
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


3060 J. Org. Chem., Vol. 42, No. 18, 1977 Burfield, Lee, and Smithers
Desiccant Efficiency in Solvent Drying. A Reappraisal by Application of a Novel Method for Solvent Water Assay
David R. Burfield,* Kim-Her Lee,’ and Roger H. Smithers*
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, West Malaysia
Received January 19,1977
The chemical literature, very inconsistent on the subject of the drying of solvents, abounds with contradictory statements as to the efficiency of even the more common desiccants. The recent advent of a novel, highly sensitive method which utilizes a tritiated water tracer for the assay of solvent water content has enabled the first compre- hensive study to be made of the efficiency of various desiccants which pertains unambiguously to solvents. Ben- zene, 1,4-dioxane, and acetonitrile, chosen as model solvents, were wetted with known amounts of tritiated water and treated with a spectrum of desiccants, and the residual water contents were then assayed. The results range from the expected to the highly surprising. Some anomalous results, obtained for benzene and acetonitrile with acidic and basic desiccants, respectively, are discussed in terms of isotopic exchange reactions.
The bench chemist is often confronted by the problem of the selection of desiccants for solvent drying, and although dry solvents are frequently required for use in both prepara- tive situations and in physicochemical studies, there is a paucity of real information in the literature. Some authors2 are content to dismiss drying with statements such as “Fre- quently a liquid can be freed from water by shaking with a drying agent such as anhydrous calcium chloride or phos- phorus pentoxide”. In the field of organic synthesis, the sit- uation is little better; different reference texts are replete with bewildering contradictions. Thus, magnesium sulfate, de- scribed as either neutral3a.b~~ or acidic,3c,e is alternately an excellent drying agent, rapid in its a c t i ~ n , ~ a , ~ , d , ~ or is ~ 1 0 ~ , 3 h removing only small amounts of water.4 Aluminum oxide is recommended mainly for use in desiccators,3f or as being preferred by many workers for ultimate solvent or reagent drying.Jp Calcium chloride is “ f a ~ t ” , ~ ~ - ~ * ~ or alternately “not rapid” sf in its action, and in any case, the consensus appears to be that calcium sulfate is to be preferred as a and a more e f f i ~ i e n t ~ ~ . ~ desiccant, even though the only existing quantitative comparison for solvents4 shows the complete reverse to be true. Metallic sodium, generally agreed upon3b.d as being efficient, but slow in its drying action, is ridiculed as a desiccant by Ple~ch,~E: who states that “the widespread use of sodium as a drying agent by organic chemists is more a ritual than an effective process”. Furthermore, there is no doubt that many literature prescriptions for desiccation rely, a t least to some extent, on the “chemical intuition” of the author, inspired perhaps by the existence of ubiquitous indices of siccative e f f i c i e n ~ y . ~ ~ ~ ? ~ ? ~ ~ , ~ These are usually based on the results of detailed studies of the comparative drying efficiency of desiccants which have been made with regard to the dryness of gases ,3h,6 and direct extrapolation to the condensed phase often gives misleading if not totally erroneous information. For example, phosphorus pentoxide, long considered the ultimate drying standard? is actually quite mediocre in cer- tain situations (vide infra). In summary, no comprehensive study of solvent drying comparable to that made for gases appears to exist, and since the efficiency of a desiccant is de- pendent on the nature of the solvent, this is a serious omis- sion.
R e ~ e n t l y , ~ an extremely sensitive method using a tritiated water tracer for the determination of solvent water content has been developed, where essentially drying efficiency is determined by addition of a specified amount of tritium- labeled water to a rigorously dried solvent and subsequent determination of the decrease in activity of the solvent after treatment with various drying agents. With the limitation that, owing to the problem of isotopic exchange, the method is not applicable to protic solvents, it provides a rapid and
extremely precise assay of solvent water content. This has prompted us to undertake a comprehensive study of the ef- ficiency of drying of a number of desiccants for the solvents benzene, l,4-dioxane, and acetonitrile, representative of a spectrum of others commonly used in the laboratory. Thus, while benzene is a model for a useful range of aromatic and hydrocarbon solvents, and dioxane exemplifies commonly used ethers and bisethers, acetonitrile probably parallels the solvent behavior of a number of other polar, and, on account of its very high dielectric constant, perhaps dipolar aprotic solvents. Although selection of drying agents was generally made on the basis of common usage, some more esoteric ex- amples which have been recommended for use in particular situations were also examined. The results have enabled us not only to present a sensible evaluation of many time-hon- ored solvent drying recipes, but also to advocate the use of novel agents in previously unfamiliar situations.
Results and Discussion Drying of Benzene. Static Drying. Benzene, despite its
carcinogenic properties, is a widely used solvent, which, be- cause of its ease of purification and relative inertness in many chemical systems, has been adopted as a secondary standard. Benzene has a zero dipole moment and on account of its low polarity has little affinity for water, the maximum solubility of water in this solvent being 0.063% wfw at 25 “C. Conse- quently, benzene is a relatively easy solvent to dry. Drying has been accomplished in the literatures with the following des- iccants: phosphorus pentoxide, metallic calcium, sodium wire, calcium hydride, and molecular sieves.
In this study benzene containing 100 ppm of water was dried with a selection of the more useful and efficient desic- cants. The results, summarized in Table I, apply to 5% w/v desiccant loadings and to static drying conditions at ambient temperatures (25-29 “C). Treatment with molecular sieves, alumina, silica gel, calcium hydride, and lithium aluminum hydride gave super-dryg solvents within 1 day. Alumina in particular was found to be an excellent desiccant for benzene, reducing the solvent water content below 0.01 ppm over this period. These findings thus corroborate earlier conclusionslO that alumina is a particularly effective drying agent for hy- drocarbons, previously exemplified by a-methylstyrene and &pinene. The apparent increase in water content after drying for 7 days is most likely due to exchange or equilibria processes which occur between trace amounts of adventitious mois- ture-released from the surface of the glass vessel or perhaps gaining entry via diffusion through the clearfit stopper seal- and labeled water adsorbed on the desiccant. In any case, it is probably unrealistic to attempt to maintain water contents of below 0.01 ppm outside of totally sealed systems.
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0438
a024
marisa
Highlight

Desiccant Efficiency in Solvent Drying J. Org. Chem., Vol. 42, No. 18, 1977 3061
Table I. Efficiency of Various Desiccants for Static Drying of BenzeneC - Residual solvent water content, ppm
Registry no. Desiccant 6 h 1 day 7 days
1344-28- 1
7789-78-8 16853-85-3 7440-23-5 1314-56-3
10043-52-4 1151-82-6
4 A molecular sieves A1203 Silica gel CaH2 LiAlHI Na pzos CaClz Na2S04
2 0.03 0.6 0.006 0.3 0.3 0.2 3 2 (0.03)" 1.5 2 ( 2 ) O
7 12 12 0.1
>28 >28
0.06 0.2 0.1 0.03 0.7
4 ( 4 ) b >28b
>28
Scintillation solution purged with nitrogen and recounted. Distilled sample. Desiccant loading 5% w/v; initial water content 100 ppm (0.01% w/w).
Table 11. Effect of Stirring on the Drying Efficiencies of Desiccants for Benzeneb
Residual solvent water content, ppm -- . -- 6 h 1 day ~~
Desiccant Static Stirred Static Stirred
CaC12 12 0.8 0.1 1 LiAlH4 3 0.7 1.6 0.3 (0.02)"
a Purged with nitrogen and reassayed. Desiccant loading 5% w/v; initial water content 100 ppm (0.01% w/w).
Calcium and lithium aluminum hydrides are both effective desiccants. The values for the complex metal hydride are apparently high through contamination of the solvent with labeled hydrogen resulting from interaction of the hydride with the labeled water. This was confirmed by recounting the sample after purging with nitrogen whereupon the apparent water content was reduced dramatically from 2 to 0.03 ppm. Interestingly, purging had little or no effect on samples dried with calcium hydride and sodium, and this parallels a quali- tative observation that lithium aluminum hydride, perhaps because of its finely divided form, appears to bind the hy- drogen, viz., gas bubbles can still be released from the desic- cant long after the solvent is essentially dry.
Sodium is observed to reduce the water content extensively within the first 6 h , but subsequently the apparent water content is seen to increase significantly. Since purging with nitrogen and distilltition do not reduce the figure it may be speculated that sodium is actually able to metalate benzene, necessarily a t an extremely low rate. Tritiation could then occur by reaction of organosodium intermediates with trace amounts of newly formed tritiated water, whose genesis would be identical with that proposed above.
Phosphorus pentoxide appears to be an ineffective drying agent. However, this conclusion must be tempered by the significant increase in apparent water content with time of drying. An increase of such magnitude can only reasonably be explained by the presence of exchange reactions. Indeed, phosphoric acid catalyzed exchange reactions have been used elsewherell for the synthesis of tritiated aromatic compounds. The presence of exchange reactions thus unfortunately pre- cludes any conclusion as to the efficiency of phosphorus pentoxide as a desiccant for benzene.
Calcium chloride is seen to be an effective drying agent, quite capable of giving super-dry benzene. In contrast, sodium sulfate is completely inept, and the samples obtained after drying were too active for direct counting, indicating little or no drying.
Effect of Stirring. The effect of stirring on rapidity of drying was investigated for calcium chloride and lithium aluminum hydride (Table 11). In both cases stirring has an
\ 3
4 8 I2 16 2 0
Durotion d drying (hr)
Figure 1. Drying of dioxane with various desiccants. Experimental conditions as for Table 111. 1, MgS04; 2, KOH pellets; 3, P205; 4, CaC12; 5 ,4 8, molecular sieves; 6, CaH2.
accelerating effect on drying. This is most likely due to breakdown of particle size which increases the effective des- iccant surface rather than diffusion control of the drying process, since finely divided silica gel is a very rapid desiccant even under static drying conditions (Table I).
Drying by Distillation. Fractionation of benzene with retention of the middle fraction, a time-honored process, has frequently been advocated as a method of drying. In this work it was found that the middle fraction, after discarding the first 20%, contained 15 ppm of water. This is significantly drier than the initial water content, but the drying pales in com- parison with static drying by the majority of desiccants (Table I).
Drying of Dioxane. Static Drying. Dioxane, although not a very polar solvent ( p = 0.45), is completely miscible with water and is consequently far more difficult to dry than ben- zene. Drying is frequently tackled in at least two stages. Pre- liminary drying agent@ include potassium hydroxide, calcium chloride, sodium hydroxide, and magnesium sulfate, whereas final drying8 has been accomplished almost exclusively with sodium and occasionally with sodium-lead alloy.
In this study dioxane with an initial water content of 2300 ppm (0.23% w/w)12 was dried with a selection of both pre- liminary and final drying agents. The initial rate of drying for a selection of desiccants is displayed in Figure 1. It is imme- diately apparent that magnesium sulfate is almost completely ineffective as a drying agent, whereas calcium hydride is both rapid and efficient. It is also interesting that, for the first 24 h at least, the speed of drying parallels desiccant efficiency. Remarkably, phosphorus pentoxide does not excel as a drying
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0043
8a02
4

3062 J . Org. Chem,., Vol. 42, No. 18, 1977 Burfield, Lee, and Smithers
Table 111. Efficiency of Various Desiccants for Static Drying of DioxaneC
Residual solvent water content, ppm Registry no. Desiccant 6 h 1 day 7 days
Na Na-K alloy CaH2 4 A molecular sieve
CaC12 p205 LiAlH4 KOH (pellets) Silica gel A1203
1118-18-9 Cas04 Na2S04
1481-88-9 MgS04
1310-58-3 KOH (ground)
132 (22)” 50
200 204 450
1050 1200 1300 1300
20 22 ( 1 l ) O
30 40
150 300 400 900
1100 1300 1700 1600 1900 2200
6 22 (6)b 23 26 14 (8Ib
300 60
1100 (340)b 200
1200 1400 1500 1800 2200
a After purging to remove H2. b After distillation from desiccant. Desiccant loading 5% w/v; initial water content 2300 ppm (0.23% w/w).
agent, being surpassed in both speed and efficiency by even calcium chloride.
Results for a wider range of desiccants and drying times are summarized in Table 111. Sodium, sodium-potassium alloy (containing 80% by weight of potassium), calcium hydride, and molecular sieves are all seen to be very effective drying agents, although at these initial water loadings none of them dry di- oxane to super-dry levels, which, however, could undoubtedly be achieved by repetitive drying. Sodium-potassium alloy, typically described as possessing a higher drying intensity than metallic s0dium,3~ has been advocated as a siccative in situations where extrcbme desiccation is required,13 where its principal advantage over sodium, i.e., its liquid state at am- bient temperatures, should expedite very efficient drying of solvents boiling below the melting point of the alkali metal. I t is somewhat remarkable therefore that the alloy is not su- perior to sodium granules under static drying conditions. Powdering of KOH pellets is seen to have a dramatic effect on the rapidity and efficiency of drying, and supports a pre- vious report3c that drying of diethyl ether and THF solely by treatment with powdered KOH gives material which is im- mediately suitable for the preparation of reactive organo- metallics. I t is striking that powdered KOH, although slightly slower in action, actually surpasses calcium hydride and mo- lecular sieves in ultimate efficiency.
LiAlH4, though described as highly effective for drying ethers,3h actually appears strangely ineffective. The very high residual water contents cannot be explained in terms of la- beled hydrogen contamination, and it is difficult to invoke any other interferences. A probable conclusion is that unlike CaH2, LiAlH4 is not effective under conditions of high initial water content, and this result, must cast some doubt on its unqual- ified recommendation as a desiccant for THF.I4
The almost complete ineffectiveness of alumina and silica gel for dioxane drying is a complete reversal of their behavior with benzene. This underscores the risks involved in extrap- olating the results of gas drying to the liquid phase.
Magnesium and sodium sulfates are again4 found to be slow and ineffective at these water concentrations. Sodium sulfate, in particular, has earlier4 been shown to be an almost com- pletely inept desiccant for ethers a t much higher water con- centration, viz., the water content of diethyl ether was reduced from 2.07% w/w to merely 1.83% w/w after a period of several weeks. The value of sodium sulfate, even as a preliminary drying agent, must therefore be questionable. In view of its unanimous recommendation, calcium sulfate also rates sur- prisingly poorly.
Effect of Stirring and Refluxing on Drying. Since sol-
Table IV. Effect of Conditions on the Drying of Dioxane” Residual solvent water content,
Drying time, ppm Desiccant h Static Refluxed Stirred
CaH2 2 200 110 6 50 29
24 30 14 4 168 23 2
Na 24 20 5 48 3
0 Desiccant loading 5% w/v; initial water content 2300 ppm (0.23% w/w).
Table V. The Use of Visual Indicators in Dioxane Drying Residual solvent
water content Desiccant- after distillation, indicator PPm
Sodium-benzophenone 20 BuLi b-triphenylmethane 22 BuLi-phenanthroline 17 Trityl fluoroborate‘ 650 (800)O
2300 ppm (0.23% w/w). b Registry no., 109-72-8. As determined by the near IR method. Initial water content
Registry no., 341-02-6.
vents are frequently dried by refluxing over desiccants such as sodium and calcium hydride, the effect of refluxing was briefly investigated. I t can be seen (Table IV) that while re- fluxing dioxane over CaHz results in moderate increases in efficiency and speed of drying, stirring is seen to be much more effective.
Refluxing over sodium (Table IV) is seen to give an im- provement in efficiency compared to static drying but pro- longed refluxing is not particularly beneficial.15 I t is worthy of note that stirring over calcium hydride a t ambient tem- peratures is as effective as refluxing over molten sodium.
Drying Agents with Visual Indication. Although desic- cation prescriptions which include a visual indication of sol- vent dryness have become fairly common in recent years, no quantitative measure of their efficiency appears to have been made. These methods generally involve the in situ generation of small amounts of colored, highly moisture-sensitive inter- mediates, often by the action of the desiccant on an added “indicator”, and the solvent presumed anhydrous when the indicator color persists. Table V, which displays a selection
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0438
a024

Desiccant Efficiency in Solvent Drying J . Org. Chem., Vol. 42, No. 18, 1977 3063
Table VI. Efficiency of Various Desiccants for Static Drying of Acetonitrile
Residual solvent water content, ppm Registry no. Desiccant 1 day 7 days
1303-86-2 584-08-7
pzo5 3 A molecular sieves Bz03 K COS 4 X molecular sieves CaC12 Silica gel A1203 CaH2 KOH (powdered) KOH (pellets) Cas04 Ph3C+-BF4
9 ( 1 2 ) Q . b 49 59 ”
250 450
1200 1300 1600 1900 2200“sb2d 2500 2500 2700“ (2800)“7c
5 27
1300 500
2200 1300 1700 1900” (1300)”,c
1300 2200
” Distilled sample. Colored residue. By near IR method. Strong amine smell in distillate. e Desiccant loading 5% w/v; initial water content 2800 ppm (0.28% w/w).
of those investigated for dioxane, reveals that although none of them give super-dry solvent (see, however, discussion below), the first three entries give comparable results to the best of those obtained for dioxane after static drying for 1 day (Table 111). The intense blue sodium ketyl of benzophenone (entry l ) , often used in the preparation of absolute diethyl ether,’6 where it presumably also serves to remove peroxides, gives similar results to butyllithium. The appearance of the red triphenylmethyl anion (entry 2) has been advocated as an indicator in the preparation of “anhydrous” THF.17 We have found that if no special precautions, e.g., anaerobic conditions, are utilized, then the amount of butyllithium required to impart a persistent color to the solvent is excessively high, owing perhaps to corisumption of the “indicator” by molecules other than water, e.g., oxygen. This shortcoming led to an experiment with 1, LO-phenanthroline (entry 3), previously suggested as an indicator in the “alcohol method” of assaying BuLi.l8 The formation of the derived rust-red complex re- quired only about half the butyllithium used in entry 2, and since the result was a slightly drier solvent, the use of this in- dicator is to be preferred. In the general context of the bu- tyllithium experimeats, i t must also be pointed out that i t is known that alkyllithiums react relatively slowly with THF,lg to give, initially, 2-lithiotetrahydrofuran, which, if the anal- ogous reaction were to occur with dioxane, may serve to label the ether, and hence raise the apparent water content, by reaction of metalated dioxane with tritiated water. While this reaction has not been reported for dioxane, and in any case would be expected to be extremely slow compared to reaction of the alkyllithium with water, some inflation of the apparent water content by this means cannot be altogether ruled out. Although trityl fluoroborate (entry 4) has not been previously used as a desiccant for ethers, it has been used to dry aceto- nitrile (vide infra), amd this experiment was run to determine its efficiency in a different solvent type. Even though, a t this solvent water concentration, the deep yellow color of the salt solution was not discharged, the recovered ether contained a surprisingly large amount of residual water, and this result was cross-checked by the near IR method. Compared to entries 1 to 3, trityl fluoroborate gives poor ultimate drying, which however, is still better than that obtainable with lithium aluminum hydride (Table 111).
Drying of Acetonitrile. Static Drying. Acetonitrile, a polar aprotic solvent ( f i = 3.44) of high solvating power and favorable physical ]properties, has been widely used as a sol- vent both in the study of chemical reactions and for physical measurements involving spectrophotometric and electro- chemical techniques. However, because of its high affinity for water it is an outstandingly difficult solvent to completely dry.
Drying is conventionally accomplished8 by treatment with preliminary drying agents such as anhydrous sodium or po- tassium carbonate, anhydrous calcium chloride, silica gel, or 3 A molecular sieves, and final drying with calcium hydride, phosphorus pentoxide, or more recently with trityl fluo- roborate.20
The results of static drying with a range of desiccants are displayed in Table VI. In contradistinction to the other sol- vents investigated phosphorus pentoxide is seen to excel in its drying efficiency, but even so super-dry acetonitrile is not obtained. I t is interesting to note that the residual water content is of similar order of magnitude to an earlier result which also utilized P Z O ~ . ~ ~ The only disadvantage to phos- phorus pentoxide drying is the partial loss of solvent through polymerization, and possible contamination by desiccant residues.22 Reasonably effective drying can also be achieved with 3 8, molecular sieves which reduce the water content to less than 30 ppm after 1 week. The relative inefficiency of 4 8, sieves emphasizes the need for careful selection of sieve pore size for effective drying. A hitherto little mentioned3f but useful desiccant is boric anhydride. Direct sampling proved impossible in this case since soluble desiccant residues in- terfered visually with the scintillant, but the sample distilled after 1 day stirring with the anhydride had a water content of 59 ppm. This reagent is advantageous compared to phos- phorus pentoxide since it does not induce polymerization of the solvent nor does it appear to be significantly volatile. It also offers advantages in its ease of handling and disposal.
Silica gel and alumina are again, as with dioxane, largely ineffective. This may reflect their rather low capacity for ef- fective drying at high water ~ontents ,~g but in any case makes them an unlikely choice for preliminary drying. Calcium sul- fate, although generally strongly recommended for efficient drying, is seen to be the least effective of the examined des- iccants, and is clearly surpassed by the underrated calcium chloride.23
The ineffectiveness of the previously excellent desiccants potassium hydroxide and calcium hydride seems anomalous. Careful examination of the results reveals that all the basic siccatives, potassium hydroxide, calcium hydride, and po- tassium carbonate, give apparently little drying. In addition the apparent water content in the presence of the weakly basic potassium carbonate increases very significantly from 251 to 1300 ppm over the course of 1 week. These observations ap- pear indicative of a base-catalysed exchange reaction, viz.
-OH CH3CN + T20 CHzTCN + HOT
Such base-catalyzed exchange reactions of the 1y hydrogens
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0438
a024

