
66
0,74
0,63
< 0,18
- 0,08 0,68 11,68 0,26
47
<
0,1
0,3
< 0,18
- - 0,3 - 0,05
28
0,09
0,75
< 0,18
- 0,47 0,42 111,9 0,55
29
0,1
-
< 0,19
- - 0,1 -
67
< 0,1
0,28
< 0,24
- - 0,28 - 0,37
68
0,16
0,28
< 0,17
- 0,08 0,22 36,36
7
0,19
0,25
< 0,15
- 0,04 0,22 18,18 0,29
11
0,54
0,34
< 0,16
- 0,14 0,44 21,82
51
0,2
0,32
< 0,2
- 0,08 0,26 30,77
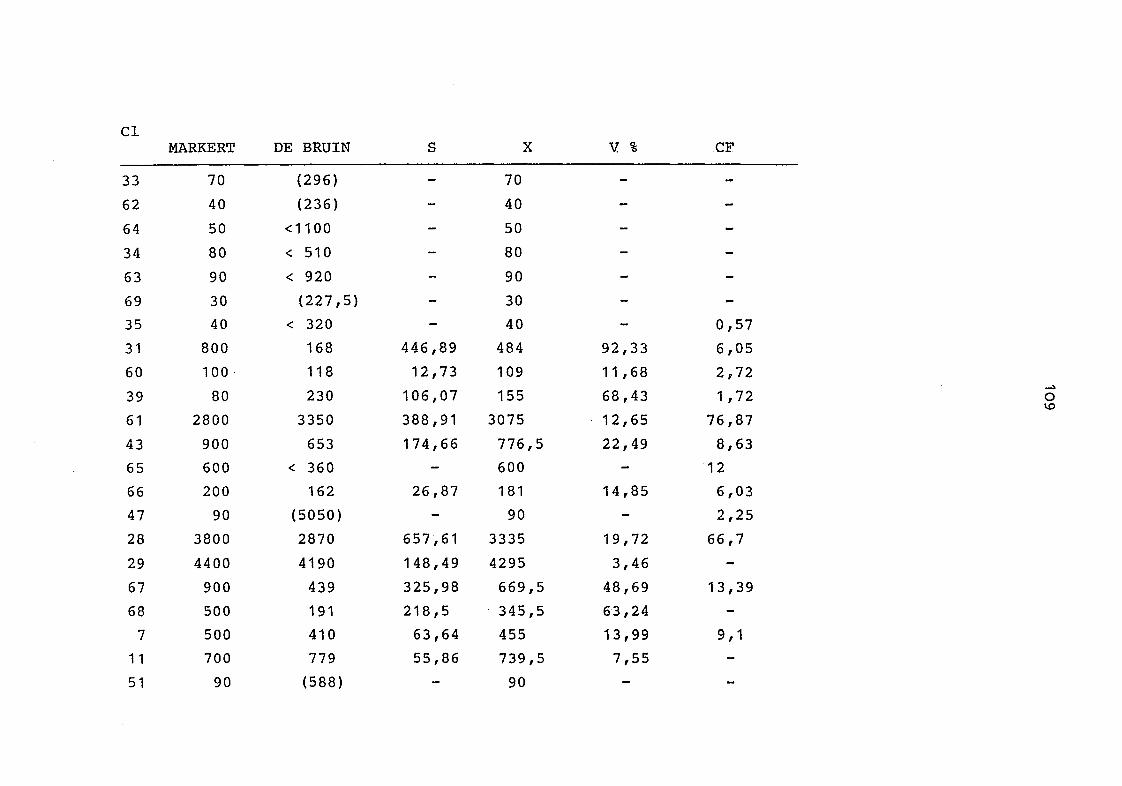
Cl
MARKE RT
DE BRUIN
S X
V. % CE
33 70
(296)
- 70
- -
62 40
(236)
- 40
- -
64 50
<1100
- 50
- -
34 80
<
510
- 80
- -
63 90
< 920
- 90
- -
69 30
(227,5)
- 30
- -
35 40
< 320
- 40
- 0,57
31 800
168
446,89 484
92,33 6,05
60 100 .118
12,73 109
11,68 2,72
39 80
230
106,07 155
68,43 1,72
61 2800
3350
388,91 3075
12,65 76,87
43 900
653
174,66 776,5
22,49 8,63
65 600
< 360
- 600
- 12
66 200
162
26,87 181
14,85 6,03
47 90
(5050)
- 90
- 2,25
28 3800
2870
657,61 3335
19,72 66,7
29 4400
4190
148,49 4295
3,46 -
67 900
439
325,98 669,5
48,69 13,39
68 500
191
218,5 345,5
63,24 -
7 500
410
63,64 455
13,99 9,1
11 700
779
55,86 . 739,5
7,55 -
51 90
(588)
- 90
- -
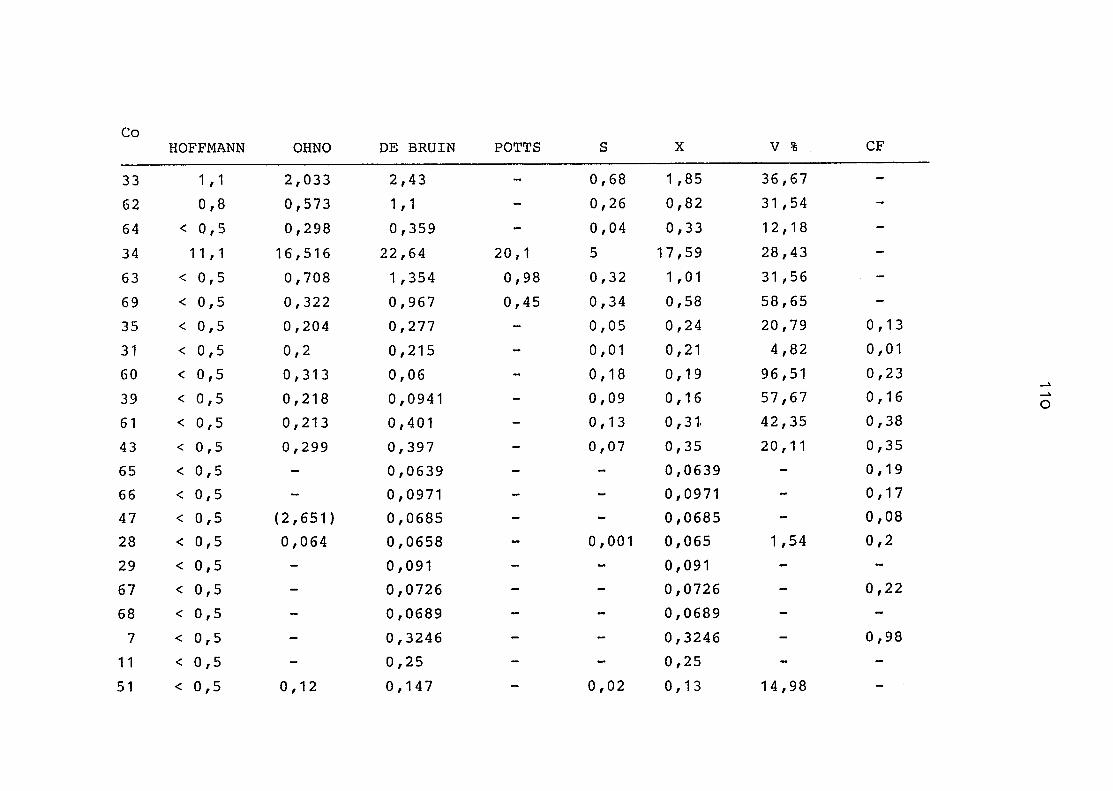
CoOHNO DE BRUIN POTTS S X V % CFHOFFMANN
33 1,1 2,033 2,43 - 0,68 1,85 36,67 -
62 0,8 0,573 1,1 - 0,26 0,82 31,54 -
64
< 0,5 0,298 0,359 - 0,04 0,33 12,18 -
34 11,1 16,516 22,64 20,1 5 17,59 28,43 -
63
< 0,5 0,708 1,354 0,98 0,32 1,01 31,56 -
69
< 0,5 0,322 0,967 0,45 0,34 0,58 58,65 -
35
< 0,5 0,204 0,277 - 0,05 0,24 20,79 0,13
31
< 0,5 0,2 0,215 - 0,01 0,21 4,82 0,01
60
< 0,5 0,313 0,06 - 0,18 0,19 96,51 0,23
39
< 0,5 0,218 0,0941 - 0,09 0,16 57,67 0,16
61
< 0,5 0,213 0,401 - 0,13 0,31 42,35 0,38
43
< 0,5 0,299 0,397 - 0,07 0,35 20,11 0,35
65
< 0,5 - 0,0639 - - 0,0639 - 0,19
66
< 0,5 - 0,0971 - - 0,0971 - 0,17
47
< 0,5 (2,651) 0,0685 - - 0,0685 - 0,08
28
< 0,5 0,064 0,0658 - 0,001 0,065 1,54 0,2
29
< 0,5 - 0,091 - - 0,091 - -
67
< 0,5 - 0,0726 - - 0,0726 - 0,22
68
< 0,5 - 0,0689 - - 0,0689 - -
7
< 0,5 - 0,3246 - - 0,3246 - 0,98
11
< 0,5 - 0,25 - - 0,25 - -
51
< 0,5 0,12 0,147 - 0,02 0,13 14,98 -
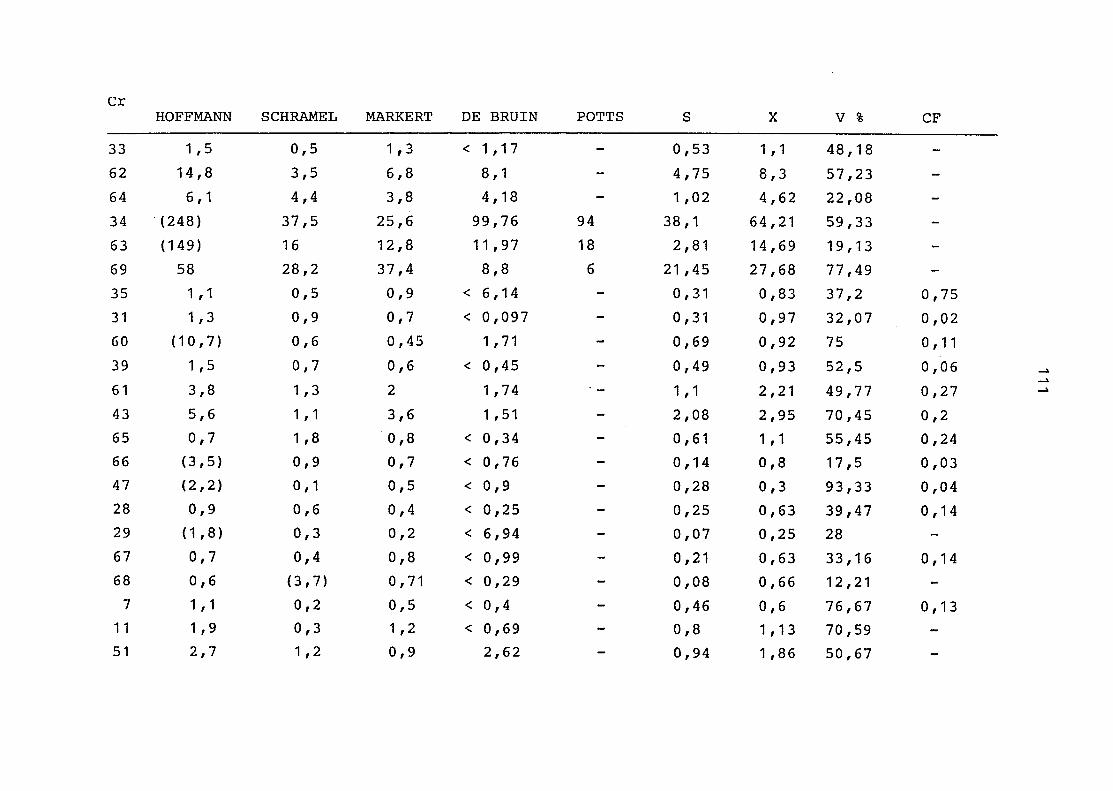
CrHOFFMANN SCHRAMEL MARKE RT
DE BRUIN
POTTS S X V % CF
33 1,5 0,5 1,3
< 1,17
- 0,53 1,1 48,18 -
62 14,8 3,5 6,8 8,1
- 4,75 8,3 57,23 -
64 6,1 4,4 3,8 4,18
- 1,02 4,62 22,08 -
34 (248) 37,5 25,6 99,76
94 38,1 64,21 59,33 -
63 (149) 16 12,8 11,97
18 2,81 14,69 19,13 -
69 58 28,2 37,4 8,8
6 21,45 27,68 77,49 -
35 1,1 0,5 0,9
< 6,14
- 0,31 0,83 37,2 0,75
31 1,3 0,9 0,7
< 0,097
- 0,31 0,97 32,07 0,02
GO (10,7) 0,6 0,45 1,71
- 0,69 0,92 75 0,11
39 1,5 0,7 0,6
< 0,45
- 0,49 0,93 52,5 0,06
61 3,8 1,3 2 1,74
- 1,1 2,21 49,77 0,27
43 5,6 1,1 3,6 1,51
- 2,08 2,95 70,45 0,2
65 0,7 1,8 '0,8
< 0,34
- 0,61 1,1 55,45 0,24
66 (3,5) 0,9 0,7
< 0,76
- 0,14 0,8 17,5 0,03
47 (2,2) 0,1 0,5
< 0,9
- 0,28 0,3 93,33 0,04
28 0,9 0,6 0,4
< 0,25
- 0,25 0,63 39,47 0,14
29 (1,8) 0,3 0,2
< 6,94
- 0,07 0,25 28 -
67 0,7 0,4 0,8
< 0,99
- 0,21 0,63 33,16 0,14
68 0,6 (3,7) 0,71
< 0,29
- 0,08 0,66 12,21 -
7 1,1 0,2 0,5
< 0,4
- 0,46 0,6 76,67 0,13
11 1,9 0,3 1,2
< 0,69
- 0,8 1,13 70,59 -
51 2,7 1,2 0,9 2,62
- 0,94 1,86 50,67 -
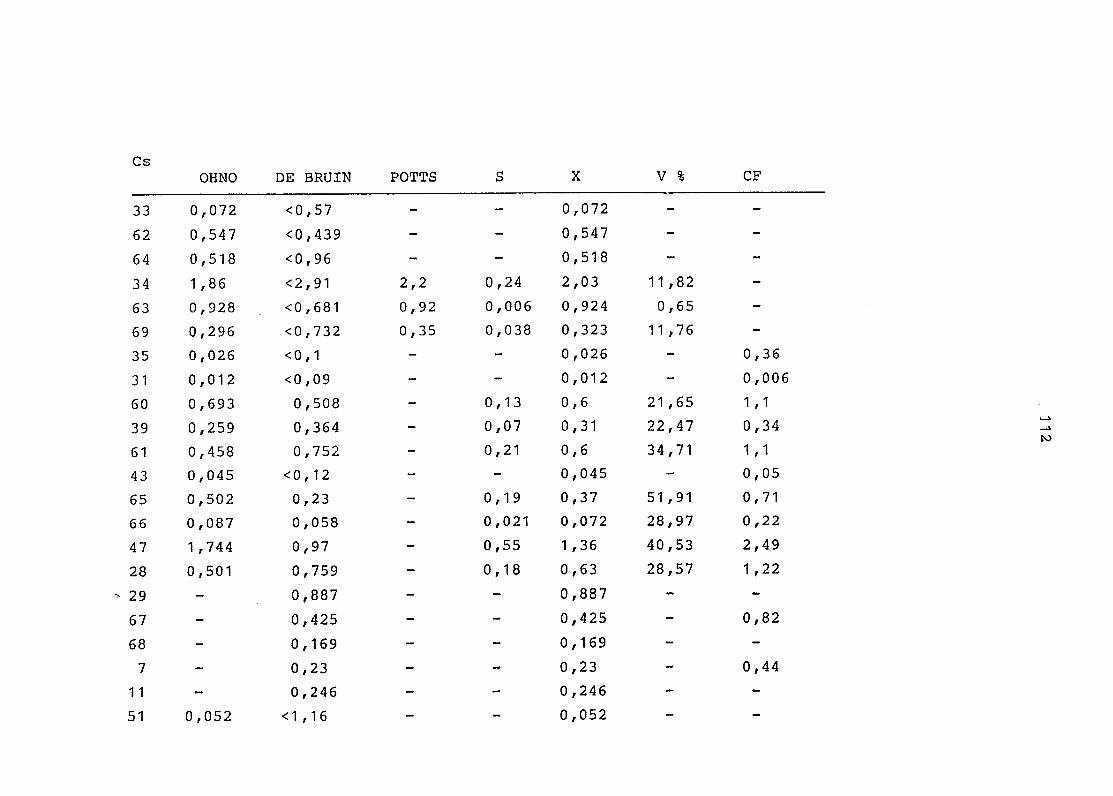
CsOHNO DE BRUIN POTTS S X V % CF
33 0,072 <0,57 - - 0,072 - -
62 0,547 <0,439 - - 0,547 - -
64 0,518 <0,96 - - 0,518 - -
34 1,86 <2,91 2,2 0,24 2,03 11,82 -
63 0,928
_ <0,681 0,92 0,006 0,924 0,65 -
69 0,296 <0,732 0,35 0,038 0,323 11,76 -
35 0,026 <0,1 - - 0,026 - 0,36
31 0,012 <0,09 - - 0,012 - 0,006
60 0,693 0,508 - 0,13 0,6 21,65 1,1
39 0,259 0,364 - 0,07 0,31 22,47 0,34
61 0,458 0,752 - 0,21 0,6 34,71 1,1
43 0,045 <0,12 - - 0,045 - 0,05
65 0,502 0,23 - 0,19 0,37 51,91 0,71
66 0,087 0,058 - 0,021 0,072 28,97 0,22
47 1,744 0,97 - 0,55 1,36 40,53 2,49
28 0,501 0,759 - 0,18 0,63 28,57 1,22
29 - 0,887 - - 0,887 - -
67 - 0,425 - - 0,425 - 0,82
68 - 0,169 - - 0,169 - -
7 - 0,23 - - 0,23 - 0,44
11 - 0,246 - - 0,246 - -
51 0,052 <1,16 - - 0,052 - -
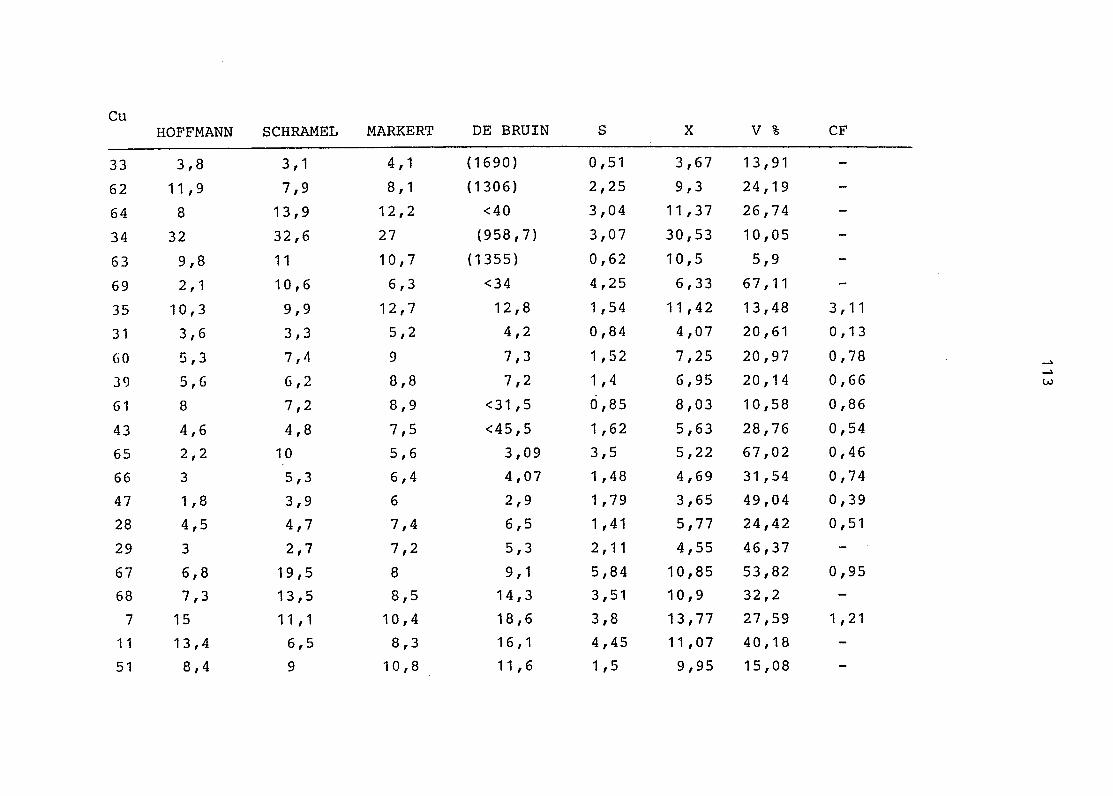
CuHOFFMANN SCHRAMEL MARKERT DE BRUIN S X V % CF
33 3,8 3,1 4,1 (1690) 0,51 3,67 13,91 -
62 11,9 7,9 8,1 (1306) 2,25 9,3 24,19 -
64 8 13,9 12,2 <40 3,04 11,37 26,74 -
34 32 32,6 27 (958,7) 3,07 30,53 10,05 -
63 9,8 11 10,7 (1355) 0,62 10,5 5,9 -
69 2,1 10,6 6,3 <34 4,25 6,33 67,11 -
35 10,3 9,9 12,7 12,8 1,54 11,42 13,48 3,11
31 3,6 3,3 5,2 4,2 0,84 4,07 20,61 0,13
60 5,3 7,4 9 7,3 1,52 7,25 20,97 0,78
39 5,6 6,2 8,8 7,2 1,4 6,95 20,14 0,66
61 8 7,2 8,9 <31,5 0,85 8,03 10,58 0,86
43 4,6 4,8 7,5 <45,5 1,62 5,63 28,76 0,54
65 2,2 10 5,6 3,09 3,5 5,22 67,02 0,46
66 3 5,3 6,4 4,07 1,48 4,69 31,54 0,74
47 1,8 3,9 6 2,9 1,79 3,65 49,04 0,39
28 4,5 4,7 7,4 6,5 1,41 5,77 24,42 0,51
29 3 2,7 7,2 5,3 2,11 4,55 46,37 -
67 6,8 19,5 8 9,1 5,84 10,85 53,82 0,95
68 7,3 13,5 8,5 14,3 3,51 10,9 32,2 -
7 15 11,1 10,4 18,6 3,8 13,77 27,59 1,21
11 13,4 6,5 8,3 16,1 4,45 11,07 40,18 -