3064 J . Org. Chem., Vol. 42, No. 18, 1977 Burfield, Lee, and Smithers
have been previous1.y encountered with b-hydroxypropioni- trile24 but rather surprisingly it has been claimed25 that ace- tonitrile itself does not exhibit similar behavior. In an attempt to confirm the presence of exchange reactions, the tracer ex- periment was cross-checked by the near IR method for the calcium hydride case. The near IR value of the water content is significantly lower, and this is suggestive of the presence of exchange reactions. Most unexpectedly, this determination also revealed that calcium hydride is largely ineffective for drying acetonitrile. 'This remarkable observation, together with the resuits for phosphorus pentoxide drying, undermines the intuitive assumption that the relative efficiencies of des- iccants should, barring chemical incompatibility, be inde- pendent of solvent type.
The interference, by exchange reactions, unfortunately makes it impossible to draw many conclusions on the effi- ciency of the basic desiccants save that potassium carbonate is clearly a reasonable desiccant, at least for preliminary drying.
I t is worthy of mention that drying with finely powdered potassium hydroxide gave rise to a colored residue and a sig- nificant amine content in the distilled fractionsz6
Trityl Fluoroborate. The use of this stable, orange car- benium ion salt as a desiccant for acetonitrile would seem to be advantageous; it can be stored in a desiccator for extended periods without deco~npos i t ion ,~~ and is used as a siccativez0 simply by adding it in small batches to the wet nitrile until a strong yellow color persists, thus furnishing a visual indication of dryness. The results obtained by using this and the IR method are displayed in Table VI, and indicate that, at these water concentrations, the carbocation salt is a spectacularly ineffective desiccant. The reason for this impotence seems obscure, although acetonitrile, by virtue of its solvation ability, has a well-known moderating influence on the stability of carbenium ions,28 and indeed, the drying of dioxane by trityl fluoroborate (vide supra) is significantly better than the present solvent. Whatever the true reason, it is clear that, as a desiccant for acetonitrile, the salt is completely worthless.
Merits of t he Study. The present study should be of con- siderable heuristic value, particularly to the bench chemist in the provision of directly relevant data, and it is also worthwhile briefly emphasising again' that for reasons which include (1) contamination of the solvent by desiccant residues and possibly labeled hydrogen, (2) exchange reactions, and (3) the kinetic isotope effect, the apparent water contents, as reported above, will always represent the upper limits of the true content. Of course, this in no way detracts from the value of the work, and, to cite an example, merely means that it is entirely possible that alumina is able to dry benzene to below 6 X lo--" ppm!
Experimental Section Radioactive samples were assayed in a scintillation solution con-
taining 0.4 g of 1,4-bis(5-phenyloxazol-2-yl)benzene (POPOP) and 4.0 g of 2,5-diphenyloxazole (PPO) per liter of toluene with a Beckman Model IJS-lOO liquid scintillation spectrometer. Determination of water content by the near IR method was performed using a Unicam SP700 spec t roph~tometer .~~ Tritium-labeled water was purchased from the Radiochemical Centre, Amersham, England, a t an initial activity of 5 Ci/mL and was diluted with appropriate quantities of inactive water.
Desiccants. Lithium aluminum hydride and phosphorus pentoxide were used as supplied; calcium hydride (99.5%) and reagent grade potassium hydroxide were rapidly powdered immediately prior to use in a mortar and a mechanical blender, respectively. Chromatographic grades of neutral alumina (activity 1) and silica gel, as well as calcium, magnesium, and sodium sulfates, calcium chloride, potassium car- bonate, and 3 and 4 8, molecular sieves were activated for 15 h at 300-320 OC before use. Since hydration occurs rapidly on cooling of these desiccants in moist air, cooling was carried out in a phosphorus pentoxide desiccator, and the samples then used immediately. Sodium
metal, whose oxide crust had previously been removed by melting under xylene, was cut into 2-mm cubes under dry petroleum ether. Sodium-potassium alloy was prepared as detailed elsewhere308 from oxide-free metals. (It is worth noting that the fire hazard associated with destroying excess alloy is completely avoided if the disposal is carried out in two steps. Addition of a little dry ethyl acetate to the alloy in dioxane smoothly consumes potassium-presumably via an acyloin reaction. Unreacted sodium can then be destroyed conven- tionally using ethanol.) Trityl fluoroborateZ7 and boric anhydride3f were respectively prepared from triphenylcarbinol and tetrafluo- roboric acid, and by high-temperature (900 "C) dehydration of boric acid.
Solvents. Benzene. AR grade reagent was stirred for 24 h with finely ground calcium hydride, refluxed, carefully fractionated (bp 80.0 "C), and stored over 4 8, molecular sieves.
1,4-Dioxane. Commercial 1,4-dioxane was purified and dried ac- cording to a method cited by F i e ~ e r , ~ * ~ whereby the glycol acetal impurity is removed by hydrolysis to acetaldehyde, which is itself voided by purging with nitrogen gas. Preliminary drying with potas- sium hydroxide pellets followed by fractionation (bp 101-101.5 "C) from sodium gave material which was stored in a dark bottle over 4 A molecular sieves.
Acetonitrile. Following well-documented procedures: reagent grade material, after being given a preliminary drying with potassium carbonate (24 h), was decanted on to phosphorus pentoxide and stirred at reflux for 2 h. Fractionation gave material of bp 81.5 "C, which was not stored, but used immediately.
Techniques. The procedure used for benzene serves as an example. A stock solution of benzene containing 100 ppm of labeled water was prepared by the addition of 18 WL of tritiated water, specific activity 40 mCi/mL, to 180 g of purified benzene; homogenization was ac- complished by stirring overnight. Aliquots of the stock solution (15.0 f 0.1 mL) were syringed directly onto 0.75 f 0.03 g of activated des- iccant contained in a 25-mL clear-fit round-bottom flask, which was then stoppered. Where appropriate samples were stirred magnetically. Samples (1.00 f 0.02 mL) were taken a t time intervals specified in the text-care was taken to avoid disturbing the desiccant-and syringed directly into the counting vials. Where possible, samples were distilled from the desiccant so as to provide a cross-check against contamination of the solvent by labeled desiccant residues. Samples were accumulated and assayed batchwise.
Similar procedures were used with dioxane and acetonitrile, except that higher water contents were examined and tritiated water of low specific activity (0.5 mCi/mL) was employed.
Registry No.-Benzene, 71-43-2; dioxane, 123-91-1; acetonitrile, 75-05-8.
References and Notes Abstracted in part from the Honours Project Work of K. H. Lee, 1975- 1976. F. Daniels, R . A. Alberty, J. W. Williams, C. D. Cornwell, P. Bender, and J. E. Harriman. "Experimental Physical Chemistry" 7th ed, McGraw-Hill, New York, N.Y., 1970, p 641. (a) T. L. Jacobs, W. E. Truce, and G. R. Robertson, "Laboratory Practice of Organic Chemistry", Macmillan, New York, N.Y., 1974; (b) R. M. Roberts, J. C. Gilbert, L. B. Rodewald, and A. S. Wingrove. "An Introduction to Modern Experimental Organic Chemistry", 2nd ed, Holt, Rinehart and Winston, New York, N.Y., 1974; (c) L. Brandsma, "Preparative Acetylenic Chemistry", Elsevier, Amsterdam, 1971; (d) R. S. Monson, "Advanced Organic Synthesis", Academic Press, New Ywk, N.Y., 1971; (e)L. F. Fieser and M. Fieser, "Reagents for Organic Synthesis", Wiley, New York, N.Y., 1967; ( f ) A. I . Vogei, "A Text-Book of Practical Organic Chemistry", 3rd ed, Longmans, London, 1964; (g) P. H. Plesch, Ed., "The Chemistry of Cationic Polymerisation", Pergamon Press, Oxford, 1963, p 682 f f ; (h) "Drying in the Laboratory", E. Merck, Darmstadt. B. D. Pearson, and J. E. Ollerenshaw, Chem. Ind., (London), 370 (1966). (a) G. Brauer, Ed.. "Handbook of Preparative inorganic Chemistry", Vol. I, 2nd ed, translated by Scripta Technica Inc., Academic Press, New York. N.Y., 1963, p 80; (b) "Handbook of Chemistry and Physics", 53rd ed, Chemical Rubber Publishing Co., Cleveland, Ohio, 1972, p E35. (a) F. Trusell and H. Diehl. Anal. Chem., 35, 674 (1963); (b) J. H . Bower, J. Res. Natl. Bur. Stand., 12, 241 (1934). D. R. Burfield, Anal. Chem., 48, 2285 (1976). See references contained in J. A. Riddlck and W. B. Bunger, "Organic Solvents", 3rd ed, Wiley-lnterscience, New York, N.Y., 1970. The term superdry has been used earlier in a qualitative sense,3f but in this context it denotes solvents containing less than 1 ppm of water. T. H. Bates, J. V. F. Best, and T. F. Williams, Nature (London). 88, 469 (1960). See E. A. Evans, "Tritium and Its Compounds", 2nd ed. Butterworths, London, 1974. This is close to the maximum water content specified for AR grade diox- ane. For example, after standing for 5 days over the alloy, both diethyl- and di-
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0043
8a02
4

Perhydrogenation of 2,8-Diaminopurine J . Org. Chem., Vol. 42, No. 18,1977 3065
methylamine are dehydrated sufficiently that spin coupling of the adjacent N-H and C-H protons is observable in their NMR spectra: K. L. Henold, Chem. Commun., 1340 (1970).
(14) H. Baumgarten, Ed., "Organic Syntheses", Collect. Vol. V, Wiley, New York. N.Y., 1973, p 976.
(15) In a peculiar sense, this result confirms an earlier statement that "One day drying over calcium hydride is found to be as effective as six months re- fluxing over sodium". See A. S. Brown, J. Chem. Phys.. 19, 1226 (1951).
(16) See, for example, G. Kobrich. W. E. Breckhoff, H. Heinemann. and A . Akhtar, J. Organornet. Chem., 3, 492 (1965).
(17) W. R. Purdum and G. .I. Bartling, J. Chem. Educ., 52, 120 (1974). (18) S. C. Watson and J. F Estham, J. Organomet. Chem.. g, 165 (1967). (19) (a) I. Fleming and T. Mah, J. Chem. Soc., Perkin Trans. 7, 964 (1975); (b)
R . B. Bates, L. M . Kroposki, and D. E. Potter, J. Org. Chem., 37, 560 (1972).
(20) Y. Pocker and W . H. VVong, J. Am. Chem. Soc., 97,7097 (1975). (21) J. F. Coetzee and G. R. Padmanabhan, J. Phys. Chem.. 66, 1708
(1962).
(22) Subsequent distillation from potassium carbonate has been recommended for removal of phosphorus pentoxide residues. See P. Walden and E. J. Birr, Z. Phys. Chem. (Leipzig), 144A, 269 (1929).
(23) Drying over calcium chloride for "at least a week" has been advocated as a method for obtaining "thoroughly dry" acetonitrile. A. H. Blatt. Ed., "Organic Syntheses", Collect. Voi. i , 2nd ed, Wiley, New Ywk. N.Y., 1946, pp 5-6.
(24) A. Lapidot. J . Reuben, and D. Samuel, J. Chem. Educ., 41, 570 (1964). (25) See ref 11, p 759. (26) These observations appear to support the earlier contention that "pre-
treatment with potassium hydroxide does more harm than good". See J . F. Coetzee, Pure Appl. Chem., 13, 429 (1966).
127) H. J. Dauben Jr., L. R. Honnen, and K . M. Harmon, J. Org. Chem., 25, 1442 (1960).
Wiley, New York, N.Y., 1968, p 140. (28) N. Lichtin in "Carbonium Ions". Vol. I , G. A. Olah and P. v. R. Schleyer, Ed.,
(29) R. L. Meeker, F. Critchfield, and E. T. Bishop, Anal. Chem., 34, 1510 (1962).
(30) (a) See ref 3e, p 1102. (b) See ref 3e, p 333.
Perhydrogenation of 2,8-Diaminopurine
Mark M. Wegner and Henry Rapoport*
Department o f Chemistry, University of California, Berkely, California 94720
Received March 7, 1977
2,8-Diaminopurine (7) can be hydrogenated over Pt02 in acidic medium to give 2-imino-4-guanidinomethyl-5- imidazolidinone (1 1) which can itself be further hydrogenated to 2-imino-4-guanidinomethylimidazolidine (12). The structures of 11 and 12 were proven by unambiguous synthesis. 2,8-Diamino-6-methylpurine (37) also can be hydrogenated in a similar manner to two analogous compounds, as isomeric mixtures, whose structures are inferred by comparison with 11 and 12. A superior method has been developed for synthesizing the diaminopurines 7 and 37, involving the condensation of the appropriate triaminopyrimidine with N-(methylmercaptochloromethy1)-p - toluenesulfonimide (20) followed by ring closure via the carbodiimide and detosylation with HF.
Saxitoxin is one of the most potent naturally occurring neurotoxins. I t is the sole toxin produced by the marine di- noflagellate Gonyauiax catenella and is a minor constituent of the toxins produced by G. tamarensis. Ingestion of these dinoflagellates by several species of normally edible shellfish is frequently responsible for their toxicity to man. X-ray crystallographic analysis of two derivatives, the bis-p-bro- mobenzenesulfonate3 and the ethyl hemiketal dihydrochlo- ride,4 have established structure 1 for crystalline saxitoxin hydrate, and 13C NMR studies have also established this structure for the molecule in s ~ l u t i o n . ~ Recently5 the major toxins of G. tamarensis, gonyautoxins I1 and 111, also existing as the hydrates, were postulated to have the closely related structures 2 and 3, respectively.
Saxitoxin and the gonyautoxins are unique among natural products in that their structures incorporate a tetrahydro- purine moiety composed of two guanidine units fused together in an azaketal linkage which remains intact under ordinary conditions. We were therefore interested in preparing a simple model of the tetrahydropurine backbone of saxitoxin, devoid of the fused ketone bearing ring and the peripheral carbamate, for both chemical and biological investigations. We chose to study the catalytic hydrogenation of 2,8-diaminopurine (7), which conceivably could lead to 2,8-diiminotetrahydropurine (10) or its tautomers, the simplest possible tetrahydropurine model of saxitoxin. We now report the results of our study of
the heterogeneous catalytic hydrogenation of 2,8-diamino- purines.
The literature relating to the catalytic reduction of purines is relatively meager. 1,6-Dihydropurine ( 5 ) has been pre- pared"' from purine and 6-chloropurine (4), and in weak acid 5 was hydrolyzed to 4(5)-aminomethyl-5(4)-aminoimidazole (6). Similarly a tetrahydropurine is claimed8 to result from catalytic reduction of 2,6,8-trichloropurine. More recently? the catalytic reduction of 2&diaminopurine (7) is reported to yield a compound whose structure was assigned as 2- amino-5-guanidino-l,4,5,6-tetrahydro-6-oxopyrimidine (8). These authors also report the preparation of 2,8-diamino- 4,5,6,9-tetrahydro-l,7,9-trimethylpurine by sodium borohy- dride reduction of 2,8-diamino-1,7,9-trimethylpurine, and claim to have electrolytically reduced 7 to 8 plus tetrahydro- purine 10, obtained as an inseparable mixture with another reduction product 9.
In contrast to that report, we have found that 7 is slowly hydrogenated with a Pt02 catalyst in hydrochloric acid (pH 1.5) a t room temperature and 20 psi pressure to give a single product, A, in quantitative yield. A could be further reduced under more drastic conditions (60 "C, 100 h) to give another product, B, also in quantitative yield. The lH NMR spectrum of A.2HC1 consisted of a doublet (2 H, J = 5 Hz) and a triplet (1 H, J = 5 Hz); its 13C NMR spectrum is tabulated in Table I.
These NMR data suggested that A was not a reduced purine with an intact bicyclic ring system but rather the five-mem- bered monocyclic imidazolidinone 11, The 13C NMR ab- sorption at 6 173 is clearly assigned to the amide carbonyl, and the simple doublet-triplet pattern of the lH NMR spectrum implies the freely rotating methylene group of 11. The alter- native six-membered ring structure 8 previously proposedg for the 2,8-diaminopurine reduction product should display
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0438
a024

J. Org. Chem. 1982,47, 3821-3824 3821
under Nz for 3.5 h in an oil bath maintained at 100 "C. The reaction mixture was poured onto an excess of ice, and after the ice had melted HzO was added to bring the total volume to 1500 mL. The solution was extracted with CHzClz (5 X 100 mL), and the combined organic layers were extracted with saturated NaHC03 untiI the extracts were colorless. The combined NaHC03 extracts were back-washed with CHzC12 (100 mL) and then acidified to pH 1 with 12 M HCl. The product was extracted into CHzClz (5 X 100 mL), the organic layer was dried (Na2SO4) and filtered, and solvent was removed on a rotary evaporator followed by a vacuum pump to yield 6.3 g of 3-hydroxy-5-methylbenzo- cyclobutenedione in 63% overall yield from dienophile 5: light yellow crys&, mp 191-193 OC (EtOAc); IR (CHzC12) 3540,1789, 1755; 'H NMR (60 MHz, acetone-ds) 6 7.29 (s, 1 H), 6.95 (8, 1 H), 2.46 (s,3 H). Anal. Calcd for CBH603: C, 66.66; H, 3.73. Found C, 66.56; H, 3.90.
Cycloaddition of l-Methoxy-3-(trimethylsiloxy)-1,3-bu- tadiene (13) to 1,4-Dichloro-3,3,4-trifluorocyclobutene (5). (A) Preparation of Enone 15. 1,4-Dichloro-3,3,4-trifluoro- cyclobutene (5; 3.08 g, 17.4 mmol) and Danishefsky's diene (13; 4.50 g, 26.1 mmol) were placed in a 3-02 Fischer-Porter pressure vessel equipped with a magnetic stirring bar, and the mixture was saturated with dry Nz for 5 min. The tube was sealed, placed in an oil bath maintained at 120 "C, and stirred for 3.5 h. After cooling to 25 OC, the tube was opened, and the contents were transferred to a 100-mL round-bottomed flask with the aid of a small amount of MeOH. To this mixture was added 100 mL of 1:l MeOH/1.2 N HC1, and the solution was stirred at room temperature for 2 h. The dark solution was poured into a sep- aratory funnel, diluted with 200 mL of HzO, and extracted with CHzClZ (3 X 75 mL), and the combined organic layers were dried (Na804), filtered, and condensed on a rotary evaporator to yield 3.11 g of crude 14 as a dark oil. The compound is contaminated with diene decomposition products as well as some enone 15, but the following spectroscopic absorptions of 14 are apparent: IR
-1 H), 3.30 (8, -3 H), 2.6-2.7 (m, -4 H). Without purification, crude 14 (3.11 g) was placed in a 250-mL round-bottomed flask and dissoved in dry benzene (175 mL). After addition of p - toluenesulfonic acid (166 mg, 0.87 mmol), the mixture was refluxed under Nz for 27 h, cooled to room temperature, transferred to a separatory funnel, washed with saturated NaHC03 (2 x 25 mL), dried (Na2SO4), filtered, and condensed on a rotary evaporator, and the residue was chromatographed on silica gel (3 cm x 0.75 m; 3 2 hexane/CHzC12) to yield enone 15: 2.25 g (53% yield from dienophile 5); white needles; mp 44-45 OC (sublimed); IR (CHzCIJ 1695; 'H NMR (60 MHz, CDC1,) 6 6.63 (d, J = 10 Hz, 1 H with smaller splittings), 6.15 (d, J = 10 Hz, 1 H with smaller splittings), 3.90-3.13 (m, 1 H), 2.71 (apparent d, J = 4 Hz, 2 H); mass spectrum, m / e (relative intensity) 244 (M'), 246 (M' + 2, 66).
(CH2ClZ) 1720; 'H NMR (60 MHz, CDC13) 6 4.72 (t, J = 5 Hz,
(B) Aromatization of Enone 15 to 2,2-Difluoro-4- hydroxybenzocyclobutenone (16). A solution of enone 15 (2.25 g, 9.2 mmol) in 60 mL of MeOH was cooled to 0 "C in a 250-mL round-bottomed flask and the solution was saturated with dry N2 for 5 min. The flask was fitted with a pressure equalizing addition funnel and a nitrogen inlet tube and the addition funnel was charged with a solution freshly prepared from sodium (0.846 g, 36.8 mmol) and MeOH (30 mL). The NaOMeIMeOH solution was added dropwise with stirring over 30 min, and after the addition was complete, the reaction mixture was allowed to warm to room temperature and stirred an additional 3 h. An equal volume of 1.2 N HCl(90 mL) was added, and the mixture was refluxed for 3 h to hydrolyze the intermediate ketal. After cooling to room temperature, the reaction mixture was diluted with 200 mL of HzO and the product extracted into CHZClz (3 x 75 mL). The combined CHzClz layers were dried (Na2S04), filtered, and evaporated to dryness on a rotary evaporator followed by a vacuum pump to give 16: 1.5 g (96%); white crystals; mp 163-164 "C (EtOAcIhexane); IR (CHZCl2) 3570, 1795, 1770; 'H NMR (60 MHz, acetone-d6) 6 9.8 (br s, 1 H), 7.70-7.16 (m, 3 H); mass spectrum, m l e 170 (M').
(C) Hydrolysis of 16 to 4-Hydroxybenzocyclobutenedione (17). The sample of 16 prepared above (1.50 g, 8.82 mmol) was placed in a 250-mL round-bottomed flask with 1:l concentrated Hfi04/HOAc (90 mL) and stirred at 90 "C under N2 for 3 h. The reaction mixture was poured onto an excess of ice and diluted with HzO to a total volume of 350 mL. The solution was extracted with Et20 (3 X 100 mL), and the combined organic layers were dried (NafiO,), filtered, and condensed to a crude solid on a rotary evaporator. This material was chromatographed on a silica gel column (3 cm X 1 m; EhO) to yield 4-hydroxybenzocyclo- butenedione: 1.10 g (84% yield, 43% overall from dienophile 5); light yellow crystals; mp 174.5-175 "C (EtOAc) (lit.7 mp 167-170 "C); IR (CHC13) 3600-3000 (br), 3575,1808 (sh), 1790,1769,1755 (sh); 'H NMR (60 MHz, CD,CN) 6 7.81 (d, J = 8 Hz, 1 H), 7.3G7.06 (m, 2 H), 6.23 (br s, 1 H); mass spectrum, m / e 148 (M+).
Acknowledgment is made to the National Cancer In- stitute, DHEW (Grant No. CA 26374), for support of this work.
Registry No. 1, 6383-11-5; 3, 3469-06-5; 4, 82431-14-9; 5, 2927- 72-2; 6a, 63383-46-0; 6b, 73912-36-4; 7a (isomer l), 82431-15-0; 7a (isomer 2), 82468-19-7; 7b (isomer l), 82431-19-4; 7b (isomer 2), 82468-20-0; 8a, 82431-16-1; 8b, 82444-39-1; Qb, 82431-20-7; 108, 82431-17-2; lob, 82431-21-8; lla, 82431-18-3; llb, 82431-22-9; 12a,
82431-25-2; 17, 75833-48-6; anthranilic acid, 118-92-3; 2-carboxy- benzenediazonium chloride, 4661-46-5; 1,l-dichloroethylene, 75-35-4; 1,l-dichlorobenzocyclobutene, 68913-13-3; 3-methyl-2-butenal, 107- 86-8; 2-methyl-3-buten-2-01, 115-18-4.
62416-21-1; 12b, 82431-23-0; 13, 59414-23-2; 15, 82431-24-1; 16,
Deperoxidation of Ethers. A Novel Application of Self-Indicating Molecular Sieves
David R. Burfield
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, West Malaysia
Received January 18, 1982
The removal of peroxides from contaminated ethers by treatment with self-indicating molecular sieves (IMS) is proposed as a safe and facile method of ether purification. Quantitative analysis of peroxide content before and after treatment with IMS show that ethers such as THF, diethyl ether, and diisopropyl ether can be readily decontaminated by an ambient-temperature or reflux process. The deperoxidation process is enhanced under nitrogen and has been safely carried out on a bulk scale and with initial peroxide contents as high as 0.5 M. IMS, in common with most other chemical reducing agents used for ether deperoxidation, are, however, ineffective for the decomposition of unreactive species such as dialkyl peroxides.
Aliphatic ethers, with their characteristic solvation abilities, excel as inert reaction media in numerous syn- thetic procedures. However, in practice this usefulness is
0022-326318211947-3821$01.25/0
often tempered by a n unfortunate proclivity to facile air oxidation at ambient temperatures which leads to peroxide formation.' T h e presence of peroxides is not only po-
0 1982 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0141
a003
marisa
Highlight

3822 J. Org. Chem., Vol. 47, No. 20, 1982 Burfield
Table I. Deperoxidation of Tetrahydrofuran with Indicating Molecular Sieves (IMS) at Ambient Temperatures
peroxide content,c mmol /L sieve scale, loading,
run mL 5% w/v initial 1 day 2 d a y s 3 days 4 days 7 days 90 days
1 50 10 7.8 0.9 0.1 N.D. 2 a 50 10 7.8 3.3 0.5 0.26 3 24 00 5 1.1 0.22 0.10 0.06 0.014 d 4 100 5 1.1 0.07 0.007 0.018 0.012 d 5 50 5 22 7.8 5.5 6 b 50 5 22 7.1 4.9 7 50 5 1 2 4 2.8 8 50 10 1 7 6 1 4 9 7 50 5 88 20 6 3 d
10 100 10 96 4
a Performed under an atmosphere of air. IMS activated at 300 "C in air oven. As determined by the spectrophoto- metric method. N o t determined.
tentially hazardous,l12 but also frequently undesirable for chemical reasons, and although possible hazards may be avoided by the routine disposal of time limit expired3 or proven peroxide contaminated4 ethers, the need for rig- orously purified solvents as well as economic and logistic' constraints may frequently necessitate peroxide removal. To this end a bewildering arrayla of safe (and not so safe!6) physical and chemical methods have been proposed.
Column chromatographic purification whereby peroxides are removed by adsorption on substrates such as alumina,1° ion-exchange resins'l or 13X molecular sieved2 have var- iously been reported, and the use of activated alumina in particular has been widely e n d o r ~ e d . ~ ~ J ~ However, such methods, though effective and generally applicable, are disadvantaged by the necessity of using substantial amounts14 of nonregenerable and relatively expensive ad-
(1) See, for example, the brief review by N. V. Steere in 'The Chem- istry of the Ether Link", S. Patai, Ed., Interscience, London, 1967.
(2) See for example: (a) A. G. Davies, J. R. Znst. Chem., 80,386 (1956); (b) "Guide for Safety in the Chemical Laboratory", 2nd Ed., Van Nost- rand-Reinhold, New York, 1972, p 302; (c) 'Safety in Academic Chem- istry Laboratories", 3rd ed., American Chemical Society, Washingtan, DC, 1979; (d) L. Bretherick, "Handbook of Reactive Chemical Hazards", 2nd ed., Butterworths, London, 1979.
(3) It has been proposed6 that unused ethers in opened containers should be disposed of within 1 week of opening (uninhibited grades) or 3 or 6 months (inhibited grades). ElsewhereZc a blanket time limit of 1 month has been suggested for ethyl or isopropyl ethers. In one labora- toe it is standard practice to use only THF from unopened bottles and to discard the remainder within 2-3 days.
(4) It has been recommended that diethyl etherJ containing more than 0.005% peroxide (yellow color with KI test) and THF containing 'larger than trace amounts of peroxiden6 should be discarded.
(5) N. V. Steere, J. Chem. Educ., 41, A575 (1964). (6) H. E. Baumgarten, 'Organic Syntheses", Collect. Vol. V, Wiley,
New York, 1973: (a) p 796, (b) p 695. (7) For laboratories situated outside the main industrial centers,
chemical delivery times exceeding 6 months are not uncommon. As such ethers from unopened bottles are frequently significantly contaminated on receipt.
(8) Potassium hydroxide, which in its finely powdered form is a pow- erful desiccant: has been advocated for peroxide removal. However, serious explosions may occur when treating impure THF with solid or concentrated aqueous potassium hydroxide.E Similarly, LiAlH,, a less efficient p g agent: has proved distinctly hazardous in the purification of ethers. (9) D. R. Burfield, K. H. Lee, and R. H. Smithers, J. Org. Chem., 42,
3060 (1977). (10) W. Dasler and C. D. Bauer, Znd. Eng. Chem. Anal. Ed., 18, 52
(1946). (11) R. N. Reinstein, J. Org. Chem., 24, 1172 (1959). (12) N. Rabjohn, "Organic Syntheses", Collect. Vol. IV, Wiley, New
York, 1963, p 475. (13) (a) A. I. Vogel, "A Text-Book of Practical Organic Chemistry", 4th
ed., Longmans, London, 1978; (b) "Purification of Solvents by Adsor- bents Woelm", Woelm Pharma, Eschwege, West Germany; (c) "Drying in the Laboratory", E. Merck, Darmstadt, West Germany; (d) D. D. Perrin, W. L. F. Armarego, and D. R. Perrin, 'Purification of Laboratory Chemicals", Pergamon, Oxford, 1980.
sorbents and the need for the subsequent safe disposal of the contaminated residue upon which the peroxides are adsorbed chemically unchanged.1°
Chemical methods encompass a wide spectrum of which effect reductive decomposition of per-
oxides. These include classical redox systems such as sulfites, bisulfites, amines, metal salts (e.g., FeS04, CuCl, Ce(OH)3, SnC12), traditional reducing couples (e.g., Sn/ HCl, Na/EtOH), and the more recently used complex metal hydrides (LiAlH,, NaBH,). Treatment with aque- oud5 ferrous s ~ l f a t e , ~ ~ * ~ J ~ * , ~ J ~ or sodium sulfite/bi- sulfite'wl6 has received approbation as a safe and effective method of deperoxidation, but their use is restricted to water-immiscible ethers,l* and in any case such procedures necessitate an additional desiccation step. For water- miscible ethers such as dioxane, diglyme, and THF, reflux over solid cuprous6b chloride, stannous ~ h l o r i d e , ~ ~ " , ~ or LiAlH4& has been advocated. The use of these procedures, however, appears to be distinctly hazardous for heavily peroxidized ethers, and preliminary small-scale purifica- tions are normally advisedS6" Interestingly, an earlier methodlg employing solid cerous hydroxide, which is ap- parently effective and widely applicable, does not appear to be generally used probably because of the necessity for preparation of fresh reagent and the inaptitude of the methodology for application to bulk purification.
In the context of these various shortcomings, we propose the use of indicating molecular sieves (IMS) as an effective, safe, and readily available reagent which is generally ap- plicable to the problem of peroxide removal from ethers.
Results and Discussion Deperoxidation of Tetrahydrofuran (THF). The
deperoxidation of THF was most thoroughly studied since of the common ethers discussed below this solvent has the highest proclivity toward peroxide formation.20 Table I
(14) The adsorbent loading required for effective deperoxidation is dependent on the grade of initial alumina and ether type, as well as on the initial water and peroxide content. However, for dry THF or dioxane of moderate peroxide content basic alumina of activity I will only purify less than an equal weight of ether.13bsc
(15) As opposed to cuprous and stannous chlorides, solid ferrous sulfate is apparent1 ineffective16 in peroxide removal, and an explosion has been reported17 on distilling THF from the solid salt. The effec- tiveness of solid inorganic reagents may perhaps be related to their sol- ubilities in ethers since both cuprous and stannous chlorides are appre- ciably more soluble than the ferrous salt.
(16) A. C. Hamstead, Znd. Eng. Chem., 56 (6), 37 (1964). (17) J. Schurz and H. Stubchen, Angew. Chem., 68, 182 (1956). (18) The use of aqueous ferrous sulfate for deperoxidation of THF,'"
is surely impractical as this ether is completely miscible with water. (19) J. B. Ramsey and F. T. Aldridge, J. Am. Chem. Soc., 77, 2561
(1955). (20) This conclusion has been drawn from the analysis of some 30
samples of stored ethers of various types and from monitoring the rate of peroxide formation in purified ether samples exposed to the atmo- sphere.
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0014
1a00
3

Deperoxidation of Ethers J. Org. Chem., Vol. 47, No. 20,1982 3823
Table 11. Deperoxidation of Various Ethers with IMS
sieve peroxide content,c mmol/L
run (I ether % w/v initial 3 h 1 day 2 days 3 days 7 days 90 days conditionsd loading,
1 dioxane 1 0 1.7 0.22 0.44 A 2 10 5.5 4 .0 0.6 A
R R A R A A A
~
3 4 5 6 7 8 9
10 11
5 10.3 0.61 5b 10.3 0.44
5 12.8 0.24 diethyl ether 5 12.8 0.05 e
diisopropyl ether l o c 53 0.3
trimethylene glycol 10 29 5.5 10 109 1.1
dimethyl ether 5 27 <0.1
dibenzyl ether 5 37 <0.15
a Scale = 100 mL except as indicated. Scale = 1500 mL. Scale = 50 mL. As m asured by sp method; A = ambient temperature, R = at reflux, and H = at ca. 110 "C. e Not determined.
summarizes some representative results for the addition of IMS to peroxide-contaminated THF obtained from a variety of sources. In every case addition of IMS leads to a reduction in the peroxide content which decreases to a negligible level over extended periods of time (runs 1, 3, 4,9). The efficiency of deperoxidation is enhanced under nitrogen. (cf. runs 1,2) but is somewhat reduced by in- creased scale (cf. runs 3, 4).
Activation of the supplied IMS by heat treatment at 300 O C only marginally increases the efficiency of deper- oxidation (runs 5,6) . However, deactivation of the IMS, by exposure to the atmosphere until the blue indicator decolorizes, renders this material completely impotent for peroxide removal.21
Deperoxidation of THF under reflux proceeds far more rapidly than by static deperoxidation at ambient tem- peratures. Thus peroxide contents as high as 500 mmol/L were dramatically reduced (95% peroxide removal) within 4 h of refluxing over 5% w/v IMS.
Deperoxidation of Various Ethers. The effects of ambient temperature and reflux treatment of various ethers with IMS are summarized in Table 11. Generally a t ambient temperatures peroxide removal from dioxane is relatively inefficient and considerably slower than for THF. On the other hand, under these conditions, de- peroxidation of diethyl ether and diisopropyl ether is facile, proceeding more readily than with THF.
At reflux temperatures the deperoxidations of the var- ious ethers are considerably accelerated, the effect being most noticeable for the higher boiling ethers. It is con- ceivable that this may in part be due to an uncatalyzed thermal decomposition. At the same time the effects of scale become less apparent (cf. runs 3, 4), and 1.5 L of dioxane is decontaminated as rapidly as 0.1 L. Refluxing diethyl ether shows little advantage over the comparable static purification, which in any case is already very effi- cient.
Nature of Peroxide Removal. Earlier studies12 have shown that ethers may be deperoxidized by selective ad- sorption with 13X (10 A) molecular sieves. Purification of THF with nonindicating sieves of varying pore sizes (Table 111) confirms the possibility of preferential peroxide adsorption for 10A and to a lesser extent for 5A sieves. However, sieves of 4-A pore size, i.e., equivalent in pore structure to the IMS, are completely ineffective. The
ctrophot
H H
metric
Table 111. Effect of Sieve Type on THF Deperoxidation peroxide content,b
mmol/L
30 days run sieve type 15 days ~
1 no added sieve 8.7 14.8 2 4A 7.6 19.6 3 5A 2.9 3.1 4 10A 0.14 0.16 5 4A (with indicator) 0.26
Scale 50 mL; sieve loading 10% w/v; at ambient tem- perature under air. photometric method. The initial content was 7.8 mmol/ L in all cases.
As determined by the spectro-
Table IV. Dialkyl Peroxide Content of Deperoxidized Ethers
peroxide content, mmol/L
after treatmentC
total sample initiala ROOHa peroxideb
THF 124 0.95 2.4 96 0.60 2.8
diisopropyl ether 109 1.1 3.3
diethyl ether -1 0.2 1.1 17
Analysis
dioxane 52 e
a Analysis by spectrophotometric method. by acid reflux. temperature. e Not determined.
Treated with 5% w/v IMS at ambient
effectiveness of IMS for deperoxidation must therefore be attributed to some alternative mechanism, and it seems not unreasonable to suppose that peroxides are decom- posed by interaction with the impregnated indicator, which is probably a cobalt salt, through redox reactions such as:
ROOH + Co2+ - RO- + -OH + Co3+ The implication of this conclusion is that in contrast to
the residues from normal chromatographic purifications, the IMS should be peroxide free and thus capable of hazard-free disposal or reactivation.
Formation, Detection, and Removal of Dialkyl Peroxides. Most methods employed for testing ether purity, including the standard acidified KI test,22 and the
(21) IMS are similarly ineffective for the treatment of grossly wet (>l% w/v water) ether samples, wherein water uptake by the molecular sieves leads to deactivation. Such samples require either predrying or addition of higher loadings (>5% w/v) of IMS to prevent saturation and deactivation of the indicator.
(22) In this test peroxides are deemed present if a faint yellow color develops on shaking 6 mL of the ether with an equal volume of 2% w/v KI solution in the presence of a few drops of dilute hydrochloric acid. (The detection limit of this test in our hands corresponds to a value of about 0.2 mmol/L as measured by the spectrophotometric method.
(23) R. D. Mair and A. J. Graupner, Anal. Chem. 36, 194 (1964).
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0014
1a00
3