51 8,4 9 10,8 11,6 1,5 9,95 15,08 -
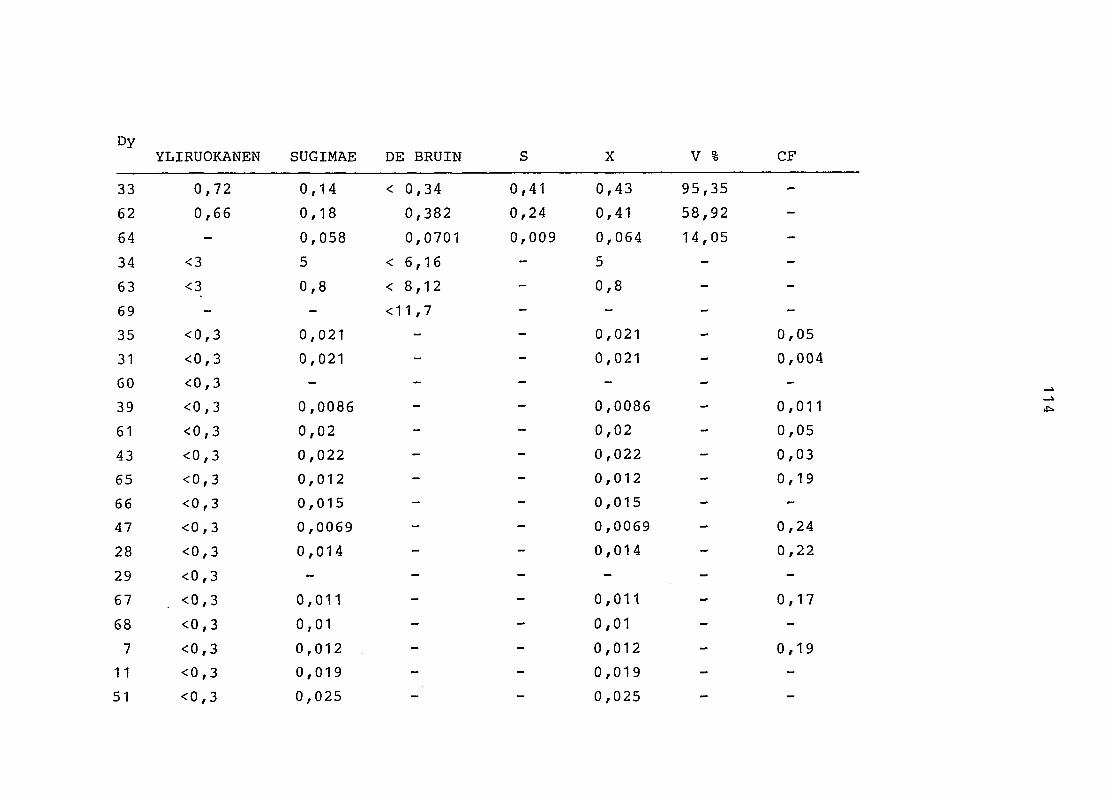
DyYLIRUOKANEN
SUGIMAE
DE BRUIN
S
X
V % CF
33 0,72
0,14
<
0,34
0,41
0,43
95,35 -
62 0,66
0,18
0,382
0,24
0,41
58,92 -
64 -
0,058
0,0701
0,009
0,064
14,05 -
34 <3
5
< 6,16
-
5
- -
63 <3
0,8
<
8,12
-
0,8
- -
69 -
-
<11,7
-
-
-
35 <0,3
0,021
-
-
0,021
- 0,05
31 <0,3
0,021
-
-
0,021
- 0,004
60 <0,3 -
39 <0,3
0,0086
-
-
0,0086
- 0,011
61 <0,3
0,02
-
-
0,02
- 0,05
43 <0,3
0,022
-
-
0,022
- 0,03
65 <0,3
0,012
-
-
0,012
- 0,19
66 <0,3
0,015
-
-
0,015
- -
47 <0,3
0,0069
-
-
0,0069
- 0,24
28 <0,3
0,014
-
-
0,014
- 0,22
29 <0,3 -
67 <0,3
0,011
-
-
0,011
- 0,17
68 <0,3
0,01
-
-
0,01
- -
7 <0,3
0,012
-
-
0,012
- 0,19
11 <0,3
0,019
-
-
0,019
- -
51 <0,3
0,025
-
-
0,025
- -
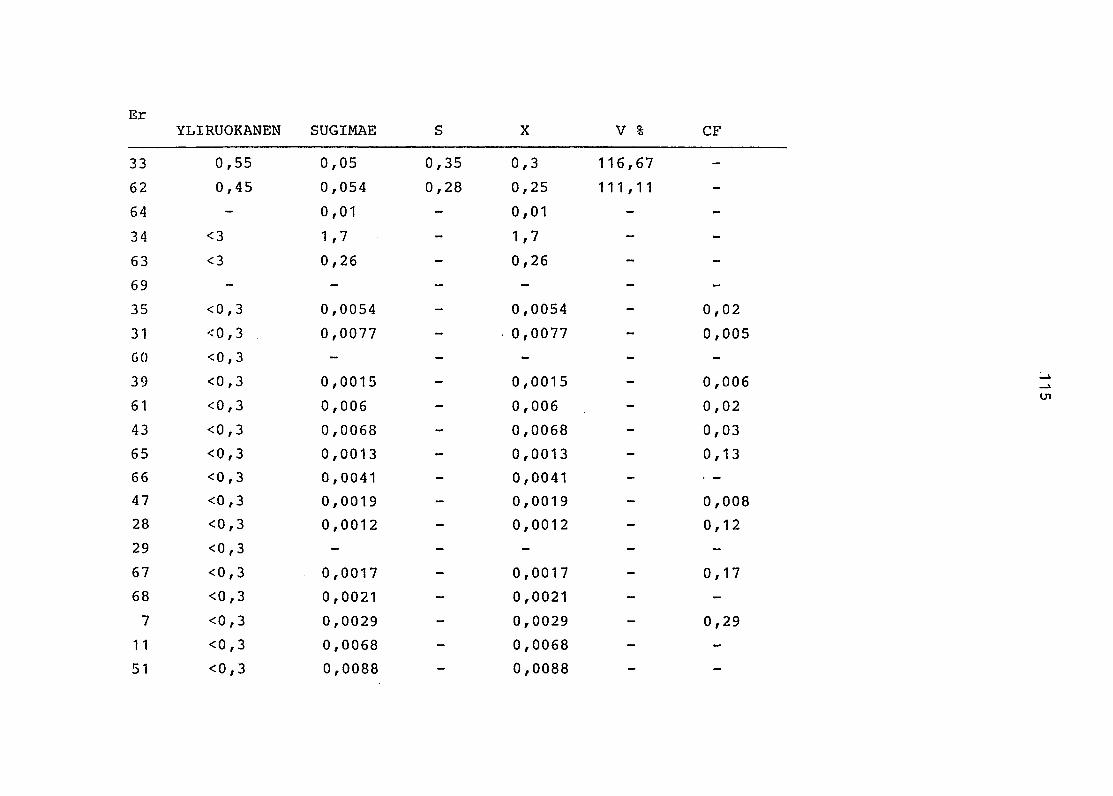
ErYLIRUOKANEN
SUGIMAE
S
X
V % CF
33
0,55
0,05
0,35
0,3
116,67 -
62
0,45
0,054
0,28
0,25
111,11 -
64
-
0,01
-
0,01
- -
34
<3
1,7
-
1,7
- -
63
<3
0,26
-
0,26
- -
69 -
35
<0,3
0,0054
-
0,0054
- 0,02
31
<0,3
0,0077
-
0,0077
- 0,005
GO
<0,3
-
-
- -
39
<0,3
0,0015
-
0,0015
- 0,006
61
<0,3
0,006
-
0,006
- 0,02
43
<0,3
0,0068
-
0,0068
- 0,03
65
<0,3
0,0013
-
0,0013
- 0,13
66
<0,3
0,0041
-
0,0041
- -
47
<0,3
0,0019
-
0,0019
- 0,008
28
<0,3
0,0012
-
0,0012
- 0,12
29
<0,3
-
-
-
- -
67
<0,3
0,0017
-
0,0017
- 0,17
68
<0,3
0,0021
-
0,0021
- -
7
<0,3
0,0029
-
0,0029
- 0,29
11
<0,3
0,0068
-
0,0068
- -
51
<0,3
0,0088
-
0,0088
- -
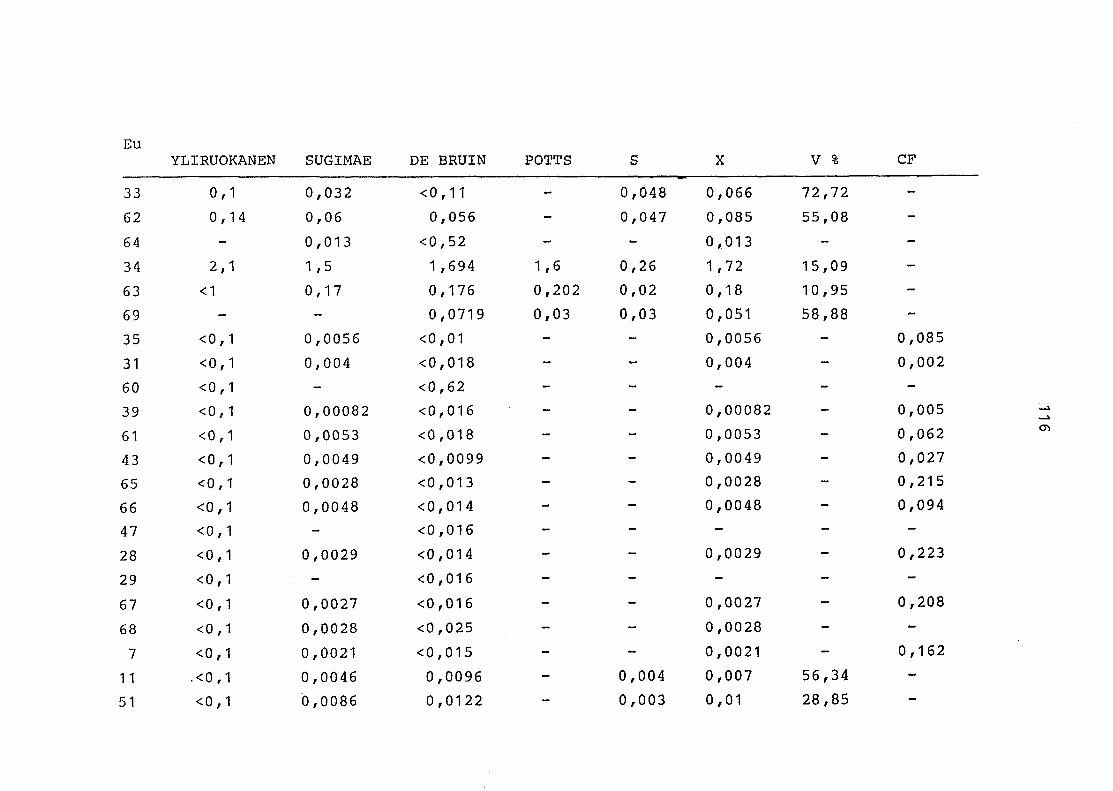
Eu
YLIRUOKANEN SUGIMAE DE BRUIN POTTS
S
X
V % CF
33 0,1 0,032 <0,11 -
0,048
0,066
72,72 -
62 0,14 0,06 0,056 -
0,047
0,085
55,08 -
64 - 0,013 <0,52 -
-
0,013
- -
34 2,1 1,5 1,694 1,6
0,26
1,72
15,09 -
63 <1 0,17 0,176 0,202
0,02
0,18
10,95 -
69 - - 0,0719 0,03
0,03
0,051
58,88 -
35 <0,1 0,0056 <0,01 -
-
0,0056
- 0,085
31 <0,1 0,004 <0,018 -
-
0,004
- 0,002
60 <0,1 - <0,62 -
-
-
- -
39 <0,1 0,00082 <0,016 -
-
0,00082
- 0,005
61 <0,1 0,0053 <0,018 -
-
0,0053
- 0,062
43 <0,1 0,0049 <0,0099 -
-
0,0049
- 0,027
65 <0,1 0,0028 <0,013 -
-
0,0028
- 0,215
66 <0,1 0,0048 <0,014 -
-
0,0048
- 0,094
47 <0,1 - <0,016 -
-
-
- -
28 <0,1 0,0029 <0,014 -
-
0,0029
- 0,223
29 <0,1 - <0,016 -
-
-
- -
67 <0,1 0,0027 <0,016 -
-
0,0027
- 0,208
68 <0,1 0,0028 <0,025 -
-
0,0028
- -
7 <0,1 0,0021 <0,015 -
0,0021
- 0,162
11 .<0,1 0,0046 0,0096 -
0,004
0,007
56,34 -
51 <0,1 0,0086 0,0122 -
0,003
0,01
28,85 -
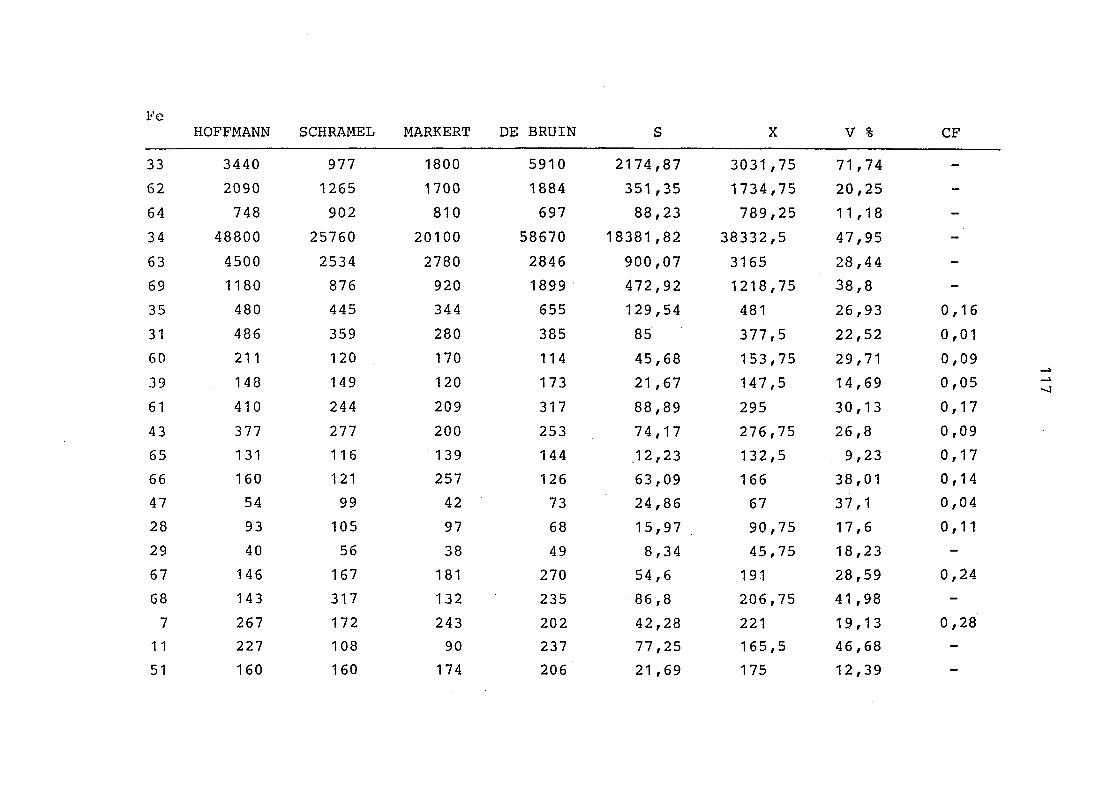
1'eHOFFMANN SCHRAMEL MARKERT DE BRUIN S X V ä CF
33 3440 977 1800 5910 2174,87 3031,75 71,74
62 2090 1265 1700 1884 351,35 1734,75 20,25
64 748 902 810 697 88,23 789,25 11,18
34 48800 25760 20100 58670 18381,82 38332,5 47,95
63 4500 2534 2780 2846 900,07 3165 28,44
69 1180 876 920 1899 472,92 1218,75 38,8
35 480 445 344 655 129,54 481 26,93 0,16
31 486 359 280 385 85 377,5 22,52 0,01
60 211 120 170 114 45,68 153,75 29,71 0,09
39 148 149 120 173 21,67 147,5 14,69 0,05
61 410 244 209 317 88,89 295 30,13 0,17
43 377 277 200 253 74,17 276,75 26,8 0,09
65 131 116 139 144 . 12,23 132,5 9,23 0,17
66 160 121 257 126 63,09 166 38,01 0,14
47 54 99 42 73 24,86 67 37,1 0,04
28 93 105 97 68 15,97 90,75 17,6 0,11
29 40 56 38 49 8,34 45,75 18,23
67 146 167 181 270 54,6 191 28,59 0,24
68 143 317 132 235 86,8 206,75 41,98
7 267 172 243 202 42,28 221 19,13 0,28
11 227 108 90 237 77,25 165,5 46,68
51 160 160 174 206 21,69 175 12,39
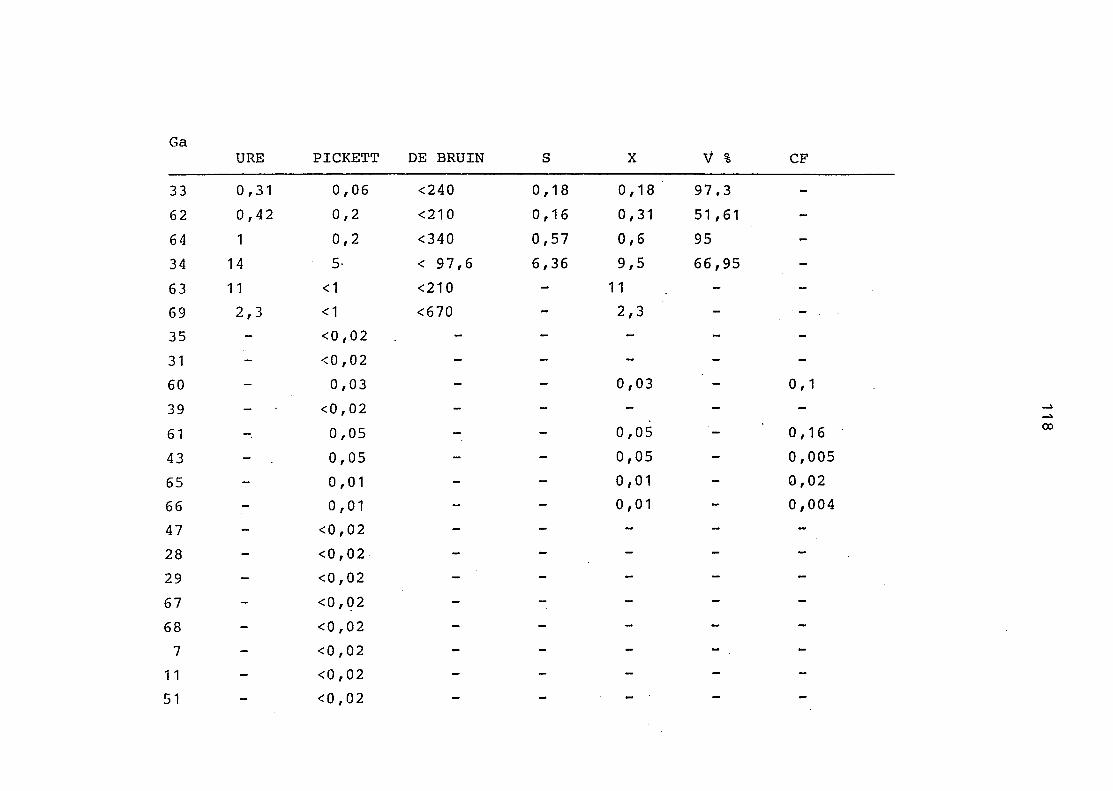
GaURE PICKETT
DE BRUIN
S
X
V % CF
33
0,31 0,06
<240
0,18
0,18
97 .3 -
62
0,42 0,2
<210
0,16
0,31
51,61 -
64
1 0,2
<340
0,57
0,6
95 -
34
14 5
<
97,6
6,36
9,5
66,95 -
63
11 <1
<210
-
11
- -
69
2,3 <1
<670
-
2,3
- -
35
- <0,02
-
-
-
- -
31
- <0,02
-
-
-
- -
60
- 0,03
-
-
0,03
- 0,1
39
- <0,02
-
-
-
- -
61 0,05
-
-
0,05 0,16
43
- 0,05
-
-
0,05
- 0,005
65
- 0,01
-
-
0,01
- 0,02
66
- 0,01
-
-
0,01
- 0,004
47
- <0,02
-
-
-
- -
28
- <0,02
-
-
-
- -
29
- <0,02
-
-
-
- -
67
- <0,02
-
-
-
- -
68
- <0,02
-
-
-
- -
7
- <0,02
-
-
-
- -
11
- <0,02
-
-
-
- -
51
- <0,02
-
-
-
- -
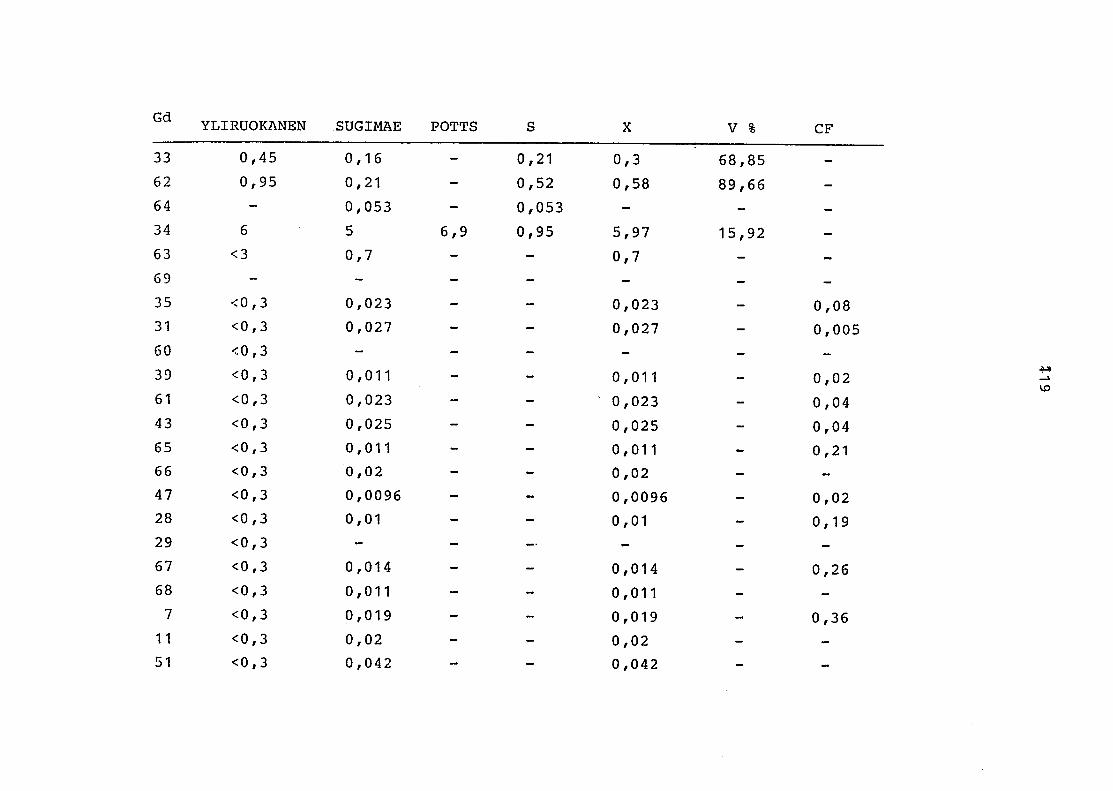
Gd YLIRUOKANEN SUGIMAE POTTS
S
X
V %
CF
33
0,45
0,16
-
0,21
0,3
68,85
-
62
0,95
0,21
-
0,52
0,58
89,66
-
64
-
0,053
-
0,053
-
-
-
34
6
5
6,9
0,95
5,97
15,92
-
63
<3
0,7
-
-
0,7
-
-
69
35
<0,3
0,023
-
-
0,023
-
0,08
31
<0,3
0,027
-
-
0,027
-
0,005
60
<0,3
39
<0,3
0,011
-
-
0,011
-
0,02
61
<0,3
0,023
-
-
0,023
-
0,04
43
<0,3
0,025
-
-
0,025
-
0,04
65
<0,3
0,011
-
-
0,011
-
0,21
66
<0,3
0,02
-
-
0,02
-
-
47
<0,3
0,0096
-
-
0,0096
-
0,02
28
<0,3
0,01
-
-
0,01
-
0,19
29
<0,3
67
<0,3
0,014
-
-
0,014
-
0,26
68
<0,3
0,011
-
-
0,011
-
-
7
<0,3
0,019
-
-
0,019
-
0,36
11
<0,3
0,02
-
-
0,02
-
-
51
<0,3
0,042
-
-
0,042
-
-
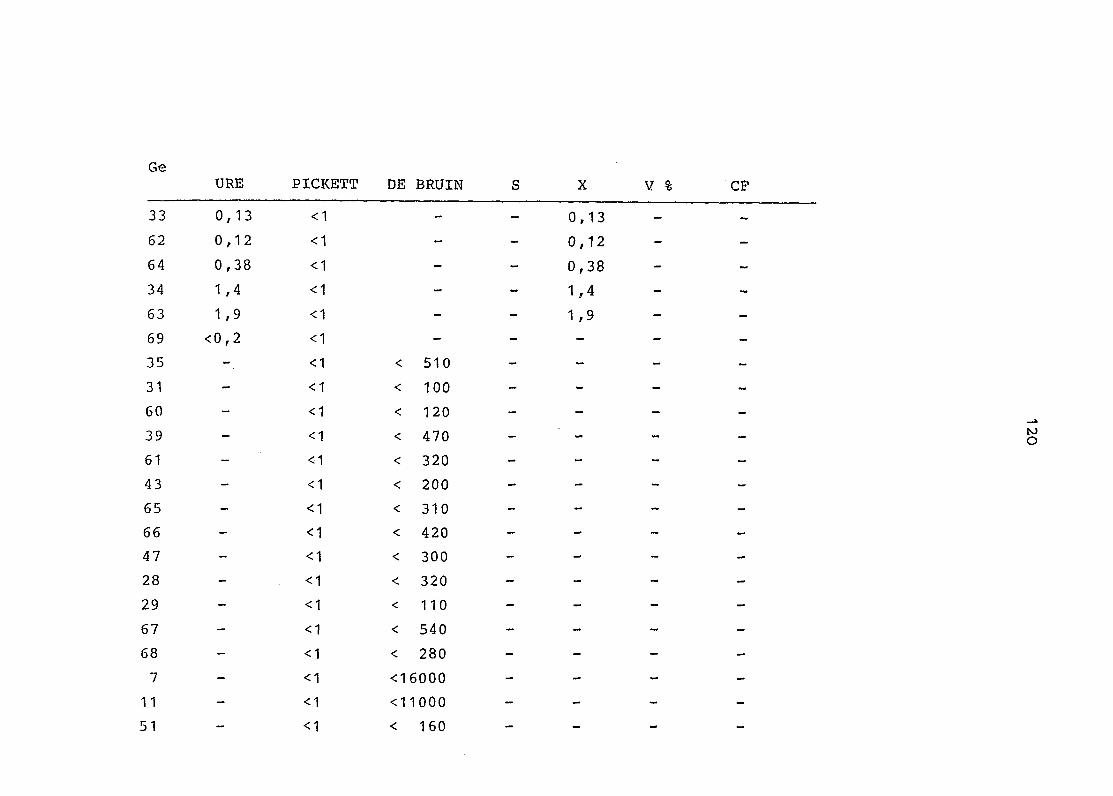
GeURE PICKETT
DE BRUIN
S
X
V %
CF
33 0,13 <1
-
-
0,13
62 0,12 <1
-
-
0,12
64 0,38 <1
-
-
0,38
34 1,4 <1
-
-
1,4
63 1,9 <1
-
-
1,9
69 <0,2 <1
-
-