3824 J. Org. Chem., Vol. 47, No. 20, 1982
spectrophotometric method used in this study are inef- fective for the detection of dialkyl peroxides. The sig- nificance of this observation is that all the methods re- ported in the literature to date for the removal of peroxides have in fact been proven only for hydroperoxides.
For evaluation of the relative importance of dialkyl peroxide formation, several badly contaminated ether samples were deperoxidized with IMS and the deconta- minated ethers analyzed for both active peroxide (hydro- peroxide) and dialkyl peroxide (Table IV). In every case, except dioxane, the total peroxide content is very much reduced by treatment with IMS. However, the acid-reflux analysis indicates the presence of small but detectable amounts of dialkyl peroxide in these samples. In the case of dioxane there is a very large concentration (>30% of original content) which is not analyzed by the spectro- photometric method and is not decomposed by IMS during the period of treatment. It would appear, therefore, that oxidation of dioxane leads to the formation of much higher concentrations of dialkyl peroxides than other ethers or to hydroperoxides which are much less reactive.
In summary, it appears that predominant contaminants of ethers such as THF and diisopropyl and diethyl ethers are active peroxides which can be readily removed by treatment with IMS. Deperoxidation of dioxane is very much less efficient. Almost certainly, the inapplicability of IMS to the problem of dialkyl peroxides is also a lim- itation with other commonly used redox purification sys- tems such as those based on CuC1, SnC14, FeS04, etc.
Applicability of IMS to Ether Deperoxidation. The prime criteria for deperoxidation methods are effectiveness and safety. Since most purifications are accompanied by distillation, which technique already allows a means of peroxide removal, the key aspect is safety. In this con- nection, IMS have proved effective in the deperoxidation of ethers with peroxide concentrations as high as 0.5 M without hazard. Compared to other methods IMS provide a relatively slow rate of peroxide decomposition, and this feature is almost certainly an important aspect of safe deperoxidation.
Since it has already been shown that molecular sieves are particularly effective in removing water24 and other polar impurities% from ethers, as applied to diethyl ether
Burfield
(24) D. R. Burfield, G. H. Gan, and R. H. Smithers, J. Appl. Chem. Biotechnol., 28, 23 (1978).
for example, the use of IMS provides a safe and effective method of concomitant deperoxidation, deethanolization, and drying of the solvent. In the case of dioxane or other ethers where the presence of significant quantities of alkyl peroxide are suspected, IMS may be used for pretreatment before more rigorous deperoxidation is executed.
Experimental Section Materials. Indicating activated Type 4A molecular sieve,26
(4-8 mesh) were kindly supplied by J. T. Baker Chemical Co. and were used as received. Ethers were typical reagent grade solvents. High peroxide levels were induced by aging purified samples in the presence of air over a period of days or weeks a t ambient temperatures.
Deperoxidation Method. ( i ) Static Deperoxidation. Ethers, in their original containers, were first deoxygenated by bubbling with oxygen-free nitrogen for about 5 min in a fume cupboard. Subsequently, 5% w/v of indicating molecular sieves was added and the ether tightly capped and set aside.
(i i) Reflux Deperoxidation. Ethers were charged into a distillation flask and deoxygenated with a slow stream of nitrogen. Subsequently, 5 % w/v of indicating molecular sieves was added and the ether brought slowly to reflux under nitrogen.
Peroxide Analysis. (i) Spectrophotometric Analysis. Hydroperoxides were analyzed by a modified spectrophotometric method based on the procedure of Wagner et This ferrous ion oxidation method permits quantitative analysis of hydro- peroxides (confirmed for tert-butyl hydroperoxide) but is com- pletely insensitive to dialkyl peroxides such as di-tert-butyl peroxide.
(ii) Total Peroxide Analysis. An acid-reflux method pro- posed by Mair and Graupnera was used and was found to provide a quantitative assay for dialkyl peroxides such as di-tert-butyl peroxide.
Acknowledgment. I acknowledge the assistance of Mr. Lee Meng Lay in conducting the peroxide analyses.
Registry No. Tetrahydrofuran, 109-99-9; dioxane, 123-91-1; di- ethyl ether, 60-29-7; diisopropyl ether, 108-20-3; trimethylene glycol dimethyl ether, 17081-21-9; dibenzyl ether, 103-50-4.
(25) D. R. Burfield and R. H. Smithers, Chem. Ind. (London), 240 (1980).
(26) Indicating molecular sieves (Type 4A) as supplied by Merck and Sigma were found to be equally effective. The product of the latter company is, however, only provided as a mixture (approximately 10% w/w) with nonindicating molecular sieves. Generally smaller bead sizes are more efficient for deperoxidation due to increased surface area.
(27) C. D. Wagner, H. L. Clever, and E. D. Peters, Anal. Chem., 19, 980 (1947).
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0014
1a00
3

J. Org. Chem. 1981,46,629-631 629
(50 mL) afforded 0.400 g (80%) of 15: mp 249-250 “C (C2H5OH); IR (KBr) v, 1709,1600 cm-’; ‘H NMR (Me2SO-ds) 6 2.68 (2 H, t , CH2), 3.04 (2 H, t , H2CC02H), 7.34-7.56 (3 H, m, Ar HI, 7.86 (1 H, s, Ar H), 7.96-8.12 (3 H, m, AI H), 8.50 (2 H, d, Ar H); UV (anhydrous C2H50H) A, 376 nm (e 3800), 357 (4400), 340 (320% 325 (1900), 255 (200000), 247 (90000).
Anal. Calcd for C1,H1402: C, 81.60; H, 5.60. Found: C, 81.33; H, 5.84. 5-(2-Anthryl)pentanoic Acid (16). Ester 6 (0.548 g, 2 mmol)
was hydrogenated over 10% Pd/C in anhydrous CZHSOH (50 mL), and the product obtained was boiled with o-chloranil (0.590 g, 1.2 mmol) for 3 h under NP Hydrolysis of the product from the above reaction with 10 alcoholic KOH solution (20 mL) afforded 0.410 g (71%) of acid 16: mp 191-192 “C (C2H50H); IR (KBr) v- 1695,1575 cm-’; ‘H NMR (DCCld 6 1.6 (4 H, m, CHJ 2.2-2.3 (2 H, m, CH2), 2.82 (2 H, m, CH2), 7.3-7.5 (3 H, m, Ar H), 7.72 (1 H, m, Ar H), 7.88 (3 H, m, Ar H), 8.00-8.32 (2 H, m, Ar H); UV (anhydrous C2H50H) A, 377 nm (e 5400), 358 (6100), 341 (4400), 327 (2500), 291 (700), 254 (237000), 249 (88900).
Anal. Calcd for C19H1802: C, 82.01; H, 6.44. Found C, 82.13; H, 6.56. 3-(9-Anthryl)propanoic Acid (22).20 Hydrogenation of acid
20 (0.496 g, 2 mmol) over 10% Pd/C (50 mg) in anhydrous C2H50H (20 mL) afforded 0.450 g (90%) of acid 22: mp 188-190 “C (C2H50H-H20, lit.20 mp 191-192 “C); IR (KBr) 1695,1600 cm-’; ‘H NMR (DCClJ 6 2.78-2.96 (2 H, br t, CH2), 3.8-4.0 (2 H, br t, CH2), 7.40-7.55 (5 H, m, Ar H); 7.9-8.1 (2 H, m, Ar H), 8.2-8.4 (2 H, m, Ar H); UV (anhydrous C2H50H) A, 386 nm (e 5000), 361 (5180), 347 (380), 332 (1450), 256 (95700). 5-(I)-Anthryl)pentanoic Acid (23). Hydrogenation of acid
21 (0.556 g, 2 mmol) in anhydrous CzH50H (30 mL) over 10% Pd/C (70 mg) afforded 0.440 g (79%) of acid 23: mp 112-113 “C (ether-petroleum ether); IR (KBr) vmar 1695, 1613 cm-’; ‘H NMR (DCClJ 6 1.70-1.98 (4 H, d, CHJ, 2.42 (2 H, m, CHJ, 3.58 (2 H, m, CH2), 7.16 (1 H, s, Ar H), 7.26-7.60 (4 H, m, Ar H), 7.94-8.20 (2 H, m, Ar H), 8.10-8.32 (1 H, m, Ar H), 11.14 (1 H, s, C0,H); UV (anhydrous C2HSOH) A,, 387 nm (e 8830), 382 (4640), 367 (8990), 348 (5420), 331 (2490), 318 (1020), 257 (169000), 250 @OW), 236 (21 600), 223 (6700).
Anal. Calcd for C l ~ l s O z : C, 82.01; H, 6.44. Found C, 81.87; H, 6.65.
Acknowledgment. We gratefully acknowledge partial support of this work from the Presidental Challenge Grant Program a t O.S.U. in the form of salary (to K.D.B.) and from the U.S.P.H.S., National Institutes of Health, via a grant from the Institute of General Medical Sciences (Grant GM 25353 to M.G.R.).
Registry No. 1, 2143-81-9; 2, 1099-45-2; 3,42997-19-3; 5,75802- 25-4; 6, 75802-26-5; 7, 75802-27-6; 8, 75802-28-7; 9, 75802-29-8; 10, 75802-30-1; 11, 75802-31-2; 12, 75802-32-3; 13, 75802-33-4; 14, 75802-34-5; 15, 75802-35-6; 16, 75802-36-7; 17,642-31-9; 18, 75802- 37-8; 19,75802-38-9; 20,5335-33-1; 21,75802-39-0; 22,41034-83-7; 23, 75802-40-3; [6-(methoxycarbonyl)hexa-2,4-dien-l-yl]triphenyl- phosphonium bromide, 75802-41-4.
Desiccant Efficiency in Solvent and Reagent Drying. 5.
David R. Burfield, Roger H. Smithem,* and Andrew Sui Chai Tan5
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, W. Malaysia
Reeeived September 9, 1980
The use of amines in synthesis can be divided into three principal areas: (i) as basic agents for the promotion of
(1) Part 1: D. R. Burfield, K. H. Lee, and R. H. Smithers, J. Org. Chem., 42, 3060 (1977).
0022-326318111946-0629$01.00/0
dehydroeliminations, (ii) as nucleophiles in simple dis- placements, and (iii) as precursors of various metalated derivatives. Because of strong N-H hydrogen bonding, in all these uses, water present in the amine system may exert damaging, i.e., yield lowering, effects by interfering with absolute basicity and nucleophilicity6 and/or reacting either as free water or hydroxide ion with unstable in- termediates or sensitive products.’ However, despite the existence of an arsenal of desiccants, the presence of water in these systems continues to be a problem for the syn- thetic chemist. This is because the recommended agents for removal of water and polar impuritiesg from other solvent and reagent typed4 may not be suitable for amines. Therefore the radiotracer method for water assay previously developed by uslo has now been applied to ob- tain quantitative data on the drying of some representative amines.
The Pyridine Group. For pyridine, and indeed gen- erally for the amine class, the traditionally recommended siccatives are the alkali and alkali earth hydroxides and 0xides.l’ Thus, literature prescriptions commonly advo- cate distillation from KOH,12a*b standing over Ba0,lZc or distillation from CaH2,13 the employment of the latter procedure reportedly yielding samples containing 18-20 ppm of residual water. The use of A1203 has also been occasionally reported.14 Our results for pyridine obtained by application of the radiotracer technique are summarized in Table I. The results are largely self-explanatory, and as can be seen, a horizontal line drawn under the entry for KOH sharply demarcates serious desiccants from those which are less efficaceous. Surprisingly perhaps, alumina is seen to be rather unimpressive; however, this ineffec- tiveness in the drying of polar reagents has been noted previously.’J It is also worth noting that the use of sodium is to be avoided; it is not particularly efficient and con- tributes to material loss by a wasteful side reaction which produces bipyridyls.
Alkylated derivatives of pyridine are more basic and often less nucleophilic than pyridine itself, and these at- tributes are considered advantageous in synthesis. We therefore thought it of interest to compare the difficulty
(2) Part 2: D. R. Burfield, G. H. Gan, and R. H. Smithers, J. Appl. Chem. Biotechnol., 28, 23 (1978).
(3) Part 3: D. R. Burfield and R. H. Smithers, J. Org. Chem., 43,3966 (1978). (4) Part 4 D. R. Burfield and R. H. Smithers, J. Chem. Technol.
Biotechnol., in press. (5) Abstracted in part from the Final Year Project of Andrew S. C.
Tan, 1978-1979. (6) As is well-known, solvation effects play a vital part in determining
both basicity and nucleophilicity; aee, for example, C. Reichardt “Solvent Effects in Organic Chemistry”, Verlag Chemie, Weinheim, Germany, 1979, pp 55-60, 148-155. (7) In a pertinent example from our own laboratories, the literature
preparation of methyl diphenylphosphinite calls for reaction between chlorcdiphenylphosphine and methanol in the presence of pyridine and gives a reported yield of 5270.8 In our hands, the use of rigorously dried pyridine and methanol increased the yield to 75%.
(8) A. E. Arbuzov and K. V. Nikonorov,, Zh. Obsch. Khim., 18,2008 (1948). (9) D. R. Burfield and R. H. Smithers, Chem. Ind. (London), 240
(1980). (10) D. R. Burfield, Anal. Chem., 48 2285 (1976). (11) See, for example: (a) D. Todd “Experimental Organic
Chemistry”, Prentice-Hall Inc., NJ, 1979; (b) R. S. Monson, “Advanced Organic Synthesis”, Academic Press, New York, 1971; (c) J. A. Riddick and W. B. bunger, “Organic Solvents”, 3rd ed., Wiley-Interscience, New York, 1970.
(12) See, for example: (a) G. A. Olah and M. Watkins, Org. Synth., 68, 75 (1978); (b) W. H. F. Sasse, “Organic Synthesis”, Collect. Vol. V, Wiley, New York, 1973, p 102; (c) R. F. Evans, H. C. Brown, H. C. Van der Plas, ibid., p 977.
(13) D. Jerchel and E. Bauer, Angew. Chem., 68, 61 (1956). (14) D. N. Glew and N. S. Rath, Can. J. Chem., 49, 837 (1971).
0 1981 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0316
a030
marisa
Highlight
marisa
Highlight
marisa
Highlight
marisa
Highlight
marisa
Highlight

630 J. Org. Chem., Vol. 46, No. 3, 1981 Notes
Table I. Desiccant Efficiency in the Dryinga,b of a PyridineC Series residual water content,d ppm
desiccant CaH, CaC, BaO 4A sieves 3A sieves benzene azeotrope KOH powder Na CaO silica gel - 4 1 2 0 3
2-methyl- pyridine pyridine 39 (14)e 84 44 ( i o j e
101 106 (0.3)f 1 1 7 125 152 388 962 926
1306
71 27
55 40
176
2,g-dimethyl- pyridine
248 (138)“ 519 360 268 (1 26) 200 (128) 20 7 325
93 5
2,4 ,g-trimethyl- pyridine
132 8
33
47 390
27
a Static drying modes unless specified otherwise. Water content assayed by the radiotracer technique. Desiccant 24-h drying times unless specified otherwise. e 168-h loading 5% w/v; initial water content 2500 ppm (0.25% w/w).
drying time. f Sequentially dried sample, 24 h.
of drying of some of the more commonly used alkylated derivatives. A survey of the literature revealed that al- though the alkylpyridines are usually subjected to similar drying procedures as pyridine itself, fractionation alone and fractionation from BF3 (!) have been advocated for 2-meth~lpyridine’~ and 2,6-dimethylpyridine,16 respec- tively. The results summarized in Table I exhibit a clear-cut trend; whereas one alkyl group at the 2-position gives results of similar overall order to pyridine, when N is flanked by two such groups (as in the lutidine) there is a marked increase in difficulty of drying, with 2-10-fold greater water levels being observed. Interestingly, the final member of series, the collidine, gave rise to some of the lowest residual water levels recorded. Not only is 2,4,6- trimethylpyridine the most basic of the series examined,17 it is also the easiest example to dry. Clearly then, there seems to be no relation between increasing basicity and drying difficulty, and instead this property appears to be determined by the interplay of water-solubility factors and of steric crowding about the N atom. Thus, for 2,6-di- methylpyridine, an infinitely water-miscible base, an ob- vious speculation is that the two flanking methyl groups present a serious impediment to the close approach of a siccative to the water-coordinating site. For trimethyl- pyridine, a drop in residual water concentrations is par- alleled by a corresponding large decrease in water solu- bility.18
Triethylamine. Triethylamine, commonly used as a mild base in dehydrohalogenations, has been dehydrated by the alkaline earth KOH,lgb CaH2,1gC and molecular sievedgd as well as by alumina and sodium metal.11c The results summarized in Table I1 require little comment beyond the fact that, despite being considerably more basic than the pyridines, triethylamine (pKb = 3.1) appears very much easier to dehydrate.
Diisopropylamine. Diisopropylamine is the precursor of lithium diisopropylamide, a powerful highly hindered
(15) L. A. Walter, “Organic Syntheses”, Collect. Vol. 111, Wiley, New York, 1955, p 757.
(16) A. N. Sharpe and S. Walker, J. Chem. SOC. 2974 (1961). (17) The relevant pKb values for pyridine, 2-methylpyridine, 2,6-di-
methylpyridine, and 2,4,6-trimethylpyridine are 8.8, 8.0, 7.3, and 6.6, respectively.
(18) While pyridine itself, as well as ita mono- and dialkyl derivatives, is essentially completely water miscible, the solubility of the trimethylated pyridine is only about 3%. See Beilsteine, 4th ed., 20, 164 (1953).
(19) (a) R. Breslow and J. Posner, “Organic Syntheses”, Collect. Vol. V, Wiley, New York, 1973, p 514; (b) H. Rinderknecht and M. Guten- stein, ibid, p 822; (e) M. E. Jung and C. A. McCombs, Org. Synth., 58, 163 (1978); (d) T. J. Atkins, R. E. Richman, and W. F. Oettle, ibid., 58, 87 (1978).
Table 11. Desiccant Efficiency in the Dryinga of Various Aminesb
residual water content,c ppm
(Me,CH),- desiccant Et,Nd NHe NH,(CH,),NH,e
KOH powder 37 ( 23)f 4A sieves 33 ( 28)h 3A sieves 34 CaH, 68 ( 34)f Na 83 BaO 89 (53)f CaC, 98 ( 80) f CaO 165 ( 56)f -4120, silica gel CaSO,
223 (223) j 451
750g 1370 (3700)g < 25 < 25 < 25 < 25 150’ 500’ < 25 150
50 1100 <25’ <25’
> 2500
a Static drying modes unless specified otherwise. Desiccant loading 5% w/v, initial water content 2500
ppm (0.25% w/w). otherwise. Water content assayed by the radiotracer method. e Water content assayed by the near-IR method. f 168-h drying time. , g Aged desiccant used. tially dried sample. t Stirred samples. 72-h drying time.
24-h drying times unless specified
Sequen-
base which is freely soluble in many organic solvents and which has achieved prominence in synthesisz0 in the con- tinuing search for potent bases of low nucleophilicity. Curiously, drying prescriptions for the parent amine are infrequently mentioned, but desiccation with KOH and storage over CaHz is probably a typical expedient.21 Because this drying problem was not amenable to inves- tigation using the radiotracer method, due to interference by isotopic exchange, the near-IR method1~6~zz was applied and yielded the results summarized in Table 11. In view of the high water solubility (-40%) and basicity (pKb = 2.9) of this amine, it may seem surprising that dehydration is so facile. However, an examination of models of the water-amine complex suggests that the avoidance of steric congestion between the water molecule and the adjacent methyl groups can only occur with the imposition of severe entropy constraints, with the probable result that the equilibrium between complex and free amine lies largely to the left.
~~ ~
(20) For usage see, for example: (a) B. M. Trost, C. D. Shuey, F. DiNinno, Jr., and S. S. McElvain, J. Am. Chem. Soc., 101, 1284 (1979); (b) R. A. Olofson and C. M. Dougherty, ibid., 95, 582 (1973).
(21) D. Enders, R. Pieter, B. Renger, and D. Seebach, Org. Synth., 58, 113 (1978).
(22) R. L. Meeker, F. Critchfield, and E. T. Bishop, Anal. Chem., 34, 1510 (1962).
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0031
6a03
0