-
35 - . <1
<
510
-
-
31 - <1
<
100
-
-
60 - <1
<
120
-
-
39 - <1
<
470
-
-
61 - <1
<
320
-
-
43 - <1
<
200
-
-
65 - <1
<
310
-
-
66 - <1
<
420
-
-
47 - <1
<
300
-
-
28 - <1
<
320
-
-
29 - <1
<
110
-
-
67 - <1
<
540
-
-
68 - <1
<
280
-
-
7 - <1
<16000
-
-
11 - <1
<11000
-
-
51 - <1
<
160
-
-
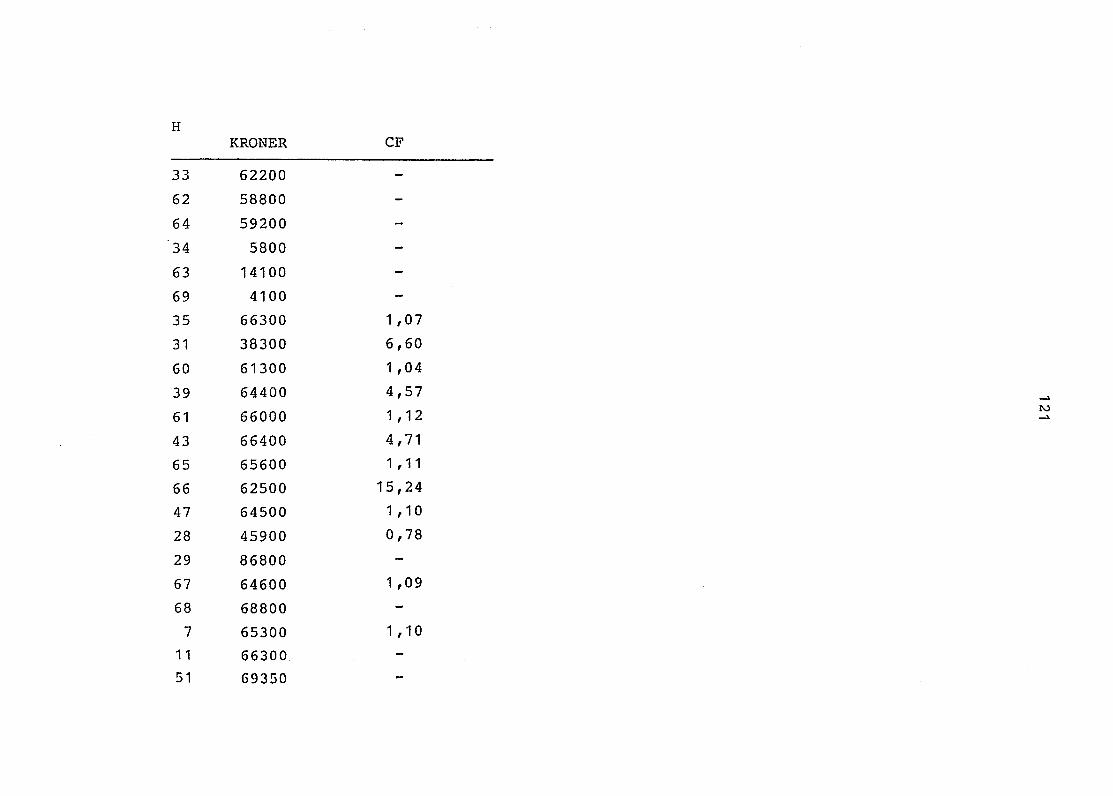
HKRONER CF
33 62200 -
62 58800 -
64 59200 -
34 5800 -
63 14100 -
69 4100 -
35 66300 1,07
31 38300 6,60
60 61300 1,04
39 64400 4,57
61 66000 1,12
43 66400 4,71
65 65600 1,11
66 62500 15,24
47 64500 1,10
28 45900 0,78
29 86800 -
67 64600 1,09
68 68800 -
7 65300 1,10
11 66300 -
51 69350 -
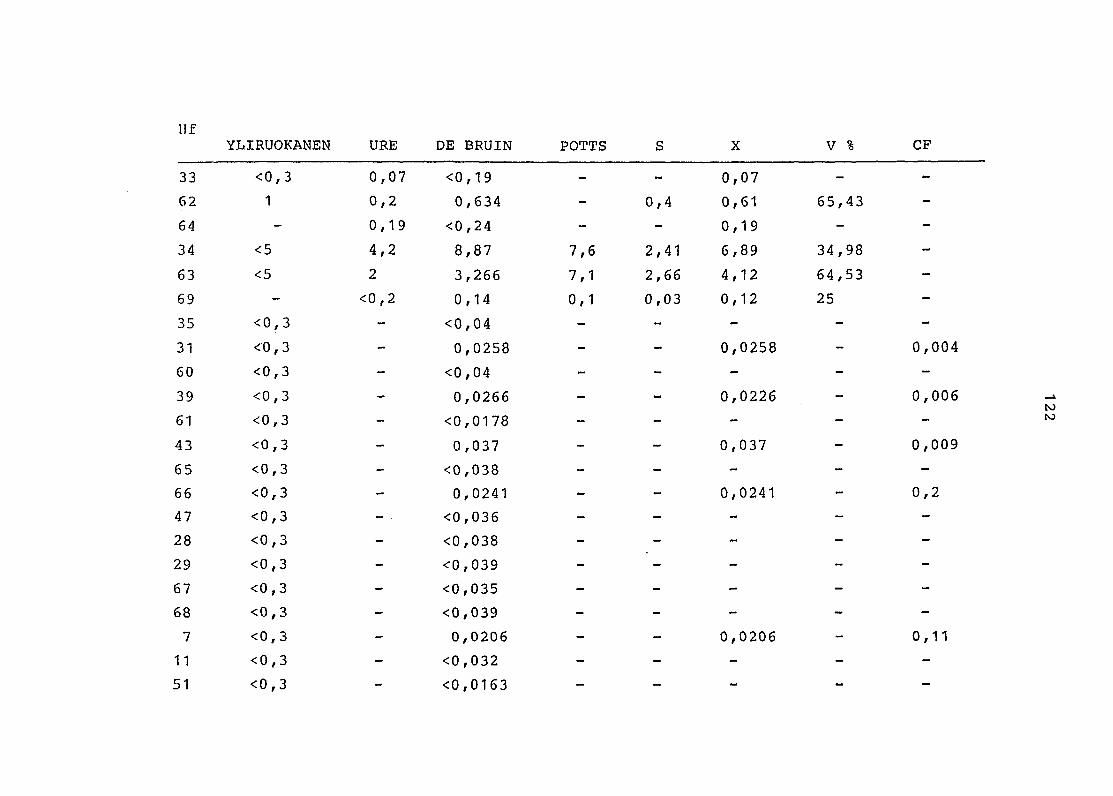
IlfYLIRUOKANEN
URE DE BRUIN
POTTS
S
X
V % CF
33 <0,3
0,07 <0,19
-
-
0,07
-
62 1
0,2 0,634
-
0,4
0,61
65,43
64 -
0,19 <0,24
-
-
0,19
-
34 <5
4,2 8,87
7,6
2,41
6,89
34,98
63 <5
2 3,266
7,1
2,66
4,12
64,53
69 -
<0,2 0,14
0,1
0,03
0,12
25
35 <0,3
- <0,04
-
-
-
-
31 <0,3
- 0,0258
-
-
0,0258
- 0,004
60 <0,3
- <0,04
-
-
-
-
39 <0,3 0,0266
-
-
0,0226
- 0,006
61 <0,3
- <0,0178
-
-
-
-
43 <0,3
- 0,037
-
-
0,037
- 0,009
65 <0,3
- <0,038
-
-
-
-
66 <0,3
- 0,0241
-
-
0,0241
- 0,2
47 <0,3 <0,036
-
-
-
-
28 <0,3
- <0,038
-
-
-
-
29 <0,3
- <0,039
-
-
-
-
67 <0,3
- <0,035
-
-
-
-
68 <0,3
- <0,039
-
-
-
-
7 <0,3
- 0,0206
-
-
0,0206
- 0,11
11 <0,3
- <0,032
-
-
-
-
51 <0,3
- <0,0163
-
-
-
-
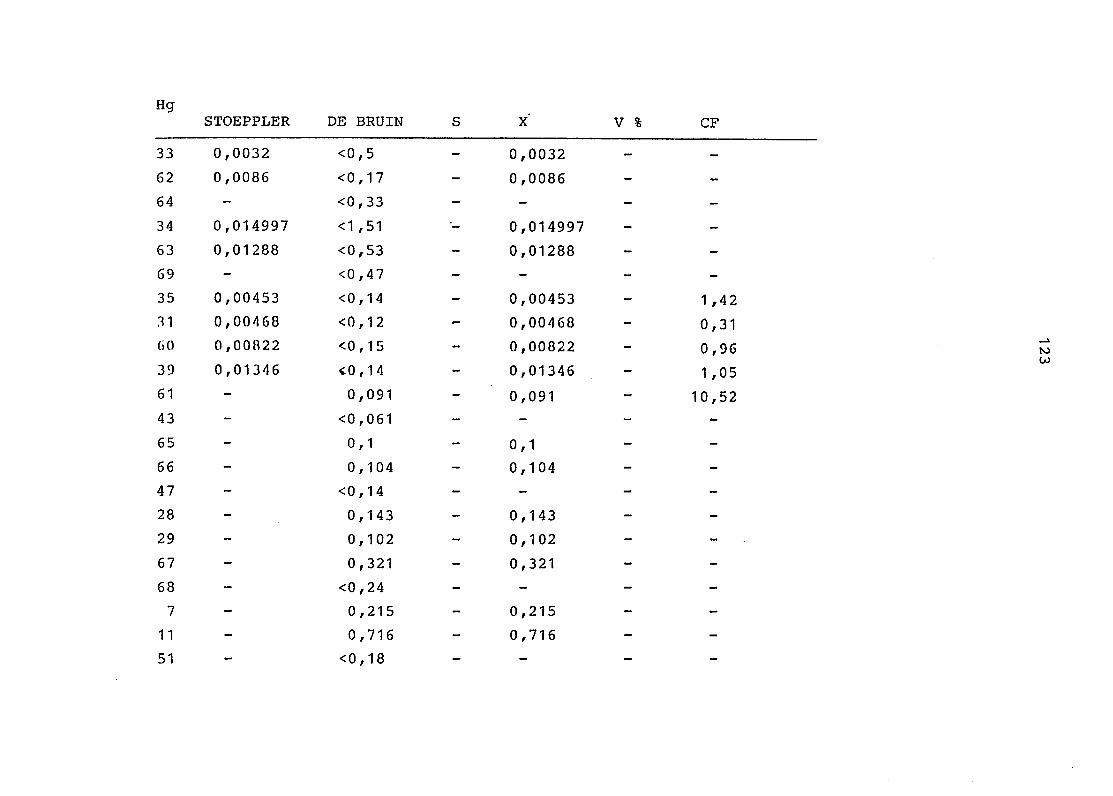
HgSTOEPPLER DE BRUIN
S
X .
V % CF
33
0,0032 <0,5
-
0,0032
- -
62
0,0086 <0,17
-
0,0086
- -
64
- <0,33
-
-
- -
34
0,014997 <1,51
0,014997
- -
63
0,01288 <0,53
-
0,01288
- -
69
- <0,47
-
-
- -
35
0,00453 <0,14
-
0,00453
- 1,42
31
0,00468 <0,12
-
0,00468
- 0,31
60
0,00822 <0,15
-
0,00822
- 0,96
39
0,01346 e0,14
-
0,01346
- 1,05
61
- 0,091
-
0,091
- 10,52
43
- <0,061
-
-
-
65
- 0,1
-
0,1
- -
66
- 0,104
-
0,104
- -
47 <0,14
-
-
- -
28
- 0,143
-
0,143
- -
29
- 0,102
-
0,102
- -
67
- 0,321
-
0,321
- -
68
- <0,24
-
-
- -
7
- 0,215
-
0,215
- -
11
- 0,716
-
0,716
- -
51 <0,18
-
-
- -
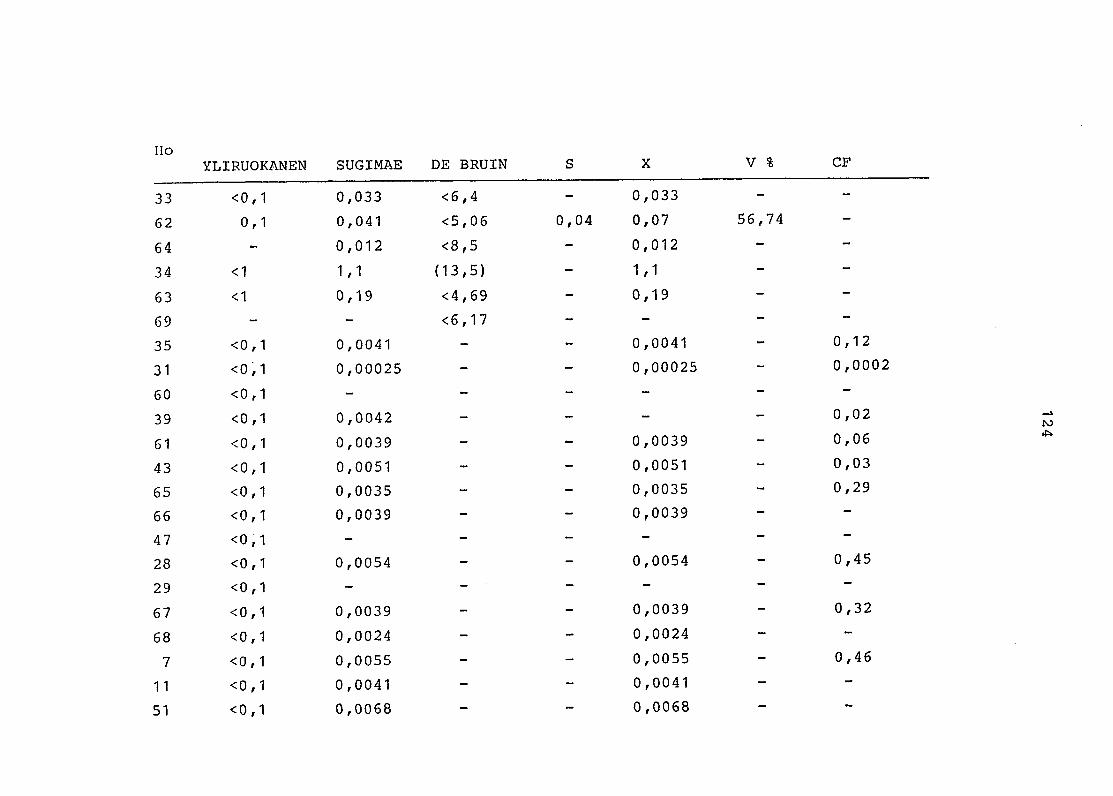
IIoYLIRUOKANEN SUGIMAE DE BRUIN
S
X
V %
CF
33
<0,1
0,033
<6,4
-
0,033
-
-
62
0,1
0,041
<5,06
0,04
0,07
56,74
-
64
-
0,012
<8,5
-
0,012
-
-
34
<1
1,1
(13,5)
-
1,1
-
-
63
<1
0,19
<4,69
-
0,19
-
-
69
-
-
<6,17
-
-
-
-
35
<0,1
0,0041
-
-
0,0041
-
0,12
31
<0,1
0,00025
-
-
0,00025
-
0,0002
60
<0,1
39
<0,1
0,0042
-
-
-
-
0,02 N
61
<0,1
0,0039
-
-
0,0039
-
0,06
43
<0,1
0,0051
-
-
0,0051
-
0,03
65
<0,1
0,0035
-
-
0,0035
-
0,29
66
<0,1
0,0039
-
-
0,0039
-
-
47
<0,1
28
<0,1
0,0054
-
-
0,0054
-
0,45
29
<0,1
67
<0,1
0,0039
-
-
0,0039
-
0,32
68
<0,1
0,0024
-
-
0,0024
-
-
7
<0,1
0,0055
-
-
0,0055
-
0,46
11
<0,1
0,0041
-
-
0,0041
-
-
51
<0,1
0,0068
-
-
0,0068
-
-
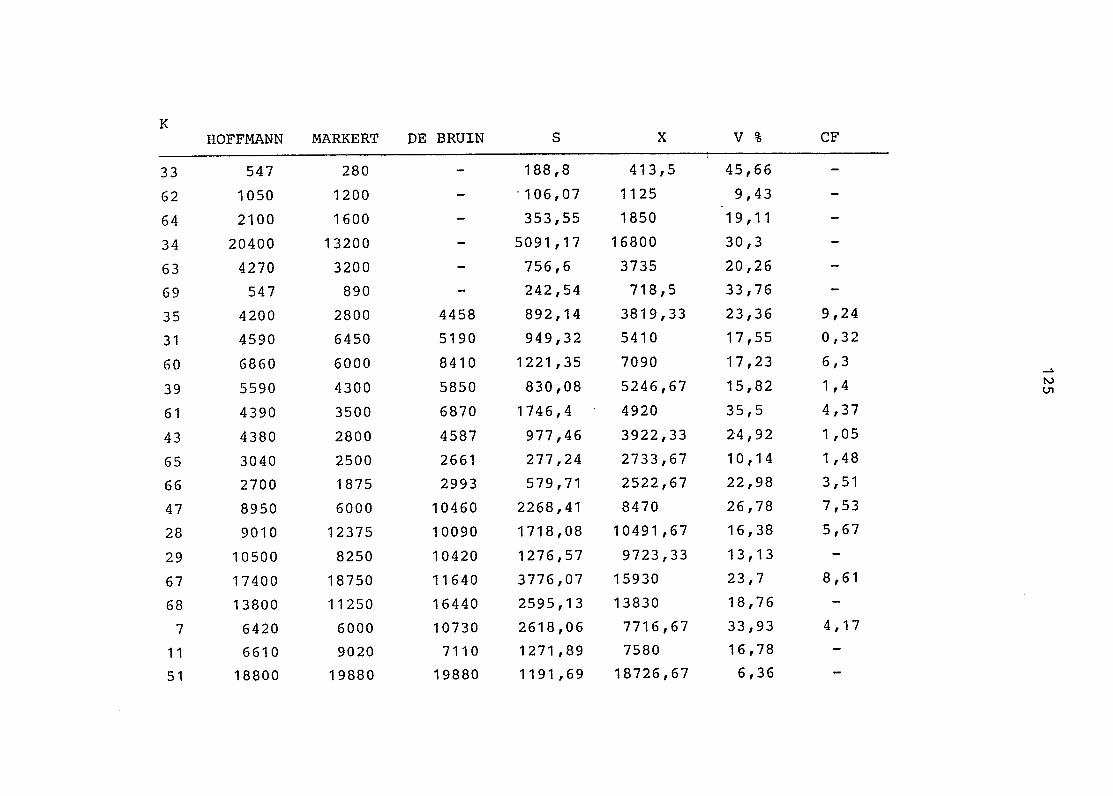
KHOFFMANN MARKE RT DE BRUIN S X V % CF
33 547 280 188,8 413,5 45,66
62 1050 1200 106,07 1125 9,43
64 2100 1600 353,55 1850 19,11
34 20400 13200 5091,17 16800 30,3
63 4270 3200 756,6 3735 20,26
69 547 890 242,54 718,5 33,76
35 4200 2800 4458 892,14 3819,33 23,36 9,24
31 4590 6450 5190 949,32 5410 17,55 0,32
60 6860 6000 8410 1221,35 7090 17,23 6,3
39 5590 4300 5850 830,08 5246,67 15,82 1,4
61 4390 3500 6870 1746,4 4920 35,5 4,37
43 4380 2800 4587 977,46 3922,33 24,92 1,05
65 3040 2500 2661 277,24 2733,67 10,14 1,48
66 2700 1875 2993 579,71 2522,67 22,98 3,51
47 8950 6000 10460 2268,41 8470 26,78 7,53
28 9010 12375 10090 1718,08 10491,67 16,38 5,67
29 10500 8250 10420 1276,57 9723,33 13,13
67 17400 18750 11640 3776,07 15930 23,7 8,61
68 13800 11250 16440 2595,13 13830 18,76
7 6420 6000 10730 2618,06 7716,67 33,93 4,17
11 6610 9020 7110 1271,89 7580 16,78
51 18800 19880 19880 1191,69 18726,67 6,36
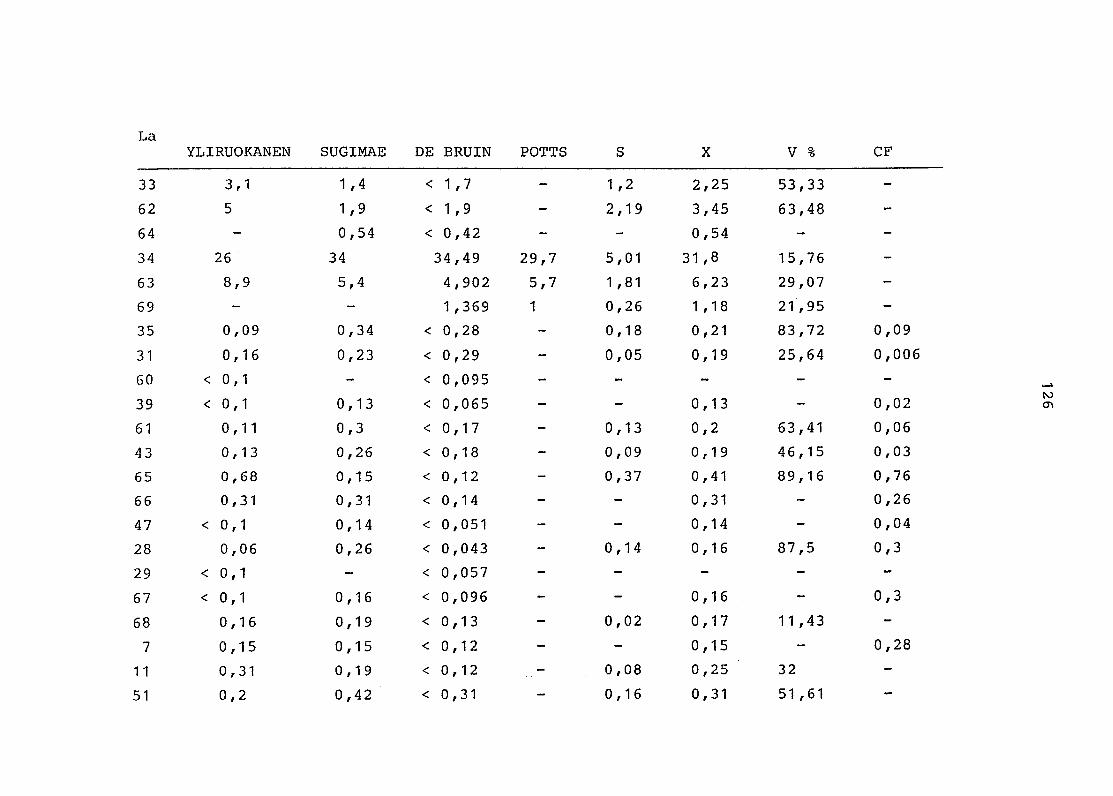
LaYLIRUOKANEN
SUGIMAE
DE BRUIN
POTTS S X V % CF
33
3,1
1,4
< 1,7
- 1,2 2,25 53,33 -
62
5
1,9
< 1,9
- 2,19 3,45 63,48 -
64
-
0,54
< 0,42
- - 0,54 - -
34
26
34 34,49
29,7 5,01 31,8 15,76 -
63
8,9
5,4 4,902
5,7 1,81 6,23 29,07 -
69
-
- 1,369
1 0,26 1,18 21,95 -
35
0,09
0,34
< 0,28
- 0,18 0,21 83,72 0,09
31
0,16
0,23
< 0,29
- 0,05 0,19 25,64 0,006
60
<
0,1
-
< 0,095
- - - - -
39
<
0,1
0,13
< 0,065
- - 0,13 - 0,02
61
0,11
0,3
< 0,17
- 0,13 0,2 63,41 0,06
43
0,13
0,26
< 0,18
- 0,09 0,19 46,15 0,03
65
0,68
0,15
< 0,12
- 0,37 0,41 89,16 0,76
66
0,31
0,31
< 0,14
- - 0,31 - 0,26
47
<
0,1
0,14
< 0,051
- - 0,14 - 0,04
28
0,06
0,26
< 0,043
- 0,14 0,16 87,5 0,3
29
< 0,1
-
< 0,057
- - - - -
67
<
0,1
0,16
< 0,096
- - 0,16 - 0,3
68
0,16
0,19
< 0,13
- 0,02 0,17 11,43 -
7
0,15
0,15
< 0,12
- - 0,15 - 0,28
11
0,31
0,19
< 0,12 0,08 0,25 32 -
51
0,2
0,42
< 0,31
- 0,16 0,31 51,61 -
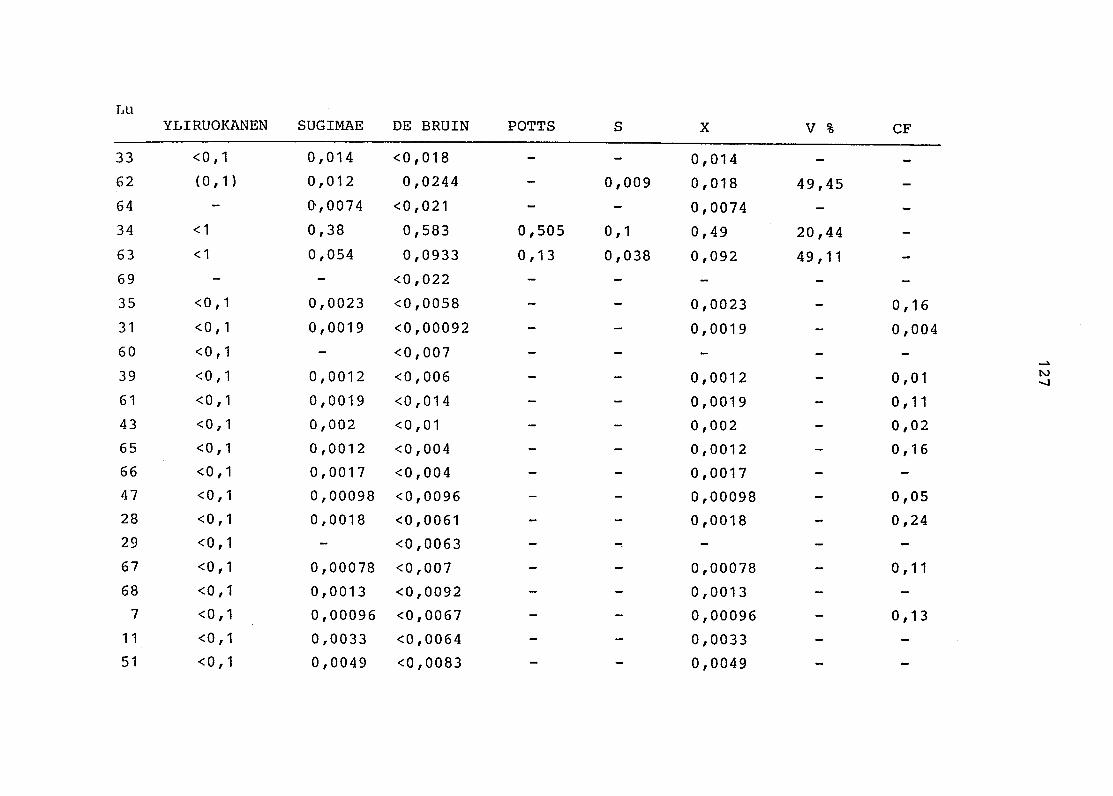
LuYLIRUOKANEN SUGIMAE DE BRUIN POTTS
S
X
V % CF
33 <0,1 0,014 <0,018 -
-
0,014
- -
62 (0,1) 0,012 0,0244 -
0,009
0,018
49,45 -
64 - 0•,0074 <0,021 -
-
0,0074
- -
34 <1 0,38 0,583 0,505
0,1
0,49
20,44 -
63 <1 0,054 0,0933 0,13
0,038
0,092
49,11 -
69 - - <0,022 -
-
-
- -
35 <0,1 0,0023 <0,0058 -
-
0,0023
- 0,16
31 <0,1 0,0019 <0,00092 -
-
0,0019
- 0,004
60 <0,1 - <0,007 -
-
-
- -
39 <0,1 0,0012 <0,006 -
-
0,0012
- 0,01
61 <0,1 0,0019 <0,014 -
-
0,0019
- 0,11
43 <0,1 0,002 <0,01 -
-
0,002
- 0,02
65 <0,1 0,0012 <0,004 -
-
0,0012
- 0,16
66 <0,1 0,0017 <0,004 -
-
0,0017
- -
47 <0,1 0,00098 <0,0096 -
-
0,00098
- 0,05
28 <0,1 0,0018 <0,0061 -
-
0,0018
- 0,24
29 <0,1 - <0,0063 -
-
-
- -
67 <0,1 0,00078 <0,007 -
-
0,00078
- 0,11
68 <0,1 0,0013 <0,0092 -
-
0,0013
- -
7 <0,1 0,00096 <0,0067 -
-
0,00096
- 0,13
11 <0,1 0,0033 <0,0064 -
-
0,0033
- -
51 <0,1 0,0049 <0,0083 -
-
0,0049
- -
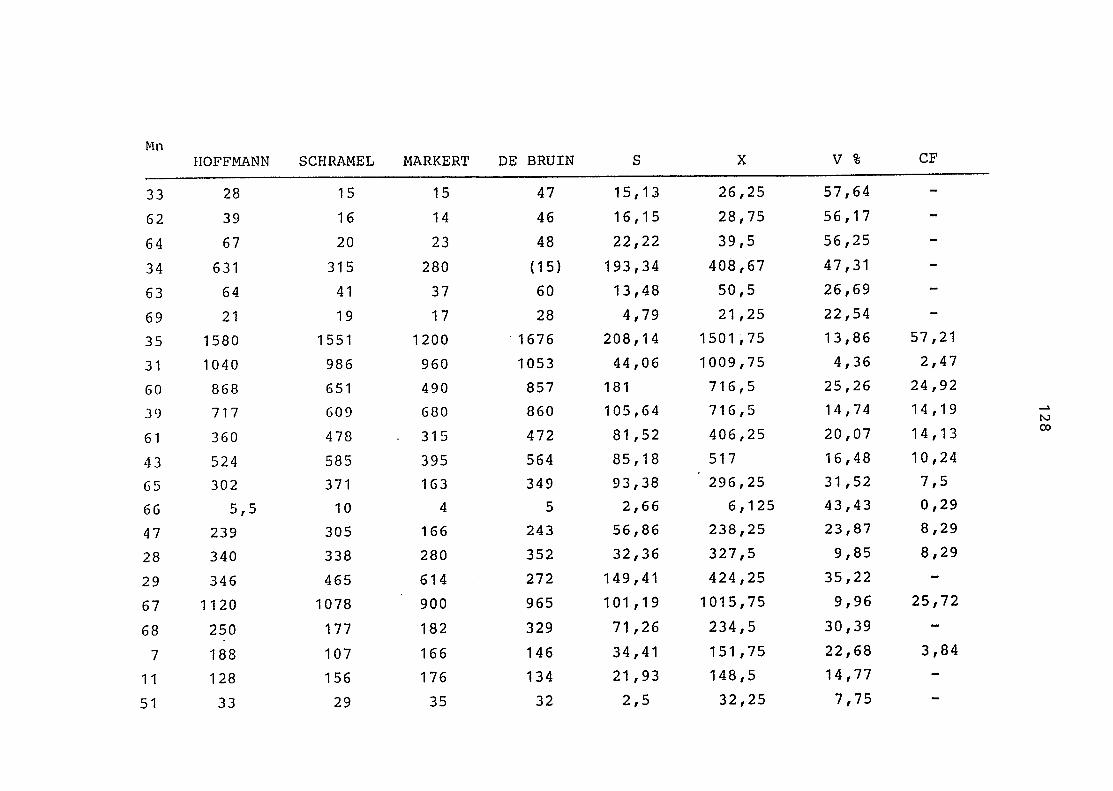
MnHOFFMANN SCHRAMEL MARKERT DE BRUIN S X V % CF
33 28 15 15 47 15,13 26,25 57,64 -
62 39 16 14 46 16,15 28,75 56,17 -
64 67 20 23 48 22,22 39,5 56,25 -
34 631 315 280 (15) 193,34 408,67 47,31 -
63 64 41 37 60 13,48 50,5 26,69 -
69 21 19 17 28 4,79 21,25 22,54 -
35 1580 1551 1200 1676 208,14 1501,75 13,86 57,21
31 1040 986 960 1053 44,06 1009,75 4,36 2,47
60 868 651 490 857 181 716,5 25,26 24,92
39 717 609 680 860 105,64 716,5 14,74 14,19
61 360 478
- 315 472 81,52 406,25 20,07 14,13
43 524 585 395 564 85,18 517 16,48 10,24
65 302 371 163 349 93,38 296,25 31,52 7,5
66 5,5 10 4 5 2,66 6,125 43,43 0,29
47 239 305 166 243 56,86 238,25 23,87 8,29
28 340 338 280 352 32,36 327,5 9,85 8,29
29 346 465 614 272 149,41 424,25 35,22 -
67 1120 1078 900 965 101,19 1015,75 9,96 25,72
68 250 177 182 329 71,26 234,5 30,39 -
7 188 107 166 146 34,41 151,75 22,68 3,84
11 128 156 176 134 21,93 148,5 14,77 -
51 33 29 35 32 2,5 32,25 7,75 -
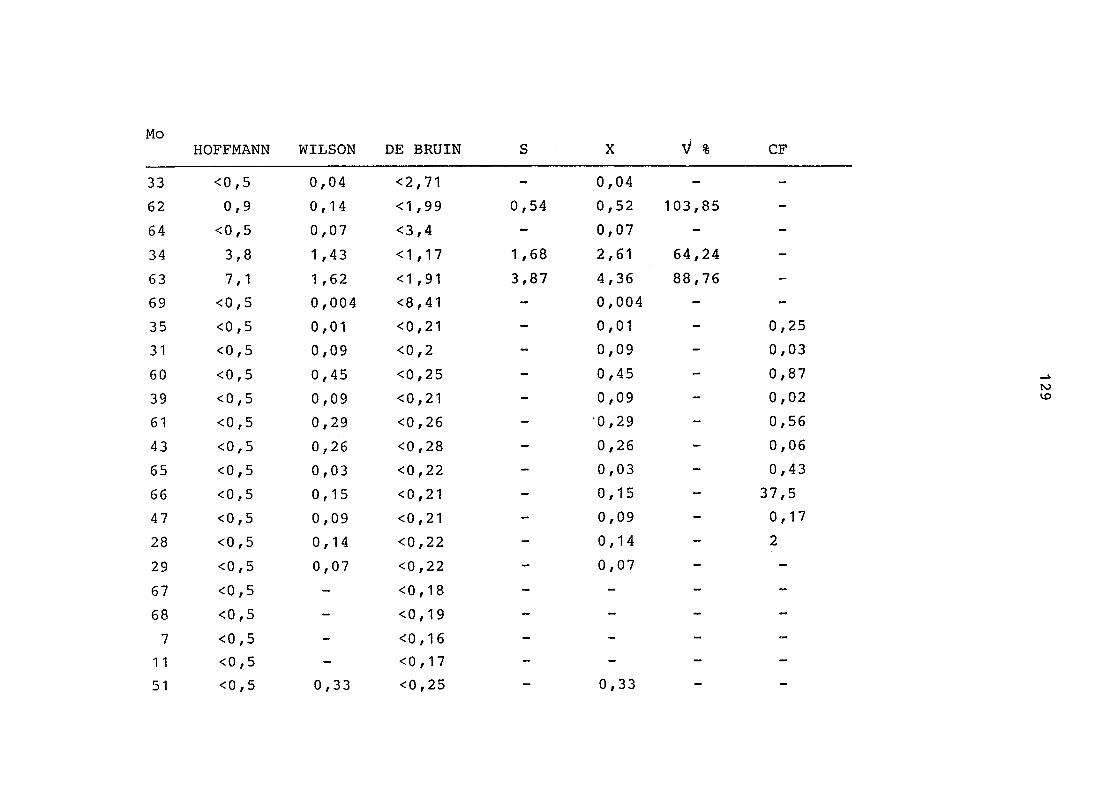
MoHOFFMANN WILSON DE BRUIN
S X
V % CF
33 <0,5 0,04 <2,71
- 0,04
- -
62 0,9 0,14 <1,99
0,54 0,52
103,85 -
64 <0,5 0,07 <3,4
- 0,07
- -
34 3,8 1,43 <1,17
1,68 2,61
64,24 -
63 7,1 1,62 <1,91
3,87 4,36
88,76 -
69 <0,5 0,004 <8,41
- 0,004
- -
35 <0,5 0,01 <0,21
- 0,01
- 0,25
31 <0,5 0,09 <0,2
- 0,09
- 0,03
60 <0,5 0,45 <0,25
- 0,45
- 0,87
39 <0,5 0,09 <0,21
- 0,09
- 0,02
61 <0,5 0,29 <0,26
- .0,29
- 0,56
43 <0,5 0,26 <0,28
- 0,26
- 0,06
65 <0,5 0,03 <0,22
- 0,03
- 0,43
66 <0,5 0,15 <0,21
- 0,15
- 37,5
47 <0,5 0,09 <0,21
- 0,09
- 0,17
28 <0,5 0,14 <0,22
- 0,14
- 2
29 <0,5 0,07 <0,22
- 0,07
- -
67 <0,5 - <0,18
- -
- -
68 <0,5 - <0,19
- -
- -
7 <0,5 - <0,16
- -
- -
11 <0,5 - <0,17
- -
- -
51 <0,5 0,33 <0,25
- 0,33
- -
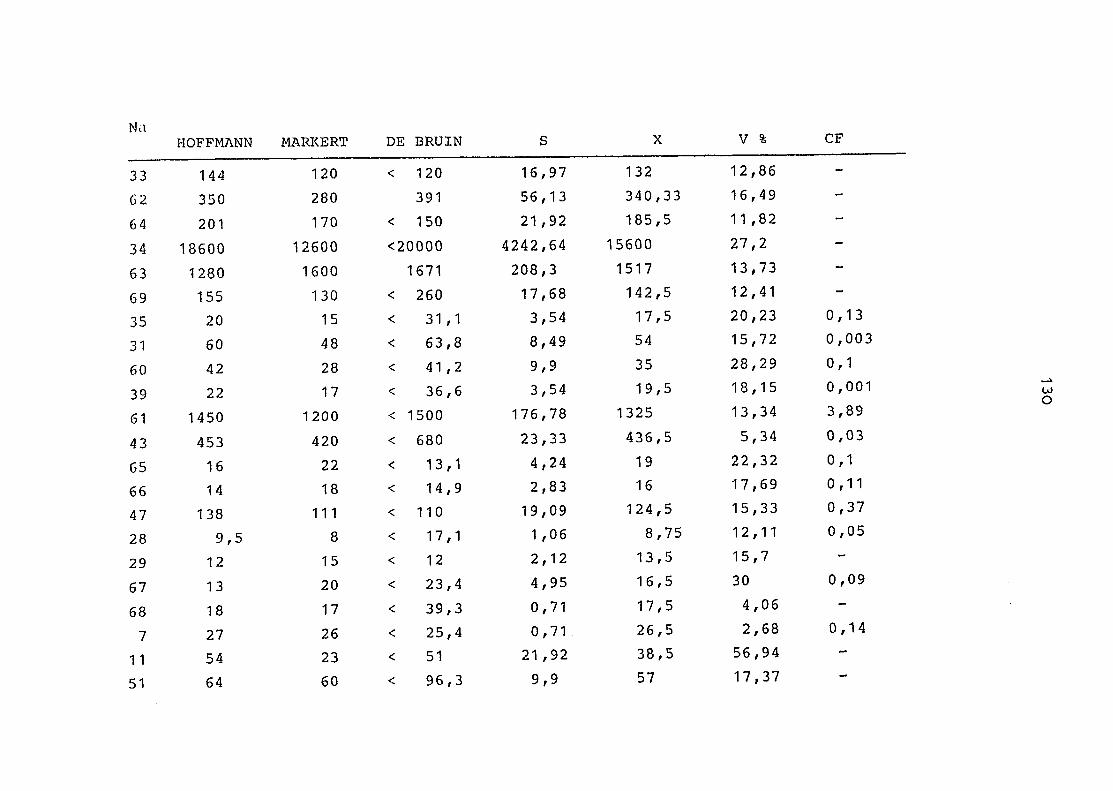
NaHOFFMANN MARKERT DE BRUIN S X V % CF
33 144 120 <
120 16,97 132 12,86
62 350 280 391 56,13 340,33 16,49
64 201 170 <
150 21,92 185,5 11,82
34 18600 12600 <20000 4242,64 15600 27,2
63 1280 1600 1671 208,3 1517 13,73
69 155 130 <
260 17,68 142,5 12,41 -
35 20 15 <
31,1 3,54 17,5 20,23 0,13
31 60 48 <
63,8 8,49 54 15,72 0,003
60 42 28 <
41,2 9,9 35 28,29 0,1
39 22 17 <
36,6 3,54 19,5 18,15 0,001
61 1450 1200 <
1500 176,78 1325 13,34 3,89
43 453 420 <
680 23,33 436,5 5,34 0,03
65 16 22 <
13,1 4,24 19 22,32 0,1
66 14 18 <
14,9 2,83 16 17,69 0,11
47 138 111 <
110 19,09 124,5 15,33 0,37
28 9,5 8 <
17,1 1,06 8,75 12,11 0,05
29 12 15 <
12 2,12 13,5 15,7
67 13 20 <
23,4 4,95 16,5 30 0,09
68 18 17 <
39,3 0,71 17,5 4,06
7 27 26 <
25,4 0,71 . 26,5 2,68 0,14
11 54 23 <
51 21,92 38,5 56,94
51 64 60 <
96,3 9,9 57 17,37
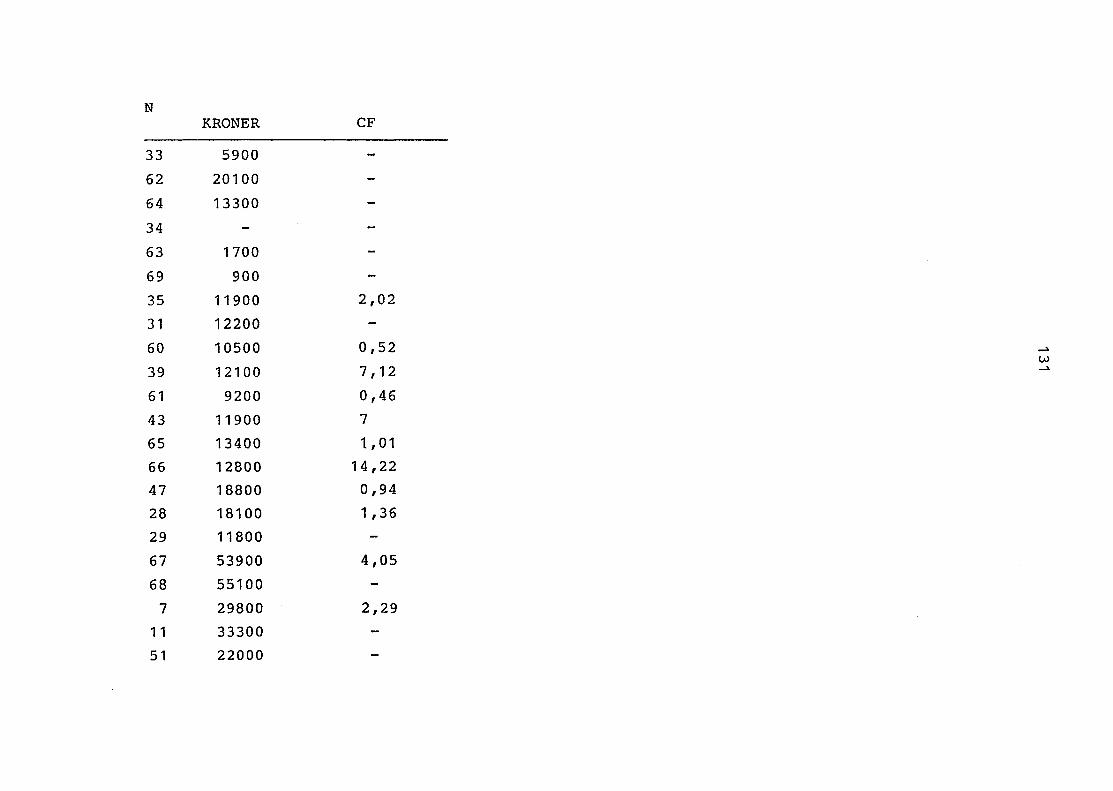
NKRONER
CF
33
5900
-
62
20100
-
64
13300
-
34
-
-
63
1700
-
69
900
-
35
11900
2,02
31
12200
-
60
10500
0,52
39
12100
7,12
61
9200
0,46
43
11900
7
65
13400
1,01
66
12800
14,22
47
18800
0,94
28
18100
1,36
29
11800
-
67
53900
4,05
68
55100
-
7
29800
2,29
11
33300
-
51
22000
-
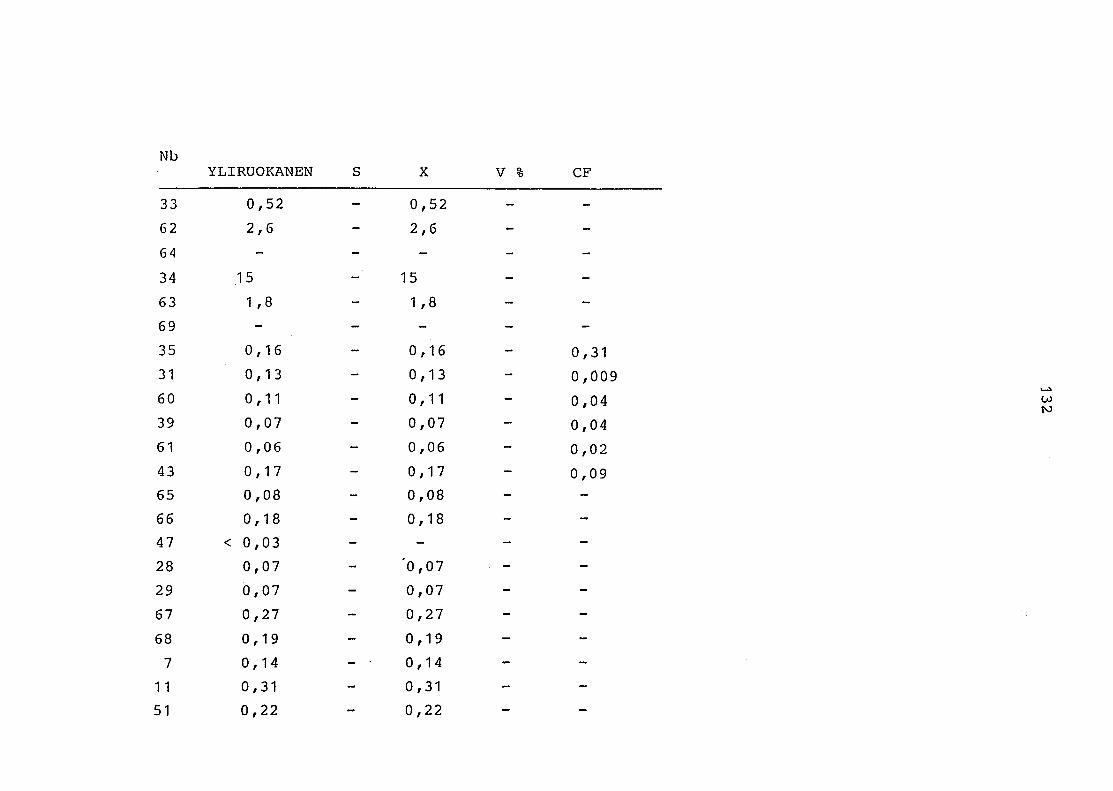
NbYLIRUOKANEN
S
X
V %
CF
33
0,52
0,52
62
2,6
-
2,6
64
-
-
-
34
15
15
63
1,8
-
1,8
69
-
-
-
35
0,16
-
0,16
31
0,13
-
0,13
60
0,11
-
0,11
39
0,07
-
0,07
61
0,06
-
0,06
43
0,17
-
0,17
65
0,08
-
0,08
66
0,18
-
0,18
47
< 0,03
-
-
28
0,07
-
' 0,07
29
0,07
-
0,07
67
0,27
-
0,27
68
0,19
-
0,19
7
0,14
-
0,14
11
0,31
-
0,31
51
0,22
-
0,22
0,31
0,009
0,04
0,04
0,02
0,09
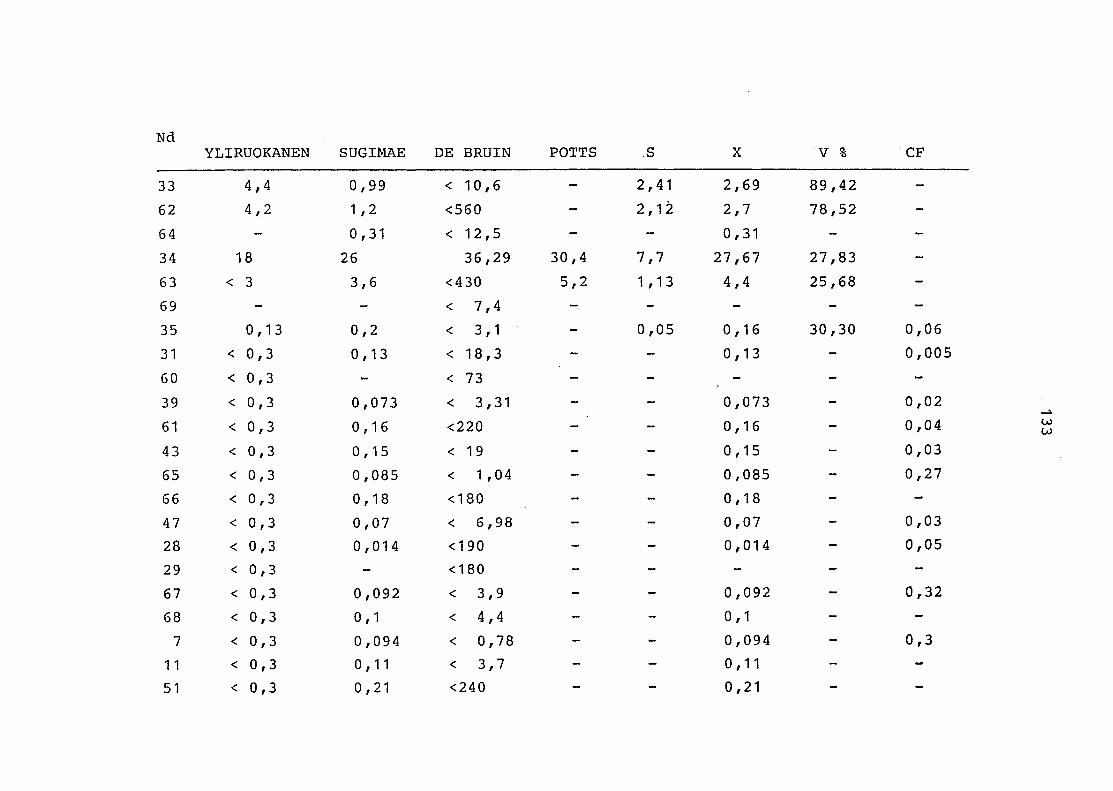
NdYLIRUOKANEN
SUGIMAE
DE BRUIN POTTS
.S
X
V % CF
33 4,4
0,99
< 10,6 -
2,41
2,69
89,42
62 4,2
1,2
<560 -
2,12
2,7
78,52
64 -
0,31
< 12,5 -
-
0,31
-
34
18 26 36,29 30,4
7,7
27,67
27,83
63
< 3
3,6
<430 5,2
1,13
4,4
25,68
69 -
-
<
7,4 -
-
-
-
35 0,13
0,2
<
3,1 -
0,05
0,16
30,30 0,06
31
< 0,3
0,13
<
18,3 -
-
0,13
- 0,005
60
< 0,3
-
<
73 -
-
-
-
39
< 0,3
0,073
<
3,31 -
-
0,073
- 0,02
61
< 0,3
0,16
<220 -
-
0,16 0,04
43
< 0,3
0,15
<
19 -
-
0,15
- 0,03
65
< 0,3
0,085
<
1,04 -
-
0,085
- 0,27
66
< 0,3
0,18
<180 -
-
0,18
-
47
< 0,3
0,07
<
6,98 -
0,07
- 0,03
28
< 0,3
0,014
<190 -
-
0,014
- 0,05
29
< 0,3
-
<180 -
-
-
67
< 0,3
0,092
<
3,9 -
-
0,092
- 0,32
68
< 0,3
0,1
<
4,4 -
-
0,1
-
7
< 0,3
0,094
<
0,78 -
-