J. Org. Chem. 1981,46, 631-632 63 1
1,3-Propanediamine. This is another example of an amine which has sprung to prominence in synthesis be- cause of unique reactions brought about by a derivative, in this case its monopotassium SalkB Desiccation has been achieved by vacuum distillation alone,% distillation from alkali metal or distillation from KOH.24c Our results using the near-IR method are displayed in Table I1 and indicate that, not surprisingly, this amine with two coordinating centers is one of the more difficult examples to dry. It is worth noting that freshly ground KOH gives a drier sample than aged material stored in a desiccator, although as may be seen, in comparison with several other siccatives, e.g., CaCz or sieves, KOH is not particularly good. In addition, although seldom recommended for use in these situations, CaS04 (Drierite) was investigated in view of some of the more extravagant claims which have been made on its behalf.25 As may be seen, the results are rather dismal.
Desiccation of Amines. General Recommendations. Perhaps not surprisingly, the results from this study lend support to the current usage of molecular sieves and CaH2 as serious and widely applicable siccatives for amines. Apart from this, it is fitting to draw attention to the high efficiency of CaC2 in these studies, which, though infre- quently prescribed as a desiccant, is often seen to surpass CaH2 in potency and is more desirable than the hydride from consideration of cost and safety in storage. On the other hand, the performance of alumina is uniformly disappointing and it cannot be advocated as a serious desiccant for amines.
Experimental Section Details of the water assay techniques as well as the source,
activation, and handling of most of the desiccants have already been described.14 Determination of water content by the near-IR method22 was carried out on a Varian Cary 17 Instrument. Calcium carbide was of industrial grade and was crushed in a mortar immediately prior to use. Appropriate venting was prc- vided for desiccants producing gases, e.g., CaC2, CaH2.
Amines were of laboratory reagent grade and purified by standard methods.llc The pyridines and triethylamine were stood over KOH for 24 h, decanted, fractionated, and stored in dark bottles over 20% w/v 3A molecular sieves. Pyridine had bp 115-116 OC, 2-methylpyridine 128-129 "C, 2,6-dimethylpyridine 145-146 "C, and 2,4,6-trimethypyridine 176-178 "C. EtaN had bp 89-89.5 "C. Diisopropylamine was first allowed to stand over 20% w/v 3A molecular sieves, decanted, stirred overnight with CaH2, fractionated, and finally stored as above, bp 84 OC. 1,3- Propanediamine was mixed with 20% v/v benzene, fractionated, stirred overnight with CaH2, and fractionated again, bp 135-136 "C.
Acknowledgment. We thank the Department of Chemistry, University of Malaya, for support of this work and Miss Lim Siew Heong for her able technical assistance.
Registry No. Pyridine, 110-86-1; 2-methylpyridine, 109-06-8; 2,6-dimethylpyridine, 108-48-5; 2,4,64rimethylpyridine, 108-75-8 EhN, 121-44-8; (Me2CH)2NH, 108-18-9; NH,(CH2)3NH2, 109-76-2; CaH2, 1789-78-8; CaC2, 15-20-1; BaO, 1304-28-5; benzene, 71-43-2; KOH, 1310-58-3; Na, 7440-23-5; CaO, 1305-78-8; A1203, 1344-28-1; CaS04, 7778-18-9.
(23) Potassium 3-aminopropylamide possesses the singular property of bringing about the conversion internal alkyne - 1-alkyne in essentially quantitative yields. See C. A. Brown and A. Yamashita, J. Am. Chem. soc., 97,891 11975).
(24) (a) A. Gero, J. Am. Chem. SOC. 76,5159 (1964); (b) L. R. Dalton, J. L. Dve. E. M. Fielden. and E. J. Hart, J. Phvs. Chem., 70,3358 (1966); (c) K. k'Badri and L. Y. Goh, Inorg. Chim. Acta, in press.
pany, Xenia, OH. (25) W. A. Hammond, "Drierite", the W. A. Hammond Drierite Com-
0022-3263/81/1946-0631$01.00/0
Trapping of Intermediates in the Thermolysis of a-Azidochalcone. Insight into the "Zwittazido
Cleavage" Reaction
Benjamin A. Belinka, Jr., Alfred Hassner,* and J. M. Hendler
Department of Chemistry, State University of New York at Binghamton, Binghamton, New York 13901
Received July 28, 1980
The "zwittazido cleavage" reaction (Scheme I) has re- cently been the subject of considerable study' because this novel rearrangement possesses both interesting mecha- nistic and advantageous synthetic characteristics. Al- though a large amount of work with cyclic and acyclic ketoazides has been reported,lV2 the mechanism of this reaction is still somewhat obscure, especially the question of whether a concerted or stepwise process (or both) occurs.
In the rearrangement of cyclic keto vinyl azides op- portunity exists for an intramolecular ring closure following an initial ring cleavage (Scheme I). In acyclic systems formation of azirines, indoles, or rearranged cyano ketones can take place.2 In transformation 1-2, the reaction has been postulated to occur via intermediate 3. An alternate pathway would involve a cleavage to ions 4 and 5, which may recombine to 2 (Scheme 11).
Our interest in the chemistry of vinyl azides3 led us to investigate this reaction and to show that even in the acyclic case cleavage can occur and that the intermediates can be trapped with alcohols or amines.
Thermolysis of a-azidochalcone (1) in o-dichlorobenzene for 24 h produced a-cyano-a-phenylacetophenone (2) in 70% yield. When the reaction was carried out in the presence of 10 equiv of either ethanol or benzyl alcohol, the yield of 2 decreased while ethyl benzoate (6) and benzyl benzoate (7), respectively, were isolated. Thermolysis with 10 equiv of benzylamine yielded N-benzylbenzamide (8). In all three trapping experiments, considerable quantities of 2 were also obtained along with benzyl cyanide (9) and polymeric materials. The results are recorded in Table I.
The formation of compounds 6-9 indicates that at least part of the thermolysis of the acyclic a-azidochalcone proceeds by a cleavage mechanism as shown in Scheme I1 which allows for an acyclic cation intermediate 4 to react with the alcohols and amine. Protonation of 5 leads to benzyl cyanide (9).
We showed that the azido ketone 1 did not react with ethanol in boiling toluene to produce ethyl ben~oa te .~ Furthermore, no ethyl benzoate (6) was detected by GC when either the cyano ketone 2 or the crude mixture re- sulting from heating 1 in o-DCB was subjected to trapping reaction ~ondi t ions.~
(1) H. W. Moore, Acc. Chem. Res., 12,125 (1979), and references cited there in.
(2) D. Knittel, Hemetsberger, R. Leipert, and H. Weidman, Tetrahe- dron Lett., 1459 (1970).
(3) For instance (a) G. L'Abb6, and A. Hassner, Angew. Chem., Int. Ed. Engl., 10, 98, (1971); (b) A. Hassner, E. S. Ferdinandi, and R. J. Isbister, J. Am. Chem. SOC., 92,1672 (1970); (c) A. Hassner, Acc. Chem. Res., 4, 9 (1971).
(4) Azidochalcone (1) reads with nucleophiles such as sodium sulfide or ylides at the azide rather than at the carbonyl function; B. A. Belinka, Jr., and A. Hassner, J. Org. Chem., 44, 4712 (1979), and unpublished results in this laboratory; G. Mathys, S. Toppet, and G. L'abbd, Chem. Ind. (London), 6,278 (1975).
(5) G. L'abbg and A. Hassner, J. Org. Chem., 36, 258 (1971). 0 1981 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0316
a030

3966 J. Org. Chem., Vol. 43, No. 20,1978 Notes
Other acetals which are commonly used as protecting groups for alcohols are the tetrahydropyranyl ether (ref 4, p 104), ethoxyethyl ether (S. Chladek and J. Smtt, Chem. Ind. (London), 1719 (1964)), 2-methoxyisopropyl ether (ref 4, p 107), and 4-methoxytetrahydropyranyl ether (ref 4, p 108). Removal of all of these groups can be accornpllshed with dilute, aqueous acid. They are prepared from the corresponding vinyl ether with acidcataiysis. The methoxymethyl ether,' as well as our tert-butoxymethyl ether, is prepared from the chloro ether with base catalysis. See, for example, (a) the synthesis of chloromethyl methyl ether: C. S. Marvel and P. K. Potter, "Organic Synthesis", Collect. Vol. I, 2nd ed, A. H. Blatt, Ed., Wiley, New York, N.Y., 1941, p 377; (b) the preparation of methoxyethyl chloromethyl ether: E. J. Corey, J.-L. Gras, and P. Ulrich, Tetrahedron Lett., 809 (1976); and (c) a review on a-halo ethers: L. Sum- mers, Chem. Rev., 55, 301 (1955). Ethers are halogenated on the (Y carbon: M. L. Poutsma in "Methods in Free Radical Chemistry", Vol. 1, E. S. Huyser, Ed., Marcel Dekker, New York, N.Y., 1969, p 137. Methods for the free-radical halogenation of organic compounds have been reviewed: E. S. Huyser, Synthesis, 7 (1970). Sulfuryl chloride has been used to chlorinate tetrahydrofuran (THF): C. G. Kruse, N. L. J. M. Broekhof, and A. van der Gen, Tetrahedron Lett., 1725 (1976). All attempts to isolate the chloro ether by concentration have led to de- composition. J. F. Norris and G. W. Rigby, J. Am. Chern. SOC., 54, 2088 (1932). There is no reaction at 0 O C after 4 h but a satisfactory reaction rate is obtained at room temperature. The water bath is used for cooling purposes only. The 'H NMR spectra of the tert-butoxymethyl ethers show singlets at 6 4.1-4.7 (2 H) and 1.2-1.25 (9 H). The corresponding l3C NMR spectra are also consistent with the proposed structures. For example, the acetal and quaternary carbons are found at 6 89.248 and 74.251, respectively, for the benzyl alcohol acetal and 6 90.038 and 74.068, respectively, for the 1-hexanol acetal.
Desiccant Efficiency in Solvent Drying. 3. Dipolar Aprotic SolventslJ
David R. Burfield* and Roger H. Smithers
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, Malaysia
Received May 2, 1978
I t is generally acknowledged that dipolar aprotic solvents are the media of choice in some reactions and are unique in facilitating others.3 The special solvent effects of molecules such as DMF and Me2SO are attributable to their large di- electric constants coupled with the absence of solvation by hydrogen bonding and typically manifest themselves in properties such as poor anion solvation, voracious cation solvation, and a marked hydrophilicity. For the chemist, this latter feature is unfortunate since small amounts of water in these systems can diminish314 their nucleophilicity and may even be hazardous to some operations.5 The drying of these solvents is thus of paramount importance, but in these cases, as previously,' the chemical literature contains little reliable quantitative data.
We have recently developed a method of solvent water assay
which utilizes a tritiated water tracer for the determination of water content.6 The method circumvents many of the problems encountered in other assay methods and has pro- vided some new correlations on the efficiency of desiccants.lS* For example, i t has been shown that, rather surprisingly, the efficiency of a given desiccant is strongly dependent upon the solvent type,' and there is thus much uncertainty in extrap- olating generalizations from one solvent type to another.
The method has now been applied to the desiccation of the dipolar aprotics acetone, DMF, MeZSO, and HMPT. Since the dielectric constants of these solvents range between 20.7 (acetone) and 46.7 (MeZSO), their rigorous desiccation is ex- pected to be difficult.
Results and Discussion Drying of Hexamethylphosphoric Triamide (HMPT).
Caution! HMPT is a suspected carcinogen. Although in re- cent years the favored desiccant for HMPT appeared to be calcium h ~ d r i d e , ~ drying has also been previously accom- plished with alkali metal^,^^^ alkali metal earth oxides,s and 4A9* and 13Xgb molecular sieves.
The results with the siccatives summarized in Table I are largely self-evident, but the following points are worth noting. The extreme resistance to desiccation is demonstrated by the impossibility of obtaining super-dry lo H M P T under any of the conditions used here. Even sequential drying,ll which was previously found to be effective with acetonitrile,2 falls short in this case. The use of sodium-potassium alloy as a drying agent seems questionable in view of the thermal instability of solutions of alkali metals in solvents of this type.lZ
Since phosphorus pentoxide causes loss of material through side reactions, the best procedure for drying HMPT appears to be distillation from calcium hydride followed by storage over molecular sieves.
Drying of Dimethylformamide One sources observes that it is doubtful whether distillation alone can remove water from this solvent and recommends a chemical method for the elimination of protonic impurities. 4A molecular sieves, alu- mina, potassium hydroxide, and calcium hydride have all been endorsed as siccativess for DMF.
The results in Table I1 indicate the powerful hydrophilicity of this solvent, although sequential drying with 3A sieves al- most achieves super-dryness. Interestingly, and contrary to an earlier suggestion,'3 while some of the basic dessiccants investigated are totally inept, e.g., alumina and potassium carbonate, others such as calcium hydride and potassium hydroxide achieve quite reasonable drying levels. Also, al- though seldom advocated for use in this circumstance, phos- phorus pentoxide is a commendable desiccant. For DMF, however, barring impurities other than water, by far the
Table I. Efficiency of Desiccants in the Drying" of HMPTb
residual solvent water content, ppm desiccant 6 h 24 h 72 h 144 h other conditions
p205 184OC 22d CaHz 1750 857 347 248 80d Bz03 190e 3A molecular sieves 1380 595 307 239 4A molecular sieves 1167 610 344 269 295 KOH (powdered) 1380 840 404 321f Na-K 162Od.g BaO 2190 1540 1040 CaO 2360 2034 1890 1380
A1203 2134
a Static drying modes unless otherwise specified.
Cas04 2080
Desiccant loading 5% w/v; initial water content 2620 ppm (0.262% w/w). Strongly Distilled sample. e Stirring for 24 h followed by distillation. f Sequentially dried sample, 72 h. Significant quantities colored solution.
of dimethylamine are released on distillation.
0022-3263/78/1943-3966$01.00/0 0 1978 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0414
a038
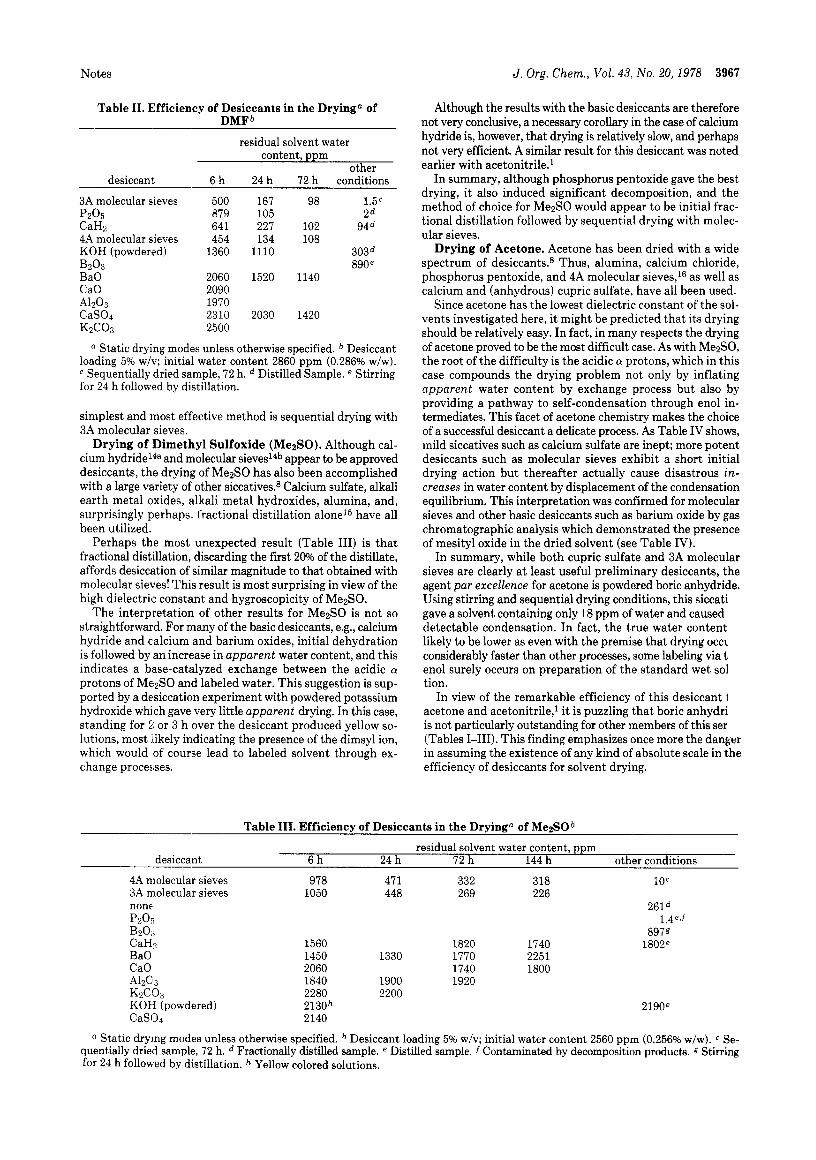
Notes J . Org. Chem., Vol. 43, No. 20,1978 3967
Table 11. Efficiency of Desiccants in the Drying" of DMFb
residual solvent water content, ppm
other desiccant 6 h 24h 72h conditions
3A molecular sieves 500 167 98 1.5c pzo6 879 105 2 d CaH2 641 227 102 94d 4A molecular sieves 454 134 108 KOH (powdered) 1360 1110 303 B203 890e BaO 2060 1520 1140 CaO 2090 A1203 1970 Cas04 2310 2030 1420 KzC03 2500
a Static drying modes unless otherwise specified. Desiccant loading 5% w/v; initial water content 2860 ppm (0.286% w/w).
Sequentially dried sample, 72 h. Distilled Sample. e Stirring for 24 h followed by distillation.
simplest and most effective method is sequential drying with 3A molecular sieves.
Drying of Dimethyl Sulfoxide (MezSO). Although cal- cium hydride14a and molecular sieves14b appear to be approved desiccants, the drying of Me2SO has also been accomplished with a large variety of other siccatives.8 Calcium sulfate, alkali earth metal oxides, alkali metal hydroxides, alumina, and, surprisingly perhaps, fractional distillation alone15 have all been utilized.
Perhaps the most unexpected result (Table 111) is that fractional distillation, discarding the first 20?h of the distillate, affords desiccation of similar magnitude to that obtained with molecular sieves! This result is most surprising in view of the high dielectric constant and hygroscopicity of MezSO.
The interpretation of other results for MezSO is not so straightforward. For many of the basic desiccants, e.g., calcium hydride and calcium and barium oxides, initial dehydration is followed by an increase in apparent water content, and this indicates a base-catalyzed exchange between the acidic a protons of Me2SO and labeled water. This suggestion is sup- ported by a desiccation experiment with powdered potassium hydroxide which gave very little apparent drying. In this case, standing for 2 or 3 h over the desiccant produced yellow so- lutions, most likely indicating the presence of the dimsyl ion, which would of course lead to labeled solvent through ex- change processes.
Although the results with the basic desiccants are therefore not very conclusive, a necessary corollary in the case of calcium hydride is, however, that drying is relatively slow, and perhaps not very efficient. A similar result for this desiccant was noted earlier with acetonitrile.'
In summary, although phosphorus pentoxide gave the best drying, i t also induced significant decomposition, and the method of choice for Me2SO would appear to be initial frac- tional distillation followed by sequential drying with molec- ular sieves.
Drying of Acetone. Acetone has been dried with a wide spectrum of desiccants.8 Thus, alumina, calcium chloride, phosphorus pentoxide, and 4A molecular sieves,16 as well as calcium and (anhydrous) cupric sulfate, have all been used.
Since acetone has the lowest dielectric constant of the sol- vents investigated here, it might be predicted that its drying should be relatively easy. In fact, in many respects the drying of acetone proved to be the most difficult case. As with MezSO, the root of the difficulty is the acidic a protons, which in this case compounds the drying problem not only by inflating apparent water content by exchange process but also by providing a pathway to self-condensation through enol in- termediates. This facet of acetone chemistry makes the choice of a successful desiccant a delicate process. As Table IV shows, mild siccatives such as calcium sulfate are inept; more potent desiccants such as molecular sieves exhibit a short initial drying action but thereafter actually cause disastrous in- creases in water content by displacement of the condensation equilibrium. This interpretation was confirmed for molecular sieves and other basic desiccants such as barium oxide by gas chromatographic analysis which demonstrated the presence of mesityl oxide in the dried solvent (see Table IV).
In summary, while both cupric sulfate and 3A molecular sieves are clearly a t least useful preliminary desiccants, the agent par excellence for acetone is powdered boric anhydride. Using stirring and sequential drying conditions, this siccati gave a solvent containing only 18 ppm of water and caused detectable condensation. In fact, the true water content likely to be lower as even with the premise that drying OCCL considerably faster than other processes, some labeling via t enol surely occurs on preparation of the standard wet sol tion.
In view of the remarkable efficiency of this desiccant f acetone and acetonitrile,l i t is puzzling that boric anhydri is not particularly outstanding for other members of this ser (Tables 1-111). This finding emphasizes once more the danger in assuming the existence of any kind of absolute scale in the efficiency of desiccants for solvent drying.
Table 111. Efficiency of Desiccants in the Dryinga of MezSOb
residual solvent water content, ppm desiccant 6 h 24 h 72 h 144 h other conditions
4A niolecular sieves 978 471 332 318 1 o c 3A molecular sieves 1050 448 269 226 none 261 P205 1.4e3f B203 897g CaH2 1560 1820 1740 1802e BaO 1450 1330 1770 2251 CaO 2060 1740 1800 A12013 1840 1900 1920
KOH (powdered) 2130h 2190e Cas04 2140
KzCQ3 2280 2200
Static drying modes unless otherwise specified. Desiccant loading 5% w/v; initial water content 2560 ppm (0.256% w/w). Se- quentially dried sample, 72 h. d Fractionally distilled sample. e Distilled sample. f Contaminated by decomposition products. g Stirring for 24 h followed by distillation. Yellow colored solutions.
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0414
a038

3968 J. Org. Chem., Vol. 43, No. 20, 1978 Notes
Table IV. Efficiency of Desiccants in the DryingD of Acetone
residual solvent water content, ppm
other desiccant 6 h 24 h 72 h conditions
Bz03 i8C.d 4 7 c . e
107f 3A molecular sieves 115 152 322g 322h CuSO4 (anhydrous) 1920 972 579 1700h
Cas04 1590 1600 BaO 1910 1870' pzo5 j 1970f KzC03 2057 2250
Desiccant loading 5% w/v; initial water content 2710 ppm (0.271% w/w), unless specified otherwise. c Initial water content 2890 ppm (0.289% w/w). d Stirred, distilled, and sequentially dried, 24 h. e Stirred for 24 h and distilled. f Dried for 24 hand then distilled. g Contamination (2%) by mesityl oxide. Fractionated sample. i Contamination (12%) by mesityl oxide. j Brown-black solu- tions.
4A molecular sieves 331 887 1720
a Static drying modes unless specified otherwise.
Experimental Section Desiccants. Details of the source, activation, and handling of most
of the desiccants have already been described.' Reagent grade cupric sulfate was activated by heating at 320 "C for 15 h before use. Barium and calcium oxides were of reagent grade, and a fresh batch was used directly without activation.
Solvents, DMF, MeZSO, and HMPT were commercial synthetic grades of 99% purity (Merck). Acetone was of analytical grade (M&B). All solvents were rigorously purified by standard methods.*
HMPT and Me2SO were treated by standing over barium oxide overnight, followed by filtration, distillation from calcium hydride, and subsequent storage over 20% w/v 4A molecular sieves. MezSO had bp 74.5-75.0 O C at 12 mmHg, and HMPT had bp 89.0-89.5 "C at -3 mmHg.
Commercial DMF was allowed to stand over 4A molecular sieves overnight and was filtered, distilled from phosphorus pentoxide (bp 55.8-56.0 "C at 20 mmHg), allowed to stand over anhydrous potas- sium carbonate, and subsequently stored over 4A molecular sieves.
Analytical grade acetone was allowed to stand over anhydrous potassium carbonate for one day and then over 4A molecular sieves overnight. Fractionation gave material, bp 56.2 OC, which was not stored but used immediately. Gas chromatographic analysis of this material showed it to be free of impurities.
Techniques. The procedure used for HMPT serves as an example. A stock solution of HMPT containing 2620 ppm of labeled water was prepared by the addition of 0.50 g of tritiated water, specific activity 0.5 mCi/mL, to the appropriate mass of purified rigorously dried HMPT. Aliquots of the stock solution (15.0 i 0.1 mL) were syringed directly onto the appropriate desiccant contained in a 25 mL clear-fit round-bottom flask, which was immediately stoppered. Experiments were conducted at ambient temperatures (26-30 "C). Where specified, samples were stirred magnetically. Aliquots (1.00 f 0.02 mL) were taken at time intervals as specified in Table I and assayed directly by liquid scintillation counting, as previously Where nec- essary, viz., in the case of colored solutions or suspected contamination by soluble desiccant residues, samples were distilled before assay. Sequential drying2 was accomplished by decanting monosiccated solvent onto a fresh charge of 5% w/v desiccant. Sampling was then effected at the time intervals given in the table footnotes.
Registry No.-HMPT, 680-31-9; DMF, 68-12-2; MeZSO, 67-68-5; acetone, 67-64-1.
References and Notes (1) Part I: D. R. Burfield, K. H. Lee, and R. H. Smithers, J. Org. Chem., 42,3060
( 1977). (2) For Part 2, see D. R. Burfield. G. H. Gan, and R. H. Smithers, J. Appl. Chem.
Biotechnol., 28, 23 (1978). (3) See, for example, Heinz Becker et ai., "Organicum, Practical Handbook
of Organic Chemistry", translated by B. J. Hazard, Pergamon Press, Braunschweig. 1973, pp 185. 190. See also L. F. Fieser and M. Fieser. "Reagents for Organic Synthesis", Vol. 1-5, Wiley, New York. N.Y., 1967,
0022-3263/78/1943-3968$01.00/0
1969, 1972, 1974and 1975. (4) For an interesting recent study involving Me2SO-H20 mixtures, see L. F.
Blackwell and J. L. Woodhead, J. Chem. Soc.. Perkin Trans. 2, 1218 (1975).
(5) The reaction of sodium hydride with Me2S0 has been reported to sometimes give rise to violent explosions: inter alia, see L. Brandsma, "Preparative Acetylenic Chemistry", Elsevier, Amsterdam, 1971, p 24. However, in our hands, the same reaction has been carried out a number of times using stringently dried solvent with no untoward effects. While the cause of these accidents remains undetermined, it is noteworthy that the equilibrium water content of Me2S0 is lo%, and it thus seems not unlikely that the origin of these mishaps may lie in the use of insufficiently dried solvents.
(6) D. R. Burfield, Anal. Chem., 48, 2285 (1976). (7) See, for example, B. M. Trost and Y. Tamaru, J. Am. Chem. Soc., 99,3101
(1977). (8) See references contained in J. A. Riddick and W. B. Bunger, "Organic
Solvents", 3rd ed., Wiley-lnterscience, New York, N.Y., 1970. (9) (a) F. Trondlin and C. Ruchardt, Chem. Ber., 110, 2949 (1977); (b) T. J.
Wallace and A. Schriesheim, Tetrahedron, 21, 2271 (1965). (10) Here, as the term super-dry denotes solvents containing less
than 1 ppm of water. (1 1) See Experimental Section. (12) C. A. Young and R. R. Dewald, J. Chem. Soc., Chem. Commun., 186
(1977). (13) See S. S. Pizey, "Synthetic Reagents", Vol. 1. Ellis Horwood Ltd., Chi-
Chester, 1974. This author reports that the use of calcium hydride and other basic desiccants in the drying of DMF could produce significant amounts of dimethylamine. However, the presence of this amine would give rise to inflated apparent water contents, and the values observed here, both for statically dried and distilled samples, suggest that this side reaction is of minor importance for these desiccants.
(14) (a) D. Martin and H. G. Hauthal, "Dimethyl Sulphoxide", translated by E. S. Halberstadt, Wiley-Halsted, New York, N.Y., 1975. (b) H. E. Baumgarten. Ed.. "Organic Syntheses", Collect. Vol. 5, Wiley, New York. N.Y., 1973, pp 243, 756.
(15) W. H. Smyrl and C. W. Tobias, J. Electrochem. Soc., 115, 33 (1968). (16) R. L. Meeker, F. Critchfield, and E. T. Bishop, Anal. Chem., 34, 1510
(1962).
3,4-Dimethyl-cis- bicyclo[ 3.3.0]-3-octene-2,8-dione: A Potentially Useful Pentalenolactone Synthon
Martha L. Quesada, Richard H. Schlessinger,* and William H. Parsons
Department of Chemistry, University of Rochester, Rochester, New York 14627
Received March 13.1978
Pentalenolactone (1) is an acidic lipophylic antibiotic iso- lated from the fermentation broth of Streptomyces UC 5319
- 1 which exhibits inhibitory activity against nucleic acid syn- thesis in bacterial cells.1t2 Both the novel structural nature of pentalenolactone together with its biological activity prompted us to consider possible routes to the synthesis of this molecule.
Inspection of the literature revealed a number of potential pentalene ~ y n t h o n s , ~ the most interesting of which was the pentalenedione 2 reported first by Stetter4 and more recently
0
3 2 - - by E a t ~ n . ~ The salient feature of both the Stetter and Eaton routes was the base-induced internal Claisen condensation of the ester 3, which in Eaton's hands gave an excellent yield of the dione 2. These data inspired us to consider the possi-
0 1978 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0414
a038

3852 J . Org. Chem. 1984,49, 3852-3854
(23.8), 94 (26.0), 93 (61.4), 80 (25.0), 79 (27.1), 31 (100.0), 29 (50.2). Anal. Calcd for CI2Hl8O: M, 176.1202. Found (high-resolution
mass spectrometry): M , 176.1201. T h e following spectra data were obtained for 14: 'H NMR
(CDCl,) S 0.8-1.3 (m, 2 H), 1.68 (s, 3 H), 1.8-3.0 (m, 9 H), 3.3 (m, 1 H), 5.28 (s,1 H); 13C NMR (CDC1,) 6 16.26 (q), 37.45 (d), 39.28
129.8 (d), 139.0 (s), 220.6 (9); IR (neat) 3040 (w), 2940 (s), 1735 (s), 1650 (w), 1440 (m), 1410 (m), 1140 (m), 1020 (m), 820 (m) cm-'; mass spectrum (70 eV), m / e (relative intensity) 176 (molecular ion, 43.6), 147 (23.4), 106 (50.2), 95 (84.3), 94 (38.9), 93 (56.9), 91 (49.4), 82 (21.7), 80 (31.8), 79 (44.4), 77 (28.7), 41 (37.9), 39 (26.2), 31 (100.0), 29 (59.5).
Anal. Calcd for Cl2HI60: M, 176.1202. Found (high-resolution mass spectrometry): M , 176.1205.
Hydrogenation of 13 and 14. A solution of either 13 or 14 (10 mg, 0.56 mmol) in ethyl acetate (2 mL) was hydrogenated over 10% palladized charcoal catalyst (2 mg) at a hydrogen pressure of 12 psig for 40 min. The reaction mixture was filtered to remove catalyst, and the filtrate was concentrated in vacuo to afford 15 (10 mg, 100%) which was purified via bulb-to-bulb distillation under reduced pressure: bp 130 "C (0.5 mm); 'H NMR (CDCl,) 6 1.2 (d, 3 H, J = 6 Hz), 1.6-3.0 (m, 15 H); IR (neat) 2945 (s), 2870 (w), 1740 (s), 1460 (m), 1410 (m), 1380 (w), 1160 (m) cm-l; mass spectrum (70 eV), m l e (relative intensity) 178 (molecular ion, 17.4), 95 (27.3), 82 (48.6), 81 (24.9), 41 (20.3), 31 (100.0), 29 (55.9).
Anal. Calcd for CI2Hl8O MI 178.1358. Found (high-resolution mass spectrometry): MI 178.1358.
Acknowledgment. Financial support of our study by the Naval Air Systems Command, the Robert A. Welch Foundation (Grant B-963), the Air Force Office of Scien- tific Research (Grant No. AFOSR-84-0085), and the North Texas State University Faculty Research Committee is gratefully acknowledged. K.S.R. gratefully acknowledges receipt of a fellowship from the CSIR, New Delhi, India. We thank Professor H.-J. Liu (University of Alberta) for kindly obtaining the high-resolution mass spectra of com- pounds 6, 7, 8, l lb , and 12b and Mr. N. Omkaram for kindly obtaining the high-resolution mass spectra of com- pounds 13-15.
Registry No. 5, 87830-51-1; 6, 91632-94-9; 7, 91632-95-0; 8, 91632-96-1; 9,91632-97-2; 10, 25282-60-4; l l a , 91632-98-3; l lb, 91632-99-4; 12a, 91633-00-0; 12b, 91633-01-1; 13,91633-02-2; 14,
(t), 39.57 (t), 42.39 (d), 44.44 (t), 44.62 (d), 45.67 (t) , 52.72 (d),
91633-03-3; 15 , 91633-04-4.
Desiccant Efficiency in Solvent and Reagent Drying.'S2 9. A Reassessment of Calcium Sulfate
as a Drying Agent
David R. Burfield
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, Malaysia
Received March 1, 1984
Anhydrous calcium sulfate (Drierite) has been applied to the problem of solvent desiccation for over 50 years3 and has received general endorsement4 as an efficient and generally applicable low cost drying agent. However, many of the extravagent claims5 made for the efficiency of this
(1) Part 7: Burfield, D. R.; Smithers, R. H. J . Org. Chem. 1983, 48, 2420.
(2) Part 8: Burfield, D. R.; Hefter, G. T.; Koh, S. P. J. Chem. Technol. Biotechnol. 1984, 34A, 187.
(3) Hammond, W. A.; Withrow, J. R. Ind. Eng. Chem. 1933,25,1112. (4) See: (a) Fieser, L. F.; Fieser, M. 'Reagents for Organic Synthesis";
Wiley; New York, 1967; p 107,1103. (b) Vogel, A. I. "Vogel's Textbook of Practical Organic Chemistry"; Longmans: London, 1978; p 139. (c) Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. "Purification of Labo- ratory Chemicals"; Pergamon: Oxford, 1980; p 23.
0022-326318411949-3852$01.50/0
Table I. Effect of Calcium Sulfate Activation Temperature on Its Efficiency in Dioxane Dryin@
residual solvent water content, activation temperature, PPm
O C l h 6 h 24 h 72 h no activation 1700 1140 540 240 100 1820 1460 1190 1130 150 2000 1230 485 240 200 1690 1080 500 250 225 1950 1200 535 250 250 1770 1290 660 310 300 1930 1430 970 970
DInitial water content = 2500 ppm; desiccant loading = 5% w/v; desiccant samples activated overnight (16 h).
Table 11. Effect of Calcium Chloride Activation TemDerature on Its Efficiency in Dioxane Dryin@
residual aolventwater content, activation temperature, PPm
O C l h 6 h 2 4 h 7 2 h no activation 1820 1020 430 350 150 1840 1230 420 290 225 1700 1090 370 290 300 1750 1040 385 325
OInitial water content = 2500 ppm; desiccant loading = 5% w/v; desiccant samples activated overnight (16 h).
product are based on very early results,3,6 and in our recent studies,7-1° where calcium sulfate has been examined alongside other desiccants it has been found to have only very modest efficiency. Thus Drierite approaches the foot of the "drying league" in a recentg evaluation of 12 desic- cants for the drying of wet ether extracts and is similarly placed in studies of the drying of benzene, dioxane, ace- tonitrile: various dipolar aprotic solvents*, and amines.1° However, in our earliest studies7sJ0 in an attempt to attain uniform and hence comparable experimental conditions desiccants were activated at 300-320 OC for 15 h before use. Since it is possible that such activation conditions may have had a deleterious effect on the activity of the calcium sulfate" and in view of the widespread utilization of this product it appeared pertinent to reevaluate its desiccant potential. This paper thus centers on a reap- praisal of Drierite as a desiccant for solvent drying with particular attention being payed to the effect of activation temperature. Comparison with two other important de- siccants, viz., calcium chloride and molecular sieves, is also made.
Results and Discussion Influence of Desiccant Activation Temperature.
Dioxane was choosen for initial tests as it is moderately ~~
(5). For example: "The last detectable traces of water are removed by Drierite from any liquid which is sufficiently fluid to make intimate contact with the porous solid" and "Regular Drierite is unsurpassed for the drying of organic liquids in the liquid or vapour phase. Moisture residue after treatment with Drierite is so low it cannot be measured, leaving the organic liquid truly anhydrous". Quoted in; Hammond, W. A. "Drierite"; W. A. Hammond Company: Xenia, OH.
(6) Thus calcium sulfate was surpassed only by barium oxide, mag- nesium perchlorate, and calcium oxide in evaluations based on gas drymg efficiency reported by: Bower, J. H. J. Res. Natl. Bur. Stand. 1934,12, 241.
(7) Burfield, D. R.; Lee, K. H.; Smithers, R. H. J. Org. Chem. 1977, 42, 3060.
(8) Burfield, D. R.; Smithers, R. H. J. Org. Chem. 1978, 43, 3966. (9) Burfield, D. R.; Smithers, R. H. J. Chem. Educ. 1982, 59, 703. (10) Burfield, D. R.; Smithers, R. H.; Tan, A. S. C. J. Org. Chem. 1981,
46, 629. (11) Thus it has been shown"* that calcium sulfate heated above 400
"C rehydrates only slowly and it has been suggested'lb that the transition to the inactive form occurs at around 313 "C. (a) Glasenapp, M. J. SOC. Chem. Ind., London 1908,27,858. (b) Chassevent, L. Liebigs Ann. Chem. 1927, 7, 43.
0 1984 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PE
NN
on
Aug
ust 4
, 200
9P
ubli
shed
on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0194
a043