0,094
- 0,3
11
< 0,3
0,11
<
3,7 -
-
0,11
-
51
< 0,3
0,21
<240 -
-
0,21
-
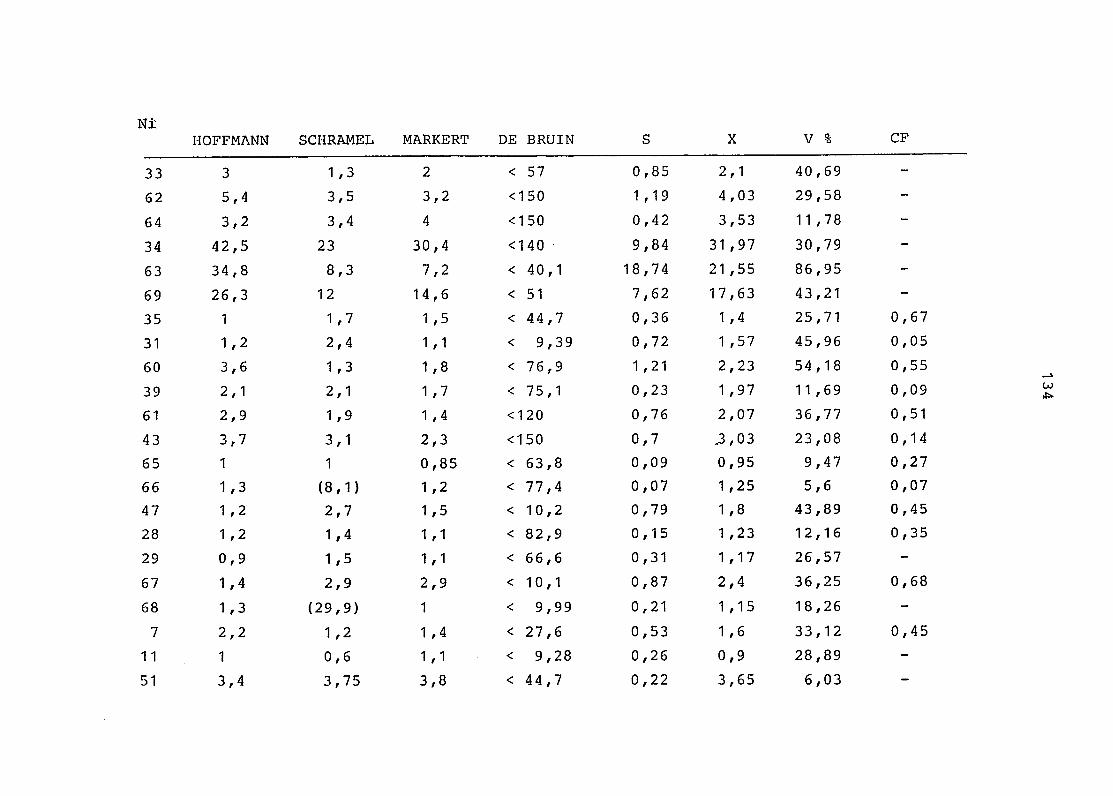
Ni.I-IOFFMANN SCHRAMEL S X V % CFMARKERT
DE BRUIN
33 3 1,3 2
< 57 0,85 2,1 40,69 -
62 5,4 3,5 3,2
<150 1,19 4,03 29,58 -
64 3,2 3,4 4
<150 0,42 3,53 11,78 -
34 42,5 23 30,4
<140 9,84 31,97 30,79 -
63 34,8 8,3 7,2
<
40,1 18,74 21,55 86,95 -
69 26,3 12 14,6
<
51 7,62 17,63 43,21 -
35 1 1,7 1,5
<
44,7 0,36 1,4 25,71 0,67
31 1,2 2,4 1,1
<
9,39 0,72 1,57 45,96 0,05
60 3,6 1,3 1,8
<
76,9 1,21 2,23 54,18 0,55
39 2,1 2,1 1,7
<
75,1 0,23 1,97 11,69 0,09
61 2,9 1,9 1,4
<120 0,76 2,07 36,77 0,51
43 3,7 3,1 2,3
<150 0,7 ,3,03 23,08 0,14
65 1 1 0,85
<
63,8 0,09 0,95 9,47 0,27
66 1,3 (8,1) 1,2
<
77,4 0,07 1,25 5,6 0,07
47 1,2 2,7 1,5
<
10,2 0,79 1,8 43,89 0,45
28 1,2 1,4 1,1
<
82,9 0,15 1,23 12,16 0,35
29 0,9 1,5 1,1
<
66,6 0,31 1,17 26,57 -
67 1,4 2,9 2,9
<
10,1 0,87 2,4 36,25 0,68
68 1,3 (29,9) 1
<
9,99 0,21 1,15 18,26 -
7 2,2 1,2 1,4
<
27,6 0,53 1,6 33,12 0,45
11 1 0,6 1,1
<
9,28 0,26 0,9 28,89 -
51 3,4 3,75 3,8
<
44,7 0,22 3,65 6,03 -
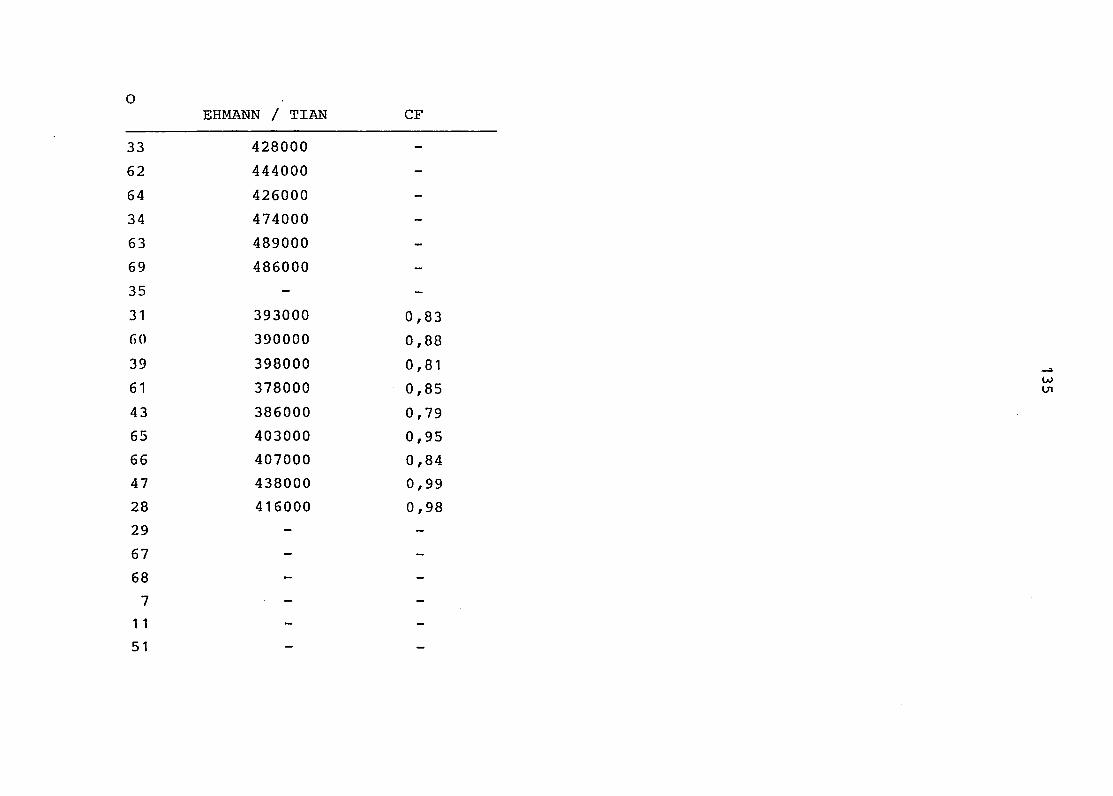
0EHMANN / TIAN CF
33 428000 -
62 444000 -
64 426000 -
34 474000 -
63 489000 -
69 486000 -
35 - -
31 393000 0,83
60 390000 0,88
39 398000 0,81
61 378000 0,85 Ui
43 386000 0,79
65 403000 0,95
66 407000 0,84
47 438000 0,99
28 416000 0,98
29 - -
67 - -
68 - -
7 - -
11 - -
51 - -
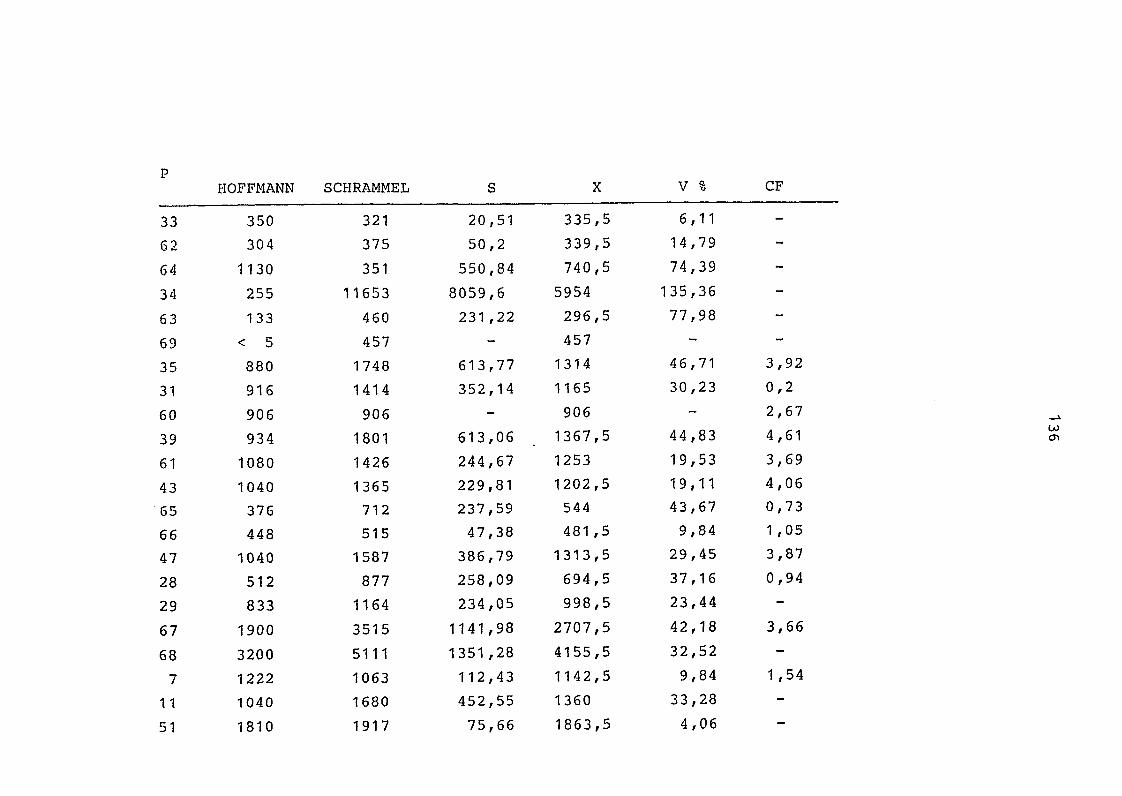
FHOFFMANN SCHRAMMEL S X V % CF
33 350 321 20,51 335,5 6,11 -
62 304 375 50,2 339,5 14,79 -
64 1130 351 550,84 740,5 74,39 -
34 255 11653 8059,6 5954 135,36 -
63 133 460 231,22 296,5 77,98 -
69 <
5 457 - 457 - -
35 880 1748 613,77 1314 46,71 3,92
31 916 1414 352,14 1165 30,23 0,2
60 906 906 - 906 - 2,67
39 934 1801 613,06 1367,5 44,83 4,61 rn
61 1080 1426 244,67 1253 19,53 3,69
43 1040 1365 229,81 1202,5 19,11 4,06
'65 376 712 237,59 544 43,67 0,73
66 448 515 47,38 481,5 9,84 1,05
47 1040 1587 386,79 1313,5 29,45 3,87
28 512 877 258,09 694,5 37,16 0,94
29 833 1164 234,05 998,5 23,44 -
67 1900 3515 1141,98 2707,5 42,18 3,66
68 3200 5111 1351,28 4155,5 32,52 -
7 1222 1063 112,43 1142,5 9,84 1,54
11 1040 1680 452,55 1360 33,28 -
51 1810 1917 75,66 1863,5 4,06 -
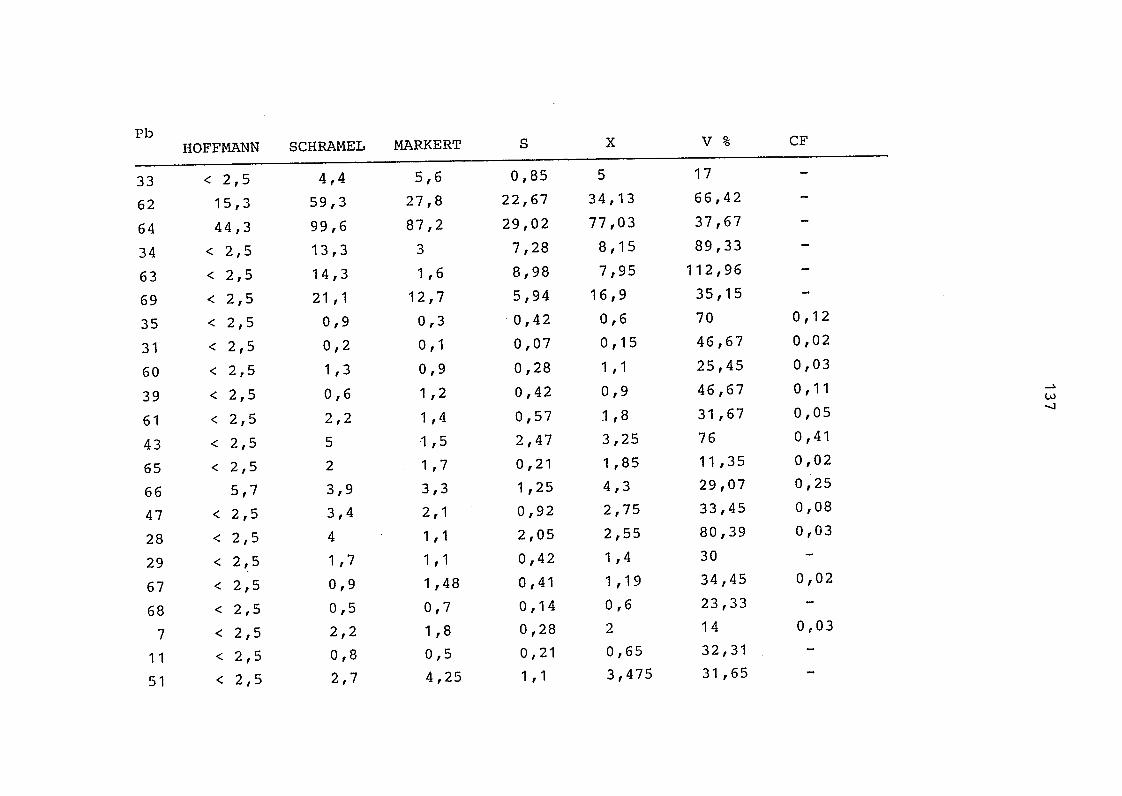
PbHOFFMANN SCHRAMEL MARKERT S X V % CF
33
< 2,5 4,4 5,6 0,85 5 17 -
62
15,3 59,3 27,8 22,67 34,13 66,42
64
44,3 99,6 87,2 29,02 77,03 37,67 -
34
< 2,5 13,3 3 7,28 8,15 89,33 -
63
< 2,5 14,3 1,6 8,98 7,95 112,96 -
69
< 2,5 21,1 12,7 5,94 16,9 35,15 -
35
< 2,5 0,9 0,3 0,42 0,6 70 0,12
31
< 2,5 0,2 0,1 0,07 0,15 46,67 0,02
60
< 2,5 1,3 0,9 0,28 1,1 25,45 0,03
39
< 2,5 0,6 1,2 0,42 0,9 46,67 0,11
61
< 2,5 2,2 1,4 0,57 1,8 31,67 0,05
43
< 2,5 5 1,5 2,47 3,25 76 0,41
65
< 2,5 2 1,7 0,21 1,85 11,35 0,02
66 5,7 3,9 3,3 1,25 4,3 29,07 0,25
47
< 2,5 3,4 2,1 0,92 2,75 33,45 0,08
28
< 2,5 4 1,1 2,05 2,55 80,39 0,03
29
< 2,5 1,7 1,1 0,42 1,4 30 -
67
< 2,5 0,9 1,48 0,41 1,19 34,45 0,02
68
< 2,5 0,5 0,7 0,14 0,6 23,33 -
7
< 2,5 2,2 1,8 0,28 2 14 0,03
11
< 2,5 0,8 0,5 0,21 0,65 32,31 -
51
< 2,5 2,7 4,25 1,1 3,475 31,65 -
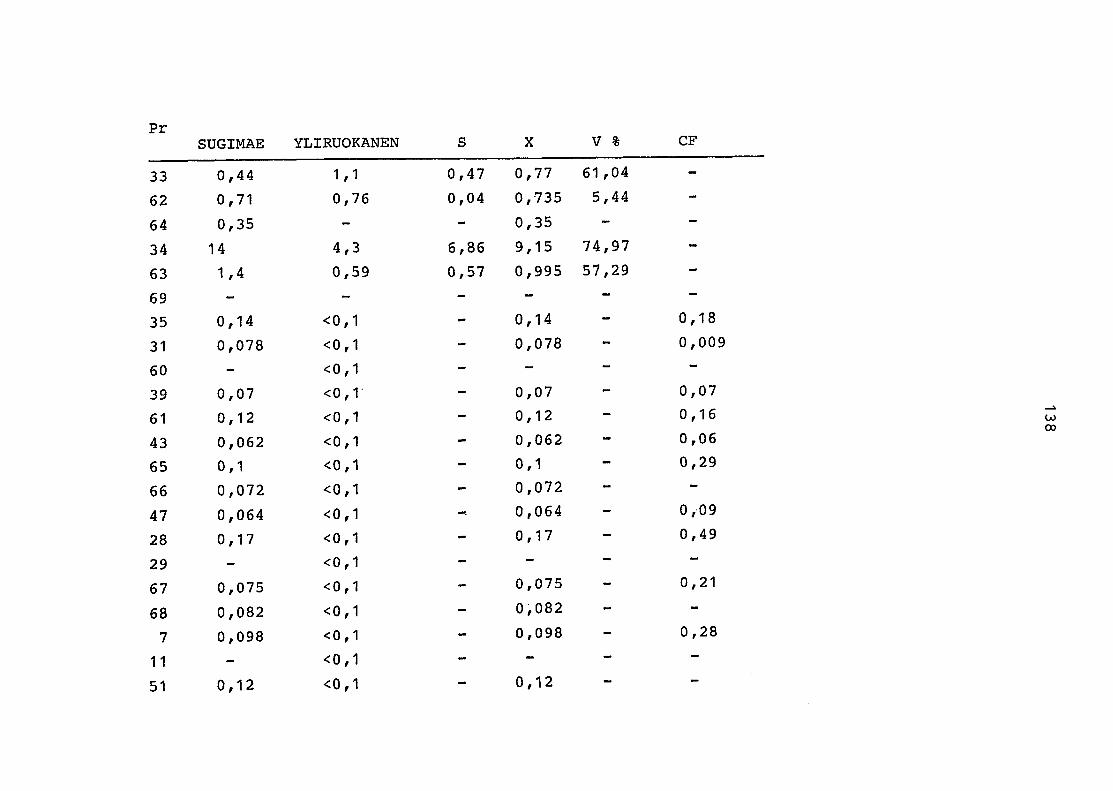
PrSUGIMAE
YLIRUOKANEN
S
X
V % CF
33
0,44
1,1
0,47
0,77
61,04 -
62
0,71
0,76
0,04
0,735
5,44 -
64
0,35
-
-
0,35
- -
34
14
4,3
6,86
9,15
74,97 -
63
1,4
0,59
0,57
0,995
57,29 -
69
35
0,14
<0,1
-
0,14
- 0,18
31
0,078
<0,1
-
0,078
- 0,009
60
-
<0,1
-
_
39
0,07
<0,1"
-
0,07
- 0,07
61
0,12
<0,1
-
0,12
- 0,16
43
0,062
<0,1
-
0,062
- 0,06
65
0,1
<0,1
-
0,1
- 0,29
66
0,072
<0,1
-
0,072
- -
47
0,064
<0,1
0,064
- 0 ;09
28
0,17
<0,1
-
0,17
- 0,49
29
-
<0,1
-
-
- -
67
0,075
<0,1
-
0,075
- 0,21
68
0,082
<0,1
-
0,082
- -
7
0,098
<0,1
-
0,098
- 0,28
11
-
<0,1
-
-
- -
51
0,12
<0,1
-
0,12
- -
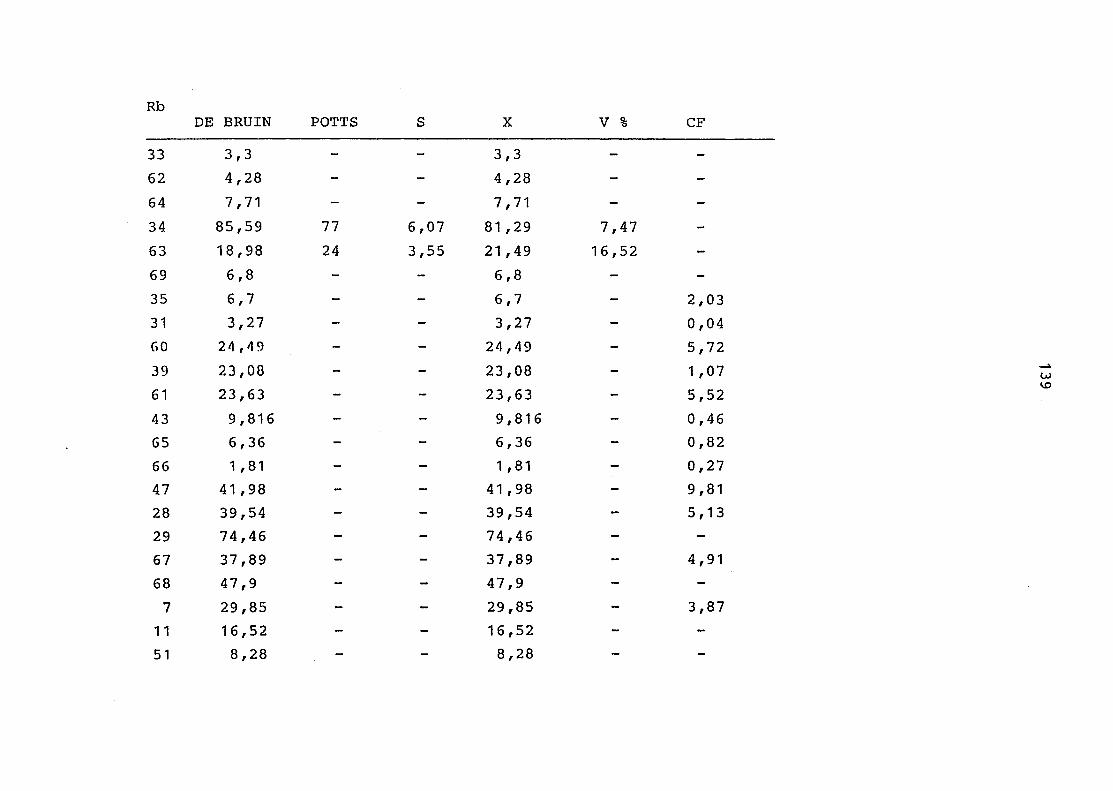
RbDE BRUIN POTTS
S X V % CF
33 3,3 -
- 3,3 - -
62 4,28 -
- 4,28 - -
64 7,71 -
- 7,71 - -
34 85,59 77
6,07 81,29 7,47 -
63 18,98 24
3,55 21,49 16,52 -
69 6,8 -
- 6,8 - -
35 6,7 -
- 6,7 - 2,03
31 3,27 -
- 3,27 - 0,04
60 24,49 -
- 24,49 - 5,72
39 23,08 -
- 23,08 - 1,07
61 23,63 -
- 23,63 - 5,52
43 9,816 -
- 9,816 - 0,46
65 6,36 -
- 6,36 - 0,82
66 1,81 -
- 1,81 - 0,27
47 41,98 -
- 41,98 - 9,81
28 39,54 -
- 39,54 - 5,13
29 74,46 -
- 74,46 - -
67 37,89 -
- 37,89 - 4,91
68 47,9 -
- 47,9 - -
7 29,85 -
- 29,85 - 3,87
11 16,52 -
- 16,52 - -
51 8,28 -
- 8,28 - -
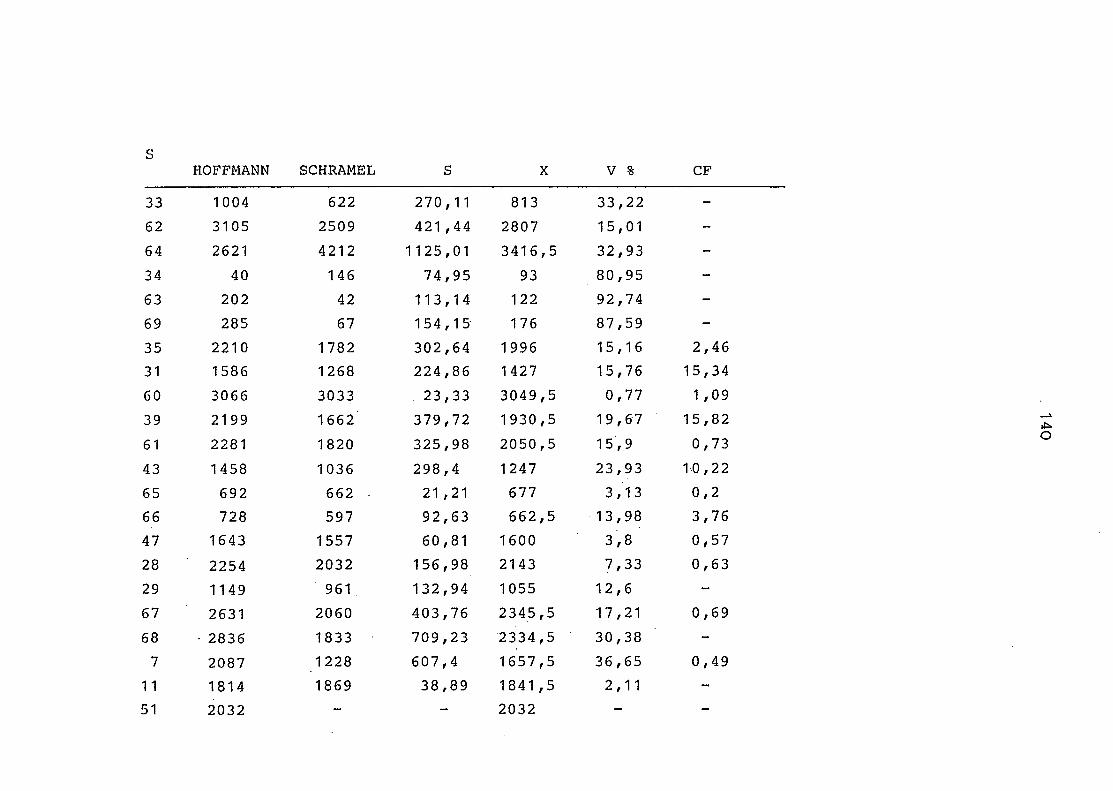
SHOFFMANN SCHRAMEL S X V % CF
33 1004 622 270,11 813 33,22 -
62 3105 2509 421,44 2807 15,01 -
64 2621 4212 1125,01 3416,5 32,93 -
34 40 146 74,95 93 80,95 -
63 202 42 113,14 122 92,74 -
69 285 67 154,15- 176 87,59 -
35 2210 1782 302,64 1996 15,16 2,46
31 1586 1268 224,86 1427 15,76 15,34
60 3066 3033 23,33 3049,5 0,77 1,09
39 2199 1662 379,72 1930,5 19,67 15,82
61 2281 1820 325,98 2050,5 15,9 0,73
43 1458 1036 298,4 1247 23,93 10,22
65 692 662 21,21 677 3,13 0,2
66 728 597 92,63 662,5 13,98 3,76
47 1643 1557 60,81 1600 3,8 0,57
28 2254 2032 156,98 2143 7,33 0,63
29 1149 961 132,94 1055 12,6 -
67 2631 2060 403,76 2345,5 17,21 0,69
68 2836 1833 709,23 2334,5 30,38 -
7 2087 1228 607,4 1657,5 36,65 0,49
11 1814 1869 38,89 1841,5 2,11 -
51 2032 - - 2032 - -
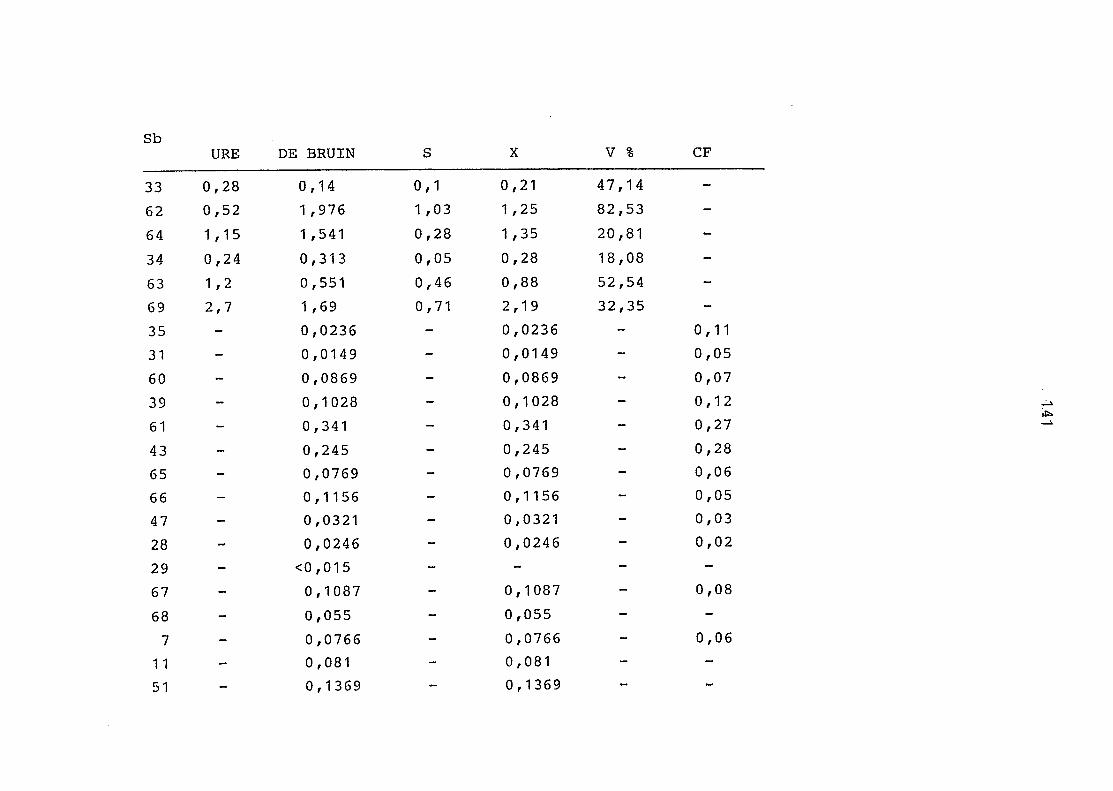
SbURE DE BRUIN
S X
V % CF
33
0,28 0,14
0,1 0,21
47,14 -
62
0,52 1,976
1,03 1,25
82,53 -
64
1,15 1,541
0,28 1,35
20,81 -
34
0,24 0,313
0,05 0,28
18,08 -
63
1,2 0,551
0,46 0,88
52,54
69
2,7 1,69
0,71 2,19
32,35 -
35
- 0,0236
- 0,0236
- 0,11
31
- 0,0149
- 0,0149 0,05
60
- 0,0869
- 0,0869
- 0,07
39
- 0,1028
- 0,1028
- 0,12
61
- 0,341
- 0,341
- 0,27
43
- 0,245
- 0,245
- 0,28
65
- 0,0769
- 0,0769
- 0,06
66
- 0,1156
- 0,1156
- 0,05
47
- 0,0321
- 0,0321
- 0,03
28
- 0,0246
- 0,0246
- 0,02
29
- <0,015
- -
- -
67
- 0,1087
- 0,1087
- 0,08
68
- 0,055
- 0,055
- -
7
- 0,0766
- 0,0766
- 0,06
11
- 0,081
- 0,081
- -
51
- 0,1369
- 0,1369
- -
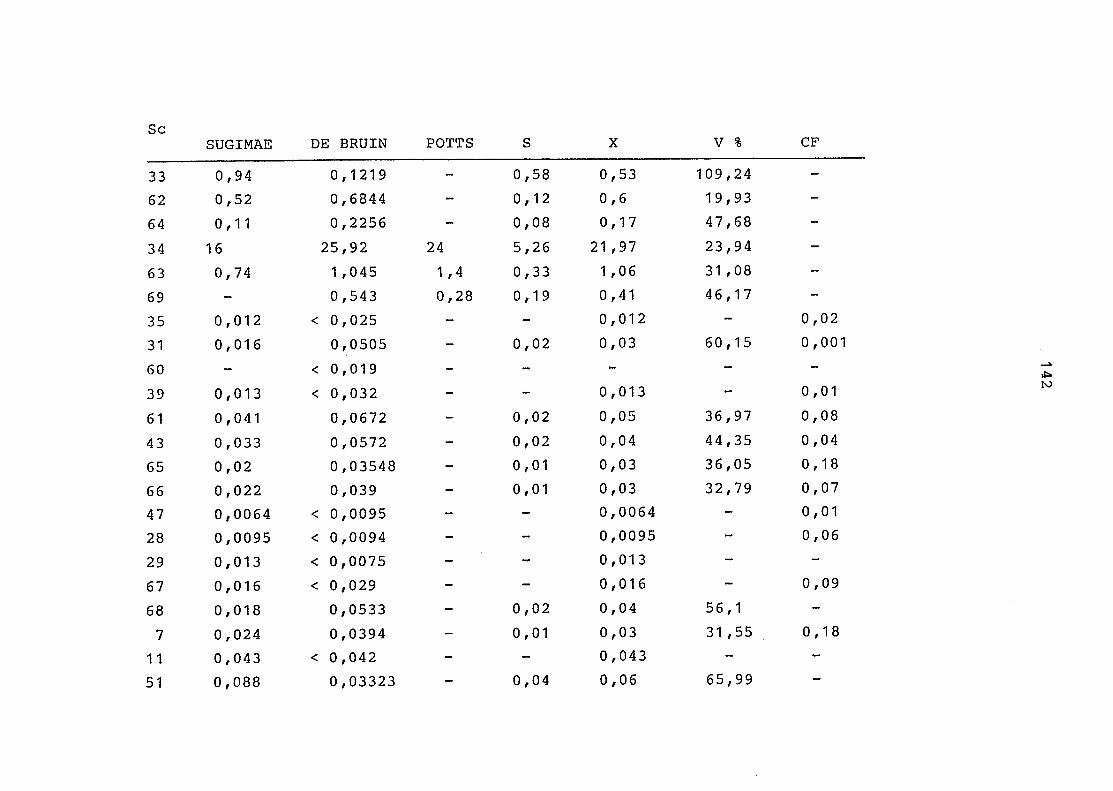
ScSUGIMAE
DE BRUIN POTTS S X V % CF
33
0,94 0,1219 - 0,58 0,53 109,24 -
62
0,52 0,6844 - 0,12 0,6 19,93 -
64
0,11 0,2256 - 0,08 0,17 47,68 -
34
16 25,92 24 5,26 21,97 23,94 -
63
0,74 1,045 1,4 0,33 1,06 31,08 -
69
- 0,543 0,28 0,19 0,41 46,17 -
35
0,012
< 0,025 - - 0,012 - 0,02
31
0,016 0,0505 - 0,02 0,03 60,15 0,001
60
-
< 0,019 - - - - -
39
0,013
< 0,032 - - 0,013 - 0,01
61
0,041 0,0672 - 0,02 0,05 36,97 0,08
43
0,033 0,0572 - 0,02 0,04 44,35 0,04
65
0,02 0,03548 - 0,01 0,03 36,05 0,18
66
0,022 0,039 - 0,01 0,03 32,79 0,07
47
0,0064
< 0,0095 - - 0,0064 - 0,01
28
0,0095
< 0,0094 - - 0,0095 - 0,06
29
0,013
< 0,0075 - - 0,013 - -
67
0,016
< 0,029 - - 0,016 - 0,09
68
0,018 0,0533 - 0,02 0,04 56,1 -
7
0,024 0,0394 - 0,01 0,03 31,55 0,18
11
0,043
< 0,042 - - 0,043
51
0,088 0,03323 - 0,04 0,06 65,99 -
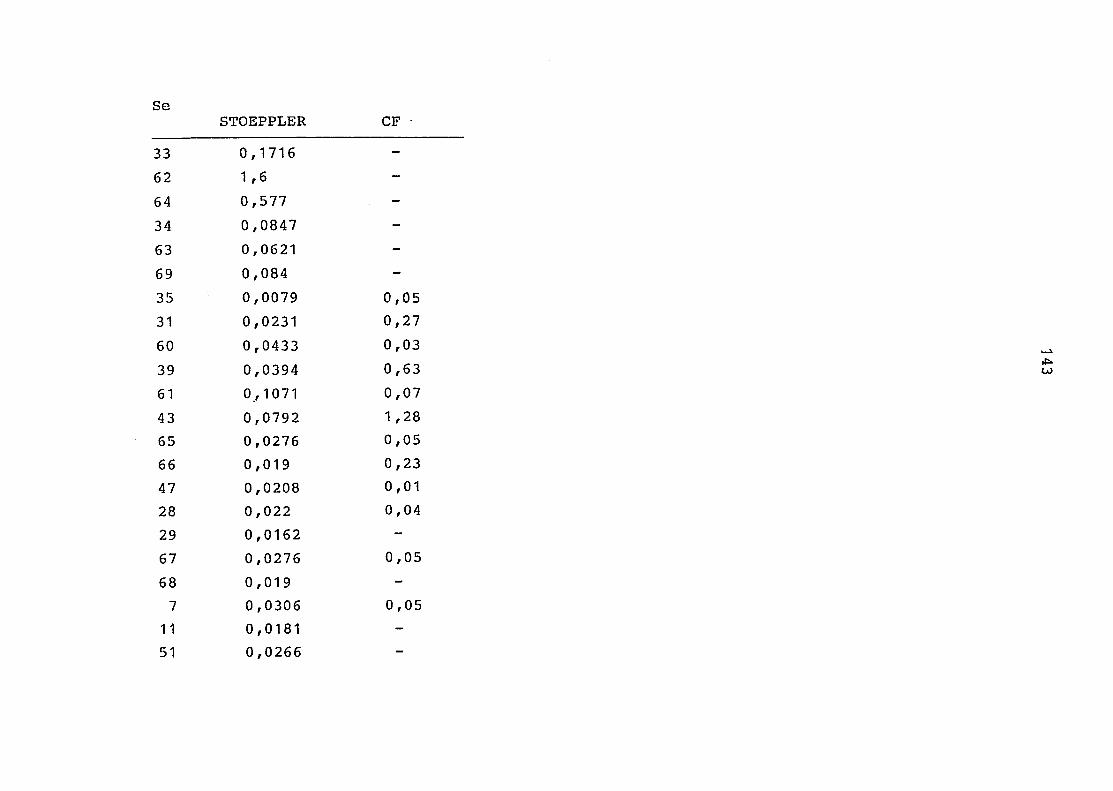
SeSTOEPPLER CF
33 0,1716 -
62 1,6 -
64 0,577 -
34 0,0847 -
63 0,0621 -
69 0,084 -
35 0,0079 0,05
31 0,0231 0,27
60 0,0433 0,03
39 0,0394 0,63 w61 0,1071 0,07
43 0,0792 1,28
65 0,0276 0,05
66 0,019 0,23
47 0,0208 0,01
28 0,022 0,04
29 0,0162 -
67 0,0276 0,05
68 0,019 -
7 0,0306 0,05
11 0,0181 -
51 0,0266 -
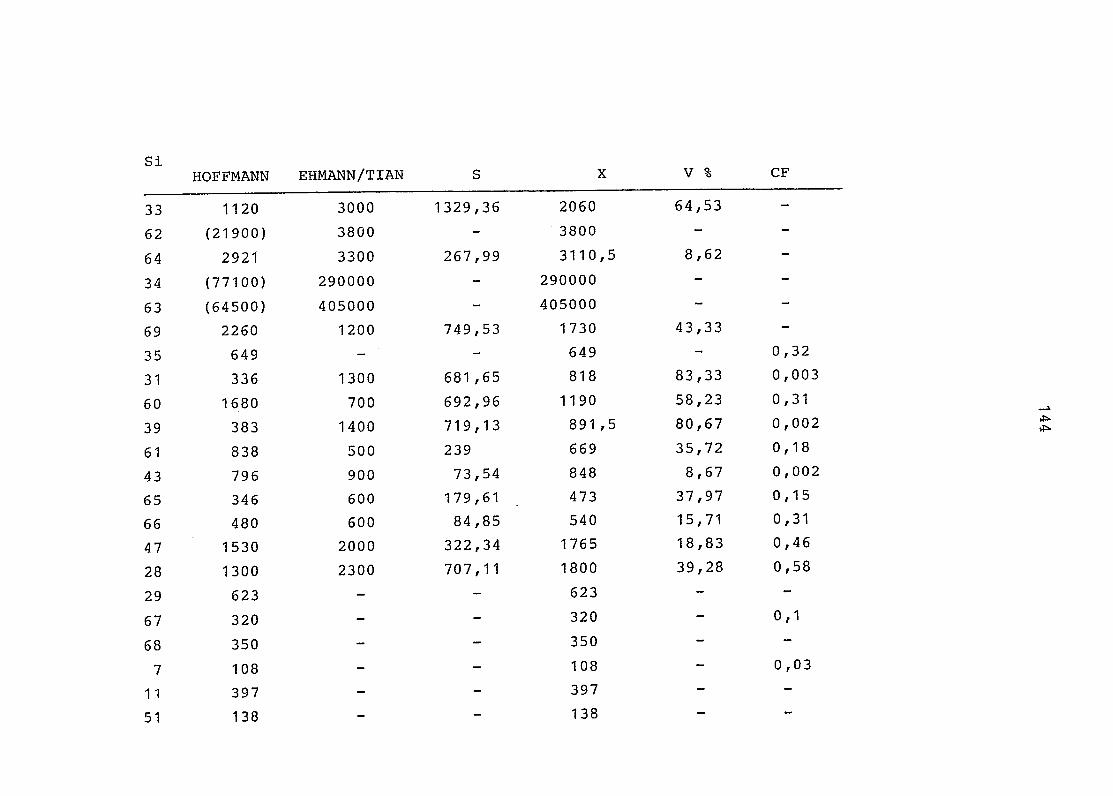
SiHOFFMANN EHMANN/TIAN S X V ö CF
33 1120 3000 1329,36 2060 64,53
62 (21900) 3800 - 3800 -
64 2921 3300 267,99 3110,5 8,62
34 (77100) 290000 - 290000 -
63 (64500) 405000 - 405000 -
69 2260 1200 749,53 1730 43,33
35 649 - - 649 - 0,32
31 336 1300 681,65 818 83,33 0,003
60 1680 700 692,96 1190 58,23 0,31
39 383 1400 719,13 891,5 80,67 0,002
61 838 500 239 669 35,72 0,18
43 796 900 73,54 848 8,67 0,002
65 346 600 179,61 473 37,97 0,15
66 480 600 84,85 540 15,71 0,31
47 1530 2000 322,34 1765 18,83 0,46
28 1300 2300 707,11 1800 39,28 0,58
29 623 - - 623 -
67 320 - - 320 - 0,1
68 350 - - 350 -
7 108 - - 108 - 0,03
11 397 - - 397 -
51 138 - - 138 -
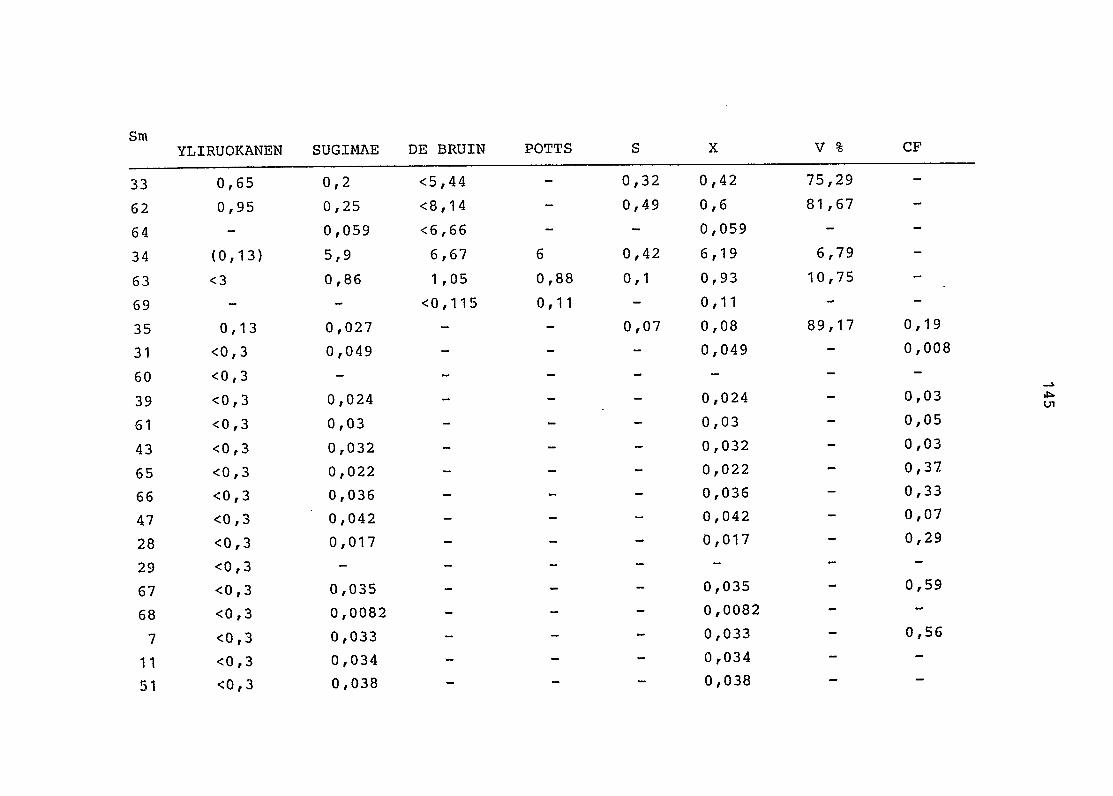
SmYLIRUOKANEN
SUGIMAE
DE BRUIN
POTTS S X
V % CF
33 0,65
0,2
<5,44
- 0,32 0,42
75,29 -
62 0,95
0,25
<8,14
- 0,49 0,6
81,67 -
64 -
0,059
<6,66
- 0,059
- -
34 (0,13)
5,9
6,67
6 0,42 6,19
6,79 -
63 <3
0,86
1,05
0,88 0,1 0,93
10,75 -
69 -
-
<0,115
0,11 - 0,11
- -
35 0,13
0,027
-
- 0,07 0,08
89,17 0,19
31 <0,3
0,049
-
- 0,049
- 0,008
60 <0,3
-
-
-
39 <0,3
0,024
-
- 0,024
- 0,03
61 <0,3
0,03
-
- 0,03
- 0,05
43 <0,3
0,032
-
- 0,032
- 0,03
65 <0,3
0,022
-
- 0,022
- 0,37
66 <0,3
0,036
-
- 0,036
- 0,33
47 <0,3
0,042
-
- 0,042
- 0,07
28 <0,3
0,017
-
- 0,017
- 0,29
29 <0,3
-
-
-
67 <0,3
0,035
-
- 0,035 0,59
68 <0,3
0,0082
-
- 0,0082
7 <0,3
0,033
-
- 0,033 0,56
11 <0,3
0,034
-
- 0,034
51 <0,3
0,038
-
- 0,038
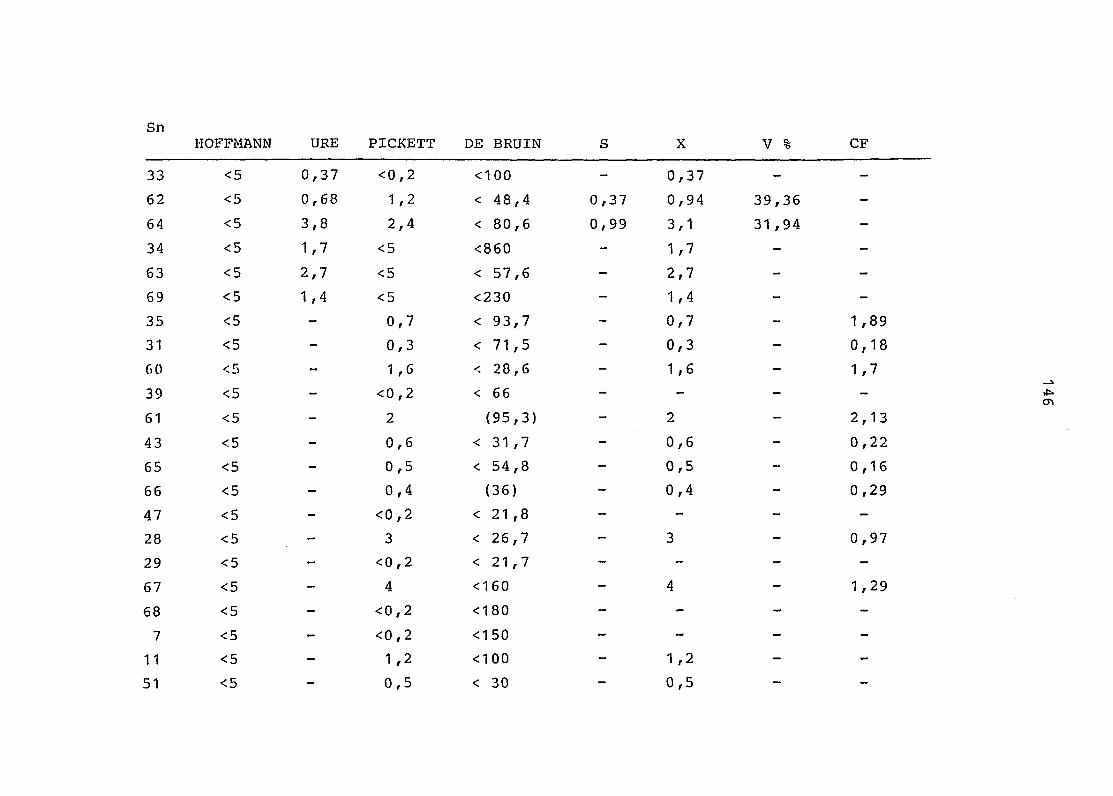
SnHOFFMANN
URE PICKETT
DE BRUIN
S
X
V % CF
33 <5
0,37 <0,2
<100
-
0,37
- -
62 <5
0,68 1,2
<
48,4
0,37
0,94
39,36 -
64 <5
3,8 2,4
<
80,6
0,99
3,1
31,94 -
34 <5
1,7 <5
<860
-
1,7
- -
63 <5
2,7 <5
< 57,6
-
2,7
- -
69 <5
1,4 <5
<230
-
1,4
- -
35 <5
- 0,7
<
93,7
-
0,7
- 1,89
31 <5
- 0,3
<
71,5
-
0,3
- 0,18
60 <5
- 1,6
<
28,6
-
1,6
- 1,7
39 <5
- <0,2
< 66
-
-
- -
61 <5
- 2
(95,3)
-
2
- 2,13
43 <5
- 0,6
<
31,7
-
0,6
- 0,22
65 <5
- 0,5
< 54,8
-
0,5
- 0,16
66 <5
- 0,4
(36)
-
0,4
- 0,29
47 <5
- <0,2
< 21,8
-
-
- -
28 <5
- 3
< 26,7
-
3
- 0,97
29 <5
- <0,2
<
21,7
-
-
- -
67 <5
- 4
<160
-
4
- 1,29
68 <5
- <0,2
<180
-
-
- -
7 <5
- <0,2
<150
-
-
- -
11 <5
- 1,2
<100
-
1,2
- -
51 <5
- 0,5
< 30
-
0,5
- -
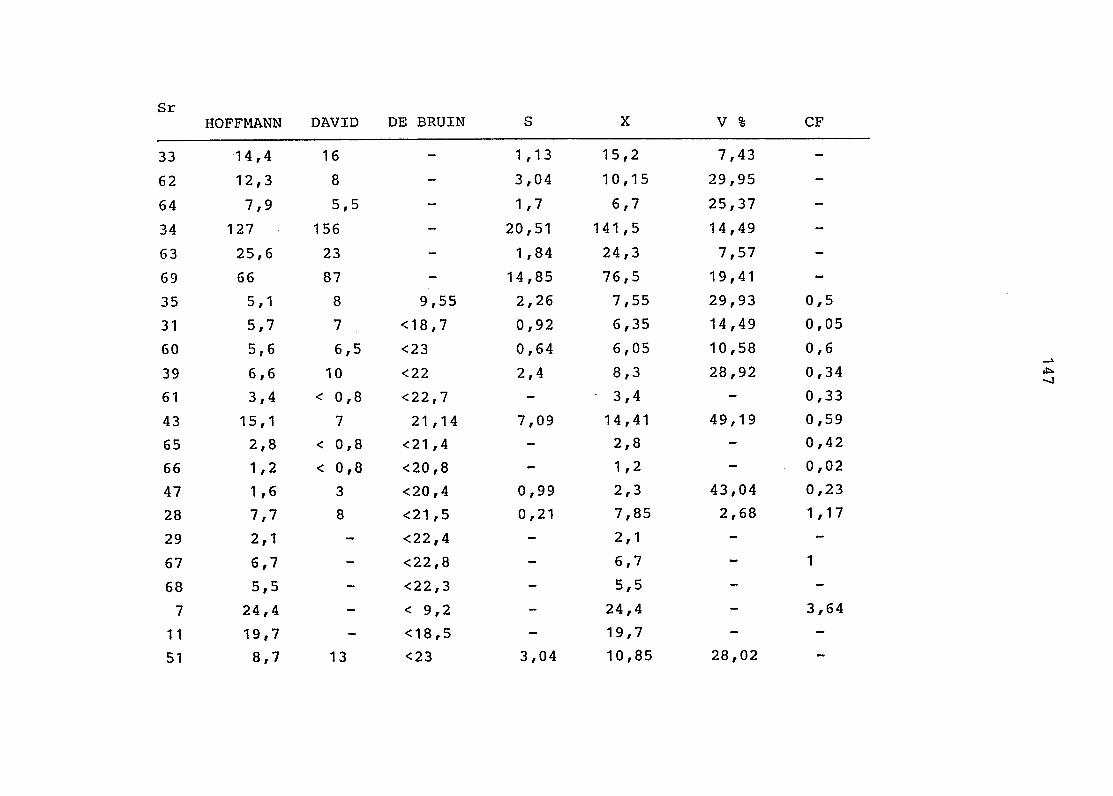
SrHOFFMANN DAVID DE BRUIN S X V % CF
33 14,4 16 - 1,13 15,2 7,43
62 12,3 8 - 3,04 10,15 29,95 -
64 7,9 5,5 - 1,7 6,7 25,37 -
34 127
- 156 - 20,51 141,5 14,49 -
63 25,6 23 - 1,84 24,3 7,57 -
69 66 87 - 14,85 76,5 19,41 -
35 5,1 8 9,55 2,26 7,55 29,93 0,5
31 5,7 7 <18,7 0,92 6,35 14,49 0,05
60 5,6 6,5 <23 0,64 6,05 10,58 0,6
39 6,6 10 <22 2,4 8,3 28,92 0,34 de.