Notes J. Org. Chem., Vol. 49, No. 20, 1984 3853
Table 111. Deuendence of Drying Efficiency on Desiccant Loading in the Drying of Grossly Wet Diethyl Ether" desiccant loading capacityd residual solvent water content, ppm
desiccant % w/v 5 min 15 min 30 min 60 min 360 min % w/w CaSO, l o b 11400 9200 10200 10700 2.8-3.9
20b 6400 3800 2100 4.5 20' 9700 7500 5800 3.1
CaCl, 5c 2100 2100 850 19.6 10c 2400 2100 1900 390 10.1 20c 2100 1400 900 4.9
"Initial water content = 14700 ppm; drying temperature = 22 "C. Activation temperature: = 220 "C. = 350 "C. dGiven by weight of water absorbed per unit of desiccant expressed as a percentage.
Table IV. Dependence of Drying Efficiency on Desiccant Loading in the Drying of Dioxane"
desiccant 1 o a d i n g
desiccant % w/v CaSOlb 1.24
2.5 5
10 20
CaC12' 1.25 2.5 5
residual solvent water content, ppm
l h 6 h 2 4 h 7 2 h 2600 2300 2020 1950 2020 1610 1220 1130 1700 1140 540 240 2140 760 150 110 1600 510 120 80 2250 1880 2320d 2500d 1940 1340 540 390 1750 1040 385 325
capacity
4.5 5.6 4.7 2.5 1.3
9.1 4.5
% w/w
" Initial water content = 2500 ppm; drying temperature u 27-30 "C. bActivated a t 225 OC. cActivated at 350 "C. dCaC12 granules observed to disintegrate.
difficult to dry and a fairly extensive set of drying data has already been accumulated for this solvent.'J2J3 Ac- tivation of the desiccant was carried out at temperatures in the range 100-300 "C. The marked influence of acti- vation temperature on drying efficiency is shown in Table I. Activation of Drierite in the temperature range 150-225 "C provides a product whose activity is little different from the as-received material. However, activation at tem- peratures of 250 "C and above is accompanied by a distinct deterioration in drying efficiency. Interestingly, a similar reduction in efficiency is observed on activation at 100 O C
and this must be ascribed to the absorption of water by the desiccant when equilibrated in an air oven at the high ambient humidities experienced in Malaysia. This sup- ports the earlier contention3 that Drierite binds water a t temperatures up to a t least 107 "C. The optimum acti- vation temperature for this material would appear to be in the range 200-225 "C.
An examination of another commonly used desiccant, calcium chloride, reveals that in this case the use of high activation temperatures does not have a deleterious effect on the drying activity (Table 11).
Since the drying activities of these two materials under optimum activation conditions appear quite similar, the earlier poor showing of calcium sulfate in our desiccant evaluation studies7v8J0 must in part be ascribed to the inactivation of the material by inappropriate high-tem- perature treatment.
Drying Capacity of Desiccant Materials. The ove- rall drying efficiency of a desiccant is determined not only by its intrinsic affinity for water but also by its capacity to contain the bound water. The drying capacity is an important parameter since it will dictate the appropriate desiccant loadings for practical use. Determination of the drying efficiency as a function of desiccant loading (Tables
(12) Burfield, D. R.; Gan, G. H.; Smithers, R. H. J. App. Chem. Bio-
(13) Burfield, D. R.; Smithers, R. H . J. Chem. Technol. Biotechnol. technol. 1978, 28, 23.
1980, 30, 491.
Table V. Comparison of Desiccant Drying Efficiency for Dioxane and Acetonitrilea
residual solvent water content, ppm
desiccant dioxane acetonitrile CaSo4* 240 180 CaC12b 290 d 3A molecular sieveC 19 52 4A molecular sievec 30 450
"Initial water content = 2500 ppm; drying time 72 h. Activation temperature: = 225 "C. = 350 "C. Drying temperature 27-30 "C. dCaC1, induces a base-catalyzed tritium exchange with aceto- nitrile which precludes determination;' desiccant loading = 5% WJV.
I11 and IV) gives a measure of the inherent capacity of the material.
In the drying of grossly wet ether samples the maximum observed water capacity is only 4.5% w/w for Drierite compared to nearly 20% w f w for calcium chloride (Table 111). Interestingly at low desiccant loadings the Drierite is observed to lose its effectiveness with increasing drying time and this may be symptomatic of a breakdown of this material in the presence of large water excesses. I t is also clear (Table 111) that activation of Drierite a t 350 "C leads to a reduction in drying capacity, and this again empha- sises the importance of moderate activation temperatures for this material.
A somewhat similar maximum drying capacity of 5.6% w/w is observed for the Drierite drying of dioxane (Table IV). In contrast, for calcium chloride the maximum ob- servable drying capacity is sharply reduced to about 9% and at low desiccant loadings the granular material is observed to break up with complete loss of water retention. Since calcium chloride appears quite stable in the presence of water-saturated diethyl ether, it would appear that some specific interaction with dioxane must occur. The some- what higher ambient temperatures (27-30 "C) encountered during the dioxane drying experiments may also have adversely affected the capacity of the calcium chloride.
Overall, it appears that calcium chloride has a signifi- cantly higher water capacity than Drierite. The optimum value for calcium chloride is quite similar to the 18-20% w/w capacity observed12 for 3A molecular sieves in the drying of dioxane.
Comparative Drying Efficiency. By employing suitably high desiccant loadings it is possible to offset the effects imposed by variable capacity and hence to probe the inherent drying efficiency of the desiccant materials. The relative efficiencies for the drying of dioxane and acetonitrile by Drierite, calcium chloride, and molecular sieves are summarized in Table V. It is apparent that calcium chloride and calcium sulfate are quite similar in drying ability but that the residual water content is of an order of magnitude higher than that attainable with mo- lecular sieves. It is of some interest however that Drierite
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0019
4a04
3

3854 J . Org. Chem. 1984,49, 3854-3856
is more efficient than 4A molecular sieves in the desicca- tion of acetonitrile. The inefficiency of the 4A sieves is brought about by competitive adsorption of acetonitrile at the water binding sites. This apparently does not occur with Drierite.
Summary Anhydrous calcium sulfate (Drierite) is a moderately
efficient desiccant for the drying of organic solvents. The material loses drying activity if heated to about 250 OC and has a limited water capacity (-5% w/w). It is therefore not appropriate for efficiently drying grossly wet solvents as earlier ob~erved .~
Experimental Section Solvent water content was determined by the radiotracer
method previously des~ribed.~J~J~ Anhydrous calcium sulfate (Drierite), 1C-20 mesh, was kindly supplied as a gift from W. A. Hammond Drierite Company. Details of solvents and other desiccants have already been described.‘
Registry No. CaSO,, 7778-18-9.
(14) Burfield, D. R. Anal. Chem. 1976, 48, 2285.
Carbon Monosulfide Chemistry in Solution. 2.’ Synthesis and Reactions of Trichloromethyl
Chlorodithioformate
Ejner K. Moltzen,2a Alexander Senning,*2a Michael P. Kramer,2b and Kenneth J. Klabunde*2b
Departments of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark, and Kansas State University,
Manhattan, Kansas 66506
Received February 3, 1984
Recently we were able to demonstrate that the insertion of CS into the sulfur-chlorine bond of sulfenyl chlorides appears to be a general and synthetically useful reaction.’ While our previous examples (1) and (2) involve highly
C,H,SCl+ CS -+ CBH,SC(S)Cl (1)
ClSSCl + 2cs - ClC(S)SSC(S)Cl (2)
reactive sulfenyl chlorides, it was not obvious that tri- chloromethanesulfenyl chloride 1 would react according to (3). In a number of reactions 1 is considerably less
(3)
reactive than “typical” aliphatic or aromatic sulfenyl chlorides; for instance, the normally rapid uncatalyzed addition of sulfenyl chlorides to ethylene does not take place in the case of l;3 moreover, 1 can be steam distilled with little decomposition4 while “typical” sulfenyl chlorides must be protected from moisture. On the other hand, 1 does react readily in the a-addition to isocyanides5 (it
C1,CSCl + cs - C1,CSC(S)C1 1 2
(1) Paper 1 in this series: Klabunde, K. J.; Kramer, M. P.; Senning,
(2) (a) Aarhus University, Denmark. (b) Kansas State University. (3) Douglass, I. B.; Martin, F. T.; Addor, R. J . Org. Chem. 1951, 16,
(4) Klason. P. Ber. 1887. 20. 2376.
A.; Moltzen, E. K. J . Am. Chem. SOC. 1984, 106, 263.
1297.
(5) Enders, E.; Kuhle, E.: Malz, H. Belg. Patent 610 175, 1960; Chem. Abstr 1962, 57, 13694.
Scheme I
S
(C,H,CH,),NC /I SCC I c13cscc12scI 9 10
/i\ s SCCI,
8
should be noted that isocyanides are isoelectronic with CS) and, according to very recent reports,6 1 and S2C12 add to certain thiocarbonyl compounds with comparable ease.
It should be noted that there are no obvious alternative synthetic pathways leading to 2.’ While the perfluoro analogue 3 is accessible by potassium fluoride catalyzed dimerization of thiocarbonyl fluoride8 and by treatment of thiocarbonyl chloride fluoride with mercury(I1) tri- fluor~methanethiolate,~ the photochemical dimerization of thiophosgene leads to 2,2,4,4-tetrachloro-1,3-dithietane (4),’O and there is no reaction between thiophosgene and
s CFBSCX I1 Cl,C<;>CCl,
- 4 3, X - F
5, X = C I
KF.” Unlike Hg(SCF,),, mercury(I1) trichloromethane- thiolate, Hg(SCC13),, is unknown because of the inherent instability of trichloromethanethio1.’2 Likewise, the re- ported synthesis of trifluoromethyl chlorodithioformate (5)13 does not lend itself to any modification leading to 2.
We can now report that reaction 3 does in fact occur readily and in good yield.14 Trichloromethyl chlorodi- thioformate (2) is a distillable liquid once preliminary silica gel chromatography has been carried out (distillation of crude 2 only gave a small amount of 10) and can be stored at room temperature for several months. It could be characterized spectroscopically and by derivatization.
As a minor byproduct some pale-yellow crystals could be isolated. According to our preliminary data this yellow solid is most likely the new thiirane 6, which probably is formed according to (4) in analogy with the formation of 2,2,3,3-tetrachlorothiirane.15 This reaction mechanism is also supported by recent results of our work on reactions between CS and thiocarbonyl compounds. The 1,4-dit- hiane structure 7 is also consistent with the analytical and spectral data, but according to the mass spectrum 6 is the most probable structure.
(6) We are very grateful to a referee who made us aware of these reports (Barany et al. J . Org. Chem. 1983,48,4750; J . Org. Chem. 1984, 49, 1043).
(7) Previously the dimerization product 4 of thiophosgene was erro- neously believed to possess structure 2 (‘‘Beilsteins Handbuch der Or- ganischen Chemie”, Val. 3,4th ed.; Springer-Verlag: Berlin, 1921; p 215).
(8) Haas, A.; Klug, W. Chem. Ber. 1968, 101, 2609. (9) Haas, A.; Klug, W.; Marsmann, H. Chern. Ber. 1972, 105, 820. (10) Schonberg, A.; Stephensen, A. Ber. 1933, 66, 567. (11) Moltzen, E. K.; Senning, A., unpublished results. (12) Senning, A. Chem. Reu. 1965, 65, 385. (13) Haas, A,; Yazdanbakhsch, M. Chem. Ber. 1976,109, 1976. (14) No effort was made to optimize the yields of the reported reac-
tions, but it is important to note the absence of significant amounts of byproducts.
(151 Seyferth, D.; Tronich, W. J . Am. Chem. SOC. 1969, 91, 2138.
0022-3263/84/1949-3854$01.50/0 0 1984 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0019
4a04
3

2420 J. Org. Chem. 1983,48, 2420-2422
2 Hz), 1.18 (d, 3 H, J = 7 Hz); mass spectrum, m l e 110 (M’).
Acknowledgment. We are grateful to Professor Sam- uel Danishefsky, Yale University, for encouragement and support through Grant No. AI 16943-03.
Registry No. 1, 85736-25-0; 2, 43124-56-7; 3, 15331-05-2; (+)-pulegone, 89-82-7.
Desiccant Efficiency in Solvent and Reagent Drying. 7. Alcohols’”
David R. Burfield and Roger H. Smithers*
Department of Chemistry, University of Malaya, Kuala Lumpur 22-11, West Malaysia
Received October 22, 1982
For many of their applications in synthesis, the lower monohydric alcohols are required to be scrupulously dry, and in contrast to alcohols containing six C atoms or more, whose desiccation does not appear difficult,’ alcohols containing one to four C atoms have long been recognized as posing a formidable drying problem. In the case of ethanol for instance, numerous synthetic procedures con- tain statements such as “traces of water depress the yield considerably”? a practical result of which is that samples dried with CaO, for example (containing up to 5000 ppm of water), are often completely una~ceptable.~ There is also no doubt that for this group, as with other cases,l* the problem of dedlccant selection has been compounded by a lack of reliable quantitative data. As an illustration, when discussing the efficiency of magnesium or alkyl phthalateNa as desiccants, many a u t h ~ r i t i e s ~ ~ continue to quote figures for water contents which were obtained in the original investigations1° and which must now be viewed with some scepticism. In the light of this, we now
(1) Part 1: Burfield, D. R.; Lee, K. H.; Smithers, R. H. J. Org. Chem. 1977, 42, 3060.
(2) Part 2: Burfield, D. R., Gan, G. H.; Smithera, R. H. J. Appl. Chem. Biotechnol. 1978, 28, 23.
(3) Part 3: Burfield, D. R.; Smithers, R. H. J. Org. Chem. 1978,43, 3966.
(4) Part 4: Burfield, D. R.; Smithers, R. H. J. Chem. Technol. Bio- technol. 1980, 30, 491.
(5) Part 5: Burfield, D. R.; Smithers, R. H.; Tan, A. S. C. J. Org. Chem. 1981,46,629.
(6) Part 6 Burfield, D. R.; Smithers, R. H. J. Chem. Educ. 1982,59, 703.
(7) The use of K2C03 as a desiccant is often considered adequate in these cases. See for example: (a) Calzada, J. G.; Hooz, J. Org. Synth. 1974,54,63. (b) Paul, R.; RiobC, 0; Maumy, M. Ibid. 1976,55, 62. (c) Vogel, A. I. “Vogel’s Textbook of Practical Organic Chemistry”; Long- mans: London, 1978; p 270. See also, however: Crandall, J. K.; Rojas, A. C. Org. Synth. 1976,55, 1.
(8) See: Weiner, N. “Organic Syntheses”; Wiley: New York, 1943; Collect. Vol. 11, p 279. Also relevant are the following quotations. (a) ‘Yields are poor if the alcohol is not completely dehydrated”; Lycan, W. H.; Puntambeker, S. V.; Marvel, C. S. “Organic Syntheses”; Wiley: New York, 1943; Collect. Vol. 11, p 319. (b) “Moisture in the reagents affects the yield seriously”: Dox, A. W. ’Organic Syntheses”; Wiley: New York, 1941; Collect, Vol. I, p 5. (c) T h e use of 98% alcohol results in a lowering of yield by 1/3”: Kaufmann, W. E.; Dreger, E. E. Ibid., p 258. (d) “The quality of absolute ethanol used has a very marked effect upon the yield”: Adams, R.; Kamm, R. M. Ibid., p 250.
(9) Thus, ‘Alcohol dried over lime gives very low yields”: Ford, S. G.; Marvel, C. S. ‘Organic Syntheses”; Wiley: New York, 1943; Collect. Vol. 11, p 373. “Ethanol dried only by a lime process gives a low yield”: Adkins, H.; Gillespie, R. H. Zbid., 1955; Collect. Vol. 111, p 672.
(IO) For the use of magnesium see: Lund, H; Bjerrum, J. Ber. Dtsch. Chem. Ges. 1931, 64, 210. For the use of a sodium-alkyl ester combi- nation see: (a) Smith, E. L. J. Chem. SOC. 1927, 1288. (b) Manske, R. H. J. Am. Chem. SOC. 1931,53, 1106.
report the results of a study of desiccant efficiency for this important group.
General Indications.7cJ1 Both chemical and absorp- tive-type desiccants have been proposed for drying alco- hols, and these i n c l ~ d e ~ ~ J ~ * ~ ~ Al, BaO, CaO, Mg, Na, and K2C03, while molecular sieves of Type 3A and 4A have been recommended for further drying.*lb CaH2 is also widely quoted,lld although some sources11a advise caution in its use with the lower alcohols.
While bearing in mind the well-known hygroscopic and hydrophilic properties of these compounds, the experi- ments reported below were not carried out by using any special techniques to obviate the entry of the atmosphere into drying systems other than those routinely used by the bench chemist.
Methanol. As one authority has observed,’ld water is the dominant impurity in this solvent, and, unless ex- traordinary care is exercised, this content increases each time MeOH is handled. It is therfore quite remarkable that a survey of the literature on the drying of EtOH and MeOH points up a curious dichotomy: while references to EtOH8vg are characterized by numerous strictures as to the importance of achieving perfectly dry solvent, in con- trast, MeOH is treated in a rather offhand manner, even though many of its applications parallel those of ita higher homologue. “All MeOH used must be anhydrous” is a typical comment,12 although information on how to realize this is lacking. The cause may lie in the belief that frac- tionation alone gives a solvent of adequate dryness, al- though the water content is still an admitted 1000 ppm.7cJ1d Where drying methods have been indicated, the use of Mg seems most often advised.”J3
The results shown in Table I certainly suggest that, on the whole, MeOH is more difficult to dry than its higher homologue. These figures require little comment, but it is worth highlighting the poor performance of 3A molecular sieve powder for MeOH in comparison with other alcohols. This is almost certainly an effect of molecular size and nonselective adsorption on the large surface area which occurs extremely rapid ly with this desiccant.14 On ac- count of ita small size, MeOH is able to compete with water for entry into the sieve pore. Similar reasons also explain the relative ineffectiveness of the 4A and 5A bead forms of molecular sieve. On the other hand, the 3A bead form constitutes a useful desiccant, whose success is presumably due to a much slower rate of absorption which occurs with greater selectivity. I t is also worth noting that a combi- nation of methods is often the most effective strategy: a 1-L sample of MeOH distilled from Mg/12 onto 10% w/v 3A molecular sieve beads and then allowed to stand 48 h had residual water content of only 12 ppm. Finally, the unimpressive results obtained with BaO and CaO tend to support an earlier assessment of these agents as “tedious and wa~teful”.’~
Ethanol. As stated above, references to the use of ethanol as a nucleophile,16 as a solvent for Bouveault-Blanc
(11) See: (a) Rickert, H.; Schwartz, H. In “Methoden der Organischen Chemie (Houben-Weyl)”; Mueller, E., Georg Thieme Verlag: Stuttgart, 1968; Band I/2, p 873. (b) Loewenthal, H. J. E. “Guide for the Perplexed Organic Experimentalist”; Heyden: London, 1980; p 50. (c) “Drying in the Laboratory”; E. Merck Co.: Darmstadt. (d) Riddick, J. A.; Bunger, W. B. “Organic Solvents”, 3rd ed.; Wiley-Interscience: New York, 1970 pp 638465.
(12) Helferich, B.; Schafer, W. “Organic Syntheses”; Wiley: New York, 1941; Collect. Vol. I, p 365.
(13) (a) Murray, J. I. ‘Organic Syntheses”; Wiley: New York, 1963; Collect. Vol. IV. p 744. (b) Baumgarten, H. E.; Petersen, J. M. Ibid., 1973; Collect. Vol. V, p 912.
(14) See ref 6 (15) See Ref l l d , p 642.
0022-3263/83/1948-2420$01.50/0 0 1983 American Chemical Society
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0016
2a02
6