61 3,4 <
0,8 <22,7 - 3,4 - 0,33
43 15,1 7 21,14 7,09 14,41 49,19 0,59
65 2,8 < 0,8 <21,4 - 2,8 - 0,42
66 1,2 < 0,8 <20,8 - 1,2 - 0,02
47 1,6 3 <20,4 0,99 2,3 43,04 0,23
28 7,7 8 <21,5 0,21 7,85 2,68 1,17
29 2,1 - <22,4 - 2,1 - -
67 6,7 - <22,8 - 6,7 - 1
68 5,5 - <22,3 - 5,5 - -
7 24,4 - < 9,2 - 24,4 - 3,64
11 19,7 - <18,5 - 19,7 - -
51 8,7 13 <23 3,04 10,85 28,02 -
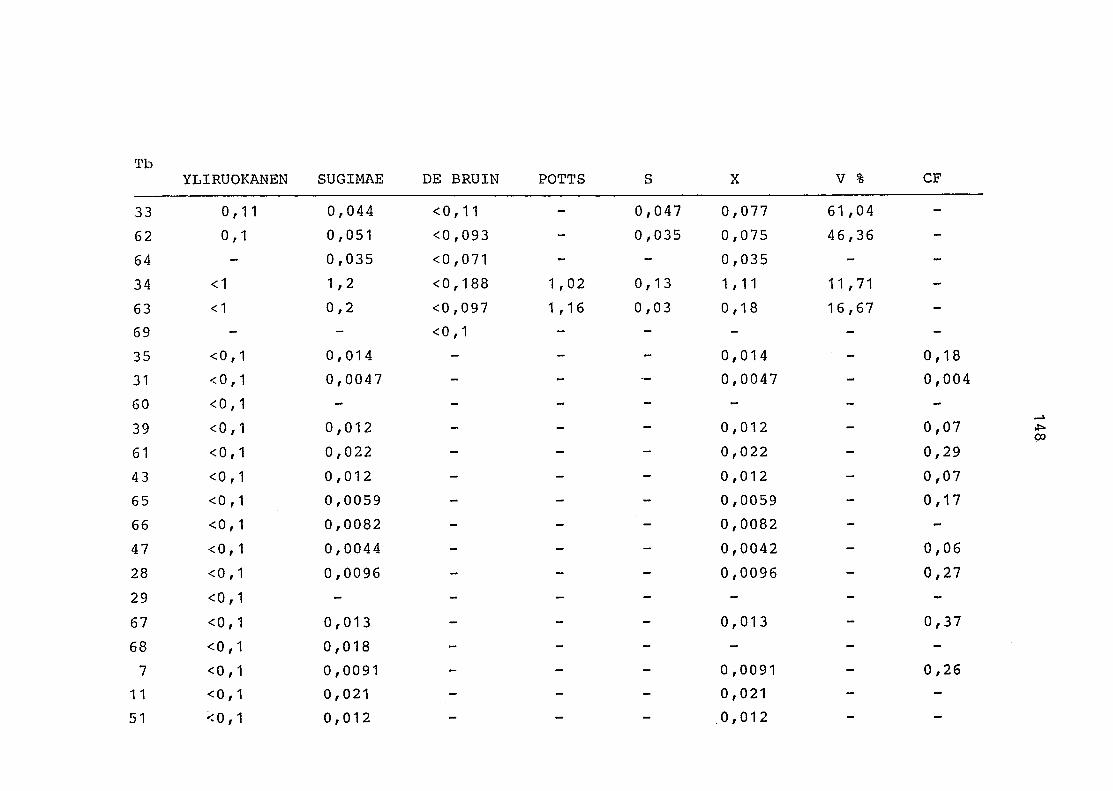
TbYLIRUOKANEN
SUGIMAE
DE BRUIN
POTTS S
X
V % CF
33
0,11
0,044
<0,11
- 0,047
0,077
61,04 -
62
0,1
0,051
<0,093
- 0,035
0,075
46,36 -
64
-
0,035
<0,071
- -
0,035
- -
34
<1
1,2
<0,188
1,02 0,13
1,11
11,71 -
63
<1
0,2
<0,097
1,16 0,03
0,18
16,67 -
69
-
-
<0,1
- -
-
- -
35
<0,1
0,014
-
- -
0,014
- 0,18
31
<0,1
0,0047
-
- --
0,0047
- 0,004
60
<0,1
-
-
- -
-
- -
39
<0,1
0,012
-
- -
0,012
- 0,07
61
<0,1
0,022
-
- -
0,022
- 0,29
43
<0,1
0,012
-
- -
0,012
- 0,07
65
<0,1
0,0059
-
- -
0,0059
- 0,17
66
<0,1
0,0082
-
- -
0,0082
- -
47
<0,1
0,0044
-
- -
0,0042
- 0,06
28
<0,1
0,0096
-
- -
0,0096
- 0,27
29
<0,1
-
-
- -
-
- -
67
<0,1
0,013
-
- -
0,013
- 0,37
68
<0,1
0,018
-
- -
-
- -
7
<0,1
0,0091
-
- -
0,0091
- 0,26
11
<0,1
0,021
-
- -
0,021
- -
51
<0,1
0,012
-
- -
.0,012
- -
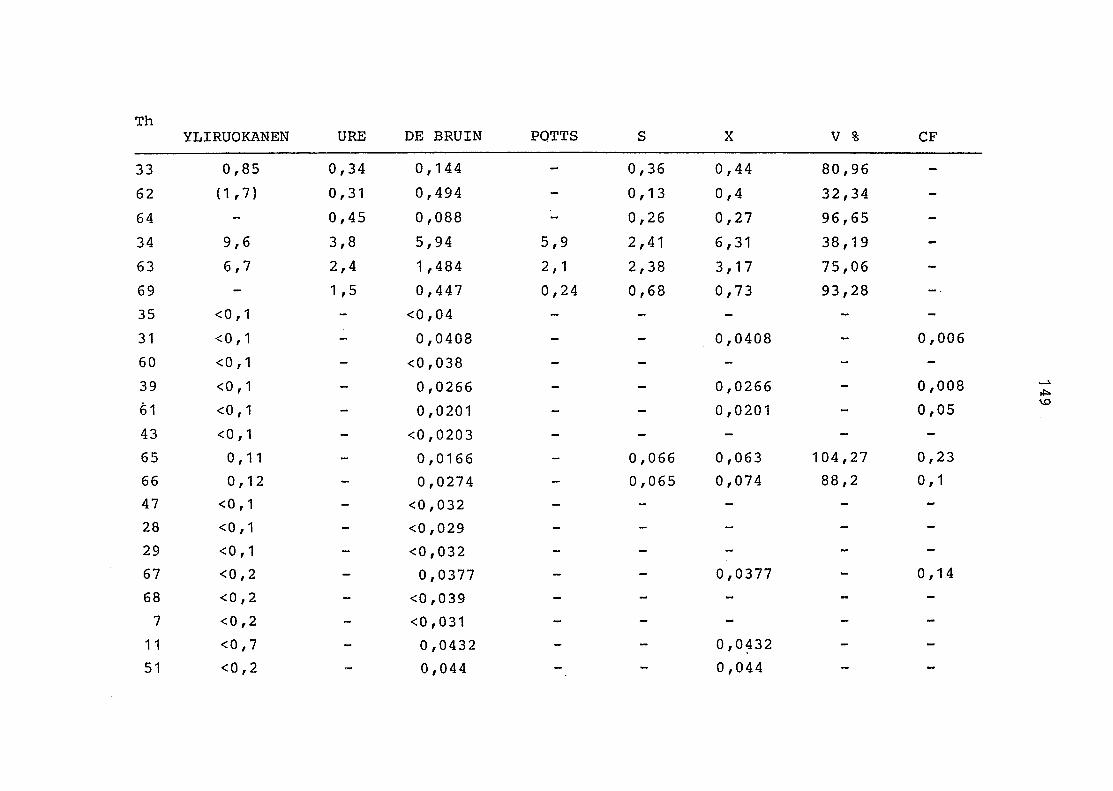
ThYLIRUOKANEN
URE DE BRUIN POTTS
S X V % CF
33 0,85
0,34 0,144 -
0,36 0,44 80,96 -
62 (1,7)
0,31 0,494 -
0,13 0,4 32,34 -
64 -
0,45 0,088 -
0,26 0,27 96,65 -
34 9,6
3,8 5,94 5,9
2,41 6,31 38,19 -
63 6,7
2,4 1,484 2,1
2,38 3,17 75,06 -
69 -
1,5 0,447 0,24
0,68 0,73 93,28
35 <0,1
- <0,04 -
- - - -
31 <0,1
- 0,0408 -
- 0,0408 - 0,006
60 <0,1
- <0,038 -
- - - -
39 <0,1
- 0,0266 -
- 0,0266 - 0,008
61 <0,1
- 0,0201 -
- 0,0201 - 0,05
43 <0,1
- <0,0203 -
- - - -
65 0,11
- 0,0166 -
0,066 0,063 104,27 0,23
66 0,12
- 0,0274 -
0,065 0,074 88,2 0,1
47 <0,1
- <0,032 -
- - - -
28 <0,1
- <0,029 -
- - - -
29 <0,1
- <0,032 -
- -
67 <0,2
- 0,0377 -
- 0,0377 - 0,14
68 <0,2
- <0,039 -
- - - -
7 <0,2
- <0,031 -
- - - -
11 <0,7
- 0,0432 -
- 0,0432 - -
51 <0,2
- 0,044 - 0,044 - -
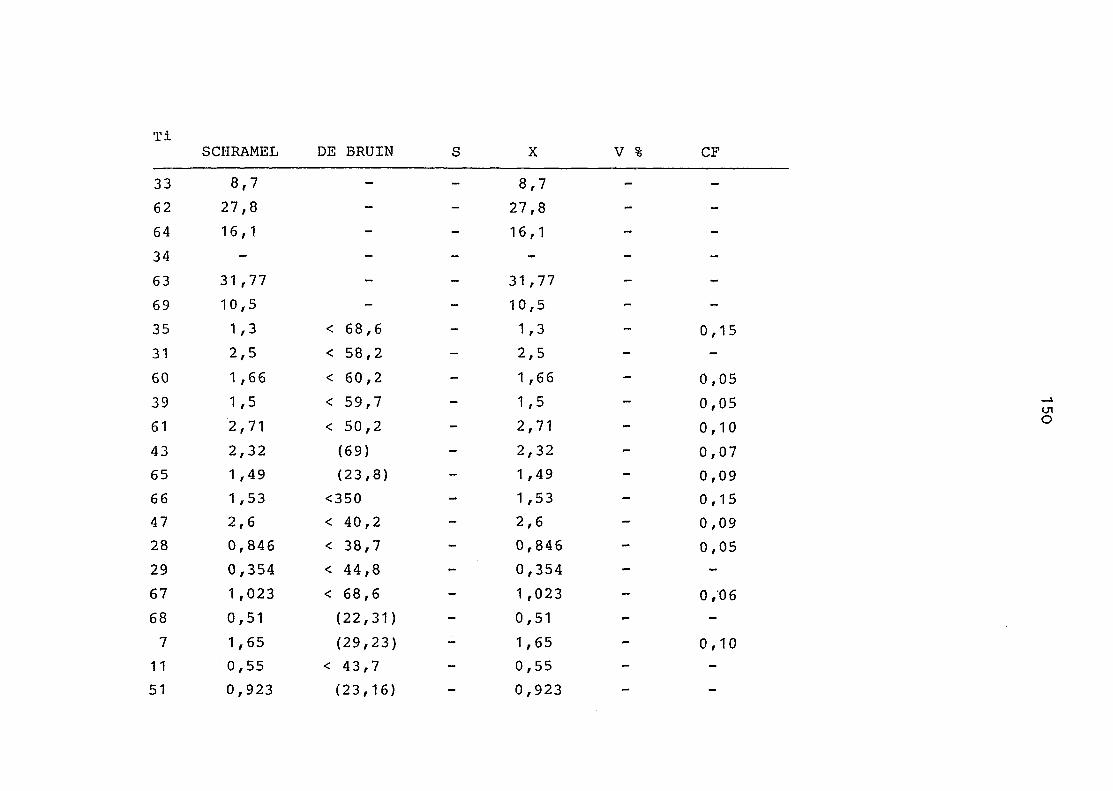
TiSCHRAMEL
DE BRUIN
S
X
V % CF
33
8,7
-
-
8,7
- -
62
27,8
-
-
27,8
- -
64
16,1
-
-
16,1
- -
34 -
63
31,77
-
-
31,77
- -
69
10,5
-
-
10,5
- -
35
1,3
<
68,6
-
1,3
- 0,15
31
2,5
<
58,2
-
2,5
- -
60
1,66
<
60,2
-
1,66
- 0,05
39
1,5
<
59,7
-
1,5
- 0,05
61
2,71
<
50,2
-
2,71
- 0,10
43
2,32
(69)
-
2,32
- 0,07
65
1,49
(23,8)
-
1,49
- 0,09
66
1,53
<350
-
1,53
- 0,15
47
2,6
<
40,2
-
2,6
- 0,09
28
0,846
< 38,7
-
0,846
- 0,05
29
0,354
< 44,8
-
0,354
- -
67
1,023
<
68,6
-
1,023
- 0,"06
68
0,51
(22,31)
-
0,51
- -
7
1,65
(29,23)
-
1,65
- 0,10
11
0,55
<
43,7
-
0,55
- -
51
0,923
(23,16)
-
0,923
- -
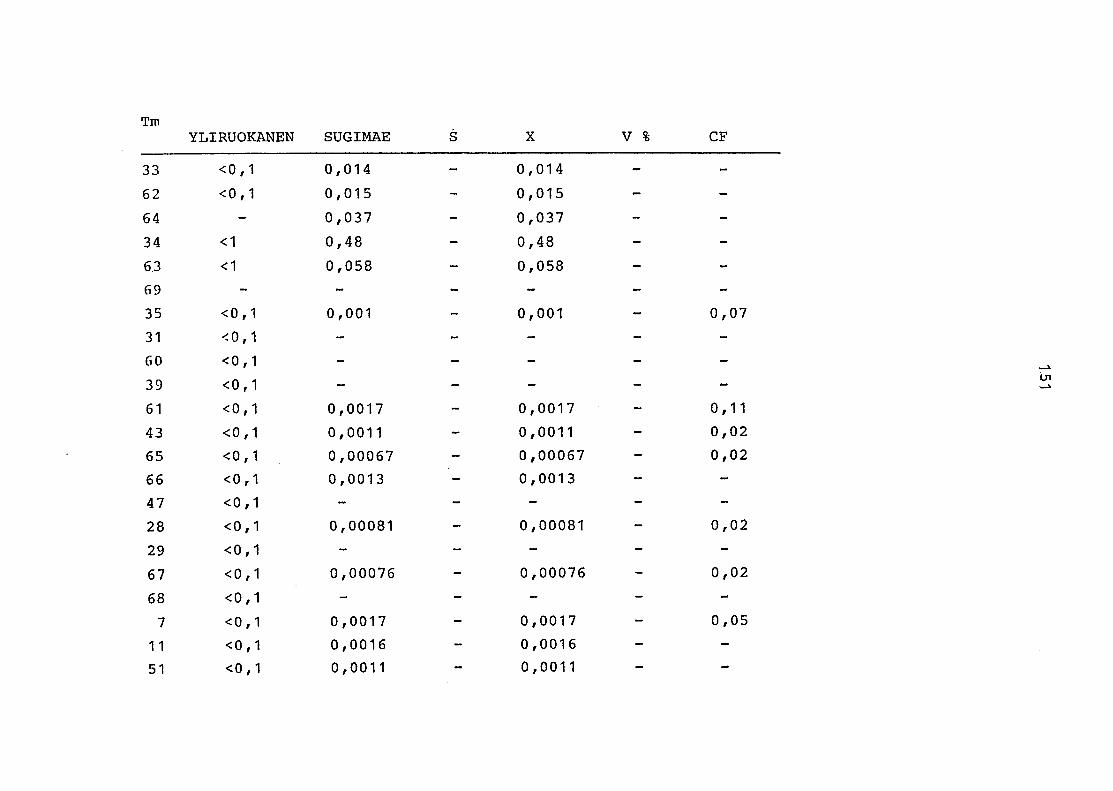
TmYLIRUOKANEN SUGIMAE
S X
V %
CF
33
<0,1 0,014
- 0,014
-
-
62
<0,1 0,015
- 0,015
-
-
64
- 0,037
- 0,037
-
-
34
<1 0,48
- 0,48
-
-
6 .3
<1 0,058
- 0,058
-
-
69
- -
- -
-
-
35
<0,1 0,001
- 0,001
-
0,07
31
<0,1 -
- -
-
-
GO
<0,1 -
- -
-
-
39
<0,1 -
- -
-
-
61
<0,1 0,0017
- 0,0017
-
0,11
43
<0,1 0,0011
- 0,0011
-
0,02
65
<0,1 0,00067
- 0,00067
-
0,02
66
<0,1 0,0013
- 0,0013
-
-
47
<0,1 -
- -
-
-
28
<0,1 0,00081
- 0,00081
-
0,02
29
<0,1 -
- -
-
-
67
<0,1 0,00076
- 0,00076
-
0,02
68
<0,1 -
- -
-
-
7
<0,1 0,0017
- 0,0017
-
0,05
11
<0,1 0,0016
- 0,0016
-
-
51
<0,1 0,0011
- 0,0011
-
-
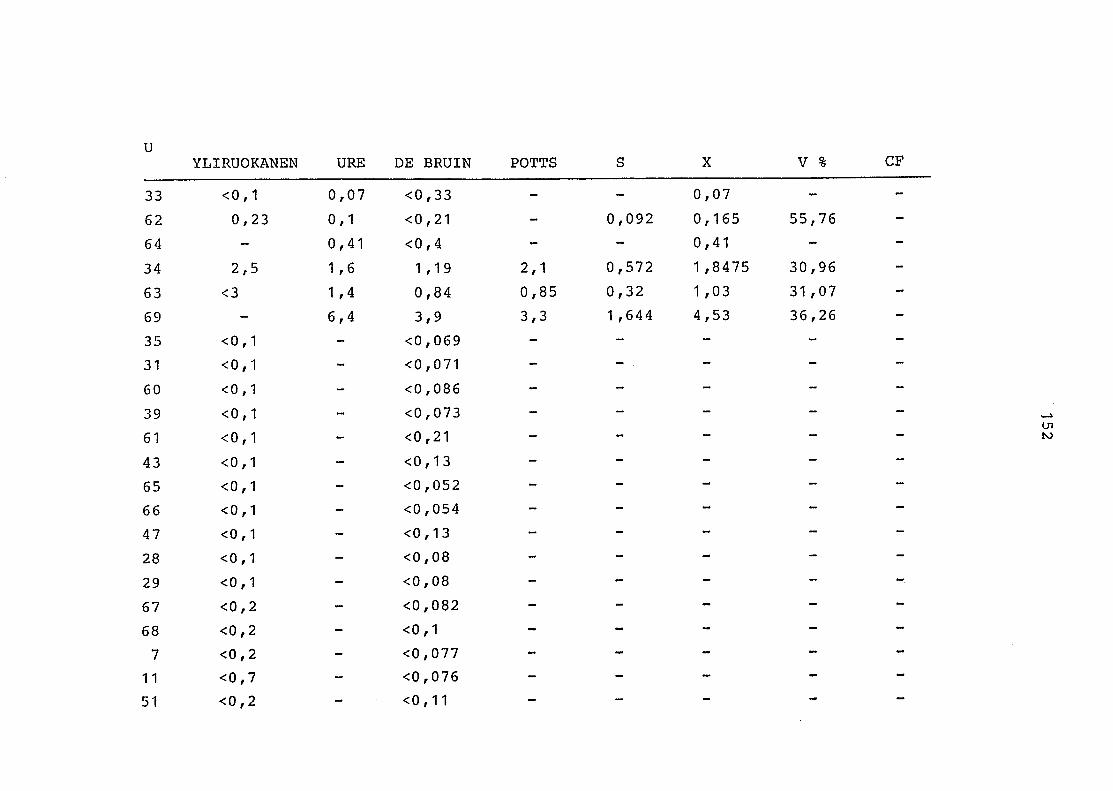
UYLIRUOKANEN
URE DE BRUIN POTTS
S
X
V %
CF
33 <0,1
0,07 <0,33 -
-
0,07
-
-
62 0,23
0,1 <0,21 -
0,092
0,165
55,76
-
64 -
0,41 <0,4 -
-
0,41
-
-
34 2,5
1,6 1,19 2,1
0,572
1,8475
30,96
-
63 <3
1,4 0,84 0,85
0,32
1,03
31,07
-
69 -
6,4 3,9 3,3
1,644
4,53
36,26
-
35 <0,1
- <0,069 -
-
-
-
-
31 <0,1
- <0,071 -
-
-
-
60 <0,1
- <0,086 -
-
-
-
-
39 <0,1
- <0,073 -
-
-
-
-cn
61 <0,1
- <0,21 -
-
-
-
- N
43 <0,1
- <0,13 -
-
-
-
-
65 <0,1
- <0,052 -
-
-
-
-
66 <0,1
- <0,054 -
-
-
-
-
47 <0,1
- <0,13 -
-
-
-
-
28 <0,1
- <0,08 -
-
-
-
-
29 <0,1
- <0,08 -
-
-
-
-
67 <0,2
- <0,082 -
-
-
-
-
68 <0,2
- <0,1 -
-
-
-
-
7 <0,2
- <0,077 -
-
-
-
-
11 <0,7
- <0,076 -
-
-
-
-
51 <0,2
- <0,11 -
-
-
-
-
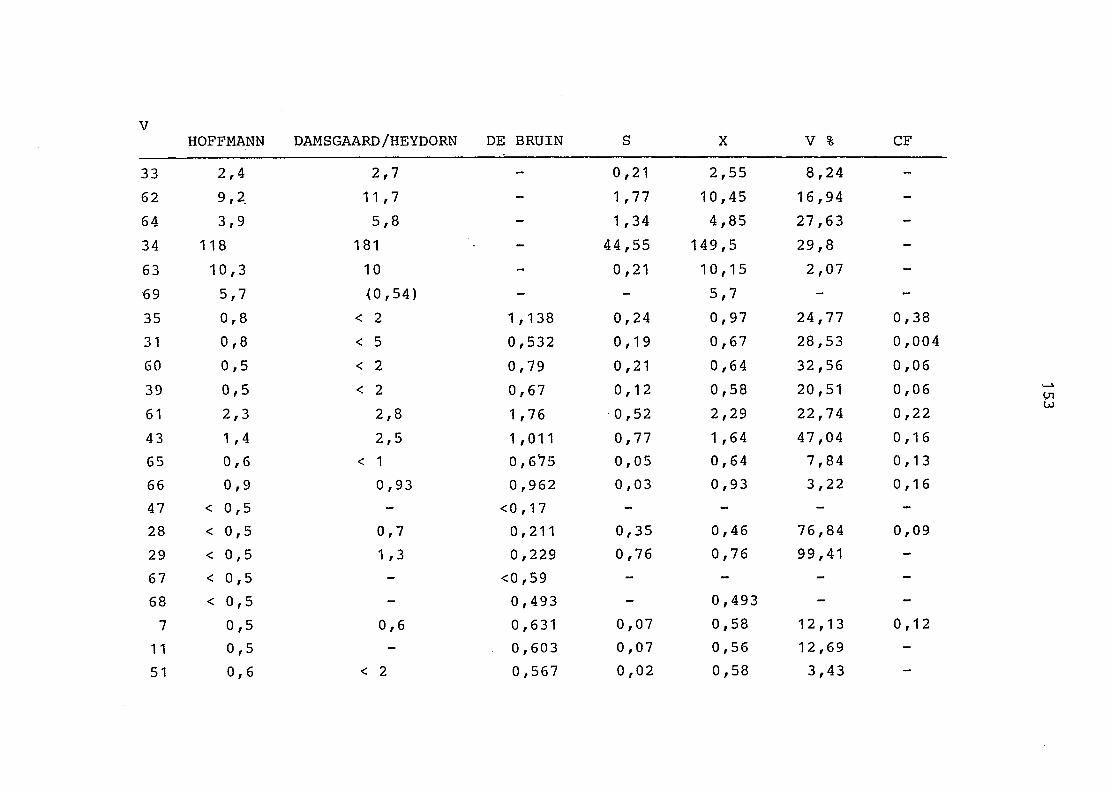
VHOFFMANN DAMSGAARD/HEYDORN DE BRUIN
S
X
V %
CF
33
2,4
2,7
-
0,21
2,55
8,24
-
62
9,2
11,7
-
1,77
10,45
16,94
-
64
3,9
5,8
-
1,34
4,85
27,63
-
34
118
181
-
44,55
149,5
29,8
-
63
10,3
10
-
0,21
10,15
2,07
-
69
5,7
{0,54)
-
-
5,7
-
-
35
0,8
< 2
1,138
0,24
0,97
24,77
0,38
31
0,8
< 5
0,532
0,19
0,67
28,53
0,004
GO
0,5
< 2
0,79
0,21
0,64
32,56
0,06
39
0,5
< 2
0,67
0,12
0,58
20,51
0,06
61
2,3
2,8
1,76
-0,52
2,29
22,74
0,22
43
1,4
2,5
1,011
0,77
1,64
47,04
0,16
65
0,6
< 1
0,615
0,05
0,64
7,84
0,13
66
0,9
0,93
0,962
0,03
0,93
3,22
0,16
47
< 0,5
-
<0,17
-
-
-
-
28
< 0,5
0,7
0,211
0,35
0,46
76,84
0,09
29
< 0,5
1,3
0,229
0,76
0,76
99,41
-
67
< 0,5
-
<0,59
-
-
-
-
68
< 0,5
-
0,493
-
0,493
-
-
7
0,5
0,6
0,631
0,07
0,58
12,13
0,12
11
0,5
-
0,603
0,07
0,56
12,69
-
51
0,6
< 2
0,567
0,02
0,58
3,43
-
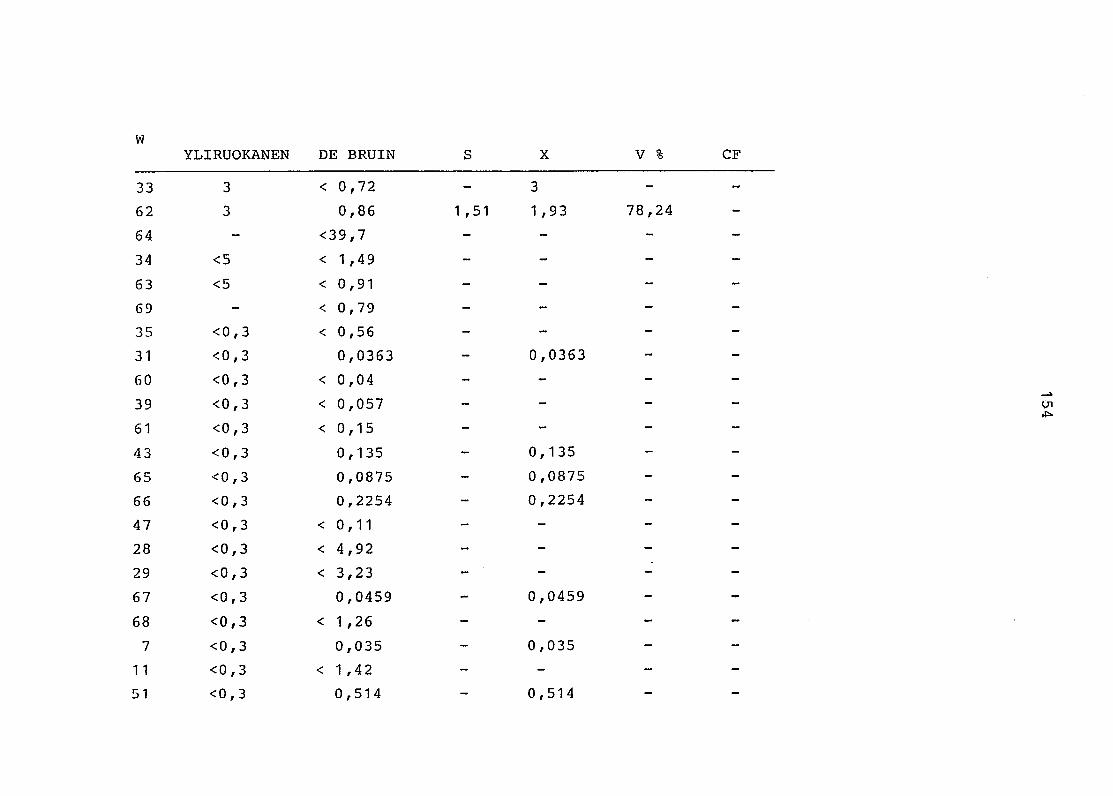
wYLIRUOKANEN
DE BRUIN
S
X
V %
CF
33 3
< 0,72
-
3
-
62 3
0,86
1,51
1,93
78,24
64 <39,7
-
-
-
34 <5
<
1,49
-
-
-
63 <5
< 0,91
-
-
-
69 -
< 0,79
-
-
-
35 <0,3
< 0,56
-
-
-
31 <0,3
0,0363
-
0,0363
-
60 <0,3
< 0,04
-
-
-
39 <0,3
< 0,057
-
-
-
61 <0,3
< 0,15
-
-
-
43 <0,3
0,135
-
0,135
-
65 <0,3
0,0875
-
0,0875
-
66 <0,3
0,2254
-
0,2254
-
47 <0,3
<
0,11
-
-
-
28 <0,3
< 4,92
-
-
-
29 <0,3
< 3,23
-
-
-
-
67 <0,3
0,0459
-
0,0459
-
-
68 <0,3
<
1,26
-
-
-
-
7 <0,3
0,035
-
0,035
-
-
11 <0,3
< 1,42
-
-
-
-
51 <0,3
0,514
-
0,514
-
-
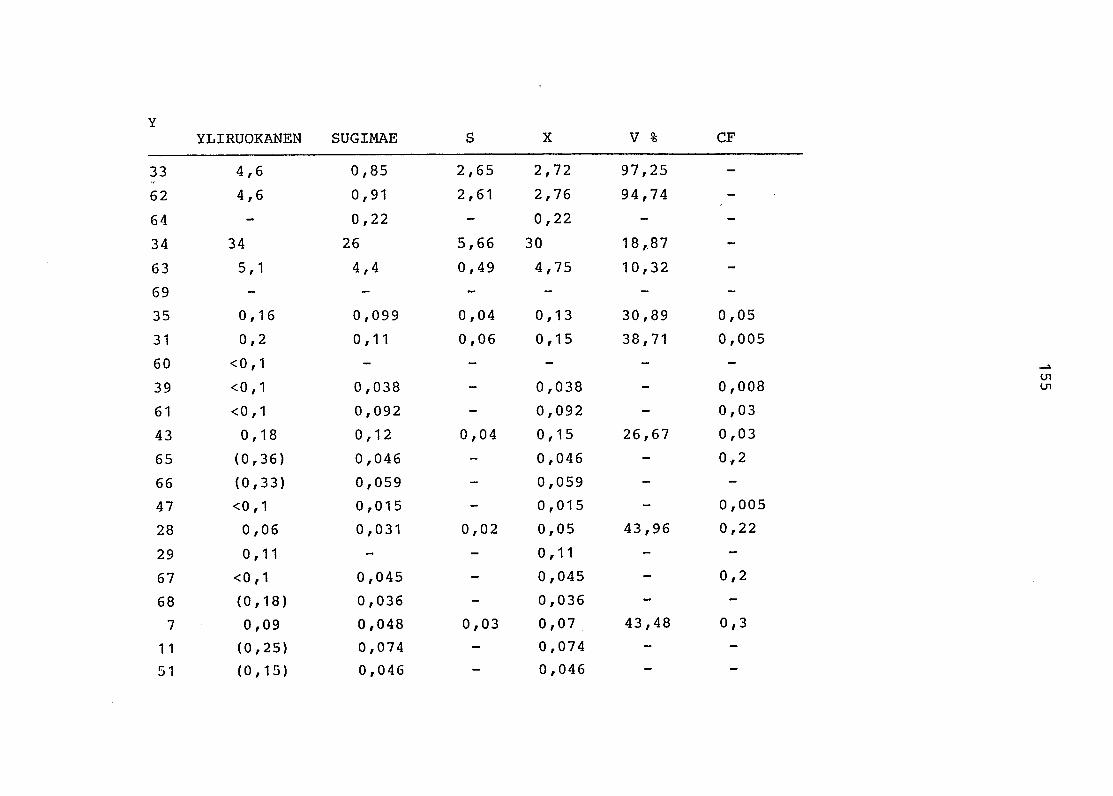
YYLIRUOKANEN SUGIMAE S X V % CF
33 4,6 0,85 2,65 2,72 97,25 -
62 4,6 0,91 2,61 2,76 94,74 -
64 - 0,22 - 0,22 - -
34 34 26 5,66 30 18,87 -
63 5,1 4,4 0,49 4,75 10,32 -
69 - - - - - -
35 0,16 0,099 0,04 0,13 30,89 0,05
31 0,2 0,11 0,06 0,15 38,71 0,005
60 <0,1 - - - - -
39 <0,1 0,038 - 0,038 - 0,008 cn
61 <0,1 0,092 - 0,092 - 0,03
43 0,18 0,12 0,04 0,15 26,67 0,03
65 (0,36) 0,046 - 0,046 - 0,2
66 (0,33) 0,059 - 0,059 - -
47 <0,1 0,015 - 0,015 - 0,005
28 0,06 0,031 0,02 0,05 43,96 0,22
29 0,11 - - 0,11 - -
67 <0,1 0,045 - 0,045 - 0,2
68 (0,18) 0,036 - 0,036 - -
7 0,09 0,048 0,03 0,07 43,48 0,3
11 (0,25) 0,074 - 0,074 - -
51 (0,15) 0,046 - 0,046 - -
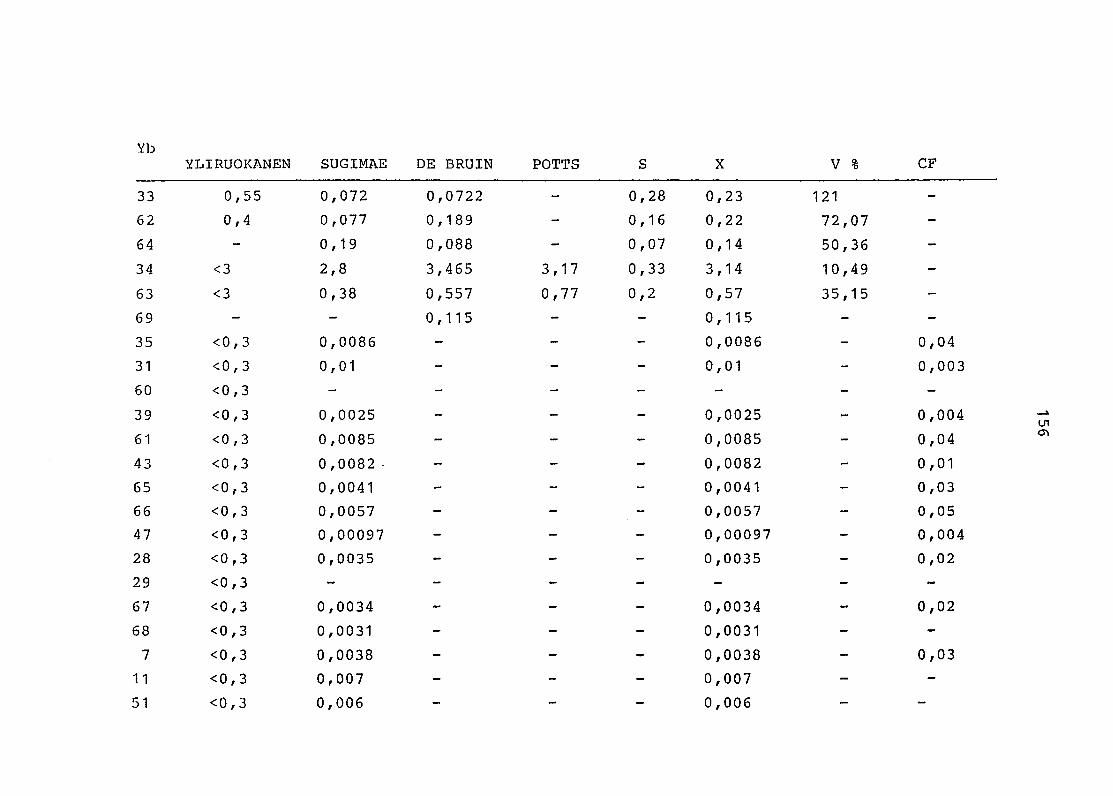
Y1)YLIRUOKANEN
SUGIMAE
DE BRUIN
POTTS
S X
V % CF
33 0,55
0,072
0,0722
-
0,28 0,23
121 -
62 0,4
0,077
0,189
-
0,16 0,22
72,07 -
64 -
0,19
0,088
-
0,07 0,14
50,36 -
34 <3
2,8
3,465
3,17
0,33 3,14
10,49 -
63 <3
0,38
0,557
0,77
0,2 0,57
35,15 -
69 -
-
0,115
-
- 0,115
- -
35 <0,3
0,0086
-
-
- 0,0086
- 0,04
31 <0,3
0,01
-
-
- 0,01
- 0,003
60 <0,3
-
-
-
- -
- -
39 <0,3
0,0025
-
-
- 0,0025
- 0,004
61 <0,3
0,0085
-
-
- 0,0085
- 0,04
43 <0,3
0,0082 .-
-
- 0,0082
- 0,01
65 <0,3
0,0041
-
-
- 0,0041
- 0,03
66 <0,3
0,0057
-
-
- 0,0057
- 0,05
47 <0,3
0,00097
-
-
- 0,00097
- 0,004
28 <0,3
0,0035
-
-
- 0,0035
- 0,02
29 <0,3
-
-
-
- -
- -
67 <0,3
0,0034
-
-
- 0,0034
- 0,02
68 <0,3
0,0031
-
-
- 0,0031
- -
7 <0,3
0,0038
-
-
- 0,0038
- 0,03
11 <0,3
0,007
-
-
- 0,007
- -
51 <0,3
0,006
-
-
- 0,006
- -
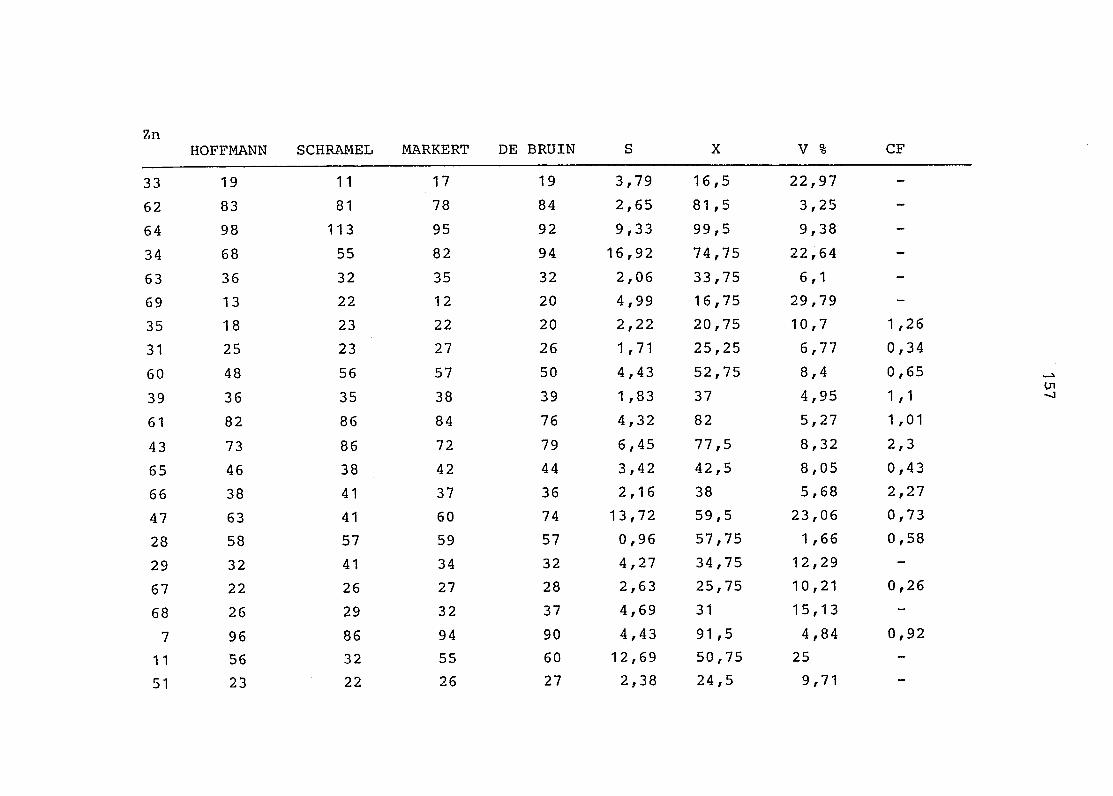
7,n
HOFFMANN SCHRAMEL MARKERT DE BRUIN S X V % CF
33 19 11 17 19 3,79 16,5 22,97 -
62 83 81 78 84 2,65 81,5 3,25 -
64 98 113 95 92 9,33 99,5 9,38 -
34 68 55 82 94 16,92 74,75 22,64 -
63 36 32 35 32 2,06 33,75 6,1 -
69 13 22 12 20 4,99 16,75 29,79 -
35 18 23 22 20 2,22 20,75 10,7 1,26
31 25 23 27 26 1,71 25,25 6,77 0,34
60 48 56 57 50 4,43 52,75 8,4 0,65
39 36 35 38 39 1,83 37 4,95 1,1
61 82 86 84 76 4,32 82 5,27 1,01
43 73 86 72 79 6,45 77,5 8,32 2,3
65 46 38 42 44 3,42 42,5 8,05 0,43
66 38 41 37 36 2,16 38 5,68 2,27
47 63 41 60 74 13,72 59,5 23,06 0,73
28 58 57 59 57 0,96 57,75 1,66 0,58
29 32 41 34 32 4,27 34,75 12,29 -
67 22 26 27 28 2,63 25,75 10,21 0,26
68 26 29 32 37 4,69 31 15,13 -
7 96 86 94 90 4,43 91,5 4,84 0,92
11 56 32 55 60 12,69 50,75 25 -
51 23 22 26 27 2,38 24,5 9,71 -
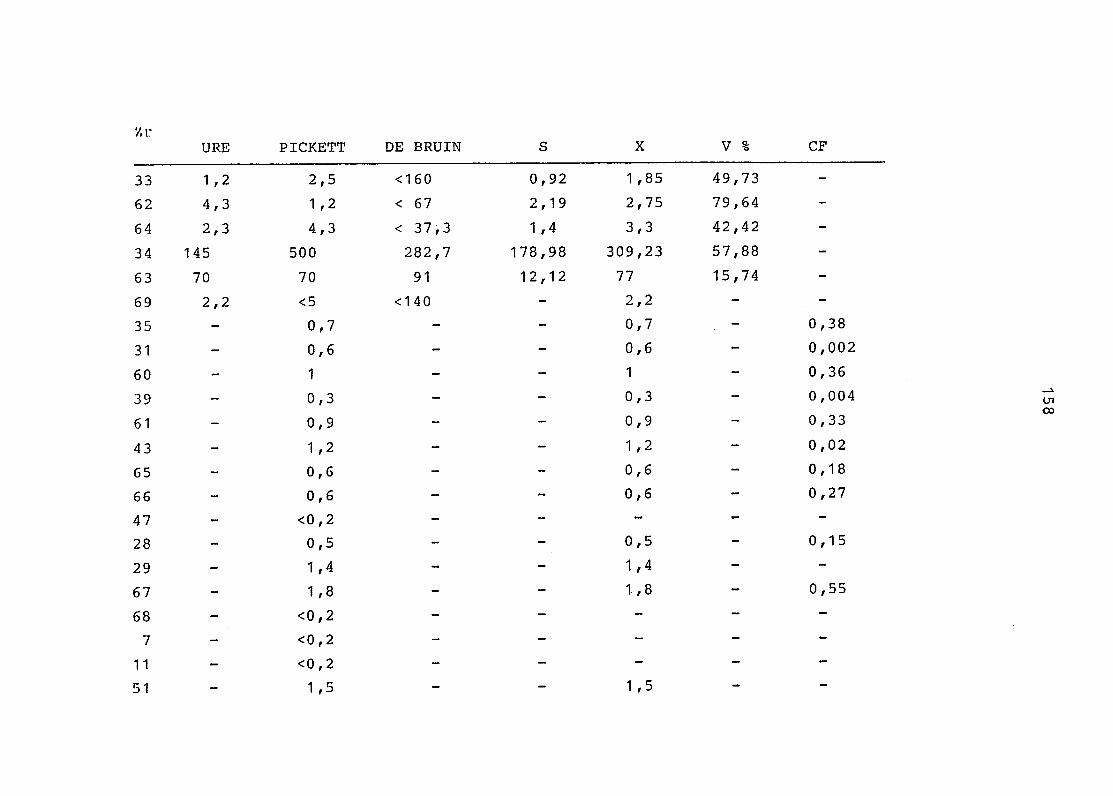
ZrURE
PICKETT
DE BRUIN
S X
V % CF
33
1,2
2,5
<160
0,92 1,85
49,73
62
4,3
1,2
<
67
2,19 2,75
79,64
64
2,3
4,3
<
37,3
1,4 3,3
42,42
34
145
500
282,7
178,98 309,23
57,88
63
70
70
91
12,12 77
15,74
69
2,2
<5
<140
- 2,2
-
35
-
0,7
-
- 0,7
- 0,38
31
-
0,6
-
- 0,6
- 0,002
60
-
1
-
- 1
- 0,36
39
-
0,3
-
- 0,3
- 0,004
61
-
0,9
-
- 0,9
- 0,33
43
-
1,2
-
- 1,2
- 0,02
65
-
0,6
-
- 0,6
- 0,18
66
-
0,6
-
- 0,6
- 0,27
47
-
<0,2
-
- -
-
28
-
0,5
-
- 0,5
- 0,15
29
-
1,4
-
- 1,4
-
67
-
1,8
-
- 1,8
- 0,55
68
-
<0,2
-
- -
-
7
-
<0,2
-
- -
-
11
-
<0,2
-
- -
-
51
-
1,5
-
- 1,5
-

159
Anhang 3
Mitarbeitende Institute

160
AUSTRALIEN
Dr . A . PinkertonCSIRO, Div . of Plant Industry,GPO Box 1600, Black Mountain, Canberra, Act 2601XRFSr
AUSTRALIEN
Dr . D . J . DavidCSIRO, Div . of Plant Industry,GPO Box 1600, Black Mountain, Canberra, ACT 2601AASSr
AUSTRALIEN
Dr . P . DuerdenAustralien Atomic Energy Commisssion Research Establishment,Private Mail Bag, Sutherland, NSW, 2232PIXE, PIGMEeinige Elemente
BRASILIEN
Prof . Dr . Gilson Brand BaptistaDepartamento de FisicaPontificia Universidade Catolica do Rio de JaneiroRio de JaneiroPIXEeinige Elemente
CANADA
Prof . Dr . S . Pawluk, Dr . M .J . DudasDepartment of Soll ScienceThe University of Alberta, Edmonton, T6G 2E3AASAl
CANADA
Dr . S .T . LamNuclear Research Centre, Department of PhysicsThe University of Alberta, Edmonton, Alberta, T6G 2N5PIXEeinige Elemente

161
CHILE
Dr . E . Cortes-ToroAnibal Aracena 565, Nunoa, SantiagoNAAeinige Elemente
DÄNEMARK
Dr . K . Heydorn, Dr . E . DamsgaardIsotope Division Ris0 National Laboratory,Postbox 49, DK-4000 RoskildeNAAV
DEUTSCHLAND
Dipl .-Ing . B .F . SchmittBundesanstalt für MaterialprüfungUnter den Eichen 87, 1000 Berlin 45PAAeinige Elemente
DEUTSCHLAND
Dr . M . StoepplerInstitut für Chemie der Kernforschungsanlage JülichInstitut 4, Angewandte physikalische ChemiePostfach 1913, 5170 JülichAASAs, Hg, Se
DEUTSCHLAND
Dr . HoffmannBayrische Landesanstalt für WasserforschungKaulbachstraße 37, 8000 München 22AES-ICPAg, Al, B, Ba, Be, Ca, Cd, Co, Cr, Cu, Fe, K, Mg, Mn, Mo, Na,Ni, P, Pb, S, Si, Sn, Sr, V, Zn
DEUTSCHLAND
Dr . J . KronerInstitut für anorganische Chemie der Universität MünchenMeierstraße 1, 8000 München 2ElementanalyseC, H, N

162
DEUTSCHLAND
Prof . Dr . LuxInstitut für RadiochemieTechnische Universität München, München-GarchingNAAeinige Elemente
DEUTSCHLAND
Dr . P . SchramelPhysikalisch Technische Abteilung, Institut für angewandtePhysik, Gesellschaft für Strahlen- und UmweltforschungIngolstädter Landstraße 1, 8042 Neuherberg, Post Oberschleiß -heimAES-ICP, AASAl, B, Ca, Cd, Cr, Cu, Fe, Mg, Mn, Ni, P, Pb, Ti, Zn
ENGLAND
Dr . N .J . BirchBiomedical Research Laboratory, School of Applied Sciences,The Polytechnic, Wolverhampton, WV 1 1 LYAASLi, A1, Rb, Cs, Co
ENGLAND
Dr . S . ParryImperial College of Science and Technology, Reactor Centre,Ascot, Berkshire S1S 7PYNAAeinige Elemente
ENGLAND
Dr . P . PottsThe Open University, Department of Earth Sciences,Walton Hall, Milton Keynes MK7 6AA, BuckinghamshireNAAeinige Elemente
FINNLAND
Dr. J . YliruokanenHelsinki University of Technology, Department of ChemistryLaboratory of Inorganic and Analytical Chemistry, Otaniemi,Otakaari 1 02150 ESPOO 15MASBi, Ce, Dy, Er, Eu, Gd, Hf, Ho, La, Lu, Nb, Nd, Pr, Sm, Tb,Th, Tm, U, W, Y, Yb

163
ITALIEN
Dr . S . CaroliIstituto Superiore di SanitaViale Regina Elena, 299-00161 RomaAES-ICPeinige Elemente
JAPAN
Dr . S . OhnoDivision of Environmental HealthNational Institute of Radiological Sciences,9-1, Anagawa-4-chome, Chiba-shi 260NAACo, Cs
JAPAN
Dr . Akiyoshi SugimaeEnvironmental Pollution Control Center, Osaka Prefecture3-62, 1-chome, Nakamichi, Uigashinari-ku, Osaka 537AES-ICPCe, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Sc, Sm, Tb, Tm, Y, Yb
MALAYSIA
Dr . Soo-Long TongUniversity Malaya, Jabatan Kinnia, Lenbah Pantai, Kuala LumpurAASeinige Elemente
NIEDERLANDE
Dr . M . de BruinInteruniversitair Reactor InstituutMekelweg 152629 Jb DelftNAANa, Cl, Ca, Sc, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, As, Se, Br, Rb,Zr, Mo, Ru, Ag, Co, In, Sn, Sb, Cs, Ba, La, Ce, Nd, Sm, Eu, Tb,Dy, Ho, Yb, Lu, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hg, Th, U
SCHOTTLAND
Dr . A .M . UreThe Macaulay Institute for Soli. ResearchDepartment of SpectrochemistryCraigiebuckler, Aberdeen AB9 2QJMASAg, Bi, Ga, Ge, Hf, Nb, Sb, Sn, Th, U, Zr

164
SOWJETUNION
Dr . DavydovaInstitut für Geologie, Petrographie, Mineralogie und GeochemieAkademie der WissenschaftenMoskauNAAeinige Elemente
SOWJETUNION
Dr . KulikowaGeologisches InstitutAkademie der WissenschaftenUlan Udeeinige Methodeneinige Elemente
SÜDAFRIKA
Dr . M . PeisachCSIRNational Accelerator CentreVan de Graaf GroupP .O . Box 72Faure 7131NAAeinige Elemente
UNGARN
Dr . Cs . M . BuczkoInstitute of Experimental PhysicsKossuth UniversityH-4001 DebrecenNAA, RFAeinige Elemente
USA
Dr . T .L . YuanUniversity of FloridaInstitute of Food and Agricultural Sciences106 Newell HallGainesvilleFlorida 32611AASSi, Al

165
USA
Dr . W .D . Ehmann, Mr . J . TianUniversity of KentuckyCollege of Arts and SciencesDepartment of ChemistryChemistry-Physics BuildingLexingtonKentucky 40506-0055
USA
Dr . G . ThompsonWoods Hole Oceanographic InstitutionDepartment of ChemistryMcLean BuildingWoods Hole, MA 02543RFAeinige Elemente
USA
Dr . E .E . PickettUniversity of Missouri-ColumbiaCollege of Agriculture and School of MedicineBiochemistry Department322-A Chemistry BuildingColumbiaMissouri 65211EmmissionsspektroskopieAg, Ga, Ge, In, Sn, Zr
USA
Dr . G . HolgrenUnited States Department of AgricultureSoll Conservation ServiceMidwest National Technical CenterFederal BuildingRoom 345100 Centennial Mali NorthLincolnNebraska 68506-3866AASeinige Elemente

166
USA
Dr . R .H . FilbyWashington State University, Nuclear Radiation CenterPullmanWashington 99164NAAeinige Elemente
USA
Dr . D .O . WilsonAgronomy DepartmentUniversity of GeorgiaGeorgia StationExperimentGeorgia 30212AASMo
VENEZUELA
Dr . J .J . La BrecqueMinisterio de Sandidad y Asistencia SocialInstituto Venezolano de Investigaciones CientificasCaracasPIXEeinige Elemente
Related Documents