Notes J. Org. Chem., Vol. 48, No. 14, 1983 2421
Table 1. Desiccant Efficiency in Dryingaib of Some Common Lower AlcoholsC
desiccant
residual water content, ppm
methanold ethanol e !&butanolf tert-butyl alcoholg 3A sieves (bead) 95 9 9 645 (9)h 428 (160)' 3A sieves (powder)] 94 0 18 1 4 13 Mg/I, 97 (12)' 50 (53)m
Nap Na/dicarboxylic acid esterQ 92" 36" 4A sieves (bead) 44 0 401 4 06 5A sieves (bead) 475 875 CaC, 490 333 (199)' 409 430" (662)O BaO 1000 s Ca 1000 s 86 0 KZCO, s s 750 CaO s s 770
ion exchange resin s 640
Desiccant loading 5% w/v with a drying period of 24 h unless specified otherwise. Initial water content 1010 ppm. e Initial water content 1500 ppm. f Initial water content 1000 ppm. g Initial water content 1030 ppm. 96-h drying period. 168-h drying period. I Analysis was performed after settling of desiccant, 3-6 h. Weight of magnesium in accord with general practice, i.e., 0.5% w/v. See text. Weight of magnesium 2% w/v. " Distilled sample. O Initial water content 1670 ppm, distilled sample. P Weight of sodium 3% w/v. See ref 32. Ratio of sodium to 2-butyl succinate for 2-BuOH and to diethyl phthalate for ethanol in accord with general practice (see ref 7c), i.e.; Na, 0.3 mol L- ' ; dicarboxylic acid ester, 0.14 mol L-l. I Stirred sample. N o apparent drying.
1 2 5 99 17" 406 ( 2 0 ) O 1800" 2400" 406 (10)'
C d Z
KOH powder s
a Static drying modes unless specified otherwise. Water content assayed by the Karl Fischer method.
or oxime type reducti0m,1~ or as a suitable medium for the preparation of metal ethoxides"' invariably include warn- ings as to the lowered yields which are obtained through the use of inadequately dried solvent. Desiccants most frequently prescribed16-18 include Ca0,lB Mg," Na,2l or Na in conjuction with a high-boiling ethyl ester such as the phthalic acid derivative. The results (Table I) show that the desiccant par excellence both from the viewpoint of residual water levels and convenience is 3A molecular sieve powder.22 Sieves of larger pore size are again seen to be inappropriate. Of the more traditional siccatives, Mg and CaH2 are both efficaceous while the performance of the alkaline earth oxides was once more disappointing. The poor result with sodium is not unexpected and is attrib- utable to the equilibrium shown between alkoxide and
NaOH + ROH H20 + RONa
hydroxide. On the other hand, the ploy of refluxing the solution obtained after sodium treatment with a high- boiling ethyl ester works well, because saponification of the ester irreversibly removes hydroxide ion.
(16) See for example: (a) Croxall, W. J.; Fegley, M. F.; Schneider, H. J. 'Organic Syntheses"; Wiley: New York, 1963; Collect. Vol. IV, p 98. (b) Moffett, R. B. Ibid., p 427. (c) Ford-Moore, A. H.; Perry, B. J. Ibid., p 955. See also ref 8b.
(17) See: Maneke, R. H. "Organic Syntheeee"; Wiler New York, 1943; Collect. Vol. 11, p 154. See also ref 8a and 9.
(18) See for example: (a) Holmes, H. L.; Trevoy, L. W. "Organic Syntheses"; Wiiey: New York, 1955, Collect. Vol. 111, p 300. (b) Meyer, K.; Bloch, H. S. Ibid., p 637. (c) Zuidema, G. D.; Van Tamelen, E.; Van Zyl, G. Ibid., 1963; Collect. Vol. IV, p 10. (c) Daeniker, H. U.; Grob, C. A. Ibid., 1973; Collect. Vol. V, p 989. See also ref 8 and 9.
(19) In addition see: (a) Kimball, R. H.; Jeffereson, G. D.; Pike, A. B. "Organic Syntheses"; Wiley New York, 1943; Collect. Vol. 11, p 284. (b) Fuson, R. C.; Wojcik, B. H. Ibid., p 260.
(20) In addition see: Lund, H.; Voigt, A. "Organic Syntheses"; Wiley New York, 1943; Collect. Vol. 11, p 694. See also ref 8 and 9.
(21) In addition see: (a) Marvel, C. S.; Hager, F. D. "Organic Syntheses"; Wiley: New York, 1941; Collect. Vol. I, p 248. (b) Levene, P. A.; Meyer, G. M. Ibid., 1943; Collect. Vol. 11, p 288. See also ref 8.
(22) Removal of the laat traces of thie desiccant from a dried solvent probably necesaitates distillation or filtration. However, if left to stand for perioda of several daye, the desiccant usually from a compacted mass at the bottom of the container, leaving a completely clear solvent layer. In many procedures, the traces of powder remaining in suspension are probably of little consequence, and dried material can be dispensed by simple decantation.
Butanols. With some notable exce~tions,2~ perfectly dry 2-butanol is probably less widely required than the other alcohols in this study but is included for purposes of comparison. In contrast, completely anhydrous tert- butyl alcohol is always required for its most common use in the preparation of the potassium derivative. An early source24 observes that "it is difficult to prepare t-BuOH free from water". Later authors recommend tackling drying by refluxing over Ca0,26a*b Na, or K25"*d or more recently by distillation from CaHz6 or 3A sieves.27 The results for both butanols on using most of the absorptive type siccatives under static conditions (Table I) indicate a very much slower rate of water absorption than with methanol or ethanol. It seems likely that this is due to the increased viscosity of the C-4 alcohols compared with their lower homologues, and thus the use of 3A sieves in the powder form (which operates as a dispersion) is able to circumvent this difficulty and again appears to be the desiccant of choice.= Interestingly, and in striking contrast to 2-butanol, the use of Na alone does efficiently remove water from tert-butyl alcohol, and this must mean that the equilibrium shown above lies predominantly to the left, presumably as a consequence of the greater disparity be- tween the acidities of the two protonated species in this case.28 I t should be emphasized, however, that sodium
(23) The "alcohol method" for the standardization of alkyllithium solutions requires perfectly dry 2-butanol. See: Watson, S. C.; Eastham, J. F. J. Organomet. Chem. 1967, 165.
(24) Norrie, J. F.; Olmated, A. "Organic Syntheses"; Wiley: New York, 1941; Collect. Vol. I, p 144.
(25) (a) Wayne, W.; Adkins, H. "Organic Syntheses"; Wiley: New York. 1955: Collect. Vol. 111. D 48. (b) Hauser. C. R.: Hudson. B. E.: AbrakoviGh, B.; Shivers, J. C.-Ibid., p 142. (c) Spassow, A. Ibid.; p 145: (d) McElvain, 5. M.; Kundiger, D. Ibid., p 506. (e) Johnson, W. S.; Schneider, W. P. Ibid., 1963; Collect. Vol. IV, p 134. (f) Raha, C. Ibid., p 264.
(26) (a) Homing, E. C.; Fmelli, A. F. 'Organic Syntheses"; Wiley: New York, 1963; Collect. Vol. IV, p 461. (b) Mikol, G. J.; Russell, G. A. Ibid., 1973; Collect. Vol. V, p 937. (c) Ireland, R. E.; Chaykovsky, M. Ibid., p 171. (d) Crowther, G. P.; Kaiser, E. M.; Woodruff, R. A.; Hauser, C. R. Org. Synth. 1971,51, 96. (e) Crockett, G.; Koch, T. H. Ibid., 1979, 59, 132.
(27) Itoh, M.; Hagiwara, D.; Kamiya, T. Org. Synth. 1979, 59, 95. (28) pK, values for EtOH and t-BuOH relative to H20 have been
reported as -1.94 and -2.6, respectively. See: (a) Lee, D. G.; Cameron, R. J . Am. Chem. SOC. 1971,93,4724. (b) Deno, N. C.; Turner, J. 0. J . Org. Chem. 1966,31, 1969.
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp://
pubs
.acs
.org
| do
i: 10
.102
1/jo
0016
2a02
6

2422 J. Org. Chem., Vol. 48, No. 14, 1983 Notes
Table 111. Relative Difficulty in Dryinga of Some Diolsb residual residual water water
content, content, d io1 diol
Table 11. Desiccant Efficiency in Dryinga*b of 1, 2-Ethanediolc
residual water content, ppm desiccant
3A sieves (bead) 3A sieves (powder) 4A sieves (powder)
CaC,
BaO CaO distillation benzene azeotrope Mg A1
1900 (1200,d 540e)
MgSO,
B203
360f' 1900 (2070)d 3600
a Static drying modes unless specified otherwise. Water content assayed by the Karl Fischer method. Dessicant loading 5% w/v with a drying period of 72 h
unless s ecified otherwise; initial water content 2700 ppm. 168-h drying period. e 600-h drying period.
8 Distilled sample, cqntamination by acetylene. 67 "C (0.1 mmHg). ' Sample contaminated by - 0.02% v/v benzene. followed by treatment with Mg/I,. drying.
does in fact react with tert-butyl alcohol under reflux at an appreciable rate, and, in our opinion, the quite severe solvent losses which can result constitute a drawback se- rious enough to discourage ita use under these conditions.
1,2-Ethanediol. Although the main thrust of this work was concerned with the lower alcohols, it was thought of interest to investigate 1,2-ethanediol as a comparison. This material does in fact possess a high solvation power for particular ions such as fluoride, and the use of dry diol is prescribed for a general synthesis of primary alkyl fluorides which utilizes this solvent property.2e It might be an- ticipated that the effective drying of ethanediol would prove more problematical than the simple monohydric alcohols, and, indeed, the powerfully hydrophilic nature of this material is indicated by a reference in the patent 1iterature3O where the diol itself is recommended as a dessicant for acetonitrile! There appears to be a paucity of methods used for desiccation, due no doubt to the fact that the use of many effective agents such as CaHz is ruled out because of their reaction with this solvent. Most in- vestigators have advocated simple di~ti l lation,~~ and, in fact, the results show this to be fairly efficaceous (Table 11). With sieves, trends similar to those noted earlier are again observed; thus, due to reasons of slowness of ab- sorption and inappropriate pore size respectively, the 3A bead form and 4A powder form are ineffective, while 3A powder once more demonstrates attributes of powerful desiccant action and simple application. Other noteworthy results were achieved with the use of the benzene azeotrope and with Mg. It would appear that a residual water level of - 70 p p m represents the lower limit for t he desiccation of this material under practical conditions.
Other Diols. Finally, i t was thought of interest t o compare the difficulty of drying 1,2-ethanediol with some selected examples of other diols where factors of a changed
Analysis was performed after settling of desiccant, - 6 h. Bp 66-
Sample size 1 L; benzene azeotroping Little apparent
(29) Vogel, A. I.; Leicester, J.; Macey, W. A. T. 'Organic Syntheses"; Wilev: New York. 1963: Collect Vol. IV. D 525.
(36) Derdulla, H. J.; Sommer, M. Ger: (East) 90 136 (Cl. CO~C), May 20,1972; Appl. WP C07c/155952, June 21,1971; Chem. Abstr. 1972, 77, 151525r.
(31) See for example: Dickinson, C. L.; Melby, L. R. 'Organic Syntheses"; Wiley: New York, 1963; Collect, Vol. IV, p 276. See also ref 28.
(32) Monson, R. S. "Advanced Organic Synthesis"; Academic Press: New York, 1971; p 141.
140 HO-OH' 220 HC
Dr ing agent was 3A MS powder at a loading of 5% w/v. i: Analysis performed by the Karl Fischer method after settling of desiccant, 3-6 h. Initial water content 3600 ppm. Initial water content 4000 ppm. e Initial water content 3700 ppm. f Initial water content 4200 PPm.
positional relationship between the two hydroxyl groups and an increased hydrophobic content might give rise to variations in residual water content. The results (Table 111) suggest that 1,2-diols are easier to dry than comparable diols where the hydroxyl groups are situated more remotely (cf. 1,2-ethanediol and 1,6butanediol), and this seems reasonable from the viewpoint of preferred intramolecular interactions in 1,2-diols rather than the intermolecular alternatives involving water which are presumably more favorable in other cases. Increasing t h e nonhydrophilic character by the inclusion of more carbon atoms results in an understandably easier drying process (cf. 1,2- ethanediol and 2,3-butanediol; l,4-butanediol and 1,5- pentanediol). Extension to higher simple diols was pre- cluded by the limit of their liquid ranges.
Experimental Section Details of the source, activation, and handling of most desic-
cants have already been described>* Materials such as CiHz and CaCz obtained in an aggregate form were crushed rapidly in a mortar and with a hammer, respectively, immediately prior to use. Where drying methods call for procedures other than simple static interaction, e.g., Mg/Iz, the customary directives were foll~wed. '~
Water contents were determined throughout by the standard Karl Fischer method with a potentiometrically determined end point.
Alcohols. Chemically pure or AFt grades of material were used without further purification. In the normal procedure the water content of a freshly opened Winchester bottle was first determined followed by wetting or by drying and wetting to obtain required water concentrations.
Drying Experiments. Investigations were carried out on 100-ml samples except where stated otherwise in the tables.
Reflux and distillation were carried out under dry nitrogen or argon, and material collected from distillations for analysis was usually a middle cut of 60-75-m1. volume. Where powdered molecular sieves were used as the desiccant, charging of the material into a flask of suitable dimensions was followed by addition of solvent, stoppering of the flask, and vigorous agitation. A clear upper layer suitable for sampling was generally obtained in 3-6 h of standing.
Acknowledgment. We thank t h e Depar tment of Chemistry and University of Malaya for their support of this work under Vote No. F124/80.
Registry No. Mg, 7439-95-4; Iz, 7553-56-2; CaHz, 7789-78-8; Na, 7440-23-5; CaC2, 75-20-7; BaO, 1304-28-5; Ca, 7440-70-2; K2C03, 584-08-7; CaO, 1305-78-8; KOH, 1310-58-3; MgS04, 7487-88-9; B203, 1303-86-2; Al, 7429-90-5; methanol, 67-56-1; ethanol, 64-17-5; 2-butanol, 78-92-2; tert-butyl alcohol, 75-65-0; 1,2-ethanediol, 107-21-1; 2,3-butanediol, 513-85-9; 1,4-butanediol, 110-63-4; 1,5-pentanediol, 111-29-5; di-2-butyl succinate, 626-31-3; diethyl phthalate, 84-66-2.
Dow
nloa
ded
by U
NIV
OF
PEN
N o
n A
ugus
t 4, 2
009
Publ
ishe
d on
May
1, 2
002
on h
ttp:
//pu
bs.a
cs.o
rg |
doi:
10.
1021
/jo0
0162
a026







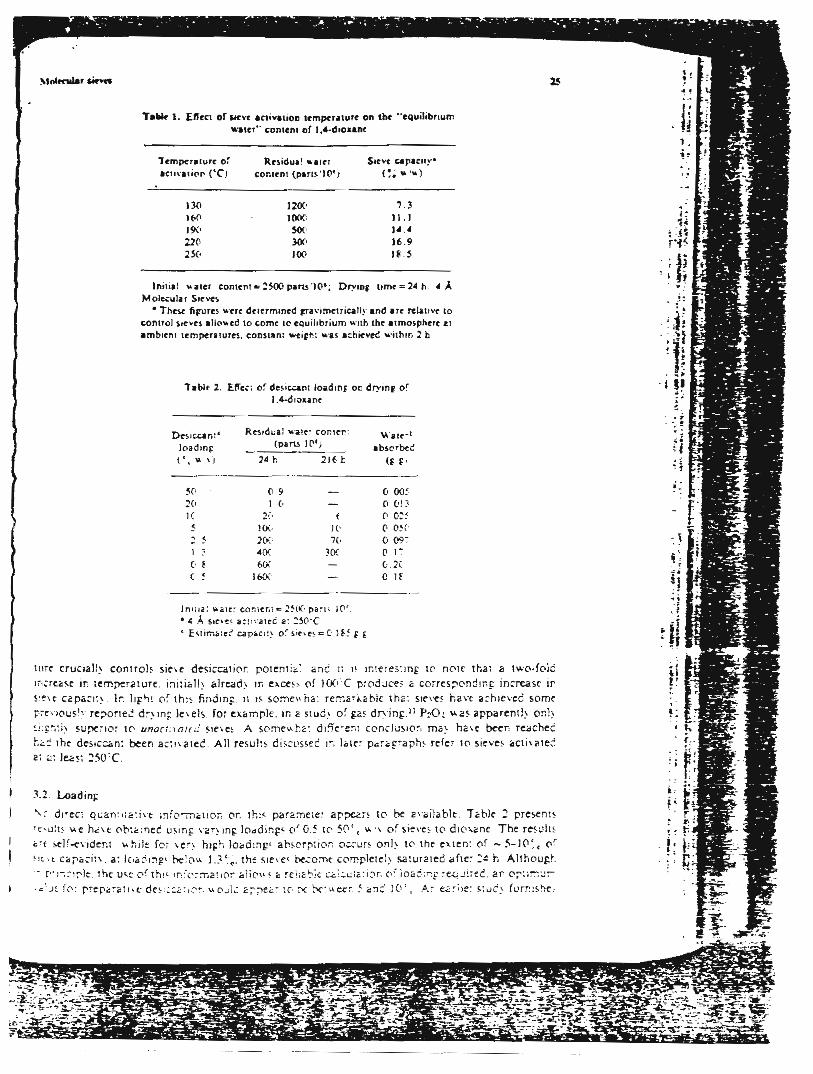





Related Documents



![[Z] Deco-Tec 5426 - Zobel Aust · 2020. 2. 26. · Zobel Australia 183 Barry Road Campbellfield VIC 3061 Phone 1300 073 074 Zobel Australia 183 Barry Road Campbellfield VIC 3061 Phone](https://static.cupdf.com/doc/110x72/611f85226e170452df39e54c/z-deco-tec-5426-zobel-aust-2020-2-26-zobel-australia-183-barry-road-campbellfield.jpg)







